WO2009117421A2 - Heterocyclic modulators of gpr119 for treatment of disease - Google Patents
Heterocyclic modulators of gpr119 for treatment of disease Download PDFInfo
- Publication number
- WO2009117421A2 WO2009117421A2 PCT/US2009/037408 US2009037408W WO2009117421A2 WO 2009117421 A2 WO2009117421 A2 WO 2009117421A2 US 2009037408 W US2009037408 W US 2009037408W WO 2009117421 A2 WO2009117421 A2 WO 2009117421A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- recited
- trifluoromethyl
- chloro
- Prior art date
Links
- SGDWFLCXNFTAPJ-UHFFFAOYSA-N C#Cc1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)nc1 Chemical compound C#Cc1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)nc1 SGDWFLCXNFTAPJ-UHFFFAOYSA-N 0.000 description 1
- XLLZKGVTAUFABE-UHFFFAOYSA-N CC(C)(C)OC(N1CCC(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)CC1)=O Chemical compound CC(C)(C)OC(N1CCC(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)CC1)=O XLLZKGVTAUFABE-UHFFFAOYSA-N 0.000 description 1
- KQMNQVKYEGLAHR-UHFFFAOYSA-N CC(C)c(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound CC(C)c(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O KQMNQVKYEGLAHR-UHFFFAOYSA-N 0.000 description 1
- IHJITRDWIOYANM-UHFFFAOYSA-N CC(C)c1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)nc1 Chemical compound CC(C)c1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)nc1 IHJITRDWIOYANM-UHFFFAOYSA-N 0.000 description 1
- GBSZHPXEZROSRP-UHFFFAOYSA-N CC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)O Chemical compound CC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)O GBSZHPXEZROSRP-UHFFFAOYSA-N 0.000 description 1
- FLVCBBMQMIBQEZ-UHFFFAOYSA-N CC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)OCc(cc1)ccc1OC(F)(F)F Chemical compound CC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)OCc(cc1)ccc1OC(F)(F)F FLVCBBMQMIBQEZ-UHFFFAOYSA-N 0.000 description 1
- DJLVZYSRFYYORW-UHFFFAOYSA-N CCN(CC)c(cc1)cc(C(F)(F)F)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 Chemical compound CCN(CC)c(cc1)cc(C(F)(F)F)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 DJLVZYSRFYYORW-UHFFFAOYSA-N 0.000 description 1
- FFDCKHRGHBWECL-UHFFFAOYSA-N CCNc(cc1)cc(C(F)(F)F)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 Chemical compound CCNc(cc1)cc(C(F)(F)F)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 FFDCKHRGHBWECL-UHFFFAOYSA-N 0.000 description 1
- PAMMATTXULNYEQ-UHFFFAOYSA-N CCc(cc(cc1)Br)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 Chemical compound CCc(cc(cc1)Br)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 PAMMATTXULNYEQ-UHFFFAOYSA-N 0.000 description 1
- RADPNXYFVVQRSI-UHFFFAOYSA-N CCc(cccc1)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 Chemical compound CCc(cccc1)c1-[n]1c(C(NCc(cc2)ccc2Oc(cc2)ccc2F)=O)ccc1 RADPNXYFVVQRSI-UHFFFAOYSA-N 0.000 description 1
- RJCNFTNKBCBQCV-YFHOEESVSA-N CN(C)/C=C(/C(c(c(Cl)c1)ccc1Cl)=O)\C(OC)=O Chemical compound CN(C)/C=C(/C(c(c(Cl)c1)ccc1Cl)=O)\C(OC)=O RJCNFTNKBCBQCV-YFHOEESVSA-N 0.000 description 1
- YZWBDJDNRYZMDE-UHFFFAOYSA-N CN(C)c(cc1)cc(C(F)(F)F)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 Chemical compound CN(C)c(cc1)cc(C(F)(F)F)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 YZWBDJDNRYZMDE-UHFFFAOYSA-N 0.000 description 1
- JICBGCDFIIKQHJ-UHFFFAOYSA-N COC(c1ccc[n]1-c(ncc(OC)c1)c1Cl)=O Chemical compound COC(c1ccc[n]1-c(ncc(OC)c1)c1Cl)=O JICBGCDFIIKQHJ-UHFFFAOYSA-N 0.000 description 1
- UZPICEUXAXVHCH-UHFFFAOYSA-N CS(NCc1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cc1)(=O)=O Chemical compound CS(NCc1ccc(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cc1)(=O)=O UZPICEUXAXVHCH-UHFFFAOYSA-N 0.000 description 1
- ZRMLXAGHPCJOAO-UHFFFAOYSA-N CS(c1cc(F)c(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cc1)(=O)=O Chemical compound CS(c1cc(F)c(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cc1)(=O)=O ZRMLXAGHPCJOAO-UHFFFAOYSA-N 0.000 description 1
- BMYYJVXEFSUBMB-UHFFFAOYSA-N Cc(cc1)cc(Cl)c1-[n]1c(C(NNc(ncc(C(F)(F)F)c2)c2Cl)=O)ccc1 Chemical compound Cc(cc1)cc(Cl)c1-[n]1c(C(NNc(ncc(C(F)(F)F)c2)c2Cl)=O)ccc1 BMYYJVXEFSUBMB-UHFFFAOYSA-N 0.000 description 1
- XGXCHEOUMMAAPO-UHFFFAOYSA-N Cc(cc1)ccc1Oc(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound Cc(cc1)ccc1Oc(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O XGXCHEOUMMAAPO-UHFFFAOYSA-N 0.000 description 1
- WKQKBZQUPZBACC-UHFFFAOYSA-N Cc(ccnc1I)c1Cl Chemical compound Cc(ccnc1I)c1Cl WKQKBZQUPZBACC-UHFFFAOYSA-N 0.000 description 1
- CQHZLXPVQOBXIV-UHFFFAOYSA-N Cc1c(C(O)=O)c(-c(ccc(C(F)(F)F)c2)c2Cl)n[o]1 Chemical compound Cc1c(C(O)=O)c(-c(ccc(C(F)(F)F)c2)c2Cl)n[o]1 CQHZLXPVQOBXIV-UHFFFAOYSA-N 0.000 description 1
- HONMVNODADDDGA-UHFFFAOYSA-N FC(c(cc1)ccc1Oc1n[o]c(CCl)n1)(F)F Chemical compound FC(c(cc1)ccc1Oc1n[o]c(CCl)n1)(F)F HONMVNODADDDGA-UHFFFAOYSA-N 0.000 description 1
- AHWQUTWZSUDAAX-UHFFFAOYSA-N N#Cc(c(C(F)(F)F)c1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound N#Cc(c(C(F)(F)F)c1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O AHWQUTWZSUDAAX-UHFFFAOYSA-N 0.000 description 1
- DKOHGMYXWJJAIA-UHFFFAOYSA-N N#Cc(cc1)cc(Cl)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 Chemical compound N#Cc(cc1)cc(Cl)c1-[n]1c(C(NNc2ncc(C(F)(F)F)cc2Cl)=O)ccc1 DKOHGMYXWJJAIA-UHFFFAOYSA-N 0.000 description 1
- UFQMYNNPZBDHLU-UHFFFAOYSA-N N#Cc(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound N#Cc(cc1)ccc1NNC(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O UFQMYNNPZBDHLU-UHFFFAOYSA-N 0.000 description 1
- OEQVEUISABTHDS-UHFFFAOYSA-N N#Cc(cc1)ccc1Oc1ccc(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)cc1 Chemical compound N#Cc(cc1)ccc1Oc1ccc(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)cc1 OEQVEUISABTHDS-UHFFFAOYSA-N 0.000 description 1
- UUVKKEUYGZLUKT-UHFFFAOYSA-N N/C(/Oc1ccc(C(F)(F)F)cc1)=N/O Chemical compound N/C(/Oc1ccc(C(F)(F)F)cc1)=N/O UUVKKEUYGZLUKT-UHFFFAOYSA-N 0.000 description 1
- VLVHEHQIMRCOBP-UHFFFAOYSA-N NCC1CCC(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)CC1 Chemical compound NCC1CCC(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)CC1 VLVHEHQIMRCOBP-UHFFFAOYSA-N 0.000 description 1
- KHFKSHYCVQZAQP-UHFFFAOYSA-N NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound NNc(ncc(C(F)(F)F)c1)c1Cl KHFKSHYCVQZAQP-UHFFFAOYSA-N 0.000 description 1
- HGAKKANATKSUCN-UHFFFAOYSA-N Nc1c(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cccc1 Chemical compound Nc1c(CNC(c2ccc[n]2-c(ncc(C(F)(F)F)c2)c2Cl)=O)cccc1 HGAKKANATKSUCN-UHFFFAOYSA-N 0.000 description 1
- FBABHUBNMLLLBH-UHFFFAOYSA-N O=C(C(CCC1)N1c(ncc(C(F)(F)F)c1)c1Cl)NNc1ncc(C(F)(F)F)cc1Cl Chemical compound O=C(C(CCC1)N1c(ncc(C(F)(F)F)c1)c1Cl)NNc1ncc(C(F)(F)F)cc1Cl FBABHUBNMLLLBH-UHFFFAOYSA-N 0.000 description 1
- BBCZYZLPZCNYTI-UHFFFAOYSA-N O=C(c1ccc[n]1-c(cc1)c(C(F)(F)F)cc1N1CCOCC1)NCc(cc1)ccc1Oc(cc1)ccc1F Chemical compound O=C(c1ccc[n]1-c(cc1)c(C(F)(F)F)cc1N1CCOCC1)NCc(cc1)ccc1Oc(cc1)ccc1F BBCZYZLPZCNYTI-UHFFFAOYSA-N 0.000 description 1
- IGGARUOSRCIKSP-UHFFFAOYSA-N O=C(c1ccc[n]1-c(cc1)ccc1Oc1ccncc1)NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound O=C(c1ccc[n]1-c(cc1)ccc1Oc1ccncc1)NNc(ncc(C(F)(F)F)c1)c1Cl IGGARUOSRCIKSP-UHFFFAOYSA-N 0.000 description 1
- JFPHNFMIYGJSSO-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCC1CCN(Cc(cc2)ccc2OC(F)(F)F)CC1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCC1CCN(Cc(cc2)ccc2OC(F)(F)F)CC1 JFPHNFMIYGJSSO-UHFFFAOYSA-N 0.000 description 1
- BHOHIYFEELWGBQ-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccc(CC(F)(F)F)cn1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccc(CC(F)(F)F)cn1 BHOHIYFEELWGBQ-UHFFFAOYSA-N 0.000 description 1
- PXCFHMOXSJAKMB-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccc(CN2CCOCC2)cc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccc(CN2CCOCC2)cc1 PXCFHMOXSJAKMB-UHFFFAOYSA-N 0.000 description 1
- XNWXVNYIHXLRJD-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1cccc(OC(F)(F)F)c1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1cccc(OC(F)(F)F)c1 XNWXVNYIHXLRJD-UHFFFAOYSA-N 0.000 description 1
- NAZPXSCMRDAWHG-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccccc1OC(F)(F)F Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccccc1OC(F)(F)F NAZPXSCMRDAWHG-UHFFFAOYSA-N 0.000 description 1
- HTXRYPCPUZABAM-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccncc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NCc1ccncc1 HTXRYPCPUZABAM-UHFFFAOYSA-N 0.000 description 1
- YJKVYCFEBSYJDP-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(cc1)ccc1Oc(cc1)ccc1Cl Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(cc1)ccc1Oc(cc1)ccc1Cl YJKVYCFEBSYJDP-UHFFFAOYSA-N 0.000 description 1
- QMALJXKGNPWOFJ-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(cccc1)c1-c1ccccc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(cccc1)c1-c1ccccc1 QMALJXKGNPWOFJ-UHFFFAOYSA-N 0.000 description 1
- YRZCWLMXVSFGRH-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cc(F)ccc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cc(F)ccc1 YRZCWLMXVSFGRH-UHFFFAOYSA-N 0.000 description 1
- OJOGJQGPUOGDPW-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1ccc(C(F)(F)F)cc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1ccc(C(F)(F)F)cc1 OJOGJQGPUOGDPW-UHFFFAOYSA-N 0.000 description 1
- XIXDOBLZRMHCPG-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cccc(-c2ccccc2)c1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cccc(-c2ccccc2)c1 XIXDOBLZRMHCPG-UHFFFAOYSA-N 0.000 description 1
- DVLOMGLEFKVGQK-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cccc(Oc2ccccc2)c1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cccc(Oc2ccccc2)c1 DVLOMGLEFKVGQK-UHFFFAOYSA-N 0.000 description 1
- GPGYUSURDJESMF-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cnccc1 Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1cnccc1 GPGYUSURDJESMF-UHFFFAOYSA-N 0.000 description 1
- UJZPJUGIVLZKBE-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1ncccc1Cl Chemical compound O=C(c1ccc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc1ncccc1Cl UJZPJUGIVLZKBE-UHFFFAOYSA-N 0.000 description 1
- ZWROIUPPWVBZKU-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCC1CN(Cc(cc2)ccc2F)CC1 Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCC1CN(Cc(cc2)ccc2F)CC1 ZWROIUPPWVBZKU-UHFFFAOYSA-N 0.000 description 1
- LZSQOEWBJOQSLE-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(c(Cl)c1)ccc1Oc(cc1)ccc1F Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(c(Cl)c1)ccc1Oc(cc1)ccc1F LZSQOEWBJOQSLE-UHFFFAOYSA-N 0.000 description 1
- USWPOPCLPNAKFA-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc1ncc(C(F)(F)F)cc1 Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc1ncc(C(F)(F)F)cc1 USWPOPCLPNAKFA-UHFFFAOYSA-N 0.000 description 1
- NCLLPTUGJPAZBA-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(cn1)ccc1Oc(cc1)ccc1F Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(cn1)ccc1Oc(cc1)ccc1F NCLLPTUGJPAZBA-UHFFFAOYSA-N 0.000 description 1
- TXGJOIAOZALQCH-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(nc1)cnc1Oc1ccc(C(F)(F)F)cc1 Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NCc(nc1)cnc1Oc1ccc(C(F)(F)F)cc1 TXGJOIAOZALQCH-UHFFFAOYSA-N 0.000 description 1
- CVVKXCHZSHKBQW-UHFFFAOYSA-N O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NNc(cc1)ccc1OC(F)(F)F Chemical compound O=C(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)NNc(cc1)ccc1OC(F)(F)F CVVKXCHZSHKBQW-UHFFFAOYSA-N 0.000 description 1
- KEQBIPQMFMRHHD-UHFFFAOYSA-N O=C(c1ccc[n]1-c(nccc1C(F)(F)F)c1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound O=C(c1ccc[n]1-c(nccc1C(F)(F)F)c1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl KEQBIPQMFMRHHD-UHFFFAOYSA-N 0.000 description 1
- IWXRXTAAJYYHJL-UHFFFAOYSA-N O=C(c1ccc[n]1-c1c(C(F)(F)F)cccc1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound O=C(c1ccc[n]1-c1c(C(F)(F)F)cccc1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl IWXRXTAAJYYHJL-UHFFFAOYSA-N 0.000 description 1
- IVXAMJUWJQCDMJ-UHFFFAOYSA-N O=C(c1ccc[n]1-c1cc(Oc2ccccc2)ccc1)NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound O=C(c1ccc[n]1-c1cc(Oc2ccccc2)ccc1)NNc(ncc(C(F)(F)F)c1)c1Cl IVXAMJUWJQCDMJ-UHFFFAOYSA-N 0.000 description 1
- BPHXYKPYRVARCY-UHFFFAOYSA-N O=C(c1ccn[n]1-c(ncc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc1cnc(C(F)(F)F)nc1 Chemical compound O=C(c1ccn[n]1-c(ncc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc1cnc(C(F)(F)F)nc1 BPHXYKPYRVARCY-UHFFFAOYSA-N 0.000 description 1
- VCJSRRWLASOCED-UHFFFAOYSA-N O=C(c1n[o]cc1-c(ccc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc(cc1)ccc1F Chemical compound O=C(c1n[o]cc1-c(ccc(Cl)c1)c1Cl)NCc(cc1)ccc1Oc(cc1)ccc1F VCJSRRWLASOCED-UHFFFAOYSA-N 0.000 description 1
- GNLQAILVEHMVHO-UHFFFAOYSA-N O=C(c1nc(cccc2)c2[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl Chemical compound O=C(c1nc(cccc2)c2[n]1-c(ncc(C(F)(F)F)c1)c1Cl)NNc(ncc(C(F)(F)F)c1)c1Cl GNLQAILVEHMVHO-UHFFFAOYSA-N 0.000 description 1
- KADSZAIDZDOMKP-UHFFFAOYSA-N OC(C1CCC(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)CC1)=O Chemical compound OC(C1CCC(CNC(c2ccc[n]2-c(ncc(Cl)c2)c2Cl)=O)CC1)=O KADSZAIDZDOMKP-UHFFFAOYSA-N 0.000 description 1
- MCFUQQHIGJRACQ-UHFFFAOYSA-N OC(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)=O Chemical compound OC(c1ccc[n]1-c(ncc(Cl)c1)c1Cl)=O MCFUQQHIGJRACQ-UHFFFAOYSA-N 0.000 description 1
- MJVRQBHLHDWWFO-UHFFFAOYSA-N OC(c1ccc[n]1Cc(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound OC(c1ccc[n]1Cc(ncc(C(F)(F)F)c1)c1Cl)=O MJVRQBHLHDWWFO-UHFFFAOYSA-N 0.000 description 1
- PCKLMOUYWSSNML-UHFFFAOYSA-N OC(c1ccn[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound OC(c1ccn[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O PCKLMOUYWSSNML-UHFFFAOYSA-N 0.000 description 1
- SAVDATFMRODONN-UHFFFAOYSA-N OC(c1ncc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O Chemical compound OC(c1ncc[n]1-c(ncc(C(F)(F)F)c1)c1Cl)=O SAVDATFMRODONN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- Obesity is a growing threat to the global health by virtue of its association with a cluster of diseases that include insulin resistance, glucose intolerance, dyslipidemia, and hypertension, collectively known as the metabolic syndrome or syndrome X. It is well documented that patients with metabolic syndrome have a higher risk for cardiovascular diseases such as coronary heart disease and stroke [Grundy S. M. et al. Circulation 112:e285-e290, 2005]. The treatment of obesity will require complex solutions, including increased public awareness to diminish food portions, improved food choices and increased physical activity. However, epidemiologic studies have shown that treating diabetes/insulin resistance in these patients can reduce the risk of cardiovascular diseases such as coronary artery disease.
- GPRl 19 modulators described herein might represent such an opportunity.
- Current therapies for diabetes mellitus include: insulin; insulin secretagogues, such as sulphonylureas, which increase insulin secretion from pancreatic ⁇ -cells; glucose-lowering effectors, such as metformin which reduce glucose production from the liver; activators of the peroxisome proliferator-activated receptor- ⁇ (PPAR- ⁇ ), such as the thiazolidinediones, which enhances insulin action; GLP-I mimetics, such as exenatide (Byetta); and ⁇ -glucosidase inhibitors which interfere with gut glucose production.
- insulin secretagogues such as sulphonylureas, which increase insulin secretion from pancreatic ⁇ -cells
- glucose-lowering effectors such as metformin which reduce glucose production from the liver
- activators of the peroxisome proliferator-activated receptor- ⁇ (PPAR- ⁇ ) such as the thiazolidinediones
- GIP and GLP-I are peptides, known as incretins, secreted from enteroendocrine K- and L-cells respectively in response to ingestion of nutrients, and have a wide variety of physiological effects that have been described in numerous publications over the past two decades. See, for example, Bojanowska, E. et al., Med. Sd. Monit., 2005, Aug 5 11(8): RA271-8; Perry, T. et al., Curr. Alzheimer Res., 2005, July 2(3): 377-85; and Meier, J.J. et al., Diabetes Metab. Res.
- GIP and GLP-I are potent stimulators of the body's ability to produce insulin in response to elevated levels of blood sugar.
- GLP- 1 glucose-lowering effects in addition to GLP- 1 's ability to stimulate glucose-dependent insulin secretion including, but not limited to, an inhibition of the release of the hormone glucagon following meals, a reduction in the rate at which nutrients are absorbed into the bloodstream, and a reduction of food intake.
- treatments to increase GLP-I may be used for a variety of conditions and disorders including but not limited to metabolic disorders, gastrointestinal disorders, inflammatory diseases, psychosomatic, depressive, and neuropsychiatric disease including but not limited to diabetes mellitus (Type 1 and Type 2), metabolic syndrome, obesity, appetite control and satiety, weight loss, stress, inflammation, myocardial ischemia/reperfusion injury, Alzheimer's Disease, and other diseases of the central nervous system.
- Type II diabetes patients display a decreased responsiveness to GIP but not GLP-I, with respect to its ability to stimulate insulin secretion.
- the mechanism behind the decreased responsiveness to GIP remains unclear since Type II diabetics retain sensitivity to a bolus administration of GIP but not to a continuous infusion (Meier et al. 2004 Diabetes 53 S220-S224).
- Moreover recent studies with a long-acting fatty-acid derivative of GIP showed beneficial effects on glucose homeostasis in ob/ob mice following 14 days of treatment (Irwin N. et al. (2006) J. Med. Chem. 49, 1047-1 054).
- a molecule which may stimulate GLPl secretion would provide a therapeutic benefit.
- a molecule which could stimulate both GLP-I secretion and insulin secretion through effects on the L-cell and direct effects on the ⁇ -cell would hold much promise for Type II diabetes therapy.
- GLP- 1 receptor agonists have proven elusive, a feature that unfortunately is characteristic of Class B GPCRs. Meanwhile, the spectrum of signaling peptides affected by inhibition of DPP-IV remains unclear and could potentially extend significantly beyond GLP-I and GIP (8, 9). It is therefore worthwhile to search for therapeutic approaches which afford both the physiological selectivity of GLP-I signaling and the opportunity for orally active treatment modalities.
- GPRl 19 was identified as a Class A, islet- enriched receptor which could potentially mediate the insulinotropic actions of lysophosphatidylcholine (LPC) observed in vitro (Soga T., et al, Biochem Biophys Res Commun, 2005, 326:744-751), but a later study suggested that oleoylethanolamide (OEA) was a more potent GPRl 19 agonist (Overton H. A., et al., Cell Metab, 2006, 3:167-175). Recently, a small molecule GPRl 19 agonist has been shown to enhance glucose-dependent insulin secretion and improve hyperglycemia in rodent models of diabetes (Chu Z.
- GPRl 19 is expressed in human gastrointestinal regions and in human islets. Activation of GPRl 19 has been demonstrated to stimulate intracellular cAMP and lead to glucose-dependent GLP-I and insulin secretion. See, T. Soga et al., Biochemical and Biophysical Research Communications 326 (2005) 744-751, herein incorporated by reference with regard to a background understanding of GPRl 19.
- GPRl 19 activation of GPRl 19 is very unlikely to induce hypoglycemia (Chu Z., et al. Endocrinology, 2007, 148:2601-2609) [Oi l] Agonists to GPRl 19 may be of therapeutic value for diabetes and associated conditions, particularly Type II diabetes, obesity, glucose intolerance, insulin resistance, metabolic syndrome X, hyperlipidemia, hypercholesterolemia, and atherosclerosis.
- GPRl 19-mediated diseases in a patient by administering the compounds.
- Xi is selected from the group consisting of N, O, S, CH 2 and CR 3 ;
- X 2 is selected from the group consisting of N, O, S, CH 2 and CR 4 ;
- X3 is selected from the group consisting of N, O, S, CH 2 , CH, and C-methyl;
- Gi is selected from the group consisting of CR 3 Rb, NR 7 , and optionally substituted N-heterocycloalkyl;
- G 2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, NRs, O and SO 2 , any of which may be optionally substituted;
- Ri is selected from the group consisting of aryl, N-containing heteroaryl; arylalkyl, and N-containing heteroarylalkyl, any of which may be optionally substituted;
- R 2 is selected from the group consisting of lower alkyl, lower alkenyl, lower alkynyl, lower hydroxyalkyl, amino, aryl, and heteroaryl, any of which may be optionally substituted; or, when Gi is substituted N-heterocycloalkyl, R 2 may be null;
- R 3 and R 4 are independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkoxy, lower perfluoroalkyl, and lower perfluoroalkoxy, or R 3 and R 4 taken together may form a 5- 6-membered aryl, cycloalkyl, heteroaryl, or heterocycloalkyl, any of which may be optionally substituted;
- Rs, Re, R7, and Rs are each independently selected from the group consisting of halogen, hydrogen, hydroxy, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, phenyloxy, acyl, carboxyl, amino, lower heteroalkyl, lower alkylthio, lower aminoalkyl, lower alkylsulfonyl, sulfonamido, lower aryl, lower arylalkyl, lower cycloalkyl, lower cycloalkylalkyl, lower haloalkyl, lower perhaloalkyl, lower perhaloalkoxy, lower heteroaryl, lower heteroarylalkyl, lower heterocycloalkyl, and lower heterocycloalkylalkyl, any of which may be optionally substituted, or R 5 and R 6 , taken together, form oxy; and
- Ra and Rb are each independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, lower hydroxyalkyl, lower perfluoroalkyl, lower perfluoroalkoxy, optionally substituted lower cycloalkyl, optionally substituted lower heterocycloalkyl, optionally substituted lower heteroaryl, and optionally substituted phenyl.
- Ri is selected from the group consisting of aryl and N-containing heteroaryl, any of which may be optionally substituted.
- Ri is selected from the group consisting of substituted phenyl and substituted, monocyclic N-containing heteroaryl.
- Ri is substituted pyridyl.
- Ri is substituted phenyl.
- Gi is NR 7 .
- Gi is NH.
- R 7 is selected from the group consisting of hydrogen, halogen, hydroxy, Ci-C 3 alkyl, Ci-C 3 hydroxyalkyl, Ci-C 3 alkynyl, perfluoromethyl, and perfluoromethoxy.
- Gi is CR a Rb,
- Gi is optionally substituted N-heterocycle.
- G 2 is a bond.
- Gi is optionally substituted with one to three substituents selected from the group consisting of lower alkyl, lower alkoxy, lower hydroxyalkyl, and hydroxy.
- Gi is optionally substituted with one to three substituents selected from the group consisting of methyl ethyl, methoxy, and hydroxy.
- G 2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, and NRs, any of which may be optionally substituted.
- G 2 is selected from the group consisting of optionally substituted C1-C3 alkyl, C 1 -C 3 heteroalkylcontaining one heteroatom selected from the group consisting of O
- Rg is selected from the group consisting of hydrogen, halogen, hydroxy, C1-C3 alkyl, C1-C3 hydroxyalkyl, C1-C3 alkynyl, perfluoromethyl, and perfluoromethoxy.
- G 2 is selected from the group consisting of NR 8 and CH 2 ;
- R 8 is hydrogen
- R 2 is selected from the group consisting of aryl and heteroaryl, either of which may be optionally substituted. [033] In certain embodiments, R 2 is substituted pyridyl. [034] In certain embodiments, R 2 is substituted phenyl. [035] In certain embodiments, compounds have structural Formula II:
- Gi is selected from the group consisting of CH 2 , and NR 7 ;
- G 2 is selected from the group consisting of a bond, CH 2 , and NRs;
- Xi is selected from the group consisting of CRio and N;
- X 2 is selected from the group consisting of CRi 2 , N, O, and S;
- X 3 is selected from the group consisting of CR 14 , N, O, and S;
- Yi is selected from the group consisting of CR 2 I, N, O, and S;
- Y 2 is selected from the group consisting of CR 22 , N, O, and S;
- Y3 is selected from the group consisting of CR 23 , N, O, and S;
- R 2 is selected from the group consisting of lower alkyl, lower amino, aryl, and heteroaryl, any of which may be optionally substituted; q is an integer from O to 3;
- R 2 is selected from the group consisting of CH, O, and N;
- R5, R 6 , R 7 , and Rs are each independently selected from the group consisting of hydrogen, hydroxy, alkoxy, aryloxy, alkyl, acyl, alkenyl, amino, alkynyl, heteroalkyl, carboxyl, alkylthio, alkylamino, alkylsulfonyl, sulfonamido, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, haloalkyl, perhaloalkyl, perhaloalkoxy, heteroaryl, heteroarylalkyl, heterocycloalkyl, and heterocycloalkylalkyl, any of which may be optionally substituted , or R 3 and R 4 , taken together, may form oxy; and
- Rio, Ri 2 , Ri4, Ri9, R 2 i, R 22 , R 2 3, R 2 7, and R 2 8 are each independently selected from the group consisting of null, hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, ary
- Yi is selected from the group consisting of CR21 and N;
- Y 2 is selected from the group consisting of CR 22 and N;
- Y3 is selected from the group consisting of CR23 and N.
- R 27 and R 2 8 taken together with R 2 to which they are attached, form cycloalkyl, heterocycloalkyl, aryl or heteroaryl, any of which may be optionally substituted.
- R 27 and R 2 8 taken together with R 2 to which they are attached, form cycloalkyl or heterocycloalkyl, either of which may be optionally substituted.
- said cycloalkyl is a C 3 -C 7 monocyclic cycloalkyl, which may be optionally substituted; and said heterocycloalkyl, is a C 4 -C 7 monocyclic heterocycloalkyl, which may be optionally substituted.
- said C 4 -C 7 monocyclic heterocycloalkyl is selected from the group consisting of pyrrolidine, piperidine, piperazine, and morpholine, any of which may be optionally substituted.
- compounds have structural Formula III:
- Gi is selected from the group consisting of CH 2 , and NR 7 ;
- G 2 is selected from the group consisting of a bond, CH 2 , and NR 8 ;
- Xi is selected from the group consisting of CH and N;
- X 2 is selected from the group consisting of CH and N;
- X3 is selected from the group consisting of CH and N;
- Yi is selected from the group consisting of CR21 and N;
- Y 2 is selected from the group consisting of CR 22 and N;
- Y 3 is selected from the group consisting of CR 23 and N;
- Zi is selected from the group consisting of CR 24 and N;
- Z 2 is selected from the group consisting of CR25 and N;
- Z 3 is selected from the group consisting of CR 26 and N;
- Z 4 is selected from the group consisting of CR 27 and N; q and r are each independently an integer from 0 to 3;
- R 7 , and Rs are each independently selected from the group consisting of hydrogen, hydroxy, lower alkoxy, lower alkyl, lower alkenyl, lower amino, lower alkynyl, lower heteroalkyl;
- Rig, Ri9, R21, R 22 , R 2 3, R 24 , R 2 5, R 2 6, and R 27 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, hetero
- Gi is selected from the group consisting of CH 2 , and NR 7 ;
- G 2 is selected from the group consisting of a bond, CH 2 , and NRs.
- Xi and X 3 are each CH;
- X 2 is N.
- Xi and X 2 are each CH;
- X 3 is N.
- Xi is N
- X 2 and X 3 are each CH.
- X 1 , X 2 and X 3 are each CH. [048] In certain embodiments,
- Y 2 is CR 22 ;
- Y 3 is N.
- Y 2 is CR 22 ;
- Y 3 is CR 23 .
- Z 3 is CR 26 ;
- R 2 6 is not hydrogen.
- Zi is N.
- G 2 is selected from the group consisting of NH and CH 2 .
- Gi is NH.
- compounds have structural Formula IV:
- Gi is selected from the group consisting of NH and CH 2 ;
- G 2 is selected from the group consisting of NH, CH 2 , SO 2 , and O;
- Zi is selected from the group consisting of CR 24 and N;
- Z 4 is selected from the group consisting of CR 27 and N; q is an integer from 1 to 3; r is an integer from 1 to 3, or r may be 0 if R 27 is not hydrogen; and each Ri 8 , each Ri 9 , R 24 , R 26 , and R 27 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalky
- G 2 is selected from the group consisting of NH and CH 2 .
- Gi is NH.
- Z 4 is CR 27 .
- Zi and Z 4 are both N.
- R 27 is not hydrogen.
- R 27 and each Ri 8 are independently selected from the group consisting of hydrogen, halogen, haloalkyl, haloalkoxy, optionally substituted arylamino, optionally substituted heteroarylamino, optionally substituted heteroaryloxy, and optionally substituted aryloxy, provided that one OfR 27 and each Ri 8 is not hydrogen.
- R 27 is selected from the group consisting of optionally substituted phenyloxy and optionally substituted 5-6 membered monocyclic heteroaryloxy.
- R 27 is selected from the group consisting of optionally substituted phenyloxy and optionally substituted pyridinyloxy.
- each Ri 9 is independently selected from the group consisting ofhalogen, haloalkyl, and haloalkoxy.
- each R19 is independently selected from the group consisting ofhalogen, perfluoromethyl, and perfluoromethoxy.
- q is 1 or 2.
- q is 2, and the two R19 groups are meta to each other.
- composition comprising a compound as disclosed herein together with a pharmaceutically acceptable carrier.
- Also provided is a method of treatment of a GPRl 19-mediated disease comprising the administration of a therapeutically effective amount of a compound as disclosed herein to a patient in need thereof.
- said disease is a metabolic disease.
- said disease is diabetes.
- Also provided is a method of treatment of a GPRl 19-mediated disease comprising the administration of: i. a therapeutically effective amount of a compound as disclosed herein; and ii. another therapeutic agent.
- said agent is selected from the group consisting of insulin, metformin, Glipizide, glyburide, Amaryl, gliclazide, meglitinides, nateglinide, repaglinide, pramlintide, PTP-112, SB-517955, SB-4195052, SB-216763, NN-57- 05441, NN-57-05445, GW-0791, AGN- 19 4 20 4, T-1095, BAY R3401, acarbose, miglitol, voglibose, Exendin-4, DPP728, LAF237, vildagliptin , BMS477118, PT-100, GSK-823093, PSN-9301, T-6666, SYR-322, SYR-619, Liraglutide, CJC-1134-PC, naliglutide, MK-0431, saxagliptin, GSK23A, pi
- Also provided is a method for achieving an effect in a patient comprising the administration of a therapeutically effective amount of a compound as disclosed herein to a patient, wherein the effect is the modulation of a metabolic disease.
- acyl refers to a carbonyl attached to an alkenyl, alkyl, aryl, cycloalkyl, heteroaryl, heterocycle, or any other moiety were the atom attached to the carbonyl is carbon.
- An “acetyl” group refers to a -C(O)C ⁇ 3 group.
- An “alkylcarbonyl” or “alkanoyl” group refers to an alkyl group attached to the parent molecular moiety through a carbonyl group. Examples of such groups include methylcarbonyl and ethylcarbonyl. Examples of acyl groups include formyl, alkanoyl and aroyl.
- alkenyl refers to a straight-chain or branched-chain hydrocarbon radical having one or more double bonds and containing from 2 to 20 carbon atoms. In certain embodiments, said alkenyl will comprise from 2 to 6 carbon atoms.
- alkoxy refers to an alkyl ether radical, wherein the term alkyl is as defined below.
- suitable alkyl ether radicals include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso- butoxy, sec-butoxy, tert-butoxy, and the like.
- alkyl refers to a straight-chain or branched-chain alkyl radical containing from 1 to 20 carbon atoms. In certain embodiments, said alkyl will comprise from 1 to 10 carbon atoms. In further embodiments, said alkyl will comprise from 1 to 6 carbon atoms. Alkyl groups may be optionally substituted as defined herein.
- alkyl radicals include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl, noyl and the like.
- alkylene refers to a saturated aliphatic group derived from a straight or branched chain saturated hydrocarbon attached at two or more positions, such as methylene (- CH 2 -). Unless otherwise specified, the term “alkyl” may include “alkylene” groups.
- alkylamino refers to an alkyl group attached to the parent molecular moiety through an amino group. Suitable alkylamino groups may be mono- or dialkylated, forming groups such as, for example, N-methylamino, N-ethylamino, N,N-dimethylamino, N,N-ethylmethylamino and the like.
- alkylidene refers to an alkenyl group in which one carbon atom of the carbon-carbon double bond belongs to the moiety to which the alkenyl group is attached.
- alkylthio refers to an alkyl thioether (R-S-) radical wherein the term alkyl is as defined above and wherein the sulfur may be singly or doubly oxidized.
- suitable alkyl thioether radicals include methylthio, ethylthio, n-propylthio, isopropylthio, n-butylthio, iso- butylthio, sec-butylthio, tert-butylthio, methanesulfonyl, ethanesulf ⁇ nyl, and the like.
- alkynyl refers to a straight-chain or branched-chain hydrocarbon radical having one or more triple bonds and containing from 2 to 20 carbon atoms. In certain embodiments, said alkynyl comprises from 2 to 6 carbon atoms. In further embodiments, said alkynyl comprises from 2 to 4 carbon atoms.
- alkynylene refers to a carbon-carbon triple bond attached at two positions such as ethynylene (-C:::C-, -C ⁇ C-).
- alkynyl radicals include ethynyl, propynyl, hydroxypropynyl, butyn-1-yl, butyn-2-yl, pentyn-1- yl, 3-methylbutyn-l-yl, hexyn-2-yl, and the like.
- alkynyl may include "alkynylene” groups.
- acylamino as used herein, alone or in combination, embraces an acyl group attached to the parent moiety through an amino group.
- acylamino is acetylamino (CH 3 C(O)NH-).
- amino refers to — NRR , wherein R and R are independently selected from the group consisting of hydrogen, alkyl, acyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl, any of which may themselves be optionally substituted. Additionally, R and R' may combine to form heterocycloalkyl, either of which may be optionally substituted.
- aminoalkyl refers to an amino group attached to the parent molecular moiety through an alkyl group.
- aryl as used herein, alone or in combination, means a carbocyclic aromatic system containing one, two or three rings wherein such polycyclic ring systems are fused together.
- aryl embraces aromatic groups such as phenyl, naphthyl, anthracenyl, and phenanthryl.
- arylalkenyl or “aralkenyl refers to an aryl group attached to the parent molecular moiety through an alkenyl group.
- arylalkoxy or “aralkoxy,” as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkoxy group.
- arylalkyl or “aralkyl,” as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkyl group.
- arylalkynyl or “aralkynyl,” as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkynyl group.
- arylalkanoyl or “aralkanoyl” or “aroyl,”as used herein, alone or in combination, refers to an acyl radical derived from an aryl-substituted alkanecarboxylic acid such as benzoyl, napthoyl, phenylacetyl, 3-phenylpropionyl
- hydrocinnamoyl 4-phenylbutyryl, (2-naphthyl)acetyl, 4-chlorohydrocinnamoyl, and the like.
- aryloxy refers to an aryl group attached to the parent molecular moiety through an oxy.
- carbamate refers to an ester of carbamic acid (-NHCOO-) which may be attached to the parent molecular moiety from either the nitrogen or acid end, and which may be optionally substituted as defined herein.
- N-carbamyl as used herein, alone or in combination, refers to a
- carbonyl when alone includes formyl [-C(O)H] and in combination is a -C(O)- group.
- carboxyl or “carboxy,” as used herein, refers to -C(O)OH or the corresponding "carboxylate” anion, such as is in a carboxylic acid salt.
- An "O-carboxy” group refers to a RC(O)O- group, where R is as defined herein.
- a “C-carboxy” group refers to a -C(O)OR groups where R is as defined herein.
- cyano as used herein, alone or in combination, refers to -CN.
- cycloalkyl or, alternatively, “carbocycle,” as used herein, alone or in combination, refers to a saturated or partially saturated monocyclic, bicyclic or tricyclic alkyl group wherein each cyclic moiety contains from 3 to 12 carbon atom ring members and which may optionally be a benzo fused ring system which is optionally substituted as defined herein.
- said cycloalkyl will comprise from 5 to 7 carbon atoms.
- cycloalkyl groups examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronapthyl, indanyl, octahydronaphthyl, 2,3-dihydro-lH-indenyl, adamantyl and the like.
- "Bicyclic” and "tricyclic” as used herein are intended to include both fused ring systems, such as decahydronaphthalene, octahydronaphthalene as well as the multicyclic (multicentered) saturated or partially unsaturated type. The latter type of isomer is exemplified in general by, bicyclo[l,l,l]pentane, camphor, adamantane, and bicyclo[3,2,l]octane.
- esters refers to a carboxy group bridging two moieties linked at carbon atoms.
- ether refers to an oxy group bridging two moieties linked at carbon atoms.
- halo or halogen
- haloalkoxy refers to a haloalkyl group attached to the parent molecular moiety through an oxygen atom.
- haloalkyl refers to an alkyl radical having the meaning as defined above wherein one or more hydrogens are replaced with a halogen. Specifically embraced are monohaloalkyl, dihaloalkyl and polyhaloalkyl radicals.
- a monohaloalkyl radical for one example, may have an iodo, bromo, chloro or fluoro atom within the radical.
- Dihalo and polyhaloalkyl radicals may have two or more of the same halo atoms or a combination of different halo radicals.
- haloalkyl radicals include fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl, dichloroethyl and dichloropropyl.
- "Haloalkylene" refers to a haloalkyl group attached at two or more positions.
- heteroalkyl refers to a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, fully saturated or containing from 1 to 3 degrees of unsaturation, consisting of the stated number of carbon atoms and from one to three heteroatoms selected from the group consisting of O, N, and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) O, N and S may be placed at any interior position of the heteroalkyl group. Up to two heteroatoms may be consecutive, such as, for example, -CH 2 -NH- OCH 3 .
- heteroaryl refers to a 3 to 7 membered unsaturated heteromonocyclic ring, or a fused monocyclic, bicyclic, or tricyclic ring system in which at least one of the fused rings is aromatic, which contains at least one atom selected from the group consisting of O, S, and N.
- said heteroaryl will comprise from 5 to 7 carbon atoms.
- heterocyclic rings are fused with aryl rings, wherein heteroaryl rings are fused with other heteroaryl rings, wherein heteroaryl rings are fused with heterocycloalkyl rings, or wherein heteroaryl rings are fused with cycloalkyl rings.
- heteroaryl groups include pyrrolyl, pyrrolinyl, imidazolyl, pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, pyranyl, furyl, thienyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, isothiazolyl, indolyl, isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, quinoxalinyl, quinazolinyl, indazolyl, benzotriazolyl, benzodioxolyl, benzopyranyl, benzoxazolyl, benzoxadiazolyl, benzothiazolyl, benzothiadiazolyl, benzofuryl, benzothienyl, chromonyl,
- Exemplary tricyclic heterocyclic groups include carbazolyl, benzidolyl, phenanthrolinyl, dibenzofuranyl, acridinyl, phenanthridinyl, xanthenyl and the like.
- heterocycloalkyl and, interchangeably, “heterocycle,” as used herein, alone or in combination, each refer to a saturated, partially unsaturated, or fully unsaturated monocyclic, bicyclic, or tricyclic heterocyclic group containing at least one heteroatom as a ring member, wherein each said heteroatom may be independently selected from the group consisting of nitrogen, oxygen, and sulfur
- said hetercycloalkyl will comprise from 1 to 4 heteroatoms as ring members.
- said hetercycloalkyl will comprise from 1 to 2 heteroatoms as ring members.
- said hetercycloalkyl will comprise from 3 to 8 ring members in each ring.
- said hetercycloalkyl will comprise from 3 to 7 ring members in each ring. In yet further embodiments, said hetercycloalkyl will comprise from 5 to 6 ring members in each ring.
- "Heterocycloalkyl” and “heterocycle” are intended to include sulfones, sulfoxides, N-oxides of tertiary nitrogen ring members, and carbocyclic fused and benzo fused ring systems; additionally, both terms also include systems where a heterocycle ring is fused to an aryl group, as defined herein, or an additional heterocycle group.
- heterocycle groups include aziridinyl, azetidinyl, 1,3- benzodioxolyl, dihydroisoindolyl, dihydroisoquinolinyl, dihydrocinnolinyl, dihydrobenzodioxinyl, dihydro[ 1 ,3]oxazolo[4,5-b]pyridinyl, benzothiazolyl, dihydroindolyl, dihy-dropyridinyl, 1,3-dioxanyl, 1 ,4-dioxanyl, 1,3-dioxolanyl, isoindolinyl, morpholinyl, piperazinyl, pyrrolidinyl, tetrahydropyridinyl, piperidinyl, thiomorpholinyl, and the like.
- the heterocycle groups may be optionally substituted unless specifically prohibited.
- hydrazinyl as used herein, alone or in combination, refers to two amino groups joined by a single bond, i.e., -N-N-.
- hydroxy refers to -OH.
- hydroxyalkyl refers to a hydroxy group attached to the parent molecular moiety through an alkyl group.
- isocyanato refers to a -NCO group.
- isothiocyanato refers to a -NCS group.
- linear chain of atoms refers to the longest straight chain of atoms independently selected from carbon, nitrogen, oxygen and sulfur.
- lower means containing from 1 to and including 6 carbon atoms.
- lower aryl as used herein, alone or in combination, means phenyl or naphthyl, which may be optionally substituted as provided.
- lower heteroaryl as used herein, alone or in combination, means either 1) monocyclic heteroaryl comprising five or six ring members, of which between one and four said members may be heteroatoms selected from the group consisting of
- each of the fused rings comprises five or six ring members, comprising between them one to four heteroatoms selected from the group consisting of O, S, and N.
- lower cycloalkyl as used herein, alone or in combination, means a monocyclic cycloalkyl having between three and six ring members. Lower cycloalkyls may be unsaturated. Examples of lower cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- lower heterocycloalkyl as used herein, alone or in combination, means a monocyclic heterocycloalkyl having between three and six ring members, of which between one and four may be heteroatoms selected from the group consisting of
- lower heterocycloalkyls examples include pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, and morpholinyl. Lower heterocycloalkyls may be unsaturated. [0132]
- R and R are independently selected from the group consisting of hydrogen, lower alkyl, and lower heteroalkyl, any of which may be optionally substituted. Additionally, the R and R' of a lower amino group may combine to form a five- or six-membered heterocycloalkyl, either of which may be optionally substituted.
- nitro refers to -NO 2 .
- perhaloalkoxy refers to an alkoxy group where all of the hydrogen atoms are replaced by halogen atoms.
- perhaloalkyl refers to an alkyl group where all of the hydrogen atoms are replaced by halogen atoms.
- sulfonate refers the -SO3H group and its anion as the sulfonic acid is used in salt formation.
- thia and thio refer to a -S- group or an ether wherein the oxygen is replaced with sulfur.
- the oxidized derivatives of the thio group namely sulfmyl and sulfonyl, are included in the definition of thia and thio.
- thiol refers to an -SH group.
- N-thiocarbamyl refers to an ROC(S)NR'- group, with R and
- O-thiocarbamyl refers to a -OC(S)NRR' , group with R and
- thiocyanato refers to a -CNS group.
- trihalomethanesulfonamido refers to a X 3 CS(O) 2 NR- group with X is a halogen and R as defined herein.
- trihalomethanesulfonyl refers to a X 3 CS(O) 2 - group where X is a halogen.
- trihalomethoxy refers to a X 3 CO- group where X is a halogen.
- trimethysilyl as used herein, alone or in combination, refers to a silicone group substituted at its three free valences with groups as listed herein under the definition of substituted amino. Examples include trimethysilyl, tert- butyldimethylsilyl, triphenylsilyl and the like.
- any definition herein may be used in combination with any other definition to describe a composite structural group.
- the trailing element of any such definition is that which attaches to the parent moiety.
- the composite group alkylamido would represent an alkyl group attached to the parent molecule through an amido group
- the term alkoxyalkyl would represent an alkoxy group attached to the parent molecule through an alkyl group.
- the term "optionally substituted” means the anteceding group may be substituted or unsubstituted.
- the substituents of an "optionally substituted” group may include, without limitation, one or more substituents independently selected from the following groups or a particular designated set of groups, alone or in combination: lower alkyl, lower alkenyl, lower alkynyl, lower alkanoyl, lower heteroalkyl, lower heterocycloalkyl, lower haloalkyl, lower haloalkenyl, lower haloalkynyl, lower perhaloalkyl, lower perhaloalkoxy, lower cycloalkyl, phenyl, aryl, aryloxy, lower alkoxy, lower haloalkoxy, oxo, lower acyloxy, carbonyl, carboxyl, lower alkylcarbonyl, lower carboxyester, lower carboxamido, cyano, hydrogen, halogen, hydroxy, amino, lower alkylcarbonyl
- Two substituents may be joined together to form a fused five-, six-, or seven-membered carbocyclic or heterocyclic ring consisting of zero to three heteroatoms, for example forming methylenedioxy or ethylenedioxy.
- An optionally substituted group may be unsubstituted (e.g., -CH 2 CH 3 ), fully substituted (e.g., -CF 2 CF 3 ), monosubstituted (e.g., -CH 2 CH 2 F) or substituted at a level anywhere in-between fully substituted and monosubstituted (e.g., -CH 2 CF 3 ).
- aryl, heterocycle, R, etc. occur more than one time in a formula or generic structure, its definition at each occurrence is independent of the definition at every other occurrence.
- certain groups may be attached to a parent molecule or may occupy a position in a chain of elements from either end as written.
- an unsymmetrical group such as -C(O)N(R)- may be attached to the parent moiety at either the carbon or the nitrogen.
- Asymmetric centers exist in the compounds disclosed herein. These centers are designated by the symbols “R” or “S,” depending on the configuration of substituents around the chiral carbon atom. It should be understood that the invention encompasses all stereochemical isomeric forms, including diastereomeric, enantiomeric, and epimeric forms, as well as d-isomers and 1-isomers, and mixtures thereof.
- Individual stereoisomers of compounds can be prepared synthetically from commercially available starting materials which contain chiral centers or by preparation of mixtures of enantiomeric products followed by separation such as conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques, direct separation of enantiomers on chiral chromatographic columns, or any other appropriate method known in the art.
- Starting compounds of particular stereochemistry are either commercially available or can be made and resolved by techniques known in the art.
- the compounds disclosed herein may exist as geometric isomers.
- the present invention includes all cis, trans, syn, anti,
- compounds may exist as tautomers; all tautomeric isomers are provided by this invention. Additionally, the compounds disclosed herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. In general, the solvated forms are considered equivalent to the unsolvated forms.
- bond refers to a covalent linkage between two atoms, or two moieties when the atoms joined by the bond are considered to be part of larger substructure.
- a bond may be single, double, or triple unless otherwise specified.
- a dashed line between two atoms in a drawing of a molecule indicates that an additional bond may be present or absent at that position.
- disease as used herein is intended to be generally synonymous, and is used interchangeably with, the terms “disorder” and “condition” (as in medical condition), in that all reflect an abnormal condition of the human or animal body or of one of its parts that impairs normal functioning, is typically manifested by distinguishing signs and symptoms, and causes the human or animal to have a reduced duration or quality of life.
- combination therapy means the administration of two or more therapeutic agents to treat a therapeutic condition or disorder described in the present disclosure. Such administration encompasses co-administration of these therapeutic agents in a substantially simultaneous manner, such as in a single capsule having a fixed ratio of active ingredients or in multiple, separate capsules for each active ingredient. In addition, such administration also encompasses use of each type of therapeutic agent in a sequential manner. In either case, the treatment regimen will provide beneficial effects of the drug combination in treating the conditions or disorders described herein.
- GPRl 19 modulator is used herein to refer to a compound that exhibits an EC50 with respect to GPRl 19 activity of no more than about 100 ⁇ M and more typically not more than about 50 ⁇ M, as measured in the cAMP production assay and glucagon- like peptide- 1 (GLP-I) secretion assays described generally hereinbelow.
- EC50 is that concentration of inhibitor which activates the activity of an enzyme (e.g., GPRl 19) to half-maximal level. Certain compounds disclosed herein have been discovered to exhibit modulatory activity against GPRl 19.
- compounds will exhibit an EC50 with respect to GPRl 19 of no more than about 10 ⁇ M; in further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of no more than about 5 ⁇ M; in yet further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of not more than about 1 ⁇ M; in yet further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of not more than about 200 nM, as measured in the GPRl 19 assay described herein.
- the phrase "therapeutically effective” is intended to qualify the amount of active ingredients used in the treatment of a disease or disorder. This amount will achieve the goal of reducing or eliminating the said disease or disorder.
- the term "therapeutically acceptable” refers to those compounds (or salts, prodrugs, tautomers, zwitterionic forms, etc.) which are suitable for use in contact with the tissues of patients without undue toxicity, irritation, and allergic response, are commensurate with a reasonable benefit/risk ratio, and are effective for their intended use.
- patient means all mammals including humans. Examples of patients include humans, cows, dogs, cats, goats, sheep, pigs, and rabbits. Preferably, the patient is a human.
- prodrug refers to a compound that is made more active in vivo.
- Certain compounds disclosed herein may also exist as prodrugs, as described in Hydrolysis in Drug and Prodrug Metabolism : Chemistry, Biochemistry, and Enzymology (Testa, Bernard and Mayer, Joachim M. Wiley- VHC A, Zurich, Switzerland 2003).
- Prodrugs of the compounds described herein are structurally modified forms of the compound that readily undergo chemical changes under physiological conditions to provide the compound.
- prodrugs can be converted to the compound by chemical or biochemical methods in an ex vivo environment. For example, prodrugs can be slowly converted to a compound when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent.
- Prodrugs are often useful because, in some situations, they may be easier to administer than the compound, or parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug.
- a wide variety of prodrug derivatives are known in the art, such as those that rely on hydrolytic cleavage or oxidative activation of the prodrug.
- An example, without limitation, of a prodrug would be a compound which is administered as an ester (the "prodrug"), but then is metabolically hydrolyzed to the carboxylic acid, the active entity. Additional examples include peptidyl derivatives of a compound.
- the compounds disclosed herein can exist as therapeutically acceptable salts.
- the present invention includes compounds listed above in the form of salts, including acid addition salts. Suitable salts include those formed with both organic and inorganic acids. Such acid addition salts will normally be pharmaceutically acceptable. However, salts of non-pharmaceutically acceptable salts may be of utility in the preparation and purification of the compound in question. Basic addition salts may also be formed and be pharmaceutically acceptable.
- Pharmaceutical Salts Properties, Selection, and Use (Stahl, P. Heinrich. Wiley- VCHA, Zurich, Switzerland, 2002).
- terapéuticaally acceptable salt represents salts or zwitterionic forms of the compounds disclosed herein which are water or oil-soluble or dispersible and therapeutically acceptable as defined herein.
- the salts can be prepared during the final isolation and purification of the compounds or separately by reacting the appropriate compound in the form of the free base with a suitable acid.
- Representative acid addition salts include acetate, adipate, alginate, L-ascorbate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, butyrate, camphorate, camphorsulfonate, citrate, digluconate, formate, fumarate, gentisate, glutarate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hippurate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethansulfonate (isethionate), lactate, maleate, malonate, DL-mandelate, mesitylenesulfonate, methanesulfonate, naphthylenesulfonate, nicotinate, 2-naphthalenesulfonate, oxalate, pamoate, pectinate, persulfate, 3-phenyl
- basic groups in the compounds disclosed herein can be quaternized with methyl, ethyl, propyl, and butyl chlorides, bromides, and iodides; dimethyl, diethyl, dibutyl, and diamyl sulfates; decyl, lauryl, myristyl, and steryl chlorides, bromides, and iodides; and benzyl and phenethyl bromides.
- acids which can be employed to form therapeutically acceptable addition salts include inorganic acids such as hydrochloric, hydrobromic, sulfuric, and phosphoric, and organic acids such as oxalic, maleic, succinic, and citric. Salts can also be formed by coordination of the compounds with an alkali metal or alkaline earth ion.
- the present invention contemplates sodium, potassium, magnesium, and calcium salts of the compounds disclosed herein, and the like.
- Basic addition salts can be prepared during the final isolation and purification of the compounds by reacting a carboxy group with a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation or with ammonia or an organic primary, secondary, or tertiary amine.
- a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation or with ammonia or an organic primary, secondary, or tertiary amine.
- the cations of therapeutically acceptable salts include lithium, sodium, potassium, calcium, magnesium, and aluminum, as well as nontoxic quaternary amine cations such as ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, diethylamine, ethylamine, tributylamine, pyridine, N,N-dimethylaniline, N- methylpiperidine, N-methylmorpholine, dicyclohexylamine, procaine, dibenzylamine, NN-dibenzylphenethylamine, 1-ephenamine, and NN-dibenzylethylenediamine.
- nontoxic quaternary amine cations such as ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, diethylamine, eth
- a salt of a compound can be made by reacting the appropriate compound in the form of the free base with the appropriate acid.
- compositions which comprise one or more of certain compounds disclosed herein, or one or more pharmaceutically acceptable salts, esters, prodrugs, amides, or solvates thereof, together with one or more pharmaceutically acceptable carriers thereof and optionally one or more other therapeutic ingredients.
- the carrier(s) must be "acceptable” in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. Proper formulation is dependent upon the route of administration chosen.
- compositions disclosed herein may be manufactured in any manner known in the art, e.g. , by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes.
- the formulations include those suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous, intraarticular, and intramedullary), intraperitoneal, transmucosal, transdermal, rectal and topical (including dermal, buccal, sublingual and intraocular) administration although the most suitable route may depend upon for example the condition and disorder of the recipient.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. Typically, these methods include the step of bringing into association a compound of the subject invention or a pharmaceutically acceptable salt, ester, amide, prodrug or solvate thereof ("active ingredient”) with the carrier which constitutes one or more accessory ingredients.
- active ingredient a pharmaceutically acceptable salt, ester, amide, prodrug or solvate thereof
- the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
- Formulations of the compounds disclosed herein suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in- water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- Pharmaceutical preparations which can be used orally include tablets, push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- Tablets may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free- flowing form such as a powder or granules, optionally mixed with binders, inert diluents, or lubricating, surface active or dispersing agents. Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein. All formulations for oral administration should be in dosages suitable for such administration.
- the push- fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- suitable liquids such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added.
- Dragee cores are provided with suitable coatings.
- concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- the compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in powder form or in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or sterile pyrogen- free water, immediately prior to use.
- sterile liquid carrier for example, saline or sterile pyrogen- free water
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Formulations for parenteral administration include aqueous and nonaqueous (oily) sterile injection solutions of the active compounds which may contain antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
- the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- compositions may take the form of tablets, lozenges, pastilles, or gels formulated in conventional manner.
- Such compositions may comprise the active ingredient in a flavored basis such as sucrose and acacia or tragacanth.
- the compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter, polyethylene glycol, or other glycerides.
- Certain compounds disclosed herein may be administered topically, that is by non-systemic administration. This includes the application of a compound disclosed herein externally to the epidermis or the buccal cavity and the instillation of such a compound into the ear, eye and nose, such that the compound does not significantly enter the blood stream.
- systemic administration refers to oral, intravenous, intraperitoneal and intramuscular administration.
- Formulations suitable for topical administration include liquid or semi- liquid preparations suitable for penetration through the skin to the site of inflammation such as gels, liniments, lotions, creams, ointments or pastes, and drops suitable for administration to the eye, ear or nose.
- the active ingredient for topical administration may comprise, for example, from 0.001% to 10% w/w (by weight) of the formulation. In certain embodiments, the active ingredient may comprise as much as 10% w/w. In other embodiments, it may comprise less than 5% w/w. In certain embodiments, the active ingredient may comprise from 2% w/w to 5% w/w. In other embodiments, it may comprise from 0.1% to 1% w/w of the formulation.
- Gels for topical or transdermal administration may comprise, generally, a mixture of volatile solvents, nonvolatile solvents, and water.
- the volatile solvent component of the buffered solvent system may include lower (Cl- C6) alkyl alcohols, lower alkyl glycols and lower glycol polymers.
- the volatile solvent is ethanol.
- the volatile solvent component is thought to act as a penetration enhancer, while also producing a cooling effect on the skin as it evaporates.
- the nonvolatile solvent portion of the buffered solvent system is selected from lower alkylene glycols and lower glycol polymers. In certain embodiments, propylene glycol is used.
- the nonvolatile solvent slows the evaporation of the volatile solvent and reduces the vapor pressure of the buffered solvent system.
- the amount of this nonvolatile solvent component, as with the volatile solvent, is determined by the pharmaceutical compound or drug being used. When too little of the nonvolatile solvent is in the system, the pharmaceutical compound may crystallize due to evaporation of volatile solvent, while an excess may result in a lack of bioavailability due to poor release of drug from solvent mixture.
- the buffer component of the buffered solvent system may be selected from any buffer commonly used in the art; in certain embodiments, water is used. A common ratio of ingredients is about 20% of the nonvolatile solvent, about 40% of the volatile solvent, and about 40% water.
- chelators and gelling agents Appropriate gelling agents can include, but are not limited to, semisynthetic cellulose derivatives (such as hydroxypropylmethylcellulose) and synthetic polymers, and cosmetic agents.
- Lotions include those suitable for application to the skin or eye.
- An eye lotion may comprise a sterile aqueous solution optionally containing a bactericide and may be prepared by methods similar to those for the preparation of drops.
- Lotions or liniments for application to the skin may also include an agent to hasten drying and to cool the skin, such as an alcohol or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil or arachis oil.
- Creams, ointments or pastes are semi-solid formulations of the active ingredient for external application. They may be made by mixing the active ingredient in finely-divided or powdered form, alone or in solution or suspension in an aqueous or non-aqueous fluid, with the aid of suitable machinery, with a greasy or non-greasy base.
- the base may comprise hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin such as almond, corn, arachis, castor or olive oil; wool fat or its derivatives or a fatty acid such as steric or oleic acid together with an alcohol such as propylene glycol or a macrogel.
- the formulation may incorporate any suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as a sorbitan ester or a polyoxyethylene derivative thereof.
- suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as a sorbitan ester or a polyoxyethylene derivative thereof.
- Suspending agents such as natural gums, cellulose derivatives or inorganic materials such as silicaceous silicas, and other ingredients such as lanolin, may also be included.
- Drops may comprise sterile aqueous or oily solutions or suspensions and may be prepared by dissolving the active ingredient in a suitable aqueous solution of a bactericidal and/or fungicidal agent and/or any other suitable preservative, and, in certain embodiments, including a surface active agent.
- the resulting solution may then be clarified by filtration, transferred to a suitable container which is then sealed and sterilized by autoclaving or maintaining at 98-100 0 C for half an hour.
- the solution may be sterilized by filtration and transferred to the container by an aseptic technique.
- bactericidal and fungicidal agents suitable for inclusion in the drops are phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01%) and chlorhexidine acetate (0.01%).
- Suitable solvents for the preparation of an oily solution include glycerol, diluted alcohol and propylene glycol.
- Formulations for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerin or sucrose and acacia.
- compounds may be conveniently delivered from an insufflator, nebulizer pressurized packs or other convenient means of delivering an aerosol spray.
- Pressurized packs may comprise a suitable propellant such as dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the compounds according to the invention may take the form of a dry powder composition, for example a powder mix of the compound and a suitable powder base such as lactose or starch.
- the powder composition may be presented in unit dosage form, in for example, capsules, cartridges, gelatin or blister packs from which the powder may be administered with the aid of an inhalator or insufflator.
- Preferred unit dosage formulations are those containing an effective dose, as herein below recited, or an appropriate fraction thereof, of the active ingredient.
- the formulations described above may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavoring agents.
- Compounds may be administered orally or via injection at a dose of from 0.1 to 500 mg/kg per day. The dose range for adult humans is generally from 5 mg to 2 g/day. Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of one or more compounds which is effective at such dosage or as a multiple of the same, for instance, units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- the compounds can be administered in various modes, e.g. orally, topically, or by injection.
- the precise amount of compound administered to a patient will be the responsibility of the attendant physician.
- the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diets, time of administration, route of administration, rate of excretion, drug combination, the precise disorder being treated, and the severity of the indication or condition being treated. Also, the route of administration may vary depending on the condition and its severity.
- one of the side effects experienced by a patient upon receiving one of the compounds herein is hypertension, then it may be appropriate to administer an anti-hypertensive agent in combination with the initial therapeutic agent.
- the therapeutic effectiveness of one of the compounds described herein may be enhanced by administration of an adjuvant (i.e., by itself the adjuvant may only have minimal therapeutic benefit, but in combination with another therapeutic agent, the overall therapeutic benefit to the patient is enhanced).
- the benefit of experienced by a patient may be increased by administering one of the compounds described herein with another therapeutic agent (which also includes a therapeutic regimen) that also has therapeutic benefit.
- increased therapeutic benefit may result by also providing the patient with another therapeutic agent for diabetes.
- the overall benefit experienced by the patient may simply be additive of the two therapeutic agents or the patient may experience a synergistic benefit.
- combination therapies include use of certain compounds disclosed herein with agents found in the following pharmacotherapeutic classifications as indicated below. These lists should not be construed to be closed, but should instead serve as illustrative examples common to the relevant therapeutic area at present.
- combination regimens may include a variety of routes of administration and should include oral, intravenous, intraocular, subcutaneous, dermal, and inhaled topical.
- compounds disclosed herein may be administered with an agent selected from the group comprising: insulin, insulin derivatives and mimetics, insulin secretagogues, insulin sensitizers, biguanide agents, alpha-glucosidase inhibitors, insulinotropic sulfonylurea receptor ligands, meglitinides, protein tyrosine phosphatase-lB (PTP-IB) inhibitors, GSK3 (glycogen synthase kinase- 3) inhibitors , GLP-I (glucagon like peptide- 1), GLP-I analogs, DPP-IV (dipeptidyl peptidase IV) inhibitors, RXR ligands, sodium-dependent glucose co-transporter (SGLT2) inhibitors, glycogen phosphorylase A inhibitors, an AGE breaker, PPAR modulators, non-glitazone type PPAR ⁇ agonist, HMG-CoA reductase inhibitors, cholesterol
- compounds disclosed herein may be administered with an agent selected from the group comprising: insulin, metformin, Glipizide, glyburide, Amaryl, gliclazide, meglitinides, nateglinide, repaglinide, amylin mimetics (for example, pramlintide), PTP-112, SB-517955, SB-4195052, SB-216763, NN-57-05441, NN-57-05445, GW-0791, AGN- 19 4 20 4, T-1095, BAY R3401, acarbose, miglitol, voglibose, Exendin-4, DPP728, LAF237, vildagliptin , BMS477118, PT-100, GSK-823093, PSN-9301, T-6666, SYR-322, SYR-619, Liraglutide, CJC-1134-PC, naliglutide, MK
- compounds disclosed herein may be administered with an agent selected from the group comprising: cholescystokinin-A (CCK-A) agonists, serotonin and norepinephrine reuptake inhibitors (for example sibutramine), dopamine agonists (for example, bromocriptine and like) sympathomimetic agents, ⁇ 3 adrenergic receptor agonists, leptin, leptin analogues, leptin receptor agonists, galanin antagonists, lipase inhibitors (for example Orlistat), Neuropeptide-Y antagonists, glucocorticoid receptor agonists or antagonists, cannabinoid 1 receptor antagonists (for example, rimonabant and like), ciliary neurotropic factors (CNTF, for example Axokine), human agouti-related proteins (AGRP), ghrelin receptor antagonists, histamine 3 receptor antagonists, appetite suppressants (for example, bupro
- CCK-A cholesc
- compounds disclosed herein may be administered with an agent selected from the group comprising: corticosteroids, non-steroidal anti-inflammatories, muscle relaxants and combinations thereof with other agents, anaesthetics and combinations thereof with other agents, expectorants and combinations thereof with other agents, antidepressants, anticonvulsants and combinations thereof; antihypertensives, opioids, topical cannabinoids, and other agents, such as capsaicin.
- an agent selected from the group comprising: corticosteroids, non-steroidal anti-inflammatories, muscle relaxants and combinations thereof with other agents, anaesthetics and combinations thereof with other agents, expectorants and combinations thereof with other agents, antidepressants, anticonvulsants and combinations thereof; antihypertensives, opioids, topical cannabinoids, and other agents, such as capsaicin.
- compounds disclosed herein may be administered with an agent selected from the group comprising: betamethasone dipropionate (augmented and nonaugemnted), betamethasone valerate, clobetasol propionate, prednisone, methyl prednisolone, diflorasone diacetate, halobetasol propionate, amcinonide, dexamethasone, dexosimethasone, fluocinolone acetononide, fluocinonide, halocinonide, clocortalone pivalate, dexosimetasone, flurandrenalide, salicylates, ibuprofen, ketoprofen, etodolac, diclofenac, meclofenamate sodium, naproxen, piroxicam, celecoxib, cyclobenzaprine, baclofen, cyclobenzaprine/lidocaine, baclof
- the multiple therapeutic agents may be administered in any order or even simultaneously. If simultaneously, the multiple therapeutic agents may be provided in a single, unified form, or in multiple forms (by way of example only, either as a single pill or as two separate pills). One of the therapeutic agents may be given in multiple doses, or both may be given as multiple doses. If not simultaneous, the timing between the multiple doses may be any duration of time ranging from a few minutes to four weeks.
- certain embodiments provide methods for treating GPRl 19-mediated disorders in a human or animal subject in need of such treatment comprising administering to said subject an amount of a compound disclosed herein effective to reduce or prevent said disorder in the subject, in combination with at least one additional agent for the treatment of said disorder that is known in the art.
- certain embodiments provide therapeutic compositions comprising at least one compound disclosed herein in combination with one or more additional agents for the treatment of GPRl 19-mediated disorders.
- diabetes type I and type II
- conditions associated with diabetic diseases which include, but are not limited to, hyperglycemia, hyperlipidemia, hyperinsulinemia, insulin resistance, inadequate glucose tolerance, impaired glucose metabolism, diabetic nephropathy, glomerulosclerosis, diabetic neuropathy, erectile dysfunction, macular degeneration, diabetic retinopathy, chronic microvascular complications, peripheral vascular disease, cataracts, stroke, foot ulcerations, renal failure, kidney disease, ketosis, metabolic acidosis, and related disorders, obesity, myocardial infarction, angina pectoris, coronary artery disease, atherosclerosis, cardiac hypertrophy, allergic diseases, fatty liver disease, nonalcoholic steatohepatitis, liver fibrosis, kidney fibrosis, anorexia nervosa, bulimia vervosa, autoimmune diseases, inflammatory diseases including rheumatoid arthritis, asthma, chronic o
- the disease is obesity and the effects to be achieved in a human or animal patient include decreasing body weight and controlling weight gain.
- topical application of GPRl 19 agonists might be useful for the treatment of cellulite and other cosmetic conditions which are characterized by subcutaneous fat accumulation.
- the disease is associated with perturbed bile acid metabolism, including, but not limited to gall bladder stones, cholecystitis, cholangitis, choledocholithiasis, jaundice, and obstetric cholestasis and the itch associated with it.
- perturbed bile acid metabolism including, but not limited to gall bladder stones, cholecystitis, cholangitis, choledocholithiasis, jaundice, and obstetric cholestasis and the itch associated with it.
- Metabolic diseases other than Type 1 and Type 2 diabetes which may be treated or prevented include, without limitation, metabolic syndrome and insulin resistance.
- the compounds disclosed herein can be used to treat insulin resistance and other metabolic disorders such as atherosclerosis that are typically associated with an exaggerated inflammatory signaling.
- the disease is a hyperproliferative condition of the human or animal body, including, but not limited to restenosis, inflammation, immune disorders, cardiac hypertrophy, atherosclerosis, pain, migraine, angiogenesis-related conditions or disorders, proliferation induced after medical conditions, including but not limited to surgery, angioplasty, or other conditions.
- compositions may be used to treat arthritis, including but not limited to rheumatoid arthritis, spondyloarthropathies, gouty arthritis, osteoarthritis, systemic lupus erythematosus, juvenile arthritis, acute rheumatic arthritis, enteropathic arthritis, neuropathic arthritis, psoriatic arthritis, and pyogenic arthritis.
- the compositions may also be used in the treatment of pulmonary inflammation, such as that associated with viral infections and cystic fibrosis.
- the particular inflammatory disease is rheumatoid arthritis.
- inflammatory diseases which may be prevented or treated include, without limitation: asthma, allergies, respiratory distress syndrome or acute or chronic pancreatitis.
- respiratory system diseases may be prevented or treated including but not limited to chronic obstructive pulmonary disease, pulmonary fibrosis, ulcerative colitis, inflammatory bowel disease, Crohn's disease, peptic ulceration, gastritis, psoriasis, and skin inflammation.
- the disease to be treated by the methods provided herein may be an ophthalmologic disorder.
- Ophthalmologic diseases and other diseases in which angiogenesis plays a role in pathogenesis may be treated or prevented and include, without limitation, dry eye (including Sjogren's syndrome), macular degeneration, closed and wide angle glaucoma, retinal ganglion degeneration, occular ischemia, retinitis, retinopathies, uveitis, ocular photophobia, and of inflammation and pain associated with acute injury to the eye tissue.
- the ophthalmologic disease to be treated is glaucomatous retinopathy and/or diabetic retinopathy.
- the ophthalmologic condition to be treated is post-operative inflammation or pain as from ophthalmic surgery such as cataract surgery and refractive surgery.
- the disease to be treated by the methods provided herein may be an autoimmune disease.
- Autoimmune diseases which may be prevented or treated include, but are not limited to: rheumatoid arthritis, inflammatory bowel disease, inflammatory pain, ulcerative colitis, Crohn's disease, periodontal disease, temporomandibular joint disease, multiple sclerosis, diabetes, glomerulonephritis, systemic lupus erythematosus, scleroderma, chronic thyroiditis, Grave's disease, hemolytic anemia, autoimmune gastritis, autoimmune neutropenia, thrombocytopenia, chronic active hepatitis, myasthenia gravis, atopic dermatitis, graft vs.
- Inflammatory diseases which may be prevented or treated include, but are not limited to: asthma, allergies, respiratory distress syndrome or acute or chronic pancreatitis.
- the particular autoimmune disease is rheumatoid arthritis.
- the compounds provided herein are also useful in treating tissue damage in such diseases as vascular diseases, migraine headaches, periarteritis nodosa, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, neuromuscular junction disease including myasthenia gravis, white matter disease including multiple sclerosis, sarcoidosis, nephritis, nephrotic syndrome, Behcet's syndrome, polymyositis, gingivitis, periodontis, hypersensitivity, swelling occurring after injury, ischemias including myocardial ischemia, cardiovascular ischemia, and ischemia secondary to cardiac arrest, and the like.
- diseases as vascular diseases, migraine headaches, periarteritis nodosa, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, neuromuscular junction disease including myasthenia gravis, white matter disease including multiple s
- the disease to be treated by the methods of the present invention may be a cardiovascular condition.
- said cardiovascular condition is selected from the group consisting of atherosclerosis, cardiac hypertrophy, idiopathic cardiomyopathies, heart failure, angiogenesis-related conditions or disorders, and proliferation induced after medical conditions, including, but not limited to restenosis resulting from surgery and angioplasty.
- the disease to be prevented or treated by the methods of the present invention may be autism.
- GIP glucose -dependent insulinotropic polypeptide
- GPRl 19 agonists increase GLP-I and GIP secretion both in vitro and in vivo
- GPRl 19 agonists in this patent might be important for the prevention or treatment of bone loss induced by age or diseases in which the normal functions of osteobalsts or osteoclasts are altered.
- Such diseases include, for example, osteopenia and osteoporosis.
- certain compounds and formulations disclosed herein may also be useful for veterinary treatment of companion animals, exotic animals and farm animals, including mammals, rodents, and the like. More preferred animals include horses, dogs, and cats.
- Reagents (a) NaH, DMF, 65 0 C, 18h. (b) AcOH, 12O 0 C, 18h. (c) NaOH, MeOH, 25- 9O 0 C, l-3h. (d) DMF-DMA, RT, 18h. (e) AcOH, reflux, 6h. (f) MeOH, reflux, 18h. (g) l-(isocyanomethylsulfonyl) -4-methylbenzene, K 2 CO 3 , EtOH, 6O 0 C, 3h.
- Reagents (a) 2,2,6,6-tetramethylheptane-3,5-dione, CuCl, Cs 2 CO 3 , NMP, 12O 0 C, 18h. (b) K 2 CO 3 , DMF, 8O 0 C, 18h. (c) BH 3 THF, reflux, 3h. (d) Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, JV-methyl morpholine),ACN, RT, 18h. (e) (i) Oxalyl Chloride, DCM, DMF (ii) Amino derivatives, TEA, O 0 C. (f) Trifluoromethyl 2,3 ,4,5,6-pentafluorobenzoate, pyridine, DMF, RT, 2.5h. (g) Ri 27 -NH-Ri 28 , DMF, RT, 18h.
- Reagents (a) 2,5-Dimethoxytetrahydrofl ⁇ ran, AcOH/l,4-dioxane, 100 0 C, 6h. (b) 2-Bromoacetyl bromide, DCM, RT, 18h. (c) Phenols, K 2 CO 3 , acetone, RT, 18h. (d) Anilines, K 2 CO 3 , acetone, 100 0 C, 18h.
- Reagents (a) NaH, DMA, 7O 0 C, 1.5 h. (b) TFA, DCM, RT, 30 min. (c) Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, N-methyl Morpholine),ACN, RT, 18h.
- Coupling reagent i.e. HATU, EDC/HOBt
- base i. e. TEA, N-methyl Morpholine
- ACN RT, 18h.
- Step 1 Methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxylate
- Methyl lH-pyrrole-2-carboxylate (11.3 g, 90.3 mmol) was added portionwise to a vigorously stirred mixture of sodium hydride (60%, 3.0 g, 75 mmol) and DMF (50 mL) at rt under N 2 . After 30 min, the reaction became homogeneous, and then 2,3-dichloro- 5-(trifluoromethyl)pyridine (7.0 mL, 50 mmol) was added. After an additional 30 min at rt, the reaction was heated at 60 0 C for 17 h, allowed to cool to rt, poured into 0.16N HCl (500 mL), and then extracted with EtOAc (100 mL x 2).
- Step 1 Methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5- carboxylate
- Step 1 (3-Chloro-4-(trifluoromethoxy)phenyl)methanamine
- Step_i tert-Butyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)piperidine-l-carboxylate
- tert-Butyl 4-( 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrole-2- carboxamido)piperidine-l-carboxylate was prepared from tert-butyl A- aminopiperidine-1-carboxylate and Intermediate 1 following the procedure outlined in
- Step 2 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(piperidin-4-yl)-lH- pyrrole-2-carboxamide
- Acetyl chloride (1 mL) was added dropwise to a solution of tert-butyl 4-(l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)- 1 H-pyrrole-2-carboxamido)piperidine- 1 -carboxylate (0.24 g, 0.50 mmol), DCM (20 mL), and MeOH (5 mL). After 15 min at rt, the reaction was concentrated to give l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N- (piperidin-4-yl)-lH-pyrrole-2-carboxamide as a yellow oil.
- Step 3 Isopropyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrr ⁇ )le-2- carboxamido)piperidine-l-carboxylate
- 3-Fluoro-2-hydrazinyl-5-(trifluoromethyl)pyridine was prepared from 2,3-difluoro-5- (trifluoromethyl)pyridine following the procedure outlined for Intermediate 3.
- the title compound was prepared from 3-fluoro-2-hydrazinyl-5-(trifluoromethyl)pyridine and Intermediate 1 following the procedure outlined in EXAMPLE 1.
- n-Butyllithium (15.0 mL, 2.5M, 37.5 mmol) was added dropwise to a solution of diisopropylamine (3.76 g, 37.2 mmol) and THF (50 mL) at -78 0 C under N 2 . After 30 min, a solution of 2,3-dichloropyridine (5.0 g, 34 mmol) and THF (50 mL) was added dropwise. After an additional hour at -78 0 C, iodine (8.6 g, 34 mmol) was added.
- Chlorotrimethylsilane (10.2 rnL, 80.1 mmol) was added to a mixture of 2,3-dichloro-4- iodopyridine (11 g, 40 mmol), sodium iodide (12 g, 80 mmol), and ACN (150 mL). The resulting mixture was refluxed overnight, allowed to cool to rt, and concentrated. Sat'd sodium thiosulfate (100 mL) was added, and the mixture was extracted with EtOAc (200 mL x 3).
- Step 4 l-(3-Chloro-4-methoxypyridin-2-yl)-N'-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide
- Step 1 (2-Fluoro-4-(methylsulfonyl)phenyl)methanamine
- Step 1 (4-chloro-3-(trifluoromethoxy)phenyl)methanamine
- Step 1 l-(2-Bromo-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid
- n-BuLi (15 mL, 2.5M) was added to a solution of diisopropylamine (3.76 g, 37.23 mmol) in THF (50 mL) cooled to -78 0 C and the resulting mixture was stirred for 30 min at -78 0 C. This was followed by the addition of a solution of 2,3-dichloropyridine (5 g, 34.01 mmol) in THF (50 mL) and the mixture was stirred at -78 0 C for 1 h. To the mixture was added I 2 (8.58 g, 33.78 mmol) and the mixture was warmed to -3O 0 C and stirred for 5 min..
- Step 1 S-Chloro-l-hydrazinyl-S-methylpyridine
- n-BuLi 52 mL, 2.5M was slowly added to a cooled (-45 0 C) solution of diisopropylamine (13.13 g, 130 mmol) in THF (50 mL) and it was stirred at this temperature for 1 h.
- the mixture was cooled to -78 0 C followed by addition of a solution of 2-chloro-3-fluoro-4-iodopyridine (25.7 g, 100 mmol) in THF (150 mL) and the resulting mixture was stirred at -78 0 C for 1.5 h.
- the reaction mixture was then quenched by the slow addition of H 2 O (100 mL).
- Step 3 NSl-BisCS-Chloro- ⁇ -Ctrifluoromethylt ⁇ yridin-l-ylHH-pyrrole-l- carbohydrazide
- Step 2 l-(3-Chloro-4-methylpyridin-2-yl)-N'-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide
- Step 1 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-2-carboxylic acid
- Step 2 N',l-Bis(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-2- carbohydrazide
- Step 1 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-indole-2-carboxylic acid
- Step 1 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2-carboxylic acid
- Step 1 l-(2-Chlorophenyl)-lH-pyrrole-2-carboxylic acid
- Step 2 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-chlorophenyl)-lH- pyrrole-2-carbohydrazide
- the title compound can be prepared as described in EXAMPLE 53 using 2,3-difluoro- 5-(trifluoromethyl)pyridine as the starting material.
- 1 H NMR 300 MHz, CDCl 3 ) ⁇ 8.83 (bs, IH), 8.59 (s, IH), 8.33 (s, IH), 7.77 (m, 3H), 7.24 (m, IH), 7.16 (m, IH), 6.43 (m, IH).
- Step 1 Methyl l-(3-methyl-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxylate
- Step 2 l-(3-Methyl-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid
- the title compound can be prepared as described in EXAMPLE 53 using 2-fluoro-5- (trifluoromethyl)pyridine and 2-hydrazinyl-5-(trifluoromethyl)pyridine as the starting materials.
- 1 H NMR 300 MHz, CD 3 OD
- LCMS 416 (M+H) + .
- the title compound can be prepared as described in EXAMPLE 53 using 3-chloro-2- fluoropyridine and 3-chloro-2-hydrazinylpyridine as the starting materials.
- Step 5 3-(2-Chloro-4-(trifluoromethyl)phenyl)-5-methylisoxazole-4-carboxylic acid
- Step 1 Methyl l-(4-nitro-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate
- Step 4 l-(4-(Diethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid
- the title compound was synthesized as described in EXAMPLE 1 using l-(4- (diethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl) methanamine hydrochloride as the starting materials.
- Step 1 l-(4-Amino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid
- Step 1 Methyl l-(4-morpholino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2- carboxylate
Abstract
The present invention relates to compounds and methods which may be useful as inhibitors of GPR119 for the treatment or prevention of metabolic, cardiovascular, and metabolic diseases.
Description
HETEROCYCLIC MODULATORS OF GPRl 19 FOR TREATMENT OF
DISEASE
[001] This application claims the benefit of United States Provisional Applications No. 61/037,239, filed March 17, 2008, and No. 61/095,362, filed September 9, 2008, the disclosures of which are hereby incorporated by reference as if written herein in their entireties.
[002] Disclosed herein are new heterocyclic compounds, compositions, and their application as pharmaceuticals for the treatment of disease. Methods of modulation of GPRl 19 activity in a human or animal subject are also provided for the treatment diseases mediated by GPRl 19.
[003] Obesity is a growing threat to the global health by virtue of its association with a cluster of diseases that include insulin resistance, glucose intolerance, dyslipidemia, and hypertension, collectively known as the metabolic syndrome or syndrome X. It is well documented that patients with metabolic syndrome have a higher risk for cardiovascular diseases such as coronary heart disease and stroke [Grundy S. M. et al. Circulation 112:e285-e290, 2005]. The treatment of obesity will require complex solutions, including increased public awareness to diminish food portions, improved food choices and increased physical activity. However, epidemiologic studies have shown that treating diabetes/insulin resistance in these patients can reduce the risk of cardiovascular diseases such as coronary artery disease. However, there remains a need for additional agents that can perhaps treat the root cause(s) of metabolic syndrome by treating obesity and diabetes. GPRl 19 modulators described herein might represent such an opportunity. [004] Current therapies for diabetes mellitus include: insulin; insulin secretagogues, such as sulphonylureas, which increase insulin secretion from pancreatic β-cells; glucose-lowering effectors, such as metformin which reduce glucose production from the liver; activators of the peroxisome proliferator-activated receptor-γ (PPAR-γ), such as the thiazolidinediones, which enhances insulin action; GLP-I mimetics, such as exenatide (Byetta); and α-glucosidase inhibitors which interfere with gut glucose production. There are, however, deficiencies associated with currently
available treatments, including hypoglycemic episodes, weight gain, loss in responsiveness to therapy over time, gastrointestinal problems, and edema. [005] There are several areas at which research is being targeted in order to bring new, more effective, therapies to the marketplace. For example, on-going research includes exploring a reduction in excessive hepatic glucose production, enhancing the pathway by which insulin transmits its signal to the cells such that they take up glucose, enhancing glucose-stimulated insulin secretion from the pancreatic β-cells, and targeting obesity and associated problems with fat metabolism and accumulation. [006] GIP and GLP-I are peptides, known as incretins, secreted from enteroendocrine K- and L-cells respectively in response to ingestion of nutrients, and have a wide variety of physiological effects that have been described in numerous publications over the past two decades. See, for example, Bojanowska, E. et al., Med. Sd. Monit., 2005, Aug 5 11(8): RA271-8; Perry, T. et al., Curr. Alzheimer Res., 2005, July 2(3): 377-85; and Meier, J.J. et al., Diabetes Metab. Res. Rev., 2005, Mar-Apr; 21(2); 91-117 (each herein incorporated by reference with regard to a background understanding of incretins). These peptides act as gut hormones which promote normoglycemia acutely by enhancing glucose-stimulated insulin release and chronically by maintaining pancreatic β-cell mass (Staffers D.A., et al., 2000, Diabetes, 49:741-748; Xu G. et al., 1999, Diabetes, 48:2270-2276; Tourrel C. et al., 2002, Diabetes, 51 :1443-1452; Wajchenberg B.L., Endocr Rev., 2007, 28(2):187-218). Although the mechanisms regulating GLP-I secretion remain unclear, the initial rapid rise in GLP-I following a meal may be a result of hormonal stimulation of neuronal afferents involving GIP. See, for example, J.N. Roberge and P.L. Brubaker, Endocrinology 133 (1993), pp. 233 — 240 (herein incorporated by reference with regard to such teaching). Later increases in GLP-I may involve direct activation of L-cells by nutrients in the distal small-intestine and the colon. GIP and GLP-I are potent stimulators of the body's ability to produce insulin in response to elevated levels of blood sugar. These effects occur via β-cell-expressed, Class B G-protein coupled receptors which in turn mediate elevated intracellular cAMP. Decreased GIP responsiveness may contribute to the pathogenesis of type 2 diabetes, resulting in mixed enthusiasm toward GIP receptor agonists as a means to treat the disease (Nauck
M.A., et al, 1993, J. Clin. Invest, 91 :301-307, Elahi, D., et al, Regul. Pept., 51 :63-74). By contrast, strategies that enhance GLP-I receptor function have shown significant therapeutic promise. These strategies have so far consisted of injectable peptidic GLP- 1 receptor agonists, or alternatively, blockade of endogenous GLP- 1 metabolism through selective inhibition of the peptidase DPP-IV (Ahren B., et al., 2000, Eur. J. Pharmacol, 404:239-245; Ahren B., et al., 2002, Diabetes Care, 25:869-875). [007] In Type II diabetes the action of GLP-I on the β-cell is maintained, although GLP-I secretion, itself, is reduced. More recently, therefore, much research has been focused on GLP-I. Studies show glucose-lowering effects in addition to GLP- 1 's ability to stimulate glucose-dependent insulin secretion including, but not limited to, an inhibition of the release of the hormone glucagon following meals, a reduction in the rate at which nutrients are absorbed into the bloodstream, and a reduction of food intake. Studies demonstrate that treatments to increase GLP-I, therefore, may be used for a variety of conditions and disorders including but not limited to metabolic disorders, gastrointestinal disorders, inflammatory diseases, psychosomatic, depressive, and neuropsychiatric disease including but not limited to diabetes mellitus (Type 1 and Type 2), metabolic syndrome, obesity, appetite control and satiety, weight loss, stress, inflammation, myocardial ischemia/reperfusion injury, Alzheimer's Disease, and other diseases of the central nervous system. In Type II diabetes, patients display a decreased responsiveness to GIP but not GLP-I, with respect to its ability to stimulate insulin secretion. The mechanism behind the decreased responsiveness to GIP remains unclear since Type II diabetics retain sensitivity to a bolus administration of GIP but not to a continuous infusion (Meier et al. 2004 Diabetes 53 S220-S224). Moreover recent studies with a long-acting fatty-acid derivative of GIP showed beneficial effects on glucose homeostasis in ob/ob mice following 14 days of treatment (Irwin N. et al. (2006) J. Med. Chem. 49, 1047-1 054).
[008] The use of exogenous GLP-I in clinical treatment is severely limited, however, due to its rapid degradation by the protease DPPIV. There are multiple GLP- 1 mimetics in development for type 2 diabetes that are reported in the literature, all are modified peptides, which display longer half- lives than endogenous GLP-I. For example, the product sold under the tradename BYETT A® is the first FDA-approved
agent of this new class of medications. These mimetics, however, require injection. An oral medication that is able to elevate GLP-I secretion is desirable. Orally available inhibitors of DPP-IV, which result in elevation in intact GLP-I, are now available, such as sitagliptin, marketed under the brand name JANUVIA®. Nevertheless, a molecule which may stimulate GLPl secretion would provide a therapeutic benefit. A molecule which could stimulate both GLP-I secretion and insulin secretion through effects on the L-cell and direct effects on the β-cell would hold much promise for Type II diabetes therapy.
[009] Orally active, small-molecule GLP- 1 receptor agonists have proven elusive, a feature that unfortunately is characteristic of Class B GPCRs. Meanwhile, the spectrum of signaling peptides affected by inhibition of DPP-IV remains unclear and could potentially extend significantly beyond GLP-I and GIP (8, 9). It is therefore worthwhile to search for therapeutic approaches which afford both the physiological selectivity of GLP-I signaling and the opportunity for orally active treatment modalities.
[010] One particular target is GPRl 19. GPRl 19 was identified as a Class A, islet- enriched receptor which could potentially mediate the insulinotropic actions of lysophosphatidylcholine (LPC) observed in vitro (Soga T., et al, Biochem Biophys Res Commun, 2005, 326:744-751), but a later study suggested that oleoylethanolamide (OEA) was a more potent GPRl 19 agonist (Overton H. A., et al., Cell Metab, 2006, 3:167-175). Recently, a small molecule GPRl 19 agonist has been shown to enhance glucose-dependent insulin secretion and improve hyperglycemia in rodent models of diabetes (Chu Z. et al. Endocrinology, 2007, 148:2601-2609). In addition to the "GPRl 19" identifier, several other identifiers exist, including but not limited to RUP 3, Snorf 25, 19 AJ, AXOR 20, GDIR, GPCR2, and PSI. GPRl 19 is expressed in human gastrointestinal regions and in human islets. Activation of GPRl 19 has been demonstrated to stimulate intracellular cAMP and lead to glucose-dependent GLP-I and insulin secretion. See, T. Soga et al., Biochemical and Biophysical Research Communications 326 (2005) 744-751, herein incorporated by reference with regard to a background understanding of GPRl 19. Moreover, activation of GPRl 19 is very unlikely to induce hypoglycemia (Chu Z., et al. Endocrinology, 2007, 148:2601-2609)
[Oi l] Agonists to GPRl 19 may be of therapeutic value for diabetes and associated conditions, particularly Type II diabetes, obesity, glucose intolerance, insulin resistance, metabolic syndrome X, hyperlipidemia, hypercholesterolemia, and atherosclerosis.
[012] Novel compounds and pharmaceutical compositions, certain of which have been found to modulate GPRl 19 have been discovered, together with methods of synthesizing and using the compounds including methods for the treatment of
GPRl 19-mediated diseases in a patient by administering the compounds.
[013] Provided herein are compounds of structural Formula I, or a salt, ester, or prodrug thereof, wherein,

Xi is selected from the group consisting of N, O, S, CH2 and CR3;
X2 is selected from the group consisting of N, O, S, CH2 and CR4;
X3 is selected from the group consisting of N, O, S, CH2, CH, and C-methyl;
Gi is selected from the group consisting of CR3Rb, NR7, and optionally substituted N-heterocycloalkyl;
G2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, NRs, O and SO2, any of which may be optionally substituted;
Ri is selected from the group consisting of aryl, N-containing heteroaryl; arylalkyl, and N-containing heteroarylalkyl, any of which may be optionally substituted;
R2 is selected from the group consisting of lower alkyl, lower alkenyl, lower alkynyl, lower hydroxyalkyl, amino, aryl, and heteroaryl, any of which may be optionally substituted; or, when Gi is substituted N-heterocycloalkyl, R2 may be null;
R3 and R4 are independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkoxy, lower perfluoroalkyl, and lower perfluoroalkoxy, or R3 and R4 taken together may form a 5-
6-membered aryl, cycloalkyl, heteroaryl, or heterocycloalkyl, any of which may be optionally substituted;
Rs, Re, R7, and Rs are each independently selected from the group consisting of halogen, hydrogen, hydroxy, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, phenyloxy, acyl, carboxyl, amino, lower heteroalkyl, lower alkylthio, lower aminoalkyl, lower alkylsulfonyl, sulfonamido, lower aryl, lower arylalkyl, lower cycloalkyl, lower cycloalkylalkyl, lower haloalkyl, lower perhaloalkyl, lower perhaloalkoxy, lower heteroaryl, lower heteroarylalkyl, lower heterocycloalkyl, and lower heterocycloalkylalkyl, any of which may be optionally substituted, or R5 and R6, taken together, form oxy; and
Ra and Rb are each independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, lower hydroxyalkyl, lower perfluoroalkyl, lower perfluoroalkoxy, optionally substituted lower cycloalkyl, optionally substituted lower heterocycloalkyl, optionally substituted lower heteroaryl, and optionally substituted phenyl.
[014] In certain embodiments, R5 and R6, taken together, form oxy. [015] In certain embodiments, Ri is selected from the group consisting of aryl and N-containing heteroaryl, any of which may be optionally substituted. [016] In certain embodiments, Ri is selected from the group consisting of substituted phenyl and substituted, monocyclic N-containing heteroaryl. [017] In certain embodiments, Ri is substituted pyridyl. [018] In certain embodiments, Ri is substituted phenyl. [019] In certain embodiments, Gi is NR7. [020] In certain embodiments, Gi is NH.
[021] In certain embodiments, R7 is selected from the group consisting of hydrogen, halogen, hydroxy, Ci-C3 alkyl, Ci-C3 hydroxyalkyl, Ci-C3 alkynyl, perfluoromethyl, and perfluoromethoxy. [022] In certain embodiments, Gi is CRaRb,
[023] In certain embodiments, Gi is optionally substituted N-heterocycle. [024] In certain embodiments, G2 is a bond.
[025] In certain embodiments, is selected from the group consisting of piperidine, piperazine, diazepine, diazepine, quinoline, thiazole, morpholine, and pyrrolidine, any of which may be optionally substituted.
[026] In certain embodiments, Gi is optionally substituted with one to three substituents selected from the group consisting of lower alkyl, lower alkoxy, lower hydroxyalkyl, and hydroxy.
[027] In certain embodiments, Gi is optionally substituted with one to three substituents selected from the group consisting of methyl ethyl, methoxy, and hydroxy.
[028] In certain embodiments,
[029] In certain embodiments, G2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, and NRs, any of which may be optionally substituted.
[030] In certain embodiments,
G2 is selected from the group consisting of optionally substituted C1-C3 alkyl, C1-C3 heteroalkylcontaining one heteroatom selected from the group consisting of O

Rg is selected from the group consisting of hydrogen, halogen, hydroxy, C1-C3 alkyl, C1-C3 hydroxyalkyl, C1-C3 alkynyl, perfluoromethyl, and perfluoromethoxy. [031 ] In certain embodiments,
G2 is selected from the group consisting of NR8 and CH2; and
R8 is hydrogen,
[032] In certain embodiments, R2 is selected from the group consisting of aryl and heteroaryl, either of which may be optionally substituted. [033] In certain embodiments, R2 is substituted pyridyl. [034] In certain embodiments, R2 is substituted phenyl. [035] In certain embodiments, compounds have structural Formula II:

Gi is selected from the group consisting of CH2, and NR7;
G2 is selected from the group consisting of a bond, CH2, and NRs;
Xi is selected from the group consisting of CRio and N;
X2 is selected from the group consisting of CRi2, N, O, and S;
X3 is selected from the group consisting of CR14, N, O, and S;
Yi is selected from the group consisting of CR2I, N, O, and S;
Y2 is selected from the group consisting of CR22, N, O, and S;
Y3 is selected from the group consisting of CR23, N, O, and S;
R2 is selected from the group consisting of lower alkyl, lower amino, aryl, and heteroaryl, any of which may be optionally substituted; q is an integer from O to 3;
R2 is selected from the group consisting of CH, O, and N;
R5, R6, R7, and Rs are each independently selected from the group consisting of hydrogen, hydroxy, alkoxy, aryloxy, alkyl, acyl, alkenyl, amino, alkynyl, heteroalkyl, carboxyl, alkylthio, alkylamino, alkylsulfonyl, sulfonamido, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, haloalkyl, perhaloalkyl, perhaloalkoxy, heteroaryl, heteroarylalkyl, heterocycloalkyl, and heterocycloalkylalkyl, any of which may be optionally substituted , or R3 and R4, taken together, may form oxy; and
Rio, Ri2, Ri4, Ri9, R2i, R22, R23, R27, and R28 are each independently selected from the group consisting of null, hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, C-amido, N-amido, carbamate, urea, N-sulfonamido, S- sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted, or R27
and R-28, taken together with R2 to which they are attached, form cycloalkyl, heterocycloalkyl, aryl or heteroaryl, any of which may be optionally substituted. [036] In certain embodiments, R5 and R6, taken together, form oxy. [037] In certain embodiments,
Yi is selected from the group consisting of CR21 and N;
Y2 is selected from the group consisting of CR22 and N; and
Y3 is selected from the group consisting of CR23 and N.
[038] In certain embodiments, R27 and R28, taken together with R2 to which they are attached, form cycloalkyl, heterocycloalkyl, aryl or heteroaryl, any of which may be optionally substituted.
[039] In certain embodiments, R27 and R28, taken together with R2 to which they are attached, form cycloalkyl or heterocycloalkyl, either of which may be optionally substituted. [040] In certain embodiments, said cycloalkyl is a C3-C7 monocyclic cycloalkyl, which may be optionally substituted; and said heterocycloalkyl, is a C4-C7 monocyclic heterocycloalkyl, which may be optionally substituted.
[041] In certain embodiments, said C4-C7 monocyclic heterocycloalkyl is selected from the group consisting of pyrrolidine, piperidine, piperazine, and morpholine, any of which may be optionally substituted. [042] In certain embodiments, compounds have structural Formula III:

Gi is selected from the group consisting of CH2, and NR7;
G2 is selected from the group consisting of a bond, CH2, and NR8;
Xi is selected from the group consisting of CH and N;
X2 is selected from the group consisting of CH and N;
X3 is selected from the group consisting of CH and N;
Yi is selected from the group consisting of CR21 and N;
Y2 is selected from the group consisting of CR22 and N;
Y3 is selected from the group consisting of CR23 and N;
Zi is selected from the group consisting of CR24 and N;
Z2 is selected from the group consisting of CR25 and N;
Z3 is selected from the group consisting of CR26 and N;
Z4 is selected from the group consisting of CR27 and N; q and r are each independently an integer from 0 to 3;
R7, and Rs are each independently selected from the group consisting of hydrogen, hydroxy, lower alkoxy, lower alkyl, lower alkenyl, lower amino, lower alkynyl, lower heteroalkyl; and
Rig, Ri9, R21, R22, R23, R24, R25, R26, and R27 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, heteroarylamino, C-amido, N-amido, carbamate, urea, N- sulfonamido, S-sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted. [043] In certain embodiments,
Gi is selected from the group consisting of CH2, and NR7; and
G2 is selected from the group consisting of a bond, CH2, and NRs.
[044] In certain embodiments,
Xi and X3 are each CH; and
X2 is N. [045] In certain embodiments,
Xi and X2 are each CH; and
X3 is N. [046] In certain embodiments,
Xi is N; and
X2 and X3 are each CH.
[047] In certain embodiments, X1, X2 and X3 are each CH. [048] In certain embodiments,
Y1 Is CR21;
Y2 is CR22; and
Y3 is N. [049] In certain embodiments,
Y1 Is CR21;
Y2 is CR22; and
Y3 is CR23. [050] In certain embodiments,
Z3 is CR26; and
R26 is not hydrogen.
[051] In certain embodiments, Zi is N.
[052] In certain embodiments, G2 is selected from the group consisting of NH and CH2. [053] In certain embodiments, Gi is NH.
[054] In certain embodiments, compounds have structural Formula IV:

Gi is selected from the group consisting of NH and CH2;
G2 is selected from the group consisting of NH, CH2, SO2, and O;
Zi is selected from the group consisting of CR24 and N;
Z4 is selected from the group consisting of CR27 and N; q is an integer from 1 to 3; r is an integer from 1 to 3, or r may be 0 if R27 is not hydrogen; and each Ri8, each Ri9, R24, R26, and R27 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, heteroarylamino, C-amido, N- amido, carbamate, urea, N-sulfonamido, S-sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted.
[055] In certain embodiments, G2 is selected from the group consisting of NH and CH2. [056] In certain embodiments, Gi is NH.
[057] In certain embodiments,
Zi is N; and
Z4 is CR27.
[058] In certain embodiments, Zi and Z4 are both N. [059] In certain embodiments, R27 is not hydrogen.
[060] The In certain embodiments, R27 and each Ri8 are independently selected from the group consisting of hydrogen, halogen, haloalkyl, haloalkoxy, optionally substituted arylamino, optionally substituted heteroarylamino, optionally substituted heteroaryloxy, and optionally substituted aryloxy, provided that one OfR27 and each Ri 8 is not hydrogen.
[061] In certain embodiments, R27 is selected from the group consisting of optionally substituted phenyloxy and optionally substituted 5-6 membered monocyclic heteroaryloxy.
[062] In certain embodiments, R27 is selected from the group consisting of optionally substituted phenyloxy and optionally substituted pyridinyloxy. [063] The compound as recited in claim 44, wherein said pyridinyloxy or aryloxy is optionally substituted with one or more substituents selected from the group consisting of halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, lower hydroxyalkyl, lower perfluoroalkyl, lower perfluoroalkoxy, optionally substituted lower cycloalkyl, optionally substituted lower heterocycloalkyl, optionally substituted lower heteroaryl, and optionally substituted phenyl.
[064] The compound as recited in claim 45, wherein said pyridinyloxy or aryloxy is optionally substituted with one or more substituents selected from the group consisting ofhalogen, hydroxy, C1-C3 alkyl, C1-C3 hydroxyalkyl, C1-C3 alkynyl, perfluoromethyl, and perfluoromethoxy.
[065] In certain embodiments, each Ri9 is independently selected from the group consisting ofhalogen, haloalkyl, and haloalkoxy.
[066] In certain embodiments, each R19 is independently selected from the group consisting ofhalogen, perfluoromethyl, and perfluoromethoxy. [067] In certain embodiments, q is 1 or 2.
[068] In certain embodiments, q is 2, and the two R19 groups are meta to each other.
[069] Also provided are Examples 1 to 640.
[070] Also provided are compounds as disclosed herein for use as a medicament.
[071] Also provided are compounds as disclosed herein for use in the manufacture of a medicament for the prevention or treatment of a disease or condition ameliorated by the modulation of GPRl 19.
[072] Also provided is a pharmaceutical composition comprising a compound as disclosed herein together with a pharmaceutically acceptable carrier.
[073] Also provided is a method of modulating GPRl 19 comprising contacting
GPRl 19 with a compound as disclosed herein.
[074] Also provided is a method of treatment of a GPRl 19-mediated disease comprising the administration of a therapeutically effective amount of a compound as disclosed herein to a patient in need thereof.
[075] In certain embodiments, said disease is a metabolic disease.
[076] In certain embodiments, said disease is diabetes.
[077] Also provided is a method of treatment of a GPRl 19-mediated disease comprising the administration of: i. a therapeutically effective amount of a compound as disclosed herein; and ii. another therapeutic agent.
[078] In certain embodiments, said agent is selected from the group consisting of insulin, metformin, Glipizide, glyburide, Amaryl, gliclazide, meglitinides, nateglinide, repaglinide, pramlintide, PTP-112, SB-517955, SB-4195052, SB-216763, NN-57- 05441, NN-57-05445, GW-0791, AGN-194204, T-1095, BAY R3401, acarbose, miglitol, voglibose, Exendin-4, DPP728, LAF237, vildagliptin , BMS477118, PT-100, GSK-823093, PSN-9301, T-6666, SYR-322, SYR-619, Liraglutide, CJC-1134-PC, naliglutide, MK-0431, saxagliptin, GSK23A, pioglitazone, rosiglitazone, AVE2268, GW869682, GSK189075, APD668, PSN-119-1, PSN-821, rosuvastatin, atrovastatin, simvastatin, lovastatin, pravastatin, fluvastatin, cerivastatin, rosuvastatin, pitavastatin, fenofibrate, benzafibrate, clofϊbrate, gemfibrozil, Ezetimibe, eflucimibe, CP-529414, CETi-I, JTT-705, cholestyramine, colestipol, niacin, implitapide, (7?)-l-{4-[5-methyl-
2-(4-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-benzenesulfonyl}2,3-dihydro-lH- indole-2-carboxylic acid, and GI-262570.
[079] Also provided is a method for achieving an effect in a patient comprising the administration of a therapeutically effective amount of a compound as disclosed herein to a patient, wherein the effect is the modulation of a metabolic disease.
[080] As used herein, the terms below have the meanings indicated.
[081] When ranges of values are disclosed, and the notation "from ni ... to n2" is used, where ni and n2 are the numbers, then unless otherwise specified, this notation is intended to include the numbers themselves and the range between them. This range may be integral or continuous between and including the end values. By way of example, the range "from 2 to 6 carbons" is intended to include two, three, four, five, and six carbons, since carbons come in integer units. Compare, by way of example, the range "from 1 to 3 μM (micromolar)," which is intended to include 1 μM, 3 μM, and everything in between to any number of significant figures (e.g., 1.255 μM, 2.1 μM,
2.9999 μM, etc.).
[082] The term "about," as used herein, is intended to qualify the numerical values which it modifies, denoting such a value as variable within a margin of error.
When no particular margin of error, such as a standard deviation to a mean value given in a chart or table of data, is recited, the term "about" should be understood to mean that range which would encompass the recited value and the range which would be included by rounding up or down to that figure as well, taking into account significant figures.
[083] The term "acyl," as used herein, alone or in combination, refers to a carbonyl attached to an alkenyl, alkyl, aryl, cycloalkyl, heteroaryl, heterocycle, or any other moiety were the atom attached to the carbonyl is carbon. An "acetyl" group refers to a -C(O)CΗ3 group. An "alkylcarbonyl" or "alkanoyl" group refers to an alkyl group attached to the parent molecular moiety through a carbonyl group. Examples of such groups include methylcarbonyl and ethylcarbonyl. Examples of acyl groups include formyl, alkanoyl and aroyl.
[084] The term "alkenyl," as used herein, alone or in combination, refers to a straight-chain or branched-chain hydrocarbon radical having one or more double bonds
and containing from 2 to 20 carbon atoms. In certain embodiments, said alkenyl will comprise from 2 to 6 carbon atoms. The term "alkenylene" refers to a carbon-carbon double bond system attached at two or more positions such as ethenylene [(-CH=CH- ),(-C::C-)]. Examples of suitable alkenyl radicals include ethenyl, propenyl, 2- methylpropenyl, 1 ,4-butadienyl and the like. Unless otherwise specified, the term "alkenyl" may include "alkenylene" groups.
[085] The term "alkoxy," as used herein, alone or in combination, refers to an alkyl ether radical, wherein the term alkyl is as defined below. Examples of suitable alkyl ether radicals include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso- butoxy, sec-butoxy, tert-butoxy, and the like.
[086] The term "alkyl," as used herein, alone or in combination, refers to a straight-chain or branched-chain alkyl radical containing from 1 to 20 carbon atoms. In certain embodiments, said alkyl will comprise from 1 to 10 carbon atoms. In further embodiments, said alkyl will comprise from 1 to 6 carbon atoms. Alkyl groups may be optionally substituted as defined herein. Examples of alkyl radicals include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl, noyl and the like. The term "alkylene," as used herein, alone or in combination, refers to a saturated aliphatic group derived from a straight or branched chain saturated hydrocarbon attached at two or more positions, such as methylene (- CH2-). Unless otherwise specified, the term "alkyl" may include "alkylene" groups. [087] The term "alkylamino," as used herein, alone or in combination, refers to an alkyl group attached to the parent molecular moiety through an amino group. Suitable alkylamino groups may be mono- or dialkylated, forming groups such as, for example, N-methylamino, N-ethylamino, N,N-dimethylamino, N,N-ethylmethylamino and the like.
[088] The term "alkylidene," as used herein, alone or in combination, refers to an alkenyl group in which one carbon atom of the carbon-carbon double bond belongs to the moiety to which the alkenyl group is attached.
[089] The term "alkylthio," as used herein, alone or in combination, refers to an alkyl thioether (R-S-) radical wherein the term alkyl is as defined above and wherein the sulfur may be singly or doubly oxidized. Examples of suitable alkyl thioether
radicals include methylthio, ethylthio, n-propylthio, isopropylthio, n-butylthio, iso- butylthio, sec-butylthio, tert-butylthio, methanesulfonyl, ethanesulfϊnyl, and the like. [090] The term "alkynyl," as used herein, alone or in combination, refers to a straight-chain or branched-chain hydrocarbon radical having one or more triple bonds and containing from 2 to 20 carbon atoms. In certain embodiments, said alkynyl comprises from 2 to 6 carbon atoms. In further embodiments, said alkynyl comprises from 2 to 4 carbon atoms. The term "alkynylene" refers to a carbon-carbon triple bond attached at two positions such as ethynylene (-C:::C-, -C≡C-). Examples of alkynyl radicals include ethynyl, propynyl, hydroxypropynyl, butyn-1-yl, butyn-2-yl, pentyn-1- yl, 3-methylbutyn-l-yl, hexyn-2-yl, and the like. Unless otherwise specified, the term "alkynyl" may include "alkynylene" groups.
[091] The terms "amido" and "carbamoyl,"as used herein, alone or in combination, refer to an amino group as described below attached to the parent molecular moiety through a carbonyl group, or vice versa. The term "C-amido" as used herein, alone or in combination, refers to a -Q=O)-NR2 group with R as defined herein. The term "N-amido" as used herein, alone or in combination, refers to a RC(=O)NH- group, with R as defined herein. The term "acylamino" as used herein, alone or in combination, embraces an acyl group attached to the parent moiety through an amino group. An example of an "acylamino" group is acetylamino (CH3C(O)NH-). [092] The term "amino," as used herein, alone or in combination, refers to — NRR , wherein R and R are independently selected from the group consisting of hydrogen, alkyl, acyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl, any of which may themselves be optionally substituted. Additionally, R and R' may combine to form heterocycloalkyl, either of which may be optionally substituted. [093] The term "aminoalkyl," as used herein, alone or in combination, refers to an amino group attached to the parent molecular moiety through an alkyl group. [094] The term "aryl," as used herein, alone or in combination, means a carbocyclic aromatic system containing one, two or three rings wherein such polycyclic ring systems are fused together. The term "aryl" embraces aromatic groups such as phenyl, naphthyl, anthracenyl, and phenanthryl.
[095] The term "arylalkenyl" or "aralkenyl," as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkenyl group.
[096] The term "arylalkoxy" or "aralkoxy," as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkoxy group.
[097] The term "arylalkyl" or "aralkyl," as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkyl group.
[098] The term "arylalkynyl" or "aralkynyl," as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an alkynyl group.
[099] The term "arylalkanoyl" or "aralkanoyl" or "aroyl,"as used herein, alone or in combination, refers to an acyl radical derived from an aryl-substituted alkanecarboxylic acid such as benzoyl, napthoyl, phenylacetyl, 3-phenylpropionyl
(hydrocinnamoyl), 4-phenylbutyryl, (2-naphthyl)acetyl, 4-chlorohydrocinnamoyl, and the like.
[0100] The term aryloxy as used herein, alone or in combination, refers to an aryl group attached to the parent molecular moiety through an oxy.
[0101] The terms "benzo" and "benz," as used herein, alone or in combination, refer to the divalent radical C6H4= derived from benzene. Examples include benzo thiophene and benzimidazole.
[0102] The term "carbamate," as used herein, alone or in combination, refers to an ester of carbamic acid (-NHCOO-) which may be attached to the parent molecular moiety from either the nitrogen or acid end, and which may be optionally substituted as defined herein.
[0103] The term "O-carbamyl" as used herein, alone or in combination, refers to a
-OC(O)NRR', group-with R and R' as defined herein.
[0104] The term "N-carbamyl" as used herein, alone or in combination, refers to a
ROC(O)NR'- group, with R and R' as defined herein.
[0105] The term "carbonyl," as used herein, when alone includes formyl [-C(O)H] and in combination is a -C(O)- group.
[0106] The term "carboxyl" or "carboxy," as used herein, refers to -C(O)OH or the corresponding "carboxylate" anion, such as is in a carboxylic acid salt. An "O-carboxy" group refers to a RC(O)O- group, where R is as defined herein. A "C-carboxy" group refers to a -C(O)OR groups where R is as defined herein. [0107] The term "cyano," as used herein, alone or in combination, refers to -CN. [0108] The term "cycloalkyl," or, alternatively, "carbocycle," as used herein, alone or in combination, refers to a saturated or partially saturated monocyclic, bicyclic or tricyclic alkyl group wherein each cyclic moiety contains from 3 to 12 carbon atom ring members and which may optionally be a benzo fused ring system which is optionally substituted as defined herein. In certain embodiments, said cycloalkyl will comprise from 5 to 7 carbon atoms. Examples of such cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronapthyl, indanyl, octahydronaphthyl, 2,3-dihydro-lH-indenyl, adamantyl and the like. "Bicyclic" and "tricyclic" as used herein are intended to include both fused ring systems, such as decahydronaphthalene, octahydronaphthalene as well as the multicyclic (multicentered) saturated or partially unsaturated type. The latter type of isomer is exemplified in general by, bicyclo[l,l,l]pentane, camphor, adamantane, and bicyclo[3,2,l]octane.
[0109] The term "ester," as used herein, alone or in combination, refers to a carboxy group bridging two moieties linked at carbon atoms.
[0110] The term "ether," as used herein, alone or in combination, refers to an oxy group bridging two moieties linked at carbon atoms.
[0111] The term "halo," or "halogen," as used herein, alone or in combination, refers to fluorine, chlorine, bromine, or iodine.
[0112] The term "haloalkoxy," as used herein, alone or in combination, refers to a haloalkyl group attached to the parent molecular moiety through an oxygen atom. [0113] The term "haloalkyl," as used herein, alone or in combination, refers to an alkyl radical having the meaning as defined above wherein one or more hydrogens are replaced with a halogen. Specifically embraced are monohaloalkyl, dihaloalkyl and polyhaloalkyl radicals. A monohaloalkyl radical, for one example, may have an iodo, bromo, chloro or fluoro atom within the radical. Dihalo and polyhaloalkyl radicals
may have two or more of the same halo atoms or a combination of different halo radicals. Examples of haloalkyl radicals include fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl, dichloroethyl and dichloropropyl. "Haloalkylene" refers to a haloalkyl group attached at two or more positions. Examples include fluoromethylene (-CFH-), difluoromethylene (-CF2 -), chloromethylene (-CHC1-) and the like. [0114] The term "heteroalkyl," as used herein, alone or in combination, refers to a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, fully saturated or containing from 1 to 3 degrees of unsaturation, consisting of the stated number of carbon atoms and from one to three heteroatoms selected from the group consisting of O, N, and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized. The heteroatom(s) O, N and S may be placed at any interior position of the heteroalkyl group. Up to two heteroatoms may be consecutive, such as, for example, -CH2-NH- OCH3.
[0115] The term "heteroaryl," as used herein, alone or in combination, refers to a 3 to 7 membered unsaturated heteromonocyclic ring, or a fused monocyclic, bicyclic, or tricyclic ring system in which at least one of the fused rings is aromatic, which contains at least one atom selected from the group consisting of O, S, and N. In certain embodiments, said heteroaryl will comprise from 5 to 7 carbon atoms. The term also embraces fused polycyclic groups wherein heterocyclic rings are fused with aryl rings, wherein heteroaryl rings are fused with other heteroaryl rings, wherein heteroaryl rings are fused with heterocycloalkyl rings, or wherein heteroaryl rings are fused with cycloalkyl rings. Examples of heteroaryl groups include pyrrolyl, pyrrolinyl, imidazolyl, pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, pyranyl, furyl, thienyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, isothiazolyl, indolyl, isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, quinoxalinyl, quinazolinyl, indazolyl, benzotriazolyl, benzodioxolyl, benzopyranyl, benzoxazolyl, benzoxadiazolyl, benzothiazolyl, benzothiadiazolyl, benzofuryl, benzothienyl, chromonyl, coumarinyl, benzopyranyl, tetrahydroquinolinyl, tetrazolopyridazinyl,
tetrahydroisoquinolinyl, thienopyridinyl, furopyridinyl, pyrrolopyridinyl and the like. Exemplary tricyclic heterocyclic groups include carbazolyl, benzidolyl, phenanthrolinyl, dibenzofuranyl, acridinyl, phenanthridinyl, xanthenyl and the like. [0116] The terms "heterocycloalkyl" and, interchangeably, "heterocycle," as used herein, alone or in combination, each refer to a saturated, partially unsaturated, or fully unsaturated monocyclic, bicyclic, or tricyclic heterocyclic group containing at least one heteroatom as a ring member, wherein each said heteroatom may be independently selected from the group consisting of nitrogen, oxygen, and sulfur In certain embodiments, said hetercycloalkyl will comprise from 1 to 4 heteroatoms as ring members. In further embodiments, said hetercycloalkyl will comprise from 1 to 2 heteroatoms as ring members. In certain embodiments, said hetercycloalkyl will comprise from 3 to 8 ring members in each ring. In further embodiments, said hetercycloalkyl will comprise from 3 to 7 ring members in each ring. In yet further embodiments, said hetercycloalkyl will comprise from 5 to 6 ring members in each ring. "Heterocycloalkyl" and "heterocycle" are intended to include sulfones, sulfoxides, N-oxides of tertiary nitrogen ring members, and carbocyclic fused and benzo fused ring systems; additionally, both terms also include systems where a heterocycle ring is fused to an aryl group, as defined herein, or an additional heterocycle group. Examples of heterocycle groups include aziridinyl, azetidinyl, 1,3- benzodioxolyl, dihydroisoindolyl, dihydroisoquinolinyl, dihydrocinnolinyl, dihydrobenzodioxinyl, dihydro[ 1 ,3]oxazolo[4,5-b]pyridinyl, benzothiazolyl, dihydroindolyl, dihy-dropyridinyl, 1,3-dioxanyl, 1 ,4-dioxanyl, 1,3-dioxolanyl, isoindolinyl, morpholinyl, piperazinyl, pyrrolidinyl, tetrahydropyridinyl, piperidinyl, thiomorpholinyl, and the like. The heterocycle groups may be optionally substituted unless specifically prohibited.
[0117] The term "hydrazinyl" as used herein, alone or in combination, refers to two amino groups joined by a single bond, i.e., -N-N-.
[0118] The term "hydroxy," as used herein, alone or in combination, refers to -OH. [0119] The term "hydroxyalkyl," as used herein, alone or in combination, refers to a hydroxy group attached to the parent molecular moiety through an alkyl group. [0120] The term "imine," as used herein, alone or in combination, refers to C=NH
[0121] The term "imino," as used herein, alone or in combination, refers to =N-.
[0122] The term "iminohydroxy," as used herein, alone or in combination, refers to
=N(OH) and =N-O-.
[0123] The phrase "in the main chain" refers to the longest contiguous or adjacent chain of carbon atoms starting at the point of attachment of a group to the compounds of any one of the formulas disclosed herein.
[0124] The term "isocyanato" refers to a -NCO group.
[0125] The term "isothiocyanato" refers to a -NCS group.
[0126] The phrase "linear chain of atoms" refers to the longest straight chain of atoms independently selected from carbon, nitrogen, oxygen and sulfur.
[0127] The term "lower," as used herein, alone or in a combination, where not otherwise specifically defined, means containing from 1 to and including 6 carbon atoms.
[0128] The term "lower aryl," as used herein, alone or in combination, means phenyl or naphthyl, which may be optionally substituted as provided.
[0129] The term "lower heteroaryl," as used herein, alone or in combination, means either 1) monocyclic heteroaryl comprising five or six ring members, of which between one and four said members may be heteroatoms selected from the group consisting of
O, S, and N, or 2) bicyclic heteroaryl, wherein each of the fused rings comprises five or six ring members, comprising between them one to four heteroatoms selected from the group consisting of O, S, and N.
[0130] The term "lower cycloalkyl," as used herein, alone or in combination, means a monocyclic cycloalkyl having between three and six ring members. Lower cycloalkyls may be unsaturated. Examples of lower cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
[0131] The term "lower heterocycloalkyl," as used herein, alone or in combination, means a monocyclic heterocycloalkyl having between three and six ring members, of which between one and four may be heteroatoms selected from the group consisting of
O, S, and N. Examples of lower heterocycloalkyls include pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, and morpholinyl. Lower heterocycloalkyls may be unsaturated.
[0132] The term "lower amino," as used herein, alone or in combination, refers to
— NRR , wherein R and R are independently selected from the group consisting of hydrogen, lower alkyl, and lower heteroalkyl, any of which may be optionally substituted. Additionally, the R and R' of a lower amino group may combine to form a five- or six-membered heterocycloalkyl, either of which may be optionally substituted.
[0133] The term "mercaptyl" as used herein, alone or in combination, refers to an
RS- group, where R is as defined herein.
[0134] The term "nitro," as used herein, alone or in combination, refers to -NO2.
[0135] The terms "oxy" or "oxa," as used herein, alone or in combination, refer to
-O-.
[0136] The term "oxo," as used herein, alone or in combination, refers to =0.
[0137] The term "perhaloalkoxy" refers to an alkoxy group where all of the hydrogen atoms are replaced by halogen atoms.
[0138] The term "perhaloalkyl" as used herein, alone or in combination, refers to an alkyl group where all of the hydrogen atoms are replaced by halogen atoms.
[0139] The terms "sulfonate," "sulfonic acid," and "sulfonic," as used herein, alone or in combination, refer the -SO3H group and its anion as the sulfonic acid is used in salt formation.
[0140] The term "sulfanyl," as used herein, alone or in combination, refers to -S-.
[0141] The term "sulfmyl," as used herein, alone or in combination, refers to
-S(O)-.
[0142] The term "sulfonyl," as used herein, alone or in combination, refers to -
S(O)2-.
[0143] The term "N-sulfonamido" refers to a RS(=O)2NR'- group with R and R' as defined herein.
[0144] The term "S-sulfonamido" refers to a -S(=O)2NRR' , group, with R and R' as defined herein.
[0145] The terms "thia" and "thio," as used herein, alone or in combination, refer to a -S- group or an ether wherein the oxygen is replaced with sulfur. The oxidized derivatives of the thio group, namely sulfmyl and sulfonyl, are included in the definition of thia and thio.
[0146] The term "thiol," as used herein, alone or in combination, refers to an -SH group.
[0147] The term "thiocarbonyl," as used herein, when alone includes thioformyl -
C(S)H and in combination is a -C(S)- group.
[0148] The term "N-thiocarbamyl" refers to an ROC(S)NR'- group, with R and
R' as defined herein.
[0149] The term "O-thiocarbamyl" refers to a -OC(S)NRR' , group with R and
R' as defined herein.
[0150] The term "thiocyanato" refers to a -CNS group.
[0151] The term "trihalomethanesulfonamido" refers to a X3CS(O)2NR- group with X is a halogen and R as defined herein.
[0152] The term "trihalomethanesulfonyl" refers to a X3CS(O)2- group where X is a halogen.
[0153] The term "trihalomethoxy" refers to a X3CO- group where X is a halogen.
[0154] The term "trisubstituted silyl," as used herein, alone or in combination, refers to a silicone group substituted at its three free valences with groups as listed herein under the definition of substituted amino. Examples include trimethysilyl, tert- butyldimethylsilyl, triphenylsilyl and the like.
[0155] Any definition herein may be used in combination with any other definition to describe a composite structural group. By convention, the trailing element of any such definition is that which attaches to the parent moiety. For example, the composite group alkylamido would represent an alkyl group attached to the parent molecule through an amido group, and the term alkoxyalkyl would represent an alkoxy group attached to the parent molecule through an alkyl group.
[0156] When a group is defined to be "null," what is meant is that said group is absent.
[0157] The term "optionally substituted" means the anteceding group may be substituted or unsubstituted. When substituted, the substituents of an "optionally substituted" group may include, without limitation, one or more substituents independently selected from the following groups or a particular designated set of groups, alone or in combination: lower alkyl, lower alkenyl, lower alkynyl, lower
alkanoyl, lower heteroalkyl, lower heterocycloalkyl, lower haloalkyl, lower haloalkenyl, lower haloalkynyl, lower perhaloalkyl, lower perhaloalkoxy, lower cycloalkyl, phenyl, aryl, aryloxy, lower alkoxy, lower haloalkoxy, oxo, lower acyloxy, carbonyl, carboxyl, lower alkylcarbonyl, lower carboxyester, lower carboxamido, cyano, hydrogen, halogen, hydroxy, amino, lower alkylamino, arylamino, amido, nitro, thiol, lower alkylthio, lower haloalkylthio, lower perhaloalkylthio, arylthio, sulfonate, sulfonic acid, trisubstituted silyl, N3, SH, SCH3, C(O)CH3, CO2CH3, CO2H, pyridinyl, thiophene, furanyl, lower carbamate, and lower urea. Two substituents may be joined together to form a fused five-, six-, or seven-membered carbocyclic or heterocyclic ring consisting of zero to three heteroatoms, for example forming methylenedioxy or ethylenedioxy. An optionally substituted group may be unsubstituted (e.g., -CH2CH3), fully substituted (e.g., -CF2CF3), monosubstituted (e.g., -CH2CH2F) or substituted at a level anywhere in-between fully substituted and monosubstituted (e.g., -CH2CF3). Where substituents are recited without qualification as to substitution, both substituted and unsubstituted forms are encompassed. Where a substituent is qualified as "substituted," the substituted form is specifically intended. Additionally, different sets of optional substituents to a particular moiety may be defined as needed; in these cases, the optional substitution will be as defined, often immediately following the phrase, "optionally substituted with."
[0158] The term R or the term R' , appearing by itself and without a number designation, unless otherwise defined, refers to a moiety selected from the group consisting of hydrogen, alkyl, cycloalkyl, heteroalkyl, aryl, heteroaryl and heterocycloalkyl, any of which may be optionally substituted. Such R and R' groups should be understood to be optionally substituted as defined herein. Whether an R group has a number designation or not, every R group, including R, R' and Rn where n = (1, 2, 3, ...n), every substituent, and every term should be understood to be independent of every other in terms of selection from a group. Should any variable, substituent, or term (e.g. aryl, heterocycle, R, etc.) occur more than one time in a formula or generic structure, its definition at each occurrence is independent of the definition at every other occurrence. Those of skill in the art will further recognize that certain groups may be attached to a parent molecule or may occupy a position in a
chain of elements from either end as written. Thus, by way of example only, an unsymmetrical group such as -C(O)N(R)- may be attached to the parent moiety at either the carbon or the nitrogen.
[0159] Asymmetric centers exist in the compounds disclosed herein. These centers are designated by the symbols "R" or "S," depending on the configuration of substituents around the chiral carbon atom. It should be understood that the invention encompasses all stereochemical isomeric forms, including diastereomeric, enantiomeric, and epimeric forms, as well as d-isomers and 1-isomers, and mixtures thereof. Individual stereoisomers of compounds can be prepared synthetically from commercially available starting materials which contain chiral centers or by preparation of mixtures of enantiomeric products followed by separation such as conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques, direct separation of enantiomers on chiral chromatographic columns, or any other appropriate method known in the art. Starting compounds of particular stereochemistry are either commercially available or can be made and resolved by techniques known in the art. Additionally, the compounds disclosed herein may exist as geometric isomers. The present invention includes all cis, trans, syn, anti, entgegen (E), and zusammen (Z) isomers as well as the appropriate mixtures thereof. Additionally, compounds may exist as tautomers; all tautomeric isomers are provided by this invention. Additionally, the compounds disclosed herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. In general, the solvated forms are considered equivalent to the unsolvated forms.
[0160] The term "bond" refers to a covalent linkage between two atoms, or two moieties when the atoms joined by the bond are considered to be part of larger substructure. A bond may be single, double, or triple unless otherwise specified. A dashed line between two atoms in a drawing of a molecule indicates that an additional bond may be present or absent at that position.
[0161] The term "disease" as used herein is intended to be generally synonymous, and is used interchangeably with, the terms "disorder" and "condition" (as in medical condition), in that all reflect an abnormal condition of the human or animal body or of
one of its parts that impairs normal functioning, is typically manifested by distinguishing signs and symptoms, and causes the human or animal to have a reduced duration or quality of life.
[0162] The term "combination therapy" means the administration of two or more therapeutic agents to treat a therapeutic condition or disorder described in the present disclosure. Such administration encompasses co-administration of these therapeutic agents in a substantially simultaneous manner, such as in a single capsule having a fixed ratio of active ingredients or in multiple, separate capsules for each active ingredient. In addition, such administration also encompasses use of each type of therapeutic agent in a sequential manner. In either case, the treatment regimen will provide beneficial effects of the drug combination in treating the conditions or disorders described herein.
[0163] "GPRl 19 modulator" is used herein to refer to a compound that exhibits an EC50 with respect to GPRl 19 activity of no more than about 100 μM and more typically not more than about 50 μM, as measured in the cAMP production assay and glucagon- like peptide- 1 (GLP-I) secretion assays described generally hereinbelow. "EC50" is that concentration of inhibitor which activates the activity of an enzyme (e.g., GPRl 19) to half-maximal level. Certain compounds disclosed herein have been discovered to exhibit modulatory activity against GPRl 19. In certain embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of no more than about 10 μM; in further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of no more than about 5 μM; in yet further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of not more than about 1 μM; in yet further embodiments, compounds will exhibit an EC50 with respect to GPRl 19 of not more than about 200 nM, as measured in the GPRl 19 assay described herein.
[0164] The phrase "therapeutically effective" is intended to qualify the amount of active ingredients used in the treatment of a disease or disorder. This amount will achieve the goal of reducing or eliminating the said disease or disorder. [0165] The term "therapeutically acceptable" refers to those compounds (or salts, prodrugs, tautomers, zwitterionic forms, etc.) which are suitable for use in contact with the tissues of patients without undue toxicity, irritation, and allergic response, are
commensurate with a reasonable benefit/risk ratio, and are effective for their intended use.
[0166] As used herein, reference to "treatment" of a patient is intended to include prophylaxis. The term "patient" means all mammals including humans. Examples of patients include humans, cows, dogs, cats, goats, sheep, pigs, and rabbits. Preferably, the patient is a human.
[0167] The term "prodrug" refers to a compound that is made more active in vivo. Certain compounds disclosed herein may also exist as prodrugs, as described in Hydrolysis in Drug and Prodrug Metabolism : Chemistry, Biochemistry, and Enzymology (Testa, Bernard and Mayer, Joachim M. Wiley- VHC A, Zurich, Switzerland 2003). Prodrugs of the compounds described herein are structurally modified forms of the compound that readily undergo chemical changes under physiological conditions to provide the compound. Additionally, prodrugs can be converted to the compound by chemical or biochemical methods in an ex vivo environment. For example, prodrugs can be slowly converted to a compound when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent. Prodrugs are often useful because, in some situations, they may be easier to administer than the compound, or parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug. A wide variety of prodrug derivatives are known in the art, such as those that rely on hydrolytic cleavage or oxidative activation of the prodrug. An example, without limitation, of a prodrug would be a compound which is administered as an ester (the "prodrug"), but then is metabolically hydrolyzed to the carboxylic acid, the active entity. Additional examples include peptidyl derivatives of a compound.
[0168] The compounds disclosed herein can exist as therapeutically acceptable salts. The present invention includes compounds listed above in the form of salts, including acid addition salts. Suitable salts include those formed with both organic and inorganic acids. Such acid addition salts will normally be pharmaceutically acceptable. However, salts of non-pharmaceutically acceptable salts may be of utility in the preparation and purification of the compound in question. Basic addition salts may
also be formed and be pharmaceutically acceptable. For a more complete discussion of the preparation and selection of salts, refer to Pharmaceutical Salts: Properties, Selection, and Use (Stahl, P. Heinrich. Wiley- VCHA, Zurich, Switzerland, 2002). [0169] The term "therapeutically acceptable salt," as used herein, represents salts or zwitterionic forms of the compounds disclosed herein which are water or oil-soluble or dispersible and therapeutically acceptable as defined herein. The salts can be prepared during the final isolation and purification of the compounds or separately by reacting the appropriate compound in the form of the free base with a suitable acid. Representative acid addition salts include acetate, adipate, alginate, L-ascorbate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, butyrate, camphorate, camphorsulfonate, citrate, digluconate, formate, fumarate, gentisate, glutarate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hippurate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethansulfonate (isethionate), lactate, maleate, malonate, DL-mandelate, mesitylenesulfonate, methanesulfonate, naphthylenesulfonate, nicotinate, 2-naphthalenesulfonate, oxalate, pamoate, pectinate, persulfate, 3-phenylproprionate, phosphonate, picrate, pivalate, propionate, pyroglutamate, succinate, sulfonate, tartrate, L-tartrate, trichloroacetate, trifluoroacetate, phosphate, glutamate, bicarbonate, para-toluenesulfonate (p-tosylate), and undecanoate. Also, basic groups in the compounds disclosed herein can be quaternized with methyl, ethyl, propyl, and butyl chlorides, bromides, and iodides; dimethyl, diethyl, dibutyl, and diamyl sulfates; decyl, lauryl, myristyl, and steryl chlorides, bromides, and iodides; and benzyl and phenethyl bromides. Examples of acids which can be employed to form therapeutically acceptable addition salts include inorganic acids such as hydrochloric, hydrobromic, sulfuric, and phosphoric, and organic acids such as oxalic, maleic, succinic, and citric. Salts can also be formed by coordination of the compounds with an alkali metal or alkaline earth ion. Hence, the present invention contemplates sodium, potassium, magnesium, and calcium salts of the compounds disclosed herein, and the like.
[0170] Basic addition salts can be prepared during the final isolation and purification of the compounds by reacting a carboxy group with a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation or with ammonia or an
organic primary, secondary, or tertiary amine. The cations of therapeutically acceptable salts include lithium, sodium, potassium, calcium, magnesium, and aluminum, as well as nontoxic quaternary amine cations such as ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, diethylamine, ethylamine, tributylamine, pyridine, N,N-dimethylaniline, N- methylpiperidine, N-methylmorpholine, dicyclohexylamine, procaine, dibenzylamine, NN-dibenzylphenethylamine, 1-ephenamine, and NN-dibenzylethylenediamine. Other representative organic amines useful for the formation of base addition salts include ethylenediamine, ethanolamine, diethanolamine, piperidine, and piperazine. [0171] A salt of a compound can be made by reacting the appropriate compound in the form of the free base with the appropriate acid.
[0172] While it may be possible for the compounds of the subject invention to be administered as the raw chemical, it is also possible to present them as a pharmaceutical formulation. Accordingly, provided herein are pharmaceutical formulations which comprise one or more of certain compounds disclosed herein, or one or more pharmaceutically acceptable salts, esters, prodrugs, amides, or solvates thereof, together with one or more pharmaceutically acceptable carriers thereof and optionally one or more other therapeutic ingredients. The carrier(s) must be "acceptable" in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. Proper formulation is dependent upon the route of administration chosen. Any of the well-known techniques, carriers, and excipients may be used as suitable and as understood in the art; e.g., in Remington's Pharmaceutical Sciences. The pharmaceutical compositions disclosed herein may be manufactured in any manner known in the art, e.g. , by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes. [0173] The formulations include those suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous, intraarticular, and intramedullary), intraperitoneal, transmucosal, transdermal, rectal and topical (including dermal, buccal, sublingual and intraocular) administration although the most suitable route may depend upon for example the condition and disorder of the
recipient. The formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. Typically, these methods include the step of bringing into association a compound of the subject invention or a pharmaceutically acceptable salt, ester, amide, prodrug or solvate thereof ("active ingredient") with the carrier which constitutes one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
[0174] Formulations of the compounds disclosed herein suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in- water liquid emulsion or a water-in-oil liquid emulsion. The active ingredient may also be presented as a bolus, electuary or paste. [0175] Pharmaceutical preparations which can be used orally include tablets, push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. Tablets may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free- flowing form such as a powder or granules, optionally mixed with binders, inert diluents, or lubricating, surface active or dispersing agents. Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein. All formulations for oral administration should be in dosages suitable for such administration. The push- fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may
be added. Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses. [0176] The compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents. The formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in powder form or in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or sterile pyrogen- free water, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
[0177] Formulations for parenteral administration include aqueous and nonaqueous (oily) sterile injection solutions of the active compounds which may contain antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions. [0178] In addition to the formulations described previously, the compounds may also be formulated as a depot preparation. Such long acting formulations may be
administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
[0179] For buccal or sublingual administration, the compositions may take the form of tablets, lozenges, pastilles, or gels formulated in conventional manner. Such compositions may comprise the active ingredient in a flavored basis such as sucrose and acacia or tragacanth.
[0180] The compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter, polyethylene glycol, or other glycerides.
[0181] Certain compounds disclosed herein may be administered topically, that is by non-systemic administration. This includes the application of a compound disclosed herein externally to the epidermis or the buccal cavity and the instillation of such a compound into the ear, eye and nose, such that the compound does not significantly enter the blood stream. In contrast, systemic administration refers to oral, intravenous, intraperitoneal and intramuscular administration.
[0182] Formulations suitable for topical administration include liquid or semi- liquid preparations suitable for penetration through the skin to the site of inflammation such as gels, liniments, lotions, creams, ointments or pastes, and drops suitable for administration to the eye, ear or nose. The active ingredient for topical administration may comprise, for example, from 0.001% to 10% w/w (by weight) of the formulation. In certain embodiments, the active ingredient may comprise as much as 10% w/w. In other embodiments, it may comprise less than 5% w/w. In certain embodiments, the active ingredient may comprise from 2% w/w to 5% w/w. In other embodiments, it may comprise from 0.1% to 1% w/w of the formulation.
[0183] Gels for topical or transdermal administration may comprise, generally, a mixture of volatile solvents, nonvolatile solvents, and water. In certain embodiments, the volatile solvent component of the buffered solvent system may include lower (Cl- C6) alkyl alcohols, lower alkyl glycols and lower glycol polymers. In further
embodiments, the volatile solvent is ethanol. The volatile solvent component is thought to act as a penetration enhancer, while also producing a cooling effect on the skin as it evaporates. The nonvolatile solvent portion of the buffered solvent system is selected from lower alkylene glycols and lower glycol polymers. In certain embodiments, propylene glycol is used. The nonvolatile solvent slows the evaporation of the volatile solvent and reduces the vapor pressure of the buffered solvent system. The amount of this nonvolatile solvent component, as with the volatile solvent, is determined by the pharmaceutical compound or drug being used. When too little of the nonvolatile solvent is in the system, the pharmaceutical compound may crystallize due to evaporation of volatile solvent, while an excess may result in a lack of bioavailability due to poor release of drug from solvent mixture. The buffer component of the buffered solvent system may be selected from any buffer commonly used in the art; in certain embodiments, water is used. A common ratio of ingredients is about 20% of the nonvolatile solvent, about 40% of the volatile solvent, and about 40% water. There are several optional ingredients which can be added to the topical composition. These include, but are not limited to, chelators and gelling agents. Appropriate gelling agents can include, but are not limited to, semisynthetic cellulose derivatives (such as hydroxypropylmethylcellulose) and synthetic polymers, and cosmetic agents.
[0184] Lotions include those suitable for application to the skin or eye. An eye lotion may comprise a sterile aqueous solution optionally containing a bactericide and may be prepared by methods similar to those for the preparation of drops. Lotions or liniments for application to the skin may also include an agent to hasten drying and to cool the skin, such as an alcohol or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil or arachis oil.
[0185] Creams, ointments or pastes are semi-solid formulations of the active ingredient for external application. They may be made by mixing the active ingredient in finely-divided or powdered form, alone or in solution or suspension in an aqueous or non-aqueous fluid, with the aid of suitable machinery, with a greasy or non-greasy base. The base may comprise hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin such as almond,
corn, arachis, castor or olive oil; wool fat or its derivatives or a fatty acid such as steric or oleic acid together with an alcohol such as propylene glycol or a macrogel. The formulation may incorporate any suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as a sorbitan ester or a polyoxyethylene derivative thereof. Suspending agents such as natural gums, cellulose derivatives or inorganic materials such as silicaceous silicas, and other ingredients such as lanolin, may also be included.
[0186] Drops may comprise sterile aqueous or oily solutions or suspensions and may be prepared by dissolving the active ingredient in a suitable aqueous solution of a bactericidal and/or fungicidal agent and/or any other suitable preservative, and, in certain embodiments, including a surface active agent. The resulting solution may then be clarified by filtration, transferred to a suitable container which is then sealed and sterilized by autoclaving or maintaining at 98-1000C for half an hour. Alternatively, the solution may be sterilized by filtration and transferred to the container by an aseptic technique. Examples of bactericidal and fungicidal agents suitable for inclusion in the drops are phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01%) and chlorhexidine acetate (0.01%). Suitable solvents for the preparation of an oily solution include glycerol, diluted alcohol and propylene glycol. [0187] Formulations for topical administration in the mouth, for example buccally or sublingually, include lozenges comprising the active ingredient in a flavored basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerin or sucrose and acacia.
[0188] For administration by inhalation, compounds may be conveniently delivered from an insufflator, nebulizer pressurized packs or other convenient means of delivering an aerosol spray. Pressurized packs may comprise a suitable propellant such as dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. Alternatively, for administration by inhalation or insufflation, the compounds according to the invention may take the form of a dry powder composition, for example a powder mix of the compound and a suitable powder base such as lactose or starch. The powder
composition may be presented in unit dosage form, in for example, capsules, cartridges, gelatin or blister packs from which the powder may be administered with the aid of an inhalator or insufflator.
[0189] Preferred unit dosage formulations are those containing an effective dose, as herein below recited, or an appropriate fraction thereof, of the active ingredient. [0190] It should be understood that in addition to the ingredients particularly mentioned above, the formulations described above may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavoring agents. [0191] Compounds may be administered orally or via injection at a dose of from 0.1 to 500 mg/kg per day. The dose range for adult humans is generally from 5 mg to 2 g/day. Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of one or more compounds which is effective at such dosage or as a multiple of the same, for instance, units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
[0192] The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
[0193] The compounds can be administered in various modes, e.g. orally, topically, or by injection. The precise amount of compound administered to a patient will be the responsibility of the attendant physician. The specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diets, time of administration, route of administration, rate of excretion, drug combination, the precise disorder being treated, and the severity of the indication or condition being treated. Also, the route of administration may vary depending on the condition and its severity. [0194] In certain instances, it may be appropriate to administer at least one of the compounds described herein (or a pharmaceutically acceptable salt, ester, or prodrug thereof) in combination with another therapeutic agent. By way of example only, if one of the side effects experienced by a patient upon receiving one of the compounds herein is hypertension, then it may be appropriate to administer an anti-hypertensive
agent in combination with the initial therapeutic agent. Or, by way of example only, the therapeutic effectiveness of one of the compounds described herein may be enhanced by administration of an adjuvant (i.e., by itself the adjuvant may only have minimal therapeutic benefit, but in combination with another therapeutic agent, the overall therapeutic benefit to the patient is enhanced). Or, by way of example only, the benefit of experienced by a patient may be increased by administering one of the compounds described herein with another therapeutic agent (which also includes a therapeutic regimen) that also has therapeutic benefit. By way of example only, in a treatment for diabetes involving administration of one of the compounds described herein, increased therapeutic benefit may result by also providing the patient with another therapeutic agent for diabetes. In any case, regardless of the disease, disorder or condition being treated, the overall benefit experienced by the patient may simply be additive of the two therapeutic agents or the patient may experience a synergistic benefit.
[0195] Specific, non- limiting examples of possible combination therapies include use of certain compounds disclosed herein with agents found in the following pharmacotherapeutic classifications as indicated below. These lists should not be construed to be closed, but should instead serve as illustrative examples common to the relevant therapeutic area at present. Moreover, combination regimens may include a variety of routes of administration and should include oral, intravenous, intraocular, subcutaneous, dermal, and inhaled topical.
[0196] For the treatment of metabolic disorders, compounds disclosed herein may be administered with an agent selected from the group comprising: insulin, insulin derivatives and mimetics, insulin secretagogues, insulin sensitizers, biguanide agents, alpha-glucosidase inhibitors, insulinotropic sulfonylurea receptor ligands, meglitinides, protein tyrosine phosphatase-lB (PTP-IB) inhibitors, GSK3 (glycogen synthase kinase- 3) inhibitors , GLP-I (glucagon like peptide- 1), GLP-I analogs, DPP-IV (dipeptidyl peptidase IV) inhibitors, RXR ligands, sodium-dependent glucose co-transporter (SGLT2) inhibitors, glycogen phosphorylase A inhibitors, an AGE breaker, PPAR modulators, non-glitazone type PPARδ agonist, HMG-CoA reductase inhibitors, cholesterol-lowering drugs and anti-obesity agents.
[0197] For the treatment of metabolic disorders, compounds disclosed herein may be administered with an agent selected from the group comprising: insulin, metformin, Glipizide, glyburide, Amaryl, gliclazide, meglitinides, nateglinide, repaglinide, amylin mimetics (for example, pramlintide), PTP-112, SB-517955, SB-4195052, SB-216763, NN-57-05441, NN-57-05445, GW-0791, AGN-194204, T-1095, BAY R3401, acarbose, miglitol, voglibose, Exendin-4, DPP728, LAF237, vildagliptin , BMS477118, PT-100, GSK-823093, PSN-9301, T-6666, SYR-322, SYR-619, Liraglutide, CJC-1134-PC, naliglutide, MK-0431, saxagliptin, GSK23A, pioglitazone, rosiglitazone, AVE2268, GW869682, GSK189075, GPRl 19 agonists including, but not limited to APD668, PSN-119-1 and PSN-821, HMG-CoA reductase inhibitors (for example, rosuvastatin, atorvastatin, simvastatin, lovastatin, pravastatin, fluvastatin, cerivastatin, rosuvastatin, pitavastatin and like), cholesterol-lowering drugs (for example, fibrates which include: fenofibrate, benzafibrate, clofϊbrate, gemfibrozil and like; cholesterol absorption inhibitors such as Ezetimibe, eflucimibe and like compounds), cholesterol ester transfer protein inhibitors (for example, CP-529414, CETi-I, JTT-705 and like compounds), bile acid sequestrants (for example, cholestyramine, colestipol, and like compounds), niacin, microsomal triglyceride transfer protein inhibitors (for example, implitapide), insulin signaling pathway modulators, like inhibitors of protein tyrosine phosphatases (PTPases) and inhibitors of glutamine-fructose-6-phosphate amidotransferase (GFAT), inhibitors of glucose-6-phosphatase (G6 Pase), inhibitors of fructose- 1,6- bisphosphatase (F-l,6-BPase), inhibitors of glycogen phosphorylase, glucagon receptor antagonists, inhibitors of phosphoenolpyruvate carboxylase (PEPCK), inhibitors of pyruvate dehydrogenase kinase, activators AMP-activated protein kinase (AMPK), (R)- 1 - {4- [5 -methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy] - benzenesulfonyl}2,3-dihydro-lH-indole-2-carboxylic acid described in the patent application WO 03/043985, as compound 19 of Example 4, and GI-262570. [0198] For the treatment of obesity, compounds disclosed herein may be administered with an agent selected from the group comprising: cholescystokinin-A (CCK-A) agonists, serotonin and norepinephrine reuptake inhibitors (for example sibutramine), dopamine agonists (for example, bromocriptine and like) sympathomimetic agents, β3 adrenergic receptor agonists, leptin, leptin analogues,
leptin receptor agonists, galanin antagonists, lipase inhibitors (for example Orlistat), Neuropeptide-Y antagonists, glucocorticoid receptor agonists or antagonists, cannabinoid 1 receptor antagonists (for example, rimonabant and like), ciliary neurotropic factors (CNTF, for example Axokine), human agouti-related proteins (AGRP), ghrelin receptor antagonists, histamine 3 receptor antagonists, appetite suppressants (for example, bupropion), urocortin binding protin antagonists, orexin receptor antagonists, and bombesin agonists.
[0199] For the treatment of inflammatory diseases , compounds disclosed herein may be administered with an agent selected from the group comprising: corticosteroids, non-steroidal anti-inflammatories, muscle relaxants and combinations thereof with other agents, anaesthetics and combinations thereof with other agents, expectorants and combinations thereof with other agents, antidepressants, anticonvulsants and combinations thereof; antihypertensives, opioids, topical cannabinoids, and other agents, such as capsaicin.
[0200] For the treatment of inflammatory diseases, compounds disclosed herein may be administered with an agent selected from the group comprising: betamethasone dipropionate (augmented and nonaugemnted), betamethasone valerate, clobetasol propionate, prednisone, methyl prednisolone, diflorasone diacetate, halobetasol propionate, amcinonide, dexamethasone, dexosimethasone, fluocinolone acetononide, fluocinonide, halocinonide, clocortalone pivalate, dexosimetasone, flurandrenalide, salicylates, ibuprofen, ketoprofen, etodolac, diclofenac, meclofenamate sodium, naproxen, piroxicam, celecoxib, cyclobenzaprine, baclofen, cyclobenzaprine/lidocaine, baclofen/cyclobenzaprine, cyclobenzaprine/lidocaine/ketoprofen, lidocaine, lidocaine/deoxy-D-glucose, prilocaine, EMLA Cream (Eutectic Mixture of Local Anesthetics (lidocaine 2.5% and prilocaine 2.5%), guaifenesin, guaifenesin/ketoprofen/cyclobenzaprine, amitryptiline, doxepin, desipramine, imipramine, amoxapine, clomipramine, nortriptyline, protriptyline, duloxetine, mirtazepine, nisoxetine, maprotiline, reboxetine, fluoxetine, fluvoxamine, carbamazepine, felbamate, lamotrigine, topiramate, tiagabine, oxcarbazepine, carbamezipine, zonisamide, mexiletine, gabapentin/clonidine, gabapentin/carbamazepine, carbamazepine/cyclobenzaprine, antihypertensives
including clonidine, codeine, loperamide, tramadol, morphine, fentanyl, oxycodone, hydrocodone, levorphanol, butorphanol, menthol, oil of wintergreen, camphor, eucalyptus oil, turpentine oil; CB1/CB2 ligands, acetaminophen, infliximab; n) nitric oxide synthase inhibitors, particularly inhibitors of inducible nitric oxide synthase; anti-TNFα agents including, but not limited to etanerecept and infliximab, and other agents, such as capsaicin.
[0201] In any case, the multiple therapeutic agents (at least one of which is a compound disclosed herein) may be administered in any order or even simultaneously. If simultaneously, the multiple therapeutic agents may be provided in a single, unified form, or in multiple forms (by way of example only, either as a single pill or as two separate pills). One of the therapeutic agents may be given in multiple doses, or both may be given as multiple doses. If not simultaneous, the timing between the multiple doses may be any duration of time ranging from a few minutes to four weeks. [0202] Thus, in another aspect, certain embodiments provide methods for treating GPRl 19-mediated disorders in a human or animal subject in need of such treatment comprising administering to said subject an amount of a compound disclosed herein effective to reduce or prevent said disorder in the subject, in combination with at least one additional agent for the treatment of said disorder that is known in the art. In a related aspect, certain embodiments provide therapeutic compositions comprising at least one compound disclosed herein in combination with one or more additional agents for the treatment of GPRl 19-mediated disorders. [0203] Specific diseases to be treated by the compounds, compositions, and methods disclosed herein include: diabetes (type I and type II) and conditions associated with diabetic diseases which include, but are not limited to, hyperglycemia, hyperlipidemia, hyperinsulinemia, insulin resistance, inadequate glucose tolerance, impaired glucose metabolism, diabetic nephropathy, glomerulosclerosis, diabetic neuropathy, erectile dysfunction, macular degeneration, diabetic retinopathy, chronic microvascular complications, peripheral vascular disease, cataracts, stroke, foot ulcerations, renal failure, kidney disease, ketosis, metabolic acidosis, and related disorders, obesity, myocardial infarction, angina pectoris, coronary artery disease, atherosclerosis, cardiac hypertrophy, allergic diseases, fatty liver disease, nonalcoholic
steatohepatitis, liver fibrosis, kidney fibrosis, anorexia nervosa, bulimia vervosa, autoimmune diseases, inflammatory diseases including rheumatoid arthritis, asthma, chronic obstructive pulmonary disease (COPD), psoriasis, ulcerative colitis, proliferative disorders, infectious diseases, angiogenic disorders, reperfusion/ischemia in stroke, vascular hyperplasia, organ hypoxia, cardiac hypertrophy, thrombin-induced platelet aggregation, and conditions associated with prostaglandin endoperoxidase synthetase-2 (COX-2).
[0204] In certain embodiments, the disease is obesity and the effects to be achieved in a human or animal patient include decreasing body weight and controlling weight gain.
[0205] In addition, topical application of GPRl 19 agonists might be useful for the treatment of cellulite and other cosmetic conditions which are characterized by subcutaneous fat accumulation.
[0206] In certain embodiments, the disease is associated with perturbed bile acid metabolism, including, but not limited to gall bladder stones, cholecystitis, cholangitis, choledocholithiasis, jaundice, and obstetric cholestasis and the itch associated with it.
[0207] Metabolic diseases other than Type 1 and Type 2 diabetes which may be treated or prevented include, without limitation, metabolic syndrome and insulin resistance. In addition, the compounds disclosed herein can be used to treat insulin resistance and other metabolic disorders such as atherosclerosis that are typically associated with an exaggerated inflammatory signaling.
[0208] In certain embodiments, the disease is a hyperproliferative condition of the human or animal body, including, but not limited to restenosis, inflammation, immune disorders, cardiac hypertrophy, atherosclerosis, pain, migraine, angiogenesis-related conditions or disorders, proliferation induced after medical conditions, including but not limited to surgery, angioplasty, or other conditions.
[0209] The compounds disclosed herein may be useful as anti-inflammatory agents with the additional benefit of having significantly less harmful side effects. The compositions may be used to treat arthritis, including but not limited to rheumatoid arthritis, spondyloarthropathies, gouty arthritis, osteoarthritis, systemic lupus erythematosus, juvenile arthritis, acute rheumatic arthritis, enteropathic arthritis,
neuropathic arthritis, psoriatic arthritis, and pyogenic arthritis. The compositions may also be used in the treatment of pulmonary inflammation, such as that associated with viral infections and cystic fibrosis. In certain embodiments, the particular inflammatory disease is rheumatoid arthritis.
[0210] Further inflammatory diseases which may be prevented or treated include, without limitation: asthma, allergies, respiratory distress syndrome or acute or chronic pancreatitis. Furthermore, respiratory system diseases may be prevented or treated including but not limited to chronic obstructive pulmonary disease, pulmonary fibrosis, ulcerative colitis, inflammatory bowel disease, Crohn's disease, peptic ulceration, gastritis, psoriasis, and skin inflammation.
[0211] In certain embodiments, the disease to be treated by the methods provided herein may be an ophthalmologic disorder. Ophthalmologic diseases and other diseases in which angiogenesis plays a role in pathogenesis, may be treated or prevented and include, without limitation, dry eye (including Sjogren's syndrome), macular degeneration, closed and wide angle glaucoma, retinal ganglion degeneration, occular ischemia, retinitis, retinopathies, uveitis, ocular photophobia, and of inflammation and pain associated with acute injury to the eye tissue. In certain embodiments, the ophthalmologic disease to be treated is glaucomatous retinopathy and/or diabetic retinopathy. In certain embodiments, the ophthalmologic condition to be treated is post-operative inflammation or pain as from ophthalmic surgery such as cataract surgery and refractive surgery.
[0212] In certain embodiments, the disease to be treated by the methods provided herein may be an autoimmune disease. Autoimmune diseases which may be prevented or treated include, but are not limited to: rheumatoid arthritis, inflammatory bowel disease, inflammatory pain, ulcerative colitis, Crohn's disease, periodontal disease, temporomandibular joint disease, multiple sclerosis, diabetes, glomerulonephritis, systemic lupus erythematosus, scleroderma, chronic thyroiditis, Grave's disease, hemolytic anemia, autoimmune gastritis, autoimmune neutropenia, thrombocytopenia, chronic active hepatitis, myasthenia gravis, atopic dermatitis, graft vs. host disease, and psoriasis. Inflammatory diseases which may be prevented or treated include, but are not limited to: asthma, allergies, respiratory distress syndrome or acute or chronic
pancreatitis. In certain embodiments, the particular autoimmune disease is rheumatoid arthritis.
[0213] The compounds provided herein are also useful in treating tissue damage in such diseases as vascular diseases, migraine headaches, periarteritis nodosa, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, neuromuscular junction disease including myasthenia gravis, white matter disease including multiple sclerosis, sarcoidosis, nephritis, nephrotic syndrome, Behcet's syndrome, polymyositis, gingivitis, periodontis, hypersensitivity, swelling occurring after injury, ischemias including myocardial ischemia, cardiovascular ischemia, and ischemia secondary to cardiac arrest, and the like. These compounds can also be used to treat allergic rhinitis, respiratory distress syndrome, endotoxic shock syndrome, and atherosclerosis. [0214] In certain embodiments, the disease to be treated by the methods of the present invention may be a cardiovascular condition. In certain embodiments, said cardiovascular condition is selected from the group consisting of atherosclerosis, cardiac hypertrophy, idiopathic cardiomyopathies, heart failure, angiogenesis-related conditions or disorders, and proliferation induced after medical conditions, including, but not limited to restenosis resulting from surgery and angioplasty. [0215] In certain embodiments, the disease to be prevented or treated by the methods of the present invention may be autism.
[0216] Glucose -dependent insulinotropic polypeptide (GIP) knock out mice showed a decreased bone size, lower bone mass (1). In contrast, GIP-overexpressing transgenic mice have increased bone mass compared to control mice (2). In addition, recent data suggested that the glucagone-like peptide- 1 (GLP-I) receptor is essential for control of bone resorption as GLP-IR (-/-) mice have cortical osteopenia and bone fragility (3). Since GPRl 19 agonists increase GLP-I and GIP secretion both in vitro and in vivo, GPRl 19 agonists in this patent might be important for the prevention or treatment of bone loss induced by age or diseases in which the normal functions of osteobalsts or osteoclasts are altered. (Xie D et al. Bone. 2005, 37: 759-769; Ding KH et al. J Bone Miner Res. 2008, 23: 536-543; Yamada C et al. Endocrinology. 2008, 149:574-9.) Such diseases include, for example, osteopenia and osteoporosis.
[0217] Besides being useful for human treatment, certain compounds and formulations disclosed herein may also be useful for veterinary treatment of companion animals, exotic animals and farm animals, including mammals, rodents, and the like. More preferred animals include horses, dogs, and cats.
[0218] All references, patents or applications, U.S. or foreign, cited in the application are hereby incorporated by reference as if written herein. Where any inconsistencies arise, material literally disclosed herein controls.
General Synthetic Methods for Preparing Compounds
Scheme 1

Reagents: (a) NaH, DMF, 650C, 18h. (b) AcOH, 12O0C, 18h. (c) NaOH, MeOH, 25- 9O0C, l-3h. (d) DMF-DMA, RT, 18h. (e) AcOH, reflux, 6h. (f) MeOH, reflux,
18h. (g) l-(isocyanomethylsulfonyl) -4-methylbenzene, K2CO3, EtOH, 6O0C, 3h. (h) K2CO3, DMF, HO0C, 18h (i) NaH, diethyl malonate, THF5O0C, 20 min then 650C, 3h. 0') MgC12, NMP, H2O, HO0C, 18h. (k) Trimethylphenylammomium tribromide, THF, RT, 4h. (1) Pyrrolidine-2-carboxylate hydrochloride, K2CO3, DMF, 12O0C, 18h.
Scheme 2

Reagents: (a) 2,2,6,6-tetramethylheptane-3,5-dione, CuCl, Cs2CO3, NMP, 12O0C, 18h. (b) K2CO3, DMF, 8O0C, 18h. (c) BH3THF, reflux, 3h. (d) Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, JV-methyl morpholine),ACN, RT, 18h. (e) (i) Oxalyl Chloride, DCM, DMF (ii) Amino derivatives, TEA, O0C. (f) Trifluoromethyl
2,3 ,4,5,6-pentafluorobenzoate, pyridine, DMF, RT, 2.5h. (g) Ri27-NH-Ri28, DMF, RT, 18h.
Scheme 3

Reagents: (a) K2CO3, DMF, 8O0C, 18h. (b) H2, Pd/C, MeOH, RT, l-2h. (c) (i) HCl, NaNO2, O0C, Ih. (ii) HCl, SnCl2.2H2O, O0C, 30 min. (d) When
 H: Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, N-methyl Morpholine), ACN, RT, 18h. When Ri4i = alkyhtrimethylaluminum, DCM, O0C to RT, Ih. (e) (i) NaH, DMF, O0C, 20 min. (ii) MeI, DMF, O0C, 20 min. (f) Hydrazine hydrate, EtOH, reflux, 3h.
Scheme 4
H: Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, N-methyl Morpholine), ACN, RT, 18h. When Ri4i = alkyhtrimethylaluminum, DCM, O0C to RT, Ih. (e) (i) NaH, DMF, O0C, 20 min. (ii) MeI, DMF, O0C, 20 min. (f) Hydrazine hydrate, EtOH, reflux, 3h.
Scheme 4

Reagents: (a) 2,5-Dimethoxytetrahydroflιran, AcOH/l,4-dioxane, 1000C, 6h. (b) 2-Bromoacetyl bromide, DCM, RT, 18h. (c) Phenols, K2CO3, acetone, RT, 18h. (d) Anilines, K2CO3, acetone, 1000C, 18h.
Scheme 5

Reagents: (a) K2CO3, DMF, 12O0C, 18h. (b) Triacetoxyborohydride, THF, RT, 6h.
Scheme 6
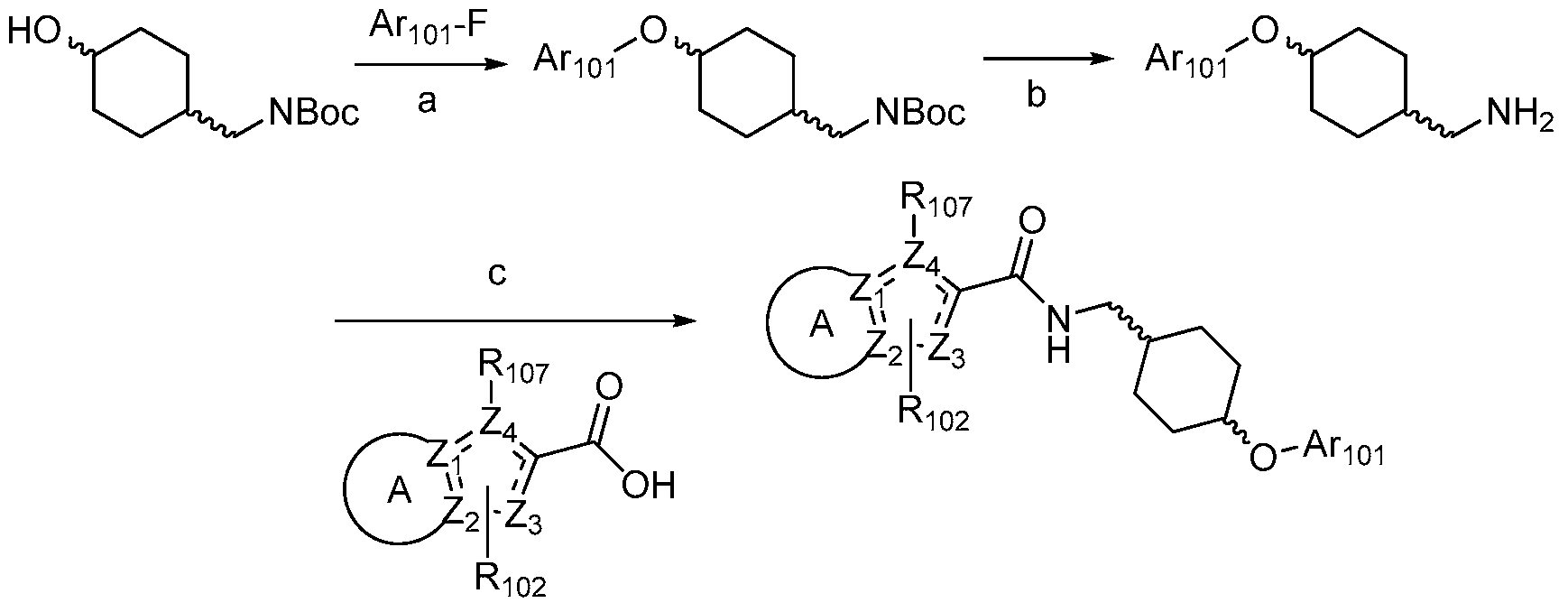
Reagents: (a) NaH, DMA, 7O0C, 1.5 h. (b) TFA, DCM, RT, 30 min. (c) Coupling reagent (i.e. HATU, EDC/HOBt), base (i. e. TEA, N-methyl Morpholine),ACN, RT, 18h.
[0219] The invention is further illustrated by the following Examples 1-640, set forth individually and in the table below, which can be prepared according to the methods of synthesis outlined above and by methods known in the art. All IUPAC nomenclature was generated using CambridgeSoft's ChemDraw 10.0.
Intermediate 1 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid

Step 1 : Methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxylate


A mixture of methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxylate (12.1 g, 39.7 mmol), MeOH (50 mL), and 1.5M NaOH (55 mL) was heated at 85 0C for 1.5 h, allowed to cool to rt, and then concentrated (~20 mL remaining). The residue was cooled to 0 0C, and 1.74N HCl (45 mL) was added with vigorous stirring. When the large clumps became free-flowing, the mixture was filtered, washed with 0 0C water (40 mL x 2), and then dried under vacuum to give 1- (3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.50 (s, IH), 8.96 (d, IH), 8.74 (d, IH), 7.31 (m, IH), 7.01 (m, IH), 6.39 (m, IH); LCMS: 273.3 (M-OH)+.
Intermediate 2 l-(3,5-Dichloropyridin-2-yl)-lH-pyrrole-2-carboxylic acid

The title compound was prepared from methyl lH-pyrrole-2-carboxylate and 3,5- dichloro-2-fluoropyridine following the procedures outlined for Intermediate 1. 1H NMR (400 MHz, DMSO-d6): δ 12.51 (s, IH), 8.58 (d, IH), 8.45 (d, IH), 7.22 (dd, IH), 6.96 (dd, IH), 6.34 (dd, IH); LCMS: 257.1 (M-OH)+.
Intemediate 3 3-chloro-2-hydrazinyl-5-(trifluoromethyl)pyridine

A mixture of 2,3-dichloro-5-(trifluoromethyl)pyridine (10 mL, 72 mmol), hydrazine (10 mL, 0.32 mol), and ethanol (100 mL) was refluxed for 4 h, allowed to cool to rt, and then concentrated. The residue was partitioned between EtOAc (100 mL) and 0.43M NaOH (175 mL). The organic layer was dried, filtered, concentrated, and dried under vacuum to give 3-chloro-2-hydrazinyl-5-(trifluoromethyl)pyridine as an off- white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.53 (s, IH), 8.35 (d, IH), 7.91 (d, IH), 4.43 (s, 2H); LCMS: 212.3 (M+H)+.
Intermediate 4 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5-carboxylic acid


Methyl 4-(dimethylamino)-2-oxobut-3-enoate (1.57 g, 10.0 mmol) was added to a solution of Intermediate 3 (2.12 g, 10.0 mmol) and acetic acid (100 mL). The reaction was refluxed for 6 h, allowed to cool to rt, concentrated, and then purified by silica gel chromatography (l :30-EtOAc/PE) to give methyl l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5-carboxylate as a red oil. Step 2: l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5-carboxylic acid

A mixture of methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5- carboxylate (420 mg, 1.37 mmol), MeOH (20 mL), and 2M NaOH (10 mL) was stirred at rt for 1 h, concentrated, and then adjusted to pH = 3 with HCl. The resulting solids were filtered and dried to give l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrazole-5-carboxylic acid as a light yellow solid. LCMS: 292 (M+H)+.
Intermediate 5 l-(3,5-Dichloropyridin-2-yl)-lH-pyrazole-5-carboxylic acid


The title compound was synthesized as described for Intermediate 3 using 3,5- dichloro-2-fluoropyridine as the starting material. Step 2: l-(3,5-Dichloropyridin-2-yl)-lH-pyrazole-5-carboxylic acid

The title compound was prepared from 3,5-dichloro-2-hydrazinylpyridine and methyl 4-(dimethylamino)-2-oxobut-3-enoate following the procedure outlined for Intermediate 4. 1H NMR (400 MHz, DMSO-d6): δ 13.65 (s, IH), 8.68 (d, IH), 8.58 (d, IH), 7.90 (d, IH), 7.09 (d, IH); LCMS: 258.1 (M+H)+.
Intermediate 6 Ethyl l-(3,5-dichloropyridin-2-yl)-lH-imidazole-2-carboxylate

A mixture of ethyl lH-imidazole-2-carboxylate (1.96 g, 14.0 mmol), 3,5-dichloro-2- fluoropyridine (1.80 g, 10.8 mmol), potassium carbonate (2.24 g, 16.2 mmol), and NMP (54 mL) was heated at 115 0C for 6 h, allowed to cool to rt, and then poured into EtOAc (200 mL). The mixture was washed with water (200 mL), washed with brine (200 mL x 2), dried, filtered, concentrated, and purified by silica gel chromatography (4:1→1 :1; hexanes: EtOAc) to give ethyl l-(3,5-dichloropyridin-2-yl)-lH-imidazole-2- carboxylate. 1H NMR (400 MHz, DMSO-d6): δ 8.68 (d, IH), 8.62 (d, IH), 7.78 (d, IH), 7.36 (d, IH), 4.15 (q, 2H), 1.13 (t, 3H); LCMS: 286.1 (M+H)+.
EXAMPLE 1
Nl,l-bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

Λ/-(3-Dimethylaminopropyl)-Λf-ethylcarbodiimide hydrochloride (0.69 g, 3.6 mmol) was added to a solution of Intermediate 1 (0.87 g, 3.0 mmol), Intermediate 3 (0.95 g, 4.5 mmol), JV-hydroxybenzotriazole hydrate (0.60 g, 3.9 mmol), 4-methylmorpholine (0.85 mL, 7.7 mmol), and CH3CN (6 mL) at rt (Note #1). After 4 h, the reaction was diluted with EtOAc (60 mL) and washed with 10% K2CO3 (40 mL x 2) followed by IN HCl (40 mL x 2). The organic layer was dried, filtered, and concentrated (Note #2) to give N', 1 -bis(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrole-2-carbohydrazide as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, IH), 9.19 (s, IH), 8.88 (d, IH), 8.64 (d, IH), 8.37 (d, IH), 8.07 (d, IH), 7.26 (m, IH), 7.24 (m, IH), 6.41 (m, IH); LCMS: 484.4 (M+H)+.
Note #1 : A variety of reaction conditions have been utilized for this coupling reaction, as well as for other coupling reactions reported herein. For example: HATU & CDI are alternate coupling agents amongst others; TEA & DIEA are alternate bases amongst others; 4-5 equiv of base are used when the starting amine or hydrazine is an HCl salt); DMF & CH2Cl2 are alternate solvents amongst others; and reaction times vary by substrate.
Note #2: In cases where the acid/base workup does not give pure product or was not utilized, compounds were purified by silica gel chromatography or reverse-phase HPLC.
EXAMPLE 2 7V-(3-Chloro-4-(trifluoromethoxy)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-
2-yl)-lH-pyrrole-2-carboxamide


A solution of 3-chloro-4-(trifluoromethoxy)benzonitrile (0.47 g, 2.1 mmol) and IM BHs'THF (10 mL, 10 mmol) was refiuxed for 2 h under N2. MeOH (1.5 mL, 37 mmol) was added slowly (caution: gas evolution) followed by IN HCl (3 mL, 3 mmol). The reaction was refiuxed for an additional 7 h, allowed to cool to rt, poured into 10% K2CO3 (40 mL), and extracted with DCM (40 mL x 2). The organic extracts were dried, filtered, and concentrated to give (3-chloro-4-
(trifluoromethoxy)phenyl)methanamine as a clear oil. LCMS: 226.1 (M+H)+. Step 2: N-(3-Chloro-4-(trifluoromethoxy)benzyl)-l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (3-chloro-4- (trifluoromethoxy)phenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.92 (t, IH), 8.89 (m, IH), 8.66 (d, IH), 7.55-7.51 (m, 2H), 7.34 (dd, IH), 7.22 (dd, IH), 7.08 (dd, IH), 6.38 (dd, IH), 4.34 (d, 2H); LCMS: 498.2 (M+H)+.
EXAMPLE 3 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((3-chloro-5- (trifluoromethyl)pyridin-2-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (3-chloro-5- (trifluoromethyl)pyridin-2-yl)methanamine hydrochloride following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.89 (d, IH), 8.86 (d, IH), 8.82 (t, IH), 8.61 (d, IH), 8.41 (d, IH), 7.18 (dd, IH), 7.12 (dd, IH), 6.36 (dd, IH), 4.08 (d, 2H); LCMS: 483.4 (M+H)+.
EXAMPLE 4 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(trifluoromethyl)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (4-(trifluoromethyl)phenyl) methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 8.92 (t, IH), 8.88 (d, IH), 8.63 (d, IH), 7.66 (d, 2H), 7.44 (d, 2H), 7.20 (dd, IH), 7.08 (dd, IH), 6.36 (dd, IH), 4.38 (d, 2H); LCMS: 448.4 (M+H)+.
EXAMPLE 5 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(pyridin-2-ylmethyl)-lH-pyrrole-
2-carboxamide

The title compound was prepared from Intermediate 1 and pyridin-2-ylmethanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.90 (t, IH), 8.88 (d, IH), 8.63 (d, IH), 8.46 (d, IH), 7.73 (td, IH), 7.23 (m, 2H), 7.19 (m, IH), 7.11 (dd, IH), 6.36 (dd, IH), 4.38 (d, 2H); LCMS: 381.3 (M+H)+.
EXAMPLE 6 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(pyridin-3-ylmethyl)-lH-pyrrole-
2-carboxamide

The title compound was prepared from Intermediate 1 and pyridin-3-ylmethanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.87 (t, IH), 8.64 (d, IH), 8.45 (d, IH), 8.42 (dd, IH), 7.61 (d, IH), 7.32 (dd, IH), 7.18 (dd, IH), 7.05 (dd, IH), 6.35 (dd, IH), 4.32 (d, 2H); LCMS: 381.3 (M+H)+.
EXAMPLE 7 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(pyridin-4-ylmethyl)-lH-pyrrole-
2-carboxamide

The title compound was prepared from Intermediate 1 and pyridin-4-ylmethanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.91 (t, IH), 8.88 (d, IH), 8.63 (d, IH), 8.46 (d, 2H), 7.22 (m, 3H), 7.09 (d, IH), 6.37 (dd, IH), 4.31 (d, 2H); LCMS: 381.3 (M+H)+.
EXAMPLE 8 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-methylbenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and o-tolylmethanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.69 (t, IH), 8.63 (d, IH), 7.17 (m, 2H), 7.12 (m, 3H), 7.09 (dd, IH), 6.35 (dd, IH), 4.27 (d, 2H), 2.22 (s, 3H); LCMS: 394.4 (M+H)+.
EXAMPLE 9 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-methylbenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and m-tolylmethanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.78 (t, IH), 8.64 (d, IH), 7.17 (m, 2H), 7.06 (dd, IH), 7.02 (m, 3H), 6.34 (dd, IH), 4.26 (d, 2H), 2.25 (s, 3H); LCMS: 394.4 (M+H)+.
EXAMPLE 10 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-methylbenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and p-tolylmethanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.76 (t, IH), 8.64 (d, IH), 7.17 (dd, IH), 7.10 (d, 4H), 7.05 (dd, IH), 6.33 (dd, IH), 4.24 (d, 2H), 2.24 (s, 3H); LCMS: 394.4 (M+H)+.
EXAMPLE 11 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-fluorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (2- fluorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.80 (t, IH), 8.64 (d, IH), 7.31-7.24 (m, 2H), 7.18 (dd, IH), 7.17-7.10 (m, 2H), 7.08 (dd, IH), 6.35 (dd, IH), 4.33 (d, 2H); LCMS: 398.3 (M+H)+.
EXAMPLE 12 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-fluorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (3- fluorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.86 (t, IH), 8.64 (d, IH), 7.33 (m, IH), 7.19 (dd, IH), 7.09-7.00 (m, 4H), 6.35 (dd, IH), 4.31 (d, 2H); LCMS: 398.3 (M+H)+.
EXAMPLE 13 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-fluorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (4- fluorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, IH), 8.82 (t, IH), 8.64 (d, IH), 7.25 (m, 2H), 7.18 (dd, IH), 7.11 (m, 2H), 7.05 (dd, IH), 6.34 (dd, IH), 4.27 (d, 2H); LCMS: 398.4 (M+H)+.
EXAMPLE 14 N-(2-Bromobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (2- bromophenyl)methanamine following the procedure outlined in EXAMPLE 1. LCMS: 458.3 (M+H)+.
EXAMPLE 15
N-(3-Bromobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (3- bromophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.86 (t, IH), 8.64 (d, IH), 7.41 (m, 2H), 7.28-7.20 (m, 2H), 7.19 (dd, IH), 7.06 (dd, IH), 6.36 (dd, IH), 4.29 (d, 2H); LCMS: 458.3 (M+H)+.
EXAMPLE 16
N-(4-Bromobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (4- bromophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.84 (t, IH), 8.64 (d, IH), 7.48 (d, 2H), 7.20-7.16 (m, 3H), 7.05 (dd, IH), 6.35 (dd, IH), 4.26 (d, 2H); LCMS: 458.3 (M+H)+.
EXAMPLE 17 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-chlorobenzyl)-lH-pyrrole-2- carboxamide
The title compound was prepared from Intermediate 1 and (2- chlorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.85 (t, IH), 8.63 (d, IH), 7.40 (d, IH), 7.33-7.23 (m, 3H), 7.20 (dd, IH), 7.12 (dd, IH), 6.37 (dd, IH), 4.35 (d, 2H); LCMS: 414.4 (M+H)+.
EXAMPLE 18 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-chlorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (3- chlorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.86 (t, IH), 8.64 (d, IH), 7.32 (dd, IH), 7.29-7.25 (m, 2H), 7.21-7.17 (m, 2H), 7.06 (dd, IH), 6.35 (dd, IH), 4.30 (d, 2H); LCMS: 414.4 (M+H)+.
EXAMPLE 19 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoromethyl)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (2-(trifluoromethyl)phenyl) methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 8.91 (t, IH), 8.88 (m, IH), 8.63 (d, IH), 7.69 (d, IH), 7.65 (t, IH), 7.49- 7.42 (m, 2H), 7.22 (dd, IH), 7.14 (dd, IH), 6.38 (dd, IH), 4.48 (d, 2H); LCMS: 448.4 (M+H)+.
EXAMPLE 20 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-(trifluoromethyl)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (3-(trifluoromethyl)phenyl) methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 8.92 (t, IH), 8.87 (m, IH), 8.62 (d, IH), 7.60-7.52 (m, 4H), 7.20 (dd, IH), 7.06 (dd, IH), 6.36 (dd, IH), 4.39 (d, 2H); LCMS: 448.5 (M+H)+.
EXAMPLE 21 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-methoxybenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (2- methoxyphenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.87 (dd, IH), 8.66 (t, IH), 8.63 (d, IH), 7.22-7.16 (m, 2H), 7.14-7.09 (m, 2H), 6.94 (d, IH), 6.88 (td, IH), 6.35 (dd, IH), 4.25 (d, 2H), 3.76 (s, 3H); LCMS: 410.4 (M+H)+.
EXAMPLE 22 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-methoxybenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (3- methoxyphenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.80 (t, IH), 8.64 (d, IH), 7.22-7.16 (m, 2H), 7.06 (dd, IH), 6.82-6.75 (m, 3H), 6.34 (dd, IH), 4.27 (d, 2H), 3.70 (s, 3H); LCMS: 410.4 (M+H)+.
EXAMPLE 23 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-methoxybenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (4- methoxyphenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.74 (t, IH), 8.64 (d, IH), 7.16 (dd, IH), 7.14 (d, 2H), 7.04 (dd, IH), 6.85 (d, 2H), 6.33 (dd, IH), 4.22 (d, 2H), 3.70 (s, 3H); LCMS: 410.4 (M+H)+.
EXAMPLE 24 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoromethoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (2- (trifluoromethoxy)phenyl) methanamine following the procedure outlined in EXAMPLE 1. LCMS: 464.4 (M+H)+.
EXAMPLE 25 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-(trifluoromethoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (3- (trifluoromethoxy)phenyl) methanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.91 (t, IH), 8.87 (m, IH), 8.63 (d, IH), 7.43 (t, IH), 7.26 (d, IH), 7.24-7.18 (m, 3H), 7.06 (dd, IH), 6.36 (dd, IH), 4.34 (d, 2H); LCMS: 464.4 (M+H)+.
EXAMPLE 26 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(trifluoromethoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (4- (trifluoromethoxy)phenyl) methanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.87 (t, IH), 8.64 (d, IH), 7.34 (d, 2H), 7.29 (d, 2H), 7.19 (m, IH), 7.06 (m, IH), 6.35 (m, IH), 4.32 (d, 2H); LCMS: 464.4 (M+H)+.
EXAMPLE 27 N-(4-tert-Butylbenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (4-tert- butylphenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (m, IH), 8.75 (t, IH), 8.64 (m, IH), 7.30 (d, 2H), 7.17 (dd, IH), 7.14 (d, 2H), 7.05 (m, IH), 6.33 (m, IH), 4.25 (d, 2H), 1.23 (s, 9H); LCMS: 436.5 (M+H)+.
EXAMPLE 28 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3,4-dimethoxybenzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and (3,4- dimethoxyphenyl)methanamine following the procedure outlined in EXAMPLE 1. LCMS: 440.5 (M+H)+.
EXAMPLE 29
N-(Benzo[d] [l,3]dioxol-5-ylmethyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and benzo[d][l,3]dioxol-5- ylmethanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 8.88 (br, IH), 8.75 (br, IH), 8.65 (br, IH), 7.17 (br, IH), 7.04 (br, IH), 6.85-6.65 br m, 3H), 6.34 (br, IH), 5.95 (br, 2H), 4.19 (br, 2H); LCMS: 424.4 (M+H)+.
EXAMPLE 30 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2,4-difluorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (2,4- difluorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (dd, IH), 8.80 (t, IH), 8.64 (dd, IH), 7.31 (m, IH), 7.22-7.14 (m, 2H), 7.08-7.00 (m, 2H), 6.34 (dd, IH), 4.29 (d, 2H); LCMS: 416.4 (M+H)+.
EXAMPLE 31 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3,4-difluorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (3,4- difluorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (br, 2H), 8.64 (br, IH), 7.35 (br, IH), 7.25 (br, IH), 7.19 (br, IH), 7.06 (br, 2H), 6.35 (br, IH), 4.27 (br, 2H); LCMS: 416.4 (M+H)+.
EXAMPLE 32 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2,4-dichlorobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and (2,4- dichlorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.88 (br, 2H), 8.63 (br, IH), 7.58 (br, IH), 7.41 (br, IH), 7.30 (br, IH), 7.21 (br, IH), 7.11 (br, IH), 6.37 (br, IH), 4.31 (br, 2H); LCMS: 448.4 (M+H)+.
EXAMPLE 33 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3,4-dichlorobenzyl)-lH-pyrrole-
2-carboxamide
The title compound was prepared from Intermediate 1 and (3,4- dichlorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.92-8.86 (m, 2H), 8.64 (d, IH), 7.56 (d, IH), 7.46 (d, IH), 7.24-7.18 (m, 2H), 7.06 (dd, IH), 6.36 (dd, IH), 4.29 (d, 2H); LCMS: 448.4 (M+H)+.
EXAMPLE 34 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2,6-dichlorobenzyl)-lH-pyrrole-
2-carboxamide

The title compound was prepared from Intermediate 1 and (2,6- dichlorophenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.90 (t, IH), 8.88 (dd, IH), 8.64 (d, IH), 7.46 (d, IH), 7.35 (dd, IH), 7.28 (d, IH), 7.23 (dd, IH), 7.11 (dd, IH), 6.38 (dd, IH), 4.33 (d, 2H); LCMS: 448.4 (M+H)+.
EXAMPLE 35
N-(2-Aminobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and 2-(aminomethyl)aniline following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.90 (m, IH), 8.87 (t, IH), 8.65 (d, IH), 8.50-6.50 (br, 2H) 7.22 (m, IH), 7.18-7.10 (m, 2H), 7.08 (dd, IH), 6.95-6.86 (m, 2H), 6.37 (dd, IH), 4.25 (d, 2H); LCMS: 395.5 (M+H)+.
EXAMPLE 36 N-(3-aminobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and 3-(aminomethyl)aniline following the procedure outlined in EXAMPLE 1. LCMS: 395.5 (M+H)+.
EXAMPLE 37
N-(4-aminobenzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and 4-(aminomethyl)aniline following the procedure outlined in EXAMPLE 1. LCMS: 395.5 (M+H)+.
EXAMPLE 38 l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(dimethylamino)benzyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from Intermediate 1 and 4-(aminomethyl)-JV,iV- dimethylaniline following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6; HCl salt): δ 12.30 (br, IH), 8.90-8.84 (m, 2H), 8.63 (d, IH), 7.44 (br, 2H) 7.32 (d, 2H), 7.17 (dd, IH), 7.07 (dd, IH), 6.34 (dd, IH), 4.28 (d, 2H), 3.03 (s, 6H); LCMS: 423.5 (M+H)+.
EXAMPLE 39
Nl-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(pyridin-4-yl)-lH-pyrrole-2- carbohydrazide

J_: Methyl l-(pyridin-4-yl)-lH-pyrrole-2-carboxylate

A mixture of methyl lH-pyrrole-2-carboxylate (1.0 g, 8.0 mmol), 4-bromopyridine (1.3 g, 8.3 mmol), K2CO3 (1.1 g, 8.0 mmol), methyl pyrrolidine -2-carboxylate (0.20 g, 1.6 mmol), CuI (0.25 g, 1.3 mmol), and DMF (50 mL) was heated at 100 0C under N2 for
15 h. The reaction was concentrated and then purified by silica gel chromatography
(l :4-EtOAc/PE) to give methyl l-(pyridin-4-yl)-lH-pyrrole-2-carboxylate as a yellow solid.
Step 2: l-(Pyridin-4-yl)-lH-pyrrole-2-carboxylic acid

The title compound was prepared from methyl l-(pyridin-4-yl)-lH-pyrrole-2- carboxylate following the procedure outlined for Intermediate 1, Step 2. Step_3.: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(pyridin-4-yl)-lH-pyrrole- 2-carbohydrazide

EXAMPLE 40
Nl-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(pyridin-3-yl)-lH-pyrrole-2- carbohydrazide
The title compound was prepared from methyl lH-pyrrole-2-carboxylate, 3- bromopyridine, and Intermediate 3 following the procedures outlined in EXAMPLE 39. LCMS: 382.5 (M+H)+.
EXAMPLE 41 Isopropyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)piperidine-l-carboxylate

Step_i: tert-Butyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)piperidine-l-carboxylate
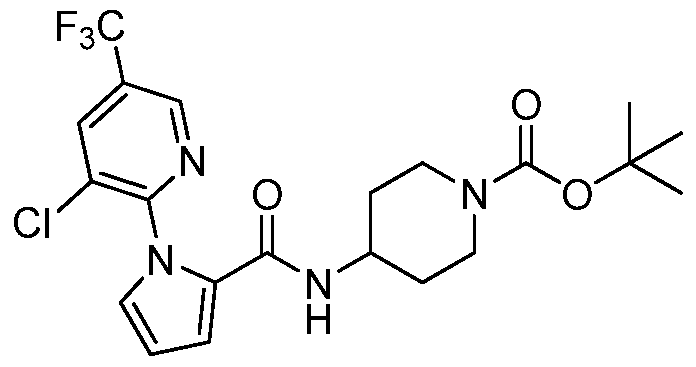
EXAMPLE 1.
Step 2: l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(piperidin-4-yl)-lH- pyrrole-2-carboxamide

Acetyl chloride (1 mL) was added dropwise to a solution of tert-butyl 4-(l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)- 1 H-pyrrole-2-carboxamido)piperidine- 1 -carboxylate (0.24 g, 0.50 mmol), DCM (20 mL), and MeOH (5 mL). After 15 min at rt, the reaction was concentrated to give l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N- (piperidin-4-yl)-lH-pyrrole-2-carboxamide as a yellow oil.
Step 3 : Isopropyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrr<)le-2- carboxamido)piperidine-l-carboxylate

Isopropyl chloroformate (85 mg, 0.70 mmol) was added to a solution of l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-N-(piperidin-4-yl)-lH-pyrrole-2-carboxamide (0.26 g, 0.71 mmol), triethylamine (0.35 g, 3.5 mmol), and DCM (20 mL). After 1 h at rt, the reaction was concentrated and purified by silica gel chromatography (1 : 5 -EtOAc: hexanes) to give isopropyl 4-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole- 2-carboxamido)piperidine-l -carboxylate as a white solid. 1H NMR (400 MHz, DMSO-de): δ 8.88 (dd, IH), 8.63 (d, IH), 8.07 (d, IH), 7.15 (dd, IH) 6.98 (dd, IH), 6.32 (dd, IH), 4.74 (septet, IH), 3.91 (br, 2H), 3.70 (m, IH), 2.76 (br, 2H), 1.68 (m, 2H), 1.32 (m, 2H), 1.16 (d, 6H); LCMS: 459.6 (M+H)+.
EXAMPLE 42 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was prepared from 3,5-dichloro-2-hydrazinylpyridine (Intermediate 5, Step 1) and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.19 (d, IH), 8.86 (m, IH), 8.66 (d, IH), 8.63 (d, IH), 8.05 (d, IH), 7.89 (d, IH), 7.25-7.20 (m, 2H), 6.39 (dd, IH); LCMS: 450.4 (M+H)+.
EXAMPLE 43 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-fluoro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

3-Fluoro-2-hydrazinyl-5-(trifluoromethyl)pyridine was prepared from 2,3-difluoro-5- (trifluoromethyl)pyridine following the procedure outlined for Intermediate 3. The title compound was prepared from 3-fluoro-2-hydrazinyl-5-(trifluoromethyl)pyridine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 10.30 (s, IH), 9.37 (s, IH), 8.67 (d, IH), 8.64 (d, IH), 8.23 (s, IH), 7.89 (dd, IH), 7.26 (dd, IH), 7.23 (dd, IH), 6.40 (dd, IH); LCMS: 468.5 (M+H)+.
EXAMPLE 44
N',l-Bis(3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from Intermediate 2 and 3,5-dichloro-2- hydrazinylpyridine (Intermediate 5, Step 1) following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 10.13 (d, IH), 8.64 (d, IH), 8.50 (d, IH), 8.37 (d, IH), 8.04 (d, IH), 7.89 (d, IH), 7.20-7.15 (m, 2H), 6.35 (dd, IH); LCMS: 416.4 (M+H)+.
EXAMPLE 45
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(isoquinolin-l-yl)-lH-pyrrole-2- carbohydrazide

The title compound was prepared from methyl lH-pyrrole-2-carboxylate, 1- iodoisoquinoline, and Intermediate 3 following the procedures outlined in EXAMPLE 39. 1H NMR (400 MHz, DMSO-d6): δ 10.25 (d, IH), 9.08 (d, IH), 8.34 (d, IH), 8.32 (m, IH), 8.04-7.98 (m, 2H), 7.89 (d, IH), 7.76 (m, IH), 7.62 (m, IH), 7.32-7.27 (m, 3H), 6.43 (dd, IH); LCMS: 432.5 (M+H)+.
EXAMPLE 46
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was prepared from methyl lH-pyrrole-2-carboxylate, l-iodo-2- (trifluoromethyl)benzene, and Intermediate 3 following the procedures outlined in EXAMPLE 39. 1H NMR (400 MHz, DMSO-d6): δ 10.12 (br, IH), 9.08 (br, IH), 8.34 (br, IH), 8.04 (br, IH), 7.77 (d, IH), 7.67 (t, IH), 7.59 (t, IH), 7.32 (d, IH), 7.15 (br, IH), 7.04 (br, IH), 6.30 (dd, IH); LCMS: 449.5 (M+H)+.
EXAMPLE 47 l-(3-Chloro-4-methoxypyridin-2-yl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbohydrazide

Step 1 : 2,3-Dichloro-4-iodopyridine


Chlorotrimethylsilane (10.2 rnL, 80.1 mmol) was added to a mixture of 2,3-dichloro-4- iodopyridine (11 g, 40 mmol), sodium iodide (12 g, 80 mmol), and ACN (150 mL). The resulting mixture was refluxed overnight, allowed to cool to rt, and concentrated. Sat'd sodium thiosulfate (100 mL) was added, and the mixture was extracted with EtOAc (200 mL x 3). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (1 :20→l :5-EtOAc/PE) to give 3-chloro- 2,4-diiodopyridine as a yellow solid. Step 3 : 3-Chloro-2-iodo-4-methoxypyridine

A solution of 3-chloro-2,4-diiodopyridine (3.06 g,5.03 mmol) and sodium methoxide (30 mL, 0.28M) was refluxed for 5 h, allowed to cool to rt, and then concentrated. The residue was diluted with water (50 mL) and extracted with EtOAc (50 mL x 3). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (l :20→l :5-EtOAc/PE) to give 3-chloro-2-iodo-4-methoxypyridine as a white solid. LCMS: 270 (M+H)+.
Step 4: l-(3-Chloro-4-methoxypyridin-2-yl)-N'-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from 3-chloro-2-iodo-4-methoxypyridine, methyl lH-pyrrole-2-carboxylate, and Intermediate 3 following the procedure outlined in EXAMPLE 39. 1H NMR (400 MHz, DMSO-d6): δ 10.19 (s, IH), 9.13 (s, IH), 8.36 (br,
IH), 8.26 (d, IH), 8.06 (d, IH), 7.26 (d, IH), 7.16 (dd, IH), 7.10 (dd, IH), 6.32 (dd, IH), 3.94 (s, 3H); LCMS: 446.5 (M+H)+.
EXAMPLE 48 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-fluoro-4-
(methylsulfonyl)benzyl)-lH-pyrrole-2-carboxamide

Step 1 : (2-Fluoro-4-(methylsulfonyl)phenyl)methanamine

Borane (3 niL, IM THF, 3mmol) was added dropwise to a solution of 2-fluoro-4- (methylsulfonyl)benzamide (0.20 g, 0.92 mmol) and THF (30 niL). The reaction was refluxed for 6 h, cooled to rt, and quenched with 6N HCl (1 rnL). The mixture was concentrated and then diluted with EtOAc (20 rnL). The resulting solid was filtered, washed with EtOAc (10 mL x 3), and dried to give (2-fluoro-4- (methylsulfonyl)phenyl)methanamine as a white solid. LCMS: 204 (M+H)+. Step 2: l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(2-fluoro-4-
(methylsulfonyl)benzyl)-lH-pyrrole-2-carboxamide

The title compound was prepared from (2-fluoro-4-
(methylsulfonyl)phenyl)methanamine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.93 (t, IH), 8.88 (dd,
IH), 8.63 (d, IH), 7.76-7.70 (m, 2H) 7.54 (t, IH), 7.20 (dd, IH), 7.10 (dd, IH), 6.37 (dd, IH), 4.40 (d, 2H), 3.22 (s, 3H); LCMS: 476.5 (M+H)+.
EXAMPLE 49
N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-5-methyl-lH-pyrrole-2- carbohydrazide

The title compound was prepared from 3-chloro-2-iodo-5-(trifluoromethyl)pyridine, methyl 5 -methyl- lH-pyrrole-2-carboxylate, and Intermediate 3 following the procedure outlined in EXAMPLE 39. 1U NMR (400 MHz, DMSO-d6): δ 10.16 (s, IH), 9.12 (s, IH), 8.91 (d, IH), 8.68 (d, IH), 8.36 (d, IH), 8.06 (d, IH), 7.16 (d, IH), 6.17 (d, IH), 1.94 (s, 3H); LCMS: 498.4 (M+H)+.
EXAMPLE 50 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-(2,2,2-trifluoroethoxy)phenyl)- lH-pyrrole-2-carbohydrazide

The title compound was prepared from Intermediate 1 and (4-(2,2,2- trifluoroethoxy)phenyl)hydrazine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.14 (d, IH), 8.87 (d, IH), 8.63 (d, IH), 7.58 (d, IH), 7.24 (dd, IH), 7.16 (dd, IH), 6.84 (d, 2H), 6.67 (d, 2H), 6.38 (dd, IH), 4.56 (q, 2H); LCMS: 479.3 (M+H)+.
EXAMPLE 51 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-cyanobenzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 1 and 4- (aminomethyl)benzonitrile hydrochloride following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.94 (t, IH), 8.90 (dd, IH), 8.65 (d, IH), 7.79 (d, 2H), 7.43 (d, 2H), 7.22 (dd, IH), 7.09 (dd, IH), 6.38 (dd, IH), 4.39 (d, 2H); LCMS: 405.3 (M+H)+.
EXAMPLE 52 N-(4-Chloro-3-(trifluoromethoxy)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-
2-yl)-lH-pyrrole-2-carboxamide


28% Ammonium hydroxide (3.0 mL, 22 mmol NH3) was added to a solution of 4- (bromomethyl)-l-chloro-2-(trifluoromethoxy)benzene (300 mg, 1.04 mmol) and DMSO (4 mL). The reaction was sealed, stirred vigorously for 30 min, vented, poured into EtOAc (40 mL), and then washed with 10% K2CO3 (100 mL x 2). Each aqueous wash was extracted with EtOAc (40 mL). The combined organic extracts were dried,
filtered, and concentrated to give impure (4-chloro-3-
(trifluoromethoxy)phenyl)methanamine as a colorless oil. LCMS: 226.1 (M+H)+. Step 2: N-(4-Chloro-3-(trifluoromethoxy)benzyl)-l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxamide

The title compound was prepared from (4-chloro-3-
(trifluoromethoxy)phenyl)methanamine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.94 (t, IH), 8.89 (dd, IH), 8.64 (d, IH), 7.64 (d, IH), 7.41 (br, IH), 7.31 (dd, IH), 7.22 (dd, IH), 7.07 (dd, IH), 6.38 (dd, IH), 4.35 (d, 2H); LCMS: 498.2 (M+H)+.
EXAMPLE 53 l-(2-Bromo-4-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

Step 1 : l-(2-Bromo-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(2-bromo-4- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and Intermediate 3 as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.18 (s, IH), 8.40 (s, IH), 8.10 (m, 2H), 7.80 (m, IH), 7.58 (m, IH), 7.20 (m, IH), 7.08 (m, IH), 6.40 (m, IH). LCMS: 527 (M+H)+.
EXAMPLE 54 N',l-Bis[3-chloro-4-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2-carbohydrazide

Step 1 : 2,3-Dichloro-4-iodopyridine

Cl CL γy / cF3
A mixture of 2,3-dichloro-4-iodopyridine (4.1 g, 15.02 mmol), KF (1.3 g, 22.41 mmol), CuI (4.3 g, 22.63 mmol), and TMS-CF3 (4.3 g, 30.28 mmol) in DMF (35 mL) was stirred at 250C for 18 h. The mixture was diluted with NH3/H2O (100 mL) and the aqueous layer was extracted with EtOAc (2 x 80 mL). The organics were combined, dried, evaporated under reduced pressure, and the residue was purified by silica gel chromatography (PE:EA) to afford 0.86 g (27%) of 2,3-dichloro-4- (trifluoromethyl)pyridine as a colorless oil. LCMS: 216 (M+H)+. Step 3 : 3-Chloro-2-hydrazinyl-4-(trifluoromethyl)pyridine
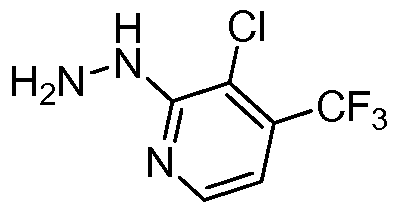
The title compound was synthesized as described for Intermediate 3 using 2,3- dichloro-4-(trifluoromethyl)pyridine as the starting material. LCMS: 212 (M+H)+. Step 4: N',l-Bis[3-chloro-4-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2,3- Dichloro-4-(trifluoromethyl)pyridine and 3 -chloro-2-hydrazinyl-4-
(trifluoromethyl)pyridine as the starting materials. 1H NMR (300 MHz, CDCl3): δ 8.90 (s, IH), 8.59 (d, IH), 8.14 (d, IH), 7.67 (d, IH), 7.20 (s, IH), 7.07 (d, IH), 6.44 (d, IH), 5.32 (m, IH), 1 NH resonance not observed. LCMS: 484 (M+H)+.
EXAMPLE 55 N',l-bis(3-chloro-5-methylpyridin-2-yl)-lH-pyrrole-2-carbohydrazide

Step 1 : S-Chloro-l-hydrazinyl-S-methylpyridine

The title compound was synthesized as described for Intermediate 3 using 2,3- dichloro-5-methylpyridine as the starting material. LCMS: 158 (M+H)+.
Step 2: N',l-Bis(3-chloro-5-methylpyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2,3-dichloro- 5-methylpyridine and 3-chloro-2-hydrazinyl-5-methylpyridine as starting materials. 1H NMR (300 MHz, CDCl3): δ 9.96 (s, IH), 8.24 (s, IH), 7.75 (s, IH), 7.64 (s, IH), 7.60 (s, IH), 7.36 (s, IH), 7.20 (d, IH), 6.92 (s, IH), 6.23 (s, IH), 2.39 (s, 3H), 2.19 (s, 3H). LCMS: 376 (M+H)+.
EXAMPLE 56
N^l-Bisβ-fluoro-S-^rifluoromethylJpyridin-l-yll-lH-pyrrole-l-carbohydrazide

Step 1 : 2-Chloro-3-fluoro-4-iodopyridine

The title compound was synthesized as described in EXAMPLE 54, Stepl using 2- chloro-3-fluoropyridine as the starting material. LCMS: 258 (M+H)+.
Step 2: 2-Chloro-3-fluoro-5-iodopyridine


The title compound was synthesized as described in EXAMPLE 54, Step 2 using 2- chloro-3-fluoro-5-iodopyridine as the starting material. LCMS: 200 (M+H)+. Step 4: 3-Fluoro-2-hydrazinyl-5-(trifluoromethyl)pyridine

The title compound was synthesized as described for Intermediate 3 using 2-chloro-3- fluoro-5-(trifluoromethyl)pyridine as the starting material. LCMS: 196 (M+H)+. Step 5: NSl-Bisβ-fluoro-S-CtrifluoromethylJpyridin-l-ylj-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2-chloro-3- fluoro-5-(trifluoromethyl)pyridine and 3-fluoro-2-hydrazinyl-5- (trifluoromethyl)pyridine as the starting materials. LCMS: 452 (M+H)+.
EXAMPLE 57
Nl,l-Bis(3-chloro-6-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide


NaNO2 (826 mg, 11.97 mmol) was added to a solution of 2-chloro-6- (trifluoromethyl)pyridin-3 -amine (2.24 g, 11.40 mmol) in cone. HCl (20 mL) cooled to O0C. The resulting solution was stirred at O0C for 1 h followed by the addition of CuCl (1.69 g, 17.09 mmol) and stirred at 4O0C for 30 min. The pH was adjusted to pH = 9 by the addition OfNHs-H2O and the resulting aqueous solution was extracted with Et2O (2 x 50 mL). The organics were combined, dried, and evaporated to dryness under reduced pressure to afford 2,3-dichloro-6-(trifluoromethyl)pyridine as red brown oil. Step 2: 3-Chloro-2-hydrazinyl-6-(trifluoromethyl)pyridine

The title compound was synthesized as described for Intermediate 3 using 2,3- dichloro-6-(trifluoromethyl)pyridine as the starting material. LCMS: 212 (M+H)+.
Step 3: NSl-BisCS-Chloro-ό-Ctrifluoromethyltøyridin-l-ylHH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2,3-dichloro- 6-(trifluoromethyl)pyridine and 3-chloro-2-hydrazinyl-6-(trifluoromethyl)pyridine as starting materials. LCMS: 484 (M+H)+.
EXAMPLE 58
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-6- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-6- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid and Intermediate 3. 1H NMR (300 MHz, CDCl3) δ 8.78 (m, IH), 8.31 (s, IH), 7.99 (m, IH), 7.70 (m, 3H), 7.09 (m, 2H), 6.43 (m, IH).
EXAMPLE 59
N'-(3-Chloro-4-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-chloro-2-hydrazinyl-4-(trifluoromethyl)pyridine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.68 (s, IH), 8.60 (bs, IH), 8.16 (m, IH), 8.05 (m, IH), 7.52 (m, IH), 7.06 (m, 3H), 6.43 (m, IH). LCMS: 484 (M+H)+.
EXAMPLE 60 l-(3-Chloro-4-methylpyridin-2-yl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbohydrazide

3-Chloro-2-iodo-4-methylpyridine

A mixture of 2,3-dichloro-4-methylpyridine (3.47 g,21.42 mmol), NaI (6.42 g,42.80 mmol), and Me3SiCl (5.4 mL) in ACN (30 mL) was refluxed for 18 h. The reaction mixture was then diluted with NaS2O3 (30 mL) and the aqueous layer was extracted with EtOAc (2 x 50 mL). The organics were combined, dried, and evaporated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/PE) to afford 3-chloro-2-iodo-4-methylpyridine as a yellow solid.
Step 2: l-(3-Chloro-4-methylpyridin-2-yl)-N'-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 39 using 3-chloro-2- iodo-4-methylpyridine in Step 1, methyl l-(3-chloro-4-methylpyridin-2-yl)-lH- pyrrole-2-carboxylate in Step 2, and l-(3-chloro-4-methylpyridin-2-yl)-lH-pyrrole-2- carboxylic acid and Intermediate 3 in Step 3. 1H NMR (300 MHz, CD3OD) δ 8.29 (s, IH), 8.23 (m, IH), 7.89 (s, IH), 7.42 (m, IH), 7.20 (m, IH), 7.10 (m, IH), 6.42 (m, IH), 2.47 (s, 3H). LCMS: 430 (M+H)+.
EXAMPLE 61
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-5-methylpyridin-2-yl)- lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- methylpyridin-2-yl)-lH-pyrrole-2-carboxylic acid and Intermediate 3 as the starting materials. 1H NMR (300 MHz, CD3OD) δ 8.29 (s, IH), 8.22 (s, IH), 7.89 (m, IH), 7.84 (m, IH), 7.20 (m, IH), 7.09 (m, IH), 6.41 (m, IH), 2.41 (s, 3H). LCMS: 430 (M+H)+.
EXAMPLE 62 NSl-Bis^-chloro-S-Ctrifluoromethyltøyridin^-ylHH-imidazole^- carbohydrazide

Step 1 : l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 using 2,3- dichloro-5-(trifluoromethyl)pyridine and lH-imidazole-2-carboxylic acid as the starting materials. LCMS: 292 (M+H)+.
Step 2: N',l-Bis(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-2- carbohydrazide
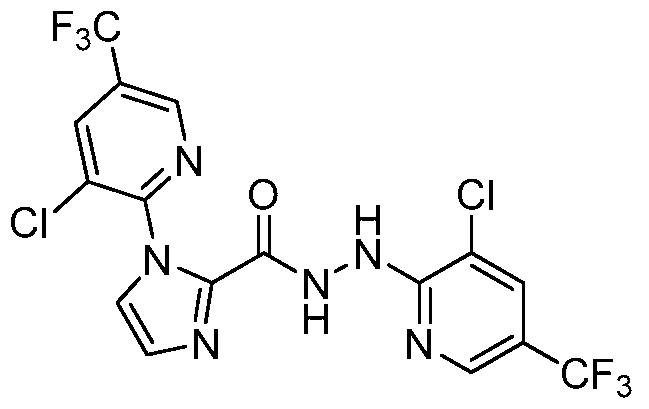
The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-imidazole-2-carboxylic acid and Intermediate 3. 1H NMR (300 MHz, CDCl3) δ 9.51 (bs, IH), 8.73 (s, IH), 8.35 (s, IH), 8.12 (m, IH), 7.73 (s, IH), 7.35 (m, 2H), 7.29 (m, IH). LCMS: 483 (M-H)+.
EXAMPLE 63 l-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-N'-p-tolyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and p-tolylhydrazine. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, IH), 8.05 (m, 2H), 7.08 (s, IH), 7.0 (m, 2H), 6.98 (m, IH), 6.81 (m, 2H), 6.41 (m, IH), 2.25 (s, 3H), 1 NH resonance not observed. LCMS: 395 (M+H)+.
EXAMPLE 64 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(trifluoromethyl)phenyl)hydrazine. 1H NMR (400 MHz, CDCl3) δ 8.66 (m, IH), 8.05 (m, IH), 7.73 (bs, IH), 7.46 (m, 2H), 7.12 (m, IH), 6.96 (m, IH), 6.89 (m, 2H), 6.45 (m, IH), 1 NH resonance not observed.
EXAMPLE 65 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-methoxyphenyl)hydrazine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.68 (s, IH), 8.05 (m, IH), 7.62 (bs, IH), 7.11 (m, IH), 6.46 (m, IH), 6.89 (m, 5H), 6.46 (m, IH), 3.86 (s, 3H). LCMS: 411 (M+H)+.
EXAMPLE 66 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (3-methoxyphenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.02 (m, IH), 7.85 (bs, IH), 7.09 (m, 2H), 6.90 (m, IH), 6.43 (m, 4H), 3.76 (s, 3H), 1 NH resonance not observed . LCMS: 411 (M+H)+.
EXAMPLE 67 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-cyanophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 4-hydrazinylbenzonitrile as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.05 (s, IH), 7.75 (bs, IH), 7.47 (m, 2H), 7.12 (m, IH), 6.96 (m, IH), 6.84 (m, 2H), 6.46 (m, IH), 1 NH resonance not observed. LCMS: 406 (M+H)+.
EXAMPLE 68 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-isopropylphenyl)-lH-pyrrole-
2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-isopropylphenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.03 (s, IH), 7.79 (bs, IH), 7.06 (m, 3H), 6.93 (bs, IH), 6.81 (m, 2H), 6.43 (m, IH), 2.79 (m, IH), 1.12 (d, 6H), 1 NH resonance not observed. LCMS: 423 (M+H)+.
EXAMPLE 69 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(quinolin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 2-hydrazinylquinoline as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, IH), 8.16 (m, 2H), 8.01 (s, IH), 7.87 (m, IH), 7.74 (m, 2H), 7.48 (m, IH), 7.19 (m, IH), 7.13 (m, IH), 7.06 (m, IH), 6.40 (m, IH), 1 NH resonance not observed. LCMS: 432 (M+H)+.
EXAMPLE 70 l-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-N'-o-tolyl-lH-pyrrole-l- carbohydrazide
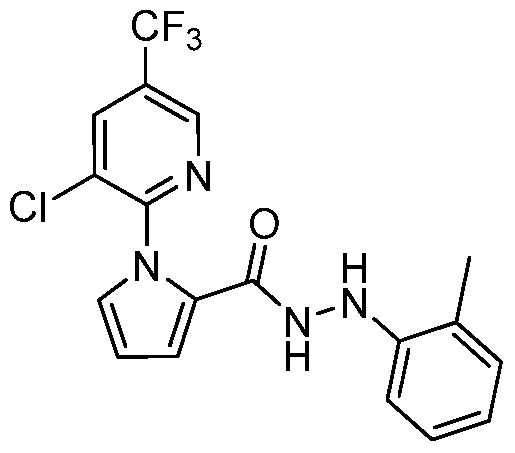
The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and o-tolylhydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, IH), 8.02 (s, IH), 7.95 (bs, IH), 7.06 (m, 3H), 6.87 (m, 3H), 6.40 (m, IH), 5.23 (bs, IH), 2.19 (s, 3H). LCMS: 395 (M+H)+.
EXAMPLE 71
N'-(4-Butylphenyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-butylphenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.03 (s, IH), 7.93 (bs, IH), 7.09 (m, IH), 7.01 (m, 2H), 6.93 (m, IH), 6.83 (m, 2H), 6.42 (m, IH), 2.49 (m, 2H), 1.56 (m, 2H), 1.32 (m, 2H), 0.95 (m, 3H), 1 NH resonance not observed. LCMS: 437 (M+H)+.
EXAMPLE 72 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2-fluoro-4- (methylsulfonyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-fluoro-4-(methylsulfonyl)phenyl)hydrazine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.67 (s, IH), 8.05 (s, IH), 7.83 (s, IH), 7.54 (m, 2H), 7.12 (m, IH), 7.02 (m, 2H), 6.49 (m, IH), 6.41 (m, IH), 3.01 (s, 3H). LCMS: 477 (M+H)+.
EXAMPLE 73 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-chloropyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-chloro-2-hydrazinylpyridine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 9.00 (bs, IH), 8.68 (s, IH), 8.04 (s, 2H), 8.04 (s, IH), 7.52 (m, IH), 7.06 (m, 2H), 6.75 (m, IH), 6.39 (m, IH). LCMS: 416 (M+H)+.
EXAMPLE 74 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 2-hydrazinyl-5-(trifluoromethyl)pyridine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.85 (bs, IH), 8.66 (s, IH), 8.35 (s, IH), 8.04 (m, IH), 7.70 (m, IH), 7.09 (m, 2H), 6.81 (m, 2H), 6.42 (m, IH). LCMS: 450 (M+H)+.
EXAMPLE 75 N-(2-Chloro-4-(trifluoromethyl)phenyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 2-chloro-4-(trifluoromethyl)aniline as the starting materials. LCMS: 468 (M+H)+.
EXAMPLE 76 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-(trifluoromethoxy)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(trifluoromethoxy)phenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, IH), 8.06 (s, IH), 7.78 (bs, IH), 7.08 (m, 3H), 6.91 (m, IH), 6.82 (m, 2H), 6.41 (m, IH), 1 NH resonance not observed.
EXAMPLE 77 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-chlorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-chlorophenyl)hydrazine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.66 (m, IH), 8.03 (s, IH), 7.65 (m, IH), 7.18 (m, 2H), 7.11 (m, IH), 6.95 (m, IH), 6.78 (m, 2H), 6.44 (m, IH), 1 NH resonance not observed. LCMS: 415 (M+H)+.
EXAMPLE 78 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and Intermediate 3 as the starting materials. 1H NMR (300 MHz, DMSO-d6) δ 10.28 (s, IH), 9.18 (s, IH), 8.51 (s, IH), 8.39 (m, 2H), 8.09 (s, IH), 7.21 (m, 2H), 6.38 (m, IH). LCMS: 450 (M+H)+.
EXAMPLE 79 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2-chlorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-chlorophenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCI3) δ 8.65 (s, IH), 8.04 (s, IH), 7.76 (bs, IH), 7.25 (m, IH), 7.14 (m, 2H), 6.67 (m, 2H), 6.81 (m, IH), 6.43 (m, IH), 1 NH resonance not observed. LCMS: 415 (M+H)+.
EXAMPLE 80 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-chlorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (3-chlorophenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCI3) δ 8.67 (m, IH), 8.04 (m, IH), 7.80 (bs, IH), 7.11 (m, 2H), 6.91 (m, IH), 6.85 (m, 2H), 6.71 (m, IH), 6.42 (m, IH), 1 NH resonance not observed. LCMS: 415 (M+H)+.
EXAMPLE 81 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-(trifluoromethyl)phenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (m, IH), 8.04 (m, IH), 7.72 (bs, IH), 7.47 (m, IH), 7.38 (m, IH), 7.10 (m, 2H), 6.93 (m, 2H), 6.43 (m, IH), 1 NH resonance not observed. LCMS: 449 (M+H)+.
EXAMPLE 82 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (3-(trifluoromethyl)phenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.63 (m, IH), 8.01 (m, IH), 7.96 (bs, IH), 7.26 (m, IH), 7.10 (m, 2H), 7.04 (m, IH), 6.94 (m, IH), 6.87 (m, IH), 6.41 (m, IH), 1 NH resonance not observed. LCMS: 449 (M+H)+.
EXAMPLE 83 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-methoxyphenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (m, IH), 8.09 (bs, IH), 8.03 (m, IH), 7.09 (m, IH), 6.94 (m, IH), 6.89 (m, 2H), 6.77 (m, 2H), 6.42 (m, IH), 3.75 (s, 3H), 1 NH resonance not observed. LCMS: 411 (M+H)+.
EXAMPLE 84 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-fluorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-fluorophenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.64 (m, IH), 8.02 (m, IH), 7.95 (bs, IH), 7.08 (m, IH), 6.86 (m, 3H), 6.79 (m, 2H), 6.41 (m, IH), 1 NH resonance not observed.
EXAMPLE 85 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(pyridin-3-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-hydrazinylpyridine as the starting materials LCMS: 382 (M+H)+.
EXAMPLE 86 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-(methylsulfonyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(methylsulfonyl)phenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.43 (m, IH), 8.05 (s, IH), 7.60 (m, 2H), 7.12 (m, 2H), 6.75 (m, 2H), 6.47 (m, IH), 5.74 (m, IH), 2.98 (s, 3H). LCMS: 459 (M+H)+.
EXAMPLE 87 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-fluorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (3-fluorophenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.03 (s, IH), 7.93 (bs, IH), 7.11 (m, 2H), 6.89 (m, IH), 6.56 (m, 3H), 6.41 (m, IH), 1 NH resonance not observed. LCMS: 399 (M+H)+.
EXAMPLE 88 l-CS-Chloro-S-CtrifluoromethyOpyridin-l-yO-N'-m-tolyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and m-tolylhydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, IH), 8.02 (s, IH), 7.95 (bs, IH), 7.08 (m, 2H), 6.89 (s, IH), 6.70 (m, IH), 6.65 (s, 2H), 6.41 (m, IH), 6.22 (s, 3H), 1 NH resonance not observed. LCMS: 395 (M+H)+.
EXAMPLE 89 l-(3-Chloro-4-(trifluoromethyl)pyridin-2-yl)-N'-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-4- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid and Intermediate 3 as the starting materials. 1U NMR (300 MHz, CDCl3) δ 8.84 (bs, IH), 8.60 (m, IH), 8.34 (m, IH), 7.78 (m, IH), 7.68 (m, IH), 7.18 (m, IH), 7.08 (m, IH), 6.48 (m, IH), 1 NH resonance not observed. LCMS: 484 (M+H)+.
EXAMPLE 90 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(pyridin-4-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 4-hydrazinylpyridine as the starting materials. LCMS: 382 (M+H)+.
EXAMPLE 91
NSl-bisCS-Chloro-S-CtrifluoromethyOpyridin-l-yO-N'-methyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-chloro-2-(l-methylhydrazinyl)-5-(trifluoromethyl)pyridine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.63 (m, IH), 8.39 (m, IH), 8.30 (bs, IH), 7.99 (m, IH), 7.73 (m, IH), 7.08 (m, IH), 6.90 (m, IH), 6.41 (m, IH), 3.40 (s, 3H).
EXAMPLE 92 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2-fluorophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-fluorophenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.04 (s, IH), 7.84 (bs, IH), 7.09 (s, IH), 6.98 (m, 4H), 6.82 (m, IH), 6.42 (s, IH), 1 NH resonance not observed.
EXAMPLE 93 Nl,l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-indole-2-carbohydrazide

Step 1 : l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-indole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 using methyl IH- indole-2-carboxylate and 2,3-dichloro-5-(trifluoromethyl)pyridine as the starting materials. LCMS: 340 (M+H)+. Step_l: N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-indole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-indole-2-carboxylic acid and Intermediate 3. 1H NMR (300 MHz, CDCl3) δ 8.78 (m, 2H), 8.33 (s, IH), 8.15 (s, IH), 7.74 (m, 2H), 7.62 (m, IH), 7.36 (m, 3H), 7.06 (m, IH). LCMS: 534 (M+H)+.
EXAMPLE 94
Nl,l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2-carbohydrazide

Step 1 : l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 using 2,3- dichloro-5-(trifluoromethyl)pyridine and methyl pyrrolidine-2-carboxylate hydrochloride as the starting materials. LCMS: 295 (M+H)+. Step 2: N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2- carbohydrazide

The title compound was synthesized as a racemic mixture as described in EXAMPLE 1 using l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2-carboxylic acid and Intermediate 3 as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.83 (bs, IH), 8.35 (s, IH), 8.25 (s, IH), 7.78 (s, IH), 7.75 (s, IH), 7.49 (bs, IH), 5.07 (m, IH), 4.26 (m, IH), 3.92 (m, IH), 2.29 (m, 3H), 2.05 (m, IH). LCMS: 488 (M+H)+.
I l l
EXAMPLE 95 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-methyl-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

2-Hydrazinyl-3-methyl-5-(trifluoromethyl)pyridine

The title compound was synthesized as described for Intermediate 3 using 2-chloro-3- methyl-5-(trifluoromethyl)pyridine as starting material. LCMS: 192 (M+H)+. Step 2: l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-methyl-5- (trifluoromethyl) pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 2-hydrazinyl-3-methyl-5-(trifluoromethyl)pyridine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.58 (bs, IH), 8.01 (s, IH), 7.10 (m, IH), 6.96 (m, IH), 6.80 (m, 2H), 6.41 (m, IH), 2.43 (s, 3H), 1 NH resonance not observed. LCMS: 464 (M+H)+.
EXAMPLE 96
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-chlorophenyl)-lH-pyrrole-2- carbohydrazide


A mixture of 2-chloroaniline (0.15 g, 1.2 mmol) and methyl 2,5- dimethoxytetrahydrofuran-2-carboxylate (0.15 g, 0.79 mmol) in AcOH (2 mL) was refluxed at 1200C for 6 h. The volatiles were removed by evaporation and the residue was further dried under high vacuum overnight. To the residue MeOH/lN NaOH (3 mL/3 mL) were added and the resulting mixture was refluxed at 900C for 3 hours. The volatiles were evaporated and the residue was partitioned between H2O (10 mL) and EtOAc(25 mL). To the aqueous layer IN HCl (10 mL) was added and it was extracted with EtOAc (45 mL). This organic layer was dried, filtered, and evaporated to dryness to afford l-(2-chlorophenyl)-lH-pyrrole-2-carboxylic acid as a yellow solid. LCMS: 222.11 (M+H)+.
Step 2: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-chlorophenyl)-lH- pyrrole-2-carbohydrazide

EXAMPLE 97 l-(Biphenyl-4-yl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using biphenyl-4- amine as starting material. LCMS: 457 (M+H)+.
EXAMPLE 98 l-(Biphenyl-2-yl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using biphenyl-2- amine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, IH), 9.10 (s, IH), 8.39 (s, IH), 8.08 (d, IH), 7.43-7.34 (m, 3H), 7.23-7.17 (m, 4H), 7.14-7.12 (m, 2H), 6.97 (dd, IH), 6.63 (dd, IH), 6.03 (dd, IH). LCMS: 457 (M+H)+.
EXAMPLE 99 l-(Biphenyl-3-yl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using biphenyl-3- amine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.20 (s, IH), 8.06 (d, IH), 7.66-7.60 (m, 2H), 7.60-7.56 (m, IH), 7.52 (t, IH), 7.50-7.40 (3H), 7.40-7.32 (m, 2H), 7.29 (dd, IH), 7.02 (dd, IH), 6.32 (dd, IH). LCMS: 457 (M+H)+.
EXAMPLE 100
N'-CS-Chloro-S-CtrifluoromethyOpyridin-l-yO-l-o-tolyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using o-toluidine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, IH), 9.10 (s, IH), 8.40 (s, IH), 8.08 (d, IH), 7.40-7.15 (m, 3H), 7.15-7.06 (m, 2H), 6.92 (dd, IH), 6.30 (dd, IH), 1.90 (s, 3H). LCMS: 395 (M+H)+.
EXAMPLE 101
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-m-tolyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using m-toluidine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.45 (s, IH), 8.10 (d, IH), 7.30-7.20 (m, IH), 7.15 (dd, IH), 7.15-7.07 (m, 3H), 6.95 (dd, IH), 6.28 (dd, IH), 2.27 (s, 3H). LCMS: 395 (M+H)+.
EXAMPLE 102
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-p-tolyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using p-toluidine as starting material. 1U NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.45 (s, IH), 8.10 (d, IH), 7.20-7.14 (m, 4H), 7.10 (dd, IH), 6.98 (dd, IH), 6.28 (dd, IH), 2.30 (s, 3H). LCMS: 395 (M+H)+.
EXAMPLE 103
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-phenyl-lH-pyrrole-l- carbohydrazide
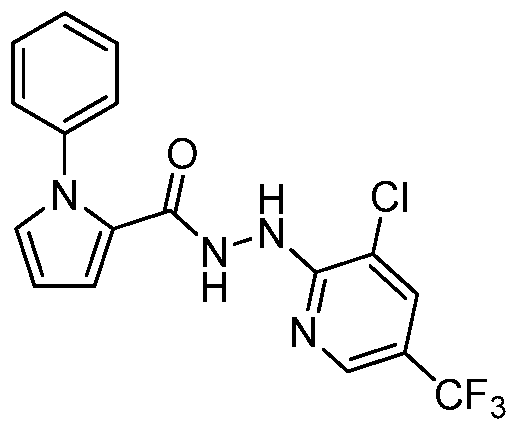
The title compound was synthesized as described in EXAMPLE 96 using aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.45 (s, IH), 8.10 (s, IH), 7.40-7.20 (m, 5H), 7.18 (s, IH), 7.00(d, IH), 6.30 (d, IH). LCMS: 381 (M+H)+.
EXAMPLE 104
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- methoxyaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, IH), 9.08 (s, IH), 8.38 (s, IH), 8.06 (d, IH), 7.32-7.26 (m, IH), 7.14 (dd, IH), 7.18-7.00 (m, 2H), 6.95-6.88 (m, 2H), 6.22 (dd, IH), 3.62 (s, 3H). LCMS: 411 (M+H)+.
EXAMPLE 105
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-Cό-CtrifluoromethylJpyridin-S-yl)- lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 6- (trifluoromethyl)pyridin-3 -amine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, IH), 9.25 (s, IH), 8.78 (d, IH), 8.44 (s, IH), 8.10 (d, IH), 8.06 (dd, IH), 7.92 (d, IH), 7.38 (dd, IH), 7.15 (dd, IH), 6.42 (dd, IH). LCMS: 450 (M+H)+.
EXAMPLE 106
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 3- methoxyaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, IH), 9.22 (s, IH), 8.45 (s, IH), 8.10 (d, IH), 7.30-7.25 (m, IH), 7.20-7.18 (dd, IH), 6.96 (dd, IH), 6.95-6.85 (m, 3H), 6.26 (dd, IH), 3.70 (s, 3H). LCMS: 411 (M+H)+.
EXAMPLE 107
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-methoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4- methoxyaniline as starting material. LCMS: 411 (M+H)+.
EXAMPLE 108
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-(trifluoromethoxy)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 3- (trifluoromethoxy) aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, IH), 9.28 (s, IH), 8.40 (s, IH), 8.10 (s, IH), 7.56-7.49 (m, IH), 7.44-7.40 (m, IH), 7.34-7.29 (2H), 7.28 (dd, IH), 6.99 (dd, IH), 6.33 (dd, IH). LCMS: 465 (M+H)+.
EXAMPLE 109 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(trifluoromethoxy)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4- (trifluoromethoxy) aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, IH), 9.20 (s, IH), 8.44 (s, IH), 8.10 (s, IH), 7.50-7.30 (m, 4H), 7.20 (dd, IH), 7.04 (dd, IH), 6.30 (dd, IH). LCMS: 465 (M+H)+.
EXAMPLE 110 l-(2-Chloro-4-(trifluoromethoxy)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-
2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-chloro-4- (trifluoromethoxy)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.40 (s, IH), 8.06 (d, IH), 7.65 (d, IH), 7.50 (d, IH), 7.40 (dd, IH), 7.20 (dd, IH), 7.10 (dd, IH), 6.38 (dd, IH). LCMS: 499 (M+H)+.
EXAMPLE 111
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-(trifluoromethoxy)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- (trifluoromethoxy) aniline as starting material. 1H NMR (400 MHz, DMSO-dβ) δ 10.20 (s, IH), 9.20 (s, IH), 8.38 (s, IH), 8.06 (d, IH), 7.50-7.45 (m, IH), 7.45-7.40 (m, 3H), 7.18 (dd, IH), 7.10 (dd, IH), 6.35 (dd, IH). LCMS: 465 (M+H)+.
EXAMPLE 112 l-(2-Bromophenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- bromoaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.40 (s, IH), 8.04 (s, IH), 7.65 (d, IH), 7.42-7.36 (m, IH), 7.34-7.26 (m, 2H), 7.16 (dd, IH), 6.99 (dd, IH), 6.32 (dd, IH). LCMS: 459 (M+H)+.
EXAMPLE 113
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-ethylphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- ethylaniline as starting material. 1U NMR (400 MHz, DMSO-d6) δ 10.08 (s, IH), 9.06 (s, IH), 8.36 (s, IH), 8.08 (s, IH), 7.32-7.24 (m, 2H), 7.22-7.16 (m, IH), 7.11 (dd, IH), 7.09 (dd, IH), 6.96 (dd, IH), 6.28 (dd, IH), 2.38-2.18 (m, 2H), 1.00-0.90 (t, 3H). LCMS: 409 (M+H)+.
EXAMPLE 114
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-cyanophenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- cyanoaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, IH), 9.18 (s, IH), 8.38 (s, IH), 8.08 (d, IH), 7.88 (d, IH), 7.75-7.70 (m, IH), 7.60-7.50 (m, IH), 7.45-7.40 (d, IH), 7.25-7.20 (m, 2H), 6.40 (dd, IH). LCMS: 406 (M+H)+.
EXAMPLE 115 l-(2-tert-Butylphenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-
2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-tert- butylaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, IH), 9.06 (s, IH), 8.30 (s, IH), 8.04 (d, IH), 7.50 (dd, IH), 7.29 (td, IH), 7.13 (td, IH), 7.10 (dd, IH), 6.98 (dd, IH), 6.84 (dd, IH), 6.24 (dd, IH), 1.10 (s, 9H). LCMS: 437 (M+H)+.
EXAMPLE 116
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-isopropylphenyl)-lH-pyrrole-
2-carbo hy dr azide

The title compound was synthesized as described in EXAMPLE 96 using 2- isopropylaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, IH), 9.06 (s, IH), 8.30 (s, IH), 8.04 (s, IH), 7.36-7.28 (m, 2H), 7.18-7.12 (m, IH), 7.11 (dd, IH), 7.06 (d, IH), 6.95 (t, IH), 6.22 (dd, IH), 2.35 (septet, IH), 1.00 (d, 6H). LCMS: 423 (M+H)+.
EXAMPLE 117 l-(2,4-bis(Trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2,4- bis(trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.16 (s, IH), 8.35 (s, IH), 8.15-8.02 (m 3H), 7.60 (d, IH), 7.20 (dd, IH), 7.15 (s, IH), 6.38 (dd, IH). LCMS: 517 (M+H)+.
EXAMPLE 118 l-(4-Chloro-2-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-chloro-2- (trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.16 (s, IH), 8.38 (s, IH), 8.08 (d, IH), 7.88 (d, IH), 7.78 (dd, IH), 7.38 (d, IH), 7.20 (dd, IH), 7.08 (s, IH), 6.32 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 119
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloropyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 2-fluoro-3- chloropyridine as the starting material. 1H NMR (300 MHz, CDCl3) δ 8.69 (s, IH), 8.48 (m, IH), 8.33 (s, IH), 7.88 (m, IH), 7.73 (s, IH), 7.69 (m, IH), 7.08 (m, 2H), 6.41 (m, IH), 1 NH resonance not observed. LCMS: 416 (M+H)+.
EXAMPLE 120 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 2-fluoro-5- (trifluoromethyl)pyridine as the starting material. 1H NMR (400 MHz, CDCl3) δ 9.45 (bs, IH), 8.76 (s, IH), 8.33 (s, IH), 7.96 (m, IH), 7.77 (s, 2H), 7.53 (d, IH), 7.40 (m, IH), 7.11 (m, IH), 6.36 (m, IH). LCMS: 450 (M+H)+.
EXAMPLE 121
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-Cpyridin-l-ylJ-lH-pyrrole-l- carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 2- fluoropyridine as the starting material. LCMS: 382 (M+H)+.
EXAMPLE 122
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-fluoro-4- (methylsulfonyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using 2-fluoro-4- (methylsulfonyl)aniline as the starting material. 1H NMR (300 MHz, CDCl3) δ 8.69 (bs, IH), 8.33 (s, IH), 7.77 (m, 3H), 7.65 (m, IH), 7.61 (m, IH), 7.14 (m, IH), 6.96 (m, IH), 6.45 (m, IH), 3.11 (s, 3H). LCMS: 477 (M+H)+.
EXAMPLE 123 l-[3-Chloro-5-(trifluoromethyl)phenyl]-N'-[3-chloro-5-(trifluoromethyl)pyridin-2- yl] - lH-pyrr ole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using 3-chloro-5- (trifluoromethyl)aniline as the starting material. 1H NMR (300 MHz, DMSO-d6) δ 10.61 (s, IH), 9.54 (s, IH), 8.66 (s, IH), 8.38 (s, IH), 8.09 (s, IH), 8.00 (s, IH), 7.88 (m, IH), 7.63 (s, IH), 7.44 (d, IH), 6.62 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 124 N',l-bis(3-chloro-5-(trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide
The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (3-chloro-5- (trifluoromethyl)phenyl) hydrazine as the starting materials.. 1H NMR (300 MHz, CDCl3) δ 7.68 (m, 2H), 7.50 (m, 2H), 7.12 (m, IH), 7.03(m, 4H), 6.41 (m, IH), 1 NH resonance not observed. LCMS: 482 (M+H)+.
EXAMPLE 125
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-fluoro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 2,3-difluoro- 5-(trifluoromethyl)pyridine as the starting material. 1H NMR (300 MHz, CDCl3) δ 8.83 (bs, IH), 8.59 (s, IH), 8.33 (s, IH), 7.77 (m, 3H), 7.24 (m, IH), 7.16 (m, IH), 6.43 (m, IH). LCMS: 468 (M+H)+.
EXAMPLE 126
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-methyl-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide


A mixture of methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxylate (1.0 g, 3.28 mmol), Pd(dppf)Cl2 (cat), and Zn(CH3)2 (1.2M, 8 mL) in 1,4-
dioxane (50 niL) was refluxed for 18 h. The mixture was concentrated by evaporation under reduced pressure and the residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford methyl l-(3-methyl-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carboxylate as a white solid. LCMS: 285 (M+H)+.
Step 2: l-(3-Methyl-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl l-(3-methyl-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylate as the starting material. LCMS: 2714 (M+H)+. Step 3: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-methyl-5-
(trifluoromethyl) pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-methyl-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid and Intermediate 3 in Step 3. 1H NMR (300 MHz, CDCl3) δ 8.63 (s, IH), 8.41 (m, IH), 8.29 (s, IH), 7.86 (s, IH), 7.71 (m, IH), 7.48 (m, IH), 7.02 (m, 2H), 6.42 (m, IH), 2.17 (s, 3H). LCMS: 464 (M+H)+.
EXAMPLE 127 N ' , 1 -Bis(py ridin-2-yl)- 1 H-py rrole-2-carbohy dr azide

The title compound can be prepared as described in EXAMPLE 53 using 2- fluoropyridine and 2-hydrazinylpyridine as the starting materials. LCMS: 280 (M+H)
EXAMPLE 128
N',l-Bis[5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 2-fluoro-5- (trifluoromethyl)pyridine and 2-hydrazinyl-5-(trifluoromethyl)pyridine as the starting materials. 1H NMR (300 MHz, CD3OD) δ 9.09 (s, IH), 8.69 (s, IH), 8.50 (d, IH), 8.18 (d, IH), 8.00 (s, IH), 7.83 (s, IH), 7.42 (s, IH), 7.26 (d, IH), 6.74 (dd, IH). LCMS: 416 (M+H)+.
EXAMPLE 129 l-IS-Chloro-S-CtrifluoromethyOpyridin-l-yll-N'-Cpyridin-l-yO-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 2-hydrazinylpyridine as the starting materials. 1H NMR (300 MHz, CDCl3) δ 8.74 (bs, IH), 8.68 (s, IH), 8.10 (t, IH), 7.52 (t, IH), 7.29 (s, IH), 7.04 (m, 2H), 6.75 (m, 2H), 6.40 (dd, IH), 1 NH resonance not observed. LCMS: 382 (M+H)+.
EXAMPLE 130 N',l-Bis(3-chloropyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 53 using 3-chloro-2- fluoropyridine and 3-chloro-2-hydrazinylpyridine as the starting materials. LCMS: 348 (M+H)+.
EXAMPLE 131
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(trifluoromethyl)benzyl)-lH- pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using (4- (trifluoromethyl)phenyl)methanamine as the starting material. 1H NMR (300 MHz, CDCl3) δ 8.58 (bs, IH), 8.23 (s, IH), 7.77 (m, IH), 7.68 (m, IH), 7.54 (m, 2H), 7.17 (m, IH), 7.01 (m, IH), 6.99 (m, IH), 6.24 (m, IH), 5.63 (s, 2H), 1 NH resonance not observed. LCMS: 463 (M+H)+.
EXAMPLE 132 l-Benzyl-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using phenylmethanamine as the starting material. 1H NMR (300 MHz, CDCl3) δ 8.35 (m, IH), 8.26 (s, IH), 7.74 (m, IH), 7.63 (m, IH), 7.31 (m, 4H), 6.91 (m, 2H), 6.20 (m, IH), 5.58 (s, 2H), 1 NH resonance not observed. LCMS: 395 (M+H)+.
EXAMPLE 133 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using 3- (trifluoromethyl)aniline as the starting material. 1H NMR (300 MHz, DMSO-d6) δ 10.33 (s, IH), 9.26 (s, IH), 8.43 (s, IH), 8.13 (s, IH), 7.64 (m, 4H), 7.31 (s, IH), 7.10 (m, IH), 6.38 (m, IH). LCMS: 449 (M+H)+.
EXAMPLE 134 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(trifluoromethyl)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound can be prepared as described in EXAMPLE 96 using 4- (trifluoromethyl)aniline as the starting material. 1H NMR (300 MHz, DMSO-d6) δ 10.37 (s, IH), 9.25 (s, IH), 8.53 (s, IH), 8.12 (s, IH), 7.97 (m, 2H), 7.58 (m, 2H), 7.31 (m, IH), 7.08 (m, IH), 6.37 (m, IH). LCMS: 449 (M+H)+.
EXAMPLE 135
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-chloro-6- (trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-chloro-6- (trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, IH), 9.12 (s, IH), 8.30 (s, IH), 8.06 (d, IH), 7.85 (d, IH), 7.77 (d, IH), 7.61 (dd, IH), 7.22 (dd, IH), 7.02 (s, IH), 6.36 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 136 l-(4-Bromo-2-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-bromo-2- (trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, IH), 9.11 (s, IH), 8.34 (s, IH), 8.07 (d, IH), 7.97 (d, IH), 7.88 (dd, IH), 7.27 (d, IH), 7.17 (d, IH), 7.08 (d, IH), 6.32 (dd, IH). LCMS: 527 (M+H)+.
EXAMPLE 137
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-isopropyl-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-isopropyl- 2-(trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, IH), 9.09 (s, IH), 8.35 (s, IH), 8.06 (d, IH), 7.58 (s, IH), 7.54 (d, IH), 7.22 (d, IH), 7.15 (d, IH), 7.02 (s, IH), 6.28 (dd, IH), 3.00 (septet, IH), 1.20 (d, 6H). LCMS: 491 (M+H)+.
EXAMPLE 138 l-(4-Bromo-2-ethylphenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-bromo-2- ethylaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, IH), 9.10 (s, IH), 8.33 (s, IH), 8.08 (d, IH), 7.76 (d, IH), 7.39 (dd, IH), 7.13 (dd, IH), 7.05 (d, IH), 76.99 (dd, IH), 6.30 (dd, IH), 2.30-2.10 (m, 2H), 0.95 (t, 3H). LCMS: 487 (M+H)+.
EXAMPLE 139
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-propylphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- propylaniline as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (d, IH), 9.08 (d, IH), 8.31 (d, IH), 8.06 (d, IH), 7.29-7.22 (m, 2H), 7.18 (td, IH), 7.11 (dd, IH), 7.08 (dd, IH), 6.95 (dd, IH), 6.28 (dd, IH), 2.30-2.20 (m, 2H), 1.40-1.20 (m, 2H), 0.70 (t, 3H). LCMS: 423 (M+H)+.
EXAMPLE 140
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-methyl-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-methyl-2- (trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, IH), 9.10 (s, IH), 8.35 (s, IH), 8.08 (s, IH), 7.57 (s, IH), 7.45 (d, IH), 7.18 (d, IH), 7.14 (dd, IH), 6.99 (s, IH), 6.28 (dd, IH), 2.40 (s, 3H). LCMS: 463 (M+H)+.
EXAMPLE 141
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(dimethylamino)-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using N τl , τNvτl - dimethyl-3-(trifluoromethyl)benzene-l,4-diamine as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, IH), 9.04 (s, IH), 8.35 (s, IH), 8.08 (s, IH), 7.10 (dd, IH), 7.05 (d, IH), 6.92 (d, IH), 6.87 (dd, IH), 6.84 (d, IH), 6.23 (dd, IH), 2.90 (s, 6H). LCMS: 492 (M+H)+.
EXAMPLE 142
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- phenoxyaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, IH), 9.10 (s, IH), 8.25 (s, IH), 8.08 (s, IH), 7.31-7.27 (m, 2H), 7.24-7.19 (m, 2H), 7.13 (t, IH), 7.05-7.02 (m, 3H), 6.90 (d, 2H), 6.83 (d, IH), 6.20 (t, IH). LCMS: 473 (M+H)+.
EXAMPLE 143 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 3- phenoxyaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.30 (s, IH), 8.08 (s, IH), 7.39-7.31 (m, 3H), 7.22 (br, IH), 7.15-7.04 (m, 3H), 6.99-6.97 (m, 3H), 6.87 (dd, IH), 6.29 (t, IH). LCMS: 473 (M+H)+.
EXAMPLE 144
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4- phenoxyaniline as starting material. LCMS: 473 (M+H)+.
EXAMPLE 145
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2,4-(iichloro-6- (trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2,4-dichloro- 6-(trifluoromethyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.15 (s, IH), 8.30 (s, IH), 8.14 (d, IH), 8.08 (d, IH), 7.90 (d, IH), 7.24 (dd, IH), 7.05 (s, IH), 6.38 (dd, IH). LCMS: 517 (M+H)+.
EXAMPLE 146 3-(2-Chloro-4-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-5-methylisoxazole-4-carbohydrazide

Step 1 : 2-Chloro-4-(trifluoromethyl)benzaldehyde

S-BuLi (84.6 mL, 1.3M) was added to a cooled solution (-780C) of l-chloro-3- (trifluoromethyl)benzene (18.1 g, 100 mmol) in THF (200 mL). DMF (10 mL) was added at -78°C and the resulting solution was stirred for 1 h at -78°C then for 30 min at
250C. The reaction was then quenched by the addition of 100 rnL OfNH4Cl (sat.) and the aqueous layer was extracted with EtOAc (200 mL). The organic layer was concentrated under reduced pressure to give 2-chloro-4-(trifluoromethyl)benzaldehyde as a yellow oil.
Step 2: 2-Chloro-4-(trifluoromethyl)benzaldehyde oxime

A mixture of 2-chloro-4-(trifluoromethyl)benzaldehyde (20.5 g, 98.1 mmol), NH2OKHCl (10.4 g, 150 mmol), NaOH (6 g, 150 mmol) in H2O (80 mL)/ EtOH (150 mL) was stirred at 250C for 1 h. The EtOH was removed and the aqueous layer was extracted with EtOAc (3 x 100 mL). The organics were combined, dried, concentrated under reduced pressure and the residue was purified by silica gel chromatography (EtOAc/PE) to afford 2-chloro-4-(trifluoromethyl)benzaldehyde oxime as a yellow oil. Step 3 : 2-Chloro-N-hydroxy-4-(trifluoromethyl)benzimidoyl chloride

A mixture of 2-chloro-4-(trifluoromethyl)benzaldehyde oxime (7.6 g, 34 mmol) and NCS (4.5 g, 34 mmol) in DMF (30 mL) was stirred at 250C for 1 h. The resulting solution was diluted with H2O (50 mL) and extracted with EtOAc (3 x 80 mL). The combined organics were washed with brine (2 x 50 mL), dried, and concentrated under reduced pressure to afford 2-chloro-4-(trifluoromethyl)benzoyl chloride oxime as a yellow oil.
Step 4: Methyl 3-(2-chloro-4-(trifluoromethyl)phenyl)-5-methylisoxazole-4- carboxylate

NaH (60%) (120 mg, 3.00 mmol) was added into MeOH (10 niL) and the resulting solution was stirred at -50C for 15 min. Methyl 3-oxobutanoate (700 mg, 3.02 mmol) and 2-chloro-4-(trifluoromethyl)benzoyl chloride oxime (650 mg, 2.47 mmol) in MeOH (5 mL) were then added and the resulting solution was stirred at -50C for 30 min. The mixture was concentrated under reduced pressure and diluted with H2O (20 mL). The aqueous layer was extracted with EtOAc (3 x 30 mL), the organics were combined, dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford methyl 3-(2-chloro-4- (trifluoromethyl)phenyl)-5-methylisoxazole-4-carboxylate as a light yellow oil. LCMS: 320 (M+H)+.
Step 5 : 3-(2-Chloro-4-(trifluoromethyl)phenyl)-5-methylisoxazole-4-carboxylic acid

The Title compound was synthesized as described for Intermediate 1, Step 2 using 3- (2-chloro-4-(trifluoromethyl)phenyl)-5-methylisoxazole-4-carboxylate as the starting material. LCMS: 306 (M+H)+.
Ste2_6: 3-(2-Chloro-4-(trifluoromethyl)phenyl)-N'-(3-chloro-5-
(trifluoromethyl)pyridin-2-yl)-5-methylisoxazole-4-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using 3-(2-chloro-4- (trifluoromethyl)phenyl)-5-methylisoxazole-4-carboxylic acid and Intermediate 3 as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (m, IH), 7.80 (s, IH), 7.80-7.70 (m, 4H), 7.25 (s, IH), 2.80 (s, 3H). LCMS: 499 (M+H)+.
EXAMPLE 147 l-(3-Chloro-2-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide
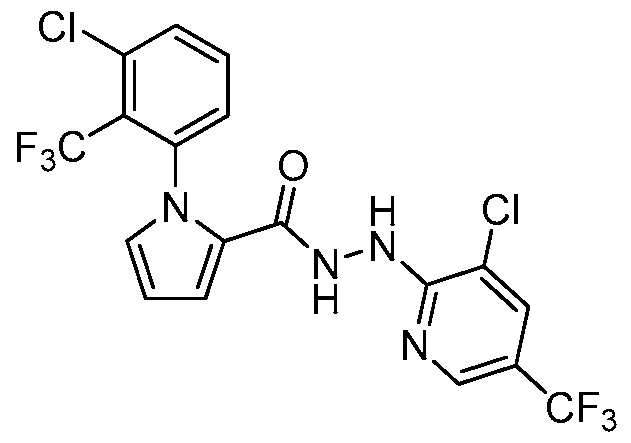
The title compound was synthesized as described in EXAMPLE 53 using l-chloro-3- fluoro-2-(trifluoromethyl)benzene as starting material. 1H NMR (400 MHz, DMSO- d6) δ 10.20 (s, IH), 9.18 (s, IH), 8.40 (s, IH), 8.10 (s, IH), 7.75 (d, IH), 7.64 (dd, IH), 7.25 (d, IH), 7.15 (dd, IH), 7.13 (dd, IH), 6.31 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 148 l-(5-Chloro-2-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 4-chloro-2- fluoro-l-(trifluoromethyl)benzene as starting material. 1H NMR (400 MHz, DMSO- d6) δ 10.20 (s, IH), 9.18 (s, IH), 8.38 (s, IH), 8.06 (s, IH), 7.80 (d, IH), 7.69 (dd, IH), 7.47 (d, IH), 7.17 (dd, IH), 7.11 (s, IH), 6.32 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 149 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(4-
(trifluoromethyl)phenoxy)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-(4- (trifluoromethyl) phenoxy)aniline as starting material. LCMS: 541 (M+H)+.
EXAMPLE 150 l-(2-Bromo-4-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 96 using 2-bromo-4- (trifluoromethyl)aniline and (4-(4-fluorophenoxy) phenyl)methanamine as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (t, IH), 8.16 (d, IH), 7.89 (dd, IH), 7.65 (d, IH), 7.34-7.24 (m, 4H), 7.13 (dd, IH), 7.12-7.06 (m, 3H), 7.04-6.98 (m, 2H), 6.39 (dd, IH), 4.38 (d, 2H). LCMS: 533 (M+H)+.
EXAMPLE 151
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(2,4,5-trichlorophenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2,4,5- trichloroaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.20 (s, IH), 8.40 (s, IH), 8.08 (d, IH), 7.98 (s, IH), 7.76 (s, IH), 7.20 (dd, IH), 7.10 (dd, IH), 6.40 (dd, IH). LCMS: 483 (M+H)+.
EXAMPLE 152 l-(2-Ethylphenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(2- ethylphenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (t, IH), 7.36-7.24 (m, 3H), 7.24-7.14 (m, 5H), 7.10 (dd, IH), 7.02-6.84 (m, 5H), 6.20 (m, IH), 4.20 (d, 2H), 2.20 (q, 2H), 1.00 (t, 3H). LCMS: 415 (M+H)+.
EXAMPLE 153 l-(4-(Dimethylamino)-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4- (dimethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl) methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.50(t, IH), 7.22-7.16 (m, 4H), 7.12-7.08 (d, IH), 7.02-6.98 (m, 2H), 6.94-6.82 (m, 6H), 6.16 (dd, IH), 4.20 (d, 2H), 3.00 (s, 6H). LCMS: 498 (M+H)+.
EXAMPLE 154 l-(2-Chloro-4-methylphenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-chloro-4- methylaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.10 (s, IH), 8.36 (s, IH), 8.08 (s, IH), 7.32 (d, IH), 7.22-7.17 (d, IH), 7.16-7.12 (m, 2H), 6.97 (dd, IH), 6.30 (dd, IH), 2.30 (s, 3H). LCMS: 429 (M+H)+.
EXAMPLE 155 l-(2-Chloro-4-methylphenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(2-chloro-4- methylphenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.60 (t, IH), 7.32 (d, IH), 7.24-7.14 (m, 6H), 7.02-6.98 (m, 2H), 6.96-6.94 (dd, IH), 6.92-6.86 (m, 3H), 6.24 (dd, IH), 4.28 (d, 2H), 2.38 (s, 3H). LCMS: 435 (M+H)+.
EXAMPLE 156 l-(2-Chloro-4-fluorophenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-chloro-4- fluoroaniline as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.10 (s, IH), 8.36 (s, IH), 8.08 (s, IH), 7.53 (dd, IH), 7.40 (dd, IH), 7.24 (td, IH), 7.17 (dd, IH), 7.03 (dd, IH), 6.32 (dd, IH). LCMS: 433 (M+H)+.
EXAMPLE 157 l-(4-Bromo-2-chlorophenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-bromo-2- chloroaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.10 (s, IH), 8.36 (s, IH), 8.08 (s, IH), 7.80 (d, IH), 7.56 (dd, IH), 7.28 (d, IH), 7.17 (dd, IH), 7.05 (dd, IH), 6.33 (dd, IH). LCMS: 493 (M+H)+.
EXAMPLE 158 l-(4-Chloro-2-methylphenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-chloro-2- methylaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.10 (s, IH), 8.36 (s, IH), 8.08 (d, IH), 7.35 (d, IH), 7.25 (dd, IH), 7.15-7.10 (m, 2H), 6.98 (dd, IH), 6.30 (dd, IH), 1.90 (s, 3H). LCMS: 429 (M+H)+.
EXAMPLE 159 l-(4-Chloro-2-methylphenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4-chloro-2- methylphenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.60 (t, IH), 7.35 (d, IH), 7.26 (dd, IH), 7.22-7.16 (m, 4H), 7.13 (d, IH), 7.02-6.96 (m, 3H), 6.94-6.86 (m, 3H), 6.24 (dd, IH), 4.20 (d, 2H), 1.84 (s, 3H). LCMS: 435 (M+H)+.
EXAMPLE 160
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-Cl^-dichlorophenylJ-lH-pyrrole-
2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2,4- dichloroaniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.10 (s, IH), 8.36 (s, IH), 8.08 (d, IH), 7.70 (d, IH), 7.45 (dd, IH), 7.36 (d, IH), 7.18 (dd, IH), 7.05 (dd, IH), 6.34 (dd, IH). LCMS: 449 (M+H)+.
EXAMPLE 161 l-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(2,4- dichlorophenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.60 (t, IH), 7.70 (d, IH), 7.46 (dd, IH), 7.39 (d, IH), 7.24- 7.16 (m, 4H), 7.02-6.88 (m, 6H), 6.27 (dd, IH), 4.22 (d, 2H). LCMS: 455 (M+H)+.
EXAMPLE 162 l-(2-Chloro-4-(methylsulfonyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-chloro-4- (methylsulfonyl)aniline as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.15 (s, IH), 8.36 (s, IH), 8.08 (d, 2H), 7.90 (dd, IH), 7.60 (d, IH), 7.22 (dd, IH), 7.12 (dd, IH), 6.38 (dd, IH), 3.30 (s, 3H). LCMS: 493 (M+H)+.
EXAMPLE 163 l-(2-Chloro-4-(methylsulfonyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(2-chloro-4- (methylsulfonyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4-fluorophenoxy) henyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO- d6) δ 8.70 (t, IH), 8.08 (d, IH), 7.92 (dd, IH), 7.65 (d, IH), 7.25-7.16 (m, 4H), 7.06 (dd, IH), 7.04-6.98 (m, 3H), 6.94-6.88 (m, 2H), 6.32 (dd, IH), 4.22 (d, 2H), 3.38 (s, 3H). LCMS: 499 (M+H)+.
EXAMPLE 164 l-(2-Chloro-4-cyanophenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 3-chloro-4- fluorobenzonitrile as starting material. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.18 (s, IH), 8.40 (s, IH), 8.18 (s, IH), 8.08 (d, IH), 7.88 (dd, IH), 7.55 (d, IH), 7.20 (dd, IH), 7.10 (dd, IH), 6.40 (dd, IH). LCMS: 440 (M+H)+.
EXAMPLE 165 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(trifluoromethyl)phenethyl)- lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-(4- (trifluoromethyl) phenyl)ethanamine as starting material. LCMS: 477 (M+H)+.
EXAMPLE 166
N-(4-(4-Fluorophenoxy)benzyl)-l-(4-nitro-2-(trifluoromethyl)phenyl)-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 53 using l-fluoro-4- nitro-2-(trifluoromethyl)benzene and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (t, IH), 8.54-8.48 (m, 2H), 7.72-7.66 (m, IH), 7.24-7.16 (m, 4H), 7.10-7.06 (m, 2H), 7.04-6.96 (m, 2H), 6.94-6.88 (m, 2H), 6.32 (dd, IH), 4.22 (d, 2H). LCMS: 500 (M+H)+.
EXAMPLE 167
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(methylsulfonyl)phenethyl)- lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2-(4- (methylsulfonyl) phenyl)ethanamine hydrochloride as starting material. LCMS: 487 (M+H)+.
EXAMPLE 168 l-(4-(Diethylamino)-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide

Step 1 : Methyl l-(4-nitro-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

The title compound was synthesized as described for Intermediate 1, Stepl using 1- fluoro-4-nitro-2-(trifluoromethyl)benzene as starting materials. LCMS: 315 (M+H)+. Step 2: Methyl l-(4-amino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(4-nitro-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate (1.0 g, 3.3 mmol), 10% Pd/C (0.1 g) in MeOH (8 mL)/DCM (5 mL) was stirred vigorously at 250C under an atmosphere of H2 for 18 h. Pd/C was removed by filtration through celite and the filtrate was concentrated to give methyl l-(4-amino-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as a grey oil. 1H NMR (400 MHz, DMSO-d6) δ 7.20-7.15 (m, IH), 6.98 (d, IH), 6.93 (dd, IH), 6.90 (d, IH), 6.76 (dd, IH), 6.22 (dd, IH), 5.86-5.72 (br, 2H), 3.58 (s, 3H). LCMS: 285.2 (M+H)+.
Step 3 : Methyl l-(4-(diethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2- carboxylate

Sodium hydride (60%, 0.32g, 8.0 mmol) was added slowly (caution: gas evolution) to a solution of methyl 1 -(4-amino-2-(trifluoromethyl)phenyl)- 1 H-pyrrole-2-carboxylate (0.14 g, 0.49 mmol) in DMSO (2 mL). The resulting mixture was stirred at 250C until the reaction became homogeneous (~30 min). Iodoethane (0.119 mL, 1.48 mmol) was added to the dark blue solution and the resulting solution was stirred at 250C for 18 h. The reaction was partitioned between brine (10 mL) and EtOAc (25 mL). The organic layer was dried, filtered, concentrated, and purified by silica gel chromatography (0 to 30% EtOAc/Hexanes) to afford methyl l-(4-(diethylamino)-2- (trifluoromethyl)phenyl)-l H-pyrrole-2-carboxylate as a white solid. LCMS: 341 (M+H)+.
Step 4: l-(4-(Diethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl 1 -(4-(diethylamino)-2-(trifluoromethyl)phenyl)- 1 H-pyrrole-2-carboxylate as the starting material. LCMS: 327 (M+H)+.
Step 5: l-(4-(Diethylamino)-2-(trifluoromethyl)phenyl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4- (diethylamino)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl) methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.50 (t, IH), 7.24-7.14 (m, 4H), 7.07 (d, IH), 7.02-6.96 (m, 2H), 6.94-6.88 (m, 3H), 6.88-6.80 (m, 2H), 6.77 (d, IH), 6.16 (dd, IH), 4.22 (d, 2H), 3.40 (q, 4H), 1.10 (t, 6H). LCMS: 526 (M+H)+.
EXAMPLE 169 l-(4-(Ethylamino)-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as a side product (from Step 3) as described in EXAMPLE 168. LCMS: 498 (M+H)+.
EXAMPLE 170 l-(4-Amino-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

Step 1 : l-(4-Amino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl l-(4-amino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as the starting material. LCMS: 271 (M+H)+. Step 2: l-(4-Amino-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4-amino-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl) methanamine as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.40 (t, IH), 7.24-7.14 (m, 5H), 7.04-6.84 (m, 8H), 6.80 (s, IH), 6.74 (dd, IH), 6.14 (dd, IH), 4.22 (d, 2H). LCMS: 470 (M+H)+.
EXAMPLE 171 l-(4-Acetamido-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4-amino-2- (trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide and acetic acid as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, IH), 8.60 (t, IH), 8.05 (d, IH), 7.75 (dd, IH), 7.28 (d, IH), 7.24-7.14 (m, 4H), 7.04- 6.95 (m, 3H), 6.95-6.85 (m, 3H), 6.20 (dd, IH), 4.22 (d, 2H), 2.10(s, 3H). LCMS: 512 (M+H)+.
EXAMPLE 172 N-(4-(4-Fluorophenoxy)benzyl)-l-(4-morpholino-2-(trifluoromethyl)phenyl)-lH- pyrrole-2-carboxamide


NaH (60%) (39 mg, 0.98 mmol) was added slowly (caution: gas evolution) to a solution of methyl 1 -(4-amino-2-(trifluoromethyl)phenyl)- 1 H-pyrrole-2-carboxylate (0.14 g, 0.49 mmol) and DMF (1 mL) and the resulting solution was stirred at 250C until the reaction became homogeneous (~30 min). 2-Bromoethyl ether (0.12 mL, 0.98 mmol) was added and the resulting mixture was heated at 1000C for 3 h and then allowed to cool to 250C. The reaction was partitioned between IN HCl (5 mL) and EtOAc (10 mL). The organic layer was concentrated under reduced pressure and purified by silica gel chromatography (0 to 35% EtOAc/hexanes) to afford methyl 1- (4-morpholino-2-(trifluoromethyl)phenyl)-l H-pyrrole-2-carboxylate as a yellow oil. LCMS: 355 (M+H)+. Step 2: l-(4-Morpholino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for INtermediate 1 , Step 2 using methyl l-(4-morpholino-2-(trifluoromethyl)phenyl)-l H-pyrrole-2-carboxylate as the starting material. LCMS: 341 (M+H)+.
Step 3 : N-(4-(4-Fluorophenoxy)benzyl)-l-(4-morpholino-2-
(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(4- morpholino-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy) phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.50 (t, IH), 7.22-7.14 (m, 7H), 7.04-6.96 (m, 2H), 6.96-6.92 (dd, IH), 6.92-6.88 (m, 2H), 6.86 (br, IH), 6.20-6.16 (m, IH), 4.22 (d, 2H), 3.78 (t, 4H), 3.20 (t, 4H). LCMS: 540 (M+H)+.
EXAMPLE 173 l-(2-Chloro-4-(dimethylamino)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

Step 1 : Methyl l-(2-chloro-4-nitrophenyl)-lH-pyrrole-2-carboxylate
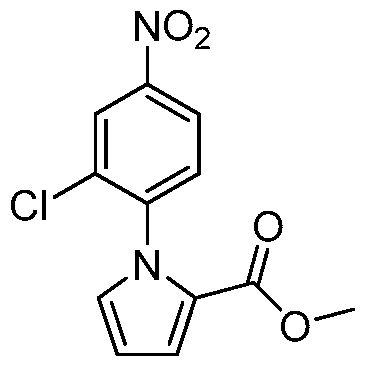
Step 2: Methyl l-(4-amino-2-chlorophenyl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(2-chloro-4-nitrophenyl)-lH-pyrrole-2-carboxylate (0.56 g, 2.0 mmol) and SnCl2.2H2O (2.26 g, 10.0 mmol) in EtOH (10 mL) was refluxed for 3 h and cooled to 250C. The reaction mixture was concentrated under reduced pressure, diluted with HCl (0.1M, 20 mL), and extracted with EtOAc (2 x 45 mL). The organics were combined, washed with brine (20 mL), dried, and concentrated under reduced pressure to afford methyl l-(4-amino-2-chlorophenyl)-lH-pyrrole-2-carboxylate. LCMS: 251 (M+H)+. Step 3 : l-(2-Chloro-4-(dimethylamino)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described in EXAMPLE 168, Step 3 and 4 using methyl l-(4-amino-2-chlorophenyl)-lH-pyrrole-2-carboxylate and iodomethane as the starting materials. LCMS: 279 (M+H)+.
Step 4: l-(2-Chloro-4-(dimethylamino)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

EXAMPLE 174
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-5-methyl-l-(5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2-fluoro-5- (trifluoromethyl)pyridine and ethyl 5 -methyl- lH-pyrrole-2-carboxylate as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, IH), 9.13 (s, IH), 8.90 (s, IH), 8.40 (s, IH), 8.30 (dd, IH), 8.06 (d, IH), 7.54 (d, IH), 7.06 (d, IH), 6.12 (d, IH), 2.10 (s, 3H). LCMS: 464 (M+H)+.
EXAMPLE 175
5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)thiazole-4-carboxamide


To a mixture of 2,4-dichlorobenzaldehyde (13 g, 75 mmol) and methyl 2,2- dichloroacetate (10.6 g, 74.6 mmol) in Et2O (300 mL) was added a solution of MeONa (5.4 g, 100 mmol) in MeOH (30 mL) dropwise with stirring at O0C. The resulting solution was stirred at O0C for 2 h then diluted with of brine (200 mL). The aqueous solution was extracted with EtOAc (3 x 100 mL), the organics were combined, dried, and concentrated under reduced pressure to afford methyl 3-chloro-3-(2,4- dichlorophenyl)-2-oxopropanoate as a yellow solid. Step 2: Methyl 2-amino-5-(2,4-dichlorophenyl)thiazole-4-carboxylate

A solution of methyl 3-chloro-3-(2,4-dichlorophenyl)-2-oxopropanoate (21 g, 71 mmol) and thiourea (6 g, 75 mmol) in MeOH (250 mL) was refluxed over night. The resulting mixture was concentrated under reduced pressure and the pH was adjusted to 7 with NH4OH (18M). The resulting solution was extracted with DCM (4 x 100 mL), the organicswere combined, dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/PE) to afford methyl 2- amino-5-(2,4-dichlorophenyl)thiazole-4-carboxylate as a yellow solid. Step 3 : Methyl 5-(2,4-dichlorophenyl)thiazole-4-carboxylate

To a solution of methyl 2-amino-5-(2,4-dichlorophenyl)thiazole-4-carboxylate (800 mg, 2.65 mmol) in H3PO2(50%, 30 mL) was added a solution OfNaNO2 (360 mg, 5.22 mmol) in H2O (10 mL) in several batches at O0C. NaOH (210 mg, 5.25 mmol) was
added and the resulting solution was stirred for 1 h at O0C. The pH of the solution was adjusted to 7 with NaOH/NaHCC>3 and the aqueous layer was extracted with EtOAc (3 x 100 mL). The organics were combined, dried, concentrated under reduced pressure, and purified by silica gel chromatography (EtOAc/PE) to afford methyl 5-(2,4- dichlorophenyl)thiazole-4-carboxylate as a yellow solid. Step 4: 5-(2,4-Dichlorophenyl)thiazole-4-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl 5-(2,4-dichlorophenyl)thiazole-4-carboxylate as the starting material. LCMS: 274 (M+H)+.
Step 5 : 5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)thiazole-4-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 5-(2,4- dichlorophenyl) thiazole-4-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 9.28 (s, IH), 9.00 (t, IH), 7.70 (d, IH), 7.50-7.40 (m, 2H), 7.30-7.15 (m, 4H), 7.05-6.95 (m, 2H), 6.94-6.88 (m, 2H), 4.30 (d, 2H). LCMS: 473 (M+H)+.
EXAMPLE 176
4-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)thiazole-5-carboxamide

Step 1 : Methyl 2-bromo-3-(2,4-dichlorophenyl)-3-oxopropanoate

A solution of methyl 3-(2,4-dichlorophenyl)-3-oxopropanoate (500 mg, 2.01 mmol), NBS (398 mg, 2.23 mmol), and AIBN (15 mg, 0.09 mmol) in CCl4 (25 mL) was stirred for 20 min at 650C. The reaction was diluted with ice/H2O (20 mL) and the aqueous layer was extracted with DCM (3 x 20 mL). The organics were combined, dried, and concentrated under reduced pressure to afford methyl 2-bromo-3-(2,4-dichlorophenyl)- 3-oxopropanoate as yellow oil. LCMS: 325 (M+H)+. Step 2: Methyl 2-amino-4-(2,4-dichlorophenyl)thiazole-5-carboxylate

A mixture of thiourea (235 mg, 3.06 mmol) and methyl 2-bromo-3-(2,4- dichlorophenyl)-3-oxopropanoate (1 g, 3.06 mmol) in EtOH (30 mL) was heated to 750C for 2 h. The resulting mixture was concentrated under reduced pressure then diluted with ice/H2O (30 mL) and the aqueous layer was extracted with DCM (3 x 20 mL). The organics were combined, dried, concentrated under reduced pressure, and the residue was purified by silica gel chromatography (EtOAc/PE) to afford methyl 2- amino-4-(2,4-dichlorophenyl)thiazole-5-carboxylate as a pale yellow solid. LCMS: 303 (M+H)+.
_3 : Methyl 4-(2,4-dichlorophenyl) thiazole-5-carboxylate

NaNO2 (69 mg, 0.99 mmol, 99%) in H2O (1 niL) was added dropwise to a solution of methyl 2-amino-4-(2,4-dichlorophenyl)thiazole-5-carboxylate (151 mg, 0.49 mmol) and H3PO2 (10 mL, 50%) cooled to O0C. The resulting mixture was stirred ay O0C for 1 h and at 250C for 3 h. The pH was adjusted to 8 with NaHCO3 and the aqueous solution was extracted with EtOAc (3 x 50 mL). The organics were combined, dried, evaporated under reduced pressure, and the residue was purified by silica gel chromatography (EtOAc/PE) to afford methyl 4-(2,4-dichlorophenyl) thiazole-5- carboxylate as a yellow solid. LCMS: 288 (M+H)+. Step 4: 4-(2,4-Dichlorophenyl)thiazole-5-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl 4-(2,4-dichlorophenyl) thiazole-5-carboxylate as the starting material. LCMS: 274 (M+H)+.
Step 5 : 4-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)thiazole-5-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 4-(2,4- dichlorophenyl) thiazole-5-carboxylic acid and (4-(4-
fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, IH), 8.70 (m, IH), 7.66 (m, IH), 7.44 (m, 2H), 7.24- 7.12 (m, 4H), 7.02 (m, 2H), 6.90 (m, 2H), 4.30 (d, 2H). LCMS: 473 (M+H)+.
EXAMPLE 177 l-(2-chloro-4-(methylamino)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as a side product as described in EXAMPLE 173. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (t, IH), 7.25-7.15 (m, 4H), 7.04-6.96 (m, 3H), 6.94-6.86 (m, 3H), 6.79 (dd, IH), 6.56 (d, IH), 6.46 (dd, IH), 6.17 (dd, IH), 4.25-4.00 (m, 3H), 2.65 (s, 3H). LCMS: 450 (M+H)+.
EXAMPLE 178 5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)-l,2,3-thiadiazole-4-
carboxamide
 Step 1 : Diethyl 2-(2,4-dichlorophenyl)-3-oxosuccinate
Step 1 : Diethyl 2-(2,4-dichlorophenyl)-3-oxosuccinate

Ethyl 2-(2,4-dichlorophenyl)acetate (22 g, 94.42 mmol) and diethyl oxalate (27.5 g, 188.36 mmol) were added to a solution of sodium ethoxide prepared from NaH (7.5 g, 187.50 mmol, 60%) in EtOH (150 mL) and the mixture was stirred for 2 h at 7O0C. The resulting mixture was concentrated under reduced pressure, diluted with EtOAc (200 mL), washed with H2O (2 x 200 mL), dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/PE) to afford diethyl 2-(2,4-dichlorophenyl)-3-oxosuccinate as a colorless oil. Step 2: Ethyl 3-(2,4-dichlorophenyl)-2-oxopropanoate

NaCl (2.3 g, 39.66 mmol) was added to a solution of diethyl 2-(2,4-dichlorophenyl)-3- oxosuccinate (13 g, 39.04 mmol) in DMSO/H2O (150/15 mL) and the resulting mixture was heated to 12O0C for 1 h. The resulting solution was diluted with H2O (200 mL), extracted with EtOAc (3 x 250 mL), the organics combined, dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/PE) to afford ethyl 3-(2,4-dichlorophenyl)-2-oxopropanoate as a white solid.
Step 3 : Ethyl 2-(3-(2,4-dichlorophenyl)-l-ethoxy-l-oxopropan-2- ylidene)hydrazine carboxylate

Ethyl carbazate (1.27 g, 12.21 mmol) was added to a solution of ethyl 3-(2,4- dichlorophenyl)-2-oxopropanoate (3.2 g, 12.26 mmol) in toluene (100 mL) and the resulting mixture was heated to 1000C for 5 h. The mixture was concentrated under reduced pressure and the residues was purified by silica gel chromatography (EtOAc/PE) to afford ethyl 2-(3-(2,4-dichlorophenyl)-l-ethoxy-l-oxopropan-2- ylidene)hydrazinecarboxylate as a yellow oil.
Step 4: Ethyl 5-(2,4-dichlorophenyl)-l,2,3-thiadiazole-4-carboxylate

Ethyl 2-(3-(2,4-dichlorophenyl)- 1 -ethoxy- 1 -oxopropan-2-ylidene)hydrazinecarboxylate (2.5 g, 7.20 mmol) was dissolved in SOCl2 (50 mL) and it was heated to 6O0C for 18 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography (EtOAc/PE) to give ethyl 5-(2,4-dichlorophenyl)- 1,2,3- thiadiazole-4-carboxylate as a white solid.
_5: 5-(2,4-Dichlorophenyl)-l,2,3-thiadiazole-4-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using ethyl 5-(2,4-dichlorophenyl)-l,2,3-thiadiazole-4-carboxylate as the starting material. LCMS: 275 (M+H)+. Step 6: 5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)-l,2,3-thiadiazole-4- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 5-(2,4- dichlorophenyl)-l,2,3-thiadiazole-4-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the tarting materials. 1H NMR (400 MHz, DMSO-de) δ 9.65 (t, IH), 7.80 (d, IH), 7.57 (d, IH), 7.52 (dd, IH), 7.30- 7.25 (m, 2H), 7.24-7.15 (m, 2H), 7.05-6.98 (m, 2H), 6.95-6.90 (m, 2H), 4.40 (d, 2H). LCMS: 474 (M+H)+.
EXAMPLE 179 tert-Butyl 4-(2-(4-(4-fluorophenoxy)benzylcarbamoyl)-lH-pyrrol-l-yl)-3- (trifluoromethyl)benzylcarbamate

Step 1 : Methyl l-(4-cyano-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

The title compound was synthesized as described for Intermediate 1 , Step 1 using 4- fluoro-3-(trifluoromethyl)benzonitrile as the starting material.
Step 2: Methyl l-(4-(aminomethyl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-
carboxylate

A mixture of methyl l-(4-cyano-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate (0.5 g, 1.7 mmol), 10% Pd/C (50 mg, 0.024 mmol), in HOAc (6 mL) was shaken vigorously at 250C under 50 PSI of H2 overnight. Pd/C was removed by filtration, the filtrate was partitioned between saturated bicarbonate solution (20 mL) and EtOAc(45 mL). The organic layer was dried, filtered, and concentrated to afford methyl l-(4- (aminomethyl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as a light dark oil. LCMS: 299 (M+H)+.
Step 3 : Methyl l-(4-((tert-butoxycarbonylamino)methyl)-2- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(4-(aminomethyl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2- carboxylate (0.52 g, 1.7 mmol), (Boc)2O (0.57 g, 2.6 mmol), and Et3N (0.36 mL, 2.6 mmol) in EtOAc (5 mL) was stirred at 250C for 18 h. The reaction mixture was partitioned between saturated bicarbonate solution (20 mL) and EtOAc (45 mL). The organic layer was dried, filtered, and concentrated to afford methyl l-(4-((tert- butoxycarbonylamino)methyl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate (-77% pure) as a yellow oil.
Step 4: l-(4-((tert-Butoxycarbonylamino)methyl)-2-(trifluoromethyl)phenyl)-lH- pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl 1 -(4-((tert-butoxycarbonylamino)methyl)-2-(trifluoromethyl)phenyl)- 1 H- pyrrole-2-carboxylate as the starting material.
Step 5 : tert-Butyl 4-(2-(4-(4-fluorophenoxy)benzylcarbamoyl)-lH-pyrrol-l-yl)-3- (trifluoromethyl)benzylcarbamate

The title compound was synthesized as described in EXAMPLE 1 using l-(4-((tert- butoxycarbonyl amino)methyl)-2-(trifluoromethyl)phenyl)- 1 H-pyrrole-2-carboxylic acid and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (br, IH), 8.55 (t, IH), 7.62-7.48 (m, 3H), 7.35- 7.15 (m, 4H), 7.05-6.85 (m, 6H), 6.28-6.20 (m, IH), 4.30-4.10 (m, 4H), 1.40 (s, 9H). LCMS: 584 (M+H)+.
EXAMPLE 180 l-(4-(lH-Tetrazol-5-yl)-2-(trifluoromethyl)phenyl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide


A mixture of methyl l-(4-cyano-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate, Bu2SnO (0.4 g, 1.7 mmol), and TMSN3 (0.45 mL, 3.4 mmol) in toluene (10 mL) was refluxed for 18 h. The mixture was cooled down, concentrated under reduced pressure, and purified by silica gel chromatography (EtOAc/hexanes) to afford methyl 1-(4-(1H- tetrazol-5-yl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as a white solid. LCMS: 338 (M+H)+.
Step 2: l-(4-(lH-Tetrazol-5-yl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2- carboxylic acid

The title compound was synthesized as described for Intermediate 1, Step 2 using l-(4- (lH-tetrazol-5-yl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as the starting material.
Step 3 : tert-Butyl 4-(2-(4-(4-fluorophenoxy)benzylcarbamoyl)-lH-pyrrol-l-yl)-3- (trifluoromethyl)benzylcarbamate

The title compound was synthesized as described in EXAMPLE 53 using 1-(4-(1H- tetrazol-5-yl)-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine as the tarting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (t, IH), 8.40 (d, 1H),8.32 (dd, IH), 7.60 (d, IH), 7.24-7.14 (m, 4H), 7.06 (d, 2H), 7.04-6.96 (m, 2H), 6.94-6.88 (m, 2H), 6.30 (dd, IH), 4.22 (d, 2H), 1 NH resonance not observed. LCMS: 523 (M+H)+.
EXAMPLE 181 3-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isothiazole-4-carboxamide

Step 1 : Ethyl 3-amino-3- (2,4-dichlorophenyl) acrylate

Zn (18.89 g, 291 mmol) was added to a solution of 2,4-dichlorobenzonitrile (10 g, 58 mmol) and methanesulfonic acid (1 niL) in THF (200 niL) at rt under N2. The solution of ethyl 2-bromoacetate (29.13 g, 174 mmol) in THF (100 mL) was then added dropwise and the resulting solution was refluxed for 1 h. MeOH (50 mL) was added to quench the reaction and the solids were removed by filtration. The filtrate was concentrated and the crude product was purified by silica gel column chromatography (EtOAc/PE) to afford ethyl 3-amino-3- (2,4-dichlorophenyl) acrylate as a yellow oil. Step 2: Ethyl 3-(2,4-dichlorophenyl) isothiazole-4-carboxylate

POCI3 (4.9 g, 32 mmol) was added in several batches to a solution of ethyl 3-amino-3- (2,4-dichlorophenyl) acrylate (6.8 g, 26 mmol) in DMF (20 mL) at -6O0C. The mixture was allowed to warm up about 250C and kept for 1 h at this temperature. It was then poured into a cool solution of Na2S.9H2O (6.28 g, 26.17 mmol) in H2O (50 mL). The resulting solution was stirred at 250C for 30 min and the solids were collected by filtration, dissolved in benzene (500 mL), and dried with CaSO4. A mixture of I2 (6 g, 24 mmol) in benzene (500 mL) was added to this mixture and stirred for 30 min at 250C. The resulting mixture was washed with 10%Na2CO3 (500 mL) and 5%Na2S2O3 (500 mL). The benzene layer was dried with CaSO4 and concentrated under reduced pressure to afford ethyl 3-(2,4-dichlorophenyl) isothiazole-4-carboxylate as a red yellow oil. LCMS: 302 (M+H)+.
_3 : 3-(2,4-Dichlorophenyl)isothiazole-4-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using ethyl 3-(2,4-dichlorophenyl) isothiazole-4-carboxylate as the starting material. LCMS: 274 (M+H)+.
Step 4: 3-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isothiazole-4-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 3-(2,4- dichlorophenyl)isothiazole-4-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 9.50 (s, IH), 9.00 (t, IH), 7.64 (d, IH), 7.46 (dd, IH), 7.40 (d, IH), 7.26-7.16 (m, 4H), 7.04-6.98 (m, 2H), 6.94-6.90 (m, 2H), 4.30 (d, 2H). LCMS: 473 (M+H)+.
EXAMPLE 182 l-(2-Ethyl-4-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-
2-carboxamide

Step 1 : ((2-Fluoro-5-(trifluoromethyl)phenyl)ethynyl)trimethylsilane

A mixture of 2-bromo-l-fluoro-4-(trifluoromethyl)benzene (1.14 mL, 10 mmol), ethynyltrimethylsilane (2.1 mL, 15 mmol), CuI (0.38 g, 2 mmol), TEA (8.3 mL, 60 mmol), and Pd(Ph3P)4 (0.58 g, 0.5 mmol) in THF (30 mL) were stirred at rt under N2 overnight. The mixture was diluted with EtOAc (45 mL) and it was washed with H2O
(2OmL), brine (20 mL), and saturated sodium bicarbonate solution (20 mL). The organics were filtered, dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (0-20% EtOAc/hexanes) to afford ((2- fluoro-5-(trifluoromethyl)phenyl)ethynyl) trimethylsilane.
Step_l: Methyl l-(4-(trifluoromethyl)-2-((trimethylsilyl)ethynyl)phenyl)-lH- pyrrole-2-carboxylate

The title compound was synthesized as described for Intermediate 1 , Step 1 using ((2- fluoro-5-(trifluoromethyl)phenyl)ethynyl)trimethylsilane, as the starting material.
Step 3 : Methyl l-(2-ethynyl-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(4-(trifluoromethyl)-2-((trimethylsilyl)ethynyl)phenyl)-lH- pyrrole-2-carboxylate (60mg, 0.16 mmol) and TBAF (0.19 mL, 0.19 mmol) in THF (1 mL) was stirred at rt for 1 h. The resulting mixture was partitioned between EtOAc (10 mL) and saturated sodium bicarbonate solution (5 mL). The organic layer was concentrated under reduced pressure to give methyl l-(2-ethynyl-4- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as a dark oil. LCMS: 294 (M+H)+. Step 4: Methyl l-(2-ethyl-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(2-ethynyl-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate (0.1 g, 0.34 mmol), 10% Pd/C (40 mg, 0.019 mmol) in EtOH (2 mL) was stirred vigorously at rt under an atmosphere of H2 (balloon) overnight. Pd/C was removed by fϊltation and the filtrate was partitioned between EtOAc (10 mL) and IN HCl (5 mL). The organic layer was concentrated under reduced pressure to give methyl l-(2-ethyl- 4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as a brown oil. LCMS: 298 (M+H)+.
Step 5 : l-(2-Ethyl-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl l-(2-ethyl-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as the starting material. LCMS: 284 (M+H)+. Step 6: l-(2-Ethyl-4-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(2-ethyl-4- (trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid and (4-(4-fluorophenoxy) phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.60 (m, IH), 7.64 (m, IH), 7.58 (m, IH), 7.33 (m, IH), 7.24-7.14 (m, 4H), 7.06-6.94 (m, 4H), 6.92-6.86 (m, 2H), 6.30 (m, IH), 4.22 (m, 2H), 2.25 (q, 2H), 0.95 (t, 3H). LCMS: 483 (M+H)+.
EXAMPLE 183 l-(4-(Aminomethyl)-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide

A mixture of tert-butyl 4-(2-(4-(4-fluorophenoxy)benzylcarbamoyl)-lH-pyrrol-l-yl)-3- rifluoromethyl)benzylcarbamate (0.17 g, 0.29 mmol) and TFA (2 rnL) in DCM (2 mL) was stirred at 250C for 1 h. It was concentrated under reduced pressure and the residue was partitioned between DCM (30 mL) and 0.3 N NaOH (10 mL). The organic layer was concentrated under reduced pressure and the residue was purified by semi- preparative chromatography (CHSCNZH2OITF A) to afford l-(4-(aminomethyl)-2- (trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- 1 H-pyrrole-2-carboxamide as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 8.60(t, IH), 8.30-8.10 (m, 2H), 7.91 (d, IH), 7.74 (dd, IH), 7.43 (d, IH), 7.24-7.14 (m, 4H), 7.04-6.96 (m, 3H), 6.95-6.85 (m, 3H), 6.26 (dd, IH), 4.28-4.24 (m, 4H). LCMS: 484 (M+H)+.
EXAMPLE 184 l-(4-(Acetamidomethyl)-2-(trifluoromethyl)phenyl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

EXAMPLE 185 l-(4-((dimethylamino)methyl)-2-(trifluoromethyl)phenyl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

A mixture of l-(4-(Aminomethyl)-2-(trifluoromethyl)phenyl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide (28 mg, 0.058 mmol), HCHO (37%, 17 uL, 0.23 mmol), and NaBH3CN ( 7 mg, 0.116 mmol) in DCM (1 mL) was stirred overnight at rt. The resulting solution was concentrated under reduced pressure and purified by semi-preparative chromatography (CH3CNZH2OITFA) to afford the title compound as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 8.60(t, IH), 8.00 (s, IH), 7.80 (dd, IH), 7.50 (d, IH), 7.25-7.15 (m, 4H), 7.06-6.88 (m, 6H), 6.28 (dd, IH), 4.40 (s, 2H), 4.22 (d, 2H), 2.80 (br, 6H). LCMS: 512 (M+H)+.
EXAMPLE 186 4-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isoxazole-3-carboxamide

J_: 2-(2,4-Dichlorophenyl)acetaldehyde

Diisopropylaluminum (64 mL, IM in Hexanes) was added dropwise to a cooled (- 780C) solution of methyl 2-(2,4-dichlorophenyl)acetate (14 g, 61.01 mmol) in DCM (300 mL) and the mixture was stirred at -780C for 2 h. HCl (300 mL, IM) was added and the resulting mixture was stirred for 1 h at rt. The resulting solution was extracted with DCM (3 x 200 mL), the organics combined, dried, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc :PE) to afford 2-(2,4-dichlorophenyl) acetaldehyde as a yellow liquid. Step 2: Ethyl 4-(2,4-dichlorophenyl)isoxazole-3-carboxylate

A mixture of 2-(2,4-dichlorophenyl)acetaldehyde (1 g, 4.79 mmol), ethyl 2-chloro-2- (hydroxyimino)acetate (400 mg, 2.65 mmol), and NaOEt (360 mg, 5.29 mmol) in THF (60 mL) was heated to 5O0C for 30 min. The reaction mixture was diluted with H2O (200 mL) and the aqueous layer was extracted with DCM (3 x 50 mL), the organics combined, dried, and concentrated under reduced pressure. The residue was purified
by silica gel chromatography (EtOAc/PE) to afford ethyl 4-(2,4- dichlorophenyl)isoxazole-3-carboxylate as a yellow solid.
Step 3 : 4-(2,4-Dichlorophenyl)isoxazole-3-carboxylic acid
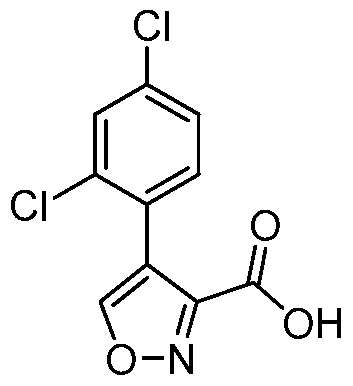
The title compound was synthesized as described for Intermediate 1 , Step 2 using ethyl 4-(2,4-dichlorophenyl)isoxazole-3-carboxylate as the starting material. LCMS: 258 (M+H)+.
Step 4: 4-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isoxazole-3-
carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 4-(2,4- dichlorophenyl) isoxazole-3-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 9.40 (t, IH), 9.28 (s, IH), 7.70 (d, IH), 7.50-7.44 (m, 2H), 7.28-7.16 (m, 4H), 7.06-7.00 (m, 2H), 6.94-6.90 (m, 2H), 4.40 (d, 2H). LCMS: 457 (M+H)+.
EXAMPLE 187
5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)oxazole-4-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 5-(2,4- dichlorophenyl)oxazole-4-carboxylic acid (commercially available) and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 9.00 (t, IH), 8.66 (s, IH), 7.80 (d, IH), 7.65 (d, IH), 7.53 (dd, IH), 7.30-7.24 (m, 2H), 7.22-7.16 (m, 2H), 7.04-6.98 (m, 2H), 6.94-6.88 (m, 2H), 4.36 (d, 2H). LCMS: 457 (M+H)+.
EXAMPLE 188
3-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isoxazole-4-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using methyl 3-(2,4- dichlorophenyl)isoxazole-4-carboxylate (J. Het. Chem. 2000, 37(1), 75-78) and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.45 (s, IH), 8.98 (t, IH), 7.76 (d, IH), 7.53 (dd, IH), 7.50 (d, IH), 7.30-7.18 (m, 4H), 7.06-6.98 (m, 2H), 6.94-6.88 (m, 2H), 4.30 (d, 2H). LCMS: 457 (M+H)+.
EXAMPLE 189
5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isoxazole-4-carboxamide

Step 1 : Methyl 2-(2,4-dichlorobenzoyl)-3-(dimethylamino)acrylate

A mixture of methyl 3-(2,4-dichlorophenyl)-3-oxopropanoate (1.8 g, 6.22 mmol) and dimethoxy-N,N-dimethylmethanamine (5 mL) was heated to 5O0C for 1 h. The reaction mixture was cooled to rt and diluted with H2O (50 mL). The aqueous layer was extracted with EtOAc (3 x 100 mL), the organics were combined, washed with brine (2 x 100 mL), dried, and evaporated under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/PE) to afford methyl 2-(2,4- dichlorobenzoyl)-3-(dimethylamino)acrylate as a yellow oil. Step 2: Methyl 5-(2,4-dichlorophenyl)isoxazole-4-carboxylate

A mixture of methyl 2-(2,4-dichlorobenzoyl)-3-(dimethylamino)acrylate (1.8 g, 5.98 mmol), sodium acetate (490 mg, 5.98 mmol), and hydroxylamine hydrochloride (412 mg, 5.97 mmol) in Et2O (50 mL)/CH30H (25 mL) was stirred for 21 h at 250C. The resulting mixture was diluted with EtOAc (50 mL), washed with brine (2 x 50 mL), dried, and concentrated under reduce pressure. The residue was purified by silica gel
chromatography (EtOAc/PE) to afford methyl 5-(2,4-dichlorophenyl)isoxazole-4- carboxylate as a white solid.
Step 3 : 5-(2,4-Dichlorophenyl)isoxazole-4-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl 5-(2,4-dichlorophenyl)isoxazole-4-carboxylate as the starting material. LCMS: 258 (M+H)+. Step 4: 5-(2,4-Dichlorophenyl)-N-(4-(4-fluorophenoxy)benzyl)isoxazole-4- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 5-(2,4- dichlorophenyl) isoxazole-4-carboxylic acid and (4-(4- fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 9.10 (s, IH), 8.95 (t, IH), 7.83 (d, IH), 7.64 (d, IH), 7.58 (dd, IH), 7.30-7.24 (m, 2H), 7.24-7.16 (m, 2H), 7.06-6.98 (m, 2H), 6.96-6.90 (m, 2H), 4.40 (d, 2H). LCMS: 457 (M+H)+.
EXAMPLE 190 l-(4-Cyano-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

Step 1 : l-(4-Cyano-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using methyl l-(4-cyano-2-(trifluoromethyl)phenyl)-lH-pyrrole-2-carboxylate as the starting material. LCMS: 281 (M+H)+. Step 2: l-(4-Cyano-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using methyl l-(4- cyano-2-(trifluoromethyl)phenyl)- 1 H-pyrrole-2-carboxylate and (4-(4- fluorophenoxy)phenyl) methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.64 (t, IH), 8.40 (d, IH), 8.20 (dd, IH), 7.60 (d, IH), 7.24- 7.14 (m, 4H), 7.08-6.98 (m, 4H), 6.94-6.86 (m, 2H), 6.30 (dd, IH), 4.22 (d, 2H). LCMS: 480 (M+H)+.
EXAMPLE 191 l-(4-Carbamoyl-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

A solution of-(4-cyano-2-(trifluoromethyl)phenyl)-N-(4-(4-fluorophenoxy)benzyl)- 1 H- pyrrole-2-carboxamide (78 mg, 0.16 mmol) in MeOH (1 niL)/ IM NaOH (1 niL) was refluxed for 1 h. It was concentrated and the residue was partitioned between EtOAc (10 rnL) and IN HCl (5 rnL). The organic layer was dried, filtered, concentrated, and purified by semi-preparative chromatography(CH3CN/H2O:TFA) to give the title compound as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (m, IH), 8.34- 8.10 (m, 3H), 7.66 (m, IH), 7.50-7.44 (m, IH), 7.24-7.14 (m, 4H), 7.06-6.86 (m, 6H), 6.28-6.24 (m, IH), 4.22 (m, 2H). LCMS: 498 (M+H)+.
EXAMPLE 192
4-(2-(4-(4-Fluorophenoxy)benzylcarbamoyl)-lH-pyrrol-l-yl)-3- (trifluoromethyl)benzoic acid

The title compound was isolated during the purification of EXAMPLE 191. LCMS: 499 (M+H)+.
EXAMPLE 193 N-(4-(4-Fluorophenoxy)benzyl)-l-(2-methylcyclohexyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 53 using 5-chloro-2- fluoro-3-methylpyridine and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1U NMR (400 MHz, DMSO-d6) δ 8.70 (t, IH), 8.32 (d, IH), 7.93 (d, IH), 7.26-7.16 (m, 4H), 7.06-6.96 (m, 4H), 6.94-6.90 (m, 2H), 6.27 (dd, IH), 4.22 (d, 2H), 1.98 (s, 3H). LCMS: 436 (M+H)+.
EXAMPLE 194
N'-(3-Chloro-5-(trifluoromethyl)pyri(iin-2-yl)-l-(2,4-(iichlorophenyl)-lH-pyrrole-
2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 2- methylcyclo hexanamine and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (t, IH), 7.30-7.26 (m, 2H), 7.24-7.16 (m, 2H), 7.09 (br, IH), 7.04-6.98 (m, 2H), 6.96-6.90 (m, 2H), 6.70 (dd, IH), 6.06 (dd, IH), 4.95-4.80 (m, IH), 4.35 (d, 2H), 1.90-1.00 (m, 9H), 0.50 (m, 3H). LCMS: 407 (M+H)+.
EXAMPLE 195
N-(4-(4-Fluorophenoxy)benzyl)-l-(l,2,3,4-tetrahydronaphthalen-l-yl)-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 96 using 1,2,3,4- tetrahydronaphthalen- 1 -amine and (4-(4-fluorophenoxy)phenyl)methanamine hydrochlorideas the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (t, IH), 7.30 (d, 2H), 7.23-7.10 (m, 4H), 7.06-6.98 (m, 3H), 6.94 (d, 2H), 6.86 (dd, IH), 6.64- 6.54 (m, 3H), 6.02 (dd, IH), 4.38 (d, 2H), 3.00-2.60 (m, 2H), 2.20-1.60 (m, 4H). LCMS: 441 (M+H)+.
EXAMPLE 196
N-(4-(4-Fluorophenoxy)benzyl)-l-(2-methyl-6-(methylsulfonyl)pyridin-3-yl)-lH- pyrrole-2-carboxamide

Step 1 : 2-Methyl-6-(methylsulfonyl)pyridin-3-amine

A solution of Fe (11.7 g, 209 mmol), NH4Cl (600 mg, 11.3 mmol), and 2-methyl-6- (methylsulfonyl)-3-nitropyridine (15 g, 69.4 mmol) in H2O (150 mL) was stirred for 30 min at 9O0C. The reaction mixture was cooled to O0C with an ice/salt bath. The solids
were collected by filtration. The crude product was purified by recrystallization from
EtOHl. This resulted in 2-methyl-6-(methylsulfonyl)pyridin-3-amine as a light yellow solid. LCMS: 187 (M+H)+.
Step 2: N-(4-(4-Fluorophenoxy)benzyl)-l-(2-methyl-6-(methylsulfonyl)pyridin-3- yl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 96 using 2-methyl-6- (methylsulfonyl)pyridin-3 -amine and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (t, IH), 8.00-7.90 (m, 2H), 7.24-7.16 (m, 4H), 7.12 (dd, IH), 7.07 (dd, IH), 7.02-6.98 (m, 2H), 6.93-6.90 (m, 2H), 6.36 (dd, IH), 4.25 (d, 2H), 3.30 (s, 3H), 2.20 (s, 3H). LCMS: 480 (M+H)+.
EXAMPLE 197 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-fluoro-5-
(trifluoromethyl)pyridin-2-yl)-5-methyl-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using 2,3-difluoro- 5-(trifluoromethyl)pyridine and ethyl 5 -methyl- lH-pyrrole-2-carboxylate as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, IH), 9.18 (s, IH), 8.80
(s, IH), 8.50 (dd, IH), 8.35 (s, IH), 8.10 (d, IH), 7.20 (d, IH), 6.10 (d, IH), 2.00 (s, 3H). LCMS: 482 (M+H)+.
EXAMPLE 198 l-(3,5-Dichloropyridin-2-yl)-N-((3-(4-(trifluoromethyl)phenoxy)-l,2,4-oxadiazol-5- yl)methyl)-lH-pyrrole-2-carboxamide

Et3N (4.1 g, 40.2 mmol) in THF (10 niL) was added dropwise to a solution of 4- (trifluoromethyl)phenol (6.4 g, 39.5 mmol) and bromoformonitrile (4.2 g, 40 mmol) in THF (100 mL) maintained at 0-50C. The resulting mixture was stirred for 2 h at O0C in an ice/salt bath. The solids were filtered out . The resulting filtrate was concentrated to afford l-cyanato-4-(trifluoromethyl)benzene as a light yellow oil. Step 2: l-Hydroxy-2-(4-(trifluoromethyl)phenyl)isourea hydrochloride

A solution of l-cyanato-4-(trifluoromethyl)benzene (7.48 g, 40 mmol) in MeOH (30 mL) was added dropwise to a solution OfNH2OH HCl (2.76 g, 40 mmol) in MeOH (100 mL) and the mixture was stirred for 2 h at rt. The resulting mixture was
concentrated and the residue was extracted with ethoxyethane (2 x 50 rnL). The solids were collected by filtration to afford crude l-hydroxy-2-(4- (trifluoromethyl)phenyl)isourea hydrochloride as a white solid. Step 3 : 4-(Trifluoromethyl)phenyl N'-l-chloroacetoxycarbamimidate

2-Chloroacetyl chloride (2.24 g, 20 mmol) in propan-2-one (10 mL) was added dropwise to a solution of l-hydroxy-2-(4-(trifluoromethyl)phenyl)isourea hydrochloride (5.12 g, 20 mmol) and Et3N (4.04 g) in propan-2-one (60 mL) at 0-50C. The resulting solution was stirred for 2 h at 0~5°C and it was concentrated under reduced pressure. The residue was partitioned between EtOAc (50 mL) and H2O (20 mL). The organic layer was concentrated, purified by silica gel chromatography (EtOAc/PE) to afford 4-(trifluoromethyl)phenyl N'-2-chloroacetoxycarbamimidate as a white solid.
Step 4: 5-(Chloromethyl)-3-(4-(trifluoromethyl)phenoxy)-l,2,4-oxadiazole

A mixture of 4-(trifluoromethyl)phenyl N'-2-chloroacetoxycarbamimidate (1.5 g, 5.07 mmol) and TsOH (cat.) in toluene (50 mL) was heated to 12O0C for 5 h. The solution was concentrated and the residue was diluted with EtOAc (100 mL). The organic layer was washed with H2O (2 x 20 mL), dried, filtered, concentrated, and purified by silica gel chromatography (EtOAc/hexane) to afford 5-(chloromethyl)-3-(4- (trifluoromethyl)phenoxy)-l,2,4-oxadiazole as a white solid.
(3-(4-(Trifluoromethyl)phenoxy)-l,2,4-oxadiazol-5-yl)methanamine

NH3(g) was bubbled into a solution of 5-(chloromethyl)-3-(4- (trifluoromethyl)phenoxy)-l,2,4-oxadiazole (1.0 g, 3.60 mmol) in MeOH (100 niL) with stirring for 4 h at rt. The resulting mixture was concentrated to afford (3-(4- (trifluoromethyl)phenoxy)-l,2,4-oxadiazol-5-yl)methanamine as a yellow oil. Step 6: l-(3,5-Dichloropyridin-2-yl)-N-((3-(4-(trifluoromethyl)phenoxy)-l,2,4- oxadiazol-5-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (3-(4-(trifluoromethyl)phenoxy)-l,2,4-oxadiazol-5-yl)methanamine as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (m, IH), 8.52 (d, IH), 8.40 (d, IH), 7.85 (d, 2H), 7.55 (d, 2H), 7.16 (m, IH), 7.06 (m, IH), 6.35 (m, IH), 4.50 (d, 2H). LCMS: 498 (M+H)+.
EXAMPLE 199 l-(3,5-Dichloropyridin-2-yl)-N-(4-(3-isopropyl-l,2,4-oxadiazol-5-yloxy)benzyl)- lH-pyrrole-2-carboxamide

Step 1 : 3-Isopropyl-5- (trichloromethyl)-l,2,4-oxadiazole

A solution of N-hydroxyisobutyramidine (7 g, 48 mmol, 70%), trichloroacetic anhydride (42 g, 137 mmol) in toluene (40 mL) was re fluxed for 1 h. The reaction was quenched by the addition of H2O (200 mL). Then the resulting solution was extracted with DCM (2 x 150 mL) and the organic layers combined, dried, filtered, and concentrated to afford 3-isopropyl-5- (trichloromethyl)-l,2,4-oxadiazole as a yellow oil.
Step 2: 3-Isopropyl-l,2,4-oxadiazol-5-ol

A solution of 3-isopropyl-5-(trichloromethyl)-l,2,4-oxadiazole (50 g, 219 mmol) and KOH (24 g, 428 mmol) in EtOH (300 mL) was stirred for 10 min. at 8O0C. The resulting mixture was concentrated and diluted with H2O (500 mL). The pH value of the solution was adjusted to 5 with HCl and the aqueous layer was extracted with EtOAc (2 x 300 mL), the organics were combined, dried, and concentrated. The crude product was re-crystallized from EtOAc :hexanes (1 :10) to afford 3-isopropyl- 1,2,4- oxadiazol-5-ol as a white solid.
Step 3 : 5-Chloro-3-isopropyl-l,2,4-oxadiazole

A solution of 3-isopropyl-l,2,4-oxadiazol-5-ol (300 mg, 2.23 mmol) , phosphoryl trichloride (1.02 g, 6.71 mmol) in pyridine (10 mL) was heated to 1000C for 1 h. The reaction was then quenched by the addition OfH2O (50 mL) and the aqueous phase was extracted with EtOAc (2 x 50 mL). The organics were combined, dried, and concentrated to afford 5-chloro-3-isopropyl-l,2,4-oxadiazole as a white solid. Step 4: 5-(Chloromethyl)-3-(4-(trifluoromethyl)phenoxy)-l,2,4-oxadiazole

A mixture of 5-chloro-3-isopropyl-l,2,4-oxadiazole (1.0 g, 6.51 mmol, 95%), CuI (53 mg, 0.28 mmol), Bu4NBr (88 mg, 0.27 mmol), K3PO4 (2.9 g, 13.68 mmol), and tert- butyl 4-hydroxybenzylcarbamate (1.52 g, 6.50 mmol) in DMF (50 mL) was stirred for 4 h at rt. The reaction was then diluted with H2O (250 mL) and extracted with EtOAc (2 x 200 mL). The organics were combined, washed with aq. NH4Cl (2 x 200 mL), dried, and concentrated. The residue was purified by silica gel chromatography (EtOAc/PE) to afford tert-butyl 4-(3-isopropyl-l,2,4-oxadiazol-5- yloxy)benzylcarbamate as a white solid. Step 5 : (4-(3-Isopropyl-l,2,4-oxadiazol-5-yloxy)phenyl)methanamine

A solution of tert-butyl 4-(3-isopropyl-l,2,4-oxadiazol-5-yloxy)benzylcarbamate (600 mg, 1.77 mmol) and 2,2,2-trifluoroacetic acid (2 mL) was stirred for 5 min at rt. The residue was purified by semi-preparative chromatography (ACN/H2O:TFA) to afford (4-(3-Isopropyl-l,2,4-oxadiazol-5-yloxy)phenyl)methanamine as a white solid. LCMS: 234 (M+H)+.
Step 6: l-(3,5-Dichloropyridin-2-yl)-N-(4-(3-isopropyl-l,2,4-oxadiazol-5- yloxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using l-(3,5- dichloropyridin-2-yl)-lH-pyrrole-2-carboxylic acid and (4-(3-isopropyl-l,2,4- oxadiazol-5-yloxy)phenyl) methanamine as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.42-7.36 (m, 2H), 7.36-7.30 (m, 2H), 7.12 (dd, IH), 7.04 (dd, IH), 6.32 (dd, IH), 4.30 (d, 2H), 2.85 (septet, IH), 1.15 (d, 6H). LCMS: 472 (M+H)+.
EXAMPLE 200 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(2,4-difluorophenyl)-lH-pyrrole-
2-carbo hy dr azide

The titled compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2,4-difluorophenyl)hydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.04 (s, IH), 7.85 (s, IH), 7.08 (m, IH), 6.92-6.86 (m, 2H), 6.79-6.69 (m, 2H), 6.40 (dd, IH), 1 NH resonance not observed. LCMS: 417 (M+H)+.
EXAMPLE 201 l-IS-Chloro-S-CtrifluoromethyOpyridin-l-yll-N'-phenyl-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and phenylhydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, IH), 8.02 (s, IH), 7.89 (s, IH), 7.19 (m, 2H), 7.08 (m, IH), 6.90-6.81 (m, 4H), 6.40 (m, IH), 1 NH resonance not observed. LCMS: 381 (M+H)+.
EXAMPLE 202 N'-(4-Chloro-2-fluorophenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH- pyrrole-2-carbohydrazide
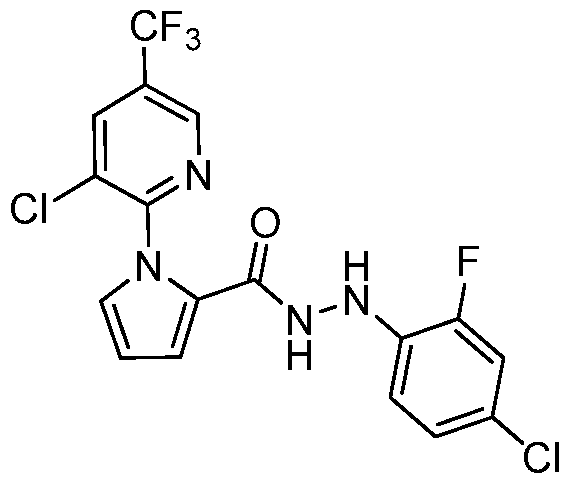
The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-chloro-2-fluorophenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.23 (s, IH), 8.87 (s, IH), 8.65 (s, IH), 7.91 (s, IH), 7.27- 7.17 (m, 3H), 7.05 (d, IH), 6.75 (dd, IH), 6.39 (m, IH). LCMS: 433 (M+H)+.
EXAMPLE 203 l-^-Chloro-S-CtrifluoromethylJpyridin-l-yll-N'-Cl^-difluorophenylJ-lH-pyrrole-
2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2,5-difluorophenyl)hydrazine as starting materials. 1H NMR (400 MHz, DMSO- d6) δ 10.25 (s, IH), 8.86 (s, IH), 8.66 (s, IH), 8.07 (s, IH), 7.28 (s, IH), 7.17 (m, IH), 7.09-7.03 (m, IH), 6.48-6.39 (m, 3H). LCMS: 417 (M+H)+.
EXAMPLE 204
N'-(4-tert-Butylphenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-
2-carbo hy dr azide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-tert-butylphenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.11 (s, IH), 8.86 (s, IH), 8.63 (s, IH), 7.47 (s, IH), 7.24 (m, IH), 7.16 (m, IH), 7.12-7.09 (m, 2H), 6.63 (d, 2H), 6.38 (dd, IH), 1.18 (s, 9H). LCMS: 437 (M+H)+.
EXAMPLE 205 l-IS-Chloro-S-CtrifluoromethyOpyridin-l-yll-N'-Cnaphthalen-l-yO-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and naphthalen-1-ylhydrazine hydrochloride as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.02 (s, IH), 7.85-7.78 (m, 3H), 7.47-7.41 (m, 3H), 7.33 (m, IH), 7.10 (s, IH), 7.00 (d, IH), 6.95 (m, IH), 6.43 (s, IH), 1 NH resonance not observed. LCMS: 431 (M+H)+.
EXAMPLE 206 N-Benzyl-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and benzylhydrazine dihydrochloride as starting materials. LCMS: 395 (M+H)+.
EXAMPLE 207 N'-(4-Bromophenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-bromophenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.19 (s, IH), 8.87 (s, IH), 8.64 (s, IH), 7.97 (s, IH), 7.25 (m, 3H), 7.16 (m, IH), 6.64 (d, 2H), 6.39 (dd, IH).
EXAMPLE 208 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(4-phenylphenyl)-lH-pyrrole-2- carbohydrazide

A mixture ofN'-(4-bromophenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH- pyrrole-2-carbohydrazide (200 mg, 0.44 mmol), phenylboronic acid (53 mg, 0.44 mmol), PdCl2(PPh3)2 (10 mg, 0.013 mmol), and K2CO3 (91 mg, 0.66 mmol) in DME/H2O (5:1, 4 mL) was heated to 15O0C in the microwave for 15 min. The reaction mixture was diluted with EtOAc (20 mL), washed with brine (2 x 15 mL), dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford of l-[3-Chloro-5-(trifluoromethyl)pyridin-
2-yl]-N'-(4-phenylphenyl)-lH-pyrrole-2-carbohydrazide as a white solid. LCMS: 457 (M+H)+.
EXAMPLE 209 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(3,4-dimethylphenyl)-lH-pyrrole-
2-carbohydrazide
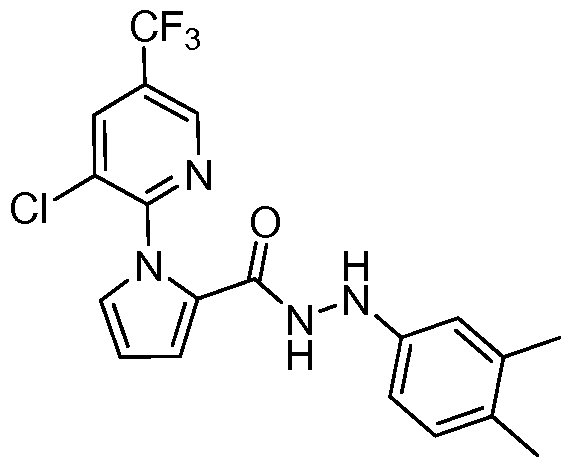
The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (3,4-dimethylphenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.02 (s, IH), 7.73 (s, IH), 7.078 (m, IH), 6.95 (d, IH), 6.90 (s, IH), 6.65-6.59 (m, 2H), 6.42 (m, IH), 5.96 (bs, IH), 2.17 (s, 3H), 2.14 (s, 3H). LCMS: 409 (M+H)+.
EXAMPLE 210 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(2,4,6-trifluorophenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2,4,6-trifluorophenyl)hydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.03 (s, IH), 7.85 (s, IH), 7.07 (m, IH), 6.82 (m, IH), 6.63 (m, 2H), 6.38 (dd, IH), 6.24 (bs, IH).
EXAMPLE 211 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(2,4,6-trichlorophenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2,4,6-trichlorophenyl)hydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.65 (s, IH), 8.02 (s, IH), 8.01 (s, IH), 7.24 (m.lH), 7.08 (m, IH), 6.86 (m, IH), 6.76 (m, IH), 6.38 (dd, IH), 1 NH resonance not observed.
EXAMPLE 212 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[2,6-dichloro-4-
(trifluoromethyl)phenyl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2,6-dichloro-4-(trifluoromethyl)phenyl)hydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, IH), 8.05 (d, IH), 8.02 (s, IH), 7.48 (s, 2H), 7.09 (m, IH), 6.89 (dd, 2H), 6.40 (dd, IH).
EXAMPLE 213 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(4-chlorophenoxy)phenyl]-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(4-chlorophenoxy)phenyl) hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.05 (d, IH), 7.78 (bs, IH), 7.22 (m, 2H), 7.10 (m, IH), 6.94 (bs, IH), 6.90-6.83 (m, 7H), 6.43 (dd, IH). LCMS: 508 (M+H)+.
EXAMPLE 214 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(3-chloro-5-methylpyridin-2-yl)- lH-pyrrole-2-carbohydrazide

3-Chloro-2-hydrazinyl-5-methylpyridine

The title compound was synthesized as described for Intermediate 3 using 2,3- dichloro-5-methylpyridine as the starting material.
Step 2: l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(3-chloro-5- methylpyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-chloro-2-hydrazinyl-5-methylpyridine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1), 8.03 (s, IH), 7.86-7.78 (m, 4H), 7.17 (dd, IH), 7.04 (dd, IH), 6.41 (dd, IH), 3.32 (s, 3H). LCMS: 430 (M+H)+.
EXAMPLE 215
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(isoquinolin-3-yl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 39 using 3- bromoisoquinoline as the starting materials. LCMS: 432 (M+H)+.
EXAMPLE 216 1- [3-Chloro-5-(trifluoromethyl)pyridin-2-yl] -N1- [4-(4-methylphenoxy)phenyl] -IH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(p-tolyloxy)phenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.16 (s, IH), 8.88 (s, IH), 8.65 (d, IH), 7.71 (bs, IH), 7.25 (m, IH), 7.16 (dd, IH), 7.08 (d, 2H), 6.79 (d, 2H), 6.76 (d, 2H), 6.71 (d, 2H), 6.37 (dd, IH), 2.22 (s, 3H). LCMS: 487 (M+H)+.
EXAMPLE 217 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-(4-phenoxyphenyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 4-phenoxyaniline as starting materials 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, IH), 8.91 (s, IH), 8.68 (d, IH), 7.58 (d, 2H), 7.33 (dd, 2H), 7.28 (m, IH), 7.23 (dd, IH), 7.07 (t, IH), 6.95-6.92 (m, 4H), 6.42 (dd, IH). LCMS: 458 (M+H)+.
EXAMPLE 218 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(pyridin-3-yloxy)phenyl]-lH- pyrrole-2-carbohydrazide

Step 1 : 3-(4-Nitrophenoxy)pyridine

A mixture of l-fluoro-4-nitrobenzene (4 g, 28.4 mmol), pyridin-3-ol (3.5 g, 37 mmol), and K2CO3 (5.8 g, 42.6 mmol) in NMP (30 mL) was heated in the microwave to 14O0C for 15 min (this reaction was done in 2 batches). The reaction mixture was diluted with EtOAc (40 mL), washed with brine (2 x 25 mL) and NaOH (IM, 2 x 25 mL), dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 3-(4-nitrophenoxy)pyridine as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (m, 2H), 8.25 (dd, 2H), 7.67 (d, IH), 7.53 (dd, IH), 7.19 (dd, 2H). Step 2: 4-(Pyridin-3-yloxy)aniline

3-(4-nitrophenoxy)pyridine (4.1 g, 18.9 mmol) was dissolved in MeOH (50 mL) and hydrogenated in the presence of a catalytic amount of Pd/C with a balloon of Hydrogen for 2.5 h. Pd/C was removed by filtration and the filtrate was evaporated to dryness under reduced pressure to afford 4-(pyridin-3-yloxy)aniline as a grey solid.
3-(4-Hydrazinylphenoxy)pyridine hydrochloride

4-(Pyridin-3-yloxy)aniline (1 g, 5.4 mmol) was suspended in HCl (cone, 10 niL) and cooled to O0C in an ice bath. Sodium nitrite (373 mg, 5.40 mmol) in H2O (2 mL) was then added dropwise and the resulting dark blue mixture was stirred at O0C for 30 min. Any insoluble particles were removed by filtration and tin dichloride dihydrate (3.6 g, 16.2 mmol) in HCl (cone, 10 mL) was added dropwise while the temperature was maintained at O0C. The yellowish precipitate was filtered, washed with H2O, and dried overnight. It afforded 3-(4-hydrazinylphenoxy)pyridine hydrochloride as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (bs, 3H), 8.52 (m, 2H), 7.82 (m, 2H), 7.15 (d, 2H), 7.09 (d, 2H).
Step 4: l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(pyridin-3- yloxy)phenyl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-(4-hydrazinylphenoxy)pyridine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 8.31 (d, IH), 8.28 (dd, IH), 7.41 (m, IH), 7.34 (m, IH), 7.26 (m, IH), 7.17 (m, IH), 6.93 (d, 2H), 6.75 (d, 2H), 6.38 (m, IH), INH resonance missing. LCMS: 474 (M+H)+.
EXAMPLE 219 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(pyridin-2-yloxy)phenyl]-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using 2-(4- nitrophenoxy)pyridine in step 2, 4-(pyridin-2-yloxy)aniline in Step 3, and Intermediate 1 and 2-(4-hydrazinylphenoxy)pyridine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, IH), 8.87 (s, IH), 8.63 (s, IH), 8.02 (bs, IH), 7.52 (dd, IH), 7.43 (td, IH), 7.26 (m, IH), 7.20 (dd, IH), 7.10 (d, 2H), 6.75 (d, 2H), 6.40 (m, 2H), 6.23 (td, IH). LCMS: 474 (M+H)+.
EXAMPLE 220
N'-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(pyridin-3-yloxy)phenyl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-(pyridin-3- yloxy)aniline as starting materials. LCMS: 474 (M+H)+.
EXAMPLE 221
Nl-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(pyridin-4-yloxy)phenyl)-lH- pyrrole-2-carbohydrazide

4-(Pyridin-4-yloxy)aniline

The title compound was synthesized as described in EXAMPLE 218 using l-fluoro-4- nitrobenzene and pyridin-4-ol in Step 1 and 4-(4-nitrophenoxy)pyridine in Step 2.
LCMS: 187 (M+H)+.
Step 2: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(4-(pyridin-4- yloxy)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 96 using 4-(pyridin-4- yloxy)aniline as starting material. LCMS: 474 (M+H)+.
EXAMPLE 222
1- [3-Chloro-5-(trifluoromethyl)pyridin-2-yl] -N1- [4-(3-methylphenoxy)phenyl] -IH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using l-methyl-3- (4-nitrophenoxy)benzene in step 2, 4-(m-tolyloxy)aniline in Step 3, and Intermediate 1 and (4-(m-tolyloxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, IH), 8.88 (s, IH), 8.65 (d, IH), 7.75 (bs, IH), 7.26 (m, IH), 7.16 (m, 2H), 6.82 (m, 3H), 6.73 (d, 2H), 6.64 (m, 2H), 6.38 (dd, IH), 2.22 (s, 3H). LCMS: 487 (M+H)+.
EXAMPLE 223 1- [3-Chloro-5-(trifluoromethyl)pyridin-2-yl] -N1- [4-(2-methylphenoxy)phenyl] -IH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using l-methyl-2- (4-nitrophenoxy)benzene in step 2, 4-(o-tolyloxy)aniline in Step 3, and Intermediate 1 and (4-(o-tolyloxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, IH), 8.87 (s, IH), 8.64 (d, IH), 7.70 (bs, IH), 7.25-7.21 (m, 2H), 7.16 (dd, IH), 7.09 (td, IH), 6.95 (td, IH), 6.77-6.69 (m, 4H), 6.65 (d, IH), 6.37 (dd, IH), 2.18 (s, 3H). LCMS: 487 (M+H)+.
EXAMPLE 224
Ethyl 4-({l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrol-2- yl} hydr azido)benzoate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and ethyl 4-hydrazinylbenzoate hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.29 (s, IH), 8.88 (s, IH), 8.65 (d, IH), 8.49 (bs, IH), 7.71 (d, 2H), 7.28 (m, IH), 7.18 (m, IH), 6.71 (d, 2H), 6.40 (dd, IH), 4.20 (q, 2H), 1.25 (t, 3H). LCMS: 452 (M+H)+.
EXAMPLE 225
N'-[2-Chloro-4-(trifluoromethyl)phenyl]-l-[3-chloro-5-(trifluoromethyl)pyridin-2- yl] - lH-pyrr ole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (2-chloro-4-(trifluoromethyl)phenyl)hydrazine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, IH), 8.05 (s, IH), 7.71 (bs, IH), 7.52 (s, IH), 7.39 (d, IH), 7.11 (m, IH), 6.99 (d, IH), 6.97 (m, IH), 6.54 (s, IH), 6.46 (dd, IH).
EXAMPLE 226 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(4-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-phenoxyphenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.17 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 7.76 (bs, IH), 7.30-7.25 (m, 4H), 7.17 (m, IH), 7.00 (t, IH), 6.86-6.83 (m, 3H), 6.72 (d, 2H), 6.38 (dd, IH).
EXAMPLE 227 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(4-methoxyphenoxy)phenyl]- lH-pyrrole-2-carbohydrazide
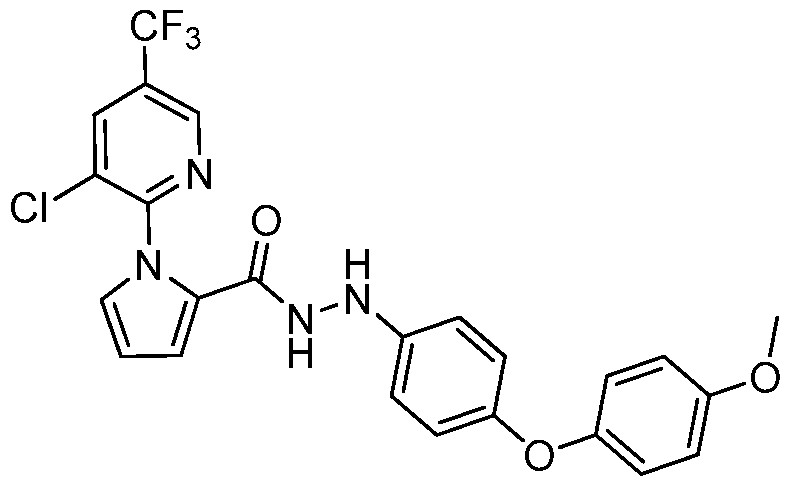
The title compound was synthesized as described in EXAMPLE 218 using 1-methoxy- 4-(4-nitrophenoxy)benzene in step 2, 4-(4-methoxyphenoxy)aniline in Step 3, and Intermediate 1 and (4-(4-methoxyphenoxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, IH), 8.87 (s, IH), 8.64 (s, IH), 7.68 (s, IH), 7.25 (m, IH), 7.16 (m, IH), 6.87-6.81 (m, 4H), 6.78 (d, 2H), 6.70 (d, 2H), 6.37 (dd, IH), 3.69 (s, 3H).
EXAMPLE 228 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(4-fluorophenoxy)phenyl]-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using l-fluoro-4- (4-nitrophenoxy)benzene in step 2, 4-(4-fluorophenoxy)aniline in Step 3, and Intermediate 1 and (4-(4-fluorophenoxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 7.76 (bs, IH), 7.26 (m, IH), 7.17 (m, IH), 7.11 (d, 2H), 6.90-6.86 (m, 2H), 6.84 (d, 2H), 6.72 (d, 2H), 6.38 (dd, IH). LCMS: 491 (M+H)+.
EXAMPLE 229 N'-(3-Chloro-4-cyanophenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH- pyrrole-2-carbohydrazide

3-Chloro-4-hydrazinylbenzonitrile hydrochloride


The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 3-chloro-4-hydrazinylbenzonitrile hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.43 (s, IH), 8.96 (s, IH), 8.89 (s, IH), 8.67 (s, IH), 7.62 (d, IH), 7.31 (m, IH), 7.18 (m, IH), 6.78 (d, IH), 6.96 (dd, IH), 6.41 (dd, IH).
EXAMPLE 230 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-cyano-3-
(trifluoromethyl)phenyl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 4-amino-2- (trifluoromethyl)benzonitrile in Step 1 and Intermediate 1 and 4-hydrazinyl-2- (trifluoromethyl)benzonitrile hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, IH), 9.15 (s, IH), 8.87 (s, IH), 8.66 (s, IH), 7.82 (d, IH), 7.31 (m, IH), 7.18 (m, IH), 7.05 (s, IH), 6.96 (d, IH), 6.42 (dd, IH). LCMS: 474 (M+H)+.
EXAMPLE 231
N'-(4-Cyanophenyl)-l-(3,5-(iichloropyri(iin-2-yl)-lH-pyrrole-2-carbohy(irazi(ie

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 4-hydrazinylbenzonitrile hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, IH), 8.63 (s, IH), 8.52 (d, IH), 8.39 (d, IH), 7.52 (dd, 2H), 7.22 (m, IH), 7.14 (m, IH), 6.73 (dd, 2H), 6.36 (dd, IH). LCMS: 372 (M+H)+.
EXAMPLE 232 l-(3,5-Dichloropyridin-2-yl)-N'-[4-(trifluoromethoxy)phenyl]-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(trifluoromethoxy)phenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, IH), 8.51 (d, IH), 8.38 (d, IH), 8.01 (s, IH), 7.19 (m, IH), 7.14 (dd, IH), 7.10 (d, 2H), 6.73 (d, 2H), 6.35 (dd, IH). LCMS: 431 (M+H)+.
EXAMPLE 233 N'-(3-chloro-5-(trifluoromethyl)phenyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 3-chloro-5- (trifluoromethyl)aniline in Step 1 and Intermediate 1 and (3-chloro-5- (trifluoromethyl)phenyl) hydrazine hydrochloride in Step 2. 1H NMR (300 MHz, CDCl3) δ 7.65 (m, 2H), 7.50 (m, 2H), 7.12 (s, IH), 7.03 (m, 4H), 6.42 (m, IH). LCMS: 482 (M-H)+.
EXAMPLE 234 NSl-bis^-Chloro-S-CtrifluoromethylJpyridin^-yll^-methyl-lH-pyrrole^- carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using ethyl 4- methyl-lH-pyrrole-2-carboxylate (J. Org. Chem. 1988, 53(2), 403) and Intermediate 3 as starting materials. 1U NMR (400 MHz, DMSO-d6) δ 10.21 (s, IH), 9.16 (s, IH), 8.83 (s, IH), 8.61 (s, IH), 8.35 (s, IH), 8.06 (s, IH), 7.04 (s, IH), 7.01 (s, IH), 2.11 (s, 3H). LCMS: 498 (M+H)+.
EXAMPLE 235 NSl-BisIS-chloro-S-CtrifluoromethylJpyridin-l-yll-S-methyl-lH-pyrrole-l- carbohydrazide


A mixture of ethyl 3 -hydroxy-3 -methyl- l-tosylpyrrolidine-2-carboxylate (Jet. Let. 1962, 921, 1.7 g, 5.23 mmol) and POCl3 (1.98 g, 13.03 mmol) in pyridine (20 mL) was stirred at 250C for 18 h. The reaction mixture was quenched by slow addition of cooled brine (30 mL). The solids were collected by filtration and dried to afford ethyl 3-methyl-l-tosyl-4,5-dihydro-lH-pyrrole-2-carboxylate as a white solid. Step 2: Ethyl 3-methyl-lH-pyrrole-2-carboxylate

A mixture of ethyl 3-methyl-l-tosyl-4,5-dihydro-lH-pyrrole-2-carboxylate (1.5 g, 4.85 mmol) and DBU (1.47 g, 9.67 mmol) in THF (20 mL) was stirred at 250C for 18 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford ethyl 3 -methyl- lH-pyrrole-2- carboxylate as a white solid.
Step 3: NSl-bisβ-Chloro-S-CtrifluoromethylJpyridin^-yll-S-methyl-lH-pyrrole^- carbohydrazide

The title compound was synthesized as described in EXAMPLE 53 using ethyl 3- methyl-lH-pyrrole-2-carboxylate and Intermediate 3 as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, IH), 9.22 (s, IH), 8.84 (s, IH), 8.65 (s, IH), 8.38 (s, IH), 8.07 (s, IH), 7.21 (d, IH), 6.22 (d, IH), 2.44 (s, 3H). LCMS: 498 (M+H)+.
EXAMPLE 236
1- [5-Chloro-4-(trifluoromethyl)pyridin-2-yl] -N1- [3-chloro-5- (trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2-carbohydrazide

Step 1 : 2-Bromo-5-chloro-4-iodopyridine

Diisopropylamine (10.1 g, 100 mmol) was added dropwise to a cooled (-450C) solution of n-BuLi (2.5M in Hexanes, 40 mL) in THF (100 mL). The resulting solution was stirred at -450C for 30 min followed by the addition of a solution of 2-bromo-5-
chloropyridine (19.2 g, 100 mmol) in THF (100 niL), while cooling to a temperature of -750C. The resulting solution was then stirred at -750C for 1 h. This was followed by the addition of a solution of I2 (25.4 g, 100 mmol) in THF (100 mL). The reaction mixture was then diluted with 150 mL of saturated Na2S2O3 and the aqueous layer was extracted with EtOAc (2 x 300 mL). The organics were combined, dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 2-bromo-5-chloro-4-iodopyridine as a light yellow solid
Step 2: 2-Bromo-5-chloro-4-(trifluoromethyl)pyridine

Tee title compound was synthesized as described in EXAMPLE 54, Step 2 using 2- bromo-5-chloro-4-iodopyridine as the starting material. Step 3: l-[5-Chloro-4-(trifluoromethyl)pyridin-2-yl]-N'-[3-chloro-5- (trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 39 using 2-bromo-5- chloro-4-(trifluoromethyl)pyridine and Intermediate 3 as starting materials. 1H NMR (400 MHz, DMSO-de) δ 10.41 (s, IH), 9.29 (s, IH), 8.82 (s, IH), 8.35 (bs, IH), 8.11 (d, IH), 7.72 (s, IH), 7.47 (m, IH), 7.14 (m, IH), 6.39 (dd, IH). LCMS: 484 (M+H)+.
EXAMPLE 237 N'-[3-Chloro-4-(trifluoromethoxy)phenyl]-l-[3-chloro-5-(trifluoromethyl)pyridin-
2-yl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 3-chloro-4- (trifluoromethoxy)aniline in Step 1 and Intermediate 1 and (3-chloro-4- (trifluoromethoxy)phenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-de) δ 10.29 (s, IH), 8.88 (s, IH), 8.66 (s, IH), 8.30 (bs, IH), 7.31 (m, IH), 7.29 (m, IH), 7.17 (dd, IH), 6.80 (d, IH), 6.70 (dd, IH), 6.40 (dd, IH). LCMS: 499 (M+H)+.
EXAMPLE 238 N'-(4-Benzylphenyl)-l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 4- benzylaniline in Step 1 and Intermediate 1 and (4-benzylphenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, IH), 8.86 (s, IH), 8.62 (d, IH), 7.25-7.12 (m, 8H), 6.95 (d, 2H), 6.62 (d, 2H), 6.37 (dd, IH), 3.76 (s, 2H).
EXAMPLE 239 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-[(4-phenoxyphenyl)methyl]-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-phenoxyphenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.89 (t, IH), 8.81 (m, IH), 8.64 (d, IH), 7.34 (dd, 2H), 7.25 (d, 2H), 7.18 (dd, IH), 7.11 (t, IH), 7.05 (dd, IH), 6.96-6.93 (m, 4H), 6.34 (dd, IH), 4.28 (d, 2H). LCMS: 472 (M+H)+.
EXAMPLE 240 l-(3,5-Dichloropyridin-2-yl)-N-[(4-phenoxyphenyl)methyl]-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-phenoxyphenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.75 (t, IH), 8.52 (d, IH), 8.37 (d, IH), 7.35 (m, 2H), 7.23 (d, 2H), 7.10 (m, 2H), 7.03 (m, IH), 6.95 (m, 4H), 6.30 (dd, IH), 4.27 (d, 2H). LCMS: 438 (M+H)+.
EXAMPLE 241 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[3-fluoro-4- (trifluoromethyl)phenyl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 3-fluoro-4- (trifluoromethyl)aniline in Step 1 and Intermediate 1 and (3-fluoro-4- (trifluoromethyl)phenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-de) δ 10.37 (s, IH), 8.88 (s, IH), 8.72 (s, IH), 8.67 (s, IH), 7.45 (m, IH), 7.30 (m, IH), 7.18 (m, IH), 6.58 (m, 2H), 6.41 (dd, IH). LCMS: 467 (M+H)+.
EXAMPLE 242 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(3-phenylphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using biphenyl-3- amine in Step 1 and Intermediate 1 and biphenyl-3-ylhydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, IH), 8.77 (s, IH), 8.62 (s, IH), 7.89 (bs, IH), 7.51 (m, 2H), 7.42 (m, 2H), 7.32 (m, IH), 7.26 (dd, IH), 7.18 (m, 2H), 6.95 (m, 2H), 6.69 (d, IH), 6.39 (dd, IH), 1 NH resonance not observed. LCMS: 457 (M+H)+.
EXAMPLE 243 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(4-propoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 4- propoxyaniline in Step 1 and Intermediate 1 and (4-propoxyphenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, IH), 8.88 (s, IH), 8.65 (d, IH), 7.25 (dd, IH), 7.18 (dd, IH), 6.74 (d, 2H), 6.65 (d, 2H), 6.39 (dd, IH), 3.79 (t, 2H), 1.66 (m, 2H), 0.94 (t, 3H), 1 NH resonance not observed. LCMS: 439 (M+H)+.
EXAMPLE 244 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-(4,5-dimethyl-l,3-thiazol-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as the undesired regioisomer as described in EXAMPLE 229 using 4,5-dimethylthiazol-2-amine hydrochloride in Step 1 and Intermediate 1 and 2-hydrazinyl-4,5-dimethylthiazole hydrochloride in Step 2. LCMS: 416 (M+H)+.
EXAMPLE 245 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 3- phenoxy aniline in Step 1 and Intermediate 1 and (3-phenoxyphenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, IH), 8.86 (s, IH), 8.63 (s, IH), 7.90 (bs, IH), 7.34 (m, 2H), 7.25 (dd, IH), 7.11-7.06 (m, 3H), 6.95 (d, 2H), 6.46 (dd, IH), 6.36 (dd, IH), 6.32 (m, IH), 6.27 (dd, IH).
EXAMPLE 246 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-(thiophen-2-ylmethyl)-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and thiophen-2-ylmethanamine as starting materials. LCMS: 386 (M+H)+.
EXAMPLE 247 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(4- fluorophenoxy)phenyl]methyl}-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(4-fluorophenoxy)phenyl)methanamine as starting materials. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, IH), 8.07 (d, IH), 7.25 (d, 2H), 7.05-6.90 (m, 7H), 6.72 (dd, IH), 6.37 (dd, IH), 6.18 (t, IH), 4.46 (d, 2H). LCMS: 490 (M+H)+.
EXAMPLE 248 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(4-fluorophenoxy)phenyl]methyl}-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(4-fluorophenoxy)phenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.74 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.24-7.17 (m, 3H), 7.10 (dd, IH), 7.02-6.98 (m, 3H), 6.91 (d, 2H), 6.30 (dd, IH), 4.25 (d, 2H). LCMS: 456 (M+H)+.
EXAMPLE 249 l-[3-chloro-5-(trifluoromethyl)pyridin-2-yl]-N-[(l-methyl-lH-imidazol-4- yl)methyl]-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (1 -methyl- lH-imidazol-4-yl)methanamine as starting materials. LCMS: 384 (M+H)+.
EXAMPLE 250 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-[(l-methyl-lH-pyrazol-3- yl)methyl]-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (l-methyl-lH-pyrazol-3-yl)methanamine as starting materials. LCMS: 384 (M+H)+.
EXAMPLE 251 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(pyridin-3- yloxy)phenyl]methyl}-lH-pyrrole-2-carboxamide

Stepl : 4-(Pyridin-3-yloxy)benzonitrile

A mixture of 4-bromobenzonitrile (1 g, 5.5 mmol), pyridin-3-ol (1 g, 11 mmol), copper(I) chloride (272 mg, 2.75 mmol), 2,2,6, 6-tetramethylheptane-3,5-dione (0.11 mL, 0.55 mmol), and Cs2CO3 (3.5 g, 11 mmol) in NMP (10 mL) was heated to 12O0C for 18 h. The reaction mixture was diluted with EtOAc (40 mL), washed with brine (2 x 25 mL) and NaOH (IM, 2 x 25 mL), dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 4-(pyridin-3-yloxy)benzonitrile as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.48 (m, 2H), 7.63 (d, 2H), 7.38 (m, 2H), 7.02 (d, 2H) Step 2: (4-(Pyridin-3-yloxy)phenyl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 4- (pyridin-3-yloxy)benzonitrile as the starting material. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 8.33 (dd, IH), 7.32-7.24 (m, 4H), 6.99 (d, 2H), 3.86 (s, 2H), 1.69 (bs, 2H).
Step 3: l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(pyridin-3- yloxy)phenyl] methyl}-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(pyridin-3-yloxy)phenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.92-8.88 (m, 2H), 8.63 (d, IH), 8.56 (d, IH), 8.50 (dd, IH), 7.81 (m, IH), 7.75 (dd, IH), 7.31 (d, 2H), 7.18 (m, IH), 7.13-7.08 (m, 3H), 6.34 (dd, IH), 4.31 (d, 2H). LCMS: 473 (M+H)+.
EXAMPLE 252 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-{4-[4-
(trifluoromethyl)phenoxy]phenyl}-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using l-nitro-4- (4-(trifluoromethyl)phenoxy)benzene in step 2, 4-(4-(trifluoromethyl)phenoxy)aniline in Step 3, and Intermediate 1 and (4-(4-(trifluoromethyl)phenoxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 7.64 (d, 2H), 7.26 (dd, IH), 7.17 (dd, IH), 6.99 (d, 2H), 6.93 (dd, 2H), 6.76 (dd, 2H), 6.38 (dd, IH), 1 NH resonance not observed.
EXAMPLE 253 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(5-cyanopyridin-2-yl)-lH- pyrrole-2-carbohydrazide

Step 1 : 6-Hydrazinylnicotinonitrile

The title compound was synthesized as described for Intermediate 3 using 6- fluoronicotinonitrile as the starting material. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, IH), 8.33 (s, IH), 7.71 (d, IH), 6.73 (bs, IH), 4.41 (bs, 2H). Step 2: l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(5-cyanopyridin-2-yl)-lH- pyrrole-2-carbohydrazide
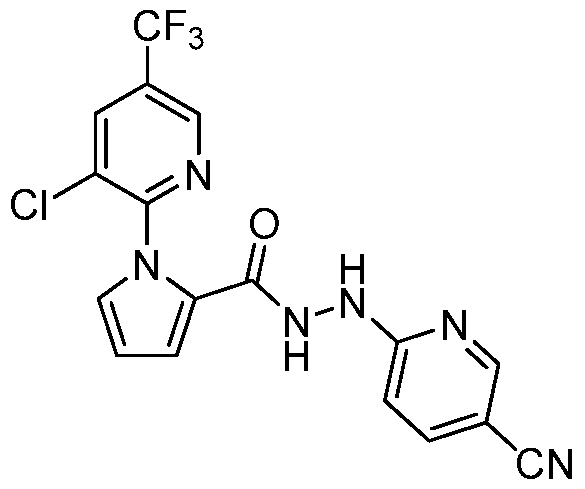
The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 6-Hydrazinylnicotinonitrile as starting materials. 1H NMR (400 MHz, DMSO-dβ) δ 10.41 (s, IH), 9.35 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 8.44 (s, IH), 7.85 (dd, IH), 7.29 (m, IH), 7.19 (m, IH), 6.59 (d, IH), 6.40 (dd, IH). LCMS: 407 (M+H)+.
EXAMPLE 254 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-({4-
[methyl(phenyl)amino] phenyl} methyl)- lH-pyr role-2-carboxamide

Step 1 : 4-(Methyl(phenyl)amino)benzonitrile

A mixture of 4-bromobenzonitrile (0.5 g, 2.75 mmol), N-methylaniline (0.6 rnL, 5.5 mmol), Pd2(dba)3 (126 mg, 0.14 mmol), rac-Binap (171 mg, 0.27 mmol), and NaOtBu (396 mg, 4.12 mmol) in Toluene (15 mL) was heated in the microwave at 15O0C for 15 min. The reaction mixture was diluted with EtOAc (20 mL), washed with brine (2 x 15 mL) and NaOH (IM, 2 x 15 mL), dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 4-(methyl(phenyl)amino)benzonitrile. LCMS: 209 (M+H)+. Step 2: 4-(Aminomethyl)-N-methyl-N-phenylaniline

The title compound was synthesized as described in EXAMPLE 2, STEP 1 using 4- (methyl(phenyl)amino)benzonitrile as the starting material. LCMS: 197 (M-NH2) .
Step 3: l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-({4-
[methyl(phenyl)amino] phenyl} methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and 4-(aminomethyl)-N-methyl-N-phenylaniline as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.88 (m, IH), 8.75 (t, IH), 8.64 (d, IH), 7.23-7.14 (m, 5H), 7.05 (dd, IH), 6.96-6.86 (m, 5H), 6.33 (dd, IH), 4.25 (d, 2H), 3.20 (s, 3H). LCMS: 485 (M+H)+.
EXAMPLE 255 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-(2-phenoxyphenyl)-lH-pyrrole-2- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 2- phenoxyaniline in Step 1 and Intermediate 1 and (2-phenoxyphenyl)hydrazine hydrochloride in Step 2. LCMS: 473 (M+H)+.
EXAMPLE 256 l-IS-Chloro-S-CtrifluoromethylJpyridin-l-yll-N'-Cl-phenylphenylJ-lH-pyrrole-l- carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using biphenyl-2- amine in Step 1 and Intermediate 1 and biphenyl-2-ylhydrazine hydrochloride in Step 2. LCMS: 457 (M+H)+.
EXAMPLE 257 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[6-(4-methylphenoxy)pyridin-3- yl] - lH-pyrr ole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using 5-nitro-2- (p-tolyloxy)pyridine in step 2, 6-(p-tolyloxy)pyridin-3 -amine in Step 3, and Intermediate 1 and 5-hydrazinyl-2-(p-tolyloxy)pyridine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, IH), 8.87 (s, IH), 8.65 (s, IH), 7.88 (d, IH), 7.61 (d, IH), 7.26 (m, IH), 7.19-7.11 (m, 4H), 6.86 (d, 2H), 6.80 (d, IH), 6.38 (dd, IH), 2.25 (s, 3H). LCMS: 488 (M+H)+.
EXAMPLE 258 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N'-[4-(3-fluorophenoxy)phenyl]-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 218 using l-fluoro-3- (4-nitrophenoxy)benzene in step 2, 4-(3-fluorophenoxy)aniline in Step 3, and Intermediate 1 and (4-(3-fluorophenoxy)phenyl)hydrazine hydrochloride in Step 4. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, IH), 8.88 (s, IH), 8.64 (s, IH), 7.82 (bs, IH), 7.31 (m, IH), 7.26 (dd, IH), 7.17 (dd, IH), 6.88 (d, 2H), 6.84 (td, IH), 6.75 (d, 2H), 6.65 (m, 2H), 6.38 (dd, IH). LCMS: 491 (M+H)+.
EXAMPLE 259 N'-[2-chloro-4-(trifluoromethoxy)phenyl]-l-[3-chloro-5-(trifluoromethyl)pyridin-
2-yl]-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 229 using 2-chloro-4- (trifluoromethoxy)aniline in Step 1 and Intermediate 1 and (2-chloro-4- (trifluoromethoxy) phenyl)hydrazine hydrochloride in Step 2. 1H NMR (400 MHz, DMSO-de) δ 10.34 (s, IH), 8.88 (s, IH), 8.65 (s, IH), 7.80 (s, IH), 7.37 (d, IH), 7.28 (dd, IH), 7.20 (m, 2H), 6.80 (d, IH), 6.40 (dd, IH). LCMS: 499 (M+H)+.
EXAMPLE 260
Nl-[2-Chloro-4-(trifluoromethoxy)phenyl]-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (2-chloro-4-(trifluoromethoxy) phenyl)hydrazine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, IH), 8.52 (d, IH), 8.39 (d, IH), 7.78 (s, IH), 7.38 (d, IH), 7.22-7.16 (m, 3H), 6.78 (d, IH), 6.36 (dd, IH). LCMS: 465 (M+H)+.
EXAMPLE 261 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(phenylamino)benzyl)-lH- pyrrole-2-carboxamide
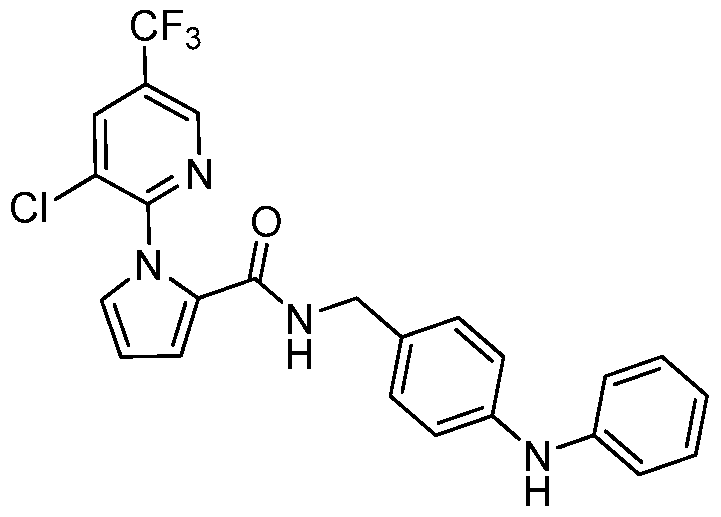
The title compound was synthesized as described in EXAMPLE 254 using aniline and 4-bromobenzonitrile in Step 1, 4-(phenylamino)benzonitrile in Step 2, and Intermediate 1 and 4-(aminomethyl)-N-phenylaniline in Step 3. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (m, IH), 8.72 (t, IH), 8.64 (d, IH), 7.20-7.15 (m, 3H), 7.09 (d, 2H), 7.05 (dd, IH), 7.01-6.96 (m, 5H), 6.76 (t, IH), 6.33 (dd, IH), 4.21 (d, 2H). LCMS: 471 (M+H)+.
EXAMPLE 262 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-(3-phenoxyphenyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1
+ and 3 -phenoxy aniline as starting materials. LCMS: 458 (M+H) .
EXAMPLE 263 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-(3-phenylphenyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and biphenyl-3 -amine as starting materials. LCMS: 442 (M+H)+.
EXAMPLE 264 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-(2,2,2-trifluoroethyl)pyridin-2- yl)methyl)-lH-pyrrole-2-carboxamide


NaBH4 (22 g, 579 mmol) was added in portion to l-(6-bromopyridin-3-yl)-2,2,2- trifluoroethanone (37 g, 103.5 mmol) in MeOH (250 mL) maintained at O0C and the mixture was stirred at O0C for 10 min. H2O (100 mL) was then slowly added and the aqueous layer was extracted with EtOAc (3 x 500 mL). The organics were combined, dried, and evaporated to dryness under reduced pressure to afford l-(6-bromopyridin- 3-yl)-2,2,2-trifluoroethanol as a yellow oil. LCMS: 256 (M+H)+. Step 2: 5-(2,2,2-Trifluoro-l-hydroxyethyl)picolinonitrile

A mixture of l-(6-bromopyridin-3-yl)-2,2,2-trifluoroethanol (3.06 g, 11.95 mmol), CuCN (2.13 g, 24 mmol) in NMP (30 mL) was stirred at 13O0C for 6 h. The reaction mixture was then diluted with NHs-H2O(15%, 100 mL) and the aqueous solution was extracted with EtOAc (5 x 100 mL). The organics were combined, dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 5-(2,2,2-trifluoro-l- hydroxyethyl)picolinonitrile as a yellow oil. Step 3 : l-(6-Cyanopyridin-3-yl)-2,2,2-trifluoroethyl 4-methylbenzenesulfonate
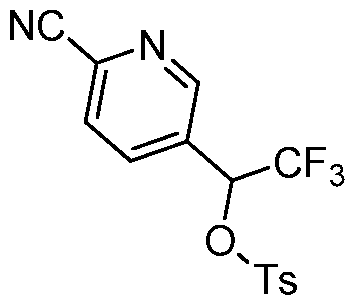
TsCl (2.7 g, 14.14 mmol) in DCM (25 mL) was added to a mixture of 5-(2,2,2- trifluoro-1 -hydroxy ethyl)picolinonitrile (2.6 g, 12.87 mmol), TEA (2.6 g, 25.74 mmol), and DMAP (30 mg, 0.25 mmol) in DCM (25 mL) maintained at O0C. The resulting solution was stirred at 250C for 30 min and quenched with ice H2O (30 mL). The two layers were separated and the organic layer was washed with brine (2 x 30 mL), dried,
and evaporated to dryness under reduced pressure to afford l-(6-cyanopyridin-3-yl)- 2,2,2-trifluoroethyl 4-methylbenzenesulfonate as a yellow solid. LCMS: 356 (M)+. Step 4: tert-Butyl (5-(2,2,2-trifluoroethyl)pyridin-2-yl)methylcarbamate

A mixture of l-(6-cyanopyridin-3-yl)-2,2,2-trifluoroethyl 4-methylbenzenesulfonate (1.78 g, 5.00 mmol), Pd/C(10%, 0.178 g, 0.1 equiv), and HCl (36%, 1.5 g) in MeOH (80 mL) was hydrogenated under an hydrogen atmosphere up to 5 atm for 3 d at 250C. The Pd/C was removed by filtration and the filtrate's pH was adjusted to 7 with NaOH. This was followed by the addition of NaOH (200 mg, 5.00 mmol) and BoC2O (1.09 g, 5.00 mmol) and the mixture was stirred at 250C for 1 h. The resulting mixture was concentrated under reduced pressure and the aqueous layer was extracted with EtOAc (3 x 50 mL)3x50 mL. The organics were combined, dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford tert-butyl (5-(2,2,2-trifluoroethyl)pyridin-2- yl)methylcarbamate as a white solid. LCMS: 291 (M+H)+. Step 5: (5-(2,2,2-Trifluoroethyl)pyridin-2-yl)methanamine hydrochloride

A solution of tert-butyl (5-(2,2,2-trifluoroethyl)pyridin-2-yl)methylcarbamate (380 mg, 1.31 mmol) in MeOH (20 mL) under an HCl atmosphere was stirred at 250C for 1 h. The solvent was removed under reduced pressure and the residue was purified by recrystallization from MeOH/Et2O to afford (5-(2,2,2-trifluoroethyl)pyridin-2- yl)methanamine hydrochloride as a yellow solid. LCMS: 191 (M+H)+.
Step 6: l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-(2,2,2- trifluoroethyl)pyridin-2-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (5-(2,2,2-trifluoroethyl)pyridin-2-yl)methanamine hydrochloride as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (t, IH), 8.87 (s, IH), 8.62 (s, IH), 8.52 (s, IH), 7.86 (d, IH), 7.36 (d, IH), 7.20 (m, IH), 7.11 (m, IH), 6.37 (dd, IH), 4.41 (d, 2H), 3.72 (q, 2H). LCMS: 463 (M+H)+.
EXAMPLE 265 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(2,4- difluorophenoxy)phenyl]methyl}-lH-pyrrole-2-carboxamide

Step 1 : 4-(2,4-Difluorophenoxy)benzonitrile

The title compound was synthesized as described in EXAMPLE 218, Step 1 using 4- fluorobenzonitrile and 2,4-difluorophenol as starting materials. 1H NMR (400 MHz, CDCl3) δ 7.60 (d, 2H), 7.18-7.12 (m, IH), 7.01-6.90 (m, 4H).
Step 2: (4-(2,4-Difluorophenoxy)phenyl)methanamine

The title compound was synthesized as described in EXAMPLE 2, STEP 1 using 4-
(2,4-difluorophenoxy)benzonitrile as the starting material.
Step 3: 1- [3-C hloro-5-(trifluor omethyl)pyridin-2-yl] -N- { [4-(2,4-difluor ophenoxy) phenyl]methyl}-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(2,4-difluorophenoxy) phenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.88 (m, IH), 8.79 (t, IH), 8.63 (d, IH), 7.45 (m, IH), 7.24- 7.16 (m, 4H), 7.09 (m, IH), 7.05 (dd, IH), 6.89 (d, 2H), 6.33 (dd, IH), 4.25 (d, 2H). LCMS: 508 (M+H)+.
EXAMPLE 266 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(2,4-difluorophenoxy)phenyl]methyl}-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(2,4-difluorophenoxy) phenyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.73 (t, IH), 8.52 (d, IH), 8.37 (d, IH), 7.45 (m, IH), 7.24-
7.18 (m, 3H), 7.12-7.06 (m, 2H), 7.01 (dd, IH), 6.89 (d, 2H), 6.29 (dd, IH), 4.25 (d, 2H). LCMS: 474 (M+H)+.
EXAMPLE 267
N-{[4-(2-Chloro-4-fluorophenoxy)phenyl]methyl}-l-[3-chloro-5- (trifluor omethyl)pyridin-2-yl] - lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4- fluorobenzonitrile and 2-chloro-4-fluorophenol in Step 1 , 4-(2-chloro-4- fluorophenoxy)benzonitrile in Step 2, and Intermediate 1 and (4-(2-chloro-4- fluorophenoxy)phenyl)methanamine in Step 3. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (m, IH), 8.79 (t, IH), 8.63 (d, IH), 7.60 (dd, IH), 7.25-7.20 (m, 3H), 7.17 (dd, IH), 7.15 (m, IH), 7.05 (dd, IH), 6.85 (d, 2H), 6.33 (dd, IH), 4.25 (d, 2H). LCMS: 524 (M+H)+.
EXAMPLE 268
N-{[4-(2-Chloro-4-fluorophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(2-chloro-4-fluorophenoxy)phenyl) methanamine as starting materials. 1H
NMR (400 MHz, DMSO-d6) δ 8.74 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.60 (dd, IH), 7.25-7.20 (m, 3H), 7.15-7.10 (m, 2H), 7.01 (dd, IH), 6.86 (d, 2H), 6.29 (dd, IH), 4.25 (d, 2H). LCMS: 490 (M+H)+.
EXAMPLE 269 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(4-fluorophenoxy)-2- methylphenyl]methyl}-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4-fluoro-2- methylbenzonitrile and 4-fluorophenol in Step 1 , 4-(4-fluorophenoxy)-2- methylbenzonitrile in Step 2, and Intermediate 1 and (4-(4-fluorophenoxy)-2- methylphenyl)methanamine in Step 3. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (s, IH), 8.66 (t, IH), 8.64 (s, IH), 7.20-7.16 (m, 4H), 7.08 (dd, IH), 7.00-7.97 (m, 2H), 6.79- 6.73 (m, 2H), 6.34 (dd, IH), 4.24 (d, 2H), 2.20 (s, 3H). LCMS: 504 (M+H)+.
EXAMPLE 270 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(4-fluorophenoxy)-2-methylphenyl]methyl}- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(4-fluorophenoxy)-2-methylphenyl)methanamine as starting materials. 1H
NMR (400 MHz, DMSO-d6) δ 8.61 (t, IH), 8.51 (d, IH), 8.38 (d, IH), 7.20-7.15 (m, 3H), 7.10 (dd, IH), 7.04 (dd, IH), 7.00-6.96 (m, 2H), 6.79-6.74 (m, 2H), 6.30 (dd, IH), 4.23 (d, 2H), 2.20 (s, 3H). LCMS: 470 (M+H)+.
EXAMPLE 271 l-[3-Chloro-5-(trifluoromethyl)pyridin-2-yl]-N-{[4-(4-fluorophenoxy)-3- methylphenyl]methyl}-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4-fluoro-3- methylbenzonitrile and 4-fluorophenol in Step 1, 4-(4-fluorophenoxy)-3- methylbenzonitrile in Step 2, and Intermediate 1 and (4-(4-fluorophenoxy)-3- methylphenyl)methanamine in Step 3. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, IH),
8.79 (t, IH), 8.64 (d, IH), 7.17-7.12 (m, 4H), 7.07-7.05 (m, 2H), 6.88-6.85 (m, 2H),
6.80 (d, IH), 6.33 (dd, IH), 4.25 (d, 2H), 2.11 (s, 3H). LCMS: 504 (M+H)+.
EXAMPLE 272 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(4-fluorophenoxy)-3-methylphenyl]methyl}- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(4-fluorophenoxy)-3-methylphenyl) methanamine as starting materials. 1H
NMR (400 MHz, DMSO-d6) δ 8.74 (t, IH), 8.52 (d, IH), 8.39 (d, IH), 7.17-7.13 (m, 3H), 7.10 (dd, IH), 7.06 (d, IH), 7.01 (dd, IH), 6.86 (m, 2H), 6.80 (d, IH), 6.30 (dd, IH), 4.25 (d, 2H), 2.11 (s, 3H). LCMS: 470 (M+H)+.
EXAMPLE 273
N-{[3-Chloro-4-(4-fluorophenoxy)phenyl]methyl}-l-[3-chloro-5- (trifluor omethyl)pyridin-2-yl] - lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 3-chloro-4- fluorobenzonitrile and 4-fluorophenol in Step 1, 3-chloro-4-(4- fluorophenoxy)benzonitrile in Step 2, and Intermediate 1 and (3-chloro-4-(4- fluorophenoxy)phenyl)methanamine in Step 3. * H NMR (400 MHz, DMSO-d6) δ 8.88 (s, IH), 8.86 (t, IH), 8.64 (d, IH), 7.42 (d, IH), 7.21-7.16 (m, 4H), 7.05 (dd, IH), 7.01 (d, IH), 6.97-6.93 (m, 2H), 6.35 (dd, IH), 4.28 (d, 2H). LCMS: 524 (M+H)+.
EXAMPLE 274 N-{[3-Chloro-4-(4-fluorophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (3-chloro-4-(4-fluorophenoxy)phenyl) methanamine as starting materials. l H
NMR (400 MHz, DMSO-d6) δ 8.81 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.41 (d, IH), 7.21-7.16 (m, 3H), 7.12 (dd, IH), 7.01 (m, 2H), 6.94 (m, 2H), 6.31 (dd, IH), 4.28 (d, 2H). LCMS: 491 (M+H)+.
EXAMPLE 275 N-{[2-Chloro-4-(4-fluorophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carboxamide

Step 1 : 2-Chloro-4-(4-fluorophenoxy)-l-methylbenzene

The title compound was synthesized as described in EXAMPLE 251 , Step 1 using 4- bromo-2-chloro-l-methylbenzene and 4-fluorophenol as starting materials. l H NMR (400 MHz, CDCl3) δ 7.16 (d, IH), 7.05-6.94 (m, 5H), 6.78 (dd, IH), 2.33 (s, 3H). Step 2: l-(Bromomethyl)-2-chloro-4-(4-fluorophenoxy)benzene

A mixture of 2-chloro-4-(4-fluorophenoxy)-l-methylbenzene (1.68 g, 7.12 mmol), NBS (1.4 g, 7.83 mmol), and benzoyl peroxide (cat.) in CCl4 (30 mL) was refluxed for 2 h. The mixture was cooled to 250C and the floating white solid was removed by filtration. The filtrate was evaporated to dryness under reduced pressure and the residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 1- (bromomethyl)-2-chloro-4-(4-fluorophenoxy)benzene as a colorless oil. l H NMR (400
MHz, CDCl3) δ 7.38 (d, IH), 7.09-6.99 (m, 4H), 6.96 (d, IH), 6.83 (dd, IH), 4.58 (s, 2H).
Step 3 : 2-(2-Chloro-4-(4-fluorophenoxy)benzyl)isoindoline-l,3-dione

A mixture of l-(bromomethyl)-2-chloro-4-(4-fluorophenoxy)benzene (712 mg, 2.26 mmol), isoindoline-l,3-dione (333 mg, 2.26 mmol), and Cs2CO3 (1.1 g, 3.39 mmol) in DMF (20 mL) was stirred at 250C for 18 h. The reaction mixture was diluted with EtOAc (50 mL), washed with brine (2 x 25 mL), dried, and evaporated to dryness under reduced pressure. The residue was purified by silica gel chromatography (EtOAc/Hexanes) to afford 2-(2-chloro-4-(4-fluorophenoxy)benzyl)isoindo line- 1,3- dione as a white solid. l H NMR (400 MHz, DMSO-d6) δ 7.91-7.84 (m, 4H), 7.28 (d, IH), 7.25-7.20 (m, 2H), 7.12-7.06 (m, 3H), 6.88 (dd, IH), 4.78 (s, 2H). Step 4: (2-Chloro-4-(4-fluorophenoxy)phenyl)methanamine

A mixture of 2-(2-chloro-4-(4-fluorophenoxy)benzyl)isoindoline-l,3-dione (590 mg, 1.55 mmol) and hydrazine (0.1 mL, 3.41 mmol) in n-butanol (10 mL) was refluxed for 50 min. The mixture was cooled to 250C and the white solid was removed by filtration. The filtrate was evaporated to dryness under reduced to afford (2-chloro-4-(4- fluorophenoxy)phenyl) methanamine as a colorless oil. * H NMR (400 MHz, DMSO- d6) δ 7.52 (d, IH), 7.25-7.20 (m, 2H), 7.09-7.05 (m, 2H), 6.99 (d, IH), 6.93 (dd, IH), 3.71 (s, 2H), 2.02 (bs, 2H).
Step 5 : N-{[2-Chloro-4-(4-fluorophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-
2-yl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (2-chloro-4-(4-fluorophenoxy)phenyl) methanamine as starting materials. l H NMR (400 MHz, DMSO-d6) δ 8.76 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.28 (d, IH), 7.25-7.20 (m, 2H), 7.12 (dd, IH), 7.09-7.05 (m, 3H), 7.02 (d, IH), 6.93 (dd, IH), 6.32 (dd, IH), 4.30 (d, 2H). LCMS: 490 (M+H)+.
EXAMPLE 276 l-(3,5-Dichloropyridin-2-yl)-N-({5-[4-(trifluoromethyl)phenoxy]pyrazin-2- yl}methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 251 using 4- (trifluoromethyl) phenol and 5-bromopyrazine-2-carbonitrile in Step 1, 5-(4- (trifluoromethyl)phenoxy)pyrazine-2-carbonitrile in Step 2, and Intermediate 2 and (5- (4-(trifluoromethyl)phenoxy)pyrazin-2-yl)methanamine hydrochloride in Step 3. 1 H NMR (400 MHz, DMSO-d6) δ 8.89 (t, IH), 8.54 (d, IH), 8.50 (d, IH), 8.37 (d, IH), 8.09 (d, IH), 7.79 (d, 2H), 7.38 (d, 2H), 7.11 (dd, IH), 7.04 (dd, IH), 6.31 (dd, IH), 4.40 (d, 2H). LCMS: 508 (M+H)+.
EXAMPLE 277
N-{[4-(4-Cyanophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-
2-carboxamide

Step 1 : 4-(p-Tolyloxy)benzonitrile

A mixture of 4-fluorobenzonitrile (2.16 g, 17.85 mmol), p-cresol (2.9 g, 26.78 mmol), and K2CO3 (5 g, 35.7 mmol) in DMF (30 mL) was heated to 8O0C for 18 h. The reaction mixture was cooled then diluted with EtOAc (50 mL), washed with brine (2 x 25 mL) and NaOH (IM, 2 x 20 mL), dried, and evaporated to dryness under reduced pressure to afford 4-(p-tolyloxy) benzonitrile as a beige oily residue (crude). Step 2: 4-(4-(Bromomethyl)phenoxy)benzonitrile

The title compound was synthesized as described in EXAMPLE 275, Step 2 using 4- (p-tolyloxy)benzonitrile as the starting material. Step 3 : 4-(4-(Aminomethyl)phenoxy)benzonitrile

A mixture of 4-(4-(bromomethyl)phenoxy)benzonitrile (250 mg, 0.87 mmol) in NH3 (2M in MeOH, 2 mL) was stirred at 250C for 2 d. The solvent was removed and the residue was used as is. LCMS: 209 (M-NH2)+.
Step 4: N-{[4-(4-Cyanophenoxy)phenyl]methyl}-l-(3,5-dichloropyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 4-(4-(aminomethyl)phenoxy) benzonitrile as starting materials. l H NMR (400 MHz, DMSO-de) δ 8.79 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.79 (dd, 2H), 7.31 (d, 2H), 7.12-7.02 (m, 6H), 6.30 (dd, IH), 4.30 (d, 2H). LCMS: 463 (M+H)+.
EXAMPLE 278 l-(3,5-Dichloropyridin-2-yl)-N-{[5-(4-fluorophenoxy)pyrazin-2-yl]methyl}-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 251 using 4- fluorophenol and 5-bromopyrazine-2-carbonitrile in Step 1, 5-(4- fluorophenoxy)pyrazine-2-carbonitrile in Step 2, and Intermediate 2 and (5-(4- fluorophenoxy)pyrazin-2-yl)methanamine hydrochloride in Step 3. 1 H NMR (400 MHz, DMSO-de) δ 8.85 (t, IH), 8.49 (d, IH), 8.44 (d, IH), 8.36 (d, IH), 8.03 (s, IH), 7.25-7.21 (m, 4H), 7.10 (m, IH), 7.02 (m, IH), 6.30 (dd, IH), 4.37 (d, 2H). LCMS: 458 (M+H)+.
EXAMPLE 279 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(pyrimidin-5-yloxy)phenyl]methyl}-lH- pyrrole-2-carboxamide

The title compound was synt esized as described in EXAMxPLE 251 using pyrimidin- 5-ol and 4-bromobenzonitrile in Step 1, 4-(pyrimidin-5-yloxy)benzonitrile in Step 2, and Intermediate 2 and (4-(pyrimidin-5-yloxy)phenyl)methanamine hydrochloride in Step 3. λ H NMR (400 MHz, DMSO-d6) δ 8.79 (t, IH), 8.59 (d, 2H), 8.53 (d, IH), 8.39 (d, IH), 7.28-7.21 (m, 3H), 7.11 (m, 3H), 7.03 (dd, IH), 6.31 (dd, IH), 4.31 (d, 2H). LCMS: 440 (M+H)+.
EXAMPLE 280 l-(3,5-Dichloropyridin-2-yl)-N-[2-(4-fluorophenoxy)ethyl]-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 2-(4-fluorophenoxy)ethanamine as starting materials. l H NMR (400 MHz, DMSO-de) δ 8.52 (d, IH), 8.41 (t, IH), 8.38 (d, IH), 7.10-7.06 (m, 3H), 6.98 (dd, IH), 6.91 (m, 2H), 6.28 (dd, IH), 3.94 (t, 2H), 3.41 (m, 2H). LCMS: 394 (M+H)+.
EXAMPLE 281 l-(3,5-Dichloropyridin-2-yl)-N-[3-(4-fluorophenoxy)propyl]-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 3-(4-fluorophenoxy)propan-l-amine as starting materials. LCMS: 408 (M+H)+.
EXAMPLE 282 l-(3,5-Dichloropyridin-2-yl)-N-[4-(4-fluorophenoxy)butyl]-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 4-(4-fluorophenoxy)butan-l -amine as starting materials. l H NMR (400 MHz, DMSO-de) δ 8.51 (d, IH), 8.36 (d, IH), 8.21 (t, IH), 7.09-7.04 (m, 3H), 6.93-6.87 (m, 3H), 6.28 (dd, IH), 3.90 (t, 2H), 3.11 (m, 2H), 1.64 (m, 2H), 1.56 (m, 2H). LCMS: 422(M+H)+.
EXAMPLE 283
4-[4-({[l-(3,5-Dichloropyridin-2-yl)-lH-pyrrol-2-yl]formamido}methyl)phenoxy]-
1-oxidopyridin-l-ium

Step 1 : 4-(4-((tert-Butoxycarbonylamino)methyl)phenoxy)pyridine 1-oxide

A mixture of tert-butyl 4-hydroxybenzylcarbamate (J. Org. Chem. 2005, 70 (20), 7956, 4.5 g, 20.2 mmol), 4-nitropyridine 1-oxide (2.35 g, 16.8 mmol), and K2CO3 (3 g, 21.9 mmol) in DMF (20 rnL) was heated to 8O0C for 18 h. The reaction mixture was cooled to 250C and the solvent was removed under reduced pressure. H2O was added and the gummy solid that crashed out was filtered, dried, and recrystallized from DCM/Hexanes to afford 4-(4-((tert-butoxycarbonylamino)methyl)phenoxy)pyridine 1- oxide as a pale orange solid. LCMS: 317(M+H)+.
Step 2: 4-(4-(Aminomethyl)phenoxy)pyridine 1-oxide 2,2,2-trifluoroacetate

A solution of 4-(4-((tert-butoxycarbonylamino)methyl)phenoxy)pyridine 1-oxide (500 mg, 1.6 mmol) in TFA (3 mL) was stirred at 250C for 10 min. TFA was removed and the residue was triturated in DCM/Hexanes, filtered, and dried to afford 4-(4- (aminomethyl)phenoxy)pyridine 1-oxide 2,2,2-trifluoroacetate as an orange solid. l H NMR (400 MHz, DMSO-d6) δ 8.51 (d, 2H), 8.23 (bs, 3H), 7.58 (d, 2H), 7.31 (d, 2H), 7.17 (d, 2H), 4.06 (m, 2H).
Step 3 4-[4-({[l-(3,5-Dichloropyridin-2-yl)-lH-pyrrol-2- yl]formamido}methyl)phenoxy]-l-oxidopyridin-l-ium

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 4-(4-(aminomethyl)phenoxy)pyridine 1 -oxide 2,2,2-trifluoroacetate as starting materials. LCMS: 455 (M+H)+.
EXAMPLE 284 l-(3,5-dichloropyridin-2-yl)-N-{[4-(pyridin-4-yloxy)phenyl]methyl}-lH-pyrrole-2- carboxamide

PBr3 (0.08 mL, 0.89 mmol) was added dropwise to a solution of 4-[4-({[l-(3,5- dichloropyridin-2-yl)- 1 H-pyrrol-2-yl]formamido } methyl)phenoxy] - 1 -oxidopyridin- 1 - ium (81 mg, 0.18 mmol) in CHCl3 (3 mL) and the resulting mixture was stirred at 250C for 30 min. MeOH was then slowly added, the solvents were removed and the residue was purified by semi preparative chromatography (ACNZH2OITFA) to afford l-(3,5- dichloropyridin-2-yl)-N- { [4-(pyridin-4-yloxy)phenyl]methyl} - 1 H-pyrrole-2- carboxamide as a white solid. l H NMR (400 MHz, DMSO-d6) δ 8.85 (t, IH), 8.70 (d, 2H), 8.52 (d, IH), 8.38 (d, IH), 7.40 (d, 2H), 7.32 (d, 2H), 7.23 (d, 2H), 7.12 (m, IH), 7.05 (m, IH), 6.32 (dd, IH), 4.35 (d, 2H). LCMS: 439 (M+H)+.
EXAMPLE 285 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(pyrimidin-2-yloxy)phenyl]methyl}-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 283 using 2- chloropyrimidine and tert-butyl 4-hydroxybenzylcarbamate (J. Org. Chem. 2005, 70 (20), 7956) in Step 1, tert-butyl 4-(pyrimidin-2-yloxy)benzylcarbamate in Step 2, and Intermediate 2 and (4-(pyrimidin-2-yloxy)phenyl)methanamine 2,2,2-trifluoroacetate in Step 3. LCMS: 440 (M+H)+.
EXAMPLE 286 l-(3,5-Dichloropyridin-2-yl)-N-{[4-(4-methylphenoxy)phenyl]methyl}-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using p-cresol and 4-fluorobenzonitrile in step 1, 4-(p-tolyloxy)benzonitrile in Step 2, and Intermediate 2 and 4(4-(p-tolyloxy)phenyl)methanamine in Step 3. 1 H NMR (400 MHz, DMSO-d6) δ 8.73 (t, IH), 8.52 (s, IH), 8.38 (s, IH), 7.21 (d, 2H), 7.15 (d, 2H), 7.13 (s, IH), 7.01 (s, IH), 6.90-6.84 (m, 4H), 6.29 (dd, IH), 4.25 (d, 2H), 2.26 (s, 3H). LCMS: 452 (M+H)+.
EXAMPLE 287 l-(3,5-Dichloropyridin-2-yl)-N-({4-[(6-methylpyridin-3-yl)oxy]phenyl}methyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4- fluorobenzonitrile and 6-methylpyridin-3-ol in Step 1, 4-(6-methylpyridin-3- yloxy)benzonitrile in Step 2, and Intermediate 2 and (4-(6-methylpyridin-3- yloxy)phenyl)methanamine in Step 3. l H NMR (400 MHz, DMSO-d6) δ 8.77 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 8.37 (s, IH), 7.57 (m, IH), 7.45 (m, IH), 7.27 (d, 2H), 7.11 (dd, IH), 7.01 (m, 3H), 6.30 (dd, IH), 4.28 (d, 2H), 2.49 (s, 3H). LCMS: 453 (M+H)+.
EXAMPLE 288 l-(3,5-Dichloropyridin-2-yl)-N-[(4-{[5-(trifluoromethyl)pyrazin-2- yl]oxy}phenyl)methyl]-lH-pyrrole-2-carboxamide


A mixture of 5-bromopyrazin-2-amine (2.3 g, 13.29 mmol), 4-hydroxybenzonitrile (3.16 g, 26.55 mmol), K2CO3 (3.66 g, 26.52 mmol), and Cu2O (370 mg, 2.61 mmol) in
pyridine (20 rnL) was heated to 12O0C for 18 h. The resulting solution was diluted with H2O (50 rnL) and extracted with EtOAc (3 x 50 mL). The organics were combined, dried, and evaporated to dryness under reduced pressure to afford 4-(5- aminopyrazin-2-yloxy) benzonitrile as a white solid. Step 2: 4-(5-Iodopyrazin-2-yloxy)benzonitrile

A mixture of 4-(5-aminopyrazin-2-yloxy)benzonitrile (4.24 g, 20 mmol), I2 (10 g, 39.7 mmol), and tert-butyl nitrite (4.04 g, 39.2 mmol) in 1-chlorobenzene (50 mL was stirred at 250C for 5 h. The resulting mixture was washed with aqueous NaHSO3 (50 mL), dried, and evaporated to dryness under reduced pressure. The residue was recrystallized from EtOAc/Hexanes to afford 4-(5-iodopyrazin-2-yloxy)benzonitrile as a light yellow solid.
Step 3 : 4-(5-(Trifluoromethyl)pyrazin-2-yloxy)benzonitrile

The title compound was synthesized as described in EXAMPLE 54, step 2 using 4-(5- iodopyrazin-2-yloxy)benzonitrile as the starting material.
Step 4: (4-(5-(Trifluoromethyl)pyrazin-2-yloxy)phenyl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 4-(5- (trifluoromethyl) pyrazin-2-yloxy)benzonitrile as the starting material.
Step 5: l-(3,5-Dichloropyridin-2-yl)-N-[(4-{[5-(trifluoromethyl)pyrazin-2- yl]oxy}phenyl) methyl]-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(5-(trifluoromethyl)pyrazin-2-yloxy)phenyl)methanamine as starting materials. 1 H NMR (400 MHz, DMSO-d6) δ 8.81 (t, IH), 8.71 (s, IH), 8.67 (s, IH), 8.53 (d, IH), 8.39 (d, IH), 7.31 (d, 2H), 7.19 (d, 2H), 7.11 (dd, IH), 7.04 (dd, IH), 6.31 (dd, IH), 4.32 (d, 2H). LCMS: 508 (M+H)+.
EXAMPLE 289 tert-Butyl 4-((l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)piperidine-l-carboxylate
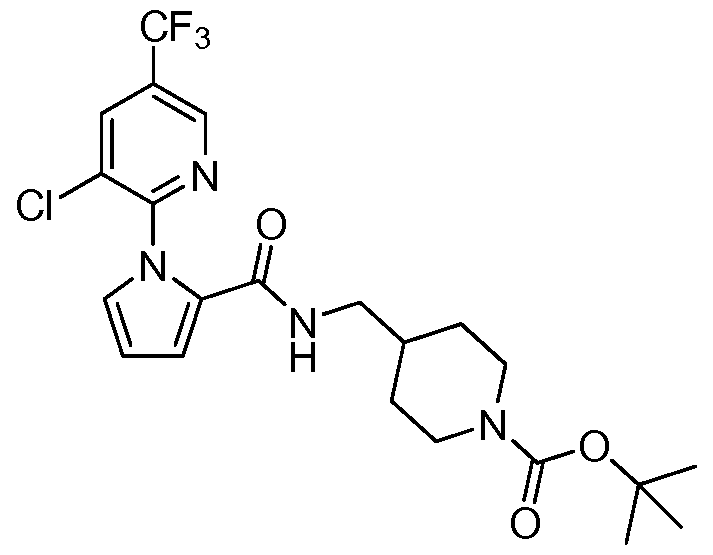
The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and tert-butyl 4-(aminomethyl)piperidine-l-carboxylate as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.88 (d, IH), 8.63 (d, IH), 8.27 (t, IH), 7.14 (m, IH), 6.98 (m, IH), 6.32 (m, IH), 3.87 (m, 2H), 2.95 (t, 2H), 2.63 (m, 2H), 1.56 (m, 3H), 1.35 (s, 9H), 0.92 (m, 2H). LCMS: 487 (M+H)+.
EXAMPLE 290 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((l-(4-
(trifluoromethoxy)benzyl)piperidin-4-yl)methyl)-lH-pyrrole-2-carboxamide

Step 1 : l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(piperidin-4-ylmethyl)-lH- pyrrole-2-carboxamide

To a solution of tert-butyl 4-((l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole- 2-carboxamido)methyl)piperidine-l-carboxylate (317 mg, 0.65 mmol) in DCM (6 mL) at 00C, was added TFA (2 mL). After 30 min at rt, the reaction mixture was partitioned between sodium bicarbonate (20 mL, IM in H2O) and EtOAc (50 mL). The aqueous layer was extracted with EtOAc (2 x 20 mL) and the organics were combined, dried, and concentrated. Purification by reverse phase HPLC (5% to 95% ACN/H2θ:TFA) gave l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(piperidin-4- ylmethyl)-lH-pyrrole-2-carboxamide. LCMS: 387 (M+H)+.
Step 2: tert-Butyl 4-((l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)piperidine-l-carboxylate

To a solution of l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(piperidin-4-ylmethyl)- lH-pyrrole-2-carboxamide (44 mg, 0.11 mmol) in THF (1.5 niL), was added DIPEA (40 uL, 2 eq), and l-(bromomethyl)-4-(trifluoromethoxy)benzene (20 μL). After 4 h at 700C, the reaction mixture was allowed to cool to rt and concentrated. Purification by reverse phase HPLC (5% to 95% ACN/H2O:TFA) gave tert-butyl 4-((l-(3-chloro-5- (trifluoromethyl)pyridin-2-y I)- 1 H-pyrrole-2-carboxamido)methyl)piperidine- 1 - carboxylate. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (m, IH), 8.63 (m, IH), 8.35 (m, IH), 7.59 (m, 2H), 7.47 (m, 2H), 7.16 (m, IH), 6.98 (m, IH), 6.32 (m, IH), 4.28 (m, 2H), 3.32 (m, 2H), 2.97 (t, 2H), 2.85 (m, 2H), 1.78 (m, 2H), 1.65 (m, IH), 1.27 (m, 2H). LCMS: 561 (M+H)+.
EXAMPLE 291 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((l-(2,2,2-trifluoroethyl)piperidin-
4-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 290, Step 2 using 2,2,2-trifluoroethyl trifluoromethanesulfonate as a starting material. 1H NMR (400 MHz, DMSO-de) δ 8.88 (m, IH), 8.63 (m, IH), 8.31 (m, IH), 7.15 (m, IH), 6.99 (m,
IH), 6.32 (m, IH), 3.60 (m, 2H), 3.15 (m, 2H), 2.97 (m, 2H), 2.60 (m, 2H), 1.70 (m, 2H), 1.58 (m, IH), 1.27 (m, 2H). LCMS: 469 (M+H)+.
EXAMPLE 292 tert-Butyl 4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)piperidine-l-carboxylate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and tert-butyl 4-(aminomethyl)piperidine-l-carboxylate as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.54 (m, IH), 8.40 (m, IH), 8.24 (t, IH), 7.09 (m, IH), 6.97 (m, IH), 6.29 (m, IH), 3.89 (m, 2H), 2.95 (t, 2H), 2.60 (m, 2H), 1.58 (m, 3H), 1.38 (s, 9H), 0.88 (m, 2H). LCMS: 453 (M+H)+.
EXAMPLE 293 l-(3,5-Dichloropyridin-2-yl)-N-((l-(4-fluorobenzyl)piperidin-4-yl)methyl)-lH- pyrrole-2-carboxamide
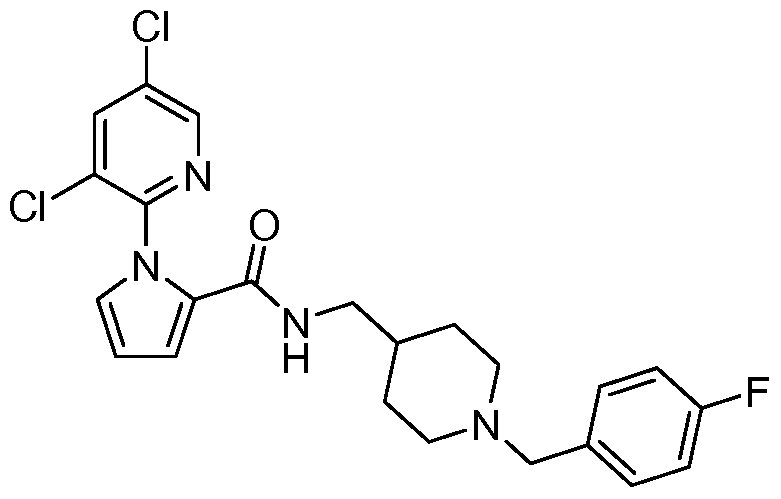
The title compound was synthesized as described in EXAMPLE 290 using tert-butyl 4- ((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carboxamido)methyl)piperidine-l- carboxylate in Step 1 and l-(3,5-dichloropyridin-2-yl)-N-(piperidin-4-ylmethyl)-lH- pyrrole-2-carboxamide and l-(bromomethyl)-4-fluorobenzene in Step 2. 1H NMR
(400 MHz, DMSO-de; TFA salt) δ 9.24 (m, IH), 8.53 (m, IH), 8.39 (m, IH), 8.25 (t, IH), 7.53 (m, 2H), 7.33 (m, 2H), 7.10 (m, IH), 6.96 (m, IH), 6.31 (m, IH), 4.28 (m, 2H), 3.43 (m, 2H), 3.00 (t, 2H), 2.88 (m, 2H), 1.78 (m, 2H), 1.68 (m, IH), 1.31 (m, 2H). LCMS: 461 (M+H)+.
EXAMPLE 294 l-(3,5-Dichloropyridin-2-yl)-N-((l-(4-(trifluoromethoxy)benzyl)piperidin-4- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 290, Step 2 using 1- (3,5-dichloropyridin-2-yl)-N-(piperidin-4-ylmethyl)-lH-pyrrole-2-carboxamide and 1- (bromomethyl)-4-(trifluoromethoxy)benzene as starting materials. 1H NMR (400 MHz, DMSO-de; TFA salt) δ 9.22 (m, IH), 8.52 (m, IH), 8.38 (m, IH), 8.24 (t, IH), 7.62 (m, 2H), 7.44 (m, 2H), 7.10 (m, IH), 6.96 (m, IH), 6.31 (m, IH), 4.60 (m, 2H), 3.47 (m, 2H), 3.00 (t, 2H), 2.88 (m, 2H), 1.78 (m, 2H), 1.68 (m, IH), 1.30 (m, 2H). LCMS: 527 (M+H)+.
EXAMPLE 295 tert-Butyl 3-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)pyrrolidine-l-carboxylate

EXAMPLE 296 l-(3,5-Dichloropyridin-2-yl)-N-((l-(4-fluorobenzyl)pyrrolidin-3-yl)methyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 290 using tert-butyl 3- (( 1 -(3 ,5 -dichloropyridin-2-yl)- 1 H-pyrrole-2-carboxamido)methyl)pyrrolidine- 1 - carboxylate in Step 1 and l-(3,5-dichloropyridin-2-yl)-N-(pyrrolidin-3-ylmethyl)-lH- pyrrole-2-carboxamide and l-(bromomethyl)-4-fluorobenzene in Step 2. 1H NMR (400 MHz, DMSO-d6; TFA salt) δ 8.42 (m, IH), 8.13 (m, IH), 7.51 (m, 2H), 7.22 (m, 2H), 7.07 (m, IH), 6.94 (m, IH), 6.36 (m, IH), 3.60-1.65 (m, 12H). LCMS: 447 (M+H)+.
EXAMPLE 297 tert-Butyl 3-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)azetidine-l-carboxylate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and tert-butyl 3-(aminomethyl)azetidine-l-carboxylate as starting materials. LCMS: 425 (M+H)+.
EXAMPLE 298 l-(3,5-Dichloropyridin-2-yl)-N-((l-(4-fluorobenzyl)azetidin-3-yl)methyl)-lH- pyrrole-2-carboxamide

1 -(3 ,5 -Dichloropyridin-2-yl)-N-(( 1 -(4-fluorobenzyl)azetidin-3 -yl)methyl)- 1 H-pyrrole- 2-carboxamide was synthesized as described in EXAMPLE 290 using tert-butyl 3-((l- (3 ,5 -dichloropyridin-2-yl)- 1 H-pyrrole-2-carboxamido)methyl)azetidine- 1 -carboxylate in Step 1 and N-(azetidin-3-ylmethyl)-l -(3, 5 -dichloropyridin-2-yl)- lH-pyrrole-2- carboxamide and l-(bromomethyl)-4-fluorobenzene in Step 2. LCMS: 433 (M+H)+.
EXAMPLE 299 N-((4-(Aminomethyl)cyclohexyl)methyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and cyclohexane-l,4-diyldimethanamine as starting materials. 1H NMR (400 MHz, DMSO-d6; TFA salt) δ 8.88 (m, IH), 8.63 (m, IH), 8.24 (m, IH), 7.62 (m, 3H), 7.14 (m, IH), 6.98 (m, IH), 6.32 (m, IH), 3.20 (t, IH), 2.91 (t, IH), 2.70 (t, IH), 2.62 (t, IH), 1.68 (m, 3H), 1.32 (m, 5H), 0.92 (m, 2H). LCMS: 415 (M+H)+.
EXAMPLE 300
Ethyl 4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carboxamido)methyl) cyclohexanecarboxylate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and ethyl 4-(aminomethyl)cyclohexanecarboxylate as starting materials. 1H NMR (400 MHz, DMSO-de) δ 8.53 (d, IH), 8.38 (d, IH), 8.19 (t, IH), 7.07 (m, IH), 6.93 (m, IH), 6.27 (m, IH), 4.00 (q, 2H), 2.88 (t, 2H), 2.18 (m, IH), 1.85 (m, 2H), 1.70 (m, 2H), 1.38 (m, IH), 1.21 (m, 2H), 1.12 (t, 3H), 0.86 (m, 2H). LCMS: 424 (M+H)+.
EXAMPLE 301 l-(3,5-dichloropyridin-2-yl)-N-((4-(4-fluorophenylcarbamoyl)cyclohexyl)methyl)- lH-pyrrole-2-carboxamide

Step 1 : 4-((l-(3,5-Dichloropyridin-2-yl)-lH-pyrrole-2-carboxamido)methyl) cyclohexanecarboxylic acid

To a solution of ethyl 4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl) cyclohexanecarboxylate (230 mg, 0.54 mmol) in EtOH (2.7 niL), was added sodium hydroxide (1.3 mL, IM in H2O). After 16 h at rt, the reaction mixture was partitioned between H2O (20 mL, adjusted to pH3) and EtOAc (50 mL). The aqueous layer was extracted with EtOAc (2 x 20 mL) and the organics were combined, dried, and concentrated. Purification by silica gel chromatography (0% to 20% MeOH/DCM) gave 4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)cyclohexanecarboxylic acid. 1H NMR (400 MHz, DMSO-dβ) δ 11.99 (s, IH), 8.53 (d, IH), 8.38 (d, IH), 8.18 (t, IH), 7.07 (m, IH), 6.96 (m, IH), 6.27 (m, IH), 2.88 (t, 2H), 2.08 (m, IH), 1.85 (m, 2H), 1.68 (m, 2H), 1.36 (m, IH), 1.21 (m, 2H), 0.85 (m, 2H). LCMS: 396 (M+H)+.
Step 2: l-(3,5-Dichloropyridin-2-yl)-N-((4-(4- fluorophenylcarbamoyl)cyclohexyl)methyl)-lH-pyrrole-2-carboxamide
The title compound was synthesized as described in EXAMPLE 1 using 4-((l-(3,5- dichloropyridin-2-yl)- 1 H-pyrrole-2-carboxamido)methyl)cy clohexanecarboxylic acid and 4-fluoroaniline as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, IH), 8.53 (d, IH), 8.38 (d, IH), 8.19 (t, IH), 7.58 (m, 2H), 7.08 (m, 3H), 6.96 (m, IH), 6.27 (m, IH), 2.94 (t, 2H), 2.21 (m, IH), 1.75 (m, 4H), 1.35 (m, 3H), 0.88 (m, 2H), 1 NH resonance not observed. LCMS: 489 (M+H)+.
EXAMPLE 302 l-(3,5-Dichloropyridin-2-yl)-N-((4-(6-(trifluoromethyl)pyridin-3- yloxyJcyclohexylJmethylJ-lH-pyrrole^-carboxamide

Step 1 : tert-Butyl (4-( -(trifluoromethyl yloxy)cyclohexyl)methylcarbamate


The title compound was synthesized as described in EXAMPLE 290, Step 1 using tert- butyl (4-(6-(trifluoromethyl)pyridin-3-yloxy)cyclohexyl)methylcarbamate as the starting material. LCMS: 275 (M+H)+.
Step 3: l-(3,5-Dichloropyridin-2-yl)-N-((4-(6-(trifluoromethyl)pyridin-3- yloxy)cyclohexyl) methyl)- lH-pyrrole-2-carboxamide

Tee title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(6-(trifluoromethyl)pyridin-3-yloxy)cyclohexyl)methanamine as starting materials. 1H NMR (400 MHz, DMSO-d6) δ 8.52 (d, IH), 8.39 (m, 2H), 8.23 (t, IH), 7.77 (m, IH), 7.61 (m, IH), 7.07 (m, IH), 6.96 (m, IH), 6.27 (m, IH), 4.47 (m, IH), 2.95 (t, 2H), 2.07 (m, 2H), 1.74 (m, 2H), 1.47 (m, IH), 1.30 (m, 2H), 1.06 (m, 2H). LCMS: 513 (M+H)+.
EXAMPLE 303 l-(3,5-Dichloropyridin-2-yl)-N-(((ls,4s)-4-(6-(trifluoromethyl)pyridin-3- yloxy)cyclohexyl) methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 302 using tert-butyl ((ls,4s)-4-hydroxycyclohexyl) methylcarbamate as starting material. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (d, IH), 8.40 (m, IH), 8.37 (d, IH), 8.22 (t, IH), 7.79 (m, IH), 7.58 (m, IH), 7.07 (m, IH), 6.95 (m, IH), 6.27 (m, IH), 4.76 (m, IH), 2.98 (t, 2H), 1.85 (m, 2H), 1.62-1.44 (m, 5H), 1.26 (m, 2H). LCMS: 513 (M+H)+.
EXAMPLE 304 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-tosyl-lH-pyrrole-2-carboxamide

Step 1. l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbonyl chloride

To a solution of Intermediate 1 (0.5 g, 1.72 mmol) in DCM (17 mL) was added oxalyl dichloride (0.295 mL, 3.44 mmol) followed by addition of 2 drops of DMF at rt. This
resulting mixture was allowed to stir at this temperature for 2 hours. The mixture was then filtered off. The filtrate was concentrated in vacuo and dried to afford a pale yellow oil that was immediately used without further purification. Step 2. l-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-N-tosyl-lH-pyrrole-l- carboxamide

To a solution of l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbonyl chloride (0.25g, 0.8 mmol) in DCM (6.7 mL) was added 4-methylbenzenesulfonamide (0.1 Ig, 0.67 mmol) followed by TEA (0.47 mL, 3.35 mmol) at O0C. This mixture was stirred at this temperature for Ih. Then it was gradually warmed up to room temperature over Ih. LCMS showed the conversion. The mixture was quenched with H2O (25 mL), rinsed into a seperatory funnel and extracted with DCM (2x50 mL). Combined organic layers were dried over Na2SO4, filtered and concentrated to give the crude product that was purified by RP C18 column chromatography eluted by 50-100 % ACN in water in the presence of 0.1 % TFA affording l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-N-tosyl-lH-pyrrole-2-carboxamide. LCMS: 444.16 (M+H)+.
EXAMPLE 305 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-chlorobenzyloxy)-lH-pyrrole-2- carboxamide

EXAMPLE 306 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((4-chlorophenyl)(imino)methyl)- lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using 4- chlorobenzimidamide hydroiodide as the starting material. LCMS: 427.22 (M+H)+.
EXAMPLE 307 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-phenoxy-lH-pyrrole-2- carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using O- phenylhydroxylamine as the starting material. LCMS: 382.17 (M+H)+.
EXAMPLE 308 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-
(trifluoromethyl)phenylsulfonyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using 4- (trifluoromethyl) benzenesulfonamide as the starting material. LCMS: 498.18 (M+H)4
EXAMPLE 309 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4- (trifluoromethoxy)phenylsulfonyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using 4- (trifluoromethoxy)benzenesulfonamide as the starting material. LCMS: 514.21 (M+H)+.
EXAMPLE 310
(S)-N-(l-(4-Bromophenyl)ethyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using (S)-I- (4-bromophenyl)ethanamine as the starting material. LCMS: 474.18 (M+H)+.
EXAMPLE 311 (R)-N-(l-(4-Bromophenyl)ethyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was prepared as described in EXAM,PLE 304, Step 2 using (R)-I- (4-bromophenyl)ethanamine as the starting material. 1H NMR (400 MHz, CDCI3) δ 8.66 (m, IH), 8.04 (m, IH), 7.44 (d, 2H), 7.20 (d, 2H), 7.01 (m, IH), 6.74 (m, IH), 6.37 (m, IH), 6.10 (d, IH), 5.12-5.05 (m, IH), 1.52 (d, 3H). LCMS: 474.18 (M+H)+.
EXAMPLE 312 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(l-(4- (trifluoromethoxy)phenyl)ethyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using l-(4- (trifluoromethoxy)phenyl)ethanamine as the starting material. 1H NMR (400 MHz, CDCl3) δ 8.66 (m, IH), 8.03 (m, IH), 7.44 (d, 2H), 7.16 (d, 2H), 7.01 (m, IH), 6.75 (m, IH), 6.38-6.36 (m, IH), 6.13 (d, IH), 5.17-5.10 (m, IH), 1.53 (d, 3H). LCMS: 478.28 (M+H)+.
EXAMPLE 313 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(l-(4-(4- fluorophenoxy)phenyl)ethyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 304, Step 2 using l-(4- (4-fluorophenoxy)phenyl)ethanamine as the starting material. 1H NMR (400 MHz, CDCl3) δ 8.67 (m, IH), 8.04 (m, IH), 7.29-7.26 (m, 2H),7.05-6.95 (m, 7H), 6.73 (m, IH), 6.37 (m, IH), 6.09 (d, IH), 5.15-5.08 (m, IH), 1.54 (d, 3H). LCMS: 504.30 (M+H)+.
EXAMPLE 314 N-(4-(3-Chloro-4-fluorophenoxy)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 3-chloro-4- fluorophenol as the starting material in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.70 (m, IH), 8.07 (m, IH), 7.28 (d, 2H), 7.09 (t, IH), 7.03 (m, 2H), 6.94 (d, 2H), 6.86 (m, IH), 6.75 (dd, IH), 6.38 (m, IH), 6.22 (t, IH), 4.48 (d, 2H). LCMS: 524.28 (M+H)+.
EXAMPLE 315 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluoro-3- (trifluoromethyl)phenoxy) benzyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 265 using 4-fluoro-3- (trifluoromethyl)phenol as the starting material in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.69 (m, IH), 8.07 (m, IH), 7.28 (d, 2H), 7.22-7.21 (m, IH), 7.16-7.14 (m, 2H), 7.02- 7.01 (m, IH), 6.97-6.94 (m, 2H), 6.75 (dd, IH), 6.38 (dd, IH), 6.25 (t, IH), 4.49 (d, 2H). LCMS: 524.28 (M+H)+
EXAMPLE 316
N-((5-(3-Chloro-4-fluorophenoxy)pyridin-2-yl)methyl)-l-(3-chloro-5- (trifluoromethyl) pyridin-2-yl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in Example 265 using 5- fluoropicolinonitrile and 3-chloro-4-fluorophenol as the starting materials in Step 1. LCMS: 525.29 (M+H)+
EXAMPLE 317
N-((5-(3-Chloro-4-fluorophenoxy)pyridin-2-yl)methyl)-l-(3,5-dichloropyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 1 using Intermediate 2 and (5-(3-chloro-4-fluorophenoxy)pyridin-2-yl)methanamine as the starting materials. LCMS: 493.21 (M+H)+.
EXAMPLE 318
N-(4-(3-Chloro-4-fluorophenoxy)benzyl)-l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-
2-carboxamide

The title compound was prepared as described in EXAMPLE 1 using Intermediate 2 and (4-(3-chloro-4-fluorophenoxy)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, IH), 7.85 (d, IH), 7.27 (d, 2H), 7.09 (t, IH), 7.02 (m, IH), 6.97-6.93 (m, 3H), 6.86 (m, IH), 6.71 (dd, IH), 6.35 (dd, IH), 6.20 (t, IH), 4.48 (d, 2H). LCMS: 492 (M+H)+.
EXAMPLE 319 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-(4-fluoro-3- (trifluoromethyl)phenoxy) pyridin-2-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 265 using 5- fluoropicolinonitrile and 4-fluoro-3-(trifluoromethyl)phenol as the starting materials in Stepl. LCMS: 559.32 (M+H)+
EXAMPLE 320 l-(3,5-Dichloropyridin-2-yl)-N-((5-(4-fluoro-3-(trifluoromethyl)phenoxy)pyridin- 2-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 1 using Intermediate 2 and (5-(4-fluoro-3-(trifluoromethyl)phenoxy)pyridin-2-yl)methanamine as the starting materials. 1H NMR (400 MHz, CD3OD) δ 8.41 (d, IH), 8.34 (d, IH), 8.11 (d, IH), 7.65 (dd, IH), 7.53 (d, IH), 7.43-7.37 (m, 3H), 7.08 (m, IH), 7.04 (m, IH), 6.38 (dd, IH), 4.54 (s, 2H), 1 NH resonance was not observed.. LCMS: 525.29 (M+H)+.
EXAMPLE 321 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluoro-3-(trifluoromethyl)phenoxy)benzyl)- lH-pyrrole-2-carboxamide

The title compound was prepared as described in Example 1 using Intermediate 2 and (4-(4-fluoro-3-(trifluoromethyl)phenoxy)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 7.85 (d, IH), 7.28 (d, 2H), 7.22 (m, IH), 7.16-7.13 (m, 2H), 6.97-6.93 (m, 3H), , 6.72 (dd, IH), 6.35 (dd, IH), 6.23 (t, IH), 4.48 (d, 2H). LCMS: 524.29 (M+H)+.
EXAMPLE 322 l-(3,5-Dichloropyridin-2-yl)-N-(4-(2-(trifluoromethyl)pyrimidin-5-yloxy)benzyl)- lH-pyrrole-2-carboxamide

Step 1. 5-Bromo-2-iodopyrimidine

A mixture of 5-bromo-2-chloropyrimidine (10 g, 52.08 mmol), NaI (60 g, 400mmol), and HI (1 rnL) in 1,4-dioxane (400 rnL) was refluxed for 4 h. The solids were filtered off. The resulting solution was diluted with 100 rnL of EtOAc, washed with brine (50 rnL), dried, and concentrated in vacuo to give the crude that was purified by silica gel column chromatography (EtOAc/PE) to afford 5-bromo-2-iodopyrimidine as a white solid. Step 2. 5-Bromo-2-(trifluoromethyl)pyrimidine

The title compound was synthesized as described in EXAMPLE 54, Step 2 using 5- bromo-2-iodopyrimidine as the starting material.
Step 3. l-(3,5-Dichloropyridin-2-yl)-N-(4-(2-(trifluoromethyl)pyrimidin-5- yloxy)benzyl)-lH-pyrrole-2-carboxamide

EXAMPLE 323 l-(3,5-Dichloropyridin-2-yl)-N-(4-(2-(trifluoromethyl)pyrimidin-5-yloxy)benzyl)- lH-pyrazole-5-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 5 and (4-(2-(trifluoromethyl)pyrimidin-5-yloxy)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, DMSO-d6) δ 9.28 (t, IH), 8.76 (s, 2H), 8.59 (d, IH), 8.49 (d, IH) 7.84 (d, IH), 7.34 (d, 2H), 7.22 (d, 2H), 7.14 (d, IH), 4.36 (d, 2H). LCMS: 509.27 (M+H)+.
EXAMPLE 324 l-(3,5-Dichloropyridin-2-yl)-N-((5-(4-fluorophenoxy)pyrimidin-2-yl)methyl)-lH- pyrrole-2-carboxamide

EXAMPLE 325 l-(3,5-Dichloropyridin-2-yl)-N-((2-(4-(trifluoromethyl)phenoxy)pyrimidin-5- yl)methyl)-lH-pyrrole-2-carboxamide

Step 1. 5-Bromo-2-(4-(trifluoromethyl)phenoxy)pyrimidine

The title compound was synthesized as described in EXAMPLE 265, Step 1 using 5- bromo-2-chloropyrimidine and 4-(trifluoromethyl)phenol as the starting materials. Step 2. 2-(4-(Trifluoromethyl)phenoxy)pyrimidine-5-carboxylic acid

Step 3. 2-(4-(Trifluoromethyl)phenoxy)pyrimidine-5-carboxamide

A solution of 2-(4-(trifluoromethyl) phenoxy)pyrimidine-5-carboxylic acid (1.1 g, 3.87 mmol) in SOCl2 (40 mL) was refluxed for 2 h, cooled down and evaporated to dryness to afford the corresponding acid chloride. 2-(4-(trifluoromethyl)phenoxy)pyrimidine- 5-carbonyl chloride was dissolved in THF (mL) and treated with NH3. H2O (80 mL, 25%), at O0C. The resulting solution was allowed to stir 0-100C for 1 h. Upon completion of the reaction, the mixture was concentrated down and taken up into EtOAc (200 ml). It was washed with H2O (3 x 10 mL), and brine, dried, filtered, and concentrated to give the crude that was recrystallized from (1 :1) Et2OZPE affording 2- (4-(trifluoromethyl)phenoxy)pyrimidine-5-carboxamide as a yellow solid. LCMS: 283 (M+H)+.
Step 4. 2-(4-(Trifluoromethyl)phenoxy)pyrimidine-5-carbonitrile

A solution of 2-(4-(trifluoromethyl)phenoxy) pyrimidine-5-carboxamide (920 mg, 3.25 mmol) in POCl3 (20 mL) was refluxed for 4 h. The reaction mixture was cooled to rt and concentrated under vacuum. Then the residue was diluted with 20 mL of H2O/ice. The pH was adjusted to 8-9 with 2N NaOH. The resulting solution was extracted with EtOAc (3 x 100 mL). Combined organics were dried, filtered, and concentrated to give the crude that was purified by silica gel column chromatography (EtOAc/PE) to afford 2-(4-(trifluoromethyl)phenoxy)pyrimidine-5-carbonitrile as a white solid.
Step 5. (2-(4-(Trifluoromethyl)phenoxy)pyrimidin-5-yl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 2-(4-
(trifluoromethyl)phenoxy)pyrimidine-5-carbonitrile as the starting material.
Step 6. l-(3,5-Dichloropyridin-2-yl)-N-((2-(4-(trifluoromethyl)phenoxy)pyrimidin-
5-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (2-(4-(trifluoromethyl)phenoxy)pyrimidin-5-yl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 2H), 8.40 (s, IH), 7.70 (m, IH), 7.72 (d, 2H), 7.33 (d, 2H), 7.00 (s, IH), 6.89 (m, 2H), 6.36 (s, IH), 4.53 (d, 2H). LCMS: 508.24 (M+H)+.
EXAMPLE 326 l-(3,5-Dichloropyridin-2-yl)-N-(4-(methylsulfonyl)benzyl)-lH-pyrrole-2- carboxamide

EXAMPLE 327 l-(3,5-Dichloropyridin-2-yl)-N-(4-(trifluoromethylthio)benzyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(trifluoromethylthio)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.38 (m, IH), 8.85 (d, IH), 7.60 (d, 2H), 7.34 (d, 2H), 7.00 (m, IH), 6.74 (m, IH), 6.37-6.32 (m, 2H), 4.52 (d, 2H). LCMS: 446.19 (M+H)+. LCMS: 446.19 (M+H)+.
EXAMPLE 328 l-(3,5-Dichloropyridin-2-yl)-N-(4-(N,N-dimethylsulfamoyl)benzyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and 4-(aminomethyl)-N,N-dimethylbenzene sulfonamide as the starting materials. LCMS: 453.23 (M+H)+.
EXAMPLE 329
4-Bromo-l-(3,5-dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-
2-carboxamide

Step 1. 4-Bromo-l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carboxylic acid
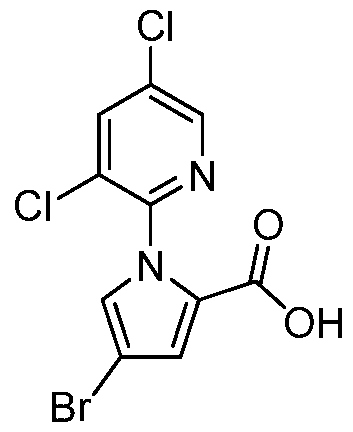
To a suspension of NaH ( 0.26g, 6.57 mmol, 60% dispersion in mineral oil) in DMSO was added 4-bromo-lH-pyrrole-2-carboxylic acid (0.5g, 2.63 mmol) at rt. This mixture was allowed to stir for 30 min. at this temperature. Then, a DMSO solution of 3,5-dichloro-2-fluoropyridine (0.44, 2.63 mmol) was added slowly and the mixture was heated to 8O0C for 2 h. Reaction mixture was cooled down to rt, quenched with H2O, and rinsed into a seperatory funnel. The pH was adjusted with IN HCl and extracted with EtOAc (3 x 100 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified on a silica gel column chromatography (EtO Ac/Hexanes) affording 4-bromo- 1 -(3 ,5 -dichloropyridin-2-yl)- 1 H-pyrrole-2- carboxylic acid.
Step 2. 4-Bromo-l-(3,5-dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using 4-bromo-l- (3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl) methanamine as the starting materials. LCMS: 536.08 (M+H)+.
EXAMPLE 330 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-4-vinyl-lH-pyrrole-2- carboxamide

A mixture of 4-bromo-l-(3,5-dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide (50 mg, 0.093 mmol), vinylboronic anhydride pyridine complex (22 mg, 0.093 mmol), Pd(PhP)2Cl2 (6.5 mg, 0.009 mmol), and K2CO3 (0.19 mL of 10 % aq. solution) in dioxane (1 mL) was degassed and purged with nitrogen three times. This mixture was then heated to 1000C for 4 h. Reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3 x 100 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified by semi preparative chromatography (ACN/H2O:TFA) to afford l-(3,5- dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-4-vinyl-lH-pyrrole-2- carboxamide. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 7.85 (d, IH), 7.24 (d, 2H),
7.05-6.91 (m, 7H), 6.83 (d, IH), 6.57 (dd, IH), 6.17 (t, IH), 5.48 (dd, IH), 5.10 (dd, IH), 4.47 (d, 2H).. LCMS: 482.17 (M+H)+.
EXAMPLE 331
4-Acetyl-l-(3,5-dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrrole-
2-carboxamide

A mixture of 4-bromo-l-(3,5-dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide (50 mg, 0.093 mmol), tributyl(l-ethoxyvinyl)stannane (33 mg, 0.093 mmol), and Pd(PhP)2Cl2 (6.5 mg, 0.009 mmol) in dioxane (1 mL) was degassed and purged with nitrogen three times. This mixture was then heated to 1000C for 4 h. It was cooled down to rt and treated with IN HCl. Reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3 x 100 mL). Combined organics were dried, filtered, and concentrated to give the crude that was purified by semi preparative chromatography (ACN/H2O:TFA) to afford 4-acetyl-l-(3,5- dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)- 1 H-pyrrole-2-carboxamide. 1H NMR (400 MHz, CDC13) δ 8.42 (d, IH), 7.89 (d, IH), 7.53 (d, IH), 7.24 (d, 2H), 7.13 (m, IH), 7.05-6.97 (m, 6H), 6.29 (t, IH), 4.46 (d, 2H), 2.45 (s, 3H). LCMS: 498.15 (M+H)+.
EXAMPLE 332 l-(3,5-Dichloropyridin-2-yl)-N-((2-(4-fluorophenoxy)pyrimidin-5-yl)methyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in 325 using 4-fluorophenol as the starting material in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 2H), 8.39 (m, IH), 7.86 (m, IH), 7.16-7.08 (m, 4H), 6.99 (dd, IH), 6.72 (dd, IH), 6.38 (t, IH), 6.35 (dd, IH), 4.44 (d, 2H). LCMS: 458.15 (M+H)+.
EXAMPLE 333 Isopropyl 4-(4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2-carboxamido)methyl) phenoxy)piperidine-l-carboxylate

Stepl. Isopropyl 4-(4-cyanophenoxy)piperidine-l-carboxylate

A solution of isopropyl 4-hydroxypiperidine-l-carboxylate (1.87 g, 10.00 mmol) was added to a solution of NaH (260 mg, 10.83 mmol) in DMF (40 mL) and the resulting solution was stirred for 5 min at rt. To the mixture 4-fluorobenzonitrile (1.21 g, 10.00 mmol) was added. The resulting solution was stirred for 30 min at rt. The reaction was
then quenched by the addition H2O (100 mL), and the mixture was extracted with EtOAc (3 x 200 mL). Combined organic layers were washed with NaOH(aq) (2 x 100 mL) and brine (2 x 100 mL), dried, filtered, concentrated, and purified by a silica gel chromatography (EtOAc/PE) to afford isopropyl 4-(4-cyanophenoxy)piperidine-l- carboxylate as a white solid. Step2. Isopropyl 4-(4-(aminomethyl)phenoxy)piperidine-l-carboxylate

A solution of isopropyl 4-(4-cyanophenoxy)piperidine-l-carboxylate (320 mg, 1.11 mmol), HC1(36%) (0.5 mL) and Pd/C (170 mg) in CH3OH (30 mL) was stirred for 4 h at rt under a H2 atmosphere. Pd/C was removed and the filtrate was concentrated under vacuum to afford the crude product that was recrystallization from EtOH/EtOAc to give isopropyl 4-(4-(aminomethyl) phenoxy)piperidine-l-carboxylate hydrochloride as a white solid.
Step3. Isopropyl 4-(4-((l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl) phenoxy)piperidine-l-carboxylate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and isopropyl 4-(4-(aminomethyl)phenoxy)piperidine-l-carboxylate as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.39 (m, IH), 7.85 (m, IH), 7.20 (d, 2H), 6.96 (dd, IH), 6.85 (d, 2H), 6.68 (dd, IH), 6.33 (dd, IH), 6.15 (t, IH), 4.95-4.89 (m, IH), 4.48-4.46 (m, IH), 4.42 (d, 2H), 3.74-3.68 (m, 2H), 3.42-3.35 (m, 2H), 1.93-1.88 (m, 2H), 1.77-1.72 (m, 2H), 1.25 (d, 6H). LCMS: 531.27 (M+H)+.
EXAMPLE 334 l-(3,5-Dichloropyridin-2-yl)-N-(4-(5-(trifluoromethyl)pyrimidin-2-yloxy)benzyl)- lH-pyrrole-2-carboxamide

The title compound was prepared as described in EXAMPLE 265 using 2-chloro-5- (trifluoromethyl)pyrimidine and 4-hydroxybenzonitrile in Step 1 and Intermediate 2 in Step 3. 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 2H), 8.39 (d, IH), 7.86 (d, IH), 7.38 (d, 2H), 7.15 (d, 2H), 6.97 (dd, IH), 6.73 (dd, IH), 6.35 (t, IH), 6.26 (t, IH), 4.53 (d, 2H). LCMS: 508.19 (M+H)+.
EXAMPLE 335 N-(l-(3,5-Dichloropyridin-2-yl)-lH-pyrazol-5-yl)-2-(4-
(trifluoromethyl)phenyl)acetamide

3,5-Dichloro-2-hydrazinylpyridine

The title compound was synthesized as described for Intermediate 3 using 2,3,5- trichloropyridine as the starting material.
Step2. Ethyl 5-amino-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-4- carboxylate

A mixture of 3,5-dichloro-2-hydrazinylpyridine (0.67 g, 3.76 mmol) and (Z)-ethyl 2- cyano-3-ethoxyacrylate (0.64 g, 3.76 mol) in EtOH (38 mL) was heated to reflux for 2 h. It was cooled down. Desired product precipitated out, filtered and washed with EtOH to afford ethyl 5 -amino- 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- IH- pyrazole-4-carboxylate as yellow solid. Step 3. 5-Amino-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-4- carboxylic acid

The title compound was synthesized as described for Intermediate 1 , Step 2 using ethyl 5 -amino- 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrazole-4-carboxylate as the starting material.
Step 4. l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazol-5-amine

A mixture of 5 -amino- 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrazole-4- carboxylic acid (0.99g, 3.6 mmol) and 6N aqueous HCl (30 mL) was refluxed for 2 h.
Then, the mixture was cooled down to rt. pH was adjusted to 9-10 with 2N NaOH. Reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3x100 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified by a silica gel chromatography (0-100 % EtOAc/hexanes) affording l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazol-5- amine.
Step 5. N-(l-(3,5-Dichloropyridin-2-yl)-lH-pyrazol-5-yl)-2-(4- (trifluoromethyl)phenyl) acetamide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrazol-5 -amine and 2-(4- (trifluoromethyl)phenyl)acetic acid as the starting materials. LCMS: 415.15 (M+H)+.
EXAMPLE 336 N-(l-(3,5-Dichloropyridin-2-yl)-lH-pyrazol-5-yl)-2-(4-
(trifluoromethoxy)phenyl)acetamide

The title compound was synthesized as described in EXAMPLE 1 using l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrazol-5 -amine and 2-(4- (trifluoromethoxy)phenyl)acetic acid as the starting materials. LCMS: 431.14 (M+H)+.
EXAMPLE 337 l-(3,5-Dichloropyridin-2-yl)-N-(4-(2-methylpyrimidin-5-yloxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 251 using 5-bromo-2- methylpyrimidine and 4-hydroxybenzonitrile in Step 1 and Intermediate 2 in Step 3. 1H NMR (400 MHz, CDCl3) δ 8.39 (m, 2H), 7.85 (d, IH), 7.30 (d, 2H), 6.99-6.95 (m, 3H), 6.72 (dd, IH), 6.35 (dd, IH), 6.26 (t, IH), 4.48 (d, 2H), 2.72 (s, 3H). LCMS: 454.13 (M+H)+.
EXAMPLE 338 tert-ButyM-^l-CS-chloro-S-CtrifluoromethylJpyridin-l-ylJ-lH-pyrrole-l- carboxamido) methyl)benzylcarbamate

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and tert-butyl 4-(aminomethyl)benzylcarbamate as the starting materials. LCMS: 408.21 (M+H)+.
EXAMPLE 339 N-(4-(Aminomethyl)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 290, Step 1 using tert- butyl 4-((l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2- carboxamido)methyl)benzyl carbamate as the starting material.. LCMS: 409.25 (M+H)+
EXAMPLE 340 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-
(methylsulfonamidomethyl)benzyl)-lH-pyrrole-2-carboxamide

To a solution of N-(4-(aminomethyl)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carboxamide (30 mg, 0.59 mmol) in ACN (0.5 mL), was added K2CO3 (24.3 mg, 0.17 mmol) followed by methanesulfonyl chloride (59 mg, 0.059 mmol) at rt. Resulting solution was allowed to stir at rt for 1 h. Reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3 x 25 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified on a silica gel chromatography (0-100 % EtOAc/hexanes) affording l-(3-
chloro-5 -(trifluoromethyl)pyridin-2-yl)-N-(4-(methylsulfonamidomethyl)benzyl)- 1 H- pyrrole-2-carboxamide. LCMS: 487.29 (M+H)+
EXAMPLE 341 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-((4-fluorophenylsulfonamido) methyl)benzyl)-lH-pyrrole-2-carboxamide

The title compound was prepared as described in Example 340 using 4- fluorophenylsulfonyl chloride as the starting material. LCMS: 567.28 (M+H)+.
EXAMPLE 342 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-ethynylpyridin-2-yl)methyl)- lH-pyrrole-2-carboxamide

Step 1. (5-Bromopyridin-2-yl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 5- bromopicolinonitrile as the starting material.
Step 2. (tert-Butyl (5-bromopyridin-2-yl)methylcarbamate

The title compound was synthesized as described in EXAMPLE 179, Step 3 using (5- bromopyridin-2-yl)methanamine as the starting material. Step 3. tert-Butyl (5-((trimethylsilyl)ethynyl)pyridin-2-yl)methylcarbamate

A mixture of tert-butyl (5-bromopyridin-2-yl)methylcarbamate (1.5 g, 5.23 mmol), Et3N (1.5 g, 14.85 mmol), CuI (100 mg, 0.53 mmol), ethynyltrimethylsilane (1.2 g, 12.24 mmol), and Pd(PPh3)2Cl2 (360 mg, 0.51 mmol) in DCM (50 mL) was heated to 4O0C for 3 h. The resulting mixture was concentrated under vacuum to give a residue that was purified by a silica gel chromatography (EtOAc/PE) to afford tert-butyl (5-(2- (trimethylsilyl)ethynyl)pyridin-2-yl)methylcarbamate as yellow oil. Step 4. tert-Butyl (5-ethynylpyridin-2-yl)methylcarbamate

A mixture of tert-butyl (5-(2-(trimethylsilyl)ethynyl)pyridin-2-yl)methylcarbamate (1.6 g, 5.26 mmol) and KOH (350 mg, 6.25 mmol) in MeOH (50 mL) was stirred at 250C for 30 min. The resulting mixture was concentrated and it was diluted with H2O (50 mL). The resulting solution was extracted with EtOAc (3 x 50 mL). Combined organics were dried, filtered, and concentrated to give a residue was purified by a silica gel chromatography (EtOAc/PE) affording of tert-butyl (5-ethynylpyridin-2- yl)methylcarbamate as a white solid.
Step 5. (5-Ethynylpyridin-2-yl)methanamine

The title compound was synthesized as described in EXAMPLE 290, Step 1 using tert- butyl (5-ethynylpyridin-2-yl)methylcarbamate as the starting material.
Step 6. l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-ethynylpyridin-2- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (5-ethynylpyridin-2-yl)methanamine as the starting materials. LCMS: 405.25 (M+H)+.
EXAMPLE 343 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-((4-methylpiperazin-l- yl)methyl)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-((4-methylpiperazin-l-yl)methyl)phenyl)methanamine as the starting materials. LCMS: 492.37 (M+H)+.
EXAMPLE 344 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-isopropylpyridin-2-yl)methyl)- lH-pyrrole-2-carboxamide

Stepl . 2-(6-Bromopyridin-3-yl)propan-2-ol


A 100-mL round-bottom flask was charged with 2-(6-bromopyridin-3-yl)propan-2-ol (5 g, 23.15 mmol, 1.00 equiv). To this was added P (6 g, 193.55 mmol, 8.36 equiv). To the mixture was added HI (50 mL). The resulting solution was allowed to react, with stirring, for 2 hours while the temperature was maintained at reflux in an oil bath. The pH was adjusted to 9 with NaOH (40 %). The resulting solution was extracted
with of ethoxyethane (5x500 mL) and the organic layers combined and dried over anhydrous sodium sulfate and concentrated under vacuum affording 2-bromo-5- isopropylpyridine as yellow oil (4.5 g, 97%). Step 3. 5-Isopropylpicolinonitrile

A mixture of 2-bromo-5-isopropylpyridine (4.5 g, 22.50 mmol) and CuCN (3 g, 33.33 mmol) in DMF (60 mL) was refluxed for 2 h and then cooled to rt. The reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3 x 25 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified on a silica gel chromatography (EtOAC/PE) affording 5- isopropylpicolinonitrile as a white solid (1.2g, 37%). Step 4. (5-Isopropylpyridin-2-yl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 5- isopropylpicolinonitrile as the starting material.
Step 5. l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-isopropylpyridin-2- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (5-isopropylpyridin-2-yl)methanamine as the starting materials. 1H NMR (400 MHz, CDC13) δ 8.69 (d, IH), 8.41 (d, IH), 8.05 (d, IH), 7.50 (dd, IH), 7.28 (t, IH),
7.17 (d, IH), 6.98 (m, IH), 6.86 (dd, IH), 6.38 (t, IH), 4.56 (d, 2H), 2.95-2.91 (m, IH), 1.26 (d, 6H). LCMS: 423.29 (M+H)+.
EXAMPLE 345 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-isobutylpyridin-2-yl)methyl)- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 344 using isobutyraldehyde as the starting material in Step 1. LCMS: 437.33 (M+H)+.
EXAMPLE 346 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(morpholinomethyl)benzyl)- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 1 and (4-(morpholinomethyl)phenyl) methanamine as the starting materials. 1H NMR (400 MHz, CDC13) δ 8.69 (m, IH), 8.07 (d, IH), 7.30-7.24 (m, 4H), 6.98 (dd, IH), 6.72 (dd, IH), 6.36 (dd, IH), 6.20 (t, IH), 4.48 (d, 2H), 3.72-3.70 (m, 4H), 3.05 (s, 2H), 2.46-2.45 (m, 4H). LCMS: 479.35 (M+H)+.
EXAMPLE 347 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(3,4-difluorophenoxy)benzyl)- lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4- fluorobenzonitrile and 3,4-difluorophenol as the starting materials in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, IH), 8.07 (s, IH), 7.26 (d, 2H), 7.05 (q, IH), 7.01 (s, IH), 6.94 (d, 2H), 6.80-6.75 (m, 3H), 6.37 (d, IH), 6.22 (t, IH), 4.48 (d, 2H). LCMS: 508.30 (M+H)+.
EXAMPLE 348 l-(3,5-Dichloropyridin-2-yl)-N-(4-(3,4-difluorophenoxy)benzyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(3,4-difluorophenoxy)phenyl) methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 7.85 (d, IH), 7.28 (d, 2H), 7.10 (q, IH), 6.97-6.93 (m, 3H), 6.83-6.80 (m IH), 6.72-6.71 (m, 2H), 6.35 (dd, IH), 6.32 (t, IH), 4.46 (d, 2H).
EXAMPLE 349 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluoro-3- methylphenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4- fluorobenzonitrile and 4-fluoro-3-methylphenol as the starting materials in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, IH), 8.07 (s, IH), 7.25-7.23 (m, 3H), 7.01-6.72 (m, 6H), 6.37 (dd, IH), 6.22 (t, IH), 4.45 (d, 2H), 2.24 (s, 3H). LCMS: 504.32 (M+H)+.
EXAMPLE 350 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluoro-3-methylphenoxy)benzyl)-lH-pyrrole-
2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(4-fluoro-3-methylphenoxy)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 7.85 (d, IH), 7.23 (d, 2H), 6.98-6.89 (m, 4H), 6.83-6.81 (m, IH), 6.78-6.75 (m, IH), 6.70 (m, IH), 6.33 (dd, IH), 6.42 (t, IH), 4.45 (d, 2H), 2.24 (s, 3H). LCMS: 470.35 (M+H)+.
EXAMPLE 351 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluorobenzyl)benzyl)-lH- pyrrole-2-carboxamide

A mixture of N-(4-bromobenzyl)- 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H- pyrrole-2-carboxamide (20 mg, 0.043 mmol), 4-fluorobenzyl zinc chloride (0.184 rnL, 0.091 mmol, 0.5 M solution in THF), CuI (1.7 mg, 0.0087 mmol), and Pd (PPh3)2Cl2 (3.1 mg, 0.004 mmol) was heated to 15O0C for 10 min in a microwave. The reaction mixture was then rinsed into a seperatory funnel and extracted with EtOAc (3 x 10 mL). Combined organics were dried, filtered, and concentrated in vacuo to give the crude that was purified by semi-preparative chromatography (ACN/H2O:TFA) to afford 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluorobenzyl)benzyl)- 1 H- pyrrole-2-carboxamide. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, IH), 8.07 (s, IH), 7.22 (d, 2H), 7.14-7.01 (m, 4H), 7.01-6.96 (m, 3H), 6.72 (dd, IH), 6.37 (dd, IH), 6.18 (t, IH), 4.47 (d, 2H), 3.93 (s, 2H). LCMS: 488.32 (M+H)+. LCMS: 437.33 (M+H)+.
EXAMPLE 352 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((6-(4-fluorophenoxy)pyridin-3- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 6- fluoronicotinonitrile and 4-fluorophenol as the starting materials in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.68 (d, IH), 8.06 (m, 2H), 7.64 (dd, IH), 7.06 (m, 4H), 7.00 (dd, IH), 6.85 (d, IH), 6.72 (dd, IH), 6.36-6.35 (m, 2H),4.42 (d, 2H). LCMS: 491.36 (M+H)+.
EXAMPLE 353 l-(3,5-Dichloropyridin-2-yl)-N-((6-(4-fluorophenoxy)pyridin-3-yl)methyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (6-(4-fluorophenoxy)pyridin-3-yl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 8.06 (s, IH), 7.85 (dd, IH), 7.65-7.63 (m, IH), 7.08- 6.97 (m, 4H), 6.96 (m, IH), 6.85 (dd, IH), 6.69 (dd, IH), 6.33-6.32 (m, IH), 6.45 (t, IH), 4.42 (d, 2H). LCMS: 457.34 (M+H)+.
EXAMPLE 354 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-((5-(4-fluorophenoxy)pyridin-2- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 5- fluoropicolino nitrile and 4-fluorophenol as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.69 (d, IH), 8.29 (dd, IH), 8.05 (dd, IH), 7.22-6.99 (m, 8H), 6.86 (dd, IH), 6.39 (dd, IH), 4.57 (d, 2H). LCMS: 491.32 (M+H)+.
EXAMPLE 355 l-(3,5-Dichloropyridin-2-yl)-N-((5-(4-fluorophenoxy)pyridin-2-yl)methyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (5-(4-fluorophenoxy)pyridin-2-yl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, IH), 8.27 (dd, IH), 7.82 (d, IH), 7.20-6.96 (m, 8H), 6.82 (dd, IH), 6.34 (dd, IH), 4.55 (d, 2H). LCMS: 457.26 (M+H)+.
EXAMPLE 356 4-Bromo-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4- fluorophenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 329 using 3-Chloro-2- fluoro-5-(trifluoromethyl) pyridine as the starting material in Step 1. 1H NMR (400 MHz, CDC13) δ 8.68 (d, IH), 8.08 (d, IH), 7.22 (d, 2H), 7.05-6.92 (m, 7H), 6.71 (d, IH), 6.14 (t, IH), 4.46 (d, 2H). LCMS: 570.24 (M+H)+.
EXAMPLE 357
4-Bromo-l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(4-(4- fluorophenoxy)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was synthesized as described in EXAMPLE 1 using 4-bromo-l-(3- chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carboxylic acid and (4-(4- fluorophenoxy)phenyl)hydrazine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 9.67 (s, IH), 8.62 (s, IH), 8.00 (s, IH), 7.38 (s, 2H), 7.12 (s, IH), 7.02-6.89 (m, 8H).
EXAMPLE 358 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(6-(trifluoromethyl)pyridin-3- yloxy) benzyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using 4- fluorobenzonitrile and 6-(trifluoromethyl)pyridin-3-ol as the starting materials in Step 1. 1H NMR (400 MHz, CDCl3) δ 8.70 (m, IH), 8.46 (d, IH), 8.05 (d, IH), 7.62 (d, IH), 7.37-7.30 (m, 3H), 7.06-7.03 (m, 3H), 6.77 (dd, IH), 6.39 (dd, IH), 6.31 (t, IH), 4.52 (d, 2H). LCMS: 541.32 (M+H)+.
EXAMPLE 359 l-(3,5-Dichloropyridin-2-yl)-N-(4-(6-(trifluoromethyl)pyridin-3-yloxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2 and (4-(6-(trifluoromethyl)pyridin-3-yloxy)phenyl)methanamine as the starting materials. 1H NMR (400 MHz, CDCl3) δ 8.45 (d, IH), 8.39 (d, IH), 7.85 (s, 1H),7.61 (d, IH), 7.35-7.29 (m, 3H), 7.03-6.97 (m, 3H), 6.73 (dd, IH), 6.35 (dd, IH), 6.30 (t, IH), 4.50 (d, 2H). LCMS: 507.28 (M+H)+.
EXAMPLE 360 l-(3,5-Dichloropyridin-2-yl)-N-((5-(4-(trifluoromethyl)phenoxy)pyrimidin-2- yl)methyl)-lH-pyrrole-2-carboxamide

The title compound can be prepared as described in EXAMPLE 251 using 4- (trifluoromethyl) phenol and 5-bromopyrimidine-2-carbonitrile as the starting materials in Step 1 and Intermediate 2 in Step 3. 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 2H), 8.38 (d, IH), 7.85 (d, IH), 7.66 (d, 2H), 7.35 (t, IH), 7.10 (d, 2H), 6.96 (m, IH), 6.89 (dd, IH), 6.37 (dd, IH), 4.75 (d, 2H). LCMS: 508.28 (M+H)+.
EXAMPLE 361 l-(3,5-Dichloropyridin-2-yl)-N-((6-(4-(trifluoromethyl)phenoxy)pyridazin-3- yl)methyl)-lH-pyrrole-2-carboxamide

Step 1. 3-Chloro-6-(4-(trifluoromethyl)phenoxy)pyridazine

The title compound was synthesized as described in EXAMPLE 265, Step 1 using 3,6- dichloropyridazine and 4-(trifluoromethyl)phenol as the starting materials.
Step 2. 6-(4-(Trifluoromethyl)phenoxy)pyridazine-3-carbonitrile

A mixture of 3-chloro-6-(4-(trifluoromethyl)phenoxy)pyridazine (2 g, 7.30 mmol), Zn(CN)2 (0.5 g, 4.27 mmol), and Pd(PPh3)4 (240 mg, 2.08 mmol) in DMF (40 mL) was heated to 1000C for 4 h. The reaction was then diluted with H2O (40 mL), extracted with EtOAc (3 x 50 mL), and the combined organic layers were washed with brine (50 mL), dried, and concentrated under vacuum to give a residue that was purified by a silica gel chromatography (EtOAc/PE) to afford 6-(4-
(trifluoromethyl)phenoxy)pyridazine-3-carbonitrile as a white solid (I g, 47%). Step 3. (6-(4-(trifluoromethyl)phenoxy)pyridazin-3-yl)methanamine

The title compound was synthesized as described in EXAMPLE 2, Step 1 using 6-(4-
(trifluoromethyl)phenoxy)pyridazine-3-carbonitrile as the starting material.
Step 4. l-(3,5-Dichloropyridin-2-yl)-N-((6-(4-(trifluoromethyl)phenoxy)pyridazin-
3-yl)methyl)-lH-pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 1 using Intermediate 2and (6-(4-(trifluoromethyl)phenoxy)pyridazin-3-yl)methanamine as the starting material. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, IH), 7.84 (d, IH), 7.69 (d, 2H), 7.51 (d, IH), 7.31 (d, 2H), 7.20 (m, 2H), 6.95 (m, IH), 6.81 (dd, IH), 6.33 (dd, IH), 4.72 (d, 2H). LCMS: 508.29 (M+H)+.
EXAMPLE 362 l-(3,5-Dichloropyridin-2-yl)-N-(4-(5-(trifluoromethyl)pyridin-2-yloxy)benzyl)-lH- pyrrole-2-carboxamide

The title compound was synthesized as described in EXAMPLE 265 using A- hydroxybenzonitrile and 2-fluoro-5-(trifluoromethyl)pyridine as starting materials in Step 1 and Intermediate 2 in Step 3. 1H NMR (400 MHz, CDCl3) δ 8.36 (m, IH), 8.32 (d, IH), 7.83 (dd, 1H),7.78 (d, IH), 7.29 (d, 2H), 7.09 (d, 2H), 6.95 (d, IH), 6.90 (dd, IH), 6.65 (dd, 1H),6.28 (dd, IH), 6.17 (t, IH), 4.44 (d, 2H). LCMS: 507.28 (M+H)+.
EXAMPLE 363 l-(3,5-Dichloropyridin-2-yl)-N-(4-(5-fluoropyridin-2-yloxy)benzyl)-lH-pyrrole-2- carboxamide

The title compound was synthesized as described in EXAMPLE 265 using A- hydroxybenzonitrile and 2,5-difluoropyridine as starting materials in Step 1 and Intermediate 2 in Step 3. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, IH), 8.02 (d, IH), 7.77 (dd, IH), 7.46-7.41 (m, IH), 7.31 (d, 2H), 7.06 (d, 2H), 6.97 (m, IH), 6.90 (dd, IH), 6.73 (dd, 1H),6.35 (dd, IH), 6.22 (t, IH), 4.48 (d, 2H). LCMS: 457.26 (M+H)+.
EXAMPLE 364 N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-CS-chloro-S-fluoropyridin-l-yl)- lH-pyrrole-2-carbohydrazide

Step 1 : Methyl l-(3-chloro-5-nitropyridin-2-yl)-lH-pyrrole-2-carboxylate

Methyl l-(3-chloro-5-nitropyridin-2-yl)-lH-pyrrole-2-carboxylate was prepared from 2,3-dichloro-5-nitropyridine and methyl lH-pyrrole-2-carboxylate following the procedure outlined in Intermediate 1, Stepl.
Step 2: Methyl l-(5-amino-3-chloropyridin-2-yl)-lH-pyrrole-2-carboxylate

In several batches, FeSO4 (10.8 g, 71.1 mmol) was added to a solution of methyl l-(3- chloro-5-nitropyridin-2-yl)-lH-pyrrole-2-carboxylate (2.0 g, 7.1 mmol) and cone, ammonium hydroxide (100 mL). After 2 h at 35°C, the reaction was extracted with EtOAc (200 mL). The extract was washed with water (100 mL x 3), dried, filtered, and concentrated to give methyl l-(5-amino-3-chloropyridin-2-yl)-lH-pyrrole-2- carboxylate as a yellow solid.
Step 3 : Methyl l-(3-chloro-5-fluoropyridin-2-yl)-lH-pyrrole-2-carboxylate

A solution of methyl l-(5-amino-3-chloropyridin-2-yl)-lH-pyrrole-2-carboxylate (1.0 g, 4.0 mmol) and 48% HBF4 (100 niL) was maintained at rt for 30 min and then cooled to 00C. A solution OfNaNO2 (300 mg, 4.35 mmol) and water (20 mL) was added dropwise. The reaction was heated at 80 0C for 1 h, cooled to rt, diluted with water (100 mL), and then extracted with EtOAc (100 mL x 2). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (l :20→l :5-EtOAc/PE) to give methyl l-(3-chloro-5-fluoropyridin-2-yl)-lH-pyrrole-2- carboxylate as a white solid. Step 4: Nl-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-5-fluoropyridin-
2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from methyl l-(3-chloro-5-fluoropyridin-2-yl)-lH- pyrrole-2-carboxylate and Intermediate 3 following the procedures outlined in Intermediate 1, Step 2 & EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.24 (s, IH), 9.16 (s, IH), 8.48 (d, IH), 8.36 (d, IH), 8.25 (dd, IH), 8.07 (d, IH), 7.20 (dd, IH), 7.17 (dd, IH), 6.35 (dd, IH); LCMS: 434.4 (M+H)+.
EXAMPLE 365
Isopropyl 4-(2-(2-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)hydrazinecarbonyl)- lH-pyrrol-l-yl)piperidine-l-carboxylate

Methyl l-(l-benzylpiperidin-4-yl)-lH-pyrrole-2-carboxylate

Methyl l-(l-benzylpiperidin-4-yl)-lH-pyrrole-2-carboxylate can be prepared from 1- benzylpiperidin-4-amine and methyl 2,5-dimethoxytetrahydrofuran-2-carboxylate following the procedure outlined in EXAMPLE 96, Step 1. LCMS: 299 (M+H)+. Step 2: Methyl l-(piperidin-4-yl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(l-benzylpiperidin-4-yl)-lH-pyrrole-2-carboxylate (500 mg, 1.68 mmol), 10% Pd/C (0.50 g, 0.47 mmol Pd), and MeOH (50 mL) was stirred vigorously at rt under an atmosphere of H2. After 6 h, the mixture was filtered through Celite, washed with MeOH (10 mL x 3), and concentrated to give methyl l-(piperidin- 4-yl)-lH-pyrrole-2-carboxylate as a white solid. LCMS: 209 (M+H)+.
_3: Isopropyl 4-(2-(methoxycarbonyl)-lH-pyrrol-l-yl)piperidine-l-carboxylate

Isopropyl 4-(2-(methoxycarbonyl)- 1 H-pyrrol- 1 -yl)piperidine- 1 -carboxylate was prepared from methyl l-(piperidin-4-yl)-lH-pyrrole-2-carboxylate following the procedure outlined in EXAMPLE 41, Step 3. LCMS: 295 (M+H)+. Step 4: Isopropyl 4-(2-(2-(3-chloro-5-(trifluoromethyl)pyridin-2- ylJhydrazinecarbonylJ-lH-pyrrol-l-ylJpiperidine-l-carboxylate

The title compound was prepared from isopropyl 4-(2-(methoxycarbonyl)-l H-pyrrol- 1- yl)piperidine-l -carboxylate and Intermediate 3 following the procedures outlined in Intermediate 1, Step 2 & EXAMPLE 1. LCMS: 474.3 (M+H)+.
EXAMPLE 366
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-5-methoxypyridin-2-yl)- lH-pyrrole-2-carbohydrazide


Sodium nitrite (443 mg, 6.42 mmol) was added to a solution of methyl l-(5-amino-3- chloropyridin-2-yl)-lH-pyrroIe-2-carboxylate (1.61 g, 6.39 mmol) and 48% HBr (40 mL) at 00C. After 2 h at 00C, CuBr (492 mg, 3.46 mmol) was added, and the reaction was allowed to warm to rt. After 4 h at rt, the reaction was diluted with water (50 mL) and filtered. The filtrate was adjusted to pH 7-8 with NaOH and extracted with EtOAc (150 mL x 3). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (l :100-EtOAc/PE) to give methyl l-(5-bromo-3- chloropyridin-2-yI)-lH-pyrrole-2-carboxylate as yellow oil. LCMS: 315 (M+H)+. Step 2: Methyl l-(3-chloro-5-methoxypyridin-2-yl)-lH-pyrrole-2-carboxylate

A mixture of methyl l-(5-bromo-3-chloropyridin-2-yl)-lH-pyrrole-2-carboxylate (1.38 g, 4.37 mmol), CuO (349 mg, 4.37 mmol), 4.37M sodium methoxide (4 mL), and DMF (10 mL) was heated at 110 0C for 2 h, allowed to cool to rt, diluted with water (30 mL), and filtered. The filtrate was extracted with EtOAc (150 mL x 3). The combined extracts were washed with water (50 mL x 2), dried, filtered, concentrated, and then purified by silica gel chromatography (1 :60-EtOAc/PE) to give methyl l-(3- chloro-5-methoxypyridin-2-yl)-lH-pyrrole-2-carboxylate as a yellow solid. LCMS: 267 (M+H)+.
Ste2J.: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3-chloro-5- methoxypyridin-2-yl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from methyl l-(3-chloro-5-methoxypyridin-2-yl)- lH-pyrrole-2-carboxylate and Intermediate 3 following the procedures outlined in Intermediate 1, Step 2 & EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.20 (s, IH), 9.15 (s, IH), 8.38 (s, IH), 8.13-8.07 (m, 2H), 7.72 (d, IH), 7.18 (m, IH), 7.11 (m, IH), 6.33 (dd, IH), 3.88 (s, 3H); LCMS: 446.2 (M+H)+.
EXAMPLE 367 N-(4-Chloro-3-(trifluoromethyl)benzyl)-l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was prepared from (4-chloro-3-
(trifluoromethyl)phenyl)methanamine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.95 (t, IH), 8.88 (m, IH), 8.64 (d, IH), 7.72 (d, IH), 7.69 (d, IH), 7.55 (dd, IH), 7.22 (dd, IH), 7.07 (dd, IH), 6.38 (dd, IH), 4.38 (d, 2H); LCMS: 482.2 (M+H)+.
EXAMPLE 368 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-(trifluoromethyl)phenethyl)- lH-pyrrole-2-carboxamide

The title compound was prepared from 2-(3-(trifluoromethyl)phenyl)ethanamine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.89 (m, IH), 8.64 (d, IH), 8.37 (t, IH), 7.58-7.49 (m, 4H), 7.16 (dd, IH), 6.94 (dd, IH), 6.33 (dd, IH), 3.35 (m, 2H), 2.85 (t, 2H); LCMS: 462.3 (M+H)+.
EXAMPLE 369 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(trifluoromethyl)phenethyl)- lH-pyrrole-2-carboxamide

The title compound was prepared from 2-(4-(trifluoromethyl)phenyl)ethanamine hydrochloride and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.90 (m, IH), 8.64 (d, IH), 8.38 (t, IH), 7.64 (d, 2H), 7.42 (d, 2H), 7.16 (dd, IH), 6.96 (dd, IH), 6.33 (dd, IH), 3.35 (m, 2H), 2.84 (t, 2H); LCMS: 462.3 (M+H)+.
EXAMPLE 370 l-(2-Chloro-4-(trifluoromethyl)phenyl)-N'-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from 2-chloro-4-(trifluoromethyl)aniline, methyl 2,5- dimethoxytetrahydrofuran-2-carboxylate, and Intermediate 3 following the procedure outlined in EXAMPLE 96. 1H NMR (400 MHz, DMSO-d6): δ 10.25 (s, IH), 9.16 (s, IH), 8.39 (m, IH), 8.09 (d, IH), 8.00 (d, IH), 7.77 (dd, IH), 7.60 (d, IH), 7.23 (dd, IH), 7.14 (dd, IH), 6.40 (dd, IH); LCMS: 483.3 (M+H)+.
EXAMPLE 371 N',l-Bis(2-chloro-4-(trifluoromethyl)phenyl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from l-(2-chloro-4-(trifluoromethyl)phenyl)-lH- pyrrole-2-carboxylic acid and (2-chloro-4-(trifluoromethyl)phenyl)hydrazine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, IH), 8.19 (s, IH), 8.00 (d, IH), 7.79 (dd, IH), 7.66-7.61 (m, 2H), 7.49 (m, IH), 7.25 (dd, IH), 7.17 (dd, IH), 6.87 (d, IH), 6.42 (dd, IH); LCMS: 482.2 (M+H)+.
EXAMPLE 372 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N'-(3-(trifluoromethyl)benzyl)-lH- pyrrole-2-carbohydrazide

The title compound was prepared from (3-(trifluoromethyl)benzyl)hydrazine and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 9.80 (d, IH), 8.91 (m, IH), 8.66 (d, IH), 7.69 (s, IH), 7.64-7.58 (m, 2H), 7.54 (t, IH), 7.20 (dd, IH), 6.93 (dd, IH), 6.32 (dd, IH), 5.45 (q, IH), 3.96 (d, 2H); LCMS: 463.3 (M+H)+.
EXAMPLE 373 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(3-(trifluoromethyl)benzyl)-lH- pyrrole-2-carbohydrazide

The title compound was isolated during the purification of EXAMPLE 372. LCMS: 463.3 (M+H)+.
EXAMPLE 374 l-(3,5-Dichloropyridin-2-yl)-N-(4-(trifluoromethoxy)benzyl)-lH-pyrrole-2- carboxamide

The title compound was prepared from Intermediate 2 and (4-(trifluoromethoxy) phenyl)methanamine following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.82 (t, IH), 8.54 (d, IH), 8.40 (d, IH), 7.36 (d, 2H), 7.31 (d, 2H), 7.14 (dd, IH), 7.04 (dd, IH), 6.33 (dd, IH), 4.33 (d, 2H); LCMS: 430.2 (M+H)+.
EXAMPLE 375
N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-((3-chloro-5- (trifluoromethyl)pyridin-2-yl)methyl)-lH-pyrrole-2-carbohydrazide

Step 1 : Ethyl 2-bromo-2-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)acetate

A solution of ethyl 2-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)acetate (10 g, 37 mmol), trimethylphenylammonium tribromide (14 g, 37 mmol), and THF (150 mL) was maintained at rt for 4 h and then filtered. The filtrate was diluted with EtOAc (150 mL), washed with water (150 mL x 2), dried, filtered, and concentrated to give ethyl 2- bromo-2-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)acetate as yellow oil. LCMS: 346 (M+H)+.
Step_2: Methyl l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-2-ethoxy-2- oxoethyl)-lH-pyrrole-2-carboxylate

Sodium hydride (60%, 1.5 g, 38 mmol) was added to a solution of methyl lH-pyrrole- 2-carboxylate (4.6 g, 37 mmol) and DMF (20 mL). After 30 min at rt, ethyl 2-bromo-2- (3-chloro-5-(trifluoromethyl)pyridin-2-yl)acetate (12.9 g, 37.2 mmol) was added, and the reaction was heated at 95 0C for 30 min, allowed to cool to rt, diluted with water (100 mL), and then extracted with EtOAc (150 mL x 3). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (l :100→l :20-EtOAc/PE) to give methyl l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-2-ethoxy-2-oxoethyl)-lH-pyrrole-2-carboxylate as yellow oil. LCMS: 391 (M+H)+. Step 3: l-((3-Chloro-5-(trifluoromethyl)pyridin-2-yl)methyl)-lH-pyrrole-2- carboxylic acid

A mixture of methyl l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-2-ethoxy-2- oxoethyl)-lH-pyrrole-2-carboxylate (10 g, 26 mmol), MeOH (80 mL), and 2M NaOH (80 mL) was refluxed for 30 min, allowed to cool to rt, and adjusted to pH = 1 with HCl. The resulting solution was heated at 40 0C for 45 min, allowed to cool to rt, and adjusted to pH = 5 with NaOH. The resulting solution was extracted with EtOAc (200 mL), and the extract was dried, filtered, and concentrated to give l-((3-chloro-5- (trifluoromethyl)pyridin-2-yl)methyl)-lH-pyrrole-2-carboxylic acid as a light brown solid. LCMS: 305 (M+H)+.
SteE4: N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-((3-chloro-5- (trifluoromethyl) pyridin-2-yl)methyl)-lH-pyrrole-2-carbohydrazide

The title compound was prepared from l-((3-chloro-5-(trifluoromethyl)pyridin-2- yl)methyl)-lH-pyrrole-2-carboxylic acid and Intermediate 3 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, IH), 9.10 (s, IH), 8.79 (d, IH), 8.42 (d, IH), 8.27 (d, IH), 8.07 (d, IH), 7.13 (dd, IH), 7.09 (dd, IH), 6.18 (dd, IH), 5.82 (s, 2H); LCMS: 498.3 (M+H)+.
EXAMPLE 376
NSl-BisCS-chloro-S-CtrifluoromethylJpyridin-l-ylJ-lH-benzoIdlimidazole-l- carbohydrazide

Stepl : Methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-benzo [d] imidazole-
2-carboxylate

Methyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-benzo[d]imidazole-2- carboxylate was prepared from methyl lH-benzo[d]imidazole-2-carboxylate and 2,3- dichloro-5-(trifluoromethyl)pyridine following the procedure outlined in Intermediate 1, Stepl.
Step2: NSl-Bis^-chloro-S-Ctrifluoromethyltøyridin-l-ylHH-benzoIdlimidazole-l- carbohydrazide

Trimethylaluminum (0.5 mL, 2M, 1 mmol) was added to a solution of Intermediate 3 (0.19 g, 0.88 mmol) and DCM (20 mL) at rt under N2. After 1 h, methyl l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-benzo[d]imidazole-2-carboxylate (50%, 0.52 g, 0.73 mmol) was added, and the reaction was warmed to 40 0C overnight. The reaction was allowed to cool to rt, quenched with 10% HCl (30 mL), stirred for 1 h, and then extracted with DCM (30 mL x 3). The combined extracts were washed with sat'd NaHCO3 (50 mL), dried, filtered, concentrated, and then purified by silica gel chromatography (l :40-EtOAc/PE) to give N',l-bis(3-chloro-5-(trifluoromethyl)pyridin- 2-yl)-lH-benzo[d]imidazole-2-carbohydrazide as a white solid. LCMS: 535.3 (M+H)+.
EXAMPLE 377
N-(3-Chloro-4-(trifluoromethoxy)benzyl)-l-(3,5-dichloropyridin-2-yl)-lH-pyrrole-
2-carboxamide

The title compound was prepared from Intermediate 2 and (3-chloro-4- (trifluoromethoxy)phenyl)methanamine following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.84 (t, IH), 8.52 (d, IH), 8.38 (d, IH), 7.53-7.49 (m, 2H), 7.31 (dd, IH), 7.14 (dd, IH), 7.02 (dd, IH), 6.32 (dd, IH), 4.31 (d, 2H); LCMS: 464.2 (M+H)+.
EXAMPLE 378 l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-2-(4- (trifluoromethoxy)phenylamino)ethanone

Step 1 : l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone

2,3-Dichloro-5-(trifluoromethyl)pyridine (18.7 g, 86 mmol) was added to a mixture of l-(lH-pyrrol-2-yl)ethanone (4.7 g, 43 mmol), cesium carbonate (13.6 g, 42 mmol), and DMF (65 mL) at 110 0C under N2. The reaction was heated at 120 0C for 4.5 h, allowed to cool to rt, poured into water (250 mL), and then extracted with EtOAc (200 mL). The extract was dried, filtered, concentrated, and purified by silica gel chromatography (10%-50% EtOAc in hexanes) to give l-(l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone as a colorless oil. LCMS: 289 (M+H)+.
Step_l: 2-Bromo-l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2- yl)ethanone

Pyrrolidone hydrotribromide (3.4 g, 6.9 mmol) was added to a solution of l-(l-(3- chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone (2.0 g, 6.9 mmol), DMF (20 mL), and 33% HBr in acetic acid (2.0 mL, 13.8 mmol). The reaction was
heated at 55 0C for 18 h, allowed to cool to rt, diluted with water (100 mL), and extracted with EtOAc (100 mL). The extract was dried, filtered, concentrated, and purified by silica gel chromatography (10%-50% EtOAc in hexanes) to give 2-bromo- l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone as an amber oil. LCMS: 367 (M+H)+.
Step 3: l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-2-(4-
(trifluoromethoxy)phenylamino)ethanone

A mixture of 2-bromo- 1 -( 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrol-2- yl)ethanone (1.0 g, 2.7 mmol), 4-(trifluoromethoxy)aniline (0.72 g, 4.1 mmol), potassium carbonate (377 mg, 2.73 mmol), and 1,4-dioxane (30 mL) was heated at 100 0C overnight and then concentrated. The residue was diluted with EtOAc (100 mL), washed with water (30 mL x2), dried, filtered, concentrated, and then purified by silica gel chromatography to give 1 -(I -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- lH-pyrrol- 2-yl)-2-(4-(trifluoromethoxy)phenylamino)ethanone as a white solid. 1H NMR (400 MHz, DMSO-de): δ 8.92 (m, IH), 8.71 (d, IH), 7.56 (dd, IH), 7.45 (dd, IH), 7.01 (d, 2H), 6.59 (d, 2H), 6.50 (dd, IH), 6.12 (t, IH), 4.47 (d, 2H); LCMS: 464.3 (M+H)+.
EXAMPLE 379
NSl-BisCS-chloro-S-CtrifluoromethyOpyridin-l-yO-N-methyl-lH-pyrrole-l- carbohydrazide

EXAMPLE 380
(S)-N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)pyrrolidine-2- carbohydrazide

The title compound was prepared from (S)-methyl pyrrolidine-2-carboxylate following the procedure outlined in EXAMPLE 94. LCMS: 488.3 (M+H)+.
EXAMPLE 381
(RJ-NSl-BisCS-chloro-S-CtrifluoromethyOpyridin-l-yOpyrrolidine-l- carbohydrazide

The title compound was prepared from (R)-methyl pyrrolidine -2-carboxylate following the procedure outlined in EXAMPLE 94. LCMS: 488.3 (M+H)+.
EXAMPLE 382 N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrazole-5-carbohydrazide

The title compound was prepared from Intermediate 4 and Intermediate 3 following the procedure outlined in EXAMPLE 1 (except: IM KH2PO4 used in place of 10% K2CO3 & IN HCl to avoid decomposition). 1U NMR (400 MHz, DMSO-d6): δ 10.86 (s, IH), 9.38 (s, IH), 8.94 (d, IH), 8.77 (d, IH), 8.40 (d, IH), 8.12 (d, IH), 7.96 (d, IH), 7.32 (d, IH); LCMS: 485.3 (M+H)+.
EXAMPLE 383
NSl-Bis^-chloro-S-CtrifluoromethyOpyridin-l-ylHH-imidazole-S- carbohydrazide

Step 1 : Ethyl 2-(3-chloro-5-(trifluoromethyl)pyridin-2-ylimino)acetate

A solution of 3-chloro-5-(trifluoromethyl)pyridin-2-amine (1.0 g, 7.9 mmol), MeOH (15 niL), and ethyl 2-oxoacetate (50% in PhMe, 9.8 niL, 49 mmol) was refluxed under N2 overnight. The reaction was allowed to cool to rt, concentrated, and then purified by silica gel chromatography (l :30-EtOAc: hexanes) to give ethyl 2-(3-chloro-5- (trifluoromethyl)pyridin-2-ylimino)acetate as a white solid. Step_2: Ethyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-5- carboxylate

Potassium carbonate (1.5 g, 11 mmol) was added to a solution of ethyl 2-(3-chloro-5- (trifluoromethyl)pyridin-2-ylimino)acetate (50 %, 1.5 g, 2.7 mmol), ethanol (50 mL), and l-(isocyanomethylsulfonyl)-4-methylbenzene (1.3 g, 6.7 mmol). The mixture was heated at 60 0C for 3 h, allowed to cool to rt, concentrated, and then purified by silica gel chromatography (l :10-EtOAc/PE) to give ethyl l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-imidazole-5-carboxylate as a light yellow solid.
Step 3: l-^-Chloro-S-^rifluoromethylJpyridin-l-ylJ-lH-imidazole-S-carboxylic acid

A mixture of ethyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-5- carboxylate (500 mg, 1.56 mmol), ethanol (30 mL), and 2M NaOH (10 rnL) was stirred at rt for 30 min, concentrated, diluted with water (50 mL), adjusted to pH = 3 with HCl, and then extracted with EtOAc (50 mL x 3). The combined extracts were dried, filtered, and concentrated to give l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH- imidazole-5-carboxylic acid as yellow oil.
Step_4: N',l-Bis(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-5- carbohydrazide

The title compound was prepared from l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-imidazole-5-carboxylic acid and Intermediate 3 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.64 (s, IH), 9.29 (s, IH), 8.95 (m, IH), 8.76 (d, IH), 8.39 (m, IH), 8.21 (d, IH), 8.10 (d, IH), 7.96 (d, IH); LCMS: 485.3 (M+H)+.
EXAMPLE 384 l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-2-(4- (trifluoromethoxy)phenoxy)ethanone

The title compound was prepared from 4-(trifluoromethoxy)phenol and 2-bromo-l-(l- (3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrol-2-yl)ethanone following the procedure outlined in EXAMPLE 378, Step 3. 1H NMR (400 MHz, DMSO-d6): δ 8.92 (m, IH), 8.71 (d, IH), 7.54 (dd, IH), 7.50 (dd, IH), 7.25 (d, 2H), 6.96 (d, 2H), 6.52 (dd, IH), 5.37 (s, 2H); LCMS: 465.3 (M+H)+.
EXAMPLE 385
N'-CS-Chloro-S-CtrifluoromethylJpyridin-l-ylJ-l-CS-CtrifluoromethylJpyridin-l-yl)- lH-pyrrole-2-carbohydrazide

The title compound was prepared from 2-fluoro-3-(trifluoromethyl)pyridine, methyl lH-pyrrole-2-carboxylate, and Intermediate 3 following the procedures outlined for Intermediate 1 and EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 10.20 (s, IH), 9.12 (s, IH), 8.70 (dd, IH), 8.34 (s, IH), 8.31 (dd, IH), 8.05 (d, IH), 7.68 (dd, IH), 7.22-7.17 (m, 2H), 6.32 (dd, IH); LCMS: 450.3 (M+H)+.
EXAMPLE 386 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(trifluoromethoxy)benzyl)-lH- pyrazole-5-carboxamide

The title compound was prepared from (4-(trifluoromethoxy)phenyl)methanamine and Intermediate 4 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-d6): δ 9.34 (t, IH), 8.95 (m, IH), 8.76 (d, IH), 7.89 (d, IH), 7.36 (d, 2H), 7.31 (d, 2H), 7.17 (d, IH), 4.36 (d, 2H); LCMS: 465.3 (M+H)+.
EXAMPLE 387 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-
(trifluoromethyl)phenoxy)benzyl)-lH-pyrrole-2-carboxamide

The title compound was prepared from (4-(4-
(trifluoromethyl)phenoxy)phenyl)methanamine hydrochloride and Intermediate 1 following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.91 (m, IH), 8.87 (t, IH), 8.67 (d, IH), 7.72 (d, 2H), 7.33 (d, 2H), 7.20 (m, IH), 7.13- 7.06 (m, 5H), 6.37 (m, IH), 4.33 (d, 2H); LCMS: 540.3 (M+H)+.
EXAMPLE 388 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-(trifluoromethyl)phenoxy)benzyl)-lH- pyrrole-2-carboxamide
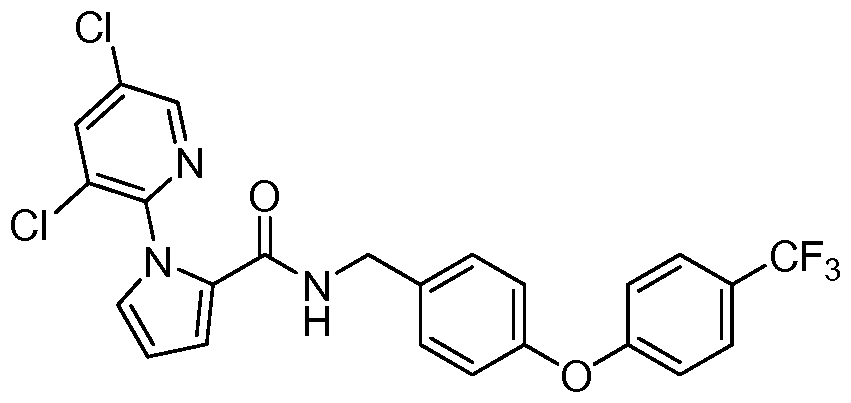
The title compound was prepared from (4-(4-
(trifluoromethyl)phenoxy)phenyl)methanamine hydrochloride and Intermediate 2 following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.81 (t, IH), 8.55 (d, IH), 8.41 (d, IH), 7.72 (d, 2H), 7.33 (d, 2H), 7.14 (dd, IH), 7.10 (d, 4H), 7.05 (dd, IH), 6.33 (dd, IH), 4.33 (d, 2H); LCMS: 506.3 (M+H)+.
EXAMPLE 389 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH- pyrazole-5-carboxamide

The title compound was prepared from (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride and Intermediate 4 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-d6): δ 9.31 (t, IH), 8.97 (s, IH), 8.79 (s, IH), 7.90 (d, IH), 7.30-7.16 (m, 5H), 7.03 (m, 2H), 6.96 (d, 2H), 4.33 (d, 2H); LCMS: 491.3 (M+H)+.
EXAMPLE 390 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4- (trifluoromethyl)phenoxy)benzyl)-lH-pyrazole-5-carboxamide

The title compound was prepared from (4-(4-
(trifluoromethyl)phenoxy)phenyl)methanamine hydrochloride and Intermediate 4 following the procedure outlined in EXAMPLE 382. 1U NMR (400 MHz, DMSO-d6): δ 9.35 (t, IH), 8.98 (s, IH), 8.79 (s, IH), 7.91 (d, IH), 7.72 (d, 2H), 7.34 (d, 2H), 7.20 (d, IH), 7.14-7.08 (m, 4H), 4.38 (d, 2H); LCMS: 541.3 (M+H)+.
EXAMPLE 391 Isopropyl 4-(2-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-2- oxoethoxy)piperidine-l-carboxylate
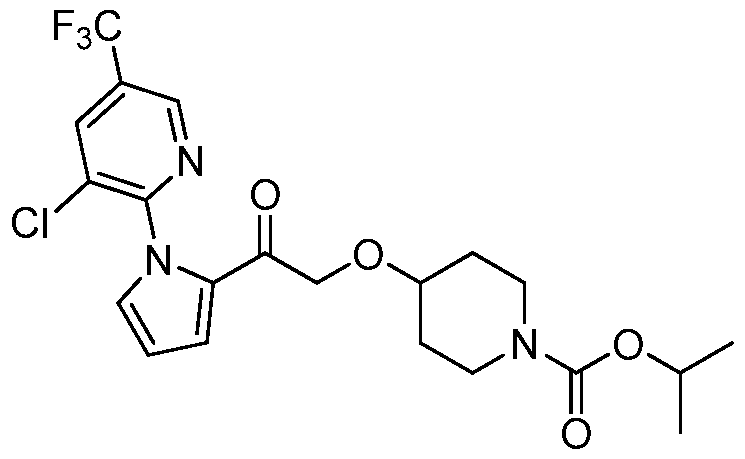

Isopropyl 4-hydroxypiperidine-l-carboxylate (5.0 g, 27 mmol) was added dropwise to a mixture of sodium hydride (60%, 5.3 g, 0.13 mol) and THF (100 mL) at rt under N2. After 30 min, 2-bromoacetic acid (11 g, 79 mmol) was added, and the reaction was stirred at rt overnight, quenched with water (20 mL), and extracted with EtOAc (50
niL). The aqueous layer was adjusted to pH = 2 with 2N HCl, and extracted with EtOAc (50 mL x 2). The combined extracts were washed with brine (50 mL), dried, filtered, and concentrated to give 2-(l-(isopropoxycarbonyl)piperidin-4-yloxy)acetic acid as a yellow oil. LCMS: 246 (M+H)+. Step 2: Isopropyl 4-(2-chloro-2-oxoethoxy)piperidine-l-carboxylate

A solution of 2-(l-(isopropoxycarbonyl)piperidin-4-yloxy)acetic acid (500 mg, 2.04 mmol) and thionyl chloride (4 mL) was refluxed for 3 h and then concentrated to give isopropyl 4-(2-chloro-2-oxoethoxy)piperidine-l-carboxylate as white oil. Step 3 : Isopropyl 4-(2-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2- yl)-2-oxoethoxy)piperidine-l-carboxylate

A mixture of isopropyl 4-(2-chloro-2-oxoethoxy)piperidine-l-carboxylate (1.2 g, 4.6 mmol) and 3-chloro-2-(lH-pyrrol-l-yl)-5-(trifluoromethyl)pyridine (1.5 g, 6.1 mmol) was heated at 150 0C for 30 min in a microwave reactor and then purified by silica gel chromatography (l :10-EtOAc/PE) to give isopropyl 4-(2-(l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)- 1 H-pyrrol-2-yl)-2-oxoethoxy)piperidine- 1 -carboxylate as a brown oil. 1H NMR (400 MHz, DMSO-d6): δ 8.93 (m, IH), 8.73 (m, IH), 7.43 (m, IH), 7.36 (dd, IH), 6.45 (m, IH), 4.72 (septet, IH), 4.63 (s, 2H), 3.65-3.55 (m, 2H), 3.50 (m, IH), 3.02 (br, 2H), 1.81-1.73 (m, 2H), 1.40-1.28 (m, 2H), 1.15 (d, 6H); LCMS: 474.3 (M+H)+.
EXAMPLE 392 N-((l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)methyl)-l-(4-
(trifluoromethoxy)phenyl)ethanamine

A solution of l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-2-carbaldehyde (668 mg, 2.44 mmol), l-(4-(trifluoromethoxy)phenyl)ethanamine (500 mg, 2.44 mmol), and THF (30 niL) was stirred at rt overnight and then heated at 45 0C for 4 h. The reaction was cooled to 00C and then sodium triacetoxylborohydride (1.03 g, 4.86 mmol) was added in several batches. After 72 h at rt, the reaction was quenched with water (20 mL) and then extracted with EtOAc (50 mL x 3). The combined extracts were washed with brine (50 mL x 2), dried, filtered, concentrated, and then purified by silica gel chromatography (l :10→l :0 DCM: petroleum ether) to give N-((l-(3-chloro- 5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrol-2-yl)methyl)- 1 -(4- (trifluoromethoxy)phenyl)-ethanamine as a yellow oil. 1H NMR (400 MHz, DMSO- d6): δ 8.83 (s, IH), 8.72 (s, IH), 7.16 (br, 4H), 7.03 (br, IH), 6.18 (br, IH), 6.14 (br, IH), 3.58-3.34 (br m, 3H), 2.25 (br, IH), 1.05 (br, 3H); LCMS: 464.3 (M+H)+.
EXAMPLE 393 l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-N-(4- (trifluoromethoxy)benzyl)methanamine

The title compound was prepared from l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrole-2-carbaldehyde and (4-(trifluoromethoxy)phenyl)methanamine following
the procedure outlined in EXAMPLE 392. 1U NMR (400 MHz, DMSO-d6; HCl salt): δ 9.54 (br, 2H), 8.80 (s, IH), 8.79 (s, IH), 7.66 (d, 2H), 7.47-7.36 (m, 3H), 6.67 (m, IH), 6.36 (m, IH), 4.15 (br, 4H); LCMS: 450.3 (M+H)+.
EXAMPLE 394 l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH- imidazole-5-carboxamide

The title compound was prepared from (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride and l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-imidazole-5- carboxylic acid following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-de): δ 9.11 (t, IH), 8.98 (m, IH), 8.78 (d, IH), 8.14 (d, IH), 7.83 (d, IH), 7.30-7.17 (m, 4H), 7.07-7.00 (m, 2H) 6.98-6.92 (m, 2H), 4.32 (d, 2H); LCMS: 491.3 (M+H)+.
EXAMPLE 395 l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-N-methyl-N-(4- (trifluoromethoxy)benzyl)methanamine

Sodium cyanoborohydride (164 mg, 2.60 mmol) was added in several portions to a solution of 1 -(I -(3-chloro-5-(trifluoromethyl)pyridin-2-yl)- lH-pyrrol-2-yl)-N-(4- (trifluoromethoxy) benzyl)methanamine (0.40 g, 0.80 mmol), formaldehyde (50%, 10 niL), and MeOH (10 mL) at 0 0C. Acetic acid (0.3 mL) was added dropwise at 0 0C,
and then the reaction was stirred at rt for 2 h, concentrated, adjusted to pH = 8 with IM NaOH, and then extracted with EtOAc (40 mL x 3). The combined extracts were washed with brine (30 mL), dried, filtered, concentrated, and then purified by silica gel chromatography (1 :50 EtOAc/PE) to give l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2- yl)-lH-pyrrol-2-yl)-N-methyl-N-(4-(trifluoromethoxy)benzyl)methanamine as a light yellow oil. LCMS: 464.1 (M+H)+.
EXAMPLE 396 l-(3,5-Dichloropyridin-2-yl)-N-(4-(trifluoromethoxy)benzyl)-lH-pyrazole-5- carboxamide

The title compound was prepared from (4-(trifluoromethoxy)phenyl)methanamine and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-de): δ 9.30 (t, IH), 8.61 (d, IH), 8.51 (d, IH), 7.86 (d, IH), 7.36 (d, 2H), 7.32 (d, 2H), 7.15 (d, IH), 4.38 (d, 2H); LCMS: 431.2 (M+H)+.
EXAMPLE 397 N-(3-Chloro-4-(trifluoromethoxy)benzyl)-l-(3,5-dichloropyridin-2-yl)-lH- pyrazole-5-carboxamide

The title compound was prepared from (3-chloro-4-
(trifluoromethoxy)phenyl)methanamine and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-d6): δ 9.34 (t, IH), 8.60 (d,
IH), 8.51 (d, IH), 7.87 (d, IH), 7.58-7.52 (m, 2H), 7.35 (dd, IH), 7.15 (d, IH), 4.38 (d, 2H); LCMS: 465.2 (M+H)+.
EXAMPLE 398 N'-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-l-(3,5-dichloropyridin-2-yl)-lH- pyrazole-5-carbohydrazide

The title compound was prepared from Intermediate 3 and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1U NMR (400 MHz, DMSO-d6): δ 10.82 (s, IH), 9.38 (s, IH), 8.60 (d, IH), 8.51 (d, IH), 8.41 (m, IH), 8.14 (d, IH), 7.93 (d, IH), 7.31 (d, IH); LCMS: 451.2 (M+H)+.
EXAMPLE 399 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-pyrazole-5- carboxamide

The title compound was prepared from (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride and Intermediate 5 following the procedure outlined in Example 382. 1H NMR (400 MHz, DMSO-d6): δ 9.25 (t, IH), 8.61 (d, IH), 8.51 (d, IH), 7.85 (d, IH), 7.28-7.18 (m, 4H), 7.15 (d, IH), 7.06-7.01 (m, 2H), 6.96 (d, 2H), 4.33 (d, 2H); LCMS: 457.3 (M+H)+.
EXAMPLE 400 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-(trifluoromethyl)phenoxy)benzyl)-lH- pyrazole-5-carboxamide

The title compound was prepared from (4-(4-
(trifluoromethyl)phenoxy)phenyl)methanamine hydrochloride and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1U NMR (400 MHz, DMSO-d6): δ 9.29 (t, IH), 8.61 (d, IH), 8.52 (d, IH), 7.86 (d, IH), 7.73 (d, 2H), 7.34 (d, 2H), 7.16 (d, IH), 7.14-7.08 (m, 4H), 4.37 (d, 2H); LCMS: 507.3 (M+H)+.
EXAMPLE 401 l-(l-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-N-(4-
(trifluoromethoxy)benzyl)ethanamine

Titanium(IV) isopropoxide (5 mL) was added to a mixture of l-(l-(3-chloro-5- (trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone (2.0 g, 6.6 mmol) and (4- (trifluoromethoxy)phenyl)methanamine (2.4 g, 12 mmol) under N2. The mixture was stirred at rt overnight. MeOH (30 mL) was added followed by sodium borohydride (1.4 g, 37 mmol) in several batches. After stirring for 4 h at rt, the mixture was concentrated, diluted with water (100 mL), and then filtered. The filtrate was extracted with EtOAc (100 mL x 3). The combined extracts were dried, filtered, concentrated and purified by silica gel chromatography [(1 :50-EtOAc/PE) then (1 : 100-MeOH:
DCM)] to give l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)-N-(4- (trifluoromethoxy)benzyl)ethanamine as a yellow oil. LCMS: 464.3 (M+H)+.
EXAMPLE 402
3-Chloro-2-(2-(l-(4-(trifluoromethoxy)benzyloxy)ethyl)-lH-pyrrol-l-yl)-5-
(trifluoromethyl)pyridine


Sodium borohydride (0.80 g, 21 mmol) was added in several portions to a solution of l-(l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrol-2-yl)ethanone (1.5 g, 5.2 mmol) and isopropanol (30 mL) at 0 0C. After 30 min at 0 0C, the reaction was allowed to warm to rt, maintained for 3.5 h, quenched with ice water (200 mL), and then filtered. The filtrate was extracted with EtOAc (100 mL x 4). The combined extracts were dried, filtered, concentrated, and then purified by silica gel chromatography (l :80-EtOAc/PE) to give l-(l-(3-chloro-5-(trifiuoromethyl)pyridin-2-yl)-lH-pyrrol-2- yl)ethanol as a yellow oil. Step_l: 3-Chloro-2-(2-(l-(4-(trifluoromethoxy)benzyloxy)ethyl)-lH-pyrrol-l-yl)-5-
(trifluoromethyl)pyridine
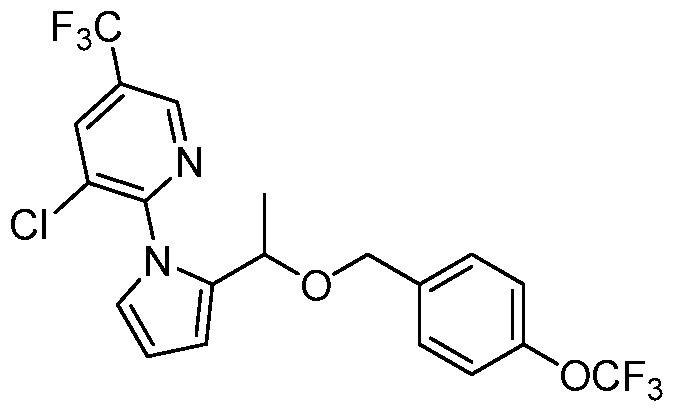
EXAMPLE 403 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-imidazole-2- carboxamide

Trimethylaluminum (0.5 mL, 2M PhMe, 1.0 mmol) was added dropwise to a suspension of (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride (254 mg, 1.0 mmol) and 1 ,2-dichloroethane (4 mL) at rt under N2, and the reaction was stirred for 40 min. Intermediate 6 (143 mg, 0.50 mmol) was added, stirring was continued for 2.3 h, and the reaction was quenched with a solution of sat' d NaK tartrate and sat'd NaHCO3 (1 : 1 , 40 mL). The resulting mixture was stirred vigorously for 2 h and then extracted with DCM (40 mL x 2). The combined extracts were dried, filtered, concentrated, and purified by silica gel chromatography (4:1→1 :1; hexanes: EtOAc) to give l-(3,5- dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-imidazole-2-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 9.25 (t, IH), 8.62 (d, IH), 8.53 (d, IH), 7.65 (d, IH), 7.28-7.17 (m, 5H), 7.06-7.00 (m, 2H), 6.93 (d, 2H), 4.29 (d, 2H); LCMS: 457.1 (M+H)+.
EXAMPLE 404 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-(trifluoromethyl)phenoxy)benzyl)-lH- imidazole-2-carboxamide

The title compound was prepared from (4-(4-
(trifluoromethyl)phenoxy)phenyl)methanamine hydrochloride and Intermediate 6 following the procedure outlined in EXAMPLE 403. 1U NMR (400 MHz, DMSO-d6): δ 9.29 (t, IH), 8.62 (d, IH), 8.53 (d, IH), 7.71 (d, 2H), 7.66 (s, IH), 7.34 (d, 2H), 7.28 (s, IH), 7.13-7.05 (m, 4H), 4.33 (d, 2H); LCMS: 507.3 (M+H)+.
EXAMPLE 405 l-(3,5-Dichloropyridin-2-yl)-N-(4-(6-(trifluoromethyl)pyridin-3-yloxy)benzyl)-lH- imidazole-2-carboxamide
The title compound was prepared from (4-(6-(trifluoromethyl)pyridin-3- yloxy)phenyl)methanamine and Intermediate 6 following the procedure outlined in EXAMPLE 403. 1H NMR (400 MHz, DMSO-d6): δ 9.31 (t, IH), 8.62 (d, IH), 8.55- 8.52 (m, 2H), 7.87 (d, IH), 7.66 (d, IH), 7.49 (dd, IH), 7.36 (d, 2H), 7.28 (d, IH), 7.15 (d, 2H), 4.34 (d, 2H); LCMS: 508.3 (M+H)+.
EXAMPLE 406 l-(3,5-Dichloropyridin-2-yl)-N-(4-(2-(trifluoromethyl)pyrimidin-5-yloxy)benzyl)- lH-imidazole-2-carboxamide

The title compound was prepared from (4-(2-(trifluoromethyl)pyrimidin-5- yloxy)phenyl)methanamine hydrochloride and Intermediate 6 following the procedure outlined in EXAMPLE 403. 1U NMR (400 MHz, DMSO-d6): δ 9.31 (t, IH), 8.78 (s, 2H), 8.62 (d, IH), 8.53 (d, IH), 7.66 (d, IH), 7.36 (d, 2H), 7.28 (d, IH), 7.23 (d, 2H), 4.35 (d, 2H); LCMS: 509.3 (M+H)+.
EXAMPLE 407 N-((3-Chloro-5-(trifluoromethyl)pyridin-2-yl)methyl)-l-(3,5-dichloropyridin-2- yl)-lH-pyrrole-2-carboxamide

The title compound was prepared from (3-chloro-5-(trifluoromethyl)pyridin-2- yl)methanamine hydrochloride and Intermediate 2 following the procedure outlined in EXAMPLE 1. 1U NMR (400 MHz, DMSO-d6): δ 8.91 (m, IH), 8.78 (t, IH), 8.52 (d, IH), 8.44 (m, IH), 8.38 (d, IH), 7.13 (dd, IH), 7.11 (dd, IH), 6.34 (dd, IH), 4.58 (d, 2H); LCMS: 449.2 (M+H)+.
EXAMPLE 408
N-((3-Chloro-5-(trifluoromethyl)pyridin-2-yl)methyl)-l-(3,5-dichloropyridin-2- yl)-lH-pyrazole-5-carboxamide

The title compound was prepared from (3-chloro-5-(trifluoromethyl)pyridin-2- yl)methanamine hydrochloride and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-d6): δ 9.32 (t, IH), 8.93 (m, IH), 8.59 (d, IH), 8.49 (d, IH), 8.46 (d, IH), 7.87 (d, IH), 7.23 (d, IH), 4.64 (d, 2H); LCMS: 450.2 (M+H)+.
EXAMPLE 409 l-(3,5-Dichloropyridin-2-yl)-N-((6-(trifluoromethyl)pyridin-3-yl)methyl)-lH- pyrrole-2-carboxamide

The title compound was prepared from (6-(trifluoromethyl)pyridin-3-yl)methanamine and Intermediate 2 following the procedure outlined in EXAMPLE 1. 1H NMR (400 MHz, DMSO-d6): δ 8.92 (t, IH), 8.65 (br, IH), 8.54 (d, IH), 8.40 (d, IH), 7.93-7.86 (m, 2H), 7.16 (dd, IH), 7.05 (dd, IH), 6.34 (dd, IH), 4.43 (d, 2H); LCMS: 415.2 (M+H)+.
EXAMPLE 410 l-(3,5-Dichloropyridin-2-yl)-N-((6-(trifluoromethyl)pyridin-3-yl)methyl)-lH- pyrazole-5-carboxamide

The title compound was prepared from (6-(trifluoromethyl)pyridin-3-yl)methanamine and Intermediate 5 following the procedure outlined in EXAMPLE 382. 1H NMR (400 MHz, DMSO-d6): δ 9.40 (t, IH), 8.67 (br, IH), 8.60 (d, IH), 8.51 (d, IH), 7.95-7.88 (m, 2H), 7.88 (d, IH), 7.16 (d, IH), 4.49 (d, 2H); LCMS: 416.3 (M+H)+.
EXAMPLE 411 l-(3,5-Dichloropyridin-2-yl)-N-(4-(6-(trifluoromethyl)pyridin-3-yloxy)benzyl)-lH- pyrazole-5-carboxamide

The title compound was prepared from Intermediate 5 and (4-(6- (trifluoromethyl)pyridin-3-yloxy)phenyl)methanamine following the procedure outlined in EXAMPLE 382. 1U NMR (400 MHz, DMSO-d6): δ 9.30 (t, IH), 8.62 (d, IH), 8.54 (d, IH), 8.52 (d, IH), 7.89 (d, IH), 7.86 (d, IH), 7.50 (dd, IH), 7.36 (d, 2H), 7.22-7.16 (m, 3H), 4.38 (d, 2H); LCMS: 508.2 (M+H)+.
EXAMPLE 412 l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-l,2,4-triazole-5- carboxamide


A mixture of 2,3,5-trichloropyridine (7.2 g, 40 mmol), lH-l,2,4-triazole (2.8 g, 41 mmol), cesium carbonate (13.0 g, 39.9 mmol), and DMF (150 mL) was heated at 130 0C for 3 h, allowed to cool to rt, concentrated, and then diluted with EtOAc (400 mL). The resulting mixture was washed with brine (200 mL x 2), dried, filtered, concentrated, and then purified by silica gel chromatography (20:l→5:l; hexanes:EtOAc) to give 3,5-dichloro-2-(lH-l,2,4-triazol-l-yl)pyridine as a light yellow solid.
Step 2: α-(3,5-Dichloropvridin-2-vl)-lH-l,2,4-triazol-5-vl)methanol Cl
N
N N // 0H
3,5-Dichloro-2-(lH-l,2,4-triazol-l-yl)pyridine (2.00 g, 8.41 mmol) and 37% formaldehyde (30 mL) was heated at 140 0C in a sealed tube overnight. The resulting solution was extracted with EtOAc (100 mL x 2). The combined extracts were washed with IN NaOH (100 mL), washed with brine (100 mL x 2), dried, filtered, concentrated, and then purified by silica gel chromatography (10:l→l:l;
hexanes:EtOAc) to give (l-(3,5-dichloropyridin-2-yl)-lH-l,2,4-triazol-5-yl)methanol as a light yellow solid.
Step 3: Methyl l-(3,5-dichloropvridin-2-vlMH-l,2,4-triazole-5-carboxylate

A mixture of (l-(3,5-dichloropyridin-2-yl)-lH-l,2,4-triazol-5-yl)methanol (1.3 g, 5.3 mmol), KCN (700 mg, 10.8 mmol), MnO2 (4.4 g, 51 mmol), tetrahydrofuran (80 niL), and MeOH (0.80 g, 25 mmol) was heated at 80 0C for 3 h, allowed to cool to rt, and then filtered. The filtrate was concentrated and then purified by silica gel chromatography (20:l→5:l; petroleum ether: EtOAc) to give methyl l-(3,5- dichloropyridin-2-yl)-lH-l,2,4-triazole-5-carboxylate as a white solid. LCMS: 273.1 (M+H)+. Step 4: l-(3,5-Dichloropyridin-2-yl)-N-(4-(4-fluorophenoxy)benzyl)-lH-l,2,4- triazole-5-carboxamide

The title compound was prepared from methyl l-(3,5-dichloropyridin-2-yl)-lH-l,2,4- triazole-5-carboxylate and (4-(4-fluorophenoxy)phenyl)methanamine hydrochloride following the procedure outlined in EXAMPLE 403. 1H NMR (400 MHz, DMSO-d6): δ 9.76 (t, IH), 8.69 (d, IH), 8.63 (d, IH), 8.49 (s, IH), 7.27 (d, 2H), 7.24-7.18 (m, 2H), 7.06-7.00 (m, 2H), 6.94 (d, 2H), 4.33 (d, 2H); LCMS: 458.2 (M+H)+.
EXAMPLES 413-640 Step 1 : Perfluorophenyl l-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)-lH-pyrrole-
2-carboxylate

Perfluorophenyl 1 -(3 -chloro-5 -(trifluoromethyl)pyridin-2-yl)- 1 H-pyrrole-2-carboxylate was prepared from Intermediate 1 using the method described in Tetrahedron Letters, 1990, 31(41), 5851-5852.
Step 2: Library synthesis

Primary and secondary amines (4 μmol) in DMF (8 μL) were transferred to each well of 384 well plate, then treated with a solution of perfluorophenyl 1 -(3 -chloro-5 - (trifluoromethyl)pyridin-2-yl)-l H-pyrrole-2-carboxylate (0.4 μmol) in DMF (10 μL). The reaction plate was heat sealed and shaken at 250C for 24 h. Solvent was removed under reduced pressure.


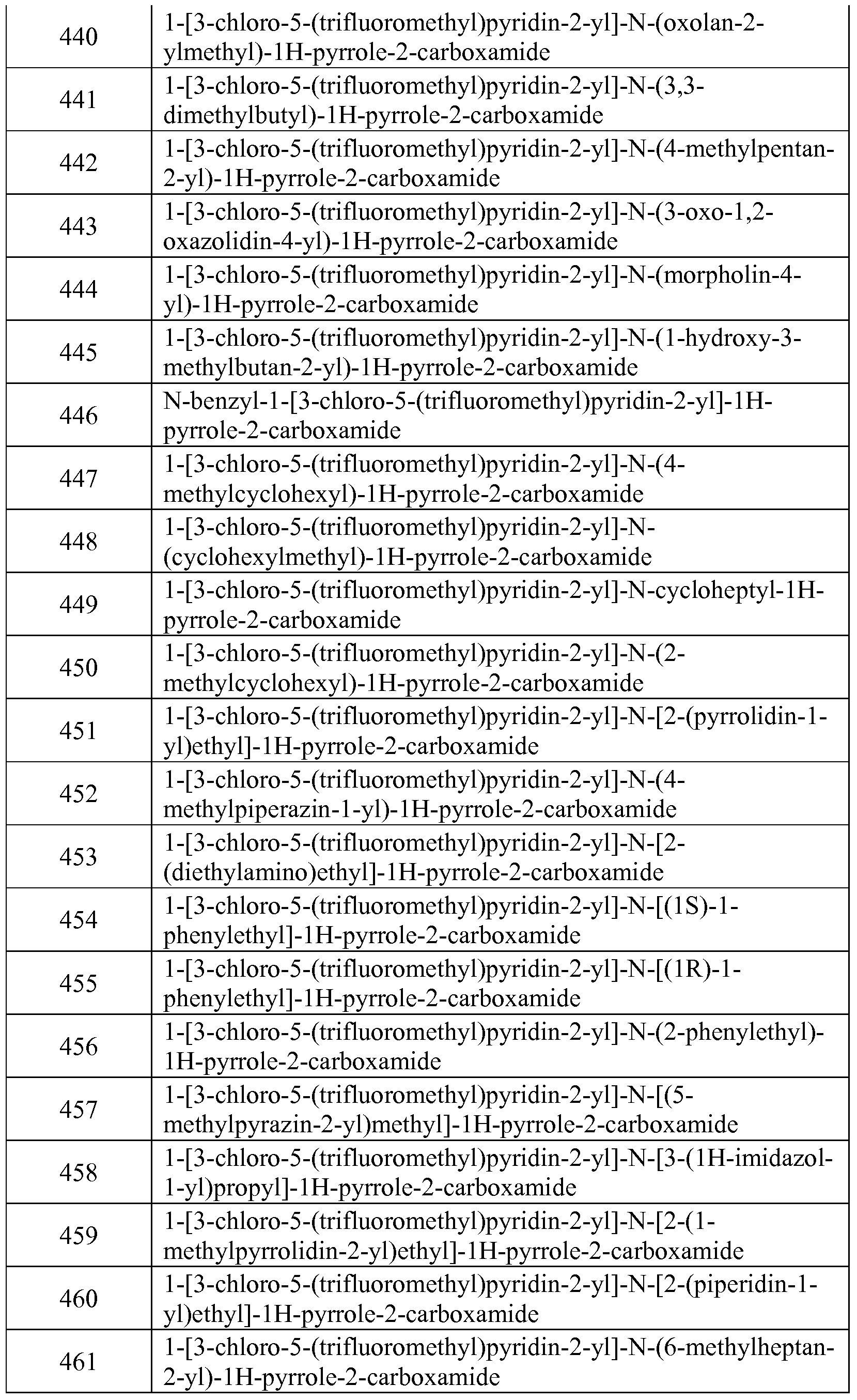









[0220] The activity of the compounds in Examples 1-640 as GPRl 19 modulators is illustrated in the following assay. The other compounds listed above, which have not yet been made and/or tested, are predicted to have activity in this assay as well.
Biological Activity Assay
cAMP Production Assay:
HEK293 cells stably expressing GPRl 19 (HEK293-GPR119) were established by stably transfecting HEK-293 cells with an expression vector (pcDNA 3.1, Invetrogen) inserted with human GPRl 19 cDNA using Fugeneβ (Roche, Indianapolis, IN) according to conventional methods. Cells were grown in DMEM (invitrogen, Carlsbad, CA) supplemented with 10% FBS, 1% penicillin/streptomycin under geneticin selection. The presence of GPRl 19 transcripts in these cells was confirmed using branched DNA (bDNA, Panomics Inc., Fremont CA) following the manufacturer's protocol and using specific probes for human GPRl 19. cAMP production assay was performed in high throughput 1536 well format using Cisbio c AMP detection kit (Cisbio Inc., Bedford, MA) according to the manufacturer's protocol. Briefly, HEK293-GPR119 cells were harvested using non-enzymatic cell dissociation buffer (Invitrogen, Carlsbad, CA), suspended in DMEM supplemented with 2% FBS at a density of 3200 cells/well dispensed in white opaque tissue culture treated Greiner 1536 well plates (USA Scientific, Inc., Ocala, FL). After an overnight incubation at 37°C in an atmosphere of 10% CO2 and 95% humidity, 1 ul of 5 mM IBMX (Sigma, St. Louis, MO) solution in DMEM was dispensed for a final concentration of 1 mM. Cells were then stimulated with test compounds for 30 minutes, after which time cAMP detection reagents were added and incubated for 1 h at room temperature. TR-FRET signal was detected using the Viewlux (Perkin Elmer Inc., Boston MA) or the Analyst GT (Molecular Devices, Sunnyvale, CA) plate reader. EC50 values for cAMP production were determined using Graph Pad Prizm analysis (Graph Pad Software, La Jolla, CA) or proprietary Kalypsys KNET software. GPRl 19-specific mechanism of action was determined by the lack of cAMP production when compounds were tested in HEK-293 cells that were transfected with an empty vector. In Table 2 below, EC50 is for cAMP production in 293-GPR119 cells. A "+" indicates an EC50 of < 10 μM, and a "-" indicates an EC50 of >10 μM.
Table 2. Biological Activity


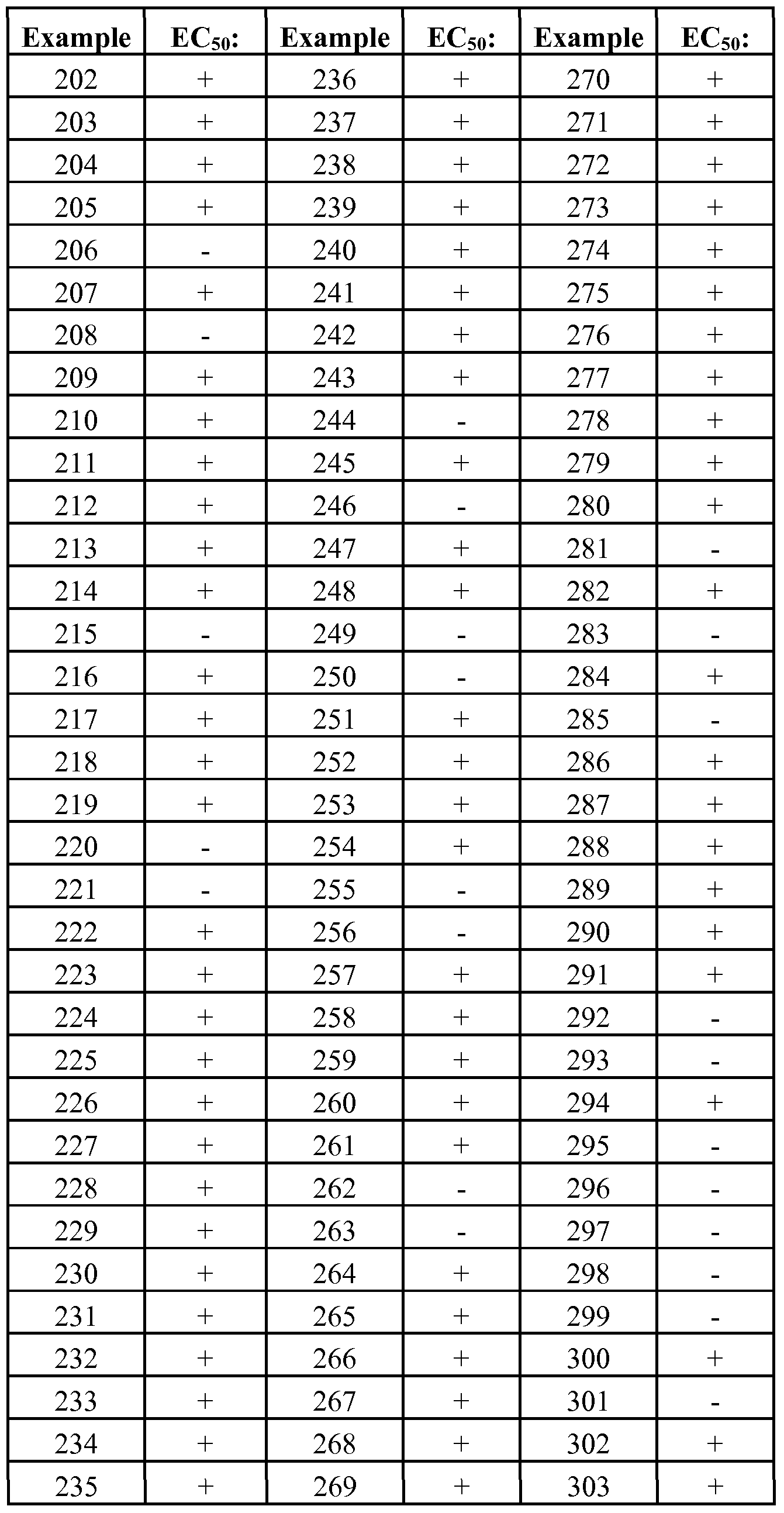


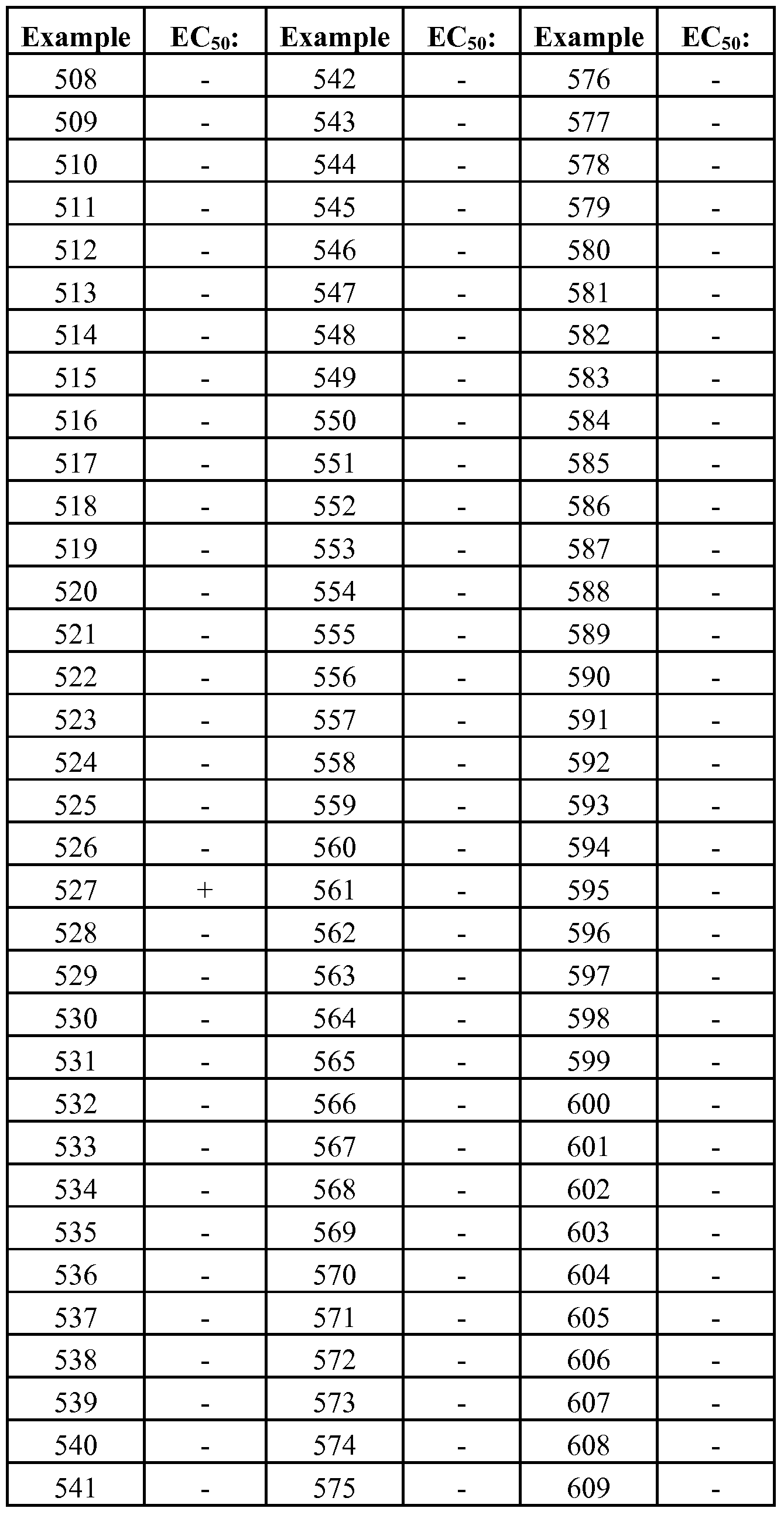

[0221] From the foregoing description, one skilled in the art can easily ascertain the essential characteristics of this invention, and without departing from the spirit and scope thereof, can make various changes and modifications of the invention to adapt it to various usages and conditions.
Claims
1. A compound of structural Formula I:

Xi is selected from the group consisting of N, O, S, CH2 and CR3;
X2 is selected from the group consisting of N, O, S, CH2 and CR4;
X3 is selected from the group consisting of N, O, S, CH2, CH, and C-methyl;
Gi is selected from the group consisting of CRaRb, NR7, and optionally substituted N-heterocycloalkyl;
G2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, NRg, O and SO2, any of which may be optionally substituted;
Ri is selected from the group consisting of aryl, N-containing heteroaryl; arylalkyl, and N-containing heteroarylalkyl, any of which may be optionally substituted;
R2 is selected from the group consisting of lower alkyl, lower alkenyl, lower alkynyl, lower hydroxyalkyl, amino, aryl, and heteroaryl, any of which may be optionally substituted; or, when Gi is substituted N-heterocycloalkyl, R2 may be null;
R3 and R4 are independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkoxy, lower perfluoroalkyl, and lower perfluoroalkoxy, or R3 and R4 taken together may form a 5-6-membered aryl, cycloalkyl, heteroaryl, or heterocycloalkyl, any of which may be optionally substituted;
R5, R6, R7, and R8 are each independently selected from the group consisting of halogen, hydrogen, hydroxy, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, phenyloxy, acyl, carboxyl, amino, lower heteroalkyl, lower alkylthio, lower aminoalkyl, lower alkylsulfonyl, sulfonamido, lower aryl, lower arylalkyl, lower cycloalkyl, lower cycloalkylalkyl, lower haloalkyl, lower perhaloalkyl, lower perhaloalkoxy, lower heteroaryl, lower heteroarylalkyl, lower heterocycloalkyl, and lower heterocycloalkylalkyl, any of which may be optionally substituted, or R5 and R6, taken together, form oxy; and
Ra and Rb are each independently selected from the group consisting of hydrogen, halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, lower hydroxyalkyl, lower perfluoroalkyl, lower perfluoroalkoxy, optionally substituted lower cycloalkyl, optionally substituted lower heterocycloalkyl, optionally substituted lower heteroaryl, and optionally substituted phenyl.
2. The compound as recited in claim 1, wherein R5 and R6, taken together, form oxy.
3. The compound as recited in claim 2, wherein Ri is selected from the group consisting of aryl and N-containing heteroaryl, any of which may be optionally substituted.
4. The compound as recited in claim 3, wherein Ri is selected from the group consisting of substituted phenyl and substituted, monocyclic N-containing heteroaryl.
5. The compound as recited in claim 4, wherein Ri is substituted pyridyl.
6. The compound as recited in claim 4, wherein Ri is substituted phenyl.
7. The compound as recited in claim 1, wherein Gi is NR7.
8. The compound as recited in claim 7, wherein Gi is NH.
9. The compound as recited in claim 7, wherein R7 is selected from the group consisting of hydrogen, halogen, hydroxy, C1-C3 alkyl, C1-C3 hydroxyalkyl, C1-C3 alkynyl, perfluoromethyl, and perfluoromethoxy.
10. The compound as recited in claim 1, wherein Gi is CR3Rb,
11. The compound as recited in claim 1 , wherein G2 is selected from the group consisting of a bond, lower alkyl, lower heteroalkyl, and NRs, any of which may be optionally substituted.
12. The compound as recited in claim 11, wherein
G2 is selected from the group consisting of optionally substituted Ci-C3 alkyl and Ci -C3 heteroalkyl containing one heteroatom selected from the group consisting of O and N, and NRs; and
Rg is selected from the group consisting of hydrogen, halogen, hydroxy, Ci-C3 alkyl, Ci-C3 hydroxyalkyl, Ci-C3 alkynyl, perfluoromethyl, and perfluoromethoxy.
13. The compound as recited in claim 12, wherein
G2 is selected from the group consisting of NRg and CH2; and Rg is hydrogen,
14. The compound as recited in claim 1, wherein R2 is selected from the group consisting of aryl and heteroaryl, either of which may be optionally substituted.
15. The compound as recited in claim 14, wherein R2 is substituted pyridyl.
16. The compound as recited in claim 14, wherein R2 is substituted phenyl.
17. The compound as recited in claim 1, having structural Formula II:

or a salt thereof, wherein:
Gi is selected from the group consisting of CH2, and NR7;
G2 is selected from the group consisting of a bond, CH2, and NRg;
Xi is selected from the group consisting of CRio and N;
X2 is selected from the group consisting of CRi2, N, O, and S;
X3 is selected from the group consisting of CR14, N, O, and S;
Yi is selected from the group consisting of CR2I, N, O, and S;
Y2 is selected from the group consisting of CR22, N, O, and S;
Y3 is selected from the group consisting of CR23, N, O, and S; R2 is selected from the group consisting of lower alkyl, lower amino, aryl, and heteroaryl, any of which may be optionally substituted; q is an integer from 0 to 3;
Rs, R6, R7, and Rs are each independently selected from the group consisting of hydrogen, hydroxy, alkoxy, aryloxy, alkyl, acyl, alkenyl, amino, alkynyl, heteroalkyl, carboxyl, alkylthio, alkylamino, alkylsulfonyl, sulfonamido, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, haloalkyl, perhaloalkyl, perhaloalkoxy, heteroaryl, heteroarylalkyl, heterocycloalkyl, and heterocycloalkylalkyl, any of which may be optionally substituted , or R3 and R4, taken together, may form oxy; and
Rio, R12, Ri4, Ri9, R21, R22, R23, R27, and R28 are each independently selected from the group consisting of null, hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, C-amido, N-amido, carbamate, urea, N- sulfonamido, S -sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted, or R27 and R28, taken together with R2 to which they are attached, form cycloalkyl, heterocycloalkyl, aryl or heteroaryl, any of which may be optionally substituted.
18. The compound as recited in claim 17, wherein R5 and R6, taken together, form oxy.
19. The compound as recited in claim 18, wherein
Yi is selected from the group consisting of CR2I and N; Y2 is selected from the group consisting of CR22 and N; and Y3 is selected from the group consisting of CR23 and N.
20. The compound as recited in claim 18, wherein R27 and R28, taken together with R2 to which they are attached, form cycloalkyl, heterocycloalkyl, aryl or heteroaryl, any of which may be optionally substituted.
21. The compound as recited in claim 20, wherein R27 and R28, taken together with R2 to which they are attached, form cycloalkyl or heterocycloalkyl, either of which may be optionally substituted.
22. The compound as recited in claim 21, wherein said cycloalkyl is a C3-C7 monocyclic cycloalkyl, which may be optionally substituted; and said heterocycloalkyl, is a C4-C7 monocyclic heterocycloalkyl, which may be optionally substituted.
23. The compound as recited in claim 22, wherein said C4-C7 monocyclic heterocycloalkyl is selected from the group consisting of pyrrolidine, piperidine, piperazine, and morpholine, any of which may be optionally substituted.
24. A compound as recited in claim 20 having structural Formula III:

Gi is selected from the group consisting of CH2, and NR7;
G2 is selected from the group consisting of a bond, CH2, and NRs;
Xi is selected from the group consisting of CH and N;
X2 is selected from the group consisting of CH and N;
X3 is selected from the group consisting of CH and N;
Yi is selected from the group consisting of CR2I and N;
Y2 is selected from the group consisting of CR22 and N;
Y3 is selected from the group consisting of CR23 and N;
Zi is selected from the group consisting of CR24 and N; Z2 is selected from the group consisting of CR25 and N;
Z3 is selected from the group consisting of CR26 and N;
Z4 is selected from the group consisting of CR29 and N; q and r are each independently an integer from 0 to 3;
R7, and Rs are each independently selected from the group consisting of hydrogen, hydroxy, lower alkoxy, lower alkyl, lower alkenyl, lower amino, lower alkynyl, lower heteroalkyl; and
Rig, Ri9, R21, R22, R23, R24, R25, R26, and R29 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, heteroarylamino, C-amido, N-amido, carbamate, urea, N-sulfonamido, S-sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted.
25. The compound as recited in claim 24, wherein
Gi is selected from the group consisting of CH2, and NR7; and G2 is selected from the group consisting of a bond, CH2, and NRs.
26. The compound as recited in claim 25, wherein 
X2 is N.
27. The compound as recited in claim 25, wherein Xi and X2 are each CH; and
X3 is N.
28. The compound as recited in claim 25, wherein Xi is N; and
X2 and X3 are each CH.
29. The compound as recited in claim 25, wherein X1, X2 and X3 are each CH.
30. The compound as recited in claim 24, wherein Y1 Is CR21;
Y2 is CR22; and Y3 is N.
31. The compound as recited in claim 25, wherein Y1 Is CR21;
Y2 is CR22; and Y3 is CR23.
32. The compound as recited in claim 30, wherein Z3 is CR26; and
R26 is not hydrogen.
33. The compound as recited in claim 32, wherein Zi is N.
34. The compound as recited in claim 25, wherein G2 is selected from the group consisting of NH and CH2.
35. The compound as recited in claim 25, wherein Gi is NH.
36. A compound as recited in claim 33 having structural Formula IV:

Gi is selected from the group consisting of NH and CH2;
G2 is selected from the group consisting of NH, CH2, SO2, and O;
Zi is selected from the group consisting of CR24 and N;
Z4 is selected from the group consisting of CR27 and N; q is an integer from 1 to 3; r is an integer from 1 to 3, or r may be 0 if R27 is not hydrogen; and each Rig, each Ri9, R24, R26, and R27 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, nitro, amino, cyano, alkyl, haloalkyl, perhaloalkyl, heteroalkyl, hydroxyalkyl, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, thioalkyl, arylalkyl, cycloalkylalkyl, heteroarylalkyl, heterocycloalkylalkyl, alkenyl, arylalkenyl , heteroarylalkenyl, heterocycloalkylalkenyl, alkynyl, arylalkynyl , heteroarylalkynyl, heterocycloalkylalkynyl, alkoxy, haloalkoxy, perhaloalkoxy, acyloxy, arylalkoxy, aryloxy, heteroaryloxy, acyl, arylalkanoyl, alkylcarbonyl, alkoxycarbonyl, carboxyl, alkylamino, arylamino, heteroarylamino, C-amido, N-amido, carbamate, urea, N-sulfonamido, S-sulfonamido, alkylsulfonyl, thiol, alkylthio, arylthio, heteroarylthio, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl, any of which may be optionally substituted.
37. The compound as recited in claim 36, wherein G2 is selected from the group consisting of NH and CH2.
38. The compound as recited in claim 37, wherein Gi is NH.
39. The compound as recited in claim 38, wherein
Zi is N; and Z4 is CR27.
40. The compound as recited in claim 38, wherein Zi and Z4 are both N.
41. The compound as recited in claim 38, wherein R27 is not hydrogen.
42. The compound as recited in claim 38, wherein R27 and each Rig are independently selected from the group consisting of hydrogen, halogen, haloalkyl, haloalkoxy, optionally substituted arylamino, optionally substituted heteroarylamino, optionally substituted heteroaryloxy, and optionally substituted aryloxy, provided that one of R27 and each Rig is not hydrogen.
43. The compound as recited in claim 42, wherein R26Is selected from the group consisting of optionally substituted phenyloxy and optionally substituted 5-6 membered monocyclic heteroaryloxy.
44. The compound as recited in claim 43, wherein R26is selected from the group consisting of optionally substituted phenyloxy and optionally substituted pyridinyloxy.
45. The compound as recited in claim 44, wherein said pyridinyloxy or aryloxy is optionally substituted with one or more substituents selected from the group consisting of halogen, cyano, hydroxy, lower amino, lower alkyl, lower alkenyl, lower alkynyl, lower alkoxy, lower hydroxyalkyl, lower perfluoroalkyl, lower perfluoroalkoxy, optionally substituted lower cycloalkyl, optionally substituted lower heterocycloalkyl, optionally substituted lower heteroaryl, and optionally substituted phenyl.
46. The compound as recited in claim 45, wherein said pyridinyloxy or aryloxy is optionally substituted with one or more substituents selected from the group consisting ofhalogen, hydroxy, Ci-C3 alkyl, Ci-C3 hydroxyalkyl, Ci-C3 alkynyl, perfluoromethyl, and perfluoromethoxy.
47. The compound as recited in claim 38, wherein each R49 is independently selected from the group consisting ofhalogen, haloalkyl, and haloalkoxy.
48. The compound as recited in claim 48, wherein each R49 is independently selected from the group consisting ofhalogen, perfluoromethyl, and perfluoromethoxy.
49. The compound as recited in claim 49, wherein q is 1 or 2.
50. The compound as recited in claim 50, wherein q is 2, and the two R19 groups are meta to each other.
51. A compound selected from the group consisting of Examples 1 to 640.
52. A compound as recited in Claim 1 for use as a medicament.
53. A compound as recited in Claim 1 for use in the manufacture of a medicament for the prevention or treatment of a disease or condition ameliorated by the modulation of GPRl 19.
54. A pharmaceutical composition comprising a compound as recited in Claim 1 together with a pharmaceutically acceptable carrier.
55. A method of modulating GPRl 19 comprising contacting GPRl 19 with a compound as recited in Claim 1.
56. A method of treatment of a GPRl 19-mediated disease comprising the administration of a therapeutically effective amount of a compound as recited in Claim 1 to a patient in need thereof.
57. The method as recited in Claim 56 wherein said disease is a metabolic disease.
58. The method as recited in Claim 57 wherein said disease is diabetes.
59. A method of treatment of a GPRl 19-mediated disease comprising the administration of: a. a therapeutically effective amount of a compound as recited in Claim 1; and b. another therapeutic agent.
60. The method as recited in Claim 59, wherein said agent is selected from the group consisting of insulin, metformin, Glipizide, glyburide, Amaryl, gliclazide, meglitinides, nateglinide, repaglinide, pramlintide, PTP-112, SB-517955, SB- 4195052, SB-216763, NN-57-05441, NN-57-05445, GW-0791, AGN-194204, T- 1095, BAY R3401, acarbose, miglitol, voglibose, Exendin-4, DPP728, LAF237, vildagliptin , BMS477118, PT-100, GSK-823093, PSN-9301, T-6666, SYR-322, SYR-619, Liraglutide, CJC- 1134-PC, naliglutide, MK-0431, saxagliptin, GSK23A, pioglitazone, rosiglitazone, AVE2268, GW869682, GSKl 89075, APD668, PSN- 119-1, PSN-821, rosuvastatin, atrovastatin, simvastatin, lovastatin, pravastatin, fluvastatin, cerivastatin, rosuvastatin, pitavastatin, fenofibrate, benzafibrate, clofibrate, gemfibrozil, Ezetimibe, eflucimibe, CP-529414, CETi-I, JTT-705, cholestyramine, colestipol, niacin, implitapide, (7?)-l-{4-[5-methyl-2-(4- trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-benzenesulfonyl}2,3-dihydro-lH- indole-2-carboxylic acid, and GI-262570.
61. A method for achieving an effect in a patient comprising the administration of a therapeutically effective amount of a compound as recited in Claim 1 to a patient, wherein the effect is the modulation of a metabolic disease.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3723908P | 2008-03-17 | 2008-03-17 | |
| US61/037,239 | 2008-03-17 | ||
| US9536208P | 2008-09-09 | 2008-09-09 | |
| US61/095,362 | 2008-09-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009117421A2 true WO2009117421A2 (en) | 2009-09-24 |
| WO2009117421A3 WO2009117421A3 (en) | 2010-01-07 |
Family
ID=41091499
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/037408 WO2009117421A2 (en) | 2008-03-17 | 2009-03-17 | Heterocyclic modulators of gpr119 for treatment of disease |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009117421A2 (en) |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102070553A (en) * | 2010-12-17 | 2011-05-25 | 西安瑞联近代电子材料有限责任公司 | Synthesis method of thiazole-4-formaldehyde |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2012530757A (en) * | 2009-06-24 | 2012-12-06 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Novel compounds, pharmaceutical compositions and methods relating thereto |
| CN102863356A (en) * | 2012-10-11 | 2013-01-09 | 常州华南化工有限公司 | Preparation method for 4-(4-methylphenoxy)cyanobenzene |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN103044393A (en) * | 2012-11-30 | 2013-04-17 | 淮阴师范学院 | Synthesis method of 1-(3-chloro-2-pyridyl)-1H-pyrazole-5-formate |
| WO2013054822A1 (en) | 2011-10-07 | 2013-04-18 | Takeda Pharmaceutical Company Limited | 1 - arylcarbonyl - 4 - oxy - piperidine compounds useful for the treatment of neurodegenerative diseases |
| CN103087041A (en) * | 2011-11-02 | 2013-05-08 | 中国中化股份有限公司 | Use of diarylether-containing pyrazole-amide compounds as agricultural bactericide |
| CN103254170A (en) * | 2012-08-15 | 2013-08-21 | 山东省联合农药工业有限公司 | Pyrazole ring-containing pesticide |
| CN103304561A (en) * | 2013-06-21 | 2013-09-18 | 天津药物研究院 | Compounds with antithrombotic effects |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| WO2014016191A1 (en) * | 2012-07-24 | 2014-01-30 | Boehringer Ingelheim International Gmbh | N-cyclopropyl-n-piperidinyl-amide derivatives and their use as gpr119 modulators |
| US8912227B1 (en) | 2014-03-07 | 2014-12-16 | Janssen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of GPR120 |
| CN104606189A (en) * | 2015-01-08 | 2015-05-13 | 苏州大学 | Application of compound to preparation of mTOR inhibitor |
| US9045454B2 (en) | 2013-03-14 | 2015-06-02 | Jansen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of GPR120 |
| US9067898B1 (en) | 2014-03-07 | 2015-06-30 | Janssen Pharmaceutica Nv | Isothiazole derivatives as GPR120 agonists for the treatment of type II diabetes |
| JP2015523404A (en) * | 2012-08-02 | 2015-08-13 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | N-cyclopropyl-N-piperidinyl-amide, pharmaceutical compositions containing them and uses thereof |
| WO2015129755A1 (en) * | 2014-02-25 | 2015-09-03 | 味の素株式会社 | Novel compound and texture improver containing said compound |
| WO2015134038A1 (en) * | 2014-03-07 | 2015-09-11 | Janssen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of gpr120 |
| US9199975B2 (en) | 2011-09-30 | 2015-12-01 | Asana Biosciences, Llc | Biaryl imidazole derivatives for regulating CYP17 |
| WO2016160985A1 (en) * | 2015-03-30 | 2016-10-06 | The General Hospital Corporation | Imaging of glycogen synthase kinase 3 |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| US9562053B2 (en) | 2013-03-14 | 2017-02-07 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| CN106748992A (en) * | 2017-01-04 | 2017-05-31 | 安徽国星生物化学有限公司 | A kind of synthetic method of the trichloromethyl pyridine of 3 chlorine, 2 diazanyl 5 |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| WO2017148902A1 (en) | 2016-03-03 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | New 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017207340A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Novel substituted benzimidazole, methods for the production thereof, pharmaceutical preparations containing same, and use thereof to produce medicaments |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| WO2017207385A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted 3-methylindazoles, methods for the production thereof, pharmaceutical preparations containing same, and use thereof for the production of medicaments |
| WO2017207386A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Use of 2-substituted indazoles for the treatment and prophylaxis of autoimmune diseases |
| WO2018060174A1 (en) | 2016-09-29 | 2018-04-05 | Bayer Pharma Aktiengesellschaft | Substituted benzimidazoles, pharmaceutical preparations containing same, and use of same to produce drugs |
| CN108017557A (en) * | 2017-12-06 | 2018-05-11 | 中国科学院兰州化学物理研究所苏州研究院 | A kind of Process for the cyanation for preparing nitrile compounds |
| US10118922B2 (en) | 2013-03-14 | 2018-11-06 | Janssen Pharmaceutica Nv | GPR120 agonists for the treatment of type II diabetes |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| WO2019195777A1 (en) | 2018-04-06 | 2019-10-10 | Aquinox Pharmaceuticals (Canada) Inc. | Indene derivatives useful in treating pain and inflammation |
| CN113024389A (en) * | 2021-05-24 | 2021-06-25 | 山东海利尔化工有限公司 | Preparation method of substituted phenoxybenzylamine compound and pyrazole carboxamide compound |
| CN113040090A (en) * | 2021-04-13 | 2021-06-29 | 南京医科大学 | Method for constructing animal model of autism and corresponding application |
| US11161850B2 (en) | 2018-07-05 | 2021-11-02 | Incyte Corporation | Fused pyrazine derivatives as A2A / A2B inhibitors |
| US11168089B2 (en) | 2018-05-18 | 2021-11-09 | Incyte Corporation | Fused pyrimidine derivatives as A2A / A2B inhibitors |
| US20210347773A1 (en) * | 2016-12-15 | 2021-11-11 | Ono Pharmaceutical Co., Ltd. | Activator of trek (twik related k+ channels) channels |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11390624B2 (en) | 2019-01-29 | 2022-07-19 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
| WO2023032872A1 (en) * | 2021-08-31 | 2023-03-09 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Production method for synthetic intermediate of monocyclic pyridine derivative |
| US11673894B2 (en) | 2018-02-27 | 2023-06-13 | Incyte Corporation | Imidazopyrimidines and triazolopyrimidines as A2A / A2B inhibitors |
| EP4238970A1 (en) * | 2022-03-03 | 2023-09-06 | InterAx Biotech AG | Modulators of ackr3 and uses thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1808168A1 (en) * | 2005-01-10 | 2007-07-18 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| WO2007116229A1 (en) * | 2006-04-06 | 2007-10-18 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2008001931A2 (en) * | 2006-06-27 | 2008-01-03 | Takeda Pharmaceutical Company Limited | Fused cyclic compounds |
| WO2008013322A1 (en) * | 2006-07-27 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | 1- (-d-glycopyranosyl) - 3 - (4-cyclopropylphenylmethyl) - 4 - halogeno indole derivatives and use thereof as sglt inhibitors. |
| WO2008025798A1 (en) * | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyridine compounds for treating gpr119 related disorders |
-
2009
- 2009-03-17 WO PCT/US2009/037408 patent/WO2009117421A2/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1808168A1 (en) * | 2005-01-10 | 2007-07-18 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| WO2007116229A1 (en) * | 2006-04-06 | 2007-10-18 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2008001931A2 (en) * | 2006-06-27 | 2008-01-03 | Takeda Pharmaceutical Company Limited | Fused cyclic compounds |
| WO2008013322A1 (en) * | 2006-07-27 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | 1- (-d-glycopyranosyl) - 3 - (4-cyclopropylphenylmethyl) - 4 - halogeno indole derivatives and use thereof as sglt inhibitors. |
| WO2008025798A1 (en) * | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyridine compounds for treating gpr119 related disorders |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012530757A (en) * | 2009-06-24 | 2012-12-06 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Novel compounds, pharmaceutical compositions and methods relating thereto |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| CN102070553B (en) * | 2010-12-17 | 2013-08-07 | 西安瑞联近代电子材料有限责任公司 | Synthesis method of thiazole-4-formaldehyde |
| CN102070553A (en) * | 2010-12-17 | 2011-05-25 | 西安瑞联近代电子材料有限责任公司 | Synthesis method of thiazole-4-formaldehyde |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9199975B2 (en) | 2011-09-30 | 2015-12-01 | Asana Biosciences, Llc | Biaryl imidazole derivatives for regulating CYP17 |
| US10273245B2 (en) | 2011-10-07 | 2019-04-30 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US8648079B2 (en) | 2011-10-07 | 2014-02-11 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds |
| WO2013054822A1 (en) | 2011-10-07 | 2013-04-18 | Takeda Pharmaceutical Company Limited | 1 - arylcarbonyl - 4 - oxy - piperidine compounds useful for the treatment of neurodegenerative diseases |
| US9193709B2 (en) | 2011-10-07 | 2015-11-24 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US11174272B2 (en) | 2011-10-07 | 2021-11-16 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US9440990B2 (en) | 2011-10-07 | 2016-09-13 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US10144743B2 (en) | 2011-10-07 | 2018-12-04 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US10717748B2 (en) | 2011-10-07 | 2020-07-21 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US9586930B2 (en) | 2011-10-07 | 2017-03-07 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| KR20140072902A (en) | 2011-10-07 | 2014-06-13 | 다케다 야쿠힌 고교 가부시키가이샤 | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US10550129B2 (en) | 2011-10-07 | 2020-02-04 | Takeda Pharmaceutical Company Limited | 1-arylcarbonyl-4-oxy-piperidine compounds useful for the treatment of neurodegenerative diseases |
| US8865717B2 (en) | 2011-10-07 | 2014-10-21 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds |
| US8871766B2 (en) | 2011-10-07 | 2014-10-28 | Takeda Pharmaceutical Co., Ltd. | Heterocyclic compounds |
| CN103087041A (en) * | 2011-11-02 | 2013-05-08 | 中国中化股份有限公司 | Use of diarylether-containing pyrazole-amide compounds as agricultural bactericide |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| US8927726B2 (en) | 2012-07-24 | 2015-01-06 | Boehringer Ingelheim International Gmbh | N-cyclopropyl-N-piperidinyl-amide derivatives, pharmaceutical compositions containing them and uses thereof |
| JP2015522633A (en) * | 2012-07-24 | 2015-08-06 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | N-cyclopropyl-N-piperidinyl-amide derivatives and their use as GPR119 modulators |
| WO2014016191A1 (en) * | 2012-07-24 | 2014-01-30 | Boehringer Ingelheim International Gmbh | N-cyclopropyl-n-piperidinyl-amide derivatives and their use as gpr119 modulators |
| JP2015523404A (en) * | 2012-08-02 | 2015-08-13 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | N-cyclopropyl-N-piperidinyl-amide, pharmaceutical compositions containing them and uses thereof |
| US9926308B2 (en) | 2012-08-02 | 2018-03-27 | Boehringer Ingelheim International Gmbh | N-cyclopropyl-N-piperidinyl-amides, pharmaceutical compositions containing them and uses thereof |
| CN103254170B (en) * | 2012-08-15 | 2014-04-30 | 山东省联合农药工业有限公司 | Pyrazole ring-containing pesticide |
| CN103254170A (en) * | 2012-08-15 | 2013-08-21 | 山东省联合农药工业有限公司 | Pyrazole ring-containing pesticide |
| CN102863356A (en) * | 2012-10-11 | 2013-01-09 | 常州华南化工有限公司 | Preparation method for 4-(4-methylphenoxy)cyanobenzene |
| CN103044393B (en) * | 2012-11-30 | 2014-07-23 | 淮阴师范学院 | Synthesis method of 1-(3-chloro-2-pyridyl)-1H-pyrazole-5-formate |
| CN103044393A (en) * | 2012-11-30 | 2013-04-17 | 淮阴师范学院 | Synthesis method of 1-(3-chloro-2-pyridyl)-1H-pyrazole-5-formate |
| US10618893B2 (en) | 2013-03-14 | 2020-04-14 | Janssen Pharmaceuticals Nv | GPR120 agonists for the treatment of type II diabetes |
| US9045454B2 (en) | 2013-03-14 | 2015-06-02 | Jansen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of GPR120 |
| US10155737B2 (en) | 2013-03-14 | 2018-12-18 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| US10118922B2 (en) | 2013-03-14 | 2018-11-06 | Janssen Pharmaceutica Nv | GPR120 agonists for the treatment of type II diabetes |
| US9562053B2 (en) | 2013-03-14 | 2017-02-07 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| US10730847B2 (en) | 2013-03-14 | 2020-08-04 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| US11254647B2 (en) | 2013-03-14 | 2022-02-22 | Janssen Pharmaceutica Nv | Benzo-fused heterocyclic derivatives useful as agonists of GPR120 |
| CN103304561A (en) * | 2013-06-21 | 2013-09-18 | 天津药物研究院 | Compounds with antithrombotic effects |
| CN103304561B (en) * | 2013-06-21 | 2016-06-01 | 天津药物研究院 | One class has the compound of anti thrombotic action |
| WO2015129755A1 (en) * | 2014-02-25 | 2015-09-03 | 味の素株式会社 | Novel compound and texture improver containing said compound |
| WO2015134038A1 (en) * | 2014-03-07 | 2015-09-11 | Janssen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of gpr120 |
| US9067898B1 (en) | 2014-03-07 | 2015-06-30 | Janssen Pharmaceutica Nv | Isothiazole derivatives as GPR120 agonists for the treatment of type II diabetes |
| US8912227B1 (en) | 2014-03-07 | 2014-12-16 | Janssen Pharmaceutica Nv | Bicyclic pyrrole derivatives useful as agonists of GPR120 |
| CN106061943A (en) * | 2014-03-07 | 2016-10-26 | 詹森药业有限公司 | Bicyclic pyrrole derivatives useful as agonists of GPR120 |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| US10793545B2 (en) | 2014-11-26 | 2020-10-06 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| EP3674298A1 (en) | 2014-11-26 | 2020-07-01 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical formulations containing them, and their use for the preparation of medicaments |
| EP4260909A2 (en) | 2014-11-26 | 2023-10-18 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical preparations containing them and their use in the preparation of drugs |
| CN104606189A (en) * | 2015-01-08 | 2015-05-13 | 苏州大学 | Application of compound to preparation of mTOR inhibitor |
| WO2016160985A1 (en) * | 2015-03-30 | 2016-10-06 | The General Hospital Corporation | Imaging of glycogen synthase kinase 3 |
| US10280152B2 (en) | 2015-03-30 | 2019-05-07 | The General Hospital Corporation | Imaging of glycogen synthase kinase 3 |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| US10435396B2 (en) | 2016-03-03 | 2019-10-08 | Bayer Pharma Aktiegesellschaft | 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| WO2017148902A1 (en) | 2016-03-03 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | New 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| WO2017157792A1 (en) | 2016-03-17 | 2017-09-21 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017207385A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted 3-methylindazoles, methods for the production thereof, pharmaceutical preparations containing same, and use thereof for the production of medicaments |
| WO2017207340A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Novel substituted benzimidazole, methods for the production thereof, pharmaceutical preparations containing same, and use thereof to produce medicaments |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| WO2017207386A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Use of 2-substituted indazoles for the treatment and prophylaxis of autoimmune diseases |
| WO2018060174A1 (en) | 2016-09-29 | 2018-04-05 | Bayer Pharma Aktiengesellschaft | Substituted benzimidazoles, pharmaceutical preparations containing same, and use of same to produce drugs |
| US20210347773A1 (en) * | 2016-12-15 | 2021-11-11 | Ono Pharmaceutical Co., Ltd. | Activator of trek (twik related k+ channels) channels |
| US11851428B2 (en) * | 2016-12-15 | 2023-12-26 | Ono Pharmaceutical Co., Ltd. | Activator of TREK (TWIK RElated K+channels) channels |
| CN106748992A (en) * | 2017-01-04 | 2017-05-31 | 安徽国星生物化学有限公司 | A kind of synthetic method of the trichloromethyl pyridine of 3 chlorine, 2 diazanyl 5 |
| CN108017557A (en) * | 2017-12-06 | 2018-05-11 | 中国科学院兰州化学物理研究所苏州研究院 | A kind of Process for the cyanation for preparing nitrile compounds |
| US11673894B2 (en) | 2018-02-27 | 2023-06-13 | Incyte Corporation | Imidazopyrimidines and triazolopyrimidines as A2A / A2B inhibitors |
| WO2019195777A1 (en) | 2018-04-06 | 2019-10-10 | Aquinox Pharmaceuticals (Canada) Inc. | Indene derivatives useful in treating pain and inflammation |
| US11168089B2 (en) | 2018-05-18 | 2021-11-09 | Incyte Corporation | Fused pyrimidine derivatives as A2A / A2B inhibitors |
| US11873304B2 (en) | 2018-05-18 | 2024-01-16 | Incyte Corporation | Fused pyrimidine derivatives as A2A/A2B inhibitors |
| US11161850B2 (en) | 2018-07-05 | 2021-11-02 | Incyte Corporation | Fused pyrazine derivatives as A2A / A2B inhibitors |
| US11884665B2 (en) | 2019-01-29 | 2024-01-30 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| US11390624B2 (en) | 2019-01-29 | 2022-07-19 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| US11851429B2 (en) | 2020-05-19 | 2023-12-26 | Kallyope, Inc. | AMPK activators |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
| CN113040090A (en) * | 2021-04-13 | 2021-06-29 | 南京医科大学 | Method for constructing animal model of autism and corresponding application |
| CN113024389A (en) * | 2021-05-24 | 2021-06-25 | 山东海利尔化工有限公司 | Preparation method of substituted phenoxybenzylamine compound and pyrazole carboxamide compound |
| JP7315805B1 (en) | 2021-08-31 | 2023-07-26 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Method for producing intermediate for synthesis of monocyclic pyridine derivative |
| WO2023032872A1 (en) * | 2021-08-31 | 2023-03-09 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Production method for synthetic intermediate of monocyclic pyridine derivative |
| EP4238970A1 (en) * | 2022-03-03 | 2023-09-06 | InterAx Biotech AG | Modulators of ackr3 and uses thereof |
| WO2023166190A1 (en) * | 2022-03-03 | 2023-09-07 | Interax Biotech Ag | Modulators of ackr3 and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009117421A3 (en) | 2010-01-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009117421A2 (en) | Heterocyclic modulators of gpr119 for treatment of disease | |
| US11767334B2 (en) | Heteroaryl inhibitors of PDE4 | |
| RU2757457C2 (en) | Pyrrolopyrimidines as cftr potentiators | |
| US8163756B2 (en) | Enzyme modulators and treatments | |
| JP5487214B2 (en) | Carbazole carboxamide compounds useful as kinase inhibitors | |
| JP4171417B2 (en) | N-substituted pyrrolidine derivatives as dipeptidyl peptidase IV inhibitors | |
| JP6346862B2 (en) | Substituted proline / piperidine as an orexin receptor antagonist | |
| WO2010048149A2 (en) | Heterocyclic modulators of gpr119 for treatment of disease | |
| MX2014015350A (en) | TRIAZOLONE COMPOUNDS AS mPGES-1 INHIBITORS. | |
| US20070155764A1 (en) | Novel substituted pyrimidinyloxy ureas useful as inhibitors of protein kinases | |
| TW200831491A (en) | N-aryl pyrazole compounds, compositions, and methods for their use | |
| US10392389B2 (en) | Heterocyclic compounds for the inhibition of PASK | |
| JP2019519484A (en) | Aromatic sulfonamide derivative | |
| US20090105124A1 (en) | Heterocyclic modulators of tgr5 | |
| JP2009544625A (en) | Benzothiophene inhibitors of RHO kinase | |
| WO2008097976A1 (en) | Heterocyclic modulators of tgr5 for treatment of disease | |
| AU2012345557A1 (en) | Novel trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease | |
| WO2007052843A1 (en) | Heterocyclic amide compound and use thereof | |
| WO2010088518A2 (en) | Heterocyclic modulators of gpr119 for treatment of disease | |
| TW201217362A (en) | Heteroaryls and uses thereof | |
| EP4192828A1 (en) | Substituted pyridine derivatives as sarm1 inhibitors | |
| ES2360935T3 (en) | PYRROLIDINE N-SUBSTITUTED DERIVATIVES AS INHIBITORS OF DIPEPTIL PEPTIDASE IV. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09721577 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09721577 Country of ref document: EP Kind code of ref document: A2 |