WO2011003870A2 - Mini-pegylated corticosteroids, compositions including same, and methods of making and using same - Google Patents
Mini-pegylated corticosteroids, compositions including same, and methods of making and using same Download PDFInfo
- Publication number
- WO2011003870A2 WO2011003870A2 PCT/EP2010/059569 EP2010059569W WO2011003870A2 WO 2011003870 A2 WO2011003870 A2 WO 2011003870A2 EP 2010059569 W EP2010059569 W EP 2010059569W WO 2011003870 A2 WO2011003870 A2 WO 2011003870A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mini
- peg
- dexamethasone
- disease
- Prior art date
Links
- 0 CC[C@@]([C@](C[C@@]([C@]1(C[C@]2NC)I)([C@]2(C(CN**(O)=C)=O)O)I)O)([C@]1(CCC(CC=C1)=CC1=O)I)N Chemical compound CC[C@@]([C@](C[C@@]([C@]1(C[C@]2NC)I)([C@]2(C(CN**(O)=C)=O)O)I)O)([C@]1(CCC(CC=C1)=CC1=O)I)N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/44—Glucocorticosteroids; Drugs increasing or potentiating the activity of glucocorticosteroids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/02—Applications for biomedical use
Definitions
- This application generally relates to corticosteroids, and more specifically to corticosteroid-based compositions for topical use.
- FIG. 1 illustrates the pathway by which corticosteroids enter the cell and interact with the glucocorticoid receptor (GR).
- GR glucocorticoid receptor
- the GR exists as a large multiunit complex in the cytoplasm, which includes 2 molecules of heat shock protein (hsp) 90, which act as chaperones.
- hsp heat shock protein
- the GR dissociates from the chaperone proteins and translocates to the nucleus.
- the GR binds as a homodimer to a specific palindromic DNA sequence, termed a GRE, located in the regulatory regions of the target genes.
- the bound GR homodimers interacts with the basal transcriptional machinery shown that includes TATA-binding protein (TBP). associated transcription factors (TAFs and TFIIs), and RNA polymerase II (pol II).
- TBP TATA-binding protein
- TFIIs associated transcription factors
- poly II RNA polymerase II
- the interaction between GR and the basal transcription complex modulates transcription of the GR target genes.
- Different model of transcription regulation by the GR-ligand complex have been described (positive and negative regulation).
- the GR may interact via protein-protein interaction with other transcription factors, e.g., activator protein (AP)-I and nuclear factor-kB (NF-kB); in this case, the gene expression is controlled by the GR without binding to DNA.
- AP activator protein
- NF-kB nuclear factor-kB
- the active hormone-GR complex has a pleiotropic effect on genes involved in several processes.
- the biological effects of corticosteroids can be summarized as anti-inflammatory/
- compositions intended for topical use may be formulated, e.g., as nasal sprays, as inhalers, as creams or ointments for dermal, ocular, or rectal administration. However, even if such compositions are applied only to a specific part of the body, the corticosteroid may be absorbed systemically.
- corticosteroid Because of its wide activity, systemic exposure to the corticosteroid may cause a variety of undesired side effects, for example, to die skeleton and muscles (muscle atrophy and osteoporosis), eyes (glaucoma), endocrine system and metabolism (Cushing's syndrome, diabetes, adrenal atrophy and hypogonadism), cardiovascular and immune systems, and gastrointestinal system.
- side effects affecting skin could range from more cosmetic aspects (e.g., teleangiectasia, hypertrichosis) to more serious disabling situations (e.g., atrophy, erythema, delayed wound healing).
- mini -PEGyI ated corticosteroids compositions including same, and methods of making and using same.
- a and B are each independently H, OH, C]-CiO alkyl, C]-C ⁇ o alkoxy, or C Cio acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
- D and E arc each independently H, C
- G, J, and K are each independently H or a halogen
- X is a non-cleavable linking group between the C 2 ] position of the corticosteroid and the mini-PEG;
- mini-PEG is polyethylene glycol having a molecular weight between about 100 and about 20,000 Da;
- Y is H, OH, Ci-C 5 alkyl, C 1 -C 5 acyl, or C,-C 5 alkoxy; and C] and C 2 are bonded together by either a single bond or a double bond.
- n is an integer between 1 and 100
- m is an integer between 0 and 100
- m+n is between 2 and 100;
- a and B are each independently H, OH, C[-C ⁇ o alkyl (e.g., CHj 1 ), C
- D and E are each independently H, C]-CiO alkyl, Ci-Qo alkoxy, or Cj-C ⁇ o acyl;
- G, J, and K are each independently H or a halogen, in some embodiments H, Cl, F,
- PEG polyethylene glycol
- the corticosteroid is selected from the group consisting of: beclomethasone, betamethasone, budesonide, clobetasol, corticosterone, cortisone, desonide, desoximetasone, desoxycorticosterone, dexamethasone, difluocortolone, flumethasone, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluticasone, fluticasone proprionate, halobetasol, hydrocortisone, methylprednisolone, prednicarbate, prednisolone, prednisone, triamcinolone, and triamcinolone acetonide.
- a pharmaceutical composition comprising a compound provided herein, and one or more pharmaceutically acceptable carriers or excipients.
- the composition is formulated as a topical dosage form.
- the topical dosage form is selected from the group consisting of emulsions, solutions, suspensions, creams, gels, oils, hydrogels, ointments, dusting powders, dressings, elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols, irrigations, sprays, suppositories, bandages, and dermal patches.
- provided herein is a method for the treatment, prevention, or amelioration of one or more symptoms of a glucocorticoid receptor-mediated disorder, disease, or condition in a subject, which comprises administering to the subject a therapeutically effective amount of a compound or composition provided herein.
- a method for modulating glucocorticoid receptor activity comprising contacting a glucocorticoid receptor with a compound or composition provided herein.
- FIG. 1 illustrates the pathway by which a corticosteroid interacts with the glucocorticoid receptor.
- FIG. 2 illustrates the results of an experiment comparing the activation of the luciferase reporter gene in HeLa/GR-luc cells by (a) dexamethasone or dexamcthasonc mini- PEGylate (Compound 1), and (b) betamethasone or betamethasone mini-PEGylare (Compound 1)
- FIG. 3 illustrates the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NFkB-luc cells by dexamethasone or dexamethasone mini-PEGylate expressed as (a) % induction and (b) luminescence.
- FIG. 4 illustrates the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NFkB-luc cells by betamethasone or betamethasone mini-PEGylate expressed as (a) % induction and (b) luminescence.
- FIG. 5 illustrates the mean plasma concentrations as a function of time after dose following a single IV administration in rats of (a) dexamethasone and (b) dexamethasone mini- PEGylate; (c) illustrates the mean plasma concentrations of (a) and (b) in a single graph.
- FIG. 6 illustrates the mean plasma concentrations as a function of time after dose following a single IV administration in rats of (a) betamethasone and (b) betamethasone mini- PEGylate; (c) illustrates the mean plasma concentrations of (a) and (b) in a single graph.
- FIG. 7 illustrates the plasma concentrations of dexamethasone as a function of time after dose following dermal application to rats of dexamethasone (limit of detection: 79 nM); there was no measurable plasma concentration following dermal application to rats of dexamethasone mini-PEGylate (limit of detection: 179 nM).
- FIG. 8 illustrates the average plasma concentration of betamethasone as a function of time after dose following dermal application to rats of betamethasone; there was no measurable plasma concentration following dermal application to rats of betamethasone mini-PEGylate (limit of detection: 37.6 nM).
- FIG. 9 illustrates the results of an experiment comparing the cytokine secretion of (a) TNF- ⁇ , (b) IFN- ⁇ , and (c) IL-6 by dexamethasone mini-PEGylate expressed as a percent inhibition of the control secretion induced by the stimulus.
- FIG. 10 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human TNF- ⁇ secretion.
- FIG. 1 1 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human IFN- ⁇ secretion.
- FIG. 12 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human IL-6 secretion.
- subject refers to an animal, including, but not limited to, a primate (e.g., human), cow, pig, sheep, goat, horse, dog, cat, rabbit, rat, guinea pig, or mouse.
- primate e.g., human
- cow, pig, sheep, goat horse
- dog cat
- rabbit rat
- guinea pig or mouse.
- patient are used interchangeably herein in reference, for example, to a mammalian subject, such as a human subject, in one embodiment, a human, in another embodiment, a non-human.
- treat '"treating
- treatment are meant to include alleviating or abrogating a disorder, disease, or condition, or one or more of the symptoms associated with the disorder, disease, or condition; or alleviating or eradicating the cause(s) of the disorder, disease, or condition itself.
- prevent are meant to include a method of delaying and/or precluding the onset of a disorder, disease, or condition, and/or its attendant symptoms; barring a subject from acquiring a disorder, disease, or condition; or reducing a subject's risk of acquiring a disorder, disease, or condition.
- terapéuticaally effective amount are meant to include the amount of a compound that, when administered, is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the disorder, disease, or condition being treated.
- therapeutically effective amount also refers to the amount of a compound that is sufficient to elicit the biological or medical response of a biological molecule (e.g., a protein, enzyme, RNA, or DNA), cell, tissue, system, animal, or human, which is being sought by a researcher, veterinarian, medical doctor, or clinician.
- pharmaceutically acceptable carrier refers to a pharmaceutically-acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, solvent, or encapsulating material.
- each component is "pharmaceutically acceptable” in the sense of being compatible with the other ingredients of a pharmaceutical formulation, and suitable for use in contact with the tissue or organ of humans and animals without excessive toxicity, irritation, allergic response, immunogenicity, or other problems or complications, commensurate with a reasonable benefit/risk ratio. See, Remington; The Science and Practice of Pharmacy, 21st Edition, Lippincott Williams & Wilkins:
- the term “about” or “approximately” means an acceptable error for a particular value as determined by one of ordinary skill in the art, which depends in part on how the value is measured or determined. In certain embodiments, the term “about” or “approximately” means within 1, 2, 3, or 4 standard deviations. In certain embodiments, the term "about” or
- “approximately” means within 50%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1 %, 0.5%, or 0.05% of a given value or range.
- active ingredient and “active substance” refer to a compound, which is administered, alone or in combination with one or more pharmaceutically acceptable excipients, to a subject for treating, preventing, or ameliorating one or more symptoms of a condition, disorder, or disease.
- drug refers to a compound, or a pharmaceutical composition thereof, which is administered to a subject for treating, preventing, or ameliorating one or more symptoms of a condition, disorder, or disease.
- mini-PEG polyethylene glycol (PEG) having a molecular weight between about 100 Da and about 20,000 Da
- X is a non-cleavable linking group between the corticosteroid and the mini-PEG. Selection of an appropriate linking group X may, in some embodiments, be based on one or more of the following factors: ease of synthesis, chemical stability, and biocompatibility, such as an expectation of low adverse effects of the linker and its metabolites.
- X is alkylene, amido, oximo, S, O, or NR, wherein R is hydrogen or alkyl.
- X is S.
- the C 21 position provides a convenient location to covalently bond the mini-PEG to the corticosteroid via linker X.
- the mini-PEG may alternatively be bonded to other positions of the corticosteroid via linker X.
- the mini- PEG has a molecular weight between about 500 and about 2,000 Da, while in other embodiments the mini-PEG has a molecular weight in one of the ranges identified herein.
- the terminal end of the mini-PEG may also be modified, as described in greater detail herein. In some embodiments, functionalizing the terminal end of the mini-PEG can reduce the reactivity of the mini-PEG and/or can enhance the convenience of synthesizing the compound.
- the linkage between the corticosteroid and the mini-PEG is non-cleavable, meaning that normal biological processes are substantially incapable of cleaving that linkage before the corticosteroid mini-PEGylate is excreted from the body.
- less than 10%, less than 5%, or less than 1% of administered corticosteroid mini-PEGylate is metabolized so as to cleave the linkage between the corticosteroid and the mini-PEG before excretion.
- Such a conjugation has numerous useful features, particularly when the corticosteroid mini-PEGylate is to be administered locally, e.g., topically.
- systemic absorption of a corticosteroid can result in a variety of undesired side effects.
- the covalently bound mini-PEG inhibits systemic absorption of the corticosteroid, which can (1) lead to an accumulation of the compound in the dermal compartment and a consequent increase in its local concentration, and (2) a reduction or elimination of undesired side effects.
- the mini-PEG allows the conjugate to sufficiently permeate the cell membranes so as to interact with the intracellular target.
- conjugates including mini-PEG having a molecular weight of less than 5,000 Da have particularly suitable physico-chemical properties, suitable pharmacological effect, and suitable capability to enter cells, for use in compositions for topical application.
- a and B are each independently H, OH, Q-Cio alkyl (e.g., CH 3 ), C]-C ⁇ o alkoxy, or Ci-Cjo acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
- D and E are each independently H, Cj-Cio alkyl, Cj-Cio alkoxy, or Cj-Cio acyl;
- G, J, and K are each independently H or a halogen, in some embodiments H, Cl, F,
- X is a non-cleavable linking group between the C 21 position of the corticosteroid and the mini-PEG, including but not limited to alkylene, amido, oximo, S, NR, or O, where R is H or alkyl, in some embodiments H; mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da, in one embodiment between about 100 Da and about 10,000 Da, in another embodiment between about 100 Da and about 5.000 Da, in another embodiment between about 200 Da and about 5,000 Da, in another embodiment between about 500 Da and about 2,500 Da, in another embodiment between about 1,500 Da and about 2,000 Da;
- Y is H, OH, C-C 5 alkyl, C 1 -C 5 acyl, or C 1 -C 5 alkoxy;
- C 1 and CT are bonded together by either a single bond or a double bond.
- X is S, NR, or O. In some embodiments, X is S. In other embodiments, X is O. In still other embodiments, X is NR.
- Y is H, OH, or C 1 -C 5 alkoxy. In some embodiments, Y is C 1 - C 5 alkoxy. In some embodiments, Y is methoxy (MeO). In other embodiments, Y is hydroxyl (HO).
- n is an integer between 1 and 100
- m is an integer between 0 and 100
- m+n is 100 or less
- a and B are each independently H, OH, C 1 -CjO alkyl (e.g., CH 3 ), C 1 -CiO alkoxy, or Ci-C ⁇ o acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
- D and E are each independently H, C I -C JO alkyl, Ci-Cio alkoxy, or C 1 -CiO acyl;
- G, J, and K are each independently H or a halogen, in some embodiments 11, Cl, F,
- X is a non-cleavable linking group between the C 21 position of the corticosteroid and the mini-PEG, including but not limited to alkylene, amido, oximo, S, NR, or O, where R is H or alkyl, in some embodiments H; mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da, in one embodiment between about 100 Da and about 10,000 Da, in another embodiment between about 100 Da and about 5,000 Da, in another embodiment between about 200 Da and about 5,000 Da, in another embodiment between about 500 Da and about 2,500 Da, in another embodiment between about 1 ,500 Da and about 2,000 Da; each Y is H, OH, C-C 5 alkyl, C-C 5 acyl, or C 1 -C 5 alkoxy; and each C) and C 2 pair is bonded together by either a single bond or a double bond.
- each of the different moieties may be selected independently of one another for each instance of the corticosteroid and for Y. That is, there may be between 1 and n different corticosteroids linked to the mini-PEG, and between 0 and m different Y groups linked to the mini-PEG, and n+m is greater than or equal to
- the mini-PEG is a linear mini-PEG (lacking branches).
- n is 1 or 2
- m is 0 or 1
- n+m equals 2.
- the corticosteroids may be the same or different, and the Y groups may be the same or different.
- the mini-PEG is a branched mini-PEG, in which between 3 and 10 PEG groups emanate from a central core group.
- n is between 1 and 10
- m is between 0 and 10
- n+m is between 3 and 10.
- corticosteroid moieties there is one corticosteroid moiety, in other embodiments two corticosteroid moieties, in other embodiments three corticosteroid moieties, in other embodiments four corticosteroid moieties, in other embodiments five corticosteroid moieties, in other embodiments six corticosteroid moieties, in other embodiments seven corticosteroid moieties, in other embodiments eight corticosteroid moieties, in other embodiments nine corticosteroid moieties, and in still other embodiments ten corticosteroid moieties.
- Any number of the corticosteroids may be the same, or different, than one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same.
- Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
- the mini-PEG is a star mini-PEG, in which between 10 and 100 PEG groups emanate from a central core group.
- n is between 10 and 100
- m is between 0 and 100
- n+m is between 10 and 100.
- corticosteroid moieties in other embodiments between 40 and 50 corticosteroid moieties, in other embodiments between 50 and 60 corticosteroid moieties, in other embodiments between 70 and 80 corticosteroid moieties, in other embodiments between 80 and 90 corticosteroid moieties, and in still other embodiments between 90 and 100 corticosteroid moieties.
- Any number of the corticosteroids may be the same or different from one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same.
- Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
- the mini-PEG is a comb mini-PEG having multiple PEG chains grafted to a polymer backbone.
- Any number of the corticosteroids may be the same or different from one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same.
- Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
- X is S, NR, or O. In some embodiments, X is S. In other embodiments, X is O. In still other embodiments, X is NR.
- Y is H, OH, or C)-Cs alkoxy. In some embodiments, Y is Q- C ⁇ alkoxy. In some embodiments, Y is methoxy (MeO). In other embodiments, Y is hydroxy] (HO).
- the mini-PEG has a molecular weight of about 100 Da, or about 200 Da, or about 300 Da, or about 400 Da, or about 500 Da, or about 600 Da, or about 700 Da, or about 800 Da, or about 900 Da, or about 1000 Da, or about 1 100 Da.
- the mini-PEG has a molecular weight between about 100 Da and about 2,000 Da, in another embodiment between about 100 Da and about 500 Da, in another embodiment between about 200 Da and about 700 Da, in another embodiment between about 300 Da and about 800 Da, in another embodiment between about 400 Da and about 900 Da, in another embodiment between about 500 Da and about 1 ,000 Da.
- corticosteroids derivatives thereof, and structural analogs thereof are suitable for use in the compounds provided herein, including, but not limited to, alclometasone, beclomethasone, betamethasone, budesonide, chloroprednisone, clobetasol, clocortolone, cloprednol, corticosterone, cortisone, desonide, desoximetasone, desoxycorticosterone, dexamethasone, diflorasone, difluocortolone, flucloronide, fludrocortisone, flumethasone, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluprednisolone,
- flurandrenolide fluticasone, fluticasone proprionate, halobetasol, halometasone, hydrocortisone, meprednisone, methylprednisolone, paramethasone, prednicarbate, prednisolone, prednisone, prednival, prednylidine, tixocortol, triamcinolone, and triamcinolone acetonide.
- Table 1 The structures of some exemplary corticosteroids that can be used in the compounds provided herein are illustrated in Table 1. Derivatives and structural analogs of these corticosteroids can also be used in the compounds provided herein.
- Such analogs may include, but arc not limited to, proprionate derivatives (in one embodiment, beclomethasone proprionate, in another embodiment, betamethasone proprionate, in another embodiment, clobetasol proprionate, in another embodiment, fluticasone proprionate); valerate derivatives (in one embodiment, betamethasone valerate); furoate derivatives (in one embodiment, fluticasone furoate); acetate derivatives (in one embodiment, hydrocortisone acetate, in another embodiment, methylprednisolone acetate, in another embodiment, prednisolone acetate); and sodium phosphate derivatives (in one embodiment, prednisolone sodium phosphate).
- proprionate derivatives in one embodiment, beclomethasone proprionate, in another embodiment, betamethasone proprionate, in another embodiment, clobetasol proprionate, in another embodiment, fluticasone proprionate
- valerate derivatives in one embodiment, betamethasone valerate
- Triamcinolone I ⁇ amcinolone Acetonide plary compound is dexamethasone mim-PEGylate, illustrated below, which nces referred to herein as Compound 1
- mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da.
- the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
- Another exemplary compound is betamethasone mini-PEGylate, illustrated below, which is in some circumstances referred to herein as Compound 2.
- Betamethasone mini-PFGylate (Compound 2) wherein mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da. In one embodiment, the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
- the compounds provided herein can be prepared or obtained by any method known to one of skill in the art.
- the compounds can be synthesized from the corresponding non-PEGylated corticosteroids according to Scheme I: miniPEG-Y wherein R is the corticosteroid residue;
- Z ' and W are selected from (i) and (ii):
- Z is a nucleophilic group, including but not limited to, R'NH, HS, HO, ONH 2 , where R' is H or alkyl, and W is an electrophilic group, including but not limited to, CF 3 SO 2 O, P-CH 3 C 6 H 4 SO 2 O, P-MePhSO 2 O, PhSO 2 O, CH 3 SO 2 O, halide, NHS ester, haloacetamido, maleimido, halotriazino, or formyl;
- W is a nucleophilic group, including but not limited to, R'NH, HS, HO, ONH 2 , where R' is H or alkyl, and Z is an electrophilic group, including but not limited to, CF 3 SO 2 O, P-CH 3 C 6 H 4 SO 2 O, p-MePhSO 2 O, PhSO 2 O, CH 3 SO 2 O, halide, NHS ester, haloacetamido, maleimido, halotriazino, or formyl; and
- compositions are as defined herein.
- compositions comprising a compound provided herein, as an active ingredient, in combination with a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a mixture thereof.
- the compounds provided herein may be administered alone, or in combination with one or more other compounds provided herein, or one or more other compounds known in the art.
- the pharmaceutical compositions that include a compound provided herein can be formulated in various dosage forms for topical administration.
- the pharmaceutical compositions can also be formulated as modified release dosage forms, including delayed-, extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated-, fast-, targeted-, and programmed- release dosage forms.
- dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art (see, Remington: The Science and Practice of Pharmacy, supra; Modified-Release Drug Delivery Technology, 2nd Edition, Rathbone et al., Eds., Marcel Dekker, Inc.: New York, NY, 2008).
- the pharmaceutical compositions are provided in a dosage form for topical administration, which comprise a compound provided herein and one or more pharmaceutically acceptable excipients or carriers.
- compositions provided herein can be provided in a unit-dosage form or multiple-dosage form.
- a unit-dosage form refers to physically discrete a unit suitable for administration to a human and animal subject, and packaged individually as is known in the art. Each unit-dose contains a predetermined quantity of an active ingredient(s) sufficient to produce the desired therapeutic effect, in association with the required
- a unit-dosage form may be administered in fractions or multiples thereof.
- a multiple-dosage form is a plurality of identical unit-dosage forms packaged in a single container to be administered in segregated unit-dosage form.
- compositions provided herein can be administered at once, or multiple times at intervals of time. It is understood that the precise dosage and duration of treatment may vary with the age, weight, and condition of the patient being treated, and may be determined empirically using known testing protocols or by extrapolation from in vivo or in vitro test or diagnostic data. It is further understood that for any particular individual, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the formulations.
- compositions provided herein can be administered topically to the skin, orifices, or mucosa.
- topical administration includes dermal, intradermal, conjunctival, intracorneal, intraocular, ophthalmic, auricular, transdermal, nasal, vaginal, urethral, respiratory, and rectal administration.
- compositions provided herein can be formulated in any dosage forms that are suitable for topical administration, including emulsions, solutions, suspensions, creams, gels, hydrogels, ointments, dusting powders, dressings, elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols, irrigations, sprays, suppositories, bandages, and dermal patches.
- the topical formulation of the pharmaceutical compositions provided herein can also comprise liposomes, micelles, microspheres, nanosystems, and mixtures thereof.
- Topical efficacy depends on the activity of the particular corticosteroid mini-PEGylate selected for the treatment, its concentration in the composition, the permeability coefficient, the vehicle and excipients, and local metabolic processes.
- the compositions herein include a compound provided herein in a concentration of between 0.001 % and 99%, in some embodiments in a concentration of between 0.01 % and 50%, in some embodiments in a concentration of between 0.1%' and 25%, in some embodiments in a concentration of between 0.1% and 10%, in some embodiments in a concentration of between 10% and 25%, in some embodiments in a concentration of between 25% and 50%, in some embodiments in a concentration of between 50% and 99%, in some embodiments in a concentration of between 1% and 5%, in some embodiments in a concentration of between 5% and 10%, in some embodiments in a concentration of between 10% and 15%, in some embodiments in a concentration of between 15% and 20%, in some embodiments in a concentration of between 20% and 25%, in some embodiments in a concentration of between 25% and 30%, in some embodiments in a concentration of between 30% and 35%, in some embodiments in a concentration of between 35% and 40%, in some embodiments in a concentration of between 40% and
- Pharmaceutically acceptable carriers and excipients suitable for use in the topical formulations provided herein include, but are not limited to, aqueous vehicles, water-miscible vehicles, non-aqueous vehicles, antimicrobial agents or preservatives against the growth of microorganisms, stabilizers, solubility enhancers, isotonic agents, buffering agents, antioxidants, local anesthetics, suspending and dispersing agents, wetting or emulsifying agents, complexing agents, sequestering or chelating agents, penetration enhancers, cryoprotectants, lyoprotectants, thickening agents, and inert gases.
- compositions can also be administered topically by
- electroporation iontophoresis, phonophoresis, sonophoresis, or microneedle or needle-free injection, such as POWDERJECTTM (Chiron Corp., Emeryville, CA), and BIOJECTTM (Bioject Medical Technologies Inc., Tualatin, OR).
- Suitable ointment vehicles include oleaginous or hydrocarbon vehicles, including lard, benzoinated lard, olive oil, cottonseed oil, and other oils, white petrolatum; emulsifiable or absorption vehicles, such as hydrophilic petrolatum, hydroxystearin sulfate, and anhydrous lanolin; water-removable vehicles, such as hydrophilic ointment; water-soluble ointment vehicles, including polyethylene glycols of varying molecular weight; emulsion vehicles, either water-in-oil (W/O) emulsions or oil-in-water (OAV) emulsions, including cetyl alcohol, glyceryl monostearate, lanolin, and stearic acid (see, Remington: The Science and Practice of Pharmacy, supra). These vehicles are emollinous or hydrocarbon vehicles, including lard, benzoinated lard, olive oil, cottonseed oil, and other oils, white petrolatum;
- Suitable cream base can be oil-in-water or water-in-oil.
- Suitable cream vehicles may be water-washable, and contain an oil phase, an emulsifier, and an aqueous phase.
- the oil phase is also called the "internal" phase, which is generally comprised of petrolatum and a fatty alcohol such as cetyl or stearyl alcohol.
- the aqueous phase usually, although not necessarily, exceeds the oil phase in volume, and generally contains a humectant.
- the emulsifier in a cream formulation may be a nonionic, anionic, cationic, or amphoteric surfactant. This formulation may further include a skin penetrating agent.
- Lotions are liquid or semiliquid preparations that contain one or more active ingredients in an appropriate vehicle.
- the lotion is a suspension of the active ingredient in an appropriate vehicle, in one embodiment water.
- the lotion includes an emulsion having at least two phases, and the active ingredient is suspended or dissolved in one or more of the phases.
- the lotion includes a solution of the active ingredient.
- the lotion may include a glycol/water mixture, and optionally also one or more surfactants, fatty acid esters, and/or fatty alcohols such as those set forth above in the discussion of a cream formulation, optionally further including stabilizers such as an antioxidant and/or other adjuvants to improve the aesthetics of the lotion.
- the lotion may include an antimicrobial preservative.
- the compounds provided herein are included in an aerosol formulation, in one embodiment a homogeneous, aqueous-alcoholic emulsion system.
- the aerosol formulation upon actuation produces a stabilized, homogeneous, expandable foam, which in some embodiments breaks easily with shear.
- the aerosol formulation may further include a skin penetrating agent, in some embodiments an alcohol such as dodecanol or oleyl alcohol; an amine such as isopropyl amine, diisopropyl amine, triethyl amine, triethanol amine, diisopropanolamine or ethylene diamine; a carboxylic acid such as oleic acid, linoleic acid or linolenic acid; an ester, such as dibutyl sebacate, dibutyl phthalate, butyl benzoate or ethyl caprate; and/or other compounds, such as Azone, N methyl pyrollidone, bile salts and urea.
- the aerosol formulation may be actuated using one or more propellants known in the pharmaceutical or cosmetic fields.
- propellants include hydrocarbons such as propane, isobutane or dimethyl ether and
- Gels are semisolid, suspension-type systems. Single-phase gels contain organic macromolecules distributed substantially uniformly throughout the liquid carrier. Suitable gelling agents include, but are not limited to, crosslinked acrylic acid polymers, such as carbomers, carboxypolyalkylenes, and CARBOPOL ® ; hydrophilic polymers, such as polyethylene oxides, polyoxyethylene-polyoxypropylene copolymers, and polyvinylalcohol; cellulosic polymers, such as hydroxypropyl cellulose, hydroxyethyl cellulose, hydroxypropyl methylcellulosc, hydroxypropyl methylcellulose phthalate, and methylcellulose; gums, such as tragacanth and xanthan gum; sodium alginate; and gelatin.
- dispersing agents such as alcohol or glycerin can be added, or the gelling agent can be
- compositions provided herein can be administered rectally, urethrally, vaginally, or perivaginally in the forms of suppositories, pessaries, bougies, poultices or cataplasm, pastes, powders, dressings, creams, plasters, contraceptives, ointments, solutions, emulsions, suspensions, tampons, gels, foams, sprays, or enemas.
- These dosage forms can be manufactured using conventional processes as described in Remington: The Science and Practice of Pharmacy, supra.
- Rectal, urethral, and vaginal suppositories are solid bodies for insertion into body orifices, which are solid at ordinary temperatures but melt or soften at body temperature to release the active ingredient(s) inside the orifices.
- Pharmaceutically acceptable carriers utilized in rectal and vaginal suppositories include bases or vehicles, such as stiffening agents, which produce a melting point in the proximity of body temperature, when formulated with the pharmaceutical compositions provided herein; and antioxidants as described herein, including bisulfite and sodium metabisulfite.
- Suitable vehicles include, but are not limited to, cocoa butter (theobroma oil), glycerin-gelatin, carbowax (polyoxyethylene glycol), spermaceti, paraffin, white and yellow wax, and appropriate mixtures of mono-, di- and triglycerides of fatty acids, and hydrogels, such as polyvinyl alcohol, hydroxyethyl methacrylate, and polyacrylic acid;. Combinations of the various vehicles can also be used. Rectal and vaginal suppositories may be prepared by compressing or molding. The typical weight of a rectal and vaginal suppository is about 2 to about 3 g.
- compositions provided herein can be administered
- the pharmaceutical compositions provided herein can be administered intranasally or by inhalation to the respiratory tract.
- the pharmaceutical compositions can be provided in the form of an aerosol or solution for delivery using a pressurized container, pump, spray, atomizer, such as an atomizer using electrohydrodynamics to produce a fine mist, or nebulizer, alone or in combination with a suitable propellanl, such as 1 , 1 , 1 ,2-tetrafluoroethane or 1,1 ,1 ,2,3,3,3- heptafluoropropane.
- the pharmaceutical compositions can also be provided as a dry powder for insufflation, alone or in combination with an inert carrier such as lactose or phospholipids; and nasal drops.
- the powder can comprise a bioadhesive agent, including chitosan or cyclodextrin.
- Solutions or suspensions for use in a pressurized container, pump, spray, atomizer, or nebulizer can be formulated to contain ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active ingredient provided herein; a propellant as solvent; and/or a surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- compositions provided herein can be micronized to a size suitable for delivery by inhalation, such as about 50 micrometers or less, or about 10 micrometers or less.
- Particles of such sizes can be prepared using a comminuting method known to those skilled in the art, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- Capsules, blisters, and cartridges for use in an inhaler or insufflator can be formulated to contain a powder mix of the pharmaceutical compositions provided herein; a suitable powder base, such as lactose or starch; and a performance modifier, such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate.
- Other suitable excipients or carriers include, but are not limited to, dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose, and trehalose.
- the pharmaceutical compositions provided herein for inhaled/intranasal administration can further comprise a suitable flavor, such as menthol and levomenthol; and/or sweeteners, such as saccharin and saccharin sodium.
- compositions provided herein for topical administration can be formulated to be immediate release or modified release, including delayed-, sustained-, pulsed-, controlled-, targeted, and programmed release.
- modified release refers to a dosage form in which the rate or place of release of the active ingredient(s) is different from that of an immediate dosage form when administered by the same route.
- Modified release dosage forms include, but are not limited to, delayed-, extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated-, fast-, targeted-, and programmed-release dosage forms.
- compositions in modified release dosage forms can be prepared using a variety of modified release devices and methods known to those skilled in the art, including, but not limited to, matrix controlled release devices, osmotic controlled release devices, multiparticulate controlled release devices, ion-exchange resins, multilayered coatings, microspheres, liposomes, and combinations thereof.
- the release rate of the active ingredient(s) can also be modified by varying the particle sizes and polymorphorism of the active
- modified release include, but are not limited to, those described in U.S. Pat. Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 5,674,533; 5,059,595;
- compositions provided herein in a modified release dosage form can be fabricated using a matrix controlled release device known to those skilled in the art (see, Takada et al. in "Encyclopedia of Controlled Drug Delivery,” Vol. 2, Mathiowitz Ed., Wiley, 1999).
- the pharmaceutical compositions provided herein in a modified release dosage form is formulated using an erodible matrix device, which is water- swellable, erodible, or soluble polymers, including, but not limited to, synthetic polymers, and naturally occurring polymers and derivatives, such as polysaccharides and proteins.
- an erodible matrix device which is water- swellable, erodible, or soluble polymers, including, but not limited to, synthetic polymers, and naturally occurring polymers and derivatives, such as polysaccharides and proteins.
- Materials useful in forming an erodible matrix include, but are not limited to, chitin, chitosan, dextran, and pullulan; gum agar, gum arabic, gum karaya, locust bean gum, gum tragacanth, carrageenans, gum ghatti, guar gum, xanthan gum, and scleroglucan; starches, such as dextrin and maltodextrin; hydrophilic colloids, such as pectin; phosphatides, such as lecithin; alginates; propylene glycol alginate; gelatin; collagen; cellulosics, such as ethyl cellulose (EC), methylethyl cellulose (MEC), carboxymethyl cellulose (CMC), CMEC, hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), cellulose acetate (CA), cellulose propionate (CP), cellulose butyrate (CB), cellulose
- copolymers of ethacrylic acid or methacrylic acid (EUDRAGIT ® , Rohm America, Inc., Piscataway, NJ); poly(2-hydroxyethyl-methacrylate); polylactides; copolymers of L-glutamic acid and ethyl-L-glutamate; degradable lactic acid-glycolic acid copolymers; poly-D-(-)-3- hydroxybutyric acid; and other acrylic acid derivatives, such as homopolymers and copolymers of butylmethacrylate, methyl methacrylate, ethyl methacrylate, ethylacrylate, (2- dimethylaminoethyl)mcthacrylate, and (trimethylaminoethyl)rnethacrylate chloride.
- EUDRAGIT ® Rohm America, Inc., Piscataway, NJ
- poly(2-hydroxyethyl-methacrylate) polylactides
- the pharmaceutical compositions provided herein are formulated with a non-erodible matrix device.
- the active ingredient(s) is dissolved or dispersed in an inert matrix and is released primarily by diffusion through the inert matrix once administered.
- Materials suitable for use as a non-erodible matrix device include, but are not limited to, insoluble plastics, such as polyethylene, polypropylene, polyisoprene,
- chlorinated polyethylene polyvinylchloride, methyl acrylate-melhyl methacrylate copolymers, ethylene- vinyl acetate copolymers, ethylene/propylene copolymers, cthylene/cthyl acrylatc copolymers, vinyl chloride copolymers with vinyl acetate, vinylidene chloride, ethylene and propylene, ionomer polyethylene terephthalate, butyl rubbers, epichlorohydrin rubbers, ethylene/vinyl alcohol copolymer, ethylene/vinyl acetate/vinyl alcohol terpolymer, ethylene/vinyloxyethanol copolymer, polyvinyl chloride, plasticized nylon, plasticized polyethylene terephthalate, natural rubber, silicone rubbers, polydimethylsiloxanes, and silicone carbonate copolymers; hydrophilic
- the desired release kinetics can be controlled, for example, via the polymer type employed, the polymer viscosity, the particle sizes of the polymer and/or the active ingredient(s), the ratio of the active ingredient(s) versus the polymer, and other excipients or carriers in the compositions.
- compositions provided herein in a modified release dosage form can be prepared by methods known to those skilled in the art, including direct compression, dry or wet granulation followed by compression, and melt-granulation followed by compression.
- compositions provided herein in a modified release dosage form can be fabricated using an osmotic controlled release device, including, but not limited to, one- chamber system, two-chamber system, asymmetric membrane technology (AMT), and extruding core system (ECS).
- AMT asymmetric membrane technology
- ECS extruding core system
- such devices have at least two components: (a) a core which contains an active ingredient; and (b) a semipermeable membrane with at least one delivery port, which encapsulates the core.
- the semipermeable membrane controls the influx of water to the core from an aqueous environment of use so as to cause drug release by extrusion through the delivery port(s).
- the core of the osmotic device optionally includes an osmotic agent, which creates a driving force for transport of water from the environment of use into the core of the device.
- osmotic agents water-swcllable hydrophilic polymers, which are also referred to as "osmopolymers” and “hydrogels.”
- Suitable water-swellable hydrophilic polymers as osmotic agents include, but are not limited to, hydrophilic vinyl and acrylic polymers, polysaccharides such as calcium alginate, polyethylene oxide (PEO), polyethylene glycol (PEG), polypropylene glycol (PPG), poly(2-hydroxyethyl methacrylatc), poly(acrylic) acid, poly(methacrylic) acid, polyvinylpyrrolidone (PVP), crosslinked PVP, polyvinyl alcohol (PVA), PVA/PVP copolymers, PV ⁇ /PVP copolymers with hydropho
- PEO polyethylene oxide
- PEG poly
- polyurethanes containing large PEO blocks sodium croscarmellose, carrageenan, hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), carboxymethyl cellulose (CMC) and carboxyethyl, cellulose (CEC), sodium alginate, polycarbophil, gelatin, xanthan gum, and sodium starch glycolate.
- the other class of osmotic agents is osmogens, which are capable of imbibing water to affect an osmotic pressure gradient across the barrier of the surrounding coating.
- Suitable osmogens include, but are not limited to, inorganic salts, such as magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, potassium phosphates, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride, and sodium sulfate; sugars, such as dextrose, fructose, glucose, inositol, lactose, maltose, mannitol, raffinose, sorbitol, sucrose, trehalose, and xylitol; organic acids, such as ascorbic acid, benzoic acid, fumaric acid, citric acid, maleic acid, sebacic acid, sorbic acid, adipic acid, edetic
- Osmotic agents of different dissolution rates can be employed to influence how rapidly the active ingredient(s) is initially delivered from the dosage form.
- amorphous sugars such as MANNOGEM EZ (SPI Pharma, Lewes, DE) can be used to provide faster delivery during the first couple of hours to promptly produce the desired therapeutic effect, and gradually and continually release of the remaining amount to maintain the desired level of therapeutic or prophylactic effect over an extended period of time.
- the active ingredient(s) is released at such a rate to replace the amount of the active ingredient metabolized and excreted.
- the core can also include a wide variety of other excipients and carriers as described herein to enhance the performance of the dosage form or to promote stability or processing.
- Materials useful in forming the semipermeable membrane include various grades of acrylics, vinyls, ethers, polyamides, polyesters, and cellulosic derivatives that are water- permeable and water-insoluble at physiologically relevant pHs, or are susceptible to being rendered water-insoluble by chemical alteration, such as crosslinking.
- Suitable polymers useful in forming the coating include plasticized, unplasticized, and reinforced cellulose acetate (CA), cellulose diacetate, cellulose triacetate, CA propionate, cellulose nitrate, cellulose acetate butyrate (CAB), CA ethyl carbamate, CAP, CA methyl carbamate, CA succinate, cellulose acetate trimellitate (CAT), CA dimethylaminoacetate, CA ethyl carbonate, CA chloroacetate, CA ethyl oxalate, CA methyl sulfonate, CA butyl sulfonate, CA p-toluene sulfonate, agar acetate, amylose triacetate, beta glucan acetate, beta glucan triacetate, acetaldehydc dimethyl acetate, triacetate of locust bean gum, hydroxylated ethylene-vinyl acetate, EC, PEG, PPG, PEG/P
- Semipermeable membrane can also be a hydrophobic microporous membrane, wherein the pores are substantially filled with a gas and are not wetted by the aqueous medium but are permeable to water vapor, as disclosed in U.S. Pat. No. 5,798.1 19.
- Such hydrophobic but water-vapor permeable membrane are typically composed of hydrophobic polymers such as polyalkenes, polyethylene, polypropylene, polytetrafluoroethylene, polyacrylic acid derivatives, polyethers, polysulfones, polyethersulfones, polystyrenes, polyvinyl halides, polyvinylidene fluoride, polyvinyl esters and ethers, natural waxes, and synthetic waxes.
- the delivery port(s) on the semipermeable membrane can be formed post-coating by mechanical or laser drilling. Delivery port(s) can also be formed in situ by erosion of a plug of water-soluble material or by rupture of a thinner portion of the membrane over an indentation in the core. In addition, delivery ports can be formed during coating process, as in the case of asymmetric membrane coatings of the type disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220.
- the total amount of the active ingredient(s) released and the release rate can substantially by modulated via the thickness and porosity of the semipermeable membrane, the composition of the core, and the number, size, and position of the delivery ports.
- compositions in an osmotic controlled-release dosage form can further comprise additional conventional excipients or carriers as described herein to promote performance or processing of the formulation.
- the osmotic controlled-release dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art (see, Remington: The Science and Practice of Pharmacy, supra; Santus and Baker, /. Controlled Release 1995, 35, 1 - 21 ; Verma et al.. Drug Development and Industrial Pharmacy 2000, 26, 695-708; Verma et al., J. Controlled Release 2002, 79. 7-27).
- the pharmaceutical compositions provided herein are formulated as AMT controlled-release dosage form, which comprises an asymmetric osmotic membrane that coats a core comprising the active ingredient(s) and other pharmaceutically acceptable excipients or carriers. See, U.S. Pat. No. 5,612,059 and WO 2002/17918.
- the AMT controlled-release dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art, including direct compression, dry granulation, wet granulation, and a dip-coating method.
- the pharmaceutical compositions provided herein are formulated as ESC controlled-release dosage form, which comprises an osmotic membrane that coats a core comprising the active ingredient(s), a hydroxylethyl cellulose, and other pharmaceutically acceptable excipients or carriers.
- compositions provided herein in a modified release dosage form can be fabricated as a multiparticulate controlled release device, which comprises a multiplicity of particles, granules, or pellets, ranging from about 10 ⁇ m to about 3 mm, about 50 ⁇ m to about 2.5 mm, or from about 100 ⁇ m to about 1 mm in diameter.
- multiparticulates can be made by the processes known to those skilled in the art, including wet-and dry-granulation, extrusion/spheronization, roller-compaction, melt-congealing, and by spray-coating seed cores. See, for example, Multiparticulate Oral Drug Delivery; Marcel Dekker: 1994; and
- the resulting particles can themselves constitute the multiparticulate device or can be coated by various film-forming materials, such as enteric coatings or water-swcllable and water-soluble polymers.
- compositions provided herein can also be formulated to be targeted to a particular tissue, receptor, or other area of the body of the subject to be treated, including liposome-, resealed erythrocyte-, and antibody-based delivery systems.
- liposome-, resealed erythrocyte-, and antibody-based delivery systems examples include, but are not limited to, those disclosed in U.S. Pat. Nos. 6,316,652; 6,274,552; 6,271,359; 6,253,872; 6,139,865; 6,131,570; 6,120,751 ; 6,071 ,495; 6,060,082; 6,048,736; 6,039,975;
- the compounds and compositions provided herein may be administered to a variety of populations of patients. In some embodiments, such populations are not able to safely receive non-conjugated corticosteroids, because of heightened risk factors associated with systemic absorption of such non-conjugated corticosteroids. Thus, in one embodiment, because the compounds and compositions provided herein reduce or eliminate systemic exposure, as described herein, they can be used in patient populations that would otherwise be cautioned against using corticosteroid compositions. In another embodiment, the compounds and compositions provided herein are used in patient populations for whom administration of corticosteroid compositions is not of significant concern.
- the compounds and compositions provided herein may be administered to a pediatric population, in one embodiment children under the age of 2, in another embodiment children under the age of 12, in another embodiment children under the age of 18, in another embodiment children between the age of 2 and 12, in another embodiment children between the age of 2 and 6, in another embodiment children between the age 6 and 12, in another embodiment children between the age of 12 and 18.
- the compounds and compositions provided herein may be administered to adults, in one embodiment adults over the age of 18, in another embodiment adults between the ages of 18 and 65, in another embodiment adults between the ages of 18 and 75, in another embodiment adults over 65 years of age, in another embodiment adults over 75 years of age.
- the compounds and compositions provided herein may be administered to pregnant women.
- the compounds and compositions provided herein may be administered to women who are trying to become pregnant. In another embodiment, the compounds and compositions provided herein may be administered to women who are nursing an infant. In another embodiment, the compounds and compositions provided herein may be administered to a patient that is already taking a systemic corticosteroid. In another embodiment, the compounds and compositions provided herein may be administered to a patient that is already using a topical corticosteroid. In another embodiment, the compounds and compositions provided herein are dermally applied and the treated area covered with an occlusive bandage.
- a method of treating, preventing, or ameliorating one or more symptoms of a disorder, disease, or condition associated with inflammation in a subject which comprises administering to the subject a therapeutically effective amount of a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II.
- the subject is a mammal.
- the subject is a human.
- the subject is an animal other than a human.
- a method of treating, preventing, or ameliorating one or more symptoms of a disorder, disease, or condition associated with the glucocorticoid receptor in a subject which comprises administering to the subject a therapeutically effective amount of a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II.
- the subject is a mammal.
- the subject is a human.
- the subject is an animal other than a human.
- inflammatory or allergic diseases including systemic anaphylaxis and hypersensitivity disorders, atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), and mastocytosis; (2) inflammatory bowel diseases, including Crohn's disease, ulcerative colitis, ileitis, and enteritis; (3) vasculitis, and Behcet's syndrome; (4) psoriasis and inflammatory dermatoses, including dermatitis, eczema, atopic dermatitis, allergic contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus, HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, aphthae,
- the disorder, disease, or condition affects the skin or mucosal tissue.
- the disorder, disease, or condition is selected from the group consisting of inflammatory or allergic diseases, including systemic anaphylaxis and
- hypersensitivity disorders atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), and mastocytosis.
- the disorder, disease or condition is selected from the group consisting of psoriasis and inflammatory dermatoses, including dermatitis, eczema (including seborrhoeic and nummular), atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus. HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, cutaneous and discoid lupus erythematosus.
- the disorder, disease, or condition treatable with a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II, is an inflammatory disease.
- the inflammatory disease is an inflammatory bowel disease.
- the inflammatory bowel disease is Crohn's disease, ulcerative colitis, or pouchitis.
- the compounds or pharmaceutical compositions provided herein can be administered by inhalation, nasal, vaginal, rectal, sublingual, dermal, transdermal, or local routes of administration and can be formulated, alone or together, in suitable dosage unit with
- compositions suitable for each route of administration. Also provided is administration of the compounds or pharmaceutical compositions provided herein in a depot formulation, in which the active ingredient is released over a predefined time period.
- the pharmaceutical compositions provided herein are sterile, and are packaged so as to maintain their sterility until use.
- inflammatory or allergic diseases including systemic anaphylaxis and hypersensitivity disorders, atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), mastocytosis, psoriasis and inflammatory dermatoses, including dermatitis, eczema (including seborrhoeic and nummular), atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus, HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, or cutaneous or discoid lupus erythematosus or other conditions, disorders or diseases associated
- the dosage can be ranging from about 0.005 to about 0.05, from about 0.05 to about 0.5, from about 0.5 to about 5.0, from about 1 to about 15, from about 1 to about 20, or from about 1 to about 50 mg/kg per day.
- the dosage level is ranging from about 0.001 to about 100 mg/kg per day.
- the dosage level is ranging from about 0.01 to about 75 mg/kg per day.
- the dosage level is ranging from about 0.1 to about 50 mg/kg per day.
- the dosage level is ranging from about 0.5 to about 25 mg/kg per day.
- the dosage level is ranging from about 1 to about 20 mg/kg per day.
- glucocorticoid receptor activity comprising contacting a glucocorticoid receptor with a compound provided herein.
- the glucocorticoid receptor is expressed by a cell.
- the compounds provided herein can be combined with one or more antibacterial agents known in the art, including, but not limited to the group including amikacin, amoxicillin, ampicillin, arsphenamine, azithromycin, aztreonam, azlocillin, bacitracin, carbenicillin, cefaclor, cefadroxil, cefamandole, cefazolin, cephalexin, cefdinir, cefditorin, cefepime, cefixime, cefoperazone, cefotaxime, cefoxitin, cefpodoxime, cefprozil, ceftazidime, ceftibuten, ceftizoxime, ceftriaxone, cefuroxime, chloramphenicol, cilastin, ciprofloxacin, clarithromycin, clindamycin, cloxacillin, colistin, dalfopristin, demeclocycline
- the compounds provided herein can be combined with one or more antifungal agents known in the art, including, but not limited to the group including amorolfine, amphotericin B, anidulafungin, bifonazole, butenafine, butoconazole, caspofungin, ciclopirox, clotrimazole, econazole, fenticonazole, filipin, fluconazole, isoconazole, itraconazole, ketoconazole, micafungin, miconazole, naftifine, natamycin, nystatin, oxyconazole,
- antifungal agents known in the art, including, but not limited to the group including amorolfine, amphotericin B, anidulafungin, bifonazole, butenafine, butoconazole, caspofungin, ciclopirox, clotrimazole, econazole, fenticonazole, filipin, fluconazole,
- the compounds provided herein can be combined with one or more non-steroidal anti-inflammatory agents known in the art, including, but not limited to, aceclofenac, acemetacin, amoxiprin, aspirin, azapropazone, benorilate, bromfenac, carprofen, celecoxib, choline magnesium salicylate, diclofenac, diflunisal, etodolac, etoricoxib, fatelamine, fenbufen, fenoprofen, flurbiprofen, ibuprofen, indometacin, ketoprofen, ketorolac, lornoxicam, loxoprofen, lumiracoxib, meclo
- ECE endothelin converting enzyme
- calcineurin inhibitors such as pimecrolimus, thromboxane receptor antagonists, such as ifetroban
- potassium channel openers thrombin inhibitors, such as hirudin
- growth factor inhibitors such as modulators of PDGF activity and nerve growth factor (NGF); platelet activating factor (PAF) antagonists
- anti-platelet agents such as GPIIb/IIIa blockers (e.g., abciximab, eptifibatide, and tirofiban), P2Y(AC) antagonists (e.g., clopidogrel, ticlopidine and CS-747), and aspirin
- anticoagulants such as warfarin
- low molecular weight heparins such as enoxaparin
- squalene synthetase inhibitors include fibrates; bile acid sequestrants, such as questran; niacin; anti-atherosclerotic agents, such as ACAT inhibitors; MTP Inhibitors; calcium channel blockers, such as amlodipine besylate;
- potassium channel activators alpha-adrenergic agents; beta-adrenergic agents, such as carvedilol and metoprolol; antiarrhythmic agents; diuretics, such as chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide,
- trichloromethiazide polythiazide, benzothiazide, ethacrynic acid, ticrynafen, chlorthalidone, furosemde, muzolimine, bumetanide, triamterene, amiloride, and spironolactone
- thrombolytic agents such as tissue plasminogen activator (tPA), recombinant tPA, streptokinase, urokinase, prourokinase.
- anti- diabetic agents such as biguanides (e.g., metformin), glucosidase inhibitors (e.g., acarbose), insulins, meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, and glipizide), thiozolidinediones (e.g., troglitazone, rosiglitazone, and pioglitazone), and PPAR- gamma agonists; mineralocorticoid receptor antagonists, such as spironolactone and eplerenone; growth hormone secretagogues; aP2 inhibitors; phosphodiesterase inhibitors, such as PDE III inhibitors (e.g., cilostazol) and PDE V inhibitors (e.g.
- biguanides e.g., metformin
- farnesyl-protein transferase inhibitors include hormonal agents, such as glucocorticoids (e.g., cortisone), estrogens/antiestrogens, androgens/antiandrogens, progestins, and luteinizing hormone-releasing hormone antagonists, and octreotide acetate: microtubule-disruptor agents, such as
- ecteinascidins ecteinascidins
- microtubule-stabilizing agents such as pacitaxel, docetaxel, and epothilones A-F
- plant-derived products such as vinca alkaloids, epipodophyllotoxins, and taxanes
- topoisomerase inhibitors prenyl-protein transferase inhibitors
- cyclosporins steroids, such as prednisone and dexamethasone
- cytotoxic drugs such as azathioprine and cyclophosphamide
- TNF-alpha inhibitors such as tenidap
- anti-TNF antibodies or soluble TNF receptor such as etanercept, rapamycin, and leflunimide
- COX-2 cyclooxygenase-2
- celecoxib and rofecoxib retinoids
- miscellaneous agents such as, hydroxyurea, procarbazine, mitot
- Such other agents, or drugs can be administered, by a route and in an amount commonly used therefor, simultaneously or sequentially with the compounds provided herein, in one embodiment a compound of Formula 1, in another embodiment a compound of Formula II.
- a pharmaceutical composition containing such other drugs in addition to the compound provided herein can be utilized, but is not required.
- the pharmaceutical compositions provided herein include those that also contain one or more other active ingredients or therapeutic agents, in addition to a compound provided herein.
- the weight ratio of a compound provided herein to the second active ingredient can be varied, and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound provided herein is combined with a NSAID, the weight ratio of the compound to the NSADD can range from about 1,000:1 to about 1: 1,000, or about 200: 1 to about 1 :200. Combinations of a compound provided herein and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
- a composition provided herein is a topical dermatological foam, cream, gel, ointment, oil, or lotion for use in treating disorders, diseases, or conditions of the skin.
- the foam, cream, gel, ointment, oil, or lotion includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II.
- the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one fluocinolone moiety, in still another embodiment at least one clobetasol moiety, in another embodiment at least one hydrocortisone moiety, in another embodiment at least one desonide moiety, in another embodiment at least one fluocinonide moiety, in yet another embodiment at least one fluticasone moiety.
- the corticosteroid moiety is derivitized, for example, betamethasone proprionate, in other embodiments betamethasone valerate, in other embodiments clobetasol proprionate, in other embodiments fluticasone proprionate, in other embodiments hydrocortisone acetate.
- the topical dermatological foam, cream, gel, ointment, oil, or lotion is administered to a patient with an inflammatory or pruritic manifestation of a corticosteroid- responsive dermatosis.
- the patient may have a disorder, disease, or condition including one or more of erysthema, scaling, pruritis, and maceration.
- the patient has atopic dermatitis.
- the patient has a fungal (mycological) infection
- the foam, cream, gel, ointment, oil, or lotion also includes an antifungal agent.
- the fungal infection is symptomatic inflammatory tinea pedis, tinea cruris, or tinea corporis.
- the antifungal agent is clotrimazole.
- the patient has a dermatosis of the scalp, in one embodiment scalp psoriasis, in another embodiment atopic dermatitis.
- the composition includes a surface or local anaesthetic, in one embodiment promoxine.
- the composition includes one or more antibacterial agents, in some embodiments one or more agents selected from the group consisting of neomycin A, neomycin B, neomycin C, polymyxin B, colistin A. colistin B, and thonzonium.
- a composition provided herein is an otic suspension for use in treating disorders, diseases, or conditions of the ear.
- the otic suspension includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II.
- the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one hydrocortisone moiety.
- the corticosteroid moiety may be derivitized, for example, hydrocortisone acetate.
- the otic suspension is administered to a patient with acute otitis media caused by the presence of a infection, in some embodiments also having a tympanostomy tube.
- the suspension also includes an antibacterial agent, in one embodiment ciprofloxacin, in other embodiments one or more of neomycin A, neomycin B, neomycin C, thonzonium, colistin A, and colistin B.
- a composition provided herein is an ophthalmic ointment or suspension for use in treating disorders, diseases, or conditions of the eye.
- the ointment or suspension includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, in other embodiments a compound of Formula II.
- the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one prednisolone moiety.
- the corticosteroid moiety is prednisolone acetate.
- the ophthalmic ointment or suspension is administered to a patient with an inflammation or edema of the palpebral and bulbar conjunctiva, cornea, or anterior segment of the globe; uveitis; or corneal injury from chemical, radiation, or thermal burns, or penetration of a foreign body.
- the ointment or suspension also includes an anti-infective drag, in one embodiment tobramycin.
- the ointment or suspension also includes an antibacterial agent, in one embodiment sulfacetamide sodium.
- a composition provided herein is an aerosol intended for oral inhalation.
- the aerosol is administered using a pressurized, metered-dose inhaler, in another embodiment using an inhalation-driven multi-dose dry powder inhaler, in another embodiment using an air driven jet nebulizer.
- the aerosol includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II.
- the compound provided herein contains at least one beclomethasone moiety, in another embodiment at least one budesonide moiety, in another embodiment at least one flunisolide moiety, in another embodiment at least one triamcinolone moiety, in still another embodiment at least one triamcinolone acetonide moiety, and in yet another embodiment at least one fluticasone moiety.
- the corticosteroid moiety is derivitized, for example beclomethasone proprionate, in another embodiment fluticasone proprionate.
- the aerosol is administered to a patient with inflammation associated with asthma.
- the composition also includes a bronchodilator such as a selective beta 2 -agonist or beta 2 - andrenoceptor, in one embodiment formoterol fumarate, in another embodiment salmeterol, in another embodiment albuterol, in still another embodiment levalbuterol.
- a bronchodilator such as a selective beta 2 -agonist or beta 2 - andrenoceptor, in one embodiment formoterol fumarate, in another embodiment salmeterol, in another embodiment albuterol, in still another embodiment levalbuterol.
- a composition provided herein is a suspension for administration via nasal spray.
- the aerosol includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II.
- the compound provided herein contains at least one beclomethasone moiety, in another embodiment at least one budesonide moiety, in another embodiment at least one triamcinolone moiety, in still another embodiment at least one triamcinolone acetonide moiety, and in yet another embodiment at least one fluticasone moiety.
- the corticosteroid moiety is derivitized, for example
- the suspension is administered to a patient with seasonal or perennial allergic or nonallergic rhinitis.
- a composition provided herein is a rectal suspension for administration via an enema.
- the suspension includes a compound provided herein.
- the compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II.
- the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, and in another embodiment at least one hydrocortisone moiety.
- the corticosteroid moiety may be derivitized, for example, hydrocortisone acetate.
- the rectal suspension is administered to a patient with ulcerative colitis.
- the ulcerative colitis may include, for example, one or more of ulcerative proctitis, ulcerative proctosigmoiditis, and left- sided ulcerative colitis.
- compositions provided herein can also be provided as an article of manufacture using packaging materials well known to those of skill in the art. See, e.g., U.S. Pat. Nos. 5,323,907; 5,052,558; and 5,033,252.
- packaging materials include, but are not limited to, blister packs, bottles, tubes, inhalers, pumps, bags, vials, containers, syringes, and any packaging material suitable for a selected formulation and intended mode of administration and treatment.
- kits which, when used by the medical practitioner, can simplify the administration of appropriate amounts of active ingredients to a subject.
- the kit provided herein includes a container and a dosage form of a compound provided herein. Kits provided herein can further include devices that are used to administer the active ingredients. Kits provided herein can further include pharmaceutically acceptable vehicles that can be used to administer one or more active ingredients.
- Betamethasone mini-PEGylate was synthesized as follows:
- Betamethasone (commercially available) Betamethasone mesylate
- Betamethasone mini-PEGylate Compound 2
- Procedure 1 synthesis of betamethasone mesylate.
- 100 mg (0.255 mmol) of betamethasone were dissolved at room temperature in 2 ml of pyridine under magnetic stirring.
- 30.41 mg (0.301 mmol) of tricthylamine were added and the reaction mixture cooled in ice bath.
- 35.0 mg (0.305 mmol) of methanesulfonyl chloride were added.
- Dexamethasone mesylate Dexamethasone mini-PEGylate (Compound 1 )
- Procedure 3 synthesis of dexamethasone mini-PEGylate (Compound 1).
- mPEG 2 ooo-SH art n° PEG 1169 from IRIS Biotech GmbH (Marktredwitz, Germany)
- 1205 ⁇ l 13.05 mg, 0.326 mmol
- ethanolic NaOH 153.7 mg (0.326 mmol) of dexamethasone-21 -mesylate were added and dissolved under gentle heating.
- the purified products were characterized by mass spectrometry (ESI-MS) and NMR spectroscopy by comparison to the unconjugated corticosteroids and/or the corticosteroid mesylates.
- Dexamethasone mini-PEGylate (Compound 1) and betamethasone mini-PEGylate (Compound 2) were prepared and purified as described in Example 1 , and were tested in the HeLa/GR-luc model in order to assess their capability to enter the cell and to bind to the intracellular glucocorticoid receptor (GR).
- HeLa/GR-luc cells are human cervical epithelial cells stably transfected with a GR-responsive luciferase reporter. The GR pathway and the mechanism of action of its ligands are illustrated in FIG. 1, which is described in greater detail above in the Background.
- HeLa/GR-luc cells were seeded in a 96- well plate with 2.5 x 10 4 cells/well in 100 ⁇ l of Complete Growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL, Hygromycin B 100 ⁇ g/mL). The plate was incubated in a humidified incubator at 37 0 C with 5% CO 2 for 48 hours. Test item solutions (Compound 1, Compound 2, dexamethasone and betamethasone) for cell treatment were freshly prepared just before use by serial dilution of 10 mM in DMSO stock solution in serum free medium (DMEM. Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL). Serum free medium was used as negative control.
- DMEM Complete Growth medium
- Test item solutions Compound 1, Compound 2, dexamethasone and betamethasone
- the cell medium was replaced with 90 ⁇ I/well of serum free medium (DMEM, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL) and 10 ⁇ l/well of test item solution at various concentrations were added. 6 wells were tested for each test item concentration. 10 ⁇ I/well of serum free medium were added to control wells. Incubation of cells with the test items was carried on at 37°C, 5% CO 2 for 6 hours.
- serum free medium DMEM, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL
- the luciferase gene transcription is stimulated only after GR activation, it can be deduced that each of the two mini-PEGylates were able to enter the cell, and bind and activate the intracellular GR.
- the activities of the mini-PEGylates were lower than those of the corticosteroids.
- the EC 50 of dexamethasone was 2.46 nM
- the EC 5 0 of dexamethasone mini-PEGylate (Compound 3) was be 0.85 ⁇ M, nearly 350 times greater.
- the EC 50 of betamethasone was 3.2 nM
- Dexamethasone mini-PEGylate (Compound 1, prepared as described in Example 1) and dexamethasone were tested in the NIH3T3/NFkB-luc model in order to assess its antiinflammatory activity, expressed as a reduction of NF-kB activity.
- NF-kB transcription factors NF-kB and AP-I.
- NF-kB is known to be a main transcriptional factor to be activated after inflammatory stimuli and is involved in the transcriptional activation of many pro-inflammatory cytokines such as TNF- ⁇ and IL-6.
- pro-inflammatory cytokines such as TNF- ⁇ and IL-6.
- NIH3T3/NFkB-luc cells are murine fibroblasts stably transfected with a NF-kB responsive luciferase reporter; NF-kB activation was induced by adding the stimulus
- NIH3T3/NF£B-luc cells were seeded in a 96-well plate with 5 x 10 4 cells/well in 90 ⁇ l of Complete Growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL, Hygromycin B 100 ⁇ g/mL) and 10 ⁇ l of test item solution (Compound 1 , Compound 2, dexamethasone or betamethasone) at the appropriate concentration were added to each well (10 ⁇ I/well of Complete Growth medium were added to control wells).
- Complete Growth medium DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL, Hygromycin B 100 ⁇ g/mL
- test item solution Compound 1 , Compound 2, dexamethasone or betamethasone
- Dexamethasone and betamethasone solutions for cell treatment were freshly prepared just before use by serial dilution of 10 niM in DMSO stock solution in complete growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL, Hygromycin B 100 ⁇ g/mL).
- DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 ⁇ g/mL, Hygromycin B 100 ⁇ g/mL complete growth medium
- Compound 1 and compound 2 10 mM starting solutions in complete growth medium were prepared just before cell treatment. These solutions were then further diluted in the same medium to obtain the desired concentrations to be tested.
- LPS 1 ng/mL solution for NIH3T3/NFkB-luc cell treatment was prepared from serial dilutions in complete growth medium of a 1 mg/mL LPS stock solution prepared in sterile dH 2 O. Complete growth medium was used as negative control.
- dexamethasone mini-PEGylate reduces the activation of the luciferase reporter gene, as evidenced in FIG. 3 (a) by a reduction in the % induction with increasing concentration, and in FIG. 3(b) by a decrease in luminescence with increasing concentration.
- dexamethasone mini-PEGylate reduces the LPS-induced NF- kB activation, although with considerably higher IC 50 as compared with the corticosteroid.
- the IC 50 of dexamethasone was 6.12 nM
- the IC50 of dexamethasone mini- PEGylate was 99.43 ⁇ m, over 16,000 times greater.
- Betamethasone mini-PEGylate (Compound 2), as prepared in Example 1, and Betamethasone were tested in the NIH3T3/NFkB-luc model to assess its anti-inflammatory activity, espressed as a reduction of NF-kB activity. The same protocols were used as described above for Compound 1.
- FIGS. 4(a) and 4(b) illustrate the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NF£B-luc cells by betamethasone or betamethasone mini-PEGylate (Compound 2) expressed as (a) % of induction and (b) luminiscence.
- Compound 2 reduces the activation of the luciferase reporter gene, as evidenced by a reduction in the % induction with increasing concentration (a), and by a decrease in luminescence with increasing concentration (b). Because the luciferase reporter gene trasnscription is stimulated only after NF-kB activation, it can be deduced that betamethasone mini-PEGylate reduces the LPS-induced NF-kB activation, although with considerably higher IC 50 as compared with the corticosteroid. Specifically, the IC 50 of betamethasone was 8.2 nM, whereas the IC 50 of betamethasone mini-PEGylate (Compound 2) was 40.81 ⁇ M, about 5,000 times greater.
- dexamethasone mini-PEGylate (Compound I) a single IV administration to male Han Wistar rats was performed. Before administration, the animals were surgically prepared for serial blood sample collection from superior vena cava (i.e., were superior vena cava cannulated).
- Dexamethasone and dexamethasone mini-PEGylate (Compound 1 ) were formulated in 8% ethanol in saline. The dexamethasone formulation was filtered due to turbidity.
- the administered doses were 1.05 mg/kg for dexamethasone, and 18 mg/kg for dexamethasone mini-PEGylate (Compound 1). The doses in this Example were not equimolar, because the theoretical equimolar dose of Dexamethasone (3 mg/kg) was near the solubility limit of this compound.
- FIG. 5(a) illustrates the mean plasma concentration over time of dexamethasone
- FIG. 5(b) illustrates the mean plasma concentration over time of dexamethasone mini-PEGylate (Compound 1).
- FIG. 5(c) illustrates the mean plasma concentration over time both of dexamethasone and Compound 1, on the same scale.
- Table 2 summarizes estimated PK parameters for dexamethasone and dexamethasone mini-PEGylate (Compound 1) based on the NCOMP and decay fitting.
- the dexamethasone mini-PEGylate (Compound 1) has different pharmacokinetic properties than dexamethasone, e.g., a higher clearance and, as a consequence, a shorter blood half-life.
- the higher solubility of dexamethasone mini-PEGylate (Compound 1) also results in a blood compartmentalization (the volume of distribution corresponds to about the mean rat blood volume) if compared to a higher V ss for dexamethasone, reflecting its wider body distribution.
- Betamethasone To evaluate IV pharmacokinetics of Compound 2 in comparison with non-PEGylated molecule Betamethasone, a single IV administration to male Sprague Dawley SD rats was performed. Betamethasone and Compound 2 were formulated in 8% ethanol in saline at 0.6 mg/mL and 3.6 mg/mL, thus resulting in a final administered dose of 3 mg/kg and 18 mg/kg respectively, which was approximately equimolar. At each sampling time, blood samples of approximately 0.8 mL were collected under light isofluorane anaesthesia from the retro-orbital sinus of 3 males of each group, each animal being sampled on each day at 3 timepoints. Blood samples in triplicate were collected at the following timepoints after treatment: 5 min, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h and 8 h, as shown in Table 3.
- Elution solvents were represented by MiIIiQ water and acetonitrile for mobile phase A and B, respectively; the starting eluent corresponded to phase A/B 80:20 with a subsequent gradient up to 100% (autosampling tray kept at 4°C).
- Plasma samples were purified before quantitation in order to eliminate any possible source of interference with the analysis itself by mean of SPE (Solid Phase Extraction) technique using the cartridges Waters Oasis ® HLB lcc/l Omg and methanol as final eluent.
- compartmentalization (the volume of distribution corresponds to about the mean rat blood volume) if compared to a higher V ss for betamethasone, reflecting its wider body distribution.
- Dexamethasone and dexamethasone mini-PEGylate were formulated in acetone/propylene glycol 50/50 to a final administered dose of 5 mg/kg and 30 mg/kg respectively.
- Blood samples were collected at 8 different timepoints after administration, and were frozen at -8O 0 C until analysis by HPLC. The blood collection, solid phase extraction, and HPLC-UV analysis were the same as described in Example 4.
- FIG. 7 illustrates the measured plasma concentration of dexamethasone after a single dermal administration to three rats (limit of detection 79 nM).
- the analysis of plasma samples from rats administered dexamethasone mini-PEGylate (Compound 1) did not reveal levels above the limit of detection (179 nM, calibration curve in rat plasma) at any timepoint. This result suggests that dexamethasone mini-PEGylate is not absorbed systemically following administration. In view of the results of Example 4, even if some dexamethasone mini-PEGylate were systemically absorbed, it would be quickly eliminated.
- Betamethasone and Compound 2 were formulated in acetone/propylene glycol 50/50 at, respectively, 12.5 mg/mL and 75 mg/niL corresponding to an administered dose of 5 mg/kg and 30 mg/kg for betamethasone and Compound 2 respectively.
- Blood samples were collected in triplicate (each animal being sampled on each day at 3 timepoints) at the following timepoints after treatment: 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h and 24 h.
- the blood collection and the SPE sample processing with subsequent quantitative analysis by HPLC-UV were carried out according to the same experimental procedure described above.
- Compound 2 would be quickly eliminated, most likely via renal filtration.
- mice were shaved on their abdominal side and sensitized by topical application of 100 ⁇ l 1.5% oxazolone in acetone.
- the ear thickness of left and right ears was measured; subsequently, the right ears were challenged by topical application of 20 ⁇ l 2.5% oxazolone in ethanol (10 ⁇ l on each side of the ear). As a control, 20 ⁇ l ethanol were applied to the left ears.
- Compound 2 was tested at three different concentration in aqueous solution: 5%, 0.5% and 0.05%. 20 ⁇ l of this solution (10 ⁇ l on either side of the ear that was challenged with oxazolone) were topically applied on challenged ears on days 4, 5, 6 and 7 (treatment on day 7 occurred 30 min after challenged). In this example, Compound 2 was not compared to betamethasone, but was tested alone. Diprosone cream (0.05% betamethasone diproprionate, Schering-Plough, Kenilworth, NJ) was used as a positive control, the treatment schedule for which was different; specifically, the diprosone cream was applied only once, on day 7 (30 min after challenge).
- Infiltrating cells were enumerated both as infiltrating cells per section (semiquantitative fashion) and as number of cells per mm 2 tissue (quantitative fashion).
- Dexamethasone mini-PEGylate (Compound 1) as prepared above in Example 1 was tested in various in vitro cytokine secretion assays in order to assess its anti-inflammatory activity; in particular, the effect of Compound 1 on the secretion of IFN- ⁇ , TNF- ⁇ and IL-6 from stimulated human PBMC was evaluated.
- IFN- ⁇ , TNF- ⁇ and IL-6 are pro-inflammatory cytokines involved in various inflammatory conditions; their gene transcription activation is under the control of one of the main transcriptional factor to be activated after pro-inflammatory stimuli, NF-kB.
- PBMC Peripheral Blood Mononuclear Cells
- FIGS. 9(a)-9(c) illustrate the results of an experiment comparing the inhibition of the cytokine secretion ((a) TNF- ⁇ , (b) IFN- ⁇ , (c) IL-6) by dexamethasone mini-PEGylate
- Compound 1 expressed as a percent inhibition of the control secretion induced by the stimulus. Compound 1 was tested at 8 different concentrations in a range between 300 nM and 1 mM, while reference compound (dexamethasone) was tested in a range between 0.1 nM and 1 ⁇ M.
- FIGS. 9(a)-9(c) illustrate, Compound 1 is able to reduce TNF- ⁇ , IFN- ⁇ and IL-6 secretion in stimulated human PBMC, showing a good concentration-response curve in this experiment. Therefore, it can be deduced that dexamethasone mini-PEGylate (Compound 1) is still able to inhibit cytokine secretion, showing an anti-inflammatory activity also after
- FIGS. 10, 1 1, and 12 illustrate the inhibition curves derived from different in vitro assays with relative IC 50 values.
- FIGS. 10(a) and 10(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for TNF- ⁇ inhibition
- FIGS. 1 l(a) and 1 l(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for IFN- ⁇ inhibition
- FIGS. 12(a) and 12(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for IL-6 inhibition.
- the results are expressed as a percent of control specific activity ((measured specific activity/control specific activity) x 100) obtained in the presence of Compound 1 or dexamethasone.
- IC 50 values concentration causing a half-maximal inhibition of control specific activity
- IC 50 of Compound 1 was: over 6,800 times greater in the case of TNF- ⁇ inhibition assay, 790 times greater in the case of IFN- ⁇ inhibition assay, and 372 times greater in the case of 1L-6 inhibition assay.
Abstract
Provided herein are mini-PEGylated corticosteroids that are useful for modulating glucocorticoid receptor activity, methods of making same, and pharmaceutical compositions thereof Also provided herein are methods for their use for treating, preventing, or ameliorating one or more symptoms of a glucocorticoid receptor-mediated disorder, disease, or condition, such as inflammation
Description
MINI-PEGYLATED CORTICOSTEROIDS, COMPOSITIONS INCLUDING SAME, AND METHODS OF MAKING AND USING SAME
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Application No. 61/223,346, filed July 6, 2009; the disclosure of which is incorporated herein by reference in its entirety.
FIELD
[0002] This application generally relates to corticosteroids, and more specifically to corticosteroid-based compositions for topical use.
BACKGROUND
[0003] Corticosteroids are potent anli-inflammalories. FIG. 1 illustrates the pathway by which corticosteroids enter the cell and interact with the glucocorticoid receptor (GR). Before ligand binding, the GR exists as a large multiunit complex in the cytoplasm, which includes 2 molecules of heat shock protein (hsp) 90, which act as chaperones. After activation by binding of the corticosteroid, the GR dissociates from the chaperone proteins and translocates to the nucleus. In the nucleus, the GR binds as a homodimer to a specific palindromic DNA sequence, termed a GRE, located in the regulatory regions of the target genes. The bound GR homodimers interacts with the basal transcriptional machinery shown that includes TATA-binding protein (TBP). associated transcription factors (TAFs and TFIIs), and RNA polymerase II (pol II). The interaction between GR and the basal transcription complex modulates transcription of the GR target genes. Different model of transcription regulation by the GR-ligand complex have been described (positive and negative regulation). In addition, the GR may interact via protein-protein interaction with other transcription factors, e.g., activator protein (AP)-I and nuclear factor-kB (NF-kB); in this case, the gene expression is controlled by the GR without binding to DNA. The active hormone-GR complex has a pleiotropic effect on genes involved in several processes. The biological effects of corticosteroids can be summarized as anti-inflammatory/
immunosuppressive, metabolic, and toxic. Anti-inflammatory and immunosuppressive effects may be useful, whereas metabolic and toxic effects are typically undesirable.
[0004] Because of their utility, a number of corticosteroid-based compositions are commercially available. However, prolonged application of steroid hormones for therapeutic purposes may involve higher risks of side effects, particularly when the administered systemically. Compositions intended for topical use may be formulated, e.g., as nasal sprays, as inhalers, as creams or ointments for dermal, ocular, or rectal administration. However, even if such compositions are applied only to a specific part of the body, the corticosteroid may be absorbed systemically. Because of its wide activity, systemic exposure to the corticosteroid may cause a variety of undesired side effects, for example, to die skeleton and muscles (muscle atrophy and osteoporosis), eyes (glaucoma), endocrine system and metabolism (Cushing's syndrome, diabetes, adrenal atrophy and hypogonadism), cardiovascular and immune systems, and gastrointestinal system. In particular, if the corticosteroid crosses the blood-brain barrier, it may adversely affect the central nervous system. Side effects affecting skin could range from more cosmetic aspects (e.g., teleangiectasia, hypertrichosis) to more serious disabling situations (e.g., atrophy, erythema, delayed wound healing).
[0005J Taken together, the side effects of corticosteroid therapy are a limiting factor for their applicability. Thus, there is a need for corticosteroid compositions that result in lower systemic exposure and/or lower side effects, while maintaining an acceptable potency.
SUMMARY
[0006] Provided herein are mini -PEGyI ated corticosteroids, compositions including same, and methods of making and using same.
[0007] In one embodiment, provided herein is a compound of Formula I:
-9-
 wherein A and B are each independently H, OH, C]-CiO alkyl, C]-Cιo alkoxy, or C Cio acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
wherein A and B are each independently H, OH, C]-CiO alkyl, C]-Cιo alkoxy, or C Cio acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
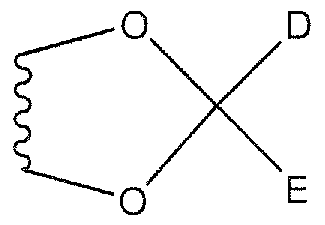
D and E arc each independently H, C|-Cio alkyl, C|-Cio alkoxy, or Ci-Cio acyl;
G, J, and K are each independently H or a halogen;
L1 and L2 arc (i) both H, or (ii) one is H and the other OH, or (iii) together form =0;
X is a non-cleavable linking group between the C2] position of the corticosteroid and the mini-PEG; mini-PEG is polyethylene glycol having a molecular weight between about 100 and about 20,000 Da;
Y is H, OH, Ci-C5 alkyl, C1-C5 acyl, or C,-C5 alkoxy; and C] and C2 are bonded together by either a single bond or a double bond.
[0008] In another embodiment, provided herein is a compound of Formula II:

A and B are each independently H, OH, C[-Cιo alkyl (e.g., CHj1), C|-Cjo alkoxy, or C]-C)O acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:

D and E are each independently H, C]-CiO alkyl, Ci-Qo alkoxy, or Cj-Cιo acyl;
G, J, and K are each independently H or a halogen, in some embodiments H, Cl, F,
Br, or I; each L1 and L2 pair is (i) both H, or (ii) one is H and the other OH, or (iii) together form =0;
each X is a non-cleavable linking group between the C2] position of the corticosteroid and the mini-PEG; mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da; each Y is H, OH, Ci-C5 alkyl, Ci-C5 acyl, or C-C5 alkoxy; and each Ci and C2 pair is bonded together by either a single bond or a double bond. [0009] In another embodiment, provided herein is a compound having the formula: Z X mini-PEG wherein Z is a corticosteroid; wherein mini-PEG is polyethylene glycol (PEG) having a molecular weight between about 100 Da and about 20,000 Da; and wherein X is S, O, or NR, where R is H or alkyl. In some embodiments, the corticosteroid is selected from the group consisting of: beclomethasone, betamethasone, budesonide, clobetasol, corticosterone, cortisone, desonide, desoximetasone, desoxycorticosterone, dexamethasone, difluocortolone, flumethasone, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluticasone, fluticasone proprionate, halobetasol, hydrocortisone, methylprednisolone, prednicarbate, prednisolone, prednisone, triamcinolone, and triamcinolone acetonide.
[0010] In another embodiment, provided herein is a pharmaceutical composition comprising a compound provided herein, and one or more pharmaceutically acceptable carriers or excipients. In some embodiments, the composition is formulated as a topical dosage form. In some embodiments, the topical dosage form is selected from the group consisting of emulsions, solutions, suspensions, creams, gels, oils, hydrogels, ointments, dusting powders, dressings, elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols, irrigations, sprays, suppositories, bandages, and dermal patches.
[0011] In another embodiment, provided herein is a method for the treatment, prevention, or amelioration of one or more symptoms of a glucocorticoid receptor-mediated disorder, disease, or condition in a subject, which comprises administering to the subject a therapeutically effective amount of a compound or composition provided herein.
[0012] In another embodiment, provided herein is a method for modulating glucocorticoid receptor activity, comprising contacting a glucocorticoid receptor with a compound or composition provided herein.
BRIEF DESCRIPTION OF DRAWINGS
[0013] FIG. 1 illustrates the pathway by which a corticosteroid interacts with the glucocorticoid receptor.
[0014] FIG. 2 illustrates the results of an experiment comparing the activation of the luciferase reporter gene in HeLa/GR-luc cells by (a) dexamethasone or dexamcthasonc mini- PEGylate (Compound 1), and (b) betamethasone or betamethasone mini-PEGylare (Compound
2).
[0015] FIG. 3 illustrates the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NFkB-luc cells by dexamethasone or dexamethasone mini-PEGylate expressed as (a) % induction and (b) luminescence.
[0016] FIG. 4 illustrates the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NFkB-luc cells by betamethasone or betamethasone mini-PEGylate expressed as (a) % induction and (b) luminescence.
[0017] FIG. 5 illustrates the mean plasma concentrations as a function of time after dose following a single IV administration in rats of (a) dexamethasone and (b) dexamethasone mini- PEGylate; (c) illustrates the mean plasma concentrations of (a) and (b) in a single graph.
[0018] FIG. 6 illustrates the mean plasma concentrations as a function of time after dose following a single IV administration in rats of (a) betamethasone and (b) betamethasone mini- PEGylate; (c) illustrates the mean plasma concentrations of (a) and (b) in a single graph.
[0019] FIG. 7 illustrates the plasma concentrations of dexamethasone as a function of time after dose following dermal application to rats of dexamethasone (limit of detection: 79 nM); there was no measurable plasma concentration following dermal application to rats of dexamethasone mini-PEGylate (limit of detection: 179 nM).
[0020] FIG. 8 illustrates the average plasma concentration of betamethasone as a function of time after dose following dermal application to rats of betamethasone; there was no measurable plasma concentration following dermal application to rats of betamethasone mini-PEGylate (limit of detection: 37.6 nM).
[0021] FIG. 9 illustrates the results of an experiment comparing the cytokine secretion of (a) TNF-α, (b) IFN-γ, and (c) IL-6 by dexamethasone mini-PEGylate expressed as a percent inhibition of the control secretion induced by the stimulus.
[0022] FIG. 10 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human TNF-α secretion.
[0023] FIG. 1 1 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human IFN-γ secretion.
[0024] FIG. 12 illustrates the results of an experiment comparing the effect of (a) dexamethasone mini-PEGylate and (b) dexamethasone on human IL-6 secretion.
DETAILED DESCRIPTION
Definitions
[0025] To facilitate understanding of the disclosure set forth herein, a number of terms are defined below.
[0026] Generally, the nomenclature used herein and the laboratory procedures in organic chemistry, medicinal chemistry, and pharmacology described herein are those well known and commonly employed in the art. Unless defined otherwise, all technical and scientific terms used herein generally have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. In the event that there is a plurality of definitions for a term used herein, those in this section prevail unless stated otherwise.
[0027J The term "subject" refers to an animal, including, but not limited to, a primate (e.g., human), cow, pig, sheep, goat, horse, dog, cat, rabbit, rat, guinea pig, or mouse. The terms '"subject" and "patient" are used interchangeably herein in reference, for example, to a
mammalian subject, such as a human subject, in one embodiment, a human, in another embodiment, a non-human.
[0028] The terms "treat," '"treating," and "treatment" are meant to include alleviating or abrogating a disorder, disease, or condition, or one or more of the symptoms associated with the disorder, disease, or condition; or alleviating or eradicating the cause(s) of the disorder, disease, or condition itself.
[0029] The terms "prevent," "preventing," and "prevention" are meant to include a method of delaying and/or precluding the onset of a disorder, disease, or condition, and/or its attendant symptoms; barring a subject from acquiring a disorder, disease, or condition; or reducing a subject's risk of acquiring a disorder, disease, or condition.
[0030] The term "therapeutically effective amount" are meant to include the amount of a compound that, when administered, is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the disorder, disease, or condition being treated. The term "therapeutically effective amount" also refers to the amount of a compound that is sufficient to elicit the biological or medical response of a biological molecule (e.g., a protein, enzyme, RNA, or DNA), cell, tissue, system, animal, or human, which is being sought by a researcher, veterinarian, medical doctor, or clinician.
[0031] The term "pharmaceutically acceptable carrier," "pharmaceutically acceptable excipient," "physiologically acceptable carrier," or "physiologically acceptable excipient" refers to a pharmaceutically-acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, solvent, or encapsulating material. In one embodiment, each component is "pharmaceutically acceptable" in the sense of being compatible with the other ingredients of a pharmaceutical formulation, and suitable for use in contact with the tissue or organ of humans and animals without excessive toxicity, irritation, allergic response, immunogenicity, or other problems or complications, commensurate with a reasonable benefit/risk ratio. See, Remington; The Science and Practice of Pharmacy, 21st Edition, Lippincott Williams & Wilkins:
Philadelphia, PA, 2005; Handbook of Pharmaceutical Excipients, 5th Edition, Rowe et al., Eds., The Pharmaceutical Press and the American Pharmaceutical Association: 2005; and Handbook of Pharmaceutical Additives, 3rd Edition, Ash and Ash Eds., Gower Publishing Company: 2007;
Pharmaceutical P reformulation and Formulation, Gibson Ed., CRC Press LLC: Boca Raton, FL, 2004.
[0032] The term "about" or "approximately" means an acceptable error for a particular value as determined by one of ordinary skill in the art, which depends in part on how the value is measured or determined. In certain embodiments, the term "about" or "approximately" means within 1, 2, 3, or 4 standard deviations. In certain embodiments, the term "about" or
"approximately" means within 50%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1 %, 0.5%, or 0.05% of a given value or range.
[0033] The terms "active ingredient" and "active substance" refer to a compound, which is administered, alone or in combination with one or more pharmaceutically acceptable excipients, to a subject for treating, preventing, or ameliorating one or more symptoms of a condition, disorder, or disease.
[0034] The terms "drug," "therapeutic agent," and "chemotherapeutic agent" refer to a compound, or a pharmaceutical composition thereof, which is administered to a subject for treating, preventing, or ameliorating one or more symptoms of a condition, disorder, or disease.
Compounds [0035] In one embodiment, compounds provided herein have the following formula:
-mini-PEG wherein Z is a corticosteroid; mini-PEG is polyethylene glycol (PEG) having a molecular weight between about 100 Da and about 20,000 Da; and X is a non-cleavable linking group between the corticosteroid and the mini-PEG. Selection of an appropriate linking group X may, in some embodiments, be based on one or more of the following factors: ease of synthesis, chemical stability, and biocompatibility, such as an expectation of low adverse effects of the linker and its metabolites. In some embodiments, X is alkylene, amido, oximo, S, O, or NR, wherein R is hydrogen or alkyl. In some embodiments, X is S.
[0036] In some embodiments, the C21 position provides a convenient location to covalently bond the mini-PEG to the corticosteroid via linker X. The mini-PEG may alternatively be
bonded to other positions of the corticosteroid via linker X. In some embodiments, the mini- PEG has a molecular weight between about 500 and about 2,000 Da, while in other embodiments the mini-PEG has a molecular weight in one of the ranges identified herein. The terminal end of the mini-PEG may also be modified, as described in greater detail herein. In some embodiments, functionalizing the terminal end of the mini-PEG can reduce the reactivity of the mini-PEG and/or can enhance the convenience of synthesizing the compound.
[0037] The linkage between the corticosteroid and the mini-PEG is non-cleavable, meaning that normal biological processes are substantially incapable of cleaving that linkage before the corticosteroid mini-PEGylate is excreted from the body. For example, less than 10%, less than 5%, or less than 1% of administered corticosteroid mini-PEGylate is metabolized so as to cleave the linkage between the corticosteroid and the mini-PEG before excretion. Such a conjugation has numerous useful features, particularly when the corticosteroid mini-PEGylate is to be administered locally, e.g., topically. As described above, systemic absorption of a corticosteroid can result in a variety of undesired side effects. Without being bound by any theory, it is believed that, the covalently bound mini-PEG inhibits systemic absorption of the corticosteroid, which can (1) lead to an accumulation of the compound in the dermal compartment and a consequent increase in its local concentration, and (2) a reduction or elimination of undesired side effects. Additionally, without being bound by any theory, it is believed that as compared to a conjugate formulated with larger PEG (e.g., PEG having a molecular weight of greater than 20,000 Da), the mini-PEG allows the conjugate to sufficiently permeate the cell membranes so as to interact with the intracellular target. Without wishing to be bound by theory, it is believed that conjugates including mini-PEG having a molecular weight of less than 5,000 Da have particularly suitable physico-chemical properties, suitable pharmacological effect, and suitable capability to enter cells, for use in compositions for topical application.
[0038] In some embodiments, compounds provided herein are described by Formula 1:
 wherein A and B are each independently H, OH, Q-Cio alkyl (e.g., CH3), C]-Cιo alkoxy, or Ci-Cjo acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:
wherein A and B are each independently H, OH, Q-Cio alkyl (e.g., CH3), C]-Cιo alkoxy, or Ci-Cjo acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula:

D and E are each independently H, Cj-Cio alkyl, Cj-Cio alkoxy, or Cj-Cio acyl; G, J, and K are each independently H or a halogen, in some embodiments H, Cl, F,
Br, or I;
L1 and L2 are (i) both H, or (ii) one is H and the other OH. or (iii) together form =0;
X is a non-cleavable linking group between the C21 position of the corticosteroid and the mini-PEG, including but not limited to alkylene, amido, oximo, S, NR, or O, where R is H or alkyl, in some embodiments H; mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da, in one embodiment between about 100 Da and about 10,000 Da, in another embodiment between about 100 Da and about 5.000 Da, in another embodiment between about
200 Da and about 5,000 Da, in another embodiment between about 500 Da and about 2,500 Da, in another embodiment between about 1,500 Da and about 2,000 Da;
Y is H, OH, C-C5 alkyl, C1-C5 acyl, or C1-C5 alkoxy; and
C1 and CT are bonded together by either a single bond or a double bond.
[0039] In some embodiments, X is S, NR, or O. In some embodiments, X is S. In other embodiments, X is O. In still other embodiments, X is NR.
[0040] In some embodiments, Y is H, OH, or C1-C5 alkoxy. In some embodiments, Y is C1- C5 alkoxy. In some embodiments, Y is methoxy (MeO). In other embodiments, Y is hydroxyl (HO).
[0041] In other embodiments, compounds provided herein are described by Formula II:

A and B are each independently H, OH, C1-CjO alkyl (e.g., CH3), C1-CiO alkoxy, or Ci-Cιo acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following
formula:

D and E are each independently H, CI -CJO alkyl, Ci-Cio alkoxy, or C1-CiO acyl; G, J, and K are each independently H or a halogen, in some embodiments 11, Cl, F,
Br, or I; each L] and L" pair is (i) both H, or (ii) one is H and the other OH, or (iii) together form =0;
X is a non-cleavable linking group between the C21 position of the corticosteroid and the mini-PEG, including but not limited to alkylene, amido, oximo, S, NR, or O, where R is H or alkyl, in some embodiments H; mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da, in one embodiment between about 100 Da and about 10,000 Da, in another embodiment between about 100 Da and about 5,000 Da, in another embodiment between about 200 Da and about 5,000 Da, in another embodiment between about 500 Da and about 2,500 Da, in another embodiment between about 1 ,500 Da and about 2,000 Da; each Y is H, OH, C-C5 alkyl, C-C5 acyl, or C1-C5 alkoxy; and each C) and C2 pair is bonded together by either a single bond or a double bond.
[0042] In some embodiments of compounds of Formula II, each of the different moieties may be selected independently of one another for each instance of the corticosteroid and for Y. That is, there may be between 1 and n different corticosteroids linked to the mini-PEG, and between 0 and m different Y groups linked to the mini-PEG, and n+m is greater than or equal to
2.
[0043] In some embodiments, the mini-PEG is a linear mini-PEG (lacking branches). In
such embodiments, n is 1 or 2, m is 0 or 1, and n+m equals 2. For example, in some embodiments there are two corticosteroids attached to the mini-PEG via linkers X, and no Y group. In other embodiments, there is one corticosteroid attached to the mini-PEG via linker X, and one Y group. The corticosteroids may be the same or different, and the Y groups may be the same or different.
[0044J In other embodiments, the mini-PEG is a branched mini-PEG, in which between 3 and 10 PEG groups emanate from a central core group. In such embodiments, n is between 1 and 10, m is between 0 and 10, and n+m is between 3 and 10. For example, in some embodiments there is one corticosteroid moiety, in other embodiments two corticosteroid moieties, in other embodiments three corticosteroid moieties, in other embodiments four corticosteroid moieties, in other embodiments five corticosteroid moieties, in other embodiments six corticosteroid moieties, in other embodiments seven corticosteroid moieties, in other embodiments eight corticosteroid moieties, in other embodiments nine corticosteroid moieties, and in still other embodiments ten corticosteroid moieties. Any number of the corticosteroids may be the same, or different, than one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same. In some embodiments, if q is the number of PEG groups emanating from the central core group of the branched mini-PEG (i.e., q is an integer between 3 and 10), then n is between 1 and q and m = q-n; that is, there are as many Y groups in the molecule as there are PEG groups that are not already linked to corticosteroids. Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
[0045J In still other embodiments, the mini-PEG is a star mini-PEG, in which between 10 and 100 PEG groups emanate from a central core group. In such embodiments, n is between 10 and 100, m is between 0 and 100, and n+m is between 10 and 100. For example, in some embodiments there are between 10 and 20 corticosteroid moieties, in other embodiments between 20 and 30 corticosteroid moieties, in other embodiments between 30 and 40
corticosteroid moieties, in other embodiments between 40 and 50 corticosteroid moieties, in other embodiments between 50 and 60 corticosteroid moieties, in other embodiments between 70 and 80 corticosteroid moieties, in other embodiments between 80 and 90 corticosteroid moieties, and in still other embodiments between 90 and 100 corticosteroid moieties. Any number of the
corticosteroids may be the same or different from one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same. In some embodiments, if q is the number of PEG groups emanating from the central core group of the star mini-PEG (i.e., q is an integer between 10 and 100), then n is between 10 and q and m = q-n; that is, there are as many Y groups in the molecule as there are PEG groups that are not already linked to corticosteroids. Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
[0046] In still other embodiments, the mini-PEG is a comb mini-PEG having multiple PEG chains grafted to a polymer backbone. As described above for the branched mini-PEG and star mini-PEG, if q is the number of PEG groups grafted to the polymer backbone, then n is between 1 and q and m = q-n. Any number of the corticosteroids may be the same or different from one another. For example, in some embodiments, all of the corticosteroids in the molecule are the same. Any number of the Y groups may be the same, or different, than one another. For example, in some embodiments, all of the Y groups are the same.
[0047] In some embodiments, X is S, NR, or O. In some embodiments, X is S. In other embodiments, X is O. In still other embodiments, X is NR.
[0048] In some embodiments, Y is H, OH, or C)-Cs alkoxy. In some embodiments, Y is Q- C^ alkoxy. In some embodiments, Y is methoxy (MeO). In other embodiments, Y is hydroxy] (HO).
[0049] In some embodiments of the compounds provided herein, the mini-PEG has a molecular weight of about 100 Da, or about 200 Da, or about 300 Da, or about 400 Da, or about 500 Da, or about 600 Da, or about 700 Da, or about 800 Da, or about 900 Da, or about 1000 Da, or about 1 100 Da. or about 1200 Da, or about 1300 Da, or about 1400 Da, or about 1500 Da, or about 1600 Da, or about 1700 Da, or about 1800 Da, or about 1900 Da, or about 2000 Da, or about 2100 Da, or about 2200 Da, or about 2300 Da, or about 2400 Da, or about 2500 Da, or about 2600 Da, or about 2700 Da, or about 2800 Da, or about 2900 Da, or about 3000 Da, or about 3100 Da, or about 3200 Da, or about 3300 Da, or about 3400 Da, or about 3500 Da, or about 3600 Da, or about 3700 Da, or about 3800 Da, or about 3900 Da, or about 4000 Da, or about 4100 Da, or about 4200 Da, or about 4300 Da, or about 4400 Da, or about 4500 Da. or
about 4600 Da, or about 4700 Da, or about 4800 Da, or about 4900 Da, or about 5000 Da.
[0050] In one embodiment of the compounds provided herein, the mini-PEG has a molecular weight between about 100 Da and about 2,000 Da, in another embodiment between about 100 Da and about 500 Da, in another embodiment between about 200 Da and about 700 Da, in another embodiment between about 300 Da and about 800 Da, in another embodiment between about 400 Da and about 900 Da, in another embodiment between about 500 Da and about 1 ,000 Da. in another embodiment between about 600 Da and about 1 ,100 Da, in another embodiment between about 700 Da and about 1,200 Da, in another embodiment between about 800 Da and about 1 ,300 Da, in another embodiment between about 900 Da and about 1,400 Da, in another embodiment between about 1,000 Da and about 1,500 Da, in another embodiment between about 1, 100 Da and about 1 ,600 Da, in another embodiment between about 1,200 Da and about 1,700 Da, in another embodiment between about 1,300 Da and about 1 ,800 Da, in another embodiment between about 1,400 Da and about 1,900 Da, in another embodiment between about 1 ,500 Da and about 2,000 Da, in another embodiment between about 1,600 Da and about 2,100 Da, in another embodiment between about 1 ,700 Da and about 2,200 Da, in another embodiment between about 1 ,800 Da and about 2,300 Da, in another embodiment between about 1 ,900 Da and about 2,400 Da, in another embodiment between about 2,000 Da and about 2,500 Da, in another embodiment between about 2,100 Da and about 2,600 Da, in another embodiment between about 2,200 Da and about 2,700 Da, in another embodiment between about 2,300 Da and about 2,800 Da, in another embodiment between about 2,400 Da and about 2,900 Da, in another embodiment between about 2,500 Da and about 3,000 Da.
[0051] A variety of corticosteroids, derivatives thereof, and structural analogs thereof are suitable for use in the compounds provided herein, including, but not limited to, alclometasone, beclomethasone, betamethasone, budesonide, chloroprednisone, clobetasol, clocortolone, cloprednol, corticosterone, cortisone, desonide, desoximetasone, desoxycorticosterone, dexamethasone, diflorasone, difluocortolone, flucloronide, fludrocortisone, flumethasone, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluprednisolone,
flurandrenolide, fluticasone, fluticasone proprionate, halobetasol, halometasone, hydrocortisone, meprednisone, methylprednisolone, paramethasone, prednicarbate, prednisolone, prednisone, prednival, prednylidine, tixocortol, triamcinolone, and triamcinolone acetonide.
[0052] The structures of some exemplary corticosteroids that can be used in the compounds provided herein are illustrated in Table 1. Derivatives and structural analogs of these corticosteroids can also be used in the compounds provided herein. Such analogs may include, but arc not limited to, proprionate derivatives (in one embodiment, beclomethasone proprionate, in another embodiment, betamethasone proprionate, in another embodiment, clobetasol proprionate, in another embodiment, fluticasone proprionate); valerate derivatives (in one embodiment, betamethasone valerate); furoate derivatives (in one embodiment, fluticasone furoate); acetate derivatives (in one embodiment, hydrocortisone acetate, in another embodiment, methylprednisolone acetate, in another embodiment, prednisolone acetate); and sodium phosphate derivatives (in one embodiment, prednisolone sodium phosphate).
Table 1

Beclomethasone Betamethasone

Budesomde
Clobetasol

Corticosterone Cortisone
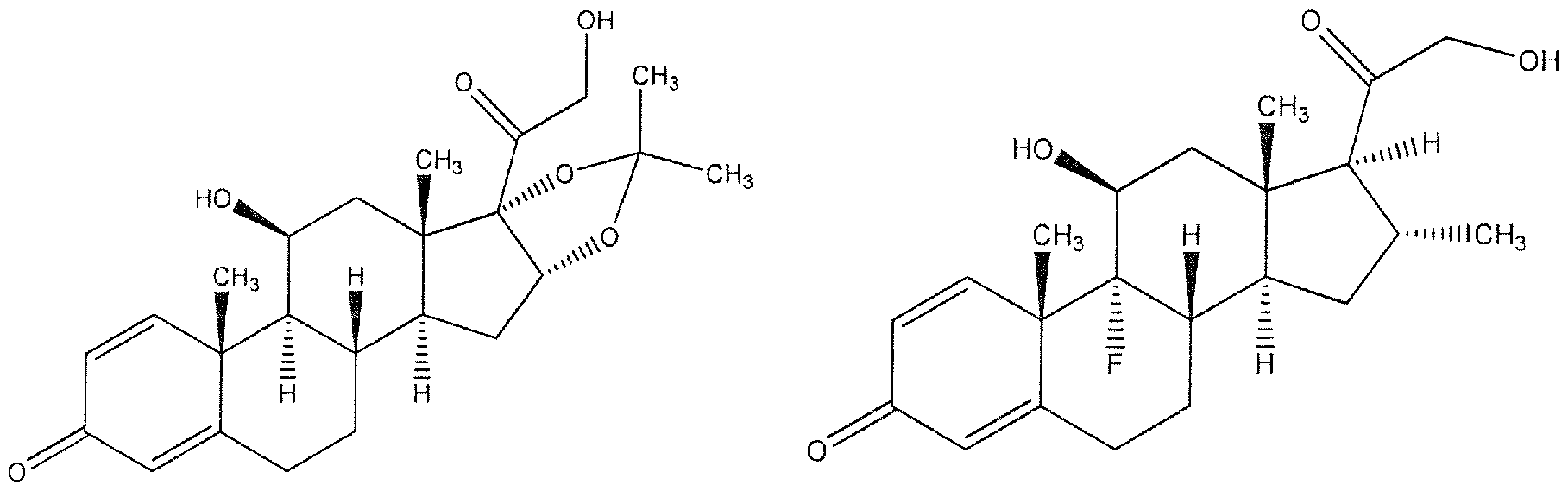
Desonide Desoximetasone

Deoxycorticosterone Dexamethasone



Fluocmomde


' Halobetasol Hydrocortisone

Prcdnicarbate
CHj Methylpiednisolone

Prednisolone Prednisone

Triamcinolone I πamcinolone Acetonide plary compound is dexamethasone mim-PEGylate, illustrated below, which nces referred to herein as Compound 1

Dexamethasone mim-PEGylate (Compound 1 )
91
wherein mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da. In one embodiment, the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
[0054] Another exemplary compound is betamethasone mini-PEGylate, illustrated below, which is in some circumstances referred to herein as Compound 2.

Betamethasone mini-PFGylate (Compound 2) wherein mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da. In one embodiment, the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
Synthesizing the Compounds
[0055] The compounds provided herein can be prepared or obtained by any method known to one of skill in the art. For an example, the compounds can be synthesized from the corresponding non-PEGylated corticosteroids according to Scheme I: miniPEG-Y
 wherein R is the corticosteroid residue;
wherein R is the corticosteroid residue;
Z' and W are selected from (i) and (ii):
(i) Z is a nucleophilic group, including but not limited to, R'NH, HS, HO, ONH2, where R' is H or alkyl, and W is an electrophilic group, including but not limited to, CF3SO2O, P-CH3C6H4SO2O, P-MePhSO2O, PhSO2O, CH3SO2O, halide, NHS ester, haloacetamido, maleimido, halotriazino, or formyl;
(ii) W is a nucleophilic group, including but not limited to, R'NH, HS, HO, ONH2, where R' is H or alkyl, and Z is an electrophilic group, including but not limited to, CF3SO2O, P-CH3C6H4SO2O, p-MePhSO2O, PhSO2O, CH3SO2O, halide, NHS ester, haloacetamido, maleimido, halotriazino, or formyl; and
Y is as defined herein. Pharmaceutical Compositions
[0056] Provided herein are pharmaceutical compositions comprising a compound provided herein, as an active ingredient, in combination with a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a mixture thereof.
[0057] The compounds provided herein may be administered alone, or in combination with one or more other compounds provided herein, or one or more other compounds known in the art. The pharmaceutical compositions that include a compound provided herein, can be formulated in various dosage forms for topical administration. The pharmaceutical compositions can also be formulated as modified release dosage forms, including delayed-, extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated-, fast-, targeted-, and programmed- release dosage forms. These dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art (see, Remington: The Science and Practice of Pharmacy, supra; Modified-Release Drug Delivery Technology, 2nd Edition, Rathbone et al., Eds., Marcel Dekker, Inc.: New York, NY, 2008).
[0058] In one embodiment, the pharmaceutical compositions are provided in a dosage form for topical administration, which comprise a compound provided herein and one or more pharmaceutically acceptable excipients or carriers.
[0059] The pharmaceutical compositions provided herein can be provided in a unit-dosage
form or multiple-dosage form. A unit-dosage form, as used herein, refers to physically discrete a unit suitable for administration to a human and animal subject, and packaged individually as is known in the art. Each unit-dose contains a predetermined quantity of an active ingredient(s) sufficient to produce the desired therapeutic effect, in association with the required
pharmaceutical carriers or excipients. A unit-dosage form may be administered in fractions or multiples thereof. A multiple-dosage form is a plurality of identical unit-dosage forms packaged in a single container to be administered in segregated unit-dosage form.
[0060] The pharmaceutical compositions provided herein can be administered at once, or multiple times at intervals of time. It is understood that the precise dosage and duration of treatment may vary with the age, weight, and condition of the patient being treated, and may be determined empirically using known testing protocols or by extrapolation from in vivo or in vitro test or diagnostic data. It is further understood that for any particular individual, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the formulations.
[0061] The pharmaceutical compositions provided herein can be administered topically to the skin, orifices, or mucosa. The topical administration, as used herein, includes dermal, intradermal, conjunctival, intracorneal, intraocular, ophthalmic, auricular, transdermal, nasal, vaginal, urethral, respiratory, and rectal administration.
[0062] The pharmaceutical compositions provided herein can be formulated in any dosage forms that are suitable for topical administration, including emulsions, solutions, suspensions, creams, gels, hydrogels, ointments, dusting powders, dressings, elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols, irrigations, sprays, suppositories, bandages, and dermal patches. The topical formulation of the pharmaceutical compositions provided herein can also comprise liposomes, micelles, microspheres, nanosystems, and mixtures thereof.
[0063] The compounds provided herein can be included in any appropriate concentration in a dosage form suitable for topical administration. Topical efficacy depends on the activity of the particular corticosteroid mini-PEGylate selected for the treatment, its concentration in the composition, the permeability coefficient, the vehicle and excipients, and local metabolic processes. For example, in some embodiments the compositions herein include a compound
provided herein in a concentration of between 0.001 % and 99%, in some embodiments in a concentration of between 0.01 % and 50%, in some embodiments in a concentration of between 0.1%' and 25%, in some embodiments in a concentration of between 0.1% and 10%, in some embodiments in a concentration of between 10% and 25%, in some embodiments in a concentration of between 25% and 50%, in some embodiments in a concentration of between 50% and 99%, in some embodiments in a concentration of between 1% and 5%, in some embodiments in a concentration of between 5% and 10%, in some embodiments in a concentration of between 10% and 15%, in some embodiments in a concentration of between 15% and 20%, in some embodiments in a concentration of between 20% and 25%, in some embodiments in a concentration of between 25% and 30%, in some embodiments in a concentration of between 30% and 35%, in some embodiments in a concentration of between 35% and 40%, in some embodiments in a concentration of between 40% and 45%, in some embodiments in a concentration of between 45% and 50%, in some embodiments a concentration of between 50% and 55%, in some embodiments in a concentration of between 55% and 60%, in some embodiments in a concentration of between 60% and 65%, in some embodiments in a concentration of between 65% and 70%, in some embodiments in a concentration of between 70% and 75%, in some embodiments in a concentration of between 75% and 80%, in some embodiments in a concentration of between 80% and 85%, in some embodiments in a concentration of between 85% and 90%, in some embodiments in a concentration of between 90% and 95%, in some embodiments a concentration of greater than 95%, in some embodiments a concentration of greater than 99%.
[0064] Pharmaceutically acceptable carriers and excipients suitable for use in the topical formulations provided herein include, but are not limited to, aqueous vehicles, water-miscible vehicles, non-aqueous vehicles, antimicrobial agents or preservatives against the growth of microorganisms, stabilizers, solubility enhancers, isotonic agents, buffering agents, antioxidants, local anesthetics, suspending and dispersing agents, wetting or emulsifying agents, complexing agents, sequestering or chelating agents, penetration enhancers, cryoprotectants, lyoprotectants, thickening agents, and inert gases.
[0065] The pharmaceutical compositions can also be administered topically by
electroporation, iontophoresis, phonophoresis, sonophoresis, or microneedle or needle-free
injection, such as POWDERJECT™ (Chiron Corp., Emeryville, CA), and BIOJECT™ (Bioject Medical Technologies Inc., Tualatin, OR).
[0066] The pharmaceutical compositions provided herein can be provided in the forms of ointments, lotions, foams, creams, and gels. Suitable ointment vehicles include oleaginous or hydrocarbon vehicles, including lard, benzoinated lard, olive oil, cottonseed oil, and other oils, white petrolatum; emulsifiable or absorption vehicles, such as hydrophilic petrolatum, hydroxystearin sulfate, and anhydrous lanolin; water-removable vehicles, such as hydrophilic ointment; water-soluble ointment vehicles, including polyethylene glycols of varying molecular weight; emulsion vehicles, either water-in-oil (W/O) emulsions or oil-in-water (OAV) emulsions, including cetyl alcohol, glyceryl monostearate, lanolin, and stearic acid (see, Remington: The Science and Practice of Pharmacy, supra). These vehicles are emollient but generally require addition of antioxidants and preservatives. This formulation may further include a skin penetrating agent.
[0067] Suitable cream base can be oil-in-water or water-in-oil. Suitable cream vehicles may be water-washable, and contain an oil phase, an emulsifier, and an aqueous phase. The oil phase is also called the "internal" phase, which is generally comprised of petrolatum and a fatty alcohol such as cetyl or stearyl alcohol. The aqueous phase usually, although not necessarily, exceeds the oil phase in volume, and generally contains a humectant. The emulsifier in a cream formulation may be a nonionic, anionic, cationic, or amphoteric surfactant. This formulation may further include a skin penetrating agent.
[0068] Lotions are liquid or semiliquid preparations that contain one or more active ingredients in an appropriate vehicle. In some embodiments, the lotion is a suspension of the active ingredient in an appropriate vehicle, in one embodiment water. In other embodiments, the lotion includes an emulsion having at least two phases, and the active ingredient is suspended or dissolved in one or more of the phases. In other embodiments, the lotion includes a solution of the active ingredient. In some embodiments, the lotion may include a glycol/water mixture, and optionally also one or more surfactants, fatty acid esters, and/or fatty alcohols such as those set forth above in the discussion of a cream formulation, optionally further including stabilizers such as an antioxidant and/or other adjuvants to improve the aesthetics of the lotion. In one
embodiment, the lotion may include an antimicrobial preservative.
[0069] In some embodiments, the compounds provided herein are included in an aerosol formulation, in one embodiment a homogeneous, aqueous-alcoholic emulsion system. The aerosol formulation upon actuation produces a stabilized, homogeneous, expandable foam, which in some embodiments breaks easily with shear. A composition of this type is sometimes referred to as a "mousse." The aerosol formulation may further include a skin penetrating agent, in some embodiments an alcohol such as dodecanol or oleyl alcohol; an amine such as isopropyl amine, diisopropyl amine, triethyl amine, triethanol amine, diisopropanolamine or ethylene diamine; a carboxylic acid such as oleic acid, linoleic acid or linolenic acid; an ester, such as dibutyl sebacate, dibutyl phthalate, butyl benzoate or ethyl caprate; and/or other compounds, such as Azone, N methyl pyrollidone, bile salts and urea. The aerosol formulation may be actuated using one or more propellants known in the pharmaceutical or cosmetic fields. Such propellants include hydrocarbons such as propane, isobutane or dimethyl ether and
chlorofluorocarbons such as P- 12, Pl 14, and a 40:60 mixture thereof. f0070] Gels are semisolid, suspension-type systems. Single-phase gels contain organic macromolecules distributed substantially uniformly throughout the liquid carrier. Suitable gelling agents include, but are not limited to, crosslinked acrylic acid polymers, such as carbomers, carboxypolyalkylenes, and CARBOPOL®; hydrophilic polymers, such as polyethylene oxides, polyoxyethylene-polyoxypropylene copolymers, and polyvinylalcohol; cellulosic polymers, such as hydroxypropyl cellulose, hydroxyethyl cellulose, hydroxypropyl methylcellulosc, hydroxypropyl methylcellulose phthalate, and methylcellulose; gums, such as tragacanth and xanthan gum; sodium alginate; and gelatin. In order to prepare a uniform gel, dispersing agents such as alcohol or glycerin can be added, or the gelling agent can be dispersed by trituration, mechanical mixing, and/or stirring.
[0071] The pharmaceutical compositions provided herein can be administered rectally, urethrally, vaginally, or perivaginally in the forms of suppositories, pessaries, bougies, poultices or cataplasm, pastes, powders, dressings, creams, plasters, contraceptives, ointments, solutions, emulsions, suspensions, tampons, gels, foams, sprays, or enemas. These dosage forms can be manufactured using conventional processes as described in Remington: The Science and Practice
of Pharmacy, supra.
[0072] Rectal, urethral, and vaginal suppositories are solid bodies for insertion into body orifices, which are solid at ordinary temperatures but melt or soften at body temperature to release the active ingredient(s) inside the orifices. Pharmaceutically acceptable carriers utilized in rectal and vaginal suppositories include bases or vehicles, such as stiffening agents, which produce a melting point in the proximity of body temperature, when formulated with the pharmaceutical compositions provided herein; and antioxidants as described herein, including bisulfite and sodium metabisulfite. Suitable vehicles include, but are not limited to, cocoa butter (theobroma oil), glycerin-gelatin, carbowax (polyoxyethylene glycol), spermaceti, paraffin, white and yellow wax, and appropriate mixtures of mono-, di- and triglycerides of fatty acids, and hydrogels, such as polyvinyl alcohol, hydroxyethyl methacrylate, and polyacrylic acid;. Combinations of the various vehicles can also be used. Rectal and vaginal suppositories may be prepared by compressing or molding. The typical weight of a rectal and vaginal suppository is about 2 to about 3 g.
[0073J The pharmaceutical compositions provided herein can be administered
ophthalmically in the forms of solutions, suspensions, ointments, emulsions, gel-forming solutions, powders for solutions, gels, ocular inserts, and implants.
[0074J The pharmaceutical compositions provided herein can be administered intranasally or by inhalation to the respiratory tract. The pharmaceutical compositions can be provided in the form of an aerosol or solution for delivery using a pressurized container, pump, spray, atomizer, such as an atomizer using electrohydrodynamics to produce a fine mist, or nebulizer, alone or in combination with a suitable propellanl, such as 1 , 1 , 1 ,2-tetrafluoroethane or 1,1 ,1 ,2,3,3,3- heptafluoropropane. The pharmaceutical compositions can also be provided as a dry powder for insufflation, alone or in combination with an inert carrier such as lactose or phospholipids; and nasal drops. For intranasal use, the powder can comprise a bioadhesive agent, including chitosan or cyclodextrin.
[0075] Solutions or suspensions for use in a pressurized container, pump, spray, atomizer, or nebulizer can be formulated to contain ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active ingredient provided herein; a
propellant as solvent; and/or a surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
[0076] The pharmaceutical compositions provided herein can be micronized to a size suitable for delivery by inhalation, such as about 50 micrometers or less, or about 10 micrometers or less. Particles of such sizes can be prepared using a comminuting method known to those skilled in the art, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
[0077] Capsules, blisters, and cartridges for use in an inhaler or insufflator can be formulated to contain a powder mix of the pharmaceutical compositions provided herein; a suitable powder base, such as lactose or starch; and a performance modifier, such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate. Other suitable excipients or carriers include, but are not limited to, dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose, and trehalose. The pharmaceutical compositions provided herein for inhaled/intranasal administration can further comprise a suitable flavor, such as menthol and levomenthol; and/or sweeteners, such as saccharin and saccharin sodium.
[0078] The pharmaceutical compositions provided herein for topical administration can be formulated to be immediate release or modified release, including delayed-, sustained-, pulsed-, controlled-, targeted, and programmed release. As used herein, the term "modified release" refers to a dosage form in which the rate or place of release of the active ingredient(s) is different from that of an immediate dosage form when administered by the same route. Modified release dosage forms include, but are not limited to, delayed-, extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated-, fast-, targeted-, and programmed-release dosage forms.
[0079] The pharmaceutical compositions in modified release dosage forms can be prepared using a variety of modified release devices and methods known to those skilled in the art, including, but not limited to, matrix controlled release devices, osmotic controlled release devices, multiparticulate controlled release devices, ion-exchange resins, multilayered coatings, microspheres, liposomes, and combinations thereof. The release rate of the active ingredient(s) can also be modified by varying the particle sizes and polymorphorism of the active
ingredient(s).
[0080] Examples of modified release include, but are not limited to, those described in U.S. Pat. Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 5,674,533; 5,059,595;
5,591,767; 5, 120,548; 5,073,543; 5,639,476; 5,354,556; 5,639,480; 5,733,566; 5,739,108: 5,891,474; 5,922,356; 5,972,891 ; 5,980,945; 5,993,855; 6,045,830; 6,087,324; 6,113,943: 6, 197,350; 6,248,363; 6,264,970; 6,267,981 ; 6,376,461 ; 6,419,961; 6,589,548; 6,613,358; and 6,699,500.
1. Matrix Controlled Release Devices
[0081] The pharmaceutical compositions provided herein in a modified release dosage form can be fabricated using a matrix controlled release device known to those skilled in the art (see, Takada et al. in "Encyclopedia of Controlled Drug Delivery," Vol. 2, Mathiowitz Ed., Wiley, 1999).
[0082] In certain embodiments, the pharmaceutical compositions provided herein in a modified release dosage form is formulated using an erodible matrix device, which is water- swellable, erodible, or soluble polymers, including, but not limited to, synthetic polymers, and naturally occurring polymers and derivatives, such as polysaccharides and proteins.
[0083J Materials useful in forming an erodible matrix include, but are not limited to, chitin, chitosan, dextran, and pullulan; gum agar, gum arabic, gum karaya, locust bean gum, gum tragacanth, carrageenans, gum ghatti, guar gum, xanthan gum, and scleroglucan; starches, such as dextrin and maltodextrin; hydrophilic colloids, such as pectin; phosphatides, such as lecithin; alginates; propylene glycol alginate; gelatin; collagen; cellulosics, such as ethyl cellulose (EC), methylethyl cellulose (MEC), carboxymethyl cellulose (CMC), CMEC, hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), cellulose acetate (CA), cellulose propionate (CP), cellulose butyrate (CB), cellulose acetate butyrate (CAB), CAP, CAT, hydroxypropyl methyl cellulose (HPMC), HPMCP, HPMCAS, hydroxypropyl methyl cellulose acetate trimellitate (HPMCAT), and ethyl hydroxyethyl cellulose (EHEC); polyvinyl pyrrolidone; polyvinyl alcohol; polyvinyl acetate; glycerol fatty acid esters; polyacrylamide; polyacrylic acid;
copolymers of ethacrylic acid or methacrylic acid (EUDRAGIT®, Rohm America, Inc., Piscataway, NJ); poly(2-hydroxyethyl-methacrylate); polylactides; copolymers of L-glutamic acid and ethyl-L-glutamate; degradable lactic acid-glycolic acid copolymers; poly-D-(-)-3-
hydroxybutyric acid; and other acrylic acid derivatives, such as homopolymers and copolymers of butylmethacrylate, methyl methacrylate, ethyl methacrylate, ethylacrylate, (2- dimethylaminoethyl)mcthacrylate, and (trimethylaminoethyl)rnethacrylate chloride.
[0084] In certain embodiments, the pharmaceutical compositions provided herein are formulated with a non-erodible matrix device. The active ingredient(s) is dissolved or dispersed in an inert matrix and is released primarily by diffusion through the inert matrix once administered. Materials suitable for use as a non-erodible matrix device include, but are not limited to, insoluble plastics, such as polyethylene, polypropylene, polyisoprene,
polyisobutylene, polybutadiene, polymethylmethacrylate, polybutylmethacrylate. chlorinated polyethylene, polyvinylchloride, methyl acrylate-melhyl methacrylate copolymers, ethylene- vinyl acetate copolymers, ethylene/propylene copolymers, cthylene/cthyl acrylatc copolymers, vinyl chloride copolymers with vinyl acetate, vinylidene chloride, ethylene and propylene, ionomer polyethylene terephthalate, butyl rubbers, epichlorohydrin rubbers, ethylene/vinyl alcohol copolymer, ethylene/vinyl acetate/vinyl alcohol terpolymer, ethylene/vinyloxyethanol copolymer, polyvinyl chloride, plasticized nylon, plasticized polyethylene terephthalate, natural rubber, silicone rubbers, polydimethylsiloxanes, and silicone carbonate copolymers; hydrophilic polymers, such as ethyl cellulose, cellulose acetate, crospovidone, and cross-linked partially hydrolyzed polyvinyl acetate; and fatty compounds, such as carnauba wax, microcrystalline wax, and triglycerides.
[0085] In a matrix controlled release system, the desired release kinetics can be controlled, for example, via the polymer type employed, the polymer viscosity, the particle sizes of the polymer and/or the active ingredient(s), the ratio of the active ingredient(s) versus the polymer, and other excipients or carriers in the compositions.
[0086] The pharmaceutical compositions provided herein in a modified release dosage form can be prepared by methods known to those skilled in the art, including direct compression, dry or wet granulation followed by compression, and melt-granulation followed by compression.
2, Osmotic Controlled Release Devices [0087] The pharmaceutical compositions provided herein in a modified release dosage form
can be fabricated using an osmotic controlled release device, including, but not limited to, one- chamber system, two-chamber system, asymmetric membrane technology (AMT), and extruding core system (ECS). In general, such devices have at least two components: (a) a core which contains an active ingredient; and (b) a semipermeable membrane with at least one delivery port, which encapsulates the core. The semipermeable membrane controls the influx of water to the core from an aqueous environment of use so as to cause drug release by extrusion through the delivery port(s).
[0088] In addition to the active ingredient(s), the core of the osmotic device optionally includes an osmotic agent, which creates a driving force for transport of water from the environment of use into the core of the device. One class of osmotic agents is water-swcllable hydrophilic polymers, which are also referred to as "osmopolymers" and "hydrogels." Suitable water-swellable hydrophilic polymers as osmotic agents include, but are not limited to, hydrophilic vinyl and acrylic polymers, polysaccharides such as calcium alginate, polyethylene oxide (PEO), polyethylene glycol (PEG), polypropylene glycol (PPG), poly(2-hydroxyethyl methacrylatc), poly(acrylic) acid, poly(methacrylic) acid, polyvinylpyrrolidone (PVP), crosslinked PVP, polyvinyl alcohol (PVA), PVA/PVP copolymers, PVΛ/PVP copolymers with hydrophobic monomers such as methyl methacrylate and vinyl acetate, hydrophilic
polyurethanes containing large PEO blocks, sodium croscarmellose, carrageenan, hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), carboxymethyl cellulose (CMC) and carboxyethyl, cellulose (CEC), sodium alginate, polycarbophil, gelatin, xanthan gum, and sodium starch glycolate.
[0089] The other class of osmotic agents is osmogens, which are capable of imbibing water to affect an osmotic pressure gradient across the barrier of the surrounding coating. Suitable osmogens include, but are not limited to, inorganic salts, such as magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, potassium phosphates, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride, and sodium sulfate; sugars, such as dextrose, fructose, glucose, inositol, lactose, maltose, mannitol, raffinose, sorbitol, sucrose, trehalose, and xylitol; organic acids, such as ascorbic acid, benzoic acid, fumaric acid, citric acid, maleic acid, sebacic acid, sorbic acid, adipic acid, edetic acid, glutamic acid, p-toluenesulfonic acid, succinic acid, and tartaric acid; urea; and mixtures thereof.
[0090] Osmotic agents of different dissolution rates can be employed to influence how rapidly the active ingredient(s) is initially delivered from the dosage form. For example, amorphous sugars, such as MANNOGEM EZ (SPI Pharma, Lewes, DE) can be used to provide faster delivery during the first couple of hours to promptly produce the desired therapeutic effect, and gradually and continually release of the remaining amount to maintain the desired level of therapeutic or prophylactic effect over an extended period of time. In this case, the active ingredient(s) is released at such a rate to replace the amount of the active ingredient metabolized and excreted.
[0091] The core can also include a wide variety of other excipients and carriers as described herein to enhance the performance of the dosage form or to promote stability or processing.
[0092] Materials useful in forming the semipermeable membrane include various grades of acrylics, vinyls, ethers, polyamides, polyesters, and cellulosic derivatives that are water- permeable and water-insoluble at physiologically relevant pHs, or are susceptible to being rendered water-insoluble by chemical alteration, such as crosslinking. Examples of suitable polymers useful in forming the coating, include plasticized, unplasticized, and reinforced cellulose acetate (CA), cellulose diacetate, cellulose triacetate, CA propionate, cellulose nitrate, cellulose acetate butyrate (CAB), CA ethyl carbamate, CAP, CA methyl carbamate, CA succinate, cellulose acetate trimellitate (CAT), CA dimethylaminoacetate, CA ethyl carbonate, CA chloroacetate, CA ethyl oxalate, CA methyl sulfonate, CA butyl sulfonate, CA p-toluene sulfonate, agar acetate, amylose triacetate, beta glucan acetate, beta glucan triacetate, acetaldehydc dimethyl acetate, triacetate of locust bean gum, hydroxylated ethylene-vinyl acetate, EC, PEG, PPG, PEG/PPG copolymers, PVP, HEC, HPC, CMC, CMEC, HPMC, HPMCP, HPMCAS, HPMCAT, poly(acrylic) acids and esters and poly-(methacrylic) acids and esters and copolymers thereof, starch, dextran, dextrin, chitosan, collagen, gelatin, polyalkenes, polyethers, polysulfones, polyethersulfones, polystyrenes, polyvinyl halides, polyvinyl esters and ethers, natural waxes, and synthetic waxes.
[0093] Semipermeable membrane can also be a hydrophobic microporous membrane, wherein the pores are substantially filled with a gas and are not wetted by the aqueous medium but are permeable to water vapor, as disclosed in U.S. Pat. No. 5,798.1 19. Such hydrophobic but
water-vapor permeable membrane are typically composed of hydrophobic polymers such as polyalkenes, polyethylene, polypropylene, polytetrafluoroethylene, polyacrylic acid derivatives, polyethers, polysulfones, polyethersulfones, polystyrenes, polyvinyl halides, polyvinylidene fluoride, polyvinyl esters and ethers, natural waxes, and synthetic waxes.
[0094] The delivery port(s) on the semipermeable membrane can be formed post-coating by mechanical or laser drilling. Delivery port(s) can also be formed in situ by erosion of a plug of water-soluble material or by rupture of a thinner portion of the membrane over an indentation in the core. In addition, delivery ports can be formed during coating process, as in the case of asymmetric membrane coatings of the type disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220.
[0095] The total amount of the active ingredient(s) released and the release rate can substantially by modulated via the thickness and porosity of the semipermeable membrane, the composition of the core, and the number, size, and position of the delivery ports.
[0096] The pharmaceutical compositions in an osmotic controlled-release dosage form can further comprise additional conventional excipients or carriers as described herein to promote performance or processing of the formulation.
[0097] The osmotic controlled-release dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art (see, Remington: The Science and Practice of Pharmacy, supra; Santus and Baker, /. Controlled Release 1995, 35, 1 - 21 ; Verma et al.. Drug Development and Industrial Pharmacy 2000, 26, 695-708; Verma et al., J. Controlled Release 2002, 79. 7-27).
[0098] In certain embodiments, the pharmaceutical compositions provided herein are formulated as AMT controlled-release dosage form, which comprises an asymmetric osmotic membrane that coats a core comprising the active ingredient(s) and other pharmaceutically acceptable excipients or carriers. See, U.S. Pat. No. 5,612,059 and WO 2002/17918. The AMT controlled-release dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art, including direct compression, dry granulation, wet granulation, and a dip-coating method.
[0099] In certain embodiments, the pharmaceutical compositions provided herein are
formulated as ESC controlled-release dosage form, which comprises an osmotic membrane that coats a core comprising the active ingredient(s), a hydroxylethyl cellulose, and other pharmaceutically acceptable excipients or carriers.
3. Multiparticulate Controlled Release Devices
[00100] The pharmaceutical compositions provided herein in a modified release dosage form can be fabricated as a multiparticulate controlled release device, which comprises a multiplicity of particles, granules, or pellets, ranging from about 10 μm to about 3 mm, about 50 μm to about 2.5 mm, or from about 100 μm to about 1 mm in diameter. Such multiparticulates can be made by the processes known to those skilled in the art, including wet-and dry-granulation, extrusion/spheronization, roller-compaction, melt-congealing, and by spray-coating seed cores. See, for example, Multiparticulate Oral Drug Delivery; Marcel Dekker: 1994; and
Pharmaceutical Pelletization Technology; Marcel Dekker: 1989.
[001011 Other excipients or carriers as described herein can be blended with the
pharmaceutical compositions to aid in processing and forming the multiparticulates. The resulting particles can themselves constitute the multiparticulate device or can be coated by various film-forming materials, such as enteric coatings or water-swcllable and water-soluble polymers.
[00102] The pharmaceutical compositions provided herein can also be formulated to be targeted to a particular tissue, receptor, or other area of the body of the subject to be treated, including liposome-, resealed erythrocyte-, and antibody-based delivery systems. Examples include, but are not limited to, those disclosed in U.S. Pat. Nos. 6,316,652; 6,274,552; 6,271,359; 6,253,872; 6,139,865; 6,131,570; 6,120,751 ; 6,071 ,495; 6,060,082; 6,048,736; 6,039,975;
6,004,534; 5,985,307; 5,972,366; 5,900,252; 5,840,674; 5,759,542; and 5,709,874.
Methods of Use
[00103] The compounds and compositions provided herein may be administered to a variety of populations of patients. In some embodiments, such populations are not able to safely receive non-conjugated corticosteroids, because of heightened risk factors associated with systemic absorption of such non-conjugated corticosteroids. Thus, in one embodiment, because the
compounds and compositions provided herein reduce or eliminate systemic exposure, as described herein, they can be used in patient populations that would otherwise be cautioned against using corticosteroid compositions. In another embodiment, the compounds and compositions provided herein are used in patient populations for whom administration of corticosteroid compositions is not of significant concern. In one embodiment, the compounds and compositions provided herein may be administered to a pediatric population, in one embodiment children under the age of 2, in another embodiment children under the age of 12, in another embodiment children under the age of 18, in another embodiment children between the age of 2 and 12, in another embodiment children between the age of 2 and 6, in another embodiment children between the age 6 and 12, in another embodiment children between the age of 12 and 18. In another embodiment, the compounds and compositions provided herein may be administered to adults, in one embodiment adults over the age of 18, in another embodiment adults between the ages of 18 and 65, in another embodiment adults between the ages of 18 and 75, in another embodiment adults over 65 years of age, in another embodiment adults over 75 years of age. In another embodiment, the compounds and compositions provided herein may be administered to pregnant women. In another embodiment, the compounds and compositions provided herein may be administered to women who are trying to become pregnant. In another embodiment, the compounds and compositions provided herein may be administered to women who are nursing an infant. In another embodiment, the compounds and compositions provided herein may be administered to a patient that is already taking a systemic corticosteroid. In another embodiment, the compounds and compositions provided herein may be administered to a patient that is already using a topical corticosteroid. In another embodiment, the compounds and compositions provided herein are dermally applied and the treated area covered with an occlusive bandage.
[00104] In one embodiment, provided is a method of treating, preventing, or ameliorating one or more symptoms of a disorder, disease, or condition associated with inflammation in a subject, which comprises administering to the subject a therapeutically effective amount of a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II. In one embodiment, the subject is a mammal. In another embodiment, the subject is a human. In another embodiment, the subject is an animal other than a human.
[00105] In another embodiment, provided is a method of treating, preventing, or ameliorating one or more symptoms of a disorder, disease, or condition associated with the glucocorticoid receptor in a subject, which comprises administering to the subject a therapeutically effective amount of a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II. In one embodiment, the subject is a mammal. In one embodiment, the subject is a human. In another embodiment, the subject is an animal other than a human.
[00106J The disorders, diseases, or conditions treatable with a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II, include, but are not limited to, (1) inflammatory or allergic diseases, including systemic anaphylaxis and hypersensitivity disorders, atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), and mastocytosis; (2) inflammatory bowel diseases, including Crohn's disease, ulcerative colitis, ileitis, and enteritis; (3) vasculitis, and Behcet's syndrome; (4) psoriasis and inflammatory dermatoses, including dermatitis, eczema, atopic dermatitis, allergic contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus, HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, aphthae, cutaneous lupus erythematosus, discoid lupus erythematosus, eczema (including seborrhoeic and nummular), irritant contact dermatitis, lichen planus, lichen sclerosus, miliaria, morphoea, parapsoriasis, pemphigoid, photoallergy, phototoxicity, pityriasis alba, pityriasis rosea, polymorphic light eruption, pompholyx, pyoderma gangrenosum, sunburns, and vitiligo; (5) asthma and respiratory allergic diseases, including allergic asthma, exercise induced asthma, allergic rhinitis, otitis media, allergic conjunctivitis, hypersensitivity lung diseases, and chronic obstructive pulmonary disease; (6) autoimmune diseases, including arthritis (including rheumatoid and psoriatic), systemic lupus erythematosus, type I diabetes, myasthenia gravis, multiple sclerosis, Graves' disease, and glomerulonephritis; (7) graft rejection (including allograft rejection and graft-v-host disease), e.g., skin graft rejection, solid organ transplant rejection, bone marrow transplant rejection; (8) fever; (9) cardiovascular disorders, including acute heart failure, hypotension, hypertension, angina pectoris, myocardial infarction, cardiomyopathy, congestive heart failure, atherosclerosis, coronary artery disease, restenosis, and vascular stenosis; (10) cerebrovascular disorders, including traumatic brain injury, stroke, ischemic reperfusion injury and aneurysm;
(1 1) cancers of the breast, skin, prostate, cervix, uterus, ovary, testes, bladder, lung, liver, larynx, oral cavity, colon and gastrointestinal tract (e.g., esophagus, stomach, pancreas), brain, thyroid, blood, and lymphatic system; (12) fibrosis, connective tissue disease, and sarcoidosis, (13) genital and reproductive conditions, including erectile dysfunction; (14) gastrointestinal disorders, including gastritis, ulcers, nausea, pancreatitis, and vomiting; ( 15) neurologic disorders, including Alzheimer's disease; (16) sleep disorders, including insomnia, narcolepsy, sleep apnea syndrome, and Pickwick Syndrome; (17) pain; (18) renal disorders; (19) ocular disorders, including glaucoma; and (20) infectious diseases, including HIV.
[00107] In some embodiments, the disorder, disease, or condition affects the skin or mucosal tissue. In certain embodiments, the disorder, disease, or condition is selected from the group consisting of inflammatory or allergic diseases, including systemic anaphylaxis and
hypersensitivity disorders, atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), and mastocytosis.
[00108] In certain other embodiments, the disorder, disease or condition is selected from the group consisting of psoriasis and inflammatory dermatoses, including dermatitis, eczema (including seborrhoeic and nummular), atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus. HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, cutaneous and discoid lupus erythematosus.
[00109] In certain embodiments, the disorder, disease, or condition treatable with a compound provided herein, in one embodiment a compound of Formula I, in another embodiment a compound of Formula II, is an inflammatory disease. In certain embodiments, the inflammatory disease is an inflammatory bowel disease. In certain embodiments, the inflammatory bowel disease is Crohn's disease, ulcerative colitis, or pouchitis.
[00110] Depending on the disorder, disease, or condition to be treated, and the subject's condition, the compounds or pharmaceutical compositions provided herein can be administered by inhalation, nasal, vaginal, rectal, sublingual, dermal, transdermal, or local routes of administration and can be formulated, alone or together, in suitable dosage unit with
pharmaceutically acceptable cxcipients, carriers, adjuvants, and vehicles appropriate for each
route of administration. Also provided is administration of the compounds or pharmaceutical compositions provided herein in a depot formulation, in which the active ingredient is released over a predefined time period.
[00111] In some embodiments, the pharmaceutical compositions provided herein are sterile, and are packaged so as to maintain their sterility until use. f 00112] In the treatment, prevention, or amelioration of one or more symptoms of inflammatory or allergic diseases, including systemic anaphylaxis and hypersensitivity disorders, atopic dermatitis, urticaria, drug allergies, insect sting allergies, food allergies (including celiac disease and the like), mastocytosis, psoriasis and inflammatory dermatoses, including dermatitis, eczema (including seborrhoeic and nummular), atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human papillomavirus, HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, or cutaneous or discoid lupus erythematosus or other conditions, disorders or diseases associated with inflammation, e.g., associated with the glucocorticoid receptor, an appropriate dosage level generally is ranging from about 0.001 to 100 mg per kg subject body weight per day (mg/kg per day), from about 0.01 to about 75 mg/kg per day, from about 0.1 to about 50 mg/kg per day, from about 0.5 to about 25 mg/kg per day, or from about 1 to about 20 mg/kg per day, which can be administered in single or multiple doses. Within this range, the dosage can be ranging from about 0.005 to about 0.05, from about 0.05 to about 0.5, from about 0.5 to about 5.0, from about 1 to about 15, from about 1 to about 20, or from about 1 to about 50 mg/kg per day. In certain embodiments, the dosage level is ranging from about 0.001 to about 100 mg/kg per day. In certain embodiments, the dosage level is ranging from about 0.01 to about 75 mg/kg per day. In certain embodiments, the dosage level is ranging from about 0.1 to about 50 mg/kg per day. In certain embodiments, the dosage level is ranging from about 0.5 to about 25 mg/kg per day. In certain embodiments, the dosage level is ranging from about 1 to about 20 mg/kg per day.
[00113] It will be understood, however, that the specific dose level and frequency of dosage for any particular patient can be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that
compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy. f 00114] Also provided herein are methods of modulating glucocorticoid receptor activity, comprising contacting a glucocorticoid receptor with a compound provided herein. In one embodiment, the glucocorticoid receptor is expressed by a cell.
[00115J In certain embodiments, the compounds provided herein can be combined with one or more antibacterial agents known in the art, including, but not limited to the group including amikacin, amoxicillin, ampicillin, arsphenamine, azithromycin, aztreonam, azlocillin, bacitracin, carbenicillin, cefaclor, cefadroxil, cefamandole, cefazolin, cephalexin, cefdinir, cefditorin, cefepime, cefixime, cefoperazone, cefotaxime, cefoxitin, cefpodoxime, cefprozil, ceftazidime, ceftibuten, ceftizoxime, ceftriaxone, cefuroxime, chloramphenicol, cilastin, ciprofloxacin, clarithromycin, clindamycin, cloxacillin, colistin, dalfopristin, demeclocycline, dicloxacillin, dirithromycin, doxycycline, erythromycin, enrofloxacin, ertepenem, ethambutol, flucloxacillin, fosfomycin, furazolidone, gatifloxacin, geldanamycin, gentamicin, herbimycin, imipenem, isoniazid. kanamycin, levofloxacin, linezolid, lomefloxacin, loracarbef, mafenide, moxifloxacin, meropenem, metronidazole, mezlocillin, minocycline, mupirocin, nafcillin, neomycin, netilmicin, nitrofurantoin, norfloxacin, ofloxacin, oxytetracycline, penicillin, piperacillin, platensimycin, polymyxin B, prontocil, pyrazinamide, quinupristine, rifampin, roxithromycin, spectinomycin, streptomycin, sulfacetamide, sulfamethizole, sulfamethoxazole, teicoplanin, telithromycin, tetracycline, ticarcillin, tobramycin, trimethoprim, troleandomycin, trovafloxacin, and vancomycin.
[00116] In certain embodiments, the compounds provided herein can be combined with one or more antifungal agents known in the art, including, but not limited to the group including amorolfine, amphotericin B, anidulafungin, bifonazole, butenafine, butoconazole, caspofungin, ciclopirox, clotrimazole, econazole, fenticonazole, filipin, fluconazole, isoconazole, itraconazole, ketoconazole, micafungin, miconazole, naftifine, natamycin, nystatin, oxyconazole,
ravuconazole, posaconazole, rimocidin, sertaconazole, sulconazole, terbinafine, terconazole. tioconazole, and voriconazole.
[00117] In certain embodiments, the compounds provided herein can be combined with one or more non-steroidal anti-inflammatory agents known in the art, including, but not limited to, aceclofenac, acemetacin, amoxiprin, aspirin, azapropazone, benorilate, bromfenac, carprofen, celecoxib, choline magnesium salicylate, diclofenac, diflunisal, etodolac, etoricoxib, faislamine, fenbufen, fenoprofen, flurbiprofen, ibuprofen, indometacin, ketoprofen, ketorolac, lornoxicam, loxoprofen, lumiracoxib, meclofenamic acid, mefenamic acid, meloxicam, metamizole, methyl salicylate, magnesium salicylate, nabumetone, naproxen, nimesulide, oxyphenbutazone, parecoxib, phenylbutazone, piroxicam, salicyl salicylate, sulindac, sulfinpyrazone, suprofen, tenoxicam, tiaprofenic acid, and tolmetin.
[00118] The compounds provided herein can also be administered in combination with other classes of compounds, including, but not limited to, endothelin converting enzyme (ECE) inhibitors, such as phosphoramidon; calcineurin inhibitors such as pimecrolimus, thromboxane receptor antagonists, such as ifetroban; potassium channel openers; thrombin inhibitors, such as hirudin; growth factor inhibitors, such as modulators of PDGF activity and nerve growth factor (NGF); platelet activating factor (PAF) antagonists; anti-platelet agents, such as GPIIb/IIIa blockers (e.g., abciximab, eptifibatide, and tirofiban), P2Y(AC) antagonists (e.g., clopidogrel, ticlopidine and CS-747), and aspirin; anticoagulants, such as warfarin; low molecular weight heparins, such as enoxaparin; Factor Vila Inhibitors and Factor Xa Inhibitors; renin inhibitors; neutral endopeptidase (NEP) inhibitors; vasopeptidase inhibitors (dual NEP-ACE inhibitors), such as omapatrilat and gemopatrilat; HMG CoA reductase inhibitors, such as pravastatin, lovastatin, atorvastatin, simvastatin, NK- 104 (a.k.a. itavastatin, nisvastatin, or nisbastatin), and ZD-4522 (also known as rosuvastatin, atavastatin, or visastatin); squalene synthetase inhibitors; fibrates; bile acid sequestrants, such as questran; niacin; anti-atherosclerotic agents, such as ACAT inhibitors; MTP Inhibitors; calcium channel blockers, such as amlodipine besylate;
potassium channel activators; alpha-adrenergic agents; beta-adrenergic agents, such as carvedilol and metoprolol; antiarrhythmic agents; diuretics, such as chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide,
trichloromethiazide, polythiazide, benzothiazide, ethacrynic acid, ticrynafen, chlorthalidone, furosemde, muzolimine, bumetanide, triamterene, amiloride, and spironolactone; thrombolytic agents, such as tissue plasminogen activator (tPA), recombinant tPA, streptokinase, urokinase, prourokinase. and anisoylated plasminogen streptokinase activator complex (APSAC); anti-
diabetic agents, such as biguanides (e.g., metformin), glucosidase inhibitors (e.g., acarbose), insulins, meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, and glipizide), thiozolidinediones (e.g., troglitazone, rosiglitazone, and pioglitazone), and PPAR- gamma agonists; mineralocorticoid receptor antagonists, such as spironolactone and eplerenone; growth hormone secretagogues; aP2 inhibitors; phosphodiesterase inhibitors, such as PDE III inhibitors (e.g., cilostazol) and PDE V inhibitors (e.g., sildenafil, tadalafil, and vardenafil); protein tyrosine kinase inhibitors; antiinflammatories; antiproliferatives, such as methotrexate, FK506 (tacrolimus), mycophenolate mofetil; chemotherapeutic agents; immunosuppressants; anticancer agents and cytotoxic agents (e.g., alkylating agents, such as nitrogen mustards, alkyl sulfonates, nitrosoureas, ethylenimines, and triazenes); antimetabolites, such as folate antagonists, purine analogues, and pyrimidine analogues; antibiotics, such as anthracyclines, bleomycins, mitomycin, dactinomycin, and plicamycin; enzymes, such as L-asparaginase;
farnesyl-protein transferase inhibitors; hormonal agents, such as glucocorticoids (e.g., cortisone), estrogens/antiestrogens, androgens/antiandrogens, progestins, and luteinizing hormone-releasing hormone antagonists, and octreotide acetate: microtubule-disruptor agents, such as
ecteinascidins; microtubule-stabilizing agents, such as pacitaxel, docetaxel, and epothilones A-F; plant-derived products, such as vinca alkaloids, epipodophyllotoxins, and taxanes; and topoisomerase inhibitors; prenyl-protein transferase inhibitors; and cyclosporins; steroids, such as prednisone and dexamethasone; cytotoxic drugs, such as azathioprine and cyclophosphamide; TNF-alpha inhibitors, such as tenidap; anti-TNF antibodies or soluble TNF receptor, such as etanercept, rapamycin, and leflunimide: and cyclooxygenase-2 (COX-2) inhibitors, such as celecoxib and rofecoxib; retinoids; and miscellaneous agents such as, hydroxyurea, procarbazine, mitotane, hexamethylmelamine, gold compounds, platinum coordination complexes, such as cisplatin, satraplatin, and carboplatin.
[00119] Such other agents, or drugs, can be administered, by a route and in an amount commonly used therefor, simultaneously or sequentially with the compounds provided herein, in one embodiment a compound of Formula 1, in another embodiment a compound of Formula II. When a compound provided herein is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound provided herein can be utilized, but is not required. Accordingly, the pharmaceutical compositions provided herein include those that also contain one or more other active ingredients or
therapeutic agents, in addition to a compound provided herein.
[00120] The weight ratio of a compound provided herein to the second active ingredient can be varied, and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound provided herein is combined with a NSAID, the weight ratio of the compound to the NSADD can range from about 1,000:1 to about 1: 1,000, or about 200: 1 to about 1 :200. Combinations of a compound provided herein and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
[00121] In one embodiment, a composition provided herein is a topical dermatological foam, cream, gel, ointment, oil, or lotion for use in treating disorders, diseases, or conditions of the skin. The foam, cream, gel, ointment, oil, or lotion includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one fluocinolone moiety, in still another embodiment at least one clobetasol moiety, in another embodiment at least one hydrocortisone moiety, in another embodiment at least one desonide moiety, in another embodiment at least one fluocinonide moiety, in yet another embodiment at least one fluticasone moiety. In some embodiments, the corticosteroid moiety is derivitized, for example, betamethasone proprionate, in other embodiments betamethasone valerate, in other embodiments clobetasol proprionate, in other embodiments fluticasone proprionate, in other embodiments hydrocortisone acetate. In some embodiments, the topical dermatological foam, cream, gel, ointment, oil, or lotion is administered to a patient with an inflammatory or pruritic manifestation of a corticosteroid- responsive dermatosis. For example, the patient may have a disorder, disease, or condition including one or more of erysthema, scaling, pruritis, and maceration. In one embodiment, the patient has atopic dermatitis. In some embodiments, the patient has a fungal (mycological) infection, and the foam, cream, gel, ointment, oil, or lotion also includes an antifungal agent. In some embodiments the fungal infection is symptomatic inflammatory tinea pedis, tinea cruris, or tinea corporis. In some embodiments, the antifungal agent is clotrimazole. In other
embodiments, the patient has a dermatosis of the scalp, in one embodiment scalp psoriasis, in
another embodiment atopic dermatitis. In some embodiments, the composition includes a surface or local anaesthetic, in one embodiment promoxine. In some embodiments, the composition includes one or more antibacterial agents, in some embodiments one or more agents selected from the group consisting of neomycin A, neomycin B, neomycin C, polymyxin B, colistin A. colistin B, and thonzonium.
[00122] In another embodiment, a composition provided herein is an otic suspension for use in treating disorders, diseases, or conditions of the ear. The otic suspension includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one hydrocortisone moiety. The corticosteroid moiety may be derivitized, for example, hydrocortisone acetate. In some embodiments, the otic suspension is administered to a patient with acute otitis media caused by the presence of a infection, in some embodiments also having a tympanostomy tube. In some embodiments, the suspension also includes an antibacterial agent, in one embodiment ciprofloxacin, in other embodiments one or more of neomycin A, neomycin B, neomycin C, thonzonium, colistin A, and colistin B.
[00123] In another embodiment, a composition provided herein is an ophthalmic ointment or suspension for use in treating disorders, diseases, or conditions of the eye. The ointment or suspension includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, in other embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, in another embodiment at least one prednisolone moiety. In some embodiments, the corticosteroid moiety is prednisolone acetate. In some embodiments, the ophthalmic ointment or suspension is administered to a patient with an inflammation or edema of the palpebral and bulbar conjunctiva, cornea, or anterior segment of the globe; uveitis; or corneal injury from chemical, radiation, or thermal burns, or penetration of a foreign body. In some embodiments, the ointment or suspension also includes an anti-infective drag, in one embodiment tobramycin. In some embodiments, the ointment or suspension also includes an antibacterial agent, in one embodiment sulfacetamide sodium.
[00124J In another embodiment, a composition provided herein is an aerosol intended for oral inhalation. In one embodiment, the aerosol is administered using a pressurized, metered-dose inhaler, in another embodiment using an inhalation-driven multi-dose dry powder inhaler, in another embodiment using an air driven jet nebulizer. The aerosol includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one beclomethasone moiety, in another embodiment at least one budesonide moiety, in another embodiment at least one flunisolide moiety, in another embodiment at least one triamcinolone moiety, in still another embodiment at least one triamcinolone acetonide moiety, and in yet another embodiment at least one fluticasone moiety. In one embodiment, the corticosteroid moiety is derivitized, for example beclomethasone proprionate, in another embodiment fluticasone proprionate. In some embodiments, the aerosol is administered to a patient with inflammation associated with asthma. In some embodiments, the composition also includes a bronchodilator such as a selective beta2-agonist or beta2- andrenoceptor, in one embodiment formoterol fumarate, in another embodiment salmeterol, in another embodiment albuterol, in still another embodiment levalbuterol.
[001251 In another embodiment, a composition provided herein is a suspension for administration via nasal spray. The aerosol includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, and in other embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one beclomethasone moiety, in another embodiment at least one budesonide moiety, in another embodiment at least one triamcinolone moiety, in still another embodiment at least one triamcinolone acetonide moiety, and in yet another embodiment at least one fluticasone moiety. In one embodiment, the corticosteroid moiety is derivitized, for example
beclomethasone proprionate, in another embodiment fluticasone furoate, in another embodiment fluticasone proprionate. In some embodiments, the suspension is administered to a patient with seasonal or perennial allergic or nonallergic rhinitis.
[00126] In another embodiment, a composition provided herein is a rectal suspension for administration via an enema. The suspension includes a compound provided herein. The compound provided herein may in some embodiments be a compound of Formula I, and in other
embodiments a compound of Formula II. In one embodiment, the compound provided herein contains at least one dexamethasone moiety, in another embodiment at least one betamethasone moiety, and in another embodiment at least one hydrocortisone moiety. The corticosteroid moiety may be derivitized, for example, hydrocortisone acetate. In some embodiments, the rectal suspension is administered to a patient with ulcerative colitis. The ulcerative colitis may include, for example, one or more of ulcerative proctitis, ulcerative proctosigmoiditis, and left- sided ulcerative colitis.
[00127] The compounds and compositions provided herein can also be provided as an article of manufacture using packaging materials well known to those of skill in the art. See, e.g., U.S. Pat. Nos. 5,323,907; 5,052,558; and 5,033,252. Examples of pharmaceutical packaging materials include, but are not limited to, blister packs, bottles, tubes, inhalers, pumps, bags, vials, containers, syringes, and any packaging material suitable for a selected formulation and intended mode of administration and treatment.
[00128] Provided herein also are kits which, when used by the medical practitioner, can simplify the administration of appropriate amounts of active ingredients to a subject. In certain embodiments, the kit provided herein includes a container and a dosage form of a compound provided herein. Kits provided herein can further include devices that are used to administer the active ingredients. Kits provided herein can further include pharmaceutically acceptable vehicles that can be used to administer one or more active ingredients.
[00129] The disclosure will be further understood by the following non-limiting examples. EXAMPLES
[00130] As used herein, the symbols and conventions used in these processes, schemes and examples, regardless of whether a particular abbreviation is specifically defined, are consistent with those used in the contemporary scientific literature, for example, the Journal of the
American Chemical Society or the Journal of Biological Chemistry. Specifically, but without limitation, the following abbreviations may be used in the examples and throughout the specification: g (grams); mg (milligrams); mL (milliliters); μL (microliters); mM (millimolar); μM (micromolar); eq. (equivalent); Hz (Hertz); MHz (megahertz); mmol (millimoles); hr or hrs
(hours); min (minutes); MS (mass spectrometry); ESI (electrospray ionization); TLC (thin layer chromatography); R1 (retention time); Rf (retardation factor); SiCb (silica): THF
(tetrahydrofuran); DCM (dichloromethane); DMSO (dimethylsulfoxide); DMS0-a?6 (deuterated dimethylsulf oxide); EtOAc (ethyl acetate); EtOH (ethanol); Et2O, (diethylether); HCl
(hydrochloric acid); K2CO3 (potassium carbonate); NaOH (sodium hydroxide); Na2SO4 (sodium sulfate); NaCl, (sodium chloride); MgSθ4 (magnesium sulfate); NaH (sodium hydride); NaHCO3 (sodium bicarbonate); TEA (triethylamine); NaNO2, (sodium nitrite); CuCl2, (cupper(II) chloride); SO2, (sulfur dioxide); Me (methyl); Et (ethyl); Z1Bu (tert-butyl); and Boc (tert- butoxylcarbonyl).
[00131] For all of the following examples, standard work-up and purification methods known to those skilled in the art can be utilized. Unless otherwise indicated, all temperatures are expressed in 0C (degrees Centigrade), All reactions conducted at room temperature unless otherwise noted. Synthetic methodologies illustrated herein are intended to exemplify the applicable chemistry through the use of specific examples and are not indicative of the scope of the disclosure.
Example 1. Preparation of Betamethasone mini-PEGylate and Dexamethasone mini- PEGylate
[00132] Dexamethasone mini-PEGylate (Compound 1) and betamethasone mini-PEGylate (Compound 2) were synthesized according to Scheme I, provided herein.
[00133] Betamethasone mini-PEGylate was synthesized as follows:

Betamethasone (commercially available) Betamethasone mesylate

Betamethasone mini-PEGylate (Compound 2)
[00134] Procedure 1 : synthesis of betamethasone mesylate. In a dry three-neck round bottom flask under nitrogen, 100 mg (0.255 mmol) of betamethasone were dissolved at room temperature in 2 ml of pyridine under magnetic stirring. Then 30.41 mg (0.301 mmol) of tricthylamine were added and the reaction mixture cooled in ice bath. After 5 minutes, 35.0 mg (0.305 mmol) of methanesulfonyl chloride were added. After 5h, TLC of the sample (eluent methylene chloride/methanol 96/4 v/v) showed the appearance of a spot at Rf~0.5 visible at UV (254 nm) (betamethasone Rf-0.3). 10 ml of NaCl 5M was then added to the reaction mixture, and an extraction then performed with ethyl acetate (5x10 ml). The organic layers were collected, dried over Na2SO4 and evaporated, yielding 140 mg of crude betamethasone mesylate. The residue was purified by flash chromatography on 4g of silica eluting with methylene chloride/methanol 96/4 (v/v). Fractions were analyzed by TLC (previous method) and those containing the spot at Rf-0.5 mixed and evaporated, yielding 92.2 mg. Further crystallization from methylene chloride gave 56.1 mg (0.119 mmol, 47%) of betamethasone mesylate.
[00135] Procedure 2: synthesis of betamethasone mini-PEGylate (Compound 2). In a dry
three-neck round bottom flask under nitrogen, 376.0 mg (0.176 mmol) of mPEGiooo-SH, product name SUNBRIGHT® ME-020SH from NOF EUROPE (Belgium) NV (Grobbendonk, Belgium; European unit of NOF CORPORATION, Shibuya-ku, Tokyo, Japan) were dissolved in 7.3 ml of absolute ethanol under magnetic stirring and gentle heating. Then 650 μl (7.0 mg, 0.176 mmol) of ethanolic NaOH were added. After 15 minutes (pale yellow solution) 83.0 mg (0.176 mmol) of betamethasone mesylate were added and dissolved under gentle heating. After 5h, a TLC of the sample (eluent methylene chloride/methanol 96/4 v/v) showed the appearance of a spot at Rf~0.05 visible at UV (254 nm) and at I2 vapors both (mPEG2ooo-SH Rf~0.05 visible only at I2 vapors, betamethasone mesylate Rf-0.5 visible mainly at UV), HPLC analysis of the reaction mixture using the method A (6.25 μl of reaction mixture diluted to 250 μl with water/acetonitrile 90/10) showed the appearance of a main peak at -18.5 min (ELS and UV detector both) and the contemporary disappearance of betamethasone mesylate. Method A was as follows:
Analytical HPLC with UV and ELS detector HPLC analytical Cl 8 column (eg.
Phenomenex Jupiter Cl 8 300A 5μm 4.6x250mm), using Acetonitrile (ACN) for HPLC and H2O for HPLC:

[00136] The reaction mixture was then evaporated under vacuum and the residue partitioned in NaCl 5 M (10 ml) and methylene chloride (5x20 ml). The organic layers were mixed, dried over Na2SOd, evaporated and precipitated with diethyl ether, yielding 460 mg of crude of reaction. Such residue was purified by HPLC yielding 210 mg (0.83 mmol, 47%) of compound
2.
[00137] Dexamethasone mini-PEGylate was synthesized as follows:

Dexamethasone mesylate Dexamethasone mini-PEGylate (Compound 1 )
(commercially available)
[00138] Procedure 3: synthesis of dexamethasone mini-PEGylate (Compound 1). In a dry three-neck round bottom flask under nitrogen, 617.5 mg (0.326 mmol) of mPEG2ooo-SH, art n° PEG 1169 from IRIS Biotech GmbH (Marktredwitz, Germany) were dissolved in 13 ml of absolute ethanol under magnetic stirring and gentle heating. Then 1205 μl (13.05 mg, 0.326 mmol) of ethanolic NaOH were added. After 10 minutes (pale yellow solution) 153.7 mg (0.326 mmol) of dexamethasone-21 -mesylate were added and dissolved under gentle heating. After 5 h, the sample for TLC (eluent methylene chloride/methanol 96/4 v/v) showed the appearance of a spot at Rf~0.05 visible at UV (254 nm) and at I2 vapors both (mPEG2Ooo-SH Rf~0.05 visible only at h vapors, dexamethasone mesylate Rf~0.5 visible mainly at UV). HPLC analysis of the crude of the reaction mixture (6.25 μl of reaction mixture diluted to 250 μl with water/acetonitrile 80/20, method A) showed appearance of 3 peaks (ELS and UV detector) and disappearance of mPEG2oϋϋ-SH (ELS only) and dexamethasone mesylate (UV only). The reaction mixture was then evaporated under vacuum and the residue partitioned in NaCl 5 M (10 ml) and methylene chloride (5x20 ml). The organic layers were mixed, dried over Na2SO4 and evaporated yielding 735 mg of crude reaction product. The crude product was purified by flash chromatography on 25 g of silica using a step elution consisting of:
250 ml of methylene chloride/methanol 96/4 100 ml of methylene chloride/methanol 94/6 200 ml of methylene chloride/methanol 90/10.
[00139] Precipitation from cold diethyl ether gave 331 mg of white powder. A further preparative HPLC purification was performed. Fractions containing Compound 1 were mixed, evaporated of the acetonitrile under reduced pressure and lyophilized, yielding 0.1 18 mmol (37%).
[00140] The purified products were characterized by mass spectrometry (ESI-MS) and NMR spectroscopy by comparison to the unconjugated corticosteroids and/or the corticosteroid mesylates.
Example 2. In Vitro Activity
[00141] Dexamethasone mini-PEGylate (Compound 1) and betamethasone mini-PEGylate (Compound 2) were prepared and purified as described in Example 1 , and were tested in the HeLa/GR-luc model in order to assess their capability to enter the cell and to bind to the intracellular glucocorticoid receptor (GR). HeLa/GR-luc cells are human cervical epithelial cells stably transfected with a GR-responsive luciferase reporter. The GR pathway and the mechanism of action of its ligands are illustrated in FIG. 1, which is described in greater detail above in the Background.
[00142] Different tests were performed with dexamethasone mini-PEGylate (Compound 1, purity -97%) and betamethasone mini-PEGylate (Compound 2, purity -100%) in comparison with the non-mini-PEGylated corticosteroids dexamethasone and betamethasone, respectively.
[00143] In this example, HeLa/GR-luc cells were seeded in a 96- well plate with 2.5 x 104 cells/well in 100 μl of Complete Growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 μg/mL, Hygromycin B 100 μg/mL). The plate was incubated in a humidified incubator at 370C with 5% CO2 for 48 hours. Test item solutions (Compound 1, Compound 2, dexamethasone and betamethasone) for cell treatment were freshly prepared just before use by serial dilution of 10 mM in DMSO stock solution in serum free medium (DMEM. Penicillin 100 units/mL, Streptomycin 100 μg/mL). Serum free medium was used as negative control.
[00144] After 48 hours incubation, the cell medium was replaced with 90 μ I/well of serum free medium (DMEM, Penicillin 100 units/mL, Streptomycin 100 μg/mL) and 10 μl/well of test item solution at various concentrations were added. 6 wells were tested for each test item
concentration. 10 μ I/well of serum free medium were added to control wells. Incubation of cells with the test items was carried on at 37°C, 5% CO2 for 6 hours.
[00145] After the proper incubation time with test items, the plate was removed from incubator and equilibrated to room temperature before adding the reagent for luciferase detection. As soon as the plate reached room temperature, 100 μl/well of reagent were added to the cells. The content of each well was pipetted several times to ensure complete cell lysis. After approximately 3 minutes luminescence was measured in a luminometer. As illustrated in FIG. 2(a), dexamethasone mini-PEGylate (Compound 1 ) was able to activate the luciferase reporter gene, as evidenced by measured luminescence (RLU), as was betamethasone mini-PEGylate (Compound 2), as illustrated in FIG. 2(b). Data are represented in bar columns as mean RLU (Relative Luminescence Unit) ± SEM (Standard Error of the Mean). EC50 values were calculated by fitting the data on a sigmoidal dose-response curve with variable slope. GraphPad Prism Software was used for data representation, statistical analysis and curve fitting. .
[00146] Because the luciferase gene transcription is stimulated only after GR activation, it can be deduced that each of the two mini-PEGylates were able to enter the cell, and bind and activate the intracellular GR. However, the activities of the mini-PEGylates were lower than those of the corticosteroids. Specifically, the EC50 of dexamethasone was 2.46 nM, whereas the EC50 of dexamethasone mini-PEGylate (Compound 3) was be 0.85 μM, nearly 350 times greater. The EC50 of betamethasone was 3.2 nM, while the EC50 of betamethasone mini-PEGylate
(Compound 2) was 3.2 μM, about 1000 times greater.
Example 3. In Vitro Anti-Inflammatory Activity of Dexamethasone mini-PEGylate and Betamethasone mini-PEGylate
[00147J Dexamethasone mini-PEGylate (Compound 1, prepared as described in Example 1) and dexamethasone were tested in the NIH3T3/NFkB-luc model in order to assess its antiinflammatory activity, expressed as a reduction of NF-kB activity.
[00148] The immunosuppressive and anti-inflammatory actions of glucocorticoid hormones are mediated by their transpression of transcription factors NF-kB and AP-I. NF-kB is known to be a main transcriptional factor to be activated after inflammatory stimuli and is involved in the
transcriptional activation of many pro-inflammatory cytokines such as TNF-α and IL-6. For further details, see PJ. Barnes, "Anti-inflammatory actions of glucocorticoids: molecular mechanisms," Clinical Science, 1998, 94(6), 557-572, the entire contents of which are incorporated by reference herein.
[00149] NIH3T3/NFkB-luc cells are murine fibroblasts stably transfected with a NF-kB responsive luciferase reporter; NF-kB activation was induced by adding the stimulus
(lipopolysaccharide (LPS) 1 ng/mL) 18 hours after cell incubation with Compound 1 or Compound 2. Specifically, NIH3T3/NF£B-luc cells were seeded in a 96-well plate with 5 x 104 cells/well in 90 μl of Complete Growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 μg/mL, Hygromycin B 100 μg/mL) and 10 μl of test item solution (Compound 1 , Compound 2, dexamethasone or betamethasone) at the appropriate concentration were added to each well (10 μ I/well of Complete Growth medium were added to control wells). The plate was then incubated overnight in a humidified incubator at 37°C with 5% CO2 and treated with LPS 1 ng/mL the following day. Dexamethasone and betamethasone solutions for cell treatment were freshly prepared just before use by serial dilution of 10 niM in DMSO stock solution in complete growth medium (DMEM 10% FBS, Penicillin 100 units/mL, Streptomycin 100 μg/mL, Hygromycin B 100 μg/mL). Compound 1 and compound 2 10 mM starting solutions in complete growth medium were prepared just before cell treatment. These solutions were then further diluted in the same medium to obtain the desired concentrations to be tested. LPS 1 ng/mL solution for NIH3T3/NFkB-luc cell treatment was prepared from serial dilutions in complete growth medium of a 1 mg/mL LPS stock solution prepared in sterile dH2O. Complete growth medium was used as negative control.
[00150] After overnight (18 hours) incubation with the test items, 10 μl/well of LPS solution (final concentration in the well: 1 ng/mL) were added to each well (except for control wells). Incubation of cells was carried on at 37°C, 5% CO2 for 6 hours after LPS treatment. After the proper incubation time with the test items, the plate was removed from incubator and
equilibrated to room temperature before adding the reagent for luciferase detection. As soon as the plate reached room temperature, 100 μl/well of reagent was added to the cells. The content of each well was pipetted several times to ensure complete cell lysis. After approximately 3 minutes luminescence was measured in a lurninometer.As illustrated in FIGS. 3(a) and 3(b),
dexamethasone mini-PEGylate (Compound 1) reduces the activation of the luciferase reporter gene, as evidenced in FIG. 3 (a) by a reduction in the % induction with increasing concentration, and in FIG. 3(b) by a decrease in luminescence with increasing concentration. Data are represented in bar columns as mean RLU (Relative Luminescence Unit) ± SEM (Standard Error of the Mean) and % induction, normalizing RLU data with negative control data mean corresponding to 0% induction and LPS 1 ng/mL data mean corresponding to 100% induction. One-way ANOVA followed by Dunnett's post test (control column data: LPS 1 ng/mL) was the statistical analysis performed. Concentration data having a post test p value < 0.05 are considered as significantly reducing LPS induced NF-kB activation. IC50 values were calculated fitting data on a sigmoidal dose-response curve with variable slope. GraphPad Prism Software was used for data representation, statistical analysis and curve fitting.
[00151] Because the luciferase reporter gene transcription was stimulated only after NF-kB activation, it can be deduced that dexamethasone mini-PEGylate reduces the LPS-induced NF- kB activation, although with considerably higher IC50 as compared with the corticosteroid. Specifically, the IC50 of dexamethasone was 6.12 nM, whereas the IC50 of dexamethasone mini- PEGylate (Compound 1) was 99.43 μm, over 16,000 times greater.
[00152] Betamethasone mini-PEGylate (Compound 2), as prepared in Example 1, and Betamethasone were tested in the NIH3T3/NFkB-luc model to assess its anti-inflammatory activity, espressed as a reduction of NF-kB activity. The same protocols were used as described above for Compound 1. FIGS. 4(a) and 4(b) illustrate the results of an experiment comparing the inhibition of the activation of the luciferase reporter gene in NIH3T3/NF£B-luc cells by betamethasone or betamethasone mini-PEGylate (Compound 2) expressed as (a) % of induction and (b) luminiscence.
[00153J As illustrated in FIGS. 4(a) and 4(b), Compound 2 reduces the activation of the luciferase reporter gene, as evidenced by a reduction in the % induction with increasing concentration (a), and by a decrease in luminescence with increasing concentration (b). Because the luciferase reporter gene trasnscription is stimulated only after NF-kB activation, it can be deduced that betamethasone mini-PEGylate reduces the LPS-induced NF-kB activation, although with considerably higher IC50 as compared with the corticosteroid. Specifically, the IC50 of
betamethasone was 8.2 nM, whereas the IC50 of betamethasone mini-PEGylate (Compound 2) was 40.81 μM, about 5,000 times greater.
[00154] As compared to Compound 1 and dexamethasone maximum activity in the same cell model, both Compound 2 and betamethasone show an almost complete ( 100%) inhibition of LPS-induced NF- kB activation, while Compound 1 and dexamethasone maximum effect is around 60-70% inhibition.
Example 4. In Vivo Pharmacokinetics of Dexamethasone mini-PEGylate and Betamethasone mini-PEGylate Following Intravenous Administration
[00155] To evaluate the bioavailability of dexamethasone mini-PEGylate (Compound 1) as compared to dexamethasone, and the bioavailability of betamethasone mini-PEGylate
(Compound 2) as compared to betamethasone, pharmacokinetic (PK) studies was performed via intravenous administration (IV).
[00156J First, in the evaluation of dexamethasone mini-PEGylate (Compound I), a single IV administration to male Han Wistar rats was performed. Before administration, the animals were surgically prepared for serial blood sample collection from superior vena cava (i.e., were superior vena cava cannulated). Dexamethasone and dexamethasone mini-PEGylate (Compound 1 ) were formulated in 8% ethanol in saline. The dexamethasone formulation was filtered due to turbidity. The administered doses were 1.05 mg/kg for dexamethasone, and 18 mg/kg for dexamethasone mini-PEGylate (Compound 1). The doses in this Example were not equimolar, because the theoretical equimolar dose of Dexamethasone (3 mg/kg) was near the solubility limit of this compound.
[00157] Blood samples from each of three animals were collected at 8 different time points following administration, and frozen at -8O0C until analyzed. Analysis of plasma levels of dexamethasone and dexamethasone mini-PEGylate (Compound 1) were performed by HPLC- UV using a Waters XTerra® RPl 8 column 3.0x50 mm, 3.5 μ, with detection by measurement of the absorbance at 214 and 242 nm, the latter wavelength representing the maximum UV-Vis absorption peak of dexamethasone (starting eluent: MiIIiQ water/acetonitrile 80:20 with a subsequent gradient to 0: 100). The plasma samples were purified before quantification with a
solid phase extraction (SPE) technique using Waters Oasis® HLB lcc/10mg cartridges and methanol as final eluent.
[00158] Mean plasma concentrations were calculated for each PK sample on a proper calibration curve acquired by the above reported processing protocol with blank rat plasma with standard solutions. FIG. 5(a) illustrates the mean plasma concentration over time of dexamethasone, and FIG. 5(b) illustrates the mean plasma concentration over time of dexamethasone mini-PEGylate (Compound 1). FIG. 5(c) illustrates the mean plasma concentration over time both of dexamethasone and Compound 1, on the same scale.
Pharmacokinetic analysis was performed by NCOMP version 3.1, and by one phase exponential decay fitting for half life evaluation. For further details, see P. B. Laub et al., "NCOMP - A Windows-based program for noncompartmental analysis of pharmacokinetic data," Journal of Pharmaceutical Sciences, 1996, 85(4), 393-395, the entire contents of which are incorporated by reference.
[00159] Table 2 summarizes estimated PK parameters for dexamethasone and dexamethasone mini-PEGylate (Compound 1) based on the NCOMP and decay fitting.
Table 2

[00160] As FIGS. 5(a)-5(c) and Table 2 illustrate, the dexamethasone mini-PEGylate (Compound 1) has different pharmacokinetic properties than dexamethasone, e.g., a higher clearance and, as a consequence, a shorter blood half-life. The higher solubility of
dexamethasone mini-PEGylate (Compound 1) also results in a blood compartmentalization (the volume of distribution corresponds to about the mean rat blood volume) if compared to a higher Vss for dexamethasone, reflecting its wider body distribution.
[00161] To evaluate IV pharmacokinetics of Compound 2 in comparison with non-PEGylated molecule Betamethasone, a single IV administration to male Sprague Dawley SD rats was performed. Betamethasone and Compound 2 were formulated in 8% ethanol in saline at 0.6 mg/mL and 3.6 mg/mL, thus resulting in a final administered dose of 3 mg/kg and 18 mg/kg respectively, which was approximately equimolar. At each sampling time, blood samples of approximately 0.8 mL were collected under light isofluorane anaesthesia from the retro-orbital sinus of 3 males of each group, each animal being sampled on each day at 3 timepoints. Blood samples in triplicate were collected at the following timepoints after treatment: 5 min, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h and 8 h, as shown in Table 3.
Table 3

[00162] Blood samples were collected from each animal into vials containing EDTA as anticoagulant from each animal and centrifuged at 40C and 3000 g for 10 min. From each tube plasma samples were recovered, put in new eppendorf tubes and frozen at -800C until HPLC analysis. Analysis of plasma levels was performed by HPLC-UV technique using a Waters
XTerra RPl 8 column 3.0x50 mm, 3.5μ. The HPLC separation was done at a flow rate of 1 mL/min, with detection by measurement of the absorbance at 214 and 242 nm (the latter wavelength representing the maximum UV- Vis absorption peak of Betamethasone and therefore used for quantitative analysis). The injection volume was 40 μL and each sample was analysed in duplicate. Elution solvents were represented by MiIIiQ water and acetonitrile for mobile phase A and B, respectively; the starting eluent corresponded to phase A/B 80:20 with a subsequent gradient up to 100% (autosampling tray kept at 4°C). Plasma samples were purified before quantitation in order to eliminate any possible source of interference with the analysis itself by mean of SPE (Solid Phase Extraction) technique using the cartridges Waters Oasis® HLB lcc/l Omg and methanol as final eluent.
[00163] Mean plasma concentrations of test items were calculated for each PK sample on a proper calibration curve acquired by the above reported processing protocol with blank rat plasma spiked with standard solutions. The PK profiles for betamethasone and Compound 2 after single IV administration in rats are shown in FIGS. 6(a) and 6(b) respectively, with FIG. 6(c) illustrating the profiles both of betamethasone and Compound 2 on the same scale.
[00164] Pharmacokinetic analysis was performed by NCOMP program and by one phase exponential decay fitting for half life evaluation. Estimated main PK parameters are listed Table 4.
Table -

* percent extrapolated AUC = 0.92%
** percent extrapolated AUC = 8.7%
[00165] The results confirm that mini-PEGylation modifies the chemico-physical properties of the parent molecule betamethasone, thus conferring higher clearance and, as consequence, shorter blood half-life. The higher solubility of Compound 2 results also in a blood
compartmentalization (the volume of distribution corresponds to about the mean rat blood volume) if compared to a higher Vss for betamethasone, reflecting its wider body distribution.
Example 5. In Vivo Pharmacokinetics of Dexamethasone mini-PEGylate and Betamethasone mini-PEGylate Following Dermal Administration
[00166] The dermal pharmacokinetics of dexamethasone as compared to dexamethasone mini-PEGylate (Compound 5), and betamethasone as compared to betamethasone mini- PEGylate, were evaluated using dermal administration in rats.
[00167] First, in the evaluation of dexamethasone and dexamethasone mini-PEGylate (Compound 1), a single dermal administration to male Han Wistar rats was performed. Before test compound administration, animals were surgically prepared for serial blood sample collection from superior vena cava (i.e., the animals were superior vena cava cannulated), and the fur from the dorsal surface of the trunk was removed by clipping (approximately 10 cm2 per animal).
[00168] Dexamethasone and dexamethasone mini-PEGylate (Compound 1) were formulated in acetone/propylene glycol 50/50 to a final administered dose of 5 mg/kg and 30 mg/kg respectively. Blood samples were collected at 8 different timepoints after administration, and were frozen at -8O0C until analysis by HPLC. The blood collection, solid phase extraction, and HPLC-UV analysis were the same as described in Example 4.
[00169] FIG. 7 illustrates the measured plasma concentration of dexamethasone after a single dermal administration to three rats (limit of detection 79 nM). The analysis of plasma samples from rats administered dexamethasone mini-PEGylate (Compound 1) did not reveal levels above the limit of detection (179 nM, calibration curve in rat plasma) at any timepoint. This result suggests that dexamethasone mini-PEGylate is not absorbed systemically following administration. In view of the results of Example 4, even if some dexamethasone mini-PEGylate were systemically absorbed, it would be quickly eliminated.
[00170J Individual concentration values (n=3 per timepoint) of the rats administered dexamethasone showed a high variability, thus making the PK parameter evaluation of limited reliability. The maximum plasma level of dexamethasone was generally reached at 3 hours after dermal administration, although the level was also observed to increase between 8 and 24 hours after administration for all of the animals. The latter increase suggests an accumulation phenomenon in the skin compartment.
[00171] To evaluate the dermal pharmacokinetics of betamethasone as compared to betamethasone mini-PEGylate (Compound 2), a single dermal administration to male Sprague Dawley SD rats was performed. One day before dosing, the fur was removed from the dorsal surfaces of the trunk over an estimated area of 10% of the total body surface, using an electric clipper with a suitable blade.
[00172] Betamethasone and Compound 2 were formulated in acetone/propylene glycol 50/50 at, respectively, 12.5 mg/mL and 75 mg/niL corresponding to an administered dose of 5 mg/kg and 30 mg/kg for betamethasone and Compound 2 respectively. Blood samples were collected in triplicate (each animal being sampled on each day at 3 timepoints) at the following timepoints after treatment: 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h and 24 h. The blood collection and the SPE sample processing with subsequent quantitative analysis by HPLC-UV were carried out according to the same experimental procedure described above.
[00173] The PK profile for betamethasone after single dermal administration in rats is shown in FIG. 8. The results are reported as mean values (n = 6) ± error (95% CI, confidence interval). The analysis of Compound 2 plasma samples at equimolar dosage did not reveal test item levels above the limit of detection (37.6 nM, calibration curve in rat plasma not shown) at any time point.
[00174] Although individual concentration values of the rats treated with betamethasone showed a high variability, a preliminary pharmacokinetic analysis was performed by NCOMP version 3.1 program and main estimated PK parameters are listed in Table 5.
Table 5

percent extrapolated AUC = 26.9%
[00175] These results confirm that mini-PEGylation modifies the chemico-physical properties of betamethasone, with the consequent absence of a detectable dermal absorption. This feature may result in an improved safety profile: even if absorbed after dermal administration,
Compound 2 would be quickly eliminated, most likely via renal filtration.
Example 6. In Vivo Efficacy Study in Murine Oxazolone-lnduced Contact Hypersensitivity Model with Betamethasone mini-PEGylate
[00176] The in vivo efficacy of betamethasone mini-PEGylate (Compound 2) as prepared above in Example 1 was tested in a murine model of contact hypersensitivity induced by repeated exposure of the ear skin of mice to oxazolone, which is a chemical allergen capable of inducing delayed type hypersensitivity after exposure.
[00177] On day 0, mice were shaved on their abdominal side and sensitized by topical application of 100 μl 1.5% oxazolone in acetone.
[00178] On day 7, the ear thickness of left and right ears was measured; subsequently, the right ears were challenged by topical application of 20 μl 2.5% oxazolone in ethanol (10 μl on each side of the ear). As a control, 20 μl ethanol were applied to the left ears.
[00179] On day 8, 24 hours after challenge with oxazolone, thickness of left and right ears was measured.
[00180] Compound 2 was tested at three different concentration in aqueous solution: 5%, 0.5% and 0.05%. 20 μl of this solution (10 μl on either side of the ear that was challenged with oxazolone) were topically applied on challenged ears on days 4, 5, 6 and 7 (treatment on day 7
occurred 30 min after challenged). In this example, Compound 2 was not compared to betamethasone, but was tested alone. Diprosone cream (0.05% betamethasone diproprionate, Schering-Plough, Kenilworth, NJ) was used as a positive control, the treatment schedule for which was different; specifically, the diprosone cream was applied only once, on day 7 (30 min after challenge).
[00181] In each individual mouse, the absolute change in thickness of left and right ears was calculated by subtracting the t=0 value from the t=24h value. In addition, these changes were expressed as a relative (%) increase. Next, the absolute and relative oxazolone-specific swelling was obtained by subtracting the value of the left (control) ear from the right (challenged) ear.
[00182] After measuring ear thickening, challenged ears were collected and fixed in formaldehyde for further immunohistochemical analysis. After embedding in paraffin, tissue sections were stained with hematoxyllin and analyzed for the presence of infiltrating cells; for each mouse, two non-serial sections of the oxazolone-challenged ears were analyzed. Likewise, two non-serial sections were analyzed for the presence of CD4+ T cells, CD8+ T cells and macrophages with the use of specific antibodies.
[00183] Infiltrating cells were enumerated both as infiltrating cells per section (semiquantitative fashion) and as number of cells per mm2 tissue (quantitative fashion).
Example 7. In Vitro Anti- Inflammatory Activity of Dexamethasone mini-PEGylate
[00184] Dexamethasone mini-PEGylate (Compound 1) as prepared above in Example 1 was tested in various in vitro cytokine secretion assays in order to assess its anti-inflammatory activity; in particular, the effect of Compound 1 on the secretion of IFN-γ, TNF-α and IL-6 from stimulated human PBMC was evaluated. IFN-γ, TNF-α and IL-6 are pro-inflammatory cytokines involved in various inflammatory conditions; their gene transcription activation is under the control of one of the main transcriptional factor to be activated after pro-inflammatory stimuli, NF-kB.
[00185] Peripheral Blood Mononuclear Cells (PBMC) suspension was pre-incubated for 30 min at 37°C with Compound 1, reference compound (dexamethasone), or culture medium (control). Thereafter, a specific stimulus (2 μg/mL PHA, phytohaemagglutinin, for the IFN-γ
detection assay: 1 μg/mL LPS, lipopolysaccharide, for the TNF-α and IL-6 detection assays) was added to cells to Induce the cytokine secretion and the mixture was incubated for 24 h at 37°C. For basal control measurement, stimulus was omitted from the incubation medium.
[00186] Following incubation, the samples were centrifuged and the supernatants collected; the amount of secreted cytokine present in the supernatant was quantified using an EIA (enzyme immunoassay) detection kit.
[00187] FIGS. 9(a)-9(c) illustrate the results of an experiment comparing the inhibition of the cytokine secretion ((a) TNF-α, (b) IFN-γ, (c) IL-6) by dexamethasone mini-PEGylate
(Compound 1 ) expressed as a percent inhibition of the control secretion induced by the stimulus. Compound 1 was tested at 8 different concentrations in a range between 300 nM and 1 mM, while reference compound (dexamethasone) was tested in a range between 0.1 nM and 1 μM.
[00188] As FIGS. 9(a)-9(c) illustrate, Compound 1 is able to reduce TNF-α, IFN-γ and IL-6 secretion in stimulated human PBMC, showing a good concentration-response curve in this experiment. Therefore, it can be deduced that dexamethasone mini-PEGylate (Compound 1) is still able to inhibit cytokine secretion, showing an anti-inflammatory activity also after
PEGylation, although with considerably higher IC50 as compared with the non-PEGylated molecule dexamethasone, as illustrated in FIGS. 10, 1 1, and 12, which illustrate the inhibition curves derived from different in vitro assays with relative IC50 values. FIGS. 10(a) and 10(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for TNF-α inhibition; FIGS. 1 l(a) and 1 l(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for IFN-γ inhibition; and FIGS. 12(a) and 12(b) respectively illustrate the inhibition curves for Compound 1 and dexamethasone for IL-6 inhibition. The results are expressed as a percent of control specific activity ((measured specific activity/control specific activity) x 100) obtained in the presence of Compound 1 or dexamethasone.
[00189] The IC50 values (concentration causing a half-maximal inhibition of control specific activity) were determined by non-linear regression analysis of the inhibition curves generated with mean replicate values using the following Hill equation curve fitting:
 where Y = specific activity, D = minimum specific activity, A = maximum specific activity, C - compound concentration, C50 = IC50 and nH = slope factor.
where Y = specific activity, D = minimum specific activity, A = maximum specific activity, C - compound concentration, C50 = IC50 and nH = slope factor.
[00190] Based on the results, one can deduce that Compound 1 is able to inhibit cytokine secretion, showing anti-inflammatory activity, although with considerably higher IC50 as compared with the non-PEGylated molecule dexamethasone. Specifically, as compared to IC50 of dexamethasone, IC50 of Compound 1 was: over 6,800 times greater in the case of TNF-α inhibition assay, 790 times greater in the case of IFN-γ inhibition assay, and 372 times greater in the case of 1L-6 inhibition assay.
Other Embodiments
[00191] While the present invention has been described with reference to specific embodiments, this application is intended to cover those various changes and substitutions that may be made by those skilled in the art without departing from the spirit and scope of the appended claims.
Claims
1. A compound of Formula I:


D and E are each independently H, CI -CJ O alkyl, Ci-C1O alkoxy, or Ci-Ci0 acyl;
G, J, and K are each independently H or a halogen;
L1 and L2 are (i) both H, or (ii) one is H and the other OH, or (iii) together form =0;
X is a non-cleavable linking group between the C2] position of the corticosteroid and the mini-PEG;
mini-PEG is polyethylene glycol having a molecular weight between about 100 and about 20,000 Da;
Y is H, OH, Ci-C5 alkyl, Ci-C5 acyl. or Ci-C5 alkoxy; and
Ci and CT are bonded together by either a single bond or a double bond.
2, A compound of Formula II:

wherein n is an integer between 1 and 100, m is an integer between 0 and 100, and m+n is between 1 and 100;
A and B are each independently H, OH, Ci-Cio alkyl (e.g., CH3), C]-CiO alkoxy, or Cj- C]0 acyl, or A and B combine to form a cyclic acetal, ketal, or orthoester of the following formula: 
D and E are each independently H, C1-Ci0 alkyl, CJ-CJO alkoxy, or Cj-C10 acyl;
G, J, and K are each independently H or a halogen, in some embodiments H, Cl, F, Br, or each L1 and L2 pair is (i) both H, or (ii) one is H and the other OH, or (iii) together form
=O;
each X is a non-cleavable linking group between the C2J position of the corticosteroid and the mini-PEG;
mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da;
each Y is H, OH, C1-C5 alkyl, Cj-C5 acyl, or C1-C5 alkoxy; and each Ci and C2 pair is bonded together by either a single bond or a double bond.
3. The compound of claim 2, wherein the mini -PEG is a linear mini-PEG, n is 1 or 2, y is 0 or 1, and n+m equals 2.
4. The compound of claim 2, wherein the mini-PEG is a branched mini-PEG having q PEG groups emanating from a central core group, q is an integer between 3 and 10, n is between 1 and q, and m = q-n.
5. The compound of claim 2, wherein the mini-PEG is a star mini-PEG having q PEG groups emanate from a central core group, q is an integer between 10 and 100, n is between 10 and q, and m = q-n.
6. The compound of claim 2, wherein the mini-PEG is a comb mini-PEG having q PEG chains grafted to a polymer backbone, n is between 1 and q, and m = q-n.
7. The compound of any of the preceding claims, wherein X is a non-cleavable linking group selected from the group consisting of: alkylene, amido, oximo, S, O, or NR, where R is hydrogen or alkyl.
8. The compound of any of the preceding claims, wherein X is S, NR, or O, where R is H or alkyl.
9. The compound of any of the preceding claims, wherein X is S.
10. The compound of any of the preceding claims, wherein the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
11. The compound of any of the preceding claims, wherein the polyethylene glycol has a molecular weight between about 1 ,500 Da and about 2,000 Da.
12. The compound of any of the preceding claims, wherein Y is OH or OCH3.
13. The compound of any of the preceding claims, wherein A is H or OH.
14. The compound of any of the preceding claims, wherein B is H, OH, or CH3.
15. The compound of any of claims 1-12, wherein A and B combine to form a cyclic acetal, wherein D is H and E is CH2-CH2-CH3.
16. The compound of any of claims 1-12, wherein A and B combine to form a cyclic ketal, wherein D and E are each CH3.
17. The compound of any of the preceding claims, wherein G is H.
18. The compound of any of the preceding claims, wherein K is H, F, or CH3.
19. The compound of any of the preceding claims, wherein J is H, Cl, or F.
20. A compound having the formula:

21. The compound of claim 20, wherein the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
22. A compound having the formula:  wherein mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da.
wherein mini-PEG is polyethylene glycol having a molecular weight between about 100 Da and about 20,000 Da.
23. The compound of claim 22, wherein the polyethylene glycol has a molecular weight between about 500 Da and about 2,500 Da.
24. A compound having the formula:
Z X mini-PEG wherein Z is a corticosteroid; wherein mini-PEG is polyethylene glycol (PEG) having a molecular weight between about 100 Da and about 20,000 Da; and wherein X is alkylene, amido, oximo, S, O, or NR, where R is H or alkyl.
25. The compound of claim 24, wherein the corticosteroid is selected from the group consisting of: beclomethasone, betamethasone, budesonide, clobetasol, corticosterone, cortisone, desonide, desoximetasone, desoxycorticosterone, dexamethasone, difluocortolone, flumethasone, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluticasone, fluticasone proprionate, halobetasol, hydrocortisone, methylprednisolone, prednicarbate, prednisolone, prednisone, triamcinolone, and triamcinolone acetonide.
26. A pharmaceutical composition comprising a compound of any of the preceding claims, and one or more pharmaceutically acceptable carriers or excipients.
27. The pharmaceutical composition of claim 26. wherein the composition is formulated as a topical dosage form.
28. The pharmaceutical composition of claim 27, wherein the topical dosage form is selected from the group consisting of emulsions, solutions, suspensions, creams, gels, oils, hydrogels, ointments, dusting powders, dressings, elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols, irrigations, sprays, suppositories, bandages, and dermal patches.
29. A method for the treatment, prevention, or amelioration of one or more symptoms of a glucocorticoid receptor-mediated disorder, disease, or condition in a subject, which comprises administering to the subject the compound of any of claims 1 to 25 or a pharmaceutical composition of any of claims 26 to 29.
30. A method for the treatment, prevention, or amelioration of one or more symptoms of a disorder, disease, or condition associated with inflammation in a subject, which comprises administering to the subject the compound of any of claims 1 to 25 or the pharmaceutical composition of any of claims 26 to 29.
31. The method of claim 30, wherein the disorder, disease, or condition is an inflammatory or allergic disease.
32. The method of claim 31 , wherein the inflammatory or allergic disease is selected from the group consisting of systemic anaphylaxis and hypersensitivity disorders, atopic dermatitis, urticaria, drug allergy, insect sting allergy, food allergy, celiac disease, and mastocytosis.
33. The method of claim 30, wherein the disorder, disease, or condition is psoriasis or an inflammatory dermatosis.
34. The method of claim 33, wherein the inflammatory dermatosis is selected from the group consisting of dermatitis, eczema, atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis, urticaria, viral cutaneous pathologies including those derived from human
papillomavirus, HIV or RLV infection, bacterial, fungal, and other parasital cutaneous pathologies, and cutaneous and discoid lupus erythematosus.
35. The method of claim 31, wherein the inflammatory disease is an inflammatory bowel disease.
36. The method of claim 35, wherein the inflammatory bowel disease is Crohn's disease, ulcerative colitis, or pouchitis.
37. A method for modulating glucocorticoid receptor activity, comprising contacting a glucocorticoid receptor with the compound of any of claims 1 to 25 or the pharmaceutical composition of any of claims 26 to 29.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22334609P | 2009-07-06 | 2009-07-06 | |
| US61/223,346 | 2009-07-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011003870A2 true WO2011003870A2 (en) | 2011-01-13 |
| WO2011003870A3 WO2011003870A3 (en) | 2011-03-31 |
Family
ID=43429598
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2010/059569 WO2011003870A2 (en) | 2009-07-06 | 2010-07-05 | Mini-pegylated corticosteroids, compositions including same, and methods of making and using same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011003870A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104387433A (en) * | 2014-12-01 | 2015-03-04 | 江西赣亮医药原料有限公司 | Preparation method of clobetasol and preparation method of clobetasol propionate |
Citations (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US5033252A (en) | 1987-12-23 | 1991-07-23 | Entravision, Inc. | Method of packaging and sterilizing a pharmaceutical product |
| US5052558A (en) | 1987-12-23 | 1991-10-01 | Entravision, Inc. | Packaged pharmaceutical product |
| US5059595A (en) | 1989-03-22 | 1991-10-22 | Bioresearch, S.P.A. | Pharmaceutical compositions containing 5-methyltetrahydrofolic acid, 5-formyltetrahydrofolic acid and their pharmaceutically acceptable salts in controlled-release form active in the therapy of organic mental disturbances |
| US5073543A (en) | 1988-07-21 | 1991-12-17 | G. D. Searle & Co. | Controlled release formulations of trophic factors in ganglioside-lipsome vehicle |
| US5120548A (en) | 1989-11-07 | 1992-06-09 | Merck & Co., Inc. | Swelling modulated polymeric drug delivery device |
| US5323907A (en) | 1992-06-23 | 1994-06-28 | Multi-Comp, Inc. | Child resistant package assembly for dispensing pharmaceutical medications |
| US5354556A (en) | 1984-10-30 | 1994-10-11 | Elan Corporation, Plc | Controlled release powder and process for its preparation |
| US5591767A (en) | 1993-01-25 | 1997-01-07 | Pharmetrix Corporation | Liquid reservoir transdermal patch for the administration of ketorolac |
| US5612059A (en) | 1988-08-30 | 1997-03-18 | Pfizer Inc. | Use of asymmetric membranes in delivery devices |
| US5639476A (en) | 1992-01-27 | 1997-06-17 | Euro-Celtique, S.A. | Controlled release formulations coated with aqueous dispersions of acrylic polymers |
| US5639480A (en) | 1989-07-07 | 1997-06-17 | Sandoz Ltd. | Sustained release formulations of water soluble peptides |
| US5674533A (en) | 1994-07-07 | 1997-10-07 | Recordati, S.A., Chemical And Pharmaceutical Company | Pharmaceutical composition for the controlled release of moguisteine in a liquid suspension |
| US5709874A (en) | 1993-04-14 | 1998-01-20 | Emory University | Device for local drug delivery and methods for using the same |
| US5733566A (en) | 1990-05-15 | 1998-03-31 | Alkermes Controlled Therapeutics Inc. Ii | Controlled release of antiparasitic agents in animals |
| US5739108A (en) | 1984-10-04 | 1998-04-14 | Monsanto Company | Prolonged release of biologically active polypeptides |
| US5759542A (en) | 1994-08-05 | 1998-06-02 | New England Deaconess Hospital Corporation | Compositions and methods for the delivery of drugs by platelets for the treatment of cardiovascular and other diseases |
| US5840674A (en) | 1990-11-01 | 1998-11-24 | Oregon Health Sciences University | Covalent microparticle-drug conjugates for biological targeting |
| US5891474A (en) | 1997-01-29 | 1999-04-06 | Poli Industria Chimica, S.P.A. | Time-specific controlled release dosage formulations and method of preparing same |
| US5900252A (en) | 1990-04-17 | 1999-05-04 | Eurand International S.P.A. | Method for targeted and controlled release of drugs in the intestinal tract and more particularly in the colon |
| US5922356A (en) | 1996-10-09 | 1999-07-13 | Sumitomo Pharmaceuticals Company, Limited | Sustained release formulation |
| US5972891A (en) | 1992-12-07 | 1999-10-26 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US5972366A (en) | 1994-11-28 | 1999-10-26 | The Unites States Of America As Represented By The Secretary Of The Army | Drug releasing surgical implant or dressing material |
| US5980945A (en) | 1996-01-16 | 1999-11-09 | Societe De Conseils De Recherches Et D'applications Scientifique S.A. | Sustained release drug formulations |
| US5985307A (en) | 1993-04-14 | 1999-11-16 | Emory University | Device and method for non-occlusive localized drug delivery |
| US5993855A (en) | 1995-09-18 | 1999-11-30 | Shiseido Company, Ltd. | Delayed drug-releasing microspheres |
| US6004534A (en) | 1993-07-23 | 1999-12-21 | Massachusetts Institute Of Technology | Targeted polymerized liposomes for improved drug delivery |
| US6039975A (en) | 1995-10-17 | 2000-03-21 | Hoffman-La Roche Inc. | Colon targeted delivery system |
| US6045830A (en) | 1995-09-04 | 2000-04-04 | Takeda Chemical Industries, Ltd. | Method of production of sustained-release preparation |
| US6048736A (en) | 1998-04-29 | 2000-04-11 | Kosak; Kenneth M. | Cyclodextrin polymers for carrying and releasing drugs |
| US6060082A (en) | 1997-04-18 | 2000-05-09 | Massachusetts Institute Of Technology | Polymerized liposomes targeted to M cells and useful for oral or mucosal drug delivery |
| US6071495A (en) | 1989-12-22 | 2000-06-06 | Imarx Pharmaceutical Corp. | Targeted gas and gaseous precursor-filled liposomes |
| US6087324A (en) | 1993-06-24 | 2000-07-11 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US6113943A (en) | 1996-10-31 | 2000-09-05 | Takeda Chemical Industries, Ltd. | Sustained-release preparation capable of releasing a physiologically active substance |
| US6120751A (en) | 1997-03-21 | 2000-09-19 | Imarx Pharmaceutical Corp. | Charged lipids and uses for the same |
| US6131570A (en) | 1998-06-30 | 2000-10-17 | Aradigm Corporation | Temperature controlling device for aerosol drug delivery |
| US6139865A (en) | 1996-10-01 | 2000-10-31 | Eurand America, Inc. | Taste-masked microcapsule compositions and methods of manufacture |
| US6197350B1 (en) | 1996-12-20 | 2001-03-06 | Takeda Chemical Industries, Ltd. | Method of producing a sustained-release preparation |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6253872B1 (en) | 1996-05-29 | 2001-07-03 | Gmundner Fertigteile Gesellschaft M.B.H & Co., Kg | Track soundproofing arrangement |
| US6264970B1 (en) | 1996-06-26 | 2001-07-24 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US6267981B1 (en) | 1995-06-27 | 2001-07-31 | Takeda Chemical Industries, Ltd. | Method of producing sustained-release preparation |
| US6271359B1 (en) | 1999-04-14 | 2001-08-07 | Musc Foundation For Research Development | Tissue-specific and pathogen-specific toxic agents and ribozymes |
| US6274552B1 (en) | 1993-03-18 | 2001-08-14 | Cytimmune Sciences, Inc. | Composition and method for delivery of biologically-active factors |
| US6316652B1 (en) | 1995-06-06 | 2001-11-13 | Kosta Steliou | Drug mitochondrial targeting agents |
| WO2002017918A2 (en) | 2000-08-30 | 2002-03-07 | Pfizer Products Inc. | Sustained release formulations for growth hormone secretagogues |
| US6419961B1 (en) | 1996-08-29 | 2002-07-16 | Takeda Chemical Industries, Ltd. | Sustained release microcapsules of a bioactive substance and a biodegradable polymer |
| US6589548B1 (en) | 1998-05-16 | 2003-07-08 | Mogam Biotechnology Research Institute | Controlled drug delivery system using the conjugation of drug to biodegradable polyester |
| US6613358B2 (en) | 1998-03-18 | 2003-09-02 | Theodore W. Randolph | Sustained-release composition including amorphous polymer |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR7604M (en) * | 1968-04-22 | 1970-01-19 | ||
| DE1768345B2 (en) * | 1968-05-03 | 1976-12-02 | Hoechst Ag, 6000 Frankfurt | DIMERIC STEROID-21 ALKYL CARBONATES AND THE METHOD OF MANUFACTURING THEREOF |
| FR7702M (en) * | 1968-07-18 | 1970-02-23 | ||
| US5681964A (en) * | 1990-10-23 | 1997-10-28 | University Of Kentucky Research Foundation | Permeable, non-irritating prodrugs of nonsteroidal and steroidal agents |
| EP1437143A4 (en) * | 2001-09-28 | 2007-05-09 | Santen Pharmaceutical Co Ltd | Injections for eye tissue containing drug bound to polyethylene glycol |
| AU2003265576A1 (en) * | 2002-08-23 | 2004-03-11 | The Mclean Hospital Corporation | Corticosteroid conjugates and uses thereof |
-
2010
- 2010-07-05 WO PCT/EP2010/059569 patent/WO2011003870A2/en active Application Filing
Patent Citations (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US5739108A (en) | 1984-10-04 | 1998-04-14 | Monsanto Company | Prolonged release of biologically active polypeptides |
| US5354556A (en) | 1984-10-30 | 1994-10-11 | Elan Corporation, Plc | Controlled release powder and process for its preparation |
| US5052558A (en) | 1987-12-23 | 1991-10-01 | Entravision, Inc. | Packaged pharmaceutical product |
| US5033252A (en) | 1987-12-23 | 1991-07-23 | Entravision, Inc. | Method of packaging and sterilizing a pharmaceutical product |
| US5073543A (en) | 1988-07-21 | 1991-12-17 | G. D. Searle & Co. | Controlled release formulations of trophic factors in ganglioside-lipsome vehicle |
| US5612059A (en) | 1988-08-30 | 1997-03-18 | Pfizer Inc. | Use of asymmetric membranes in delivery devices |
| US5698220A (en) | 1988-08-30 | 1997-12-16 | Pfizer Inc. | Asymmetric membranes in delivery devices |
| US5059595A (en) | 1989-03-22 | 1991-10-22 | Bioresearch, S.P.A. | Pharmaceutical compositions containing 5-methyltetrahydrofolic acid, 5-formyltetrahydrofolic acid and their pharmaceutically acceptable salts in controlled-release form active in the therapy of organic mental disturbances |
| US5639480A (en) | 1989-07-07 | 1997-06-17 | Sandoz Ltd. | Sustained release formulations of water soluble peptides |
| US5120548A (en) | 1989-11-07 | 1992-06-09 | Merck & Co., Inc. | Swelling modulated polymeric drug delivery device |
| US6071495A (en) | 1989-12-22 | 2000-06-06 | Imarx Pharmaceutical Corp. | Targeted gas and gaseous precursor-filled liposomes |
| US5900252A (en) | 1990-04-17 | 1999-05-04 | Eurand International S.P.A. | Method for targeted and controlled release of drugs in the intestinal tract and more particularly in the colon |
| US5733566A (en) | 1990-05-15 | 1998-03-31 | Alkermes Controlled Therapeutics Inc. Ii | Controlled release of antiparasitic agents in animals |
| US5840674A (en) | 1990-11-01 | 1998-11-24 | Oregon Health Sciences University | Covalent microparticle-drug conjugates for biological targeting |
| US5639476A (en) | 1992-01-27 | 1997-06-17 | Euro-Celtique, S.A. | Controlled release formulations coated with aqueous dispersions of acrylic polymers |
| US5323907A (en) | 1992-06-23 | 1994-06-28 | Multi-Comp, Inc. | Child resistant package assembly for dispensing pharmaceutical medications |
| US5972891A (en) | 1992-12-07 | 1999-10-26 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US5591767A (en) | 1993-01-25 | 1997-01-07 | Pharmetrix Corporation | Liquid reservoir transdermal patch for the administration of ketorolac |
| US6274552B1 (en) | 1993-03-18 | 2001-08-14 | Cytimmune Sciences, Inc. | Composition and method for delivery of biologically-active factors |
| US5709874A (en) | 1993-04-14 | 1998-01-20 | Emory University | Device for local drug delivery and methods for using the same |
| US5985307A (en) | 1993-04-14 | 1999-11-16 | Emory University | Device and method for non-occlusive localized drug delivery |
| US6376461B1 (en) | 1993-06-24 | 2002-04-23 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US6087324A (en) | 1993-06-24 | 2000-07-11 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US6004534A (en) | 1993-07-23 | 1999-12-21 | Massachusetts Institute Of Technology | Targeted polymerized liposomes for improved drug delivery |
| US5674533A (en) | 1994-07-07 | 1997-10-07 | Recordati, S.A., Chemical And Pharmaceutical Company | Pharmaceutical composition for the controlled release of moguisteine in a liquid suspension |
| US5759542A (en) | 1994-08-05 | 1998-06-02 | New England Deaconess Hospital Corporation | Compositions and methods for the delivery of drugs by platelets for the treatment of cardiovascular and other diseases |
| US5972366A (en) | 1994-11-28 | 1999-10-26 | The Unites States Of America As Represented By The Secretary Of The Army | Drug releasing surgical implant or dressing material |
| US6316652B1 (en) | 1995-06-06 | 2001-11-13 | Kosta Steliou | Drug mitochondrial targeting agents |
| US6267981B1 (en) | 1995-06-27 | 2001-07-31 | Takeda Chemical Industries, Ltd. | Method of producing sustained-release preparation |
| US6045830A (en) | 1995-09-04 | 2000-04-04 | Takeda Chemical Industries, Ltd. | Method of production of sustained-release preparation |
| US5993855A (en) | 1995-09-18 | 1999-11-30 | Shiseido Company, Ltd. | Delayed drug-releasing microspheres |
| US6039975A (en) | 1995-10-17 | 2000-03-21 | Hoffman-La Roche Inc. | Colon targeted delivery system |
| US5980945A (en) | 1996-01-16 | 1999-11-09 | Societe De Conseils De Recherches Et D'applications Scientifique S.A. | Sustained release drug formulations |
| US6253872B1 (en) | 1996-05-29 | 2001-07-03 | Gmundner Fertigteile Gesellschaft M.B.H & Co., Kg | Track soundproofing arrangement |
| US6264970B1 (en) | 1996-06-26 | 2001-07-24 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| US6419961B1 (en) | 1996-08-29 | 2002-07-16 | Takeda Chemical Industries, Ltd. | Sustained release microcapsules of a bioactive substance and a biodegradable polymer |
| US6139865A (en) | 1996-10-01 | 2000-10-31 | Eurand America, Inc. | Taste-masked microcapsule compositions and methods of manufacture |
| US5922356A (en) | 1996-10-09 | 1999-07-13 | Sumitomo Pharmaceuticals Company, Limited | Sustained release formulation |
| US6113943A (en) | 1996-10-31 | 2000-09-05 | Takeda Chemical Industries, Ltd. | Sustained-release preparation capable of releasing a physiologically active substance |
| US6699500B2 (en) | 1996-10-31 | 2004-03-02 | Takeda Chemical Industries, Ltd. | Sustained-release preparation capable of releasing a physiologically active substance |
| US6197350B1 (en) | 1996-12-20 | 2001-03-06 | Takeda Chemical Industries, Ltd. | Method of producing a sustained-release preparation |
| US5891474A (en) | 1997-01-29 | 1999-04-06 | Poli Industria Chimica, S.P.A. | Time-specific controlled release dosage formulations and method of preparing same |
| US6120751A (en) | 1997-03-21 | 2000-09-19 | Imarx Pharmaceutical Corp. | Charged lipids and uses for the same |
| US6060082A (en) | 1997-04-18 | 2000-05-09 | Massachusetts Institute Of Technology | Polymerized liposomes targeted to M cells and useful for oral or mucosal drug delivery |
| US6613358B2 (en) | 1998-03-18 | 2003-09-02 | Theodore W. Randolph | Sustained-release composition including amorphous polymer |
| US6048736A (en) | 1998-04-29 | 2000-04-11 | Kosak; Kenneth M. | Cyclodextrin polymers for carrying and releasing drugs |
| US6589548B1 (en) | 1998-05-16 | 2003-07-08 | Mogam Biotechnology Research Institute | Controlled drug delivery system using the conjugation of drug to biodegradable polyester |
| US6131570A (en) | 1998-06-30 | 2000-10-17 | Aradigm Corporation | Temperature controlling device for aerosol drug delivery |
| US6271359B1 (en) | 1999-04-14 | 2001-08-07 | Musc Foundation For Research Development | Tissue-specific and pathogen-specific toxic agents and ribozymes |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| WO2002017918A2 (en) | 2000-08-30 | 2002-03-07 | Pfizer Products Inc. | Sustained release formulations for growth hormone secretagogues |
Non-Patent Citations (5)
| Title |
|---|
| P.B. LAUB ET AL.: "NCOMP - A Windows-based program for noncompartmental analysis of pharmacokinetic data", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 85, no. 4, 1996, pages 393 - 395 |
| P.J. BARNES: "Anti-inflammatory actions of glucocorticoids: molecular mechanisms", CLINICAL SCIENCE, vol. 94, no. 6, 1998, pages 557 - 572 |
| SANTUS; BAKER, 1. CONTROLLED RELEASE, vol. 35, 1995, pages L-21 |
| VERMA ET AL., DRUG DEVELOPMENT AND INDUSTRIAL PHARMACY, vol. 26, 2000, pages 695 - 708 |
| VERMA ET AL., J. CONTROLLED RELEASE, vol. 79, 2002, pages 7 - 27 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104387433A (en) * | 2014-12-01 | 2015-03-04 | 江西赣亮医药原料有限公司 | Preparation method of clobetasol and preparation method of clobetasol propionate |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011003870A3 (en) | 2011-03-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3569237B1 (en) | Sodium nitrite-containing pharmaceutical compositions | |
| US9187425B2 (en) | 2,5-disubstituted arylsulfonamide CCR3 antagonists | |
| EP2547655B1 (en) | Arylsulfonamide ccr3 antagonists | |
| AU2011313906B2 (en) | Salts of arylsulfonamide CCR3 antagonists | |
| SG175277A1 (en) | Arylsulfonamide ccr3 antagonists | |
| WO2011003870A2 (en) | Mini-pegylated corticosteroids, compositions including same, and methods of making and using same | |
| US9637460B2 (en) | Isotopically enriched arylsulfonamide CCR3 antagonists | |
| AU2022202914A1 (en) | Sodium nitrite-containing pharmaceutical compositions | |
| TWI485139B (en) | Isotopically enriched arylsulfonamide ccr3 antagonists | |
| AU2019200140B8 (en) | Sodium nitrite-containing pharmaceutical compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10732906 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 32PN | Ep: public notification in the ep bulletin as address of the adressee cannot be established |
Free format text: NOTING OF LOSS OF RIGHTS PURSUANT TO RULE 112(1) EPC (EPO FORM 1205A DATED 10.04.2012.) |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10732906 Country of ref document: EP Kind code of ref document: A2 |