WO2015087187A1 - Anti-sclerostin antibodies - Google Patents
Anti-sclerostin antibodies Download PDFInfo
- Publication number
- WO2015087187A1 WO2015087187A1 PCT/IB2014/066354 IB2014066354W WO2015087187A1 WO 2015087187 A1 WO2015087187 A1 WO 2015087187A1 IB 2014066354 W IB2014066354 W IB 2014066354W WO 2015087187 A1 WO2015087187 A1 WO 2015087187A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- antibody
- sequence
- sost
- bone
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/22—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against growth factors ; against growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- the present invention relates to antibodies, e.g., full length antibodies or antigen binding fragments thereof, that specifically bind to sclerostin.
- the invention further relates to compositions comprising antibodies to sclerostin, and methods of using such anti-sclerostin antibodies as a medicament.
- the anti-sclerostin antibodies are useful for treating and preventing a sclerostin-associated condition or disorder (e.g., a bone related disorder or cancer), such as multiple myeloma and osteoporosis.
- Methods of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density as well as methods of inducing canonical Wnt signaling activity, methods of preventing or reducing tumor burden, methods of inhibiting or preventing tumor growth or progression, and methods of inhibiting metastasis of cancer cells or tumors or delaying tumor growth or progression are also provided.
- myeloma also known as myeloma, plasma cell myeloma, or Kahler's disease
- myeloma cells are malignant plasma cells (myeloma cells) and is associated with bone loss attributed to an imbalance of bone metabolism.
- myeloma cells directly or indirectly interact with the bone surface
- SOST sclerostin
- SOST amino acid sequence of SOST (SEQ ID NO: 1 ), as reported by Brunkow et al., Am. J. Hum. Genet., 68:577-589, 2001 .
- SOST sclerosteosis
- sclerosteosis a genetic disease characterized by progressive overgrowth of bone tissue See, e.g., Brunkow et al., 2001 , supra; Balemans et al., Hum. Mol. Genet., 10:537-543, 2001 ; and Beighton, J. Med. Genet., 25:200-203, 1988.
- SOST is a secreted, cystine-knot protein expressed primarily in osteocytes, the most abundant cell type in bone. Beside expression in osteocytes, SOST has also been found to express in osteoblasts and chondrocyte, but not osteoclasts.
- the invention disclosed herein is directed to antibodies and fragments thereof
- the anti-SOST antibody of the present invention can significantly improve bone density, bone strength, bone quality, increase bone formation, bone volume, and bone mass, reduce/prevent/restore bone loss in mammals, such as in ovariectomized mice and rats, and prevent tumor burden in a syngeneic murine model of multiple myeloma.
- the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GX-
- the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide, wherein the antibody comprises: a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 14; and/or
- the VH region comprises (i) a VH CDR1 comprising the sequence GHTFSDYWMQ (SEQ ID NO: 63), DYWMQ (SEQ ID NO: 56), or GHTFSDY (SEQ ID NO: 64); (ii) a VH CDR2 comprising the sequence YPGDGD (SEQ ID NO: 57) or AIYPGDGDTRYNQKFKD (SEQ ID NO: 58); and (iii) a VH CDR3 comprising the sequence SMDYW (SEQ ID NO: 65).
- the VL region comprises (i) a VL CDR1 comprising the sequence RASKTVDSYGNSFMH (SEQ ID NO: 60); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQTIEFPYT (SEQ ID NO: 67).
- the antibody comprises (a) heavy chain CDRs comprising (i) a VH CDR1 comprising the sequence GHTFSDYWMQ (SEQ ID NO: 63), DYWMQ (SEQ ID NO: 56), or GHTFSDY (SEQ ID NO: 64); (ii) a VH CDR2 comprising the sequence YPGDGD (SEQ ID NO: 57) or AIYPGDGDTRYNQKFKD (SEQ ID NO: 58); and (iii) a VH CDR3 comprising the sequence SMDYW (SEQ ID NO: 65) and b) light chain CDRs comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGNSFMH (SEQ ID NO: 60); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQTIEFPYT (SEQ ID NO: 67
- the VH region comprises the sequence shown in SEQ ID NO: 14 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 18 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR.
- the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 88 and/or a heavy chain comprising the sequence shown in SEQ ID NO: 87.
- the antibody comprises a VH region produced by the expression vector with ATCC Accession No. PTA-12710.
- the antibody comprises a VL region produced by the expression vector with ATCC Accession No. PTA-1271 1 .
- the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST polypeptide), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence Xi ISGGDTYTYYADSVKG, wherein ⁇ is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising (i
- the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST polypeptide), wherein the antibody comprises: a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 6; and/or a VL region comprising VL CDR1 , VL CDR2, and VL CDR3 of the VL sequence shown in SEQ ID NO: 7.
- a SOST polypeptide e.g. , human SOST polypeptide
- the VH region comprises (i) a VH CDR1 comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), or GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence LISGGDTYTYYADSVKG (SEQ ID NOs: 52) or SGGDTY (SEQ ID NO: 46); and (iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48).
- the VL region comprises (i) a VL CDR1 comprising the sequence RSSQSLLDNDGETYLN (SEQ ID NO: 53); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
- the antibody comprises (a) heavy chain CDRs comprising (i) a VH CDR1 comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), or GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence LISGGDTYTYYADSVKG (SEQ ID NOs: 52) or SGGDTY (SEQ ID NO: 46); and (iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48) and (b) light chain CDRs comprising (i) a VL CDR1 comprising the sequence RSSQSLLDNDGETYLN (SEQ ID NO: 53); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
- the VH region comprises the sequence shown in SEQ ID NO: 6 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 7 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR.
- the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 90 and/or a heavy chain comprising the sequence shown in SEQ ID NO: 89.
- the antibody can be a human antibody, a humanized antibody, or a chimeric antibody.
- the antibody is a monoclonal antibody.
- the antibody comprises a constant region. In some embodiments, the antibody is of the human lgG1 , lgG2 or lgG2Aa, lgG3, or lgG4 subclass. In some embodiments, the antibody comprises a glycosylated constant region. In some embodiments, the antibody comprises a constant region having increased binding affinity to a human Fc gamma receptor.
- the invention also provides an isolated antibody, or an antigen binding fragment thereof, which competes for binding to the anti-SOST antibodies described herein.
- the invention also provides a conjugate of the anti-SOST antibody or the antigen binding fragment as described herein, wherein the antibody or the antigen binding fragment is conjugated to an agent, wherein the agent is selected from the group consisting of a chemotherapeutic agent (e.g., cytotoxic agent), an immunomodulating agent, an imaging agent, a therapeutic protein, a biopolymer, and an oligonucleotide.
- a chemotherapeutic agent e.g., cytotoxic agent
- an immunomodulating agent e.g., an immunomodulating agent
- an imaging agent e.g., a therapeutic protein, a biopolymer, and an oligonucleotide.
- the invention also provides pharmaceutical compositions comprising any of the anti-SOST antibodies described herein.
- the pharmaceutical composition comprises a pharmaceutically acceptable carrier.
- the invention also provides cell lines that recombinantly produce any of the anti- SOST antibodies described herein.
- the invention also provides nucleic acids encoding any of the anti-SOST antibodies described herein.
- the invention also provides nucleic acids encoding a heavy chain variable region and/or a light chain variable region of any of the anti-SOST antibodies described herein.
- a method of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies described herein.
- a method of treating or preventing a bone related disorder in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies described herein, wherein the bone related disorder is selected from the group consisting of osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, rheumatoid arthritis, osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss, metastatic bone malignancy, multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease (
- a method of activating canonical Wnt signaling activity in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies as described herein.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- a SOST polypeptide or a pharmaceutical composition thereof comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide or a pharmaceutical composition thereof.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- a SOST polypeptide or a pharmaceutical composition comprising thereof.
- a method of inhibiting metastasis of cancer cells or tumors comprising administering to the patient an effective amount of an anti- SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide or a pharmaceutical composition comprising thereof.
- an anti- SOST antibody e.g., the anti-SOST antibodies as described herein
- the patient can be a human.
- the individual can be a mammal, such as a cow, a cat, a mouse, a rat, a monkey, or a dog.
- the anti-SOST antibodies described herein can be administered in combination with other therapeutic agents (e.g., chemotherapeutic agents, osteoclast activity inhibiting agents, osteoblast activity enhancing agents, and dietary supplements as described herein).
- other therapeutic agents e.g., chemotherapeutic agents, osteoclast activity inhibiting agents, osteoblast activity enhancing agents, and dietary supplements as described herein.
- Figure 1 shows the dose-dependent inhibition of Wnt-10B signaling by recombinant mouse SOST protein (panel A) or recombinant human SOST protein (Panel B).
- Figure 2 shows that anti-SOST antibodies DP99, DP1 , DM99, DM21 , and DM4 neutralized recombinant human SOST activity in vitro.
- FIG. 4 depicts that anti-SOST antibodies of the present invention increased bone mass and prevented bone loss in ovariectomized (OVX) mice.
- Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle, DP99, or DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries.
- Total volumetric bone mineral content (BMC), bone mineral density (BMD) and bone area (AREA) were measured by pQCT on distal femurs (A, B, C, respectively) and on femoral diaphyses (D, E, F, respectively). Data are expressed as mean ⁇ SEM. a : p ⁇ 0.05 vs.
- Figure 5 shows the representative Micro-Computed Tomography ( ⁇ ) images of distal femurs (A) and the mean values of cancellous bone volume (B) from Sham, OVX, DP99, and DM99 treated mice groups.
- Figure 6 shows representative CT images of midshaft femurs from sham surgery or OVX mice treated with indicated mAbs (IgG control, DP99, DP1 , and DM1 1 at 25 mg/kg) 1 x/wk for 6 wks.
- Figures 7A-7B show that anti-SOST antibodies DP99, DP1 , and DM1 1 are efficacious in preventing ovariectomy-induced bone loss in mice in dose-response manner.
- Figure 8 shows representative ⁇ images of distal femurs from sham surgery or ovariectomized (OVX) mice treated with indicated mAbs (IgG control, DP99, DP1 , and DM1 1 at 25 mg/kg) 1 x/wk for 6 wks.
- indicated mAbs IgG control, DP99, DP1 , and DM1 1 at 25 mg/kg
- Figures 9A-9D show that anti-SOST antibodies DP99, DP1 , and DM1 1 are efficacious in preventing ovariectomy-induced bone loss in mice in dose-response manner.
- BV/TV bone volume/tissue volume; Tb.
- N trabecular number.
- Figure 10 shows the results of CT analyses of L4 lumbar vertebra (A), or distal femurs (B), and pQCT analyses of the femoral diaphysis (C).
- BV/TV bone volume/tissue volume
- BS/BV bone surface/bone volume
- Tb. N trabecular number
- Tb.Th trabecular thickness
- Tb.Sp trabecular spacing.
- Figure 1 1 shows that anti-SOST antibody DP99 prevents tumor burden in multiple myeloma (5TGM1 ) model in comparison to the IgG control group and the Bortezomib treated group (positive control). Data shown are based on 2 way ANOVA, Boneferroni post test (*** p ⁇ 0.001 ).
- Figure 12 also shows that anti-SOST antibody DP99 inhibits progression of multiple myeloma in the multiple myeloma (5TGM1 ) model in comparison to the IgG control group and the Bortezomib treated group (positive control).
- Figure 13 shows that anti-SOST antibodies DP99 and DM1 delays growth of the progressing tumor as represented by total flux.
- Figure 14 shows that anti-SOST antibodies DP99 and DM1 delays growth of the progressing tumor as represented by percent survival.
- the invention disclosed herein provides antibodies (e.g., antagonistic antibodies) that specifically bind to SOST (e.g., human SOST such as SEQ ID NO: 1 ).
- SOST e.g., human SOST such as SEQ ID NO: 1
- the invention also provides polynucleotides encoding these antibodies, compositions comprising these antibodies, and methods of making and using these antibodies.
- the invention further provides methods of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density using the anti- SOST antibodies as described herein.
- Methods of treating a SOST-associated condition/disorder/disease such as bone related disorder (e.g., osteoporosis (such as postmenopausal osteoporosis), osteopenia, Paget's Disease, bone fracture, multiple myeloma, acute myeloid leukemia (AML), cancer, or bone loss), inhibiting or reducing tumor burden, inhibiting or preventing tumor growth or progression, inhibiting metastasis of cancer cells or tumors or delaying tumor growth or progression, and inducing canonical Wnt signaling activity are also provided.
- bone related disorder e.g., osteoporosis (such as postmenopausal osteoporosis), osteopenia, Paget's Disease, bone fracture, multiple myeloma, acute myeloid leukemia (AML), cancer, or bone loss
- AML acute myeloid leukemia
- the term "isolated molecule" (where the molecule is, for example, a polypeptide, a polynucleotide, or an antibody) is a molecule that by virtue of its origin or source of derivation (1 ) is not associated with naturally associated components that accompany it in its native state, (2) is substantially free of other molecules from the same species (3) is expressed by a cell from a different species, or (4) does not occur in nature.
- a molecule that is chemically synthesized, or expressed in a cellular system different from the cell from which it naturally originates will be "isolated" from its naturally associated components.
- a molecule also may be rendered substantially free of naturally associated components by isolation, using purification techniques well known in the art.
- Molecule purity or homogeneity may be assayed by a number of means well known in the art.
- the purity of a polypeptide sample may be assayed using polyacrylamide gel electrophoresis and staining of the gel to visualize the polypeptide using techniques well known in the art.
- higher resolution may be provided by using HPLC or other means well known in the art for purification.
- an “antibody” is an immunoglobulin molecule capable of specific binding to a target, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc., through at least one antigen recognition site, located in the variable region of the immunoglobulin molecule.
- a target such as a carbohydrate, polynucleotide, lipid, polypeptide, etc.
- the term encompasses not only intact polyclonal or monoclonal antibodies, but also, unless otherwise specified, any antigen binding portion thereof that competes with the intact antibody for specific binding, fusion proteins comprising an antigen binding portion, and any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site.
- Antigen binding portions include, for example, Fab, Fab', F(ab') 2 , Fd, Fv, domain antibodies (dAbs, e.g., shark and camelid antibodies), fragments including complementarity determining regions (CDRs), single chain variable fragment antibodies (scFv), maxibodies, minibodies, intrabodies, diabodies, triabodies, tetrabodies, v-NAR and bis-scFv, and polypeptides that contain at least a portion of an immunoglobulin that is sufficient to confer specific antigen binding to the polypeptide.
- An antibody includes an antibody of any class, such as IgG, IgA, or IgM (or sub-class thereof), and the antibody need not be of any particular class.
- immunoglobulins can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgG-i , lgG 2 , lgG 3 , lgG 4 , IgA-i and lgA 2 .
- the heavy-chain constant regions that correspond to the different classes of immunoglobulins are called alpha, delta, epsilon, gamma, and mu, respectively.
- the subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
- An antibody, an antibody conjugate, or a polypeptide that "preferentially binds” or “specifically binds” (used interchangeably herein) to a target is a term well understood in the art, and methods to determine such specific or preferential binding are also well known in the art.
- a molecule is said to exhibit "specific binding” or “preferential binding” if it reacts or associates more frequently, more rapidly, with greater duration and/or with greater affinity with a particular cell or substance than it does with alternative cells or substances.
- An antibody “specifically binds” or “preferentially binds” to a target if it binds with greater affinity, avidity, more readily, and/or with greater duration than it binds to other substances.
- an antibody that specifically or preferentially binds to a SOST epitope is an antibody that binds this epitope with greater affinity, avidity, more readily, and/or with greater duration than it binds to other SOST epitopes or non-SOST epitopes. It is also understood that by reading this definition, for example, an antibody (or moiety or epitope) that specifically or preferentially binds to a first target may or may not specifically or preferentially bind to a second target. As such, “specific binding” or “preferential binding” does not necessarily require (although it can include) exclusive binding. Generally, but not necessarily, reference to binding means preferential binding.
- variable region refers to the variable region of the antibody light chain or the variable region of the antibody heavy chain, either alone or in combination.
- the variable regions of the heavy and light chains each consist of four framework regions (FRs) connected by three complementarity determining regions (CDRs) also known as hypervariable regions, and contribute to the formation of the antigen binding site of antibodies.
- variants of a subject variable region are desired, particularly with substitution in amino acid residues outside of a CDR region (i.e., in the framework region), appropriate amino acid substitution, preferably, conservative amino acid substitution, can be identified by comparing the subject variable region to the variable regions of other antibodies which contain CDR1 and CDR2 sequences in the same canonical class as the subject variable region (Chothia and Lesk, J Mol Biol 196(4): 901 -917, 1987).
- a "constant region" of an antibody refers to the constant region of the antibody light chain or the constant region of the antibody heavy chain, either alone or in combination.
- a "complementary determining region" or “CDR" of a variable domain are the amino acid residues within the variable region that are identified in accordance with the definitions of the Kabat, Chothia, the accumulation of both Kabat, Chothia, extended, AbM, contact, and/or conformational definitions or any method of CDR determination well known in the art.
- Antibody CDRs may be identified as the hypervariable regions originally defined by Kabat et al. See, e.g., Kabat et al., 1992, Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, NIH, Washington D.C. The positions of the CDRs may also be identified as the structural loops originally described by Chothia and others.
- CDR identification includes the "AbM definition,” which is a compromise between Kabat and Chothia and is derived using Oxford Molecular's AbM antibody modeling software (now ACCELRYS®), or the "contact definition" of CDRs based on observed antigen contacts, set forth in MacCallum et al., 1996, J. Mol. Biol., 262:732- 745.
- the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding.
- CDR may refer to CDRs defined by any approach known in the art, including combinations of approaches. The methods used herein may utilize CDRs defined according to any of these approaches. For any given embodiment containing more than one CDR, the CDRs may be defined in accordance with any of Kabat, Chothia, extended, AbM, contact, and/or conformational definitions.
- mAb monoclonal antibody
- mAb refers to an antibody that is derived from a single copy or clone, including e.g., any eukaryotic, prokaryotic, or phage clone, and not the method by which it is produced.
- a monoclonal antibody of the invention exists in a homogeneous or substantially homogeneous population.
- humanized antibody refers to forms of non-human (e.g. murine) antibodies that are chimeric immunoglobulins, immunoglobulin chains, or fragments thereof (such as Fv, Fab, Fab', F(ab') 2 or other antigen-binding subsequences of antibodies) that contain minimal sequence derived from non-human immunoglobulin.
- humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a CDR of the recipient are replaced by residues from a CDR of a non-human species (donor antibody) such as mouse, rat, or rabbit having the desired specificity, affinity, and capacity.
- a "human antibody” is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen binding residues.
- chimeric antibody is intended to refer to antibodies in which the variable region sequences are derived from one species and the constant region sequences are derived from another species, such as an antibody in which the variable region sequences are derived from a mouse antibody and the constant region sequences are derived from a human antibody.
- epitope refers to that portion of a molecule capable of being recognized by and bound by an antibody at one or more of the antibody's antigen- binding regions. Epitopes often consist of a surface grouping of molecules such as amino acids or sugar side chains and have specific three-dimensional structural characteristics as well as specific charge characteristics.
- the epitope can be a protein epitope. Protein epitopes can be linear or conformational. In a linear epitope, all of the points of interaction between the protein and the interacting molecule (such as an antibody) occur linearly along the primary amino acid sequence of the protein.
- a “nonlinear epitope” or “conformational epitope” comprises noncontiguous polypeptides (or amino acids) within the antigenic protein to which an antibody specific to the epitope binds.
- the term "antigenic epitope” as used herein, is defined as a portion of an antigen to which an antibody can specifically bind as determined by any method well known in the art, for example, by conventional immunoassays. Once a desired epitope on an antigen is determined, it is possible to generate antibodies to that epitope, e.g., using the techniques described in the present specification. Alternatively, during the discovery process, the generation and characterization of antibodies may elucidate information about desirable epitopes.
- SOST or "sclerostin” includes, for example, murine and human native forms of SOST.
- Exemplary SOST protein and nucleotides are disclosed, e.g., by Brunkow et al., 2001 , supra (amino acid sequence is disclosed as SEQ ID NO: 1 ).
- the term also includes variants of such native sequences that are immunologically cross-reactive with these native proteins. These proteins can inhibit the interaction between LRP5 and/or LRP6 with Wnt, which is required for canonical Wnt signaling activity.
- the term can also refer to a fragment of a native or variant form of SOST that contains an epitope to which an antibody can specifically bind.
- antagonist antibody refers to an antibody that binds to a target and prevents or reduces the biological effect of that target.
- the term can denote an antibody that prevents the target, e.g., SOST, to which it is bound from performing a biological function.
- an "SOST antagonist antibody” or “anti-SOST antagonist antibody” refers to an antibody that is able to inhibit SOST biological activity and/or downstream events(s) mediated by SOST.
- Anti-SOST antagonist antibodies encompass antibodies that block, antagonize, suppress, or reduce (to any degree including significantly) SOST biological activity, including downstream events mediated by SOST, such as cell surface interaction, receptor binding and downstream signaling (e.g., canonical Wnt signaling).
- an anti-SOST antagonist antibody encompasses all the previously identified terms, titles, and functional states and characteristics whereby the SOST itself, an SOST biological activity (including but not limited to its ability to bind a cell and bind a receptor), or the consequences of the biological activity, are substantially nullified, decreased, or neutralized in any meaningful degree.
- an anti-SOST antibody binds SOST and prevents SOST from interacting with LRP5 and/or LRP6, thus antagonizing the SOST-mediated inhibition of canonical Wnt signaling pathway.
- an anti-SOST antibody binds SOST and activates canonical Wnt signaling pathway.
- the antagonist ability is described in terms of an IC50 or EC50 value.
- polypeptide oligopeptide
- peptide protein
- the terms “polypeptide”, “oligopeptide”, “peptide” and “protein” are used interchangeably herein to refer to chains of amino acids of any length.
- the chain may be linear or branched, it may comprise modified amino acids, and/or may be interrupted by non-amino acids.
- the terms also encompass an amino acid chain that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component.
- polypeptides containing one or more analogs of an amino acid including, for example, unnatural amino acids, etc.
- the polypeptides can occur as single chains or associated chains.
- polynucleotide or “nucleic acid,” as used interchangeably herein, refer to chains of nucleotides of any length, and include DNA and RNA.
- the nucleotides can be deoxyribonucleotides, ribonucleotides, modified nucleotides or bases, and/or their analogs, or any substrate that can be incorporated into a chain by DNA or RNA polymerase.
- a polynucleotide may comprise modified nucleotides, such as methylated nucleotides and their analogs. If present, modification to the nucleotide structure may be imparted before or after assembly of the chain.
- the sequence of nucleotides may be interrupted by non-nucleotide components.
- a polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component.
- Other types of modifications include, for example, "caps", substitution of one or more of the naturally occurring nucleotides with an analog, internucleotide modifications such as, for example, those with uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates, carbamates, etc.) and with charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, for example, proteins (e.g., nucleases, toxins, antibodies, signal peptides, poly-L-lysine, etc.), those with intercalators (e.g., acridine, psoralen, etc.), those containing chelators (e.g., metal
- any of the hydroxyl groups ordinarily present in the sugars may be replaced, for example, by phosphonate groups, phosphate groups, protected by standard protecting groups, or activated to prepare additional linkages to additional nucleotides, or may be conjugated to solid supports.
- the 5' and 3' terminal OH can be phosphorylated or substituted with amines or organic capping group moieties of from 1 to 20 carbon atoms.
- Other hydroxyls may also be derivatized to standard protecting groups.
- Polynucleotides can also contain analogous forms of ribose or deoxyribose sugars that are generally known in the art, including, for example, 2'-O-methyl- 2'-O-allyl, 2'-fluoro- or 2'-azido-ribose, carbocyclic sugar analogs, alpha- or beta-anomeric sugars, epimeric sugars such as arabinose, xyloses or lyxoses, pyranose sugars, furanose sugars, sedoheptuloses, acyclic analogs and abasic nucleoside analogs such as methyl riboside.
- One or more phosphodiester linkages may be replaced by alternative linking groups.
- linking groups include, but are not limited to, embodiments wherein phosphate is replaced by P(O)S("thioate"), P(S)S ("dithioate"), (O)NR 2 ("amidate"), P(O)R, P(O)OR', CO or CH 2 ("formacetal"), in which each R or R' is independently H or substituted or unsubstituted alkyl (1 -20 C) optionally containing an ether (-O-) linkage, aryl, alkenyl, cycloalkyl, cycloalkenyl or araldyl. Not all linkages in a polynucleotide need be identical. The preceding description applies to all polynucleotides referred to herein, including RNA and DNA.
- substantially pure refers to material which is at least 50% pure (i.e., free from contaminants), more preferably, at least 90% pure, more preferably, at least 95% pure, yet more preferably, at least 98% pure, and most preferably, at least 99% pure.
- a "host cell” includes an individual cell or cell culture that can be or has been a recipient for vector(s) for incorporation of polynucleotide inserts.
- Host cells include progeny of a single host cell, and the progeny may not necessarily be completely identical (in morphology or in genomic DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation.
- a host cell includes cells transfected in vivo with a polynucleotide(s) of this invention.
- the term "Fc region” is used to define a C-terminal region of an immunoglobulin heavy chain.
- the "Fc region” may be a native sequence Fc region or a variant Fc region.
- the human IgG heavy chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl- terminus thereof.
- the numbering of the residues in the Fc region is that of the EU index as in Kabat. Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md., 1991 .
- the Fc region of an immunoglobulin generally comprises two constant domains, CH2 and CH3. As is known in the art, an Fc region can be present in dimer or monomeric form.
- Fc receptor and “FcR” describe a receptor that binds to the
- the preferred FcR is a native sequence human FcR.
- a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the FcyRI, FcyRII, and FcyRIII subclasses, including allelic variants and alternatively spliced forms of these receptors.
- FcyRII receptors include FcyRIIA (an “activating receptor") and FcyRIIB (an "inhibiting receptor”), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof. FcRs are reviewed in Ravetch and Kinet, 1991 , Ann. Rev.
- FcR also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al., 1976, J. Immunol., 1 17:587; and Kim et al., 1994, J. Immunol., 24:249).
- ADCC antibody-dependent cell-mediated cytotoxicity
- FcRs Fc receptors
- NK natural killer cells
- macrophages e.g. natural killer cells, neutrophils, and macrophages
- ADCC activity of a molecule of interest can be assessed using an in vitro ADCC assay, such as that described in U.S. Patent No. 5,500,362 or 5,821 ,337.
- Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and NK cells.
- PBMC peripheral blood mononuclear cells
- ADCC activity of the molecule of interest may be assessed in vivo, e.g., in an animal model such as that disclosed in Clynes et al., PNAS (USA), 95:652-656, 1998.
- “Complement dependent cytotoxicity” or “CDC” refers to the lysing of a target in the presence of complement.
- the complement activation pathway is initiated by the binding of the first component of the complement system (C1 q) to a molecule (e.g. an antibody) complexed with a cognate antigen.
- a CDC assay e.g. as described in Gazzano-Santoro et al., J. Immunol. Methods, 202: 163, 1996, may be performed
- Compet means that a first antibody, or an antigen-binding portion thereof, binds to an epitope in a manner sufficiently similar to the binding of a second antibody, or an antigen-binding portion thereof, such that the result of binding of the first antibody with its cognate epitope is detectably decreased in the presence of the second antibody compared to the binding of the first antibody in the absence of the second antibody.
- the alternative, where the binding of the second antibody to its epitope is also detectably decreased in the presence of the first antibody can, but need not be the case. That is, a first antibody can inhibit the binding of a second antibody to its epitope without that second antibody inhibiting the binding of the first antibody to its respective epitope.
- each antibody detectably inhibits the binding of the other antibody with its cognate epitope or ligand, whether to the same, greater, or lesser extent, the antibodies are said to "cross-compete" with each other for binding of their respective epitope(s).
- Both competing and cross-competing antibodies are encompassed by the present invention. Regardless of the mechanism by which such competition or cross-competition occurs (e.g., steric hindrance, conformational change, or binding to a common epitope, or portion thereof), the skilled artisan would appreciate, based upon the teachings provided herein, that such competing and/or cross-competing antibodies are encompassed and can be useful for the methods disclosed herein.
- a “functional Fc region” possesses at least one effector function of a native sequence Fc region.
- exemplary “effector functions” include C1 q binding; complement dependent cytotoxicity; Fc receptor binding; antibody-dependent cell-mediated cytotoxicity; phagocytosis; down-regulation of cell surface receptors (e.g. B cell receptor), etc.
- Such effector functions generally require the Fc region to be combined with a binding domain (e.g. an antibody variable domain) and can be assessed using various assays known in the art for evaluating such antibody effector functions.
- a “native sequence Fc region” comprises an amino acid sequence identical to the amino acid sequence of an Fc region found in nature.
- a “variant Fc region” comprises an amino acid sequence which differs from that of a native sequence Fc region by virtue of at least one amino acid modification, yet retains at least one effector function of the native sequence Fc region.
- the variant Fc region has at least one amino acid substitution compared to a native sequence Fc region or to the Fc region of a parent polypeptide, e.g. from about one to about ten amino acid substitutions, and preferably, from about one to about five amino acid substitutions in a native sequence Fc region or in the Fc region of the parent polypeptide.
- the variant Fc region herein will preferably possess at least about 80% sequence identity with a native sequence Fc region and/or with an Fc region of a parent polypeptide, and most preferably, at least about 90% sequence identity therewith, more preferably, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99% sequence identity therewith.
- beneficial or desired clinical results include, but are not limited to, one or more of the following: increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density, decreasing symptoms resulting from a SOST associated disease (e.g., a bone related disorder), increasing the quality of life of those suffering from a SOST-associated disease (e.g., a bone related disorder), decreasing the dose of other medications required to treat a SOST associated disease (e.g., a bone related disorder), delaying the progression or onset of a SOST associated disease (e.g., a bone related disorder), curing a SOST associated disease (e.g., a bone related disorder), and/or prolong survival of patients having a SOST associated disease (e.g., a bone related disorder).
- Treatment may be prophylactic (e.g., to prevent or delay the onset of the disease, or to prevent the manifestation of clinical or subclinical symptoms
- Reducing incidence means any of reducing severity (which can include reducing need for and/or amount of (e.g., exposure to) other drugs and/or therapies generally used for this condition), reducing duration, and/or reducing frequency.
- individuals may vary in terms of their response to treatment, and, as such, for example, a "method of reducing incidence” reflects administering the anti-SOST antibody based on a reasonable expectation that such administration may likely cause such a reduction in incidence in that particular individual.
- “Ameliorating” means a lessening or improvement of one or more symptoms as compared to not administering an anti-SOST antibody. “Ameliorating” also includes shortening or reduction in duration of a symptom.
- an "effective dosage” or “effective amount” of drug, compound, or pharmaceutical composition is an amount sufficient to effect any one or more beneficial or desired results.
- an effective amount prevents, alleviates or ameliorates symptoms of disease, and/or prolongs the survival of the subject being treated.
- beneficial or desired results include eliminating or reducing the risk, lessening the severity, or delaying the outset of the disease, including biochemical, histological and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease.
- beneficial or desired results include clinical results such as reducing one or more symptoms of a bone-related disease, decreasing the dose of other medications required to treat the disease, enhancing the effect of another medication, and/or delaying the progression of the disease of patients.
- An effective dosage can be administered in one or more administrations.
- an effective dosage of drug, compound, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly.
- an effective dosage of a drug, compound, or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition.
- an "effective dosage" may be considered in the context of administering one or more therapeutic agents, and a single agent may be considered to be given in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved.
- mammals also include, but are not limited to, farm animals (e.g., cows, pigs, horses, chickens, etc.), sport animals, pets, primates, horses, dogs, cats, mice and rats.
- farm animals e.g., cows, pigs, horses, chickens, etc.
- sport animals e.g., pets, primates, horses, dogs, cats, mice and rats.
- the individual is considered to be at risk for a bone related disorder disease (e.g., osteoporosis, osteopenia, Paget's Disease, bone loss, bone fracture, and multiple myeloma).
- a bone related disorder disease e.g., osteoporosis, osteopenia, Paget's Disease, bone loss, bone fracture, and multiple myeloma.
- Such individuals include, but are not limited to, an individual who is hospitalized or will be hospitalized, an individual who is or will be put in an intensive care unit, an individual who will undergo surgery (e.g., bone related surgery such as implant surgery), an individual who will be anesthetized or under general anesthesia, an individual on dialysis, an individual with an indwelling catheter, an individual over the age of 65, an individual with a compromised immune system, a pediatric individual, an individual who is or may be put on a respirator or other mechanical ventilator, an individual in whom an endotracheal tube will or has been placed, an individual who is or will be immobilized, an individual who will undergo, is undergoing, or has undergone chemotherapy or myeloablative therapy, an individual who will take, is taking, or has taken one or more immunosuppressants, particularly for a significant period of time (longer than a month); an individual who is at risk of bone fracture (e.g., vertebral and/or non vertebral bone fracture).
- the individuals may be male
- vector means a construct, which is capable of delivering, and, preferably, expressing, one or more gene(s) or sequence(s) of interest in a host cell.
- vectors include, but are not limited to, viral vectors, naked DNA or RNA expression vectors, plasmid, cosmid or phage vectors, DNA or RNA expression vectors associated with cationic condensing agents, DNA or RNA expression vectors encapsulated in liposomes, and certain eukaryotic cells, such as producer cells.
- expression control sequence means a nucleic acid sequence that directs transcription of a nucleic acid.
- An expression control sequence can be a promoter, such as a constitutive or an inducible promoter, or an enhancer.
- the expression control sequence is operably linked to the nucleic acid sequence to be transcribed.
- pharmaceutically acceptable carrier or “pharmaceutical acceptable excipient” includes any material which, when combined with an active ingredient, allows the ingredient to retain biological activity and is non-reactive with the subject's immune system.
- examples include, but are not limited to, any of the standard pharmaceutical carriers such as a phosphate buffered saline solution, water, emulsions such as oil/water emulsion, and various types of wetting agents.
- Preferred diluents for aerosol or parenteral administration are phosphate buffered saline (PBS) or normal (0.9%) saline.
- Compositions comprising such carriers are formulated by well known conventional methods (see, for example, Remington's Pharmaceutical Sciences, 18th edition, A. Gennaro, ed., Mack Publishing Co., Easton, PA, 1990; and Remington, The Science and Practice of Pharmacy 21 th Ed. Mack Publishing, 201 1 ).
- k on refers to the rate constant for association of an antibody to an antigen. Specifically, the rate constants (k on and k 0 ff) and equilibrium dissociation constants are measured using full-length antibodies and/or Fab antibody fragments and SOST.
- k 0 ff refers to the rate constant for dissociation of an antibody from the antibody/antigen complex.
- K D refers to the equilibrium dissociation constant of an antibody-antigen interaction.
- references to "about” a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. For example, description referring to "about X” includes description of "X.” Numeric ranges are inclusive of the numbers defining the range.
- the present invention encompasses not only the entire group listed as a whole, but each member of the group individually and all possible subgroups of the main group, but also the main group absent one or more of the group members.
- the present invention also envisages the explicit exclusion of one or more of any of the group members in the claimed invention.
- the present invention provides an antibody (e.g., an antagonist antibody) that binds to SOST and blocks, suppresses, or reduces (including significantly reduces) SOST biological activity.
- the anti-SOST antibody of the present invention should exhibit any one or more of the following characteristics: (a) binds to SOST; (b) neutralizes, decreases, and/or downregulates the protein expression of SOST; (c) increases bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density (e.g., bone mineral density) in a subject; (d) treats, prevents, or ameliorates one or more SOST associated disorder(s) (e.g., bone related disorder, including but not limited to osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, rheumatoid arthritis, osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss, metastatic bone malignancy
- the anti-SOST antibodies of the present invention have at least two or more of these characteristics. In some embodiments, the anti-SOST antibodies have at least three or more of the characteristics.
- an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide e.g.
- the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GXiTFX 2 DYWMQ, wherein X ⁇ is F or H, X 2 is T or S (SEQ ID NO: 81 ) or GXiTFX 2 DY, wherein X ⁇ is F or H, and X 2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX 2 X 3 KX 4 , wherein X is A or N, X 2 is S or K, X 3 is V or F, and X 4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SX-i DYW, wherein X ⁇ is F or M (SEQ ID NO: 84); and/or (b) a light chain variable (VH) region complementary
- an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide e.g. , human SOST polypeptide
- the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence X1 ISGGDTYTYYADSVKG, wherein X is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising
- the antibodies useful in the present invention can encompass monoclonal antibodies, polyclonal antibodies, antibody fragments (e.g. , Fab, Fab', F(ab') 2 , Fv, Fc, etc.), chimeric antibodies, bispecific antibodies, heteroconjugate antibodies, single chain (ScFv), mutants thereof, fusion proteins comprising an antibody portion (e.g., a domain antibody), humanized antibodies, and any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site of the required specificity, including glycosylation variants of antibodies, amino acid sequence variants of antibodies, and covalently modified antibodies.
- the antibodies may be murine, rat, human, or any other origin (including chimeric or humanized antibodies).
- the anti-SOST antibody as described herein is a monoclonal antibody.
- the anti-SOST antibody is a humanized monoclonal antibody or a chimeric monoclonal antibody.
- the binding affinity (K D ) of the anti-SOST antibodies of the present invention can be about 0.0005 to about 100 nM.
- the binding affinity is any of about 100 nM, about 50 nM, about 10 nM, about 7.5 nM, about 5 nM, about 3.7 nM, about 1 nM, about 500 pM, about 250 pM, about 130 pM, about 100 pM, about 61 pM, about 60 pM, about 50 pM, about 21 pM, about 20 pM, about 19 pM, about 15 pM, about 10 pM, about 9 pM, about 5 pM, about 2 pM, or about 1 pM.
- the binding affinity is less than any of about 200 nM, about 150 nM, about 100 nM, about 50 nM, about 10 nM, about 5 nM, about 1 nM, about 500 pM, about 100 pM, about 75 pM, about 50 pM, about 25 pM, about 20 pM, about 10 pM, about 5 pM, or about 2 pM.
- the binding affinity of the anti-SOST antibodies as described herein is about 4 nM or less as measured by surface plasmon resonance at 37°C. In some embodiments, the binding affinity of the antibodies as described herein is about 100 pM or less as measured by surface plasmon resonance at room temperature (e.g., 20°C to 26 °C).
- Binding affinity may be determined using Kinexa Biosensor, scintillation proximity assays, Enzyme-linked Immunosorbent Assay (ELISA), ORIGEN immunoassay (IGEN), fluorescence quenching, fluorescence transfer, and/or yeast display. Binding affinity may also be screened using a suitable bioassay.
- One way of determining binding affinity of antibodies to SOST is by measuring binding affinity of monofunctional Fab fragments of the antibody. To obtain
- an antibody for example, IgG
- an antibody can be cleaved with papain or expressed recombinantly.
- the affinity of a SOST Fab fragment of an antibody can be determined by surface plasmon resonance (BiacoreTM3000TM surface plasmon resonance (SPR) system, BiacoreTM, INC, Piscataway NJ) equipped with pre- immobilized streptavidin sensor chips (SA) or anti-mouse Fc or anti-human Fc using HBS-EP running buffer (0.01 M HEPES, pH 7.4, 0.15 NaCI, 3 mM EDTA, 0.005% v/v Surfactant P20).
- Biotinylated or Fc fusion human SOST can be diluted into HBS-EP buffer to a concentration of less than 0.5 pg/mL and injected across the individual chip channels using variable contact times, to achieve two ranges of antigen density, either 50-200 response units (RU) for detailed kinetic studies or 800-1 ,000 RU for screening assays.
- Regeneration studies have shown that 25 mM NaOH in 25% v/v ethanol effectively removes the bound Fab while keeping the activity of SOST on the chip for over 200 injections.
- serial dilutions spanning concentrations of 0.1 -1 Ox estimated K D
- purified Fab samples are injected for 1 min at 100 ⁇ _/ ⁇ ⁇ and dissociation times of up to 2 hours are allowed.
- the concentrations of the Fab proteins are determined by ELISA and/or SDS-PAGE electrophoresis using a Fab of known concentration (as determined by amino acid analysis) as a standard.
- Kinetic association rates (k on ) and dissociation rates (k 0ff ) are obtained simultaneously by fitting the data globally to a 1 : 1 Langmuir binding model (Karlsson, R. Roos, H. Fagerstam, L.
- compositions comprising antibodies described herein or made by the methods and having the characteristics described herein.
- compositions comprise one or more antibodies that bind to SOST, and/or one or more polynucleotides comprising sequences encoding one or more these antibodies.
- compositions may further comprise suitable excipients, such as pharmaceutically acceptable excipients including buffers, which are well known in the art.
- the invention provides an antibody or compositions (including pharmaceutical compositions) comprising an antibody having a partial light chain sequence and/or a partial heavy chain sequence as found in Table 1 , or variants thereof.
- Table 1 the underlined sequences are CDR sequences according to Kabat, and the sequences in bold are CDR sequences according to Chothia. Table 1
- GGGTKLEIK (SEQ ID NO: 3)
- GGGTKLEIK (SEQ ID NO: 1 1 )
- GQGTKLEIK (SEQ ID NO: 16) mAb Light Chain Heavy Chain
- GQGTKLEIK (SEQ ID NO: 31 )
- GQGTKLEIK (SEQ ID NO: 24) mAb Light Chain Heavy Chain
- GQGTKLEIK (SEQ ID NO: 41 )
- GQGTKLEIK (SEQ ID NO: 41 )
- CDRs can be a combination of the Kabat and Chothia CDR (also termed “combined CDRs” or “extended CDRs”).
- the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., Journal of Biological Chemistry, 283: 1 156-1 166, 2008.
- “conformational CDRs” include the residue positions in the Kabat CDRs and Vernier zones which are constrained in order to maintain proper loop structure for the antibody to bind a specific antigen. Determination of conformational CDRs is well within the skill of the art.
- the CDRs are the Kabat CDRs.
- the CDRs are the Chothia CDRs.
- the CDRs are the extended, AbM, conformational, or contact CDRs.
- the CDRs may be any of Kabat, Chothia, extended, AbM, conformational, contact CDRs or combinations thereof.
- Table 2 provides examples of CDR sequences of anti-SOST antibodies provided herein.
- nsus is T or S (SEQ ID NOs: A or N, X 2 is S or K, 84)
- X-i is S wherein X ! is T or S, X 2 or N, and X 2 is N or S is E or D, X 3 is H, D, F, (SEQ ID NO: 85) or E, and X 4 is H, P, or
- the present invention provides an antibody that binds to SOST and competes with the antibody as described herein, such as DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, and DM34.
- an antibody that binds to SOST and competes with the antibody as described herein, such as DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 ,
- the present invention provides an antibody or an antigen binding fragment, which specifically binds to SOST, wherein the antibody comprises a VH region comprising a sequence shown in SEQ ID NO: 14; and/or a VL region comprising a sequence shown in SEQ ID NO: 18.
- the antibody comprises a light chain comprising the sequence DIVMTQSPDSLAVSLGERATINCRASKTVDSYGNSFMHWFQQKPGQPPKLLIHHSSNL ESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCLQTIEFPYTFGQGTKLEIKRTVAAP SVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKD STYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
- the present invention provides an antibody or an antigen binding fragment, which specifically bind to SOST, wherein the antibody comprises a VH region comprising a sequence shown in SEQ ID NO: 6; and/or a VL region comprising a sequence shown in SEQ ID NO: 7.
- the antibody comprises a light chain comprising the sequence
- the invention also provides CDR portions of antibodies to anti-SOST antibodies based on CDR contact regions.
- CDR contact regions are regions of an antibody that imbue specificity to the antibody for an antigen.
- CDR contact regions include the residue positions in the CDRs and Vernier zones which are constrained in order to maintain proper loop structure for the antibody to bind a specific antigen. See, e.g., Makabe et al., J. Biol. Chem., 283: 1 156-1 166, 2007. Determination of CDR contact regions is well within the skill of the art.
- anti-SOST antibodies as described herein may be made by any method known in the art.
- the route and schedule of immunization of the host animal are generally in keeping with established and
- any mammalian subject including humans or antibody producing cells therefrom can be manipulated to serve as the basis for production of mammalian, including human and hybridoma cell lines.
- the host animal is inoculated intraperitoneally, intramuscularly, orally, subcutaneously, intraplantar, and/or intradermally with an amount of immunogen, including as described herein.
- Hybridomas can be prepared from the lymphocytes and immortalized myeloma cells using the general somatic cell hybridization technique of Kohler, B. and Milstein, C, Nature 256:495-497, 1975 or as modified by Buck, D. W., et al., In Vitro, 18:377-381 , 1982.
- Available myeloma lines including but not limited to X63-Ag8.653 and those from the Salk Institute, Cell Distribution Center, San Diego, Calif., USA, may be used in the hybridization.
- the technique involves fusing myeloma cells and lymphoid cells using a fusogen such as polyethylene glycol, or by electrical means well known to those skilled in the art.
- the cells are separated from the fusion medium and grown in a selective growth medium, such as hypoxanthine-aminopterin-thymidine (HAT) medium, to eliminate unhybridized parent cells.
- a selective growth medium such as hypoxanthine-aminopterin-thymidine (HAT) medium
- HAT hypoxanthine-aminopterin-thymidine
- Any of the media described herein, supplemented with or without serum, can be used for culturing hybridomas that secrete monoclonal antibodies.
- EBV immortalized B cells may be used to produce the anti-SOST monoclonal antibodies of the subject invention.
- hybridomas are expanded and subcloned, if desired, and supernatants are assayed for anti-immunogen activity by conventional immunoassay procedures (e.g., radioimmunoassay, enzyme immunoassay, or fluorescence
- Hybridomas that may be used as source of antibodies encompass all derivatives, progeny cells of the parent hybridomas that produce monoclonal antibodies specific for SOST, or a portion thereof.
- Hybridomas that produce such antibodies may be grown in vitro or in vivo using known procedures.
- the monoclonal antibodies may be isolated from the culture media or body fluids, by conventional immunoglobulin purification procedures such as ammonium sulfate precipitation, gel electrophoresis, dialysis, chromatography, and ultrafiltration, if desired.
- Undesired activity, if present, can be removed, for example, by running the preparation over adsorbents made of the immunogen attached to a solid phase and eluting or releasing the desired antibodies off the immunogen.
- Immunization of a host animal with SOST e.g., human SOST
- SOST e.g., human SOST
- a fragment containing the target amino acid sequence conjugated to a protein that is immunogenic in the species to be immunized
- the anti-SOST antibody (monoclonal or polyclonal) of interest may be sequenced and the polynucleotide sequence may then be cloned into a vector for expression or propagation.
- the sequence encoding the antibody of interest may be maintained in vector in a host cell and the host cell can then be expanded and frozen for future use.
- Production of recombinant monoclonal antibodies in cell culture can be carried out through cloning of antibody genes from B cells by means known in the art. See, e.g. Tiller et al., J. Immunol. Methods 329, 1 12, 2008; U.S. Pat. No. 7,314,622.
- polynucleotide sequence may be used for genetic editing
- the constant region may be engineered to more nearly resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. It may be desirable to genetically manipulate the antibody sequence to obtain greater affinity to SOST and greater efficacy in inhibiting SOST.
- humanized molecules are designed to minimize unwanted immunological response toward rodent anti-human antibody molecules which limits the duration and
- the antibody constant region can be engineered such that it is immunologically inert (e.g., does not trigger complement lysis). See, e.g. PCT Publication No.
- humanized antibodies discussed above are also applicable to customizing antibodies for use, for example, in dogs, cats, primate, equines, and bovines. Further, one or more aspects of humanizing an antibody described herein may be combined, e.g., CDR grafting, framework mutation and CDR mutation.
- Fully human antibodies may be obtained by using commercially available mice that have been engineered to express specific human immunoglobulin proteins.
- Transgenic animals that are designed to produce a more desirable (e.g., fully human antibodies) or more robust immune response may also be used for generation of humanized or human antibodies. Examples of such technology are XenomouseTM from Abgenix, Inc. (Fremont, CA) and HuMAb-Mouse® and TC MouseTM from Medarex, Inc. (Princeton, NJ).
- antibodies may be made recombinantly and expressed using any method known in the art. In another alternative, antibodies may be made
- phage display technology can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from unimmunized donors.
- V domain genes are cloned in-frame into either a major or minor coat protein gene of a filamentous bacteriophage, such as M13 or fd, and displayed as functional antibody fragments on the surface of the phage particle.
- the filamentous particle contains a single- stranded DNA copy of the phage genome, selections based on the functional properties of the antibody also result in selection of the gene encoding the antibody exhibiting those properties.
- the phage mimics some of the properties of the B cell.
- Phage display can be performed in a variety of formats; for review see, e.g., Johnson, Kevin S. and Chiswell, David J., Current Opinion in Structural Biology 3:564-571 , 1993.
- Several sources of V-gene segments can be used for phage display.
- Gene shuffling can also be used to derive human antibodies from rodent antibodies, where the human antibody has similar affinities and specificities to the starting rodent antibody.
- this method which is also referred to as "epitope imprinting"
- the heavy or light chain V domain gene of rodent antibodies obtained by phage display technique is replaced with a repertoire of human V domain genes, creating rodent-human chimeras.
- Selection on antigen results in isolation of human variable regions capable of restoring a functional antigen binding site, i.e., the epitope governs (imprints) the choice of partner.
- the process is repeated in order to replace the remaining rodent V domain, a human antibody is obtained (see PCT Publication No. WO 93/06213).
- this technique provides completely human antibodies, which have no framework or CDR residues of rodent origin.
- Antibodies may be made recombinantly by first isolating the antibodies and antibody producing cells from host animals, obtaining the gene sequence, and using the gene sequence to express the antibody recombinantly in host cells (e.g., CHO cells). Another method which may be employed is to express the antibody sequence in plants (e.g., tobacco) or transgenic milk. Methods for expressing antibodies recombinantly in plants or milk have been disclosed. See, for example, Peeters, et al. Vaccine 19:2756, 2001 ; Lonberg, N. and D. Huszar Int. Rev. Immunol 13:65, 1995; and Pollock, et al., J Immunol Methods 231 : 147, 1999. Methods for making derivatives of antibodies, e.g., humanized, single chain, etc. are known in the art.
- Immunoassays and flow cytometry sorting techniques such as fluorescence activated cell sorting (FACS) can also be employed to isolate antibodies that are specific for SOST.
- FACS fluorescence activated cell sorting
- DNA encoding the monoclonal antibodies is readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies).
- the hybridoma cells serve as a preferred source of such DNA.
- the DNA may be placed into expression vectors (such as expression vectors disclosed in PCT Publication No. WO 87/04462), which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells.
- expression vectors such as expression vectors disclosed in PCT Publication No. WO 87/04462
- host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells
- the DNA also may be modified, for example, by substituting the coding sequence for human heavy and light chain constant regions in place of the homologous murine sequences, Morrison et al., Proc. Nat. Acad. Sci.
- chimeric or “hybrid” antibodies are prepared that have the binding specificity of an anti- SOST monoclonal antibody herein.
- the anti-SOST antibodies as described herein can be identified or characterized using methods known in the art, whereby reduction of SOST expression levels is detected and/or measured.
- an anti-SOST antibody is identified by incubating a candidate agent with SOST and monitoring binding and/or attendant reduction of SOST expression levels.
- the binding assay may be performed with purified SOST polypeptide(s), or with cells naturally expressing, or transfected to express, SOST polypeptide(s).
- the binding assay is a competitive binding assay, where the ability of a candidate antibody to compete with a known SOST antibody for SOST binding is evaluated.
- the assay may be performed in various formats, including the ELISA format.
- bioassays known to test the targeted biological activities.
- bioassays can be used to screen candidates directly.
- Anti-SOST antibodies may be characterized using methods well known in the art. For example, one method is to identify the epitope to which it binds, or "epitope mapping.” There are many methods known in the art for mapping and characterizing the location of epitopes on proteins, including solving the crystal structure of an antibody-antigen complex, competition assays, gene fragment expression assays, and synthetic peptide-based assays, as described, for example, in Chapter 1 1 of Harlow and Lane, Using Antibodies, a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999. In an additional example, epitope mapping can be used to determine the sequence to which an anti-SOST antibody binds.
- Epitope mapping is commercially available from various sources, for example, Pepscan Systems (Edelhertweg 15, 8219 PH Lelystad, The Netherlands).
- the epitope can be a linear epitope, i.e., contained in a single stretch of amino acids, or a conformational epitope formed by a three-dimensional interaction of amino acids that may not necessarily be contained in a single stretch.
- Peptides of varying lengths e.g., at least 4-6 amino acids long
- the epitope to which the anti-SOST antibody binds can be determined in a systematic screening by using overlapping peptides derived from the SOST sequence and determining binding by the anti-SOST antibody.
- the open reading frame encoding SOST is fragmented either randomly or by specific genetic constructions and the reactivity of the expressed fragments of SOST with the antibody to be tested is determined.
- the gene fragments may, for example, be produced by PCR and then transcribed and translated into protein in vitro, in the presence of radioactive amino acids. The binding of the antibody to the radioactively labeled SOST fragments is then determined by immunoprecipitation and gel electrophoresis.
- Certain epitopes can also be identified by using large libraries of random peptide sequences displayed on the surface of phage particles (phage libraries). Alternatively, a defined library of
- overlapping peptide fragments can be tested for binding to the test antibody in simple binding assays.
- mutagenesis of an antigen binding domain can be performed to identify residues required, sufficient, and/or necessary for epitope binding.
- domain swapping experiments can be performed using a mutant SOST in which various fragments of the SOST protein have been replaced (swapped) with sequences from SOST from another species (e.g., mouse), or a closely related, but antigenically distinct protein.
- Yet another method which can be used to characterize an anti-SOST antibody is to use competition assays with other antibodies known to bind to the same antigen, i.e., various fragments on SOST, to determine if the anti-SOST antibody binds to the same epitope as other antibodies.
- Competition assays are well known to those of skill in the art.
- the antibody of the present invention can be produced by proteolytic or other degradation of the antibodies, by recombinant methods (i.e., single or fusion polypeptides) as described above or by chemical synthesis.
- Polypeptides of the antibodies, especially shorter polypeptides up to about 50 amino acids, are conveniently made by chemical synthesis. Methods of chemical synthesis are known in the art and are commercially available.
- an antibody could be produced by an automated polypeptide synthesizer employing the solid phase method. See also, U.S. Pat. Nos. 5,807,715; 4,816,567; and 6,331 ,415.
- the antibodies can be made recombinantly using procedures that are well known in the art.
- a polynucleotide comprises a sequence encoding the heavy chain and/or the light chain variable regions of antibody DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, and DM34.
- the sequence encoding the antibody of interest may be maintained in a vector in a host cell and the host cell can then be expanded and frozen for future use. Vectors (including expression vectors) and host cells are further described herein.
- the invention also encompasses scFv of antibodies of this invention.
- Single chain variable region fragments are made by linking light and/or heavy chain variable regions by using a short linking peptide (Bird et al., Science 242:423-426, 1988).
- An example of a linking peptide is (GGGGS ⁇ (SEQ ID NO: 97), which bridges approximately 3.5 nm between the carboxy terminus of one variable region and the amino terminus of the other variable region.
- Linkers of other sequences have been designed and used (Bird et al., 1988, supra). Linkers should be short, flexible polypeptides and preferably comprised of less than about 20 amino acid residues.
- Linkers can in turn be modified for additional functions, such as attachment of drugs or attachment to solid supports.
- the single chain variants can be produced either recombinantly or synthetically.
- an automated synthesizer can be used for synthetic production of scFv.
- a suitable plasmid containing polynucleotide that encodes the scFv can be introduced into a suitable host cell, either eukaryotic, such as yeast, plant, insect or mammalian cells, or prokaryotic, such as E. coli.
- Polynucleotides encoding the scFv of interest can be made by routine manipulations such as ligation of polynucleotides.
- the resultant scFv can be isolated using standard protein purification techniques known in the art.
- Diabodies are bivalent, bispecific antibodies in which heavy chain variable (VH) and light chain variable (VL) domains are expressed on a single polypeptide chain, but using a linker that is too short to allow for pairing between the two domains on the same chain, thereby forcing the domains to pair with complementary domains of another chain and creating two antigen binding sites (see e.g., Holliger, P., et al., Proc. Natl. Acad Sci. USA 90:6444-6448, 1993; Poljak, R. J., et al. , Structure 2:1 121 -1 123, 1994).
- VH heavy chain variable
- VL light chain variable
- bispecific antibodies monoclonal antibodies that have binding specificities for at least two different antigens
- Methods for making bispecific antibodies are known in the art (see, e.g., Suresh et al., Methods in Enzymology 121 :210, 1986).
- the recombinant production of bispecific antibodies was based on the coexpression of two immunoglobulin heavy chain-light chain pairs, with the two heavy chains having different specificities (Millstein and Cuello, Nature 305, 537-539, 1983).
- antibody variable domains with the desired binding specificities are fused to immunoglobulin constant region sequences.
- the fusion preferably is with an immunoglobulin heavy chain constant region, comprising at least part of the hinge, CH2 and CH3 regions. It is preferred to have the first heavy chain constant region (CH1 ), containing the site necessary for light chain binding, present in at least one of the fusions.
- DNAs encoding the immunoglobulin heavy chain fusions and, if desired, the immunoglobulin light chain are inserted into separate expression vectors, and are cotransfected into a suitable host organism.
- the bispecific antibodies are composed of a hybrid immunoglobulin heavy chain with a first binding specificity in one arm, and a hybrid immunoglobulin heavy chain-light chain pair (providing a second binding specificity) in the other arm.
- This asymmetric structure with an immunoglobulin light chain in only one half of the bispecific molecule, facilitates the separation of the desired bispecific compound from unwanted immunoglobulin chain combinations. This approach is described in PCT Publication No. WO 94/04690.
- the bispecific antibodies are composed of amino acid modification in the first hinge region in one arm, and the substituted/replaced amino acid in the first hinge region has an opposite charge to the corresponding amino acid in the second hinge region in another arm.
- the CH3 region in one or both arms may also include one or more amino acid modification(s). This approach is described in PCT Publication No. WO201 1/143545.
- the bispecific antibodies can be generated using a glutamine-containing peptide tag engineered to the antibody directed to an epitope (e.g., SOST) in one arm and another peptide tag (e.g., a Lys-containing peptide tag or a reactive endogenous Lys) engineered to a second antibody directed to a second epitope in another arm in the presence of transglutaminase.
- an epitope e.g., SOST
- another peptide tag e.g., a Lys-containing peptide tag or a reactive endogenous Lys
- the bispecific antibody has a first binding specificity to SOST in one arm, and a second binding specificity to DKK-1 (Dickkopf 1 ) in the other arm.
- Heteroconjugate antibodies comprising two covalently joined antibodies, are also within the scope of the invention. Such antibodies have been used to target immune system cells to unwanted cells (U.S. Pat. No. 4,676,980), and for treatment of HIV infection (PCT Publication Nos. WO 91/00360 and WO 92/200373; EP 03089). Heteroconjugate antibodies may be made using any convenient cross-linking methods. Suitable cross-linking agents and techniques are well known in the art, and are described in U.S. Pat. No. 4,676,980.
- Chimeric or hybrid antibodies also may be prepared in vitro using known methods of synthetic protein chemistry, including those involving cross-linking agents.
- immunotoxins may be constructed using a disulfide exchange reaction or by forming a thioether bond.
- suitable reagents for this purpose include iminothiolate and methyl-4-mercaptobutyrimidate.
- the Fey portion can be modified to avoid interaction with Fey receptor and the complement and immune systems.
- the techniques for preparation of such antibodies are described in WO 99/58572.
- the constant region may be engineered to more resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. See, for example, U.S. Pat. Nos. 5,997,867 and 5,866,692.
- the invention encompasses modifications to the antibodies and polypeptides of the invention variants shown in Table 3, including functionally equivalent antibodies which do not significantly affect their properties and variants which have enhanced or decreased activity and/or affinity.
- the amino acid sequence may be mutated to obtain an antibody with the desired binding affinity to SOST.
- Modification of polypeptides is routine practice in the art and need not be described in detail herein. Examples of modified polypeptides include polypeptides with conservative substitutions of amino acid residues, one or more deletions or additions of amino acids which do not significantly deleteriously change the functional activity, or which mature (enhance) the affinity of the polypeptide for its ligand, or use of chemical analogs.
- Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues.
- terminal insertions include an antibody with an N-terminal methionyl residue or the antibody fused to an epitope tag.
- Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the antibody of an enzyme or a polypeptide which increases the half-life of the antibody in the blood circulation.
- Substitution variants have at least one amino acid residue in the antibody molecule removed and a different residue inserted in its place.
- the sites of greatest interest for substitutional mutagenesis include the hypervariable regions, but FR alterations are also contemplated.
- Conservative substitutions are shown in Table 3 under the heading of "conservative substitutions.” If such substitutions result in a change in biological activity, then more substantial changes, denominated "exemplary substitutions" in Table 3, or as further described below in reference to amino acid classes, may be introduced and the products screened.
- Substantial modifications in the biological properties of the antibody are accomplished by selecting substitutions that differ significantly in their effect on maintaining (a) the structure of the polypeptide backbone in the area of the substitution, for example, as a sheet or helical conformation, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the bulk of the side chain.
- Naturally occurring residues are divided into groups based on common side-chain properties:
- Non-conservative substitutions are made by exchanging a member of one of these classes for another class.
- cysteine residue not involved in maintaining the proper conformation of the antibody also may be substituted, generally with serine, to improve the oxidative stability of the molecule and prevent aberrant cross-linking.
- cysteine bond(s) may be added to the antibody to improve its stability, particularly where the antibody is an antibody fragment such as an Fv fragment.
- Amino acid modifications can range from changing or modifying one or more amino acids to complete redesign of a region, such as the variable region. Changes in the variable region can alter binding affinity and/or specificity. In some embodiments, no more than one to five conservative amino acid substitutions are made within a CDR domain. In other embodiments, no more than one to three conservative amino acid substitutions are made within a CDR domain. In still other embodiments, the CDR domain is CDR H3 and/or CDR L3.
- Modifications also include glycosylated and nonglycosylated polypeptides, as well as polypeptides with other post-translational modifications, such as, for example, glycosylation with different sugars, acetylation, and phosphorylation.
- Antibodies are glycosylated at conserved positions in their constant regions (Jefferis and Lund, Chem. Immunol. 65: 1 1 1 -128, 1997; Wright and Morrison, TibTECH 15:26-32, 1997).
- the oligosaccharide side chains of the immunoglobulins affect the protein's function (Boyd et al., Mol. Immunol. 32: 131 1 -1318, 1996; Wittwe and Howard, Biochem.
- Oligosaccharides may also serve to target a given glycoprotein to certain molecules based upon specific recognition structures. Glycosylation of antibodies has also been reported to affect antibody-dependent cellular cytotoxicity (ADCC).
- CHO cells with tetracycline-regulated expression of ⁇ (1 ,4)-N-acetylglucosaminyltransferase III (GnTIII), a glycosy transferase catalyzing formation of bisecting GlcNAc, was reported to have improved ADCC activity (Umana et al., Mature Biotech. 17: 176-180, 1999).
- N-linked refers to the attachment of the carbohydrate moiety to the side chain of an asparagine residue.
- the tripeptide sequences asparagine-X-serine, asparagine-X-threonine, and asparagine-X-cysteine, where X is any amino acid except proline, are the recognition sequences for enzymatic attachment of the carbohydrate moiety to the asparagine side chain.
- O-linked glycosylation refers to the attachment of one of the sugars N-acetylgalactosamine, galactose, or xylose to a hydroxyamino acid, most commonly serine or threonine, although 5-hydroxyproline or 5-hydroxylysine may also be used.
- glycosylation sites to the antibody is conveniently accomplished by altering the amino acid sequence such that it contains one or more of the above- described tripeptide sequences (for N-linked glycosylation sites).
- the alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the original antibody (for O-linked glycosylation sites).
- glycosylation pattern of antibodies may also be altered without altering the underlying nucleotide sequence. Glycosylation largely depends on the host cell used to express the antibody. Since the cell type used for expression of recombinant glycoproteins, e.g. antibodies, as potential therapeutics is rarely the native cell, variations in the glycosylation pattern of the antibodies can be expected (see, e.g. Hse et al., J. Biol. Chem. 272:9062-9070, 1997).
- factors that affect glycosylation during recombinant production of antibodies include growth mode, media formulation, culture density, oxygenation, pH, purification schemes and the like.
- Various methods have been proposed to alter the glycosylation pattern achieved in a particular host organism including introducing or overexpressing certain enzymes involved in oligosaccharide production (U.S. Pat. Nos. 5,047,335; 5,510,261 and 5,278,299).
- Glycosylation or certain types of glycosylation, can be enzymatically removed from the glycoprotein, for example, using endoglycosidase H (Endo H), N-glycosidase F, endoglycosidase F1 , endoglycosidase F2, endoglycosidase F3.
- Endo H endoglycosidase H
- N-glycosidase F N-glycosidase F
- endoglycosidase F1 endoglycosidase F2
- endoglycosidase F3 endoglycosidase F3
- the recombinant host cell can be genetically engineered to be defective in processing certain types of polysaccharides.
- Modifications include using coupling techniques known in the art, including, but not limited to, enzymatic means, oxidative substitution and chelation. Modifications can be used, for example, for attachment of labels for immunoassay. Modified polypeptides are made using established procedures in the art and can be screened using standard assays known in the art, some of which are described below and in the Examples.
- the antibody comprises a modified constant region, such as a constant region that increased potential for provoking an immune response (e.g., increased affinity to a human Fc gamma receptor such as, e.g., FcyRI, FcyRIIA, or Fcylll), is immunologically inert or partially inert, e.g., does not trigger complement mediated lysis, does not stimulate antibody-dependent cell mediated cytotoxicity (ADCC), or does not activate macrophages; or has reduced activities (compared to the unmodified antibody) in any one or more of the following: triggering complement mediated lysis, or stimulating antibody-dependent cell mediated cytotoxicity (ADCC).
- a modified constant region such as a constant region that increased potential for provoking an immune response (e.g., increased affinity to a human Fc gamma receptor such as, e.g., FcyRI, FcyRIIA, or Fcylll)
- ADCC antibody-dependent cell mediated cytotoxicity
- the constant region may be used to achieve optimal level and/or combination of effector functions. See, for example, Morgan et al., Immunology 86:319-324, 1995; Lund et al., J. Immunology 157:4963-9 157:4963-4969, 1996; Idusogie et al., J. Immunology 164:4178-4184, 2000; Tao et al., J. Immunology 143: 2595-2601 , 1989; and Jefferis et al., Immunological Reviews 163:59-76, 1998.
- the constant region is modified as described in Eur. J. Immunol., 1999, 29:2613-2624; PCT Application No.
- the Fc can be human lgG1 , human lgG2, human lgG3, or human lgG4.
- the antibodies of the present invention comprise a human heavy chain lgG2 constant region comprising the following mutations: A330P331 to S330S331 (amino acid numbering with reference to the wild type lgG2 sequence). Eur. J. Immunol., 1999, 29:2613-2624.
- the antibodies of the present invention comprises a constant region of lgG 4 comprising the following mutations (Armour et al., Molecular Immunology 40 585-593, 2003): E233F234L235 to P233V234A235 (lgG4Ac), in which the numbering is with reference to wild type lgG4.
- the Fc is human lgG4 E233F234L235 to P233V234A235 with deletion G236 (lgG4Ab).
- the Fc is any human lgG4 Fc (lgG4, lgG4Ab or lgG4Ac) containing hinge stabilizing mutation S228 to P228 (Aalberse et al., Immunology 105, 9-19, 2002).
- the Fc can be aglycosylated Fc.
- the constant region is aglycosylated for N-linked glycosylation.
- the constant region is aglycosylated for N-linked glycosylation by mutating the glycosylated amino acid residue or flanking residues that are part of the N- glycosylation recognition sequence in the constant region.
- N-glycosylation site N297 may be mutated to A, Q, K, or H. See, Tao et al., J. Immunology 143: 2595- 2601 , 1989; and Jefferis et al., Immunological Reviews 163:59-76, 1998.
- the constant region may be aglycosylated for N-linked glycosylation enzymatically (such as removing carbohydrate by enzyme PNGase), or by expression in a glycosylation deficient host cell.
- Other antibody modifications include antibodies that have been modified as described in PCT Publication No. WO 99/58572.
- These antibodies comprise, in addition to a binding domain directed at the target molecule, an effector domain having an amino acid sequence substantially homologous to all or part of a constant region of a human immunoglobulin heavy chain. These antibodies are capable of binding the target molecule without triggering significant complement dependent lysis, or cell-mediated destruction of the target.
- the effector domain is capable of specifically binding FcRn and/or FcYRIIb. These are typically based on chimeric domains derived from two or more human immunoglobulin heavy chain C H 2 domains. Antibodies modified in this manner are particularly suitable for use in chronic antibody therapy, to avoid inflammatory and other adverse reactions to conventional antibody therapy.
- a modification or mutation may also be made in a framework region or constant region to increase the half-life of an SOST antibody. See, e.g., PCT Publication No. WO 00/09560.
- a mutation in a framework region or constant region can also be made to alter the immunogenicity of the antibody, to provide a site for covalent or non-covalent binding to another molecule, or to alter such properties as complement fixation, FcR binding and antibody-dependent cell-mediated cytotoxicity.
- a single antibody may have mutations in any one or more of the CDRs or framework regions of the variable domain or in the constant region.
- VH and VL sequences can be mutated to match those found naturally in germline VH and V L sequences.
- the amino acid sequences of the framework regions in the V H and VL sequences can be mutated to match the germline sequences to reduce the risk of immunogenicity when the antibody is administered.
- Germline DNA sequences for human VH and VL genes are known in the art (see e.g., the "Vbase” human germline sequence database; see also Kabat, E. A., et al., 1991 , Sequences of Proteins of Immunological Interest, Fifth Edition, U.S. Department of Health and Human Services, NIH Publication No. 91 -3242; Tomlinson et al. , J. Mol. Biol. 227:776-798, 1992; and Cox et al., Eur. J. Immunol. 24:827-836, 1994).
- Another type of amino acid substitution that may be made is to remove potential proteolytic sites in the antibody. Such sites may occur in a CDR or framework region of a variable domain or in the constant region of an antibody. Substitution of cysteine residues and removal of proteolytic sites may decrease the risk of heterogeneity in the antibody product and thus increase its homogeneity.
- Another type of amino acid substitution is to eliminate asparagine-glycine pairs, which form potential deamidation sites, by altering one or both of the residues.
- the C-terminal lysine of the heavy chain of an anti-SOST antibody of the invention can be cleaved.
- the heavy and light chains of the SOST antibodies may optionally include a signal sequence.
- DNA fragments encoding the VH and VL segments of the present invention are obtained, these DNA fragments can be further manipulated by standard
- variable region genes are operatively linked to another DNA fragment encoding another protein, such as an antibody constant region or a flexible linker.
- operatively linked is intended to mean that the two DNA fragments are joined such that the amino acid sequences encoded by the two DNA fragments remain in-frame.
- the isolated DNA encoding the VH region can be converted to a full-length heavy chain gene by operatively linking the VH-encoding DNA to another DNA molecule encoding heavy chain constant regions (CH1 , CH2 and CH3).
- CH1 , CH2 and CH3 The sequences of human heavy chain constant region genes are known in the art (see e.g., Kabat, E. A., et al., 1991 , Sequences of Proteins of Immunological Interest, Fifth Edition, U.S.
- the heavy chain constant region can be an lgG1 , lgG2, lgG3, lgG4, IgA, IgE, IgM or IgD constant region, but most preferably is an lgG1 or lgG2 constant region.
- the IgG constant region sequence can be any of the various alleles or allotypes known to occur among different individuals, such as Gm(1 ), Gm(2), Gm(3), and
- the V H -encoding DNA can be operatively linked to another DNA molecule encoding only the heavy chain CH1 constant region.
- the CH1 heavy chain constant region may be derived from any of the heavy chain genes.
- the isolated DNA encoding the VL region can be converted to a full-length light chain gene (as well as a Fab light chain gene) by operatively linking the VL-encoding DNA to another DNA molecule encoding the light chain constant region, CL.
- the sequences of human light chain constant region genes are known in the art (see e.g., Kabat, E.
- the light chain constant region can be a kappa or lambda constant region.
- the kappa constant region may be any of the various alleles known to occur among different individuals, such as lnv(1 ), lnv(2), and lnv(3).
- the lambda constant region may be derived from any of the three lambda genes.
- the VH- and VL-encoding DNA fragments are operatively linked to another fragment encoding a flexible linker, e.g., encoding the amino acid sequence (Gly 4 -Ser) 3 , (SEQ ID NO: 98) such that the VH and VL sequences can be expressed as a contiguous single-chain protein, with the VL and VH regions joined by the flexible linker (See e.g., Bird et al., 1988, Science 242:423-426; Huston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., 1990, Nature 348:552- 554.
- a flexible linker e.g., encoding the amino acid sequence (Gly 4 -Ser) 3 , (SEQ ID NO: 98) such that the VH and VL sequences can be expressed as a contiguous single-chain protein, with the VL and VH regions joined
- the single chain antibody may be monovalent, if only a single VH and VL are used, bivalent, if two VH and VL are used, or polyvalent, if more than two VH and VL are used. Bispecific or polyvalent antibodies may be generated that bind specifically to SOST and to another molecule.
- a fusion antibody or immunoadhesin may be made that comprises all or a portion of an anti-SOST antibody of the invention linked to another polypeptide.
- only the variable domains of the SOST antibody are linked to the polypeptide.
- the VH domain of an SOST antibody is linked to a first polypeptide, while the VL domain of an SOST antibody is linked to a second polypeptide that associates with the first polypeptide in a manner such that the VH and VL domains can interact with one another to form an antigen binding site.
- the VH domain is separated from the VL domain by a linker such that the VH and VL domains can interact with one another.
- VH-linker- VL antibody is then linked to the polypeptide of interest.
- fusion antibodies can be created in which two (or more) single-chain antibodies are linked to one another. This is useful if one wants to create a divalent or polyvalent antibody on a single polypeptide chain, or if one wants to create a bispecific antibody.
- modified antibodies may be prepared using SOST antibody encoding nucleic acid molecules.
- SOST antibody encoding nucleic acid molecules.
- “Kappa bodies” III et al., Protein Eng. 10:949-57, 1997), “Minibodies” (Martin et al., EMBO J., 13:5303-9, 1994), “Diabodies” (Holliger et al., Proc. Natl. Acad. Sci. USA 90:6444-6448, 1993), or
- affinity matured antibodies can be produced by procedures known in the art (Marks et al., Bio/Technology, 10:779-783, 1992; Barbas et al., Proc Nat. Acad. Sci, USA 91 :3809- 3813, 1994; Schier et al., Gene, 169: 147-155, 1995; Yelton et al., J. Immunol., 155:1994-2004, 1995; Jackson et al., J. Immunol., 154(7):3310-9, 1995, Hawkins et al., J. Mol. Biol., 226:889-896, 1992; and PCT Publication No. WO2004/058184).
- library scanning mutagenesis One way of characterizing a CDR of an antibody and/or altering (such as improving) the binding affinity of a polypeptide, such as an antibody, termed "library scanning mutagenesis".
- library scanning mutagenesis works as follows. One or more amino acid positions in the CDR are replaced with two or more (such as 3, 4, 5, 6, 7, 8, 9, 10, 1 1 , 12, 13, 14, 15, 16, 17, 18, 19, or 20) amino acids using art recognized methods. This generates small libraries of clones (in some embodiments, one for every amino acid position that is analyzed), each with a complexity of two or more members (if two or more amino acids are substituted at every position).
- the library also includes a clone comprising the native (unsubstituted) amino acid.
- a small number of clones, e.g., about 20-80 clones (depending on the complexity of the library), from each library are screened for binding affinity to the target polypeptide (or other binding target), and candidates with increased, the same, decreased, or no binding are identified.
- Methods for determining binding affinity are well-known in the art. Binding affinity may be determined using BiacoreTM surface plasmon resonance analysis, which detects differences in binding affinity of about 2-fold or greater. BiacoreTM is particularly useful when the starting antibody already binds with a relatively high affinity, for example a K D of about 10 nM or lower. Screening using BiacoreTM surface plasmon resonance is described in the Examples, herein.
- every amino acid position in a CDR is replaced (in some embodiments, one at a time) with all 20 natural amino acids using art recognized mutagenesis methods (some of which are described herein). This generates small libraries of clones (in some embodiments, one for every amino acid position that is analyzed), each with a complexity of 20 members (if all 20 amino acids are substituted at every position).
- the library to be screened comprises substitutions in two or more positions, which may be in the same CDR or in two or more CDRs.
- the library may comprise substitutions in two or more positions in one CDR.
- the library may comprise substitution in two or more positions in two or more CDRs.
- the library may comprise substitution in 3, 4, 5, or more positions, said positions found in two, three, four, five or six CDRs.
- the substitution may be prepared using low redundancy codons. See, e.g., Table 2 of Balint et al., Gene 137(1 ): 109-18, 1993.
- the CDR may be CDRH3 and/or CDRL3.
- the CDR may be one or more of CDRL1 , CDRL2, CDRL3, CDRH1 , CDRH2, and/or CDRH3.
- the CDR may be a Kabat CDR, a Chothia CDR, or an extended CDR.
- Candidates with improved binding may be sequenced, thereby identifying a CDR substitution mutant which results in improved affinity (also termed an "improved" substitution).
- Candidates that bind may also be sequenced, thereby identifying a CDR substitution which retains binding.
- candidates each comprising an amino acid substitution at one or more position of one or more CDR
- candidates with improved binding are also useful for the design of a second library containing at least the original and substituted amino acid at each improved CDR position (i.e., amino acid position in the CDR at which a substitution mutant showed improved binding).
- Preparation, and screening or selection of this library is discussed further below.
- Library scanning mutagenesis also provides a means for characterizing a CDR, in so far as the frequency of clones with improved binding, the same binding, decreased binding or no binding also provide information relating to the importance of each amino acid position for the stability of the antibody-antigen complex. For example, if a position of the CDR retains binding when changed to all 20 amino acids, that position is identified as a position that is unlikely to be required for antigen binding. Conversely, if a position of CDR retains binding in only a small percentage of substitutions, that position is identified as a position that is important to CDR function.
- the library scanning mutagenesis methods generate information regarding positions in the CDRs that can be changed to many different amino acids (including all 20 amino acids), and positions in the CDRs which cannot be changed or which can only be changed to a few amino acids.
- Candidates with improved affinity may be combined in a second library, which includes the improved amino acid, the original amino acid at that position, and may further include additional substitutions at that position, depending on the complexity of the library that is desired, or permitted using the desired screening or selection method.
- adjacent amino acid position can be randomized to at least two or more amino acids. Randomization of adjacent amino acids may permit additional conformational flexibility in the mutant CDR, which may in turn, permit or facilitate the introduction of a larger number of improving mutations.
- the library may also comprise substitution at positions that did not show improved affinity in the first round of screening.
- the second library is screened or selected for library members with improved and/or altered binding affinity using any method known in the art, including screening using BiacoreTM surface plasmon resonance analysis, and selection using any method known in the art for selection, including phage display, yeast display, and ribosome display.
- fusion proteins comprising one or more fragments or regions from the antibodies of this invention.
- a fusion polypeptide is provided that comprises at least 10 contiguous amino acids of the variable light chain region shown in SEQ ID NOs: 3, 7, 1 1 , 15, 16, 17, 18, 19, 20, 22, 23, 24, 28, 29, 30, 31 , 32, 33, and 41 and/or at least 10 amino acids of the variable heavy chain region shown in SEQ ID NOs: 2, 6, 10, 14, 21 , 25, 34, 35, 36, 37, 38, 39, and 40.
- a fusion polypeptide comprises at least about 10, at least about 15, at least about 20, at least about 25, or at least about 30 contiguous amino acids of the variable light chain region and/or at least about 10, at least about 15, at least about 20, at least about 25, or at least about 30 contiguous amino acids of the variable heavy chain region.
- the fusion polypeptide comprises a light chain variable region and/or a heavy chain variable region, as shown in any of the sequence pairs selected from among SEQ ID NOs: 3 and 2, 7 and 6, 1 1 and 10, 15 and 14, 16 and 14, 17 and 14, 18 and 14, 19 and 14, 20 and 14, 16 and 21 , 20 and 21 , 22 and 14, 23 and 21 , 24 and 25, 28 and 25, 29 and 25, 23 and 25, 15 and 25, 30 and 25, 31 and 25, 32 and 25, 33 and 25, 24 and 34, 29 and 34, 24 and 35, 24 and 36, 29 and 36, 24 and 37, 24 and 38, 24 and 39, 24 and 40, 29 and 14, 23 and 14, 41 and 14, 29 and 21 , 17 and 21 , and 41 and 21 .
- the fusion polypeptide comprises one or more CDR(s).
- the fusion polypeptide comprises CDR H3 (VH CDR3) and/or CDR L3 (VL CDR3).
- a fusion protein contains one or more antibodies and another amino acid sequence to which it is not attached in the native molecule, for example, a heterologous sequence or a homologous sequence from another region.
- heterologous sequences include, but are not limited to a "tag" such as a FLAG tag or a 6His tag. Tags are well known in the art.
- a fusion polypeptide can be created by methods known in the art, for example, synthetically or recombinantly.
- the fusion proteins of this invention are made by preparing an expressing a polynucleotide encoding them using recombinant methods described herein, although they may also be prepared by other means known in the art, including, for example, chemical synthesis.
- compositions comprising antibodies conjugated (for example, linked) to an agent that facilitate coupling to a solid support (such as biotin or avidin).
- a solid support such as biotin or avidin.
- Conjugation generally refers to linking these components as described herein.
- the linking (which is generally fixing these components in proximate association at least for administration) can be achieved in any number of ways. For example, a direct reaction between an agent and an antibody is possible when each possesses a substituent capable of reacting with the other.
- a nucleophilic group such as an amino or sulfhydryl group
- a carbonyl-containing group such as an anhydride or an acid halide, or with an alkyl group containing a good leaving group (e.g., a halide) on the other.
- Carriers can be active and/or inert.
- Examples of well-known carriers include polypropylene, polystyrene, polyethylene, dextran, nylon, amylases, glass, natural and modified celluloses, polyacrylamides, agaroses, and magnetite.
- the nature of the carrier can be either soluble or insoluble for purposes of the invention. Those skilled in the art will know of other suitable carriers for binding antibodies, or will be able to ascertain such, using routine experimentation.
- the carrier comprises a moiety that targets the myocardium.
- This invention also provides a conjugate (or immunoconjugate) of the anti-SOST antibody as described herein, or the antigen binding fragment thereof, wherein the antibody or the antigen binding fragment is conjugate to a pharmaceutical agent for targeted immunotherapy (e.g., antibody-drug conjugates).
- a pharmaceutical agent for targeted immunotherapy e.g., antibody-drug conjugates.
- the pharmaceutical agents that can be conjugated to the anti-SOST antibodies or the antigen binding fragments of the present invention include, but are not limited to, chemotherapeutic agents (e.g., cytotoxic agents), immunomodulating agents, imaging agents (e.g., labeling agents), therapeutic proteins, biopolymers, or oligonucleotides.
- the agent is an imaging agent (e.g., a fluorophore or a
- PET Pulsitron Emission Tomography
- SPECT Single-Photon Emission
- Computed Tomography label such as fluorescein, rhodamine, lanthanide phosphors, and their derivatives thereof and various isotopes.
- fluorophores examples include, but are not limited to, fluorescein
- FITC isothiocyanate
- FAM fluorescein amidite
- eosin carboxyfluorescein
- Alexa Fluor ® e.g., Alexa 350, 405, 430, 488, 500, 514, 532, 546, 555, 568, 594, 610, 633, 647, 660, 680, 700, or 750
- Alexa Fluor ® e.g., Alexa 350, 405, 430, 488, 500, 514, 532, 546, 555, 568, 594, 610, 633, 647, 660, 680, 700, or 750
- TAMRA carboxytetramethylrhodamine
- TMR tetramethylrhodamine
- SR sulforhodamine
- therapeutic or diagnostic radioisotopes or other labels are provided.
- radioisotope or other labels examples include, but are not limited to, 3 H, 11 C, 13 N, 14 C, 15 N, 15 O,
- the invention also provides polynucleotides encoding any of the antibodies (e.g., antagonistic antibodies), including antibody fragments and modified antibodies described herein, such as, e.g., antibodies having impaired effector function. Accordingly, the invention provides polynucleotides or compositions, including pharmaceutical compositions, comprising polynucleotides, encoding any of the following: DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, or DM34. In some embodiments, the antibodies or any fragment
- the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 4 and SEQ ID NO: 5 below:
- the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 8 and SEQ ID NO: 9 below
- the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 12 and SEQ ID NO: 13 below
- the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 27 and SEQ ID NO: 26 below
- the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 95 and SEQ ID NO: 96 below
- Polynucleotides complementary to any such sequences are also encompassed by the present invention.
- Polynucleotides may be single-stranded (coding or antisense) or double-stranded, and may be DNA (genomic, cDNA or synthetic) or RNA molecules.
- RNA molecules include HnRNA molecules, which contain introns and correspond to a DNA molecule in a one-to-one manner, and mRNA molecules, which do not contain introns. Additional coding or non-coding sequences may, but need not, be present within a polynucleotide of the present invention, and a polynucleotide may, but need not, be linked to other molecules and/or support materials.
- Representative materials of the present invention were deposited in the American Type Culture Collection (ATCC) on March 28, 2012.
- Vector having ATCC Accession No. PTA-12710 is a polynucleotide encoding a humanized SOST antibody heavy chain variable region
- vector having ATCC Accession No. PTA-1271 1 is a polynucleotide encoding a humanized SOST antibody light chain variable region.
- the deposits were made under the provisions of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purpose of Patent Procedure and Regulations thereunder (Budapest Treaty). This assures maintenance of a viable culture of the deposit for 30 years from the date of deposit.
- the deposit will be made available by ATCC under the terms of the Budapest Treaty, and subject to an agreement between Pfizer, Inc./Rinat Neuroscience Corp. and ATCC, which assures permanent and unrestricted availability of the progeny of the culture of the deposit to the public upon issuance of the pertinent U.S. patent or upon laying open to the public of any U.S. or foreign patent application, whichever comes first, and assures availability of the progeny to one determined by the U.S. Commissioner of Patents and Trademarks to be entitled thereto according to 35 U.S.C. Section 122 and the Commissioner's rules pursuant thereto (including 37 C.F.R. Section 1 .14 with particular reference to 886 OG 638).
- Polynucleotides may comprise a native sequence (i.e., an endogenous sequence that encodes an antibody or a fragment thereof) or may comprise a variant of such a sequence.
- Polynucleotide variants contain one or more substitutions, additions, deletions and/or insertions such that the immunoreactivity of the encoded polypeptide is not diminished, relative to a native immunoreactive molecule.
- the effect on the immunoreactivity of the encoded polypeptide may generally be assessed as described herein.
- Variants preferably exhibit at least about 70% identity, more preferably, at least about 80% identity, yet more preferably, at least about 90% identity, and most preferably, at least about 95% identity to a polynucleotide sequence that encodes a native antibody or a fragment thereof.
- Two polynucleotide or polypeptide sequences are said to be “identical” if the sequence of nucleotides or amino acids in the two sequences is the same when aligned for maximum correspondence as described below. Comparisons between two sequences are typically performed by comparing the sequences over a comparison window to identify and compare local regions of sequence similarity.
- a “comparison window” as used herein refers to a segment of at least about 20 contiguous positions, usually 30 to about 75, or 40 to about 50, in which a sequence may be compared to a reference sequence of the same number of contiguous positions after the two sequences are optimally aligned.
- the "percentage of sequence identity” is determined by comparing two optimally aligned sequences over a window of comparison of at least 20 positions, wherein the portion of the polynucleotide or polypeptide sequence in the comparison window may comprise additions or deletions (i.e., gaps) of 20 percent or less, usually 5 to 15 percent, or 10 to 12 percent, as compared to the reference sequences (which does not comprise additions or deletions) for optimal alignment of the two sequences.
- the percentage is calculated by determining the number of positions at which the identical nucleic acid bases or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the reference sequence (i.e. the window size) and multiplying the results by 100 to yield the percentage of sequence identity.
- Variants may also, or alternatively, be substantially homologous to a native gene, or a portion or complement thereof.
- Such polynucleotide variants are capable of hybridizing under moderately stringent conditions to a naturally occurring DNA sequence encoding a native antibody (or a complementary sequence).
- Suitable “moderately stringent conditions” include prewashing in a solution of 5 X SSC, 0.5% SDS, 1 .0 mM EDTA (pH 8.0); hybridizing at 50°C-65°C, 5 X SSC, overnight; followed by washing twice at 65°C for 20 minutes with each of 2X, 0.5X and 0.2X SSC containing 0.1 % SDS.
- highly stringent conditions or “high stringency conditions” are those that: (1 ) employ low ionic strength and high temperature for washing, for example 0.015 M sodium chloride/0.0015 M sodium citrate/0.1 % sodium dodecyl sulfate at 50°C; (2) employ during hybridization a denaturing agent, such as formamide, for example, 50% (v/v) formamide with 0.1 % bovine serum albumin/0.1 % Ficoll/0.1 % polyvinylpyrrolidone/50 mM sodium phosphate buffer at pH 6.5 with 750 mM sodium chloride, 75 mM sodium citrate at 42°C; or (3) employ 50% formamide, 5 x SSC (0.75 M NaCI, 0.075 M sodium citrate), 50 mM sodium phosphate (pH 6.8), 0.1 % sodium pyrophosphate, 5 x Denhardt's solution, sonicated salmon sperm DNA (50 pg/ml), 0.1 % SDS,
- formamide for example
- Alleles are endogenous genes that are altered as a result of one or more mutations, such as deletions, additions and/or substitutions of nucleotides.
- the resulting mRNA and protein may, but need not, have an altered structure or function. Alleles may be identified using standard techniques (such as hybridization, amplification and/or database sequence comparison).
- polynucleotides of this invention can be obtained using chemical synthesis, recombinant methods, or PCR. Methods of chemical polynucleotide synthesis are well known in the art and need not be described in detail herein. One of skill in the art can use the sequences provided herein and a commercial DNA synthesizer to produce a desired DNA sequence.
- a polynucleotide comprising a desired sequence can be inserted into a suitable vector, and the vector in turn can be introduced into a suitable host cell for replication and amplification, as further discussed herein.
- Polynucleotides may be inserted into host cells by any means known in the art. Cells are transformed by introducing an exogenous polynucleotide by direct uptake, endocytosis, transfection, F-mating or electroporation. Once introduced, the exogenous polynucleotide can be maintained within the cell as a non-integrated vector (such as a plasm id) or integrated into the host cell genome.
- the polynucleotide so amplified can be isolated from the host cell by methods well known within the art. See, e.g., Sambrook et al., 1989.
- PCR allows reproduction of DNA sequences.
- PCR technology is well known in the art and is described in U.S. Patent Nos. 4,683,195, 4,800, 159, 4,754,065 and 4,683,202, as well as PCR: The Polymerase Chain Reaction, Mullis et al. eds., Birkauswer Press, Boston, 1994.
- RNA can be obtained by using the isolated DNA in an appropriate vector and inserting it into a suitable host cell. When the cell replicates and the DNA is transcribed into RNA, the RNA can then be isolated using methods well known to those of skill in the art, as set forth in Sambrook et al., 1989, supra, for example.
- Suitable cloning vectors may be constructed according to standard techniques, or may be selected from a large number of cloning vectors available in the art. While the cloning vector selected may vary according to the host cell intended to be used, useful cloning vectors will generally have the ability to self-replicate, may possess a single target for a particular restriction endonuclease, and/or may carry genes for a marker that can be used in selecting clones containing the vector.
- Suitable examples include plasmids and bacterial viruses, e.g., pUC18, pUC19, Bluescript (e.g., pBS SK+) and its derivatives, mp18, mp19, pBR322, pMB9, ColE1 , pCR1 , RP4, phage DNAs, and shuttle vectors such as pSA3 and pAT28.
- Bluescript e.g., pBS SK+
- shuttle vectors such as pSA3 and pAT28.
- Expression vectors are further provided.
- Expression vectors generally are replicable polynucleotide constructs that contain a polynucleotide according to the invention. It is implied that an expression vector must be replicable in the host cells either as episomes or as an integral part of the chromosomal DNA. Suitable expression vectors include but are not limited to plasmids, viral vectors, including adenoviruses, adeno-associated viruses, retroviruses, cosmids, and expression vector(s) disclosed in PCT Publication No. WO 87/04462.
- Vector components may generally include, but are not limited to, one or more of the following: a signal sequence; an origin of replication; one or more marker genes; suitable transcriptional controlling elements (such as promoters, enhancers and terminator). For expression (i.e., translation), one or more translational controlling elements are also usually required, such as ribosome binding sites, translation initiation sites, and stop codons.
- the vectors containing the polynucleotides of interest can be introduced into the host cell by any of a number of appropriate means, including electroporation, transfection employing calcium chloride, rubidium chloride, calcium phosphate, DEAE- dextran, or other substances; microprojectile bombardment; lipofection; and infection (e.g., where the vector is an infectious agent such as vaccinia virus).
- electroporation employing calcium chloride, rubidium chloride, calcium phosphate, DEAE- dextran, or other substances
- microprojectile bombardment e.g., where the vector is an infectious agent such as vaccinia virus.
- infection e.g., where the vector is an infectious agent such as vaccinia virus.
- the choice of introducing vectors or polynucleotides will often depend on features of the host cell.
- the invention also provides host cells comprising any of the polynucleotides described herein. Any host cells capable of over-expressing heterologous DNAs can be used for the purpose of isolating the genes encoding the antibody, polypeptide or protein of interest.
- mammalian host cells include but not limited to COS, HeLa, and CHO cells. See also PCT Publication No. WO 87/04462.
- Suitable non-mammalian host cells include prokaryotes (such as E. coli or B. subtillis) and yeast (such as S. cerevisae, S. pombe; or K. lactis).
- the host cells express the cDNAs at a level of about 5 fold higher, more preferably, 10 fold higher, even more preferably, 20 fold higher than that of the corresponding endogenous antibody or protein of interest, if present, in the host cells.
- Screening the host cells for a specific binding to SOST or an SOST domain is effected by an immunoassay or FACS.
- a cell overexpressing the antibody or protein of interest can be identified.
- An expression vector can be used to direct expression of the anti-SOST antibody of the present invention.
- One skilled in the art is familiar with administration of expression vectors to obtain expression of an exogenous protein in vivo. See, e.g., U.S. Patent Nos. 6,436,908; 6,413,942; and 6,376,471.
- Administration of expression vectors includes local or systemic administration, including injection, oral administration, particle gun or catheterized administration, and topical administration.
- the expression vector is administered directly to the sympathetic trunk or ganglion, or into a coronary artery, atrium, ventrical, or pericardium.
- Targeted delivery of therapeutic compositions containing an expression vector, or subgenomic polynucleotides can also be used.
- Receptor-mediated DNA delivery techniques are described in, for example, Findeis et al., Trends Biotechnol., 1993, 1 1 :202; Chiou et al., Gene Therapeutics: Methods And Applications Of Direct Gene Transfer, J.A. Wolff, ed., 1994; Wu et al., J. Biol. Chem., 1988, 263:621 ; Wu et al., J. Biol. Chem., 1994, 269:542; Zenke et al., Proc. Natl. Acad. Sci.
- compositions containing a polynucleotide are administered in a range of about 100 ng to about 200 mg of DNA for local administration in a gene therapy protocol. Concentration ranges of about 500 ng to about 50 mg, about 1 g to about 2 mg, about 5 g to about 500 g, and about 20 g to about 100 g of DNA can also be used during a gene therapy protocol.
- the therapeutic polynucleotides and polypeptides can be delivered using gene delivery vehicles.
- the gene delivery vehicle can be of viral or non-viral origin (see generally, Jolly, Cancer Gene Therapy, 1994, 1 :51 ; Kimura, Human Gene Therapy, 1994, 5:845; Connelly, Human Gene Therapy, 1995, 1 : 185; and Kaplitt, Nature Genetics, 1994, 6: 148). Expression of such coding sequences can be induced using endogenous mammalian or heterologous promoters. Expression of the coding sequence can be either constitutive or regulated.
- Viral-based vectors for delivery of a desired polynucleotide and expression in a desired cell are well known in the art.
- Exemplary viral-based vehicles include, but are not limited to, recombinant retroviruses (see, e.g., PCT Publication Nos. WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/1 1230; WO 93/10218; WO 91/02805; U.S. Patent Nos. 5, 219,740 and 4,777, 127; GB Patent No. 2,200,651 ; and EP Patent No.
- alphavirus-based vectors e.g., Sindbis virus vectors, Semliki forest virus (ATCC VR-67; ATCC VR-1247), Ross River virus (ATCC VR-373; ATCC VR-1246) and Venezuelan equine encephalitis virus (ATCC VR-923; ATCC VR-1250; ATCC VR 1249; ATCC VR-532)
- AAV adeno-associated virus
- Non-viral delivery vehicles and methods can also be employed, including, but not limited to, polycationic condensed DNA linked or unlinked to killed adenovirus alone (see, e.g., Curiel, Hum. Gene Then, 1992, 3: 147); ligand-linked DNA (see, e.g., Wu, J. Biol. Chem., 1989, 264: 16985); eukaryotic cell delivery vehicles cells (see, e.g., U.S. Patent No. 5,814,482; PCT Publication Nos. WO 95/07994; WO 96/17072; WO 95/30763; and WO 97/42338) and nucleic charge neutralization or fusion with cell membranes. Naked DNA can also be employed.
- Exemplary naked DNA introduction methods are described in PCT Publication No. WO 90/1 1092 and U.S. Patent No. 5,580,859.
- Liposomes that can act as gene delivery vehicles are described in U.S. Patent No. 5,422, 120; PCT Publication Nos. WO 95/13796; WO 94/23697; WO 91/14445; and EP 0524968. Additional approaches are described in Philip, Mol. Cell Biol., 1994, 14:241 1 , and in Woffendin, Proc. Natl. Acad. Sci., 1994, 91 : 1581 .
- the antibodies of the present invention are useful in various applications including, but are not limited to, therapeutic treatment methods and diagnostic treatment methods.
- the invention provides a method of increasing bone formation, bone mass, bone mineralization, bone volume, bone quality, bone strength, and/or bone density (e.g., in vertebral and/or non-vertebral bone) in a patient in need thereof.
- the method of increasing bone formation, bone mass, bone mineralization, bone volume, bone quality, bone strength, and/or bone density in a patient in need thereof comprises administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- the invention provides a method of inducing canonical Wnt signaling activity in a patient in need thereof.
- the method of inducing canonical Wnt signaling activity in a patient in need thereof comprises administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- the invention provides a method of treating or preventing a SOST-associated condition, disease, or disorder (e.g., a bone related disorder) that is responsive to the inhibition of SOST activity or canonical Wnt signaling.
- a SOST-associated condition, disease, or disorder e.g., a bone related disorder
- a method of treating or preventing a bone related disorder in a patient in need thereof comprising administering to the patient an effective amount of the anti- SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- a "bone related disorder” includes, but is not limited to, a bone condition/disorder/disease, a bone condition/disorder/disease associated with renal failure or cancer, and nutritional, gastrointestinal and/or hepatic associated bone conditions/disorders/diseases.
- a bone related disorder examples include, but are not limited to, osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, arthritis (e.g., rheumatoid arthritis or psoriatic arthritis), osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss (e.g., due to immobilization), metastatic bone malignancy (e.g., lytic bone metastases), multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease, hemochromatosis, celiac sprue
- arthritis e.g
- provided is a method of treating or preventing osteoporosis in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- a composition e.g., pharmaceutical composition
- osteoporosis can be treated using the anti-SOST antibodies as described herein, including but not limited to, primary osteoporosis (type 1 or postmenopausal osteoporosis; type 2 or senile osteoporosis) and secondary osteoporosis.
- secondary osteoporosis is glucocorticoid induced osteoporosis, osteoporosis induced after transplantation, osteoporosis associated with chemotherapy (i.e., chemotherapy induced osteoporosis), immobilization induced osteoporosis, osteoporosis due to mechanical unloading, or osteoporosis associated with anticonvulsant use.
- Different forms of arthritis can also be treated using the anti-SOST antibodies as described herein, including but not limited to, osteoarthritis and rheumatoid arthritis.
- patients may benefit by perilesional or intralesional injections of the subject antibodies or fragments thereof.
- the anti-SOST antibodies can be injected adjacent to or directly into an inflamed joint, thus stimulating repair of damaged bone at the site.
- any cancer that can metastasize to bone or any bone tumor/cancer can also be treated using the anti-SOST antibodies as described herein.
- certain cancers such as breast and prostate cancer
- Certain other cancers such as multiple myeloma, arise in bone marrow, and is associated with bone loss due to increased osteoclast activation in localized regions of the bone. Accordingly, reducing SOST activity by administering the subject antibodies thereof can result in an increase in osteoblast activity that serves to counteract the excessive osteoclast activity, thereby reducing the severity of the aforementioned disorders, reducing bone erosion, and inducing new bone formation in the multiple myeloma patient.
- a method of treating multiple myeloma in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- anti-SOST antibodies of the present inventions can also be used for preventing or reducing tumor burden, inhibiting or preventing tumor growth or progression, inhibiting metastasis of cancer cells or tumors, or delaying tumor growth or progression in a subject (or patient) having cancer.
- cancers include, but are not limited to, a solid cancer (such as bladder, breast, cervical, choriocarcinoma, colon, esophageal, gastric, glioblastoma, head and neck, kidney, liver, lung (e.g., Non Small Cell Lung Cancer (NSCLC)), oral, ovarian, pancreatic, prostate, and skin cancer); and a liquid cancer (such as acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML), hairy cell leukemia (HCL), T-cell prolymphocytic leukemia (T-PLL), large granular lymphocytic leukemia, multiple myeloma, and adult T-cell leukemia).
- ALL acute lymphoblastic leukemia
- CLL chronic lymphocytic leukemia
- AML acute myelogenous leukemia
- CML chronic my
- a method of preventing or reducing tumor burden in a patient in need thereof comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition (e.g., pharmaceutical composition) comprising thereof.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- SOST polypeptide e.g., SEQ ID NO: 1
- a composition e.g., pharmaceutical composition
- a method of inhibiting or preventing tumor growth or progression in a patient in need thereof comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition (e.g., pharmaceutical composition) comprising thereof.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- SOST polypeptide e.g., SEQ ID NO: 1
- a composition e.g., pharmaceutical composition
- a method of inhibiting metastasis of cancer cells or tumors comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition comprising thereof.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- SOST polypeptide e.g., SEQ ID NO: 1
- a method of inducing tumor regression in a subject in need thereof, in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition comprising thereof.
- an anti-SOST antibody e.g., the anti-SOST antibodies as described herein
- SOST polypeptide e.g., SEQ ID NO: 1
- the anti-SOST antibody can comprise (1 )(a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GX 1 TFX 2 DYWMQ, wherein X ⁇ is F or H, X 2 is T or S (SEQ ID NO: 81 ) or GXiTFX 2 DY, wherein X ⁇ is F or H, and X 2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX 2 X 3 KX 4 , wherein X is A or N, X 2 is S or K, X 3 is V or F, and X 4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SXi DYW, wherein X ⁇ is F or M (SEQ ID NO: 84); and/
- GFTFSIYAMS SEQ ID NO: 43
- I YAMS SEQ ID NO: 45
- GFTFSIY SEQ ID NO: 44
- a VH CDR2 comprising the sequence Xi ISGGDTYTYYADSVKG, wherein X ⁇ is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence X1 SSQSLLDNDGETYLN, wherein X is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51
- the antibody is DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, DM34, AMG167, or AMG785 (Romosozumab).
- Anti-SOST antibodies of the present inventions can also be used in various bone repair applications.
- the anti-SOST antibodies can retard wear debris osteolysis associated with artificial joints, accelerating the repair of bone fractures, and enhancing the incorporation of bone grafts or fixation of implants into the surrounding living bone into which they have been engrafted or implanted.
- a method of treating bone fracture in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g. , pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- a method of enhancing or strengthening implant fixation e.g., by improving and/or accelerating bone formation
- a method of enhancing or strengthening implant fixation comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
- Anti-SOST antibodies of the present invention can also be used to treat diseases in which it is desirable to promote stem cell renewal.
- diseases include, but are not limited to, diabetes (e.g., type 1 diabetes mellitus, type 2 diabetes mellitus, gestational diabetes, steroid diabetes, congenital diabetes, cystic fibrosis-related diabetes, or monogenic diabetes), chronic heart failure and various diseases of the muscle [e.g., disuse atrophy resulting, for instance, from immobilization or bed-rest); aging frailty (sarcopenia of the elderly); muscular dystrophies; cachexia associated with cancer, Acquired Immune Deficiency Syndrome (AIDS) or inflammation; protein-energy malnutrition in renal failure/uremia, and muscle wasting in obesity.
- AIDS Acquired Immune Deficiency Syndrome
- inflammatory diseases can also be treated, including, for instance, inflammatory bowel disease (e.g., Crohn's disease or ulcerative colitis).
- inflammatory bowel disease e.g., Crohn's disease or ulcerative colitis.
- Different renal diseases e.g., end stage renal disease, chronic renal disease, glomerulonephritis, tubulointerstitial nephritis, renal osteodystrophy, and IgA nephropathy
- pulmonary diseases e.g., chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and cystic fibrosis
- skin disorders including dermal and epidermal diseases.
- Examples of skin disorders that can be treated include damaged intestinal epithelium (e.g., chemotherapy induced damage), and other diseases in which it is desirable to stimulate growth and survival of the intestinal epithelium.
- Various congenital and/or hereditary bone diseases can also be treated using anti-SOST antibodies, such as fibrous dysplasia, achondroplasia, osteogenesis imperfecta, osteopetrosis (marble bone disease or osteosclerosis), hereditary multiple exotosis (osteochondromatosis), enchondromatosis (Ollier's Disease), congenital osteopenia or osteoporosis.
- a method of detecting, diagnosing, and/or monitoring a condition associated with SOST expression in a biological sample e.g., overexpression or underexpression of SOST in tissues or cells.
- the anti- SOST antibodies as described herein can be labeled with a detectable moiety such as an imaging agent and an enzyme-substrate label.
- the antibodies as described herein can also be used for in vivo diagnostic assays, such as in vivo imaging (e.g., PET or SPECT), or a staining reagent.
- a method of screening for a molecule that binds to SOST For example, a SOST molecule or fragment thereof is contacted with the anti-SOST antibody disclosed herein together with another molecule (e.g., a candidate molecule). A reduction in binding between the anti-SOST antibody and SOST is an indication that the candidate molecule binds SOST. Binding of the antibody can be detected using a variety of methods, e.g., an ELISA. Detection of binding to the anti-SOST antibody can be simplified by detectably labeling the antibody. In some methods, a molecule that exhibits binding in the initial screen is further analyzed to determine whether it inhibits SOST activity (e.g., whether the molecule activates Wnt signaling).
- SOST activity e.g., whether the molecule activates Wnt signaling
- the methods described herein further comprise a step of treating a patient in need thereof with an additional form of therapy.
- the additional form of therapy is one or more therapeutic agent, such as chemotherapeutic agents (e.g., cytotoxic agents), osteoclast activity inhibiting agents, osteoblast activity enhancing agents (e.g., bone growth anabolic agents and bone anti- resorptive agents), and dietary supplements (e.g., calcium, vitamin D, and vitamin K).
- chemotherapeutic agents e.g., cytotoxic agents
- osteoclast activity inhibiting agents e.g., osteoblast activity enhancing agents
- osteoblast activity enhancing agents e.g., bone growth anabolic agents and bone anti- resorptive agents
- dietary supplements e.g., calcium, vitamin D, and vitamin K
- Chemotherapeutic agents include, but are not limited to, a second antibody (e.g., an anti-VEGF (Vascular Endothelial Growth Factor) antibody (e.g., AVASTIN ® ), an anti-HER2 antibody (e.g., HERCEPTIN ® ), an anti-CD25 antibody, an anti-CD33 antibody, an anti-CD20 antibody (e.g., RITUXAN ® ), an anti-mucin-like glycoprotein antibody, an anti- TNF antibody, and/or an epidermal growth factor receptor (EGFR) antibody (e.g., ERBITUX ® )), an angiogenesis inhibitor, a cytotoxic agent (e.g., anthracyclines (e.g., daunorubicin, doxorubicin, epirubicin, idarubicin, valrubicin, and mitoxantrone), taxane (e.g., paclitaxel and docetaxel), do
- a method of treating multiple myeloma comprising administering to a patient need thereof an effective amount of a composition comprising the anti-SOST antibodies as described herein and one other therapeutic agent such as a chemotherapeutic agent or thalidomide or its derivative thereof (e.g., lenalidomide).
- a composition comprising the anti-SOST antibodies as described herein and one other therapeutic agent such as a chemotherapeutic agent or thalidomide or its derivative thereof (e.g., lenalidomide).
- the one other therapeutic agent is selecting from the group consisting of bortezomib (e.g., VELCADE ® ), melphalan, prednisone, doxorubicin, lenalidomide, thalidomide, prednisone, carmustine, etoposide, cisplatin, cyclophosphamide, and vincristine.
- the other therapeutic agent is bortezomib (e.g., VELCADE ® ), melphalan, or prednisone.
- the patient is relapsing or refractory to previous multiple myeloma therapy.
- Osteoblast activity enhancing agents include, but are not limited to, bone morphogenic factors designated BMP-1 to BMP-12, transforming growth factor (TGFJ- ⁇ and TGF- ⁇ family members, fibroblast growth factors (FGF)-1 to FGF-10, interleukin (IL)-1 inhibitors (including IL-1 ra, antibodies to IL-1 and antibodies to IL-1 receptors), Tumor Necrosis Factor (TNF)a inhibitors (including etanercept, adalimumab and infliximab), RANK ligand inhibitors (including soluble RANK, osteoprotegerin and antagonistic antibodies that specifically bind RANK or RANK ligand (e.g., PROLIA ® ), parathyroid hormone (e.g., FORTEO ® ), E series prostaglandins, bisphosphonates (e.g., FOSAMAX ), transforming growth factor (TGFJ- ⁇ and TGF- ⁇ family members, fibroblast growth factors (F
- IL-1 receptor antagonist suitable for such combination treatment is described in WO89/1 1540 and a suitable soluble TNF receptor-1 is described in WO98/01555.
- exemplary RANK ligand antagonists are disclosed, for example, in WO 03/086289, WO 03/002713, U.S. Pat. Nos.: 6,740,51 1 and 6,479,635.
- the anti-SOST antibodies as described herein can be administered to an individual via any suitable route. It should be understood by persons skilled in the art that the examples described herein are not intended to be limiting but to be illustrative of the techniques available. Accordingly, in some embodiments, the anti-SOST antibody is administered to an individual in accord with known methods, such as intravenous administration, e.g., as a bolus or by continuous infusion over a period of time, by intramuscular, intraperitoneal, intracerebrospinal, transdermal, subcutaneous, intraarticular, sublingually, intrasynovial, insufflation, intrathecal, oral, inhalation, or topical routes.
- intravenous administration e.g., as a bolus or by continuous infusion over a period of time
- intramuscular, intraperitoneal, intracerebrospinal transdermal, subcutaneous, intraarticular, sublingually, intrasynovial, insufflation, intrathecal, oral, in
- Administration can be systemic, e.g., intravenous administration, or localized.
- Commercially available nebulizers for liquid formulations including jet nebulizers and ultrasonic nebulizers are useful for administration.
- Liquid formulations can be directly nebulized and lyophilized powder can be nebulized after reconstitution.
- the anti-SOST antibody can be aerosolized using a fiuorocarbon formulation and a metered dose inhaler, or inhaled as a lyophilized and milled powder.
- the anti-SOST antibody is administered via site-specific or targeted local delivery techniques.
- site-specific or targeted local delivery techniques include various implantable depot sources of the SOST antibody or local delivery catheters, such as infusion catheters, indwelling catheters, or needle catheters, synthetic grafts, adventitial wraps, shunts and stents or other implantable devices, site specific carriers, direct injection, or direct application. See, e.g., PCT Publication No. WO 00/5321 1 and U.S. Pat. No. 5,981 ,568.
- the anti-SOST antibody may be administered neat.
- the anti-SOST antibody and a pharmaceutically acceptable excipient may be in various formulations.
- Pharmaceutically acceptable excipients are known in the art, and are relatively inert substances that facilitate administration of a pharmacologically effective substance.
- an excipient can give form or consistency, or act as a diluent.
- Suitable excipients include but are not limited to stabilizing agents, wetting and emulsifying agents, salts for varying osmolarity, encapsulating agents, buffers, and skin penetration enhancers. Excipients as well as formulations for parenteral and
- these agents are formulated for administration by injection (e.g., intraperitoneally, intravenously, subcutaneously, intramuscularly, etc.).
- these agents can be combined with pharmaceutically acceptable vehicles such as saline, Ringer's solution, dextrose solution, and the like.
- pharmaceutically acceptable vehicles such as saline, Ringer's solution, dextrose solution, and the like.
- the particular dosage regimen i.e., dose, timing and repetition, will depend on the particular individual and that individual's medical history.
- the anti-SOST antibody as described herein can be administered using any suitable method, including by injection (e.g., intraperitoneally, intravenously,
- the anti-SOST antibody can also be any anti-SOST antibody.
- an initial candidate dosage can be about 2 mg/kg.
- a typical daily dosage might range from about any of 3 g/kg to 30 g/kg to 300 g/kg to 3 mg/kg, to 30 mg/kg, to 100 mg/kg or more, depending on the factors mentioned above. For example, dosage of about 1 mg/kg, about 2.5 mg/kg, about 5 mg/kg, about 10 mg/kg, and about 25 mg/kg may be used.
- An exemplary dosing regimen comprises administering an initial dose of about 2 mg/kg, followed by a weekly maintenance dose of about 1 mg/kg of the anti-SOST antibody, or followed by a maintenance dose of about 1 mg/kg every other week.
- Other exemplary dosing regimen comprises administering increasing doses (e.g., initial dose of 1 mg/kg and gradual increase to one or more higher doses every week or longer time period).
- Other dosage regimens may also be useful, depending on the pattern of pharmacokinetic decay that the practitioner wishes to achieve. For example, in some embodiments, dosing from one to four times a week is contemplated. In other embodiments dosing once a month or once every other month or every three months is contemplated. The progress of this therapy is easily monitored by
- the dosing regimen (including the anti-SOST antibody used) can vary over time.
- an anti-SOST antibody for the purpose of the present invention, the appropriate dosage of an anti-SOST antibody will depend on the anti-SOST antibody (or compositions thereof) employed, the type and severity of symptoms to be treated, whether the agent is administered for therapeutic purposes, previous therapy, the patient's clinical history and response to the agent, the patient's clearance rate for the administered agent, and the discretion of the attending physician.
- the clinician will administer an anti-SOST antibody until a dosage is reached that achieves the desired result. Dose and/or frequency can vary over course of treatment. Empirical considerations, such as the half-life, generally will contribute to the determination of the dosage.
- antibodies that are compatible with the human immune system may be used to prolong half-life of the antibody and to prevent the antibody being attacked by the host's immune system.
- Frequency of administration may be determined and adjusted over the course of therapy, and is generally, but not necessarily, based on treatment and/or suppression and/or amelioration and/or delay of symptoms, e.g., tumor growth inhibition or delay, etc.
- sustained e.g., chronic obstructive pulmonary disease, etc.
- dosages for an anti-SOST antibody may be determined empirically in individuals who have been given one or more administration(s) of the anti- SOST antibody. Individuals are given incremental dosages of an anti-SOST antibody or an SOST antagonist. To assess efficacy, an indicator of the disease can be followed.
- Administration of an anti-SOST antibody in accordance with the method in the present invention can be continuous or intermittent, depending, for example, upon the recipient's physiological condition, whether the purpose of the administration is therapeutic or prophylactic, and other factors known to skilled practitioners.
- the administration of an anti-SOST antibody may be essentially continuous over a preselected period of time or may be in a series of spaced doses.
- more than one anti-SOST antibody may be present. At least one, at least two, at least three, at least four, at least five different or more anti- SOST antibody can be present. Generally, those anti-SOST antibodies may have complementary activities that do not adversely affect each other. For example, one or more of the following anti-SOST antibody may be used: a first anti-SOST antibody directed to one epitope on SOST and a second anti-SOST antibody directed to a different epitope on SOST.
- Therapeutic formulations of the anti-SOST antibody used in accordance with the present invention are prepared for storage by mixing an antibody having the desired degree of purity with optional pharmaceutically acceptable carriers, excipients or stabilizers (Remington, The Science and Practice of Pharmacy 21 st Ed. Mack
- Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations employed, and may comprise buffers such as phosphate, citrate, and other organic acids; salts such as sodium chloride; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride;
- alkyl parabens such as methyl or propyl paraben
- catechol resorcinol; cyclohexanol; 3-pentanol; and m-cresol
- low molecular weight polypeptides include proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins;
- chelating agents such as EDTA
- sugars such as sucrose, mannitol, trehalose or sorbitol
- salt-forming counter-ions such as sodium
- metal complexes e.g. Zn-protein complexes
- non-ionic surfactants such as TWEENTM, PLURONICSTM or polyethylene glycol (PEG).
- Liposomes containing the anti-SOST antibody are prepared by methods known in the art, such as described in Epstein, et al., Proc. Natl. Acad. Sci. USA 82:3688, 1985; Hwang, et al., Proc. Natl Acad. Sci. USA 77:4030, 1980; and U.S. Pat. Nos. 4,485,045 and 4,544,545. Liposomes with enhanced circulation time are disclosed in U.S. Pat. No. 5,013,556. Particularly useful liposomes can be generated by the reverse phase evaporation method with a lipid composition comprising phosphatidylcholine, cholesterol and PEG-derivatized phosphatidylethanolamine (PEG-PE). Liposomes are extruded through filters of defined pore size to yield liposomes with the desired diameter.
- PEG-PE PEG-derivatized phosphatidylethanolamine
- the active ingredients may also be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example, hydroxymethylcellulose or gelatin-microcapsules and poly-(methylmethacrylate) microcapsules, respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in macroemulsions.
- colloidal drug delivery systems for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules
- Sustained-release preparations may be prepared. Suitable examples of sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the antibody, which matrices are in the form of shaped articles, e.g. films, or microcapsules. Examples of sustained-release matrices include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or 'poly(vinylalcohol)), polylactides (U.S. Pat. No.
- copolymers of L-glutamic acid and 7 ethyl-L- glutamate copolymers of L-glutamic acid and 7 ethyl-L- glutamate, non-degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOTTM (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), sucrose acetate isobutyrate, and poly-D-(-)-3-hydroxybutyric acid.
- LUPRON DEPOTTM injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate
- sucrose acetate isobutyrate sucrose acetate isobutyrate
- poly-D-(-)-3-hydroxybutyric acid poly-D-(-)-3-hydroxybutyric acid.
- compositions to be used for in vivo administration must be sterile. This is readily accomplished by, for example, filtration through sterile filtration membranes.
- Therapeutic SOST antibody compositions are generally placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle.
- compositions according to the present invention may be in unit dosage forms such as tablets, pills, capsules, powders, granules, solutions or suspensions, or suppositories, for oral, parenteral or rectal administration, or administration by inhalation or insufflation.
- the principal active ingredient is mixed with a pharmaceutical carrier, e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g. water, to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention, or a non-toxic pharmaceutically acceptable salt thereof.
- a pharmaceutical carrier e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g. water
- a pharmaceutical carrier e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium
- This solid preformulation composition is then subdivided into unit dosage forms of the type described above containing from 0.1 to about 500 mg of the active ingredient of the present invention.
- the tablets or pills of the novel composition can be coated or otherwise compounded to provide a dosage form affording the advantage of prolonged action.
- the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former.
- the two components can be separated by an enteric layer that serves to resist disintegration in the stomach and permits the inner component to pass intact into the duodenum or to be delayed in release.
- enteric layers or coatings such materials including a number of polymeric acids and mixtures of polymeric acids with such materials as shellac, cetyl alcohol and cellulose acetate.
- Suitable surface-active agents include, in particular, non-ionic agents, such as polyoxyethylenesorbitans (e.g. TweenTM 20, 40, 60, 80 or 85) and other sorbitans (e.g. SpanTM 20, 40, 60, 80 or 85).
- non-ionic agents such as polyoxyethylenesorbitans (e.g. TweenTM 20, 40, 60, 80 or 85) and other sorbitans (e.g. SpanTM 20, 40, 60, 80 or 85).
- Suitable emulsions may be prepared using commercially available fat emulsions, such as IntralipidTM, LiposynTM, InfonutrolTM, LipofundinTM and LipiphysanTM.
- the active ingredient may be either dissolved in a pre-mixed emulsion composition or alternatively it may be dissolved in an oil (e.g. soybean oil, safflower oil, cottonseed oil, sesame oil, corn oil or almond oil) and an emulsion formed upon mixing with a phospholipid (e.g. egg phospholipids, soybean phospholipids or soybean lecithin) and water.
- an oil e.g. soybean oil, safflower oil, cottonseed oil, sesame oil, corn oil or almond oil
- a phospholipid e.g. egg phospholipids, soybean phospholipids or soybean lecithin
- other ingredients may be added, for example glycerol or glucose, to adjust the tonicity of the emulsion.
- Suitable emulsions will typically contain up to 20% oil, for example, between 5 and 20%.
- the fat emulsion can comprise fat droplets between 0.1 and 1 .0 pm, particularly 0.1 and 0.5 pm, and have a pH in the range of 5.5 to 8.0.
- the emulsion compositions can be those prepared by mixing an anti-SOST antibody or an anti-SOST antibody conjugate with IntralipidTM or the components thereof (soybean oil, egg phospholipids, glycerol and water).
- compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders.
- the liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as set out above.
- the compositions are administered by the oral or nasal respiratory route for local or systemic effect.
- Compositions in preferably sterile pharmaceutically acceptable solvents may be nebulised by use of gases. Nebulised solutions may be breathed directly from the nebulising device or the nebulising device may be attached to a face mask, tent or intermittent positive pressure breathing machine.
- Solution, suspension or powder compositions may be administered, preferably orally or nasally, from devices which deliver the formulation in an appropriate manner.
- compositions used in the methods of the invention comprise an effective amount of an anti-SOST antibody as described herein. Examples of such compositions, as well as how to formulate, are also described in an earlier section and below.
- the composition comprises one or more anti-SOST antibodies.
- an anti-SOST antibody recognizes human SOST.
- the anti-SOST antibody is a human antibody, a humanized antibody, or a chimeric antibody.
- the anti-SOST antibody comprises a constant region that is capable of triggering a desired immune response, such as antibody-mediated lysis or ADCC.
- the anti-SOST antibody comprises a constant region that does not trigger an unwanted or undesirable immune response, such as antibody- mediated lysis or ADCC.
- the anti-SOST antibody comprises one or more CDR(s) of the antibody (such as one, two, three, four, five, or, in some embodiments, all six CDRs).
- compositions can comprise more than one anti-SOST antibody (e.g., a mixture of anti-SOST antibodies that recognize different epitopes of SOST).
- Other exemplary compositions comprise more than one anti-SOST antibodies that recognize the same epitope(s), or different species of anti-SOST antibodies that bind to different epitopes of SOST (e.g., human SOST).
- composition used in the present invention can further comprise
- Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations, and may comprise buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin
- kits of the invention include one or more containers comprising an anti-SOST antibody described herein and instructions for use in accordance with any of the methods of the invention described herein. Generally, these instructions comprise a description of administration of the anti-SOST antibody for the above described therapeutic treatments.
- kits are provided for producing a single- dose administration unit.
- the kit can contain both a first container having a dried protein and a second container having an aqueous formulation.
- kits containing single and multi-chambered pre-filled syringes are included.
- the antibody is a human antibody. In some embodiments, the antibody is a humanized antibody. In some embodiments, the antibody is a monoclonal antibody.
- the instructions relating to the use of an anti-SOST antibody generally include information as to dosage, dosing schedule, and route of administration for the intended treatment.
- the containers may be unit doses, bulk packages (e.g., multi-dose packages) or sub-unit doses. Instructions supplied in the kits of the invention are typically written instructions on a label or package insert (e.g., a paper sheet included in the kit), but machine-readable instructions (e.g., instructions carried on a magnetic or optical storage disk) are also acceptable.
- kits of this invention are in suitable packaging.
- suitable packaging includes, but is not limited to, vials, bottles, jars, flexible packaging (e.g., sealed Mylar or plastic bags), and the like.
- packages for use in combination with a specific device such as an inhaler, nasal administration device (e.g., an atomizer) or an infusion device such as a minipump.
- a kit may have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- the container may also have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- At least one active agent in the composition is an anti-SOST antibody.
- the container may further comprise a second pharmaceutically active agent, such as a chemotherapeutic agent, osteoclast activity inhibiting agents, osteoblast activity enhancing agents (e.g., bone growth anabolic agents and bone anti-resorptive agents), and dietary supplements (e.g., calcium, vitamin D, and vitamin K) as described herein.
- a chemotherapeutic agent such as a chemotherapeutic agent, osteoclast activity inhibiting agents, osteoblast activity enhancing agents (e.g., bone growth anabolic agents and bone anti-resorptive agents), and dietary supplements (e.g., calcium, vitamin D, and vitamin K) as described herein.
- Kits may optionally provide additional components such as buffers and interpretive information.
- the kit comprises a container and a label or package insert(s) on or associated with the container.
- the invention also provides diagnostic kits comprising any or all of the antibodies described herein.
- the diagnostic kits are useful for, for example, detecting the presence of SOST in a sample.
- a diagnostic kit can be used to determine whether an individual is at risk for developing a SOST-associated disorder, e.g., a bone related disorder, such as osteoporosis, osteopenia, bone fracture, bone loss, Paget's Disease, or multiple myeloma.
- a bone related disorder such as osteoporosis, osteopenia, bone fracture, bone loss, Paget's Disease, or multiple myeloma.
- Diagnostic kits of the invention include one or more containers comprising an anti-SOST antibody described herein and instructions for use in accordance with any of the methods of the invention described herein.
- these instructions comprise a description of use of the anti-SOST antibody to detect the presence of in individuals at risk for, or suspected of having, a SOST-associated conditions, disorder/disease (e.g., a bone related disorder).
- an exemplary diagnostic kit can be configured to contain reagents such as, for example, an anti-SOST antibody, a negative control sample, a positive control sample, and directions for using the kit.
- Piscataway NJ was used to determine the binding affinities of anti-SOST antibodies to recombinant human SOST (rhSOST). See Table 4.
- An anti-human Fc sensor chip was prepared by activating all flow cells of a Biacore CM4 sensor chip with a 1 : 1 (v/v) mixture of 400 mM EDC (1 -Ethyl-3-[3-dimethylaminopropyl]-carbodiimide) and 100 mM N-hydroxysuccinimide for 7 minutes, at a flow rate of 10 pL/min.
- a goat F(AB') 2 fragment-anti-human IgG Fc (Cappel Catalog) was diluted to 60 pg/mL in 10 mM Sodium Acetate at pH 5.0 and injected on all flow cells for 7 minutes at 20 pL/min. All flow cells were blocked with 100 mM ethylenediamine in 150 mM Borate buffer pH 8.5 for 7 minutes at 10 pL/min. Antibodies were captured onto downstream flow cells (flow cells 2, 3 and 4) at 2 pg/mL at a flow rate of 10 pL/min for 1 minute. Different antibodies were captured on each flow cell. Flow cell 1 was used as a reference surface.
- analyte (buffer, or rhSOST) was injected at 30 pL/min on all flow cells for two minutes. After the analyte injection, dissociation was monitored for 30 minutes followed by regeneration of all flow cells with two 30-second injections of 75 mM Phosphoric Acid. A 3-membered dilution series of rhSOST was analyzed using this method, where the top concentration was 15 nM and the dilution factor was 3-fold. Buffer cycles were collected for each captured antibody for double-referencing purposes (double-referencing as described in Myszka, J. Mol. Recognit. 12:279-284, 1999).
- the double-referenced sensorgrams were fit globally to a simple 1:1 Langmuir with mass transport binding model.
- the experiments were performed at 37°C using a running buffer of 10 mM HEPES, 150 mM NaCI, 0.05% (v/v) Tween-20, pH 7.4, 1 mg/ml_ BSA.
- Table 4 shows the affinity constants of human IgGs binding to rhSOST at 37°C.
- Canonical Wnt signaling leads to stabilization and nuclear translocation of intracellular ⁇ -catenin.
- ⁇ -catenin interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) and this complex binds to the Wnt response element in the promoters of target genes leading to their transcriptional upregulation.
- TCF/LEF T-cell factor/lymphoid enhancer factor
- Canonical Wnt/ ⁇ - catenin signaling plays a fundamental role in osteoblastogenesis and control of bone mass. Activation of the Wnt ⁇ -catenin signaling pathway facilitates osteoblast specification from mesenchymal progenitor cells at the expense of adipogenesis, stimulates osteoblast proliferation, and prolongs the survival of osteoblasts and osteocytes leading to enhanced bone mass and bone strength.
- SOST is a secreted glycoprotein localized almost exclusively to osteocytes, and a negative regulator of Wnt ⁇ -catenin signaling.
- an in vitro cell culture system for the quantitation of Wnt signaling was developed.
- U-2OS osteosarcoma cells were maintained at 37°C, 5% CO2 in McCoy's 5A media (MediaTech, Manassas VA) supplemented with 10% dialyzed fetal bovine serum, 100U/ml Pen/Strep, and 2mM glutamine.
- Anti-SOST antibodies (DP99, DP1 , DM99, DM21 , and DM4) were produced as human lgG2Aa/kappa isotype, and purified by Protein-A affinity chromatography. The antibodies were concentrated to ⁇ 1 mg/ml in phosphate-buffered saline, pH 7.4. An irrelevant isotype control antibody IgG which does not recognize any mammalian proteins was expressed and affinity-purified.
- Recombinant human (rh)SOST (SEQ ID NO: 1 (excluding the secretory signal sequence) with a carboxy terminal 10-His tag was produced from bacculovirus and purified by Ni-NTA affinity chromatography (Qiagen, Valencia, CA). Purified rhSOST was quantitated by ELISA, and stored at a concentration of 0.13 mg/ml in 0.4 M NaCI, 10% glycerol in aliquots at - 80°C.
- U-2OS-TF-Wnt10B cells were plated in regular growth media in poly-D-lysine coated 96-well tissue culture plates (BD Biosciences, Beford, MA) at a density of 15,000 cells/well and incubated overnight at 37°C.
- Cells were treated with 1 ⁇ g/ml recombinant human or mouse SOST protein (R&D Systems, Minneapolis, MN) or vehicle (PBS) and varying concentrations of anti-SOST antibodies or isotype control antibody (26H6) in OptiMEM (Gibco, Grand Island NY) in a final volume of 10 ⁇ .
- the SOST protein and the antibody to be tested were incubated at room temperature for 15 minutes before adding to the cells.
- Figure 1 shows does-dependent inhibition of Wnt-10B signaling by recombinant mouse SOST and human SOST proteins.
- the IC50 for mouse or rhSOST inhibition of Wntl OB activity was calculated to be 10.4 nM or 13.3 nM, respectively.
- Figure 2 shows in vitro neutralization of rhSOST activity by anti-SOST antibodies of the present invention.
- Anti-SOST antibodies DP99, DP1 , DM99, DM21 , and DM4 dose-dependently blocked the SOST-mediated inhibition of Wnt-10B activity in U-2OS-TF-Wnt1 OB cells.
- Various concentrations of anti-SOST antibodies or isotype control mAb were preincubated with rhSOST (1 .5 pg/rnl, approximately 65nM) for 15 minutes at room temperature, then added to cells. Luciferase activity was measured after 24 hours. All conditions were tested in triplicate.
- responses were normalized between plates, such that the100% response represents cells without added SOST or mAb (a condition tested in triplicate on each plate).
- the calculated EC 50 values (in Mg/ml) for the anti-SOST mAbs were: DP99, 5.95; DP1 , 22; DM99, 1 .95; DM21 , 5.53 and DM4, 2.07.
- the 15-week (wk) old female Sprague-Dawley rats were ovariectomized (OVX) and maintained at the animal facility for 8 weeks after surgery to allow for bone loss to occur.
- rats were weighed and randomized into groups.
- baseline Sham and baseline OVX groups were sacrificed, whereas dosing began for all other groups.
- Dosing consisted of subcutaneous injection of vehicle or antibody (irrelevant isotype control at 20mg/kg or anti-SOST mAb DP99 at 2 or 20 mg/kg) 1x/week for 6 weeks, or Parathyroid Hormone (PTH) (40 g/kg) 5x/wk for 6 wks.
- PTH Parathyroid Hormone
- animals were sacrificed, and the left femurs and lumbar vertebra L4-6 were collected, wrapped in saline-soaked gauze, frozen at -20°C and analyzed by bone densiometry and biomechanical strength testing.
- Peripheral Quantitative Computed Tomography was performed on the excised left femurs using a Stratec XCT-RM instrument and associated software (Stratectechnik GmbH, Pforzheim, Germany; software version 5.40). The scan was performed at the midshaft femur. The positions were verified using scout views and one 0.5-mm slice perpendicular to the long axis of the femoral shaft was acquired from each site. The scans were analyzed using a threshold for delineation of the external boundary. Cortical bone mineral content and bone mineral density, endosteal and periosteal circumference, average cortical thickness and axial moment of inertia were reported. Summarized data is shown in Table 6.
- the anterior to posterior diameter at the midpoint of the femoral shaft was taken with an electronic caliper and recorded.
- the femur was then placed on the lower supports of a three point bending fixture with the anterior side facing downward in an Instron Mechanical Testing instrument (Instron 4465 retrofitted to 5500).
- the span between the two lower supports was set at 14mm.
- the upper loading device was aligned to the center of the femoral shaft.
- the load was applied at a constant displacement rate of 6 mm/min until the femur broke.
- the locations of maximum load, stiffness and energy absorbed were selected from the load and displacement curve and values calculated by instrument software (Bluehill v2.5, Instron).
- the intrinsic properties, ultimate strength, elastic modulus and toughness, were calculated from maximum load, stiffness, energy absorbed, anterior-posterior diameter, length between two supports and axial area moment of inertia using the following equations:
- treated groups (3-8) were compared to all other treated groups using an ANOVA with a post hoc Duncan Test, n.s.; non-significant.
- the proximal half of the femur was used in the cantilever compression test of the femoral neck.
- the proximal half of the femur was placed firmly in an anchoring platform where the greater trochanter was lodged in a notch cut in the platform.
- the test was conducted with an Instron
- each de-fleshed vertebrae sample (L5) the posterior pedicle arch and spinous process were removed using a low-speed diamond saw. Additionally, the cranial and caudal ends of each vertebral body were cut off with the same diamond saw to obtain a vertebral body specimen with two parallel surfaces and a height
- Width in the medial-lateral and anterior-posterior directions at both the cranial and caudal ends was measured using digital calipers.
- Ovariectomized rats treated with 20 mg/kg antibody DP99 or PTH showed significantly improved bone density and strength over the OVX animals treated with vehicle or irrelevant antibody isotype control, and in some cases, results were significantly higher than vehicle-treated Sham controls. Significant increases were seen in cortical bone mineral content, cortical thickness at the midshaft femur, maximum load and energy at the femoral neck, and energy, ultimate strength, and toughness
- Example 4 Anti-SOST Antibodies Stimulate Bone Formation and Increases Bone Mass in Ovariectomized Mice
- U2OS cell line was obtained from ATCC and stably transfected with TOPFIash plasmid (Upstate; Millipore, Millerica, MA), a TCF-luciferase reporter construct. Cells were maintained at 37°C, 5% CO 2 in McCoys 5A Media supplemented with 10% fetal calf serum, 1 % Pen Strep, and 2mM Glutamine.
- U2OS TOPFIash cells were plated in regular growth media at a density of 37,500 cells/cm 2 and incubated overnight Cells were transfected using Lipofectamine Plus (Invitrogen, Grand Island, NY) with plasmids carrying Wnt10b and MesDC2 cDNAs (Origene, Rockville, MD) and then incubated in regular growth media for 24 hours. Transfected cells were treated with 1 ⁇ g/ml rhSOST protein (R & D Systems, Minneapolis, MN) or vehicle (PBS, Gibco, Grand Island, NY) and varying concentrations of SOST mAbs in Optimem (Gibco, Grand Island, NY). After an overnight incubation, cells were lysed with Reporter Lysis Buffer (Promega, Madison, Wl) and luciferase expression was quantified using Luciferase Reagent (Promega, Madison, WA).
- Lipofectamine Plus Invitrogen, Grand Island, NY
- mice were subcutaneously injected with demeclocycline at 20 mg/kg prior to the first dose, and then with xylenal orange at 90 mg/kg or calcein at 5 mg/kg (Sigma Chemical Co., St. Louis, MO) on -9 and -2 days before euthanasia. All mice were euthanized by CO2 asphyxiation followed by exsanguination. Serum, both right and left femurs, right tibia, and lumbar spine were harvested for further analyses. The experiments were conducted according to Pfizer animal care-approved protocols, and animals were maintained in accordance with the ILAR (Institute of Laboratory Animal Research) Guide for the Care and Use of Laboratory Animals.
- ILAR Institute of Laboratory Animal Research
- Serum concentrations of anti-SOST mouse antibodies were determined by an ELISA method. Samples and anti-SOST mouse antibody calibration and quality control standards were diluted in a PBS buffer. The 96-well immunosorbent assay plates were overnight coated with recombinant mouse SOST (R&D Systems, Minneapolis, MN) and then blocked with Superblock T20 (Thermo Scientific, Rockford, IL) after washing with PBS buffer. The diluted samples and standards were added to plates and incubated at room temperature for 1 hour. The plates were then washed, followed by incubation with a horseradish peroxidase-conjugated anti-mouse lgG1 secondary antibody (Southern Biotech, Birmingham, AL) for 1 hour.
- a horseradish peroxidase-conjugated anti-mouse lgG1 secondary antibody Southern Biotech, Birmingham, AL
- the anti-SOST mouse antibody calibration standards were used to construct a standard curve using 4-parameter fitting with uniform weighting in SoftMax Pro 4.8. Serum concentrations of anti-SOST mouse antibody in unknown samples were interpolated from this standard curve.
- Total SOST protein levels were measured in mouse serum using the Meso Scale Diagnostics (MSD) Human Sclerostin (SOST) ELISA kit (catalog number: K1 1 1 HGC-2), which cross-reacted with mouse SOST protein.
- Recombinant mouse SOST protein was purchased from R & D Systems (catalog number 1589-ST) and used to generate the standard for the MSD SOST assay.
- Mouse recombinant SOST was reconstituted in PBS containing 0.1 % fetal bovine serum and stored at -20 °C.
- the recombinant mouse SOST was diluted to 25 ng/ml immediately prior to running the MSD SOST assay and subjected to 3-fold serial dilutions to prepare the mouse SOST standard (the SOST standards ranged from 25 ng/ml to 0.006 ng/ml).
- the MSD SOST assay was run according to manufacturer's instructions.
- Mouse serum osteocalcin was measured using a two-side immunoradiometric assay (ALPOCO Diagnostics, Windham, NH). Serum C-telopeptide (CTX) was determined by RatLaps ELISA (Nordic Bioscience, Diagnositics, Herlev, Denmark). pQCT measurement of right femurs
- the right femurs were scanned by peripheral quantitative computed tomography (pQCT, Stratec XCT Research M; Norland Medical Systems, Fort Atkinson, Wl, USA) with software version 5.40 as described in Ke et al., J. Musculoskel. Neuron Interact, 2:479-488, 2002.
- a 1 -mm-thick cross-section of each distal femoral metaphysis was taken at 2.5 mm proximal to the distal end ( ⁇ 1 .5 mm to the growth plate, a cancellous bone enriched site), and 1 -mm-thick cross-section of each femoral diaphysis was taken at 8 mm proximal from the distal end (a cortical bone enriched site) with a voxel size of 0.10 mm.
- Volumetric total bone content (BMC), density (BMD) and area were determined at the distal femurs and femoral diaphyses.
Abstract
The present disclosure provides antagonizing antibodies that specifically bind sclerostin. The disclosure further provides a method of obtaining such antibodies and antibody encoding nucleic acids. The disclosure further relates to therapeutic methods for use of these antibodies for the treatment and/or prevention of bone related disorders, including, for example, multiple myeloma, osteoporosis, osteopenia, Paget's Disease, bone fracture, or bone loss. Methods of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density as well as methods of inducing canonical Wnt signaling activity and methods of preventing or reducing tumor burden, methods of inhibiting or preventing tumor growth or progression, methods of inhibiting metastasis of cancer cells or tumors or delaying tumor growth or progression are also provided.
Description
ANTI-SCLEROSTIN ANTIBODIES
Related Application
This application claims the benefits of U.S. Provisional Application No.
61/914,094, filed December 10, 2013, which is hereby incorporated by reference in its entirety.
Field
The present invention relates to antibodies, e.g., full length antibodies or antigen binding fragments thereof, that specifically bind to sclerostin. The invention further relates to compositions comprising antibodies to sclerostin, and methods of using such anti-sclerostin antibodies as a medicament. The anti-sclerostin antibodies are useful for treating and preventing a sclerostin-associated condition or disorder (e.g., a bone related disorder or cancer), such as multiple myeloma and osteoporosis. Methods of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density as well as methods of inducing canonical Wnt signaling activity, methods of preventing or reducing tumor burden, methods of inhibiting or preventing tumor growth or progression, and methods of inhibiting metastasis of cancer cells or tumors or delaying tumor growth or progression are also provided.
Background
Multiple myeloma (also known as myeloma, plasma cell myeloma, or Kahler's disease) is a type of cancer formed by malignant plasma cells (myeloma cells) and is associated with bone loss attributed to an imbalance of bone metabolism. In multiple myeloma, myeloma cells directly or indirectly interact with the bone surface
microenvironment resulting in an apparent increase in osteoclast (bone resorbing) activity and a decrease in osteoblast (bone forming) activity.
Current treatment for multiple myeloma is focused on plasma cells apoptosis and/or decreasing osteoclast activity (e.g., chemotherapy, thalidomide, lenalidomide, bisphosphonates, and/or proteasome inhibitors such as bortezomib (VELCADE®)).
However, a significant number of patients have developed resistance to these agents and eventually relapse. Accordingly, an alternative treatment to multiple myeloma, such as increasing osteoblast activity, would make a superior therapeutic agent.
In recent years, novel targets with anabolic potential have emerged from the studies of human genetic diseases, which are characterized by high bone mass associated with increased bone formation. One of such targets is sclerostin (SOST) and its encoding gene, SOST. See, e.g., amino acid sequence of SOST (SEQ ID NO: 1 ), as reported by Brunkow et al., Am. J. Hum. Genet., 68:577-589, 2001 . Loss of SOST resulting from inactivating mutations in SOST is linked to sclerosteosis, a genetic disease characterized by progressive overgrowth of bone tissue See, e.g., Brunkow et al., 2001 , supra; Balemans et al., Hum. Mol. Genet., 10:537-543, 2001 ; and Beighton, J. Med. Genet., 25:200-203, 1988. SOST is a secreted, cystine-knot protein expressed primarily in osteocytes, the most abundant cell type in bone. Beside expression in osteocytes, SOST has also been found to express in osteoblasts and chondrocyte, but not osteoclasts. Winkler et al., EMBO J., 22:6267-6276, 2003. It inhibits osteoblast proliferation and differentiation, decreases osteoblast activity and promotes the apoptosis of osteoblasts in vitro. See e.g., van Benooijen et al., J. Exp. Med., 199:805- 814 and Sutherland et al., Bone, 35:828-835, 2004. Overexpression of SOST in mice led to decrease of alkaline phosphatase activity in mesenchymal cells, low bone mineral content with impaired bone strength, and decrease of osteoblast surface and bone formation. Winkler et al., 2003, supra. In contrast, targeted deletion of the SOST gene {SOST KO) in mice resulted in increased bone formation, bone mass, and bone strength. Li et al., J. Bone Miner Res. 23:860-869, 2008. The molecular mechanism of the action of SOST on bone is not entirely clear. However, recent studies have found that SOST antagonized canonical Wnt signaling by binding to the Wnt co-receptor Low Density Lipoprotein Receptor-Related Protein (LRP) 5 and LRP6. Zhang et al., J. Biol. Chem., 280:19883-19887, 2005. Collectively, the human and mouse genetic and phenotypic data have pointed to the potential of SOST inhibition as a novel mechanism for treatment of bone-related conditions/diseases/disorders, such as multiple myeloma. As a secreted protein, SOST poses an excellent opportunity to be neutralized by antibody. However, the fact that SOST acts at bone and is locally secreted by osteocytes, the cells encased within mineralized bone, may pose a challenge for targeting with a circulating antibody. Although various anti-SOST antibodies have been disclosed (see, e.g., U.S. Pat. Nos. 7,592,429, 7,872, 106, 7,879,322, 7,381 ,409, and 7744874), it has been exceedingly difficult to identify antagonistic monoclonal anti- SOST antibodies having high affinity and specifity and which are suitable for use with
human patients for the treatment of bone related conditions/diseases/disorders or SOST-associated conditions/diseases/disorders, such as multiple myeloma or cancer.
Summary
The invention disclosed herein is directed to antibodies and fragments thereof
(e.g. , antigen binding fragment) that selectively bind to a SOST polypeptide (e.g. , human SOST polypeptide). The inventors have discovered that the anti-SOST antibody of the present invention can significantly improve bone density, bone strength, bone quality, increase bone formation, bone volume, and bone mass, reduce/prevent/restore bone loss in mammals, such as in ovariectomized mice and rats, and prevent tumor burden in a syngeneic murine model of multiple myeloma.
In one aspect, the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GX-|TFX2DYWMQ, wherein X-i is F or H, X2 is T or S (SEQ ID NO: 81 ) or GXiTFX2DY, wherein X^ is F or H, and X2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX2X3KX4, wherein X is A or N, X2 is S or K, X3 is V or F, and X4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SXi DYW, wherein X^ is F or M (SEQ ID NO: 84); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGXiX2FMH, wherein X is S or N, and X2 is N or S (SEQ ID NO: 85); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQX1 IX2X3X4YT , wherein X^ is T or S, X2 is E or D, X3 is H, D, F, or E, and X4 is H, P, or S (SEQ ID NO: 86).
In another aspect, the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide, wherein the antibody comprises: a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 14; and/or
a VL region comprising VL CDR1 , VL CDR2, and VL CDR3 of the VL sequence shown in SEQ ID NO: 18. In some embodiments, the VH region comprises (i) a VH CDR1 comprising the sequence GHTFSDYWMQ (SEQ ID NO: 63), DYWMQ (SEQ ID NO: 56),
or GHTFSDY (SEQ ID NO: 64); (ii) a VH CDR2 comprising the sequence YPGDGD (SEQ ID NO: 57) or AIYPGDGDTRYNQKFKD (SEQ ID NO: 58); and (iii) a VH CDR3 comprising the sequence SMDYW (SEQ ID NO: 65). In some embodiments, the VL region comprises (i) a VL CDR1 comprising the sequence RASKTVDSYGNSFMH (SEQ ID NO: 60); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQTIEFPYT (SEQ ID NO: 67). In some embodiments, the antibody comprises (a) heavy chain CDRs comprising (i) a VH CDR1 comprising the sequence GHTFSDYWMQ (SEQ ID NO: 63), DYWMQ (SEQ ID NO: 56), or GHTFSDY (SEQ ID NO: 64); (ii) a VH CDR2 comprising the sequence YPGDGD (SEQ ID NO: 57) or AIYPGDGDTRYNQKFKD (SEQ ID NO: 58); and (iii) a VH CDR3 comprising the sequence SMDYW (SEQ ID NO: 65) and b) light chain CDRs comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGNSFMH (SEQ ID NO: 60); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQTIEFPYT (SEQ ID NO: 67). In some embodiments, the VH region comprises the sequence shown in SEQ ID NO: 14 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 18 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR. In some embodiments, the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 88 and/or a heavy chain comprising the sequence shown in SEQ ID NO: 87. In some embodiments, the antibody comprises a VH region produced by the expression vector with ATCC Accession No. PTA-12710. In some embodiments, the antibody comprises a VL region produced by the expression vector with ATCC Accession No. PTA-1271 1 .
In another aspect, the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST polypeptide), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence Xi ISGGDTYTYYADSVKG, wherein ^ is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary
determining regions comprising (i) a VL CDR1 comprising the sequence XiSSQSLLDNDGETYLN, wherein ^ is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
In another aspect, the invention provides an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST polypeptide), wherein the antibody comprises: a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 6; and/or a VL region comprising VL CDR1 , VL CDR2, and VL CDR3 of the VL sequence shown in SEQ ID NO: 7. In some embodiments, the VH region comprises (i) a VH CDR1 comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), or GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence LISGGDTYTYYADSVKG (SEQ ID NOs: 52) or SGGDTY (SEQ ID NO: 46); and (iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48). In some embodiments, the VL region comprises (i) a VL CDR1 comprising the sequence RSSQSLLDNDGETYLN (SEQ ID NO: 53); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ). In some embodiments, the antibody comprises (a) heavy chain CDRs comprising (i) a VH CDR1 comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), or GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence LISGGDTYTYYADSVKG (SEQ ID NOs: 52) or SGGDTY (SEQ ID NO: 46); and (iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48) and (b) light chain CDRs comprising (i) a VL CDR1 comprising the sequence RSSQSLLDNDGETYLN (SEQ ID NO: 53); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ). In some embodiments, the VH region comprises the sequence shown in SEQ ID NO: 6 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 7 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR. In some embodiments, the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 90 and/or a heavy chain comprising the sequence shown in SEQ ID NO: 89.
In some embodiments, the antibody can be a human antibody, a humanized antibody, or a chimeric antibody. In some embodiments, the antibody is a monoclonal antibody.
In some embodiments, the antibody comprises a constant region. In some embodiments, the antibody is of the human lgG1 , lgG2 or lgG2Aa, lgG3, or lgG4 subclass. In some embodiments, the antibody comprises a glycosylated constant region. In some embodiments, the antibody comprises a constant region having increased binding affinity to a human Fc gamma receptor.
The invention also provides an isolated antibody, or an antigen binding fragment thereof, which competes for binding to the anti-SOST antibodies described herein.
The invention also provides a conjugate of the anti-SOST antibody or the antigen binding fragment as described herein, wherein the antibody or the antigen binding fragment is conjugated to an agent, wherein the agent is selected from the group consisting of a chemotherapeutic agent (e.g., cytotoxic agent), an immunomodulating agent, an imaging agent, a therapeutic protein, a biopolymer, and an oligonucleotide.
The invention also provides pharmaceutical compositions comprising any of the anti-SOST antibodies described herein. In some embodiments, the pharmaceutical composition comprises a pharmaceutically acceptable carrier.
The invention also provides cell lines that recombinantly produce any of the anti- SOST antibodies described herein.
The invention also provides nucleic acids encoding any of the anti-SOST antibodies described herein. The invention also provides nucleic acids encoding a heavy chain variable region and/or a light chain variable region of any of the anti-SOST antibodies described herein.
The invention also provides kits comprising an effective amount of any of the anti-
SOST antibodies described herein.
In another aspect, provided is a method of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies described herein.
In another aspect, provided is a method of treating or preventing a bone related disorder in a patient in need thereof comprising administering to the patient an effective
amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies described herein, wherein the bone related disorder is selected from the group consisting of osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, rheumatoid arthritis, osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss, metastatic bone malignancy, multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease (e.g., Crohn's disease or ulcerative colitis), hemochromatosis, celiac sprue (or celiac disease), renal tubular acidosis, renal osteodystrophy, hypercalciuria, fibrous dysplasia, and diabetes. In some embodiments, the bone related disorder is osteoporosis. In other embodiments, the bone related disorder is multiple myeloma.
In another aspect, provided is a method of activating canonical Wnt signaling activity in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or the pharmaceutical composition comprising the anti-SOST antibodies as described herein.
In another aspect, provided is a method of preventing or reducing tumor burden in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide or a pharmaceutical composition thereof.
In another aspect, provided is a method of inhibiting or preventing tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide or a pharmaceutical composition comprising thereof.
In another aspect, provided is a method of inhibiting metastasis of cancer cells or tumors (e.g., solid or liquid tumors), or delaying tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti- SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide or a pharmaceutical composition comprising thereof.
In some embodiments, the patient can be a human. In some embodiments, the individual can be a mammal, such as a cow, a cat, a mouse, a rat, a monkey, or a dog.
In some embodiments, the anti-SOST antibodies described herein can be administered in combination with other therapeutic agents (e.g., chemotherapeutic agents, osteoclast activity inhibiting agents, osteoblast activity enhancing agents, and dietary supplements as described herein).
Brief Description of the Figures/Drawings
Figure 1 shows the dose-dependent inhibition of Wnt-10B signaling by recombinant mouse SOST protein (panel A) or recombinant human SOST protein (Panel B).
Figure 2 shows that anti-SOST antibodies DP99, DP1 , DM99, DM21 , and DM4 neutralized recombinant human SOST activity in vitro.
Figure 3 (A): A U2OS cell line stably expressing TOPFIash TCF-luciferase reporter construct was stimulated by transiently transfecting with Wnt10b. Addition of 1 μg/mL recombinant human SOST protein to the media inhibited the WNT stimulation. SOST-mediated inhibition of Wnt signaling, in turn, was reversed by adding 30 nM SOST neutralizing antibody DM99. 30 nM of a nonspecific IgG control had no effect on the suppression of WNTI Ob activity by SOST. Error bars represent Standard Error with n=6. (B): Anti-SOST antibodies (DP99 and DM99) dose dependently inhibited the ability of SOST to suppress WNT stimulation in U2OS TopFlash cells. The 100% control was set to the value of with WNTI Ob stimulation, and the 0% control was set to the value of WNTI Ob plus 1 μg/mL exogenous SOST.
Figure 4 depicts that anti-SOST antibodies of the present invention increased bone mass and prevented bone loss in ovariectomized (OVX) mice. Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle, DP99, or DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries. Total volumetric bone mineral content (BMC), bone mineral density (BMD) and bone area (AREA) were measured by pQCT on distal femurs (A, B, C, respectively) and on femoral diaphyses (D, E, F, respectively). Data are expressed as mean ± SEM. a: p < 0.05 vs. Sham; and b: p < 0.05 vs. OVX.
Figure 5 shows the representative Micro-Computed Tomography (μΟΎ) images of distal femurs (A) and the mean values of cancellous bone volume (B) from Sham, OVX, DP99, and DM99 treated mice groups.
Figure 6 shows representative CT images of midshaft femurs from sham surgery or OVX mice treated with indicated mAbs (IgG control, DP99, DP1 , and DM1 1 at 25 mg/kg) 1 x/wk for 6 wks.
Figures 7A-7B show that anti-SOST antibodies DP99, DP1 , and DM1 1 are efficacious in preventing ovariectomy-induced bone loss in mice in dose-response manner.
Figure 8 shows representative μΟΤ images of distal femurs from sham surgery or ovariectomized (OVX) mice treated with indicated mAbs (IgG control, DP99, DP1 , and DM1 1 at 25 mg/kg) 1 x/wk for 6 wks.
Figures 9A-9D show that anti-SOST antibodies DP99, DP1 , and DM1 1 are efficacious in preventing ovariectomy-induced bone loss in mice in dose-response manner. BV/TV: bone volume/tissue volume; Tb. N: trabecular number.
Figure 10 shows the results of CT analyses of L4 lumbar vertebra (A), or distal femurs (B), and pQCT analyses of the femoral diaphysis (C). BV/TV: bone volume/tissue volume; BS/BV: bone surface/bone volume; Tb. N: trabecular number; Tb.Th: trabecular thickness; and Tb.Sp: trabecular spacing. Data shown are means ± SEM for N=10 per group, except for biomechanical measurements in which N= 6-10 per group; *p < 0.05, **p<0.01 , ***p < 0.001 vs. IgG control.
Figure 1 1 shows that anti-SOST antibody DP99 prevents tumor burden in multiple myeloma (5TGM1 ) model in comparison to the IgG control group and the Bortezomib treated group (positive control). Data shown are based on 2 way ANOVA, Boneferroni post test (*** p<0.001 ).
Figure 12 also shows that anti-SOST antibody DP99 inhibits progression of multiple myeloma in the multiple myeloma (5TGM1 ) model in comparison to the IgG control group and the Bortezomib treated group (positive control).
Figure 13 shows that anti-SOST antibodies DP99 and DM1 delays growth of the progressing tumor as represented by total flux.
Figure 14 shows that anti-SOST antibodies DP99 and DM1 delays growth of the progressing tumor as represented by percent survival.
Detailed Description
The invention disclosed herein provides antibodies (e.g., antagonistic antibodies) that specifically bind to SOST (e.g., human SOST such as SEQ ID NO: 1 ). The invention also provides polynucleotides encoding these antibodies, compositions comprising these antibodies, and methods of making and using these antibodies. The invention further provides methods of increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density using the anti- SOST antibodies as described herein. Methods of treating a SOST-associated condition/disorder/disease such as bone related disorder (e.g., osteoporosis (such as postmenopausal osteoporosis), osteopenia, Paget's Disease, bone fracture, multiple myeloma, acute myeloid leukemia (AML), cancer, or bone loss), inhibiting or reducing tumor burden, inhibiting or preventing tumor growth or progression, inhibiting metastasis of cancer cells or tumors or delaying tumor growth or progression, and inducing canonical Wnt signaling activity are also provided.
General Techniques
The practice of the present invention will employ, unless otherwise indicated, conventional techniques of molecular biology (including recombinant techniques), microbiology, cell biology, biochemistry and immunology, which are within the skill of the art. Such techniques are explained fully in the literature, such as, Molecular Cloning: A Laboratory Manual, second edition (Sambrook et al., 1989) Cold Spring Harbor Press; Oligonucleotide Synthesis (M.J. Gait, ed., 1984); Methods in Molecular Biology, Humana Press; Cell Biology: A Laboratory Notebook (J.E. Cellis, ed., 1998) Academic Press; Animal Cell Culture (R. I. Freshney, ed., 1987); Introduction to Cell and Tissue Culture (J. P. Mather and P.E. Roberts, 1998) Plenum Press; Cell and Tissue Culture: Laboratory Procedures (A. Doyle, J.B. Griffiths, and D.G. Newell, eds., 1993-1998) J. Wiley and Sons; Methods in Enzymology (Academic Press, Inc.); Handbook of Experimental Immunology (D.M. Weir and C.C. Blackwell, eds.); Gene Transfer Vectors for Mammalian Cells (J.M. Miller and M.P. Calos, eds., 1987); Current Protocols in Molecular Biology (F.M. Ausubel et al., eds., 1987); PCR: The Polymerase Chain Reaction, (Mullis et al., eds., 1994); Current Protocols in Immunology (J.E. Coligan et al., eds., 1991 ); Short Protocols in Molecular Biology (Wiley and Sons, 1999); Immunobiology (C.A. Janeway and P. Travers, 1997); Antibodies (P. Finch, 1997);
Antibodies: a practical approach (D. Catty., ed., IRL Press, 1988-1989); Monoclonal antibodies: a practical approach (P. Shepherd and C. Dean, eds., Oxford University Press, 2000); Using antibodies: a laboratory manual (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999); The Antibodies (M. Zanetti and J.D. Capra, eds., Harwood Academic Publishers, 1995).
Definitions
The following terms, unless otherwise indicated, shall be understood to have the following meanings: the term "isolated molecule" (where the molecule is, for example, a polypeptide, a polynucleotide, or an antibody) is a molecule that by virtue of its origin or source of derivation (1 ) is not associated with naturally associated components that accompany it in its native state, (2) is substantially free of other molecules from the same species (3) is expressed by a cell from a different species, or (4) does not occur in nature. Thus, a molecule that is chemically synthesized, or expressed in a cellular system different from the cell from which it naturally originates, will be "isolated" from its naturally associated components. A molecule also may be rendered substantially free of naturally associated components by isolation, using purification techniques well known in the art. Molecule purity or homogeneity may be assayed by a number of means well known in the art. For example, the purity of a polypeptide sample may be assayed using polyacrylamide gel electrophoresis and staining of the gel to visualize the polypeptide using techniques well known in the art. For certain purposes, higher resolution may be provided by using HPLC or other means well known in the art for purification.
An "antibody" is an immunoglobulin molecule capable of specific binding to a target, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc., through at least one antigen recognition site, located in the variable region of the immunoglobulin molecule. As used herein, the term encompasses not only intact polyclonal or monoclonal antibodies, but also, unless otherwise specified, any antigen binding portion thereof that competes with the intact antibody for specific binding, fusion proteins comprising an antigen binding portion, and any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site. Antigen binding portions include, for example, Fab, Fab', F(ab')2, Fd, Fv, domain antibodies (dAbs, e.g., shark and camelid antibodies), fragments including complementarity determining
regions (CDRs), single chain variable fragment antibodies (scFv), maxibodies, minibodies, intrabodies, diabodies, triabodies, tetrabodies, v-NAR and bis-scFv, and polypeptides that contain at least a portion of an immunoglobulin that is sufficient to confer specific antigen binding to the polypeptide. An antibody includes an antibody of any class, such as IgG, IgA, or IgM (or sub-class thereof), and the antibody need not be of any particular class. Depending on the antibody amino acid sequence of the constant region of its heavy chains, immunoglobulins can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgG-i , lgG2, lgG3, lgG4, IgA-i and lgA2. The heavy-chain constant regions that correspond to the different classes of immunoglobulins are called alpha, delta, epsilon, gamma, and mu, respectively. The subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
An antibody, an antibody conjugate, or a polypeptide that "preferentially binds" or "specifically binds" (used interchangeably herein) to a target (e.g., SOST protein) is a term well understood in the art, and methods to determine such specific or preferential binding are also well known in the art. A molecule is said to exhibit "specific binding" or "preferential binding" if it reacts or associates more frequently, more rapidly, with greater duration and/or with greater affinity with a particular cell or substance than it does with alternative cells or substances. An antibody "specifically binds" or "preferentially binds" to a target if it binds with greater affinity, avidity, more readily, and/or with greater duration than it binds to other substances. For example, an antibody that specifically or preferentially binds to a SOST epitope is an antibody that binds this epitope with greater affinity, avidity, more readily, and/or with greater duration than it binds to other SOST epitopes or non-SOST epitopes. It is also understood that by reading this definition, for example, an antibody (or moiety or epitope) that specifically or preferentially binds to a first target may or may not specifically or preferentially bind to a second target. As such, "specific binding" or "preferential binding" does not necessarily require (although it can include) exclusive binding. Generally, but not necessarily, reference to binding means preferential binding.
A "variable region", "VH", or "VL" of an antibody refers to the variable region of the antibody light chain or the variable region of the antibody heavy chain, either alone or in combination. As known in the art, the variable regions of the heavy and light
chains each consist of four framework regions (FRs) connected by three complementarity determining regions (CDRs) also known as hypervariable regions, and contribute to the formation of the antigen binding site of antibodies. If variants of a subject variable region are desired, particularly with substitution in amino acid residues outside of a CDR region (i.e., in the framework region), appropriate amino acid substitution, preferably, conservative amino acid substitution, can be identified by comparing the subject variable region to the variable regions of other antibodies which contain CDR1 and CDR2 sequences in the same canonical class as the subject variable region (Chothia and Lesk, J Mol Biol 196(4): 901 -917, 1987).
As known in the art, a "constant region" of an antibody refers to the constant region of the antibody light chain or the constant region of the antibody heavy chain, either alone or in combination.
A "complementary determining region" or "CDR" of a variable domain are the amino acid residues within the variable region that are identified in accordance with the definitions of the Kabat, Chothia, the accumulation of both Kabat, Chothia, extended, AbM, contact, and/or conformational definitions or any method of CDR determination well known in the art. Antibody CDRs may be identified as the hypervariable regions originally defined by Kabat et al. See, e.g., Kabat et al., 1992, Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, NIH, Washington D.C. The positions of the CDRs may also be identified as the structural loops originally described by Chothia and others. See, e.g., Chothia et al., 1989, Nature 342:877-883. Other approaches to CDR identification include the "AbM definition," which is a compromise between Kabat and Chothia and is derived using Oxford Molecular's AbM antibody modeling software (now ACCELRYS®), or the "contact definition" of CDRs based on observed antigen contacts, set forth in MacCallum et al., 1996, J. Mol. Biol., 262:732- 745. In another approach, referred to herein as the "conformational definition" of CDRs, the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., 2008, Journal of Biological Chemistry, 283: 1 156-1 166. Still other CDR boundary definitions may not strictly follow one of the above approaches, but will nonetheless overlap with at least a portion of the Kabat CDRs, although they may be shortened or lengthened in light of prediction or experimental findings that particular residues or groups of residues do not significantly impact antigen binding. As used herein, a CDR may refer to CDRs defined by any
approach known in the art, including combinations of approaches. The methods used herein may utilize CDRs defined according to any of these approaches. For any given embodiment containing more than one CDR, the CDRs may be defined in accordance with any of Kabat, Chothia, extended, AbM, contact, and/or conformational definitions.
As used herein, "monoclonal antibody" (mAb) refers to an antibody that is derived from a single copy or clone, including e.g., any eukaryotic, prokaryotic, or phage clone, and not the method by which it is produced. Preferably, a monoclonal antibody of the invention exists in a homogeneous or substantially homogeneous population.
As used herein, "humanized" antibody refers to forms of non-human (e.g. murine) antibodies that are chimeric immunoglobulins, immunoglobulin chains, or fragments thereof (such as Fv, Fab, Fab', F(ab')2 or other antigen-binding subsequences of antibodies) that contain minimal sequence derived from non-human immunoglobulin. Preferably, humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a CDR of the recipient are replaced by residues from a CDR of a non-human species (donor antibody) such as mouse, rat, or rabbit having the desired specificity, affinity, and capacity.
A "human antibody" is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen binding residues.
The term "chimeric antibody" is intended to refer to antibodies in which the variable region sequences are derived from one species and the constant region sequences are derived from another species, such as an antibody in which the variable region sequences are derived from a mouse antibody and the constant region sequences are derived from a human antibody.
The term "epitope" refers to that portion of a molecule capable of being recognized by and bound by an antibody at one or more of the antibody's antigen- binding regions. Epitopes often consist of a surface grouping of molecules such as amino acids or sugar side chains and have specific three-dimensional structural characteristics as well as specific charge characteristics. In some embodiments, the epitope can be a protein epitope. Protein epitopes can be linear or conformational. In a linear epitope, all of the points of interaction between the protein and the interacting
molecule (such as an antibody) occur linearly along the primary amino acid sequence of the protein. A "nonlinear epitope" or "conformational epitope" comprises noncontiguous polypeptides (or amino acids) within the antigenic protein to which an antibody specific to the epitope binds. The term "antigenic epitope" as used herein, is defined as a portion of an antigen to which an antibody can specifically bind as determined by any method well known in the art, for example, by conventional immunoassays. Once a desired epitope on an antigen is determined, it is possible to generate antibodies to that epitope, e.g., using the techniques described in the present specification. Alternatively, during the discovery process, the generation and characterization of antibodies may elucidate information about desirable epitopes. From this information, it is then possible to competitively screen antibodies for binding to the same epitope. An approach to achieve this is to conduct competition and cross-competition studies to find antibodies that compete or cross-compete with one another for binding to SOST, e.g., the antibodies compete for binding to the antigen.
As used herein, the term "SOST" or "sclerostin" includes, for example, murine and human native forms of SOST. Exemplary SOST protein and nucleotides are disclosed, e.g., by Brunkow et al., 2001 , supra (amino acid sequence is disclosed as SEQ ID NO: 1 ). The term also includes variants of such native sequences that are immunologically cross-reactive with these native proteins. These proteins can inhibit the interaction between LRP5 and/or LRP6 with Wnt, which is required for canonical Wnt signaling activity. The term can also refer to a fragment of a native or variant form of SOST that contains an epitope to which an antibody can specifically bind.
The term "antagonist antibody" refers to an antibody that binds to a target and prevents or reduces the biological effect of that target. For example, the term can denote an antibody that prevents the target, e.g., SOST, to which it is bound from performing a biological function.
As used herein, an "SOST antagonist antibody" or "anti-SOST antagonist antibody" refers to an antibody that is able to inhibit SOST biological activity and/or downstream events(s) mediated by SOST. Anti-SOST antagonist antibodies encompass antibodies that block, antagonize, suppress, or reduce (to any degree including significantly) SOST biological activity, including downstream events mediated by SOST, such as cell surface interaction, receptor binding and downstream signaling (e.g., canonical Wnt signaling). For purposes of the present invention, it will be explicitly
understood that the term "anti-SOST antagonist antibody" (interchangeably termed "antagonist SOST antibody", "antagonist anti-SOST antibody" or "SOST antagonist antibody") encompasses all the previously identified terms, titles, and functional states and characteristics whereby the SOST itself, an SOST biological activity (including but not limited to its ability to bind a cell and bind a receptor), or the consequences of the biological activity, are substantially nullified, decreased, or neutralized in any meaningful degree. In some embodiments, an anti-SOST antibody binds SOST and prevents SOST from interacting with LRP5 and/or LRP6, thus antagonizing the SOST-mediated inhibition of canonical Wnt signaling pathway. In some embodiments, an anti-SOST antibody binds SOST and activates canonical Wnt signaling pathway. In some embodiments, the antagonist ability is described in terms of an IC50 or EC50 value.
The terms "polypeptide", "oligopeptide", "peptide" and "protein" are used interchangeably herein to refer to chains of amino acids of any length. The chain may be linear or branched, it may comprise modified amino acids, and/or may be interrupted by non-amino acids. The terms also encompass an amino acid chain that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component. Also included within the definition are, for example, polypeptides containing one or more analogs of an amino acid (including, for example, unnatural amino acids, etc.), as well as other modifications known in the art. It is understood that the polypeptides can occur as single chains or associated chains.
As known in the art, "polynucleotide," or "nucleic acid," as used interchangeably herein, refer to chains of nucleotides of any length, and include DNA and RNA. The nucleotides can be deoxyribonucleotides, ribonucleotides, modified nucleotides or bases, and/or their analogs, or any substrate that can be incorporated into a chain by DNA or RNA polymerase. A polynucleotide may comprise modified nucleotides, such as methylated nucleotides and their analogs. If present, modification to the nucleotide structure may be imparted before or after assembly of the chain. The sequence of nucleotides may be interrupted by non-nucleotide components. A polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component. Other types of modifications include, for example, "caps", substitution of one or more of the naturally occurring nucleotides with an analog, internucleotide
modifications such as, for example, those with uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates, carbamates, etc.) and with charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, for example, proteins (e.g., nucleases, toxins, antibodies, signal peptides, poly-L-lysine, etc.), those with intercalators (e.g., acridine, psoralen, etc.), those containing chelators (e.g., metals, radioactive metals, boron, oxidative metals, etc.), those containing alkylators, those with modified linkages (e.g., alpha anomeric nucleic acids, etc.), as well as unmodified forms of the polynucleotide(s). Further, any of the hydroxyl groups ordinarily present in the sugars may be replaced, for example, by phosphonate groups, phosphate groups, protected by standard protecting groups, or activated to prepare additional linkages to additional nucleotides, or may be conjugated to solid supports. The 5' and 3' terminal OH can be phosphorylated or substituted with amines or organic capping group moieties of from 1 to 20 carbon atoms. Other hydroxyls may also be derivatized to standard protecting groups. Polynucleotides can also contain analogous forms of ribose or deoxyribose sugars that are generally known in the art, including, for example, 2'-O-methyl- 2'-O-allyl, 2'-fluoro- or 2'-azido-ribose, carbocyclic sugar analogs, alpha- or beta-anomeric sugars, epimeric sugars such as arabinose, xyloses or lyxoses, pyranose sugars, furanose sugars, sedoheptuloses, acyclic analogs and abasic nucleoside analogs such as methyl riboside. One or more phosphodiester linkages may be replaced by alternative linking groups. These alternative linking groups include, but are not limited to, embodiments wherein phosphate is replaced by P(O)S("thioate"), P(S)S ("dithioate"), (O)NR2 ("amidate"), P(O)R, P(O)OR', CO or CH2 ("formacetal"), in which each R or R' is independently H or substituted or unsubstituted alkyl (1 -20 C) optionally containing an ether (-O-) linkage, aryl, alkenyl, cycloalkyl, cycloalkenyl or araldyl. Not all linkages in a polynucleotide need be identical. The preceding description applies to all polynucleotides referred to herein, including RNA and DNA.
As used herein, "substantially pure" refers to material which is at least 50% pure (i.e., free from contaminants), more preferably, at least 90% pure, more preferably, at least 95% pure, yet more preferably, at least 98% pure, and most preferably, at least 99% pure.
A "host cell" includes an individual cell or cell culture that can be or has been a recipient for vector(s) for incorporation of polynucleotide inserts. Host cells include
progeny of a single host cell, and the progeny may not necessarily be completely identical (in morphology or in genomic DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation. A host cell includes cells transfected in vivo with a polynucleotide(s) of this invention.
As known in the art, the term "Fc region" is used to define a C-terminal region of an immunoglobulin heavy chain. The "Fc region" may be a native sequence Fc region or a variant Fc region. Although the boundaries of the Fc region of an immunoglobulin heavy chain might vary, the human IgG heavy chain Fc region is usually defined to stretch from an amino acid residue at position Cys226, or from Pro230, to the carboxyl- terminus thereof. The numbering of the residues in the Fc region is that of the EU index as in Kabat. Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md., 1991 . The Fc region of an immunoglobulin generally comprises two constant domains, CH2 and CH3. As is known in the art, an Fc region can be present in dimer or monomeric form.
As used in the art, "Fc receptor" and "FcR" describe a receptor that binds to the
Fc region of an antibody. The preferred FcR is a native sequence human FcR. Moreover, a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the FcyRI, FcyRII, and FcyRIII subclasses, including allelic variants and alternatively spliced forms of these receptors. FcyRII receptors include FcyRIIA (an "activating receptor") and FcyRIIB (an "inhibiting receptor"), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof. FcRs are reviewed in Ravetch and Kinet, 1991 , Ann. Rev. Immunol., 9:457-92; Capel et al., 1994, Immunomethods, 4:25-34; and de Haas et al., 1995, J. Lab. Clin. Med., 126:330-41 . "FcR" also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al., 1976, J. Immunol., 1 17:587; and Kim et al., 1994, J. Immunol., 24:249).
As used herein "antibody-dependent cell-mediated cytotoxicity" or "ADCC" refers to a cell-mediated reaction in which nonspecific cytotoxic cells that express Fc receptors (FcRs) (e.g. natural killer (NK) cells, neutrophils, and macrophages) recognize bound antibody on a target cell and subsequently cause lysis of the target cell. ADCC activity of a molecule of interest can be assessed using an in vitro ADCC assay, such as that described in U.S. Patent No. 5,500,362 or 5,821 ,337. Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and NK cells. Alternatively,
or additionally, ADCC activity of the molecule of interest may be assessed in vivo, e.g., in an animal model such as that disclosed in Clynes et al., PNAS (USA), 95:652-656, 1998.
"Complement dependent cytotoxicity" or "CDC" refers to the lysing of a target in the presence of complement. The complement activation pathway is initiated by the binding of the first component of the complement system (C1 q) to a molecule (e.g. an antibody) complexed with a cognate antigen. To assess complement activation, a CDC assay, e.g. as described in Gazzano-Santoro et al., J. Immunol. Methods, 202: 163, 1996, may be performed
The term "compete", as used herein with regard to an antibody, means that a first antibody, or an antigen-binding portion thereof, binds to an epitope in a manner sufficiently similar to the binding of a second antibody, or an antigen-binding portion thereof, such that the result of binding of the first antibody with its cognate epitope is detectably decreased in the presence of the second antibody compared to the binding of the first antibody in the absence of the second antibody. The alternative, where the binding of the second antibody to its epitope is also detectably decreased in the presence of the first antibody, can, but need not be the case. That is, a first antibody can inhibit the binding of a second antibody to its epitope without that second antibody inhibiting the binding of the first antibody to its respective epitope. However, where each antibody detectably inhibits the binding of the other antibody with its cognate epitope or ligand, whether to the same, greater, or lesser extent, the antibodies are said to "cross-compete" with each other for binding of their respective epitope(s). Both competing and cross-competing antibodies are encompassed by the present invention. Regardless of the mechanism by which such competition or cross-competition occurs (e.g., steric hindrance, conformational change, or binding to a common epitope, or portion thereof), the skilled artisan would appreciate, based upon the teachings provided herein, that such competing and/or cross-competing antibodies are encompassed and can be useful for the methods disclosed herein.
A "functional Fc region" possesses at least one effector function of a native sequence Fc region. Exemplary "effector functions" include C1 q binding; complement dependent cytotoxicity; Fc receptor binding; antibody-dependent cell-mediated cytotoxicity; phagocytosis; down-regulation of cell surface receptors (e.g. B cell receptor), etc. Such effector functions generally require the Fc region to be combined
with a binding domain (e.g. an antibody variable domain) and can be assessed using various assays known in the art for evaluating such antibody effector functions.
A "native sequence Fc region" comprises an amino acid sequence identical to the amino acid sequence of an Fc region found in nature. A "variant Fc region" comprises an amino acid sequence which differs from that of a native sequence Fc region by virtue of at least one amino acid modification, yet retains at least one effector function of the native sequence Fc region. Preferably, the variant Fc region has at least one amino acid substitution compared to a native sequence Fc region or to the Fc region of a parent polypeptide, e.g. from about one to about ten amino acid substitutions, and preferably, from about one to about five amino acid substitutions in a native sequence Fc region or in the Fc region of the parent polypeptide. The variant Fc region herein will preferably possess at least about 80% sequence identity with a native sequence Fc region and/or with an Fc region of a parent polypeptide, and most preferably, at least about 90% sequence identity therewith, more preferably, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99% sequence identity therewith.
As used herein, "treatment" is an approach for obtaining beneficial or desired clinical results. For purposes of this invention, beneficial or desired clinical results include, but are not limited to, one or more of the following: increasing bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density, decreasing symptoms resulting from a SOST associated disease (e.g., a bone related disorder), increasing the quality of life of those suffering from a SOST-associated disease (e.g., a bone related disorder), decreasing the dose of other medications required to treat a SOST associated disease (e.g., a bone related disorder), delaying the progression or onset of a SOST associated disease (e.g., a bone related disorder), curing a SOST associated disease (e.g., a bone related disorder), and/or prolong survival of patients having a SOST associated disease (e.g., a bone related disorder). Treatment may be prophylactic (e.g., to prevent or delay the onset of the disease, or to prevent the manifestation of clinical or subclinical symptoms thereof) or therapeutic suppression or alleviation of symptoms after the manifestation of the disease.
"Reducing incidence" means any of reducing severity (which can include reducing need for and/or amount of (e.g., exposure to) other drugs and/or therapies generally used for this condition), reducing duration, and/or reducing frequency. As is understood by those skilled in the art, individuals may vary in terms of their response to
treatment, and, as such, for example, a "method of reducing incidence" reflects administering the anti-SOST antibody based on a reasonable expectation that such administration may likely cause such a reduction in incidence in that particular individual.
"Ameliorating" means a lessening or improvement of one or more symptoms as compared to not administering an anti-SOST antibody. "Ameliorating" also includes shortening or reduction in duration of a symptom.
As used herein, an "effective dosage" or "effective amount" of drug, compound, or pharmaceutical composition is an amount sufficient to effect any one or more beneficial or desired results. In more specific aspects, an effective amount prevents, alleviates or ameliorates symptoms of disease, and/or prolongs the survival of the subject being treated. For prophylactic use, beneficial or desired results include eliminating or reducing the risk, lessening the severity, or delaying the outset of the disease, including biochemical, histological and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease. For therapeutic use, beneficial or desired results include clinical results such as reducing one or more symptoms of a bone-related disease, decreasing the dose of other medications required to treat the disease, enhancing the effect of another medication, and/or delaying the progression of the disease of patients. An effective dosage can be administered in one or more administrations. For purposes of this invention, an effective dosage of drug, compound, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly. As is understood in the clinical context, an effective dosage of a drug, compound, or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition. Thus, an "effective dosage" may be considered in the context of administering one or more therapeutic agents, and a single agent may be considered to be given in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved.
An "individual", a "subject", or a "patient" is a mammal, more preferably, a human. Mammals also include, but are not limited to, farm animals (e.g., cows, pigs, horses, chickens, etc.), sport animals, pets, primates, horses, dogs, cats, mice and rats. For example, the individual is considered to be at risk for a bone related disorder disease (e.g., osteoporosis, osteopenia, Paget's Disease, bone loss, bone fracture, and multiple
myeloma). Such individuals include, but are not limited to, an individual who is hospitalized or will be hospitalized, an individual who is or will be put in an intensive care unit, an individual who will undergo surgery (e.g., bone related surgery such as implant surgery), an individual who will be anesthetized or under general anesthesia, an individual on dialysis, an individual with an indwelling catheter, an individual over the age of 65, an individual with a compromised immune system, a pediatric individual, an individual who is or may be put on a respirator or other mechanical ventilator, an individual in whom an endotracheal tube will or has been placed, an individual who is or will be immobilized, an individual who will undergo, is undergoing, or has undergone chemotherapy or myeloablative therapy, an individual who will take, is taking, or has taken one or more immunosuppressants, particularly for a significant period of time (longer than a month); an individual who is at risk of bone fracture (e.g., vertebral and/or non vertebral bone fracture). The individuals may be male or female. For example, the individual may be a female who is at risk for or having post-menopausal osteoporosis.
As used herein, "vector" means a construct, which is capable of delivering, and, preferably, expressing, one or more gene(s) or sequence(s) of interest in a host cell. Examples of vectors include, but are not limited to, viral vectors, naked DNA or RNA expression vectors, plasmid, cosmid or phage vectors, DNA or RNA expression vectors associated with cationic condensing agents, DNA or RNA expression vectors encapsulated in liposomes, and certain eukaryotic cells, such as producer cells.
As used herein, "expression control sequence" means a nucleic acid sequence that directs transcription of a nucleic acid. An expression control sequence can be a promoter, such as a constitutive or an inducible promoter, or an enhancer. The expression control sequence is operably linked to the nucleic acid sequence to be transcribed.
As used herein, "pharmaceutically acceptable carrier" or "pharmaceutical acceptable excipient" includes any material which, when combined with an active ingredient, allows the ingredient to retain biological activity and is non-reactive with the subject's immune system. Examples include, but are not limited to, any of the standard pharmaceutical carriers such as a phosphate buffered saline solution, water, emulsions such as oil/water emulsion, and various types of wetting agents. Preferred diluents for aerosol or parenteral administration are phosphate buffered saline (PBS) or normal (0.9%) saline. Compositions comprising such carriers are formulated by well known
conventional methods (see, for example, Remington's Pharmaceutical Sciences, 18th edition, A. Gennaro, ed., Mack Publishing Co., Easton, PA, 1990; and Remington, The Science and Practice of Pharmacy 21 th Ed. Mack Publishing, 201 1 ).
The term "kon", as used herein, refers to the rate constant for association of an antibody to an antigen. Specifically, the rate constants (kon and k0ff) and equilibrium dissociation constants are measured using full-length antibodies and/or Fab antibody fragments and SOST.
The term "k0ff ", as used herein, refers to the rate constant for dissociation of an antibody from the antibody/antigen complex.
The term " KD ", as used herein, refers to the equilibrium dissociation constant of an antibody-antigen interaction.
Reference to "about" a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. For example, description referring to "about X" includes description of "X." Numeric ranges are inclusive of the numbers defining the range.
It is understood that wherever embodiments are described herein with the language "comprising," otherwise analogous embodiments described in terms of "consisting of" and/or "consisting essentially of" are also provided.
Where aspects or embodiments of the invention are described in terms of a Markush group or other grouping of alternatives, the present invention encompasses not only the entire group listed as a whole, but each member of the group individually and all possible subgroups of the main group, but also the main group absent one or more of the group members. The present invention also envisages the explicit exclusion of one or more of any of the group members in the claimed invention.
Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. In case of conflict, the present specification, including definitions, will control. Throughout this specification and claims, the word "comprise," or variations such as "comprises" or "comprising" will be understood to imply the inclusion of a stated integer or group of integers but not the exclusion of any other integer or group of integers. Unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular. Any example(s) following the term "e.g." or "for example" is not meant to be exhaustive or limiting.
Exemplary methods and materials are described herein, although methods and materials similar or equivalent to those described herein can also be used in the practice or testing of the present invention. The materials, methods, and examples are illustrative only and not intended to be limiting.
SOST Antibodies and Methods of Making Thereof
The present invention provides an antibody (e.g., an antagonist antibody) that binds to SOST and blocks, suppresses, or reduces (including significantly reduces) SOST biological activity. The anti-SOST antibody of the present invention should exhibit any one or more of the following characteristics: (a) binds to SOST; (b) neutralizes, decreases, and/or downregulates the protein expression of SOST; (c) increases bone formation, bone mass, bone volume, bone mineralization, bone quality, bone strength, or bone density (e.g., bone mineral density) in a subject; (d) treats, prevents, or ameliorates one or more SOST associated disorder(s) (e.g., bone related disorder, including but not limited to osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, rheumatoid arthritis, osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss, metastatic bone malignancy, multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease (e.g., Crohn's disease or ulcerative colitis), hemochromatosis, celiac sprue (or celiac disease), renal tubular acidosis, renal osteodystrophy, hypercalciuria, fibrous dysplasia, or diabetes); (e) blocks binding of SOST to LRP5 and/or LRP6 and reverses the inhibition of canonical Wnt activity by SOST; and activates canonical Wnt signaling activity (e.g., stimulating osteoblast proliferation, prolonging osteoblast and osteocyte survival, and enhancing bone mass and bone strength through activation of Wnt/p-catenin signaling pathway); and (f) blocks SOST interaction with other yet to be identified factors in the canonical Wnt signaling pathway. In some embodiments, the anti-SOST antibodies of the present invention have at least two or more of these characteristics. In some embodiments, the anti-SOST antibodies have at least three or more of the characteristics.
In one aspect, provided is an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GXiTFX2DYWMQ, wherein X^ is F or H, X2 is T or S (SEQ ID NO: 81 ) or GXiTFX2DY, wherein X^ is F or H, and X2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX2X3KX4, wherein X is A or N, X2 is S or K, X3 is V or F, and X4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SX-i DYW, wherein X^ is F or M (SEQ ID NO: 84); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGXiX2FMH, wherein X is S or N, and X2 is N or S (SEQ ID NO: 85); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQX1 IX2X3X4YT , wherein X^ is T or S, X2 is E or D, X3 is H, D, F, or E, and X4 is H, P, or S (SEQ ID NO: 86).
In another aspect, provided is an isolated antibody, or an antigen binding fragment thereof, which specifically binds to a SOST polypeptide (e.g. , human SOST polypeptide), wherein the antibody comprises (a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence X1 ISGGDTYTYYADSVKG, wherein X is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence X-iSSQSLLDNDGETYLN, wherein X^ is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
The antibodies useful in the present invention can encompass monoclonal antibodies, polyclonal antibodies, antibody fragments (e.g. , Fab, Fab', F(ab')2, Fv, Fc, etc.), chimeric antibodies, bispecific antibodies, heteroconjugate antibodies, single chain (ScFv), mutants thereof, fusion proteins comprising an antibody portion (e.g., a domain antibody), humanized antibodies, and any other modified configuration of the
immunoglobulin molecule that comprises an antigen recognition site of the required specificity, including glycosylation variants of antibodies, amino acid sequence variants of antibodies, and covalently modified antibodies. The antibodies may be murine, rat, human, or any other origin (including chimeric or humanized antibodies). In some embodiments, the anti-SOST antibody as described herein is a monoclonal antibody. In some embodiments, the anti-SOST antibody is a humanized monoclonal antibody or a chimeric monoclonal antibody.
In some embodiments, the binding affinity (KD) of the anti-SOST antibodies of the present invention can be about 0.0005 to about 100 nM. In some embodiments, the binding affinity is any of about 100 nM, about 50 nM, about 10 nM, about 7.5 nM, about 5 nM, about 3.7 nM, about 1 nM, about 500 pM, about 250 pM, about 130 pM, about 100 pM, about 61 pM, about 60 pM, about 50 pM, about 21 pM, about 20 pM, about 19 pM, about 15 pM, about 10 pM, about 9 pM, about 5 pM, about 2 pM, or about 1 pM. In some embodiments, the binding affinity is less than any of about 200 nM, about 150 nM, about 100 nM, about 50 nM, about 10 nM, about 5 nM, about 1 nM, about 500 pM, about 100 pM, about 75 pM, about 50 pM, about 25 pM, about 20 pM, about 10 pM, about 5 pM, or about 2 pM.
In some embodiments, the binding affinity of the anti-SOST antibodies as described herein is about 4 nM or less as measured by surface plasmon resonance at 37°C. In some embodiments, the binding affinity of the antibodies as described herein is about 100 pM or less as measured by surface plasmon resonance at room temperature (e.g., 20°C to 26 °C).
Binding affinity may be determined using Kinexa Biosensor, scintillation proximity assays, Enzyme-linked Immunosorbent Assay (ELISA), ORIGEN immunoassay (IGEN), fluorescence quenching, fluorescence transfer, and/or yeast display. Binding affinity may also be screened using a suitable bioassay.
One way of determining binding affinity of antibodies to SOST is by measuring binding affinity of monofunctional Fab fragments of the antibody. To obtain
monofunctional Fab fragments, an antibody (for example, IgG) can be cleaved with papain or expressed recombinantly. The affinity of a SOST Fab fragment of an antibody can be determined by surface plasmon resonance (Biacore™3000™ surface plasmon resonance (SPR) system, Biacore™, INC, Piscataway NJ) equipped with pre- immobilized streptavidin sensor chips (SA) or anti-mouse Fc or anti-human Fc using
HBS-EP running buffer (0.01 M HEPES, pH 7.4, 0.15 NaCI, 3 mM EDTA, 0.005% v/v Surfactant P20). Biotinylated or Fc fusion human SOST can be diluted into HBS-EP buffer to a concentration of less than 0.5 pg/mL and injected across the individual chip channels using variable contact times, to achieve two ranges of antigen density, either 50-200 response units (RU) for detailed kinetic studies or 800-1 ,000 RU for screening assays. Regeneration studies have shown that 25 mM NaOH in 25% v/v ethanol effectively removes the bound Fab while keeping the activity of SOST on the chip for over 200 injections. Typically, serial dilutions (spanning concentrations of 0.1 -1 Ox estimated KD) of purified Fab samples are injected for 1 min at 100 μΙ_/Γη ίηυίβ and dissociation times of up to 2 hours are allowed. The concentrations of the Fab proteins are determined by ELISA and/or SDS-PAGE electrophoresis using a Fab of known concentration (as determined by amino acid analysis) as a standard. Kinetic association rates (kon) and dissociation rates (k0ff) are obtained simultaneously by fitting the data globally to a 1 : 1 Langmuir binding model (Karlsson, R. Roos, H. Fagerstam, L.
Petersson, B. (1994). Methods Enzymology 6. 99-1 10) using the BIAevaluation program. Equilibrium dissociation constant (KD) values are calculated as k0fl/kon. This protocol is suitable for use in determining binding affinity of an antibody to any SOST, including human SOST, SOST of another mammal (such as mouse SOST, rat SOST, or primate SOST), as well as different forms of SOST (e.g., glycosylated SOST). Binding affinity of an antibody is generally measured at 37°C, but can also be measured at room temperature or 25°C.
In some embodiments, the invention encompasses compositions, including pharmaceutical compositions, comprising antibodies described herein or made by the methods and having the characteristics described herein. As used herein, compositions comprise one or more antibodies that bind to SOST, and/or one or more polynucleotides comprising sequences encoding one or more these antibodies. These compositions may further comprise suitable excipients, such as pharmaceutically acceptable excipients including buffers, which are well known in the art.
Accordingly, the invention provides an antibody or compositions (including pharmaceutical compositions) comprising an antibody having a partial light chain sequence and/or a partial heavy chain sequence as found in Table 1 , or variants thereof. In Table 1 , the underlined sequences are CDR sequences according to Kabat, and the sequences in bold are CDR sequences according to Chothia.
Table 1
mAb Light Chain Heavy Chain
DP99 DVVLTQIPLTLSVTIGQPATIS EVMLAESGGGLVQPGGSLQLSCAASGFT
CKSSQSLLDNDGETYLNWLL FSIYAMSWVRQTPEKRLEWVATISGGDTY
VRPGQSPKRLIYQVSKLDSG TYYADSVKGRFTISRDNAKNTLYLHMTGL
FPERFTGGGSGTDFTLKISRV RSEDTALYYCARHGYDDFDYWGLGTTLT EAEDLGIYYCWQGTHFPHTF VSS (SEQ ID NO: 2)
GGGTKLEIK (SEQ ID NO: 3)
DP1 DVVMTQSPLSLPVTLGQPASI EVQLVESGGGLVQPGGSLRLSCAASGFT
SCRSSQSLLDNDGETYLNW FSIYAMSWVRQAPGKGLEWVALISGGDT
FQQRPGQSPRRLIYQVSKLD YTYYADSVKGRFTISRDNAKNSLYLQMNS
SGVPDRFSGSGSGTDFTLKI LRAEDTAVYYCARHGYDDFDYWGQGTLV S RVEAE DVGVYYCWQGTH F TVSS (SEQ ID NO: 6)
PHTFGGGTKVEIK (SEQ ID
NO: 7)
DM99 DIVLTQSPASLAVSLGQRAFM QGQLQQSGAELARPGASVKLSCKSSGHT
SCRASKTVDSYGNSFMHWF FTDYWMQWVKQRPGQGLEWIGAIYPGD
QQKAGQPPKLLIHHSSNLES GDTRYNQKFKDKATLTADTFSNTAYMQL
GIPARFSGSGSRTDFTLTIDP N N LASE DSAVYYCARSFDYWAQGTTLTV VEADDVAIYYCLQSIEDPYTF SS (SEQ ID NO: 10)
GGGTKLEIK (SEQ ID NO: 1 1 )
DM1 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 15)
DM2 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 14)
mAb Light Chain Heavy Chain
GQGTKLEIK (SEQ ID NO: 16)
DM3 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 17)
DM4 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAEDVAVYYCLQTIEFPYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 18)
DM5 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAEDVAVYYCLQTIEFPYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 19)
DM6 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAEDVAVYYCLQTIEFPYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 20)
DM7 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 16)
mAb Light Chain Heavy Chain
DM8 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAEDVAVYYCLQTIEFPYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 20)
DM9 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
N C RAS KTVDSYGS N FM HWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 22)
DM10 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI ED PYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 23)
DM1 1 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI ED PYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 24)
DM12 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI ED PYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 28)
DM13 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
mAb Light Chain Heavy Chain
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 29)
DM14 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 23)
DM15 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 15)
DM16 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E DH YTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 30)
DM17 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E DSYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 31 )
DM18 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
mAb Light Chain Heavy Chain
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTIDEPYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO: 32)
DM19 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAEDVAVYYCLQTIEFHYTF S (SEQ ID NO: 25)
GQGTKLEIK (SEQ ID NO:33)
DM20 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FSDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 34)
GQGTKLEIK (SEQ ID NO: 24)
DM21 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FSDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 34)
GQGTKLEIK (SEQ ID NO: 29)
DM22 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 35)
GQGTKLEIK (SEQ ID NO: 24)
mAb Light Chain Heavy Chain
DM23 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWF FSDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 36)
GQGTKLEIK (SEQ ID NO: 24)
DM24 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWF FSDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 36)
GQGTKLEIK (SEQ ID NO: 29)
DM25 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWVGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 37)
GQGTKLEIK (SEQ ID NO: 24)
DM26 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWISAIYPGDG
QQKPGQPPKLLIHHSSNLES DTRYNQKFKDRFTISRDNSKNTLYLQMNS
GVPDRFSGSGSGTDFTLTISS LRAEDTAVYYCARSMDYWGQGTLVTVSS LQAE DVAVYYC LQTI EDPYTF (SEQ ID NO: 38)
GQGTKLEIK (SEQ ID NO: 24)
DM27 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWVSAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 39)
GQGTKLEIK (SEQ ID NO: 24)
DM28 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
mAb Light Chain Heavy Chain
NCRASKTVDSYGNSFMHWF FTDYWMQWVRQAPGKGLEWIGAIYPGD
QQKPGQPPKLLIHHSSNLES GDTRYAQSVKGRFTISRDNSKNTLYLQM
GVPDRFSGSGSGTDFTLTISS NS LRAE DTAVYYCARSM DYWGQGTLVTV LQAE DVAVYYC LQTI EDPYTF SS (SEQ ID NO: 40)
GQGTKLEIK (SEQ ID NO: 24)
DM29 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 29)
DM30 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 23)
DM31 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGHT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E DSYTF S (SEQ ID NO: 14)
GQGTKLEIK (SEQ ID NO: 41 )
DM32 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWF FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI EDPYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 29)
DM33 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWY FS DYWM Q WVRQ AP G KG L E WVGA I YPG D
mAb Light Chain Heavy Chain
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E H PYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 17)
DM34 D IVMTQSPDS LAVS LGE RATI EVQLLESGGGLVQPGGSLRLSCAASGFT
NCRASKTVDSYGNSFMHWY FS DYWM QWVRQ AP G KG L EWVGA I YPG D
QQKPGQPPKLLIYHSSNLES GDTRYNQKFKDRFTISRDNSKNTLYLQMN
GVPDRFSGSGSGTDFTLTISS S LRAE DTAVYYCARSM DYWGQGTLVTVS LQAE DVAVYYC LQTI E DSYTF S (SEQ ID NO: 21 )
GQGTKLEIK (SEQ ID NO: 41 )
The invention also provides CDR portions of antibodies to SOST. Determination of CDR regions is well within the skill of the art. It is understood that in some embodiments, CDRs can be a combination of the Kabat and Chothia CDR (also termed "combined CDRs" or "extended CDRs"). In another approach, referred to herein as the "conformational definition" of CDRs, the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., Journal of Biological Chemistry, 283: 1 156-1 166, 2008. In general, "conformational CDRs" include the residue positions in the Kabat CDRs and Vernier zones which are constrained in order to maintain proper loop structure for the antibody to bind a specific antigen. Determination of conformational CDRs is well within the skill of the art. In some embodiments, the CDRs are the Kabat CDRs. In other embodiments, the CDRs are the Chothia CDRs. In other embodiments, the CDRs are the extended, AbM, conformational, or contact CDRs. In other words, in embodiments with more than one CDR, the CDRs may be any of Kabat, Chothia, extended, AbM, conformational, contact CDRs or combinations thereof.
Table 2 provides examples of CDR sequences of anti-SOST antibodies provided herein.
Table 2. Anti-SOST antibodies and antigen-binding CDR sequences according to Kabat (underlined) and Chothia (bold)
MAb CDR1 CDR2 CDR3
MAb CDR1 CDR2 CDR3
DP99 HC GFTFSIYAMS (SEQ TISGGDTYTYYADSV HGYDDFDY (SEQ ID
ID NOs: KG (SEQ ID NOs: 46 NO: 48)
43 (extended), 44 and and 47)
45)
LC KSSQSLLDNDGETY QVSKLDS (SEQ ID WQGTHFPHT (SEQ
LN (SEQ ID NO: 49) NO: 50) ID NO: 51)
DP1 HC GFTFSIYAMS (SEQ LISGGDTYTYYADSV HGYDDFDY (SEQ ID
ID NOs: KG (SEQ ID NOs: 46 NO: 48)
43 (extended), 44 and and 52)
45)
LC RSSQSLLDNDGETY QVSKLDS (SEQ ID WQGTHFPHT (SEQ
LN (SEQ ID NO: 53) NO: 50) ID NO: 51)
DP HC Xi ISGGDTYTYYADS
Conse VKG, wherein X1 is T
nsus or L (SEQ ID NO: 79)
LC XiSSQSLLDNDGETY
LN, wherein Xi is K or
R (SEQ ID NO: 80)
DM99 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SFDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 59)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQSIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 62)
DM1 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 66)
DM2 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
MAb CDR1 CDR2 CDR3
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 66)
DM3 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 66)
DM4 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEFPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 67)
DM5 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEFPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 67)
DM6 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEFPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 67)
DM7 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
MAb CDR1 CDR2 CDR3
H (SEQ ID NO: 60) NO: 61) NO: 66)
DM8 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEFPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 67)
DM9 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGSNFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 70) NO: 61) NO: 71)
DM10 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM11 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM12 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM13 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
MAb CDR1 CDR2 CDR3
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM14 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM15 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM16 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDHYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 72)
DM17 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDSYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 73)
DM18 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIDEPYT (SEQ ID
MAb CDR1 CDR2 CDR3
H (SEQ ID NO: 60) NO: 61) NO: 74)
DM19 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEFHYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 75)
DM20 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM21 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM22 HC GFTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
76 (extended), 77 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM23 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM24 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
MAb CDR1 CDR2 CDR3
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM25 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM26 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM27 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 66)
54 (extended), 55 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM28 HC GHTFTDYWMQ (SEQ AIYPGDGDTRYAQS SMDYW (SEQ ID NO:
ID NOs: VKG (SEQ ID NOs: 57 66)
54 (extended), 55 and and 78)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM29 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
MAb CDR1 CDR2 CDR3
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM30 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM31 HC GHTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
63 (extended), 64 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDSYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 73)
DM32 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 71)
DM33 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEHPYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 66)
DM34 HC GFTFSDYWMQ (SEQ AIYPGDGDTRYNQK SMDYW (SEQ ID NO:
ID NOs: FKD (SEQ ID NOs: 57 65)
69 (extended), 68 and and 58)
56)
LC RASKTVDSYGNSFM HSSNLES (SEQ ID LQTIEDSYT (SEQ ID
H (SEQ ID NO: 60) NO: 61) NO: 73)
DM HC GX1TFX2DYWMQ, AIYPGDGDTRYXiQX SX1DYW, wherein Xi Conse wherein X! is F or H, X2 2XsKX4, wherein Xi is is F or M (SEQ I D NO:
MAb CDR1 CDR2 CDR3
nsus is T or S (SEQ ID NOs: A or N, X2 is S or K, 84)
81 (extended) and 82 X3 is V or F, and X4 is
G or D (SEQ ID NO:83)
LC RASKTVDSYGX1X2F LQX1 IX2X3X4YT ,
MH, wherein X-i is S wherein X! is T or S, X2 or N, and X2 is N or S is E or D, X3 is H, D, F, (SEQ ID NO: 85) or E, and X4 is H, P, or
S (SEQ ID NO: 86)
In some embodiments, the present invention provides an antibody that binds to SOST and competes with the antibody as described herein, such as DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, and DM34.
In some embodiments, the present invention provides an antibody or an antigen binding fragment, which specifically binds to SOST, wherein the antibody comprises a VH region comprising a sequence shown in SEQ ID NO: 14; and/or a VL region comprising a sequence shown in SEQ ID NO: 18. In some embodiments, the antibody comprises a light chain comprising the sequence DIVMTQSPDSLAVSLGERATINCRASKTVDSYGNSFMHWFQQKPGQPPKLLIHHSSNL ESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCLQTIEFPYTFGQGTKLEIKRTVAAP SVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKD STYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
(SEQ ID NO: 88) and a heavy chain comprising the sequence EVQLLESGGGLVQPGGSLRLSCAASGHTFSDYWMQVWRQAPGKGLEVWGAIYPGD GDTRYNQKFKDRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARSMDYWGQGTLVTVS SASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPV AGPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPRE EQFNSTFRWSVLTWHQDWLNGKEYKCKVSNKGLPSSIEKTISKTKGQPREPQVYTL PPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYS KLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 87).
In some embodiments, the present invention provides an antibody or an antigen binding fragment, which specifically bind to SOST, wherein the antibody comprises a VH region comprising a sequence shown in SEQ ID NO: 6; and/or a VL region comprising a sequence shown in SEQ ID NO: 7. In some embodiments, the antibody comprises a light chain comprising the sequence
DWMTQSPLSLPVTLGQPASISCRSSQSLLDNDGETYLNWFQQRPGQSPRRLIYQVSK LDSGVPDRFSGSGSGTDFTLKISRVEAEDVGVYYCWQGTHFPHTFGGGTKVEIKRTVA APSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDS KDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 90) and a heavy chain comprising the sequence
EVQLVESGGGLVQPGGSLRLSCAASGFTFSIYAMSVWRQAPGKGLEVWALISGGDTY TYYADSVKGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARHGYDDFDYWGQGTLVT VSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPA VLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPP VAGPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPR EEQFNSTFRWSVLTVVHQDWLNGKEYKCKVSNKGLPSSIEKTISKTKGQPREPQVYT LPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYS KLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 89).
In some embodiments, the invention also provides CDR portions of antibodies to anti-SOST antibodies based on CDR contact regions. CDR contact regions are regions of an antibody that imbue specificity to the antibody for an antigen. In general, CDR contact regions include the residue positions in the CDRs and Vernier zones which are constrained in order to maintain proper loop structure for the antibody to bind a specific antigen. See, e.g., Makabe et al., J. Biol. Chem., 283: 1 156-1 166, 2007. Determination of CDR contact regions is well within the skill of the art.
The anti-SOST antibodies as described herein may be made by any method known in the art. For the production of hybridoma cell lines, the route and schedule of immunization of the host animal are generally in keeping with established and
conventional techniques for antibody stimulation and production, as further described herein. General techniques for production of human and mouse antibodies are known in the art and/or are described herein.
It is contemplated that any mammalian subject including humans or antibody producing cells therefrom can be manipulated to serve as the basis for production of
mammalian, including human and hybridoma cell lines. Typically, the host animal is inoculated intraperitoneally, intramuscularly, orally, subcutaneously, intraplantar, and/or intradermally with an amount of immunogen, including as described herein.
Hybridomas can be prepared from the lymphocytes and immortalized myeloma cells using the general somatic cell hybridization technique of Kohler, B. and Milstein, C, Nature 256:495-497, 1975 or as modified by Buck, D. W., et al., In Vitro, 18:377-381 , 1982. Available myeloma lines, including but not limited to X63-Ag8.653 and those from the Salk Institute, Cell Distribution Center, San Diego, Calif., USA, may be used in the hybridization. Generally, the technique involves fusing myeloma cells and lymphoid cells using a fusogen such as polyethylene glycol, or by electrical means well known to those skilled in the art. After the fusion, the cells are separated from the fusion medium and grown in a selective growth medium, such as hypoxanthine-aminopterin-thymidine (HAT) medium, to eliminate unhybridized parent cells. Any of the media described herein, supplemented with or without serum, can be used for culturing hybridomas that secrete monoclonal antibodies. As another alternative to the cell fusion technique, EBV immortalized B cells may be used to produce the anti-SOST monoclonal antibodies of the subject invention. The hybridomas are expanded and subcloned, if desired, and supernatants are assayed for anti-immunogen activity by conventional immunoassay procedures (e.g., radioimmunoassay, enzyme immunoassay, or fluorescence
immunoassay).
Hybridomas that may be used as source of antibodies encompass all derivatives, progeny cells of the parent hybridomas that produce monoclonal antibodies specific for SOST, or a portion thereof.
Hybridomas that produce such antibodies may be grown in vitro or in vivo using known procedures. The monoclonal antibodies may be isolated from the culture media or body fluids, by conventional immunoglobulin purification procedures such as ammonium sulfate precipitation, gel electrophoresis, dialysis, chromatography, and ultrafiltration, if desired. Undesired activity, if present, can be removed, for example, by running the preparation over adsorbents made of the immunogen attached to a solid phase and eluting or releasing the desired antibodies off the immunogen. Immunization of a host animal with SOST (e.g., human SOST), or a fragment containing the target amino acid sequence conjugated to a protein that is immunogenic in the species to be immunized, e.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or
soybean trypsin inhibitor using a bifunctional or derivatizing agent, for example, maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine residues), N- hydroxysuccinimide (through lysine residues), glutaraldehyde, succinic anhydride, SOC , or R1 N=C=NR, where R and R1 are different alkyl groups, can yield a population of antibodies (e.g., monoclonal antibodies).
If desired, the anti-SOST antibody (monoclonal or polyclonal) of interest may be sequenced and the polynucleotide sequence may then be cloned into a vector for expression or propagation. The sequence encoding the antibody of interest may be maintained in vector in a host cell and the host cell can then be expanded and frozen for future use. Production of recombinant monoclonal antibodies in cell culture can be carried out through cloning of antibody genes from B cells by means known in the art. See, e.g. Tiller et al., J. Immunol. Methods 329, 1 12, 2008; U.S. Pat. No. 7,314,622.
In an alternative, the polynucleotide sequence may be used for genetic
manipulation to "humanize" the antibody or to improve the affinity, or other
characteristics of the antibody. For example, the constant region may be engineered to more nearly resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. It may be desirable to genetically manipulate the antibody sequence to obtain greater affinity to SOST and greater efficacy in inhibiting SOST.
There are four general steps to humanize a monoclonal antibody. These are: (1 ) determining the nucleotide and predicted amino acid sequence of the starting antibody light and heavy variable domains (2) designing the humanized antibody, i.e., deciding which antibody framework region to use during the humanizing process (3) the actual humanizing methodologies/techniques and (4) the transfection and expression of the humanized antibody. See, for example, U.S. Pat. Nos. 4,816,567; 5,807,715;
5,866,692; 6,331 ,415; 5,530, 101 ; 5,693,761 ; 5,693,762; 5,585,089; and 6, 180,370.
A number of "humanized" antibody molecules comprising an antigen binding site derived from a non-human immunoglobulin have been described, including chimeric antibodies having rodent or modified rodent V regions and their associated CDRs fused to human constant regions. See, for example, Winter et al. Nature 349:293-299, 1991 , Lobuglio et al. Proc. Nat. Acad. Sci. USA 86:4220-4224, 1989, Shaw et al. J Immunol. 138:4534-4538, 1987, and Brown et al. Cancer Res. 47:3577-3583, 1987. Other references describe rodent CDRs grafted into a human supporting framework region
(FR) prior to fusion with an appropriate human antibody constant region. See, for example, Riechmann et al. Nature 332:323-327, 1988, Verhoeyen et al. Science
239:1534-1536, 1988, and Jones et al. Nature 321 :522-525, 1986. Another reference describes rodent CDRs supported by recombinantly engineered rodent framework regions. See, for example, European Patent Publication No. 0519596. These
"humanized" molecules are designed to minimize unwanted immunological response toward rodent anti-human antibody molecules which limits the duration and
effectiveness of therapeutic applications of those moieties in human recipients. For example, the antibody constant region can be engineered such that it is immunologically inert (e.g., does not trigger complement lysis). See, e.g. PCT Publication No.
PCT/GB99/01441 ; UK Patent Application No. 9809951 .8. Other methods of humanizing antibodies that may also be utilized are disclosed by Daugherty et al., Nucl. Acids Res. 19:2471 -2476, 1991 , and in U.S. Pat. Nos. 6,180,377; 6,054,297; 5,997,867; 5,866,692; 6,210,671 ; and 6,350,861 ; and in PCT Publication No. WO 01/27160.
The general principles related to humanized antibodies discussed above are also applicable to customizing antibodies for use, for example, in dogs, cats, primate, equines, and bovines. Further, one or more aspects of humanizing an antibody described herein may be combined, e.g., CDR grafting, framework mutation and CDR mutation.
In one variation, fully human antibodies may be obtained by using commercially available mice that have been engineered to express specific human immunoglobulin proteins. Transgenic animals that are designed to produce a more desirable (e.g., fully human antibodies) or more robust immune response may also be used for generation of humanized or human antibodies. Examples of such technology are Xenomouse™ from Abgenix, Inc. (Fremont, CA) and HuMAb-Mouse® and TC Mouse™ from Medarex, Inc. (Princeton, NJ).
In an alternative, antibodies may be made recombinantly and expressed using any method known in the art. In another alternative, antibodies may be made
recombinantly by phage display technology. See, for example, U.S. Pat. Nos.
5,565,332; 5,580,717; 5,733,743; and 6,265,150; and Winter et al., Annu. Rev.
Immunol. 12:433-455, 1994. Alternatively, the phage display technology (McCafferty et al., Nature 348:552-553, 1990) can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from
unimmunized donors. According to this technique, antibody V domain genes are cloned in-frame into either a major or minor coat protein gene of a filamentous bacteriophage, such as M13 or fd, and displayed as functional antibody fragments on the surface of the phage particle. Because the filamentous particle contains a single- stranded DNA copy of the phage genome, selections based on the functional properties of the antibody also result in selection of the gene encoding the antibody exhibiting those properties. Thus, the phage mimics some of the properties of the B cell. Phage display can be performed in a variety of formats; for review see, e.g., Johnson, Kevin S. and Chiswell, David J., Current Opinion in Structural Biology 3:564-571 , 1993. Several sources of V-gene segments can be used for phage display. Clackson et al., Nature 352:624-628, 1991 , isolated a diverse array of anti-oxazolone antibodies from a small random combinatorial library of V genes derived from the spleens of immunized mice. A repertoire of V genes from unimmunized human donors can be constructed and antibodies to a diverse array of antigens (including self-antigens) can be isolated essentially following the techniques described by Mark et al., J. Mol. Biol. 222:581 -597, 1991 , or Griffith et al., EMBO J. 12:725-734, 1993. In a natural immune response, antibody genes accumulate mutations at a high rate (somatic hypermutation). Some of the changes introduced will confer higher affinity, and B cells displaying high-affinity surface immunoglobulin are preferentially replicated and differentiated during
subsequent antigen challenge. This natural process can be mimicked by employing the technique known as "chain shuffling." (Marks et al., Bio/Technol. 10:779-783, 1992). In this method, the affinity of "primary" human antibodies obtained by phage display can be improved by sequentially replacing the heavy and light chain V region genes with repertoires of naturally occurring variants (repertoires) of V domain genes obtained from unimmunized donors. This technique allows the production of antibodies and antibody fragments with affinities in the pM-nM range. A strategy for making very large phage antibody repertoires (also known as "the mother-of-all libraries") has been described by Waterhouse et al., Nucl. Acids Res. 21 :2265-2266, 1993. Gene shuffling can also be used to derive human antibodies from rodent antibodies, where the human antibody has similar affinities and specificities to the starting rodent antibody. According to this method, which is also referred to as "epitope imprinting", the heavy or light chain V domain gene of rodent antibodies obtained by phage display technique is replaced with a repertoire of human V domain genes, creating rodent-human chimeras. Selection on
antigen results in isolation of human variable regions capable of restoring a functional antigen binding site, i.e., the epitope governs (imprints) the choice of partner. When the process is repeated in order to replace the remaining rodent V domain, a human antibody is obtained (see PCT Publication No. WO 93/06213). Unlike traditional humanization of rodent antibodies by CDR grafting, this technique provides completely human antibodies, which have no framework or CDR residues of rodent origin.
Antibodies may be made recombinantly by first isolating the antibodies and antibody producing cells from host animals, obtaining the gene sequence, and using the gene sequence to express the antibody recombinantly in host cells (e.g., CHO cells). Another method which may be employed is to express the antibody sequence in plants (e.g., tobacco) or transgenic milk. Methods for expressing antibodies recombinantly in plants or milk have been disclosed. See, for example, Peeters, et al. Vaccine 19:2756, 2001 ; Lonberg, N. and D. Huszar Int. Rev. Immunol 13:65, 1995; and Pollock, et al., J Immunol Methods 231 : 147, 1999. Methods for making derivatives of antibodies, e.g., humanized, single chain, etc. are known in the art.
Immunoassays and flow cytometry sorting techniques such as fluorescence activated cell sorting (FACS) can also be employed to isolate antibodies that are specific for SOST.
DNA encoding the monoclonal antibodies is readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies). The hybridoma cells serve as a preferred source of such DNA. Once isolated, the DNA may be placed into expression vectors (such as expression vectors disclosed in PCT Publication No. WO 87/04462), which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. See, e.g., PCT Publication No. WO 87/04462. The DNA also may be modified, for example, by substituting the coding sequence for human heavy and light chain constant regions in place of the homologous murine sequences, Morrison et al., Proc. Nat. Acad. Sci.
81 :6851 , 1984, or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non-immunoglobulin polypeptide. In that manner,
"chimeric" or "hybrid" antibodies are prepared that have the binding specificity of an anti- SOST monoclonal antibody herein.
The anti-SOST antibodies as described herein can be identified or characterized using methods known in the art, whereby reduction of SOST expression levels is detected and/or measured. In some embodiments, an anti-SOST antibody is identified by incubating a candidate agent with SOST and monitoring binding and/or attendant reduction of SOST expression levels. The binding assay may be performed with purified SOST polypeptide(s), or with cells naturally expressing, or transfected to express, SOST polypeptide(s). In one embodiment, the binding assay is a competitive binding assay, where the ability of a candidate antibody to compete with a known SOST antibody for SOST binding is evaluated. The assay may be performed in various formats, including the ELISA format.
Following initial identification, the activity of a candidate anti-SOST antibody can be further confirmed and refined by bioassays, known to test the targeted biological activities. Alternatively, bioassays can be used to screen candidates directly. Some of the methods for identifying and characterizing anti-SOST antibodies are described in detail in the Examples.
Anti-SOST antibodies may be characterized using methods well known in the art. For example, one method is to identify the epitope to which it binds, or "epitope mapping." There are many methods known in the art for mapping and characterizing the location of epitopes on proteins, including solving the crystal structure of an antibody-antigen complex, competition assays, gene fragment expression assays, and synthetic peptide-based assays, as described, for example, in Chapter 1 1 of Harlow and Lane, Using Antibodies, a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999. In an additional example, epitope mapping can be used to determine the sequence to which an anti-SOST antibody binds. Epitope mapping is commercially available from various sources, for example, Pepscan Systems (Edelhertweg 15, 8219 PH Lelystad, The Netherlands). The epitope can be a linear epitope, i.e., contained in a single stretch of amino acids, or a conformational epitope formed by a three-dimensional interaction of amino acids that may not necessarily be contained in a single stretch. Peptides of varying lengths (e.g., at least 4-6 amino acids long) can be isolated or synthesized (e.g., recombinantly) and used for binding assays with an anti-SOST antibody. In another example, the epitope to which the anti-SOST
antibody binds can be determined in a systematic screening by using overlapping peptides derived from the SOST sequence and determining binding by the anti-SOST antibody. According to the gene fragment expression assays, the open reading frame encoding SOST is fragmented either randomly or by specific genetic constructions and the reactivity of the expressed fragments of SOST with the antibody to be tested is determined. The gene fragments may, for example, be produced by PCR and then transcribed and translated into protein in vitro, in the presence of radioactive amino acids. The binding of the antibody to the radioactively labeled SOST fragments is then determined by immunoprecipitation and gel electrophoresis. Certain epitopes can also be identified by using large libraries of random peptide sequences displayed on the surface of phage particles (phage libraries). Alternatively, a defined library of
overlapping peptide fragments can be tested for binding to the test antibody in simple binding assays. In an additional example, mutagenesis of an antigen binding domain, domain swapping experiments and alanine scanning mutagenesis can be performed to identify residues required, sufficient, and/or necessary for epitope binding. For example, domain swapping experiments can be performed using a mutant SOST in which various fragments of the SOST protein have been replaced (swapped) with sequences from SOST from another species (e.g., mouse), or a closely related, but antigenically distinct protein. By assessing binding of the antibody to the mutant SOST, the importance of the particular SOST fragment to antibody binding can be assessed.
Yet another method which can be used to characterize an anti-SOST antibody is to use competition assays with other antibodies known to bind to the same antigen, i.e., various fragments on SOST, to determine if the anti-SOST antibody binds to the same epitope as other antibodies. Competition assays are well known to those of skill in the art.
The antibody of the present invention can be produced by proteolytic or other degradation of the antibodies, by recombinant methods (i.e., single or fusion polypeptides) as described above or by chemical synthesis. Polypeptides of the antibodies, especially shorter polypeptides up to about 50 amino acids, are conveniently made by chemical synthesis. Methods of chemical synthesis are known in the art and are commercially available. For example, an antibody could be produced by an automated polypeptide synthesizer employing the solid phase method. See also, U.S. Pat. Nos. 5,807,715; 4,816,567; and 6,331 ,415.
In another alternative, the antibodies can be made recombinantly using procedures that are well known in the art. In one embodiment, a polynucleotide comprises a sequence encoding the heavy chain and/or the light chain variable regions of antibody DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, and DM34. The sequence encoding the antibody of interest may be maintained in a vector in a host cell and the host cell can then be expanded and frozen for future use. Vectors (including expression vectors) and host cells are further described herein.
The invention also encompasses scFv of antibodies of this invention. Single chain variable region fragments are made by linking light and/or heavy chain variable regions by using a short linking peptide (Bird et al., Science 242:423-426, 1988). An example of a linking peptide is (GGGGS^ (SEQ ID NO: 97), which bridges approximately 3.5 nm between the carboxy terminus of one variable region and the amino terminus of the other variable region. Linkers of other sequences have been designed and used (Bird et al., 1988, supra). Linkers should be short, flexible polypeptides and preferably comprised of less than about 20 amino acid residues. Linkers can in turn be modified for additional functions, such as attachment of drugs or attachment to solid supports. The single chain variants can be produced either recombinantly or synthetically. For synthetic production of scFv, an automated synthesizer can be used. For recombinant production of scFv, a suitable plasmid containing polynucleotide that encodes the scFv can be introduced into a suitable host cell, either eukaryotic, such as yeast, plant, insect or mammalian cells, or prokaryotic, such as E. coli. Polynucleotides encoding the scFv of interest can be made by routine manipulations such as ligation of polynucleotides. The resultant scFv can be isolated using standard protein purification techniques known in the art.
Other forms of single chain antibodies, such as diabodies are also encompassed. Diabodies are bivalent, bispecific antibodies in which heavy chain variable (VH) and light chain variable (VL) domains are expressed on a single polypeptide chain, but using a linker that is too short to allow for pairing between the two domains on the same chain, thereby forcing the domains to pair with complementary domains of another chain and creating two antigen binding sites (see e.g., Holliger, P., et al., Proc. Natl. Acad Sci. USA 90:6444-6448, 1993; Poljak, R. J., et al. , Structure 2:1 121 -1 123, 1994).
For example, bispecific antibodies, monoclonal antibodies that have binding specificities for at least two different antigens, can be prepared using the antibodies disclosed herein. Methods for making bispecific antibodies are known in the art (see, e.g., Suresh et al., Methods in Enzymology 121 :210, 1986). Traditionally, the recombinant production of bispecific antibodies was based on the coexpression of two immunoglobulin heavy chain-light chain pairs, with the two heavy chains having different specificities (Millstein and Cuello, Nature 305, 537-539, 1983).
According to one approach to making bispecific antibodies, antibody variable domains with the desired binding specificities (antibody-antigen combining sites) are fused to immunoglobulin constant region sequences. The fusion preferably is with an immunoglobulin heavy chain constant region, comprising at least part of the hinge, CH2 and CH3 regions. It is preferred to have the first heavy chain constant region (CH1 ), containing the site necessary for light chain binding, present in at least one of the fusions. DNAs encoding the immunoglobulin heavy chain fusions and, if desired, the immunoglobulin light chain, are inserted into separate expression vectors, and are cotransfected into a suitable host organism. This provides for great flexibility in adjusting the mutual proportions of the three polypeptide fragments in embodiments when unequal ratios of the three polypeptide chains used in the construction provide the optimum yields. It is, however, possible to insert the coding sequences for two or all three polypeptide chains in one expression vector when the expression of at least two polypeptide chains in equal ratios results in high yields or when the ratios are of no particular significance.
In one approach, the bispecific antibodies are composed of a hybrid immunoglobulin heavy chain with a first binding specificity in one arm, and a hybrid immunoglobulin heavy chain-light chain pair (providing a second binding specificity) in the other arm. This asymmetric structure, with an immunoglobulin light chain in only one half of the bispecific molecule, facilitates the separation of the desired bispecific compound from unwanted immunoglobulin chain combinations. This approach is described in PCT Publication No. WO 94/04690.
In another approach, the bispecific antibodies are composed of amino acid modification in the first hinge region in one arm, and the substituted/replaced amino acid in the first hinge region has an opposite charge to the corresponding amino acid in the second hinge region in another arm. The CH3 region in one or both arms may also
include one or more amino acid modification(s). This approach is described in PCT Publication No. WO201 1/143545.
In another approach, the bispecific antibodies can be generated using a glutamine-containing peptide tag engineered to the antibody directed to an epitope (e.g., SOST) in one arm and another peptide tag (e.g., a Lys-containing peptide tag or a reactive endogenous Lys) engineered to a second antibody directed to a second epitope in another arm in the presence of transglutaminase. This approach is described in International Patent Application No. PCT/IB201 1/054899.
In some embodiments, the bispecific antibody has a first binding specificity to SOST in one arm, and a second binding specificity to DKK-1 (Dickkopf 1 ) in the other arm.
Heteroconjugate antibodies, comprising two covalently joined antibodies, are also within the scope of the invention. Such antibodies have been used to target immune system cells to unwanted cells (U.S. Pat. No. 4,676,980), and for treatment of HIV infection (PCT Publication Nos. WO 91/00360 and WO 92/200373; EP 03089). Heteroconjugate antibodies may be made using any convenient cross-linking methods. Suitable cross-linking agents and techniques are well known in the art, and are described in U.S. Pat. No. 4,676,980.
Chimeric or hybrid antibodies also may be prepared in vitro using known methods of synthetic protein chemistry, including those involving cross-linking agents. For example, immunotoxins may be constructed using a disulfide exchange reaction or by forming a thioether bond. Examples of suitable reagents for this purpose include iminothiolate and methyl-4-mercaptobutyrimidate.
In the recombinant humanized antibodies, the Fey portion can be modified to avoid interaction with Fey receptor and the complement and immune systems. The techniques for preparation of such antibodies are described in WO 99/58572. For example, the constant region may be engineered to more resemble human constant regions to avoid immune response if the antibody is used in clinical trials and treatments in humans. See, for example, U.S. Pat. Nos. 5,997,867 and 5,866,692.
The invention encompasses modifications to the antibodies and polypeptides of the invention variants shown in Table 3, including functionally equivalent antibodies which do not significantly affect their properties and variants which have enhanced or decreased activity and/or affinity. For example, the amino acid sequence may be
mutated to obtain an antibody with the desired binding affinity to SOST. Modification of polypeptides is routine practice in the art and need not be described in detail herein. Examples of modified polypeptides include polypeptides with conservative substitutions of amino acid residues, one or more deletions or additions of amino acids which do not significantly deleteriously change the functional activity, or which mature (enhance) the affinity of the polypeptide for its ligand, or use of chemical analogs.
Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues. Examples of terminal insertions include an antibody with an N-terminal methionyl residue or the antibody fused to an epitope tag. Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the antibody of an enzyme or a polypeptide which increases the half-life of the antibody in the blood circulation.
Substitution variants have at least one amino acid residue in the antibody molecule removed and a different residue inserted in its place. The sites of greatest interest for substitutional mutagenesis include the hypervariable regions, but FR alterations are also contemplated. Conservative substitutions are shown in Table 3 under the heading of "conservative substitutions." If such substitutions result in a change in biological activity, then more substantial changes, denominated "exemplary substitutions" in Table 3, or as further described below in reference to amino acid classes, may be introduced and the products screened.
Table 3: Amino Acid Substitutions
Conservative
Original Residue Substitutions Exemplary Substitutions
Ala (A) Val Val; Leu; lie
Arg (R) Lys Lys Gin; Asn
Asn (N) Gin Gin His; Asp, Lys; Arg
Asp (D) Glu Glu Asn
Cys (C) Ser Ser Ala
Gin (Q) Asn Asn; Glu
Glu (E) Asp Asp; Gin
Gly (G) Ala Ala
Conservative
Original Residue Substitutions Exemplary Substitutions
His (H) Arg Asn; Gin; Lys; Arg
Leu; Val; Met; Ala; Phe;
He (1) Leu
Norleucine
Norleucine; lie; Val; Met;
Leu (L) He
Ala; Phe
Lys (K) Arg Arg; Gin; Asn
Met (M) Leu Leu; Phe; lie
Phe (F) Tyr Leu; Val; lie; Ala; Tyr
Pro (P) Ala Ala
Ser (S) Thr Thr
Thr (T) Ser Ser
Trp (W) Tyr Tyr; Phe
Tyr (Y) Phe Trp; Phe; Thr; Ser
lie; Leu; Met; Phe; Ala;
Val (V) Leu
Norleucine
Substantial modifications in the biological properties of the antibody are accomplished by selecting substitutions that differ significantly in their effect on maintaining (a) the structure of the polypeptide backbone in the area of the substitution, for example, as a sheet or helical conformation, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the bulk of the side chain. Naturally occurring residues are divided into groups based on common side-chain properties:
(1 ) Non-polar: Norleucine, Met, Ala, Val, Leu, lie;
(2) Polar without charge: Cys, Ser, Thr, Asn, Gin;
(3) Acidic (negatively charged): Asp, Glu;
(4) Basic (positively charged): Lys, Arg;
(5) Residues that influence chain orientation: Gly, Pro; and
(6) Aromatic: Trp, Tyr, Phe, His.
Non-conservative substitutions are made by exchanging a member of one of these classes for another class.
Any cysteine residue not involved in maintaining the proper conformation of the antibody also may be substituted, generally with serine, to improve the oxidative stability of the molecule and prevent aberrant cross-linking. Conversely, cysteine bond(s) may
be added to the antibody to improve its stability, particularly where the antibody is an antibody fragment such as an Fv fragment.
Amino acid modifications can range from changing or modifying one or more amino acids to complete redesign of a region, such as the variable region. Changes in the variable region can alter binding affinity and/or specificity. In some embodiments, no more than one to five conservative amino acid substitutions are made within a CDR domain. In other embodiments, no more than one to three conservative amino acid substitutions are made within a CDR domain. In still other embodiments, the CDR domain is CDR H3 and/or CDR L3.
Modifications also include glycosylated and nonglycosylated polypeptides, as well as polypeptides with other post-translational modifications, such as, for example, glycosylation with different sugars, acetylation, and phosphorylation. Antibodies are glycosylated at conserved positions in their constant regions (Jefferis and Lund, Chem. Immunol. 65: 1 1 1 -128, 1997; Wright and Morrison, TibTECH 15:26-32, 1997). The oligosaccharide side chains of the immunoglobulins affect the protein's function (Boyd et al., Mol. Immunol. 32: 131 1 -1318, 1996; Wittwe and Howard, Biochem. 29:4175-4180, 1990) and the intramolecular interaction between portions of the glycoprotein, which can affect the conformation and presented three-dimensional surface of the glycoprotein (Jefferis and Lund, supra; Wyss and Wagner, Current Opin. Biotech. 7:409-416, 1996). Oligosaccharides may also serve to target a given glycoprotein to certain molecules based upon specific recognition structures. Glycosylation of antibodies has also been reported to affect antibody-dependent cellular cytotoxicity (ADCC). In particular, CHO cells with tetracycline-regulated expression of β(1 ,4)-N-acetylglucosaminyltransferase III (GnTIII), a glycosy transferase catalyzing formation of bisecting GlcNAc, was reported to have improved ADCC activity (Umana et al., Mature Biotech. 17: 176-180, 1999).
Glycosylation of antibodies is typically either N-linked or O-linked. N-linked refers to the attachment of the carbohydrate moiety to the side chain of an asparagine residue. The tripeptide sequences asparagine-X-serine, asparagine-X-threonine, and asparagine-X-cysteine, where X is any amino acid except proline, are the recognition sequences for enzymatic attachment of the carbohydrate moiety to the asparagine side chain. Thus, the presence of either of these tripeptide sequences in a polypeptide creates a potential glycosylation site. O-linked glycosylation refers to the attachment of one of the sugars N-acetylgalactosamine, galactose, or xylose to a hydroxyamino acid,
most commonly serine or threonine, although 5-hydroxyproline or 5-hydroxylysine may also be used.
Addition of glycosylation sites to the antibody is conveniently accomplished by altering the amino acid sequence such that it contains one or more of the above- described tripeptide sequences (for N-linked glycosylation sites). The alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the original antibody (for O-linked glycosylation sites).
The glycosylation pattern of antibodies may also be altered without altering the underlying nucleotide sequence. Glycosylation largely depends on the host cell used to express the antibody. Since the cell type used for expression of recombinant glycoproteins, e.g. antibodies, as potential therapeutics is rarely the native cell, variations in the glycosylation pattern of the antibodies can be expected (see, e.g. Hse et al., J. Biol. Chem. 272:9062-9070, 1997).
In addition to the choice of host cells, factors that affect glycosylation during recombinant production of antibodies include growth mode, media formulation, culture density, oxygenation, pH, purification schemes and the like. Various methods have been proposed to alter the glycosylation pattern achieved in a particular host organism including introducing or overexpressing certain enzymes involved in oligosaccharide production (U.S. Pat. Nos. 5,047,335; 5,510,261 and 5,278,299). Glycosylation, or certain types of glycosylation, can be enzymatically removed from the glycoprotein, for example, using endoglycosidase H (Endo H), N-glycosidase F, endoglycosidase F1 , endoglycosidase F2, endoglycosidase F3. In addition, the recombinant host cell can be genetically engineered to be defective in processing certain types of polysaccharides. These and similar techniques are well known in the art.
Other methods of modification include using coupling techniques known in the art, including, but not limited to, enzymatic means, oxidative substitution and chelation. Modifications can be used, for example, for attachment of labels for immunoassay. Modified polypeptides are made using established procedures in the art and can be screened using standard assays known in the art, some of which are described below and in the Examples.
In some embodiments of the invention, the antibody comprises a modified constant region, such as a constant region that increased potential for provoking an immune response (e.g., increased affinity to a human Fc gamma receptor such as, e.g.,
FcyRI, FcyRIIA, or Fcylll), is immunologically inert or partially inert, e.g., does not trigger complement mediated lysis, does not stimulate antibody-dependent cell mediated cytotoxicity (ADCC), or does not activate macrophages; or has reduced activities (compared to the unmodified antibody) in any one or more of the following: triggering complement mediated lysis, or stimulating antibody-dependent cell mediated cytotoxicity (ADCC). Different modifications of the constant region may be used to achieve optimal level and/or combination of effector functions. See, for example, Morgan et al., Immunology 86:319-324, 1995; Lund et al., J. Immunology 157:4963-9 157:4963-4969, 1996; Idusogie et al., J. Immunology 164:4178-4184, 2000; Tao et al., J. Immunology 143: 2595-2601 , 1989; and Jefferis et al., Immunological Reviews 163:59-76, 1998. In some embodiments, the constant region is modified as described in Eur. J. Immunol., 1999, 29:2613-2624; PCT Application No. PCT/GB 99/01441 ; and/or UK Patent Application No. 9809951 .8. The Fc can be human lgG1 , human lgG2, human lgG3, or human lgG4. In some embodiments, the antibodies of the present invention comprise a human heavy chain lgG2 constant region comprising the following mutations: A330P331 to S330S331 (amino acid numbering with reference to the wild type lgG2 sequence). Eur. J. Immunol., 1999, 29:2613-2624. In some embodiments, the antibodies of the present invention comprises a constant region of lgG4 comprising the following mutations (Armour et al., Molecular Immunology 40 585-593, 2003): E233F234L235 to P233V234A235 (lgG4Ac), in which the numbering is with reference to wild type lgG4. In yet another embodiment, the Fc is human lgG4 E233F234L235 to P233V234A235 with deletion G236 (lgG4Ab). In another embodiment the Fc is any human lgG4 Fc (lgG4, lgG4Ab or lgG4Ac) containing hinge stabilizing mutation S228 to P228 (Aalberse et al., Immunology 105, 9-19, 2002). In another embodiment, the Fc can be aglycosylated Fc. In other embodiments, the constant region is aglycosylated for N-linked glycosylation. In some embodiments, the constant region is aglycosylated for N-linked glycosylation by mutating the glycosylated amino acid residue or flanking residues that are part of the N- glycosylation recognition sequence in the constant region. For example, N-glycosylation site N297 may be mutated to A, Q, K, or H. See, Tao et al., J. Immunology 143: 2595- 2601 , 1989; and Jefferis et al., Immunological Reviews 163:59-76, 1998. The constant region may be aglycosylated for N-linked glycosylation enzymatically (such as removing carbohydrate by enzyme PNGase), or by expression in a glycosylation deficient host cell.
Other antibody modifications include antibodies that have been modified as described in PCT Publication No. WO 99/58572. These antibodies comprise, in addition to a binding domain directed at the target molecule, an effector domain having an amino acid sequence substantially homologous to all or part of a constant region of a human immunoglobulin heavy chain. These antibodies are capable of binding the target molecule without triggering significant complement dependent lysis, or cell-mediated destruction of the target. In some embodiments, the effector domain is capable of specifically binding FcRn and/or FcYRIIb. These are typically based on chimeric domains derived from two or more human immunoglobulin heavy chain CH2 domains. Antibodies modified in this manner are particularly suitable for use in chronic antibody therapy, to avoid inflammatory and other adverse reactions to conventional antibody therapy.
A modification or mutation may also be made in a framework region or constant region to increase the half-life of an SOST antibody. See, e.g., PCT Publication No. WO 00/09560. A mutation in a framework region or constant region can also be made to alter the immunogenicity of the antibody, to provide a site for covalent or non-covalent binding to another molecule, or to alter such properties as complement fixation, FcR binding and antibody-dependent cell-mediated cytotoxicity. According to the invention, a single antibody may have mutations in any one or more of the CDRs or framework regions of the variable domain or in the constant region.
In a process known as "germ lining", certain amino acids in the VH and VL sequences can be mutated to match those found naturally in germline VH and VL sequences. In particular, the amino acid sequences of the framework regions in the VH and VL sequences can be mutated to match the germline sequences to reduce the risk of immunogenicity when the antibody is administered. Germline DNA sequences for human VH and VL genes are known in the art (see e.g., the "Vbase" human germline sequence database; see also Kabat, E. A., et al., 1991 , Sequences of Proteins of Immunological Interest, Fifth Edition, U.S. Department of Health and Human Services, NIH Publication No. 91 -3242; Tomlinson et al. , J. Mol. Biol. 227:776-798, 1992; and Cox et al., Eur. J. Immunol. 24:827-836, 1994).
Another type of amino acid substitution that may be made is to remove potential proteolytic sites in the antibody. Such sites may occur in a CDR or framework region of a variable domain or in the constant region of an antibody. Substitution of cysteine
residues and removal of proteolytic sites may decrease the risk of heterogeneity in the antibody product and thus increase its homogeneity. Another type of amino acid substitution is to eliminate asparagine-glycine pairs, which form potential deamidation sites, by altering one or both of the residues. In another example, the C-terminal lysine of the heavy chain of an anti-SOST antibody of the invention can be cleaved. In various embodiments of the invention, the heavy and light chains of the SOST antibodies may optionally include a signal sequence.
Once DNA fragments encoding the VH and VL segments of the present invention are obtained, these DNA fragments can be further manipulated by standard
recombinant DNA techniques, for example to convert the variable region genes to full- length antibody chain genes, to Fab fragment genes, or to a scFv gene. In these manipulations, a VL- or VH-encoding DNA fragment is operatively linked to another DNA fragment encoding another protein, such as an antibody constant region or a flexible linker. The term "operatively linked", as used in this context, is intended to mean that the two DNA fragments are joined such that the amino acid sequences encoded by the two DNA fragments remain in-frame.
The isolated DNA encoding the VH region can be converted to a full-length heavy chain gene by operatively linking the VH-encoding DNA to another DNA molecule encoding heavy chain constant regions (CH1 , CH2 and CH3). The sequences of human heavy chain constant region genes are known in the art (see e.g., Kabat, E. A., et al., 1991 , Sequences of Proteins of Immunological Interest, Fifth Edition, U.S.
Department of Health and Human Services, NIH Publication No. 91 -3242) and DNA fragments encompassing these regions can be obtained by standard PCR
amplification. The heavy chain constant region can be an lgG1 , lgG2, lgG3, lgG4, IgA, IgE, IgM or IgD constant region, but most preferably is an lgG1 or lgG2 constant region. The IgG constant region sequence can be any of the various alleles or allotypes known to occur among different individuals, such as Gm(1 ), Gm(2), Gm(3), and
Gm(17). These allotypes represent naturally occurring amino acid substitution in the lgG1 constant regions. For a Fab fragment heavy chain gene, the VH-encoding DNA can be operatively linked to another DNA molecule encoding only the heavy chain CH1 constant region. The CH1 heavy chain constant region may be derived from any of the heavy chain genes.
The isolated DNA encoding the VL region can be converted to a full-length light chain gene (as well as a Fab light chain gene) by operatively linking the VL-encoding DNA to another DNA molecule encoding the light chain constant region, CL. The sequences of human light chain constant region genes are known in the art (see e.g., Kabat, E. A., et al., 1991 , Sequences of Proteins of Immunological Interest, Fifth Edition, U.S. Department of Health and Human Services, NIH Publication No. 91 -3242) and DNA fragments encompassing these regions can be obtained by standard PCR amplification. The light chain constant region can be a kappa or lambda constant region. The kappa constant region may be any of the various alleles known to occur among different individuals, such as lnv(1 ), lnv(2), and lnv(3). The lambda constant region may be derived from any of the three lambda genes.
To create a scFv gene, the VH- and VL-encoding DNA fragments are operatively linked to another fragment encoding a flexible linker, e.g., encoding the amino acid sequence (Gly4 -Ser)3, (SEQ ID NO: 98) such that the VH and VL sequences can be expressed as a contiguous single-chain protein, with the VL and VH regions joined by the flexible linker (See e.g., Bird et al., 1988, Science 242:423-426; Huston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., 1990, Nature 348:552- 554. The single chain antibody may be monovalent, if only a single VH and VL are used, bivalent, if two VH and VL are used, or polyvalent, if more than two VH and VL are used. Bispecific or polyvalent antibodies may be generated that bind specifically to SOST and to another molecule.
In another embodiment, a fusion antibody or immunoadhesin may be made that comprises all or a portion of an anti-SOST antibody of the invention linked to another polypeptide. In another embodiment, only the variable domains of the SOST antibody are linked to the polypeptide. In another embodiment, the VH domain of an SOST antibody is linked to a first polypeptide, while the VL domain of an SOST antibody is linked to a second polypeptide that associates with the first polypeptide in a manner such that the VH and VL domains can interact with one another to form an antigen binding site. In another preferred embodiment, the VH domain is separated from the VL domain by a linker such that the VH and VL domains can interact with one another. The VH-linker- VL antibody is then linked to the polypeptide of interest. In addition, fusion antibodies can be created in which two (or more) single-chain antibodies are linked to
one another. This is useful if one wants to create a divalent or polyvalent antibody on a single polypeptide chain, or if one wants to create a bispecific antibody.
In other embodiments, other modified antibodies may be prepared using SOST antibody encoding nucleic acid molecules. For instance, "Kappa bodies" (III et al., Protein Eng. 10:949-57, 1997), "Minibodies" (Martin et al., EMBO J., 13:5303-9, 1994), "Diabodies" (Holliger et al., Proc. Natl. Acad. Sci. USA 90:6444-6448, 1993), or
"Janusins" (Traunecker et al., EMBO J. 10:3655-3659, 1991 and Traunecker et al., Int. J. Cancer (Suppl.) 7:51 -52, 1992) may be prepared using standard molecular biological techniques following the teachings of the specification.
The invention includes affinity matured embodiments. For example, affinity matured antibodies can be produced by procedures known in the art (Marks et al., Bio/Technology, 10:779-783, 1992; Barbas et al., Proc Nat. Acad. Sci, USA 91 :3809- 3813, 1994; Schier et al., Gene, 169: 147-155, 1995; Yelton et al., J. Immunol., 155:1994-2004, 1995; Jackson et al., J. Immunol., 154(7):3310-9, 1995, Hawkins et al., J. Mol. Biol., 226:889-896, 1992; and PCT Publication No. WO2004/058184).
The following methods may be used for adjusting the affinity of an antibody and for characterizing a CDR. One way of characterizing a CDR of an antibody and/or altering (such as improving) the binding affinity of a polypeptide, such as an antibody, termed "library scanning mutagenesis". Generally, library scanning mutagenesis works as follows. One or more amino acid positions in the CDR are replaced with two or more (such as 3, 4, 5, 6, 7, 8, 9, 10, 1 1 , 12, 13, 14, 15, 16, 17, 18, 19, or 20) amino acids using art recognized methods. This generates small libraries of clones (in some embodiments, one for every amino acid position that is analyzed), each with a complexity of two or more members (if two or more amino acids are substituted at every position). Generally, the library also includes a clone comprising the native (unsubstituted) amino acid. A small number of clones, e.g., about 20-80 clones (depending on the complexity of the library), from each library are screened for binding affinity to the target polypeptide (or other binding target), and candidates with increased, the same, decreased, or no binding are identified. Methods for determining binding affinity are well-known in the art. Binding affinity may be determined using Biacore™ surface plasmon resonance analysis, which detects differences in binding affinity of about 2-fold or greater. Biacore™ is particularly useful when the starting antibody already binds with a relatively high affinity, for example a KD of about 10 nM or lower.
Screening using Biacore™ surface plasmon resonance is described in the Examples, herein.
In some embodiments, every amino acid position in a CDR is replaced (in some embodiments, one at a time) with all 20 natural amino acids using art recognized mutagenesis methods (some of which are described herein). This generates small libraries of clones (in some embodiments, one for every amino acid position that is analyzed), each with a complexity of 20 members (if all 20 amino acids are substituted at every position).
In some embodiments, the library to be screened comprises substitutions in two or more positions, which may be in the same CDR or in two or more CDRs. Thus, the library may comprise substitutions in two or more positions in one CDR. The library may comprise substitution in two or more positions in two or more CDRs. The library may comprise substitution in 3, 4, 5, or more positions, said positions found in two, three, four, five or six CDRs. The substitution may be prepared using low redundancy codons. See, e.g., Table 2 of Balint et al., Gene 137(1 ): 109-18, 1993.
The CDR may be CDRH3 and/or CDRL3. The CDR may be one or more of CDRL1 , CDRL2, CDRL3, CDRH1 , CDRH2, and/or CDRH3. The CDR may be a Kabat CDR, a Chothia CDR, or an extended CDR.
Candidates with improved binding may be sequenced, thereby identifying a CDR substitution mutant which results in improved affinity (also termed an "improved" substitution). Candidates that bind may also be sequenced, thereby identifying a CDR substitution which retains binding.
Multiple rounds of screening may be conducted. For example, candidates (each comprising an amino acid substitution at one or more position of one or more CDR) with improved binding are also useful for the design of a second library containing at least the original and substituted amino acid at each improved CDR position (i.e., amino acid position in the CDR at which a substitution mutant showed improved binding). Preparation, and screening or selection of this library is discussed further below.
Library scanning mutagenesis also provides a means for characterizing a CDR, in so far as the frequency of clones with improved binding, the same binding, decreased binding or no binding also provide information relating to the importance of each amino acid position for the stability of the antibody-antigen complex. For example, if a position of the CDR retains binding when changed to all 20 amino acids, that position is
identified as a position that is unlikely to be required for antigen binding. Conversely, if a position of CDR retains binding in only a small percentage of substitutions, that position is identified as a position that is important to CDR function. Thus, the library scanning mutagenesis methods generate information regarding positions in the CDRs that can be changed to many different amino acids (including all 20 amino acids), and positions in the CDRs which cannot be changed or which can only be changed to a few amino acids.
Candidates with improved affinity may be combined in a second library, which includes the improved amino acid, the original amino acid at that position, and may further include additional substitutions at that position, depending on the complexity of the library that is desired, or permitted using the desired screening or selection method. In addition, if desired, adjacent amino acid position can be randomized to at least two or more amino acids. Randomization of adjacent amino acids may permit additional conformational flexibility in the mutant CDR, which may in turn, permit or facilitate the introduction of a larger number of improving mutations. The library may also comprise substitution at positions that did not show improved affinity in the first round of screening.
The second library is screened or selected for library members with improved and/or altered binding affinity using any method known in the art, including screening using Biacore™ surface plasmon resonance analysis, and selection using any method known in the art for selection, including phage display, yeast display, and ribosome display.
The invention also encompasses fusion proteins comprising one or more fragments or regions from the antibodies of this invention. In one embodiment, a fusion polypeptide is provided that comprises at least 10 contiguous amino acids of the variable light chain region shown in SEQ ID NOs: 3, 7, 1 1 , 15, 16, 17, 18, 19, 20, 22, 23, 24, 28, 29, 30, 31 , 32, 33, and 41 and/or at least 10 amino acids of the variable heavy chain region shown in SEQ ID NOs: 2, 6, 10, 14, 21 , 25, 34, 35, 36, 37, 38, 39, and 40. In other embodiments, a fusion polypeptide is provided that comprises at least about 10, at least about 15, at least about 20, at least about 25, or at least about 30 contiguous amino acids of the variable light chain region and/or at least about 10, at least about 15, at least about 20, at least about 25, or at least about 30 contiguous amino acids of the variable heavy chain region. In another embodiment, the fusion
polypeptide comprises a light chain variable region and/or a heavy chain variable region, as shown in any of the sequence pairs selected from among SEQ ID NOs: 3 and 2, 7 and 6, 1 1 and 10, 15 and 14, 16 and 14, 17 and 14, 18 and 14, 19 and 14, 20 and 14, 16 and 21 , 20 and 21 , 22 and 14, 23 and 21 , 24 and 25, 28 and 25, 29 and 25, 23 and 25, 15 and 25, 30 and 25, 31 and 25, 32 and 25, 33 and 25, 24 and 34, 29 and 34, 24 and 35, 24 and 36, 29 and 36, 24 and 37, 24 and 38, 24 and 39, 24 and 40, 29 and 14, 23 and 14, 41 and 14, 29 and 21 , 17 and 21 , and 41 and 21 . In another embodiment, the fusion polypeptide comprises one or more CDR(s). In still other embodiments, the fusion polypeptide comprises CDR H3 (VH CDR3) and/or CDR L3 (VL CDR3). For purposes of this invention, a fusion protein contains one or more antibodies and another amino acid sequence to which it is not attached in the native molecule, for example, a heterologous sequence or a homologous sequence from another region. Exemplary heterologous sequences include, but are not limited to a "tag" such as a FLAG tag or a 6His tag. Tags are well known in the art.
A fusion polypeptide can be created by methods known in the art, for example, synthetically or recombinantly. Typically, the fusion proteins of this invention are made by preparing an expressing a polynucleotide encoding them using recombinant methods described herein, although they may also be prepared by other means known in the art, including, for example, chemical synthesis.
This invention also provides compositions comprising antibodies conjugated (for example, linked) to an agent that facilitate coupling to a solid support (such as biotin or avidin). For simplicity, reference will be made generally to antibodies with the understanding that these methods apply to any of the anti-SOST antibody embodiments described herein. Conjugation generally refers to linking these components as described herein. The linking (which is generally fixing these components in proximate association at least for administration) can be achieved in any number of ways. For example, a direct reaction between an agent and an antibody is possible when each possesses a substituent capable of reacting with the other. For example, a nucleophilic group, such as an amino or sulfhydryl group, on one may be capable of reacting with a carbonyl-containing group, such as an anhydride or an acid halide, or with an alkyl group containing a good leaving group (e.g., a halide) on the other.
The antibodies as described herein can be bound to many different carriers. Carriers can be active and/or inert. Examples of well-known carriers include
polypropylene, polystyrene, polyethylene, dextran, nylon, amylases, glass, natural and modified celluloses, polyacrylamides, agaroses, and magnetite. The nature of the carrier can be either soluble or insoluble for purposes of the invention. Those skilled in the art will know of other suitable carriers for binding antibodies, or will be able to ascertain such, using routine experimentation. In some embodiments, the carrier comprises a moiety that targets the myocardium.
This invention also provides a conjugate (or immunoconjugate) of the anti-SOST antibody as described herein, or the antigen binding fragment thereof, wherein the antibody or the antigen binding fragment is conjugate to a pharmaceutical agent for targeted immunotherapy (e.g., antibody-drug conjugates). The pharmaceutical agents that can be conjugated to the anti-SOST antibodies or the antigen binding fragments of the present invention include, but are not limited to, chemotherapeutic agents (e.g., cytotoxic agents), immunomodulating agents, imaging agents (e.g., labeling agents), therapeutic proteins, biopolymers, or oligonucleotides.
In some embodiments, the agent is an imaging agent (e.g., a fluorophore or a
PET (Positron Emission Tomography) label or SPECT (Single-Photon Emission
Computed Tomography) label), such as fluorescein, rhodamine, lanthanide phosphors, and their derivatives thereof and various isotopes.
Examples of fluorophores include, but are not limited to, fluorescein
isothiocyanate (FITC) (e.g., 5-FITC), fluorescein amidite (FAM) (e.g., 5-FAM), eosin, carboxyfluorescein, erythrosine, Alexa Fluor® (e.g., Alexa 350, 405, 430, 488, 500, 514, 532, 546, 555, 568, 594, 610, 633, 647, 660, 680, 700, or 750),
carboxytetramethylrhodamine (TAMRA) (e.g., 5,-TAMRA), tetramethylrhodamine (TMR), and sulforhodamine (SR) (e.g., SR101 ).
In some embodiments, therapeutic or diagnostic radioisotopes or other labels
(e.g., PET or SPECT labels) can be incorporated in the agent for conjugation to the anti- SOST antibodies or the antigen binding fragments as described herein. Examples of a radioisotope or other labels include, but are not limited to, 3H, 11C, 13N, 14C, 15N, 15O,
35S 18F 32p 33p 47^ 51 ^ 57^ 58^ 59^ 62^ 64^ 67^ 67^ 68^ 75^ 76^ 77Br, 86Y, 89Zr, 90Y, 94Tc, 95Ru, 97Ru, 99Tc, 103Ru, 105Rh, 105Ru, 107Hg, 109Pd, 111Ag, 111 ln, 113ln, 121Te, 122Te, 123l, 124l, 125l, 125Te, 126l, 3 1, 131 In, 133l, 142Pr, 143Pr, 153Pb, 153Sm, 161Tb, 165Tm, 166Dy, 166H, 167Tm, 168Tm, 169Yb, 177Lu, 186Re, 188Re, 189Re, 197Pt, 198Au, 199Au,
201T| 203H g 21 1At 212B. 212p b 213β. 223Rg 224^ gnd 225^
Polynucleotides, vectors, and host cells
The invention also provides polynucleotides encoding any of the antibodies (e.g., antagonistic antibodies), including antibody fragments and modified antibodies described herein, such as, e.g., antibodies having impaired effector function. Accordingly, the invention provides polynucleotides or compositions, including pharmaceutical compositions, comprising polynucleotides, encoding any of the following: DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, or DM34. In some embodiments, the antibodies or any fragment or part thereof have the ability to bind and antagonize SOST.
In some embodiments, the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 4 and SEQ ID NO: 5 below:
DP99 heavy chain variable region
GAAGTGATGCTGGCAGAGTCTGGGGGAGGCTTAGTGCAGCCTGGAGGGTCCCTG CAACTCTCCTGTGCAGCCTCTGGATTCACTTTCAGTATCTATGCCATGTCTTGGGTT CGCCAGACTCCGGAAAAGAGACTGGAGTGGGTCGCGACCATTAGTGGTGGTGATA CTTACACCTACTATGCAGACAGTGTGAAGGGACGATTCACCATCTCCAGAGACAAT GCCAAGAACACCCTGTACCTGCACATGACCGGTCTGAGGTCTGAGGACACGGCCC TGTATTATTGTGCAAGACATGGGTACGACGATTTTGACTATTGGGGCCTAGGCACC ACTCTCACAGTCTCCTCA DP99 light chain variable region
GATGTTGTGCTGACCCAAATCCCACTGACCTTGTCGGTCACCATTGGACAACCAGC CACCATCTCTTGCAAGTCAAGTCAGAGCCTCTTAGATAATGATGGAGAGACATATTT G AATTG GTTGTTAGTG AG G C CAG G C C AGTCTC C AAAG C G C CTAATCTATC AG GTGT CTAAACTGGACTCTGGATTCCCTGAGAGGTTCACTGGCGGTGGATCAGGGACAGA TTTCACACTGAAAATCAGCAGAGTGGAGGCTGAGGATTTGGGAATTTATTATTGCT G G C AAG GTAC AC ATTTTC CTC AC AC GTTC G GAG G G G G G AC C AAG CTG G AAATAAA A
In some embodiments, the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 8 and SEQ ID NO: 9 below
DP1 heavy chain variable region
GAGGTGCAGCTGGTGGAATCCGGCGGAGGACTGGTGCAGCCTGGCGGCTCCCTG AGACTGTCTTGCGCCGCCTCCGGCTTCACCTTCTCCATCTACGCCATGTCCTGGGT CCGACAGGCCCCTGGCAAGGGCCTGGAATGGGTGGCCCTGATCTCTGGCGGCGA CACCTACACCTACTACGCCGACTCCGTGAAGGGCCGGTTCACCATCTCCCGGGAC AACGCCAAGAACTCCCTGTACCTGCAGATGAACTCCCTGCGGGCCGAGGACACCG CCGTGTACTACTGCGCCAGACACGGCTACGACGACTTCGACTACTGGGGCCAGGG CACCCTGGTCACCGTCTCCTCA DP1 light chain variable region
GACGTGGTCATGACCCAGTCCCCCCTGTCCCTGCCTGTGACCCTGGGCCAGCCTG CCTCCATCTCCTGCCGGTCCTCCCAGTCCCTGCTGGACAACGACGGCGAGACATA CCTGAACTGGTTCCAGCAGCGGCCTGGCCAGTCCCCTCGGCGGCTGATCTACCAG GTGTCCAAGCTGGACTCCGGCGTGCCCGACAGATTCTCCGGCTCTGGCTCCGGCA CCGACTTCACCCTGAAGATCTCCAGAGTGGAAGCCGAGGACGTGGGCGTGTACTA CTGCTGGCAGGGCACCCACTTCCCCCACACCTTCGGCGGAGGCACCAAGGTGGA AATCAAA
In some embodiments, the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 12 and SEQ ID NO: 13 below
DM99 heavy chain variable region
CAGGGCCAACTTCAGCAGTCTGGGGCTGAACTGGCAAGACCTGGGGCATCAGTGA AGTTGTCCTGCAAGTCTTCTGGCCACACCTTTACTGATTACTGGATGCAGTGGGTA AAACAGAGGCCTGGACAGGGTCTGGAATGGATTGGGGCTATTTATCCTGGAGATG GTGATACTAGATACAATCAGAAGTTTAAGGACAAGGCCACATTGACTGCAGATACA TTCTCCAACACAGCCTACATGCAACTCAACAATTTGGCATCTGAGGACTCTGCGGT CTATTATTGTGCAAGAAGTTTTGACTATTGGGCCCAAGGCACCACTCTCACAGTCTC CTCG
DM99 light chain variable region
GACATTGTGCTGACACAGTCTCCTGCCTCTTTGGCTGTGTCTCTAGGGCAGAGGG CCTTCATGTCCTGCAGAGCCAGTAAAACTGTTGATAGTTATGGTAATAGTTTTATGC ACTGGTTCCAGCAGAAAGCAGGACAGCCACCCAAACTCCTCATCCATCATTCATCC AACCTAGAATCTGGGATCCCTGCCAGGTTCAGTGGCAGTGGGTCCAGGACAGACT TCACCCTCACCATTGATCCTGTGGAGGCTGATGATGTTGCAATCTATTACTGTCTTC AAAGTATTGAAGATCCGTACACGTTCGGAGGGGGGACCAAGCTGGAAATAAAA
In some embodiments, the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 27 and SEQ ID NO: 26 below
DM11 heavy chain variable region
GAAGTTCAGCTGCTGGAGAGCGGCGGCGGCCTGGTTCAGCCAGGTGGGTCCCTG CGGCTGTCATGTGCCGCAAGTGGCCACACCTTTACCGACTACTGGATGCAGTGGG TGCGCCAGGCACCGGGCAAAGGACTGGAATGGATTGGCGCCATTTACCCGGGGG ATGGCGATACGCGGTATAATCAGAAATTTAAGGATCGCTTCACCATTAGCCGCGAC AATTCTAAAAATACCCTGTACCTGCAAATGAACAGCCTGCGGGCAGAAGATACGGC CGTCTATTACTGCGCACGCTCTATGGATTACTGGGGGCAGGGCACGCTGGTCACC GTCTCCTCA
DM1 1 light chain variable region
GATATTGTGATGACCCAGAGCCCCGATTCCCTGGCCGTGAGCCTGGGCGAACGCG CAACCATCAACTGTCGCGCATCCAAAACCGTTGATAGCTATGGGAACAGTTTCATG CATTGGTTCCAGCAGAAACCGGGTCAGCCACCCAAGCTGCTGATTCATCATTCCAG CAACCTGGAGTCAGGCGTGCCTGATCGCTTCTCTGGCAGTGGTAGTGGCACTGAT TTCACCCTGACAATTAGTAGCCTGCAGGCAGAGGACGTCGCCGTTTATTATTGCCT GCAGACCATCGAAGATCCATATACTTTCGGCCAGGGTACGAAACTGGAGATTAAA
In some embodiments, the invention provides a composition comprising either or both of the polynucleotides shown in SEQ ID NO: 95 and SEQ ID NO: 96 below
DM4 heavy chain variable region
GAAGTTCAGCTGCTGGAGAGCGGCGGCGGCCTGGTTCAGCCAGGTGGGTCCCTG CGGCTGTCATGTGCCGCAAGTGGCCACACCTTTTCCGACTACTGGATGCAGTGGG TGCGCCAGGCACCGGGCAAAGGACTGGAATGGGTCGGCGCCATTTACCCGGGGG ATGGCGATACGCGGTATAATCAGAAATTTAAGGATCGCTTCACCATTAGCCGCGAC AATTCTAAAAATACCCTGTACCTGCAAATGAACAGCCTGCGGGCAGAAGATACGGC CGTCTATTACTGCGCACGCTCTATGGATTACTGGGGGCAGGGCACGCTGGTCACC GTCTCCTCA
DM4 light chain variable region
GATATTGTGATGACCCAGAGCCCCGATTCCCTGGCCGTGAGCCTGGGCGAACGCG CAACCATCAACTGTCGCGCATCCAAAACCGTTGATAGCTATGGGAACAGTTTCATG CATTGGTTCCAGCAGAAACCGGGTCAGCCACCCAAGCTGCTGATTCATCATTCCAG CAACCTGGAGTCAGGCGTGCCTGATCGCTTCTCTGGCAGTGGTAGTGGCACTGAT
TTCACCCTGACAATTAGTAGCCTGCAGGCAGAGGACGTCGCCGTTTATTATTGCCT GCAGACCATCGAATTTCCATATACTTTCGGCCAGGGTACGAAACTGGAGATTAAA
Polynucleotides complementary to any such sequences are also encompassed by the present invention. Polynucleotides may be single-stranded (coding or antisense) or double-stranded, and may be DNA (genomic, cDNA or synthetic) or RNA molecules. RNA molecules include HnRNA molecules, which contain introns and correspond to a DNA molecule in a one-to-one manner, and mRNA molecules, which do not contain introns. Additional coding or non-coding sequences may, but need not, be present within a polynucleotide of the present invention, and a polynucleotide may, but need not, be linked to other molecules and/or support materials.
Representative materials of the present invention were deposited in the American Type Culture Collection (ATCC) on March 28, 2012. Vector having ATCC Accession No. PTA-12710 is a polynucleotide encoding a humanized SOST antibody heavy chain variable region, and vector having ATCC Accession No. PTA-1271 1 is a polynucleotide encoding a humanized SOST antibody light chain variable region. The deposits were made under the provisions of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purpose of Patent Procedure and Regulations thereunder (Budapest Treaty). This assures maintenance of a viable culture of the deposit for 30 years from the date of deposit. The deposit will be made available by ATCC under the terms of the Budapest Treaty, and subject to an agreement between Pfizer, Inc./Rinat Neuroscience Corp. and ATCC, which assures permanent and unrestricted availability of the progeny of the culture of the deposit to the public upon issuance of the pertinent U.S. patent or upon laying open to the public of any U.S. or foreign patent application, whichever comes first, and assures availability of the progeny to one determined by the U.S. Commissioner of Patents and Trademarks to be entitled thereto according to 35 U.S.C. Section 122 and the Commissioner's rules pursuant thereto (including 37 C.F.R. Section 1 .14 with particular reference to 886 OG 638). The assignee of the present application has agreed that if a culture of the materials on deposit should die or be lost or destroyed when cultivated under suitable conditions, the materials will be promptly replaced on notification with another of the same. Availability of the deposited material is not to be construed as a license to practice the invention in contravention of the rights granted under the authority of any government in accordance with its patent laws.
Polynucleotides may comprise a native sequence (i.e., an endogenous sequence that encodes an antibody or a fragment thereof) or may comprise a variant of such a sequence. Polynucleotide variants contain one or more substitutions, additions, deletions and/or insertions such that the immunoreactivity of the encoded polypeptide is not diminished, relative to a native immunoreactive molecule. The effect on the immunoreactivity of the encoded polypeptide may generally be assessed as described herein. Variants preferably exhibit at least about 70% identity, more preferably, at least about 80% identity, yet more preferably, at least about 90% identity, and most preferably, at least about 95% identity to a polynucleotide sequence that encodes a native antibody or a fragment thereof.
Two polynucleotide or polypeptide sequences are said to be "identical" if the sequence of nucleotides or amino acids in the two sequences is the same when aligned for maximum correspondence as described below. Comparisons between two sequences are typically performed by comparing the sequences over a comparison window to identify and compare local regions of sequence similarity. A "comparison window" as used herein, refers to a segment of at least about 20 contiguous positions, usually 30 to about 75, or 40 to about 50, in which a sequence may be compared to a reference sequence of the same number of contiguous positions after the two sequences are optimally aligned.
Optimal alignment of sequences for comparison may be conducted using the
MEGALIGN® program in the LASERGENE® suite of bioinformatics software (DNASTAR®, Inc., Madison, Wl), using default parameters. This program embodies several alignment schemes described in the following references: Dayhoff, M.O., 1978, A model of evolutionary change in proteins - Matrices for detecting distant relationships. In Dayhoff, M.O. (ed.) Atlas of Protein Sequence and Structure, National Biomedical Research Foundation, Washington DC Vol. 5, Suppl. 3, pp. 345-358; Hein J., 1990, Unified Approach to Alignment and Phylogenes pp. 626-645 Methods in Enzymology vol. 183, Academic Press, Inc., San Diego, CA; Higgins, D.G. and Sharp, P.M., 1989, CABIOS 5: 151 -153; Myers, E.W. and Muller W., 1988, CABIOS 4: 1 1 -17; Robinson, E.D., 1971 , Comb. Theor. 1 1 : 105; Santou, N., Nes, M., 1987, Mol. Biol. Evol. 4:406-425; Sneath, P.H.A. and Sokal, R. R., 1973, Numerical Taxonomy the Principles and Practice of Numerical Taxonomy, Freeman Press, San Francisco, CA; Wilbur, W.J. and Lipman, D.J., 1983, Proc. Natl. Acad. Sci. USA 80:726-730.
Preferably, the "percentage of sequence identity" is determined by comparing two optimally aligned sequences over a window of comparison of at least 20 positions, wherein the portion of the polynucleotide or polypeptide sequence in the comparison window may comprise additions or deletions (i.e., gaps) of 20 percent or less, usually 5 to 15 percent, or 10 to 12 percent, as compared to the reference sequences (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid bases or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the reference sequence (i.e. the window size) and multiplying the results by 100 to yield the percentage of sequence identity.
Variants may also, or alternatively, be substantially homologous to a native gene, or a portion or complement thereof. Such polynucleotide variants are capable of hybridizing under moderately stringent conditions to a naturally occurring DNA sequence encoding a native antibody (or a complementary sequence).
Suitable "moderately stringent conditions" include prewashing in a solution of 5 X SSC, 0.5% SDS, 1 .0 mM EDTA (pH 8.0); hybridizing at 50°C-65°C, 5 X SSC, overnight; followed by washing twice at 65°C for 20 minutes with each of 2X, 0.5X and 0.2X SSC containing 0.1 % SDS.
As used herein, "highly stringent conditions" or "high stringency conditions" are those that: (1 ) employ low ionic strength and high temperature for washing, for example 0.015 M sodium chloride/0.0015 M sodium citrate/0.1 % sodium dodecyl sulfate at 50°C; (2) employ during hybridization a denaturing agent, such as formamide, for example, 50% (v/v) formamide with 0.1 % bovine serum albumin/0.1 % Ficoll/0.1 % polyvinylpyrrolidone/50 mM sodium phosphate buffer at pH 6.5 with 750 mM sodium chloride, 75 mM sodium citrate at 42°C; or (3) employ 50% formamide, 5 x SSC (0.75 M NaCI, 0.075 M sodium citrate), 50 mM sodium phosphate (pH 6.8), 0.1 % sodium pyrophosphate, 5 x Denhardt's solution, sonicated salmon sperm DNA (50 pg/ml), 0.1 % SDS, and 10% dextran sulfate at 42°C, with washes at 42°C in 0.2 x SSC (sodium chloride/sodium citrate) and 50% formamide at 55°C, followed by a high-stringency wash consisting of 0.1 x SSC containing EDTA at 55°C. The skilled artisan will recognize how to adjust the temperature, ionic strength, etc. as necessary to accommodate factors such as probe length and the like.
It will be appreciated by those of ordinary skill in the art that, as a result of the degeneracy of the genetic code, there are many nucleotide sequences that encode a polypeptide as described herein. Some of these polynucleotides bear minimal homology to the nucleotide sequence of any native gene. Nonetheless, polynucleotides that vary due to differences in codon usage are specifically contemplated by the present invention. Further, alleles of the genes comprising the polynucleotide sequences provided herein are within the scope of the present invention. Alleles are endogenous genes that are altered as a result of one or more mutations, such as deletions, additions and/or substitutions of nucleotides. The resulting mRNA and protein may, but need not, have an altered structure or function. Alleles may be identified using standard techniques (such as hybridization, amplification and/or database sequence comparison).
The polynucleotides of this invention can be obtained using chemical synthesis, recombinant methods, or PCR. Methods of chemical polynucleotide synthesis are well known in the art and need not be described in detail herein. One of skill in the art can use the sequences provided herein and a commercial DNA synthesizer to produce a desired DNA sequence.
For preparing polynucleotides using recombinant methods, a polynucleotide comprising a desired sequence can be inserted into a suitable vector, and the vector in turn can be introduced into a suitable host cell for replication and amplification, as further discussed herein. Polynucleotides may be inserted into host cells by any means known in the art. Cells are transformed by introducing an exogenous polynucleotide by direct uptake, endocytosis, transfection, F-mating or electroporation. Once introduced, the exogenous polynucleotide can be maintained within the cell as a non-integrated vector (such as a plasm id) or integrated into the host cell genome. The polynucleotide so amplified can be isolated from the host cell by methods well known within the art. See, e.g., Sambrook et al., 1989.
Alternatively, PCR allows reproduction of DNA sequences. PCR technology is well known in the art and is described in U.S. Patent Nos. 4,683,195, 4,800, 159, 4,754,065 and 4,683,202, as well as PCR: The Polymerase Chain Reaction, Mullis et al. eds., Birkauswer Press, Boston, 1994.
RNA can be obtained by using the isolated DNA in an appropriate vector and inserting it into a suitable host cell. When the cell replicates and the DNA is transcribed
into RNA, the RNA can then be isolated using methods well known to those of skill in the art, as set forth in Sambrook et al., 1989, supra, for example.
Suitable cloning vectors may be constructed according to standard techniques, or may be selected from a large number of cloning vectors available in the art. While the cloning vector selected may vary according to the host cell intended to be used, useful cloning vectors will generally have the ability to self-replicate, may possess a single target for a particular restriction endonuclease, and/or may carry genes for a marker that can be used in selecting clones containing the vector. Suitable examples include plasmids and bacterial viruses, e.g., pUC18, pUC19, Bluescript (e.g., pBS SK+) and its derivatives, mp18, mp19, pBR322, pMB9, ColE1 , pCR1 , RP4, phage DNAs, and shuttle vectors such as pSA3 and pAT28. These and many other cloning vectors are available from commercial vendors such as BioRad, Strategene, and Invitrogen.
Expression vectors are further provided. Expression vectors generally are replicable polynucleotide constructs that contain a polynucleotide according to the invention. It is implied that an expression vector must be replicable in the host cells either as episomes or as an integral part of the chromosomal DNA. Suitable expression vectors include but are not limited to plasmids, viral vectors, including adenoviruses, adeno-associated viruses, retroviruses, cosmids, and expression vector(s) disclosed in PCT Publication No. WO 87/04462. Vector components may generally include, but are not limited to, one or more of the following: a signal sequence; an origin of replication; one or more marker genes; suitable transcriptional controlling elements (such as promoters, enhancers and terminator). For expression (i.e., translation), one or more translational controlling elements are also usually required, such as ribosome binding sites, translation initiation sites, and stop codons.
The vectors containing the polynucleotides of interest can be introduced into the host cell by any of a number of appropriate means, including electroporation, transfection employing calcium chloride, rubidium chloride, calcium phosphate, DEAE- dextran, or other substances; microprojectile bombardment; lipofection; and infection (e.g., where the vector is an infectious agent such as vaccinia virus). The choice of introducing vectors or polynucleotides will often depend on features of the host cell.
The invention also provides host cells comprising any of the polynucleotides described herein. Any host cells capable of over-expressing heterologous DNAs can be used for the purpose of isolating the genes encoding the antibody, polypeptide or
protein of interest. Non-limiting examples of mammalian host cells include but not limited to COS, HeLa, and CHO cells. See also PCT Publication No. WO 87/04462. Suitable non-mammalian host cells include prokaryotes (such as E. coli or B. subtillis) and yeast (such as S. cerevisae, S. pombe; or K. lactis). Preferably, the host cells express the cDNAs at a level of about 5 fold higher, more preferably, 10 fold higher, even more preferably, 20 fold higher than that of the corresponding endogenous antibody or protein of interest, if present, in the host cells. Screening the host cells for a specific binding to SOST or an SOST domain is effected by an immunoassay or FACS. A cell overexpressing the antibody or protein of interest can be identified.
An expression vector can be used to direct expression of the anti-SOST antibody of the present invention. One skilled in the art is familiar with administration of expression vectors to obtain expression of an exogenous protein in vivo. See, e.g., U.S. Patent Nos. 6,436,908; 6,413,942; and 6,376,471. Administration of expression vectors includes local or systemic administration, including injection, oral administration, particle gun or catheterized administration, and topical administration. In another embodiment, the expression vector is administered directly to the sympathetic trunk or ganglion, or into a coronary artery, atrium, ventrical, or pericardium.
Targeted delivery of therapeutic compositions containing an expression vector, or subgenomic polynucleotides can also be used. Receptor-mediated DNA delivery techniques are described in, for example, Findeis et al., Trends Biotechnol., 1993, 1 1 :202; Chiou et al., Gene Therapeutics: Methods And Applications Of Direct Gene Transfer, J.A. Wolff, ed., 1994; Wu et al., J. Biol. Chem., 1988, 263:621 ; Wu et al., J. Biol. Chem., 1994, 269:542; Zenke et al., Proc. Natl. Acad. Sci. USA, 1990, 87:3655; Wu et al., J. Biol. Chem., 1991 , 266:338. Therapeutic compositions containing a polynucleotide are administered in a range of about 100 ng to about 200 mg of DNA for local administration in a gene therapy protocol. Concentration ranges of about 500 ng to about 50 mg, about 1 g to about 2 mg, about 5 g to about 500 g, and about 20 g to about 100 g of DNA can also be used during a gene therapy protocol. The therapeutic polynucleotides and polypeptides can be delivered using gene delivery vehicles. The gene delivery vehicle can be of viral or non-viral origin (see generally, Jolly, Cancer Gene Therapy, 1994, 1 :51 ; Kimura, Human Gene Therapy, 1994, 5:845; Connelly, Human Gene Therapy, 1995, 1 : 185; and Kaplitt, Nature Genetics, 1994, 6: 148). Expression of such coding sequences can be induced using endogenous mammalian or
heterologous promoters. Expression of the coding sequence can be either constitutive or regulated.
Viral-based vectors for delivery of a desired polynucleotide and expression in a desired cell are well known in the art. Exemplary viral-based vehicles include, but are not limited to, recombinant retroviruses (see, e.g., PCT Publication Nos. WO 90/07936; WO 94/03622; WO 93/25698; WO 93/25234; WO 93/1 1230; WO 93/10218; WO 91/02805; U.S. Patent Nos. 5, 219,740 and 4,777, 127; GB Patent No. 2,200,651 ; and EP Patent No. 0 345 242), alphavirus-based vectors (e.g., Sindbis virus vectors, Semliki forest virus (ATCC VR-67; ATCC VR-1247), Ross River virus (ATCC VR-373; ATCC VR-1246) and Venezuelan equine encephalitis virus (ATCC VR-923; ATCC VR-1250; ATCC VR 1249; ATCC VR-532)), and adeno-associated virus (AAV) vectors (see, e.g., PCT Publication Nos. WO 94/12649, WO 93/03769; WO 93/19191 ; WO 94/28938; WO 95/1 1984 and WO 95/00655). Administration of DNA linked to killed adenovirus as described in Curiel, Hum. Gene Then, 1992, 3: 147 can also be employed.
Non-viral delivery vehicles and methods can also be employed, including, but not limited to, polycationic condensed DNA linked or unlinked to killed adenovirus alone (see, e.g., Curiel, Hum. Gene Then, 1992, 3: 147); ligand-linked DNA (see, e.g., Wu, J. Biol. Chem., 1989, 264: 16985); eukaryotic cell delivery vehicles cells (see, e.g., U.S. Patent No. 5,814,482; PCT Publication Nos. WO 95/07994; WO 96/17072; WO 95/30763; and WO 97/42338) and nucleic charge neutralization or fusion with cell membranes. Naked DNA can also be employed. Exemplary naked DNA introduction methods are described in PCT Publication No. WO 90/1 1092 and U.S. Patent No. 5,580,859. Liposomes that can act as gene delivery vehicles are described in U.S. Patent No. 5,422, 120; PCT Publication Nos. WO 95/13796; WO 94/23697; WO 91/14445; and EP 0524968. Additional approaches are described in Philip, Mol. Cell Biol., 1994, 14:241 1 , and in Woffendin, Proc. Natl. Acad. Sci., 1994, 91 : 1581 .
Methods of Using the Anti-SOST Antibodies
The antibodies of the present invention are useful in various applications including, but are not limited to, therapeutic treatment methods and diagnostic treatment methods.
In one aspect, the invention provides a method of increasing bone formation, bone mass, bone mineralization, bone volume, bone quality, bone strength, and/or bone
density (e.g., in vertebral and/or non-vertebral bone) in a patient in need thereof. In some embodiments, the method of increasing bone formation, bone mass, bone mineralization, bone volume, bone quality, bone strength, and/or bone density in a patient in need thereof comprises administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
In another aspect, the invention provides a method of inducing canonical Wnt signaling activity in a patient in need thereof. In some embodiments, the method of inducing canonical Wnt signaling activity in a patient in need thereof comprises administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
In another aspect, the invention provides a method of treating or preventing a SOST-associated condition, disease, or disorder (e.g., a bone related disorder) that is responsive to the inhibition of SOST activity or canonical Wnt signaling. Accordingly, provided is a method of treating or preventing a bone related disorder in a patient in need thereof comprising administering to the patient an effective amount of the anti- SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein. As used herein, a "bone related disorder" includes, but is not limited to, a bone condition/disorder/disease, a bone condition/disorder/disease associated with renal failure or cancer, and nutritional, gastrointestinal and/or hepatic associated bone conditions/disorders/diseases. Examples of a "bone related disorder", as used herein, include, but are not limited to, osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, arthritis (e.g., rheumatoid arthritis or psoriatic arthritis), osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss (e.g., due to immobilization), metastatic bone malignancy (e.g., lytic bone metastases), multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease, hemochromatosis, celiac sprue (or celiac disease), renal tubular acidosis, renal osteodystrophy, hypercalciuria, fibrous dysplasia, or diabetes. In
some embodiments, provided is a method of treating or preventing osteoporosis in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
Different forms of osteoporosis can be treated using the anti-SOST antibodies as described herein, including but not limited to, primary osteoporosis (type 1 or postmenopausal osteoporosis; type 2 or senile osteoporosis) and secondary osteoporosis. In some embodiments, secondary osteoporosis is glucocorticoid induced osteoporosis, osteoporosis induced after transplantation, osteoporosis associated with chemotherapy (i.e., chemotherapy induced osteoporosis), immobilization induced osteoporosis, osteoporosis due to mechanical unloading, or osteoporosis associated with anticonvulsant use.
Different forms of arthritis can also be treated using the anti-SOST antibodies as described herein, including but not limited to, osteoarthritis and rheumatoid arthritis. In treating arthritis, patients may benefit by perilesional or intralesional injections of the subject antibodies or fragments thereof. For example, the anti-SOST antibodies can be injected adjacent to or directly into an inflamed joint, thus stimulating repair of damaged bone at the site.
Any cancer that can metastasize to bone or any bone tumor/cancer can also be treated using the anti-SOST antibodies as described herein. For example, certain cancers, such as breast and prostate cancer, can metastasize to bone readily, increase osteoclast activity, and induce bone resorption. Certain other cancers, such as multiple myeloma, arise in bone marrow, and is associated with bone loss due to increased osteoclast activation in localized regions of the bone. Accordingly, reducing SOST activity by administering the subject antibodies thereof can result in an increase in osteoblast activity that serves to counteract the excessive osteoclast activity, thereby reducing the severity of the aforementioned disorders, reducing bone erosion, and inducing new bone formation in the multiple myeloma patient. In some embodiments, provided is a method of treating multiple myeloma in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
In one variation, anti-SOST antibodies of the present inventions can also be used for preventing or reducing tumor burden, inhibiting or preventing tumor growth or progression, inhibiting metastasis of cancer cells or tumors, or delaying tumor growth or progression in a subject (or patient) having cancer. As used herein, cancers include, but are not limited to, a solid cancer (such as bladder, breast, cervical, choriocarcinoma, colon, esophageal, gastric, glioblastoma, head and neck, kidney, liver, lung (e.g., Non Small Cell Lung Cancer (NSCLC)), oral, ovarian, pancreatic, prostate, and skin cancer); and a liquid cancer (such as acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML), hairy cell leukemia (HCL), T-cell prolymphocytic leukemia (T-PLL), large granular lymphocytic leukemia, multiple myeloma, and adult T-cell leukemia).
Accordingly, in some embodiments, provided is a method of preventing or reducing tumor burden in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition (e.g., pharmaceutical composition) comprising thereof. In some embodiments, provided is a method of inhibiting or preventing tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition (e.g., pharmaceutical composition) comprising thereof. In other
embodiments, provided is a method of inhibiting metastasis of cancer cells or tumors (e.g., solid or liquid tumors), or delaying tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition comprising thereof. In other embodiments, provided is a method of inducing tumor regression in a subject in need thereof, in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody (e.g., the anti-SOST antibodies as described herein) that binds specifically to a SOST polypeptide (e.g., SEQ ID NO: 1 ) or a composition comprising thereof. For example, in some embodiments, the anti-SOST antibody can comprise (1 )(a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the
sequence GX1TFX2DYWMQ, wherein X^ is F or H, X2 is T or S (SEQ ID NO: 81 ) or GXiTFX2DY, wherein X^ is F or H, and X2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX2X3KX4, wherein X is A or N, X2 is S or K, X3 is V or F, and X4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SXi DYW, wherein X^ is F or M (SEQ ID NO: 84); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGXiX2FMH, wherein X is S or N, and X2 is N or S (SEQ ID NO: 85); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQX^ IX2X3X4YT , wherein X is T or S, X2 is E or D, X3 is H, D, F, or E, and X4 is H, P, or S (SEQ ID NO: 86); or (2)(a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence
GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence Xi ISGGDTYTYYADSVKG, wherein X^ is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1 comprising the sequence X1 SSQSLLDNDGETYLN, wherein X is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ). In other embodiments, the antibody is DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, DM34, AMG167, or AMG785 (Romosozumab).
Anti-SOST antibodies of the present inventions can also be used in various bone repair applications. For example, the anti-SOST antibodies can retard wear debris osteolysis associated with artificial joints, accelerating the repair of bone fractures, and enhancing the incorporation of bone grafts or fixation of implants into the surrounding living bone into which they have been engrafted or implanted. Accordingly, in some embodiments, provided is a method of treating bone fracture in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g. , pharmaceutical composition) comprising the anti-SOST antibodies as described herein. In other embodiments, provided is a method of
enhancing or strengthening implant fixation (e.g., by improving and/or accelerating bone formation) in a patient in need thereof comprising administering to the patient an effective amount of the anti-SOST antibodies or a composition (e.g., pharmaceutical composition) comprising the anti-SOST antibodies as described herein.
Anti-SOST antibodies of the present invention can also be used to treat diseases in which it is desirable to promote stem cell renewal. Such diseases include, but are not limited to, diabetes (e.g., type 1 diabetes mellitus, type 2 diabetes mellitus, gestational diabetes, steroid diabetes, congenital diabetes, cystic fibrosis-related diabetes, or monogenic diabetes), chronic heart failure and various diseases of the muscle [e.g., disuse atrophy resulting, for instance, from immobilization or bed-rest); aging frailty (sarcopenia of the elderly); muscular dystrophies; cachexia associated with cancer, Acquired Immune Deficiency Syndrome (AIDS) or inflammation; protein-energy malnutrition in renal failure/uremia, and muscle wasting in obesity. Various inflammatory diseases can also be treated, including, for instance, inflammatory bowel disease (e.g., Crohn's disease or ulcerative colitis). Different renal diseases (e.g., end stage renal disease, chronic renal disease, glomerulonephritis, tubulointerstitial nephritis, renal osteodystrophy, and IgA nephropathy) and various pulmonary diseases (e.g., chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and cystic fibrosis) and skin disorders, including dermal and epidermal diseases can also be treated with the antibodies of the present invention. Examples of skin disorders that can be treated include damaged intestinal epithelium (e.g., chemotherapy induced damage), and other diseases in which it is desirable to stimulate growth and survival of the intestinal epithelium. Various congenital and/or hereditary bone diseases can also be treated using anti-SOST antibodies, such as fibrous dysplasia, achondroplasia, osteogenesis imperfecta, osteopetrosis (marble bone disease or osteosclerosis), hereditary multiple exotosis (osteochondromatosis), enchondromatosis (Ollier's Disease), congenital osteopenia or osteoporosis.
In another aspect, provided is a method of detecting, diagnosing, and/or monitoring a condition associated with SOST expression in a biological sample (e.g., overexpression or underexpression of SOST in tissues or cells). For example, the anti- SOST antibodies as described herein can be labeled with a detectable moiety such as an imaging agent and an enzyme-substrate label. The antibodies as described herein
can also be used for in vivo diagnostic assays, such as in vivo imaging (e.g., PET or SPECT), or a staining reagent.
In some embodiments, provided is a method of screening for a molecule that binds to SOST. For example, a SOST molecule or fragment thereof is contacted with the anti-SOST antibody disclosed herein together with another molecule (e.g., a candidate molecule). A reduction in binding between the anti-SOST antibody and SOST is an indication that the candidate molecule binds SOST. Binding of the antibody can be detected using a variety of methods, e.g., an ELISA. Detection of binding to the anti-SOST antibody can be simplified by detectably labeling the antibody. In some methods, a molecule that exhibits binding in the initial screen is further analyzed to determine whether it inhibits SOST activity (e.g., whether the molecule activates Wnt signaling).
In some embodiments, the methods described herein further comprise a step of treating a patient in need thereof with an additional form of therapy. In some embodiments, the additional form of therapy is one or more therapeutic agent, such as chemotherapeutic agents (e.g., cytotoxic agents), osteoclast activity inhibiting agents, osteoblast activity enhancing agents (e.g., bone growth anabolic agents and bone anti- resorptive agents), and dietary supplements (e.g., calcium, vitamin D, and vitamin K).
Chemotherapeutic agents include, but are not limited to, a second antibody (e.g., an anti-VEGF (Vascular Endothelial Growth Factor) antibody (e.g., AVASTIN®), an anti- HER2 antibody (e.g., HERCEPTIN®), an anti-CD25 antibody, an anti-CD33 antibody, an anti-CD20 antibody (e.g., RITUXAN®), an anti-mucin-like glycoprotein antibody, an anti- TNF antibody, and/or an epidermal growth factor receptor (EGFR) antibody (e.g., ERBITUX®)), an angiogenesis inhibitor, a cytotoxic agent (e.g., anthracyclines (e.g., daunorubicin, doxorubicin, epirubicin, idarubicin, valrubicin, and mitoxantrone), taxane (e.g., paclitaxel and docetaxel), dolastatin, duocarmycin, enediyne, geldanamycin, maytansine, puromycin, vinca alkaloid (e.g., vincristine), a topoisomerase inhibitor (e.g., etoposide), tubulysin, a pyrimidine analog (e.g., fluorouracil), platinum-containing agents (e.g., cisplatin, carboplatin, and oxaliplatin), alkylating agents (e.g., melphalan, cyclophosphamide, or carmustine) and hemiasterlin), immunomodulating agent (e.g., prednisone), an anti-inflammatory agent (e.g., dexamethasone), an aromatase inhibitor (e.g., anastrozole, exemestane, letrozole, vorozole, formestane, or testolactone), a proteasome inhibitor (e.g., bortezomib such as VELCADE® ([(1 R)-3-methyl-1 -[[(2S)-1 -
oxo-3-phenyl-2-[(pyrazinylcarbonyl)amino]propy- l]amino]butyl] boronic acid), and other agents such as tamoxifen.
For example, in some embodiments, provided is a method of treating multiple myeloma comprising administering to a patient need thereof an effective amount of a composition comprising the anti-SOST antibodies as described herein and one other therapeutic agent such as a chemotherapeutic agent or thalidomide or its derivative thereof (e.g., lenalidomide). In some embodiments, the one other therapeutic agent is selecting from the group consisting of bortezomib (e.g., VELCADE®), melphalan, prednisone, doxorubicin, lenalidomide, thalidomide, prednisone, carmustine, etoposide, cisplatin, cyclophosphamide, and vincristine. In some embodiments, the other therapeutic agent is bortezomib (e.g., VELCADE®), melphalan, or prednisone. In some embodiments, the patient is relapsing or refractory to previous multiple myeloma therapy.
Osteoblast activity enhancing agents (e.g., bone growth promoting (anabolic) agents or bone anti-resorptive agents) include, but are not limited to, bone morphogenic factors designated BMP-1 to BMP-12, transforming growth factor (TGFJ-β and TGF-β family members, fibroblast growth factors (FGF)-1 to FGF-10, interleukin (IL)-1 inhibitors (including IL-1 ra, antibodies to IL-1 and antibodies to IL-1 receptors), Tumor Necrosis Factor (TNF)a inhibitors (including etanercept, adalimumab and infliximab), RANK ligand inhibitors (including soluble RANK, osteoprotegerin and antagonistic antibodies that specifically bind RANK or RANK ligand (e.g., PROLIA®), parathyroid hormone (e.g., FORTEO®), E series prostaglandins, bisphosphonates (e.g., FOSAMAX® ACTONEL® and BONIVA®) and bone-enhancing minerals such as fluoride and calcium, insulin-like growth factor (IGF) (wherein the agent is preferably complexed with an IGF binding protein), strontium ranelate, estrogen analogs, raloxifene, and calcitonin. An IL-1 receptor antagonist suitable for such combination treatment is described in WO89/1 1540 and a suitable soluble TNF receptor-1 is described in WO98/01555. Exemplary RANK ligand antagonists are disclosed, for example, in WO 03/086289, WO 03/002713, U.S. Pat. Nos.: 6,740,51 1 and 6,479,635.
The anti-SOST antibodies as described herein can be administered to an individual via any suitable route. It should be understood by persons skilled in the art that the examples described herein are not intended to be limiting but to be illustrative of the techniques available. Accordingly, in some embodiments, the anti-SOST antibody is
administered to an individual in accord with known methods, such as intravenous administration, e.g., as a bolus or by continuous infusion over a period of time, by intramuscular, intraperitoneal, intracerebrospinal, transdermal, subcutaneous, intraarticular, sublingually, intrasynovial, insufflation, intrathecal, oral, inhalation, or topical routes. Administration can be systemic, e.g., intravenous administration, or localized. Commercially available nebulizers for liquid formulations, including jet nebulizers and ultrasonic nebulizers are useful for administration. Liquid formulations can be directly nebulized and lyophilized powder can be nebulized after reconstitution. Alternatively, the anti-SOST antibody can be aerosolized using a fiuorocarbon formulation and a metered dose inhaler, or inhaled as a lyophilized and milled powder.
In one embodiment, the anti-SOST antibody is administered via site-specific or targeted local delivery techniques. Examples of site-specific or targeted local delivery techniques include various implantable depot sources of the SOST antibody or local delivery catheters, such as infusion catheters, indwelling catheters, or needle catheters, synthetic grafts, adventitial wraps, shunts and stents or other implantable devices, site specific carriers, direct injection, or direct application. See, e.g., PCT Publication No. WO 00/5321 1 and U.S. Pat. No. 5,981 ,568.
Various formulations of the anti-SOST antibody may be used for administration. In some embodiments, the anti-SOST antibody may be administered neat. In some embodiments, the anti-SOST antibody and a pharmaceutically acceptable excipient may be in various formulations. Pharmaceutically acceptable excipients are known in the art, and are relatively inert substances that facilitate administration of a pharmacologically effective substance. For example, an excipient can give form or consistency, or act as a diluent. Suitable excipients include but are not limited to stabilizing agents, wetting and emulsifying agents, salts for varying osmolarity, encapsulating agents, buffers, and skin penetration enhancers. Excipients as well as formulations for parenteral and
nonparenteral drug delivery are set forth in Remington, The Science and Practice of Pharmacy 21 st Ed. Mack Publishing, 2005.
In some embodiments, these agents are formulated for administration by injection (e.g., intraperitoneally, intravenously, subcutaneously, intramuscularly, etc.).
Accordingly, these agents can be combined with pharmaceutically acceptable vehicles such as saline, Ringer's solution, dextrose solution, and the like. The particular dosage
regimen, i.e., dose, timing and repetition, will depend on the particular individual and that individual's medical history.
The anti-SOST antibody as described herein can be administered using any suitable method, including by injection (e.g., intraperitoneally, intravenously,
subcutaneously, intramuscularly, etc.). The anti-SOST antibody can also be
administered via inhalation, as described herein. Generally, for administration of an anti-SOST antibody, an initial candidate dosage can be about 2 mg/kg. For the purpose of the present invention, a typical daily dosage might range from about any of 3 g/kg to 30 g/kg to 300 g/kg to 3 mg/kg, to 30 mg/kg, to 100 mg/kg or more, depending on the factors mentioned above. For example, dosage of about 1 mg/kg, about 2.5 mg/kg, about 5 mg/kg, about 10 mg/kg, and about 25 mg/kg may be used. For repeated administrations over several days or longer, depending on the condition, the treatment is sustained until a desired suppression of symptoms occurs or until sufficient therapeutic levels are achieved, for example, to inhibit or delay tumor growth/progression or metastasis of cancer cells. An exemplary dosing regimen comprises administering an initial dose of about 2 mg/kg, followed by a weekly maintenance dose of about 1 mg/kg of the anti-SOST antibody, or followed by a maintenance dose of about 1 mg/kg every other week. Other exemplary dosing regimen comprises administering increasing doses (e.g., initial dose of 1 mg/kg and gradual increase to one or more higher doses every week or longer time period). Other dosage regimens may also be useful, depending on the pattern of pharmacokinetic decay that the practitioner wishes to achieve. For example, in some embodiments, dosing from one to four times a week is contemplated. In other embodiments dosing once a month or once every other month or every three months is contemplated. The progress of this therapy is easily monitored by
conventional techniques and assays. The dosing regimen (including the anti-SOST antibody used) can vary over time.
For the purpose of the present invention, the appropriate dosage of an anti-SOST antibody will depend on the anti-SOST antibody (or compositions thereof) employed, the type and severity of symptoms to be treated, whether the agent is administered for therapeutic purposes, previous therapy, the patient's clinical history and response to the agent, the patient's clearance rate for the administered agent, and the discretion of the attending physician. Typically the clinician will administer an anti-SOST antibody until a dosage is reached that achieves the desired result. Dose and/or frequency can vary
over course of treatment. Empirical considerations, such as the half-life, generally will contribute to the determination of the dosage. For example, antibodies that are compatible with the human immune system, such as humanized antibodies or fully human antibodies, may be used to prolong half-life of the antibody and to prevent the antibody being attacked by the host's immune system. Frequency of administration may be determined and adjusted over the course of therapy, and is generally, but not necessarily, based on treatment and/or suppression and/or amelioration and/or delay of symptoms, e.g., tumor growth inhibition or delay, etc. Alternatively, sustained
continuous release formulations of anti-SOST antibodies may be appropriate. Various formulations and devices for achieving sustained release are known in the art.
In one embodiment, dosages for an anti-SOST antibody may be determined empirically in individuals who have been given one or more administration(s) of the anti- SOST antibody. Individuals are given incremental dosages of an anti-SOST antibody or an SOST antagonist. To assess efficacy, an indicator of the disease can be followed.
Administration of an anti-SOST antibody in accordance with the method in the present invention can be continuous or intermittent, depending, for example, upon the recipient's physiological condition, whether the purpose of the administration is therapeutic or prophylactic, and other factors known to skilled practitioners. The administration of an anti-SOST antibody may be essentially continuous over a preselected period of time or may be in a series of spaced doses.
In some embodiments, more than one anti-SOST antibody may be present. At least one, at least two, at least three, at least four, at least five different or more anti- SOST antibody can be present. Generally, those anti-SOST antibodies may have complementary activities that do not adversely affect each other. For example, one or more of the following anti-SOST antibody may be used: a first anti-SOST antibody directed to one epitope on SOST and a second anti-SOST antibody directed to a different epitope on SOST.
Therapeutic formulations of the anti-SOST antibody used in accordance with the present invention are prepared for storage by mixing an antibody having the desired degree of purity with optional pharmaceutically acceptable carriers, excipients or stabilizers (Remington, The Science and Practice of Pharmacy 21 st Ed. Mack
Publishing, 2005), in the form of lyophilized formulations or aqueous solutions.
Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages
and concentrations employed, and may comprise buffers such as phosphate, citrate, and other organic acids; salts such as sodium chloride; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride;
phenol, butyl or benzyl alcohol; alkyl parabens, such as methyl or propyl paraben;
catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins;
chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes (e.g. Zn-protein complexes); and/or non-ionic surfactants such as TWEEN™, PLURONICS™ or polyethylene glycol (PEG).
Liposomes containing the anti-SOST antibody are prepared by methods known in the art, such as described in Epstein, et al., Proc. Natl. Acad. Sci. USA 82:3688, 1985; Hwang, et al., Proc. Natl Acad. Sci. USA 77:4030, 1980; and U.S. Pat. Nos. 4,485,045 and 4,544,545. Liposomes with enhanced circulation time are disclosed in U.S. Pat. No. 5,013,556. Particularly useful liposomes can be generated by the reverse phase evaporation method with a lipid composition comprising phosphatidylcholine, cholesterol and PEG-derivatized phosphatidylethanolamine (PEG-PE). Liposomes are extruded through filters of defined pore size to yield liposomes with the desired diameter.
The active ingredients may also be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example, hydroxymethylcellulose or gelatin-microcapsules and poly-(methylmethacrylate) microcapsules, respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in macroemulsions. Such techniques are disclosed in Remington, The Science and Practice of Pharmacy 21 st Ed. Mack Publishing, 2005.
Sustained-release preparations may be prepared. Suitable examples of sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the antibody, which matrices are in the form of shaped articles, e.g. films, or microcapsules. Examples of sustained-release matrices include polyesters,
hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or 'poly(vinylalcohol)), polylactides (U.S. Pat. No. 3,773,919), copolymers of L-glutamic acid and 7 ethyl-L- glutamate, non-degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOT™ (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), sucrose acetate isobutyrate, and poly-D-(-)-3-hydroxybutyric acid.
The formulations to be used for in vivo administration must be sterile. This is readily accomplished by, for example, filtration through sterile filtration membranes. Therapeutic SOST antibody compositions are generally placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle.
The compositions according to the present invention may be in unit dosage forms such as tablets, pills, capsules, powders, granules, solutions or suspensions, or suppositories, for oral, parenteral or rectal administration, or administration by inhalation or insufflation.
For preparing solid compositions such as tablets, the principal active ingredient is mixed with a pharmaceutical carrier, e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g. water, to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention, or a non-toxic pharmaceutically acceptable salt thereof. When referring to these preformulation compositions as homogeneous, it is meant that the active ingredient is dispersed evenly throughout the composition so that the composition may be readily subdivided into equally effective unit dosage forms such as tablets, pills and capsules. This solid preformulation composition is then subdivided into unit dosage forms of the type described above containing from 0.1 to about 500 mg of the active ingredient of the present invention. The tablets or pills of the novel composition can be coated or otherwise compounded to provide a dosage form affording the advantage of prolonged action. For example, the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former. The two components can be separated by an enteric layer that serves to resist disintegration in the stomach and permits the inner component to pass intact into the duodenum or to be delayed in release. A variety of materials can be used for such
enteric layers or coatings, such materials including a number of polymeric acids and mixtures of polymeric acids with such materials as shellac, cetyl alcohol and cellulose acetate.
Suitable surface-active agents include, in particular, non-ionic agents, such as polyoxyethylenesorbitans (e.g. Tween™ 20, 40, 60, 80 or 85) and other sorbitans (e.g. Span™ 20, 40, 60, 80 or 85). Compositions with a surface-active agent will
conveniently comprise between 0.05 and 5% surface-active agent, and can be between 0.1 and 2.5%. It will be appreciated that other ingredients may be added, for example mannitol or other pharmaceutically acceptable vehicles, if necessary.
Suitable emulsions may be prepared using commercially available fat emulsions, such as Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ and Lipiphysan™. The active ingredient may be either dissolved in a pre-mixed emulsion composition or alternatively it may be dissolved in an oil (e.g. soybean oil, safflower oil, cottonseed oil, sesame oil, corn oil or almond oil) and an emulsion formed upon mixing with a phospholipid (e.g. egg phospholipids, soybean phospholipids or soybean lecithin) and water. It will be appreciated that other ingredients may be added, for example glycerol or glucose, to adjust the tonicity of the emulsion. Suitable emulsions will typically contain up to 20% oil, for example, between 5 and 20%. The fat emulsion can comprise fat droplets between 0.1 and 1 .0 pm, particularly 0.1 and 0.5 pm, and have a pH in the range of 5.5 to 8.0.
The emulsion compositions can be those prepared by mixing an anti-SOST antibody or an anti-SOST antibody conjugate with Intralipid™ or the components thereof (soybean oil, egg phospholipids, glycerol and water).
Compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders. The liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as set out above. In some embodiments, the compositions are administered by the oral or nasal respiratory route for local or systemic effect. Compositions in preferably sterile pharmaceutically acceptable solvents may be nebulised by use of gases. Nebulised solutions may be breathed directly from the nebulising device or the nebulising device may be attached to a face mask, tent or intermittent positive pressure breathing machine. Solution, suspension or powder
compositions may be administered, preferably orally or nasally, from devices which deliver the formulation in an appropriate manner.
Compositions
The compositions used in the methods of the invention comprise an effective amount of an anti-SOST antibody as described herein. Examples of such compositions, as well as how to formulate, are also described in an earlier section and below. In some embodiments, the composition comprises one or more anti-SOST antibodies. For example, an anti-SOST antibody recognizes human SOST. In some embodiments, the anti-SOST antibody is a human antibody, a humanized antibody, or a chimeric antibody. In some embodiments, the anti-SOST antibody comprises a constant region that is capable of triggering a desired immune response, such as antibody-mediated lysis or ADCC. In other embodiments, the anti-SOST antibody comprises a constant region that does not trigger an unwanted or undesirable immune response, such as antibody- mediated lysis or ADCC. In other embodiments, the anti-SOST antibody comprises one or more CDR(s) of the antibody (such as one, two, three, four, five, or, in some embodiments, all six CDRs).
It is understood that the compositions can comprise more than one anti-SOST antibody (e.g., a mixture of anti-SOST antibodies that recognize different epitopes of SOST). Other exemplary compositions comprise more than one anti-SOST antibodies that recognize the same epitope(s), or different species of anti-SOST antibodies that bind to different epitopes of SOST (e.g., human SOST).
The composition used in the present invention can further comprise
pharmaceutically acceptable carriers, excipients, or stabilizers (Remington: The
Science and practice of Pharmacy 21 st Ed., 2005, Lippincott Williams and Wilkins, Ed. K. E. Hoover), in the form of lyophilized formulations or aqueous solutions. Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations, and may comprise buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as
serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrans; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes (e.g. Zn-protein complexes); and/or non-ionic surfactants such as TWEEN™, PLURONICS™ or polyethylene glycol (PEG). Pharmaceutically acceptable excipients are further described herein Kits
The invention also provides kits comprising any or all of the antibodies described herein. Kits of the invention include one or more containers comprising an anti-SOST antibody described herein and instructions for use in accordance with any of the methods of the invention described herein. Generally, these instructions comprise a description of administration of the anti-SOST antibody for the above described therapeutic treatments. In some embodiments, kits are provided for producing a single- dose administration unit. In certain embodiments, the kit can contain both a first container having a dried protein and a second container having an aqueous formulation. In certain embodiments, kits containing single and multi-chambered pre-filled syringes (e.g., liquid syringes and lyosyringes) are included.
In some embodiments, the antibody is a human antibody. In some embodiments, the antibody is a humanized antibody. In some embodiments, the antibody is a monoclonal antibody. The instructions relating to the use of an anti-SOST antibody generally include information as to dosage, dosing schedule, and route of administration for the intended treatment. The containers may be unit doses, bulk packages (e.g., multi-dose packages) or sub-unit doses. Instructions supplied in the kits of the invention are typically written instructions on a label or package insert (e.g., a paper sheet included in the kit), but machine-readable instructions (e.g., instructions carried on a magnetic or optical storage disk) are also acceptable.
The kits of this invention are in suitable packaging. Suitable packaging includes, but is not limited to, vials, bottles, jars, flexible packaging (e.g., sealed Mylar or plastic bags), and the like. Also contemplated are packages for use in combination with a specific device, such as an inhaler, nasal administration device (e.g., an atomizer) or an
infusion device such as a minipump. A kit may have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). The container may also have a sterile access port (for example the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). At least one active agent in the composition is an anti-SOST antibody. The container may further comprise a second pharmaceutically active agent, such as a chemotherapeutic agent, osteoclast activity inhibiting agents, osteoblast activity enhancing agents (e.g., bone growth anabolic agents and bone anti-resorptive agents), and dietary supplements (e.g., calcium, vitamin D, and vitamin K) as described herein.
Kits may optionally provide additional components such as buffers and interpretive information. Normally, the kit comprises a container and a label or package insert(s) on or associated with the container.
The invention also provides diagnostic kits comprising any or all of the antibodies described herein. The diagnostic kits are useful for, for example, detecting the presence of SOST in a sample. In some embodiments, a diagnostic kit can be used to determine whether an individual is at risk for developing a SOST-associated disorder, e.g., a bone related disorder, such as osteoporosis, osteopenia, bone fracture, bone loss, Paget's Disease, or multiple myeloma.
Diagnostic kits of the invention include one or more containers comprising an anti-SOST antibody described herein and instructions for use in accordance with any of the methods of the invention described herein. Generally, these instructions comprise a description of use of the anti-SOST antibody to detect the presence of in individuals at risk for, or suspected of having, a SOST-associated conditions, disorder/disease (e.g., a bone related disorder). In some embodiments, an exemplary diagnostic kit can be configured to contain reagents such as, for example, an anti-SOST antibody, a negative control sample, a positive control sample, and directions for using the kit.
The following examples are offered for illustrative purposes only, and are not intended to limit the scope of the present invention in any way. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art from the foregoing description and fall within the scope of the appended claims.
Examples
Example 1 : Determination of Kinetics and Affinity of Recombinant Human SOST/human IgG Interactions
a. Determination of Binding Kinetics and Affinity Constants of Human IgGs binding to Recombinant Human SOST at 37°C
A Biacore T200 surface Plasmon resonance biosensor (GE Lifesciences,
Piscataway NJ) was used to determine the binding affinities of anti-SOST antibodies to recombinant human SOST (rhSOST). See Table 4. An anti-human Fc sensor chip was prepared by activating all flow cells of a Biacore CM4 sensor chip with a 1 : 1 (v/v) mixture of 400 mM EDC (1 -Ethyl-3-[3-dimethylaminopropyl]-carbodiimide) and 100 mM N-hydroxysuccinimide for 7 minutes, at a flow rate of 10 pL/min. A goat F(AB')2 fragment-anti-human IgG Fc (Cappel Catalog) was diluted to 60 pg/mL in 10 mM Sodium Acetate at pH 5.0 and injected on all flow cells for 7 minutes at 20 pL/min. All flow cells were blocked with 100 mM ethylenediamine in 150 mM Borate buffer pH 8.5 for 7 minutes at 10 pL/min. Antibodies were captured onto downstream flow cells (flow cells 2, 3 and 4) at 2 pg/mL at a flow rate of 10 pL/min for 1 minute. Different antibodies were captured on each flow cell. Flow cell 1 was used as a reference surface. Following capture of antibodies, analyte (buffer, or rhSOST) was injected at 30 pL/min on all flow cells for two minutes. After the analyte injection, dissociation was monitored for 30 minutes followed by regeneration of all flow cells with two 30-second injections of 75 mM Phosphoric Acid. A 3-membered dilution series of rhSOST was analyzed using this method, where the top concentration was 15 nM and the dilution factor was 3-fold. Buffer cycles were collected for each captured antibody for double-referencing purposes (double-referencing as described in Myszka, J. Mol. Recognit. 12:279-284, 1999). The double-referenced sensorgrams were fit globally to a simple 1:1 Langmuir with mass transport binding model. The experiments were performed at 37°C using a running buffer of 10 mM HEPES, 150 mM NaCI, 0.05% (v/v) Tween-20, pH 7.4, 1 mg/ml_ BSA. Table 4 shows the affinity constants of human IgGs binding to rhSOST at 37°C.
Table 4. Affinity Constants of human IgGs Binding to rhSOST at 37°C mAb Hvar Lvar ka (1/Ms) KD (1/s) KD (nM)
DM1 mH10 ml_4 1 .310E+6 7.41 OE-4 0.566
DM2 mH10 ml_14 2.483E+6 7.972E-4 0.321
DM3 mH10 mL15 1 .552E+6 2.853E-3 1 .838
DM4 mH10 mL13 3.907E+6 5.077E-4 0.130
DM5 mH10 ml_16 3.909E+6 7.535E-4 0.193
DM6 mH10 mL17 2.125E+6 2.508E-3 1 .180
DM7 mH1 1 mL14 4.162E+6 4.022E-3 0.966
DM8 mH1 1 mL17 2.390E+6 7.000E-3 2.928
DM9 mH10 mL18 1 .870E+6 4.340E-3 2.321
DM1 1 wt wt 1 .70E+07 <2.8E-05 <0.0016
DM12 wt ml_1 6.55E+06 5.62E-05 0.009
DM13 wt mL2 1 .72E+07 <2.8E-05 < 0.0016
DM14 wt mL3 5.22E+06 8.31 E-05 0.016
DM15 wt mL4 3.72E+06 2.97E-04 0.080
DM16 wt ml_5 3.94E+06 8.17E-04 0.207
DM17 wt mL6 4.70E+06 2.07E-04 0.044
DM18 wt mL8 5.81 E+06 1 .04E-03 0.179
DM19 wt mL10 4.06E+06 3.88E-04 0.096
DM20 mH1 wt 7.83E+06 1 .20E-04 0.015
DM21 mH1 ml_2 8.86E+06 7.41 E-05 0.008
DM22 mH2 wt 1 .04E+07 1 .52E-04 0.015
DM23 mH3 wt 1 .39E+07 1 .34E-04 0.010
DM24 mH3 mL2 6.90E+06 2.39E-04 0.035
DM25 mH4 wt 5.81 E+06 1 .01 E-04 0.017
DM26 mH5 wt 4.20E+06 2.22E-04 0.053
DM28 mH8 wt 3.52E+06 3.50E-04 0.099
DM29 mH10 ml_2 7.41 E+06 1 .01 E-04 0.014
DM30 mH10 mL3 5.34E+06 2.39E-04 0.045
DM31 mH10 mL19 2.10E+06 1 .71 E-03 0.816
DM32 mH1 1 ml_2 7.75E+06 4.19E-04 0.054
DM33 mH1 1 mL15 6.03E+-6 5.63E-04 0.093
DM34 mH1 1 mL19 2.16E+06 3.56E-03 1 .651
DP1 pH1 pL1 1 .80E+06 6.67E-03 3.700
DP99 pH99 pL99 2.90E+07 3.00E-03 0.103 b. Determination of Binding Kinetics and Affinity Constants of Human IgGs Binding to rhSOST at Room Temperature Solution affinities of antigen-antibody interactions were measured on a KinExA
3000 instrument (Sapidyne Inc, Boise, ID) and expressed in the form of equilibrium dissociation constants (KD values; see: Darling et al., Assay and Drug Development Technologies, 2(6):647-657, 2005). All experiments were performed at room temperature (e.g., 22°C) using PBS pH 7.4 with 0.01 % (v/v) Tween-20 as running buffer. Samples were prepared in running buffer supplemented with 1 mg/ml_ BSA. In each experiment, a series of samples with a fixed concentration of rhSOST (R&D systems, Minneapolis, MN) was titrated with a dilution series of the antibody to be tested
and allowed to equilibrate at room temperature. Equilibrated samples were probed for free, not antibody-bound, SOST with the KinExA instrument. This was accomplished by capturing free SOST on polymethylmethacrylate beads adsorption-coated with an antibody that bound the same epitope as the tested antibody. Captured SOST was then detected by fluorescence-labeled anti-His antibody (R&D systems, Minneapolis, MN). The measured titration curves were fit to a standard bimolecular binding equation to obtain the KD value of the interaction (See Table 5), assuming that the antibody was 100% active. If several experiments were performed for a given interaction, the titration curves were fit together using the n-curve analysis tool implemented in the KinExA instrument software.
Table 5. Binding kinetics in solution, of human IgGs to rhSOST at room temperature

Example 2: Anti-SOST Antibodies Neutralizes SOST activity In Vitro
This example illustrates that the anti-SOST antibodies of the present invention neutralized the inhibitory activity of SOST on Wnt/ β-catenin signaling.
Canonical Wnt signaling leads to stabilization and nuclear translocation of intracellular β-catenin. In the nucleus, β-catenin interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) and this complex binds to the Wnt response element in the promoters of target genes leading to their transcriptional upregulation. Canonical Wnt/β- catenin signaling plays a fundamental role in osteoblastogenesis and control of bone mass. Activation of the Wnt^-catenin signaling pathway facilitates osteoblast specification from mesenchymal progenitor cells at the expense of adipogenesis, stimulates osteoblast proliferation, and prolongs the survival of osteoblasts and osteocytes leading to enhanced bone mass and bone strength. SOST is a secreted glycoprotein localized almost exclusively to osteocytes, and a negative regulator of Wnt^-catenin signaling. To assess the ability of the anti-SOST antibodies of the present invention to neutralize the inhibitory activity of SOST on Wnt signaling, an in vitro cell culture system for the quantitation of Wnt signaling was developed. U-2OS osteosarcoma cells were maintained at 37°C, 5% CO2 in McCoy's 5A media (MediaTech, Manassas VA) supplemented with 10% dialyzed fetal bovine serum,
100U/ml Pen/Strep, and 2mM glutamine. Cells were stably transfected with a TOPFIash (TF) reporter plasmid (Upstate Biotechnology, Billerica MA) encoding a luciferase gene under the control of a thymidine kinase promoter downstream of 6 Wnt-responsive TCF/LEF binding sites; the resulting cells were then stably co-transfected with a human Wnt-10B expression plasmid (Origene, accession NM_003394). These cells, named U- 2OS-TF-Wnt10B, were maintained in growth media containing 250pg/ml Geneticin (Gibco, Grand Island NY). Anti-SOST antibodies (DP99, DP1 , DM99, DM21 , and DM4) were produced as human lgG2Aa/kappa isotype, and purified by Protein-A affinity chromatography. The antibodies were concentrated to ~1 mg/ml in phosphate-buffered saline, pH 7.4. An irrelevant isotype control antibody IgG which does not recognize any mammalian proteins was expressed and affinity-purified. Recombinant human (rh)SOST (SEQ ID NO: 1 (excluding the secretory signal sequence) with a carboxy terminal 10-His tag was produced from bacculovirus and purified by Ni-NTA affinity chromatography (Qiagen, Valencia, CA). Purified rhSOST was quantitated by ELISA, and stored at a concentration of 0.13 mg/ml in 0.4 M NaCI, 10% glycerol in aliquots at - 80°C.
For the in vitro activity assay, U-2OS-TF-Wnt10B cells were plated in regular growth media in poly-D-lysine coated 96-well tissue culture plates (BD Biosciences, Beford, MA) at a density of 15,000 cells/well and incubated overnight at 37°C. Cells were treated with 1 μg/ml recombinant human or mouse SOST protein (R&D Systems, Minneapolis, MN) or vehicle (PBS) and varying concentrations of anti-SOST antibodies or isotype control antibody (26H6) in OptiMEM (Gibco, Grand Island NY) in a final volume of 10ΟμΙ. The SOST protein and the antibody to be tested were incubated at room temperature for 15 minutes before adding to the cells. All conditions were tested in triplicate or greater. After an overnight incubation, treated cells were washed once with Dulbecco's PBS, without Ca+2 and Mg+2 (MediaTech, Manassas VA), and lysed with Ι ΟΟμΙ/well Reporter Lysis Buffer (Promega, Madison, Wl). Fifty μΙ of each lysate was aliquoted into the wells of a white 96-well plate (Greiner Bio-One), 50μΙ of Luciferase Reagent (Promega, Madison, Wl) was added per well, and luminescence was recorded using a Victor3V multilabel counter (Perkin Elmer, Waltham, MA). Results were calculated as the mean ± S.E.M. of n=3-6 samples for each condition tested. GraphPad Prism 5 software was used for the dose-response curves and determination of IC50 and EC50 values.
Figure 1 shows does-dependent inhibition of Wnt-10B signaling by recombinant mouse SOST and human SOST proteins. Various concentrations of mouse SOST (A) or human SOST (B) were added to U-2OS-TF-Wnt1 OB cells, and luciferase reporter activity was measured after 24 hours. N=6 for each condition tested. The IC50 for mouse or rhSOST inhibition of Wntl OB activity was calculated to be 10.4 nM or 13.3 nM, respectively. Figure 2 shows in vitro neutralization of rhSOST activity by anti-SOST antibodies of the present invention. Anti-SOST antibodies DP99, DP1 , DM99, DM21 , and DM4 dose-dependently blocked the SOST-mediated inhibition of Wnt-10B activity in U-2OS-TF-Wnt1 OB cells. Various concentrations of anti-SOST antibodies or isotype control mAb were preincubated with rhSOST (1 .5 pg/rnl, approximately 65nM) for 15 minutes at room temperature, then added to cells. Luciferase activity was measured after 24 hours. All conditions were tested in triplicate. To allow for direct comparison between antibodies tested in different 96-well plates, responses were normalized between plates, such that the100% response represents cells without added SOST or mAb (a condition tested in triplicate on each plate). The calculated EC50 values (in Mg/ml) for the anti-SOST mAbs were: DP99, 5.95; DP1 , 22; DM99, 1 .95; DM21 , 5.53 and DM4, 2.07.
Example 3: Anti-SOST Antibodies Significantly Improved Bone Density and Strength Over the Ovariectomized Rats
This example illustrates that ant-SOST antibodies of the present invention significantly improved bone density and strength over the ovariectomized rats.
The 15-week (wk) old female Sprague-Dawley rats were ovariectomized (OVX) and maintained at the animal facility for 8 weeks after surgery to allow for bone loss to occur. At 8 weeks post-surgery, rats were weighed and randomized into groups. At this time, baseline Sham and baseline OVX groups were sacrificed, whereas dosing began for all other groups. Dosing consisted of subcutaneous injection of vehicle or antibody (irrelevant isotype control at 20mg/kg or anti-SOST mAb DP99 at 2 or 20 mg/kg) 1x/week for 6 weeks, or Parathyroid Hormone (PTH) (40 g/kg) 5x/wk for 6 wks. At the end of the treatment period, animals were sacrificed, and the left femurs and lumbar vertebra L4-6 were collected, wrapped in saline-soaked gauze, frozen at -20°C and analyzed by bone densiometry and biomechanical strength testing.
Densitometry
Peripheral Quantitative Computed Tomography (pQCT) was performed on the excised left femurs using a Stratec XCT-RM instrument and associated software (Stratec Medizintechnik GmbH, Pforzheim, Germany; software version 5.40). The scan was performed at the midshaft femur. The positions were verified using scout views and one 0.5-mm slice perpendicular to the long axis of the femoral shaft was acquired from each site. The scans were analyzed using a threshold for delineation of the external boundary. Cortical bone mineral content and bone mineral density, endosteal and periosteal circumference, average cortical thickness and axial moment of inertia were reported. Summarized data is shown in Table 6. Data shown are the mean ± SEM for each group (N=10). Statistically significant differences between groups (p < 0.05) are shown in the last two rows. Untreated baseline Sham and OVX groups (1 and 2) were compared to each other using an unpaired t-test; treated groups (3-8) were compared to all other treated groups using an ANOVA with a post hoc Duncan Test, n.s.; nonsignificant.
Table 6. Summary Data for pQCT at Midshaft Femur

vs. 2) n.s. n.s. n.s. n.s. n.s. n.s. n.s.
Duncan
Test 7,8 > 4,5 7 > 4,5 n.s. n.s. n.s. n.s. n.s.
Mechanical Testing a. Three Point Bending Test of the Femoral Shaft
Upon completion of the pQCT scanning, the anterior to posterior diameter at the midpoint of the femoral shaft was taken with an electronic caliper and recorded. The femur was then placed on the lower supports of a three point bending fixture with the anterior side facing downward in an Instron Mechanical Testing instrument (Instron 4465 retrofitted to 5500). The span between the two lower supports was set at 14mm. The upper loading device was aligned to the center of the femoral shaft. The load was applied at a constant displacement rate of 6 mm/min until the femur broke. The locations of maximum load, stiffness and energy absorbed were selected from the load and displacement curve and values calculated by instrument software (Bluehill v2.5, Instron). The intrinsic properties, ultimate strength, elastic modulus and toughness, were calculated from maximum load, stiffness, energy absorbed, anterior-posterior diameter, length between two supports and axial area moment of inertia using the following equations:
Instrument measurements:
Maximum load (units: N) (Fu)
Stiffness (units: N/mm) (S)
Energy absorbed (units: mJ) (W)
Electronic caliper and pQCT measurements:
Anterior-posterior diameter (units: mm) (a)
Axial area moment of inertia, IX_CRT_A from pQCT database at the midshaft (units: mm4) (I)
Constants:
Length between two supports (units: mm) (L)
Derived parameters:
Ultimate Strength (units: N/mm2) (s)
Formula: s = ((Fu * L *(a/2)) / (4* I)
Elastic modulus (units: MPa) (E)
Formula: E = (S) * (L)3 / (48 * I)
Toughness (units: MJ/m3) (T)
Formula: T = 3 * (W) * (a/2)2 / (L* I)
Summarized data are shown in Table 7, which indicates that anti-SOST antibody DP99 significantly improved bone strength over the ovariectomized rats. Data shown in Table 7 are the mean ± SEM for each group (N=10). Statistically significant differences between groups (p < 0.05) are shown in the last two rows. Untreated baseline Sham and OVX groups (1 and 2) were compared to each other using an unpaired t-test;
treated groups (3-8) were compared to all other treated groups using an ANOVA with a post hoc Duncan Test, n.s.; non-significant.
Table 7. Summary Data for Three Point Bending Test of Femoral Shaft

After three point bend testing of the femoral shaft, the proximal half of the femur was used in the cantilever compression test of the femoral neck. The proximal half of the femur was placed firmly in an anchoring platform where the greater trochanter was lodged in a notch cut in the platform. The test was conducted with an Instron
Mechanical Testing Instrument (Instron 4465 retrofitted to 5500). The load was applied to the femoral head with a stainless steel probe, parallel to the femoral shaft at a constant displacement rate of 6 mm/min until failure. The locations of maximum load,
stiffness and energy absorbed were selected manually from the load and displacement curve and calculated by instrument software (Bluehill v2.5, Instron).
Instrument measurements:
Maximum load (units: N) (Fu)
Stiffness (units: N/mm) (S)
Energy absorbed (units: mJ) (W)
Summarized data are shown in Table 8, which indicates that anti-SOST antibody DP99 significantly improved bone strength over the ovariectomized rats. Data shown are the mean ± SEM for each group (N=10). Statistically significant differences between groups (p < 0.05) are shown in the last two rows. Untreated baseline Sham and OVX groups (1 and 2) were compared to each other using an unpaired t-test; treated groups (3-8) were compared to all other treated groups using an ANOVA with a post hoc Duncan Test, n.s.; non-significant. Table 8. Summary Data for Cantilever Compression Test of Femoral Neck

3. Compression Test of the Lumbar Vertebral Body
For each de-fleshed vertebrae sample (L5), the posterior pedicle arch and spinous process were removed using a low-speed diamond saw. Additionally, the cranial and caudal ends of each vertebral body were cut off with the same diamond saw to obtain a vertebral body specimen with two parallel surfaces and a height
approximately equal to 2 mm. Width in the medial-lateral and anterior-posterior directions at both the cranial and caudal ends was measured using digital calipers.
Values obtained from the two ends were recorded and the average value used in the calculation of cross-sectional area. The height of the vertebral body specimen was also taken with electronic calipers and recorded. The specimens were then placed between two platens and a load applied at a constant displacement rate of 0.6 mm/min until failure in an Instron Mechanical Testing Instrument (Instron 4465 retrofitted to 5500). The load and displacement curve was recorded by instrument software (Bluehill 2 v. 2.5, Instron). The locations for maximum load at failure, stiffness and energy absorbed were selected manually from the load and displacement curve and calculated by instrument software (Bluehill 2 v. 2.5, Instron). The intrinsic properties, ultimate strength, elastic modulus and toughness, were calculated from maximum load, stiffness, energy absorbed, cross-sectional area and height using the following equations:
Instrument measurements:
Maximum load (units: N) (Fu)
Stiffness (units: N/mm) (S)
Energy (units: mJ) (W)
Electronic caliper measurements:
Width in the anterior-posterior direction (units: mm) (a)
Width in the medial-lateral direction (units: mm) (b)
Height of the vertebral body in the cranio-caudal direction (units: mm) (h)
Derived parameters:
Cross sectional area (units: mm2) (CSA)
Formula: CSA =Π * a * b / 4
Ultimate Strength (units: N/mm2) (o)
Formula: σ = Fu / CSA
Elastic modulus (units: MPa) (E)
Formula: E = S / (CSA / h)
Toughness (units: MJ/m3) (T)
Formula: T = W / (CSA * h)
Summarized data are shown in Table 9, which indicates that anti-SOST antibody DP99 significantly improved bone strength over the ovariectomized rats. Data shown are the mean ± SEM for each group (N=10). Statistically significant differences between groups (p < 0.05) are shown in the last two rows. Untreated baseline Sham and OVX groups (1 and 2) were compared to each other using an unpaired t-test; treated groups (3-8) were compared to all other treated groups using an ANOVA with a post hoc Duncan Test, n.s.; non-significant.
Table 9. Summary Data for Vertegral compression Test of L5 Vertebra
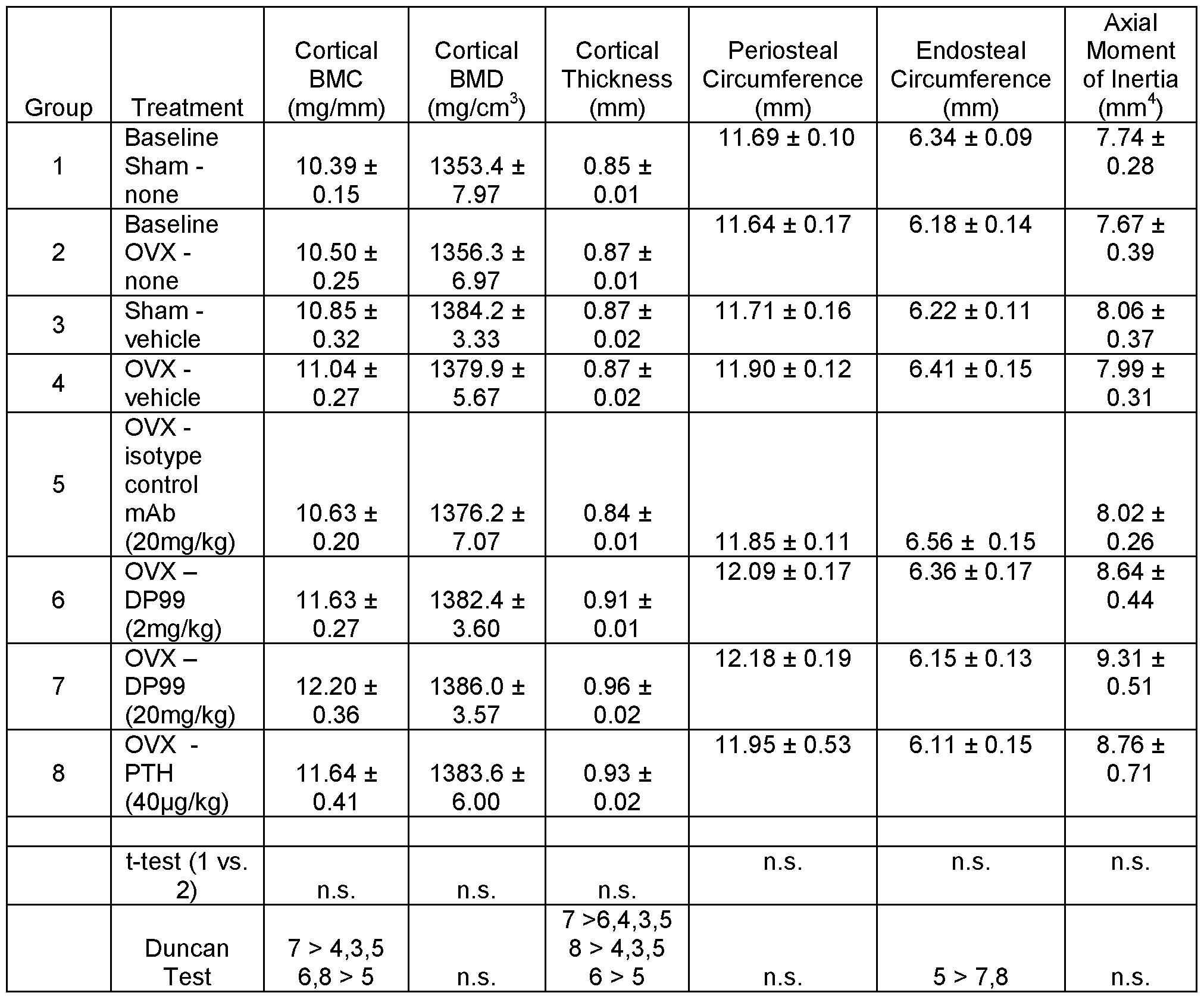
Ovariectomized rats treated with 20 mg/kg antibody DP99 or PTH showed significantly improved bone density and strength over the OVX animals treated with vehicle or irrelevant antibody isotype control, and in some cases, results were significantly higher than vehicle-treated Sham controls. Significant increases were seen
in cortical bone mineral content, cortical thickness at the midshaft femur, maximum load and energy at the femoral neck, and energy, ultimate strength, and toughness
associated with vertebral compression. Example 4: Anti-SOST Antibodies Stimulate Bone Formation and Increases Bone Mass in Ovariectomized Mice
This example illustrates that neutralization of SOST by antibodies of the present invention increased cancellous and cortical bone mass and prevented bone loss induced by estrogen deficiency in ovariectomized mice and that the antibodies as described herein reversed the inhibition of Wnt activity by SOST in vitro. a. Methods
Determination of the effect of mAb in Wnt activity in U20S TOPFIash cells
U2OS cell line was obtained from ATCC and stably transfected with TOPFIash plasmid (Upstate; Millipore, Millerica, MA), a TCF-luciferase reporter construct. Cells were maintained at 37°C, 5% CO2 in McCoys 5A Media supplemented with 10% fetal calf serum, 1 % Pen Strep, and 2mM Glutamine. U2OS TOPFIash cells were plated in regular growth media at a density of 37,500 cells/cm2 and incubated overnight Cells were transfected using Lipofectamine Plus (Invitrogen, Grand Island, NY) with plasmids carrying Wnt10b and MesDC2 cDNAs (Origene, Rockville, MD) and then incubated in regular growth media for 24 hours. Transfected cells were treated with 1 μg/ml rhSOST protein (R & D Systems, Minneapolis, MN) or vehicle (PBS, Gibco, Grand Island, NY) and varying concentrations of SOST mAbs in Optimem (Gibco, Grand Island, NY). After an overnight incubation, cells were lysed with Reporter Lysis Buffer (Promega, Madison, Wl) and luciferase expression was quantified using Luciferase Reagent (Promega, Madison, WA).
Animals and experimental Protocol
Fifty C57BL/6 female mice were randomized into 5 groups with 10 per group and subjected to sham (n=10) or ovariectomized (OVX) surgeries (n=40) under general anesthesia with ketamine/xylazine. Starting the day after surgery, the sham and one group of OVX rats were subcutaneously injected with vehicle (PBS) 2 times per week for 6 weeks. The remaining OVX rats were subcutaneously injected with 25 mg/kg of DP99 or DM99, 2 times per week for 6 weeks. The animals were housed at 24°C with a 12h
light/12h dark cycle and allowed free access to water and a commercial diet (Purina laboratory Rodent Chow 5001 , Purina-Mills, St. Louis, MO) containing 0.95% calcium, 0.67% phosphorus, and 4.5 lU/g vitamin D3. To label the active bone formation surfaces, all mice were subcutaneously injected with demeclocycline at 20 mg/kg prior to the first dose, and then with xylenal orange at 90 mg/kg or calcein at 5 mg/kg (Sigma Chemical Co., St. Louis, MO) on -9 and -2 days before euthanasia. All mice were euthanized by CO2 asphyxiation followed by exsanguination. Serum, both right and left femurs, right tibia, and lumbar spine were harvested for further analyses. The experiments were conducted according to Pfizer animal care-approved protocols, and animals were maintained in accordance with the ILAR (Institute of Laboratory Animal Research) Guide for the Care and Use of Laboratory Animals.
Measurement of serum concentration of anti-SOST Antibodies
Serum concentrations of anti-SOST mouse antibodies were determined by an ELISA method. Samples and anti-SOST mouse antibody calibration and quality control standards were diluted in a PBS buffer. The 96-well immunosorbent assay plates were overnight coated with recombinant mouse SOST (R&D Systems, Minneapolis, MN) and then blocked with Superblock T20 (Thermo Scientific, Rockford, IL) after washing with PBS buffer. The diluted samples and standards were added to plates and incubated at room temperature for 1 hour. The plates were then washed, followed by incubation with a horseradish peroxidase-conjugated anti-mouse lgG1 secondary antibody (Southern Biotech, Birmingham, AL) for 1 hour. After washing, the plates were developed by color reaction for ~10 min. The anti-SOST mouse antibody calibration standards were used to construct a standard curve using 4-parameter fitting with uniform weighting in SoftMax Pro 4.8. Serum concentrations of anti-SOST mouse antibody in unknown samples were interpolated from this standard curve.
Measurement of serum concentration of total SOST
Total SOST protein levels were measured in mouse serum using the Meso Scale Diagnostics (MSD) Human Sclerostin (SOST) ELISA kit (catalog number: K1 1 1 HGC-2), which cross-reacted with mouse SOST protein. Recombinant mouse SOST protein was purchased from R & D Systems (catalog number 1589-ST) and used to generate the standard for the MSD SOST assay. Mouse recombinant SOST was reconstituted in
PBS containing 0.1 % fetal bovine serum and stored at -20 °C. The recombinant mouse SOST was diluted to 25 ng/ml immediately prior to running the MSD SOST assay and subjected to 3-fold serial dilutions to prepare the mouse SOST standard (the SOST standards ranged from 25 ng/ml to 0.006 ng/ml). The MSD SOST assay was run according to manufacturer's instructions.
Measurement of serum osteocalcin and CTX
Mouse serum osteocalcin was measured using a two-side immunoradiometric assay (ALPOCO Diagnostics, Windham, NH). Serum C-telopeptide (CTX) was determined by RatLaps ELISA (Nordic Bioscience, Diagnositics, Herlev, Denmark). pQCT measurement of right femurs
The right femurs were scanned by peripheral quantitative computed tomography (pQCT, Stratec XCT Research M; Norland Medical Systems, Fort Atkinson, Wl, USA) with software version 5.40 as described in Ke et al., J. Musculoskel. Neuron Interact, 2:479-488, 2002. A 1 -mm-thick cross-section of each distal femoral metaphysis was taken at 2.5 mm proximal to the distal end (~1 .5 mm to the growth plate, a cancellous bone enriched site), and 1 -mm-thick cross-section of each femoral diaphysis was taken at 8 mm proximal from the distal end (a cortical bone enriched site) with a voxel size of 0.10 mm. Volumetric total bone content (BMC), density (BMD) and area were determined at the distal femurs and femoral diaphyses.
Histomorphometric measurements of the right distal femurs and tibial diaphyses
The right distal femurs and tibial diaphyses were processed for
histomorphometric assessment on cancellous and cortical bone, respectively, as described, e.g., Baron et al., In: Recker RR (ed.) Bone Histomorphometry: Techniques and Interpretation, CRC Press, Boca Raton, FL, USA, 13-35, 1983; Parfitt et al., J. Bone Miner. Res., 2:595-610, 1987; and Jee et al., In: Takahashi N (ed) Handbook of Bone Morphology, Nishimusa, Niigata City, Japan 87-1 12, 1997. Briefly, the bones were dehydrated in graded concentrations of ethanol and embedded undecalcified in methyl methacrylate. Longitudinal frontal sections of the distal femur were cut at 4- and 10-μΓη thickness using a Reichert-Jung Polycut S microtome (Leica Corp., Heidelberg,
Germany). The 4-μΓη sections were stained with a modified Masson's Trichrome stain
for the measurements of static parameters and the 10-μΓη sections remained unstained for the fluorochrome-based dynamic parameters. Cross-sections of tibial diaphyses were cut at the tibial-fibular junction using a Saw Microtome (Leica SP1600, Leica Corp., Heidelberg, Germany) and remained unstained for static and dynamic
parameters. All histomorphometric measurements were performed by using an image analysis system (Osteomeasure, Inc., Atlanta, GA).
Biomechanical measurements of left femurs
Femurs were tested in 3 point bending using the materials testing machine (TestResources, model 100R250, Shakopee, MN) with 50 lb. load cell. The span between the lower supports was 8.5 mm. Load was applied at a rate of 20 mm/min and data were collected at 15/sec to define a load-displacement curve. The energy to failure, which reflects how much energy the bone absorbs before it breaks, was calculated as the area under the curve. The ultimate force or strength of the bone was calculated as the maximum height of the curve and the stiffness was maximum slope of the curve. See, e.g., Turner and Burr, Bone, 14:595-608, 1993, and Turner et al., J. Bone Miner. Res. 16:206-213, 2001 .
Statistical analysis
Data are expressed as the mean ± SEM for each group. Statistical differences between the anti-SOST antibodies and vehicle-treated or among antibody-treated groups were evaluated with one-way ANOVA followed by the Fisher's PLSD. Statistical differences between sham and OVX controls were determined by unpaired Student's t- test. Probabilities (p) less than 0.05 were considered significant. b. Results
Characterization of monoclonal antibodies to SOST
The functional ability of antibodies DP99 and DM99 to neutralize SOST activity was examined in the osteosarcoma U2OS TOPFIash cell line (Figure 3(A) and 3(B)). In this cell system, WNTI Ob stimulated a luciferase reporter gene and recombinant SOST protein inhibited the WNTI Ob induced stimulation. The ability of these antibodies to neutralize SOST was determined by their ability to bring the luciferase activity back to the levels of WNTI Ob stimulation observed in the absence of SOST protein. Dose
response studies demonstrated that DP99 and DM99 were very potent at restoring WNT10b activity (EC50s 37 nM and 15 nM, respectively). In addition, differences were observed between the antibodies in that DM99 fully restored the WNT activity and DP99 was intermediate.
Changes in body weight and levels of serum mAbs, total SOST and bone biomarkers
To test whether inhibition of SOST increases bone mass and prevent bone loss in an estrogen deficient condition, the antibodies DP99 and DM99 were administered to OVX mice for 6 weeks starting the day after surgeries. At 6 weeks post surgery, all OVX mice were heavier than sham controls with no difference between the antibodies and vehicle-treated OVX mice. High concentrations of antibodies DP99 and DM99 were detected even at 72 hours after last dose of each of them, confirming systemic exposure of the treated mAbs. To confirm whether the tested mAbs neutralize SOST in vivo, an ELISA assay was established to detect the serum total SOST level. A very low level of total SOST was detected in the vehicle-treated sham and OVX mice. However, the mice treated mAbs had significant higher serum total SOST levels than sham and OVX controls. The elevated level of serum total SOST in the mice treated with DP99 was significantly higher than those treated with DM99. Serum osteocalcin, a bone formation biomarker, was increased in the mAb-treated rats compared with vehicle-treated sham and OVX controls. An increase in serum CTX, a bone resorption biomarker, was seen only in the mice treated with DM99.
Table 10. Body weight and serum levels of mAb, total SOST, osteocalcin, and CTX
Parameters Sham OVX OVX+DP99 OVX+DM99
Body weight (g) 25.9±0.4 27.8±0.7a 28.4±0.7a 30.8±0.9a
Serum mAb (ng/ml) NA NA 876±260 674±125
Serum total SOST (ng/ml) 0.003±0.001 0.004±0.001 1 .53±0.14ab 0.19±0.1 1 abc
Serum osteocalcin (ng/ml) 77.6±3.8 70.5±6.4 108.8±6.9ab 1 16.2±10.2ab
ab
Serum CTX (ng/ml) 39.4±4.4 42.1 ±3.8 57.9±5.0 62.4±4.2
Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle or DP99, or DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries. Data are expressed as mean ± SEM. Serum mAb levels were 72 hours after last dose. NA: non-applicable. Statistical analyses were not performed for serum mAb values. a: p < 0.05 vs. Sham; b: p < 0.05 vs. OVX; c: p < 0.05 vs. DP99.
Increased bone mass and suppressed bone loss In OVX mice by antl-SOST antibodies The effects of anti-SOST antibody treatment on bone mass were assessed by pQCT measurements on distal femur, a cancellous bone site and femoral diaphyses, a cortical bone site. At distal femurs, OVX mice treated with vehicle exhibited decreased bone mass as evidenced by lower total bone mineral content (BMC) and total bone mineral density (BMD) than sham controls. However, DP99 and DM99 increased BMC, BMD, and total bone area in OVX mice to the levels that were higher than the sham and OVX groups. See Figures 4A, 4B, and 4C. At femoral diaphyses, BMC and BMD were decreased whereas total bone area was increased in vehicle-treated OVX mice compared with sham controls. See Figures 4D, 4E, and 4F. Both DP99 and DM99 increased BMC, BMD, and bone area in comparison with both sham and OVX controls. There was no significant difference between DP99 and DM99 groups. Consistent with pQCT data, 3-D analysis showed that vehicle-treated OVX mice had significantly lower cancellous bone than sham controls. See Figure 5A. It was significantly increased by the treatment with DP99 and DM99 to the levels above sham and OVX controls. See Figure 5B.
Increased bone formation In cancellous bone by mAb treatment at distal femurs
To understand the mechanism underlying the observed increase of bone mass by anti-SOST antibody treatment at the tissue level, standard cancellous bone histomorphometric analysis was performed on distal femurs. At 6 weeks post surgery, despite an increase in osteoclast surface and number on the cancellous surface of distal femur, OVX caused no significant differences in parameters reflecting bone mass and bone formation. Osteoclast surface and number in the mice treated with each of the mAb were significantly lower than that of OVX controls but maintained at the sham control levels. Treatment of OVX mice with DP99 increased cancellous bone volume, mineralizing surface, surface referent bone formation rate, and tissue referent bone
formation compared with vehicle treated sham and OVX mice. Cancellous bone volume and mineral apposition rate in these mice were also higher than those treated with sham controls. Treatment of OVX mice with DM99 increased cancellous bone volume, mineralizing surface, mineral apposition rate, surface referent bone formation rate, and tissue referent bone formation compared with vehicle-treated sham and OVX mice. None of the mAb treatments altered bone volume referent bone formation rate. See Table 1 1 .
Table 1 1 . Cancellous bone histomorphometric parameters of distal femurs
Parameters Sham OVX OVX+DP99 OVX+DM99
Cancellous bone volume (%) 6.3±0.9 5.8±0.7 1 1 .3±0.8a 9.9±0.6a
Osteoclast surface (%) 0.66±0.09 1 .20±0.1 1 a 0.67±0.09 0.46±0.05
Osteoclast number (#/mm) 0.32±0.04 0.48±0.06a 0.28±.03 0.24±0.02
Mineralizing surface (%) 13.2±1 .6 15.1 ±2.0 22.0±1 .3a 22.9±2.3a
MAR (μΓΠ/d) 1 .24±0.09 1 .28±0.07 1 .48±0.08a 1 .53±0.10a
BFR/BS (μΐτι2/μηι/α:) 0.33±0.23 0.35±0.03 0.49±0.04a 0.53±0.05a
BFR/BV (%/year) 2.01 ±0.19 1 .96±0.10 2.00±0.17 2.03±0.19
BFR/TV (%/year) 0.13±0.02 0.1 1 ±0.02 0.23±0.03a 0.20±0.03a
Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle, DP99, or DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries.
Data are expressed as mean ± SEM. a: p < 0.05 vs. Sham; b: p < 0.05 vs. OVX.
MAR: mineral apposition rate; BFR/BS: bone surface referent bone formation rate;
BFR/BA: bone area referent bone formation rate; BFR/TV: tissue volume referent bone formation rate.
Increased bone formation in cortical bone by anti-SOST antibody treatment at femoral diaphyses Standard cortical bone histomorphometric analysis was performed on tibial diaphyses. In vehicle-treated OVX mice, total tissue area, marrow area, periosteal mineral apposition rate and bone formation rate were higher whereas endocortical mineralizing surface and bone formation rate were lower than sham controls. Both
DP99 and DM99 had very similar effects on cortical bone. They increased total tissue area, cortical area and thickness, periosteal mineral apposition rate and bone formation compared with treatment of sham and OVX mice with vehicle. In addition, periosteal mineralizing surface was elevated by DP99 and DM99 relative to vehicle treatment for sham and OVX mice. Furthermore, endocortical mineral apposition rate was higher in the mice treated with DP99 and DM99 than those in sham groups. Endocortical eroded surface was observed in 5 out of 10, 8 out of 9 (one mouse died prematurely), 8 out of 10, and 2 out of 10 in sham, OVX, DP99, and DM99 group, respectively. See Table 12. Due to the lack of sufficient number of mice with observed eroded surface, statistical analysis was not conducted for this parameter.
Table 12. Cortical bone histomorphometric parameters of tibial diaphyses
Parameters Sham OVX OVX+DP99 OVX+DM99
!b
Total tissue area (mm2) 0.89±0.01 0.93±0.01 a 1 .03±0.02a 1 2±0.02ί
!b
Cortical bone area (mm2) 0.61 ±0.01 0.61 ±0.01 0.79±0.01 a o.79±o.or
!b
Marrow area (mm2) 0.28±0.01 0.32±0.01 a 0.25±0.01 a o.23±o.or
!b
Cortical thickness (mm) 227.0±2.3 224.4±6.0 287.0±4.7a 291 .0±3.2ί
Periosteal surface
Mineralizing surface (%) 19.1 ±2.9 26.7±4.2 50.9±5.2a 41 .8±4.3a
MAR ^m/d) 0.65±0.05 0.90±0.09a 1 .27±0.06a 1 .40±0.06a
BFR/BS (μΠΊ2/μΐτι/α!) 0.14±0.02 0.23±0.03a 0.63±0.06a 0.56±0.06a Endocortical surface
Mineralizing surface (%) 45.5±2.9 31 .0±3.1 a 51 .2±2.1 43.4±3.5 MAR ^m/d) 0.82±0.07 0.67±0.09 0.57±0.06a 0.57±0.06a
BFR/BS (μΐτι2/μΐτιΛ1) 0.39±0.04 0.23±0.06a 0.29±0.03 0.26±0.04
Eroded surface* (%) 5.05±1 .01 6.76±1 .65 6.08±0.06 2.21
Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle, DP99, or DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries.
Data are expressed as mean ± SEM. a: p < 0.05 vs. Sham; b: p < 0.05 vs. OVX.
MAR: mineral apposition rate; BFR/BS: surface referent bone formation rate.
*: Number of mice with observed eroded surface was varied: 5 out of 10, 8 out of 9 (one died prematurely), 8 out of 10, and 2 out of 10 in sham, OVX, DP99, and DM99 group, respectively. Values were the mean from the mice with observed eroded surface. Due to the lack of sufficient number of mice with observed eroded surface, statistical analysis was not conducted for this parameter.
Improved biomechanical properties by anti-SOST antibody treatment in femur and lumbar vertebral body To evaluate the biomechanical properties of the bones from the mice treated with anti-SOST antibodies compared with vehicle treatment, the moments of inertia were generated prior to a 3-point bending test performed on the left femurs from the same mice. There was no difference between vehicle-treated sham and OVX mice in biomechanical properties of femur. All of these parameters in the mice treated with DP99 or DM99 were higher than vehicle-treated controls. The moments of inertia generated from μΟΤ measurements at the femoral diaphyses were similar to the aforementioned data generated by 3-point bending test.
Table 13. Biomechanical parameters of femurs
Parameters Sham OVX OVX+DP99 OVX+DM99
3-point bending test
Energy to failure (mJ) 9.5±0.6 9.2±0.9 16.9±0.9a 17.3±0.7a
Stiffness (N/mm) 49.9±2.2 48.3±3.0 61 .8±2.8a 62.4±0.8a
Ultimate force (N) 18.5±0.7 17.9±0.7 25.7±0.8a 24.8±1 .0a
Postyield displacement (mm) 0.64±0.06 0.59±0.04 0.90±0.05a 0.94±0.03a ΟΤ measurement at femoral diaphyses
Max moment of inertia (mm4) 0.21 ±0.01 0.21 ±0.01 0.31 ±0.01 a 0.30±0.02a
Min moment of inertia (mm4) 0.15±0.01 0.13±0.01 0.18±0.01 a 0.20±0.01 a
Polar moment of inertia (mm4) 0.36±0.02 0.34±0.01 0.49±0.02a 0.50±0.02a
Female C57BL/6 mice were subjected to sham or OVX at 4 months of age. They were subcutaneously injected with vehicle, DP99, and DM99 at 25 mg/kg, twice per week for 6 weeks starting the date after surgeries.
Data are expressed as mean ± SEM. a: p < 0.05 vs. Sham; b: p < 0.05 vs. OVX.
Results from the current study showed that neutralization of SOST by anti-SOST antibodies (e.g., DP99 and DM99) of the present invention augmented both cancellous and cortical bone mass in OVX mice, an animal model of estrogen deficiency induced bone loss. The effect of increasing cancellous bone mass by these mAbs was evidenced by the increases in volumetric BMC and BMD as assessed by pQCT and the increase in cancellous bone volume as assessed by histomorphometry at the distal femurs, a cancellous rich bone site. Regarding the tissue level mechanism underlying such efficacy, the significant increases in surface and tissue referent bone formation rates indicate a stimulation of bone formation by mAb treatment. Both the mineralizing surface, a measurement reflecting active osteoblast number, and mineral apposition rate, a parameter reflecting osteoblast activity, were increased by mAb treatment, suggesting that the higher bone formation rate resulted from a promotion of osteoblast differentiation and proliferation. The effects of the tested mAbs on increasing cortical bone mass were
demonstrated by the increases in BMC and BMD at the femoral diaphyses and the increases in cortical bone area and cortical width at the tibial diaphyses. On the periosteal surface, mAb treatment increased mineralizing surface, mineral apposition rate and bone formation rate. These data indicated that the tested mAbs stimulated periosteal bone formation via increasing osteoblast recruitment and activity. The increased periosteal bone formation resulted in greater bone deposition on the periosteal surface as illustrated by the increased total tissue area of the tibial diaphyses from the mice treated with mAbs. On the endocortical bone surface, mAb treatment of OVX mice also produced greater bone mass as demonstrated by the decrease in marrow area of tibial diaphyses. However, which process, bone formation or resorption, contributed to the increased bone mass on this surface was not clear and it appeared to be different among the tested mAbs. In the DP99 treated group, mineralizing surface was significantly higher whereas mineral apposition rate and the number of the mice with observed eroded surface were lower than the vehicle-treated OVX mice. These
data suggested that the combination of increased osteoblast number and decreased resorption induced more bone on endocortical surface. In the DM99 treated group, mineral apposition rate was lower than the vehicle-treated mice and only one mouse had observed eroded surface, signifying a suppression of bone resorption by DM99. Dynamic histomorphometric analysis revealed the anabolic effects of anti-SOST antibodies on bone formation, which was supported by a significant increase in serum osteocalcin, one of the biomarkers of global bone formation, in the mice treated with both the DP99 and DM99. This finding is consistent with the increased bone formation rate in sclerosteosis patient (Stein et al., Neurology, 33:267-277, 1983), in SOST KO mice (Li et al., J. Bone Miner. Res. 23:860-869, 2008) and in OVX rats treated with a SOST mAb (Li et al., 2008, supra).
Based on the greatly improved bone mass phenotype of scleroteosis patients and SOST KO mice, the same result can therefore be expected for the mouse bones treated with mAbs. Biomechanical testing of the femurs from the mice treated with the anti- SOST antibodies of the present invention in this study confirmed the increased bone mass induced by mAb treatment was biomechanically sound. The anti-SOST antibodies treated femurs were stronger and stiffen Importantly, all of the anti-SOST antibodies used in this study improved bone quality. The energy required to break the bone was increased and the brittleness was decreased as evidenced by increased post- yield displacement. The maximum, minimum, and polar moments of inertia of the femurs were increased by the anti-SOST antibody treatment indicating that the treated bones were more structurally robust.
The changes in serum total SOST levels were also measured using an ELISA kit. The two tested anti-SOST antibodies caused a highly significant increase in serum total SOST levels. Although the change in serum total SOST did not appear to be correlated with bone efficacy, it served as a surrogate for the confirmation of SOST inhibition in vivo.
This study demonstrated that administration of anti-SOST monoclonal antibodies of the present invention can effectively stimulate bone formation on cancellous, periosteal and endocortical surfaces, resulting in markedly increased cancellous and cortical bone mass as well as bone strength.
Example 5: Anti-SOST Antibodies are Efficacious in Preventing Bone Loss in Ovariectomized Mice
Three anti-SOST mAbs were compared in a 4-week mouse OVX prevention study for their efficacy to prevent OVX-induced bone loss. One hundred and ten C57/BI6 female mice aged 18 weeks were subject to ovariectomy or sham surgery, and the dosing was begun the day after surgery. Antibodies were administered by subcutaneous injection at a dose of 1 , 5 or 25 mg/kg once per week for 4 weeks. DP1 and DM1 1 mAbs were expressed and affinity-purified as recombinant chimeric antibodies with humanized variable regions and mouse lgG1 /mouse kappa constant regions. DP99 mAb is a recombinant lgG1 /kappa mAb, and 26H6 is an irrelevant isotype control mAb. Mouse femurs and L4-L5 vertebra were harvested 6 weeks after the initiation of dosing.
Micro-Computed Tomography (μΟΤ)
The femur and lumbar vertebrae were used for μΟΤ analyses of cancellous and cortical bone. Ex vivo μΟΤ analysis was performed on distal femoral metaphysis or lumbar vertebral body for cancellous bone and on the femoral mid-dyaphysis for cortical bone using a Viva μΟΤ-40® computed tomography system (Scanco Medical, Bassersdorf, Switzerland) (see, e.g., Bagi et al., Bone, 38:136, 2006). For scanning, the proximal tibia sample was oriented vertically in a 16.5-mm sample holder with the epiphyseal head facing downward. A control file, or measurement protocol, was created to define the scanning parameters such as source energy, sample size and desired image resolution. Parameters selected for this study included a source voltage of 55 kVp and / of 145 / mA to obtain the best contrast between bone and soft tissues. The sample area selected for the three-dimensional structural analysis of cancellous bone was a 2.5-mm length of the metaphyseal secondary spongiosa, originating 0.5 mm below the epiphyseal growth plate and extending cranially. The standard resolution mode (21 mm) was used to scan 1 19 slices in which the region of interest (ROI) was drawn; the cortical bone was intentionally excluded. Two- and three-dimensional analyses were then performed on the ROI in each bone, and the following cancellous bone parameters were measured: tissue volume (TV), bone volume (BV), bone volume/tissue volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th),
trabecular separation (Tb.Sp), connective density (Conn.D) and bone mineral density (BMD).
Cortical bone parameters were evaluated by CT using previously described methods (see, e.g., Hanson and Bagi, Bon, 35:326, 2004. In brief, CT images of the cortical bone were obtained approximately 1 .5 cm proximal from the distal end of the femur. Sample measurements (scans) were performed on this region using medium resolution settings. Each scan yielded a data set of 25 images of 1024mm x 1024mm 2D axial slices through the mid-shaft region of the femur. The calculation was performed on 25 slices (1 slice = 10.5 mm), using the average for the final calculation. The total area (TA) was calculated by counting all voxels within the counter, the bone area (BA) was calculated by counting all voxels that were segmented as bone and the bone marrow area (MA) was calculated as TA-BA. The outer and inner perimeters of the cortical mid-shaft were determined using a 3D triangulation of the bone surface (BS) of the 25 slices, and cortical thickness was calculated using the formula C.Th = ½ * BS/BV. Figures 6-9 show that anti-SOST antibodies DP99, DP1 , and DM1 1 are efficacious in preventing OVX-induced bone loss.
Example 6: Restoration of Ovariectomv-lnduced Bone Loss in Rat Using Anti-SOST Antibodies
Two anti-Sost mAbs (DP1 and DM21 ) were tested in a 6-week rat OVX (ovariectomy) bone restoration study for their efficacy to restore the bone loss induced by ovariectomy. Sixty female Sprague Dawley rats aged 3.5 months were subject to OVX or sham surgery, and allowed to recover for 6 weeks prior to the initiation of treatment. Following surgery, rats were restricted to 15g rat chow/rat/day to prevent excessive weight gain. Antibodies were administered by subcutaneous injection at a dose of 3 or 30 mg/kg once per week for 6 weeks. DM21 , DP1 and the negative isotype control mAb were expressed and affinity-purified as recombinant chimeric antibodies, with humanized variable regions and rat lgG2b/rat kappa constant regions. Necropsy was performed 1 week after the completion of dosing. Left and right femurs and L4 vertebrae were collected, cleaned of soft tissue, wrapped in saline soaked gauze and frozen at -20°C. L4 vertebra and left femoral metphysis or mid-diaphysis were analyzed by CT or pQCT. Right femurs were subjected to 3-point bending analysis using an Instron Figures 10A-10C show that anti-SOST antibodies DP1 and DM21 were efficacious to restore or significantly reverse the ovariectomy-induced bone loss in the
cancellous-rich bone sites of the lumbar vertebra (10A) and distal femur (10B), and in the cortical-rich bone site of the midshaft femur (10C).
Example 7: Therapeutic Effects of Anti-SOST Antibodies in a Syngenic Murine Model of Multiple Myeloma
This example illustrates that the anti-SOST antibodies of the present invention have therapeutic effects in multiple myeloma.
The 6-week (wk) old female C57BL/KaLwRijHsd mice were inoculated with 5TGM1 luciferase (luc) transfected cells (syngeneic multiple myeloma cell line derived from spontaneously occurring multiple myeloma disease in aging C57BL/KaLwRijHsd mice transfected with luciferase) (106 cells in 0.1 ml) intravenously at tail vein or with phosphate buffered saline (PBS) (naive group) subcutaneously. In the 5TGM1 treated mice, 1 ) 30 mg/kg of IgG control (N=10), 2) 30 mg/kg of DP99 (N=10), and 3) 1 mg/kg of Bortezomib ((N=9) LC Laboratories (Woburn, MA), cat# B-1408) were administered either subcutaneously once per week (IgG control and DP99) or Intraperitoneally (Bortezomib) three times per week throughout study. Intraperitoneal injection of luciferin (150 ug/mL: at 200uL per animal) and subsequent bioluminescent imaging was then conducted on the 5TGM1 and PBS treated mice at days 18, 21 , 24, 28 and 32 (IgG, DP99, and Bortezomib mice) or days 21 and 28 (naive mice). Data were expressed as average total flux in photons/second (p/s), which was a measure of the bioluminescence emitted from the luciferase expressing tumor cells exposed to the system ically delivered luciferin substrate and was thus proportional to tumor burden. As shown in Figure 1 1 , DP99 prevented tumor burden in the multiple myeloma 5TGM1 model. More specifically, the average total flux (p/s) in the 5TGM1 treated mice receiving DP99 was comparable to the 5TGM1 treated mice receiving Bortezomib (positive control).
Example 8: Therapeutic Effects of Anti-SOST Antibodies in a Syngenic Murine Model of Multiple Myeloma
This example further illustrates that the anti-SOST antibodies of the present invention have therapeutic effects in multiple myeloma.
The 6-week (wk) old female C57BL/Kal_wRijHsd mice were inoculated with 5TGM1 luciferase (luc) transfected cells (syngeneic multiple myeloma cell line derived from spontaneously occurring multiple myeloma disease in aging C57BL/Kal_wRijHsd
mice) (106 cells in 0.1 ml) intravenously at tail vein or with phosphate buffered saline (PBS) (naive group). Intraperitoneal injection of luciferin (150 ug/mL: at 200uL per animal) and subsequent bioluminescent imaging were then conducted on the mice every 3 days starting at day 18 post-5TGM1 -luc inoculation. When an animal reached a tumor burden level represented by a total flux level of 1 .0e6 photons/sec (total flux level represents a measure of luminescence of tumor cells as a result of luciferase-luciferin reaction and is proportional to the number of tumor cells present), it was recruited into the study and randomly distributed to one of 4 dose groups (1 ) 30 mg/kg of IgG control 2) 30 mg/kg of DP99, and 3) 1 mg/kg of Bortezomib ((N=9) LC Laboratories (Woburn, MA), cat# B-14084); and 4) Vehicle-phosphate buffered saline, N=6/group). Animals were observed on a daily basis and sacrificed upon demonstrating hindlimb paralysis, which was recognized in this model as representative of final stages of excessive tumor burden. As shown in Figure 12, DP99 treatment delayed progression to hindlimb paralysis in the multiple myeloma 5TGM1 model. More specifically, the percent demonstrating hindlimb paralysis in the 5TGM1 inoculated mice receiving DP99 was 100 percent at day 27 in comparison to the 5TGM1 treated mice receiving Bortezomib (66%, positive control).
This study demonstrates that treatment with an anti-SOST antibody inhibits progression of multiple myeloma.
Example 9: Therapeutic treatment with Anti-SOST Antibodies
This example illustrates the effect of anti-SOST antibodies on tumor growth in a mouse model of multiple myeloma.
For the mouse multiple myeloma model, six weeks (wk) old female BALB/c mice were inoculated with MOPC315.BM.Luc mouse myeloma cells (see, e.g., Hofgaard et al., PLoS One, 7(12): e51892, 2012) in 2e5 cells/1 OOul intravenously at tail vein. Intraperitoneal injection of luciferin (150 ug/mL: at 200uL per animal) and subsequent bioluminescent imaging were then conducted on the mice twice a week starting at day 1 1 post inoculation to monitor tumor growth progression. On day 22 post cell inoculation, animals reached a tumor burden level represented by a total flux level of 1 .0e6 photons/sec flux units (total flux level represents a measure of luminescence of tumor cells as a result of luciferase-luciferin reaction and is proportional to the number of tumor cells present) and were randomly distributed to one of 3 dose groups: 1 ) 30 mg/kg
of IgG control (n=9); 2) 30 mg/kg of DP99 (n=10); or 3) 30 mg/kg of DM1 (n=10). Antibodies were administered subcutaneously once per week starting on day 22 and continuing throughout study. As shown in Figure 13, both DP99 and DM1 treatment delayed growth of the progressing tumor. Table 14 lists the mean total flux value in p/s as well as standard deviation and standard error of mean for each treatment group at various time points. More specifically, tumor growth was first established in all groups of MOPC315.BM.Luc inoculated mice (an increase from day 1 1 -420665 p/s to day 21 - 804866 p/s) before treatment began (day 22). By day 32, DP99 and DM1 treated mice were at a lower mean total flux value than IgG control treated mice (2.3 x 106 p/s DP99, 3.6 x 106 p/s DM1 vs. 6.6x 106 p/s IgG control).
Table 14

These results demonstrate treatment with anti-SOST antibody delays tumor growth and progression. Example 10: Therapeutic treatment with Anti-SOST Antibodies
This example illustrates treatment of multiple myeloma with anti-SOST antibodies.
Six weeks (wk) old female BALB/c mice are inoculated with MOPC315.BM.Luc myeloma cells (see, e.g., Hofgaard et al., PLoS One, 7(12): e51892, 2012) in 2e5 cells/100 ul intravenously at tail vein. Intraperitoneal injection of luciferin (150 ug/mL at 200 uL per animal) and subsequent bioluminescent imaging are then conducted on the mice twice a week starting at day 1 1 post inoculation to monitor tumor growth progression. On approximately day 22 post cell inoculation, when animals reach a tumor burden level represented by a total flux level of 1.0e6 photons/sec flux units, the animals are randomly distributed to one of 2 dose groups: 1 ) 30 mg/kg of IgG control (n=9); pr 2) 30 mg/kg of romosozumab (n=10). Antibodies are administered subcutaneously once per week starting on day 22 and continuing throughout the study.
Although the disclosed teachings have been described with reference to various applications, methods, kits, and compositions, it will be appreciated that various changes and modifications can be made without departing from the teachings herein and the claimed invention below. The foregoing examples are provided to better illustrate the disclosed teachings and are not intended to limit the scope of the teachings presented herein. While the present teachings have been described in terms of these exemplary embodiments, the skilled artisan will readily understand that numerous variations and modifications of these exemplary embodiments are possible without undue experimentation. All such variations and modifications are within the scope of the current teachings.
Further, the foregoing description and Examples detail certain specific
embodiments of the invention and describes the best mode contemplated by the inventors. It will be appreciated, however, that no matter how detailed the foregoing may appear in text, the invention may be practiced in many ways and the invention should be construed in accordance with the appended claims and any equivalents thereof.
All references cited herein, including patents, patent applications, papers, text books, and the like, and the references cited therein, to the extent that they are not already, are hereby incorporated by reference in their entirety. In the event that one or more of the incorporated literature and similar materials differs from or contradicts this application, including but not limited to defined terms, term usage, described techniques, or the like, this application controls.
Claims
1 . An isolated antibody, or an antigen binding fragment thereof, which specifically binds to a sclerostin (SOST) polypeptide, wherein the antibody comprises
(a) a heavy chain variable (VH) region complementary determining regions
comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GX1TFX2DYWMQ, wherein X^ is F or H, X2 is T or S (SEQ ID NO: 81 ) or GXiTFX2DY, wherein X^ is F or H, and X2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX2X3KX4, wherein X is
A or N, X2 is S or K, X3 is V or F, and X4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SXi DYW, wherein X^ is F or M (SEQ ID NO: 84); and/or
(b) a light chain variable region (VL) region complementary determining regions
comprising (i) a VL CDR1 comprising the sequence RASKTVDSYGX1X2FMH, wherein X^ is S or N, and X2 is N or S (SEQ ID NO: 85); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQXi IX2X3X4YT , wherein Xi is T or S, X2 is E or D, X3 is H, D, F, or E, and X4 is H, P, or S (SEQ ID NO: 86).
2. An isolated antibody, or an antigen binding fragment thereof, which specifically binds to a sclerostin polypeptide, wherein the antibody comprises: a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 14; and/or a VL region comprising VL CDR1 , VL CDR2, and VL CDR3 of the VL sequence shown in SEQ ID NO: 18.
3. The antibody or the antigen binding fragment of claim 2, wherein the VH region comprises (i) a VH CDR1 comprising the sequence GHTFSDYWMQ (SEQ ID NO: 63), DYWMQ (SEQ ID NO: 56), or GHTFSDY (SEQ ID NO: 64); (ii) a VH CDR2 comprising the sequence YPGDGD (SEQ ID NO: 57) or AIYPGDGDTRYNQKFKD (SEQ ID NO: 58); and (iii) a VH CDR3 comprising the sequence SMDYW (SEQ ID NO: 65).
4. The antibody or antigen binding fragment of claim 3, wherein the VL region comprises (i) a VL CDR1 comprising the sequence RASKTVDSYGNSFMH (SEQ ID NO: 60); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQTIEFPYT (SEQ ID NO: 67).
5. The antibody or the antigen binding fragment of claim 4, wherein the VH region comprises the sequence shown in SEQ ID NO: 14 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 18 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR.
6. The antibody or the antigen binding fragment of claim 5, wherein the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 88 and a heavy chain comprising the sequence shown in SEQ ID NO: 87.
7. An isolated antibody, or an antigen binding fragment thereof, which specifically binds to a sclerostin polypeptide, wherein the antibody comprises
(a) a heavy chain variable (VH) region complementary determining regions
comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45),
GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence
X ISGGDTYTYYADSVKG, wherein X^ is T or L (SEQ ID NO: 79) or SGGDTY
(SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or
(b) a light chain variable region (VL) region complementary determining regions
comprising (i) a VL CDR1 comprising the sequence Xi SSQSLLDNDGETYLN, wherein X is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence
QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
8. An isolated antibody, or an antigen binding fragment thereof, which specifically binds to a sclerostin polypeptide, wherein the antibody comprises:
a VH region comprising a VH CDR1 , VH CDR2, and VH CDR3 of the VH sequence shown in SEQ ID NO: 6; and/or a VL region comprising VL CDR1 , VL CDR2, and VL CDR3 of the VL sequence shown in SEQ ID NO: 7.
9. The antibody or the antigen binding fragment of claim 8, wherein the VH region comprises (i) a VH CDR1 comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), or GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence LISGGDTYTYYADSVKG (SEQ ID NOs: 52) or SGGDTY (SEQ ID NO: 46); and (iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48).
10. The antibody or antigen binding fragment of claim 9, wherein the VL region comprises (i) a VL CDR1 comprising the sequence RSSQSLLDNDGETYLN (SEQ ID NO: 53); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
1 1 . The antibody or the antigen binding fragment of claim 10, wherein the VH region comprises the sequence shown in SEQ ID NO: 6 or a variant with one or several conservative amino acid substitutions in residues that are not within a CDR and/or the VL region comprises the amino acid sequence shown in SEQ ID NO: 7 or a variant thereof with one or several amino acid substitutions in amino acids that are not within a CDR.
12. The antibody or the antigen binding fragment of claim 1 1 , wherein the antibody comprises a light chain comprising the sequence shown in SEQ ID NO: 90 and a heavy chain comprising the sequence shown in SEQ ID NO: 89.
13. A pharmaceutical composition comprising a therapeutically effective amount of the antibody of any one of claims 1 -12 and a pharmaceutically acceptable carrier.
14. An isolated polynucleotide comprising a nucleotide sequence encoding the antibody of any one of claims 1 -12.
15. A vector comprising the polynucleotide of claim 14.
16. An isolated host cell that recombinantly produces the antibody of any one of claims 1 -12.
17. A method of producing an antibody, comprising culturing the host cell of claim 16 under conditions that result in production of the antibody, and isolating the antibody from the host cell or culture.
18. A method of increasing bone formation, bone mass, bone mineralization, bone quality, bone volume, bone strength, or bone density in a patient in need thereof, comprising administering to the patient an effective amount of the pharmaceutical composition of claim 13 or the antibody in any one of claims 1 -12.
19. A method of treating or preventing a bone related disorder in a patient in need thereof, comprising administering to the patient an effective amount of the pharmaceutical composition of claim 13 or the antibody of any one of claims 1 -12, wherein the bone disorder is selected from the group consisting of osteoporosis, osteopenia, osteomalacia, osteogenesis imperfect, Paget's Disease, periodontitis, rheumatoid arthritis, osteoarthritis, pain associated with osteoarthritis, avascular necrosis, bone fracture, implant fixation, bone loss, metastatic bone malignancy, multiple myeloma, acute myeloid leukemia (AML), costochondritis, polychondritis, achondroplasia, spinal disc herniation, ankylosing spondylitis, hypophosphatemia, hypophophatasia, Vitamin D resistance, hyperparathyroidism, mastocytosis, Gaucher's disease, osteogenesis imperfecta, Marian's syndrome, inflammatory bowel disease, hemochromatosis, celiac sprue, renal tubular acidosis, renal osteodystrophy,
hypercalciuria, fibrous dysplasia, and diabetes.
20. The method of claim 19, wherein the bone disorder is multiple myeloma.
21 . A method of activating canonical Wnt signaling activity in a patient in need thereof, comprising administering to the patient an effective amount of the
pharmaceutical composition of claim 13 or the antibody or fragment of any one of claims 1 -12.
22. A method of preventing or reducing tumor burden in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST
antibody that binds specifically to a SOST polypeptide or a pharmaceutical composition comprising thereof.
23. A method of inhibiting or preventing tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody that binds specifically to a SOST polypeptide or a
pharmaceutical composition comprising thereof.
24. A method of inhibiting metastasis of cancer cells or tumors (e.g. , solid or liquid tumors), or delaying tumor growth or progression in a patient in need thereof, comprising administering to the patient an effective amount of an anti-SOST antibody that binds specifically to a SOST polypeptide or a pharmaceutical composition comprising thereof.
25. The method of any one of claims 22-24, wherein the antibody comprises: (1 )(a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GX1TFX2DYWMQ, wherein X^ is F or H, X2 is T or S (SEQ ID NO: 81 ) or GXiTFX2DY, wherein X^ is F or H, and X2 is T or S (SEQ ID NO: 82); (ii) a VH CDR2 comprising the sequence AIYPGDGDTRYXi QX2X3KX4, wherein X is A or N, X2 is S or K, X3 is V or F, and X4 is G or D (SEQ ID NO:83), and iii) a VH CDR3 comprising the sequence SXi DYW, wherein X^ is F or M (SEQ ID NO: 84); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL
CDR1 comprising the sequence RASKTVDSYGXiX2FMH, wherein X is S or N, and X2 is N or S (SEQ ID NO: 85); (ii) a VL CDR2 comprising the sequence HSSNLES (SEQ ID NO: 61 ); and (iii) a VL CDR3 comprising the sequence LQX1 IX2X3X4YT , wherein X is T or S, X2 is E or D, X3 is H, D, F, or E, and X4 is H, P, or S (SEQ ID NO: 86); or
(2)(a) a heavy chain variable (VH) region complementary determining regions comprising (i) a VH complementary determining region one (CDR1 ) comprising the sequence GFTFSIYAMS (SEQ ID NO: 43), I YAMS (SEQ ID NO: 45), GFTFSIY (SEQ ID NO: 44); (ii) a VH CDR2 comprising the sequence Xi ISGGDTYTYYADSVKG, wherein X^ is T or L (SEQ ID NO: 79) or SGGDTY (SEQ ID NO: 46 ); and iii) a VH CDR3 comprising the sequence HGYDDFDY (SEQ ID NO: 48); and/or (b) a light chain variable region (VL) region complementary determining regions comprising (i) a VL CDR1
comprising the sequence XiSSQSLLDNDGETYLN, wherein ^ is K or R (SEQ ID NO: 80); (ii) a VL CDR2 comprising the sequence QVSKLDS (SEQ ID NO: 50); and (iii) a VL CDR3 comprising the sequence WQGTHFPHT (SEQ ID NO: 51 ).
26. The method of any one of claims 22-24, wherein the antibody is DP99, DP1 , DM99, DM1 , DM2, DM3, DM4, DM5, DM6, DM7, DM8, DM9, DM10, DM1 1 , DM12, DM13, DM14, DM15, DM16, DM17, DM18, DM19, DM20, DM21 , DM22, DM23, DM24, DM25, DM26, DM27, DM28, DM29, DM30, DM31 , DM32, DM33, DM34, AMG167, or AMG785 (Romosozumab).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361914094P | 2013-12-10 | 2013-12-10 | |
| US61/914,094 | 2013-12-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015087187A1 true WO2015087187A1 (en) | 2015-06-18 |
Family
ID=52273365
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2014/066354 WO2015087187A1 (en) | 2013-12-10 | 2014-11-26 | Anti-sclerostin antibodies |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015087187A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020002673A1 (en) * | 2018-06-29 | 2020-01-02 | Mereo Biopharma 3 Limited | Use of a sclerostin antagonist |
| US10961305B2 (en) | 2016-12-21 | 2021-03-30 | Mereo Biopharma 3 Limited | Use of anti-sclerostin antibodies in the treatment of osteogenesis imperfecta |
Citations (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| EP0003089A1 (en) | 1978-01-06 | 1979-07-25 | Bernard David | Drier for silkscreen printed sheets |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| WO1987004462A1 (en) | 1986-01-23 | 1987-07-30 | Celltech Limited | Recombinant dna sequences, vectors containing them and method for the use thereof |
| US4754065A (en) | 1984-12-18 | 1988-06-28 | Cetus Corporation | Precursor to nucleic acid probe |
| GB2200651A (en) | 1987-02-07 | 1988-08-10 | Al Sumidaie Ayad Mohamed Khala | A method of obtaining a retrovirus-containing fraction from retrovirus-containing cells |
| US4777127A (en) | 1985-09-30 | 1988-10-11 | Labsystems Oy | Human retrovirus-related products and methods of diagnosing and treating conditions associated with said retrovirus |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| WO1989011540A1 (en) | 1988-05-27 | 1989-11-30 | Synergen, Inc. | Interleukin-1 inhibitors |
| EP0345242A2 (en) | 1988-06-03 | 1989-12-06 | Smithkline Biologicals S.A. | Expression of gag proteins from retroviruses in eucaryotic cells |
| WO1990007936A1 (en) | 1989-01-23 | 1990-07-26 | Chiron Corporation | Recombinant therapies for infection and hyperproliferative disorders |
| WO1990011092A1 (en) | 1989-03-21 | 1990-10-04 | Vical, Inc. | Expression of exogenous polynucleotide sequences in a vertebrate |
| WO1991000360A1 (en) | 1989-06-29 | 1991-01-10 | Medarex, Inc. | Bispecific reagents for aids therapy |
| WO1991002805A2 (en) | 1989-08-18 | 1991-03-07 | Viagene, Inc. | Recombinant retroviruses delivering vector constructs to target cells |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5047335A (en) | 1988-12-21 | 1991-09-10 | The Regents Of The University Of Calif. | Process for controlling intracellular glycosylation of proteins |
| WO1991014445A1 (en) | 1990-03-21 | 1991-10-03 | Research Development Foundation | Heterovesicular liposomes |
| WO1992020373A1 (en) | 1991-05-14 | 1992-11-26 | Repligen Corporation | Heteroconjugate antibodies for treatment of hiv infection |
| EP0519596A1 (en) | 1991-05-17 | 1992-12-23 | Merck & Co. Inc. | A method for reducing the immunogenicity of antibody variable domains |
| WO1993003769A1 (en) | 1991-08-20 | 1993-03-04 | THE UNITED STATES OF AMERICA, represented by THE SECRETARY, DEPARTEMENT OF HEALTH AND HUMAN SERVICES | Adenovirus mediated transfer of genes to the gastrointestinal tract |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993010218A1 (en) | 1991-11-14 | 1993-05-27 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Vectors including foreign genes and negative selective markers |
| WO1993011230A1 (en) | 1991-12-02 | 1993-06-10 | Dynal As | Modified mammalian stem cell blocking viral replication |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| WO1993019191A1 (en) | 1992-03-16 | 1993-09-30 | Centre National De La Recherche Scientifique | Defective recombinant adenoviruses expressing cytokines for use in antitumoral treatment |
| WO1993025234A1 (en) | 1992-06-08 | 1993-12-23 | The Regents Of The University Of California | Methods and compositions for targeting specific tissue |
| WO1993025698A1 (en) | 1992-06-10 | 1993-12-23 | The United States Government As Represented By The | Vector particles resistant to inactivation by human serum |
| US5278299A (en) | 1991-03-18 | 1994-01-11 | Scripps Clinic And Research Foundation | Method and composition for synthesizing sialylated glycosyl compounds |
| WO1994003622A1 (en) | 1992-07-31 | 1994-02-17 | Imperial College Of Science, Technology & Medicine | D-type retroviral vectors, based on mpmv |
| WO1994004690A1 (en) | 1992-08-17 | 1994-03-03 | Genentech, Inc. | Bispecific immunoadhesins |
| WO1994012649A2 (en) | 1992-12-03 | 1994-06-09 | Genzyme Corporation | Gene therapy for cystic fibrosis |
| WO1994023697A1 (en) | 1993-04-22 | 1994-10-27 | Depotech Corporation | Cyclodextrin liposomes encapsulating pharmacologic compounds and methods for their use |
| WO1994028938A1 (en) | 1993-06-07 | 1994-12-22 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy sponsorship |
| WO1995000655A1 (en) | 1993-06-24 | 1995-01-05 | Mc Master University | Adenovirus vectors for gene therapy |
| WO1995007994A2 (en) | 1993-09-15 | 1995-03-23 | Viagene, Inc. | Recombinant alphavirus vectors |
| WO1995011984A2 (en) | 1993-10-25 | 1995-05-04 | Canji, Inc. | Recombinant adenoviral vector and methods of use |
| WO1995013796A1 (en) | 1993-11-16 | 1995-05-26 | Depotech Corporation | Vesicles with controlled release of actives |
| US5422120A (en) | 1988-05-30 | 1995-06-06 | Depotech Corporation | Heterovesicular liposomes |
| WO1995030763A2 (en) | 1994-05-09 | 1995-11-16 | Chiron Viagene, Inc. | Retroviral vectors having a reduced recombination rate |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5510261A (en) | 1991-11-21 | 1996-04-23 | The Board Of Trustees Of The Leland Stanford Juniot University | Method of controlling the degradation of glycoprotein oligosaccharides produced by cultured Chinese hamster ovary cells |
| WO1996017072A2 (en) | 1994-11-30 | 1996-06-06 | Chiron Viagene, Inc. | Recombinant alphavirus vectors |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5580859A (en) | 1989-03-21 | 1996-12-03 | Vical Incorporated | Delivery of exogenous DNA sequences in a mammal |
| US5580717A (en) | 1990-05-01 | 1996-12-03 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1997042338A1 (en) | 1996-05-06 | 1997-11-13 | Chiron Corporation | Crossless retroviral vectors |
| WO1998001555A2 (en) | 1996-07-09 | 1998-01-15 | Amgen Inc. | Truncated soluble tumor necrosis factor type-i and type-ii receptors |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5807715A (en) | 1984-08-27 | 1998-09-15 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and transformed mammalian lymphocyte cells for producing functional antigen-binding protein including chimeric immunoglobulin |
| US5814482A (en) | 1993-09-15 | 1998-09-29 | Dubensky, Jr.; Thomas W. | Eukaryotic layered vector initiation systems |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5866692A (en) | 1991-09-18 | 1999-02-02 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing humanized chimera antibody |
| US5981568A (en) | 1993-01-28 | 1999-11-09 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| WO1999058572A1 (en) | 1998-05-08 | 1999-11-18 | Cambridge University Technical Services Limited | Binding molecules derived from immunoglobulins which do not trigger complement mediated lysis |
| US5997867A (en) | 1991-07-16 | 1999-12-07 | Waldmann; Herman | Method of using humanized antibody against CD18 |
| WO2000009560A2 (en) | 1998-08-17 | 2000-02-24 | Abgenix, Inc. | Generation of modified molecules with increased serum half-lives |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| WO2000053211A2 (en) | 1999-03-09 | 2000-09-14 | University Of Southern California | Method of promoting myocyte proliferation and myocardial tissue repair |
| US6180377B1 (en) | 1993-06-16 | 2001-01-30 | Celltech Therapeutics Limited | Humanized antibodies |
| US6210671B1 (en) | 1992-12-01 | 2001-04-03 | Protein Design Labs, Inc. | Humanized antibodies reactive with L-selectin |
| WO2001027160A1 (en) | 1999-10-14 | 2001-04-19 | Applied Molecular Evolution, Inc. | Methods of optimizing antibody variable region binding affinity |
| US6265150B1 (en) | 1995-06-07 | 2001-07-24 | Becton Dickinson & Company | Phage antibodies |
| US6350861B1 (en) | 1992-03-09 | 2002-02-26 | Protein Design Labs, Inc. | Antibodies with increased binding affinity |
| US6376471B1 (en) | 1997-10-10 | 2002-04-23 | Johns Hopkins University | Gene delivery compositions and methods |
| US6413942B1 (en) | 1989-03-21 | 2002-07-02 | Vical, Inc. | Methods of delivering a physiologically active polypeptide to a mammal |
| US6436908B1 (en) | 1995-05-30 | 2002-08-20 | Duke University | Use of exogenous β-adrenergic receptor and β-adrenergic receptor kinase gene constructs to enhance myocardial function |
| US6479635B1 (en) | 1996-12-23 | 2002-11-12 | Immunex Corporation | Receptor activator of NF-κB |
| WO2003002713A2 (en) | 2001-06-26 | 2003-01-09 | Abgenix, Inc. | Antibodies to opgl |
| WO2003086289A2 (en) | 2002-04-05 | 2003-10-23 | Amgen Inc. | Human anti-opgl neutralizing antibodies as selective opgl pathway inhibitors |
| US6740511B1 (en) | 1999-08-27 | 2004-05-25 | Transgene S.A. | Modified adenoviral fibre and uses thereof |
| WO2004058184A2 (en) | 2002-12-24 | 2004-07-15 | Rinat Neuroscience Corp. | Anti-ngf antibodies and methods using same |
| US7314622B2 (en) | 2005-04-15 | 2008-01-01 | Neogenix Oncology, Inc. | Recombinant monoclonal antibodies and corresponding antigens for colon and pancreatic cancers |
| US7381409B2 (en) | 2003-06-16 | 2008-06-03 | Celltech R&D, Inc. | Antibodies specific for sclerostin and methods of screening and use therefor |
| WO2008133722A2 (en) * | 2006-11-10 | 2008-11-06 | Ucb Pharma S.A. | Anti human sclerostin antibodies |
| WO2009047356A1 (en) * | 2007-10-12 | 2009-04-16 | Novartis Ag | Compositions and methods for use for antibodies against sclerostin |
| US7592429B2 (en) | 2005-05-03 | 2009-09-22 | Ucb Sa | Sclerostin-binding antibody |
| US7744874B2 (en) | 2007-03-20 | 2010-06-29 | Eli Lilly And Company | Anti-sclerostin antibodies |
| WO2011143545A1 (en) | 2010-05-14 | 2011-11-17 | Rinat Neuroscience Corporation | Heterodimeric proteins and methods for producing and purifying them |
| WO2013019954A1 (en) * | 2011-08-04 | 2013-02-07 | Amgen Inc. | Method for treating bone gap defects |
-
2014
- 2014-11-26 WO PCT/IB2014/066354 patent/WO2015087187A1/en active Application Filing
Patent Citations (95)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| EP0003089A1 (en) | 1978-01-06 | 1979-07-25 | Bernard David | Drier for silkscreen printed sheets |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US6331415B1 (en) | 1983-04-08 | 2001-12-18 | Genentech, Inc. | Methods of producing immunoglobulins, vectors and transformed host cells for use therein |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| US5807715A (en) | 1984-08-27 | 1998-09-15 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and transformed mammalian lymphocyte cells for producing functional antigen-binding protein including chimeric immunoglobulin |
| US4754065A (en) | 1984-12-18 | 1988-06-28 | Cetus Corporation | Precursor to nucleic acid probe |
| US4683202B1 (en) | 1985-03-28 | 1990-11-27 | Cetus Corp | |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4777127A (en) | 1985-09-30 | 1988-10-11 | Labsystems Oy | Human retrovirus-related products and methods of diagnosing and treating conditions associated with said retrovirus |
| WO1987004462A1 (en) | 1986-01-23 | 1987-07-30 | Celltech Limited | Recombinant dna sequences, vectors containing them and method for the use thereof |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683195B1 (en) | 1986-01-30 | 1990-11-27 | Cetus Corp | |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| GB2200651A (en) | 1987-02-07 | 1988-08-10 | Al Sumidaie Ayad Mohamed Khala | A method of obtaining a retrovirus-containing fraction from retrovirus-containing cells |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| WO1989011540A1 (en) | 1988-05-27 | 1989-11-30 | Synergen, Inc. | Interleukin-1 inhibitors |
| US5422120A (en) | 1988-05-30 | 1995-06-06 | Depotech Corporation | Heterovesicular liposomes |
| EP0345242A2 (en) | 1988-06-03 | 1989-12-06 | Smithkline Biologicals S.A. | Expression of gag proteins from retroviruses in eucaryotic cells |
| US5047335A (en) | 1988-12-21 | 1991-09-10 | The Regents Of The University Of Calif. | Process for controlling intracellular glycosylation of proteins |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5693761A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Polynucleotides encoding improved humanized immunoglobulins |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US6180370B1 (en) | 1988-12-28 | 2001-01-30 | Protein Design Labs, Inc. | Humanized immunoglobulins and methods of making the same |
| US5693762A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| WO1990007936A1 (en) | 1989-01-23 | 1990-07-26 | Chiron Corporation | Recombinant therapies for infection and hyperproliferative disorders |
| WO1990011092A1 (en) | 1989-03-21 | 1990-10-04 | Vical, Inc. | Expression of exogenous polynucleotide sequences in a vertebrate |
| US6413942B1 (en) | 1989-03-21 | 2002-07-02 | Vical, Inc. | Methods of delivering a physiologically active polypeptide to a mammal |
| US5580859A (en) | 1989-03-21 | 1996-12-03 | Vical Incorporated | Delivery of exogenous DNA sequences in a mammal |
| WO1991000360A1 (en) | 1989-06-29 | 1991-01-10 | Medarex, Inc. | Bispecific reagents for aids therapy |
| WO1991002805A2 (en) | 1989-08-18 | 1991-03-07 | Viagene, Inc. | Recombinant retroviruses delivering vector constructs to target cells |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| WO1991014445A1 (en) | 1990-03-21 | 1991-10-03 | Research Development Foundation | Heterovesicular liposomes |
| EP0524968A1 (en) | 1990-03-21 | 1993-02-03 | Res Dev Foundation | Heterovesicular liposomes. |
| US5580717A (en) | 1990-05-01 | 1996-12-03 | Affymax Technologies N.V. | Recombinant library screening methods |
| US5278299A (en) | 1991-03-18 | 1994-01-11 | Scripps Clinic And Research Foundation | Method and composition for synthesizing sialylated glycosyl compounds |
| WO1992020373A1 (en) | 1991-05-14 | 1992-11-26 | Repligen Corporation | Heteroconjugate antibodies for treatment of hiv infection |
| EP0519596A1 (en) | 1991-05-17 | 1992-12-23 | Merck & Co. Inc. | A method for reducing the immunogenicity of antibody variable domains |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5997867A (en) | 1991-07-16 | 1999-12-07 | Waldmann; Herman | Method of using humanized antibody against CD18 |
| WO1993003769A1 (en) | 1991-08-20 | 1993-03-04 | THE UNITED STATES OF AMERICA, represented by THE SECRETARY, DEPARTEMENT OF HEALTH AND HUMAN SERVICES | Adenovirus mediated transfer of genes to the gastrointestinal tract |
| US5866692A (en) | 1991-09-18 | 1999-02-02 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing humanized chimera antibody |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993010218A1 (en) | 1991-11-14 | 1993-05-27 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Vectors including foreign genes and negative selective markers |
| US5510261A (en) | 1991-11-21 | 1996-04-23 | The Board Of Trustees Of The Leland Stanford Juniot University | Method of controlling the degradation of glycoprotein oligosaccharides produced by cultured Chinese hamster ovary cells |
| WO1993011230A1 (en) | 1991-12-02 | 1993-06-10 | Dynal As | Modified mammalian stem cell blocking viral replication |
| US6350861B1 (en) | 1992-03-09 | 2002-02-26 | Protein Design Labs, Inc. | Antibodies with increased binding affinity |
| WO1993019191A1 (en) | 1992-03-16 | 1993-09-30 | Centre National De La Recherche Scientifique | Defective recombinant adenoviruses expressing cytokines for use in antitumoral treatment |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1993025234A1 (en) | 1992-06-08 | 1993-12-23 | The Regents Of The University Of California | Methods and compositions for targeting specific tissue |
| WO1993025698A1 (en) | 1992-06-10 | 1993-12-23 | The United States Government As Represented By The | Vector particles resistant to inactivation by human serum |
| WO1994003622A1 (en) | 1992-07-31 | 1994-02-17 | Imperial College Of Science, Technology & Medicine | D-type retroviral vectors, based on mpmv |
| WO1994004690A1 (en) | 1992-08-17 | 1994-03-03 | Genentech, Inc. | Bispecific immunoadhesins |
| US6210671B1 (en) | 1992-12-01 | 2001-04-03 | Protein Design Labs, Inc. | Humanized antibodies reactive with L-selectin |
| WO1994012649A2 (en) | 1992-12-03 | 1994-06-09 | Genzyme Corporation | Gene therapy for cystic fibrosis |
| US5981568A (en) | 1993-01-28 | 1999-11-09 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| WO1994023697A1 (en) | 1993-04-22 | 1994-10-27 | Depotech Corporation | Cyclodextrin liposomes encapsulating pharmacologic compounds and methods for their use |
| WO1994028938A1 (en) | 1993-06-07 | 1994-12-22 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy sponsorship |
| US6180377B1 (en) | 1993-06-16 | 2001-01-30 | Celltech Therapeutics Limited | Humanized antibodies |
| WO1995000655A1 (en) | 1993-06-24 | 1995-01-05 | Mc Master University | Adenovirus vectors for gene therapy |
| US5814482A (en) | 1993-09-15 | 1998-09-29 | Dubensky, Jr.; Thomas W. | Eukaryotic layered vector initiation systems |
| WO1995007994A2 (en) | 1993-09-15 | 1995-03-23 | Viagene, Inc. | Recombinant alphavirus vectors |
| WO1995011984A2 (en) | 1993-10-25 | 1995-05-04 | Canji, Inc. | Recombinant adenoviral vector and methods of use |
| WO1995013796A1 (en) | 1993-11-16 | 1995-05-26 | Depotech Corporation | Vesicles with controlled release of actives |
| WO1995030763A2 (en) | 1994-05-09 | 1995-11-16 | Chiron Viagene, Inc. | Retroviral vectors having a reduced recombination rate |
| WO1996017072A2 (en) | 1994-11-30 | 1996-06-06 | Chiron Viagene, Inc. | Recombinant alphavirus vectors |
| US6436908B1 (en) | 1995-05-30 | 2002-08-20 | Duke University | Use of exogenous β-adrenergic receptor and β-adrenergic receptor kinase gene constructs to enhance myocardial function |
| US6265150B1 (en) | 1995-06-07 | 2001-07-24 | Becton Dickinson & Company | Phage antibodies |
| WO1997042338A1 (en) | 1996-05-06 | 1997-11-13 | Chiron Corporation | Crossless retroviral vectors |
| WO1998001555A2 (en) | 1996-07-09 | 1998-01-15 | Amgen Inc. | Truncated soluble tumor necrosis factor type-i and type-ii receptors |
| US6479635B1 (en) | 1996-12-23 | 2002-11-12 | Immunex Corporation | Receptor activator of NF-κB |
| US6376471B1 (en) | 1997-10-10 | 2002-04-23 | Johns Hopkins University | Gene delivery compositions and methods |
| WO1999058572A1 (en) | 1998-05-08 | 1999-11-18 | Cambridge University Technical Services Limited | Binding molecules derived from immunoglobulins which do not trigger complement mediated lysis |
| WO2000009560A2 (en) | 1998-08-17 | 2000-02-24 | Abgenix, Inc. | Generation of modified molecules with increased serum half-lives |
| WO2000053211A2 (en) | 1999-03-09 | 2000-09-14 | University Of Southern California | Method of promoting myocyte proliferation and myocardial tissue repair |
| US6740511B1 (en) | 1999-08-27 | 2004-05-25 | Transgene S.A. | Modified adenoviral fibre and uses thereof |
| WO2001027160A1 (en) | 1999-10-14 | 2001-04-19 | Applied Molecular Evolution, Inc. | Methods of optimizing antibody variable region binding affinity |
| WO2003002713A2 (en) | 2001-06-26 | 2003-01-09 | Abgenix, Inc. | Antibodies to opgl |
| WO2003086289A2 (en) | 2002-04-05 | 2003-10-23 | Amgen Inc. | Human anti-opgl neutralizing antibodies as selective opgl pathway inhibitors |
| WO2004058184A2 (en) | 2002-12-24 | 2004-07-15 | Rinat Neuroscience Corp. | Anti-ngf antibodies and methods using same |
| US7381409B2 (en) | 2003-06-16 | 2008-06-03 | Celltech R&D, Inc. | Antibodies specific for sclerostin and methods of screening and use therefor |
| US7314622B2 (en) | 2005-04-15 | 2008-01-01 | Neogenix Oncology, Inc. | Recombinant monoclonal antibodies and corresponding antigens for colon and pancreatic cancers |
| US7592429B2 (en) | 2005-05-03 | 2009-09-22 | Ucb Sa | Sclerostin-binding antibody |
| US7872106B2 (en) | 2005-05-03 | 2011-01-18 | Amgen Inc. | Sclerostin-binding antibodies |
| WO2008133722A2 (en) * | 2006-11-10 | 2008-11-06 | Ucb Pharma S.A. | Anti human sclerostin antibodies |
| US7744874B2 (en) | 2007-03-20 | 2010-06-29 | Eli Lilly And Company | Anti-sclerostin antibodies |
| WO2009047356A1 (en) * | 2007-10-12 | 2009-04-16 | Novartis Ag | Compositions and methods for use for antibodies against sclerostin |
| US7879322B2 (en) | 2007-10-12 | 2011-02-01 | Novartis Ag | Compositions and methods for use for antibodies against sclerostin |
| WO2011143545A1 (en) | 2010-05-14 | 2011-11-17 | Rinat Neuroscience Corporation | Heterodimeric proteins and methods for producing and purifying them |
| WO2013019954A1 (en) * | 2011-08-04 | 2013-02-07 | Amgen Inc. | Method for treating bone gap defects |
Non-Patent Citations (146)
| Title |
|---|
| "Methods in Enzymology", ACADEMIC PRESS, INC. |
| "Methods in Molecular Biology", HUMANA PRESS |
| "REMINGTON The Science and Practice of Pharmacy 21 th Ed.", 2011, MACK PUBLISHING |
| "Remington: The Science and practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS AND WILKINS |
| "Short Protocols in Molecular Biology", 1999, WILEY AND SONS |
| A. DOYLE, J.B. GRIFFITHS, AND D.G. NEWELL,: "Tissue Culture: Laboratory Procedures", 1993, J. WILEY AND SONS |
| A. GENNARO,: "Remington's Pharmaceutical Sciences, 18th edition,", 1990, MACK PUBLISHING CO. |
| AALBERSE ET AL., IMMUNOLOGY, vol. 105, 2002, pages 9 - 19 |
| ARMOUR ET AL., MOLECULAR IMMUNOLOGY, vol. 40, 2003, pages 585 - 593 |
| BAGI ET AL., BONE, vol. 38, 2006, pages 136 |
| BALEMANS ET AL., HUM. MOL. GENET., vol. 10, 2001, pages 537 - 543 |
| BALINT ET AL., GENE, vol. 137, no. 1, 1993, pages 109 - 18 |
| BARBAS ET AL., PROC NAT. ACAD. SCI, USA, vol. 91, 1994, pages 3809 - 3813 |
| BARON ET AL.: "Bone Histomorphometry: Techniques and Interpretation", CRC PRESS, pages: 13 - 35 |
| BEIGHTON, J. MED. GENET., vol. 25, 1988, pages 200 - 203 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| BOYD ET AL., MOL. IMMUNOL., vol. 32, 1996, pages 1311 - 1318 |
| BROWN ET AL., CANCER RES., vol. 47, 1987, pages 3577 - 3583 |
| BRUNKOW ET AL., AM. J. HUM. GENET., vol. 68, 2001, pages 577 - 589 |
| BUCK, D. W. ET AL., IN VITRO, vol. 18, 1982, pages 377 - 381 |
| C.A. JANEWAY; P. TRAVERS, IMMUNOBIOLOGY, 1997 |
| CAPEL ET AL., IMMUNOMETHODS, vol. 4, 1994, pages 25 - 34 |
| CHIOU ET AL.: "Gene Therapeutics: Methods And Applications Of Direct Gene Transfer", 1994 |
| CHOTHIA ET AL., NATURE, vol. 342, 1989, pages 877 - 883 |
| CHOTHIA; LESK, J MOL BIOL, vol. 196, no. 4, 1987, pages 901 - 917 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLYNES ET AL., PNAS (USA, vol. 95, 1998, pages 652 - 656 |
| CONNELL, HUMAN GENE THERAPY, vol. 1, 1995, pages 185 |
| COX ET AL., EUR. J. IMMUNOL., vol. 24, 1994, pages 827 - 836 |
| CURIEL, HUM. GENE THER., vol. 3, 1992, pages 147 |
| D. CATTY.,: "Antibodies: a practical approach", 1988, IRL PRESS |
| D.M. WEIR AND C.C. BLACKWELL,: "Handbook of Experimental Immunology", 1986 |
| DARLING ET AL., ASSAY AND DRUG DEVELOPMENT TECHNOLOGIES, vol. 2, no. 6, 2005, pages 647 - 657 |
| DAUGHERTY ET AL., NUCL. ACIDS RES., vol. 19, 1991, pages 2471 - 2476 |
| DAYHOFF, M.O.: "Atlas of Protein Sequence and Structure", vol. 5, 1978, NATIONAL BIOMEDICAL RESEARCH FOUNDATION, article "A model of evolutionary change in proteins - Matrices for detecting distant relationships", pages: 345 - 358 |
| DE HAAS ET AL., J. LAB. CLIN. MED., vol. 126, 1995, pages 330 - 41 |
| E. HARLOW; D. LANE: "Using antibodies: a laboratory manual", 1999, COLD SPRING HARBOR LABORATORY PRESS |
| EPSTEIN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 82, 1985, pages 3688 |
| EUR. J. IMMUNOL., vol. 29, 1999, pages 2613 - 2624 |
| F.M. AUSUBEL ET AL.,: "Current Protocols in Molecular Biology", 1987 |
| FINDEIS ET AL., TRENDS BIOTECHNOL., vol. 11, 1993, pages 202 |
| GAZZANO-SANTORO ET AL., J. IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| GRIFFITH ET AL., EMBO J., vol. 12, 1993, pages 725 - 734 |
| GUYER ET AL., J. IMMUNOL., vol. 117, 1976, pages 587 |
| H. SEE, TAO ET AL., J. IMMUNOLOGY, vol. 143, 1989, pages 2595 - 2601 |
| HANSON; BAGI, BON, vol. 35, 2004, pages 326 |
| HARLOW AND LANE,: "Using Antibodies, a Laboratory Manual", 1999, COLD SPRING HARBOR LABORATORY PRESS, article "Chapter 11" |
| HAWKINS ET AL., J. MOL. BIOL., vol. 226, 1992, pages 889 - 896 |
| HEIN J.: "Methods in Enzymology", vol. 183, 1990, ACADEMIC PRESS, INC., article "Unified Approach to Alignment and Phylogenes", pages: 626 - 645 |
| HIGGINS, D.G.; SHARP, P.M., CABIOS, vol. 5, 1989, pages 151 - 153 |
| HOFGAARD ET AL., PLOS ONE, vol. 7, no. 12, 2012, pages E51892 |
| HOLLIGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOLLIGER, P. ET AL., PROC. NATL. ACAD SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HSE ET AL., J. BIOL. CHEM., vol. 272, 1997, pages 9062 - 9070 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 5879 - 5883 |
| HWANG ET AL., PROC. NATL ACAD. SCI. USA, vol. 77, 1980, pages 4030 |
| IDUSOGIE ET AL., J. IMMUNOLOGY, vol. 164, 2000, pages 4178 - 4184 |
| III ET AL., PROTEIN ENG., vol. 10, 1997, pages 949 - 57 |
| J.E. CELLIS,: "Cell Biology: A Laboratory Notebook", 1998, ACADEMIC PRESS |
| J.E. COLIGAN ET AL.,: "Current Protocols in Immunology", 1991 |
| J.M. MILLER AND M.P. CALOS,: "Gene Transfer Vectors for Mammalian Cells", 1987 |
| J.P. MATHER; P.E. ROBERTS: "Introduction to Cell and Tissue Culture", 1998, PLENUM PRESS |
| JACKSON ET AL., J. IMMUNOL., vol. 154, no. 7, 1995, pages 3310 - 9 |
| JEE ET AL.: "Handbook of Bone Morphology", 1997, NISHIMUSA, NIIGATA CITY, pages: 87 - 112 |
| JEFFERIS ET AL., IMMUNOLOGICAL REVIEWS, vol. 163, 1998, pages 59 - 76 |
| JEFFERIS; LUND, CHEM. IMMUNOL., vol. 65, 1997, pages 111 - 128 |
| JOHNSON, KEVIN S.; CHISWELL, DAVID J., CURRENT OPINION IN STRUCTURAL BIOLOGY, vol. 3, 1993, pages 564 - 571 |
| JOLLY, CANCER GENE THERAPY, vol. 1, 1994, pages 51 |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest, 5th Ed.", 1991, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest, 5th Ed.", 1992, NIH |
| KABAT, E. A. ET AL.: "Sequences of Proteins of Immunological Interest, Fifth Edition,", 1991, NIH PUBLICATION NO. 91-3242 |
| KAPLITT, NATURE GENETICS, vol. 6, 1994, pages 148 |
| KARLSSON, R. ROOS, H.; FAGERSTAM, L.; PETERSSON, B., METHODS ENZYMOLOGY, vol. 6, 1994, pages 99 - 110 |
| KE ET AL., J. MUSCULOSKEL. NEURON INTERACT, vol. 2, 2002, pages 479 - 488 |
| KIM ET AL., J. IMMUNOL., vol. 24, 1994, pages 249 |
| KIMURA, HUMAN GENE THERAPY, vol. 5, 1994, pages 845 |
| KOHLER, B.; MILSTEIN, C., NATURE, vol. 256, 1975, pages 495 - 497 |
| LI ET AL., J. BONE MINER RES., vol. 23, 2008, pages 860 - 869 |
| LI ET AL., J. BONE MINER. RES., vol. 23, 2008, pages 860 - 869 |
| LOBUGLIO ET AL., PROC. NAT. ACAD. SCI. USA, vol. 86, 1989, pages 4220 - 4224 |
| LONBERG, N.; D. HUSZAR, INT. REV. LMMUNO, vol. 13, 1995, pages 65 |
| LUND ET AL., J. IMMUNOLOGY, vol. 157, 1996, pages 4963 - 4969 |
| M. ZANETTI AND J.D. CAPRA,: "The Antibodies", 1995, HARWOOD ACADEMIC PUBLISHERS |
| M.J. GAIT,: "Oligonucleotide Synthesis", 1984 |
| MACCALLUM ET AL., J. MOL. BIOL., vol. 262, 1996, pages 732 - 745 |
| MAKABE ET AL., J. BIOL. CHEM., vol. 283, 2007, pages 1156 - 1166 |
| MAKABE ET AL., JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 283, 2008, pages 1156 - 1166 |
| MARK ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 - 597 |
| MARKS ET AL., BIO/TECHNOL., vol. 10, 1992, pages 779 - 783 |
| MARKS ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MARTIN ET AL., EMBO J., vol. 13, 1994, pages 5303 - 9 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 553 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 554 |
| MILLSTEIN; CUELLO, NATURE, vol. 305, 1983, pages 537 - 539 |
| MORGAN ET AL., IMMUNOLOGY, vol. 86, 1995, pages 319 - 324 |
| MORRISON ET AL., PROC. NAT. ACAD. SCI., vol. 81, 1984, pages 6851 |
| MULLIS ET AL.,: "PCR: The Polymerase Chain Reaction", 1994 |
| MULLIS ET AL.: "PCR: The Polymerase Chain Reaction", 1994, BIRKAUSWER PRESS |
| MYERS, E.W.; MULLER W., CABIOS, vol. 4, 1988, pages 11 - 17 |
| MYSZKA, J. MOL. RECOGNIT., vol. 12, 1999, pages 279 - 284 |
| P. FINCH, ANTIBODIES, 1997 |
| P. SHEPHERD AND C. DEAN,: "Monoclonal antibodies: a practical approach", 2000, OXFORD UNIVERSITY PRESS |
| PARFITT ET AL., J. BONE MINER. RES., vol. 2, 1987, pages 595 - 610 |
| PEETERS ET AL., VACCINE, vol. 19, 2001, pages 2756 |
| PHILIP, MOL. CELL BIOL., vol. 14, 1994, pages 2411 |
| POLJAK, R. J. ET AL., STRUCTURE, vol. 2, 1994, pages 1121 - 1123 |
| POLLOCK ET AL., J IMMUNOL METHODS, vol. 231, 1999, pages 147 |
| R.I. FRESHNEY,: "Animal Cell Culture", 1987 |
| RAVETCH; KINET, ANN. REV. IMMUNOL., vol. 9, 1991, pages 457 - 92 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| ROBINSON, E.D., COMB. THEOR., vol. 11, 1971, pages 105 |
| SAMBROOK ET AL.,: "Molecular Cloning: A Laboratory Manual, second edition", 1989, COLD SPRING HARBOR PRESS |
| SANTOU, N.; NES, M., MOL. BIOL. EVOL., vol. 4, 1987, pages 406 - 425 |
| SCHIER ET AL., GENE, vol. 169, 1995, pages 147 - 155 |
| SHAW ET AL., J IMMUNOL., vol. 138, 1987, pages 4534 - 4538 |
| SNEATH, P.H.A.; SOKAL, R.R.: "Numerical Taxonomy the Principles and Practice of Numerical Taxonomy", 1973, FREEMAN PRESS |
| STEIN ET AL., NEUROLOGY, vol. 33, 1983, pages 267 - 277 |
| SURESH ET AL., METHODS IN ENZYMOLOGY, vol. 121, 1986, pages 210 |
| SUTHERLAND ET AL., BONE, vol. 35, 2004, pages 828 - 835 |
| TAO ET AL., J. IMMUNOLOGY, vol. 143, 1989, pages 2595 - 2601 |
| TILLER ET AL., J. IMMUNOL. METHODS, vol. 329, 2008, pages 112 |
| TOMLINSON ET AL., J. MOL. BIOL., vol. 227, 1992, pages 776 - 798 |
| TRAUNECKER ET AL., EMBO J., vol. 10, 1991, pages 3655 - 3659 |
| TRAUNECKER ET AL., INT. J. CANCER, vol. 7, 1992, pages 51 - 52 |
| TURNER ET AL., J. BONE MINER. RES., vol. 16, 2001, pages 206 - 213 |
| TURNER; BURR, BONE, vol. 14, 1993, pages 595 - 608 |
| UMANA ET AL., MATURE BIOTECH., vol. 17, 1999, pages 176 - 180 |
| VAN BENOOIJEN ET AL., J. EXP. MED., vol. 199, pages 805 - 814 |
| VERHOEYEN ET AL., SCIENCE, vol. 239, 1988, pages 1534 - 1536 |
| WATERHOUSE ET AL., NUCL. ACIDS RES., vol. 21, 1993, pages 2265 - 2266 |
| WILBUR, W.J.; LIPMAN, D.J., PROC. NATL. ACAD. SCI. USA, vol. 80, 1983, pages 726 - 730 |
| WINKLER ET AL., EMBO J., vol. 22, 2003, pages 6267 - 6276 |
| WINTER ET AL., ANNU. REV. IMMUNOL., vol. 12, 1994, pages 433 - 455 |
| WINTER ET AL., NATURE, vol. 349, 1991, pages 293 - 299 |
| WITTWE; HOWARD, BIOCHEM., vol. 29, 1990, pages 4175 - 4180 |
| WOFFENDIN, PROC. NATL. ACAD. SCI., vol. 91, 1994, pages 1581 |
| WRIGHT; MORRISON, TIBTECH, vol. 15, 1997, pages 26 - 32 |
| WU ET AL., J. BIOL. CHEM., vol. 263, 1988, pages 621 |
| WU ET AL., J. BIOL. CHEM., vol. 266, 1991, pages 338 |
| WU ET AL., J. BIOL. CHEM., vol. 269, 1994, pages 542 |
| WU, J. BIOL. CHEM., vol. 264, 1989, pages 16985 |
| WYSS; WAGNER, CURRENT OPIN. BIOTECH., vol. 7, 1996, pages 409 - 416 |
| YELTON ET AL., J. IMMUNOL., vol. 155, 1995, pages 1994 - 2004 |
| ZENKE ET AL., PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 3655 |
| ZHANG ET AL., J. BIOL. CHEM., vol. 280, 2005, pages 19883 - 19887 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10961305B2 (en) | 2016-12-21 | 2021-03-30 | Mereo Biopharma 3 Limited | Use of anti-sclerostin antibodies in the treatment of osteogenesis imperfecta |
| WO2020002673A1 (en) * | 2018-06-29 | 2020-01-02 | Mereo Biopharma 3 Limited | Use of a sclerostin antagonist |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2389960T3 (en) | Procedures for treating osteoarthritis pain by administering a nerve growth factor antagonist and compositions containing the same | |
| US9175093B2 (en) | PCSK9 antagonists | |
| ES2697876T3 (en) | Anti-NGF antibodies and methods of using them | |
| CA2854720C (en) | Antibodies specific for trop-2 and their uses | |
| AU2011219488B2 (en) | Antagonist anti-IL-7 receptor antibodies and methods | |
| JP2021121637A (en) | Antagonist antibodies against calcitonin gene related peptide and use method thereof | |
| US9249224B2 (en) | Human growth hormone receptor antagonist antibodies and methods of use thereof | |
| WO2015087187A1 (en) | Anti-sclerostin antibodies | |
| AU2011201157B2 (en) | Anti-NGF antibodies and methods using same | |
| AU2015200427B2 (en) | PCSK9 antagonists | |
| AU2014201648B2 (en) | Antagonist anti-il-7 receptor antibodies and methods |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14821284 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14821284 Country of ref document: EP Kind code of ref document: A1 |