WO2011060195A2 - Topical eutectic formulation - Google Patents
Topical eutectic formulation Download PDFInfo
- Publication number
- WO2011060195A2 WO2011060195A2 PCT/US2010/056419 US2010056419W WO2011060195A2 WO 2011060195 A2 WO2011060195 A2 WO 2011060195A2 US 2010056419 W US2010056419 W US 2010056419W WO 2011060195 A2 WO2011060195 A2 WO 2011060195A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition
- pharmaceutical composition
- etoricoxib
- choline
- urea
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/186—Quaternary ammonium compounds, e.g. benzalkonium chloride or cetrimide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
Definitions
- Osteoarthritis is a chronic joint disease characterized by progressive degeneration of articular cartilage. Symptoms include joint pain and impaired movement. OA is one of the leading causes of disability worldwide and a major financial burden to health care systems. It is estimated to affect over 15 million adults in the United States alone. See Boh, L.E.; Osteoarthritis. In: DiPiro, J.T.; Talbert, R.L.; Yee, G.C. et al. editors.
- An OA treatment's efficacy is generally assessed by three outcome measures: pain, physical function, and a patient global assessment. See Bellamy, N.; Kirwan, J.; Boers, M.; Brooks, P.; Strand, V.; Tugwell, P. et al. Recommendations for a core set of outcome measures for future Phase III clinical trials in knee, hip and hand osteoarthritis. Consensus development at OMERACT III., J Rheumatol, 24:799-802 (1997). To be suitable for chronic use, a therapy must generally show efficacy on these three variables over a sustained period of time. In the U.S., the Food and Drug Administration (FDA) has required OA therapies to show superiority over placebo over a twelve- week period before approval of a new drug application.
- FDA Food and Drug Administration
- NSAIDs Oral non-steroidal anti-inflammatory drugs
- COX cyclooxygenase
- the COX enzyme has two isoforms, COX-1 and COX-2.
- Traditional NSAIDs inhibit both isoforms of the COX enzyme, while the selective COX-2 (coxib) class of NSAIDs preferentially inhibits COX-2.
- NSAIDs have analgesic, anti-inflammatory, and antipyretic effects and are useful in reducing pain and inflammation. They are, however, associated with serious potential side effects including nausea, vomiting, peptic ulcer disease, and gastrointestinal (GI)
- selective COX-2 inhibitors produce fewer gastrointestinal side effects, they may increase the risk of thrombotic events (e.g., stroke or heart attack). Because of this potential side effect, most of the selective COX-2 inhibitors have been withdrawn from the U.S. market.
- Topical NSAIDs offer the possibility of achieving local therapeutic benefit while reducing or eliminating the risk of systemic side effects.
- data supporting the efficacy of topical NSAIDs in the treatment of OA is limited.
- RCT's placebo controlled trials
- Efficacy of topical non-steroidal anti-inflammatory drugs in the treatment of osteoarthritis metaanalysis of randomized controlled trials, BMJ, doi:10.1136/bmj.38159.639028.7C (2004).
- Pennsaid Gel is a topical formulation comprising diclofenac sodium that overcomes disadvantages of prior art NSAID formulations.
- U.S. Patent Publication No. 2008/0300311 PennsaidTM solution has been shown in clinical trials to be effective for treating the pain and symptoms of osteoarthritis, and it has been approved for use in Canada, the U.S., and several European countries.
- a topical formulation containing a COX-2 selective inhibitor would offer patients an attractive new treatment modality. Such a formulation could minimize systemic exposure to the active pharmaceutical ingredient by localizing the drug at the site of action. At the same time a topical coxib might have even better GI safety profile than topical formulations containing traditional NSAIDs, making it particularly suitable for patients at risk of GI bleeds.
- the dearth of options with robust efficacy data for topical NSAID treatment of OA partially arises from the difficulty associated with delivering a molecule through the skin both in a sufficient quantity to exert a therapeutic effect and in a manner that makes the treatment itself tolerable. It is generally believed that for topical OA treatments, clinical efficacy requires absorption of the active ingredient and its penetration in sufficient quantities into underlying inflamed tissues including the synovium and synovial fluid of joints. See
- Naito demonstrates significant variability in penetration among topical NSAID formulations simply by changing the gelling agent used in the compositions.
- Naito et al. Percutaneous absorption of diclofenac sodium ointment, Int. Jour, of
- the pH of the vehicle can influence penetration from a gel dosage form.
- the pH value affects the balance between ionized and non-ionized forms of the drag, which typically have different permeation properties. Obata, International Journal of ' Pharmaceutics, 89: 191-198 (1993).
- the viscosity can affect diffusion of the drug through the gel matrix and release of the drug from the vehicle into the skin.
- the solubility of the drag in the vehicle will affect the partition coefficient of the drug between the composition and the recipient membrane or tissue. Ho, Id.
- the skin barrier can be compromised by several physical methods, such as iontophoresis, ultrasound, electroporation, heat, and microneedles.
- Molecular penetration enhancers are a preferred means for reversibly lowering the skin barrier. At least 400 chemicals have been identified as skin permeability enhancers.
- General categories of MPETMs include pyrrolidones, fatty acids, fatty acid esters, fatty acid alcohols, sulfoxides, essential oils, terpenes, oxazolidines, surfactants, polyols, azone and derivatives, and epidermal enzymes.
- MPETMs have the challenge with use of MPETMs. This is because an MPETM's disruption of the skin barrier can potentially cause skin irritation. With increased disruption, skin irritation is expected to become a greater issue. This is particularly problematic with topical OA treatments where the goal is to have the active penetrate deeply into joint tissue and where the drug must be used on a long-term basis due to the nature of the disease.
- a eutectic mixture is often undesired, but it can be a useful property of drug combinations or formulations.
- formulations comprising eutectic mixtures have been viewed as unstable, the melting point depression of a eutectic mixture can be accompanied by an increase in the mixture's dermal solubility and transdermal permeation, which can increase a drug's bioavailability. See, e.g., Barry, B. W. Eur. J.
- EMLA eutectic mixture of the local anesthetics lidocaine and prilocaine
- the present invention provides pharmaceutical compositions, methods for preparation, and methods of treatment comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water.
- the selective COX-2 inhibitor is etoricoxib.
- the compositions enhance permeability and bioavailability, and they are useful for topical treatment of pain, inflammation, or both.
- the method of treatment is directed to pain associated with OA.
- the present invention provides a pharmaceutical composition for topical administration, the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water.
- the composition comprises 0.1% to 5% (w/w) etoricoxib, 3% to 21% (w/w) of a eutectic melt consisting essentially of choline and urea at least one lower alcohol, and water.
- the composition is a gel.
- the composition is a low- viscosity gel.
- the composition comprises 1% to 3% (w/w) of a selective COX-2 inhibitor. Still more preferably, the composition comprises 1% (w/w) of a selective COX-2 inhibitor. Alternatively, the composition comprises 2% (w/w) of a selective COX-2 inhibitor. Alternatively or yet still more preferably, the selective COX-2 inhibitor is etoricoxib.
- the eutectic melt consists essentially of choline (e.g., a choline salt) and urea. Still more preferably, the eutectic melt or the pharmaceutical composition comprises from a 3:1 ratio to a 1 :2 ratio of urea to choline.
- the eutectic melt comprises a 2:1 ratio of urea to choline.
- the composition comprises 2.5% (w/w) of choline.
- the composition comprises 5% (w/w) urea.
- the composition comprises ethanol and at least a second lower alcohol. More preferably, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). Alternatively, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). Alternatively, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). Alternatively, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). Alternatively, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). Alternatively, the second lower alcohol is a member selected from is
- composition comprises 3% to 10% (w/w) of the second lower alcohol. More preferably, the composition comprises 10% (w/w) of the second lower alcohol. Still more preferably, the composition further comprises 3% to 10% (w/w) of a third lower alcohol. Yet still more preferably, the composition comprises 10% (w/w) of the third lower alcohol. Alternatively or more preferably, at least one, at least two, or at least three of the lower alcohols are monohydric alcohols.
- the composition comprises at least one molecular penetration enhancer. More preferably, the molecular penetration enhancer is a fatty acid ester or a terpene. Still more preferably, the composition comprises about 0.1 to 10% (w/w), about 3% to 5% (w/w), or about 3% to 10% (w/w) of the fatty acid ester. Yet still more preferably, the fatty acid ester is selected from glycerol monolaurate and isopropyl myristate. Alternatively, the composition comprises 3% to 5% (w/w) of the terpene. More preferably, the terpene is limonene. Alternatively or yet still more preferably, the composition further comprises a second molecular penetration enhancer; more preferably, 3% to 5% (w/w) or from 3% to 10% (w/w) of the second molecular penetration enhancer.
- the composition further comprises an alpha- hydroxy acid. More preferably, the composition comprises 2% (w/w) of the alpha-hydroxy acid. Alternatively or still more preferably, the alpha-hydroxy acid is lactic acid. [0026] In another preferred aspect, the composition further comprises a fatty acid ester.
- the composition comprises about 0.1 to 10% (w/w), about 3% to 5% (w/w), or about 3% to 10% (w/w) of the fatty acid ester.
- the fatty acid ester is selected from glycerol monolaurate and isopropyl myristate.
- the composition further comprises a nonionic surfactant. More preferably, the composition comprises 1.5 to 5% (w/w) of the nonionic surfactant, and still more preferably 3% (w/w). Alternatively or yet still more preferably, the nonionic surfactant is polysorbate 20 (Tween ® 20).
- the composition further comprises a thickening agent. More preferably, the composition comprises 2% (w/w) of the thickening agent. Still more preferably, the thickening agent is a cellulosic thickening agent, and yet still more preferably, the thickening agent is hydroxypropyl cellulose.
- the present invention provides a pharmaceutical composition for topical administration, the composition consisting of, consisting essentially of, or comprising etoricoxib, urea, choline, a lower alcohol, and water; wherein choline and urea form a eutectic melt.
- the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially eutectic. More preferably, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic.
- the present invention provides a pharmaceutical composition for topical administration, the composition comprising etoricoxib, urea, choline, a lower alcohol and water; the composition prepared according to the method of: (a) mixing urea and choline together and, optionally, heating to form a eutectic melt; (b) mixing the etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) adding the lower alcohol and water to the composition of (b) and mixing thoroughly.
- the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially eutectic. More preferably, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic.
- the present invention provides a pharmaceutical composition for topical administration, the composition comprising etoricoxib, urea, choline, a lower alcohol and water; the composition prepared according to the method of: (a) mixing urea
- the present invention provides a method for topically treating pain in a subject, the method comprising topically applying a pharmaceutical composition to treat pain in the subject; the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water.
- the composition comprises 0.1% to 5% (w/w) etoricoxib, 3% to 21% (w/w) of a eutectic melt consisting essentially of or comprising choline and urea, at least one molecular penetration enhancer, at least one lower alcohol, and water.
- the pain is associated with OA.
- the present invention provides a use of a composition in the manufacture of a medicament for the topical treatment of pain; the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one molecular penetration enhancer, at least one lower alcohol, and water. More preferably, the composition comprises 0.1% to 5% (w/w) etoricoxib, about 3.0% to 21% (w/w) of a eutectic melt consisting essentially of or comprising choline and urea, at least one lower alcohol, and water. Alternatively or more preferably, the pain is associated with OA. Preferably, the composition further comprises at least one molecular penetration enhancer.
- FIG. 1 illustrates etoricoxib permeation through porcine skin from a first series of topical formulations (Table 1) at 4, 20, and 24 hours after application (25 ⁇ dosing).
- FIG. 2 illustrates etoricoxib permeation through porcine skin from a second series of topical formulations (Table 2) at 4, 21, and 26 hours after application (25 ⁇ dosing).
- FIG. 3 illustrates etoricoxib permeation through porcine skin from a third series of topical formulations (Table 3) at 4, 21, and 24 hours after application (25 ⁇ dosing).
- FIG. 4 illustrates etoricoxib permeation through porcine skin from a fourth series of topical formulations (Table 4) at 4, 21, and 26 hours after application (25 ⁇ dosing).
- FIG. 5 illustrates etoricoxib permeation through porcine skin from a fifth series of topical formulations (Table 5) at 4, 21, and 26 hours after application (50 ⁇ dosing).
- FIG. 6 illustrates etoricoxib permeation through porcine skin from a sixth series of topical formulations (Table 6) at 4, 21, and 26 hours after application ( 25 ⁇ dosing).
- FIG. 7A illustrates etoricoxib permeation through porcine skin from a seventh series of topical formulations (Table 7) at 4, 21, and 26 hours after application.
- FIG. 7B illustrates skin retention of etoricoxib from the seventh series of topical formulations (Table 7) after 26 hours of application (25 ⁇ dosing).
- FIG. 8 illustrates etoricoxib permeation through porcine skin from an eighth series of topical formulations (Table 8) at 4, 21, and 26 hours after application (10 and 5 ⁇ dosing).
- FIG. 9 illustrates etoricoxib permeation through porcine skin from a ninth series of topical formulations (Table 9) at 4, 21, and 24 hours after application (25 ⁇ dosing).
- FIG. 10 illustrates etoricoxib permeation through porcine skin from a tenth series of topical formulations (Table 10) at 4, 21, and 24 hours after application (10 ⁇ dosing).
- FIG. 11 illustrates etoricoxib permeation through porcine skin from an eleventh series of topical formulations (Table 11) at 4, 21, and 26 hours after application (10 ⁇ dosing).
- FIG. 12 illustrates etoricoxib permeation through human cadaver skin from a twelfth series of topical formulations (Table 12) at 4, 8, 12, 16, 20, and 24 hours after application (5 ⁇ dosing).
- FIG. 13 illustrates etoricoxib permeation through human cadaver skin from a thirteenth series of topical formulations (Table 13) at 4, 8, 14, 24, 36, and 48 hours after application (5 ⁇ dosing).
- FIG. 14 illustrates etoricoxib permeation through human cadaver skin from the fourteenth series of topical formulations (Table 14) at 4, 8, 14, 24, 36, and 48 hours after application (5 ⁇ dosing).
- FIG. 15 illustrates etoricoxib permeation through human cadaver skin from a fifteenth series of topical formulations (Table 15) at 4, 8, 14, 24, 36, and 48 hours after application (5 ⁇ dosing).
- a not only include aspects with one member, but also include aspects with more than one member.
- an embodiment including “a cellulosic thickening agent and a lower alcohol” should be understood to present certain aspects with two or more cellulosic thickening agents, two or more lower alcohols, or both.
- the term "about” as used herein includes a close (i.e., narrow) range around the explicit value for a variable. For example, in certain instances the term about includes 5 %- 10% higher or 5-10% lower than the value given. For example, “about 10" includes the range of values from 9.5 to 10.5 or from 9 to 11.
- agent indicates a compound or mixture of compounds that, when added to a pharmaceutical composition, tend to produce a particular effect on the composition's properties.
- a composition comprising a thickening agent is likely to be more viscous than an otherwise identical comparative composition that lacks the thickening agent.
- Alpha-hydroxy acid indicates an organic compound comprising at least one carbon substituted with a hydroxyl group and a carboxylic acid group (i.e., a C(OH)(C0 2 H) group).
- alpha-hydroxy acids include citric acid, glycolic acid, aldonic acids (e.g., gluconic acid), 2-hydroxycaproic acid, 2-hydroxycaprylic acid, 2- hydroxypropionic acid, lactic acid, malic acid, mandelic acid, tartaric acid, and the like, as well as mixtures thereof.
- Cellulosic thickening agent includes a thickening agent that is a natural or synthetic polymeric carbohydrate (e.g., cellulose, pharmaceutically acceptable vegetable gums) or a polymeric or oligomeric derivative of a polymeric carbohydrate that is produced by chemical modification (e.g., hydroxypropyl cellulose, hydroxypropyl methyl cellulose, hydroxyethyl cellulose).
- Representative cellulosic thickening agents include cellulose, hydroxypropyl cellulose ("HPC"), hydroxypropyl methyl cellulose, hydroxyethyl cellulose, methyl cellulose, carboxymethyl cellulose, and the like.
- chiral compounds described herein include the racemic form or the D- or L- enantiomer thereof (e.g., D-lactic acid or L-lactic acid).
- Choline indicates a compound consisting essentially of the cation (2-hydroxyethyl)trimethylammonium and an accompanying anion.
- Examples of “choline” include (2-hydroxyethyl)trimethylammonium chloride, (2-hydroxyethyl)trimethylammonium hydroxide, (2-hydroxyethyl)trimethylammonium tartrate, and the like.
- choline is (2-hydroxyethyl)trimethylammonium chloride. Where specific weight percentages or weight ratios are provided in this application, they are expressed in terms of (2- hydroxyethyl)trimethylammonium chloride .
- ER Enhancement ratio
- a test result e.g. , ug/cm 2 accumulated dose of product
- a selective COX-2 inhibitor e.g., etoricoxib
- Erutectic agent includes a compound that, in combination with at least a second eutectic agent, forms a eutectic melt.
- Representative eutectic agents include menthol, phenol, 2-amino-2-methylpropanol, choline chloride, urea, panthenol, niacinamide, citric acid, betaine, arginine, resorcinol, butylated hydroxytoluene, 4-chloroxylenol, camphor, glycerin monolaurate, lauryl alcohol, certain pharmaceutical agents (e.g., allylamine antifungal agents such as terbinafme and butenafine; local anesthetics such as lidocaine and prilocaine), and the like.
- allylamine antifungal agents such as terbinafme and butenafine
- local anesthetics such as lidocaine and prilocaine
- compounds A, B, and C can each be a eutectic agent as defined herein even if compounds A and C cannot be combined with each other to form a eutectic melt.
- a compound is a eutectic agent in a specific embodiment of a composition, it is able to form a eutectic melt with at least a second compound contained in that embodiment of the composition.
- Eutectic melt includes a combination of two or more eutectic agents that are solids before they are combined, but that when combined, form a substantially homogeneous liquid that is stable at one or more temperatures between about 10 °C and about 100 °C (e.g., between about 10 °C, 15 °C, 20 °C, 25 °C, 30 °C, 35 °C, 40 °C, 45 °C, 50 °C, 55 °C, 60 °C, 65 °C, 70 °C, 75 °C, 80 °C, 85 °C, 90 °C, 95 °C, 100 °C, and the temperatures in between).
- °C and about 100 °C e.g., between about 10 °C, 15 °C, 20 °C, 25 °C, 30 °C, 35 °C, 40 °C, 45 °C, 50 °C, 55 °C, 60 °C, 65 °C, 70 °C
- the eutectic melt is further characterized in that the melting point of the eutectic melt is lower than the individual melting points of the eutectic agents that comprise the melt.
- the eutectic melt comprises two eutectic agents, that when combined, form a substantially homogeneous liquid, the eutectic melt having a melting point that is lower than that of both of the eutectic agents when in purified or non-mixed form.
- both the solid and liquid phases have about the same proportional composition of eutectic agents.
- the melting point of a eutectic melt is lower than the melting point of other mixtures containing the same components, but with different proportions of the same eutectic agents.
- at least one of the liquid components of the eutectic melt is a solid when in purified or non-mixed form at the same temperature, and the component's change from solid to liquid state is caused by its melting point depression upon mixing with the other eutectic agent.
- the eutectic melt comprises two eutectic agents, both solids in purified or non-mixed form at the same temperature, and the agents' change from solid to liquid state is caused by their melting point depression upon mixing.
- the melting point of at least one, more than one, or all of the eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C. In one embodiment, the melting point of all of the eutectic agent components of the eutectic melt is greater than 10 °C. In another embodiment, the melting point of the eutectic melt is lower than 24 °C, and the melting point of at least one, more than one, or all of the eutectic agent components of the eutectic melt is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C. In a preferred
- the melting point of two eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C, and the melting point of a eutectic melt comprising the two eutectic agents is greater than 10 °C.
- the melting point of a eutectic melt comprising two eutectic agents is lower than 24 °C, and the melting point of the two eutectic agents in purified or non-mixed form is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C.
- a mixture is heated until the components are liquid or substantially liquid to form a eutectic melt.
- a mixture is heated to a temperature above 50 °C to form a eutectic melt (e.g., 60 °C, 65 °C, 70 °C, 70 °C, 80 °C, 85 °C, 90 °C, 95 °C, or 100 °C).
- Finite dosing as used herein generally includes an application of a limited reservoir of an active agent.
- the the active agent in the reservoir is depleted with time, leading to a tapering off of the absorption rate of the active agent after a maximum absorption rate is reached.
- composition as used herein are equivalent terms referring to a composition of matter suitable for pharmaceutical use.
- Infinite dosing as used herein generally includes an application of a large reservoir of an active agent.
- the active agent in the reservoir is not significantly depleted with time, thereby providing protracted, continuous, steady-state absorption of the active.
- “Lower alcohol” as used herein includes straight- or branched-chain alkyl alcohols of 1 to 6 carbon atoms.
- Representative lower monohydric alcohols include methanol, ethanol, n-propanol, isopropanol (i.e., isopropyl alcohol or ⁇ ), n-butanol, t-butanol, n- pentanol, 3-pentanol, 2-methoxyethanol, propylene glycol, and the like.
- Melt composition as used herein includes a mixture of at least two compounds.
- the “melting point” as used herein is the temperature at which a solid changes from solid to liquid phase under standard pressure (i.e., 1 atm or 101.325 kPa). If a solid melts over a temperature range, the melting point is the temperature at which the liquid phase first appears under standard pressure.
- “Monohydric alcohol” as used herein includes straight- or branched-chain alkyl alcohols with a single hydroxyl group.
- Representative monohydric alcohols include methanol, ethanol, n-propanol, isopropanol, n-butanol, t-butanol, n-pentanol, 3-pentanol, 2- methoxyethanol, 2-(2-ethoxyethoxy)ethanol, oleyl alcohol, and the like.
- Non-eutectic and “essentially non-eutectic” as used interchangeably herein include compositions that may initially contain or be a eutectic melt (e.g., choline and urea), but when additional components are added, the final composition is not a eutectic melt. For example, mixing urea and chloine and optionally heating can form a eutectic melt. When further components are added, however, the final composition may not be a eutectic melt (i.e., it is non-eutectic). Therefore, a composition can be non-eutectic, but still comprise or once have contained a eutectic melt.
- a eutectic melt e.g., choline and urea
- Nonionic surfactant indicates a surface-active agent that is uncharged under the conditions of the formulation.
- nonionic surfactants include the polysorbates (e.g., polysorbate 20) and fatty acid esters (e.g., isopropyl myristate).
- compositions comprising A or B would typically present an aspect with a composition comprising both A and B.
- Or should, however, be construed to exclude those aspects presented that cannot be combined without contradiction (e.g., a composition pH that is between 9 and 10 or between 7 and 8).
- a percentage range when a percentage range is taught, it incorporates all full or partial percentages in between (i.e. , within the bounds of the range). For example, a percentage range of 15 to 25% would also teach inter alia the specific values of 17.36% and 21%. A percentage range of about 13% to 17% would also teach inter alia the specific values of 12.97%, 16%, and 17.1%.
- Penetration enhancer includes an agent or a combination of agents that improves the transport of molecules such as a pharmaceutically or cosmetically active agent into or through a natural membrane such as skin or nail.
- a pharmaceutically or cosmetically active agent into or through a natural membrane such as skin or nail.
- Various conditions may occur at different sites in the body, either in the skin or below the skin, creating a need to target delivery of compounds. For example, in a treatment for osteoarthritis, delivery of the active agent to the underlying tissue surrounding the joint may be necessary to achieve therapeutic benefit.
- An MPETM may be used to assist in the delivery of an active agent i) directly into the skin, or nail; ii) locally, or regionally, into tissue(s) underlying or near to the skin or nail; or iii) indirectly via systemic distribution to the site of the disease. If systemic distribution of an active agent (e.g., etoricoxib) would be likely to produce side effects [e.g., etoricoxib), an MPETM is preferably selected to maximize direct delivery and to minimize systemic distribution.
- An MPETM may be a pure substance or may comprise, consist essentially of, or consist of a mixture of different chemical entities.
- Ratio as it pertains to comparative flux values described herein, are calculated based on the cumulative amount of active (e.g., etoricoxib) delivered through the skin over a period of 4-60 hrs, preferably 24 hrs.
- active e.g., etoricoxib
- Selective COX-2 inhibitor as used herein should in general be construed to mean the selective COX-2 (coxib) class of NSAIDs that preferentially inhibits COX-2, as well as the pharmaceutically acceptable derivatives or salts thereof.
- etoricoxib as used herein, includes pharmaceutically acceptable derivatives or salts thereof.
- substantially referes to a considerable amount, size, or physical state.
- a golf ball would be a “substantially spherical” object.
- a “substantially liquid” composition may have up to 5% or even 10% (w/w) of a solid, but generally has the physical properties of an liquid.
- substantially anhydrous and “essentially free of water” as used interchangeably herein include compositions without deliberately added water.
- a substantially anhydrous composition can contain up to 5% w/w water, which may be adventitiously incorporated from impurities in the starting materials, side products from reactions or manufacturing processes, or air absorption.
- substantially homogeneous designates a composition that includes at least 90%, 95%, or 99% by weight of a single phase, although it may include small amounts of a different phase (e.g., a liquid phase containing a small amount of immiscible liquid or a solid).
- substantially liquid designates a composition that includes at least 90%, 95%, or 99% by weight of a liquid phase, although it may include small amounts of at least one solid phase.
- Substituted phenol as used herein includes hydroxybenzenes and
- dihydroxybenzenes with from 1 to 3 additional substituents independently selected from the group of acyl, alkyl, alkenyl, alkoxy, amido, amino, aryl, carboxy, and halo substituents.
- Thickening agent includes an agent or combination of agents that increases the viscosity of a composition.
- a thickening agent may be a pure substance, or it may comprise, consist essentially of, or consist of a mixture of different chemical entities.
- Exemplary thickening agents include cellulose polymers, carbomer polymers, carbomer derivatives, cellulose derivatives, polyvinyl alcohol, poloxamers, polysaccharides, and the like, as well as mixtures thereof.
- Topical formulation includes a composition that is suitable for topical application to the skin, a nail, or a mucosa.
- a topical formulation may, for example, be used to confer a therapeutic or cosmetic benefit to its user.
- Specific topical formulations can be used for topical, local, regional, or transdermal application of substances.
- Transdermal as used herein includes a process that occurs through the skin.
- the terms “transdermal,” “percutaneous,” and “transcutaneous” can be used interchangeably.
- “transdermal” may also include epicutaneous.
- Transdermal application as used herein includes administration through the skin. Transdermal application can be used for systemic delivery of an active agent; however, it is also useful for delivery of an active agent to tissues underlying the skin with minimal systemic absorption. In certain embodiments, “transdermal application” may also include epicutaneous application.
- the unit prefix "u” as used herein is equivalent to " ⁇ " or "micro.” For example, “ul” is equivalent to " ⁇ " or "microliters.”
- Rea as used herein includes urea and pharmaceutically acceptable derivatives or salts thereof.
- w/w or "wt/wt” means a percentage expressed in terms of weight of the ingredient or agent over the total weight of the composition multiplied by 100.
- the present invention provides a pharmaceutical composition comprising, consisting essentially of, or consisting of a selective COX-2 inhibitor.
- the selective COX-2 inhibitor is selected from the group of celecoxib, etoricoxib, lumiracoxib, parecoxib, rofecoxib, valdecoxib, and a combination thereof. More preferably, the selective COX-2 inhibitor is selected from the group of celecoxib, etoricoxib, and rofecoxib. Still more preferably, the selective COX-2 inhibitor is etoricoxib.
- the pharmaceutical composition comprises 0.1% to 5% (w/w) of etoricoxib, more preferably about 1% to 3% (w/w), and still more preferably about 1 % or 2% (w/w).
- a composition permits delivery of a selective COX-2 inhibitor daily dosage of about 0.01 mg to about 120 mg, preferably about 0.1 mg to 60 mg, preferably about 1 mg to about 30 mg, and most preferably about 1 mg to about 10 mg.
- the formulation permits delivery of a daily dosage of about 3 mg.
- the concentration is such that this dosage amount can be provided by application of the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm 2 , preferably about 1200 to 1800 cm 2 (750 cm 2 /knee).
- the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm 2 , preferably about 1200 to 1800 cm 2 (750 cm 2 /knee).
- the concentration is such that this dosage amount can be provided by application of the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm 2 , preferably about 1200 to 1800 cm 2 (750 cm 2 /knee).
- composition can be applied to a skin area of about 1 to 50 cm , about 50 to 250 cm , about 100 to 500 cm 2 , about 200 to 800 cm 2 , or about 800 to 1200 cm 2 .
- the dosage and application area will vary on and can be tailored to the area being treated (e.g., knees, fingers, toes, back, and the like).
- a single knee is treated and the application area is about 750 cm 2 .
- both knees of an individual are treated and the application area is about 1500 cm (about 750 cm per knee).
- the formulation of the present invention provides a total or a systemic dose that is less than 50% of the systemic daily dose of the maximum approved oral dose; preferably less than 25%, more preferably less than 10%, and most preferably less than 5%, yet provides local or regional delivery levels sufficient for therapeutic benefit.
- the concentration is such that this dosage amount can be provided by application of the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm 2 , preferably about 1200 to 1800 cm 2 (750 cm 2 /knee).
- the composition can be applied to a skm area of about 1 to 50 cm , about 50 to 250 cm 2 , about 100 to 500 cm 2 , about 200 to 800 cm 2 , or about 800 to 1200 cm 2 .
- the pharmaceutical composition comprising etoricoxib provides better flux (as determined by the Franz cell procedure, e.g., the method of Example 2) than an analogous comparative formulation comprising a selective COX-2 inhibitor.
- this comparative formulation comprises etoricoxib. More preferably, the flux of etoricoxib is at least 1.5 times greater than the flux of the comparative formulation's active. In other words, the ratio of (i) the composition's etoricoxib flux to (ii) the comparative formulation's coxib flux is preferably greater than 1.0, and more preferably at least about 1.5.
- the composition has an etoricoxib flux that is at least 2.0 times greater than the comparative formulation's coxib flux. Yet still more preferably, the composition has an etoricoxib flux that is at least 4.0 times greater than the comparative formulation's coxib flux.
- the composition has a selective COX-2 inhibitor flux equal to or greater than the selective COX-2 inhibitor flux from a known comparative formulation with the same selective COX-2 inhibitor.
- the selective COX-2 inhibitor flux is greater than the flux of the comparative formulation with the same selective COX-2 inhibitor. More preferably, the selective COX-2 inhibitor flux is at least 1.5 times greater than the flux of a comparative formulation with the same selective COX-2 inhibitor.
- the ratio of (i) the selective COX-2 inhibitor flux of the composition to (ii) the selective COX-2 inhibitor flux from a comparative formulation with the same selective COX-2 inhibitor is preferably greater than 1.0, and more preferably at least about 1.5.
- the composition has a selective COX-2 inhibitor flux that is at least 2.0 times greater than the selective COX-2 inhibitor flux from a known comparative formulation with the same selective COX-2 inhibitor. Yet still more preferably, the composition has a selective COX-2 inhibitor flux that is at least 4.0 times greater than the selective COX-2 inhibitor flux from a comparative formulation with the same selective COX- 2 inhibitor.
- the present invention provides a composition comprising etoricoxib and having an etoricoxib flux (as determined by the Franz cell procedure of Example 2) of at least 0.1 ⁇ g/hr/cm 2 at 24 hours, preferably at least 0.2 ⁇ g/hr/cm 2 at 24 hours.
- the composition comprising etoricoxib has an enhancement ratio (ER) of at least 2. Yet still more preferably, the composition comprising etoricoxib has an ER of at least 5.0. Yet still more preferably, the composition comprising etoricoxib has an ER that is at least 10.0.
- ER enhancement ratio
- the pharmaceutical composition comprises a eutectic melt.
- the eutectic melt comprises, consists essentially of, or consists of choline and urea. More preferably, the eutectic melt or the pharmaceutical composition comprises from a 3: 1 ratio to a 1 :2 ratio of urea to choline. Even more preferably, the eutectic melt comprises a 1 : 1 or 2: 1 ratio of urea to choline. Alternatively, the composition comprises about 3.0% to 21% (w/w) of a eutectic melt. Preferably, the composition comprises about 2% to 10%> (w/w) of urea and about 1 % to 10% (w/w) of choline (e.g., a. choline salt). More preferably, the composition comprises about 2.5% (w/w) of choline and about 5% (w/w) urea.
- a homogeneous composition can be prepared by first combining choline and urea in a eutectic melt and then adding additional components of the composition.
- the present invention provides a method of preparing a pharmaceutical composition comprising the steps of: (a) mixing urea and choline together and, optionally, heating to form a eutectic melt; (b) mixing etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) adding a lower alcohol and water to the composition of (b) and mixing thoroughly.
- the steps of combining ingredients can be suitably rearranged, so long as preparing a eutectic melt comprises one step.
- both the solid and liquid phases have about the same proportional composition of eutectic agents.
- At least one of the liquid components of the eutectic melt is a solid when in purified or non-mixed form at the same temperature, and the component's change from solid to liquid state is caused by its melting point depression upon mixing with the other component or components.
- the melting point of at least one, more than one, or all of the eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, 50 °C, 100 °C, or 150 °C.
- the melting point of all of the eutectic agent components of the eutectic melt is greater than 10 °C. More preferably, they are greater than 50 °C, and still more preferably, they are greater than 100 °C.
- the melting point of the eutectic melt is lower than 24 °C, and the melting point of at least one, more than one, or all of the eutectic agent components of the eutectic melt is greater than 24 °C, 30 °C, 36 °C, 40 °C, 50 °C, or 100 °C.
- a mixture is heated until the components are liquid or substantially liquid to form a eutectic melt.
- a mixture is heated to a temperature above 50 °C to form a eutectic melt (e.g., 60 °C, 65 °C, 70 °C, 70 °C, 80 °C, 85 °C, 90 °C, 95 °C, 100 °C, 115 °C, or 130 °C).
- the composition further comprises at least one molecular penetration enhancer.
- the molecular penetration enhancer is selected from terpenes, fatty acid esters, and fatty acid alcohols. More preferably, the molecular penetration enhancer is a terpene.
- Examples include D-limonene, limonene oxide, geraniol, a-pinene, -pinene oxide, thymol, menthone, menthol, neomenthol, 3-carene, L-cavol, carvone, carveol, 1,8-cineole (eucalyptol), citral, dihydrocarveol, dihydrocarvone, 4- terpinenol, fenthone, menthone, pulegone, pulegol, isopulegol, piperitone, camphor, a- terpineol, terpinen-4-ol, linalool, carvacrol, trans-anethole, ascaridole, safrole, racemic mixtures thereof (e.g., DL-limonene), and pharmaceutically acceptable isomers thereof.
- D-limonene limonene oxide, geraniol, a-pinene, -pin
- a second molecular penetration enhancer can be present (e.g., a fatty acid ester and a terpene).
- the composition of the present invention comprises limonene.
- the composition comprises about 0.1% to 5% (w/w) of limonene or geraniol, such as about 0.1 , 0.5, 1, 2, 3, 4 or 5% (w/w), and more preferably about 3% to 5% (w/w).
- the terpene molecular penetration enhancer can be included within an essential oil.
- Essential oils that include a substantial proportion of at least one terpene molecular penetration enhancer include oils of peppermint, eucalyptus, chenopodium, anise, and yling-yling.
- a fatty acid ester or fatty alcohol ester is used as a an MPETM in the composition.
- preferred fatty acid ester MPETMs are glyceryl monoesters and isopropyl myristate. More preferably, the MPETM is glyceryl monolaurate. Still more preferably, the MPETM is isopropyl myristate .
- the composition comprises about 0.1% to 10% (w/w) of the fatty acid ester or fatty alcohol ester , such as about 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10% (w/w).
- the composition comprises about 1 to 3% (w/w) (e.g. about 1%; about 3% (w/w)), about 0.1% to 10% (w/w), or about 3 to 5% (w/w).
- fatty acid esters include butyl acetate, caproyl glycolate, cetyl lactate, cocoyl glycolate, decyl N,N-dimethylamino acetate, decyl N,N-dimethylamino isopropionate, diethyleneglycol oleate, diethyl sebacate, diisopropyl sebacate, dodecyl N, V- dimethylamino acetate, dodecyl N,N-dimethylamino butyrate, dodecyl N,N-dimethylamino isopropionate, dodecyl 2-(N,N-dimethylamino)propionate, EO-5-oleyl ester, ethyl acetate, ethyl acetoacetate, ethyl propionate, glyceryl dilaurate, glyceryl dioleate, glycerol monoethers, glyceryl dil
- MPETMs include fatty acids, lactic acid, fatty alcohols (e.g., oleyl alcohol, stearyl alcohol, decanol), fatty alcohol ethers, hexahydro-l-dodecyl-2H-azepin-2-one (e.g., laurocapram, AzoneTM) and derivatives thereof, dimethylsulfoxide (DMSO) and related sulfoxides (e.g., n-decyl methylsulfoxide), salicylic acid and alkyl esters thereof (e.g., methyl salicylate), ⁇ , ⁇ -dimethylacetamide, dimethylformamide, ⁇ , ⁇ -dimethyltoluamide, 2- pyrrolidinone and N-alkyl derivatives thereof (e.g., N-methyl-2-pyrrolidone (NMP) and N- octyl-2-pyrrolidinone), and 2-nonyl-l,3-diox
- the composition comprises a mixture of a lower alcohol and water. More preferably, the lower alcohol is a monohydric lower alcohol, and still more preferably, the lower alcohol is ethanol, isopropanol, or 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol ® ). In certain aspects, the composition comprises a second lower alcohol. In certain other aspects, the composition comprises a third lower alcohol.
- the composition comprises at least about 3, 5, 7, 9.5, 10, 10.5, 11, 11.5, 12, 14, 15, 20, 25, 30, 31, 31.5, 32, 32.5, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises at least about 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises at least about 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises at most about 3, 5, 7, 9.5, 10, 10.5, 11, 11.5, 12, 14, 15, 20, 25, 30, 31, 31.5, 32, 32.5, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises at most about 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises at most about 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
- the composition comprises 35 to 75% (w/w) of a lower alcohol.
- the composition comprises the same or differing amounts of a first and at least one additional lower alcohol. More preferably, the composition comprises 40% to 65 % (w/w) of ethanol and 10% (w/w) of isopropanol. In another more preferred aspect, the composition comprises 40% to 65% (w/w) of ethanol, 10% (w/w) of isopropanol, and 3% to 10% (w/w) of 2-(2-ethoxyethoxy)ethanol.
- the first alcohol is about 35 to 75% (w/w)
- the at least one additional alcohol is between about 0.1 to 10% (w/w) (e.g., about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10% (w/w)) and the at least one more additional alcohol is between about 0.1 to 10 % (w/w) (e.g., about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10% (w/w); about 35% to 95% (w/w) total alcohol).
- the first alcohol is about 35 to 75% (w/w)
- the second lower alcohol is about 3% to 10%(w/w).
- the composition further comprises about 3 to 10% (w/w) of a third lower alcohol.
- the lower alcohol is a diol.
- the composition further comprises a diol.
- Suitable diols include, but are not limited to, propylene glycol, butanediol, butynediol, pentanediol, hexanediol, octanediol, neopentyl glycol, 2-methyl-l,3-propanediol, diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, dibutylene glycol, propylene glycol, and a combination thereof.
- the formulation comprises about 0.1% to 15% (w/w) of propylene glycol, such as about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15% (w/w), and preferably about 0.1 to 5% (w/w).
- composition further comprises at least one
- the surfactant is a nonionic surfactant. More preferably, the surfactant is a polysorbate surfactant. Still more preferably, the surfactant is polysorbate 20.
- nonionic surfactants include (but are not limited to) cetomacrogol 1000, cetostearyl alcohol, cetyl alcohol, cocoamide diethanolamine, cocoamide monoethanolamine, decyl glucoside, glyceryl laurate, lauryl glucoside, polyoxyethylene ethers of fatty acids such as cetyl alcohol or stearyl alcohol, narrow-range ethoxylates, octyl glucoside, oleyl alcohol, poloxamers, polyethylene glycol, sorbitan monolaurate, polyoxyethylene sorbitan
- the composition further comprises at least one thickening agent, preferably a cellulosic thickening agent.
- suitable cellulosic thickening agents include, but are not limited to, hydroxypropyl cellulose (HPC) of various grades, hydroxypropyl methyl cellulose, hydroxyethyl cellulose, hydroxyethyl methyl cellulose, ethyl cellulose, methyl cellulose, carboxymethyl cellulose, dextran, guar gum, pectin, starch, cellulose, and the like. More preferably, the cellulosic thickening agent is HPC.
- the composition comprises about 1% to 5% (w/w) of a cellulosic thickening agent, such as about 1, 2, 3, 4, or 5% (w/w). More preferably, the composition comprises from 1% to 2% (w/w) of a cellulosic thickening agent. Still more preferably, the composition comprises 1% (w/w) of a cellulosic thickening agent. Alternatively, the composition comprises ?% (w/w) of a cellulosic thickening agent. [0127] In one aspect, the composition further comprises an anti-oxidant.
- Preferred antioxidants for use in the present invention include butylated hydroxytoluene, butylated hydroxyanisole, ascorbyl linoleate, ascorbyl dipalmitate, ascorbyl tocopherol maleate, calcium ascorbate, carotenoids, kojic acid and its pharmaceutically acceptable salts, thioglycolic acid and its pharmaceutically acceptable salts (e.g., ammonium), tocopherol, tocopherol acetate, tocophereth-5, tocophereth-12, tocophereth-18, tocophereth-80, and the like.
- composition further comprises a chelating agent.
- Preferred chelating agents include ethylenediamine tetraacetic acid (EDTA), diamnionium EDTA, dipotassium EDTA, calcium disodium EDTA, H-EDTA, tetraethylammonium (TEA- ) EDTA, tetrasodium EDTA, tnpotassium EDTA, tnsodium phosphate, diamnionium citrate, galactaric acid, galacturonic acid, gluconic acid, glucuronic acid, humic acid, cyclodextrin, sodium citrate, potassium citrate, the sodium salt of ethylenediamine -tetra (methylene phosphonic acid) (EDTMP), potassium EDTMP, and the like.
- EDTA ethylenediamine tetraacetic acid
- EDTMP diamnionium EDTA
- dipotassium EDTA calcium disodium EDTA
- H-EDTA te
- compositions of the invention optionally include a buffer or a pH-adjusting agent (e.g., in addition, the topical formulations of the present invention can also comprise a pH-adjusting agent).
- the pH- adjusting agent is a base.
- Suitable pH-adjusting bases include bicarbonates, carbonates, hydroxides (such as alkali or alkaline earth metal hydroxide as well as transition metal hydroxides), and the like.
- suitable pH-adjusting bases include amines, such as diethanolamine, triethanolamine, or aminopropanol; bicarbonates;
- the pH-adjusting agent can also be an acid, an acid salt, or mixtures thereof.
- the pH adjusting agent can be present in an amount sufficient to adjust the pH of the composition to between about pH 4.0 to about 10.0, more preferably about pH 7.0 to about 9.5.
- the unadjusted pH of the admixed components is between 8 and 10, such as 9, without the need for the addition of any pH adjusting agents.
- the pH-adjusting agent is sodium hydroxide, hydrochloric acid, or a combination of both, and is present in an amount sufficient to adjust the pH of the
- composition to between about pH 4.0 to 8.5, more preferably, to between about pH 5.5 to 7.0, such as about 6.0 or 6.5. Even more preferably, the pH is adjusted to about 4.0, 4.2, 4.4, 4.6, 4.8, 5.0, 5.2, 5.4, 5.6, 5.8, 6.0, 6.2, 6.3, 6.4, 6.6, 6.8, 7.0, 7.2, 7.4, 7.6, 7.8, 8.0, 8.4, 8.5, or any fraction in-between.
- a small amount of acid or base is included in the formulation.
- amounts of acid or base that may be included in the formulation are about 0.000001%, 0.00001%, 0.0001%, 0.001%, 0.0012%, 0.01%, 0.012%, 0.1%, or 1.0%. Preferably, this amount is about 0.0001%.
- the pH-adjusting agent can also be a buffer.
- the pH of the composition of the invention can be adjusted or stabilized with a buffer.
- Suitable buffers include citrate/citric acid buffers, acetate/acetic acid buffers, phosphate/phosphoric acid buffers, formate/formic acid buffers, propionate/propionic acid buffers, lactate/lactic acid buffers, carbonate/carbonic acid buffers, ammonium/ammonia buffers, and the like.
- the buffer is an acidic buffer system such as, for example, benzocaine.
- the acidic acid buffer system is citric acid or a citric acid salt.
- the buffer is present at a concentration of about
- this amount is about 0.0010 M, 0.0015 M, 0.002 M, 0.003 M, 0.004 M, 0.005 M, 0.006 M, 0.007 M, 0.008 M, 0.009 M, 0.01 M. 0.012 M, or 0.02 M. Alternatively and preferably, this amount is about 0.001 M.
- this amount is about 0.10 M, 0.11 M, 0.12 M, 0.13 M, 0.14 M, 0.15 M, ' 0.16 M, 0.17 M, 0.18 M, 0.19 M, 0.20 M, 0.21 M, 0.22 M, 0.23 M, 0.24 M, 0.25 M, 0.26 M, 0.27 M, 0.28 M, 0.29 M, 0.30 M, 0.31 M, 0.32 M, 0.33 M, 0.34 M, 0.35 M, 0.36 M, 0.37 M, 0.38 M, 0.39 M, 0.40 M, 0.41 M, 0.42 M, 0.43 M, 0.44 M, 0.45 M, 0.46 M, 0.47 M, 0.48 M, 0.49 M, 0.50 M, 0.55 M, 0.60 M, 0.65 M, 0.7 M, 0.75 M, 0.8 M, 0.85 M, 0.9 M, 0.95 M, or 1.0 M.
- the inventive formulation includes a buffer, and a second pH-adjusting agent (e.g., sodium hydroxide or hydrochloric acid) to adjust the pH of the composition to a desired pH.
- a second pH-adjusting agent e.g., sodium hydroxide or hydrochloric acid
- the second pH-adjusting agent comprises two agents (e.g., sodium hydroxide and hydrochloric acid) that are included as needed to adjust the pH of the composition to a desired pH.
- composition of the present invention comprises a
- preservative such as propyl paraben or methyl paraben, or combinations thereof.
- the formulation may be made bacteriostatic by the addition of preservatives.
- a composition can contain about 0.001 to 8% (w/w); preferably, about 0.01 to 6% (w/w); and more preferably, about 0.05 to 5% (w/w) of a preservative or a combination of preservatives.
- preservatives include, but not limited to, benzoic acid, benzyl alcohol, benzylhemiformal, benzylparaben, 5-bromo-5-nitro-l,3-dioxane, 2-bromo-2- nitropropane-l,3-diol, butyl paraben, phenoxyethanol, methyl paraben, propyl paraben, diazolidinyl urea, calcium benzoate, calcium propionate, captan, chlorhexidine diacetate, chlorhexidine digluconate, chlorhexidine dihydrochloride, chloroacetamide, chlorobutanol, p- chloro-m-cresol, chlorophene, chlorothymol, chloroxylenol, m-cresol, o-cresol, diethylene glycol dimethyl ether ("DEDM”) hydantoin, DEDM hydantoin dilaurate, dehydroacetic acid,
- DEDM di
- the composition is selected from the group of a gel, a foam, a cream, an emulsion, a micro emulsion, a lotion, an organogel, an ointment, a solution, and a transdermal patch. More preferably, the composition is a gel, and still more preferably, a low-viscosity gel. Alternatively, the composition is a solution.
- the composition is more viscous than water at standard temperature and pressure (STP).
- STP standard temperature and pressure
- the composition has a kinematic viscosity of more than about 1 centistokes (cSt) or a dynamic viscosity of more than about 1 centipoise (cP).
- the dynamic viscosity of the composition is at least about 2, 3, 4, 5, 7, 10, 12, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 75, 80, 90, 100, 150, 200, 250, 500, 1000, 2000, 3000, 5000, 10,000 cP at STP.
- the composition is thixotropic (i.e., it decreases in viscosity upon being stirred or shaken).
- the composition's viscosity can be adjusted by the addition of a cellulosic thickening agent, such as
- hydroxypropyl cellulose or other thickening agents.
- the composition is acidic.
- the composition has a pH of below 7.5, of below 6.5, of below 5.5, of below 4.5, of below 3.5, or of below 2.5.
- the pH of the composition may range from about 1.5 to about 7, about 2 to about 7, about 3 to about 7, about 4 to about 7, or about 5 to about 7.
- the pH of the composition may range from about 1.5 to about 5.5, about 2.5 to about 5.5, about 3.5 to about 5.5, or about 4.5 to about 5.5.
- the composition is basic. In certain aspects, the composition has a pH of above 7, of above 8, of above 9, of above 10, of above 11, or of above 12. In certain other aspects, the pH of the composition may range from about 7 to about 12.5, about 7 to about 11.5, about 7 to about 10.5, about 7 to about 9.5, or about 7 to about 8.5. In still other aspects, the pH of the composition may range from about 9 to about 12.5, about 9 to about 11.5, about 9 to about 10.5, or about 8.5 to about 10. [0141] In still yet another aspect, the composition is neutral. In certain aspects, the composition has a pH of about 7. In certain other aspects, the composition has a pH from about 6 to about 8.5, from about 5.5 to about 8, about 6 to about 8, about 6.5 to about 8.5, or from about 6.5 to about 7.5.
- a composition is designed for high penetration, for high retention in the skin, or for both high penetration and high retention.
- the optimal composition will have a balance between penetration and retention, enabling an effective amount of the active ingredient to pass through the skin, but also enabling it to stay in the target area for a sufficient duration to alleviate the patient's pain or other symptoms.
- a composition is designed for topical efficacy with minimal systemic distribution of the coxib through the body by the circulatory system (e.g., the cardiovascular system). Without being bound by theory, it is believed that minimization of systemic distribution would decrease the side effects of the composition, especially the side effect of adverse cardiovascular events.
- the optimal composition will have low systemic bioavailability, but will effectively treat pain (or other symptoms) associated with the site of application.
- a formulation provides the advantage of favorable stability at six months, as reflected in the lack of any substantial changes in viscosity, the absence of phase separation and crystallization at low temperatures, and a low level of impurities.
- a formulation comprising etoricoxib provides additional advantages in comparison to previously described etoricoxib compositions. Such advantages may include one or more of the following: adhering well to the skin, spreading easily, drying more quickly, and showing greater in vivo absorption. In some more preferred aspects, the drying rate results in a residue of at most 50% of a starting amount after 24 hours.
- the transdermal selective COX-2 inhibitor ⁇ e.g., and still more preferably, etoricoxib) flux as determined by Franz cell procedure at finite dosing or at infinite dosing is at least 1.5 times that of a comparative liquid formulation.
- an "acceptable time period" is at least about 1 day; preferably, at least about 30 days; more preferably, at least about six months; still more preferably, at least about one year; and yet still more preferably, at least about two years.
- the present invention provides a formulation that degrades by less than 1% over the course of 6 months at room temperature. More preferably, the rate of degradation is less than about 0.9, 0.8, 0.7, 0.6, 0.5, 0.4, 0.3, 0.2, or less than 0.1 %, and all fractions in between, over the course of six months at room temperature.
- the pharmaceutical composition is formulated as a cream, an emulsion, a microemulsion, a gel (e.g., a hydrogel, an organogel, or an inorganic or silica gel), a lotion, a lacquer, an ointment, a solution (e.g. , a moderate to highly viscous solution), or a transdermal patch.
- the pharmaceutical composition may also be prepared so that it may be applied to the skin as a foam.
- the composition is a gel, and more preferably, a low-viscosity gel.
- the pharmaceutical composition is formulated as a solution.
- the pharmaceutical composition is formulated as a transdermal patch.
- the invention describes a method for treating pain comprising the step of applying a topical, selective COX-2 inhibitor composition to a subject.
- the pharmaceutical composition is applied to the skin of the subject.
- the selective COX-2 inhibitor is delivered locally to the skin with minimal systemic absorption.
- the selective COX-2 inhibitor is delivered to and through the skin with minimal systemic absorption.
- the selective COX-2 inhibitor is delivered to the tissue surrounding or under the area of skin application with minimal systemic absorption.
- the subject is a human.
- the subject is a non-human mammal.
- the treatment is continued for at least 12 weeks. More preferably, the treatment is continued for at least six months.
- compositions of the invention may be useful to alleviate acute pain, chronic pain, or both.
- Compositions of the invention are particularly suited for use in treating OA chronically. They may also be useful for the treatment of other chronic joint diseases characterized by joint pain, degeneration of articular cartilage, impaired movement, and stiffness. Suitable joints include the knee, elbow, hand, wrist and hip.
- the compositions of the invention may also be useful for the treatment of other pain-associated disorders, including (but not limited to) muscle pain, lower back pain, neck pain, rheumatoid arthritis, fibromyalgia, myofascial pain, gout, sprains, strains, contusions, and neuropathic pain conditions.
- compositions of the present invention can be administered at lower dosing than previously described etoricoxib formulations.
- the compositions of the invention can be used at twice-a-day or once-a-day dosing in the treatment of OA. This would represent a significant improvement as lower dosing is associated with better patient compliance, an important factor in treating chronic conditions.
- compositions of the present invention may, if desired, be presented in a bottle, jar, or other container-closure system approved by the FDA or other regulatory authority, which may provide one or more dosages containing the active ingredient.
- the package or dispenser may also be accompanied by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, the notice indicating approval by the agency.
- urea/choline eutectic melt composition For a typical urea/choline eutectic melt composition, the urea and (2-hydroxyethyl)- trimethylammonium chloride were mixed by vortex, heating gently (e.g., 80-90°C) if necessary for liquification. The etoricoxib was then added and mixed by vortex with optional heating. The other components were then added except for alpha-hydroxy acids (if present) and some of the monohydric alcohol and water. The resulting suspension or solution was thoroughly mixed by vortex until a clear solution was obtained. [0159] Finally, any alpha-hydroxy acid (e.g., lactic acid) was added, followed by the remaining monohydric alcohol and water. The resulting suspension or solution was thoroughly mixed by vortexing for about 30 min or until a clear and homogeneous solution was obtained.
- any alpha-hydroxy acid e.g., lactic acid
- Example 2 General Procedure for Skin Permeation Measurement: [0161] Comparisons were mainly performed using porcine skin, but in some cases, human cadaver skin was used (i.e., Tables 12 to 15). The permeation of etoricoxib through porcine skin or human cadaver skin from each of the present formulations was measured using Franz diffusion cells ("FDC's).
- FDC's Franz diffusion cells
- Porcine skin pieces were obtained from Lampire Biological Laboratories, Inc., Pipersville, PA. Porcine skins were collected immediately following animal sacrifice, and the hairs were trimmed with clippers. Larger pieces of excess fat were removed with a filet knife. The skin was then trimmed to a set thickness of some 2 mm, cut into individual pieces, wrapped in aluminum foil, frozen, shipped, and stored at -78 °C.
- the skin pieces Prior to use, the skin pieces were allowed to thaw, in air, to room temperature.
- the skin was dermatomed to a thickness of 0.5 to 1 mm and cut into circular pieces of an appropriate size prior to mounting in the FDC.
- the FDCs had a 3-ml receptor well volume, that was filled with isotonic phosphate buffered saline ("PBS") doped with 0.01% sodium azide.
- PBS isotonic phosphate buffered saline
- the flanges of the FDCs were coated with vacuum grease to ensure a complete seal and were clamped together with uniform pressure using a pinch clamp (SS #18 VWR 80073-350 from VW Scientific, West Chester PA).
- the porcine skin was optionally allowed to pre-hydrate for 45 min with isotonic PBS.
- Isotonic PBS was then removed and formulation was applied to the donor well or directly to the skin surface, depending on the amount of formulation applied.
- the receptor wells were maintained at 37 °C (temperature on the surface of the skin is about 30 °C) in a stirring block with continual agitation via a stir bar.
- the initial applied dose was 100 ul. This application dose was gradually reduced to 50 ul, 25 ul, 10 ul, and 5 ul levels.
- transdermal studies it is common practice to use a hydro alcoholic control, since this combination appears to provide optimal dissolution for various drug compounds.
- the flux rates were calculated using the approximate area of each donor well (0.55 cm 2 ). Samples were drawn from the receptor wells at various times, as provided in the examples that follow. Franz diffusion cell measurements were typically made in five- to tenfold replicates for each formulation, based on availability of skin and number of formulations tested. The concentrations of etoricoxib in the samples were measured using HPLC analysis using a CI 8 column and acetonitrile and water as the mobile phase. Generally, in the examples that follow, permeation data were reported by plotting a curve showing the cumulative amount of etoricoxib that permeates across the skin as a function of time. The flux rate can be computed as the time derivative of this curve.
- FIG. 7 A shows the delivery of etoricoxib through the skin
- FIG. 7B shows the retention of etoricoxib in the skin.
- a formulation with isopropyl myristate (F55) showed similar behavior to a formulation containing DL-Limonene (F57).
- F58 A choline/urea mixture incorporated into the formulation with and without heating (F52 vs. F58).
- F58 is a version of F52 without any application of heat during formulation.
- F58 was prepared without eutectic formation, and it was obtained by simple addition of ingredients.
- Both F52 and F58 provided similar delivery of etoricoxib.
- F58 was not homogeneous the next day, whereas F52, which was prepared with eutectic formation first, was homogeneous.
- Example 11 Etoricoxib Formulations VIII
- Glycerin mono laurate 3 [0194] The results for the formulations described in Table 8 are shown in FIG. 8. The results are for finite dosing. The first set of results in FIG. 8 is for 10 ⁇ doses; the second set, for 5 ⁇ doses.
- the optimal level of urea appears to be between 2.5% to 5% (F76 vs. F77, F78, and F79).
- the highest level of etoricoxib delivery was observed with a formulation containing a 2:1 urea/choline chloride ratio (F82), which was consistent with the results from the previous studies.
- Transcutol ® further enhanced the etoricoxib permeation (F87 vs. F98, F99).
- Example 15 General Procedure for Flow-Through
- Flow-through experiments were essentially performed using an automated version of the Franz Cell methodology of Example 2, with the exception that the surface area was 1 cm and the receptor volume was 1 ml. Human cadaver skin was not dermatoned.
- the etoricoxib concentrations used were 0.5, 1.0, 2.0, 3.0, and 5.0%.
- the etoricoxib dosing studies included 48-hour, two-dose studies (dosing at 0 and 8 hours); 48-hour, four-dose studies (dosing at 0, 9, 21 and 29 hours); and 48-hour studies after pretreatment w/placebo with 1 and 2 % actives (dosing at 0, 21 and 29 hours).
- the placebo acts as a conditioning agent and also may act as a permeation enhancer.
- the Enhancement Ratio (ER) of the test formulation which is defined above, is provided for certain embodiments.
- Example 18 48-h Dosing Studies: Dosing at 0, 9, 21 and 29 h
- Table 14A 48-h Dosing Studies: Dosing at 0, 9, 21 and 29 h
- Example 19 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h
- Table 15A 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h
- Table 15B 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h
- PCT/US2010/052111 (filed October 9, 2010), and PCT/US2010/044036 (filed July 30, 2010).
Abstract
The present invention provides topical pharmaceutical compositions, methods for preparation, and methods of treatment comprising a selective COX-2 inhibitor and useful for the treatment of pain, particularly pain associated with osteoarthritis. The compositions can provide good permeability and bioavailability at the target site. In certain preferred embodiments, the invention provides a pharmaceutical composition comprising etoricoxib, a eutectic melt, at least one lower alcohol, and water.
Description
TOPICAL EUTECTIC FORMULATION
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Patent Application No.
61/260,351, filed November 11, 2009. The contents of this priority document are
incorporated herein in their entirety for all purposes.
BACKGROUND OF THE INVENTION
[0002] Osteoarthritis (OA) is a chronic joint disease characterized by progressive degeneration of articular cartilage. Symptoms include joint pain and impaired movement. OA is one of the leading causes of disability worldwide and a major financial burden to health care systems. It is estimated to affect over 15 million adults in the United States alone. See Boh, L.E.; Osteoarthritis. In: DiPiro, J.T.; Talbert, R.L.; Yee, G.C. et al. editors.
Pharmacotherapy: a pathophysiological approach. 4th ed. Norwalk (CT): Appleton & Lange, pp. 1441-59 (1999).
[0003] An OA treatment's efficacy is generally assessed by three outcome measures: pain, physical function, and a patient global assessment. See Bellamy, N.; Kirwan, J.; Boers, M.; Brooks, P.; Strand, V.; Tugwell, P. et al. Recommendations for a core set of outcome measures for future Phase III clinical trials in knee, hip and hand osteoarthritis. Consensus development at OMERACT III., J Rheumatol, 24:799-802 (1997). To be suitable for chronic use, a therapy must generally show efficacy on these three variables over a sustained period of time. In the U.S., the Food and Drug Administration (FDA) has required OA therapies to show superiority over placebo over a twelve- week period before approval of a new drug application.
[0004] Oral non-steroidal anti-inflammatory drugs (NSAIDs) are a mainstay in the management of OA. These drugs are thought to exert their analgesic effect by impeding the production of signaling molecules called prostaglandins through inhibition of the
cyclooxygenase ("COX") enzyme. The COX enzyme has two isoforms, COX-1 and COX-2. Traditional NSAIDs inhibit both isoforms of the COX enzyme, while the selective COX-2 (coxib) class of NSAIDs preferentially inhibits COX-2.
[0005] NSAIDs have analgesic, anti-inflammatory, and antipyretic effects and are useful in reducing pain and inflammation. They are, however, associated with serious potential side
effects including nausea, vomiting, peptic ulcer disease, and gastrointestinal (GI)
hemorrhage. Although selective COX-2 inhibitors produce fewer gastrointestinal side effects, they may increase the risk of thrombotic events (e.g., stroke or heart attack). Because of this potential side effect, most of the selective COX-2 inhibitors have been withdrawn from the U.S. market.
[0006] Topical NSAIDs offer the possibility of achieving local therapeutic benefit while reducing or eliminating the risk of systemic side effects. There has been widespread interest in this approach to treating OA, but data supporting the efficacy of topical NSAIDs in the treatment of OA is limited. For instance, a study of thirteen randomized placebo controlled trials (RCT's) of various topical NSAIDs tested specifically for use in the treatment of OA concluded that they were not generally efficacious for chronic use in OA. Lin et al, Efficacy of topical non-steroidal anti-inflammatory drugs in the treatment of osteoarthritis: metaanalysis of randomized controlled trials, BMJ, doi:10.1136/bmj.38159.639028.7C (2004).
[0007] Pennsaid Gel is a topical formulation comprising diclofenac sodium that overcomes disadvantages of prior art NSAID formulations. U.S. Patent Publication No. 2008/0300311. Pennsaid™ solution has been shown in clinical trials to be effective for treating the pain and symptoms of osteoarthritis, and it has been approved for use in Canada, the U.S., and several European countries.
[0008] A topical formulation containing a COX-2 selective inhibitor would offer patients an attractive new treatment modality. Such a formulation could minimize systemic exposure to the active pharmaceutical ingredient by localizing the drug at the site of action. At the same time a topical coxib might have even better GI safety profile than topical formulations containing traditional NSAIDs, making it particularly suitable for patients at risk of GI bleeds. [0009] The dearth of options with robust efficacy data for topical NSAID treatment of OA partially arises from the difficulty associated with delivering a molecule through the skin both in a sufficient quantity to exert a therapeutic effect and in a manner that makes the treatment itself tolerable. It is generally believed that for topical OA treatments, clinical efficacy requires absorption of the active ingredient and its penetration in sufficient quantities into underlying inflamed tissues including the synovium and synovial fluid of joints. See
Rosenstein, Topical agents in the treatment of rheumatic disorders, Rheum. Dis. Clin North Am., 25: 899-918 (1999).
[0010] Various factors can affect the absorption rates and penetration depth of topical pharmaceutical preparations, including the nature of the active ingredient, the nature of the vehicle, the pH, and the relative solubility of the active in the vehicle versus the skin.
Ostrenga J. et al, Significance of vehicle composition I: relationship between topical vehicle composition, skin penetrability, and clinical efficacy, Journal of Pharmaceutical Sciences, 60: 1175-1179 (1971). More specifically, drug attributes such as solubility, size and charge, as well as vehicle attributes such as the drug dissolution rate, spreadability, adhesion, and ability to alter the membrane permeability can each have significant effects on permeability.
[0011] Seemingly minor variations in formulations can produce significant changes in their performance. For instance, Naito demonstrates significant variability in penetration among topical NSAID formulations simply by changing the gelling agent used in the compositions. Naito et al. , Percutaneous absorption of diclofenac sodium ointment, Int. Jour, of
Pharmaceutics, 24: 115-124 (1985). Similarly, Ho noted significant variability in penetration by changing the proportions of alcohol, propylene glycol, and water. Ho et al, The influence of cosolvents on the in-vitro percutaneous penetration of diclofenac sodium from a gel system, J. Pharm. Pharmacol, 46:636-642 (1994). It was noted that the changes affected three distinct variables: (i) the solubility of the drug in the vehicle, (ii) the partition coefficient of the drug between the vehicle and the skin, and (iii) the alteration of skin structure. Id. [0012] Ho et al. also noted that (i) the pH of the vehicle, (ii) the drug solubility, and (iii) the viscosity of a gel matrix can influence penetration from a gel dosage form. Id. The pH value affects the balance between ionized and non-ionized forms of the drag, which typically have different permeation properties. Obata, International Journal of 'Pharmaceutics, 89: 191-198 (1993). The viscosity can affect diffusion of the drug through the gel matrix and release of the drug from the vehicle into the skin. The solubility of the drag in the vehicle will affect the partition coefficient of the drug between the composition and the recipient membrane or tissue. Ho, Id.
[0013] The skin barrier can be compromised by several physical methods, such as iontophoresis, ultrasound, electroporation, heat, and microneedles. Molecular penetration enhancers (MPE™s) are a preferred means for reversibly lowering the skin barrier. At least 400 chemicals have been identified as skin permeability enhancers. General categories of MPE™s include pyrrolidones, fatty acids, fatty acid esters, fatty acid alcohols, sulfoxides, essential oils, terpenes, oxazolidines, surfactants, polyols, azone and derivatives, and epidermal enzymes.
[0014] The mechanisms by which MPE™s reduce the skin barrier function are not well understood {see Williams and Barry "Penetration Enhancers" Advanced Drug Delivery Reviews 56: 603-618 (2004)), although it has been proposed that the mechanisms can be grouped into three broad categories: lipid disruption, increasing comeocyte permeability, and promoting partitioning of the drug into the tissue.
[0015] The challenge with use of MPE™s is that few seem to induce a significant or therapeutic enhancement of drug transport at tolerable levels. This is because an MPE™'s disruption of the skin barrier can potentially cause skin irritation. With increased disruption, skin irritation is expected to become a greater issue. This is particularly problematic with topical OA treatments where the goal is to have the active penetrate deeply into joint tissue and where the drug must be used on a long-term basis due to the nature of the disease.
[0016] The formation of a eutectic mixture is often undesired, but it can be a useful property of drug combinations or formulations. Although formulations comprising eutectic mixtures have been viewed as unstable, the melting point depression of a eutectic mixture can be accompanied by an increase in the mixture's dermal solubility and transdermal permeation, which can increase a drug's bioavailability. See, e.g., Barry, B. W. Eur. J.
Pharm. Sci. 2001, 14, 101-114; Benson, H. A. E. Curr. Drug Del. 2005, 2, 23-33; and U.S. Patent No. 6,368,618.
[0017] One example of using eutectic mixtures to enhance topical drug delivery is the eutectic mixture of the local anesthetics lidocaine and prilocaine (EMLA). See U.S. Patent No. 4,529,601. EMLA's lidocaine-prilocaine binary eutectic system is said to produce high thermodynamic activity and a high driving force for transdermal permeation of lidocaine.
[0018] In light of the foregoing, there is a considerable need for the development of topical NSAED formulations suitable for long-term use in the treatment of OA, and especially for topical formulations containing coxibs. The challenge has been to develop an optimal composition which will deliver the active agent to the underlying tissue in sufficient concentration to treat OA on a long-term basis, while reducing or minimizing the incidence of intolerable skin irritation caused by disrupting the skin barrier and while providing a composition and dosage that leads to and encourages patient compliance. The present invention seeks to satisfy these and other needs.
BRIEF SUMMARY OF THE INVENTION
[0019] The present invention provides pharmaceutical compositions, methods for preparation, and methods of treatment comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water. In a preferred embodiment, the selective COX-2 inhibitor is etoricoxib. The compositions enhance permeability and bioavailability, and they are useful for topical treatment of pain, inflammation, or both. In a preferred embodiment, the method of treatment is directed to pain associated with OA.
[0020] As such, in one embodiment, the present invention provides a pharmaceutical composition for topical administration, the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water. In a preferred aspect, the composition comprises 0.1% to 5% (w/w) etoricoxib, 3% to 21% (w/w) of a eutectic melt consisting essentially of choline and urea at least one lower alcohol, and water. More preferably, the composition is a gel. Still more preferably, the composition is a low- viscosity gel. [0021] In a more preferred aspect, the composition comprises 1% to 3% (w/w) of a selective COX-2 inhibitor. Still more preferably, the composition comprises 1% (w/w) of a selective COX-2 inhibitor. Alternatively, the composition comprises 2% (w/w) of a selective COX-2 inhibitor. Alternatively or yet still more preferably, the selective COX-2 inhibitor is etoricoxib. [0022] In another more preferred aspect, the eutectic melt consists essentially of choline (e.g., a choline salt) and urea. Still more preferably, the eutectic melt or the pharmaceutical composition comprises from a 3:1 ratio to a 1 :2 ratio of urea to choline. In one aspect, the eutectic melt comprises a 2:1 ratio of urea to choline. Alternatively, the composition comprises 2.5% (w/w) of choline. Alternatively or more preferably, the composition comprises 5% (w/w) urea.
[0023] In yet another preferred aspect, the composition comprises ethanol and at least a second lower alcohol. More preferably, the second lower alcohol is a member selected from isopropanol and 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol®). Alternatively, the
composition comprises 3% to 10% (w/w) of the second lower alcohol. More preferably, the composition comprises 10% (w/w) of the second lower alcohol. Still more preferably, the composition further comprises 3% to 10% (w/w) of a third lower alcohol. Yet still more preferably, the composition comprises 10% (w/w) of the third lower alcohol. Alternatively or
more preferably, at least one, at least two, or at least three of the lower alcohols are monohydric alcohols.
[0024] In another aspect, the composition comprises at least one molecular penetration enhancer. More preferably, the molecular penetration enhancer is a fatty acid ester or a terpene. Still more preferably, the composition comprises about 0.1 to 10% (w/w), about 3% to 5% (w/w), or about 3% to 10% (w/w) of the fatty acid ester. Yet still more preferably, the fatty acid ester is selected from glycerol monolaurate and isopropyl myristate. Alternatively, the composition comprises 3% to 5% (w/w) of the terpene. More preferably, the terpene is limonene. Alternatively or yet still more preferably, the composition further comprises a second molecular penetration enhancer; more preferably, 3% to 5% (w/w) or from 3% to 10% (w/w) of the second molecular penetration enhancer.
[0025] In still yet another preferred aspect, the composition further comprises an alpha- hydroxy acid. More preferably, the composition comprises 2% (w/w) of the alpha-hydroxy acid. Alternatively or still more preferably, the alpha-hydroxy acid is lactic acid. [0026] In another preferred aspect, the composition further comprises a fatty acid ester.
More preferably, the composition comprises about 0.1 to 10% (w/w), about 3% to 5% (w/w), or about 3% to 10% (w/w) of the fatty acid ester. Alternatively or still more preferably, the fatty acid ester is selected from glycerol monolaurate and isopropyl myristate.
[0027] In still another preferred aspect, the composition further comprises a nonionic surfactant. More preferably, the composition comprises 1.5 to 5% (w/w) of the nonionic surfactant, and still more preferably 3% (w/w). Alternatively or yet still more preferably, the nonionic surfactant is polysorbate 20 (Tween® 20).
[0028] In yet another preferred aspect, the composition further comprises a thickening agent. More preferably, the composition comprises 2% (w/w) of the thickening agent. Still more preferably, the thickening agent is a cellulosic thickening agent, and yet still more preferably, the thickening agent is hydroxypropyl cellulose.
[0029] In one preferred embodiment, the present invention provides a pharmaceutical composition for topical administration, the composition consisting of, consisting essentially of, or comprising etoricoxib, urea, choline, a lower alcohol, and water; wherein choline and urea form a eutectic melt. In one aspect, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially eutectic. More preferably, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic.
[0030] In another preferred embodiment, the present invention provides a pharmaceutical composition for topical administration, the composition comprising etoricoxib, urea, choline, a lower alcohol and water; the composition prepared according to the method of: (a) mixing urea and choline together and, optionally, heating to form a eutectic melt; (b) mixing the etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) adding the lower alcohol and water to the composition of (b) and mixing thoroughly. In one aspect, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially eutectic. More preferably, the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic. [0031] In still another preferred embodiment, the present invention provides a
pharmaceutical composition for topical administration, the composition consisting of, consisting essentially of, or comprising 1% to 2% (w/w) etoricoxib, 5% (w/w) urea, 2.5% (w/w) choline, at least 3% (w/w) of at least one molecular penetration enhancer, 10% (w/w) isopropanol, and water. [0032] In yet another embodiment, the present invention provides a method for topically treating pain in a subject, the method comprising topically applying a pharmaceutical composition to treat pain in the subject; the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one lower alcohol, and water. More preferably, the composition comprises 0.1% to 5% (w/w) etoricoxib, 3% to 21% (w/w) of a eutectic melt consisting essentially of or comprising choline and urea, at least one molecular penetration enhancer, at least one lower alcohol, and water. Alternatively or more preferably, the pain is associated with OA.
[0033] In yet still another embodiment, the present invention provides a use of a composition in the manufacture of a medicament for the topical treatment of pain; the composition consisting of, consisting essentially of, or comprising a selective COX-2 inhibitor, a eutectic melt, at least one molecular penetration enhancer, at least one lower alcohol, and water. More preferably, the composition comprises 0.1% to 5% (w/w) etoricoxib, about 3.0% to 21% (w/w) of a eutectic melt consisting essentially of or comprising choline and urea, at least one lower alcohol, and water. Alternatively or more preferably, the pain is associated with OA. Preferably, the composition further comprises at least one molecular penetration enhancer.
[0034] These and other objects, aspects and embodiments and will become more apparent when read with the following detailed description and drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
[0035] FIG. 1 illustrates etoricoxib permeation through porcine skin from a first series of topical formulations (Table 1) at 4, 20, and 24 hours after application (25 μΐ dosing).
[0036] FIG. 2 illustrates etoricoxib permeation through porcine skin from a second series of topical formulations (Table 2) at 4, 21, and 26 hours after application (25 μΐ dosing).
[0037] FIG. 3 illustrates etoricoxib permeation through porcine skin from a third series of topical formulations (Table 3) at 4, 21, and 24 hours after application (25 μΐ dosing).
[0038] FIG. 4 illustrates etoricoxib permeation through porcine skin from a fourth series of topical formulations (Table 4) at 4, 21, and 26 hours after application (25 μΐ dosing). [0039] FIG. 5 illustrates etoricoxib permeation through porcine skin from a fifth series of topical formulations (Table 5) at 4, 21, and 26 hours after application (50 μΐ dosing).
[0040] FIG. 6 illustrates etoricoxib permeation through porcine skin from a sixth series of topical formulations (Table 6) at 4, 21, and 26 hours after application ( 25 μΐ dosing).
[0041] FIG. 7A illustrates etoricoxib permeation through porcine skin from a seventh series of topical formulations (Table 7) at 4, 21, and 26 hours after application. FIG. 7B illustrates skin retention of etoricoxib from the seventh series of topical formulations (Table 7) after 26 hours of application (25 μΐ dosing).
[0042] FIG. 8 illustrates etoricoxib permeation through porcine skin from an eighth series of topical formulations (Table 8) at 4, 21, and 26 hours after application (10 and 5 μΐ dosing). [0043] FIG. 9 illustrates etoricoxib permeation through porcine skin from a ninth series of topical formulations (Table 9) at 4, 21, and 24 hours after application (25 μΐ dosing).
[0044] FIG. 10 illustrates etoricoxib permeation through porcine skin from a tenth series of topical formulations (Table 10) at 4, 21, and 24 hours after application (10 μΐ dosing).
[0045] FIG. 11 illustrates etoricoxib permeation through porcine skin from an eleventh series of topical formulations (Table 11) at 4, 21, and 26 hours after application (10 μΐ dosing).
[0046] FIG. 12 illustrates etoricoxib permeation through human cadaver skin from a twelfth series of topical formulations (Table 12) at 4, 8, 12, 16, 20, and 24 hours after application (5 μΐ dosing).
[0047] FIG. 13 illustrates etoricoxib permeation through human cadaver skin from a thirteenth series of topical formulations (Table 13) at 4, 8, 14, 24, 36, and 48 hours after application (5 μΐ dosing).
[0048] FIG. 14 illustrates etoricoxib permeation through human cadaver skin from the fourteenth series of topical formulations (Table 14) at 4, 8, 14, 24, 36, and 48 hours after application (5 μΐ dosing).
[0049] FIG. 15 illustrates etoricoxib permeation through human cadaver skin from a fifteenth series of topical formulations (Table 15) at 4, 8, 14, 24, 36, and 48 hours after application (5 μΐ dosing). DETAILED DESCRIPTION OF THE INVENTION
I. Definitions
[0050] The terms "a," "an," and "the" as used herein not only include aspects with one member, but also include aspects with more than one member. For example, an embodiment including "a cellulosic thickening agent and a lower alcohol" should be understood to present certain aspects with two or more cellulosic thickening agents, two or more lower alcohols, or both.
[0051] The term "about" as used herein includes a close (i.e., narrow) range around the explicit value for a variable. For example, in certain instances the term about includes 5 %- 10% higher or 5-10% lower than the value given. For example, "about 10" includes the range of values from 9.5 to 10.5 or from 9 to 11.
[0052] When "about" is applied to the beginning of a numerical range, it applies to both ends of the range. Thus, "from about 5 to 20%" is equivalent to "from about 5% to about 20%o." When "about" is applied to the first value of a set of values, it applies to all values in that set. Thus, "about 7, 9, or 11%" is equivalent to "about 7%, about 9%, or about 1 [0053] In compositions comprising an "additional" or "second" component, the second component as used herein is chemically different from the other components or first component. A "third" component is different from the other, first , and second components, and further enumerated or "additional" components are similarly different.
[0054] The term "agent" as used herein indicates a compound or mixture of compounds that, when added to a pharmaceutical composition, tend to produce a particular effect on the composition's properties. For example, a composition comprising a thickening agent is likely
to be more viscous than an otherwise identical comparative composition that lacks the thickening agent.
[0055] "Alpha-hydroxy acid" as used herein indicates an organic compound comprising at least one carbon substituted with a hydroxyl group and a carboxylic acid group (i.e., a C(OH)(C02H) group). Examples of alpha-hydroxy acids include citric acid, glycolic acid, aldonic acids (e.g., gluconic acid), 2-hydroxycaproic acid, 2-hydroxycaprylic acid, 2- hydroxypropionic acid, lactic acid, malic acid, mandelic acid, tartaric acid, and the like, as well as mixtures thereof.
[0056] "Cellulosic thickening agent" as used herein includes a thickening agent that is a natural or synthetic polymeric carbohydrate (e.g., cellulose, pharmaceutically acceptable vegetable gums) or a polymeric or oligomeric derivative of a polymeric carbohydrate that is produced by chemical modification (e.g., hydroxypropyl cellulose, hydroxypropyl methyl cellulose, hydroxyethyl cellulose). Representative cellulosic thickening agents include cellulose, hydroxypropyl cellulose ("HPC"), hydroxypropyl methyl cellulose, hydroxyethyl cellulose, methyl cellulose, carboxymethyl cellulose, and the like.
[0057] In general, chiral compounds described herein (e.g., lactic acid), include the racemic form or the D- or L- enantiomer thereof (e.g., D-lactic acid or L-lactic acid).
[0058] "Choline" as used herein indicates a compound consisting essentially of the cation (2-hydroxyethyl)trimethylammonium and an accompanying anion. Examples of "choline" include (2-hydroxyethyl)trimethylammonium chloride, (2-hydroxyethyl)trimethylammonium hydroxide, (2-hydroxyethyl)trimethylammonium tartrate, and the like. In a preferred aspect, choline is (2-hydroxyethyl)trimethylammonium chloride. Where specific weight percentages or weight ratios are provided in this application, they are expressed in terms of (2- hydroxyethyl)trimethylammonium chloride . [0059] "Enhancement ratio" ("ER") as used herein is the ratio of a test result (e.g. , ug/cm2 accumulated dose of product) from a formulation comprising a selective COX-2 inhibitor (e.g., etoricoxib) to the corresponding test result from a hydroalcoholic control solution comprising the same selective COX-2 inhibitor at the same concentration.
[0060] In general, the "error bars" on the graphs provided in the figures represent the standard error of the mean value, whereas the top of the solid, shaded, or patterned bar represents a single data value, which is the mean value of the distribution of data values.
[0061] "Eutectic agent" as used herein includes a compound that, in combination with at least a second eutectic agent, forms a eutectic melt. Representative eutectic agents include menthol, phenol, 2-amino-2-methylpropanol, choline chloride, urea, panthenol, niacinamide, citric acid, betaine, arginine, resorcinol, butylated hydroxytoluene, 4-chloroxylenol, camphor, glycerin monolaurate, lauryl alcohol, certain pharmaceutical agents (e.g., allylamine antifungal agents such as terbinafme and butenafine; local anesthetics such as lidocaine and prilocaine), and the like.
[0062] In certain aspects, if a mixture of compounds A and B or compounds B and C can form a eutectic melt, compounds A, B, and C can each be a eutectic agent as defined herein even if compounds A and C cannot be combined with each other to form a eutectic melt. However, if a compound is a eutectic agent in a specific embodiment of a composition, it is able to form a eutectic melt with at least a second compound contained in that embodiment of the composition.
[0063] "Eutectic melt" as used herein includes a combination of two or more eutectic agents that are solids before they are combined, but that when combined, form a substantially homogeneous liquid that is stable at one or more temperatures between about 10 °C and about 100 °C (e.g., between about 10 °C, 15 °C, 20 °C, 25 °C, 30 °C, 35 °C, 40 °C, 45 °C, 50 °C, 55 °C, 60 °C, 65 °C, 70 °C, 75 °C, 80 °C, 85 °C, 90 °C, 95 °C, 100 °C, and the temperatures in between). The eutectic melt is further characterized in that the melting point of the eutectic melt is lower than the individual melting points of the eutectic agents that comprise the melt. In one embodiment, the eutectic melt comprises two eutectic agents, that when combined, form a substantially homogeneous liquid, the eutectic melt having a melting point that is lower than that of both of the eutectic agents when in purified or non-mixed form. [0064] In certain aspects, at the melting point of a eutectic melt, both the solid and liquid phases have about the same proportional composition of eutectic agents.
[0065] In certain aspects, the melting point of a eutectic melt is lower than the melting point of other mixtures containing the same components, but with different proportions of the same eutectic agents. [0066] In certain aspects, at least one of the liquid components of the eutectic melt is a solid when in purified or non-mixed form at the same temperature, and the component's change from solid to liquid state is caused by its melting point depression upon mixing with the other eutectic agent. In one embodiment, the eutectic melt comprises two eutectic agents,
both solids in purified or non-mixed form at the same temperature, and the agents' change from solid to liquid state is caused by their melting point depression upon mixing.
[0067] In certain aspects, the melting point of at least one, more than one, or all of the eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C. In one embodiment, the melting point of all of the eutectic agent components of the eutectic melt is greater than 10 °C. In another embodiment, the melting point of the eutectic melt is lower than 24 °C, and the melting point of at least one, more than one, or all of the eutectic agent components of the eutectic melt is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C. In a preferred
embodiment, the melting point of two eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C, and the melting point of a eutectic melt comprising the two eutectic agents is greater than 10 °C. In another preferred embodiment, the melting point of a eutectic melt comprising two eutectic agents is lower than 24 °C, and the melting point of the two eutectic agents in purified or non-mixed form is greater than 24 °C, 30 °C, 36 °C, 40 °C, or 50 °C.
[0068] In certain embodiments, a mixture is heated until the components are liquid or substantially liquid to form a eutectic melt. In still other embodiments, a mixture is heated to a temperature above 50 °C to form a eutectic melt (e.g., 60 °C, 65 °C, 70 °C, 70 °C, 80 °C, 85 °C, 90 °C, 95 °C, or 100 °C).
[0069] "Finite dosing" as used herein generally includes an application of a limited reservoir of an active agent. The the active agent in the reservoir is depleted with time, leading to a tapering off of the absorption rate of the active agent after a maximum absorption rate is reached.
[0070] "Formulation," "pharmaceutical composition," and "composition" as used herein are equivalent terms referring to a composition of matter suitable for pharmaceutical use.
[0071] "Infinite dosing" as used herein generally includes an application of a large reservoir of an active agent. The active agent in the reservoir is not significantly depleted with time, thereby providing protracted, continuous, steady-state absorption of the active.
[0072] "Lower alcohol" as used herein includes straight- or branched-chain alkyl alcohols of 1 to 6 carbon atoms. Representative lower monohydric alcohols include methanol, ethanol, n-propanol, isopropanol (i.e., isopropyl alcohol or ΓΡΑ), n-butanol, t-butanol, n- pentanol, 3-pentanol, 2-methoxyethanol, propylene glycol, and the like.
[0073] "Melt composition" as used herein includes a mixture of at least two compounds.
[0074] The "melting point" as used herein is the temperature at which a solid changes from solid to liquid phase under standard pressure (i.e., 1 atm or 101.325 kPa). If a solid melts over a temperature range, the melting point is the temperature at which the liquid phase first appears under standard pressure. [0075] "Monohydric alcohol" as used herein includes straight- or branched-chain alkyl alcohols with a single hydroxyl group. Representative monohydric alcohols include methanol, ethanol, n-propanol, isopropanol, n-butanol, t-butanol, n-pentanol, 3-pentanol, 2- methoxyethanol, 2-(2-ethoxyethoxy)ethanol, oleyl alcohol, and the like.
[0076] "Non-eutectic" and "essentially non-eutectic" as used interchangeably herein include compositions that may initially contain or be a eutectic melt (e.g., choline and urea), but when additional components are added, the final composition is not a eutectic melt. For example, mixing urea and chloine and optionally heating can form a eutectic melt. When further components are added, however, the final composition may not be a eutectic melt (i.e., it is non-eutectic). Therefore, a composition can be non-eutectic, but still comprise or once have contained a eutectic melt.
[0077] "Nonionic surfactant" as used herein indicates a surface-active agent that is uncharged under the conditions of the formulation. Examples of nonionic surfactants include the polysorbates (e.g., polysorbate 20) and fatty acid esters (e.g., isopropyl myristate).
[0078] The term "or" as used herein should in general be construed non- exclusively. For example, an embodiment of "a composition comprising A or B" would typically present an aspect with a composition comprising both A and B. "Or" should, however, be construed to exclude those aspects presented that cannot be combined without contradiction (e.g., a composition pH that is between 9 and 10 or between 7 and 8).
[0079] Generally, when a percentage range is taught, it incorporates all full or partial percentages in between (i.e. , within the bounds of the range). For example, a percentage range of 15 to 25% would also teach inter alia the specific values of 17.36% and 21%. A percentage range of about 13% to 17% would also teach inter alia the specific values of 12.97%, 16%, and 17.1%.
[0080] "Penetration enhancer," "molecular penetration enhancer," or "MPE™" as used herein includes an agent or a combination of agents that improves the transport of molecules such as a pharmaceutically or cosmetically active agent into or through a natural membrane such as skin or nail. Various conditions may occur at different sites in the body, either in the
skin or below the skin, creating a need to target delivery of compounds. For example, in a treatment for osteoarthritis, delivery of the active agent to the underlying tissue surrounding the joint may be necessary to achieve therapeutic benefit. An MPE™ may be used to assist in the delivery of an active agent i) directly into the skin, or nail; ii) locally, or regionally, into tissue(s) underlying or near to the skin or nail; or iii) indirectly via systemic distribution to the site of the disease. If systemic distribution of an active agent (e.g., etoricoxib) would be likely to produce side effects [e.g., etoricoxib), an MPE™ is preferably selected to maximize direct delivery and to minimize systemic distribution. An MPE™ may be a pure substance or may comprise, consist essentially of, or consist of a mixture of different chemical entities. [0081] "Ratio", as it pertains to comparative flux values described herein, are calculated based on the cumulative amount of active (e.g., etoricoxib) delivered through the skin over a period of 4-60 hrs, preferably 24 hrs.
[0082] "Selective COX-2 inhibitor" as used herein should in general be construed to mean the selective COX-2 (coxib) class of NSAIDs that preferentially inhibits COX-2, as well as the pharmaceutically acceptable derivatives or salts thereof. By extension, the term
"etoricoxib" as used herein, includes pharmaceutically acceptable derivatives or salts thereof.
[0083] "Substantially" or "essentially" as used interchangeably herein indicates a quality that is considerable and largely characteristic of its subject. In certain instances,
"substantially" referes to a considerable amount, size, or physical state. For example, a golf ball would be a "substantially spherical" object. A "substantially liquid" composition may have up to 5% or even 10% (w/w) of a solid, but generally has the physical properties of an liquid.
[0084] "Substantially anhydrous" and "essentially free of water" as used interchangeably herein include compositions without deliberately added water. A substantially anhydrous composition can contain up to 5% w/w water, which may be adventitiously incorporated from impurities in the starting materials, side products from reactions or manufacturing processes, or air absorption.
[0085] "Substantially homogeneous" as used herein designates a composition that includes at least 90%, 95%, or 99% by weight of a single phase, although it may include small amounts of a different phase (e.g., a liquid phase containing a small amount of immiscible liquid or a solid).
[0086] "Substantially liquid" as used herein designates a composition that includes at least 90%, 95%, or 99% by weight of a liquid phase, although it may include small amounts of at least one solid phase.
[0087] "Substituted phenol" as used herein includes hydroxybenzenes and
dihydroxybenzenes with from 1 to 3 additional substituents independently selected from the group of acyl, alkyl, alkenyl, alkoxy, amido, amino, aryl, carboxy, and halo substituents.
[0088] "Thickening agent" as used herein includes an agent or combination of agents that increases the viscosity of a composition. A thickening agent may be a pure substance, or it may comprise, consist essentially of, or consist of a mixture of different chemical entities. Exemplary thickening agents include cellulose polymers, carbomer polymers, carbomer derivatives, cellulose derivatives, polyvinyl alcohol, poloxamers, polysaccharides, and the like, as well as mixtures thereof.
[0089] "Topical formulation" as used herein includes a composition that is suitable for topical application to the skin, a nail, or a mucosa. A topical formulation may, for example, be used to confer a therapeutic or cosmetic benefit to its user. Specific topical formulations can be used for topical, local, regional, or transdermal application of substances.
[0090] "Transdermal" as used herein includes a process that occurs through the skin. The terms "transdermal," "percutaneous," and "transcutaneous" can be used interchangeably. In certain embodiments, "transdermal" may also include epicutaneous. [0091] "Transdermal application" as used herein includes administration through the skin. Transdermal application can be used for systemic delivery of an active agent; however, it is also useful for delivery of an active agent to tissues underlying the skin with minimal systemic absorption. In certain embodiments, "transdermal application" may also include epicutaneous application. [0092] In general, the unit prefix "u" as used herein is equivalent to "μ" or "micro." For example, "ul" is equivalent to "μΐ" or "microliters."
[0093] "Urea" as used herein includes urea and pharmaceutically acceptable derivatives or salts thereof.
[0094] The term "w/w" or "wt/wt" means a percentage expressed in terms of weight of the ingredient or agent over the total weight of the composition multiplied by 100.
II. Selective COX-2 Inhibitor
[0095] The present invention provides a pharmaceutical composition comprising, consisting essentially of, or consisting of a selective COX-2 inhibitor. In a preferred aspect, the selective COX-2 inhibitor is selected from the group of celecoxib, etoricoxib, lumiracoxib, parecoxib, rofecoxib, valdecoxib, and a combination thereof. More preferably, the selective COX-2 inhibitor is selected from the group of celecoxib, etoricoxib, and rofecoxib. Still more preferably, the selective COX-2 inhibitor is etoricoxib. In another preferred aspect, the pharmaceutical composition comprises 0.1% to 5% (w/w) of etoricoxib, more preferably about 1% to 3% (w/w), and still more preferably about 1 % or 2% (w/w). [0096] In one aspect, a composition permits delivery of a selective COX-2 inhibitor daily dosage of about 0.01 mg to about 120 mg, preferably about 0.1 mg to 60 mg, preferably about 1 mg to about 30 mg, and most preferably about 1 mg to about 10 mg. Yet still more preferably, the formulation permits delivery of a daily dosage of about 3 mg. Preferably the concentration is such that this dosage amount can be provided by application of the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm2, preferably about 1200 to 1800 cm2 (750 cm2/knee). Alternatively, the
2 2
composition can be applied to a skin area of about 1 to 50 cm , about 50 to 250 cm , about 100 to 500 cm2, about 200 to 800 cm2, or about 800 to 1200 cm2.
[0097] A person skilled in the art will appreciate that the dosage and application area will vary on and can be tailored to the area being treated (e.g., knees, fingers, toes, back, and the like). In one preferred aspect, a single knee is treated and the application area is about 750 cm2. In another preferred aspect, both knees of an individual are treated and the application area is about 1500 cm (about 750 cm per knee).
[0098] In another aspect, the formulation of the present invention provides a total or a systemic dose that is less than 50% of the systemic daily dose of the maximum approved oral dose; preferably less than 25%, more preferably less than 10%, and most preferably less than 5%, yet provides local or regional delivery levels sufficient for therapeutic benefit.
Preferably the concentration is such that this dosage amount can be provided by application of the composition from one to four times a day, preferably one to two times a day, to a skin area of up to about 2500 cm2, preferably about 1200 to 1800 cm2 (750 cm2/knee).
* · 2
Alternatively, the composition can be applied to a skm area of about 1 to 50 cm , about 50 to 250 cm2, about 100 to 500 cm2, about 200 to 800 cm2, or about 800 to 1200 cm2.
[0099] In still another aspect, the pharmaceutical composition comprising etoricoxib provides better flux (as determined by the Franz cell procedure, e.g., the method of Example 2) than an analogous comparative formulation comprising a selective COX-2 inhibitor.
Preferably, this comparative formulation comprises etoricoxib. More preferably, the flux of etoricoxib is at least 1.5 times greater than the flux of the comparative formulation's active. In other words, the ratio of (i) the composition's etoricoxib flux to (ii) the comparative formulation's coxib flux is preferably greater than 1.0, and more preferably at least about 1.5.
[0100] Still more preferably, the composition has an etoricoxib flux that is at least 2.0 times greater than the comparative formulation's coxib flux. Yet still more preferably, the composition has an etoricoxib flux that is at least 4.0 times greater than the comparative formulation's coxib flux.
[0101] In an alternative aspect, the composition has a selective COX-2 inhibitor flux equal to or greater than the selective COX-2 inhibitor flux from a known comparative formulation with the same selective COX-2 inhibitor. Preferably, the selective COX-2 inhibitor flux is greater than the flux of the comparative formulation with the same selective COX-2 inhibitor. More preferably, the selective COX-2 inhibitor flux is at least 1.5 times greater than the flux of a comparative formulation with the same selective COX-2 inhibitor. In other words, the ratio of (i) the selective COX-2 inhibitor flux of the composition to (ii) the selective COX-2 inhibitor flux from a comparative formulation with the same selective COX-2 inhibitor is preferably greater than 1.0, and more preferably at least about 1.5.
[0102] Still more preferably, the composition has a selective COX-2 inhibitor flux that is at least 2.0 times greater than the selective COX-2 inhibitor flux from a known comparative formulation with the same selective COX-2 inhibitor. Yet still more preferably, the composition has a selective COX-2 inhibitor flux that is at least 4.0 times greater than the selective COX-2 inhibitor flux from a comparative formulation with the same selective COX- 2 inhibitor.
[0103] In another alternative aspect, the present invention provides a composition comprising etoricoxib and having an etoricoxib flux (as determined by the Franz cell procedure of Example 2) of at least 0.1 μg/hr/cm2 at 24 hours, preferably at least 0.2 μg/hr/cm2 at 24 hours.
[0104] Still more preferably, the composition comprising etoricoxib has an enhancement ratio (ER) of at least 2. Yet still more preferably, the composition comprising etoricoxib has
an ER of at least 5.0. Yet still more preferably, the composition comprising etoricoxib has an ER that is at least 10.0.
III. Eutectic Melt
[0105] In one aspect, the pharmaceutical composition comprises a eutectic melt.
Preferably, the eutectic melt comprises, consists essentially of, or consists of choline and urea. More preferably, the eutectic melt or the pharmaceutical composition comprises from a 3: 1 ratio to a 1 :2 ratio of urea to choline. Even more preferably, the eutectic melt comprises a 1 : 1 or 2: 1 ratio of urea to choline. Alternatively, the composition comprises about 3.0% to 21% (w/w) of a eutectic melt. Preferably, the composition comprises about 2% to 10%> (w/w) of urea and about 1 % to 10% (w/w) of choline (e.g., a. choline salt). More preferably, the composition comprises about 2.5% (w/w) of choline and about 5% (w/w) urea.
[0106] Adding choline and urea separately to the formulation may not result in a homogeneous composition (e.g. , Example 10). Surprisingly, a homogeneous composition can be prepared by first combining choline and urea in a eutectic melt and then adding additional components of the composition. Accordingly, in one preferred embodiment, the present invention provides a method of preparing a pharmaceutical composition comprising the steps of: (a) mixing urea and choline together and, optionally, heating to form a eutectic melt; (b) mixing etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) adding a lower alcohol and water to the composition of (b) and mixing thoroughly.
Alternatively, the steps of combining ingredients can be suitably rearranged, so long as preparing a eutectic melt comprises one step.
[0107] In certain aspects, at the melting point of a eutectic melt, both the solid and liquid phases have about the same proportional composition of eutectic agents.
[0108] [0109] In still other aspects, at least one of the liquid components of the eutectic melt is a solid when in purified or non-mixed form at the same temperature, and the component's change from solid to liquid state is caused by its melting point depression upon mixing with the other component or components.
[0110] In yet still other aspects, the melting point of at least one, more than one, or all of the eutectic agents is greater than 24 °C, 30 °C, 36 °C, 40 °C, 50 °C, 100 °C, or 150 °C.
Alternatively, the melting point of all of the eutectic agent components of the eutectic melt is greater than 10 °C. More preferably, they are greater than 50 °C, and still more preferably,
they are greater than 100 °C. Alternatively, the melting point of the eutectic melt is lower than 24 °C, and the melting point of at least one, more than one, or all of the eutectic agent components of the eutectic melt is greater than 24 °C, 30 °C, 36 °C, 40 °C, 50 °C, or 100 °C.
[0111] In certain embodiments, a mixture is heated until the components are liquid or substantially liquid to form a eutectic melt. In still other embodiments, a mixture is heated to a temperature above 50 °C to form a eutectic melt (e.g., 60 °C, 65 °C, 70 °C, 70 °C, 80 °C, 85 °C, 90 °C, 95 °C, 100 °C, 115 °C, or 130 °C).
IV. Molecular Penetration Enhancer
[0112] In one embodiment, the composition further comprises at least one molecular penetration enhancer. In certain aspects, the molecular penetration enhancer is selected from terpenes, fatty acid esters, and fatty acid alcohols. More preferably, the molecular penetration enhancer is a terpene. Examples include D-limonene, limonene oxide, geraniol, a-pinene, -pinene oxide, thymol, menthone, menthol, neomenthol, 3-carene, L-cavol, carvone, carveol, 1,8-cineole (eucalyptol), citral, dihydrocarveol, dihydrocarvone, 4- terpinenol, fenthone, menthone, pulegone, pulegol, isopulegol, piperitone, camphor, a- terpineol, terpinen-4-ol, linalool, carvacrol, trans-anethole, ascaridole, safrole, racemic mixtures thereof (e.g., DL-limonene), and pharmaceutically acceptable isomers thereof. In certain preferred aspects, a second molecular penetration enhancer can be present (e.g., a fatty acid ester and a terpene). [0113] In one specific embodiment, the composition of the present invention comprises limonene. In one aspect, the composition comprises about 0.1% to 5% (w/w) of limonene or geraniol, such as about 0.1 , 0.5, 1, 2, 3, 4 or 5% (w/w), and more preferably about 3% to 5% (w/w).
[0114] In certain aspects, the terpene molecular penetration enhancer can be included within an essential oil. Essential oils that include a substantial proportion of at least one terpene molecular penetration enhancer include oils of peppermint, eucalyptus, chenopodium, anise, and yling-yling.
[0115] In addition, or alternatively, a fatty acid ester or fatty alcohol ester is used as a an MPE™ in the composition. Examples of preferred fatty acid ester MPE™s are glyceryl monoesters and isopropyl myristate. More preferably, the MPE™ is glyceryl monolaurate. Still more preferably, the MPE™ is isopropyl myristate . In one aspect, the composition comprises about 0.1% to 10% (w/w) of the fatty acid ester or fatty alcohol ester , such as
about 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10% (w/w). Preferably, the composition comprises about 1 to 3% (w/w) (e.g. about 1%; about 3% (w/w)), about 0.1% to 10% (w/w), or about 3 to 5% (w/w).
[0116] Other examples of fatty acid esters include butyl acetate, caproyl glycolate, cetyl lactate, cocoyl glycolate, decyl N,N-dimethylamino acetate, decyl N,N-dimethylamino isopropionate, diethyleneglycol oleate, diethyl sebacate, diisopropyl sebacate, dodecyl N, V- dimethylamino acetate, dodecyl N,N-dimethylamino butyrate, dodecyl N,N-dimethylamino isopropionate, dodecyl 2-(N,N-dimethylamino)propionate, EO-5-oleyl ester, ethyl acetate, ethyl acetoacetate, ethyl propionate, glyceryl dilaurate, glyceryl dioleate, glycerol monoethers, glycerol monooleate, glycerol monolinoleate, isopropyl isostearate, isopropyl laurate, isopropyl linoleate, isopropyl palmitate, isostearoyl glycolate, lauroyl glycolate, methyl acetate, methyl caprate, methyl laurate, methyl oleate, methyl propionate, methyl valerate, 1-monocaproyl glycerol, medium-chain-length monoglycerides, benzyl or substituted benzyl nicotinate, octyl acetate, octyl N,N-dimethylamino acetate, oleyl oleate, n- pentyl N-acetylprolinate, propylene glycol monolaurate, sodium lauroyl glycolate, tetradecyl N,N-dimethylamino acetate, tromethamine lauroyl glycolate, and the like. Still other examples include sunscreens such as Padimate-O, homosalate, cinnamate esters, octocrylene, and the like.
[0117] Other MPE™s include fatty acids, lactic acid, fatty alcohols (e.g., oleyl alcohol, stearyl alcohol, decanol), fatty alcohol ethers, hexahydro-l-dodecyl-2H-azepin-2-one (e.g., laurocapram, Azone™) and derivatives thereof, dimethylsulfoxide (DMSO) and related sulfoxides (e.g., n-decyl methylsulfoxide), salicylic acid and alkyl esters thereof (e.g., methyl salicylate), Ν,Ν-dimethylacetamide, dimethylformamide, Ν,Ν-dimethyltoluamide, 2- pyrrolidinone and N-alkyl derivatives thereof (e.g., N-methyl-2-pyrrolidone (NMP) and N- octyl-2-pyrrolidinone), and 2-nonyl-l,3-dioxolane. See Osborne, D.W.; Henke, J. J. "Skin Penetration Enhancers Cited in the Technical Literature," Pharmaceut. Tech. 58-66 (Nov. 1997).
V. Lower Alcohol
[0118] In one preferred aspect, the composition comprises a mixture of a lower alcohol and water. More preferably, the lower alcohol is a monohydric lower alcohol, and still more preferably, the lower alcohol is ethanol, isopropanol, or 2-(2-ethoxyethoxy)ethanol (i.e., Transcutol®). In certain aspects, the composition comprises a second lower alcohol. In certain other aspects, the composition comprises a third lower alcohol.
[0119] In another aspect, the composition comprises at least about 3, 5, 7, 9.5, 10, 10.5, 11, 11.5, 12, 14, 15, 20, 25, 30, 31, 31.5, 32, 32.5, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol. More preferably, the composition comprises at least about 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol. Still more preferably, the composition comprises at least about 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
[0120] In other aspects, the composition comprises at most about 3, 5, 7, 9.5, 10, 10.5, 11, 11.5, 12, 14, 15, 20, 25, 30, 31, 31.5, 32, 32.5, 33, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol. More preferably, the composition comprises at most about 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 44.5, 45, 46, 46.5, 47, 47.5, 48, 48.5, 49, 49.5, 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol. Still more preferably, the composition comprises at most about 50, 50.5, 51, 51.5, 52, 52.5, 53, 53.5, 54, 54.5, 55, 55.5, 56, 56.5, 57, 58, 59, 60, 61, 62, 63, 64, ,65, 66, 67, 68, 69, 70, 71, 72, 73, 74, or 75% (w/w) of a lower alcohol.
[0121] In another aspect, the composition comprises 35 to 75% (w/w) of a lower alcohol. In still another aspect, the composition comprises the same or differing amounts of a first and at least one additional lower alcohol. More preferably, the composition comprises 40% to 65 % (w/w) of ethanol and 10% (w/w) of isopropanol. In another more preferred aspect, the composition comprises 40% to 65% (w/w) of ethanol, 10% (w/w) of isopropanol, and 3% to 10% (w/w) of 2-(2-ethoxyethoxy)ethanol. In certain instances, the first alcohol is about 35 to 75% (w/w), the at least one additional alcohol is between about 0.1 to 10% (w/w) (e.g., about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10% (w/w)) and the at least one more additional alcohol is between about 0.1 to 10 % (w/w) (e.g., about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10% (w/w); about 35% to 95% (w/w) total alcohol). In one aspect, the first alcohol is about 35 to 75% (w/w), and the second lower alcohol is about 3% to 10%(w/w). In certain aspects, the composition further comprises about 3 to 10% (w/w) of a third lower alcohol.
9
[0122] In another aspect, the lower alcohol is a diol. Alternatively, the composition further comprises a diol. Suitable diols include, but are not limited to, propylene glycol, butanediol, butynediol, pentanediol, hexanediol, octanediol, neopentyl glycol, 2-methyl-l,3-propanediol, diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, dibutylene glycol, propylene glycol, and a combination thereof. In one aspect, the formulation comprises about 0.1% to 15% (w/w) of propylene glycol, such as about 0.1, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15% (w/w), and preferably about 0.1 to 5% (w/w).
VI. Other Components
[0123] In yet another aspect, the composition further comprises at least one
pharmaceutically acceptable surfactant. Preferably, the surfactant is a nonionic surfactant. More preferably, the surfactant is a polysorbate surfactant. Still more preferably, the surfactant is polysorbate 20.
[0124] Other nonionic surfactants include (but are not limited to) cetomacrogol 1000, cetostearyl alcohol, cetyl alcohol, cocoamide diethanolamine, cocoamide monoethanolamine, decyl glucoside, glyceryl laurate, lauryl glucoside, polyoxyethylene ethers of fatty acids such as cetyl alcohol or stearyl alcohol, narrow-range ethoxylates, octyl glucoside, oleyl alcohol, poloxamers, polyethylene glycol, sorbitan monolaurate, polyoxyethylene sorbitan
monolaurate, sorbitan dioleate, sorbitan trilaurate, sorbitan monopalmitate, polyoxyethylene (20) sorbitan monopalmitate, sorbitan monostearate, sorbitan tristearate, polyoxyethylene (20) sorbitan monostearate, sorbitan monooleate, sorbitan trioleate, polyoxyethylene (20) sorbitan monooleate, stearyl alcohol, sucrose coconut fatty ester mixtures, and sucrose monolaurate.
[0125] In still yet another aspect, the composition further comprises at least one thickening agent, preferably a cellulosic thickening agent. Suitable cellulosic thickening agents include, but are not limited to, hydroxypropyl cellulose (HPC) of various grades, hydroxypropyl methyl cellulose, hydroxyethyl cellulose, hydroxyethyl methyl cellulose, ethyl cellulose, methyl cellulose, carboxymethyl cellulose, dextran, guar gum, pectin, starch, cellulose, and the like. More preferably, the cellulosic thickening agent is HPC.
[0126] In an alternative or preferred aspect, the composition comprises about 1% to 5% (w/w) of a cellulosic thickening agent, such as about 1, 2, 3, 4, or 5% (w/w). More preferably, the composition comprises from 1% to 2% (w/w) of a cellulosic thickening agent. Still more preferably, the composition comprises 1% (w/w) of a cellulosic thickening agent. Alternatively, the composition comprises ?% (w/w) of a cellulosic thickening agent.
[0127] In one aspect, the composition further comprises an anti-oxidant. Preferred antioxidants for use in the present invention include butylated hydroxytoluene, butylated hydroxyanisole, ascorbyl linoleate, ascorbyl dipalmitate, ascorbyl tocopherol maleate, calcium ascorbate, carotenoids, kojic acid and its pharmaceutically acceptable salts, thioglycolic acid and its pharmaceutically acceptable salts (e.g., ammonium), tocopherol, tocopherol acetate, tocophereth-5, tocophereth-12, tocophereth-18, tocophereth-80, and the like.
[0128] In still another aspect, the composition further comprises a chelating agent.
Preferred chelating agents include ethylenediamine tetraacetic acid (EDTA), diamnionium EDTA, dipotassium EDTA, calcium disodium EDTA, H-EDTA, tetraethylammonium (TEA- ) EDTA, tetrasodium EDTA, tnpotassium EDTA, tnsodium phosphate, diamnionium citrate, galactaric acid, galacturonic acid, gluconic acid, glucuronic acid, humic acid, cyclodextrin, sodium citrate, potassium citrate, the sodium salt of ethylenediamine -tetra (methylene phosphonic acid) (EDTMP), potassium EDTMP, and the like. [0129] In certain preferred aspects, the compositions of the invention optionally include a buffer or a pH-adjusting agent (e.g., in addition, the topical formulations of the present invention can also comprise a pH-adjusting agent). In one particular embodiment, the pH- adjusting agent is a base. Suitable pH-adjusting bases include bicarbonates, carbonates, hydroxides (such as alkali or alkaline earth metal hydroxide as well as transition metal hydroxides), and the like. In an alternative aspect, suitable pH-adjusting bases include amines, such as diethanolamine, triethanolamine, or aminopropanol; bicarbonates;
carbonates; and hydroxides, such as ammonium hydroxide, alkali or alkaline earth metal hydroxide, or transition metal hydroxides. Alternatively, the pH-adjusting agent can also be an acid, an acid salt, or mixtures thereof. [0130] The pH adjusting agent can be present in an amount sufficient to adjust the pH of the composition to between about pH 4.0 to about 10.0, more preferably about pH 7.0 to about 9.5. In certain embodiments, the unadjusted pH of the admixed components is between 8 and 10, such as 9, without the need for the addition of any pH adjusting agents.
[0131] Preferably, the pH-adjusting agent is sodium hydroxide, hydrochloric acid, or a combination of both, and is present in an amount sufficient to adjust the pH of the
composition to between about pH 4.0 to 8.5, more preferably, to between about pH 5.5 to 7.0, such as about 6.0 or 6.5. Even more preferably, the pH is adjusted to about 4.0, 4.2, 4.4, 4.6,
4.8, 5.0, 5.2, 5.4, 5.6, 5.8, 6.0, 6.2, 6.3, 6.4, 6.6, 6.8, 7.0, 7.2, 7.4, 7.6, 7.8, 8.0, 8.4, 8.5, or any fraction in-between.
[0132] In certain preferred aspects, a small amount of acid or base is included in the formulation. Non-limiting examples of amounts of acid or base that may be included in the formulation are about 0.000001%, 0.00001%, 0.0001%, 0.001%, 0.0012%, 0.01%, 0.012%, 0.1%, or 1.0%. Preferably, this amount is about 0.0001%. 0.0002%, 0.0003%, 0.0004%, 0.0005%, 0.0006%, 0.0007%, 0.0008%, 0.0009%, 0.0010%, 0.0011%, 0.0012%, 0.0015%, 0.0016%, 0.0017%, 0.0018%, 0.0019%, 0.002%, 0.003%, 0.004%, 0.005%, 0.006%, 0.007%, 0.008%, 0.009%, 0.01%, 0.012%, or 0.02% (w/w). More preferably, this amount is about 0.001%. 0.002%, 0.003%, 0.004%, 0.005%, 0.006%, 0.007%, 0.008%, 0.009%, 0.010%,
0.011%, 0.012%, 0.015%, 0.016%, 0.017%, 0.018%, 0.019%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1% (w/w), or as needed to adjust the formulation to the desired pH.
[0133] Further, the pH-adjusting agent can also be a buffer. Preferably, the pH of the composition of the invention can be adjusted or stabilized with a buffer. Suitable buffers include citrate/citric acid buffers, acetate/acetic acid buffers, phosphate/phosphoric acid buffers, formate/formic acid buffers, propionate/propionic acid buffers, lactate/lactic acid buffers, carbonate/carbonic acid buffers, ammonium/ammonia buffers, and the like. In certain instances, the buffer is an acidic buffer system such as, for example, benzocaine. In more preferred instances, the acidic acid buffer system is citric acid or a citric acid salt.
[0134] In certain preferred aspects, the buffer is present at a concentration of about
0.000001 M, 0.00001 M, 0.0001 M, 0.001 M, 0.0012 M, 0.01 M, 0.012 M, 0.1 M, or 1.0 M. Preferably, this amount is about 0.0010 M, 0.0015 M, 0.002 M, 0.003 M, 0.004 M, 0.005 M, 0.006 M, 0.007 M, 0.008 M, 0.009 M, 0.01 M. 0.012 M, or 0.02 M. Alternatively and preferably, this amount is about 0.001 M. 0.002 M, 0.003 M, 0.004 M, 0.005 M, 0.006 M, 0.007 M, 0.008 M, 0.009 M, 0.010 M, 0.011 M, 0.012 M, 0.015 M, 0.016 M, 0.017 M, 0.018 M, 0.019 M, 0.02 M, 0.025 M, 0.03 M, 0.035 M, 0.04 M, 0.045 M, 0.05 M, 0.055 M, 0.06 M, 0.065 M, 0.07 M, 0.075 M, 0.08 M, 0.085 M, 0.09 M, 0.095 M, or 0.1 M. Alternatively and preferably, this amount is about 0.10 M, 0.11 M, 0.12 M, 0.13 M, 0.14 M, 0.15 M,' 0.16 M, 0.17 M, 0.18 M, 0.19 M, 0.20 M, 0.21 M, 0.22 M, 0.23 M, 0.24 M, 0.25 M, 0.26 M, 0.27 M, 0.28 M, 0.29 M, 0.30 M, 0.31 M, 0.32 M, 0.33 M, 0.34 M, 0.35 M, 0.36 M, 0.37 M, 0.38 M, 0.39 M, 0.40 M, 0.41 M, 0.42 M, 0.43 M, 0.44 M, 0.45 M, 0.46 M, 0.47 M, 0.48 M, 0.49 M, 0.50 M, 0.55 M, 0.60 M, 0.65 M, 0.7 M, 0.75 M, 0.8 M, 0.85 M, 0.9 M, 0.95 M, or 1.0 M.
[0135] In certain preferred aspects, the inventive formulation includes a buffer, and a second pH-adjusting agent (e.g., sodium hydroxide or hydrochloric acid) to adjust the pH of the composition to a desired pH. More preferably, the second pH-adjusting agent comprises two agents (e.g., sodium hydroxide and hydrochloric acid) that are included as needed to adjust the pH of the composition to a desired pH.
[0136] In certain aspects, the composition of the present invention comprises a
preservative, such as propyl paraben or methyl paraben, or combinations thereof. The formulation may be made bacteriostatic by the addition of preservatives. For example, a composition can contain about 0.001 to 8% (w/w); preferably, about 0.01 to 6% (w/w); and more preferably, about 0.05 to 5% (w/w) of a preservative or a combination of preservatives. A variety of preservatives are suitable, including, but not limited to, benzoic acid, benzyl alcohol, benzylhemiformal, benzylparaben, 5-bromo-5-nitro-l,3-dioxane, 2-bromo-2- nitropropane-l,3-diol, butyl paraben, phenoxyethanol, methyl paraben, propyl paraben, diazolidinyl urea, calcium benzoate, calcium propionate, captan, chlorhexidine diacetate, chlorhexidine digluconate, chlorhexidine dihydrochloride, chloroacetamide, chlorobutanol, p- chloro-m-cresol, chlorophene, chlorothymol, chloroxylenol, m-cresol, o-cresol, diethylene glycol dimethyl ether ("DEDM") hydantoin, DEDM hydantoin dilaurate, dehydroacetic acid, dibromopropamidine diisethionate, and l,3-bis(hydroxymethyl)-5,5-dimethylimidazolidine- 2,4-dione ("DMDM") hydantoin. In certain aspects, the formulations herein may be (i) sterile or essentially free from microorganisms such as bacteria and viruses that can cause infection and (ii) optionally preservative-free.
VII. Other Properties
[0137] In still yet another aspect, the composition is selected from the group of a gel, a foam, a cream, an emulsion, a micro emulsion, a lotion, an organogel, an ointment, a solution, and a transdermal patch. More preferably, the composition is a gel, and still more preferably, a low-viscosity gel. Alternatively, the composition is a solution.
[0138] In yet another alternative aspect, the composition is more viscous than water at standard temperature and pressure (STP). Alternatively, the composition has a kinematic viscosity of more than about 1 centistokes (cSt) or a dynamic viscosity of more than about 1 centipoise (cP). In certain aspects, the dynamic viscosity of the composition is at least about 2, 3, 4, 5, 7, 10, 12, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 75, 80, 90, 100, 150, 200, 250, 500, 1000, 2000, 3000, 5000, 10,000 cP at STP. In yet other aspects, the composition is thixotropic (i.e., it decreases in viscosity upon being stirred or shaken). The composition's
viscosity can be adjusted by the addition of a cellulosic thickening agent, such as
hydroxypropyl cellulose, or other thickening agents.
[0139] In another aspect, the composition is acidic. In certain aspects, the composition has a pH of below 7.5, of below 6.5, of below 5.5, of below 4.5, of below 3.5, or of below 2.5. In certain other aspects, the pH of the composition may range from about 1.5 to about 7, about 2 to about 7, about 3 to about 7, about 4 to about 7, or about 5 to about 7. In still other aspects, the pH of the composition may range from about 1.5 to about 5.5, about 2.5 to about 5.5, about 3.5 to about 5.5, or about 4.5 to about 5.5.
[0140] In yet another aspect, the composition is basic. In certain aspects, the composition has a pH of above 7, of above 8, of above 9, of above 10, of above 11, or of above 12. In certain other aspects, the pH of the composition may range from about 7 to about 12.5, about 7 to about 11.5, about 7 to about 10.5, about 7 to about 9.5, or about 7 to about 8.5. In still other aspects, the pH of the composition may range from about 9 to about 12.5, about 9 to about 11.5, about 9 to about 10.5, or about 8.5 to about 10. [0141] In still yet another aspect, the composition is neutral. In certain aspects, the composition has a pH of about 7. In certain other aspects, the composition has a pH from about 6 to about 8.5, from about 5.5 to about 8, about 6 to about 8, about 6.5 to about 8.5, or from about 6.5 to about 7.5.
[0142] In certain other aspects, a composition is designed for high penetration, for high retention in the skin, or for both high penetration and high retention. The optimal composition will have a balance between penetration and retention, enabling an effective amount of the active ingredient to pass through the skin, but also enabling it to stay in the target area for a sufficient duration to alleviate the patient's pain or other symptoms.
[0143] In another aspect, a composition is designed for topical efficacy with minimal systemic distribution of the coxib through the body by the circulatory system ( e.g., the cardiovascular system). Without being bound by theory, it is believed that minimization of systemic distribution would decrease the side effects of the composition, especially the side effect of adverse cardiovascular events. The optimal composition will have low systemic bioavailability, but will effectively treat pain (or other symptoms) associated with the site of application.
[0144] In a preferred aspect, a formulation provides the advantage of favorable stability at six months, as reflected in the lack of any substantial changes in viscosity, the absence of phase separation and crystallization at low temperatures, and a low level of impurities.
[0145] In another preferred aspect, a formulation comprising etoricoxib provides additional advantages in comparison to previously described etoricoxib compositions. Such advantages may include one or more of the following: adhering well to the skin, spreading easily, drying more quickly, and showing greater in vivo absorption. In some more preferred aspects, the drying rate results in a residue of at most 50% of a starting amount after 24 hours. In other more preferred aspects, the transdermal selective COX-2 inhibitor {e.g., and still more preferably, etoricoxib) flux as determined by Franz cell procedure at finite dosing or at infinite dosing is at least 1.5 times that of a comparative liquid formulation.
[0146] Despite the fact that formulations comprising eutectic mixtures have typically been viewed as unstable, the formulations of the instant invention have the advantage of maintaining chemical and/or physical stability over time in accordance with certain aspects of the application. Accordingly, in one embodiment of the invention,provides a composition that remains stable for an acceptable time period between preparation and use when stored in a closed container at normal ambient temperature. Preferably, an "acceptable time period" is at least about 1 day; preferably, at least about 30 days; more preferably, at least about six months; still more preferably, at least about one year; and yet still more preferably, at least about two years.
[0147] In an alternative aspect, the present invention provides a formulation that degrades by less than 1% over the course of 6 months at room temperature. More preferably, the rate of degradation is less than about 0.9, 0.8, 0.7, 0.6, 0.5, 0.4, 0.3, 0.2, or less than 0.1 %, and all fractions in between, over the course of six months at room temperature. VIII. Methods of Preparation
[0148] In one aspect, the pharmaceutical composition is formulated as a cream, an emulsion, a microemulsion, a gel (e.g., a hydrogel, an organogel, or an inorganic or silica gel), a lotion, a lacquer, an ointment, a solution (e.g. , a moderate to highly viscous solution), or a transdermal patch. The pharmaceutical composition may also be prepared so that it may be applied to the skin as a foam. In a preferred aspect, the composition is a gel, and more preferably, a low-viscosity gel. Alternatively, the pharmaceutical composition is formulated as a solution. Alternatively, the pharmaceutical composition is formulated as a transdermal patch.
IX. Methods of Treatment
[0149] In certain embodiments, the invention describes a method for treating pain comprising the step of applying a topical, selective COX-2 inhibitor composition to a subject. In one aspect, the pharmaceutical composition is applied to the skin of the subject. [0150] In another aspect, the selective COX-2 inhibitor is delivered locally to the skin with minimal systemic absorption. In yet another aspect, the selective COX-2 inhibitor is delivered to and through the skin with minimal systemic absorption. In a still yet another aspect, the selective COX-2 inhibitor is delivered to the tissue surrounding or under the area of skin application with minimal systemic absorption. [0151] In other aspects, the subject is a human. Alternatively, the subject is a non-human mammal.
[0152] In still other aspects, the treatment is continued for at least 12 weeks. More preferably, the treatment is continued for at least six months.
[0153] The compositions of the invention may be useful to alleviate acute pain, chronic pain, or both. Compositions of the invention are particularly suited for use in treating OA chronically. They may also be useful for the treatment of other chronic joint diseases characterized by joint pain, degeneration of articular cartilage, impaired movement, and stiffness. Suitable joints include the knee, elbow, hand, wrist and hip. The compositions of the invention may also be useful for the treatment of other pain-associated disorders, including (but not limited to) muscle pain, lower back pain, neck pain, rheumatoid arthritis, fibromyalgia, myofascial pain, gout, sprains, strains, contusions, and neuropathic pain conditions.
[0154] Due to the properties of higher flux and greater in vivo absorption, it is believed that the formulations of the present invention can be administered at lower dosing than previously described etoricoxib formulations. In particular, it is expected that the compositions of the invention can be used at twice-a-day or once-a-day dosing in the treatment of OA. This would represent a significant improvement as lower dosing is associated with better patient compliance, an important factor in treating chronic conditions.
[0155] Compositions of the present invention may, if desired, be presented in a bottle, jar, or other container-closure system approved by the FDA or other regulatory authority, which may provide one or more dosages containing the active ingredient. The package or dispenser may also be accompanied by a notice associated with the container in a form prescribed by a
governmental agency regulating the manufacture, use, or sale of pharmaceuticals, the notice indicating approval by the agency.
X. Examples
[0156] Below, the present invention will be described by way of examples, which are provided for illustrative purposes only. Accordingly, they are not to be construed as limiting the scope of the present invention as defined by the appended claims. Unless otherwise specified, the percentage specified is a weight/weight percentage.
[0157] Example 1: General Procedure for Formulation Preparation
[0158] For a typical urea/choline eutectic melt composition, the urea and (2-hydroxyethyl)- trimethylammonium chloride were mixed by vortex, heating gently (e.g., 80-90°C) if necessary for liquification. The etoricoxib was then added and mixed by vortex with optional heating. The other components were then added except for alpha-hydroxy acids (if present) and some of the monohydric alcohol and water. The resulting suspension or solution was thoroughly mixed by vortex until a clear solution was obtained. [0159] Finally, any alpha-hydroxy acid (e.g., lactic acid) was added, followed by the remaining monohydric alcohol and water. The resulting suspension or solution was thoroughly mixed by vortexing for about 30 min or until a clear and homogeneous solution was obtained.
[0160] Example 2: General Procedure for Skin Permeation Measurement: [0161] Comparisons were mainly performed using porcine skin, but in some cases, human cadaver skin was used (i.e., Tables 12 to 15). The permeation of etoricoxib through porcine skin or human cadaver skin from each of the present formulations was measured using Franz diffusion cells ("FDC's).
[0162] Porcine skin pieces were obtained from Lampire Biological Laboratories, Inc., Pipersville, PA. Porcine skins were collected immediately following animal sacrifice, and the hairs were trimmed with clippers. Larger pieces of excess fat were removed with a filet knife. The skin was then trimmed to a set thickness of some 2 mm, cut into individual pieces, wrapped in aluminum foil, frozen, shipped, and stored at -78 °C.
[0163] Prior to use, the skin pieces were allowed to thaw, in air, to room temperature.
Before use, the skin was dermatomed to a thickness of 0.5 to 1 mm and cut into circular pieces of an appropriate size prior to mounting in the FDC. The FDCs had a 3-ml receptor
well volume, that was filled with isotonic phosphate buffered saline ("PBS") doped with 0.01% sodium azide. The flanges of the FDCs were coated with vacuum grease to ensure a complete seal and were clamped together with uniform pressure using a pinch clamp (SS #18 VWR 80073-350 from VW Scientific, West Chester PA). After the FDCs were assembled, the porcine skin was optionally allowed to pre-hydrate for 45 min with isotonic PBS.
Isotonic PBS was then removed and formulation was applied to the donor well or directly to the skin surface, depending on the amount of formulation applied. The receptor wells were maintained at 37 °C (temperature on the surface of the skin is about 30 °C) in a stirring block with continual agitation via a stir bar. [0164] During exploratory studies, the initial applied dose was 100 ul. This application dose was gradually reduced to 50 ul, 25 ul, 10 ul, and 5 ul levels. In transdermal studies, it is common practice to use a hydro alcoholic control, since this combination appears to provide optimal dissolution for various drug compounds.
[0165] The flux rates were calculated using the approximate area of each donor well (0.55 cm2). Samples were drawn from the receptor wells at various times, as provided in the examples that follow. Franz diffusion cell measurements were typically made in five- to tenfold replicates for each formulation, based on availability of skin and number of formulations tested. The concentrations of etoricoxib in the samples were measured using HPLC analysis using a CI 8 column and acetonitrile and water as the mobile phase. Generally, in the examples that follow, permeation data were reported by plotting a curve showing the cumulative amount of etoricoxib that permeates across the skin as a function of time. The flux rate can be computed as the time derivative of this curve.
[0166] Example 3: General Method for Skin Retention Studies
[0167] At the end of the permeation study, skin samples were removed from the Franz cells for skin retention studies. Any excess of formulation was carefully wiped away, first with cotton swabs and then with lint-free paper. The skin samples were quickly washed with cold water and ethanol, and the skin samples were then dried for 1 h at room temperature. The formulation contact area of corresponding skin pieces were punched, and the other areas were discarded. The punched skin portions were cut into small pieces with a pair of stainless steel scissors, the samples were transferred into 5 -ml scintillation vials, and 2 ml of absolute ethanol was added. The samples were allowed to incubate for 24 h at room temperature. The liquid phase was then filtered through 9 mm diameter disposable syringe filters (0.45 μηι, Acrodisc®). The filtrate, after appropriate dilution, was assayed by HPLC.
[0168] Example 4: Etoricoxib Formulations I
[0169] Table 1: Etoricoxib Formulations I

[0170] Permeation performance of the formulations listed in Table 1 were tested using the Franz cell methodology of Example 2. The results are shown in FIG. 1. Inclusion of choline and urea enhanced the delivery of etoricoxib (F9).
[0171] Example 5: Etoricoxib Formulations II
[0172] Table 2: Etoricoxib Formulations II

[0175] Table 3: Etoricoxib Formulations III

[0176] Reduction of the amount of urea/choline from 15% (10:5) to 7.5% (5:2.5) improved the delivery of etoricoxib (F21 vs. F22). Results are shown in FIG. 3.
[0177] Example 7: Etoricoxib Formulations IV
[0178] Table 4: Etoricoxib Formulations IV
Formulations
Ingredients F28 F29 F30 F31 F32 F33 F34 F35 F26-con
Etoricoxib 2 2 2 2 2 2 2 2 2
Ethanol 57 47 52 48 59.5 49.5 55.5 45.5 48
Water 5 15 14 15 10 10 15 15 50
Isopropanol 10 10 10 10 10 10 10 10
Choline 5 5 5 5 2.5 2.5 2.5 2.5
chloride
Urea 10 10 10 10 5 5 5 5
Glycerin 3 3 3 . 3
monolaurate
DL-Limonene 3 3 3 3
Polysorbate 20 3 3 3 3 3 3 3
Isopropyl 5 5 5 5
myristate
2-(2-Ethoxy- 10 10
ethoxy)ethanol
9

[0179] Variations of the formulations were tested. The water component was increased (F28 vs. F29 to F35), and the eutectic melt content was decreased (F28-F31 vs. F32-F35). Results are shown in FIG. 4. [0180] Example 8: Etoricoxib Formulations V
[0181] Table 5: Etoricoxib Formulations V

[0182] Reversing the proportion of urea to choline chloride affected the delivery of etoricoxib (F37 vs. F39). The incorporation of DMSO into the formulation further reduced the amount of etoricoxib delivered (F44). Results are shown in FIG. 5.
[0183] Example 9: Etoricoxib Formulations VI
[0184] Table 6: Etoricoxib Formulations VI

[0185] The incorporation of propylene glycol into the formulation provided a physically appealing composition (F46). The results are shown in FIG. 6.
[0186] Example 10: Etoricoxib Formulations VII
[0187] Table 7: Etoricoxib Formulations VII
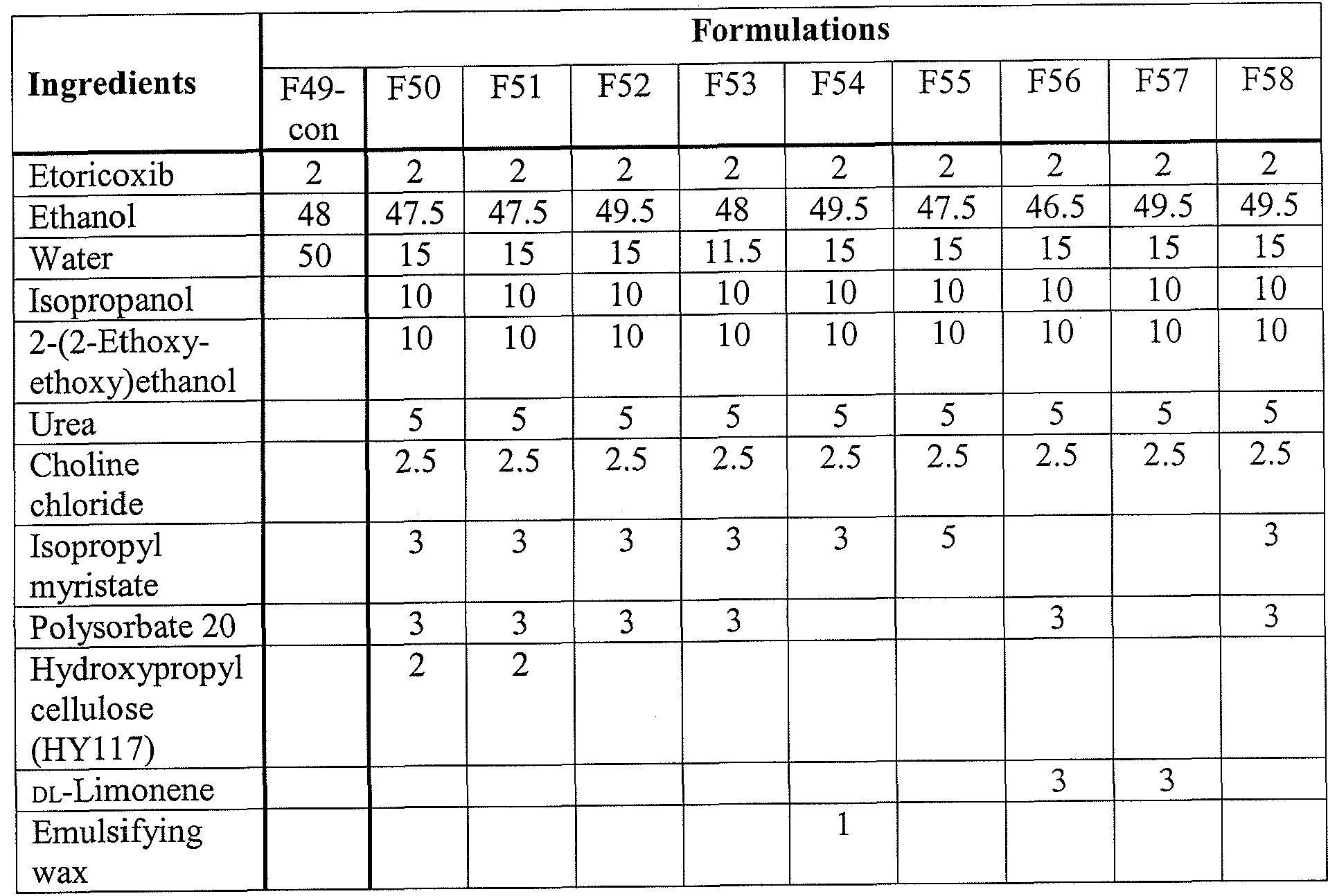

[0188] FIG. 7 A shows the delivery of etoricoxib through the skin, and FIG. 7B shows the retention of etoricoxib in the skin.
[0189] Incorporation of propylene glycol into the formulation provided a physically appealing composition (F53), and its delivery of etoricoxib was comparable to that of F55 and F57.
[0190] A formulation with isopropyl myristate (F55) showed similar behavior to a formulation containing DL-Limonene (F57).
[0191] A choline/urea mixture incorporated into the formulation with and without heating (F52 vs. F58). F58 is a version of F52 without any application of heat during formulation. F58 was prepared without eutectic formation, and it was obtained by simple addition of ingredients. Both F52 and F58 provided similar delivery of etoricoxib. However, F58 was not homogeneous the next day, whereas F52, which was prepared with eutectic formation first, was homogeneous. [0192] Example 11: Etoricoxib Formulations VIII
[0193] Table 8: Etoricoxib Formulations VIII
Ingredients Formulations
F49-con F59 F64
Etoricoxib 2 2 2
Ethanol 48 58.5 49.5
Isopropanol 10 10
Isopropyl myristate 5
Choline chloride 2.5 2.5
Urea 5 5
Water 50 15 15
HPC HY117 2
Transcutol® 10
Limonene 3
Glycerin mono laurate 3
[0194] The results for the formulations described in Table 8 are shown in FIG. 8. The results are for finite dosing. The first set of results in FIG. 8 is for 10 μΐ doses; the second set, for 5 μΐ doses.
[0195] Example 12: Optimization Study at Constant Ethanol Concentration
(Etoricoxib Formulations IX)
[0196] Table 9: Optimization Study at Constant Ethanol Concentration (Etoricoxib Formulations IX)

*F69, F70 and F72 were not homogeneous and not included in the porcine skin experiment. [0197] Results
[0198] The results for the formulations described in Table 9 are shown in FIG. 9.
[0199] The amount of etoricoxib delivered over 24 hours was reduced in the absence of IPM (F66). The optimal concentration of isopropyl myristate (IPM) appears to be at the 5% level; etoricoxib delivery was slightly less with ΓΡΜ at 2.5 % or 10% concentration (F67 vs. F68). In the absence of both choline chloride and urea, the formulations were not homogenous (F70 and F72). Formulations with 2.5 % urea (F71, F73) seem to show similar effectiveness to other urea-containing formulations (F74 and F75) in delivering etoricoxib across skin. The Formulations containing choline chloride and urea at a 1 :2 ratio (F71) or a 1 : 1 ratio (F73) seemed to provide the best results.
[0200] Example 13: Optimization Study at Constant Water Concentration
(Etoricoxib Formulations X)
[0201] Table 10: Optimization Study at Constant Water Concentration (Etoricoxib Formulations X)

*F79 and F81 were not homogeneous and not included in the porcine skin experiment. [0202] Results
[0203] The results for the formulations described in Table 10 are shown in FIG. 10. [0204] Isopropyl myristate appears to function as a permeation enhancer in this formulation chassis, as in the absence of IPM, the etoricoxib delivery was reduced. In this study, formulations with 5 or 2.5% IPM concentrations appeared to be similarly effective in delivering etoricoxib across porcine skin.
[0205] The optimal level of urea appears to be between 2.5% to 5% (F76 vs. F77, F78, and F79). The highest level of etoricoxib delivery was observed with a formulation containing a 2:1 urea/choline chloride ratio (F82), which was consistent with the results from the previous studies.
[0206] Overall, the variation of alcohol concentrations appeared to have more impact on the etoricoxib permeation than the variation of other ingredients. [0207] Example 14: Optimization Study with Addition of Transcutol® and Redution of Ethanol Concentrations (Etoricoxib Formulations XI)
[0208] Table 11: Optimization Study with Addition of Transcutol® and Redution of Ethanol Concentrations (Etoricoxib Formulations XI)

*F91 and F92 were not homogeneous and not included in the porcine skin experiment.
[0209] The results for the formulations described in Table 11 are shown in FIG. 11
[0210] In this series of formulation experiments, the inclusion of isopropyl myristate had less impact on etoricoxib permeation (F87 vs. F88, F89, F90, and F96); however, the incorporation of both ΓΡΜ and Transcutol® (i.e., 2-(2-ethoxy-ethoxy)ethanol) appeared to stabilize the formulation, producing an esthetically pleasing formulation feel on skin.
[0211] In the absence of choline chloride and urea, the formulation was not physically stable (F91). Choline chloride and urea at 5:5% (w/w) provided the highest etoricoxib permeation (F95 vs. F96);
[0212] The addition of Transcutol® further enhanced the etoricoxib permeation (F87 vs. F98, F99).
[0213] Example 15: General Procedure for Flow-Through
[0214] Flow-through experiments were essentially performed using an automated version of the Franz Cell methodology of Example 2, with the exception that the surface area was 1 cm and the receptor volume was 1 ml. Human cadaver skin was not dermatoned. [0215] In certain embodiments below, the etoricoxib concentrations used were 0.5, 1.0, 2.0, 3.0, and 5.0%. The etoricoxib dosing studies included 48-hour, two-dose studies (dosing at 0 and 8 hours); 48-hour, four-dose studies (dosing at 0, 9, 21 and 29 hours); and 48-hour studies after pretreatment w/placebo with 1 and 2 % actives (dosing at 0, 21 and 29 hours). In the latter study, the placebo acts as a conditioning agent and also may act as a permeation
enhancer. The Enhancement Ratio (ER) of the test formulation, which is defined above, is provided for certain embodiments.
[0216] Example 16: Concentration Variations of Etoricoxib
[0217] Table 12 A: Concentration Variations of Etoricoxib

[0218] Table 12B: Concentration Variations of Etoricoxib:

[0219] The results for the formulations described in Tables 12A and 12B are shown in FIG. 12. [0220] Example 17: 48-h Dosing Studies: Dosing at 0 and 8 h
[0221] Table 13A: 48-h Dosing Studies: Dosing at 0 and 8 h

[0222] Table 13B: 48-h Dosing Studies: Dosing at 0 and 8 h

[0223] The results for the formulations described in Tables 13 A and 13B are shown in FIG. 13.
[0224] Example 18: 48-h Dosing Studies: Dosing at 0, 9, 21 and 29 h
[0225] Table 14A: 48-h Dosing Studies: Dosing at 0, 9, 21 and 29 h

[0226] Table 14B: 48-h Studies: Dosing at 0, 9, 21 and 29 h

[0228] Example 19: 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h
[0229] Table 15A: 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h
Formulations:

[0230] Table 15B: 48-h Dosing Studies with 1 and 2% Active Formulations After Pretreatment with Placebo: Dosing at 0, 21 and 29 h

[0231] The results for the formulations described in Tables 15A and 15B are shown in FIG. 15.
[0232] Conclusions for Dosing Studies
[0233] Twice-daily application of either 1% or 2% formulation was effective to further increase etoricoxib delivery by approximately 30 to 60% over the single-dosing regimen.
[0234] With a once-daily dosing regimen, the highest etoricoxib delivery was observed from a 2% API-containing formulation. Twice-daily application of 1% formulation,
however, was as effective as the 2% formulation in delivery etoricoxib across intact human skin.
[0235] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims. All publications, patents, and patent applications cited herein are hereby incorporated by reference in their entirety for all purposes, including U.S. Patent Applications 61/247,222 (filed September 30, 2009), 61/250,452 (filed October 9, 2009), and 61/230,473 (filed July 31, 2009) as well as international applications PCT/US2010/051001 (filed September 30, 2010),
PCT/US2010/052111 (filed October 9, 2010), and PCT/US2010/044036 (filed July 30, 2010).
Claims
WHAT IS CLAIMED IS: 1. A pharmaceutical composition for topical administration, the composition comprising:
etoricoxib;
urea;
choline;
a lower alcohol; and
water
wherein the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic.
2. A pharmaceutical composition for topical administration, the composition comprising:
about 0.1% to 5% (w/w) etoricoxib;
about 3% to 21% (w/w) of a eutectic melt comprising urea and choline; a lower alcohol; and
water.
3. The pharmaceutical composition of claim 1 or 2, wherein the composition comprises about 1% to 3% (w/w) etoricoxib.
4. The pharmaceutical composition of claim 3, wherein the composition comprises about 1% (w/w) etoricoxib.
5. The pharmaceutical composition of claim 3, wherein the composition comprises about 2% (w/w) etoricoxib.
6. The pharmaceutical composition of claim 1 or 2, wherein the eutectic melt comprises about 2% to 10% (w/w) of urea and about 1% to 10% (w/w) of choline.
7. The pharmaceutical composition of claim 1 or 2, wherein the eutectic melt comprises about a 3: 1 ratio to a 1 :2 ratio of urea to choline.
8. The pharmaceutical composition of claim 7, wherein the eutectic melt comprises a 2: 1 ratio of urea to choline.
9. The pharmaceutical composition of claim 7, wherein the composition comprises about 5% (w/w) urea.
10. The pharmaceutical composition of any one of claims 1, 3, 4, 5, 5, 7, 8, or 9, wherein choline is a choline salt.
11. The pharmaceutical composition of claim 10, wherein the composition comprises 2.5% (w/w) of the choline salt.
12. The pharmaceutical composition of claim 1 or 2, wherein the composition comprises about 35% to 65% of the lower alcohol.
13. The pharmaceutical composition of claim 12, wherein the lower alcohol is ethanol, and wherein the composition further comprises a second lower alcohol.
14. The pharmaceutical composition of claim 13, wherein the second lower alcohol is a member selected from the group consisting of isopropanol and 2-(2- ethoxyethoxy)ethanol.
15. The pharmaceutical composition of any one of claims 12, 13,or 14, wherein the composition comprises about 1% to 25% (w/w) of the second lower alcohol.
16. The pharmaceutical composition of claim 15, wherein the composition comprises about 10% of the second lower alcohol.
17. The pharmaceutical composition of claim 15, wherein the composition further comprises about 1% to 25% of a third lower alcohol.
18. The pharmaceutical composition of claim 17, wherein the composition comprises about 10% of a third lower alcohol.
19. The pharmaceutical composition of claim 1 or 2, wherein the composition is a low-viscosity gel.
20. The pharmaceutical composition of claim 1 or 2, wherein the composition is a high-viscosity gel.
21. The pharmaceutical composition of claim 1 or 2, wherein the composition is a solution.
22. The pharmaceutical composition of claim 1 or 2, wherein the composition further comprises an alpha-hydroxy acid.
23. The pharmaceutical composition of claim 22, wherein the composition comprises about 0.5% to 5%(w/w) of the alpha-hydroxy acid.
24. The pharmaceutical composition of claim 23, wherein the alpha- hydroxy acid is lactic acid.
25. The pharmaceutical composition of claim 1 or 2, wherein the composition further comprises a molecular penetration enhancer.
26. The pharmaceutical composition of claim 25, wherein the molecular penetration enhancer is a member selected from the group consisting of a fatty acid ester and a terpene.
27. The pharmaceutical composition of claim 26, wherein the composition further comprises about 3% to 10% (w/w) of the fatty acid ester.
28. The pharmaceutical composition of claim 27, wherein the composition comprises about 5% of the fatty acid ester.
29. The pharmaceutical composition of claim 27 or 28, wherein the fatty acid ester is a member selected from the group consisting of glycerol monolaurate and isopropyl myristate.
30. The pharmaceutical composition of claim 26, wherein the composition comprises about 0.1% to 10%> (w/w) of the terpene.
31. The pharmaceutical composition of claim 30, wherein the terpene is limonene.
32. The pharmaceutical composition of any one of claims 26, 30, or 31, wherein the composition comprises about 3% to 5% (w/w) of the terpene.
33. The pharmaceutical composition of any one of claims 1, 2, 27, 29, 30, or 31, wherein the composition further comprises about 3% to 5% (w/w) of a second molecular penetration enhancer.
34. The pharmaceutical composition of claim 1, 2, or 25, wherein the composition further comprises a fatty acid ester.
35. The pharmaceutical composition of claim 34, wherein the composition comprises about 0.1% to 10% (w/w) of the fatty acid ester
36. The pharmaceutical composition of claim 35, wherein the fatty acid ester is isopropyl myristate.
37. The pharmaceutical composition of claim 35, wherein the composition comprises about 0.1% to 5% (w/w) of the fatty acid ester.
38. The pharmaceutical composition of claim 37, wherein the additional fatty acid ester is glycerol monolaurate.
39. The pharmaceutical composition of claim 1 or 2, wherein the composition further comprises a nonionic surfactant.
40. The pharmaceutical composition of claim 39, wherein the composition comprises about 0.1% to 15% (w/w) of the nonionic surfactant.
41. The pharmaceutical composition of claim 40, wherein the composition comprises about 3% (w/w) of the nonionic surfactant.
42. The pharmaceutical composition of claim 40, wherein the nonionic surfactant is polysorbate 20.
43. The pharmaceutical composition of claim 1 or 2, wherein the composition further comprises a thickening agent.
44. The pharmaceutical composition of claim 43, wherein the composition comprises about 0.1% to 5% (w/w) of the thickening agent.
45. The pharmaceutical composition of claim 44, wherein the composition comprises about 2% (w/w) of the thickening agent.
46. The pharmaceutical composition of any one of claims 43, 44, or 45, wherein the thickening agent is hydroxypropyl cellulose.
47. A pharmaceutical solution for topical administration, the solution comprising:
about 0.1 % to 5% (w/w) etoricoxib; about 2% to 10% urea;
about 2% to 10% choline;
a lower alcohol; and
water.
48. The pharmaceutical solution of claim 47, the solution comprising: about 1% to 2% (w/w) of etoricoxib;
about 5% urea;
about 2.5% choline;
at least 3% of at least one molecular penetration enhancer;
about 10% isopropanol; and
water.
49. A method for topically treating pain in a subject, the method comprising:
topically applying a pharmaceutical composition comprising:
about 0.1% to 5% (w/w) etoricoxib;
about 2% to 10% urea;
about 2% to 10% choline;
a lower alcohol; and
water.
50. The method of claim 49, the method comprising:
topically applying a pharmaceutical composition comprising:
about 0.1 % to 5% (w/w) of etoricoxib;
about 3% to 21% of a eutectic melt of choline and urea;
at least one molecular penetration enhancer;
at least one lower alcohol; and
water.
51. The method of claim 49 or 50, wherein the pain is associated with osteoarthritis.
52. The use of the pharmaceutical composition of any one of claims 1-48 in the manufacture of a medicament for the treatment of pain.
53. The use of claim 52, wherein the pain is associated with osteoarthritis.
54. A pharmaceutical composition for topical administration, the composition comprising etoricoxib, urea, choline, a lower alcohol and water, wherein the composition is prepared according to the method of:
(a) mixing the urea and choline together and, optionally, heating to form a eutectic melt;
(b) mixing the etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) admixing the lower alcohol and water to the composition of (b) to form the pharmaceutical composition.
55. The composition of claim 54, wherein the choline and urea form a eutectic melt, but the pharmaceutical composition is essentially non-eutectic 56. A method of preparing a pharmaceutical composition comprising the steps of:
(a) mixing urea and choline together and, optionally, heating to form a eutectic melt; (b) mixing etoricoxib with the eutectic melt of (a) and, optionally, heating; and (c) admixing a lower alcohol and water to the composition of (b) to form the pharmaceutical composition.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10779189A EP2498819A2 (en) | 2009-11-11 | 2010-11-11 | Topical etoricoxib formulation comprsing an eutectic mixture of permeation enhancers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26035109P | 2009-11-11 | 2009-11-11 | |
| US61/260,351 | 2009-11-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011060195A2 true WO2011060195A2 (en) | 2011-05-19 |
| WO2011060195A3 WO2011060195A3 (en) | 2011-07-21 |
Family
ID=43807135
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/056419 WO2011060195A2 (en) | 2009-11-11 | 2010-11-11 | Topical eutectic formulation |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP2498819A2 (en) |
| WO (1) | WO2011060195A2 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011149645A1 (en) * | 2010-05-28 | 2011-12-01 | Nuvo Research Inc. | Topical etoricoxib formulation |
| US9144553B2 (en) | 2012-12-21 | 2015-09-29 | Teikoku Pharma Usa, Inc. | Compositions and methods for transdermal delivery of hormones and other medicinal agents |
| WO2016207871A1 (en) * | 2015-06-26 | 2016-12-29 | University Of The Witwatersrand, Johannesburg | An oral pharmaceutical dosage form for the delivery of a peptide and/or protein |
| WO2020028471A1 (en) * | 2018-08-01 | 2020-02-06 | Bullseyetx Llc | Systems and methods for delivery of drugs and other substances comprising deep eutectic solvents |
| CN112427047A (en) * | 2019-08-10 | 2021-03-02 | 石家庄搏澳增塑材料科技有限公司 | Method for large-scale preparation of urea-choline chloride green catalyst and solvent |
| CN112839633A (en) * | 2018-08-01 | 2021-05-25 | 诺维拉制药有限公司 | Eutectic solvent containing medicament and manufacturing method and application thereof |
| CN113382753A (en) * | 2018-10-24 | 2021-09-10 | 瑟拉诺沃有限公司 | Deep eutectic solvent platform for oral pharmaceutical formulations |
| CN114555124A (en) * | 2019-08-19 | 2022-05-27 | 瑟拉诺沃控股有限公司 | Pharmaceutical co-crystal salt formulation |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4529601A (en) | 1977-12-01 | 1985-07-16 | Astra Lakemedel Aktiebolag | Local anesthetic mixture for topical application and method for obtaining local anesthesia |
| US6368618B1 (en) | 1999-07-01 | 2002-04-09 | The University Of Georgia Research Foundation, Inc. | Composition and method for enhanced transdermal absorption of nonsteroidal anti-inflammatory drugs |
| US20080300311A1 (en) | 2006-10-17 | 2008-12-04 | Nuvo Research | Diclofenac gel |
| US20100044036A1 (en) | 2003-12-31 | 2010-02-25 | Pierre Mouget | Tracer injector tool for well investigation |
| US20100052111A1 (en) | 2008-08-26 | 2010-03-04 | Kabushiki Kaisha Toshiba | Stacked-chip device |
| US20100051001A1 (en) | 2008-08-27 | 2010-03-04 | Timothy Webb | Exhaust Gas Recirculation (EGR) System |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005044227A1 (en) * | 2003-11-05 | 2005-05-19 | Glenmark Pharmaceuticals Limited | Topical pharmaceutical compositions |
-
2010
- 2010-11-11 EP EP10779189A patent/EP2498819A2/en not_active Withdrawn
- 2010-11-11 WO PCT/US2010/056419 patent/WO2011060195A2/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4529601A (en) | 1977-12-01 | 1985-07-16 | Astra Lakemedel Aktiebolag | Local anesthetic mixture for topical application and method for obtaining local anesthesia |
| US6368618B1 (en) | 1999-07-01 | 2002-04-09 | The University Of Georgia Research Foundation, Inc. | Composition and method for enhanced transdermal absorption of nonsteroidal anti-inflammatory drugs |
| US20100044036A1 (en) | 2003-12-31 | 2010-02-25 | Pierre Mouget | Tracer injector tool for well investigation |
| US20080300311A1 (en) | 2006-10-17 | 2008-12-04 | Nuvo Research | Diclofenac gel |
| US20100052111A1 (en) | 2008-08-26 | 2010-03-04 | Kabushiki Kaisha Toshiba | Stacked-chip device |
| US20100051001A1 (en) | 2008-08-27 | 2010-03-04 | Timothy Webb | Exhaust Gas Recirculation (EGR) System |
Non-Patent Citations (12)
| Title |
|---|
| "Pharmacotherapy: a pathophysiological approach", 1999, APPLETON & LANGE, pages: 1441 - 59 |
| BARRY, B. W., EUR. J. PHARM. SCI., vol. 14, 2001, pages 101 - 114 |
| BELLAMY, N.; KIRWAN, J.; BOERS, M.; BROOKS, P.; STRAND, V.; TUGWELL, P. ET AL.: "Recommendations for a core set of outcome measures for future Phase III clinical trials in knee, hip and hand osteoarthritis. Consensus development at OMERACT m.", JRHEUMATOL, vol. 24, 1997, pages 799 - 802 |
| BENSON, H. A. E., CURR. DRUG DEL, vol. 2, 2005, pages 23 - 33 |
| HO ET AL.: "The influence of cosolvents on the in-vitro percutaneous penetration of diclofenac sodium from a gel system", J. PHARM. PHARMACOL., vol. 46, 1994, pages 636 - 642 |
| NAITO ET AL.: "Percutaneous absorption of diclofenac sodium ointment", INT. JOUR. OF PHARMACEUTICS, vol. 24, 1985, pages 115 - 124 |
| OA. LIN ET AL.: "Efficacy of topical non-steroidal anti-inflammatory drugs in the treatment of osteoarthritis: meta- analysis of randomized controlled trials", BMJ, 2004 |
| OBATA, INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 89, 1993, pages 191 - 198 |
| OSBORNE, D.W.; HENKE, J. J.: "Skin Penetration Enhancers Cited in the Technical Literature", PHARMACEUT. TECH, November 1997 (1997-11-01), pages 58 - 66 |
| OSTRENGA J. ET AL.: "Significance of vehicle composition I: relationship between topical vehicle composition, skin penetrability, and clinical efficacy", JOURNAL OFPHARMACEUTICAL SCIENCES, vol. 60, 1971, pages 1175 - 1179 |
| ROSENSTEIN: "Topical agents in the treatment of rheumatic disorders", RHEUM. DIS. CLIN NORTH AM., vol. 25, 1999, pages 899 - 918 |
| WILLIAMS; BARRY: "Penetration Enhancers", ADVANCED DRUG DELIVERY REVIEWS, vol. 56, 2004, pages 603 - 618 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011149645A1 (en) * | 2010-05-28 | 2011-12-01 | Nuvo Research Inc. | Topical etoricoxib formulation |
| US9144553B2 (en) | 2012-12-21 | 2015-09-29 | Teikoku Pharma Usa, Inc. | Compositions and methods for transdermal delivery of hormones and other medicinal agents |
| US9579296B2 (en) | 2012-12-21 | 2017-02-28 | Teikoku Pharma Usa, Inc. | Compositions and methods for transdermal delivery of hormones and other medicinal agents |
| WO2016207871A1 (en) * | 2015-06-26 | 2016-12-29 | University Of The Witwatersrand, Johannesburg | An oral pharmaceutical dosage form for the delivery of a peptide and/or protein |
| US10973766B2 (en) | 2015-06-26 | 2021-04-13 | University Of The Witwatersrand, Johannesburg | Oral pharmaceutical dosage form for the delivery of a peptide and/or protein |
| WO2020028471A1 (en) * | 2018-08-01 | 2020-02-06 | Bullseyetx Llc | Systems and methods for delivery of drugs and other substances comprising deep eutectic solvents |
| CN112839633A (en) * | 2018-08-01 | 2021-05-25 | 诺维拉制药有限公司 | Eutectic solvent containing medicament and manufacturing method and application thereof |
| CN112839634A (en) * | 2018-08-01 | 2021-05-25 | 诺维拉制药有限公司 | Systems and methods for delivering drugs and other substances including deep eutectic solvents |
| CN113382753A (en) * | 2018-10-24 | 2021-09-10 | 瑟拉诺沃有限公司 | Deep eutectic solvent platform for oral pharmaceutical formulations |
| CN112427047A (en) * | 2019-08-10 | 2021-03-02 | 石家庄搏澳增塑材料科技有限公司 | Method for large-scale preparation of urea-choline chloride green catalyst and solvent |
| CN114555124A (en) * | 2019-08-19 | 2022-05-27 | 瑟拉诺沃控股有限公司 | Pharmaceutical co-crystal salt formulation |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011060195A3 (en) | 2011-07-21 |
| EP2498819A2 (en) | 2012-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8470886B2 (en) | Topical ibuprofen formulations | |
| WO2011060195A2 (en) | Topical eutectic formulation | |
| US9084754B2 (en) | Highly permeating terbinafine formulation | |
| US9168305B2 (en) | Diclofenac topical formulation | |
| US10646441B2 (en) | Foamable formulation | |
| WO2011014850A2 (en) | Topical eutectic-based formulations | |
| WO2011041609A2 (en) | Topical formulations | |
| WO2011149645A1 (en) | Topical etoricoxib formulation | |
| US20150342871A1 (en) | Highly permeating terbinafine formulation | |
| EP2485730A1 (en) | Topical formulation comprising etoricoxib and a zwitterionic surfactant |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10779189 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010779189 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010779189 Country of ref document: EP |