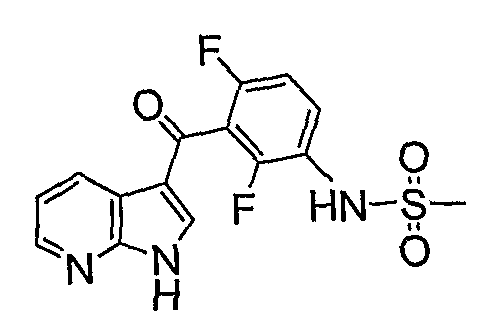
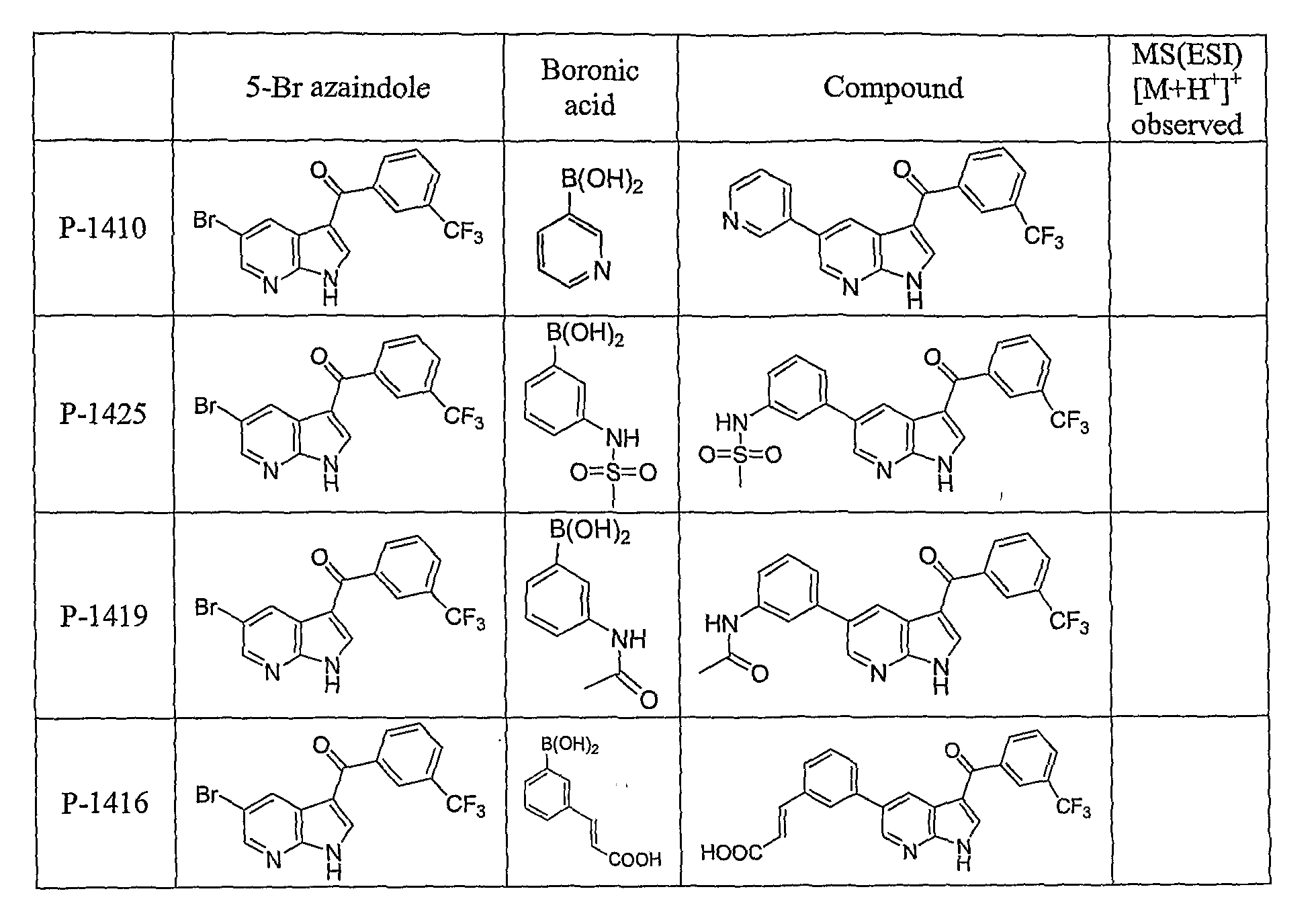
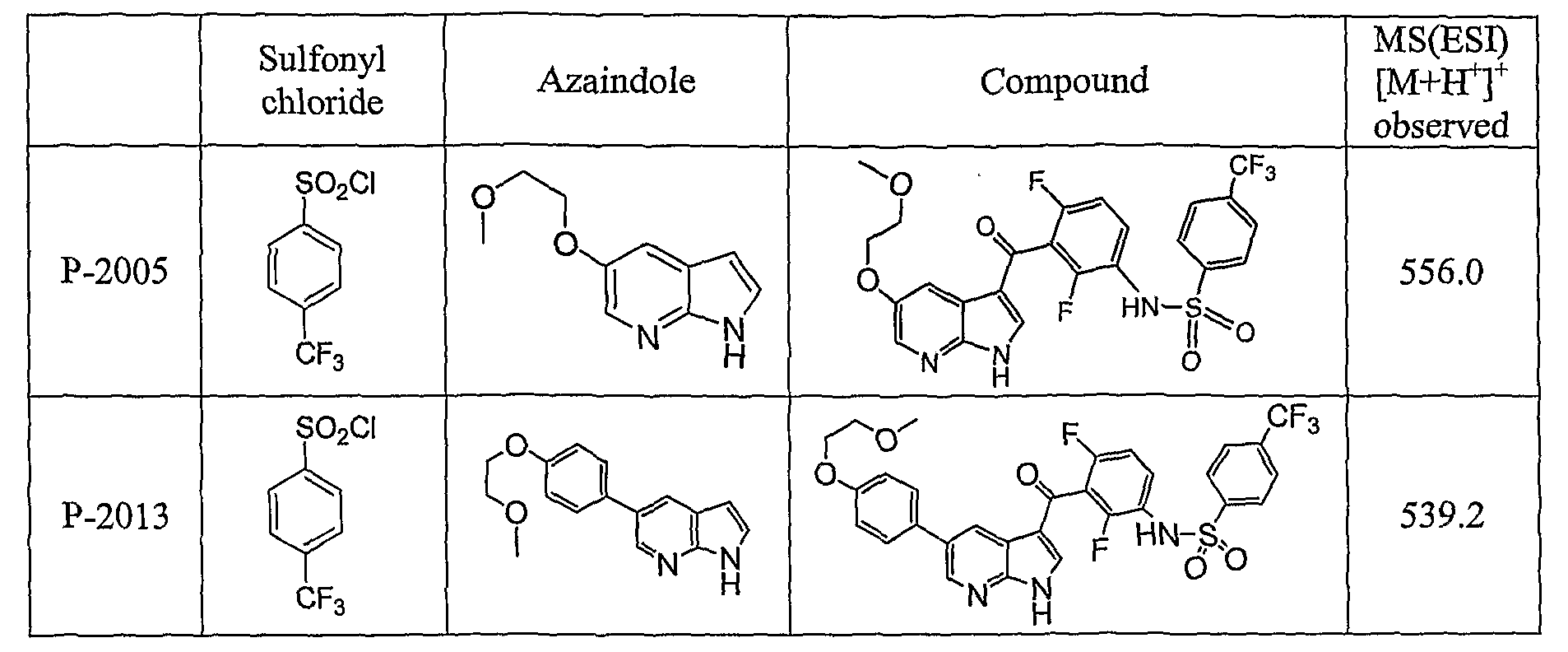
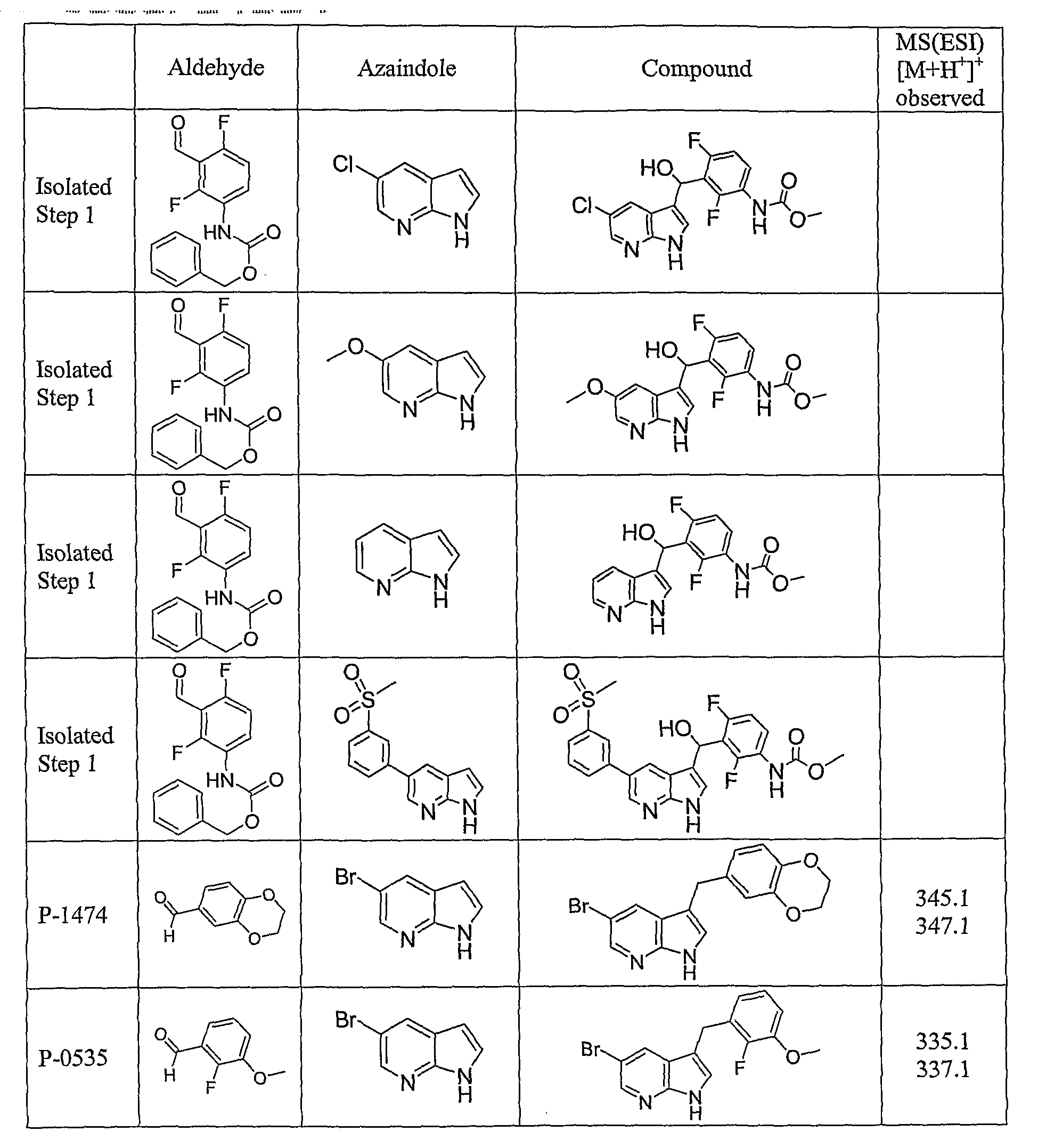
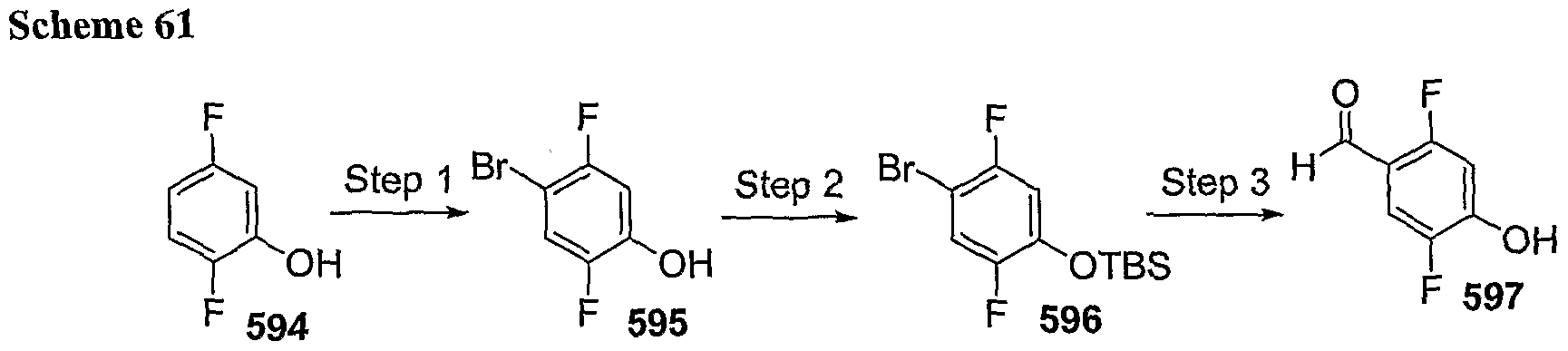
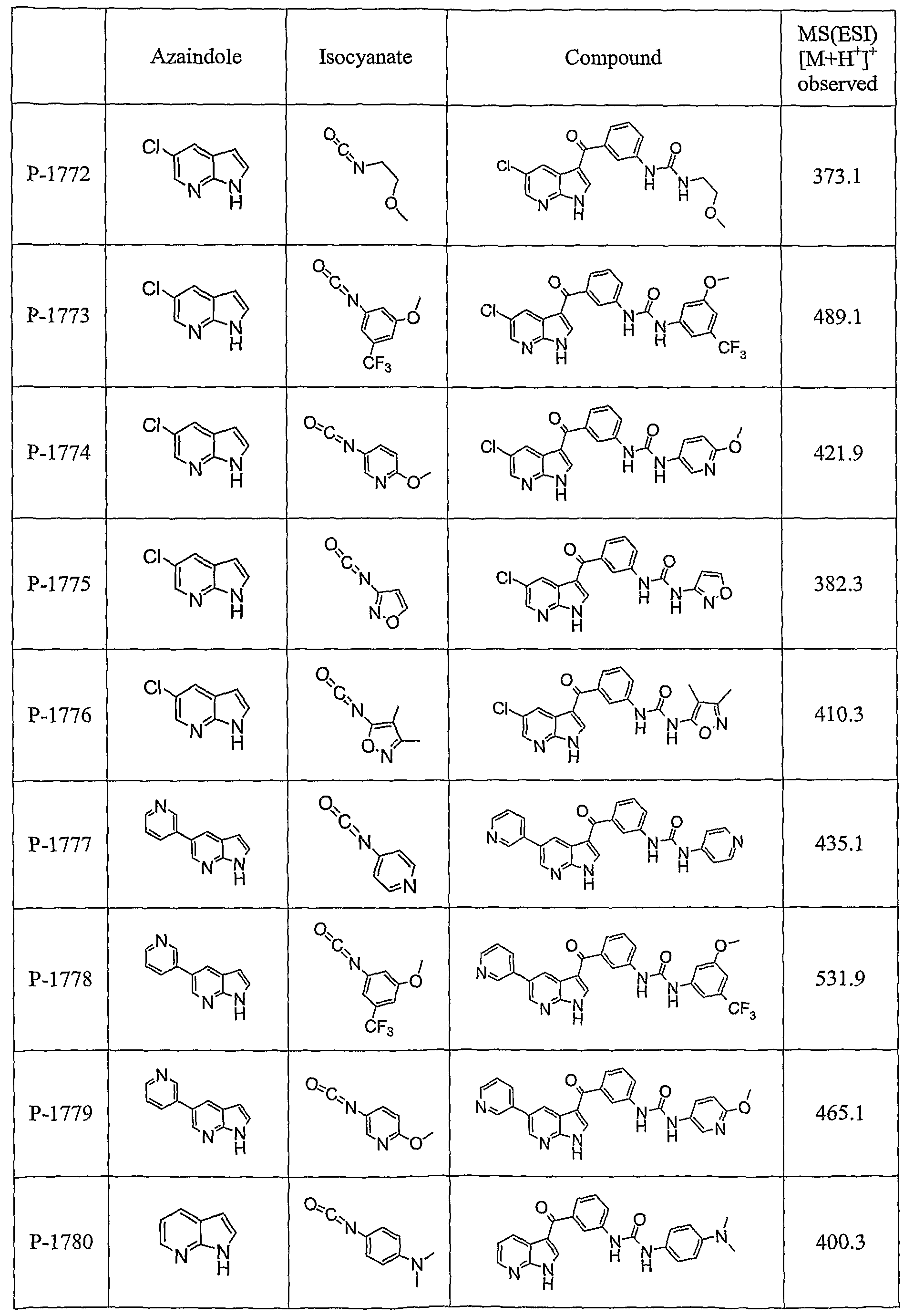
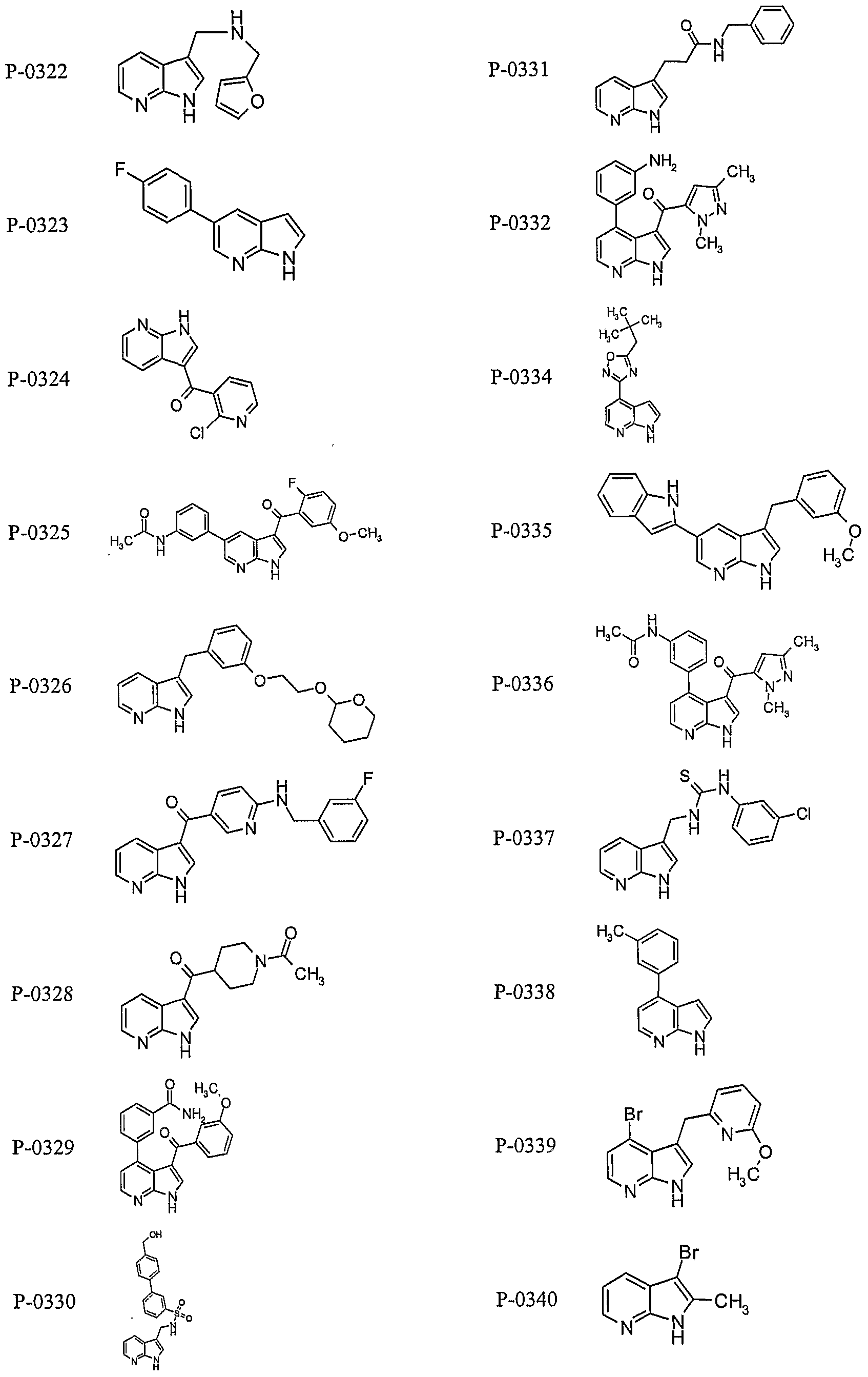
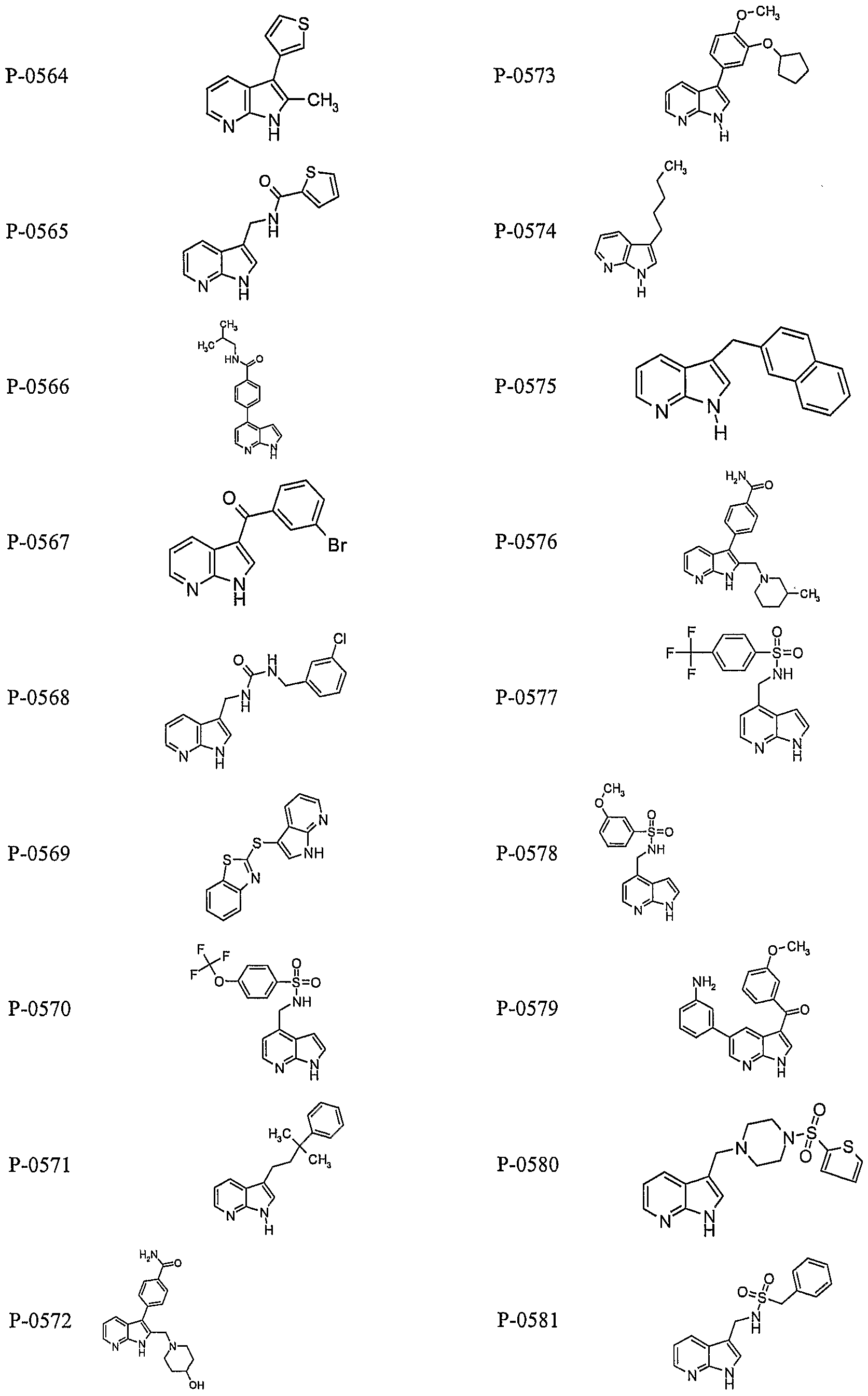
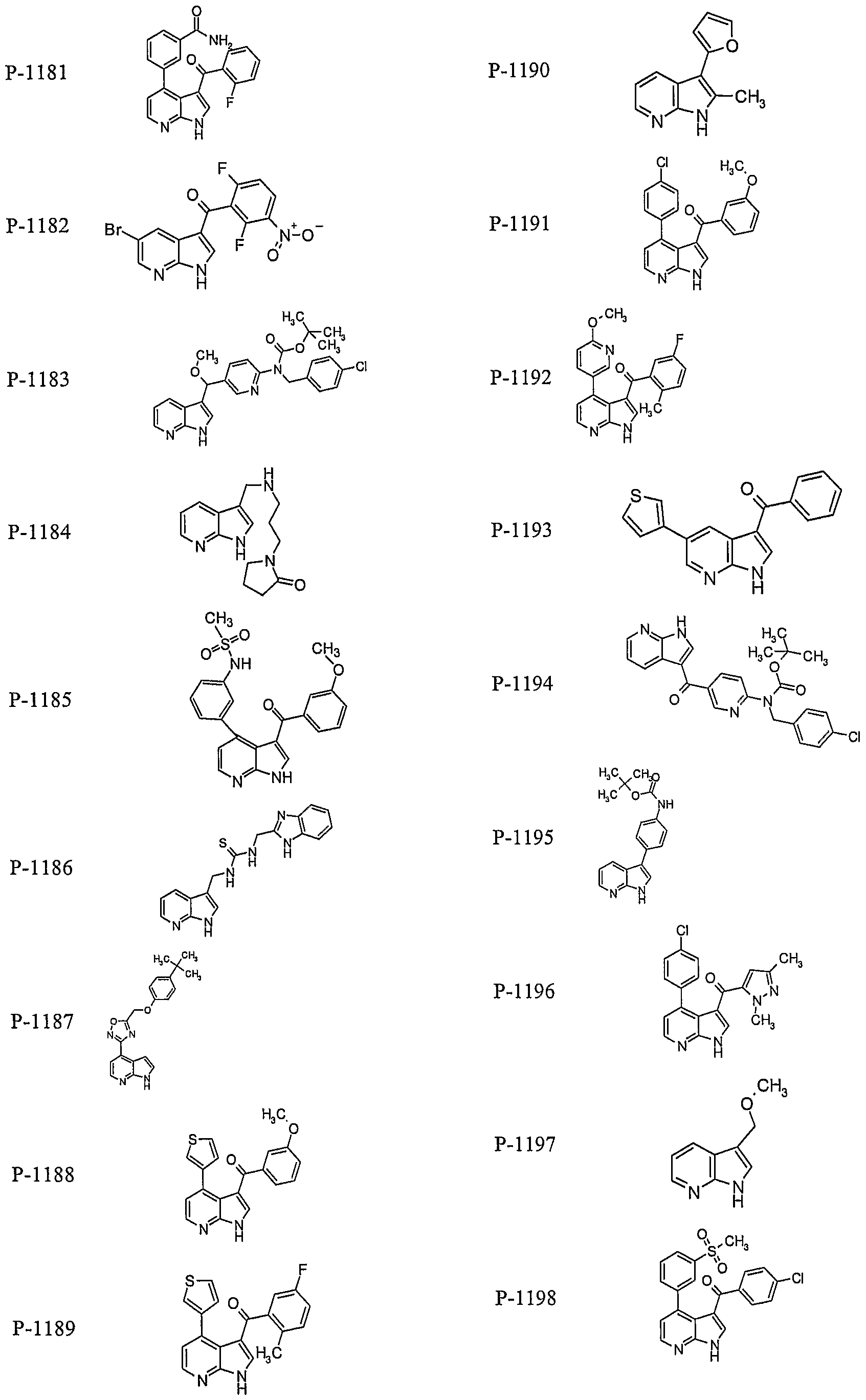
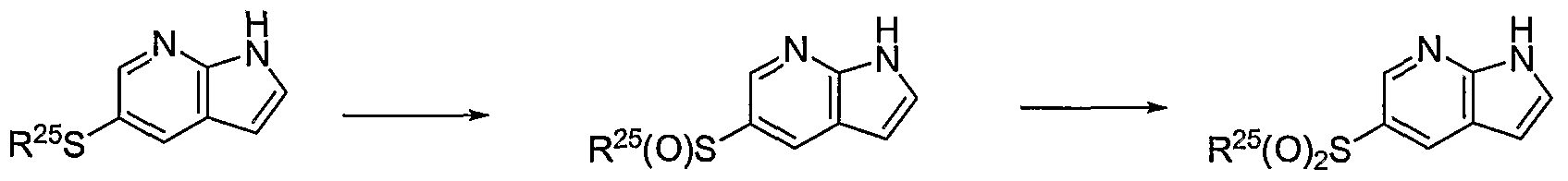
PYRROLO [2 , 3-B] PYRIDINE DERIVATIVES AS PROTEIN KINASE INHIBITORS
RELATED PATENT APPLICATIONS
[0001] This application claims the benefit of U.S. Prov. App. No. 60/692,960, filed June 22, 2005, and U.S. Prov. App. No. 60/731,528, filed October 28, 2005, both of which are incorporated herein by reference in their entireties and for all purposes.
FIELD OF THE INVENTION
[0002] The present invention relates to kinases and compounds which modulate kinases, and uses therefor. Particular embodiments contemplate disease indications which are amenable to treatment by modulation of kinase activity by the compounds of the present invention.
BACKGROUND OF THE INVENTION
[0003] The information provided herein is intended solely to assist the understanding of the reader. None of the information provided nor references cited is admitted to be prior art to the present invention. Each of the references cited herein is incorporated in its entirety.
[0004] Receptor protein kinases regulate key signal transduction cascades that control or are involved in the control of a plethora of physiological functions including cellular growth and proliferation, cell differentiation, cellular development, cell division, cell adhesion, stress response, short-range contact-mediated axonal guidance, transcription regulation, aberrant mitogenesis, angiogenesis, abnormal endothelial cell-cell or cell-matrix interactions during vascular development, inflammation, lymphohematopoietic stem cell activity, protective immunity against specific bacteria, allergic asthma, aberrant tissue-specific responses to the activation of the JNK signal transduction pathway, cell transformation, memory, apoptosis, competitive activity-dependent synapse modification at the neuromuscular synapse, immunological mediation of disease, and calcium regulation.
[0005] Specific disease states associated with aberrant regulation of protein kinases include, for example without limitation, acrocephalosyndactyly type I, acute myeloid leukemia, AIDS-induced non-Hodgkin's lymphoma, Alzheimer's disease, amyotrophic lateral sclerosis, arthritis, asthma, atherosclerosis, atopic dermatitis, autoimmune diseases, bacterial infection, bladder cancer, cancer of the breast, cancer of the central nervous system, cancer of the colon, cancer of the endometrium, cancer of the fallopian tube, cancer of the gastrointestinal tract, cancer of the ovary, heart failure, chronic myeloid leukemia, colon carcinoma, colorectal cancer, chronic obstructive pulmonary disease (COPD), Crouzon Syndrome, diabetes, diabetic nephropathy, emphysema, endometriosis, epidermoid
cancer, fibrotic disorders, gastrointestinal stromal tumor (GIST), glomerulonephritis, Graves' disease, head injury, hepatocellular carcinoma, Hirschsprung's disease, human gliomas, immunodeficiency diseases, inflammatory disorders, ischemic stroke, Jackson- Weiss syndrome, leiomyosarcoma, leukemias, lupus nephritis, malignant melanoma, malignant nephrosclerosis, mastocytosis, mast cell tumors, melanoma of the colon, MEN2 syndromes, metabolic disorders, migraine, multiple sclerosis, myeloproliferative disorders, nephritis, neurodegenerative diseases, neurotraumatic diseases, non small cell lung cancer, organ transplant rejection, osteoporosis, pain, Parkinson's disease, Pfeiffer Syndrome, polycystic kidney disease, primary lymphoedema, prostate cancer, psoriasis, vascular restenosis, rheumatoid arthritis, dermal and tissue scarring, selective T-cell defect (STD), severe combined immunodeficiency (SCID), small cell lung cancer, spinal cord injury, squamous cell carcinoma, systemic lupus erythematosis, testicular cancer, thrombotic microangiopathy syndromes, Wegener's granulomatosis, X-linked agammaglobulinemia, viral infection, diabetic retinopathy, alopecia, erectile dysfunction, macular degeneration, chronic lymphocytic leukemia (CLL), myelodysplastic syndrome (MDS), neurofibromatosis, and tuberous sclerosis. Accordingly, there is a need in the art for additional compounds and methods of use thereof for the modulation of receptor protein kinases.
[0006] This application is related to the following published patent applications: WO 2004024895, US 20040142864, WO 2004078923, US 20050170431, WO 2005028624, US 20050164300, and WO 2005062795, each of which are hereby incorporated by reference herein in their entireties including all specifications, figures, and tables, and for all purposes.
SUMMARY OF THE INVENTION
[0007] The present invention concerns compounds active on protein kinases in general, including, but not limited to, AbI, Aktl , Akt2, Akt3, ALK, Alk5, B-Raf, Brk, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, FGFRl, FGFR2, FGFR3, FGFR4, FItI, Flt3, Flt4, Fms, Frk, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kdr, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDGFRA, PDGFRB, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, Plkl, Pyk2, Ret, ROCKl, ROCK2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, and/or Zap70, including any mutations of these kinases, and the use thereof in treating disease and conditions associated with regulation of the activity of the kinase. In particular, the invention concerns compounds of Formula I as described below. Thus, the invention provides novel use of compounds for therapeutic methods involving modulation of protein kinases, as well as novel compounds that can be used for therapeutic methods involving modulation of protein kinases.
[0008] The compounds of Formula I have the following structure:
Formula I all salts, prodrugs, tautomers, and isomers thereof, wherein:
R2, R4, R5, and R6 are independently selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -NO2, -CRaRbR26, and -LR26;
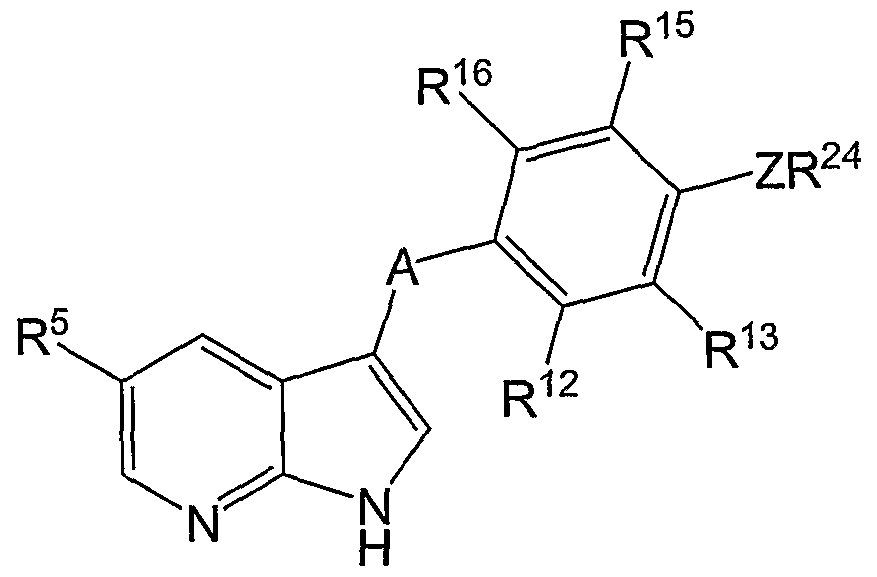
R3 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -NO2, -CRaRbR26, -LR26 and -A-Ar-L1-R24;
A is selected from the group consisting of -O-, -S-, -CRaRb-, -NR1-, -C(O)-, -C(S)-, -S(O)-, and -S(O)2-;
R1 is selected from the group consisting of hydrogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -C(O)R7, -C(S)R7, -S(O)2R7, -C(O)NHR7, -C(S)NHR7, and -S(O)2NHR7, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro, further provided, however, that when R1 is lower alkyl, any substitution on the lower alkyl carbon bound to the N of -NR1- is fluoro, and wherein cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R7 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, lower alklylthio, mono-alkylamino, di-alkylamino, and -KR8R9, provided, however, that any substitution of the alkyl carbon bound to the N of -C(O)NHR7, -C(S)NHR7 or -S(O)2NHR7 is fluoro, wherein the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro, and wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
Ar is selected from the group consisting of optionally substituted arylene and optionally substituted heteroarylene;
L at each occurrence is independently selected from the group consisting of -(alk)a-S-(alk)b-, -(alk)a-O-(alk)b-, -(alk)a-NR25-(alk)b-, -(alk)a~C(O)-(alk)b-, -(alk)a-C(S)-(alk)b-, -(aUc)a-S(O)-(alk)b-, -(alk)a-S(O)2-(alk)b-, -(alk)a-OC(O)-(alk)b-, -(alk)a-C(O)O-(alk)b-, -(alk)a-OC(S)-(alk)b-, -(alk)a-C(S)O-(alk)b-, -(alk)a-C(O)NR25-(alk)b-, -(alk)a-C(S)NR25-(alk)b-, -(alk)a-S(O)2NR25-(alk)b-, -(alk)a-NR25C(O)-(alk)b-, -(alk)a-NR25C(S)-(alk)b-, -(alk)a-NR25S(O)2-(alk)b-, -(alk)a-NR25C(O)O-(alk)b-, -(alk)a-NR25C(S)O-(alk)b-, -(alk)a-OC(O)NR25-(alk)b-, -(alk)a-OC(S)NR25-(alk)b-, -(alk)a-NR25C(O)NR25-(alk)b-, -(alk)a-NR25C(S)NR25-(alk)b-, and -(alk)a-NR25S(O)2NR25-(alk)b-; a and b are independently O or 1 ; alk is Cj-3 alkylene or Q-3 alkylene substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein lower alkyl or the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro;
L1 is -(CRaRb)v- or L, wherein v is 1, 2, or 3;
wherein Ra and Rb at each occurrence are independently selected from the group consisting of hydrogen, fluoro, -OH, -NH2, lower alkyl, lower alkoxy, lower alklylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein the alkyl chain(s) of lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro; or any two of Ra and Rb on the same or different carbons combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl and any others of Ra and Rb are independently selected from the group consisting of hydrogen, fluoro, -OH, -NH2, lower alkyl, lower alkoxy, lower alklylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein the alkyl chain(s) of lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro, and wherein the 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono- alkylamino, di-alkylamino, and cycloalkylamino;
R8 and R9 combine with the nitrogen to which they are attached to form a 5-7 membered heterocycloalkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, and fluoro substituted lower alkylthio;
R25 at each occurrence is independently selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl; and
R24 and R26 at each occurrence are independently selected from the group consisting of hydrogen, provided, however, that hydrogen is not bound to any of S(O), S(O)2, C(O) or C(S) of L or Li , optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R24 or R26 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of L or L1, optionally substituted lower alkynyl, provided, however, that when R24 or R26 is optionally substituted lower alkynyl, no alkyne
carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of L or L1, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl.
[0009] The description above of substituents in Formula I includes descriptions of each combination of the specified substituents, R2, R3, R4, R5, and R6. In some embodiments, at least one of R2, R3, R4, R5 and R6 is other than hydrogen.
[0010] In some embodiments involving compounds of Formula I, R2 and R6 are hydrogen, or R2 and R5 are hydrogen, or R2 and R4 are hydrogen, or R2 and R3 are hydrogen, or R3 and R6 are hydrogen, or R3 and R5 are hydrogen, or R3 and R4 are hydrogen, or R4 and R6 are hydrogen, or R4 and R5 are hydrogen, or R5 and R6 are hydrogen, wherein the substitutions at the other positions are non- hydrogen. In some embodiments, R2, R3 and R4 are hydrogen, or R2, R3 and R5 are hydrogen, or R2, R3 and R6 are hydrogen, or R2, R4 and R5 are hydrogen, or R2, R4 and R6 are hydrogen, or R2, R5 and R6 are hydrogen, or R3, R4 and R5 are hydrogen, or R3, R4 and R6 are hydrogen, or R3, R5 and R6 are hydrogen, or R4, R5 and R6 are hydrogen, wherein the substitutions at the other positions are non- hydrogen. In some embodiments, the compounds are mono-substituted with non-hydrogen at one of R2, R3, R4, R5 or R6 (i.e. hydrogen at the other four positions) . In some embodiments, compounds of Formula I have non-hydrogen substitution at R3; non-hydrogen substitution at R4; non-hydrogen substitution at R5; non-hydrogen substitution at R3 and R4; non-hydrogen substitution at R3 and R5. In some embodiments, the substitutions as listed are the only substitutions; the substitutions as listed are combined with R2 and R6 as H; the substitutions as listed are combined with substitution at one other of the substitution positions shown in Formula I. The compounds of Formula I, and all sub- embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0011] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ia:
Formula Ia all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 is a bond, -CRaRb-> -A-Ar-Lr, or L, and Y5 is a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3 or Y5 is a
bond, or R26, provided, however, that neither of Y3R27 and Y5R27 are hydrogen, wherein Ra, Rb, L, L1, A, Ar and R26 are as defined with reference to Formula I.
[0012] In some embodiments of compounds of Formula Ia, Y3 and Y5 are bonds. In some embodiments, Y3 and Y5 are independently -CRaRb- or L. In some embodiments, Y3 and Y5 are independently L. In some embodiments, Y3 and Ys are independently -CRaRb-. In some embodiments, Y3 is a bond, and Y5 is -CRaRb- or L. In some embodiments, Y3 is a bond, and Y5 is L. In some embodiments, Y3 is a bond, and Y5 is -CRaRb-. In some embodiments, Y5 is a bond, and Y3 is -CRaRb- or L. In some embodiments, Y5 is a bond, and Y3 is L. In some embodiments, Y5 is a bond, and Y3 is -CRaRb-.
[0013] In some embodiments of any of the above embodiments of compounds of Formula Ia, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3 or Ys is a bond. In some embodiments, Y5 is a bond, -CRaRb-, or L, Y3 is -CRaRb-, -O-, -S-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2-, wherein R25 is as defined for Formula I, and each R27 is independently R26 or Y5R27 is halogen; in further embodiments, Y3 is -CRaRb- or -C(O)-; in further embodiments, -CRaRb- is -CH2-. In some embodiments, Y5 is a bond, -CRaRb-, or L, R27 bound to Y5 is R26 or Y5R27 is halogen, Y3 is -CRaRb- or -C(O)-, and R27 bound to Y3 is optionally substituted aryl or optionally substituted heteroaryl.
[0014] In some embodiments of any of the above embodiments of compounds of Formula Ia, Y3R27 is -A-Ar-Li-R24, wherein A, Ar, and Li are as defined for Formula I, and R24 is substituted methyl, optionally substituted C2-6 alkyl, optionally substituted lower alkenyl, provided, however, that when R24 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of Li, optionally substituted lower alkynyl, provided, however, that when R24 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of Li, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, provided, however, that -A-Ar-Li-R24 is not
indicates the point of attachment to the 3 position of the azaindole ring; in further embodiments, R
24 is optionally substituted C
2.β alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or substituted methyl, wherein methyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, when Ar is optionally substituted heteroarylene, the heteroarylene
nng is not a five or six membered ring having the structure
v '
0-' wherein
indicates the point of attachment to A and " indicates the point of attachment to L1, and wherein the indicated N is either =N- or -N=, and wherein each Q is independently a heteroaryl ring atom that may be optionally substituted. The term "heteroaryl ring atom" refers to any atom that can be part of a heteroaryl ring structure (i.e., C, N, O, or S).
[0015] In some embodiments of any of the above embodiments of compounds of Formula Ia, Y3 and Y5 are independently -O-, -S-, -CRaRb-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2-, where Ra, Rb and R25 are as defined in Formula I, and each R27 is independently optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, Y5 is -O-, -NR25-, or -S(O)2-, preferably wherein R25 is hydrogen or lower alkyl, and R27 bound to Y3 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, Y3 is -CRaRb-, or -C(O)-, preferably -CH2- or -C(O)-, Y5 is -O-, -NR25-, or -S(O)2-, preferably -NR25-, wherein R25 is hydrogen or lower alkyl, and each R27 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl,
provided, however, that Y
3R
27 is not
0H , wherein ^ indicates the bond to the 3 position
of the 7-azaindole ring, and Y
5R
27 is not
'
<• « — , wherein indicates the bond to the 5 position of the 7-azaindole ring, i.e. the compound is not (3-hydroxy-phenyl)-(5-phenylamino-lH- pyrrolo[2,3-b]pyridin-3-yl)-methanone; in further embodiments, when R
27 bound to Y
3 is optionally substituted heteroaryl, the heteroaryl ring is not a five or six membered ring having the structure
wherein -I >— indicates the point of attachment to Y
3, and wherein the indicated N is either =N- or -N=, and wherein each Q is independently a heteroaryl ring atom that may be appropriately optionally substituted and wherein R
200 is other than hydrogen. The term "other than hydrogen" and like terms refer to substituents contemplated herein which are not hydrogen. For example without limitation, if substituent R
ex were defined as selected from the group consisting of hydrogen and optionally substituted lower alkyl, then the phrase "R
ex is other than hydrogen" would
contemplate only optionally substituted lower alkyl, i.e., all options of the substituent, excluding hydrogen.
[0016] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ib:
Formula Ib all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 and Y4 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3 or Y4 is a bond, or R26, provided, however, that neither of Y3R27 and Y4R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0017] In some embodiments of compounds of Formula Ib, Y3 and Y4 are bonds. In some embodiments, Y3 and Y4 are independently -CRaRb- or L. In some embodiments, Y3 and Y4 are independently L. In some embodiments, Y3 and Y4 are independently -CRaRb-. In some embodiments, Y3 is a bond and Y4 is -CRaRb- or L. In some embodiments, Y3 is a bond and Y4 is L. In some embodiments, Y3 is a bond and Y4 is -CRaRb-. In some embodiments, Y4 is a bond, and Y3 is -CRaRb- or L. In some embodiments, Y4 is a bond and Y3 is L. In some embodiments, Y4 is a bond and Y3 is -CRaRb-.
[0018] In some embodiments of any of the above embodiments of compounds of Formula Ib, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3 or Y4 is a bond. In some embodiments, Y4 is a bond, -CRaRb-, or L, Y3 is -CRaRb-, -O-, -S-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2-, wherein R25 is as defined for Formula I, and R27 is independently R26 or Y4R27 is halogen; in further embodiments, Y3 is -CRaRb- or -C(O)-; in further embodiments, -CRaRb- is -CH2-. In some embodiments, Y4 is a bond, -CRaRb-, or L, R27 bound to Y4 is R26 or Y4R27 is halogen, Y3 is -CRaRb- or -C(O)-, and R27 bound to Y3 is optionally substituted aryl or optionally substituted heteroaryl.
[0019] In some embodiments of any of the above embodiments of compounds of Formula Ib, Y3 and Y4 are independently -O-, -S-, -CRaRb-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2-, where Ra, Rb and R25 are as defined in Formula I, and each R27 is independently optionally substituted lower alkyl,
optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in a further embodiment Y4 is -O-, -NR25-, or -S(O)2-, preferably wherein R25 is hydrogen or lower alkyl, and R27 bound to Y3 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl; in a further embodiment Y3 is -CRaRb-, or -C(O)-, preferably -CH2- or -C(O)-, Y4 is -O-, -NR25-, or -S(O)2-, preferably -NR25-, wherein R25 is hydrogen or lower alkyl, and each R27 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl.
[0020] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ic:
Formula Ic all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 and Y6 are independently a bond, -CRaRb- , or L, and each R27 is independently halogen, provided that Y3 or Y6 is a bond, or R26, provided, however, that neither of Y3R27 and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0021] In some embodiments of compounds of Formula Ic, Y3 and Y6 are bonds. In some embodiments, Y3 and Y6 are independently -CRaRb- or L. In some embodiments, Y3 and Y6 are independently L. In some embodiments, Y3 and Y6 are independently -CRaRb-. In some embodiments, Y3 is a bond, and Y6 is -CRaRb- or L. hi some embodiments, Y3 is a bond, and Y6 is L. In some embodiments, Y3 is a bond, and Y6 is -CRaRb-. In some embodiments, Y6 is a bond, and Y3 is -CRaRb- or L. In some embodiments, Y6 is a bond, and Y3 is L. hi some embodiments, Y6 is a bond, and Y3 is -CRaRb-.
[0022] hi some embodiments of any of the above embodiments of compounds of Formula Ic, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3 or Y6 is a bond.
[0023] In some embodiments of any of the above embodiments of compounds of Formula Ia, Y3 and Y6 are independently -O-, -S-, -CRaRb-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2-, where Ra, Rb and R25 are as defined in Formula I, and each R27 is independently optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, Y6 is -O-, -NR25-, or -S(O)2-, preferably wherein R25 is hydrogen or lower alkyl, and R27 bound to Y3 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, Y3 is -CRaRb-, or -C(O)-, preferably -CH2- or -C(O)-, Y6 is -O-, -NR25-, or -S(O)2-, preferably -NR25-, wherein R25 is hydrogen or lower alkyl, and each R27 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, preferably optionally substituted aryl, or optionally substituted heteroaryl.
[0024] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Id:
Formula Id all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 and Y2 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3 or Y2 is a bond, or R26, provided, however, that neither of Y2R27 and Y3R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0025] In some embodiments of compounds of Formula Id, Y3 and Y2 are bonds. In some embodiments, Y3 and Y2 are independently -CRaRb- or L. In some embodiments, Y3 and Y2 are independently L. In some embodiments, Y3 and Y2 are independently -CRaRb-. In some embodiments, Y3 is a bond, and Y2 is -CRaRb- or L. In some embodiments, Y3 is a bond, and Y2 is L. In some embodiments, Y3 is a bond, and Y2 is -CRaRb-. In some embodiments, Y2 is a bond, and Y3 is -CRaRb- or L. In some embodiments, Y2 is a bond, and Y3 is L. In some embodiments, Y2 is a bond, and Y3 is -CRaRb-.
[0026] In some embodiments of any of the above embodiments of compounds of Formula Id, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl,
optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3 or Y2 is a bond.
[0027] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ie:
Formula Ie all salts, prodrugs, tautomers, and isomers thereof, wherein Y4 and Y2 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y4 or Y2 is a bond, or R26, provided, however, that neither of Y2R27 and Y4R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0028] In some embodiments of compounds of Formula Ie, Y4 and Y2 are bonds. In some embodiments, Y4 and Y2 are independently -CRaRb- or L. In some embodiments, Y4 and Y2 are independently L. In some embodiments, Y4 and Y2 are independently -CRaRb-. In some embodiments, Y4 is a bond, and Y2 is -CRaRb- or L. In some embodiments, Y4 is a bond, and Y2 is L. In some embodiments, Y4 is a bond, and Y2 is -CRaRb-. In some embodiments, Y2 is a bond and Y4 is -CRaRb- or L. In some embodiments, Y2 is a bond, and Y4 is L. In some embodiments, Y2 is a bond, and Y4 Is -CR3R13-.
[0029] In some embodiments of any of the above embodiments of compounds of Formula Id, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y4 or Y2 is a bond.
[0030] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula If:
Formula If
all salts, prodrugs, tautomers, and isomers thereof, wherein Y5 and Y2 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y5 or Y2 is a bond, or R26, provided, however, that neither of Y2R27 and Y5R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0031] In some embodiments of compounds of Formula If, Y5 and Y2 are bonds. In some embodiments, Y5 and Y2 are independently -CRaRb- or L. In some embodiments, Y5 and Y2 are independently L. In some embodiments, Y5 and Y2 are independently -CRaRb-. In some embodiments, Y5 is a bond, and Y2 is -CRaRb- or L. In some embodiments, Y5 is a bond, and Y2 is L. In some embodiments, Y5 is a bond, and Y2 is -CRaRb-. In some embodiments, Y2 is a bond, and Y5 is -CRaRb- or L. In some embodiments, Y2 is a bond, and Y5 is L. In some embodiments, Y2 is a bond, and Y5 is -CRaRb-.
[0032] In some embodiments of any of the above embodiments of compounds of Formula If, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y5 or Y2 is a bond.
[0033] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ig:
Formula Ig all salts, prodrugs, tautomers, and isomers thereof, wherein Y6 and Y2 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y6 or Y2 is a bond, or R26, provided, however, that neither of Y2R27 and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0034] In some embodiments of compounds of Formula Ig, Y6 and Y2 are bonds. In some embodiments, Y6 and Y2 are independently -CRaRb- or L. In some embodiments, Y6 and Y2 are independently L. In some embodiments, Y6 and Y2 are independently -CRaRb-. In some embodiments, Y6 is a bond, and Y2 is -CRaRb- or L. m some embodiments, Y6 is a bond, and Y2 is L. In some embodiments, Y6 is a bond, and Y2 is -CRaRb-. In some embodiments, Y2 is a bond, and Y6 is -CRaRb- or L. In some embodiments, Y2 is a bond, and Y6 is L. In some embodiments, Y2 is a bond, and Y6 is -CRaRb-.
[0035] In some embodiments of any of the above embodiments of compounds of Formula Ig, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y6 or Y2 is a bond.
[0036] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ih:
all salts, prodrugs, tautomers, and isomers thereof, wherein Y and Y are independently a bond, -CR
aR
b-, or L, and each R
27 is independently halogen, provided that Y
6 or Y
4 is a bond, or R
26, provided, however, that neither of Y
4R
27 and Y
6R
27 are hydrogen, wherein R
a, R
b, L and R
26 are as defined with reference to Formula I.
[0037] In some embodiments of compounds of Formula Ih, Y6 and Y4 are bonds. In some embodiments, Y6 and Y4 are independently -CRaRb- or L. In some embodiments, Y6 and Y4 are independently L. In some embodiments, Y6 and Y4 are independently -CRaRb-. In some embodiments, Y6 is a bond, and Y4 is -CRaRb- or L. In some embodiments, Y6 is a bond, and Y4 is L. In some embodiments, Y6 is a bond, and Y4 is -CRaRb-. In some embodiments, Y4 is a bond and Y6 is -CRaRb- or L. In some embodiments, Y4 is a bond, and Y6 is L. In some embodiments, Y4 is a bond, and Y6 is -CRaRb-.
[0038] In some embodiments of any of the above embodiments of compounds of Formula Ih, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y6 or Y4 is a bond.
[0039] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ii:
all salts, prodrugs, tautomers, and isomers thereof, wherein Y
6 and Y
5 are independently a bond, -CR
aR
b-, or L, and each R
27 is independently halogen, provided that Y
6 or Y
5 is a bond, or R
26, provided, however, that neither of Y
5R
27 and Y
6R
27 are hydrogen, wherein R
a, R
b, L and R
26 are as defined with reference to Formula I.
[0040] In some embodiments of compounds of Formula Ii, Y6 and Y5 are bonds. In some embodiments, Y6 and Y5 are independently -CRaRb- or L. In some embodiments, Y6 and Y5 are independently L. In some embodiments, Y6 and Y5 are independently -CRaRb-. In some embodiments, Y6 is a bond, and Y5 is -CRaRb- or L. In some embodiments, Y6 is a bond, and Y5 is L. In some embodiments, Y6 is a bond, and Y5 is -CRaRb-. In some embodiments, Y5 is a bond, and Y6 is -CRaRb- or L. In some embodiments, Y5 is a bond, and Y6 is L. In some embodiments, Y5 is a bond, and Y6 is -CRaRb-.
[0041] In some embodiments of any of the above embodiments of compounds of Formula Ii, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y6 or Y5 is a bond.
[0042] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ij:
Formula Ij all salts, prodrugs, tautomers, and isomers thereof, wherein Y4 and Y5 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y4 or Y5 is a bond, or R26, provided, however, that neither of Y4R27 and Y5R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0043] In some embodiments of compounds of Formula Ij, Y4 and Y5 are bonds. LQ some embodiments, Y4 and Y5 are independently -CRaRb- or L. In some embodiments, Y4 and Y5 are independently L. In some embodiments, Y4 and Y5 are independently -CRaRb-. In some embodiments, Y4 is a bond, and Y5 is -CRaRb- or L. In some embodiments, Y4 is a bond, and Y5 is L. In some embodiments, Y4 is a bond, and Y5 is -CRaRb-. In some embodiments, Y5 is a bond, and Y4 is
-CRaRb- or L. In some embodiments, Y5 is a bond, and Y4 is L. La some embodiments, Y5 is a bond, and Y4 is -CRaRb-.
[0044] In some embodiments of any of the above embodiments of compounds of Formula Ij , each
R is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y4 or Y5 is a bond.
[0045] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula Dc:
Formula Ik all salts, prodrugs, tautomers, and isomers thereof, wherein R26 is as defined in Formula I, and Y is selected from the group consisting of a bond, -CRaRb- and L, where Ra, Rb and L are as defined with reference to Formula I, provided that YR26 is not hydrogen.
[0046] In some embodiments of compounds of Formula Ik, Y is -(alk)a-S-(alk)b-, -(alk)a-O-(alk)b-, -(alk)a-OC(O)-(alk)b-, -(alk)a-C(O)O-(alk)b-, -(alk)a-OC(S)-(alk)b-, -(alk)a-C(S)O-(alk)b-, -(alk)a-C(O)-(alk)b-, -(alk)a-C(S)-(alk)b-, -(alk)a-C(O)NR25-(alk)b-, -(alk)a-OC(O)NR25-(alk)b-, -(alk)a-OC(S)NR25-(alk)b-, -(alk)a-C(S)NR2S-(alk)b-, -(alk)a-S(O)-(alk)b-, -(alk)a-S(O)2-(alk)b-, -(alk)a-S(O)2NR25-(alk)b-, -(alk)a-NR25-(alk)b-, -(alk)a-NR25C(O)-(alk)b-, -(alk)a-NR25C(S)-(alk)b-, -(alk)a-NR25C(O)NR25-(alk)b-, -(alk)a-NR25C(S)NR25-(alk)b-, -(alk)a-NR25C(O)O-(alk)b-, -(alk)a-NR25C(S)O-(alk)b-, -(alk)a-NR25S(O)2-(alk)b-, or -(alk)a-NR25S(O)2NR25-(alk)b-, wherein alk, a, b, and R25 are as defined for Formula I.
[0047] In some embodiments of compounds of Formula Dc, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alky, -C(0)O(alk)b-, -OC(S>(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0048] In some embodiments of compounds of Formula Dc, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -OC(S)-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-,
S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2TSπR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0049] In some embodiments of compounds of Formula Dc, R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -O-, -S-, -NR25-, -C(O)-, -C(S)-, -S(O)-, or -S(O)2- and R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -NR25-, preferably wherein R25 is hydrogen or lower alkyl, preferably wherein Y is -NH-; in further embodiments, R26 is optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, lower alkyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, heteroaryl is monocyclic. In some embodiments, Y is -NH-; in further embodiments, R26 is substituted phenyl or optionally substituted heteroaryl, provided that heteroaryl is monocyclic.
[0050] In some embodiments of any of the above embodiments of compounds of Formula Ik, YR26 is not optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -C s€CH2N(CH3)2, -C(O)H, -CH2N(CH3)2ϊ -C(O)OCH3,
-CH
2OH, -OH, -OCH
3, -NHCH
2CH=CH
2, -NHCH
3, -NHCH
2CH
3, -NHphenyl,
,
indicates the bond attached to the 5-position of the 7-azaindole ring; and when Y is -O- or -NR
25-, then R
26 is not
, which is optionally substituted, wherein
? indicates the bond attached to Y; and
when Y is -O-, then R
26 is not
w wVhiiircVhh i iss n onpttiinonnaailllvy s sπuHbssttiittiuitteedd,.
■ wwhheerreeiinn ^ ^ i innrdiiircates the bond attached Y.
[0051] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula Im:
Formula Im all salts, prodrugs, tautomers, and isomers thereof, wherein R26 and Y are as defined for Formula Ik.
[0052] In some embodiments of compounds of Formula Im, Y is -OC(O)-(alk)b-, -C(O)O-(alk)b-, -OC(S)-(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, S(O)2NR25-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0053] In other embodiments of compounds of Formula Im, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -C(O)O-(alk)b-, -OC(S)-(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0054] In other embodiments of compounds of Formula Im, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -OC(S)-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR2SC(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0055] In some embodiments of compounds of Formula Im, R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -O-, -S-, -NR25-, -C(O)-, -C(S)-; -S(O)-, or -S(O)2- and R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -NR25-, preferably wherein R25 is hydrogen or lower alkyl, preferably wherein Y is -NH-; in further embodiments, R26 is optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, lower alkyl is
substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, heteroaryl is monocyclic.
[0056] In some embodiments of any of the above embodiments of compounds of Formula Im, when Y is -CH2NH-, then R26 is not optionally substituted thiophene or optionally substituted pyridine; when Y is -O- or -NH-, then R26 is not optionally substituted bicyclic heteroaryl; when Y is -O-, then R26 is not optionally substituted phenyl; when Y is -NH- or -N(CH3)- and R26 is substituted phenyl, the phenyl is not substituted by halogen ortho to Y and optionally substituted amine para to Y, Y is not -NH-C(O)- or -C(O)-NH-, and YR26 is not optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted -CH=CH2, -C(O)OH, -C(O)OCH3, -C(O)OtBu, -OH, -OCH3, -NHCH2CH=CH2, -N(CH3)2, -NH2,
-NHCH
2C(O)OCH
2CH
3, -N(CH
3)phenyl,
indicates the bond attached to the 4-position of the 7- azaindole ring.
[0057] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula In:
Formula In all salts, prodrugs, tautomers, and isomers thereof, wherein R26 and Y are as defined for Formula Ik.
[0058] In other embodiments of compounds of Formula In, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -C(O)O-(alk)b-, -OC(S)-(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR2S-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-,
-NR25C(O)NR2s-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0059] In other embodiments of compounds of Formula In, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -OC(S)-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0060] In some embodiments of compounds of Formula In, R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -NR25-, preferably wherein R25 is hydrogen or lower alkyl, preferably wherein Y is -NH-; in further embodiments, R26 is optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, lower alkyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl.
[0061] In some embodiments of any of the above embodiments of compounds of Formula In,
compounds are excluded where Y is -O- and R
26 is optionally substituted ^ ' , wherein ^
indicates the bond attached to Y; and where YR is -NH
2, -CH
3, -OC(O)phenyl,
indicates the bond attached to the 6-position of the
7-azaindole ring.
[0062] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula Io:
Formula Io all salts, prodrugs, tautomers, and isomers thereof, wherein R26 and Y are as defined for Formula Ik.
[0063] In other embodiments of compounds of Formula Io, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-CaIkV, -CCO)O-CaIkV, -OC(S)-(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR2S-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0064] In other embodiments of compounds of Formula Io, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -OC(S)-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0065] In some embodiments of compounds of Formula Io, R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -NR25-, preferably wherein R25 is hydrogen or lower alkyl, preferably wherein Y is -NH-; in further embodiments, R26 is optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, lower alkyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl.
[0066] In some embodiments of any of the above embodiments of compounds of Formula Io, compounds are excluded when -YR26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CH2NR5R", wherein NR'R" is optionally substituted heterocycloalkyl or optionally substituted heteroaryl, -C(O)NR'R", wherein NR'R" is optionally substituted heterocycloalkyl or optionally substituted heteroaryl, or R' is H and R" is optionally substituted cycloalkyl, optionally substituted aryl or optionally substituted heteroaryl, optionally substituted -CH=CH2, -CH3, -CH2CH2NHCH3, -CH2CH(NH2)C(O)OH,
-CH2CH(NH2)C(O)OCH3, -CH2CH(C(O)OH)NHCH3, -C(O)C(O)OCH3, -CH2C(O)OCH2CH3, -CH2C(O)NH2, -CH2CN, -NH2, -N(CH3)2, -SCH3, -N=C(CH3)NHOAc, -C(O)CCl3, -C(O)OCH3, -C(O)CH2Br, -C(O)NH2, -C(S)NH2, -CH2NH-thiophene, wherein thiophene is optionally substituted,
D
2 and D
3,
wherein the ring is optionally substituted at Di, D
2 and D
3,
when R is H, 4-F, 4-CF
3 or 3-F, or
when R' is OCH
3 or CH
3 and R is CF
3, or when R' is CH
3 and R
is Cl, wherein ^ indicates the bond attached to the 3-position of the 7-azaindole ring.
[0067] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula Ip:
Formula Ip all salts, prodrugs, tautomers, and isomers thereof, wherein R26 and Y are as defined for Formula Ik.
[0068] In other embodiments of compounds of Formula Ip, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-(alk)b-, -C(O)O-(alk)b-, -OC(S)-(alk)b-, -C(S)O-(alk)b-, -C(O)-(alk)b-, -C(S)-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-5
-NR25C(O)NR25-(alk)b-, -NR25C(S)NR2S-(alk)b-, -NR2sC(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0069] In other embodiments of compounds of Formula Ip, Y is -S-(alk)b-, -O-(alk)b-, -OC(O)-CaIkV, -OC(S)-CaIkV, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -S(O)-(alk)b-, -S(O)2-(alk)b-, -S(O)2NR25-(alk)b-, -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR2s-(alk)b-, wherein alk, b and R25 are as defined for Formula I.
[0070] In some embodiments of compounds of Formula Ip, R26 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, Y is -NR25-, preferably wherein R25 is hydrogen or lower alkyl, preferably wherein Y is -NH-; in further embodiments, R26 is optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl; in further embodiments, lower alkyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl.
[0071] In some embodiments of any of the above embodiments of compounds of Formula Ip above, compounds are excluded when YR26 is -CH3, optionally substituted aryl, optionally substituted
indicates the bond attached to the 2-position of the 7-azaindole ring.
[0072] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iq:
Formula Iq all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y3 and Y4 are independently a bond, -CRaRb- or L, and each R27 is independently halogen, provided that Y2, Y3 or Y4 is a bond, or R26
provided, however, that none of Y2R27, Y3R27, and Y4R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0073] In some embodiments of compounds of Formula Iq, Y2, Y3 and Y4 are bonds. In some embodiments, Y2, Y3 and Y4 are independently -CRaRb- or L. In some embodiments, Y2, Y3 and Y4 are independently L. In some embodiments, one of Y2, Y3 and Y4 is a bond, and the others are independently -CRaRb- or L. In some embodiments, one of Y2, Y3 and Y4 is a bond, and the others are independently L. In some embodiments, two of Y2, Y3 and Y4 are bonds, and the other is -CRaRb- or L. In some embodiments, two of Y2, Y3 and Y4 are bonds and the other is L.
[0074] In some embodiments of any of the above embodiments of compounds of Formula Iq, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y3 or Y4 is a bond.
[0075] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ir:
Formula Ir all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y3 and Y5 are independently a bond, -CRaRb- or L, and each R27 is independently halogen, provided that Y2, Y3 or Y5 is a bond, or R26, provided, however, that none of Y2R27, Y3R27, and Y5R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0076] In some embodiments of compounds of Formula Ir, Y2, Y3 and Y5 are bonds. In some embodiments, Y2, Y3 and Y5 are independently -CRaRb- or L. In some embodiments, Y2, Y3 and Y5 are independently L. In some embodiments, any one of Y2, Y3 and Y5 is a bond, and the remaining of Y2, Y3 and Y5 are independently -CRaRb- or L. In some embodiments, any one of Y2, Y3 and Y5 is a bond, and the remaining of Y2, Y3 and Y5 are independently L. In some embodiments, any two of Y2, Y3 and Y5 are bonds, and the remaining of Y2, Y3 and Y5 is -CRaRb- or L. In some embodiments, any two of Y2, Y3 and Y5 are bonds, and the remaining of Y2, Y3 and Y5 is L.
[0077] In some embodiments of any of the above embodiments of compounds of Formula Ir, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl,
optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y3 or Y5 is a bond.
[0078] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Is:
Formula Is all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y3 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y2, Y3 or Y6 is a bond, or R26, provided, however, that none of Y2R27, Y3R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0079] In some embodiments of compounds of Formula Is, Y2, Y3 and Y6 are bonds. In some embodiments, Y2, Y3 and Y6 are independently -CRaRb- or L. In some embodiments, Y2, Y3 and Y6 are independently L. In some embodiments, any one of Y2, Y3 and Y6 is a bond, and the remaining of Y2, Y3 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y2, Y3 and Y6 is a bond, and the remaining of Y2, Y3 and Y6 are independently L. In some embodiments, any two of Y2, Y3 and Y6 are bonds, and the remaining of Y2, Y3 and Y6 is -CRaRb- or L. In some embodiments, any two ofY2, Y3 and Y6 are bonds, and the remaining of Y2, Y3 and Y6 is L.
[0080] In some embodiments of any of the above embodiments of compounds of Formula Is, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y3 or Y6 is a bond.
[0081] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula It:
Formula It
all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y4 and Y5 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y2, Y4 or Y5 is a bond, or R26, provided, however, that none of Y2R27, Y4R27, and Y5R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0082] In some embodiments of compounds of Formula It, Y2, Y4 and Y5 are bonds. In some embodiments, Y2, Y4 and Y5 are independently -CRaRb- or L. In some embodiments, Y2, Y4 and Y5 are independently L. In some embodiments, any one of Y2, Y4 and Y5 is a bond, and the remaining of Y2, Y4 and Y5 are independently -CRaRb- or L. hi some embodiments, any one of Y2, Y4 and Y5 is a bond, and the remaining of Y2, Y4 and Y5 are independently L. In some embodiments, any two of Y2, Y4 and Y5 are bonds, and the remaining of Y2, Y4 and Y5 is -CRaRb- or L. In some embodiments, any two of Y2, Y4 and Y5 are bonds, and the remaining of Y2, Y4 and Y5 is L.
[0083] In some embodiments of any of the above embodiments of compounds of Formula It, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y4 or Y5 is a bond.
[0084] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iu:
Formula Iu all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y4 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y2, Y4 or Y6 is a bond, or R26, provided, however, that none of Y2R27, Y4R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0085] In some embodiments of compounds of Formula Iu, Y2, Y4 and Y6 are bonds. In some embodiments, Y2, Y4 and Y6 are independently -CRaRb- or L. In some embodiments, Y2, Y4 and Y6 are independently L. In some embodiments, any one of Y2, Y4 and Y6 is a bond, and the remaining of Y2, Y4 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y2, Y4 and Y6 is a bond, and the remaining of Y2, Y4 and Y6 are independently L. In some embodiments, any two of Y2,
Y4 and Y6 are bonds, and the remaining of Y2, Y4 and Y6 is -CRaRb- or L. In some embodiments, any two ofY2, Y4 and Y6 are bonds, and the remaining of Y2, Y4 and Y6 is L.
[0086] In some embodiments of any of the above embodiments of compounds of Formula Iu, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y4 or Y6 is a bond.
[0087] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iv:
Formula Iv all salts, prodrugs, tautomers, and isomers thereof, wherein Y2 Y5 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y2, Y5 or Y6 is a bond, or R26, provided, however, that none of Y2R27, Y5R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0088] In some embodiments of compounds of Formula Iv, Y2, Y5 and Y6 are bonds. In some embodiments, Y2, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, Y2, Y5 and Y6 are independently L. In some embodiments, any one of Y2, Y5 and Y6 is a bond, and the remaining of Y2, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y2, Y5 and Y6 is a bond, and the remaining of Y2, Y5 and Y6 are independently L. In some embodiments, any two of Y2, Y5 and Y6 are bonds, and the remaining of Y2, Y5 and Y6 is -CRaRb- or L. In some embodiments, any two ofY2, Y5 and Y6 are bonds, and the remaining of Y2, Y5 and Y6 is L.
[0089] In some embodiments of any of the above embodiments of compounds of Formula Iv, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y2, Y5 or Y6 is a bond.
[0090] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iw:
Formula Iw all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 Y4 and Y5 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3, Y4 or Y5 is a bond, or R26, provided, however, that none of Y3R27, Y4R27, and Y5R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0091] In some embodiments of compounds of Formula Iw, Y3, Y4 and Y5 are bonds. In some embodiments, Y3, Y4 and Y5 are independently -CRaRb- or L. In some embodiments, Y3, Y4 and Y5 are independently L. In some embodiments, any one of Y3, Y4 and Y5 is a bond, and the remaining of Y3, Y4 and Y5 are independently -CRaRb- or L. In some embodiments, any one of Y3, Y4 and Y5 is a bond, and the remaining of Y3, Y4 and Y5 are independently L. In some embodiments, any two of Y3, Y4 and Y5 are bonds, and the remaining of Y3, Y4 and Y5 is -CRaRb- or L. In some embodiments, any two ofY3, Y4 and Y5 are bonds, and the remaining of Y3, Y4 and Y5 is L.
[0092] In some embodiments of any of the above embodiments of compounds of Formula Iw, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3, Y4 or Y5 is a bond.
[0093] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Ix:
Formula Ix all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 Y4 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3, Y4 or Y6 is a bond, or R26, provided, however, that none of Y3R27, Y4R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0094] In some embodiments of compounds of Formula Ix, Y3, Y4 and Y6 are bonds. In some embodiments, Y3, Y4 and Y6 are independently -CRaRb- or L. In some embodiments, Y3, Y4 and Y6 are independently L. In some embodiments, any one of Y3, Y4 and Y6 is a bond, and the remaining of Y3, Y4 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y3, Y4 and Y6 is a bond, and the remaining of Y3, Y4 and Y6 are independently L. In some embodiments, any two of Y3, Y4 and Y6 are bonds, and the remaining of Y3, Y4 and Y6 is -CRaRb- or L. In some embodiments, any two ofY3, Y4 and Y6 are bonds, and the remaining of Y3, Y4 and Y6 is L.
[0095] In some embodiments of any of the above embodiments of compounds of Formula Ix, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3, Y4 or Y6 is a bond.
[0096] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iy:
Formula Iy all salts, prodrugs, tautomers, and isomers thereof, wherein Y3 Y5 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y3, Y5 or Y6 is a bond, or R26, provided, however, that none of Y3R27, Y5R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0097] In some embodiments of compounds of Formula Iy, Y3, Y5 and Y6 are bonds. In some embodiments, Y3, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, Y3, Y5 and Y6 are independently L. In some embodiments, any one of Y3, Y5 and Y6 is a bond, and the remaining of Y3, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y3, Y5 and Y6 is a bond, and the remaining of Y3, Y5 and Y6 are independently L. In some embodiments, any two of Y3, Y5 and Y6 are bonds, and the remaining of Y3, Y5 and Y6 is -CRaRb- or L. In some embodiments, any two ofY3, Y5 and Y6 are bonds, and the remaining of Y3, Y5 and Y6 is L.
[0098] In some embodiments of any of the above embodiments of compounds of Formula Iy, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl,
optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y3, Y5 or Y6 is a bond.
[0099] In some embodiments, the compound of Formula I has a structure according to the following sub-generic structure Formula Iz:
Formula Iz all salts, prodrugs, tautomers, and isomers thereof, wherein Y4 Y5 and Y6 are independently a bond, -CRaRb-, or L, and each R27 is independently halogen, provided that Y4, Y5 or Y6 is a bond, or R26, provided, however, that none of Y4R27, Y5R27, and Y6R27 are hydrogen, wherein Ra, Rb, L and R26 are as defined with reference to Formula I.
[0100] In some embodiments of compounds of Formula Iz, Y4, Y5 and Y6 are bonds. In some embodiments, Y4, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, Y4, Y5 and Y6 are independently L. In some embodiments, any one of Y4, Y5 and Y6 is a bond, and the remaining of Y4, Y5 and Y6 are independently -CRaRb- or L. In some embodiments, any one of Y4, Y5 and Y6 is a bond, and the remaining of Y4, Y5 and Y6 are independently L. In some embodiments, any two of Y4, Y5 and Y6 are bonds, and the remaining of Y4, Y5 and Y6 is -CRaRb- or L. Ih some embodiments, any two of Y4, Y5 and Y6 are bonds, and the remaining of Y4, Y5 and Y6 is L.
[0101] In some embodiments, of any of the above embodiments of compounds of Formula Iz, each R27 is independently optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R27 is halogen, provided that Y4, Y5 or Y6 is a bond.
[0102] The compounds of Formulae Ia-Iz, and all sub-embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0103] In some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula II:
Formula II all salts, prodrugs, tautomers and isomers thereof, wherein:
A, R4, R5 and R6 are as defined with reference to Formula I; and
R12, R13, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -NO2, -CRaRbR24, and -LR24, where L and R24 are as defined for Formula I.
[0104] In some embodiments of compounds of Formula π, at least one of R12, R13, R14, R15 and R16 is other than hydrogen. In some embodiments, at least two of R12, R13, R14, R15 and R16 are other than hydrogen. In some embodiments, at least three of R12, R13, R14, R15 and R16 are other than hydrogen. In some embodiments, at least four of R12, R13, R14, R15 and R16 are other than hydrogen. In some embodiments, R12, R13, R14, R15 and R16 are other than hydrogen.
[0105] In some embodiments of compounds of Formula π, one of R12, R13, R14, R15 and R16 is other than hydrogen, and the others of R12, R13, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fiuoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 is other than hydrogen and R13, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 is other than hydrogen, and R12, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R14 is other than hydrogen, and R12, R13, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0106] In some embodiments of compounds of Formula II, any two of R12, R13, R14, R15 and Ri6 are independently other than hydrogen, and the others of R12, R13, R14, R15 and R16 are each independently
hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R13 are each independently other than hydrogen, and R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R14 are each independently other than hydrogen, and R13, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R15 are each independently other than hydrogen, and R13, RM and Rlδ are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R16 are each independently other than hydrogen, and R13, R14 and R15 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, Ri3 and R14 are each independently other than hydrogen, and R12, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 and R15 are each independently other than hydrogen, and R12, R14 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0107] In some embodiments of any of the above embodiments of compounds of Formula II, wherein any of R12, R13, R14, R15, and R16 is designated as other than hydrogen, each such R12, R13, R14, R15, or R16 is independently -LR24.
[0108] In some embodiments of any of the above embodiments of compounds of Formula II, R5 is other than hydrogen, and R4 and R6 are hydrogen, or R4 is other than hydrogen, and R5 and R6 are hydrogen, or R6 is other than hydrogen, and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen, and R6 is hydrogen, or R4 and R6 are other than hydrogen, and R5 is hydrogen, or R5 and R6 are other than hydrogen, and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0109] In some embodiments of compounds of Formula II, the following compounds are excluded: R4, R5, R6, R12, R13, R14, R15 and R16 are hydrogen and A is -CH2-, -S- or -S(O)2-; R4, R6, R12, R13, R14, R15 and R16 are hydrogen, R5 is -Br or thiophen-3-yl, and A is -C(O)-;
R4 is 3, 5 di-fluorophenyl, -NH2, or -NO2, R5, R6, R12, R13, R14, R15 and R16 are hydrogen, and A is -C(O)-;
R4 is NO2, R5 is Br, R12, R13, R14, R15 and R16 are hydrogen, and A is -C(O)-;
R5 is -Br, R4, R6, R12, R13, R14, R15 and R16 are hydrogen, and A is -S(O)-;
R12 is -CH3 or -F, R6, R13, R14, R15 and R16 are hydrogen, and A is -C(O)-;
R12 is -OH, R4, R6, R13, R14, R15 and R16 are hydrogen, R5 is thiophen-2-yl, and A is -C(O)-;
R12 is -CF3, R4, R5, R6, R13, R14, R15 and R16 are hydrogen, and A is -S(O)2-;
R13 is -OH or -OCH3, R4, R6, R12, R14, R15 and R16 are hydrogen, and A is -CH2-;
R13 is -OCH3, R5, R6, R12, R14, R15 and R16 are hydrogen, R4 is -Br, and A is -CH2-;
R13 is -OH or -OCH3, R4, R6, R12, R14, R15 and R16 are hydrogen, R5 is thiophen-2-yl, and A is -CHOH-;
R
13 is
indicates the bond to the phenyl ring, R
4, R
6, R
12, R
14,
R15 and R16 are hydrogen, R5 is thiophen-3-yl, and A is -CH2-;
R13 is -F, -OH or -OCH3, R6, R12, R14, R15 and R16 are hydrogen, and A is -C(O)-;
R is -NO
2, -NH
2, or
1^ , wherein
*> indicates the bond to the phenyl ring, R
4, R
5, R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -C(O)-; R
13 is -F, -Cl, or -CF
3, R
4, R
5, R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -S(O)
2-; R
13 and R
14 are -OH, R
4, R
6, R
12, R
15 and R
16 are hydrogen, R
5 is thiophen-3-yl, and A is -CH
2-;
R14 is -OH or -OCH3, R4, R6, R12, R13, R15 and R16 are hydrogen, R5 is thiophen-2-yl, and A is -C(O)-;
R14 is -OCH3, R4, R5, R6, R12, R13, R15 and R16 are hydrogen, and A is -C(O)-; R14 is -Cl, R6, R12, R13, R15 and R16 are hydrogen, and A is -C(O)-;
R
14 is
indicates the bond to the phenyl ring, R
4, R
5,
R6, R12, R13, R15 and R16 are hydrogen, and A is -CH2-;
R14 is -F, R4, R5, R6, R12, R13, R15 and R16 are hydrogen, and A is -S- or -S(O)2-;
R14 is -CH3, R5, R6, R12, R13, R15 and R16 are hydrogen, R4 is 3-(hydroxymethyl)phenyl, and A is -S-;
R12 and R16 are -F3 R5, R6, R13, R14 and R15 are hydrogen, R4 is 3,5 difluorophenyl, and A is -C(O)-;
R12 is -Cl, R13 is -Cl, R6, R14, R15 and R16 are hydrogen, and A is -C(O)-;
R12 is -F, R13 is -F, R5, R6, R14, R15 and R16 are hydrogen, R4 is 3,5 difluorophenyl, and A is -C(O)-;
R12 is -F, R13 is -OH or -OCH3, R4, R6, R14, RIS and R16 are hydrogen, R5 is thiophen-2-yl, and A is -C(O)-;
R12 is -F, R13 is -OCH3, R4, R6, R14, R15 and R16 are hydrogen, R5 is -Br, and A is -C(O)-;
R12 and R14 are -F, R5, R6, R13, R15 and R16 are hydrogen, R4 is 3,5 difluorophenyl, and A is -C(O)-;
R12 is -CH3, R15 is -F, R6, R13, R14 and R16 are hydrogen and A is -C(O)-;
R12 is -F, R15 is -Cl, R4, R6, R13, R14 and R16 are hydrogen, R5 is thiophen-2-yl and A is -C(O)-;
R12 and R15 are -F, R6, R13, R14 and R16 are hydrogen, and A is -C(O)-;
R12 is halogen, R15 is -OH or -OCH3, R6, R13, R14 and R16 are hydrogen and A is -C(O)-;
R12 is -F, R15 is -NHS(O)2CH3, R4, R5, R6, R13, R14 and R16 are hydrogen and A is -C(O)-;
R13 and R15 are -OCH3, R4, R5, R6, R12, R14 and R16 are hydrogen, and A is -CH2-;
R13 and R15 are -Cl, R4, R5, R6, R12, R14 and R16 are hydrogen, and A is -S(O)2-;
R13 is -OH and R15 is -OH or -OCH3, R4, R6, R12, R14 and R16 are hydrogen, R5 is thiophen-3-yl, and A is -CH2-;
, or wherein "> indicates the bond to the 5- position of the 7-azaindole ring;
wherein ' indicates the bond to the 4-position of the 7-azaindole
R
4, R
5, R
12, R
13, R
14, R
IS and R
16 are hydrogen, A is -S(O)
2-, and R
6 is
, or
wherein
s indicates the bond to the 6-position of the 7-azaindole ring;
R
13 is -CN, R
4, R
6, R
12, R
14, R
15 and R
16 are hydrogen, A is -S(O)
2-, and R
5 is
wherein
*> indicates the bond to the 5-position of the 7-azaindole ring; R
12 is -Cl, R
14 is -F or hydrogen, R
4, R
6, R
13, R
15 and R
16 are hydrogen, A is -S(O)
2-, and R
5 is
^ ^ ^ wherein -I $— indicates the bond to the 5-position of the 7-azaindole
R
14 is -NH
2, R
4, R
6, R
12, R
13, R
15 and R
16 are hydrogen, A is -S(O)
2-, and R
5 is
wherein ^ indicates the bond to the 5-position of the 7-azaindole ring;
R
6, R
12, R
13, R
14, R
15 and R
16 are hydrogen, A is -S(O)
2-, R
4 is -Cl, and R
5 is
wherein
' indicates the bond to the 5-position of the 7-azaindole ring; R13 is -F, R4, R6, R12, R14, R15 and R16 are hydrogen, A is -CH2-, and R5 is 3-hydroxy-phenyl;
R14 is -N(CH3)2, R4, R6, R12, R13, R15 and R16 are hydrogen, A is -CH2-, and R5 is 3-hydroxy-phenyl;
R14 is hydrogen or -Br, R4, R6, R12, R13, R15 and R16 are hydrogen, A is -S-, and R5 is 3-hydroxy-phenyl; and
R4, R6, R!2, R13, R14, R15 and R16 are hydrogen, A is -C(O)-, and R5 is 3-hydroxy-phenyl.
[0110] In some embodiments of compounds of Formula II, at least one of R
12, R
13, R
14, R
15 and R
16 is -LR
24, wherein R
24 is substituted methyl, optionally substituted C
2-6 alkyl, optionally substituted lower alkenyl, provided, however, that when R
24 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)
2, C(O) or C(S) of L, optionally substituted lower alkynyl, provided, however, that when R
24 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)
2, C(O) or C(S) of L, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted
heteroaryl, provided, however, that R
13 is not
that R is not
, wherein ' indicates the bond to the phenyl ring; in further embodiments, R
24 is optionally substituted C
2-6 alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or substituted methyl, wherein methyl is substituted with optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, R
5 is other than hydrogen; in further embodiments, R
4 and R
6 are hydrogen.
[0111] In some embodiments of compounds of Formula II, R12 is other than hydrogen. In some embodiments, R12 is -LR24. In some embodiments, R12 is other than hydrogen and R13, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 is -LR24 and R13, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 is -LR24, any three of R13, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R14, R15 and R16 is hydrogen. In some embodiments, R12 is -LR24, any two of R13, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R14, R15 and R16 are hydrogen. In some embodiments, R12 is -LR24, any one of R13, R14, R15 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R14, R15 and R16 are hydrogen. In some embodiments, R12 is -LR24, and R13, R14, R15 and R16 are hydrogen. In some embodiments, R12 is other than hydrogen, and R13, R14, RIS and R16 are hydrogen.
[0112] In other embodiments of compounds of Formula II, wherein R12 is other than hydrogen, when R13, R14, R15 and R16 are hydrogen and A is -C(O)-, then R12 is not -CH3, -F, or -OH; and when R4, R5, R6, R13, R14, R15 and R16 are hydrogen, and A is -S(O)2-, then R12 is not -CF3; and when R12 and R16 are :F, R5, R6, R13, R14 and R15 are hydrogen, and A is -C(O)-, R4 is not 3,5 difluorophenyl; and when R12 is halogen, R6, R14, R15 and R16 are hydrogen, and A is -C(O)-, R13 is not halogen, -OH, or -OCH3; and when R12 and R14 are -F, R5, R6, R13, R15 and R16 are hydrogen, and A is -C(O)-, R4 is not 3,5 difluorophenyl; and when R15 is halogen, -OH or -OCH3, R6, R13, R14 and R16 are hydrogen,
and A is -C(O)-, R12 is not halogen; and when R15 is -F, R6, R13, R14 and R16 are hydrogen and A is -C(O)-, R12 is not -CH3; and when R12 is -F, R4, R5, R6, R13, R14 and R16 are hydrogen and A is -C(O)-, R15 is not -NHS(O)2CH3; and when R12 is -Cl, R14 is -F or hydrogen, R4, R6, R13, R15 and R16 are
hydrogen, and A is -S(O)
2-, R is not
wherein "> indicates the bond to the 5- position of the 7-azaindole ring.
[0113] In some embodiments of any of the above embodiments of compounds of Formula II wherein R12 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen; or R4 is other than hydrogen and R5 and R6 are hydrogen; or R6 is other than hydrogen and R4 and R5 are hydrogen; or R4 and R5 are other than hydrogen and R6 is hydrogen; or R4 and R6 are other than hydrogen and R5 is hydrogen; or R5 and R6 are other than hydrogen and R4 is hydrogen; or R4, R5 and R6 are all other than hydrogen.
[0114] In some embodiments of compounds of Formula II, R13 is other than hydrogen. In some embodiments, R13 is -LR24. In some embodiments, R13 is other than hydrogen and R12, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 is -LR24 and R12, R14, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 is -LR24, any three of R12, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14, R15 and R16 is hydrogen. In some embodiments, R13 is -LR24, any two of R12, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the the remaining of R12, R14, R15 and R16 are hydrogen. In some embodiments, R13 is -LR24, any one of R12, R14, R15 and R16 is independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R12, R14, R15 and R16 are hydrogen. In some embodiments, R13 is -LR24, and R12, R14, R15 and R16 are hydrogen. In some embodiments, R13 is other than hydrogen, and R12, R14, R15 and R16 are hydrogen.
[0115] In other embodiments of compounds of Formula II, wherein R13 is other than hydrogen, when R4, R6, R12, R14, R15 and R16 are hydrogen, and A is -CH2- or -CH(OH)-, then R13 is not -OH or -OCH3; and when R5, R6, R12, R14, R15 and R16 are hydrogen, and A is -CH2-, then R13 is not -OCH3;
and when R
13 is
, wherein ' indicates the bond to the phenyl ring, R
4, R
6, R
12,
R
14, R
15 and R
16 are hydrogen, and A is -CH
2-, then R
5 is not thiophen-3-yl; and when R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -C(O)-, then R
13 is not -F, -OH, or -OCH
3; and when R
4, R
5, R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -C(O)-, then R
13 is not -NO
2, -NH
2, or
, wherein ' indicates the bond to the phenyl ring; and when R
4, R
5, R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -S(O)
2-, then R
13 is not -F, -Cl, or -CF
3; and when R
13 and R
14 are -OH, R
4, R
6, R
12, R
15 and R
16 are hydrogen and A is -CH
2-, R
5 is not thiophen-3-yl; and when R
6, R
14, R
15 and R
16 are hydrogen and A is -C(O)-, then R
13 and R
12 are not both -Cl; and when R
12 and R
13 are both -F, R
5, R
6, R
14, R
15 and R
16 are hydrogen and A is -C(O)-, then R
4 is not 3,5-difluorophenyl; and when R
13 is -OH or -OCH
3, R
12 is -F, R
4, R
6, R
14, R
15 and R
16 are hydrogen, and A is -C(O)-, then R
5 is not thiophen-2-yl; and when R
13 is -OCH
3, R
12 is -F, R
4, R
6, R
14, R
15 and R
16 are hydrogen, and A is -C(O)-, then R
5 is not -Br; and when R
6, R
12, R
14 and R
15 are hydrogen, R
16 is -CH
3 and A is -C(O)-, then R
13 is not -F; and when R
13 is -Cl, R
16 is -F, R
4, R
6, R
12, R
14 and R
15 are hydrogen, and A is -C(O)-, then R
5 is not thiophen-2-yl; and when R
6, R
12, R
14 and R
15 are hydrogen and A is -C(O)-, then R
13 and R
16 are not both -F; and when R
6, R
12, R
14 and R
15 are hydrogen, R
16 is halogen, and A is -C(O)-, then R
13 is not -OH or -OCH
3; and when R
4, R
5, R
6, R
12, R
14 and R
15 are hydrogen, R
13 is -NHS(O)
2CH
3, and A is -C(O)-, then R
16 is not -F; and when R
4, R
5, R
6, R
12, R
14 and R
16 are hydrogen, and A is -CH
2-, then R
13 and R
15 are not both -OCH
3; and when R
4, R
5, R
6, R
12, R
14 and R
16 are hydrogen, and A is -S(O)
2-, then R
13 and R
15 are not both -Cl; and when R
13 is -OH, R
15 is -OH or -OCH
3, R
4, R
6, R
12, R
14 and R
16 are hydrogen, and A is -CH
2-, then R
5 is not thiophen-3-yl; and when R
13 is -F, R
4, R
6, R
12, R
14, R
15 and R
16 are hydrogen, and A is -CH
2-, then R
5 is not 3-hydroxy-phenyl; and when R
13 is -CN, R
4, R
6, R
12, R
14, R
15 and R
16 are hydrogen, A is -S(O)
2-, then
R
5 is not
wherein
*> indicates the bond to the 5-position of the 7-azaindole ring.
[0116] In some embodiments of any of the above embodiments of compounds of Formula II wherein R13 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0117] In some embodiments of compounds of Formula II, R14 is other than hydrogen. In some embodiments, R14 is -LR24. In some embodiments, R14 is other than hydrogen and R12, R13, R15 and R16 are each independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some
embodiments, R14 is -LR24, and R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. Ih some embodiments, R14 is -LR24, any three of R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R12, R13, R15 and R16 is hydrogen. In some embodiments, R14 is -LR24, any two of R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R12, R13, R15 and R16 are hydrogen. In some embodiments, R14 is -LR24, any one of R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R12, R13, R15 and R16 are hydrogen. In some embodiments, R14 is -LR24, and R12, R13, R15 and R16 are hydrogen. In some embodiments, R14 is other than hydrogen, and R12, R13, R15 and R16 are hydrogen.
[0118] In other embodiments of compounds of Formula II, wherein R14 is other than hydrogen, when R4, R6, R12, R13, R15 and R16 are hydrogen, R5 is thiophen-2-yl and A is -C(O)-, then R14 is not -OH, or -OCH3; and when R4, R5, R6, R12, R13, R15 and R16 are hydrogen and A is -C(O)-, then R14 is not -OCH3; and when R6, R12, R13, R15 and R16 are hydrogen and A is -C(O)-, then R14 is not -Cl; and when R4, R5, R6, R12, R13, R15 and R16 are hydrogen and A is -CH2-, then R14 is not
, wherein ' indicates the bond to the phenyl ring; and when R
4, R
5,
R6, R12, R13, R15 and R16 are hydrogen and A is -S- or -S(O)2-, then R14 is not -F; and when R5, R6, R12, R13, R15 and R16 are hydrogen, R4 is 3-(hydroxymethyl)phenyl and A is -S-, then R14 is not -CH3; and when R5, R6, R12, R13, R15 and R16 are hydrogen, R4 is 3,5-difluorophenyl and A is -C(O)-, R12 and R14 are not both -F; and when R14 is -N(CH3)2, R4, R6, R12, R13, R15 and R16 are hydrogen, and A is -CH2-, then R5 is not 3 -hydroxy-phenyl; and when R14 is -Br, R4, R6, R12, R13, R15 and R16 are hydrogen, and A is -S-, then R5 is not 3 -hydroxy-phenyl; and when R12 is -Cl, R14 is -F, R4, R6, R13,
R
15 and R
16 are hydrogen, and A is -S(O)
2-, then R
5 is not
, wherein "
> indicates the bond to the 5-position of the 7-azaindole ring; and when R
14 is -NH
2, R
4, R
6, R
12, R
13, R
15 and R
16 I—
, wherein -
ς indicates the bond to the 5- position of the 7-azaindole ring; and when R
4, R
6, R
12, R
15 and R
16 are hydrogen, R
13 and R
14 are -OH, and A is -CH
2-, then R
5 is not thiophen-3-yl.
[0119] In some embodiments of any of the above embodiments of compounds of Formula II wherein R
14 is other than hydrogen, R
5 is other than hydrogen and R
4 and R
6 are hydrogen, or R
4 is other than hydrogen and R
5 and R
6 are hydrogen, or R
6 is other than hydrogen and R
4 and R
5 are hydrogen, or R
4 and R
5 are other than hydrogen and R
6 is hydrogen, or R
4 and R
6 are other than hydrogen and R
5 is hydrogen, or R
5 and R
6 are other than hydrogen and R
4 is hydrogen, or R
4, R
5 and R
6 are other than hydrogen.
[0120] In some embodiments of compounds of Formula II, R12 and R16 are other than hydrogen. In some embodiments, R12 and R16 are independently -LR24. In some embodiments, R12 and R16 are other than hydrogen and R13, R14 and R15 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R16 are independently -LR24 and R13, R14 and R15 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R16 are independently -LR24, any two of R13, R14 and R15 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R13, R14 and R15 is hydrogen. In some embodiments, R12 and R16 are independently -LR24, any one of R13, R14 and R15 is independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R13, R14 and R15 are hydrogen. In some embodiments, R12 and R16 are independently -LR24, and R13, R14 and R15 are hydrogen. In some embodiments, R12 and R16 are other than hydrogen, and R13, R14 and R15 are hydrogen. In other embodiments, wherein R12 and R16 are other than hydrogen, when R5, R6, R13, R14 and R15 are hydrogen and A is -C(O)-, then R12 and R16 are not both -F.
[0121] In some embodiments of any of the above embodiments of compounds of Formula II wherein R12 and RIδ is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and Rδ are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0122] In some embodiments of compounds of Formula II, R12 and R13 are other than hydrogen. In some embodiments, R12 and R13 are independently -LR24. In some embodiments, R12 and R13 are other than hydrogen and R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R13 are independently -LR24, and R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro
substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R13 are independently -LR24, any two of R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R14, R15 and R16 is hydrogen. In some embodiments, R12 and R13 are independently -LR24, any one of R14, R15 and R16 is independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R14, R15 and R16 are hydrogen. In some embodiments, R12 and R13 are independently -LR24, and R14, R15 and R16 are hydrogen. In some embodiments, R12 and R13 are other than hydrogen and R14, R15 and R16 are hydrogen. In other embodiments, wherein R12 and R13 are other than hydrogen, when R6, R14, R15 and R16 are hydrogen, A is -C(O)-, and R13 is halogen, -OH, or -OCH3, then R12 is not halogen. In alternate embodiments, when R12 and R13 are other than hydrogen, R6, R14, R15 and R16 are hydrogen and A is -C(O)-, then both R12 and R13 are not halogen; and when R12 and R13 are other than hydrogen, R4, R6, R14, R15 and R16 are hydrogen, and A is -C(O)-, R12 is -F and R13 is -OH or -OCH3, then R5 is not -Br or thiophen-2-yl.
[0123] In some embodiments of any of the above embodiments of compounds of Formula II wherein R12 and R13 is other than hydrogen, R3 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0124] In some embodiments of compounds of Formula II, R12 and R14 are other than hydrogen. In some embodiments, R12 and R14 are independently -LR24. In some embodiments, R12 and R14 are other than hydrogen and R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R14 are independently -LR24, and R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R14 are independently -LR24, and any one of R13, R15 and R16 is independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R13, R15 and R16 is hydrogen. In some embodiments, R12 and R14 are independently -LR24, and any two of R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R15 and R16 is hydrogen. In some embodiments, R12 and R14 are independently -LR24 and R13, RIS and R16 are
hydrogen. In some embodiments, R12 and R14 are other than hydrogen and R13, R15 and R16 are hydrogen. In some embodiments, wherein R12 and R14 are other than hydrogen, when R5, R6, R13, R15 and R16 are hydrogen, A is -C(O)-, and R4 is 3,5 difluorophenyl, then R12 and R14 are not both -F; and when R12 is -Cl, R14 is -F, R4, R6, R13, R15 and R16 are hydrogen, and A is -S(O)2-, then R5 is not
indicates the bond to the 5-position of the 7-azaindole ring.
[0125] In some embodiments of any of the above embodiments of compounds of Formula II wherein R12 and R14 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0126] In some embodiments of compounds of Formula II, R12 and R15 are other than hydrogen. In some embodiments, R12 and R15 are independently -LR24. In some embodiments, R12 and R15 are other than hydrogen and R13, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R15 are independently -LR24 and R13, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R12 and R15 are independently -LR24, any two of R13, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R14 and R16 is hydrogen. In some embodiments, R12 and R15 are independently -LR24, any one of R13, R14 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R13, R14 and R16 are hydrogen. In some embodiments, R12 and R15 are independently -LR24 and R13, R14 and R16 are hydrogen. In some embodiments, R12 and R15 are other than hydrogen and R13, R14 and R16 are hydrogen. In other embodiments, wherein R12 and R15 are other than hydrogen, when R6, R13, R14 and R16 are hydrogen and A is -C(O)-, then R12 and R15 are not both halogen; and when R6, R13, R14 and R16 are hydrogen, A is -C(O)-, and R12 is -CH3, then R15 is not -F; and when R6, R13, R14 and R16 are hydrogen, A is -C(O)-, and R12 is halogen, then R15 is not -OH or -OCH3; and when R4, R5, R6, R13, R14 and R16 are hydrogen, A is -C(O)-, and R12 is -F then R15 is not NHS(O)2CH3.
[0127] In some embodiments of any of the above embodiments of compounds of Formula II wherein R12 and R15 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or
R" is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0128] In some embodiments of compounds of Formula II, R13 and R14 are other than hydrogen. In some embodiments, R13 and R14 are independently -LR24. In some embodiments, R13 and R14 are other than hydrogen and R12, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 and R14 are independently -LR24 and R12, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 and R14 are independently -LR24, any two of R12, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R15 and R16 is hydrogen. In some embodiments, R13 and R14 are independently -LR24, any one of R12, R15 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R15 and R16 are hydrogen. In some embodiments, R13 and R14 are independently -LR24 and R12, R15 and R16 are hydrogen. In some embodiments, R13 and R14 are other than hydrogen and R12, R15 and R16 are hydrogen. In other embodiments, wherein R13 and R14 are other than hydrogen, when R4, R6, R12, R15 and R16 are hydrogen, A is -CH2-, and R5 is thiophen-3-yl, then R13 and R14 are not both -OH.
[0129] In some embodiments of any of the above embodiments of compounds of Formula II wherein R13 and R14 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0130] In some embodiments of compounds of Formula II, R13 and R15 are other than hydrogen. In some embodiments, R13 and R15 are independently -LR24. In some embodiments, R13 and R15 are other than hydrogen and R12, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 and R15 are independently -LR24 and R12, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, R13 and R15 are independently -LR24, two of R12, R14 and R16 are independently hydrogen, halogen,
optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14 and R16 is hydrogen. In some embodiments, R13 and R15 are independently -LR24, one of R12, R14 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14 and R16 are hydrogen. In some embodiments, R13 and R15 are independently -LR24 and R12, R14 and R16 are hydrogen, some embodiments, R13 and R15 are other than hydrogen and R12, R14 and R16 are hydrogen. Ih other embodiments, wherein R13 and R15 are other than hydrogen, when R4, R5, R6, R12, R14 and R16 are hydrogen and A is -CH2-, then R13 and R15 are not both -OCH3; and when R4, R5, R6, R12, R14 and R16 are hydrogen and A is -S(O)2-, then R13 and R15 are not both -Cl; and when R4, R6, R12, R14 and R16 are hydrogen, A is -CH2-, R13 is -OH and R15 is -OH or -OCH3, then Rs is not thiophen-3-yl.
[0131] In some embodiments of any of the above embodiments of compounds of Formula II wherein R13 and R15 is other than hydrogen, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0132] In some embodiments of compounds of Formula II, R13 is -LR24, provided, however, that when A is -CH2-, -CH(OH)- or -C(O)-, and R12, R14, R15, and R16 are hydrogen, then R13 is not -OH,
-OCH
3, or
, wherein * indicates the bond to the phenyl ring; and when R
4,
R5, R6, R12, R14, R15, and R16 are hydrogen and A is -C(O)-, R13 is not NH2, or
indicates the bond to the phenyl ring; and when R
4, R
5, R
6, R
12, R
14 and R
16 are hydrogen and A is -CH
2-, R
13 and R
15 are not both -OCH
3; and when R
4, R
6, R
12, R
14 and R
16 are hydrogen, A is -CH
2-, R
5 is thiophen-3-yl, R
13 is -OH, R
15 is not -OH or -OCH
3; and when R
4, R
6, R
12, R
15 and R
16 are hydrogen, A is -CH
2-, and R
5 is thiophen-3-yl, R
13 and R
14 are not both -OH; and when R
4, R
6, R
14, R
15 and R
16 are hydrogen and A is -C(O)-, R
12 is -F, R
5 is -Br or thiophen-2-yl, R
13 is not -OH or -OCH
3; and when R
12, R
14, and R
15 are hydrogen, A is -C(O)-, and R
16 is halogen, R
13 is not -OH or -OCH
3; and when R
4, R
5, R
6, R
12, R
14, and R
IS are hydrogen, A is -C(O)-, and R
16 is -F, R
13 is not -NHS(O)
2CH
3. In further embodiments, R
13 is -LR
24 and L is -NHS(O)
2CH
2-, -0-, or -0-CH
2-, R
24 is not H, or when L is -NHS(O)
2- or -0-, R
24 is not CH
3. In further embodiments, where R
13 is -LR
24, one of R
12, R
14, R
15, and R
16 is other than hydrogen and the remaining of R
12, R
14, R
15, and R
16 are hydrogen. In further embodiments, where R
13 is -LR
24, two of
R , R
14, R
IS, and R
16 are independently other than hydrogen and the remaining two of R
12, R
14, R
15, and R
16 are hydrogen. In some embodiments where R
13 is -LR
24, R
14 and R
15 are hydrogen, and at least one of R
12 and R
16 is other than hydrogen. In further embodiments, where R
13 is -LR
24, R
14 and R
15 are hydrogen, and at least one of R
12 and R
16 is other than hydrogen, L is -(alk)
a-NR
25-(alk)
b-, -(alk)
a-C(O)NR
25-(alk)
b-, -(alk)
a-OC(O)NR
25-(alk)
b-, -(alk)
a-OC(S)NR
25-(alk)
b-, -(alk)
a-C(S)NR
25-(alk)
b-, -(alk)
a-S(O)
2NR
25-(alk)
b-, -(alk)
a-NR
25C(O)-(alk)
b-, -(alk)
a-NR
25C(S)-(alk)
b-, -(alk)
a-NR
25C(O)NR
25-(alk)
b-, -(alk)
a-NR
25C(S)NR
25-(alk)
b-, -(alk)
a-NR
2sC(O)O-(alk)
b-
J -(alk)
a-NR
25C(S)O-(alk)
b-, -(alk)
a-NR
25S(O)
2-(alk)
b-, or -(alk)
a-NR
25S(O)
2NR
25-(alk)
b-, wherein a, b, R
25 and alk are as defined for Formula I.
[0133] In further embodiments of compounds of Formula II, where R13 is -LR24, R14 and R15 are hydrogen, and at least one of R12 and R16 is other than hydrogen, L is -NR25-, -NR25-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR2s-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)2NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25 -(alk)b-, -NR25C(S)NR2s-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-.
[0134] In further embodiments of compounds of Formula II, where R13 is -LR24, R14 and R15 are hydrogen, and at least one of R12 and R16 is other than hydrogen, L is -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-. In further embodiments where R13 is -LR24, R14 and R15 are hydrogen, and at least one of R12 and R16 is other than hydrogen, L is -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25S(O)2-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, or -NR25S(O)2NR25-(alk)b-. In further embodiments where R13 is -LR24, R14 and R15 are hydrogen, and at least one of R12 and R16 is other than hydrogen, L is -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -OC(O)NR25-(alk)b-, or -OC(S)NR25-(alk)b-. In some embodiments of any of the above embodiments where R13 is -LR24, R5 is other than hydrogen and R4 and R6 are hydrogen, or R4 is other than hydrogen and R5 and R6 are hydrogen, or R6 is other than hydrogen and R4 and R5 are hydrogen, or R4 and R5 are other than hydrogen and R6 is hydrogen, or R4 and R6 are other than hydrogen and R5 is hydrogen, or R5 and R6 are other than hydrogen and R4 is hydrogen, or R4, R5 and R6 are other than hydrogen.
[0135] In some embodiments of compounds of Formula II, A is -CRaRb- or -C(O)-, R13 is -LR24, R14 and R15 are hydrogen, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, wherein one of R12 and R16 is other than hydrogen, and wherein L is -NR25-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)2NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein b
and R^ are as defined for Formula I, alk is Cj-3 alkylene optionally substituted with fluoro or optionally fluoro substituted lower alkyl, and R24 is hydrogen, provided, however, that said hydrogen would not be attached to S(O)2-, optionally fluoro substituted lower alkyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and Ra and Rb are independently hydrogen, halogen, optionally fluoro substituted lower alkyl or optionally fluoro substituted lower alkoxy, provided, however, that when R4, R5, R6, R12, R14, and R15 are hydrogen, A is -C(O)-, and R16 is -F, then R13 is not -NHS(O)2CH3. In other embodiments, R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In other embodiments, A is -CH2-. In other embodiments, A is -C(O)-. In other embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, A is -CH2-, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, A is -C(O)-, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, A is -CH2-, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, A is -C(O)-, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, A is -CH2-, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, A is -C(O)-, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, R4 and R5 are other than hydrogen and R6 is hydrogen. In other embodiments, R4 and R6 are other than hydrogen, and R5 is hydrogen. In other embodiments, R4 and R5 are hydrogen, and R6 is other than hydrogen. In other embodiments, R4, R5, and R6 are other than hydrogen.
[0136] In some embodiments of compounds of Formula II, A is -CRaRb- or -C(O)-, R13 is -LR24, R14 and R15 are hydrogen, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, wherein one of R12 and R16 is other than hydrogen, and wherein L is -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NTt25-(alk)b-, wherein b and R25 are as defined for Formula I, alk is Ci-3 alkylene optionally substituted with fluoro or optionally fluoro substituted lower alkyl, R24 is hydrogen provided, however, that said hydrogen would not be attached to S(O)2-, optionally fluoro substituted lower alkyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, and optionally fluoro substituted lower alkylthio, Ra and Rb are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, or optionally fluoro
substituted lower alkoxy, one of R4 and R5 is other than hydrogen and R6 is hydrogen. In other embodiments, R16 is hydrogen, hi other embodiments, R12 is hydrogen, m other embodiments, R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In other embodiments, A is -CH2-. In other embodiments, A is -C(O)-. In other embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, A is -CH2-, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, A is -C(O)-, R4 is other than hydrogen and R5 and R6 are hydrogen. In other embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, A is -CH2-, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, -C(O)-, R5 is other than hydrogen and R4 and R6 are hydrogen. In other embodiments, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, A is -CH2-, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, A is -C(O)-, R6 is other than hydrogen and R4 and R5 are hydrogen. In other embodiments, R4 and R5 are other than hydrogen and R6 is hydrogen. In other embodiments, R4 and R6 are other than hydrogen and R5 is hydrogen. In other embodiments, R4 and R5 are hydrogen and R6 is other than hydrogen. In other embodiments, R4, R5, and R6 are other than hydrogen.
[0137] In some embodiments of compounds of Formula II, A is -CRaRb- or -C(O)-, R13 is -LR24, R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, one of R14, R15 and R16 are halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the other two are hydrogen, L is -NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR25C(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-) wherein b and R25 are as defined for Formula I, alk is C!-3 alkylene optionally substituted with fluoro or optionally fluoro substituted lower alkyl, R24 is hydrogen provided, however, that said hydrogen would not be attached to S(O) or S(O)2, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, Ra and Rb are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy, one of R4 and R5 is other than hydrogen and R6 is hydrogen. In other embodiments, R4 is hydrogen and R5 is other than hydrogen. In other embodiments, R5 is hydrogen and R4 is other than hydrogen. In other embodiments, A is -CH2-, R5 is hydrogen and R4 is other than hydrogen. In other embodiments, A is -C(O)-, R5 is hydrogen and R4 is other than hydrogen. In other embodiments, A is -CH2-, R4 is hydrogen and R5 is other than hydrogen. In other embodiments, A is -C(O)-, R4 is hydrogen and R5 is other than hydrogen.
[0138] In some embodiments of compounds of Formula II, A is -CRaRb- or -C(O)-, R13 is -LR24, R , R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, where at least one of R12, R14, R15 and R16 is other than hydrogen, L is -NR25C(O)-(aIk)b-, -NR25C(S)-(alk)b-, -NR25S(O)2-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR2S-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein b and R25 are as defined for Formula I, alk is Ci-3 alkylene optionally substituted with fluoro or optionally fluoro substituted lower alkyl, R24 is hydrogen provided, however, that said hydrogen would not be attached to S(O)2, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, Ra and Rb are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy, provided, however, that when R4, R5, R6, R12, R14, and R15 are hydrogen, A is -C(O)-, and R16 is -F, then R13 is not -NHS(O)2CH3. In further embodiments, R12 is other than hydrogen and R14, R15 and R16 are hydrogen. In further embodiments, R16 is other than hydrogen and R12, R14 and R15 are hydrogen. In further embodiments, at least two of R12, R14, R15 and R16 are other than hydrogen. In further embodiments, R12 and R16 are other than hydrogen and R14 and R15 are hydrogen. In further embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In further embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen.
[0139] In some embodiments of compounds of Formula II, A is -CRaRb- or -C(O)-, R13 is -LR24, R12, R14, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, where at least one of R12, R14, R15 and R16 is other than hydrogen, L is -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -OC(O)NR25-(alkV, or -OC(S)Ml25-(alk)b-, wherein b and R25 are as defined for Formula I, alk is Cj-3 alkylene optionally substituted with fluoro or optionally fluoro substituted lower alkyl, R24 is hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, Ra and Rb are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, and optionally fluoro substituted lower alkoxy. In further embodiments, R12 is other than hydrogen and R14, R15 and R16 are hydrogen. In further embodiments, R16 is other than hydrogen and R12, R15 and R16 are hydrogen. In further embodiments, at least two of R12, R14, RIS and R16 are other than hydrogen. In further embodiments, R12 and R16 are other than hydrogen and R14 and R15 are hydrogen. In further embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In further embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen.
[0140] In some embodiments of any of the above embodiments of compounds of Formula II
wherein R
13 is -LR
24, -LR
24 is not -NH
2, -OH, -OCH
3, -NHS(O)
2CH
3,
, or
wherein "> indicates the bond to the phenyl ring.
[0141] In some embodiments of compounds of Formula II, R14 is -LR24, provided, however, that when R4, R6, R12, R13, R15 and R16 are hydrogen, R5 is hydrogen or thiophen-2-yl and A is -C(O)-, then R14 is not -OH, or -OCH3; and when R4, R5, R6, R12, R13, R15 and R16 are hydrogen and A is -CH2-, R14
is not
indicates the bond to the phenyl ring, when R
4,
R6, R12, R15 and R16 are hydrogen, A is -CH2-, and Rs is thiophen-3-yl, R13 and R14 are not both -OH; and when R5, R6, R12, R13, R15 and R16 are hydrogen and A is -S-, R14 is not -CH3; and when R14 is -N(CHs)2, R4, R6, R12, R13, R15 and R16 are hydrogen, and A is -CH2-, then R5 is not 3-hydroxy-phenyl; and when R14 is -NH2, R4, R6, R12, R13, R15 and R16 are hydrogen, and A is -S(O)2-, then R5 is not
, wherein ' indicates the bond to the 5-position of the 7-azaindole ring. In further embodiments, when L is -O-, R
24 is not H or CH
3; and when L is -OCH
2-, R
24 is not H. In some embodiments, one of R
12, R
13, R
15, and R
16 is other than hydrogen and the others are hydrogen. In some embodiments, at least one of R
12, R
13, R
15, and R
16 are other than hydrogen. In some embodiments, two of R
12, R
13, R
15, and R
16 are other than hydrogen and the others are hydrogen. In some embodiments, at least two of R
12, R
13, R
15, and R
16 are other than hydrogen. In a some embodiments, at least three of R
12, R
13, R
15, and R
16 are other than hydrogen. In some embodiments, R
12, R
13, R
15, and R
16 are other than hydrogen. In some embodiments, one of R
12, R
13, R
15 and R
16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the others of R
12, R
13, R
15 and R
16 are hydrogen. In some embodiments, any two of R
12, R
13, R
15 and R
16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R
12, R
13, R
15 and R
16 are hydrogen. In some embodiments, R
14 is -OR
24, where R
24 is optionally substituted C
2-6 alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R
14 is -O-alk-R
24, where R
24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, R
14 is -SR
24, where R
24 is
hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R
14 is -S-alk-R
24, where R
24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, R
14 is -NHR
24, where R
24 is hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R
14 is -NH-alk-R
24, where R
24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, A is -CH
2- or -C(O)-. In some embodiments, R
5 is other than hydrogen and R
4 and R
6 are hydrogen. In some embodiments, R
4 is other than hydrogen and R
s and R
6 are hydrogen. In some embodiments, R
6 is other than hydrogen and R
4 and R
5 are hydrogen. In some embodiments, A is -CH
2-, R
5 is other than hydrogen and R
4 and R
6 are hydrogen. Ih some embodiments, A is -CH
2-, R
4 is other than hydrogen and R
5 and R
6 are hydrogen. In some embodiments, A is -CH
2-, R
6 is other than hydrogen and R
4 and R
5 are hydrogen. In some embodiments, A is -C(O)-, R
5 is other than hydrogen and R
4 and R
6 are hydrogen. In some embodiments, A is -C(O)-, R
4 is other than hydrogen and R
5 and R
6 are hydrogen. In some embodiments, A is -C(O)-, R
6 is other than hydrogen and R
4 and R
5 are hydrogen.
[0142] In some embodiments of compounds of Formula II, R14 is -OR24 where R24 is optionally substituted C2_6 alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R14 is -SR24 where R24 is hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R14 is -NHR24, where R24 is hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, any one of R12, R13, R15 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R13, R15 and R16 are hydrogen. In some embodiments, two of R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining two of R12, R13, R15 and R16 are hydrogen. In some embodiments, R12 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and R13, R15 and R16 are hydrogen. In some embodiments, A is -CH2- or -C(O)-. In some embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, R6 is other than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -CH2-, R5 is other
than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -CH2-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -CH2-, R6 is other than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -C(O)-, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -C(O)-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -C(O)-, R6 is other than hydrogen and R4 and R5 are hydrogen.
[0143] In some embodiments of compounds of Formula II, R14 is -LR24, where L is -NR25-(alk)b-, -C(O)NR25-(alk)b-, -OC(O)NR25-(alk)b-, -OC(S)NR25-(alk)b-, -C(S)NR25-(alk)b-, -S(O)2NR25-(alk)b-, -NR25C(O)-(alk)b-, -NR2sC(S)-(alk)b-, -NR25C(O)NR25-(alk)b-, -NR25C(S)NR25-(alk)b-, -NR25C(O)O-(alk)b-, -NR25C(S)O-(alk)b-, -NR25S(O)2-(alk)b-, or -NR25S(O)2NR25-(alk)b-, wherein b, alk and R25 are as defined for Formula I, and R24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, any one of R12, R13, R15, and R16 is other than hydrogen and the remaining of R12, R13, R15, and R16 are hydrogen. In some embodiments, at least one of R12, R13, R15, and R16 are other than hydrogen. In some embodiments, any two of R12, R13, R15, and R16 are other than hydrogen and the remaining of R12, R13, R15, and R16 are hydrogen. In some embodiments, at least two of R12, R13, R15, and R16 are other than hydrogen. In a some embodiments, at least three of R12, R13, R15, and R16 are other than hydrogen. In a some embodiments, R12, R13, R15, and R16 are other than hydrogen. In some embodiments, any one of R12, R13, R15 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the remaining of R12, R13, R15, and R16 are hydrogen. In some embodiments, two of R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the others of R12, R13, R15 and R16 are hydrogen. In some embodiments, A is selected from -CH2- and -C(O)-. In some embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, R6 is other than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -CH2-, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -CH2-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -CH2-, R6 is other than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -C(O)-, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -C(O)-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -C(O)-, R6 is other than hydrogen and R4 and R5 are hydrogen.
[0144] In some embodiments of any of the above embodiments of compounds of Formula II
wherein R14 is -LR24, -LR24 is not -NH2, -OH, -OCH3, -N(CH3)*
or
wherein ^ indicates the bond to the phenyl ring.
[0145] In some embodiments of compounds of Formula II, R13 and R15 are halogen, optionally fluoro substituted lower alkyl, -OR24, where R24 is optionally substituted C2-6 alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, -O-alk-R24, where R24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, -SR24, where R24 is hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, -S-alk-R24, where R24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, -NHR24, where R24 is hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or -NH-alk-R24, where R24 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, at least one of R12, R14 and R16 is other than hydrogen. In some embodiments, at least two of R12, R14 and R16 are other than hydrogen. In some embodiments, R12, R14 and R16 are other than hydrogen. In some embodiments, any one of R12, R14 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14 and R16 are hydrogen. In some embodiments, any two of R12, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14 and R16 is hydrogen. In some embodiments, R12, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2- or -C(O)-. In some embodiments, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, R6 is other than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -CH2-, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -CH2-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -CH2-, R6 is other
than hydrogen and R4 and R5 are hydrogen. In some embodiments, A is -C(O)-, R5 is other than hydrogen and R4 and R6 are hydrogen. In some embodiments, A is -C(O)-, R4 is other than hydrogen and R5 and R6 are hydrogen. In some embodiments, A is -C(O)-, R6 is other than hydrogen and R4 and R5 are hydrogen.
[0146] The compounds of Formula II, and all sub-embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0147] Li some embodiments, compounds of Formula II have the structure according to the following sub-generic structure Formula Ila:
Formula Ha all salts, prodrugs, tautomers and isomers thereof, wherein A, R4, R5, R6, R!2, R16 and R24 are as defined for Formula II, provided, however, that when A is C(O), R4, R5, R6, and R12 are hydrogen and R16 is fluoro, then R24 is not CH3.
[0148] In some embodiments of compounds of Formula Ila, A is -CH2- or -C(O)-.
[0149] In other embodiments of compounds of Formula Ha, A is -CH2- or -C(O)- and R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted Ci-3alkyl, optionally fluoro substituted Ci-3alkoxy, or optionally fluoro substituted C]-3alkylthio. In some embodiments, A is -CH2- or -C(O)-, and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 and R16 are independently halogen, optionally fluoro substituted Ci-3alkyl, optionally fluoro substituted C1-3alkoxy, or optionally fluoro substituted Ci.3alkylthio.
[0150] In other embodiments of compounds of Formula Ila, A is -CH2-, R12 is hydrogen and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R16 is halogen, optionally fluoro
substituted C^alkyl, optionally fluoro substituted Ci-3alkoxy, or optionally fluoro substituted Ci-3alkylthio. In some embodiments, A is -CH2-, R16 is hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 is halogen, optionally fluoro substituted Ci-3alkyl, optionally fluoro substituted Ci_3alkoxy, or optionally fluoro substituted Ci-3alkylthio.
[0151] In some embodiments of compounds of Formula Ila, A is -CH2- and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 and R16 are independently halogen, optionally fluoro substituted C1-3alkyl, optionally fluoro substituted C]-3alkoxy, and optionally fluoro substituted Ci-3alkylthio.
[0152] In other embodiments of compounds of Formula Ila, A is -C(O)-, R12 is hydrogen and RIδ is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R16 is halogen, optionally fluoro substituted Ci-3alkyl, optionally fluoro substituted Cj.3alkoxy, or optionally fluoro substituted Ci-3alkylthio. In some embodiments, A is -C(O)-, R!6 is hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 is halogen, optionally fluoro substituted Ci-3alkyl, optionally fluoro substituted C1-3alkoxy, or optionally fluoro substituted C1-3alkylthio.
[0153] In some embodiments of compounds of Formula Ha, A is -C(O)- and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio; in further embodiments, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted Cμ3alkyl, optionally fluoro substituted Ci-3alkoxy, or optionally fluoro substituted Ci-3alkylthio.
[0154] In some embodiments of any of the above embodiments of compounds of Formula Ila, R and R6 are hydrogen, or R5 and R6 are hydrogen, or R4 and R5 are hydrogen.
[0155] In some embodiments of any of the above embodiments of compounds of Formula Ila, R24 is substituted methyl, optionally substituted C2-β alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; in further embodiments, R24 is C2-β lower alkyl, aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with halogen, lower alkyl or lower alkoxy; in further embodiments, R24 is n-propyl, i-propyl, or phenyl, wherein phenyl is optionally substituted with halogen, lower alkyl or lower alkoxy.
[0156] In some embodiments, compounds of Formula II have the structure according to the following sub-generic structure Formula lib:
Formula lib all salts, prodrugs, tautomers and isomers thereof, wherein A, R4, R5, R6, R12, R14, R15 and R16, are as defined for Formula II, R28 is hydrogen, lower alkyl, or lower alkyl substituted with fluoro, hydroxyl, lower alkoxy, thiol, lower alkylthio, or -NR8R9, wherein R8 and R9 are as defined for Formula I, provided, however, that when R28 is substituted lower alkyl, any substitution of the alkyl carbon bound to the nitrogen of NR28 is fluoro, and R29 is hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that nitrogen of NR29 is not bound to any alkene carbon thereof, optionally substituted lower alkynyl, provided, however, that nitrogen of NR29 is not bound to any alkyne carbon thereof, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R28 and R29 combine with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl. In some embodiments of compounds of Formula lib, A is -CH2- or -C(O)-, R12, R14, R15, and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, R28 is hydrogen or lower alkyl and R29 is optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or R28 and R29 combine with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl.
[0157] In some embodiments of compounds of Formula lib, A is -CH2-, one of R12, R14, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14, R15 and R16 are hydrogen.
[0158] In other embodiments of compounds of Formula lib, A is -CH2-, R12, R14 and R15 are hydrogen and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0159] In other embodiments of compounds of Formula lib, A is -CH2-, R14, R15 and R16 are hydrogen and R12 is halogen, optionally fiuoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0160] In some embodiments of compounds of Formula lib, A is -CH2-, two of R12, R14, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the other two of R12, R14, R15 and R15 are hydrogen.
[0161] In some embodiments of compounds of Formula lib, A is -CH2-, R14 and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0162] In some embodiments of compounds of Formula lib, A is -C(O)-, one of R12, R14, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the others of R12, R14, R15 and R16 are hydrogen.
[0163] In some embodiments of compounds of Formula lib, A is -C(O)-, R12, R14 and R15 are hydrogen and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0164] In some embodiments of compounds of Formula lib, A is -C(O)-, R14, R15 and R16 are hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0165] In some embodiments of compounds of Formula lib, A is -C(O)-, two of R12, R14, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the other two of R12, R14, R15 and R16 are hydrogen.
[0166] In some embodiments of compounds of Formula Hb, A is -C(O)-, R14and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0167] In some embodiments of any of the above embodiments of compounds of Formula lib, R4 and R6 are hydrogen, or R5 and R6 are hydrogen, or R4 and R5 are hydrogen. In some embodiments of any of the above embodiments of compounds of Formula lib, R28 and R29 are both lower alkyl, or R28 and R29 combine with the nitrogen to which they are attached to form optionally substituted 5-7 membered heterocycloalkyl, further wherein the heterocycloalkyl is pyrrolidine, piperidine, piperazine or morpholine.
[0168] In some embodiments, compounds of Formula II have the structure according to the following sub-generic structures Formulae Hc, lid, He, Ilf, or Iig:
Formula Hc
Formula Hd
Formula He
Formula Hg all salts, prodrugs, tautomers and isomers thereof, wherein A, X, R4, R5, R6, R12, R14, R15, R16 and R24 are as defined for Formula π. In some embodiments of compounds of Formulae lie, lid, He, Hf, or Hg, A is -CH2- or -C(O)-. In some embodiments, any one of R12, R14, R15, and R16 is other than hydrogen. In some embodiments, any two of R12, R14, R15, and R16 are other than hydrogen. In some embodiments, any three of R12, R14, R15, and R16 are other than hydrogen. In some embodiments, R12, R14, R15, and R16 are other than hydrogen. In some embodiments, A is -CH2- or -C(O)- and R12, R14, R15, and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, any one of R12, R14, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14, R15 and R16 are hydrogen. In some embodiments, A is -CH2-, R12, R14 and R15 are hydrogen and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, R14, R15 and R16 are hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, any two of R12, R14, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining two of R12, R14, R15 and R16 are hydrogen. In some embodiments, A is -CH2-, R14 and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, any one of R12, R14, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R14, R15 and R16 are hydrogen. In some embodiments, A is -C(O)-, R12, R14 and R15 are hydrogen and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, R14, R15 and R16 are hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, any two of R12, R14, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl,
optionally rluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining two of R12, R14, R15 and RIδ are hydrogen. In some embodiments, A is -C(O)-, R14 and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments of any of the above embodiments of compounds of Formula lie, lid, He, Hf, and Hg, R4 and R6 are hydrogen or R5 and R6 are hydrogen, or R4 and R5 are hydrogen.
[0169] In some embodiments, compounds of Formula II have the structure according to the following sub-generic structure Formula IDi:
Formula EDb. all salts, prodrugs, tautomers and isomers thereof, wherein A, R5, R12, R13, R15, R16 and R24 are as defined for Formula II, and Z is -O-, -S- or -NH-. In some embodiments of compounds of Formula πh, R24 is hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, R24 is hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl and at least one of R12, R13, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the others of R12, R13, R15 and R16 are hydrogen; in further embodiments, R13 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and R12, R15 and R16 are hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0170] In some embodiments of compounds of Formula IHi, Z is -O- or -NH-, R24 is hydrogen, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or lower alkyl optionally substituted with optionally substituted aryl or optionally substituted heteroaryl, and at least one of R12, R13, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and the others of R12, R13, R15 and R16 are hydrogen; in further embodiments, R13 is halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and R12, R15 and R16 are hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0171] In some embodiments of compounds of Formula IDi, Z is -O- or -NH-, R24 is hydrogen, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or lower alkyl optionally substituted with optionally substituted aryl or optionally substituted heteroaryl, R13 is halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and one of R12, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and the others of R12, R15 and
> 16
R are hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0172] In some embodiments of compounds of Formula IDi, Z is -O- or -NH-, R24 is hydrogen, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or lower alkyl optionally substituted with optionally substituted aryl or optionally substituted heteroaryl, R13 is halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and two of R12, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy and the other of R12, R15 and R16 is hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0173] In some embodiments of compounds of Formula IDi, Z is -O- or -NH-, R24 is hydrogen, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, or lower alkyl optionally substituted with optionally substituted aryl or optionally substituted heteroaryl, R12, R13, R15 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, or optionally fluoro substituted lower alkoxy; in further embodiments, A is -CH2- or -C(O)-.
[0174] Di some embodiments, compounds of Formula II have the structure according to the following sub-generic structures Formulae Ili, Hj, IDc, Ilm, or IDi:
Formula Hj
Formula Ilk
Formula urn, or
Formula IIn all salts, prodrugs, tautomers and isomers thereof, wherein A, X, R4, R5, R6, R12, R13, R15, R16 and R24 are as defined for Formula II. In some embodiments of compounds of Formulae Hi, Hj, Hk, Hm, or Hn, A is -CH2- or -C(O)-. In some embodiments, one of R12, R13, R15, and R16 is other than hydrogen. In some embodiments, two of R12, R13, R15, and R16 are other than hydrogen. In one embodiment, three of R12, R13, R15, and R16 are other than hydrogen. In some embodiments, R12, R13, R15, and R16 are other than hydrogen.
[0175] In some embodiments of compounds of Formulae Hi, Hj, Hk, Hm, or Hn, A is -CH2- or -C(O)-, R12, R13, R15, and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, any one of R12, R13, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R13, R15 and R16 are hydrogen. In some embodiments, A is -CH2-, R13, R15 and R16 are hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, R12, R15 and R16 are hydrogen and R13 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, any two of R12, R13, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining two of R12, R13, R15 and R16 are hydrogen. In some embodiments, A is -CH2-, R15 and R16 are hydrogen and R12 and R13 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, R13 and R16 are hydrogen and R12 and R15 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, R13 and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -CH2-, R12 and R16 are hydrogen and R13 and R15 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio.
[0176] In some embodiments of compounds of Formulae Ili, Hj, Ilk, urn, or Hn, A is -C(O)-, any one of R12, R13, R15 and R16 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining of R12, R13, R15 and R16 are hydrogen. In some embodiments, A is -C(O)-, R13, R15 and R16 are hydrogen and R12 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, R12, R15 and R!6 are hydrogen and R13 is halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, two of R12, R13, R15 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and the remaining two of R12, R13, R15 and R16 are hydrogen. In some embodiments, A is -C(O)-, R15 and R16 are hydrogen and R12 and R13 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some
embodiments, A is -C(O)-, R13 and R16 are hydrogen and R12 and R15 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, R13 and R15 are hydrogen and R12 and R16 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments, A is -C(O)-, R12 and R16 are hydrogen and R13 and R15 are independently halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio. In some embodiments of any of the above embodiments of compounds of Formula Ila, R4 and R6 are hydrogen or R5 and R6 are hydrogen, or R4 and R5 are hydrogen.
[0177] In some embodiments, compounds of Formula II have the structure according to the following sub-generic structure Formula IIo:
Formula Ho all salts, prodrugs, tautomers and isomers thereof, wherein A, R5, R12, R14, R16 and R24 are as defined for Formula II, where each R24 is selected independently, and each Z is independently -O-, -S- or -NH-. In some embodiments of compounds of Formula Ho, each R24 is independently hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl. In some embodiments, each R24 is independently hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl and R12, R14 and R16 are hydrogen.
[0178] In some embodiments of compounds of Formula IIo, each R24 is independently hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl and at least one of R12, R14 and R16 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the others of R12, R14 and R16 are hydrogen; in further embodiments, R12 is hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and R14 and R16 are hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0179] In some embodiments of compounds of Formula IIo, each R24 is independently hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl and at least two of R12, R14 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio, and the other of R12, R14 and R16 is hydrogen; in further embodiments, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and R14 is hydrogen; in further embodiments, A iS -CH2- Or -C(O)-.
[0180] In some embodiments of compounds of Formula Ho, L is -O- or -NH-, each R24 is independently hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, R12 and R16 are independently hydrogen, halogen, optionally fluoro substituted lower alkyl, optionally fluoro substituted lower alkoxy, or optionally fluoro substituted lower alkylthio and R14 is hydrogen; in further embodiments, A is -CH2- or -C(O)-.
[0181] The compounds of Formulae Ha-IIo, and all sub-embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0182] hi some embodiments, compounds of Formula I have the structure according to the following sub-generic structure Formula III:
Formula m
all salts, prodrugs, tautomers, and isomers thereof, wherein: Q has a structure selected from the group consisting of
in which ? indicates the attachment point of Q to A of Formula III;
Z2 is N or CR12; Z4 is N or CR14; Z5 is N or CR15; Z5 is N or CR16;
L2 is selected from the group consisting of -(CR10R11VNR25^CR10R11V,
-(CR
10R
11)
p-O-(CR
10R
n)
q-, -(CR
10R
11)
p-S-(CR
10R
π)
q-, -(CR
10R
11VC(O)-(CR
10R
11V, -(CR
10R
11VC(S)-(CR
10R
11V, -(CR
1V)
P-S(O)-(CR
10R
11V, -(CR
10R
1VS(O)
2-(CR
10R
11V, -(CR
10R
11)
p-C(O)NR
25-(CR
10R
n)
q-, -(CR
10R
11)
p-C(S)NR
25-(CR
10R
11)
q-,
-(CR
10R
11VNR
25C(S)-(CR
10R
11V and -(CR
10R
11VNR
25S(O)
2-(CR
10R
11),-; p and q are independently O, 1, or 2 provided, however, that at least one of p and q is O; s is 1 or 2;
X is O or S;
A is selected from the group consisting of -0-, -S-, -CRaRb-, -NR1-, -C(O)-, -C(S)-, -S(O)-, and -S(O)2-;
Ra and Rb at each occurrence are independently selected from the group consisting of hydrogen, fluoro, -OH, -NH2, lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein the alkyl chain(s) of lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro; or
Ra and Rb combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl are
61
optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R1 is selected from the group consisting of hydrogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -C(O)R7, -C(S)R7, -S(O)2R7, -C(O)NHR7, -C(S)NHR7, and -S(O)2NHR7, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro, further provided that when R1 is lower alkyl, any substitution on the lower alkyl carbon bound to the N of -NR1- is fluoro, and wherein cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R7 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, provided, however, that any substitution of the alkyl carbon bound to the N of -C(O)NHR7, -C(S)NHR7 or -S(O)2NHR7 is fluoro, wherein the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino, or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro, and wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R4, R5, R6, R12, R14, R15, R16, R42, R43, R45, R46 and R47 are independently selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted
lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -NO2, -CRaRbR26, and -LR26;
L at each occurrence is independently selected from the group consisting of -(alk)a-S-(alk)b-, -(alk)a-O-(alk)b-, -(alk)a-NR25-(alk)b-, -(alk)a-C(O)-(alk)b-, -(alk)a-C(S)-(alk)b-, -(alk)a-S(O)-(alk)b-, -(alk)a-S(O)2-(alk)b-, -(alk)a-OC(O)-(alk)b-, -(alk)a-C(O)O-(alk)b-, -(alk)a-OC(S)-(alk)b-, -(alk)a-C(S)O-(alk)b-, -(alk)a-C(O)NR25-(alk)b-, -(alk)a-C(S)NR25-(alk)b-, -(alk)a-S(O)2NR25-(alk)b-J -(alk)a-NR25C(O)-(alk)b-, -(alk)a-NR25C(S)-(alk)b-, -(alk)a-NR25S(O)2-(alk)b-, -(alk)a-NR25C(O)O-(alk)b-, -(alk)a-NR25C(S)O-(alk)b-, -(alk)a-OC(O)NR25-(alk)b-, -(alk)a-OC(S)NR2S-(alk)b-, -(alk)a-NR25C(O)NR25-(alk)b-, -(alk)a-NR25C(S)NR25-(alk)b-, and -(alk)a-NR25S(O)2NR25-(alk)bs a and b are independently 0 or 1 ; alk is C]-3 alkylene or Ci-3 alkylene substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkyl, lower alkoxy, lower alkylthio, mono-alkylamino, di-alkylamino, and -NR8R9, wherein lower alkyl or the alkyl chain(s) of lower alkoxy, lower alkylthio, mono-alkylamino or di-alkylamino are optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution of the alkyl chain carbon bound to O of alkoxy, S of thioalkyl or N of mono- or di-alkylamino is fluoro;
R25 at each occurrence is independently selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R26 at each occurrence is independently selected from the group consisting of hydrogen, provided, however, that hydrogen is not bound to any of S(O), S(O)2, C(O) or C(S) of L, optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R26 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O1 S(O), S(O)2, C(O) or C(S) of L, optionally substituted lower alkynyl, provided, however, that when R26 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of L, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R10 and R11 at each occurrence are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, and lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted
lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di- alkylamino, and cycloalkylamino; or any two of R10 and R11 on the same or adjacent carbon atoms combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, and any others of R10 and R11 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, and lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, and wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl are optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R8 and R9 combine with the nitrogen to which they are attached to form a 5-7 membered heterocycloalkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, and fluoro substituted lower alkylthio;
R17 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl and -OR18;
R31 and R33 are independently selected from the group consisting of optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, and optionally substituted heterocycloalkyl;
R36 is selected from the group consisting of substituted methyl, optionally substituted C2-6 alkyl, optionally substituted lower alkenyl, provided, however, that when R36 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to the S(O)2 of S(O)2R36, optionally substituted lower alkynyl, provided, however, that when R36 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to the S(O)2 of S(O)2R36, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR19R20;
R19, R20, R34, R35, R37, and R38 are independently selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R19, R20, R34, R35, R37, or R38 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to the N OfNR19R20, NR34R35 or NR37R38, optionally substituted lower alkynyl, provided, however, that when R19, R20, R34, R35, R37, or R38 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to the N OfNR19R20, NR34R35 or NR37R38, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl and optionally substituted heteroaryl; or
R34 and R35 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl; or R37 and R38 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl; R32 is selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, and -OR18; R82 is selected from hydrogen or lower alkyl; and R18 is hydrogen or optionally substituted lower alkyl; provided, however, that the compound is not 3-{3-[2-(tetrahydropyran-2-yloxy)-ethoxy]-benzyl}-
5-thiophen-3-yl-lH-pyrrolo[2,3-b]pyridine, which has the structure
4-(4-Methyl-piperazin-l-ylmethyl)-N-[4-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]- benzamide, which has the structure
[0183] The compounds of Formula III, and all sub-embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0184] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula Ilia:
all salts, prodrugs, tautomers and isomers thereof, wherein A, L
2, Z
2, Z
6, R
4, R
5, R
6, R
15, R
17 and R
31 are as defined for Formula III.
[0185] In some embodiments of compounds of Formula Ilia, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, R17 is selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fiuoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, and R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0186] In some embodiments of compounds of Formula Ilia, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, R17 is selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, Z2 is N or CR12, Z6 is N or CR16, R12 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino.
[0187] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula HIb:
Formula mb all salts, prodrugs, tautomers, and isomers thereof, wherein:
A, is -O-, -CR
40R
41-, -C(O)- or -NR
48-;
Z16 is N or CR56;
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
L3 is selected from the group consisting of -NR48-, -S-, -O-, -NR48CH(R49)-, -SCH(R49)-, -OCH(R49)-, -C(O)NR48-, -S(O)2NR48-, -CH(R49)NR48-, -CH(R49)O-, -CH(R49)S-, -NR48C(O)-, and -NR48S(O)2-;
R53 and R55 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino or cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro;
R52 and R56 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy;
R49 is selected from the group consisting of hydrogen, lower alkyl, and fluoro substituted lower alkyl;
Cy is selected from the group consisting of aryl, heteroaryl, cycloalkyl, and heterocycloalkyl;
R39 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, aryl, heteroaryl, and NR50R51, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally
substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, and wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R50 is hydrogen or lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino;
R51 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R23 at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R23, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R57 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro
substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono- alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R 8 at each occurrence is independently hydrogen or lower alkyl; and t is O, 1, 2, or 3.
[0188] In some embodiments of compounds of Formula IHb, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-. In some embodiments, A1 is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, and R53 and R55 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy. In some embodiments, L3 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably -OCH(R49)-. In some embodiments, A1 is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, and L3 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably -OCH(R49)-.
[0189] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula IHp:
Formula IHp all salts, prodrugs, tautomers, and isomers thereof, wherein:
A1 is -0-, -CR40R41-, -C(O)- or -NR48-;
Z22 is N or CR62;
Z26 is N or CR66; r is O, 1, or 2;
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower
alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R62, R63, R65 and R66 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro;
Cy is selected from the group consisting of aryl, heteroaryl, cycloalkyl, and heterocycloalkyl;
R39 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, aryl, heteroaryl, and NR50R51, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, and wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R50 is hydrogen or lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino;
R51 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R23 at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R23, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R57 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino,
cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R48 at each occurrence is independently hydrogen or lower alkyl; and t is O, 1, 2, or 3.
[0190] In some embodiments of compounds of Formula Mp, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-. In some embodiments, A1 is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, and R62, R64, R65 and R66 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0191] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula IHc:
Formula Etc all salts, prodrugs, tautomers and isomers thereof, wherein A, s, Z2, Z6, R4, R5, R6, R15, R17, and R32 are as defined for Formula III.
[0192] In some embodiments of compounds of Formula IIIc, R4 and R6 are hydrogen, A is -0-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, R17 is selected from
the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, and R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0193] In some embodiments of compounds of Formula IIIc, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, R17 is selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, Z2 is N or CR12, Z6 is N or CR16, R12 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and -NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, further wherein R32 is optionally substituted lower alkyl or -OR18, where R18 is as defined for Formula III.
[0194] In some embodiments, compounds of Formula UI have the structure according to the following sub-generic structure Formula HIn:
Formula IHn all salts, prodrugs, tautomers and isomers thereof, wherein A, s, Z
2, Z
6, R
4, R
5, R
6, R
15, and R
32 are as defined for Formula III.
[0195] In some embodiments of compounds of Formula IHn, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, and R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0196] In some embodiments of compounds of Formula IIIc, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, R15 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, Z2 is N or CR12, Z6 is N or CR16, R12 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and -NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, further wherein R32 is optionally substituted lower alkyl or -OR18, where R18 is as defined for Formula III.
[0197] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula HIo:
Formula HIo all salts, prodrugs, tautomers and isomers thereof, wherein A, L2, Z2, Z4, Z5, Z6, R4, R5, R6, and R33 are as defined for Formula III.
[0198] In some embodiments of compounds of Formula IHo, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, and R12, R14, R15 and R16 are independently selected from the group consisting
19
of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro.
[0199] In some embodiments of compounds of Formula IIIo, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino.
[0200] In some embodiments of compounds of Formula HIo, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino.
[0201] In some embodiments, compounds of Formula HI have the structure according to the following sub-generic structure Formula ELIq:
Formula Illq all salts, prodrugs, tautomers, and isomers thereof, wherein:
A1 is -O-, -CR40R41-, -C(O)- or -NR48
Zi2 is N or CR52;
Z14 is N or CR54;
Z15 is N or CR55;
Z16 is N or CR56:
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
L3 is selected from the group consisting of -NR48-, -S-, -O-, -NR48CH(R49)-, -SCH(R49)-, -OCH(R49)-, -C(O)NR48-, -S(O)2NR48-, -CH(R49)NR48-, -CH(R49)O-, -CH(R49)S-, -NR48C(O)-, and -NR48S(O)2-;
R54 and R55 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino or cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro;
R52 and R56 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy;
R49 is selected from the group consisting of hydrogen, lower alkyl, and fluoro substituted lower alkyl;
Cy is selected from the group consisting of aryl, heteroaryl, cycloalkyl, and heterocycloalkyl;
R^ is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, aryl, heteroaryl, andNR50Rsl, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, and wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R50 is hydrogen or lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino;
R51 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R23 at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R23, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R57 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono- alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R48 at each occurrence is independently hydrogen or lower alkyl; and t is O, 1, 2, or 3.
[0202] In some embodiments of compounds of Formula IHq, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-. In some embodiments, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, and R54 and R55 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy. In some embodiments, L3 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably -OCH(R49)-. In some embodiments, A1 is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -CH2-, and L3 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably -OCH(R49)-.
[0203] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula IHd:
Formula md all salts, prodrugs, tautomers and isomers thereof, wherein A, Z2, Z4, Z5, Z6, R4, R5, R6, R10, R11 and R33 are as defined for Formula III, and r is O, 1, or 2.
[0204] In some embodiments of compounds of Formula md, R4 and R6 are hydrogen, A is -0-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, and R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro.
[0205] In some embodiments of compounds of Formula HId, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, R10 and R11 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, and fluoro substituted lower alkyl, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino.
[0206] In some embodiments of compounds of Formula Eld, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, R10 and R11 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, and fluoro substituted lower alkyl, and R5 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R5, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino.
[0207] In some embodiments, compounds of Formula m have the structure according to the following sub-generic structure Formula IHe:
Formula Hie all salts, prodrugs, tautomers, and isomers thereof, wherein:
A1 is -O-, -CR40R41-, -C(O)- or -NR48-;
Z22 is N or CR62;
Z24 is N or CR64;
Z25 is N or CR65;
Z26 is N or CR66; r is O, 1, or 2;
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R62, R64, R65 and R66 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro;
Cy is selected from the group consisting of aryl, heteroaryl, cycloalkyl, and heterocycloalkyl;
R39 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, aryl, heteroaryl, and NR50R51, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, and wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R ° is hydrogen or lower alkyl optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino;
R51 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more independent substituents R23;
R23 at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R23, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R57 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R48 at each occurrence is independently hydrogen or lower alkyl; and t is 0, 1, 2, or 3.
[0208] In some embodiments of compounds of Formula HIe, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-. In some embodiments, A1 is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, and R62, R64, R65 and R66 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0209] In some embodiments, compounds of Formula HI have the structure according to the following sub-generic structure Formula HIf:
Formula IHf all salts, prodrugs, tautomers and isomers thereof, wherein A, Z2, Z4, Z5, Z6, X, R4, R5, R6, R34, R35 and R82 are as defined for Formula III.
[0210] In some embodiments of compounds of Formula fflf, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, and R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro.
[0211] In some embodiments of compounds of Formula HHv R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, Z6 is N or CR16, R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, and one of R34 and R35 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl and optionally
substituted heteroaryl, and the other of R34 and R35 is hydrogen or lower alkyl, or R34 and R35 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl.
[0212] In some embodiments, compounds of Formula HI have the structure according to the following sub-generic structure Formula nig:
Formula lug all salts, prodrugs, tautomers, and isomers thereof, wherein:
Ai is -O-, -CR40R41-, -C(O)- or -NR48-;
Z32 is N or CR72;
Z34 is N or CR74;
Z35 is N or CR75;
Z36 is N or CR76;
X is O or S;
R48 at each occurrence is independently hydrogen or lower alkyl;
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R72, R74, R75 and R76 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl and lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro;
R67 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -C(S)NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, and -S(O)2R68; one of R60 and R61 is lower alkyl, fluoro substituted lower alkyl, or -(CH2V2R70, and the other of R60 and R61 is hydrogen or lower alkyl; or R60 and R61 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl;
R68 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R68 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted lower alkynyl, provided, however, that when R68 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O)5 S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R69 is hydrogen or optionally substituted lower alkyl;
R70 is selected from the group consisting of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl; and
R82 is hydrogen or lower alkyl.
[0213] In some embodiments of compounds of Formula HIg, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-.
[0214] In some embodiments of compounds of Formula lug, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, and R67 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -S(O)2NH2, -C(O)NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69S(O)2R68, -S(O)R68, and -S(O)2R68.
[0215] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula Illh:
Formula EDi all salts, prodrugs, tautomers and isomers thereof, wherein A, X, R4, R5, R6, R37, R38, R42, R43, R45, R46, and R47 are as defined for Formula III.
[0216] In some embodiments of compounds of Formula Illh, R4 and R6 are hydrogen, A is -0-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, and R42, R43, R45, R46 and R47 are independently selected from the group consisting of hydrogen, halogen, -OH, -CN, -NO2, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkyl, mono-alkylamino, di-alkylamino, and cycloalkylamino, further wherein at least one of, at least two of, at least three of, at least four of, or preferably all of R42, R43, R45, R46 and R47 are hydrogen.
[0217] In some embodiments, compounds of Formula in have the structure according to the following sub-generic structure Formula iπi:
Formula IIIi all salts, prodrugs, tautomers, and isomers thereof, wherein:
A1 is -O-, -CR40R41-, -C(O)- or -NR48-;
X is O or S;
R48 at each occurrence is independently hydrogen or lower alkyl;
R40 and R41 are independently selected from the group consisting of hydrogen, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino; or
R40 and R41 combine to form a 3-7 membered monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl, wherein the monocyclic cycloalkyl or monocyclic heterocycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
R67 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -C(S)NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, and -S(O)2R68; one of R60 and R61 is lower alkyl, fluoro substituted lower alkyl, or -(CH2V2R70 and the other of R60 and R61 is hydrogen or lower alkyl; or R60 and R61 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl;
R68 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R68 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted lower alkynyl, provided, however, that when R68 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted
cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R69 is hydrogen or optionally substituted lower alkyl; and R70 is selected from the group consisting of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl.
[0218] In some embodiments of compounds of Formula IHi, A] is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-.
[0219] In some embodiments of compounds of Formula HIi, A] is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, and R67 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -S(O)2NH2, -C(O)NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69S(O)2R68, -S(O)R68, and -S(O)2R68.
[0220] In some embodiments of compounds of Formula IHi, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -C(O)-, R67 is selected from the group consisting of hydrogen, halogen, lower alkyl, lower alkoxy, optionally substituted aryl, optionally substituted heteroaryl, and NR21R22, wherein R21 is hydrogen or lower alkyl, and R22 is hydrogen, lower alkyl, optionally substituted aryl or optionally substituted heteroaryl, and wherein the alkyl chain of R67, R21 or R22, when lower alkyl, or the alkyl chain of lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, and one of R60 and R61 is lower alkyl or fluoro substituted lower alkyl, and the other of R60 and R61 is hydrogen or lower alkyl. In some embodiments, A] is -C(O)-, R67 is optionally substituted aryl or optionally substituted heteroaryl, and one of R60 and R61 is lower alkyl or fluoro substituted lower alkyl, and the other of R60 and R61 is hydrogen or lower alkyl.
[0221] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula HIj :
Formula HIj
all salts, prodrugs, tautomers and isomers thereof, wherein A, R4, R5, R6, R12, R14, R15, R16, and R36 are as defined for Formula HI.
[0222] In some embodiments of compounds of Formula I-Ij, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, and R12, R14, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, preferably wherein R14 and R15 are hydrogen, more preferably wherein R12 is fluoro, R16 is hydrogen, fluoro or chloro, and R14 and R15 are hydrogen.
[0223] In some embodiments of compounds of Formula HIj, R4 and R6 are hydrogen, A is -O-, -CRaRb-, -NR1-, or -C(O)-, preferably -CH2- or -C(O)-, R12, RM, R15 and R16 are independently selected from the group consisting of hydrogen, halogen, lower alkyl and lower alkoxy, wherein the alkyl chain of lower alkyl or lower alkoxy is optionally substituted with one or more substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino and cycloalkylamino, provided, however, that any substitution on the alkyl carbon bound to the -O- of lower alkoxy is fluoro, preferably wherein R14 and R15 are hydrogen, more preferably wherein R12 is fluoro, R16 is hydrogen, fluoro or chloro, and R14 and R15 are hydrogen, and R36 is selected from the group consisting of optionally substituted C2-6 alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR19R20, where R19 and R20 are as defined for Formula III, further wherein one of R19 and R20 is hydrogen or optionally substituted lower alkyl, and the other of R19 and R20 is selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl and optionally substituted heteroaryl.
[0224] In some embodiments, compounds of Formula HI have the structure according to the following sub-generic structure Formula IIDc:
Formula IIDc
all salts, prodrugs, tautomers and isomers thereof, wherein:
A1 is -O-, -CR40R41-, -C(O)- or -NR48-;
R81 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -C(S)NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R65, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, and -S(O)2R68;
R71 and R78 are independently selected from the group consisting of hydrogen, halogen, C1-3 alkyl, and fluoro substituted Cj-3 alkyl;
R77 is selected from the group consisting of substituted methyl, optionally substituted C2-6 alkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR79R80, wherein methyl is substituted with one or more substituents selected from the group consisting of optionally substituted aryl and optionally substituted heteroaryl;
R68 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R68 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted lower alkynyl, provided, however, that when R68 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R69 is hydrogen or optionally substituted lower alkyl; and
R79 and R80 are independently hydrogen or optionally substituted lower alkyl, or R79 and R80 combine with the nitrogen to which they are attached to form optionally substituted 5-7 membered heterocycloalkyl.
[0225] Ia some embodiments of compounds of Formula ink, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -C(O)-.
[0226] In some embodiments of compounds of Formula HIk, Ai is -CR40R41- or -C(O)-, preferably -CH2- or -C(O)-, more preferably -C(O)-, and R81 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -S(O)2NH2, -C(O)NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69S(O)2R68, -S(O)R68, and -S(O)2R68.
[0227] In some embodiments, compounds of Formula III have the structure according to the following sub-generic structure Formula Illm:
Formula IHm all salts, prodrugs, tautomers and isomers thereof, wherein:
R81 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -C(S)NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, and -S(O)2R68;
R83 is selected from the group consisting of hydrogen, fluoro and chloro;
R112 is selected from the group consisting of optionally substituted C2-6 alkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR79R80;
R68 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R68 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2,
-NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted lower alkynyl, provided, however, that when R68 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to N, S, O, S(O), S(O)2, C(O) or C(S) of -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)OR68, -C(O)NR69R68, -C(S)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69C(S)R68, -NR69S(O)2R68, -NR69C(O)NH2, -NR69C(O)NR69R68, -NR69C(S)NH2, -NR69C(S)NR69R68, -NR69S(O)2NH2, -NR69S(O)2NR69R68, -S(O)R68, or -S(O)2R68, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl;
R69 is selected from the group consisting of hydrogen and optionally substituted lower alkyl; and R79 and R80 are independently hydrogen or optionally substituted lower alkyl, or R79 and R80 combine with the nitrogen to which they are attached to form optionally substituted 5-7 membered heterocycloalkyl.
[0228] In some embodiments of compounds of Formula IHm, R81 is selected from the group consisting of hydrogen, halogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, -CN, -S(O)2NH2, -C(O)NH2, -OR68, -SR68, -NR69R68, -C(O)R68, -C(S)R68, -C(O)NR69R68, -S(O)2NR69R68, -NR69C(O)R68, -NR69S(O)2R68, -S(O)R68, and -S(O)2R68.
[0229] The compounds of Formulae HIa-IIIq, and all sub-embodiments detailed herein, may be used to treat a subject suffering from or at risk for any of the protein kinase mediated diseases or conditions contemplated herein.
[0230] In one aspect, the present invention includes compounds that are useful as intermediates in the preparation of compounds of Formula III, the compounds having a structure selected from the group consisting of Formula W, Formula V, Formula VI, Formula VII, Formula VIH, and Formula IX as follows:
Z2, Z4, Z5, Z6, L2, X, s, R15, R17, R31, R32, R33, R37, R38, R42, R43, R45, R46, and R47 are as defined for Formula III;
R108 is selected from the group consisting Of -C(O)R84, -CH2I, -CH2Cl, -CH2Br, -CH2OH, and -CH2OS(O)2R109;
R109 is selected from the group consisting of lower alkyl and aryl;
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl;
R85 is selected from the group consisting of hydrogen, a nitrogen protecting group, -S(O)2R87, -C(O)NR88R89, and -C(S)NR88R89;
R86 is selected from the group consisting of hydrogen, lower alkyl, and a nitrogen protecting group;
R87 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted lower alkenyl, provided, however, that when R87 is optionally substituted lower alkenyl, no alkene carbon thereof is bound to S(O)2, optionally substituted lower alkynyl, provided, however, that when R87 is optionally substituted lower alkynyl, no alkyne carbon thereof is bound to S(O)2, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR90R91; and
R88, R89, R90 and R91 are independently selected from the group consisting of hydrogen, optionally substituted lower alkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl and optionally substituted heteroaryl; or
R88 and R89 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl.
[0231] In some embodiments of compounds of Formulae IV, V, VI, VII, or VIH, R108 is -C(O)R84, preferably wherein R84 is hydrogen. In some embodiments of compounds of Formulae IV, V, VI, VII, or Vπi, Z2 is N or CR12, Z4 is N or CR14, Z5 is N or CR15, and Z6 is N or CR16 and R12, R14, R15, R16, R , R42, R43, R45, R and R47 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0232] In some embodiments, compounds of Formula IV have the structure according to the following sub-generic structure Formula IVa:
Formula IVa wherein:
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl;
R92, R93, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy;
L4 is selected from the group consisting of -NR48-, -S-, -O-, -NR48CH(R49)-, -SCH(R49)-, -OCH(R49)-, -C(O)NR48-, -S(O)2NR48-, -CH(R49)NR48-, -CH(R49)O-, -CH(R49)S-, -NR48C(O)-, and -NR48S(O)2-;
Cy is selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl and heteroaryl;
R97 at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R97, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R49 is selected from the group consisting of hydrogen, lower alkyl, and fluoro substituted lower alkyl;
RS7 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino; and
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono- alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R48 at each occurrence is independently hydrogen or lower alkyl; and u is O, 1, 2, or 3.
[0233] In one embodiment of compounds of Formula IVa, at least two of R92, R93, R95 and R96 are hydrogen. In one embodiment, at least two of R92, R93, R95 and R96 are hydrogen, L4 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably L4 is -OCH2-, Cy is aryl or heteroaryl, and each R97 is independently selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0234] In some embodiments, compounds of Formula IV have the structure according to the following sub-generic structure Formula IVb:
Formula IVb wherein R84, R92, R93, R95, R96, R97, Cy and u are as defined for Formula IVa and r is 0, 1 or 2.
[0235] In some embodiments of compounds of Formula IVb, at least two of R92, R93, R95 and R96 are hydrogen. In some embodiments, at least two of R92, R93, R95 and R96 are hydrogen, Cy is aryl or heteroaryl, and each R97 is independently selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0236] In some embodiments, compounds of Formula V have the structure according to the following sub-generic structure Formula Va:
Formula Va wherein:
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R92, R93, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; R98 is selected from the group consisting of hydrogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; and s is 0, 1 , or 2;
[0237] In some embodiments, compounds of Formula VI have the structure according to the following sub-generic structure Formula Via:
Formula Via wherein:
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R92, R94, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; L4 is selected from the group consisting of -NR48-, -S-, -O-, -NR48CH(R49)-, -SCH(R49)-,
-OCH(R49)-, -C(O)NR48-, -S(O)2NR48-, -CH(R49)NR48-, -CH(R49)O-, -CH(R49)S-,
-NR48C(O)-, and -NR48S(O)2-; Cy is selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl and heteroaryl;
Ryy at each occurrence is independently selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR57, -SR57, -NR48R57, -NR48C(O)R57, -NR48S(O)2R57, -S(O)2R57, -C(O)R57, -C(O)OR57, -C(O)NR48R57, -S(O)2NR48R57, halogen, lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R97, or as substituents of lower alkyl, are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R49 is selected from the group consisting of hydrogen, lower alkyl, and fluoro substituted lower alkyl;
R57 is selected from the group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR57, -SR57, -NR48R57, -C(O)OR57, -C(O)NR48R57, or -S(O)2NR48R57 is fluoro, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R57 or as substituents of lower alkyl are optionally substituted with one or more substituents selected from the group consisting of -OH, -NH2, -CN, -NO2, -C(O)OH, -S(O)2NH2, -C(O)NH2, -OR58, -SR58, -NR48R58, -NR48C(O)R58, -NR48S(O)2R58, -S(O)2R58, -C(O)R58, -C(O)OR58, -C(O)NR48R58, -S(O)2NR48R58, halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
R58 at each occurrence is independently selected from the group consisting of lower alkyl, heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted with one or more substituents selected from the group consisting of fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono- alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -OR58, -SR58, -NR48R58, -C(O)OR58, -C(O)NR48R58, or -S(O)2NR48R58 is fluoro;
R48 at each occurrence is independently hydrogen or lower alkyl; and u is O, 1, 2 or 3.
[0238] In some embodiments of compounds of Formula Via, at least two of R92, R94, R95 and R96 are hydrogen. In some embodiments, at least two of R92, R94, R95 and R96 are hydrogen, L4 is -NR48CH(R49)-, -SCH(R49)-, or -OCH(R49)-, preferably L4 is -OCH2-, Cy is aryl or heteroaryl, and each R97 is independently selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0239] Ia some embodiments, compounds of Formula VI have the structure according to the following sub-generic structure Formula VIb:
Formula VIb wherein R84, R92, R94, R95, R96, R97, Cy and u are as defined for Formula Via and r is 0, 1 or 2.
[0240] In some embodiments of compounds of Formula VIb, at least two of R92, R94, R95 and R96 are hydrogen. In some embodiments, at least two of R92, R94, R95 and R96 are hydrogen, Cy is aryl or heteroaryl, and each R97 is independently selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0241] In some embodiments, compounds of Formula VII have the structure according to the following sub-generic structure Formula Vila:
Formula Vila wherein:
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R92, R94, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; R99 is selected from the group consisting of optionally substituted lower alkyl, optionally substituted aryl, optionally substituted heteroaryl, and -NR79R80; and R79 and R80 are independently hydrogen or optionally substituted lower alkyl, or R79 and R80 combine with the nitrogen to which they are attached to form optionally substituted 5-7 membered heterocycloalkyl.
[0242] In some embodiments of compounds of Formula Vila, one of R92 and R96 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy, and the other of R92 and R96 is selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; in further embodiments, one of R92 and R96 is hydrogen, fluoro or chloro, and the other of R92 and R96 is fluoro or chloro; in further embodiments, R92 is fluoro and R96 is hydrogen, fluoro, or chloro; in further embodiments, R92 and R96 are both fluoro.
[0243] In some embodiments of compounds of Formula Vila, R94 and R95 are hydrogen; in further embodiments, one of R92 and R96 is selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy and the other of R92 and R96 is selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; in further embodiments, one of R92 and R96 is selected from hydrogen, fluoro or chloro and the other of R92 and R96 is selected from fluoro or chloro; in further embodiments, R92 is fluoro and R96 is selected from hydrogen, fluoro, or chloro; in further embodiments, R92 and R96 are both fluoro.
[0244] In some embodiments, compounds of Formula VII have the structure according to the following sub-generic structure Formula VIIb:
Formula VIIb wherein: X is O or S;
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R92, R94, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; R82 is hydrogen or lower alkyl; one of R100 and R101 is lower alkyl, fluoro substituted lower alkyl, or -(CH2)O-2R70, and the other of
R100 and R101 is hydrogen or lower alkyl; or R100 and R101 together with the nitrogen to which they are attached form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl; and
R70 is selected from the group consisting of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl.
[0245] In some embodiments of compounds of Formula VIIb, at least two of R92, R94, R95 and R96 are hydrogen. In some embodiments, at least two of R92, R94, R95 and R96 are hydrogen, and R70 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0246] In some embodiments, compounds of Formula VIII have the structure according to the following sub-generic structure Formula Villa:
Formula Villa wherein: X is O or S;
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R102, R103, R105, R106, and R107 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; one ofR100 and R101 is lower alkyl, fluoro substituted lower alkyl, or -(CH2)o-2R7° and the other of
R100 and R101 is hydrogen or lower alkyl; or R100 and R101 together with the nitrogen to which they are attached form optionally substituted
5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl; and R70 is selected from the group consisting of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl.
[0247] In some embodiments of compounds of Formula Villa, at least two, also at least three, also at least four, or all of R102, R103, R105, R106, and R107 are hydrogen. In some embodiments, at least two, also at least three, also at least four, or all of R102, R103, R105, R106, and R107 are hydrogen, and R70 is aryl or heteroaryl, wherein aryl and heteroaryl are optionally substituted with one or more substituents selected from the group consisting of halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy.
[0248] In some embodiments, compounds of Formula EX have the structure according to the following sub-generic structure Formula EXa:
Formula EXa wherein:
R84 is selected from the group consisting of hydrogen, lower alkoxy, -OH, and -Cl; R92, R95, and R96 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; R98 is selected from the group consisting of hydrogen, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, and fluoro substituted lower alkoxy; and s is 0, 1, or 2;
[0249] In some embodiments of any of the above embodiments of compounds of Formula EVa, EVb, Va, Via, VIb, Vila, VIIb, Villa, or EXa, R84 is hydrogen.
[0250] In some embodiments of the above compounds, compounds are excluded where N (except where N is a heteroaryl ring atom), O, or S is bound to a carbon that is also bound to N (except where N is a heteroaryl ring atom), O, or S; or where N (except where N is a heteroaryl ring atom), O, C(S), C(O), or S(O)n (n is 0-2) is bound to an alkene carbon of an alkenyl group or bound to an alkyne carbon of an alkynyl group; accordingly, in some embodiments compounds which include linkages such as the following are excluded from the present invention: -NR-CH2-NR-, -0-CH2-NR-, -S-CH2-NR-, -NR-CH2-O-, -0-CH2-O-, -S-CH2-O-, -NR-CH2-S-, -0-CH2-S-, -S-CH2-S-, -NR-CH=CH-, -CH=CH-NR-, -NR-C ≡€-, -C sC-NR-, -0-CH=CH-, -CH=CH-O-, -O-C ≡C-, -C sC-O-, -S(OV2-CH=CH-, -CH=CH-S(O)0-2-, -S(O)0.2-C ≡€-, -C HC-S(O)0-2-, -C(O)-CH=CH-, -CH=CH-C(O)-, -C ≡€-C(O)-, or -C(O)-C sC-, -C(S)-CH=CH-, -CH=CH-C(S)-, -Cs€-C(S)-, or -C(S)-C sC-.
[0251] In reference to compounds herein, specification of a compound or group of compounds includes pharmaceutically acceptable salts of such compound(s) unless clearly indicated to the contrary, prodrug(s), and all stereoisomers. In reference to compositions, kits, methods of use, etc. of compounds of Formula I described herein, it is understood that a compound of Formula I includes compounds of Formulae Ia-Iz, and all sub-embodiments thereof, compounds of Formula II, including Formulae ECa-IIo, and all sub-embodiments thereof, and compounds of Formula IEt, including Formulae EtIa-IIIq, and all sub-embodiments thereof, unless indicated otherwise. In reference to
compositions, kits, methods of use, etc. of compounds of Formula II described herein, it is understood that this includes compounds of Formulae Ila-IIo, and all sub-embodiments thereof, unless indicated otherwise. In reference to compositions, kits, methods of use, etc. of compounds of Formula III described herein, it is understood that this includes compounds of Formulae IHa-IIIq, and all sub- embodiments thereof, unless indicated otherwise.
[0252] In one aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula I. The terms "treat," "therapy," and like terms refer to the administration of material, e.g., compound of Formula I, in an amount effective to prevent, alleviate, or ameliorate one or more symptoms of a disease or condition, i.e., indication, and/or to prolong the survival of the subject being treated. The term "protein kinase mediated disease or condition" refers to a disease or condition in which the biological function of a protein kinase affects the development and/or course of the disease or condition, and/or in which modulation of the protein kinase alters the development, course, and/or symptoms of the disease or condition. A protein kinase mediated disease or condition includes a disease or condition for which modulation provides a therapeutic benefit, e.g. wherein treatment with protein kinase inhibitors, including compounds described herein, provides a therapeutic benefit to the subject suffering from or at risk of the disease or condition. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula I in combination with one or more other therapies for the disease or condition.
[0253] In one aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of any one or more of Formula Ia through Formula Iz, and all sub- embodiments thereof.
[0254] In another aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula II,
[0255] In another aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of any one or more of Formula Ha through Formula IIo, and all sub- embodiments thereof.
[0256] In another aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula III.
[0257] In another aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of any one or more of Formula IHa through Formula πio, and all sub-embodiments thereof.
[0258] In one aspect, the invention provides methods for treating a Raf protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula I. The terms "Raf protein kinase mediated disease or condition," "Raf mediated disease or condition," and the like refer to a disease or condition in which the biological function of a Raf kinase, including any mutations thereof, affects the development and/or course of the disease or condition, and/or in which modulation of the Raf protein kinase alters the development, course, and/or symptoms of the disease or condition. The Raf protein kinase includes, but is not limited to, B-Raf, mutations of B-Raf, c-Raf-1 and mutations of c-Raf-1. In some embodiments, the Raf protein kinase is B-Raf mutation V600E. In further embodiments, the disease or condition is a cancer that is amenable to treatment by an inhibitor of the V600E mutant B-Raf. A Raf protein kinase mediated disease or condition includes a disease or condition for which Raf inhibition provides a therapeutic benefit, e.g. wherein treatment with Raf inhibitors, including compounds described herein, provides a therapeutic benefit to the subject suffering from or at risk of the disease or condition. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula I in combination with one or more other therapies for the disease or condition.
[0259] In one aspect, the invention provides methods for treating a Raf protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula IIa or EI. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula IIa or HI in combination with one or more other therapies for the disease, further wherein the compound is of Formula I-Ij, IHk, or IHm. The Raf protein kinase includes, but is not limited to, B-Raf, mutations of B-Raf, c-Raf- 1 and mutations of c-Raf-1. In some embodiments, the Raf protein kinase is B-Raf mutation V600E. In further embodiments, the disease or condition is a cancer that is amenable to treatment by an inhibitor of the V600E mutant B-Raf.
[0260] In one aspect, the invention provides methods for treating a Fms protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula I. The terms "Fms protein kinase mediated disease or condition," "Fms mediated disease or condition," and the like refer to a disease or condition in which the biological function of a Fms protein kinase, including any mutations thereof, affects the development and/or course of the disease or condition, and/or in which modulation of Fms alters the
development, course, and/or symptoms of the disease or condition. A Fms mediated disease or condition includes a disease or condition for which Fms inhibition provides a therapeutic benefit, e.g. wherein treatment with Fms inhibitors, including compounds described herein, provides a therapeutic benefit to the subject suffering from or at risk of the disease or condition. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula I in combination with one or more other therapies for the disease or condition.
[0261] In one aspect, the invention provides methods for treating a Kit protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula I. The terms "Kit mediated disease or condition," "Kit protein kinase mediated disease or condition," and the like refer to a disease or condition in which the biological function of a Kit protein kinase, including any mutation thereof, affects the development and/or course of the disease or condition, and/or in which modulation of Kit alters the development, course, and/or symptoms of the disease or condition. A Kit mediated disease or condition includes a disease or condition for which Kit inhibition provides a therapeutic benefit, e.g. wherein treatment with Kit inhibitors, including compounds described herein, provides a therapeutic benefit to the subject suffering from or at risk of the disease or condition. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula I in combination with one or more other therapies for the disease or condition.
[0262] In one aspect, the invention provides methods for treating a Jnk protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a compound of Formula I. The terms "Jnk mediated disease or condition," "Jnk protein kinase mediated disease or condition," and the like refer to a disease or condition in which the biological function of a Jnk kinase, e.g. Jnkl, Jnk2, Jnk3, or any mutation thereof, affects the development and/or course of the disease or condition, and/or in which modulation of the Jnk kinase alters the development, course, and/or symptoms of the disease or condition. A Jnk mediated disease or condition includes a disease or condition for which Jnk inhibition provides a therapeutic benefit, e.g. wherein treatment with Jnk inhibitors, including compounds described herein, provides a therapeutic benefit to the subject suffering from or at risk of the disease or condition. In one aspect, the method involves administering to the subject an effective amount of a compound of Formula I in combination with one or more other therapies for the disease or condition. The Jnk protein kinase includes, but is not limited to, Jnkl, Jnk2, or Jnk3.
[0263] In some embodiments, a compound of Formula I will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted kinase activity assay. In some embodiments, a compound of any of Formula I will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20
nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of AbI, Aktl, Akt2, Akt3, ALK, Alk5, B-Raf, Brk, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, FGFRl, FGFR2, FGFR3, FGFR4, Fltl, Flt3, Flt4, Fms, Frk, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kdr, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDGFRA, PDGFRB, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, Plkl, Pyk2, ROCKl, ROCK2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, and Zaρ70, including any mutations thereof.
[0264] In some embodiments, a compound of Formula I will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of AbI, Akt 1, Akt2, Akt3, ALK, Alk5, B-Raf, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, Fms, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, Plkl, Pyk2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, and Zap70, including any mutations thereof.
[0265] In some embodiments, a compound of Formula I will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of AbI, B-Raf, Btk, c-Raf-1, EGFR, EphB2, Erk2, Fak, FGFRl, Fltl, Flt3, Flt4, Fms, Irak4, Jnkl, Jnk2, Jnk3, Kdr, Kit, MAP2K1, MAPKAP kinase 2, Met, p38, PDGFRB, Piml, PKC theta, Pyk2, Ret, Src, Stk6, Yes, and Zap70, including any mutations thereof.
[0266] In some embodiments, a compound of Formula I will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of AbI, B-Raf, Btk, c-Raf-1, EGFR, EphB2, Erk2, Fak, Fms, Irak4, Jnkl, Jnk2, Jnk3, Kit, MAP2K1, MAPKAP kinase 2, Met, p38, Piml, PKC theta, Pyk2, Src, Stk6, Yes, and Zaρ70, including any mutations thereof.
[0267] In some embodiments, a compound of Formula II will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of B-Raf, B-Raf V600E mutant, c-Raf-1, FGFRl, FGFR2, FGFR3, FGFR4, Jnkl, Jnk2, Jnk3, Met, Piml, Pim2, Pim3, Pyk2, Kdr and Ret, including any mutations thereof.
[0268] Ih some embodiments, a compound of Formula III will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of B-Raf, c-Raf-1, Fms, Jnkl, Jnk2, Jnk3, and Kit, and any mutations thereof. In some embodiments, a compound of any of Formula HI will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to at least one kinase selected from the group consisting of B-Raf, B-Raf V600E mutant, c-Raf-1, Fms, Jnkl, Jnk2, Jnk3, and Kit, preferably B-Raf, B-Raf V600E mutant or c-Raf-1.
[0269] In some embodiments, a compound of Formula III is an inhibitor of a Raf kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Raf kinase activity assay. In some embodiments, a compound of Formula III will have an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM with respect to B-Raf, c-Raf-1, or B-Raf V600E mutant. In some embodiments, a compound of Formula III will selectively inhibit one Raf kinase relative to one or more other Raf kinases. In some embodiments, the compound of Formula III will selectively inhibit a mutation of the Raf kinase relative to the wild type kinase, for example B-Raf V600E relative to wild type B-Raf.
[0270] In some embodiments, a compound of Formula III is an inhibitor of a Fms kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Fms kinase activity assay. In some embodiments, a compound of Formula III will selectively inhibit Fms kinase relative to Kit kinase.
[0271] In some embodiments, a compound of Formula III is an inhibitor of a Kit kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Kit kinase activity assay.
[0272] In some embodiments, a compound of Formula III is an inhibitor of a Jnk kinase and has an ICso of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Jnk kinase activity assay. In some embodiments, a compound of Formula III is an inhibitor of a Jnkl kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Jnkl kinase activity assay. In some embodiments, a compound of Formula III is an inhibitor of a Jnk2 kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Jnk2 kinase activity assay. In some embodiments, a compound of
Formula HI is an inhibitor of a Jnk3 kinase and has an IC50 of less than 500 nm, less than 100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM, or less than 1 nM as determined in a generally accepted Jnk3 kinase activity assay. In some embodiments, a compound of Formula HI will selectively inhibit one Jnk kinase relative to one or more other Jnk kinases, such as selectively inhibiting Jnk 1 relative to Jnk 2 and/or Jnk3, selectively inhibiting Jnk2 relative to Jnk3 and/or Jnkl, or selectively inhibiting Jnk3 relative to Jnkl and/or Jnk 2.
[0273] Further to any of the above mentioned embodiments, a compound of the invention will also inhibit the effects of a mutation of the kinase, including, but not limited to, a mutation that is related to a disease state, such as a cancer. For example, B-Raf V600E mutant is present in a high percentage of some cancers, such as melanoma, and compounds of the invention will inhibit the kinase activity of this mutant.
[0274] Further to any of the above embodiments, a compound of the invention may selectively inhibit one kinase relative to one or more other kinases, where preferably inhibition is selective with respect to any of the other kinases, whether a kinase discussed herein, or other kinases. In some embodiments, the compound may selectively inhibit the effects of a mutation of the kinase relative to the wild type kinase, for example B-Raf V600E relative to wild type B-Raf. In some embodiments, the compound may selectively inhibit Fms relative to Kit. Selective inhibition of one kinase relative to another is such that the IC50 for the one kinase may be at least about 2-fold, also 5-fold, also 10-fold, also 20-fold, also 50-fold, or at least about 100-fold less than the IC50 for any of the other kinases as determined in a generally accepted kinase activity assay.
[0275] In another aspect, the invention provides compositions that include a therapeutically effective amount of a compound of Formula I and at least one pharmaceutically acceptable carrier, excipient, and/or diluent. The composition can include a plurality of different pharmacologically active compounds, which can include a plurality of compounds of Formula I. In another aspect, the composition can include one or more compounds of Formula I, Formula II, or Formula III along with one or more compounds that are therapeutically effective for the same disease indication. In one aspect, the composition includes one or more compounds of Formula I, Formula II, or Formula III along with one or more compounds that are therapeutically effective for the same disease indication, wherein the compounds have a synergistic effect on the disease indication.
[0276] In another aspect, the invention provides compositions that include a therapeutically effective amount of at least one compound of Formula III and at least one pharmaceutically acceptable carrier, excipient, and/or diluent. The composition can include a plurality of different pharmacologically active compounds, which can include a plurality of compounds of Formula III, or can include at least one compound of Formula UI along with at least one compound that is
therapeutically effective for the same disease indication. In one aspect, the at least one compound of Formula HI and the at least one compound that is therapeutically effective for the same disease indication have a synergistic effect on the disease indication. In one aspect, the composition includes one or more compounds of Formula III effective in treating a cancer and one or more other compounds that are effective in treating the cancer, further wherein the compounds are synergistically effective in treating the cancer.
[0277] In another aspect, the invention provides a method for modulating the activity of a protein kinase selected from the group consisting of AbI, Aktl, Akt2, Akt3, ALK, Alk5, B-Raf, Brk, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, FGFRl, FGFR2, FGFR3, FGFR4, Fltl, Flt3, Flt4, Fms, Frk, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, KK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kdr, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDGFRA, PDGFRB, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, PM, Pyk2, ROCKl, R0CK2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, or Zap70 by contacting the protein kinase with an effective amount of a compound of Formula I.
[0278] In another aspect, the invention provides methods for treating a protein kinase mediated disease or condition in an animal subject, wherein the method involves administering to the subject an effective amount of a composition including a compound of Formula I.
[0279] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of AbI, Akt 1, Akt2, Akt3, ALK, Alk5, B-Raf, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, FGFRl, FGFR2, FGFR3, FGFR4, Fltl, Flt3, Flt4, Fms, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kdr, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDGFRA, PDGFRB, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, Plkl, Pyk2, ROCKl, ROCK2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, and Zap70 by administering to the subject an effective amount of a composition including a compound of Formula I.
[0280] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of AbI, Akt 1, Akt2, Akt3, ALK, Alk5, B-Raf, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, Fms, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, Plkl, Pyk2, Ron,
Src, Stk6, Syk, TEC, Tie23 TrkA, Yes, and Zap70 by administering to the subject an effective amount of a composition including a compound of Formula I.
[0281] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of AbI, B-Raf, Btk, c-Raf-1, EGFR, EphB2, Erk2, Fak, FGFRl, Fltl, Flt3, Flt4, Fms, Irak4, Juki, Jnk2, Jnk3, Kdr, Kit, MAP2K1, MAPKAPK2, Met, p38, PDGFRB, Piml, PKC theta, Pyk2, Ret, Src, Stkό, Yes, and Zap70 by administering to the subject an effective amount of a composition including a compound of Formula I.
[0282] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of AbI, B-Raf, Btk, c-Raf-1, EGFR, EphB2, Erk2, Fak, Fms, Irak4, Juki, Jnk2, Jnk3, Kit, MAP2K1, MAPKAPK2, Met, p38, Piml, PKC theta, Pyk2, Src, Stkό, Yes, and Zap70 by administering to the subject an effective amount of a composition including a compound of Formula I.
[0283] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of B-Raf, B-Raf V600E mutant, c-Raf-1, FGFRl, FGFR2, FGFR3, FGFR4, Juki, Jnk2, Jnk3, Met, Piml, Pim2, Pim3, Pyk2, Kdr and Ret by administering to the subject an effective amount of a composition including a compound of Formula I.
[0284] In one aspect, the invention provides methods for treating a disease or condition mediated by a protein kinase selected from the group consisting of B-Raf, c-Raf-1, Fms, Md, Jnk2, Jnk3, and Kit, and any mutations thereof, by administering to the subject an effective amount of a composition including a compound of Formula in.
[0285] In one aspect, the invention provides methods for treating a disease or condition mediated by B-Raf, c-Raf-1, or B-Raf V600E by administering to the subject an effective amount of a composition including a compound of Formula II or Formula III, where in further embodiments, the compound is of Formula Ha or Formula III. In one aspect, the invention provides methods for treating a disease or condition mediated by B-Raf, c-Raf-1, or B-Raf V600E by administering to the subject an effective amount of a composition including a compound of Formula fflj, IIUc, or IHm in combination with one or more other suitable therapies for treating the disease. In one aspect, the invention provides methods for treating a cancer mediated by B-Raf V600E mutant by administering to the subject an effective amount of a composition of Formula IIIj, UIk, or IIIm in combination with one or more suitable anticancer therapies, such as one or more chemotherapeutic drugs.
[0286] In one aspect, the invention provides a method of treating a cancer by administering to the subject an effective amount of a composition including a compound of Formula I, or where the compound is of Formula III, or where the compound is of Formula IIIj, HIk, or IHm, in combination
with one or more other therapies or medical procedures effective in treating the cancer. Other therapies or medical procedures include suitable anticancer therapy (e.g. drug therapy, vaccine therapy, gene therapy, photodynamic therapy) or medical procedure (e.g. surgery, radiation treatment, hyperthermia heating, bone marrow or stem cell transplant). In one aspect, the one or more suitable anticancer therapies or medical procedures is selected from treatment with a chemotherapeutic agent (e.g. chemotherapeutic drug), radiation treatment (e.g. x-ray, γ-ray, or electron, proton, neutron, or α particle beam), hyperthermia heating (e.g. microwave, ultrasound, radiofrequency ablation), Vaccine therapy (e.g. AFP gene hepatocellular carcinoma vaccine, AFP adenoviral vector vaccine, AG-858, allogeneic GM-CSF-secretion breast cancer vaccine, dendritic cell peptide vaccines), gene therapy (e.g. Ad5CMV-p53 vector, adenovector encoding MDA7, adenovirus 5-tumor necrosis factor alpha), photodynamic therapy (e.g. aminolevulinic acid, motexafm lutetium), surgery, and bone marrow and stem cell transplantation.
[0287] In a preferred embodiment, the invention provides a method of treating a cancer by administering to the subject an effective amount of a composition including a compound of Formula I, or wherein the compound is of Formula HI, or wherein the compound is of Formula IHj, Illk, or Him, in combination with one or more suitable chemotherapeutic agents. In one aspect, the one or more suitable chemotherapeutic agents is selected from an alkylating agent, including, but not limited to, adozelesin, altretamine, bizelesin, busulfan, carboplatin, carboquone, carmustine, chlorambucil, cisplatin, cyclophosphamide, dacarbazine, estramustine, fotemustine, hepsulfam, ifosfamide, improsulfan, irofulven, lomustine, mechlorethamine, melphalan, oxaliplatin, piposulfan, semustine, streptozocin, temozolomide, thiotepa, and treosulfan; an antibiotic, including, but not limited to, bleomycin, dactinomycin, daunorubicin, doxorubicin, epirubicin, idarubicrn, menogaril, mitomycin, mitoxantrone, neocarzinostatin, pentostatin, and plicamycin; an antimetabolite, including, but not limited to, azacitidine, capecitabine, cladribine, clofarabine, cytarabine, decitabine, floxuridine, fludarabine, 5-fluorouracil, ftorafur, gemcitabine, hydroxyurea, mercaptopurine, methotrexate, nelarabine, pemetrexed, raltitrexed, thioguanine, and trimetrexate; an immunotherapy, including, but not limited to, alemtuzumab, bevacizumab, cetuximab, galiximab, gemtuzumab, panitumumab, pertuzumab, riτuximab, tositumomab, trastuzumab, and 90 Y ibritumomab tiuxetan; a hormone or hormone antagonist, including, but not limited to, anastrozole, androgens, buserelin, diethylstilbestrol, exemestane, flutamide, fulvestrant, goserelin, idoxifene, letrozole, leuprolide, magestrol, raloxifene, tamoxifen, and toremifene; a taxane, including, but not limited to, DJ-927, docetaxel, TPI 287, paclitaxel and DHA-paclitaxel; a retinoid, including, but not limited to, alitretinoin, bexarotene, fenretinide, isotretinoin, and tretinoin; an alkaloid, including, but not limited to, etoposide, homoharringtonine, teniposide, vinblastine, vincristine, vindesine, and vinorelbine; an antiangiogenic agent, including, but not limited to, AE-941 (GW786034, Neovastat), ABT-510, 2-methoxyestradiol, lenalidomide, and thalidomide; a topoisomerase inhibitor, including, but not limited to, amsacrine,
edotecarin, exatecan, irinotecan (also active metabolite SN-38 (7-ethyl-lO-hydroxy-camptothecin)), rubitecan, topotecan, and 9-aminocamptothecin; a kinase inhibitor, including, but not limited to, erlotinib, gefϊtinib, flavopiridol, imatinib mesylate, lapatinib, sorafenib, sunitinib malate, AEE-788, AG-013736, AMG 706, AMN107, BMS-354825, BMS-599626, UCN-01 (7-hydroxystaurosporine), and vatalanib; a targeted signal transduction inhibitor including, but not limited to bortezomib, geldanamycin, and rapamycin; a biological response modifier, including, but not limited to, imiquimod, interferon-α, and interleukin-2; and other chemotherapeutics, including, but not limited to 3-AP (3-amino-2-carboxyaldehyde thiosemicarbazone), aminoglutethimide, asparaginase, bryostatin- 1, cilengitide, E7389, ixabepilone, procarbazine, sulindac, temsirolimus, tipifarnib. Preferably, the method of treating a cancer involves administering to the subject an effective amount of a composition of Formula I (more preferably Formula III, and even more preferably Formulae IHj, TTTIc, or Illm) in combination with a chemotherapeutic agent selected from 5-fluorouracil, carboplatin, dacarbazine, gefitinib, oxaliplatin, paclitaxel, SN-38, temozolomide, vinblastine, bevacizumab, cetuximab, or erlotinib.
[0288] In another aspect, the invention provides a method of treating or prophylaxis of a disease or condition in a mammal, by administering to the mammal a therapeutically effective amount of a compound of Formula I, a prodrug of such compound, or a pharmaceutically acceptable salt of such compound or prodrug. The compound can be alone or can be part of a composition. In another aspect, the invention provides a method of treating or prophylaxis of a disease or condition in a mammal, by administering to the mammal a therapeutically effective amount of a compound of Formula HJ, a prodrug of such compound, or a pharmaceutically acceptable salt of such compound or prodrug in combination with one or more other suitable therapies for the disease or condition.
[0289] In another aspect, the invention provides compositions that include a therapeutically effective amount of a compound of Formula III and at least one pharmaceutically acceptable carrier, excipient, and/or diluent. The composition can include a plurality of different pharmacologically active compounds, which can include a plurality of compounds of Formula III.
[0290] In a related aspect, the invention provides kits that include a composition as described herein. In some embodiments, the composition is packaged, e.g., in a vial, bottle, flask, which may be further packaged, e.g., within a box, envelope, or bag; the composition is approved by the U.S. Food and Drug Administration or similar regulatory agency for administration to a mammal, e.g., a human; the composition is approved for administration to a mammal, e.g., a human, for a protein kinase mediated disease or condition; the invention kit includes written instructions for use and/or other indication that the composition is suitable or approved for administration to a mammal, e.g., a human, for a protein kinase-mediated disease or condition; and the composition is packaged in unit dose or single dose form, e.g., single dose pills, capsules, or the like.
[0291] In aspects involving treatment or prophylaxis of a disease or condition with the compounds of Formula I, the disease or condition is, for example without limitation, neurologic diseases such as ischemic stroke, cerebrovascular ischemia, multi-infarct dementia, head injury, spinal cord injury, Alzheimer's disease (AD), Parkinson's disease, amyotrophic lateral sclerosis, dementia, senile chorea, and Huntington's disease; neoplastic diseases and associated complications, including chemotherapy- induced hypoxia, gastrointestinal stromal tumors (GISTs), prostate rumors, mast cell tumors (including canine mast cell tumors), acute myeloid leukemia, acute lymphocytic leukemia, chronic myeloid leukemia, chronic lymphocytic leukemia, multiple myeloma, melanoma, mastocytosis, gliomas, glioblastoma, astrocytoma, neuroblastoma, sarcomas (e.g. sarcomas of neuroectodermal origin, leiomyosarcoma), carcinomas (e.g. lung, breast, pancreatic, colon, hepatocellular, renal, female genital tract, squamous cell, carcinoma in situ), lymphoma (e.g. histiocytic lymphoma, non-Hodgkin's lymphoma), MEN2 syndromes, neurofibromatosis (including Schwann cell neoplasia), myelodysplastic syndrome, leukemia, tumor angiogenesis, and cancers of the thyroid, liver, bone, skin, brain, central nervous system, pancreas, lung (e.g. small cell lung cancer, non small cell lung cancer), breast, colon, bladder, prostate, gastrointestinal tract, endometrium, fallopian tube, testes and ovary; pain of neuropathic or inflammatory origin, including acute pain, chronic pain, and migraine; cardiovascular diseases including heart failure, cardiac hypertrophy, thrombosis (e.g. thrombotic microangiopathy syndromes), atherosclerosis, reperfusion injury and ischemia (e.g. cerebrovascular ischemia, liver ischemia); inflammation including, but not limited to, polycystic kidney disease (PKD), age-related macular degeneration, rheumatoid arthritis, allergic rhinitis, inflammatory bowel disease (IBD), ulcerative colitis, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, Wegener's granulomatosis, psoriasis, scleroderma, chronic thyroiditis, Grave's disease, myasthenia gravis, multiple sclerosis, osteoarthritis, endometriosis, scarring (e.g. dermal, tissue), vascular restenosis, fibrotic disorders, hypereosinophilia, CNS inflammation, pancreatitis, nephritis, atopic dermatitis, and hepatitis; immunodeficiency diseases (e.g. severe combined immunodeficiency (SCID)), organ transplant rejection, graft versus host disease; renal or prostatic diseases including diabetic nephropathy, nephrosclerosis, glomerulonephritis, interstitial nephritis, Lupus nephritis, prostate hyperplasia, chronic renal failure, tubular necrosis, diabetes-associated renal complications, and hypertrophy; metabolic diseases including type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity, hepatic steatosis, insulin resistance, hyperglycemia, lipolysis and obesity; infection, including, but not limited to Helicobacter pylori and Influenza virus, fever, sepsis; pulmonary diseases including chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), asthma, allergy, bronchitis, emphysema, and pulmonary fibrosis; genetic developmental diseases such as Noonan's syndrome, Crouzon syndrome, acrocephalo-syndactyly type I, Pfeiffer's syndrome, Jackson-Weiss syndrome, Costello syndrome, (faciocutaneoskeletal syndrome), leopard syndrome, cardio-faciocutaneous syndrome (CFC) and neural crest syndrome abnormalities causing cardiovascular, skeletal, intestinal, skin, hair and endocrine diseases; disorders
of bone structure or mineralization, including osteoporosis, increased risk of fracture, hypercalcemia, and bone metastases; Grave's disease; Hirschsprung's disease; lymphoedema; selective T-cell defect (STD); X-linked agammaglobulinemia; diabetic retinopathy; alopecia; erectile dysfunction; and tuberous sclerosis.
[0292] In a related aspect, compounds of Formula I, further where the compound is of Formula IH, can be used in the preparation of a medicament for the treatment of a B-Raf-mediated disease or condition, selected from the group consisting of neurologic diseases such as ischemic stroke, multi- infarct dementia, head injury, spinal cord injury, Alzheimer's disease (AD), Parkinson's disease; neoplastic diseases including, but not limited to, melanoma, glioma, sarcoma, carcinoma (e.g. lung, breast, pancreatic, renal), lymphoma (e.g. histiocytic lymphoma) and cancer of the thyroid, lung (e.g. small cell lung cancer), liver, breast, ovary and colon, neurofibromatosis, myelodysplastic syndrome, leukemia, tumor angiogenesis; pain of neuropathic or inflammatory origin, including acute pain, chronic pain, and migraine; cardiovascular diseases including heart failure, cardiac hypertrophy, thrombosis (e.g. thrombotic microangiopathy syndromes), atherosclerosis, reperfusion injury; inflammation including, but not limited to, psoriasis, polycystic kidney disease (PKD), arthritis and autoimmune diseases and conditions, osteoarthritis, endometriosis, scarring, vascular restenosis, fibrotic disorders, rheumatoid arthritis, inflammatory bowel disease (IBD); immunodeficiency diseases, organ transplant rejection, graft versus host disease; renal or prostatic diseases including diabetic nephropathy, nephrosclerosis, glomerulonephritis, prostate hyperplasia; metabolic disorders, obesity; infection, including, but not limited to Helicobacter pylori and Influenza virus, fever, sepsis; pulmonary diseases including chronic obstructive pulmonary disease (COPD) and acute respiratory distress syndrome (ARDS); genetic developmental diseases such as Noonan's syndrome, Costello syndrome, (faciocutaneoskeletal syndrome), leopard syndrome, cardio-faciocutaneous syndrome (CFC), and neural crest syndrome abnormalities causing cardiovascular, skeletal, intestinal, skin, hair and endocrine diseases.
[0293] In a related aspect, compounds of Formula in, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a c-Raf-1 -mediated disease or condition selected from the group consisting of colorectal, ovarian, lung and renal cell carcinoma, acute myeloid leukemia, myelodysplastic syndromes, tumor angiogenesis, and neuroendocrine tumors such as medullary thyroid cancer, carcinoid, small cell lung cancer and pheochromocytoma.
[0294] In a related aspect, compounds of Formula III, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a Fms-mediated disease or condition selected from the group consisting of immune disorders, including rheumatoid arthritis, systemic lupus erythematosis (SLE), Wegener's granulomatosis, and transplant rejection, inflammatory diseases including Chronic Obstructive Pulmonary Disease (COPD), emphysema, and
atherosclerosis, metabolic disorders, including insulin resistance, hyperglycemia, and lipolysis, disorders of bone structure or mineralization, including osteoporosis, increased risk of fracture, hypercalcemia, and bone metastases, kidney diseases, including nephritis (e.g. glomerulonephritis, interstitial nephritis, Lupus nephritis), tubular necrosis, diabetes-associated renal complications, and hypertrophy and cancers, including multiple myeloma, acute myeloid leukemia, chronic myeloid leukemia (CML), breast cancer, and ovarian cancer.
[0295] In a related aspect, compounds of Formula III, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a Jnk-mediated disease or condition selected from the group consisting of Metabolic diseases including type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity, and hepatic steatosis; cardiovascular diseases such as atherosclerosis, ischemia (e.g. cerebrovascular ischemia, liver ischemia), reperfusion injury, cardiac hypertrophy; renal diseases such as chronic renal failure; neoplastic diseases and associated complications, including chemotherapy-induced hypoxia, prostate tumors, myeloid leukemia and cancers of the liver, bone, skin, brain, pancreas, lung breast, colon, prostate and ovary; transplant rejection; pain of neuropathic or inflammatory origin including acute and chronic pain; inflammatory and autoimmune diseases including age-related macular degeneration, rheumatoid arthritis, inflammatory bowel disease, ulcerative colitis, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, psoriasis, scleroderma, chronic thyroiditis, Grave's disease, myasthenia gravis, and multiple sclerosis, and inflammation in other organs including CNS inflammation, pancreatitis, nephritis, atopic dermatitis, and hepatitis; airway inflammatory diseases such as asthma, allergy, bronchitis, pulmonary fibrosis, chronic obstructive pulmonary disease; neurologic diseases such as stroke, cerebrovascular ischemia, neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, dementia, senile chorea, head and spinal cord trauma, and Huntington's disease.
[0296] In a related aspect, compounds of Formula III, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a Jnkl -mediated disease or condition selected from the group consisting of type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity and hepatic steatosis.
[0297] In a related aspect, compounds of Formula III, further where the compound is of Formula in, can be used in the preparation of a medicament for the treatment of a Jnk2-mediated disease or condition, such as atherosclerosis.
[0298] In a related aspect, compounds of Formula III, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a Jnk3 -mediated disease or condition selected from the group consisting of inflammatory diseases including autoimmune diseases
such as rheumatoid arthritis, inflammatory bowel syndrome, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, psoriasis and multiple sclerosis, airway inflammatory diseases such as asthma, allergy, pulmonary fibrosis, and chronic obstructive pulmonary disease, and inflammation in other organs, such as CNS inflammation, pancreatitis, nephritis, and hepatitis; neurologic diseases such as stroke, cerebrovascular ischemia, and neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, and Huntington's disease; and neoplastic diseases such as prostate tumors and myeloid leukemia.
[0299] In a related aspect, compounds of Formula III, further where the compound is of Formula III, can be used in the preparation of a medicament for the treatment of a Kit-mediated disease or condition selected from the group consisting of malignancies, including mast cell tumors, small cell lung cancer, testicular cancer, gastrointestinal stromal tumors (GISTs), glioblastoma, astrocytoma, neuroblastoma, carcinomas of the female genital tract, sarcomas of neuroectodermal origin, colorectal carcinoma, carcinoma in situ, Schwann cell neoplasia associated with neurofibromatosis, acute myelocytic leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, mastocytosis, melanoma, and canine mast cell tumors, and inflammatory diseases, including asthma, rheumatoid arthritis, allergic rhinitis, multiple sclerosis, inflammatory bowel syndrome, transplant rejection, and hypereosinophilia.
[0300] The compounds of Formula I with kinase activity IC50 less than 10 μM as determined in a standard assay described herein can be used to treat protein kinase mediated diseases and conditions related to the following protein kinases, for example without limitation:
AbI, related to chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML); Aktl, related to gastric, prostate, colorectal, ovarian, pancreatic and breast cancer, glioblastoma and leukemia, as well as schizophrenia and bipolar disorders, and also use in combination with other chemotherapeutic drugs; Akt25 related to hyperglycemia due to peripheral insulin resistance and nonsuppressible hepatic glucose production accompanied by inadequate compensatory hyperinsulinemia, also related to pancreatic, ovarian and breast cancer; Akt3, related to melanoma, prostate and breast cancer; ALK, related to non-Hodgkin lymphomas such as diffuse large B-cell lymphoma and anaplastic large cell lymphoma;
Alk5, related to pancreatic and biliary cancers, and cutaneous T-cell lymphoma; B-Raf. related to neurologic diseases such as ischemic stroke, multi-infarct dementia, head injury, spinal cord injury, Alzheimer's disease (AD), Parkinson's disease; neoplastic diseases including, but not limited to, melanoma, glioma, sarcoma, carcinoma (e.g. lung, breast,
pancreatic, renal), lymphoma (e.g. histiocytic lymphoma) and cancer of the thyroid, lung (e.g. small cell lung cancer), liver, breast, ovary and colon, neurofibromatosis, myelodysplastic syndrome, leukemia, tumor angiogenesis; pain of neuropathic or inflammatory origin, including acute pain, chronic pain, and migraine; cardiovascular diseases including heart failure, cardiac hypertrophy, thrombosis (e.g. thrombotic microangiopathy syndromes), atherosclerosis, reperfusion injury; inflammation including, but not limited to, psoriasis, polycystic kidney disease (PKD), arthritis and autoimmune diseases and conditions, osteoarthritis, endometriosis, scarring, vascular restenosis, fibrotic disorders, rheumatoid arthritis, inflammatory bowel disease (IBD); immunodeficiency diseases, organ transplant rejection, graft versus host disease; renal or prostatic diseases including diabetic nephropathy, nephrosclerosis, glomerulonephritis, prostate hyperplasia; metabolic disorders, obesity; infection, including, but not limited to Helicobacter pylori and Influenza virus, fever, sepsis; pulmonary diseases including chronic obstructive pulmonary disease (COPD) and acute respiratory distress syndrome (ARDS); genetic developmental diseases such as Noonan's syndrome, Costello syndrome, (faciocutaneoskeletal syndrome), leopard syndrome, cardio- faciocutaneous syndrome (CFC), and neural crest syndrome abnormalities causing cardiovascular, skeletal, intestinal, skin, hair and endocrine diseases; c-Raf-1, related to colorectal, ovarian, lung and renal cell carcinoma, acute myeloid leukemia, myelodysplastic syndromes, tumor angiogenesis, and neuroendocrine tumors such as medullary thyroid cancer, carcinoid, small cell lung cancer and pheochromocytoma;
Brk, related to breast and colon cancer, and head and neck squamous cell carcinoma;
Btk, related to X-linked agammaglobulinemia, acute lymphocytic leukemia, autoimmune diseases such as multiple sclerosis, systemic lupus erythematosis, rheumatoid arthritis, and Graves' disease, immune suppression in organ transplant, and drug sensitivity of B-lineage cells;
Cdk2, related to prostate, breast, colorectal and ovarian cancer;
Cdk4, related to glioblastoma (e.g. glioblastoma multiforme), anaplastic astrocytoma, and breast cancer;
Cdk5, related to Alzheimer's disease, amyotrophic lateral sclerosis and Lewy body disease;
Cdk6, related to glioblastoma multiforme, non-Hodgkin's lymphoma, splenic marginal zone lymphoma, T-cell lymphoblastic lymphoma (T-LBL) and T-cell acute lymphoblastic leukemia (T-ALL);
CHKl, related to DNA damage repair, sensitizes cells to chemotherapeutic agents;
Csk, related to colon and pancreatic carcinomas and autoimmune pathology such as type 1 diabetes, rheumatoid arthritis and systemic lupus erythematosus;
EGFR, related to breast, colorectal, bladder, prostate and non small cell lung cancer, squamous cell carcinomas of the head and neck cancer, oral cavity, and esophagus, and glioblastoma multiforme;
EphAl, related to head and neck squamous cell carcinoma, hepatoma and lung cancer; EphA2, related to aberrant short-range contact-mediated axonal guidance, bladder, breast, prostate, colon, skin, cervical, ovarian, pancreatic and lung cancers, and metastatic melanoma; EphB2, related to angiogenesis disorder (e.g. ocular angiogenesis disease such as retinopathy), and cancer (e.g. glioblastoma, breast and liver cancer); EphB4, related to colorectal cancer (CRC), head and neck squamous cell carcinoma, and tumours of the prostate, breast, endometrium, and bladder; Erk2, related to aberrant proliferation, differentiation, transcription regulation and development, and may be useful in treating inflammation, for example inflammation associated with Lyme neuroborreliosis, and in treating cancers, such as gastric cancer; Fak, related to colon and breast tumors, and is also related to esophageal squamous cell carcinoma, melanoma, anaplastic astrocytoma, glioblastoma, ductal carcinoma in situ, prostate and hepatocellular carcinoma, and tumor metastases, and may also provide synergistic effects when used with other chemotherapeutic drugs; FGFRl, related to 8pll myeloproliferative syndrome; FGFR2, related to Crouzon Syndrome, Jackson-Weiss Syndrome, Apert Syndrome, craniosynostosis, Pfeiffer Syndrome, acrocephalo syndactyly type V, and Beare-Stevenson
Cutis Gyrata Syndrome; FGFR3, related to angiogenesis, wound healing, achondroplasia, Muenke craniosynostosis,
Crouzon syndrome, acanthosis nigricans, thanatophoric dysplasia, bladder carcinomas, and multiple myeloma; FGFR4, related to cancer of the breast, lung, colon, medullary thyroid, pancreas, ovary, prostate, endometrium, and fallopian tube, head and neck squamous cell carcinomas and leiomyosarcoma;
Fltl, related to non-small cell lung carcinoma, prostate carcinoma, and colorectal cancer; Flt3, related to acute myeloid leukemia, myelodysplastic syndrome, acute lymphoblastic leukemia;
Flt4, related to primary lymphoedema; Fms, related to immune disorders, including rheumatoid arthritis, systemic lupus erythematosis
(SLE), Wegener's granulomatosis, and transplant rejection, inflammatory diseases including
Chronic Obstructive Pulmonary Disease (COPD), emphysema, and atherosclerosis, metabolic disorders, including insulin resistance, hyperglycemia, and lipolysis, disorders of bone structure or mineralization, including osteoporosis, increased risk of fracture, hypercalcemia, and bone metastases, kidney diseases, including nephritis (e.g. glomerulonephritis, interstitial nephritis, Lupus nephritis), tubular necrosis, diabetes-associated renal complications, and hypertrophy and cancers, including multiple myeloma, acute myeloid leukemia, chronic myeloid leukemia (CML), breast cancer, and ovarian cancer;
Frk, related to acute myeloid leukemia and type 1 diabetes;
Fyn, related to Alzheimer's disease, schizophrenia and prevention of metastases, e.g. in melanoma and squamous cell carcinoma;
GSK3 (Gsk3α and/or Gsk3β), related to CNS disorders such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, diabetes type II, bipolar disorders, stroke, cancer, chronic inflammatory disease, leucopenia, schizophrenia, chronic pain, neuropathic pain, and traumatic head injury;
HCK, related to chronic myelogenous leukemia and acute lymphocytic leukemia;
Her2/Erbb2, related to prostate and breast cancer;
Her4/Erbb4, related to childhood medulloblastoma;
IGFlR, related to prostate cancer, hepatocellular carcinoma;
IKK beta, related to leukemia of T-cells, necrosis, insulin resistance, and malignant neoplasms;
Irak4, related to bacterial infections, immunodeficiency syndrome, Crohn's disease, ulcerative colitis, asthma, chronic bronchitis, cardio hypertrophy, and kidney hypertension;
Itk, related to allergic asthma;
Jakl, related to Hepatitis C virus infection;
Jak2, related to myeloproliferative disorders such as polycythaemia vera, myelofibrosis, essential thrombocythemia, myeloid metaplasia and leukemias, including acute lymphoblastic leukemia, chronic neutrophilic leukemia, juvenile myelomonocytic leukemia, CMML, Philadelphia chromosome-negative CML, megakaryocytic leukemia, and acute erythroid leukemia;
Jak3, related to X-linked severe combined immunodeficiency, myeloproliferative disorders, transplant rejection and autoimmune diseases such as rheumatoid arthritis, inflammatory bowel syndrome, Crohn's disease, systemic lupus erythematosis, ulcerative colitis, psoriasis and multiple sclerosis;
Jnk (Jnkl, Jnk2, Jnk3), related to metabolic diseases including type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity, and hepatic steatosis; cardiovascular diseases such as atherosclerosis, ischemia (e.g. cerebrovascular ischemia, liver ischemia), reperfusion injury, cardiac hypertrophy; renal diseases such as chronic renal failure; neoplastic diseases and associated complications, including chemotherapy-induced hypoxia, prostate tumors, myeloid leukemia and cancers of the liver, bone, skin, brain, pancreas, lung breast, colon, prostate and ovary; transplant rejection; pain of neuropathic or inflammatory origin including acute and chronic pain; inflammatory and autoimmune diseases including age-related macular degeneration, rheumatoid arthritis, inflammatory bowel disease, ulcerative colitis, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, psoriasis, scleroderma, chronic thyroiditis, Grave's disease, myasthenia gravis, and multiple sclerosis, and inflammation in other organs including CNS inflammation, pancreatitis, nephritis, atopic dermatitis, and
hepatitis; airway inflammatory diseases such as asthma, allergy, bronchitis, pulmonary fibrosis, chronic obstructive pulmonary disease; neurologic diseases such as stroke, cerebrovascular ischemia, neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, dementia, senile chorea, head and spinal cord trauma, and Huntington's disease. More particularly, Md is related to type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity and hepatic steatosis, Jnk2 is related to atherosclerosis, and Jnk3 is related to inflammatory diseases including autoimmune diseases such as rheumatoid arthritis, inflammatory bowel syndrome, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, psoriasis and multiple sclerosis, airway inflammatory diseases such as asthma, allergy, pulmonary fibrosis, and chronic obstructive pulmonary disease, and inflammation in other organs, such as CNS inflammation, pancreatitis, nephritis, and hepatitis; neurologic diseases such as stroke, cerebrovascular ischemia, and neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, and Huntington's disease; and neoplastic diseases such as prostate tumors and myeloid leukemia;
Kdr, related to anti-angiogenesis for treating solid tumor growth (e.g. ovarian, lung, breast, prancreatic, prostate, colon, gastrointestinal stromal tumor, non small cell lung cancer, and epidermoid cancer), metastasis, psoriasis, rheumatoid arthritis, diabetic retinopathy and age related macular degeneration;
Kit, related to malignancies, including mast cell tumors, small cell lung cancer, testicular cancer, gastrointestinal stromal tumors (GISTs), glioblastoma, astrocytoma, neuroblastoma, carcinomas of the female genital tract, sarcomas of neuroectodermal origin, colorectal carcinoma, carcinoma in situ, Schwann cell neoplasia associated with neurofibromatosis, acute myelocytic leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, mastocytosis, melanoma, and canine mast cell tumors, and inflammatory diseases, including asthma, rheumatoid arthritis, allergic rhinitis, multiple sclerosis, inflammatory bowel syndrome, transplant rejection, and hypereosinophilia;
LCK, related to acute lymphoblastic leukemia, T-cell lymphoma, lymphopenia, renal carcinoma, colon carcinoma, severe combined immunodeficiency, multiple sclerosis, inflammatory bowel and type I diabetes;
MAP2K1, related to acute myeloid leukemia, breast, ovarian and liver cancer;
MAP2K2, related to cancer and inflammation;
MAP4K4, related to cancer and tumor metastasis, diabetes and metabolic syndrome;
MAPKAPK2, cancer (e.g. prostate, breast), stroke, menengitis, and inflammatory disorders;
Met, related to kidney, breast, bladder, non-small-cell lung, colorectal, and bladder cancers, and hepatocellular carcinoma;
Mnkl, related to conditions associated with heat shock, nutrient deprivation, oxidative or osmotic stress, and infection of mammalian cells (e.g. with viruses such as adenovirus (Ad) or influenza virus), and autoimmune diseases;
MLKl, related to neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease, and inflammatory disorders; p38, related to acute coronary syndrome, stroke, atherosclerosis, and inflammatory autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease, and Crohn's disease;
PDGFR (PDGFRA, PDGFRB), related to idiopathic hypereosinophilic syndrome, chronic eosinophilic leukemia, glioma, gastrointestinal stromal tumors (GISTs), juvenile myelomonocytic leukemia, metastatic medulloblastoma, atherogenesis, and restenosis. More particularly, PDGFRA related to idiopathic hypereosinophilic syndrome, chronic eosinophilic leukemia, glioma, gastrointestinal stromal tumors (GISTs), juvenile myelomonocytic leukemia, metastatic medulloblastoma, atherogenesis, and restenosis, and PDGFRB related to idiopathic hypereosinophilic syndrome, chronic eosinophilic leukemia, juvenile myelomonocytic leukemia, and metastatic medulloblastoma;
PDPKl, related to cancer and diabetes;
Piml, related to cancers such as hematopoietic (e.g. acute myeloid and acute lymphoid leukemias) and prostate cancers, and non-Hodgkin's lymphomas;
Pim2, related to lymphomas;
Pim3, related to hepatocellular carcinoma;
PKC alpha, related to pituitary tumors and prefrontal cortical dysfunction such as distractibility, impaired judgment, impulsivity, and thought disorder, also maybe used to sensitize chemotherapy in breast, colon, and non small cell lung cancers;
PKC beta, related to diabetic retinopathy;
PKC-theta, related to insulin resistance, T-cell lymphoma;
Plkl, related to cancers (e.g. lymphoma of the thyroid, non-Hodgkin's lymphomas, colorectal cancers, leukemias and melanoma), also useful as sensitizer in chemotherapy;
Pyk2, related to inflammation (e.g. osteoporosis, polycystic kidney disease, rheumatoid arthiritis and inflammatory bowel disease), CNS disease (e.g. Parkinson's disease and Alzheimer's disease), stroke and cancers (e.g. gliomas, breast cancer, and pancreatic cancer);
Ret, related to cancer of the thyroid, neuroblastoma, familial medullary thyroid carcinoma (FMTC), multiple endocrine neoplasia type HA and HB (MEN2A, MEN2B), and neurodegenerative disorders (e.g. Hirschsprung's disease, Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis);
ROCK (ROCK-I, ROCK-2), related to cancers (e.g. ovarian cancer, hepatocellular carcinoma, pancreatic cancer), ocular disease (e.g. glaucoma), cardiac hypertrophy, improved renal perfusion, transplant rejection, and acute respiratory distress syndrome;
Ron, related to cancer and inflammation;
Src, related to cancer and osteoporosis;
Stk6, related to gastric, bladder, breast, lung, CNS, ovarian, kidney, colon, prostate, pancreas, and cervical cancers, melanoma, leukemia, and neuroblastoma;
Syk, related to lymphomas (e.g. mantle cell lymphoma);
TEC, related to sepsis, septic shock, inflammation, rheumatoid arthritis, Crohn's disease, irritable bowel disease (IBD), and ulcerative colitis;
Tie2 (TEK), related to cancer, arthritis (e.g. rheumatoid arthritis), and atherosclerosis;
TrkA, related to pain (e.g. chronic pain, neuropathic pain), cancer, arthritis, diabetic retinopathy, macular degeneration and psoriasis;
Yes, related to various cancers including esophageal squamous cell carcinoma; and
Zap70, related to ABDS, systemic lupus erythematosus, myasthenia gravis, atherosclerosis, rejection of transplanted organs or tissues, allograft rejection including acute and chronic allograft rejection, graft versus host disease, rheumathoid arthritis, psoriasis, systemic sclerosis, atopic dermatitis, eczematous dermatitis, alopecia, and inflammation of the nasal mucus membrane, including all forms of rhinitis.
[0301] Additional aspects and embodiments will be apparent from the following Detailed Description and from the claims.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
As used herein the following definitions apply unless clearly indicated otherwise:
[0302] "Halogen" refer to all halogens, that is, chloro (Cl), fluoro (F), bromo (Br), or iodo (I).
[0303] "Hydroxyl" or "hydroxy" refer to the group -OH.
[0304] "Thiol" refers to the group -SH.
[0305] "Lower alkyl" alone or in combination means an alkane-derived radical containing from 1 to 6 carbon atoms (unless specifically defined) that includes a straight chain alkyl or branched alkyl. The straight chain or branched alkyl group is attached at any available point to produce a stable
compound. In many embodiments, a lower alkyl is a straight or branched alkyl group containing from 1-6, 1-4, or 1-2, carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, t-butyl, and the like. A "substituted lower alkyl" denotes lower alkyl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of -F, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPRC, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Re, -Rf, and -Rg. Furthermore, possible substitutions include subsets of these substitutions, such as are indicated herein, for example, in the description of compounds of Formula III, attached at any available atom to produce a stable compound. For example "fluoro substituted lower alkyl" denotes a lower alkyl group substituted with one or more fluoro atoms, such as perfluoroalkyl, where preferably the lower alkyl is substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms. While it is understood that substitutions are attached at any available atom to produce a stable compound, when optionally substituted alkyl is an R group of a moiety such as -OR (e.g. alkoxy), -SR (e.g. thioalkyl), -NHR (e.g. alkylamino), -C(O)NHR, and the like, substitution of the alkyl R group is such that substitution of the alkyl carbon bound to any O, S, or N of the moiety (except where N is a heteroaryl ring atom) excludes substituents that would result in any O, S, or N of the substituent (except where N is a heteroaryl ring atom) being bound to the alkyl carbon bound to any O, S, or N of the moiety. "C2-6 alkyl" denotes lower alkyl containing 2-6 carbon atoms. A "substituted C2-6 alkyl" denotes optionally substituted lower alkyl containing 2-6 carbon atoms. A "substituted methyl" denotes methyl that is independently substituted, unless indicated otherwise, with 1, 2, or 3 substituents, wherein the substituents are selected as per optionally substituted lower alkyl.
[0306] "Ci-3 alkylene" refers to a divalent alkane-derived radical containing 1-3 carbon atoms, straight chain or branched, from which two hydrogen atoms are taken from the same carbon atom or from different carbon atoms. Ci.3 alkylene includes methylene -CH2-, ethylene -CH2CH2-, propylene -CH2CH2CH2-, and isopropylene -CH(CH3)CH2- or -CH2CH(CH3)-. C1-3 alkylene substituted with one or more substituents indicates Ci-3 alkylene that is independently substituted, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents as indicated, attached at any available atom to produce a stable compound.
[0307] "Lower alkenyl" alone or in combination means a straight or branched hydrocarbon containing 2-6 carbon atoms (unless specifically defined) and at least one, preferably 1-3, more preferably 1-2, most preferably one, carbon to carbon double bond. Carbon to carbon double bonds may be either contained within a straight chain or branched portion. Examples of lower alkenyl groups include ethenyl, propenyl, isopropenyl, butenyl, and the like. A "substituted lower alkenyl" denotes lower alkenyl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of -F, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPR°, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Rf, and -Rg. Further, possible substitutions include subsets of these substitutions, such as are indicated herein, for example, in the description of compounds of Formula m, attached at any available atom to produce a stable compound. For example "fiuoro substituted lower alkenyl" denotes a lower alkenyl group substituted with one or more fiuoro atoms, where preferably the lower alkenyl is substituted with 1, 2, 3, 4 or 5 fiuoro atoms, also 1, 2, or 3 fiuoro atoms. While it is understood that substitutions are attached at any available atom to produce a stable compound, substitution of alkenyl groups are such that -F, -C(O)-, -C(S)-, -C(NH)-, -S(O)-, -S(O)2-, -O-, -S-, or N (except where N is a heteroaryl ring atom), are not bound to an alkene carbon thereof. Further, where alkenyl is a substituent of another moiety or an R group of a moiety such as -OR, -NHR, -C(O)R, and the like, substitution of the moiety is such that any -C(O)-, -C(S)-, -S(O)-, -S(O)2-, -0-, -S-, or N thereof (except where N is a heteroaryl ring atom) are not bound to an alkene carbon of the alkenyl substituent or R group. Further, where alkenyl is a substituent of another moiety or an R group of a moiety such as -OR, -NHR, -C(O)NHR, and the like, substitution of the alkenyl R group is such that substitution of the alkenyl carbon bound to any O, S, or N of the moiety (except where N is a heteroaryl ring atom) excludes substituents that would result in any O, S, or N of the substituent (except where N is a heteroaryl ring atom) being bound to the alkenyl carbon bound to any O, S, or N of the moiety. An "alkenyl carbon" refers to any carbon within an alkenyl group, whether saturated or part of the carbon to carbon double bond. An "alkene carbon" refers to a carbon within an alkenyl group that is part of a carbon to carbon double bond.
[0308] "Lower alkynyl" alone or in combination means a straight or branched hydrocarbon containing 2-6 carbon atoms (unless specifically defined) containing at least one, preferably one,
carbon to carbon triple bond. Examples of alkynyl groups include ethynyl, propynyl, butynyl, and the like. A "substituted lower alkynyl" denotes lower alkynyl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of -F, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPR°, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Re, and -Rε. Further, possible substitutions include subsets of these substitutions, such as are indicated herein, for example, in the description of compounds of Formula III, attached at any available atom to produce a stable compound. For example "fluoro substituted lower alkynyl" denotes a lower alkynyl group substituted with one or more fluoro atoms, where preferably the lower alkynyl is substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms. While it is understood that substitutions are attached at any available atom to produce a stable compound, substitution of alkynyl groups are such that -F, -C(O)-, -C(S)-, -C(NH)-, -S(O)-, -S(O)2-, -0-, -S-, or N (except where N is a heteroaryl ring atom) are not bound to an alkyne carbon thereof. Further, where alkynyl is a substituent of another moiety or an R group of a moiety such as -OR, -NHR, -C(O)R, and the like, substitution of the moiety is such that any -C(O)-, -C(S)-,-S(O)-, -S(O)2-, -0-, -S-, or N thereof (except where N is a heteroaryl ring atom) are not bound to an alkyne carbon of the alkynyl substituent or R group. Further, where alkynyl is a substituent of another moiety or an R group of a moiety such as -OR, -NHR, -C(O)NHR, and the like, substitution of the alkynyl R group is such that substitution of the alkynyl carbon bound to any O, S, or N of the moiety (except where N is a heteroaryl ring atom) excludes substituents that would result in any O, S, or N of the substituent (except where N is a heteroaryl ring atom) being bound to the alkynyl carbon bound to any O, S, or N of the moiety. An "alkynyl carbon" refers to any carbon within an alkynyl group, whether saturated or part of the carbon to carbon triple bond. An "alkyne carbon" refers to a carbon within an alkynyl group that is part of a carbon to carbon triple bond.
[0309] "Cycloalkyl" refers to saturated or unsaturated, non-aromatic monocyclic, bicyclic or tricyclic carbon ring systems of 3-10, also 3-8, more preferably 3-6, ring members per ring, such as cyclopropyl, cyclopentyl, cyclohexyl, adamantyl, and the like. A "substituted cycloalkyl" is a cycloalkyl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of halogen, -OH, -NH2,
-NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPR°, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Re, -Rf, and -Rg.
[0310] "Heterocycloalkyl" refers to a saturated or unsaturated non-aromatic cycloalkyl group having from 5 to 10 atoms in which from 1 to 3 carbon atoms in the ring are replaced by heteroatoms of O, S or N, and are optionally fused with benzo or heteroaryl of 5-6 ring members. Heterocycloalkyl is also intended to include oxidized S or N, such as sulfinyl, sulfonyl and N-oxide of a tertiary ring nitrogen. Heterocycloalkyl is also intended to include compounds in which a ring carbon may be oxo substituted, i.e. the ring carbon is a carbonyl group, such as lactones and lactams. The point of attachment of the heterocycloalkyl ring is at a carbon or nitrogen atom such that a stable ring is retained. Examples of heterocycloalkyl groups include, but are not limited to, morpholino, tetrahydrofuranyl, dihydropyridinyl, piperidinyl, pyrrolidinyl, pyrrolidonyl, piperazinyl, dihydrobenzofuryl, and dihydroindolyl. A "substituted heterocycloalkyl" is a heterocycloalkyl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPRC, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Re, -Rf, and -Rg.
[0311] "Aryl" alone or in combination refers to a monocyclic or bicyclic ring system containing aromatic hydrocarbons such as phenyl or naphthyl, which may be optionally fused with a cycloalkyl of preferably 5-7, more preferably 5-6, ring members. "Arylene" is a divalent aryl. A "substituted aryl" is an aryl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of halogen, -OH, -NH2,
-NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPR°, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Re, -Rf, and -Rg. A "substituted aiylene" is a divalent substituted aryl,
[0312] "Heteroaryl" alone or in combination refers to a monocyclic aromatic ring structure containing 5 or 6 ring atoms, or a bicyclic aromatic group having 8 to 10 atoms, containing one or more, preferably 1-4, more preferably 1-3, even more preferably 1-2, heteroatoms independently selected from the group consisting of O, S, and N. Heteroaryl is also intended to include oxidized S or N, such as sulfinyl, sulfonyl and N-oxide of a tertiary ring nitrogen. A carbon or nitrogen atom is the point of attachment of the heteroaryl ring structure such that a stable compound is produced. Examples of heteroaryl groups include, but are not limited to, pyridinyl, pyridazinyl, pyrazinyl, quinaoxalyl, indolizinyl, benzo[b]thienyl, quinazolinyl, purinyl, indolyl, quinolinyl, pyrimidinyl, pyrrolyl, pyrazolyl, oxazolyl, thiazolyl, thienyl, isoxazolyl, oxathiadiazolyl, isothiazolyl, tetrazolyl, imidazolyl, triazolyl, furanyl, benzofuryl, and indolyl. "Nitrogen containing heteroaryl" refers to heteroaryl wherein any heteroatoms are N. "Heteroarylene" is a divalent heteroaryl. A "substituted heteroaryl" is a heteroaryl that is independently substituted, unless indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents, attached at any available atom to produce a stable compound, wherein the substituents are selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -OR0, -SR0, -OC(O)R0, -OC(S)R0, -C(O)R0, -C(S)R0, -C(O)OR0, -C(S)OR0, -S(O)R0, -S(O)2R0, -C(O)NHR0, -C(S)NHR0, -C(O)NR0R0, -C(S)NR0R0, -S(O)2NHR0, -S(O)2NR0R0, -C(NH)NHR0, -C(NH)NRPR°, -NHC(O)R0, -NHC(S)R0, -NR0C(O)R0, -NR0C(S)R0, -NHS(O)2R0, -NR0S(O)2R0, -NHC(O)NHR0, -NHC(S)NHR0, -NR0C(O)NH2, -NR0C(S)NH2, -NR0C(O)NHR0, -NR0C(S)NHR0, -NHC(O)NR0R0, -NHC(S)NR0R0, -NR0C(O)NR0R0, -NR0C(S)NR0R0, -NHS(O)2NHR0, -NR0S(O)2NH2, -NR0S(O)2NHR0, -NHS(O)2NR0R0, -NR0S(O)2NR0R0, -NHR0, -NR0R0, -Rd, -Re, -Rf, and -Rg. "Substituted heteroarylene" is a divalent substituted heteroaryl.
[0313] The variables R0, Rp, R°, Rd, Re, Rf and Rs as used in the description of optional substituents for alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl are defined as follows:
each R0, Rp, and Rc are independently selected from the group consisting of Rd, Re, Rf, and Rg, or Rp and Rc combine with the nitrogen to which they are attached to form a 5-7 membered heterocycloalkyl or a 5 or 7 membered nitrogen containing heteroaryl, wherein the 5-7 membered heterocycloalkyl or 5 or 7 membered nitrogen containing heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -NO2, -CN, -OH, -NH2, -ORU, -SRU, -NHRU, -NRURU, -Rx, and -Ry;
each Rd is independently lower alkyl, wherein lower alkyl is optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of fluoro, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rk, -SRk, -0C(0)Rk, -OC(S)Rk, -C(0)Rk, -C(S)Rk, -C(O)ORk, -C(S)ORk, -S(O)Rk, -S(O)2Rk, -C(0)NHRk, -C(S)NHRk, -C(0)NRkRk, -C(S)NRkRk, -S(0)2NHRk, -S(0)2NRkRk, -C(NH)NHRk, -C(NH)NRmRn, -NHC(O)Rk, -NHC(S)Rk, -NRkC(0)Rk, -NRkC(S)Rk, -NHS(0)2Rk, -NRkS(0)2Rk, -NHC(0)NHRk, -NHC(S)NHRk, -NRkC(0)NH2, -NRkC(S)NH2, -NRkC(O)NHRk, -NRkC(S)NHRk, -NHC(0)NRkRk, -NHC(S)NRkRk, -NRkC(0)NRkRk, -NRkC(S)NRkRk, -NHS(0)2NHRk, -NRkS(O)2NH2, -NRkS(0)2NHRk, -NHS(O)2NRkRk, -NRkS(O)2NRkRk, -NHRk, -NRkRk, -R1, and -Rj;
each R
eis independently lower alkenyl, wherein lower alkenyl is optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of fluoro, -OH, -NH
2, -NO
2, -CN, -C(O)OH, -C(S)OH, -C(O)NH
2, -C(S)NH
2, -S(O)
2NH
2, -NHC(O)NH
2, -NHC(S)NH
2, -NHS(O)
2NH
2, -C(NH)NH
2, -0R
k, -SR
k, -0C(0)R
k, -OC(S)R
k, -C(0)R
k, -C(S)R
k, -C(0)0R\ -C(S)OR
k, -S(O)R
k, -S(O)
2R\ -C(0)NHR
k, -C(S)NHR
k, -C(O)NR
kR
k, -C(S)NR
kR
k, -S(0)
2NHR
k, -S(0)
2NR
kR
k, -C(NH)NHR
k, -C(NH)NR
mR
n, -NHC(0)R
k, -NHC(S)R
k, -NR
kC(0)R
k, -NR
kC(S)R
k, -NHS(O)
2R
k, -NR
kS(0)
2R
k, -NHC(O)NHR
k, -NHC(S)NHR
k, -NR
kC(0)NH
2, -NR
kC(S)NH
2, -NR
kC(0)NHR
k, -NR
kC(S)NHR
k, -NHC(0)NR
kR
k, -NHC(S)NR
kR
k, -NR
kC(0)NR
kR
k, -NR
kC(S)NR
kR
k, -NHS(0)
2NHR
k, -NR
kS(O)
2NH
2, -NR
kS(0)
2NHR
k, -NHS(0)
2NR
kR
k, -NR
kS(0)
2NR
kR
k,
-NR
kR
k, -R
h, and -R
j;
each Rf is independently lower alkynyl, wherein lower alkynyl is optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of fluoro, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rk, -SRk, -OC(O)Rk, -OC(S)Rk, -C(0)Rk, -C(S)Rk, -C(0)0Rk, -C(S)ORk, -S(O)Rk, -S(O)2Rk, -C(0)NHRk, -C(S)NHRk, -C(0)NRkRk, -C(S)NRkRk, -S(0)2NHRk, -S(O)2NRkRk, -C(NH)NHRk, -C(NH)NR171R", -NHC(0)Rk, -NHC(S)Rk, -NRkC(0)Rk, -NRkC(S)Rk, -NHS(0)2Rk, -NRkS(0)2Rk, -NHC(0)NHRk, -NHC(S)NHRk, -NRkC(0)NH2, -NRkC(S)NH2, -NRkC(0)NHRk, -NRkC(S)NHRk, -NHC(0)NRkRk, -NHC(S)NRkRk, -NRkC(0)NRkRk,
-INK"C(S)NRΛR% -NHS(O)2NHR", -MTS(O)2NH2, -NRKS(O)2NHRK, -NHS(O)2NRKRK, -NRkS(O)2NRkRk, -NHRk, -NRkRk, -Rh, and -Rj;
each Rg is independently selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH5 -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rk, -SRk, -OC(O)Rk, -OC(S)Rk, -C(O)Rk, -C(S)Rk, -C(O)ORk, -C(S)ORk, -S(O)Rk, -S(O)2Rk, -C(0)NHRk, -C(S)NHRk, -C(0)NRkRk, -C(S)NRkRk, -S(0)2NHR\ -S(0)2NRkRk, -C(NH)NHRk, -C(NH)NRmRn, -NHC(0)Rk, -NHC(S)Rk, -NRkC(O)Rk, -NRkC(S)Rk, -NHS(0)2Rk, -NRkS(O)2Rk, -NHC(0)NHRk, -NHC(S)NHRk, -NRkC(0)NH2) -NRkC(S)NH2, -NRkC(0)NHRk, -NRkC(S)NHRk, -NHC(0)NRkRk, -NHC(S)NRkRk, -NRkC(0)NRkRk, -NRkC(S)NRkRk, -NHS(0)2NHRk, -NRkS(O)2NH2, -NRkS(0)2NHRk, -NHS(0)2NRkRk, -NRkS(0)2NRkRk, -NHRk, -NRkRk, -Rh, -Rf, and -R1;
wherein Rk, Rm, and Rn at each occurrence are independently selected from the group consisting of Rh, R1, and RJ, or Rm and Rn combine with the nitrogen to which they are attached form a 5-7 membered heterocycloalkyl or a 5 or 7 membered nitrogen containing heteroaryl, wherein the 5-7 membered heterocycloalkyl or 5 or 7 membered nitrogen containing heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -NO2, -CN, -OH, -NH2, ORU, -SRU, -NHR", -NRURU, -Rx, and -Ry;
wherein each Rhis independently lower alkyl optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of fluoro, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rr, -SRr, -0C(0)Rr, -OC(S)Rr, -C(0)Rr, -C(S)Rr, -C(O)ORr, -C(S)ORr, -S(O)Rr, -S(O)2Rr, -C(0)NHRr, -C(S)NHR1, -C(O)NR1R/, -C(S)NR1R1, -S(O)2NHRr, -S(O)2NR1R', -C(NH)NHRr, -C(NH)NR8R1, -NHC(0)Rr, -NHC(S)Rr, -NRrC(0)Rr, -NRrC(S)Rr, -NHS(O)2Rr, -NRrS(0)2Rr, -NHC(0)NHRr, -NHC(S)NHRr, -NRrC(0)NH2, -NRrC(S)NH2, -NRrC(0)NHRr, -NRrC(S)NHRr, -NHC(0)NRrRr, -NHC(S)NRrRr, -NRrC(0)NRrRr, -NRrC(S)NRrRr, -NHS(0)2NHRr, -NRrS(O)2NH2, -NRrS(0)2NHRr, -NHS(0)2NRrRr, -NRrS(0)2NRrRr, -NHRr, -NR"Rr, -R\ and -Rj;
wherein each R1 is independently selected from the group consisting of lower alkenyl and lower alkynyl, wherein lower alkenyl or lower alkynyl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of fluoro, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rr, -SRr, -OC(O)Rr, -OC(S)R1, -C(0)Rr, -C(S)Rr,
-C(O)ORr, -C(S)ORr, -S(O)Rr, -S(O)2Rr, -C(O)NHRr, -C(S)NHRr, -C(O)NR1R1, -C(S)NRrRr, -S(O)2NHRr, -S(O)2NR1R1, -C(NH)NHRr, -C(NH)NR8R', -NHC(O)Rr, -NHC(S)Rr, -NRrC(O)Rr, -NRrC(S)Rr, -NHS(O)2Rr, -NRrS(O)2Rr, -NHC(O)NHRr, -NHC(S)NHRr, -NRrC(O)NH2, -NRrC(S)NH2, -NRrC(O)NHRr, -NRrC(S)NHRr, -NHC(O)NR1Rr, -NHC(S)NR1R', -NRrC(O)NRrRr, -NRrC(S)NR1Rr, -NHS(O)2NHRr, -NRrS(O)2NH2, -NRrS(O)2NHRr, -NHS(O)2NR1Rr, -NRrS(O)2NR1R', -NHRr, -NRrRr, and -Rj;
wherein each RJ is independently selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2 or 3 substituents selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, -C(O)OH, -C(S)OH, -C(O)NH2, -C(S)NH2, -S(O)2NH2, -NHC(O)NH2, -NHC(S)NH2, -NHS(O)2NH2, -C(NH)NH2, -0Rr, -SRr, -0C(0)Rr, -OC(S)Rr, -C(O)Rr, -C(S)Rr, -C(O)ORr, -C(S)ORr, -S(O)Rr, -S(O)2Rr, -C(O)NHRr, -C(S)NHRr, -C(O)NRrRr, -C(S)NRrRr, -S(O)2NHRr, -S(O)2NR1Rr, -C(NH)NHRr, -C(NH)NR8R', -NHC(O)Rr, -NHC(S)Rr, -NRrC(O)Rr, -NRrC(S)Rr, -NHS(O)2Rr, -NRrS(O)2Rr, -NHC(0)NHRr, -NHC(S)NHRr, -NRrC(O)NH2, -NRrC(S)NH2, -NRrC(O)NHRr, -NRrC(S)NHRr, -NHC(O)NRrRr, -NHC(S)NRrRr, -NRrC(O)NRrRr, -NRrC(S)NRrRr, -NHS(O)2NHRr, -NRrS(O)2NH2, -NRrS(O)2NHRr, -NHS(O)2NRrRr, -NRrS(O)2NRrRr, -NHRr, -NRrRr, cycloalkylamino, and -Rx;
wherein each Rr, Rs, and Rt at each occurrence are independently selected from the group consisting of lower alkyl, C3-6 alkenyl, C3-6 alkynyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein lower alkyl is optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of -Ry, fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the lower alkyl carbon bound to any O, S, or N, of -ORr, -SRr, -C(O)ORr, -C(S)ORr, -C(0)NHRr, -C(S)NHRr, -C(O)NRrRr, -C(S)NRrRr, -S(O)2NHRr, -S(O)2NRrRr, -C(NH)NHRr, -NRrC(O)Rr, -NRrC(S)Rr, -NRrS(O)2Rr, -NHC(O)NHRr, -NHC(S)NHRr, -NRrC(O)NH2, -NRrC(S)NH2, -NRrC(O)NHRr, -NRrC(S)NHRr, -NHC(O)NRrRr, -NHC(S)NRrRr, -NRrC(O)NR1Rr, -NRrC(S)NRrRr, -NHS(O)2NHRr, -NRrS(O)2NH2, -NRrS(O)2NHRr, -NHS(O)2NRrRr, -NRrS(O)2NRrRr, -NHRr, or -NRrRr is selected from the group consisting of fluoro and -Ry, and wherein C3-6 alkenyl or C3-e alkynyl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of -Ry, fluoro, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any
substitution of the C3-6 alkenyl or C3-6 alkynyl carbon bound to any O, S, or N, of -ORr, -SRr, -C(O)ORr, -C(S)ORr, -C(O)NHR', -C(S)NHRr, -C(O)NR1R', -C(S)NR1R', -S(O)2NHR', -S(O)2NR1R', -C(NH)NHR', -NRrC(O)Rr, -NRrC(S)Rr, -NR'S(0)2Rr, -NHC(0)NHRr, -NHC(S)NHR', -NRrC(0)NH2, -NR1C(S)NH2, -NRrC(0)NHRr, -NRrC(S)NHRr, -NHC(O)NR1R1, -NHC(S)NRIRr, -NR^(O)NR1R', -NRrC(S)NRrRr, -NHS(0)2NHRr, -NRrS(O)2NH2, -NRrS(0)2NHRr, -NHS(O)2NR1R', -NRrS(O)2NR1R', -NHRr, or -NR1R1 Is selected from the group consisting of fluoro, lower alkyl, fluoro substituted lower alkyl, and -Ry, and wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, or Rs and R' combine with the nitrogen to which they are attached form a 5-7 membered heterocycloalkyl or a 5 or 7 membered nitrogen containing heteroaryl, wherein the 5-7 membered heterocycloalkyl or 5 or 7 membered nitrogen containing heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -NO2, -CN, -OH, -NH2, ORU, -SRU, -NHRU, -NRURU, -Rx, and -Ry;
wherein each Ru is independently selected from the group consisting of lower alkyl, C3-6 alkenyl, C3-6 alkynyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is optionally substituted with one or more, preferably 1 , 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of-Ry, fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the lower alkyl carbon bound to the O of ~ORU, S of -SRU, or N of -NHR" is fluoro or -Ry, and wherein C3-6 alkenyl or C3-6 alkynyl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of -Ry, fluoro, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, provided, however, that any substitution of the C3-6 alkenyl or C3-6 alkynyl carbon bound to the O of -ORU, S of -SRU, or N of -NHRU is fluoro, lower alkyl, fluoro substituted lower alkyl, or -Ry, and wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
wherein each Rx is selected from the group consisting of lower alkyl, lower alkenyl and lower alkynyl, wherein lower alkyl is optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of -Ry, fhioro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino, and wherein lower alkenyl or lower alkynyl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of -Ry, fluoro, -OH, -NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
wherein each Ry is selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl are optionally substituted with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents selected from the group consisting of halogen, -OH, -NH2, -NO2, -CN, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino.
[0314] In some embodiments, all occurrences of optionally substituted lower alkyl, optionally substituted C2-6 alkyl, optionally substituted lower alkenyl, or optionally substituted lower alkynyl are optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -NO2, -CN, -ORla, -SRla, -NRlaRla, -OC(O)RIa, -OC(S)Rla, -C(O)Rla, -C(S)Rla, -C(O)OR13, -C(S)ORIa, -C(0)NRlaRla, -C(S)NRlaRla, -S(O)2NRlaRla, -C(NH)NRlaRla, -NRlaC(O)Rla, -NRlaC(S)Rla, -NRlaS(O)2RIa, -NRlaC(O)NRlaRla, -NRlaC(S)NRlaRla, -NRlaS(O)2NRlaRla, -S(O)Rla, -S(O)2Rla, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of halogen, -NO2, -CN3 -0Rla, -SRla, -NRIaRla, -OC(O)Rla, -OC(S)Rla, -C(O)RIa, -C(S)Rla, -C(O)ORla, -C(S)ORla, -C(0)NRlaRla, -C(S)NRlaRIa, -S(O)2NRlaRla, -C(NH)NRlaRla, -NRlaC(0)RIa, -NRIaC(S)Rla, -NRlaS(O)2Rla, -NRlaC(0)NRlaRla, -NRlaC(S)NRlaRla, -NRlaS(O)2NRlaRla, -S(O)Rla, -S(O)2Rla, -Rlb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -Rlb, and all occurrences of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted 5-7 membered heterocycloalkyl, optionally substituted aryl, optionally substituted arylene, optionally substituted heteroaryl, optionally substituted heteroarylene, or optionally substituted 5 or 7 membered nitrogen containing heteroaryl are optionally substituted with one or more, also 1, 2, or 3 groups or substituents selected from the group consisting of halogen, -NO2, -CN, -0Rla, -SRla, -NRlaRla,
-OC(O)R13, -OC(S)Rla, -C(O)Rla, -C(S)Rla, -C(O)ORIa, -C(S)ORla, -C(O)NRlaRla, -C(S)NRlaRla, -S(O)2NRlaRla, -C(NH)NRlaRla, -NRlaC(O)Rla, -NRIaC(S)Rla, -NRlaS(O)2Rla, -NRlaC(O)NRlaRla, -NRIaC(S)NRlaRla, -NRlaS(O)2NRIaRla, -S(O)Rla, -S(O)2R13, -Rlb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -Rlb, wherein Rla is selected from the group consisting of hydrogen, provided, however, that hydrogen is not bound to any of C(S), C(O), S(O), or S(O)2 of -OC(O)Rla, -OC(S)Rla, -C(O)RIa, -C(S)Rla, -NRlaC(O)Rla, -NRlaC(S)Rla, -NRlaS(O)2Rla, -S(O)RIa, or -S(O)2Rla, -Rlb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -RIb, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -0RIa, -SRla, -NRlaRla, -C(O)ORla, -C(S)ORla, -C(O)Ml13R1 \ -C(S)NRlaRIa, -S(0)2NRlaRla, -C(NH)NRlaRla, -NRlaC(0)Rla, -NRlaC(S)Rla, -NRlaS(O)2Rla, -NRlaC(0)NRlaRla, -NRlaC(S)NRlaRla, or -NRlaS(0)2NRlaRla, is fluoro or -Rlb, and wherein -Rlb is selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of halogen, -CN, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino.
[0315] In some embodiments, all occurrences of optionally substituted lower alkyl, optionally substituted C2-6 alkyl, optionally substituted lower alkenyl, or optionally substituted lower alkynyl are optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -CN, -0Rla, -SRla, -NRlaRla, -C(O)Rla, -C(S)Rla, -C(O)ORla, -C(0)NRlaRla, -C(S)NRlaRla, -S(O)2NR13R13, -NRIaC(0)Rla, -NRlaC(S)R!a, -NRIaS(O)2Rla, -S(O)Rla, -S(O)2Rla, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more, also 1 , 2 or 3 groups or substituents selected from the group consisting of halogen, -CN, -0Rla, -SRla, -NRlaRla, -C(O)Rla, -C(S)Rla, -C(O)ORla, -C(O)NRlaRla, -C(S)NRlaRla, -S(0)2NRlaRla, -NRlaC(O)Rla, -NRlaC(S)Rla, -NRlaS(O)2Rla, -S(O)Rla, -S(O)2Rla, -Rlb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -Rlb, and all occurrences of optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted 5-7 membered heterocycloalkyl, optionally substituted aryl, optionally substituted arylene, optionally substituted heteroaryl, optionally substituted heteroarylene, or optionally substituted 5 or 7 membered nitrogen containing heteroaryl are optionally substituted
with one or more, also 1, 2, or 3 groups or substituents selected from the group consisting of halogen, -CN, -ORla, -SRla, -NRlaRla, -C(O)Rla, -C(S)Rla, -C(O)ORla, -C(O)NRlaRla, -C(S)NRlaRla, -S(O)2NRlaRla, -NRlaC(O)Rla, -NRlaC(S)Rla, -NRlaS(O)2Rla, -S(O)Rla, -S(O)2Rla, -Rlb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -Rlb, wherein Rla is selected from the group consisting of hydrogen, provided, however, that hydrogen is not bound to any of C(S), C(O), S(O), or S(O)2 of -C(O)Rla, -C(S)Rla, -NRlaC(0)Rla, -NRlaC(S)Rla, -NRlaS(0)2Rla, -S(O)Rla, or -S(O)2Rla, -RIb, and lower alkyl optionally substituted with one or more, also 1, 2 or 3 groups or substituents selected from the group consisting of fluoro, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and -Rlb, provided, however, that any substitution of the alkyl carbon bound to O, S, or N of -0Rla, -SRla, -NRlaRla, -C(O)OR13, -C(0)NRlaRla, -C(S)NRlaRla, -S(0)2NRIaRIa, -NRlaC(O)Rla, -NRlaC(S)Rla, or -NRlaS(O)2Rla, is fluoro or -Rlb, and wherein -Rlb is selected from the group consisting of cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally substituted with one or more, also 1 , 2 or 3 groups or substituents selected from the group consisting of halogen, -CN, -OH, -NH2, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino.
[0316] "Lower alkoxy" denotes the group -ORZ, where Rz is lower alkyl. "Substituted lower alkoxy" denotes lower alkoxy in which Rz is lower alkyl substituted with one or more substituents as indicated herein, for example, in the description of compounds of Formula III, including descriptions of substituted cycloalkyl, cycloheteroalkyl, aryl and heteroaryl, attached at any available atom to produce a stable compound. Preferably, substitution of lower alkoxy is with 1, 2, 3, 4, or 5 substituents, also 1, 2, or 3 substituents. For example "fluoro substituted lower alkoxy" denotes lower alkoxy in which the lower alkyl is substituted with one or more fluoro atoms, where preferably the lower alkoxy is substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms. While it is understood that substitutions on alkoxy are attached at any available atom to produce a stable compound, substitution of alkoxy is such that O, S, or N (except where N is a heteroaryl ring atom), are not bound to the alkyl carbon bound to the alkoxy O. Further, where alkoxy is described as a substituent of another moiety, the alkoxy oxygen is not bound to a carbon atom that is bound to an O, S, or N of the other moiety (except where N is a heteroaryl ring atom), or to an alkene or alkyne carbon of the other moiety.
[0317] "Lower alkylthio" denotes the group -SRaa, where Raa is lower alkyl. "Substituted lower alkylthio" denotes lower alkylthio in which Raa is lower alkyl substituted with one or more
substituents as indicated herein, for example, in the description of compounds of Formula IE, including descriptions of substituted cycloalkyl, cycloheteroalkyl, aryl and heteroaryl, attached at any available atom to produce a stable compound. Preferably, substitution of lower alkylthio is with 1, 2, 3, 4, or 5 substituents, also 1, 2, or 3 substituents. For example "fluoro substituted lower alkylthio" denotes lower alkylthio in which the lower alkyl is substituted with one or more fluoro atoms, where preferably the lower alkylthio is substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms. While it is understood that substitutions on alkylthio are attached at any available atom to produce a stable compound, substitution of alkylthio is such that O, S, or N (except where N is a heteroaryl ring atom), are not bound to the alkyl carbon bound to the alkylthio S. Further, where alkylthio is described as a substituent of another moiety, the alkylthio sulfur is not bound to a carbon atom that is bound to an O, S, or N of the other moiety (except where N is a heteroaryl ring atom), or to an alkene or alkyne carbon of the other moiety.
[0318] "Amino" or "amine" denotes the group -NH2. "Mono-alkylamino" denotes the group -NHRbb where Rbb is lower alkyl. "Di-alkylamino" denotes the group -NRbbRcc, where Rbb and R00 are independently lower alkyl. "Cycloalkylamino" denotes the group -NRddRee, where Rdd and Ree combine with the nitrogen to form a 5-7 membered heterocycloalkyl, where the heterocycloalkyl may contain an additional heteroatom within the ring, such as O, N, or S, and may also be further substituted with lower alkyl. Examples of 5-7 membered heterocycloalkyl include, but are not limited to, piperidine, piperazine, 4-methylpiperazine, morpholine, and thiomorpholine. While it is understood that when mono-alkylamino, di-alkylamino, or cycloalkylamino are substituents on other moieties that are attached at any available atom to produce a stable compound, the nitrogen of mono-alkylamino, di-alkylamino, or cycloalkylamino as substituents is not bound to a carbon atom that is bound to an O, S, or N of the other moiety.
[0319] A "nitrogen protecting group" is a chemical group covalently bound to a nitrogen atom of a compound that is used to protect the nitrogen from reaction during a synthetic step. The nitrogen protecting group may be added to a compound and removed in a subsequent step by methods known to those of skill in the art. Nitrogen protecting groups include, without limitation, carbamates, amides, N-sulfonyl derivatives, groups of formula -C(O)OR, wherein R is, for example, methyl, ethyl, t-butyl, benzyl, phenylethyl, CH2=CHCH2-, and the like, groups of the formula -C(O)R', wherein R' is, for example, methyl, phenyl, trifluoromethyl, and the like, groups of the formula -SO2R", wherein R" is, for example, tolyl, phenyl, trifluoromethyl, 2,2,5,7,8-pentamethylchroman-6-yl, 2,3,6-trimethyl- 4-methoxyphenyl, and the like, and silanyl containing groups, such as 2-trimethylsilylethoxymethyl, t-butyldimethylsilyl, triisopropylsilyl, and the like. Other suitable nitrogen protecting groups may be found in texts such as T. W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991.
[0320] As used herein, the term "composition" refers to a formulation suitable for administration to an intended animal subject for therapeutic purposes that contains at least one pharmaceutically active compound and at least one pharmaceutically acceptable carrier or excipient.
[0321] The term "pharmaceutically acceptable" indicates that the indicated material does not have properties that would cause a reasonably prudent medical practitioner to avoid administration of the material to a patient, taking into consideration the disease or conditions to be treated and the respective route of administration. For example, it is commonly required that such a material be essentially sterile, e.g., for injectibles.
[0322] In the present context, the term "therapeutically effective" or "effective amount" indicates that the materials or amount of material is effective to prevent, alleviate, or ameliorate one or more symptoms of a disease or medical condition, and/or to prolong the survival of the subject being treated.
[0323] In the present context, the terms "synergistically effective" or "synergistic effect" indicate that two or more compounds that are therapeutically effective, when used in combination, provide improved therapeutic effects greater than the additive effect that would be expected based on the effect of each compound used by itself.
[0324] As used herein, the terms "ligand" and "modulator" are used equivalently to refer to a compound that changes (i.e., increases or decreases) the activity of a target biomolecule, e.g., an enzyme such as a kinase. Generally a ligand or modulator will be a small molecule, where "small molecule refers to a compound with a molecular weight of 1500 daltons or less, or preferably 1000 daltons or less, 800 daltons or less, or 600 daltons or less. Thus, an "improved ligand" is one that possesses better pharmacological and/or pharmacokinetic properties than a reference compound, where "better" can be defined by one skilled in the relevant art for a particular biological system or therapeutic use.
[0325] In the context of binding compounds and ligands, the term "derivative" or "derivative compound" refers to a compound having a chemical structure that contains a common core chemical structure as a parent or reference compound, but differs by having at least one structural difference, e.g., by having one or more substituents added and/or removed and/or replaced, and/or by having one or more atoms replaced with different atoms. Unless clearly indicated to the contrary, the term "derivative" does not mean that the derivative is synthesized using the parent compound as a starting material or as an intermediate, although in some cases, the derivative may be synthesized from the parent.
10326] Thus, the term "parent compound" refers to a reference compound having structural features also found in the derivative compound. Often but not always, a parent compound has a simpler chemical structure than the derivative.
[0327] By "chemical structure" or "chemical substructure" is meant any definable atom or group of atoms that constitute an individually identifiable portion of a molecule, such as a substituent moiety, a core which is optionally substituted, and the like. Normally, chemical substructures of a ligand can have a role in binding of the ligand to a target molecule, or can influence the three-dimensional shape, electrostatic charge, and/or conformational properties of the ligand.
[0328] The term "binds" in connection with the interaction between a target and a potential binding compound indicates that the potential binding compound associates with the target to a statistically significant degree as compared to association with proteins generally (i. e. , non-specific binding). Thus, the term "binding compound" refers to a compound that has a statistically significant association with a target molecule. Preferably a binding compound interacts with a specified target with a dissociation constant (KQ) of 1 mM or less, 1 μM or less, 100 nM or less, 10 nM or less, or 1 nM or less.
[0329] In the context of compounds binding to a target, the terms "greater affinity" and "selective" indicates that the compound binds more tightly than a reference compound, or than the same compound in a reference condition, i.e., with a lower dissociation constant. In some embodiments, the greater affinity is at least 2, 3, 4, 5, 8, 10, 50, 100, 200, 400, 500, 1000, or 10,000-fold greater affinity.
[0330] As used herein in connection with compounds of the invention, the term "synthesizing" and like terms means chemical synthesis from one or more precursor materials. Further, by "assaying" is meant the creation of experimental conditions and the gathering of data regarding a particular result of the experimental conditions. For example, enzymes can be assayed based on their ability to act upon a detectable substrate. A compound or ligand can be assayed based on its ability to bind to a particular target molecule or molecules.
[0331] As used herein, the term "modulating" or "modulate" refers to an effect of altering a biological activity, especially a biological activity associated with a particular biomolecule such as a protein kinase. For example, an agonist or antagonist of a particular biomolecule modulates the activity of that biomolecule, e.g., an enzyme, by either increasing (e.g. agonist, activator), or decreasing (e.g. antagonist, inhibitor) the activity of the biomolecule, such as an enzyme. Such activity is typically indicated in terms of an inhibitory concentration (IC50) or excitation concentration
(Ji(J5O) of the compound for an inhibitor or activator, respectively, with respect to, for example, an enzyme.
[0332] Jn the context of the use, testing, or screening of compounds that are or may be modulators, the term "contacting" means that the compound(s) are caused to be in sufficient proximity to a particular molecule, complex, cell, tissue, organism, or other specified material that potential binding interactions and/or chemical reaction between the compound and other specified material can occur.
[0333] As used herein in connection with amino acid or nucleic acid sequence, the term "isolate" indicates that the sequence is separated from at least a portion of the amino acid and/or nucleic acid sequences with which it would normally be associated.
[0334] In connection with amino acid or nucleic sequences, the term "purified" indicates that the subject molecule constitutes a significantly greater proportion of the biomolecules in a composition than the proportion observed in a prior composition, e.g., in a cell culture. The greater proportion can be 2-fold, 5-fold, 10-fold, or more than 10-fold, with respect to the proportion found in the prior composition.
I. General
[0335] The present invention concerns compounds of Formula I and all sub-generic formulae, compounds of Formula II and all sub-generic formulae, and compounds of Formula III and all sub- generic formulae that are modulators of protein kinases, for example without limitation, the compounds are modulators of at least one of the kinases selected from the group consisting of AbI, Aktl, Akt2, Akt3, ALK, Alk5, B-Raf, Brk, Btk, Cdk2, CDK4, CDK5, CDK6, CHKl, c-Raf-1, Csk, EGFR, EphAl, EphA2, EphB2, EphB4, Erk2, Fak, FGFRl, FGFR2, FGFR3, FGFR4, Fltl, Flt3, Flt4, Fms, Frk, Fyn, Gsk3α, Gsk3β, HCK, Her2/Erbb2, Her4/Erbb4, IGFlR, IKK beta, Irak4, Itk, Jakl, Jak2, Jak3, Jnkl, Jnk2, Jnk3, Kdr, Kit, LCK, MAP2K1, MAP2K2, MAP4K4, MAPKAPK2, Met, Mnkl, MLKl, p38, PDGFRA, PDGFRB, PDPKl, Piml, Pim2, Pim3, PKC alpha, PKC beta, PKC theta, PM, Pyk2, Ret, ROCKl, ROCK2, Ron, Src, Stk6, Syk, TEC, Tie2, TrkA, Yes, and Zap70, and the use of such compounds in the treatment of diseases or conditions.
II. Kinase targets and indications of the invention
[0336] Protein kinases play key roles in propagating biochemical signals in diverse biological pathways. More than 500 kinases have been described, and specific kinases have been implicated in a wide range of diseases or conditions (i.e., indications), including for example without limitation, cancer, cardiovascular disease, inflammatory disease, neurological disease, and other diseases. As
such, kinases represent important control points for small molecule therapeutic intervention. Description of specific target protein kinases contemplated by the present invention follow:
[0337] AbI: Target AbI (i.e., Abelson Murine Leukemia Viral Oncogene Homolog 1) is a 122.9 kDa tyrosine kinase encoded by chromosome 9q34.1 (symbol: ABLl.) The mature protein comprises SH3 (i.e., Src homology region 3) and SH2 (i.e., Src homology region 2) domains and the TK (i.e., tyrosine kinase) domain.
[0338] OMIM indicates AbI is expressed ubiquitously and can be localized to the nucleus where it binds DNA. Accordingly, AbI has been implicated in processes of cell differentiation, cell division, cell adhesion, and stress response. Alterations of the gene ABLl by a t(9;22)(q34;ql 1) chromosomal rearrangement or viral transduction lead to malignant transformation, as in chronic myeloid leukemia (CML). The kinase activity of AbI is negatively regulated by the constituent SH3 domain, and deletion of the SH3 domain turns ABLl into an oncogene. The t(9;22) translocation occurs in greater than 90% of chronic myelogeneous leukemia, 25 to 30% of adult and 2 to 10% of childhood acute lymphoblastic leukemia (ALL), and rare cases of acute myelogenous leukemia. The translocation results in the head-to-tail fusion of the BCR and ABL genes (Chissoe et al., Genomics 1995, 27: 67- 82). The DNA-binding activity of AbI is regulated by CDC2-mediated phosphorylation suggesting a cell cycle function for ABL. Welch & Wang (Cell 1993, 75: 779-790) showed that the tyrosine kinase activity of nuclear AbI is regulated in the cell cycle through a specific interaction with the retinoblastoma protein RBl. A domain in the C-terminus of RBl binds to the ATP-binding lobe of AbI, resulting in kinase inhibition. The RB-ABL interaction is not affected by the viral oncoproteins that bind to RB. Hyperphosphorylation of RB correlates with release of AbI and activation of AbI in S phase cells. In the nucleus AbI can enhance transcription, and this activity is inhibited by RB. Thus, nuclear AbI is an S phase-activated tyrosine kinase that might participate directly in the regulation of transcription. (Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MM Number: 189980: 12/20/2005: . World Wide Web URL: http ://www3.ncbi.nlm.nih. gov/omim/) . AbI inhibitors may be useful in treating leukemia, including chronic myelogeneous leukemia, acute lymphoblastic leukemia, and acute myelogenous leukemia.
[0339] Aktl: Target kinase Aktl (i.e., v-akt murine thymoma viral oncogene homolog 1) is a 55.7 kDa STK encoded by chromosome 14q32.32 (symbol: AKTl). Aktl is also known as protein kinase B-alpha, or PKB-alpha. OMIM indicates phosphoinositide 3-kinases (i.e., PDKs) generate specific inositol lipids implicated in the regulation of cell growth, proliferation, survival, differentiation, and cytoskeletal changes. One of the best characterized targets of PI3K lipid products is the protein kinase AKT, or protein kinase B (PKB). In quiescent cells, PKB resides in the cytosol in a low- activity conformation. Upon cellular stimulation, PKB is activated through recruitment to cellular membranes by PI3K lipid products and by phosphorylation by 3-prime phosphoinositide-dependent
kinase-1. Most proliferating cells are programmed to undergo apoptosis unless specific survival signals are provided. PDGF promotes cellular proliferation, inhibits apoptosis, and activates the RAS/PIK3/AKT1/IKBKA/NFKB1 pathway (Romashkova and Makarov, Nature 1999, 401: 86-90). In this pathway, NFKBl does not induce c-myc and apoptosis, but instead induces putative antiapoptotic genes. In response to PDGF, Aktl transiently associates with IKBK and induces IKBK activation (OMM MM Number: 164730: 10/26/2005)Aberrant Aktl activity is correlated with) including gastric and prostate tumors, colorectal, ovarian, pancreatic, and breast cancer, glioblastoma and leukemia. Sun et al., Am J Pathol 2001, 159(2):431-7, report that significantly increased AKTl kinase activity was detected in primary carcinomas of prostate (16 of 30), breast (19 of 50), and ovary (11 of 28). Tanno et al., Cancer Res 2001, 61(2):589-93, provide evidence for a link between AKT signaling and the regulation of IGF-IR expression and demonstrate that active AKT promotes the invasiveness of pancreatic cancer cells through the up-regulation of IGF-IR expression. Neri et al., MoI Cancer Res 2003, l(3):234-46, suggest that an up-regulation of the PI3K/AKT1 pathway might be one of the survival mechanisms responsible for the onset of resistance to chemotherapeutic and differentiating therapy in patients with acute leukemia. Aktl activity is also important in schizophrenia and bipolar disorders. Emamian et al., Nat Genet 2004, 36(2): 115-6, present convergent evidence for a decrease in AKTl protein levels and levels of phosphorylation of GSK3beta at Ser9 in the peripheral lymphocytes and brains of individuals with schizophrenia; a significant association between schizophrenia and an AKTl haplotype associated with lower AKTl protein levels; and a greater sensitivity to the sensorimotor gating-disruptive effect of amphetamine, conferred by AKTl deficiency. Aktl inhibitors may be useful in treating cancer, including gastric, prostate, colorectal, ovarian, pancreatic and breast cancer, glioblastoma and leukemia, and also for use in combination with other chemotherapeutic drugs.
[0340] Akt2: Target kinase Akt2 (i.e., v-akt murine thymoma viral oncogene homolog 2) is a 55.8 kDa STK encoded by chromosome 19q 13.1-13.2 (symbol: AKT2). Akt2 is also known as protein kinase B beta, PKB-beta. OMDvI indicates that the Akt2 isoform of Akt (see e.g., Aktl and Akt3) is enriched in insulin-responsive tissues and has been implicated in the metabolic actions of insulin. Glucose homeostasis depends on insulin responsiveness in target tissues, most importantly, muscle and liver. The critical initial steps in insulin action include phosphorylation of scaffolding proteins and activation of phosphatidylinositol 3-kinase. These early events lead to activation of the serine- threonine protein kinase Akt, also known as protein kinase B. Cho et al., Science 2001, 292:1728- 1731, showed that mice deficient in Akt2 are impaired in the ability of insulin to lower blood glucose because of defects in the action of the hormone on liver and skeletal muscle. Ablation of Akt2 in mice results in a mild but statistically significant fasting hyperglycemia due to peripheral insulin resistance and nonsuppressible hepatic glucose production accompanied by inadequate compensatory hyperinsulinemia (OMM MM Number: 164731: 10/26/2005). Arboleda et al., Cancer Res 2003,
63(1): 196-206 showed that AKT2 transfected breast and ovarian cancer cells demonstrated increased adhesion and invasion through collagen IV because of up-regulation of betal integrins and that AKT2 cells were more metastatic than control cells in vivo. Yamamoto et al., Clin Cancer Res 2004, 10(8):2846-50 studied the prognostic significance of Akt2 and activated Akt expression in pancreatic ductal adenocarcinoma (PDAC), concluding that p-Akt expression is a significant prognostic indicator for PDAC and inhibition of Akt is a possible molecular approach for treatment of PDAC. Yuan et al., Oncogene 2000, 19(19):2324-30, demonstrate that activation of AKT2 is a common occurrence in human ovarian cancer and that the PI 3 -kinase/ Akt pathway may be an important target for ovarian cancer intervention. Yuan et al., J Biol Chem 2003, 278(26):23432-40, demonstrate that constitutively active AKT2 renders cisplatin-sensitive A2780S ovarian cancer cells resistant to cisplatin, whereas phosphatidylinositol 3-kinase inhibitor or dominant negative AKT2 sensitizes A2780S and cisplatm-resistant A2780CP cells to cisplatin-induced apoptosis through regulation of the ASKl/JNK/p38 pathway. Akt2 inhibitors may be useful in treating cancer, including ovarian and pancreatic cancers, pancreatic ductal adenocarcinoma, and metastases of breast and ovarian cancer, and also for use in combination with other chemotherapeutic drugs, where such use sensitizes the tumor cells to the effects of the other chemotherapeutics.
[0341] Akt3: Target kinase Akt3 (i.e., v-akt murine thymoma viral oncogene homolog 3) is a 55.8 kDa STK encoded by chromosome Iq43-q44 (symbol:AKT3); Akt3 is also known as PKB gamma. Akt3 was identified as a protein kinase with high homology with the protein kinases A and C, hence, the name PKB. Akt3 comprises a PH domain that preferentially binds PtdIns(3,4,5)P3 and PtdIns(3,4)P2 over other phosphatidyl inositols (PIs). In quiescent cells, PKB resides in the cytosol in a low-activity conformation.
[0342] Upon cellular stimulation, PKB is activated through recruitment to cellular membranes by PI3K lipid products and phosphorylation by 3 '-phosphoinositide-dependent kinase-1 (PDKl). Active PKB then appears to detach from the plasma membrane and to translocate through the cytosol to the nucleus.
[0343] Stahl et al. have found that selective activation of the Akt3 protein promotes cell survival and tumor development in 43 to 60% of nonfamilial melanomas. The predominant Akt isoform active in melanomas was identified by showing that small interfering RNA (siRNA) against only Akt3, and not Aktl or Akt2, lowered the amount of phosphorylated (active) Akt in melanoma cells. The amount of active Akt3 increased progressively during melanoma tumor progression with highest levels present in advanced-stage metastatic melanomas. Mechanisms of Akt3 deregulation occurred through a combination of overexpression of Akt3 accompanying copy number increases of the gene and decreased PTEN protein function occurring through loss or haploinsufficiency of the PTEN gene. Targeted reduction of Akt3 activity with siRNA or by expressing active PTEN protein stimulated
apoptotic signaling, which reduced cell survival by increasing apoptosis rates thereby inhibiting melanoma tumor development. Therefore, Akt3 is a selective target in melanoma cells which provides therapeutic opportunities for subjects in advanced stages of the disease (Stahl et al., Cancer Res. 2004, 64:7002-10). Nakatani et al., J Biol Chem 1999, 274(31):21528-32 showed that in estrogen receptor-deficient breast cancer cells and androgen-insensitive prostate cells, the amount of Akt3 enzymatic activity was approximately 20-60-fold higher than in cells that were estrogen- or androgen- responsive. These and other results indicate that Akt3 may contribute to the more aggressive clinical phenotype of the estrogen receptor-negative breast cancers and androgen-insensitive prostate carcinomas and inhibitors may provide therapeutic benefits in treating these cancers. Akt3 inhibitors may be useful in treating cancer, including estrogen receptor-negative breast cancers, androgen- insensitive prostate carcinomas, and melanomas.
[0344] ALK: Target kinase ALK (i.e., anaplastic lymphoma kinase) is a 176.4 kDa receptor tyrosine kinase encoded by chromosome 2p23 (symbol: ALK). ALK appears to play a role in the development of the central nervous system. Perkins, et al., show that systemic ALCL is highly associated with anaplastic lymphoma kinase (ALK) gene translocations with over-expression of ALK protein. Anaplastic large cell lymphoma (ALCL) comprises 10-15% of childhood non-Hodgkin lymphomas (NHL) (Perkins et al., Br J Haematol 2005, 131(5):624-7). Marzec et al., states aberrant expression of the ALK tyrosine kinase as a chimeric protein with nucleophosmin (NPM) and other partners plays a key role in malignant cell transformation of T-lymphocytes and other cells. Further, studies with inhibitors of NPM/ALK enzyme activity suggest ALK as an attractive therapeutic target in T-cell lymphomas and other malignancies that express the kinase in an active form (Marzec et al., Lab Invest 2005, 85(12):1544-54). ALK inhibitors may be useful in treating cancer, including anaplastic large cell lymphoma and other T cell lymphomas.
[0345] Alk5: Target kinase Alk5 (i.e., Activin receptor-like kinase 5) is a 56.0 kDa STK encoded by chromosome 9q22 (symbol: TGFBRl). Alk5, the gene for which was isolated by Franzen et al (Cell 1993, 75: 681-692), is also known as transforming growth factor-beta receptor, type I, from which term the gene symbol derives. Among other activities, Alk5 mediates the induction of multiple genes involved in cell-matrix interactions. Alk5 inhibitors may be useful in treating pancreatic and biliary cancers and cutaneous T-cell lymphoma.
[0346] B-Raf: Target kinase B-Raf (i.e., v-raf murine sarcoma viral oncogene homolog Bl) is a 84.4 kDa serine/threonine kinase encoded by chromosome 7q34 (symbol: BRAF). The mature protein comprises RBD (i.e., Ras binding domain), Cl (i.e., protein kinase C conserved region 1) and STK (i.e., serine/threonine kinase) domains.
[0347] Target kinase B-Raf is involved in the transduction of mitogenic signals from the cell membrane to the nucleus and may play a role in the postsynaptic responses of hippocampal neurons. As such, genes of the RAF family encode kinases that are regulated by Ras and mediate cellular responses to growth signals. Indeed, B-Raf kinase is a key component of the RAS->Raf->MEK- >ERK/MAP kinase signaling pathway, which plays a fundamental role in the regulation of cell growth, division and proliferation, and, when constitutively activated, causes tumorigenesis. Among several isoforms of Raf kinase, the B-type, or B-Raf, is the strongest activator of the downstream MAP kinase signaling.
[0348] The BRAF gene is frequently mutated in a variety of human tumors, especially in malignant melanoma and colon carcinoma. The most common reported mutation was a missense thymine (T) to adenine (A) transversion at nucleotide 1796 (T1796A; amino acid change in the B-Raf protein is Val<600> to Glu<600> ) observed in 80% of malignant melanoma tumors. Functional analysis reveals that this transversion is the only detected mutation that causes constitutive activation of B-Raf kinase activity, independent of RAS activation, by converting B-Raf into a dominant transforming protein.
[0349] Niihori et al., report that in 43 individuals with cardio-facio-cutaneous (CFC) syndrome, they identified two heterozygous KRAS mutations in three individuals and eight BRAF mutations in 16 individuals, suggesting that dysregulation of the RAS-RAF-ERK pathway is a common molecular basis for the three related disorders (Niihori et al., Nat Genet. 2006, 38(3):294-6).
[0350] B-Raf inhibitors may be useful in treating neurologic diseases such as ischemic stroke, multi-infarct dementia, head injury, spinal cord injury, Alzheimer's disease (AD), Parkinson's disease; neoplastic diseases including, but not limited to, melanoma, glioma, sarcoma, carcinoma (e.g. lung, breast, pancreatic, renal), lymphoma (e.g. histiocytic lymphoma) and cancer of the thyroid, lung (e.g. small cell lung cancer), liver, breast, ovary and colon, neurofibromatosis, myelodysplastic syndrome, leukemia, tumor angiogenesis; pain of neuropathic or inflammatory origin, including acute pain, chronic pain, and migraine; cardiovascular diseases including heart failure, cardiac hypertrophy, thrombosis (e.g. thrombotic microangiopathy syndromes), atherosclerosis, reperfusion injury; inflammation including, but not limited to, psoriasis, polycystic kidney disease (PKD), arthritis and autoimmune diseases and conditions, osteoarthritis, endometriosis, scarring, vascular restenosis, fibrotic disorders, rheumatoid arthritis, inflammatory bowel disease (IBD); immunodeficiency diseases, organ transplant rejection, graft versus host disease; renal or prostatic diseases including diabetic nephropathy, nephrosclerosis, glomerulonephritis, prostate hyperplasia; metabolic disorders, obesity; infection, including, but not limited to Helicobacter pylori and Influenza virus, fever, sepsis; pulmonary diseases including chronic obstructive pulmonary disease (COPD) and acute respiratory distress syndrome (ARDS); genetic developmental diseases such as Noonan's syndrome, Costello
syndrome, (faciocutaneoskeletal syndrome), leopard syndrome, cardio-faciocutaneous syndrome (CFC), and neural crest syndrome abnormalities causing cardiovascular, skeletal, intestinal, skin, hair and endocrine diseases.
[0351] c-Raf-1: Target kinase c-Raf-1 (i.e., v-raf murine sarcoma viral oncogene homolog 1) is a 73.0 kDa STK encoded by chromosome 3p25 (symbol: RAFl). c-Raf-1 can be targeted to to the mitochondria by BCL2 (i.e., oncogene B-cell leukemia 2) which is a regulator of apoptotic cell death. Active c-Raf-1 improves BCL2-mediated resistance to apoptosis, and c-Raf-1 phosphorylates BAD (i.e., BCL2-binding protein). c-Raf-1 is implicated in carcinomas, including colorectal, ovarian, lung and renal cell carcinoma. C-Raf-1 is also implicated as an important mediator of tumor angiogenesis (Hood, J.D. et al., 2002, Science 296, 2404). C-Raf-1 inhibitors may also be useful for the treatment of acute myeloid leukemia and myelodysplastic syndromes (Crump, Curr Pharm Des 2002, 8(25):2243-8). Raf-1 activators may be useful as treatment for neuroendocrine tumors, such as medullary thyroid cancer, carcinoid, small cell lung cancer and pheochromocytoma (Kunnimalaiyaan et al., Anticancer Drugs 2006, 17(2): 139-42). C-Raf-1 inhibitors may be useful in treating colorectal, ovarian, lung and renal cell carcinoma, acute myeloid leukemia, myelodysplastic syndromes, tumor angiogenesis, and neuroendocrine tumors such as medullary thyroid cancer, carcinoid, small cell lung cancer and pheochromocytoma.
[0352] Brk: Target kinase Brk (i.e. breast tumor kinase, also known as PTK6) is a 51.8 IcDa nonreceptor tyrosine kinase encoded by human chromosome 20ql3.3 (symbol: BRK). The kinase has an SH3 domain, an SH2 domain, and a catalytic domain. In normal tissues, the expression of Brk (breast tumor kinase) is restricted to differentiating epithelial cells of the skin and gastrointestinal tract. According to Harvey and Crompton, Brk is expressed in over 60% of breast carcinoma tissue samples and breast tumour cell lines, but not normal mammary tissue or benign lesions. They used RNA interference to efficiently and specifically downregulate Brk protein levels in breast carcinoma cells, and determined that this results in a significant suppression of their proliferation (Harvey and Crompton, Oncogene, 2003, 22(32): 5006-5010). Lin et al. identified protein-tyrosine kinases that may be involved in the development and progression of head and neck squamous cell carcinoma (HNSCC), and their findings suggest that the signaling pathways mediated through EphAl, Brk, and Ron may be involved in the development and progression of HNSCC (Lin et al., Arch Otolaryngol Head Neck Surg. 2004, 130(3):311-6). Llor et al. examined BRK expression in the normal gastrointestinal tract, colon tumor cell lines, and primary colon tumor samples and showed BRK is expressed in normal epithelial cells of the gastrointestinal tract that are undergoing terminal differentiation. BRK expression also increased during differentiation of the Caco-2 colon adenocarcinoma cell line. Modest increases in BRK expression were detected in primary colon tumors by RNase protection, in situ hybridization, and immunohistochemical assays (Llor et al., Clin
Cancer Res. 1999, 5(7): 1767-77). Brk inhibitors may be useful in treating cancer, such as breast and colon cancer, and head and neck squamous cell carcinoma.
[0353] Btk: Target kinase Btk (i.e., Bruton's tyrosine kinase) is a 76.3 kDa tyrosine kinase encoded by chromosome Xq21.33-q22 (symbol: BTK). The mature kinase comprises a PH (i.e., Pleckstrin homology) domain, a BTK (i.e., Bruton's tyrosine kinase motif) motif, two SH3 domains, and a TK domain. Mao et al. determined the X-ray crystal structure of the Btk kinase domain in its unphosphorylated state to 2.1-angstrom resolution (Mao et al., J. Biol. Chem., 2001, 276:41435).
[0354] As a member of the BTK/Tec family of protein tyrosine kinases (i.e., PTKs), cytoplasmic Btk is involved in signal transduction pathways regulating growth and differentiation of B-lineage lymphoid cells (Rawlings, D. J., and Witte, O. N., 1994. Immunol. Rev. 138:105-119; Kurosaki, T., 1997, Curr Opin. Immunol 9:309-318; Uckun, F. M., 1998, Biochemical Pharmacology 56:683-691). As such, Btk participates in signal transduction pathways initiated by the binding of a variety of extracellular ligands to their cell surface receptors. For example, following ligation of B cell antigen receptors (BCR), Btk activation by the concerted actions of the PTKs Lyn and Syk (Kurosaki, T. (1997) Curr Opin. Immunol. 9, 309-318) is required for induction of phospholipase C-[gamma]2 mediated calcium mobilization (Kurosaki, T., 1997, Curr Opin. Immunol. 9:309-318). Furthermore, Btk regulates B cell antigen receptor-mediated JNKl response through Racl and phospholipase C- gamma2 activation.
[0355] Mutations in the human BTK gene are the cause of X-linked agammaglobulinemia (XLA), a male immune deficiency disorder characterized by a lack of mature, immunoglobulin producing, peripheral B cells (Tsukada, S., et al. (1993) Cell 72, 279-290; and Vetrie, D., et al. (1993) Nature 361, 226-233) and associated with a failure of Ig heavy chain rearrangement. Patients are unusually prone to bacterial infection but not to viral infection. A clinical picture resembling rheumatoid arthritis develops in many. Before antibiotics, death occurred in the first decade. In the more usual X-linked form of the disease, plasma cells are lacking. A rarer form of agammaglobulinemia (Hitzig, W. H et al. 1961, Med. Wschr., 91:1625), which is inherited as an autosomal recessive, shows marked depression of the circulating lymphocytes, and lymphocytes are absent from the lymphoid tissue. Mensink et al. (Clin. Genet., 1987, 31 :91) mapped XLA to Xq21 ,3-q22. Schwaber (Clin. Invest., 1992, 89:2053) presented direct evidence that of a failure OfV(D)J recombination which causes arrest in the transition from pre-B cell to B lymphocyte. XLA patients have been classified in 2 general groups: those presenting at an early age with particularly severe infections and those with less severe disease in which production of immunoglobulin is sustained at low-to-normal levels well into the first decade of life. In the latter cases, an oncogenetic change may occur in which the defective tyrosine kinase no longer can sustain the B-cell population, and a progressive reduction in immunoglobulin production occurs (Ohta, Y et al., Proc. Nat. Acad. Sci., 1994, 91:9062)
[0356] Btk is an inhibitor of the Fas/APO-1 death inducing signaling complex (DISC) in B-lineage lymphoid cells (Vassilev, A., et al, 1998, J. Biol. Chem., 274:1646-1656). Additionally, Btk prevents ceramide- and vincristine-induced apoptosis. The fate of leukemia/lymphoma cells may reside in the balance between the opposing proapoptotic effects of caspases activated by DISC and an upstream anti-apoptotic regulatory mechanism involving Btk and/or its substrates (Vassilev, A., et al., 1998, J. Biol. Chem. 274:1646-1656).
[0357] Accordingly, inhibitors of Btk are likely to enhance the drug sensitivity of B-lineage (e.g. leukemia/lymphoma such as acute lymphocytic leukemia) cells. Thus, pharmacological agents with Btk-modulatory activity can be used as chemosensitizing agents for treating Btk-expressing malignancies or diseases caused by proliferation and antibody production of BTK-expressing B-cells, and as B-cell reconstituting agents in humoral immunodeficiencies with decreased numbers or absence of B-cells. Furthermore, Btk modulating agents are useful as immunosuppressive agents for prevention of hyperacute rejection of organs in transplantation, which is directed by B-cells, autoimmune diseases, and conversion of immunity to drugs (e.g. antibodies or biologicals) or blood products (e.g. coagulation factors such as Factor VIII) in patients who develop antibodies to such agents.
[0358] Significant additional research has defined the role of Btk in the cell. For example, Cheng et al. (Proc. Nat. Acad. Sci., 1994, 91:8152) showed that Btk interacts with the SH3 domains of Fyn, Lyn, and Hck, all of which are activated upon stimulation of B- and T-cell receptors. These findings extended the range of interactions mediated by SH3 domains and provide indication of a link between Btk and previously established signaling pathways in B lymphocytes. Further, linkage studies involving 1,114 progeny backcross revealed colocalization of X-linked immunodeficiency (xid) mutation in mice with the BTK gene for (Thomas, J. D. et al., Science 1993, 261 :355). And further, Uckun et al. (Uckun, F. M et al. 1996, Science 273: 1096) reported that DT-40 lymphoma B cells rendered Btk deficient through targeted disruption of the BTK gene did not undergo radiation-induced apoptosis. Finally, Btk plays a key role in endotoxin-induced TNFα release from monocytes (Horwood, NJ. et al., J. Exp. Med. 2003, 197:1603).
[0359] Btk inhibitors may be useful in treating multiple sclerosis, systemic lupus erythematosis, rheumatoid arthritis, and Graves' disease, in addition to the diseases discussed above.
[0360] Cy clin dependent kinases: The cyclin dependent kinases (Cdk) play a major role in the signaling of cell cycles. The Cdk binds cyclin protein to form complex involved in the various stages of the cell cycle. Aberrant cell cycle progression occurs in cancers, often involving the activity of Cdk, such that inhibitors of Cdk are potential anti-cancer therapeutics.
[0361] Cdk2: Target kinase Cdk2 (i.e., Cyclin dependent kinase 2) is a 33.9 kDa serine/threonine kinase (STK) encoded by chromosome 12ql3 (symbol: CDK2). Cdk2 is also known as p33 protein kinase, and cell division protein kinase 2. De Bondt et al. reported the X-ray crystallographic structure of Cdk2 (De Bondt et al., Nature 1993, 363: 595-602). Cdk2 is involved in control of the human cell cycle. Cdk2 activation requires association with cyclins and leads to cell proliferation. Further, inhibition of cellular proliferation occurs upon association of inhibitors (e.g., cyclin- dependent kinase inhibitor IB) with the cyclin-Cdk2 complex. Cyclin E/Cdk2 complexes play a role in carcinogenesis, for example, Cdk2 amplication is associated with concurrent Cyclin E amplification in several cancers, including colorectal and ovarian cancer (Kitahara et al., Int. J. Cancer, 1995, 53: 1986-1989; Marone et al., Int. J. Cancer, 1998, 75: 31-39). Teixeira et al. show that retinoic acid inhibits cell cycle progression of MCF-7 human breast cancer cells by inhibiting cdk2 mKNA and protein production and by decreasing cdk2 activity (Teixeira et al., MoI Endocrinol 1997, 11(9): 1191-202). Cipriano and Chen studied the TGF-betal effect on normal human prostate and carcinoma cells, and showed that normal cells were sensitive to growth inhibition, whereas tumor cells were not or only minimally inhibited regardless of the concentration of TGF-betal, and correlated these results to Cdk2 activity. Their results indicate that a lack of inhibition of the Cdk2 activity correlates with insensitivity to TGF-betal in prostate tumor cells (Cipriano and Chen, Oncogene 1998, 17(12):1549-56). Cdk2 inhibitors may be useful in treating cancer, including prostate, breast, colorectal and ovarian cancer.
[0362] Cdk4: Target kinase Cdk4 (i.e., Cyclin dependent kinase 4) is a 33 kDa STK encoded by chromosome 12ql4 (symbol: CDK4). Lam et al. reported expression of CDK4 and CDK6 was elevated relative to matched normal brain tissue in eight of 18 glioblastoma multiforme (GBM) tumours (44%). Their data attests to the functional importance of both CDK4 and CDK6 in astrocytic tumourigenesis, particularly during the later stages of tumour progression (Lam et al., Br J Neurosurg 2000, 14(l):28-32). Backlund et al. found that loss of both wild-type copies of any of the three tumour suppressor genes CDKN2A, CDKN2B and RBl or gene amplification of CDK4, disrupting the RbI pathway, were associated with shorter survival in anaplastic astrocytoma patients (Backlund et al., Br J Cancer 2005, 93(l):124-30). Yu et al. report that the ability of cyclin Dl to activate cyclin- dependent kinase CDK4 underlies the critical role for cyclin Dl in breast cancer formation. They also found that the continued presence of CDK4-associated kinase activity is required to maintain breast tumorigenesis (Yu et al., Cancer Cell 2006, 9(l):23-32). Cdk4 inhibitors may be useful in treating
cancer, including glioblastoma (e.g. glioblastoma multiforme), anaplastic astrocytoma, and breast cancer.
[0363] Cdk5: Target kinase Cdk5 (i.e., Cyclin dependent kinase 5) is a 33.3 kDa STK encoded by chromosome 7q36 (symbol: CDK5). Cruz et al. state that proteolytic cleavage of p35 generates p25, leading to aberrant Cdk5 activation. The accumulation of p25 is implicated in several neurodegenerative diseases. Their findings provide compelling evidence that in vivo deregulation of Cdk5 by p25 plays a causative role in neurodegeneration and the development of neurofibrillary pathology (Cruz et al., Neuron 2003, 40(3):471-83). Takahashi et al. investigated the Cdk5 distribution pattern in diffuse Lewy body disease brains using immunohistochemistry. Their data suggest that Cdk5 may be associated with Lewy body formation (Takahashi et al., Brain Res 2000, 862(1 -2):253-6). Cdk5 inhibitors may be useful in treating neurodegenerative disorders, including Alzheimer's disease, amyotrophic lateral sclerosis and Lewy body disease.
[0364] Cdk6: Target kinase Cdk6 (i.e., Cyclin dependent kinase 6) is a 36.9 kDa STK encoded by chromosome 7q21-q22 (symbol: CDK6). Lam et al. reported expression of CDK4 and CDK6 was elevated relative to matched normal brain tissue in eight of 18 glioblastoma multiforme (GBM) tumours (44%). Their data attests to the functional importance of both CDK4 and CDK6 in astrocytic tumourigenesis, particularly during the later stages of tumour progression (Lam et al., Br J Neurosurg 2000, 14(l):28-32). Costello et al., applied restriction landmark genomic scanning to matched samples of glioma and normal brain DNA and found tumor-specific amplification of the gene encoding cyclin-dependent kinase 6 (CDK6). They also corroborated this finding by identifying both amplification-associated and amplification-independent increases in CDK6 protein levels in gliomas relative to matched normal brain samples (Costello et al., Cancer Res 1997, 57(7):1250-4). Corcoran et al. found in two samples from patients with splenic lymphoma with villous lymphocytes (SLVL), the CDK6 protein was markedly over expressed. They suggest that dysregulation of CDK6 gene expression contributes to the pathogenesis of SLVL and splenic marginal zone lymphoma (SMZL) (Oncogene 1999, 18(46): 6271-7). Chilosi et al. provide evidence that CDK6 is abnormally expressed in T-cell lymphoblastic lymphoma/leukemia (T-LBL/ALL) and may be involved in the pathogenesis of this malignancy (Chilosi et al., Am J Pathol 1998, 152(l):209-17). Cram et al. show that Indole-3- carbinol (I3C) can induce a G(I) cell cycle arrest of human MCF-7 breast cancer cells that is accompanied by the selective inhibition of cyclin-dependent kinase 6 (CDK6) expression (Cram et al., J Biol Chem 2001, 276(25): 22332-40). Cdk6 inhibitors may be useful in treating cancer, including glioblastoma multiforme, non-Hodgkin's lymphoma, splenic marginal zone lymphoma, T-cell lymphoblastic lymphoma (T-LBL) and T-cell acute lymphoblastic leukemia (T-ALL).
[0365] CHKl; Target kinase Chkl (i.e., Checkpoint kinase) is a 54.4 kDa STK encoded by chromosome 1 Iq24 (symbol: CHEKl, CHKl). CHKl is involved in DNA damage checkpoint.
Carassa et al., to understand the role of Chkl and Chk2 in the cellular response to different anticancer agents, knocked down the expression of each protein or simultaneously of both proteins by using the small interfering RNA technique in a HCT-116 colon carcinoma cell line and in its isogenic systems in which p53 and p21 had been inactivated by targeted homologous recombination. They show that inhibition of Chkl but not of Chk2 in p21(-/-) and p53(-/-) cells caused a greater abrogation of G(2) block induced by ionizing radiation and cis-diamine-dichloroplatinum treatments and a greater sensitization to the same treatments than in the parental cell line with p53 and p21 wild type proteins. Their data further emphasise the role of Chkl as a molecular target to inhibit in tumors with a defect in the G(I) checkpoint with the aim of increasing the selectivity and specificity of anticancer drug treatments (Carrassa et al., Cell Cycle 2004, 3(9):1177-81). Similarly, studies by Hirose et al. focused on the mechanism by which Temozolomide (TMZ) induces G(2)-M arrest and on whether inhibition of such G(2)-M arrest might sensitize glioma cells to TMZ-induced toxicity. U87MG glioma cells treated with TMZ underwent G(2)-M arrest associated with Chkl activation and phosphorylation of both cdc25C and cdc2. These TMZ-induced effects were inhibited by the Chkl kinase inhibitor UCN-Ol. Although not in itself toxic, UCN-01 increased the cytotoxicity of TMZ 5-fold, primarily by inhibiting cellular senescence and increasing the percentage of cells bypassing G(2)-M arrest and undergoing mitotic catastrophe. In addition to enhancing TMZ-induced cytotoxicity in p53-proficient cells, UCN-01 also blocked TMZ-induced Chkl activation and transient G(2)-M arrest in p53- defϊcient U87MG-E6 cells and similarly enhanced TMZ-induced mitotic catastrophe and cell death. Taken together, their results indicate that Chkl links TMZ-induced DNA mismatch repair to G(2)-M arrest. Furthermore, inhibition of the cytoprotective G(2) arrest pathway sensitizes cells to TMZ- induced cytotoxicity and may represent a novel, mechanism-based means of increasing TMZ efficacy in both p53 wild-type and p53 mutant glioma cells. (Hirose et al., Cancer Res 2001, 61(15):5843-9). As such, CHKl inhibitors may be used in combination therapy to improve the therapeutic efficacy of chemotherapeutic drugs. CHKl inhibitors may be useful in combination with chemotherapeutic drugs in treating cancer.
[0366] Csk: Target kinase Csk (i.e., c Src kinase) is a 50.7 kDa tyrosine kinase encoded by chromosome 15q23-q25 (symbol: CSK). Csk, cloned by Partanen et al. (Oncogene 1991, 6: 2013- 2018), is a cytoplasmic tyrosine kinase that downregulates the tyrosine kinase activity of the Src oncoprotein through tyrosine phosphorylation of the Src carboxy terminus. Activation of Csk may be therapeutic for cancers in which Src is activated, such as in colon and pancreatic carcinomas (Lutz et al., Biochem Biophys Res Commun 1998, 243(2):503-8; Cam et al., Cancer 2001, 92(l):61-70). Zheng and She state that the lymphoid-specific phosphatase (LYP) encoded by PTPN22 is involved in preventing spontaneous T-cell activation by dephosphorylating and inactivating T-cell receptor- associated Csk kinase. They genotyped 396 type 1 diabetic patients and 1,178 control subjects of Caucasian descent from north central Florida and report a strong association between type 1 diabetes
and a polymorphism (R620W) in the PTPN22 gene. In vitro experiments have shown that the mutant 620W LYP protein (1858T) does not bind Csk. Together with previous reports of the association between PTPN22 and type 1 diabetes, as well as rheumatoid arthritis and systemic lupus erythematosus, these results provide compelling evidence that LYP is a critical player in multiple autoimmune disorders (Zheng and She, Diabetes 2005, 54(3):906-8). Csk modulators, including inhibitors, may be useful in treating autoimmune diseases, including type 1 diabetes, rheumatoid arthritis and systemic lupus erythematosus.
[0367] EGFR; Target kinase EGFR (i.e., Epidermal Growth Factor Receptor) is a 134.3 kDa transmembrane tyrosine kinase coded by chromosome 7pl2.3-pl2.1 (symbol: EGFR). OMIM indicates that EGF enhances phosphorylation of several endogenous membrane proteins, including EGFR. EGFR has 2 components of different molecular weight; both contain phosphotyrosine and phosphothreonine but only the higher molecular weight form contains phosphoserine (Carlin and Knowles, Proc. Nat. Acad. Sci. 1982, 79: 5026-5030.). Carlin et al. (Cytogenet. Cell Genet. 1982, 32: 256) showed that the specific cell surface antigen previously called SA7 (Aden and Knowles, Imrnunogenetics 1976, 3: 209-211) is identical to EGFR. EGFR signaling involves small GTPases of the Rho family, and EGFR trafficking involves small GTPases of the Rab family. Lanzetti et al. (Nature 2000, 408: 374-377) reported that the EPS8 protein connects these signaling pathways. EPS8 is a substrate of EGFR that is held in a complex with SOSl by the adaptor protein E3B1, thereby mediating activation of RAC. Through its SH3 domain, EPS8 interacts with RNTRE. Further, Lanzetti et al. (ibid) showed that RNTRE is a RAB5 GTPase-activating protein whose activity is regulated by EGFR. By entering in a complex with EPS8, RNTRE acts on RAB5 and inhibits internalization of EGFR. Furthermore, RNTRE diverts EPS8 from its RAC-activating function, resulting in the attenuation of RAC signaling. Thus, depending on its state of association with E3B1 or RNTRE, EPS8 participates in both EGFR signaling through RAC and EGFR trafficking through RAB5 (OMM MM Number: 131550: 12/16/2005).
[0368] EGFR is implicated in breast cancer, colorectal, and bladder cancer, and modulation of EGFR activity is a therapeutic route to amelioration of these pathologic states (Xu et al., MoI Cancer Ther 2005, 4(3):435-42). An important unmet need has emerged in non small cell lung cancer patients who initially respond to treatment with EGFR inhibitors but then develop resistance to the initial drug (Koboyashi, S. et al. N Engl J Med. 2005, 352:786-92). EGFR is also a possible target for treating glioblastoma multiforme (Raizer, J Neurooncol 2005, 74(l):77-86), and squamous cell carcinomas, for example in the esophagus (Hanawa et al., Int J Cancer 2006, 118(5): 1173-80), head and neck (Hambek et al., Anticancer Res 2004, 24(6):3881-6), and oral cavity and tongue (Ekberg et al., Int J Oncol 2005, 26(5):1177-85). UnIu and Leake studied the effect of epidermal growth factor (EGF) and a specific inhibitor of EGFR1 on the growth and invasiveness of the prostate cancer cell
lines PC3 and DU145. Their results indicate that EGF is a potent stimulative agent for both growth and invasion in prostate cancer cells, and that targeting the EGFR function inhibits not only tumor growth but also invasiveness (UnIu and Leake, Int J Biol Markers 2003, 18(2): 139-46). EGFR inhibitors may be useful in treating cancer, including breast, colon, bladder, prostate and non small cell lung cancer, squamous cell carcinomas of the head and neck, oral cavity, and esophagus, and glioblastoma multiforme.
[0369] EphAl; Target kinase EphAl (i.e., Ephrin Receptor Al) is a 108.1 kDa tyrosine kinase encoded by chromosome 7q32-q34 (symbol: EPHAl). OMM indicates that the EPH and EPH- related receptors comprise the largest subfamily of receptor protein-tyrosine kinases. They have been implicated in mediating developmental events, particularly in the nervous system. Receptors in the Eph subfamily typically have a single kinase domain and an extracellular region containing a Cys-rich domain and 2 fibronectin type III repeats. The ligands for Eph receptors have been named "ephrins" by the Eph Nomenclature Committee (Cell 1997, 90: 403-404). Based on their structures and sequence relationships, ephrins are divided into the ephrin-A (EFNA) class, which are anchored to the membrane by a glycosylphosphatidylinositol linkage, and the ephrin-B (EFNB) class, which are transmembrane proteins. The Eph family of receptors are divided into 2 groups based on the similarity of their extracellular domain sequences and their affinities for binding ephrin-A and ephrin-B ligands. The Eph Nomenclature Committee (ibid.) proposed that Eph receptors interacting preferentially with ephrin-A proteins be called EphA and Eph receptors interacting preferentially with ephrin-B proteins be called EphB. Maru et al. (1988) reported characterization of the novel receptor tyrosine kinase gene, EPH. The splicing points of kinase domain-encoding exons were completely distinct from those of other protein tyrosine kinase genes, suggesting that this is the earliest evolutionary split within this family. In Northern blot analysis, EPH gene mRNA was detected in liver, lung, kidney, and testes of rat; screening of 25 human cancers of various cell types showed preferential expression in cells of epithelial origin. Overexpression of EPH mRNA was found in a hepatoma and a lung cancer without gene amplification. Southern blot analysis of DNAs from human-mouse hybrid clones with an EPH probe showed that this gene is present on human chromosome 7. Two other receptor tyrosine kinase genes, MET and EGFR, are on the same chromosome. By in situ hybridization, Yoshida et al. (1989) assigned the EPH locus to 7q32-q36. Although ephrins foπn a high-affinity multivalent complex with their receptors present on axons, axons can be rapidly repelled rather than being bound (OMM MM Number: 179610: 9/5/2000). Lin et al. identified 5 PTKs that were overexpressed in head and neck squamous cell carcinoma (HNSCC) using a reverse transcriptase-PCR technique and confirmed the overexpression of 3 known PTKs in some of the 8 archival HNSCC specimens studied. Their finding suggests that the signaling pathways mediated through EphAl, Brk, and Ron may be involved in the development and progression of HNSCC (Lin et al., Arch Otolaryngol Head Neck Surg 2004, 130(3):311-6). EphAl inhibitors may
be useful in treating cancer, including liver and lung cancer, and head and neck squamous cell carcinoma.
[0370] EphA2: Target kinase EphA2 (i.e., Eplirin Receptor A2) is a 108.3 kDa tyrosine kinase encoded by chromosome Ip36.1 (symbol: EPHA2). EphA2, similar to other ephrin receptors, is found in epithelial, lymphoid, and especially neuron tissue where EphA2 is critically involved in short-range contact-mediated axonal guidance. Further, EphA2 is highly expressed in metastatic melanoma cells. Ephrin Al, a ligand for EphA2, was shown to be up regulated during melanoma progression (Easty et al., Int J Cancer 1999, 84(5):494-501). Hattori et al. showed that ephrin-A2 forms a stable complex with the metalloproteinase Kuzbanian, involving interactions outside the cleavage region and the protease domain. Eph receptor binding triggered ephrin-A2 cleavage in a localized reaction specific to the cognate ligand. The cleavage-inhibiting mutation in ephrin- A2 delayed axon withdrawal. Hattori et al. (ibid) concluded that their studies reveal mechanisms for protease recognition and control of cell surface proteins, and, for ephrin- A2, they may provide a means for efficient axon detachment and termination of signaling (Hattori et al., Science 2000, 289: 1360-1365). Ireton and Chen review EphA2 as a promising target for cancer therapeutics, indicating EphA2 is overexpressed in breast, prostate, lung, and colon cancers (Ireton and Chen, Curr Cancer Drug Targets 2005, 5(3):149-57). Landen et al. state that EphA2 is involved in many processes crucial to malignant progression, such as migration, invasion, metastasis, proliferation, survival and angiogenesis. Inducing EphA2 downregulation by any one of several mechanisms (antibody- mediated inhibition of signalling, antibody-mediated downregulation of total EphA2 expression and siRNA-mediated inhibition of expression) has been shown to decrease tumour growth, prolong survival and inhibit angiogenesis in multiple preclinical models of ovarian, breast and pancreatic cancer. Targeting EphA2 is especially attractive in ovarian cancer, in which overexpression is present in > 75% of cases (Landen et al., Expert Opin Ther Targets 2005, 9(6): 1179-87). Abraham et al. state that the clinical significance of the expression of EpliA2 was observed in breast, prostate, colon, skin, cervical, ovarian, and lung cancers. They studied EphA2 to determine the expression of EphA2 and its ligand, Ephrin A-I, and E-cadherin in carcinoma of the urinary bladder, and determine EphA2 as a new target for therapy in bladder cancer. They conclude EphA2 may serve as a novel target for bladder cancer therapy, (Abraham et al., Clin Cancer Res 2006, 12(2):353-60;). EphA2 inhibitors may be useful in treating cancer, including bladder, breast, prostate, colon, skin, cervical, ovarian, pancreatic, and lung cancer and melanoma.
[0371] EphB2: Target kinase EphB2 (i.e., Ephrin Receptor B2) is a 117.5 kDa transmembrane tyrosine kinase encoded by chromosome Ip36.1-p35 (symbol: EPHB2). Mann et al. state that forward and reverse signaling mediated by EphB tyrosine kinase receptors and their transmembrane ephrin-B ligands play important roles in axon pathfmding. In their investigations of growth cones
from the ventral (EphB receptor-bearing) and dorsal (ephrin-B -bearing) embryonic Xenopus retina designed to investigate the signaling mechanisms in both forward and reverse directions, it is reported that unclustered, but not clustered, EphB2 ectodomains trigger fast (5-10 min) transient collapse responses in growth cones. This collapse response is mediated by low levels of intracellular cyclic GMP and requires proteasome function. In contrast, clustered, but not unclustered, ephrin-B 1 ectodomains cause slow (30-60 min) growth cone collapse that depends on high cGMP levels and is insensitive to inhibition of the proteasomal pathway. Upon receptor-ligand binding, endocytosis occurs in the reverse direction (EphB2-Fc into dorsal retinal growth cones), but not the forward direction, and is also sensitive to proteasomal inhibition. Endocytosis is functionally important because blocking of EphB2 internalization inhibits growth cone collapse. They state their data reveals that distinct signaling mechanisms exist for B-type Eph/ephrin-mediated growth cone guidance and suggest that endocytosis provides a fast mechanism for switching off signaling in the reverse direction (Mann et al., J Neurobiol 2003, 57(3):323-36). Nakada et al. demonstrate that migrating glioblastoma cells overexpress EphB2 in vitro and in vivo; glioma migration and invasion are promoted by activation of EphB2 or inhibited by blocking EphB2. Dysregulation of EphB2 expression or function may underlie glioma invasion (Nakada et al., Cancer Res 2004, 64(9):3179-85). Wu et al., investigated the expression of EphB2 and EphB4 in breast carcinomas. Clinicopathological and survival correlations were statistically analyzed in a series of 94 breast carcinomas, 9 normal specimens and 4 breast carcinoma cell lines. Both EphB2 and EphB4 RTPCR products could be detected in all specimens. Increased EphB2 protein expression was negatively associated with overall survival (Wu et al., Pathol Oncol Res 2004, 10(l):26-33). Hafner et al. studied expression profiles of 12 different Eph receptors and 8 ephrins inhuman lung, colorectal, kidney, liver, and brain cancers. They report EρhB2 was up-regulated 9-fold in hepatocellular carcinoma (Hafner et al., Clinical Chemistry 2004, 50:490-99). Umeda et al. studied the expression of ephrinB2 and EphB receptors within fϊbroproliferative membranes in patients with ocular angiogenic diseases collected during vitrectomy. EphB2 and EphB3 expression was observed on fibroproliferative membranes that were harvested from patients with proliferative diabetic retinopathy (EphB2, 90.0%; EphB3, 70.0%) and retinopathy of prematurity (EphB2, 35.0%; EphB3, 45.0%). Their data suggest that the ephrinB2- EphB2/B3 system may play an important role in ocular angiogenesis (Umeda et al., Am J Ophthalmol 2004, 138(2):270-9). EphB2 inhibitors may be useful in treating cancer, including breast cancer, hepatocellular carcinoma and glioblastoma, and for use in treating ocular angiogenesis diseases, including retinopathy (e.g. retinopathy of prematurity and proliferative diabetic retinopathy).
[0372] EphB4: Target kinase EphB4 (i.e., Ephrin Receptor B4) is a 108.3 kDa transmembrane tyrosine kinase encoded by chromosome 7q22 (symbol: EPHB4). EphB4 belongs to the Eph family of receptor tyrosine kinases. Developmental studies have shown that the Eph receptors, by regulating cell adhesion and cell movement in the embryo, are important for the proper organization and
integrity of tissues. Because tissue disorganization and abnormal cell adhesion, movement, and survival characterize the more advanced stages of cancer, the inappropriate functioning of an Eph receptor in breast tumor cells enhances malignancy. Xia et al. studied the biological function of the receptor tyrosine kinase EphB4 in bladder cancer. All of nine bladder cancer cell lines examined expressed EphB4. Further, they showed EphB4 knockdown using specific siRNA and antisense oligodeoxynucleotides molecules led to a profound inhibition in cell viability associated with apoptosis via activation of caspase-8 pathway and downregulation of antiapoptotic factor, bcl-xl. Furthermore, EphB4 knockdown significantly inhibited tumor cell migration and invasion. EphB4 knockdown in an in vivo murine tumor xenograft model led to a nearly 80% reduction in tumor volume associated with reduced tumor proliferation, increased apoptosis and reduced tumor microvasculature (Xia et al., Oncogene 2006, 25(5):769-80). Xia et al. also studied the expression and biological role of EphB4 in prostate cancer. They found EphB4 mRNA is expressed in 64 of 72 (89%) prostate tumor tissues assessed and EphB4 protein expression is found in the majority (41 of 62, 66%) of tumors, and 3 of 20 (15%) normal prostate tissues. They also showed knockdown of the EphB4 protein using EphB4 short interfering RNA or antisense oligodeoxynucleotide significantly inhibits cell growth/viability, migration, and invasion, and induces apoptosis in prostate cancer cell lines (Xia et al., Cancer Res 2005, 65(11):4623-32). Lee et al. used RT-PCR, western blotting and immunohistochemical techniques to examine EphB4 expression and protein levels in human prostate cancer cell lines LNCaP, DUl 45 and PC3. Immunohistochemistry was also used to examine localisation of EphB4 in tissue samples from 15 patients with prostate carcinomas. All three prostate cancer cell lines expressed the EphB4 gene and protein. EphB4 immunoreactivity in vivo was significantly greater in human prostate cancers as compared with matched normal prostate epithelium and there appeared to be a trend towards increased expression with higher grade disease (Lee et al., BMC Cancer 2005, 5:119). Stephenson et al. used commercially available cDNA arrays to identify EphB4 as a gene that is up-regulated in colon cancer tissue when compared with matched normal tissue from the same patient (Stephenson et al., BMC MoI Biol 2001, 2:15). Takai et al. used fluorescent immunohistochemistry to analyze serial frozen sections of 20 endometrial carcinomas and 20 normal endometria for EphB4 and ephrin-B2 protein expression. Further, they analyzed the relationship between the patient's characteristics and the percentages of EphB4- and ephrm-B2-stamed cells. They indicate the results demonstrate that increased EphB4 and ephrin-B2 expression may reflect or induce in endometrial carcinomas increased potential for growth and tumorigenicity (Takai et al., Oncol Rep 2001, 8(3):567-73). Sinha et al., studied expression of EphB4 in six men with primary squamous cell carcinoma of the head and neck (HNSCC) that had metastasized to the cervical lymph nodes. They obtained specimens of the primary tumor, the nodal metastasis, and the adjacent normal mucosa, and performed immunocytochemistry on each. They observed EphB4 expression in all primary and metastatic tumors and no expression in the normal tissue. In each of the six patients, expression was greater in the metastatic tumor than in the primary tumor (Sinha et al., Ear Nose
Throat J 2003, 82(11):866, 869-70, 887). EphB4 inhibitors may be useful in treating cancer, including breast, bladder, prostate, colon, and endometrial cancers and head and neck squamous cell carcinoma.
[0373] Erk2: Target kinase Erk2 (i.e., extracellular signal-regulated kinase 2) is a 41.4 kDa dual function serine/threonine-tyrosine kinase encoded by chromosome 22ql 1.2 (symbol: MAPKl). Erk2 is a member of the mitogen-activated protein (MAP) kinase family and is alternatively known as mitogen-activated protein kinase 1 (i.e., MAPKl). MAP kinases act as an integration point for multiple biochemical signals, and are involved in a wide variety of cellular processes such as proliferation, differentiation, transcription regulation and development.
[0374] The activation of Erk2 requires phosphorylation by upstream kinases. Upon activation, Erk2 translocates to the nucleus of the stimulated cells, where it phosphorylates nuclear targets, in addition to other targets including microtubule associated protein 2, myelin basic protein and ELKl. MacKenzie et al. state that the cAMP-specific phosphodiesterase family 4, subfamily D, isoform 3 (i.e., PDE4D3) is shown to have FQF (i.e., Phe-Gln-Phe) and KIM (i.e., Kinase Interaction Motif) docking sites for Erk2. These sites straddle the Ser(579) target residue for Erk2 phosphorylation of PDE4D3. Mutation of either or both of these docking sites prevent Erk2 from being co- immunoprecipitated with PDE4D3, ablate the ability of epidermal growth factor (EGF) to inhibit PDE4D3 through Erk2 action in transfected COS cells, and attenuate the ability of Erk2 to phosphorylate PDE4D3 in vitro. The two conserved NH(2)-terminal blocks of sequence, called upstream conserved regions 1 and 2 (i.e., UCRl and UCR2), that characterize PDE4 long isoforms, are proposed to amplify the small, inherent inhibitory effect that Erk2 phosphorylation exerts on the PDE4D catalytic unit. In contrast to this, the lone intact UCR2 region found in PDE4D1 directs COOH-terminal Erk2 phosphorylation to cause the activation of this short isoform. From the analysis of PDE4D3 truncates, it is suggested that UCRl and UCR2 provide a regulatory signal integration module that serves to orchestrate the functional consequences of Erk2 phosphorylation. The PDE4D gene thus encodes a series of isoenzymes that are either inhibited or activated by Erk2 phosphorylation and thereby offers the potential for ERK2 activation either to increase or decrease cAMP levels in cellular compartments (MacKenzie et al., J Biol Chem 2000, 275(22): 16609-17).
[0375] According to OMM, Pleschka et al. (Nature Cell Biol., 2001 , 3 : 301 -305) proposed that Erk2 regulates a cellular factor involved in the viral nuclear export protein function. They suggested that local application of MEK inhibitors may have only minor toxic effects on the host while inhibiting viral replication without giving rise to drug-resistant virus variants (OMIM MIM Number: 176948: 10/27/2005). Erk2 is involved in cytokine signaling and is a target for treating inflammation. Ramesh and Philipp state that lipoproteins are the key inflammatory molecule type of Borrelia burgdorferi, the spirochete that causes Lyme disease. They investigated whether specific inhibition of
p^δ ana ϋriu/z MAJfJβ. woum inniDit UN^-aipna ana JJL-O proauuuuu tuiu inus asirocyie apυpiυsis, and proliferation, respectively. Lipoprotein-stimulated IL-6 production was unaffected by the MAPK inhibitors. In contrast, inhibition of both p38 and Erkl/2 significantly diminished TNF-alpha production, and totally abrogated production of this cytokine when both MAPK pathways were inhibited simultaneously. MAPK inhibition thus may be considered as a strategy to control inflammation and apoptosis in Lyme neuroborreliosis (Ramesh and Philipp, Neurosci Lett 2005, 384(l-2):112-6). The role of Erk2 in signaling of cell differentiation, proliferation and survival suggests that inhibition of Erk2 may be therapeutic for several types of cancer. Husain et al. studied the effect of NSAIDs on MAPK activity and phosphorylation in gastric cancer. They conclude that NS-398 (a selective COX-2 inhibitor) and indomethacin (a non-selective NSAED) significantly inhibit proliferation and growth of human gastric cancer cell line MKN28. This effect is mediated by NSAID-induced inhibition of MAPK (ERK2) kinase signaling pathway, essential for cell proliferation (Husain et al., Life Sci 2001, 69(25-6):3045-54). Erk2 inhibitors may be useful in treating cancer, including gastric cancer, and in treating inflammation, including control of inflammation and apoptosis in Lyme neuroborreliosis.
[0376] Fak: Target kinase Fak (i.e., Focal adhesion kinase 1 , aka protein tyrosine kinase 2, PTK2) is a 119.2 kDa tyrosine kinase encoded by chromosome 8q24.3 (symbol: PTK2). The structure of Fak comprises a B41 (i.e., Band 4.1 homology) domain in addition to the TK domain. Fak and a related protein Pyk2/CAK-beta are cytoplasmic non-receptor protein tyrosine kinases. Localization of Fak via its C-terminal Focal Adhesion Targeting domain to focal complexes/adhesions (sites of integrin receptor clustering) is a prerequisite for Fak activation. Auto- or trans-phosphorylation of Fak on Tyr397 allows for docking of the Src family kinases, among other molecules. Src kinases phosphorylate Fak not only on the Tyr residues in the activation loop of the FAK kinase, but also create binding sites for downstream signaling components. Moreover, FAK can be activated by other receptors, linking them to the integrin signaling pathway. Activation of FAK can promote cell spreading, locomotion, survival and anchorage-dependent growth.
[0377] FAK-related non-kinase (FRNK), the C-terminal portion of FAK is expressed in some cell types by activating transcription from an alternative promoter. FRNK is proposed to function as an endogenous inhibitor of Fak signaling.
[0378] The role of Fak in signaling of cell proliferation, migration, adhesion and survival may be targeted in therapeutics for several types of cancer. Lightfoot et al. used immunohistochemical techniques to assess FAK expression in patients with fibrocystic disease (FCD), atypical ductal hyperplasia (ADH), ductal carcinoma in situ (DCIS) and infiltrating ductal carcinoma (IDC). The pattern of FAK expression in DCIS was significantly higher than ADH (p < 0.0001) and IDC (p = 0.02). They conclude that FAK overexpression in preinvasive, DCIS tumors precedes tumor cell
invasion or metastasis, suggesting that FAK may function as a survival signal and be an early event in breast tumorigenesis (Lightfoot et al., Breast Cancer Res Treat 2004, 88(2): 109-16). Miyazaki et al. performed an immunohistochemical analysis of FAK protein expression to determine the relationship between FAK overexpression and clinicopathological factors in oesophageal squamous cell carcinoma (ESCC). They concluded that FAK overexpression of ESCC was related to cell differentiation, tumour invasiveness, and lymph node metastasis. Consequently, patients with ESCC who had FAK overexpression had a poor prognosis (Miyazaki et al., Br J Cancer 2003, 89(1): 140-5). Smith et al. tested antisense oligonucleotide inhibitors of FAK, in combination with 5-fluorouracil (5- FU), to increase its sensitivity in human melanoma cell lines. They conclude their data show that the downregulation of FAK by antisense oligonucleotide combined with 5-FU chemotherapy results in a greater loss of adhesion and greater apoptosis in melanoma cells than treatment with either agent alone, suggesting that the combination may be a potential therapeutic agent for human melanoma in vivo (Smith et al., Melanoma Res 2005, 15(5):357-62). Li a review, Natarajan et al. summarize data that has demonstrated 1) elevated FAK expression in anaplastic astrocytoma and glioblastoma tumor biopsy samples, 2) a role for FAK in the promotion of glioblastoma cell proliferation, survival and migration in vitro, and 3) a role for FAK in the promotion of glioblastoma cell proliferation in vivo in an animal model (Natarajan et al., Cancer J 2003, 9(2): 126-33). Rovin et al. investigated FAK expression in human prostate specimens by using immunohistochemistry. In their conclusion, they suggest that the sustained elevated levels of FAK expression during prostate tumor cell progression is consistent with a role for FAK in the development and maintenance of prostate carcinoma (Rovin et al., Prostate 2002, 53(2): 124-32). Itoh et al. investigated whether focal adhesion kinase (FAK) is involved in the progression of human hepatocellular carcinoma (HCC). They conclude their data suggests that FAK plays an important role in promoting tumor progression, especially vascular invasion, in HCC (Itoh et al., Clin Cancer Res 2004, 10(8):2812-7). von Sengbusch et al. state that organ-specific tumor cell adhesion to extracellular matrix (ECM) components and cell migration into host organs often involve integrin-mediated cellular processes that can be modified by environmental conditions acting on metastasizing tumor cells, such as shear forces within the blood circulation. Since the focal adhesion kinase (FAK) appears to be essential for the regulation of the integrin- mediated adhesive and migratory properties of tumor cells, they investigated its role in early steps of the metastatic cascade using in vitro and in vivo approaches. They summarize that FAK appears to be involved in early events of integrin-mediated adhesion of circulating carcinoma cells under fluid flow in vitro and in vivo. This kinase may take part in the establishment of definitive adhesive interactions that enable adherent tumor cells to resist fluid shear forces, resulting in an organ-specific formation of distant metastases (von Sengbusch et al., Am J Pathol 2005, 166(2):585-96).
[0379] Westhoff et al., note that elevated expression of the nonreceptor tyrosine kinases Src and Fak correlates with malignancy potential and poor clinical prognosis in colon and breast tumors. They
also state that recent studies monitoring focal adhesion dynamics in cells deficient for Fak and Src implicate Src and Fak as critical mediators of integrin adhesion turnover that promote cell migration. Cells devoid of FAK exhibit impaired migration and have large peripheral focal adhesion structures, while cells lacking the three ubiquitous Src family members Src, Fyn, and Yes also demonstrate altered distribution of focal adhesions and impaired cell migration. Src kinase activity is clearly necessary for focal adhesion turnover and cell motility, presumably by tyrosine phosphorylation of key focal adhesion substrates, such as FAK. The extracellular regulated kinase (ERK)/mitogen- activated protein kinase (MAPK) pathway is also important in regulating focal adhesion dynamics during cell motility and it is likely that ERK/MAPK contributes to Src-induced focal adhesion turnover. Further, Westhoff et al. have recently reported that ERK/MAPK, which is recruited to focal adhesions following v-Src activation, is required for maximal activity of the protease calpain 2 promoting focal adhesion turnover and migration of v-Src-transformed cells. ERK/MAPK-induced activation of calpain 2 is also required for epidermal growth factor-induced substrate deadhesion and cell motility. Six major tyrosine phospho-acceptor sites have been identified on Fak, at positions 397, 407, 576, 577, 861, and 925. Tyr 397 becomes phosphorylated (presumed by auto-phosphorylation) upon integrin engagement. This leads to the formation of a consensus binding site for the Src SH2 domain, promoting association between Src and FAK. Phosphorylation of the remaining tyrosine residues on Fak is considered to be Src-dependent. Westhoff et al., generated a FAK mutant (4-9F- Fak) in which each of the putative Src-induced tyrosine phosphorylation sites (Tyr 407, 576, 577, 861, and 925) has been mutated to a phenylalanine (Phe). They found that v-Src-induced phosphorylation of FAK on tyrosine residues is necessary to enhance the adaptor function of Fak with regard to assembly of the calpain 2/FAK/ρ42ERK complex. Src-induced phosphorylation of Fak is also required for Fak to undergo proteolytic cleavage by calpain in v-Src-transformed cells and is necessary for calpain-mediated focal adhesion turnover during transformation and cell migration. In addition, they show that Src-induced phosphorylation of Fak also regulates F-actin assembly and cell spreading. We further demonstrate a role for Src-induced tyrosine phosphorylation of Fak in survival and anchorage-independent growth of transformed cells (Westhoff et al., Molecular and Cellular Biology, 2004, 24: 8113-8133).
[0380] Fak inhibitors may be useful in treating cancer, including colon and breast cancers, melanoma, ductal carcinoma in situ, oesophageal squamous cell carcinoma, anaplastic astrocytoma and glioblastoma, and human hepatocellular and prostate carcinomas, as well as in reducing tumor metastasis. They may also be used in combination with other chemotherapeutic drugs to provide synergistic effects in treating cancers such as melanoma.
[0381] FGFR kinase family: The FGFRs (i.e., Fibroblast Growth Factor Receptors) comprise a family of related but individually distinct tyrosine kinase receptors. They have a similar protein
structure, with 3 immunoglobulin-like domains in the extracellular region, a single membrane spanning segment, and a cytoplasmic tyrosine kinase domain. The fibroblast growth factor receptors that have been identified are FGFRl, FGFR2, FGFR3, which is mutant in achondroplasia; and FGFR4. Sequence analysis of the 4.5-kb human FGFR2 gene shows an open reading frame encoding the typical membrane-spanning, tyrosine kinase receptor structure of the FGFR gene family. A discussion of FGFRl, FGFR2, FGFR3, and FGFR4 follows.
FGFRl
[0382] FGFRl: Target FGFRl (i.e., Fibroblast Growth Factor Receptor 1) is a 91.9 kDa transmembrane tyrosine kinase encoded by chromosome 8pl 1.2pl 1.1 (symbol: FGFRl). FGFRl is also known as FMS-like tyrosine kinase 2 (i.e., Flt2). FGFRl is implicated in a variety of cancers (e.g. 8pl 1 syndrome, Braun & Shannon, Cancer Cell 2004, 5:203). Additionally, FGFRl is an important mediator of tumor angiogenesis (Compagni et al., Cancer Res. 2000, 60:7163). The X-ray crystallographic structure of FGFRl bound to fibroblast growth factor 2 has been reported by Plotnikov et al {Cell 1999, 98: 641-650). Rossi et al report an FGFRl P252R mutation in families affected by Pfeiffer syndrome (Rossi et al., Clin Dysmorphol. 2003, 12(4):269-74). FGFRl inhibitors may be useful in treating 8pl 1 myeloproliferative syndrome.
[0383] FGFR2: Target FGFR2 (i.e., Fibroblast Growth Factor Receptor 2) is a 92 kDa transmembrane tyrosine kinase encoded by chromosome 10q26 (symbol: FGFR2). According to OMIM, from a human tumor cDNA library, Houssaint et al. (Proc. Nat. Acad. Sci.1990, 87: 8180- 8184) isolated a gene encoding a putative receptor-like protein-tyrosine kinase that they called TKl 4. The amino acid sequence was closely related to that of the mouse protein bek (bacterially expressed kinase), and more distantly related to the sequences of a chicken basic fibroblast growth factor receptor (73% sequence homology) and its presumed human equivalent, the FLG protein. Overexpression of the TK14 protein by transfection of COS-I cells led to the appearance of new cell- surface binding sites for both acidic and basic fibroblast growth factors (OMlM MIM Number: 176943: 04/06/2006).
[0384] Sequence analysis of the 4.5-kbp human FGFR2 gene shows an open reading frame encoding the typical membrane-spanning, tyrosine kinase receptor structure of the FGFR gene family. Two alternative gene products have been characterized: KGFR and BEK. These two isoforms are identical except for a 49-amino acid sequence spanning the second half of the third Ig loop in the extracellular region. This local diversity is due to the presence of alternative exons within FGFR2, exon B being expressed in the BEK product and exon K26 in KGFR. Control of these alternative splice sites is thought to involve transacting factors (Gilbert et al., Molec. Cell. Biol. 1993, 13: 5461- 5468). The variation in expressed gene product is highly significant because the ligand-binding
characteristics of KGFR and BEK are quite distinct. Furthermore, they have different patterns of expression in murine embryogenesis. Whereas KGFR appears to have a role in skin development, BEK is preferentially expressed in osteogenesis. BEK transcripts are concentrated in the frontal bones, maxilla, mandibula, and ossicles of the middle ear.
[0385] To elucidate the structural determinants governing specificity in FGF signaling, Plotnikov et al. (Cell 2000, 101: 413-424) determined the crystal structures of FGFl and FGF2 complexed with the immunoglobulin-like ligand-binding domains 2 and 3 (D2 and D3) of FGFRl and FGFR2, respectively. They found that highly conserved FGF-D2 and FGF-linker (between D2 and D3) interfaces define a general binding site for all FGF-FGFR complexes. Specificity is achieved through interactions between the N-terminal and central regions of FGFs and 2 loop regions in D3 that are subject to alternative splicing. These structures provide a molecular basis for FGFl as a universal FGFR ligand and for modulation of FGF-FGFR specificity through primary sequence variations and alternative splicing (OMIM MIM Number: 176943: 04/06/2006).
[0386] Defects in FGFR2 are a cause of Crouzon Syndrome (CS); also called craniofacial dysostosis type I (CFDl). CS is an autosomal dominant syndrome characterized by craniosynostosis (premature fusion of the skull sutures), hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism (OMIM MIM Number: #123500: 05/04/2000).
[0387] Further, defects in FGFR2 are a cause of Jackson-Weiss syndrome (JWS). JWS is an autosomal dominant craniosynostosis syndrome characterized by craniofacial abnormalities and abnormality of the feet including broad great toes with medial deviation and tarsal-metatarsal coalescence (OMJM MM Number: 176943: 04/06/2006).
[0388] Further, defects in FGFR2 are a cause of Apert Syndrome, also known as acrocephalosyndactyly type I (ACSl), which is characterized by craniosynostosis (premature fusion of the skull sutures) and severe syndactyly (cutaneous and bony fusion of the digits), and is autosomal dominant.
[0389] Further, defects in FGFR2 are a cause of Pfeiffer Syndrome (PS), also known as acrocephalosyndactyly type V (ACS5), which is characterized by craniosynostosis with deviation and enlargement of the thumbs and great toes, brachymesophalangy, with phalangeal ankylosis and a varying degree of soft tissue syndactyly. Three subtypes of PS have been described: mild autosomal dominant form (type 1); cloverleaf skull, elbow ankylosis, early death, sporadic (type 2); craniosynostosis, early demise, sporadic (type 3).
[0390] Further, defects in FGFR2 are the cause of beare-stevenson cutis gyrata syndrome (BSCGS) which is an autosomal dominant condition characterized by the furrowed skin disorder of cutis gyrata,
acanthosis nigricans, craniosynostosis, craniofacial dysmorphism, digital anomalies, umbilical and anogenital abnormalities and early death (OMM MM Number: 176943: 04/06/2006).
[0391] Further, defects in FGFR2 are the cause of Antley-Bixler syndrome which is characterized by trapezoidocephaly, midface hypoplasia, humeroradial synostosis, bowing of femora, fractures and other abnormalities (OMM MM Number: #207410: 11/29/2005). FGFR2 inhibitors may be useful in treating Crouzon Syndrome, Jackson-Weiss Syndrome, Apert Syndrome, craniosynostosis, Pfeiffer Syndrome, acrocephalo syndactyly type V, and Beare-Stevenson Cutis Gyrata Syndrome.
[0392] FGFR3: Target kinase FGFR3 (i.e., Fibroblast Growth Factor Receptor 3) is a 87.7 kDa transmembrane tyrosine kinase encoded by chromosome 4pl6.3 (symbol: FGFR3). As a member of the fibroblast growth factor family, FGFR3 is involved in a variety of activities, including mitogenesis, angiogenesis, and wound healing. Furthermore, FGFR3 plays a role in the development and maintenance of bone and brain tissue. FGFR3 regulates bone growth by limiting the formation of bone from cartilage, particularly in the long bones. In addition, FGFR3 is activated by translocation in approximately 15% of multiple myeloma (Trudel, S., Blood 2004, 103:3521). FGFR3 inhibitors may be useful in treating angiogenesis disorders, wounds, achondroplasia, Muenke craniosynostosis, Crouzon syndrome, acanthosis nigricans, thanatophoric dysplasia, bladder carcinomas, and multiple myeloma.
[0393] FGFR4: Target kinase FGFR4 (i.e., Fibroblast Growth Factor Receptor 4) is a 88.0 kDa transmembrane tyrosine kinase encoded by chromosome 5q35.3 (symbol: FGFR4). According to OMM, Partanen et al. (EMBO J. 1991, 10: 1347-1354) reported the cDNA cloning and analysis of a novel member of the fibroblast growth factor receptor (FGFR) gene family expressed in K562 erythroleukemia cells. Its deduced amino acid sequence was 55% identical with the previously characterized FGFRs, FLG (FGFRl) and BEK, and had the structural characteristics of an FGFR family member including 3 immunoglobulin-like domains in its extracellular part. The expression pattern of FGFR4 was found to be distinct from that of FLG and BEK and also distinct from that of FGFR3, which had also cloned from K562 erythroleukemia cells. To elucidate further the physiologic relevance of protein-tyrosine kinases and to search for additional members of the gene family as possible factors in carcinogenesis, Holtrich et al. (Proc. Nat. Acad. Sci. 1991, 88: 10411- 10415) amplified mRNA from lung tissue by the polymerase chain reaction (PCR) using PTK- specific primers followed by sequencing of the clones. They identified a novel protein-tyrosine kinase, which they called TKF (tyrosine kinase related to fibroblast growth factor receptor). Among a wide variety of cells and tissues tested, including human lymphocytes and macrophages, TKF was found to be expressed only in lung and in some tumors of lung origin as well as in malignancies not derived from lung tissues. Sequence comparison has demonstrated that TKF is identical to FGFR4 (OMM MM Number: 134935: 05/03/2002).
[0394] The FGFR4 protein interacts with specific growth factors to conduct signals from the environment outside the cell to the nucleus. Animal studies indicate that the FGFR4 gene is involved in muscle development and the maturation of bone cells in the skull. FGFR4 may also play a role in the development and maintenance of specialized cells (called foveal cones) in the light-sensitive layer (retina) at the back of the eye. Aberrant expression of FGFR4 is correlated with cancer of the breast, ovary, endometrium, and fallopian tube, and with leiomyosarcoma. FGFR4 inhibitors may be useful in treating cancer of the breast, lung, colon, medullary thyroid, pancreas, ovary, prostate, endometrium, and fallopian tube, head and neck squamous cell carcinomas and leiomyosarcoma.
[0395] FItI: Target kinase Fltl (i.e., Fms like tyrosine kinase 1) is a 150.7 kDa transmembrane tyrosine kinase encoded by chromosome 13ql2 (symbol: FLTl), also known as VEGFRl (i.e., Vascular Endothelial Growth Factor Receptor 1). According to OMM, oncogene FLT belongs to the SRC gene family and is related to oncogene ROS. Like other members of this family, it shows tyrosine protein kinase activity that is important for the control of cell proliferation and differentiation via interaction with PLC-gammas, PTPNl 1, GRB2, CRK, NCKl and other proteins. The name is due to the resemblance of the sequence structure of the FLT gene to that of the FMS gene. VEGF and its high-affinity binding receptors, the tyrosine kinases Flkl and Fltl , are thought to be important for the development of embryonic vasculature. Studying transgenic mice in whom the Flkl gene was disrupted, Shalaby et al. (Nature 1995, 376: 62-65) demonstrated a total failure of embryonic mice to develop blood vessels and failure of blood island formation in the yolk sac. Fong et al. (Nature 1995, 376: 65-69) reported that in mice Fltl is essential for the organization of embryonic vasculature, but is not essential for endothelial cell differentiation. Transgenic mouse embryos homozygous for a targeted mutation in the Fltl locus formed endothelial cells in both embryonic and extraembryonic regions, but assembled these cells into abnormal vascular channels and died in utero at mid-somite stages. At earlier stages, the blood islands of homozygous mice were abnormal, with angioblasts in the interior as well as on the periphery. Fong et al. (ibid.) suggested that the Fltl signaling pathway may regulate normal endothelial cell-cell or cell-matrix interactions during vascular development (OMIM MIM Number: 165070: 03/27/2006). Fit 1 inhibitors may be useful in treating non-small cell lung carcinoma, prostate carcinoma, and colorectal cancer.
[0396] Flt3: Target kinase Flt3 (i.e., Fms-like tyrosine kinase 3) is a transmembrane tyrosine kinase of 112.8 kDa encoded by chromosome 13ql2 (symbol: FLT3). According to OMIM, Rosnet et al. (Genomics 1991, 9: 380-385) isolated a novel member of the class 3 receptors discussed above. They demonstrated that this gene of the tyrosine kinase family, called FLT3, has strong sequence similarities with other members of the group. Lymphohematopoietic stem cells serve as a reservoir for virtually all blood cells but make up only approximately 0.01% of human or murine marrow cells. The ability to isolate and expand this population has clinical applications in bone marrow
transplantations for cancer and genetic diseases. Small et al. (Proc. Nat. Acad. Sci. 1994, 91: 459-463) cloned the cDNA for stem cell tyrosine kinase 1, the human homolog of murine Flk2/Flt3, from a CD34+ hematopoietic stem cell-enriched library. The cDNA encoded a protein of 993 amino acids with 85% identity and 92% similarity to the murine homolog. STKl, which is identical to FLT3, is a member of the type III receptor tyrosine kinase family that includes KIT, FMS, and platelet-derived growth factor receptor. STKl expression in human blood and marrow is restricted to CD34+ cells, a population greatly enriched by stem/progenitor cells. Antisense oligonucleotides directed against STKl sequences inhibited hematopoietic colony formation, most strongly in long-term bone marrow cultures. The data suggested that STKl may function as a growth factor receptor on hematopoietic stem and/or progenitor cells (OMM MIM Number: 136351: 03/03/2005).
[0397] Levis et al, state that Internal tandem duplication (ITD) mutations of the receptor tyrosine kinase FLT3 have been found in 20% to 30% of patients with acute myeloid leukemia (AML). These mutations constitutively activate the receptor and appear to be associated with a poor prognosis. In their study, dose-response cytotoxic assays were performed with AGl 295, a tyrosine kinase inhibitor active against FLT3, on primary blasts from patients with AML, and they found that AG 1295 was specifically cytotoxic to AML blasts harboring FLT3/ITD mutations. They suggest that these mutations contribute to the leukemic process and that the FLT3 receptor represents a therapeutic target in AML (Levis et al., Blood 2001, 98:885-887). Flt3 inhibitors may be useful in treating acute myeloid leukemia, myelodysplastic syndrome, acute lymphoblastic leukemia.
[0398] Flt4: Target kinase Flt4 (i.e., Fms-like tyrosine kinase 4) is a transmembrane tyrosine kinase of 145.6 kDa encoded by chromosome 5q35.3 (symbol: FLT4). Flt4 is also known as VEGFR3 (i.e., Vascular Endothelial Growth Factor Receptor 3). According to OMM, by screening a placenta cDNA library with a mouse Flt3 probe, Galland et al. (Genomics 1992, 13: 475-478) isolated a human gene encoding a putative receptor-type tyrosine kinase, FLT4. The deduced amino acid sequence of the intracellular portion of the molecule showed that it was strongly related to FLTl and KDR and to a lesser degree to members of the class 3 receptor-type tyrosine kinases: FMS, PDGFR, KIT, and FLT3. Primary lymphoedema, a rare, autosomal dominant disorder that leads to a disabling and disfiguring swelling of the extremities and, when untreated, tends to worsen with time, has been linked to the FLT4 locus (Karkkainen et al., Nat. Genet. 2000, 25: 153-9). All disease-associated alleles analyzed had missense mutations and encoded proteins with an inactive tyrosine kinase, preventing downstream gene activation (OMM MM Number: 136352: 11/19/2003). Flt4 inhibitors may be useful in treating primary lymphoedema.
[0399] Fms: Target kinase Fms (i.e., feline McDonough sarcoma) is a member of the family of genes originally isolated from the Susan McDonough strain of feline sarcoma viruses. Fms is a transmembrane tyrosine kinase of 108.0 kDa coded by chromosome 5q33.2-q33.3 (symbol: CSFlR).
The structure of the transmembrane receptor Fms comprises two Ig-like domains, a IgC2-like domain, two additional Ig-like domains, a TM domain, and the TK domain.
[0400] Fms is the receptor for the macrophage colony-stimulating factor (M-CSF), and is crucial for the growth and differentiation of the monocyte-macrophage lineage. Upon binding of M-CSF to the extracellular domain of Fms, the receptor dimerizes and trans-autophosphorylates cytoplasmic tyrosine residues.
[0401] M-CSF, first described by Robinson and co-workers (Blood. 1969, 33:396-9), is a cytokine that controls the production, differentiation, and function of macrophages. M-CSF stimulates differentiation of progenitor cells to mature monocytes, and prolongs the survival of monocytes. Furthermore, M-CSF enhances cytotoxicity, superoxide production, phagocytosis, chemotaxis, and secondary cytokine production of additional factors in monocytes and macrophages. Examples of such additional factors include granulocyte colony stimulating factor (G-CSF), interleukin-6 (IL-6), and interleukin-8 (IL-8). M-CSF stimulates hematopoiesis, promotes differentiation and proliferation of osteoclast progenitor cells, and has profound effects on lipid metabolism. Furthermore, M-CSF is important in pregnancy. Physiologically, large amounts of M-CSF are produced in the placenta, and M-CSF is believed to play an essential role in trophoblast differentiation (Motoyoshi, Int J Hematol. 1998, 67: 109-22). The elevated serum M-CSF levels of early pregnancy may participate in the immunologic mechanisms responsible for the maintenance of the pregnancy (Flanagan & Lader, Curr Opin Hematol. 1998, 5:181-5).
[0402] Aberrant expression and/or activation of Fms has been implicated in acute myeloid leukemia, AML (Ridge et al, Proc. Nat. Acad. Sci., 1990, 87:1377-1380). Mutations at codon 301 are believed to lead to neoplastic transformation by ligand independence and constitutive tyrosine kinase activity of the receptor. The tyrosine residue at codon 969 has been shown to be involved in a negative regulatory activity, which is disrupted by amino acid substitutions. Accordingly, Fms mutations are most prevalent (20%) in chronic myelomonocytic leukemia and AML type M4 (23%), both of which are characterized by monocytic differentiation.
[0403] A condition related to AML is chronic myeloid leukemia (CML). During the myeloid blast crisis (BC) of CML, non-random additional chromosome abnormalities occur in over 80% of patients. However, these cytogenetic changes have been reported to precede the clinical signs of CML-BC by several months to years suggesting that other biological events may participate in the multistep process of acute transformation of CML. The autocrine production of growth factors has been shown to occur in several hematological malignancies and particularly in AML. Specchia et al [Br J Haematol. 1992 Mar; 80(3):310-6] have demonstrated that DL-I beta gene is expressed in almost all cases of CML in myeloid blast crisis, and that a high proportion of cases showed constitutive
expression of the M-CSF gene. Many of the same patients in the Specchia et al study demonstrated simultaneous co-expression of Fms. After exposure of leukemic cells to phorbol myristate acetate (PMA), release of M-CSF protein was documented in three of five patients studied; however, no significant interleukin-3 (IL-3), granulocyte-macrophage colony-stimulating factor (GM-CSF) or granulocyte colony-stimulating factor (G-CSF), was detected in these patients. This demonstrates that different patterns of growth factors secretion exist in AML and CML, and that distinct molecular events are likely involved in the control of leukemic proliferation.
[0404] The observation that production of M-CSF, the major macrophage growth factor, is increased in tissues during inflammation (Le Meur et al, J. Leukocyte Biology. 2002;72:530-537) provides a role for Fms in certain diseases. For example, COPD is characterized by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases. The chronic inflammation of COPD is observed through the airways, parenchyma, and pulmonary vasculature. The inflammatory cell population consists of neutrophils, macrophages, and T lymphocytes, along with eosinophils in some patients. Macrophages are postulated to play an orchestrating role in COPD inflammation by releasing mediators such as TNF-a, IL-8 and LTB4, which are capable of damaging lung structures and/or sustaining neutrophilic inflammation.
[0405] Further, M-CSF/fms signaling is critical to osteoclast formation and survival of osteoclast precursors. For example, estrogen loss in menopause results in increased M-CSF and thus increased osteoclast number and bone resorption which leads to increased risk of fracture and osteoporosis. Accordingly, blockage of this signal is a target for the inhibition of bone resorption (Teitelbaum, Science. 2000;289:1504; Rohan, Science. 2000;289:1508).
[0406] Atherosclerosis, an inflammatory disease of the vessel walls, is associated with significant morbidity and mortality. A effect for Fms inhibition in the treatment and prevention of atherosclerosis depends on several observations (Libby, Nature. 2002;420: 868-874). First, monocytes resident in the arterial intima increase expression of scavenger receptors and internalize modified lipoproteins. The resulting lipid-laden macrophages develop into foam cells characteristic of the atherosclerotic lesion. Macrophages in atheroma secrete cytokines and growth factors involved in lesion progression. Additionally, macrophages replicate within the intima. Through Fms, M-CSF activates the transition from monocyte to lipid-laden macrophage and augments expression of scavenger receptor A. Indeed, atherosclerotic plaques over-express M-CSF which is critical for atherosclerotic progression. Mice deficient in M-CSF have been found to experience less severe atherosclerosis than mice with normal M-CSF (Rajavashisth, et. al., J. Clin. Invest. 1998;101:2702- 2710; Qiao, et. al., Am. J. Path. 1997;150:1687-1699). Accordingly, inhibitors of Fms disrupt M-CSF
signaling, compromising monocyte to macrophage foam cell progression, macrophage survival and replication, and cytokine signaling that participates in lesion progression.
[0407] Wegener's granulomatosis, also known as vasculitis, is characterized by granulomatous inflammation of the blood vessels with necrosis. This inflammation limits blood flow to organs with consequent damage. Although the disease can involve any organ system, Wegener's granulomatosis mainly affects the respiratory tract (i.e., sinuses, nose, trachea, and lungs) and the kidneys. The endothelium plays a central role in the immunopathology of several vascular disorders in many inflammatory conditions such as Wegener's granulomatosis in which use of intravenous immunoglobulin (IV Ig) has been shown to be beneficial (see e.g., Basta et al, J Clin Invest 1994, 94:1729-1735). It has been reported (Xu et al, Am. J. Path., 1998;153:1257-1266) that IV Ig inhibits endothelial cell proliferation in a dose- and time-dependent manner and down-regulates the expression of adhesion molecule mJRNA (ICAM-I and VCAM-I), chemokine mRNA (MCP-I, M- CSF, and GM-CSF), and proinflammatory cytokine mRNA (TNF-α, IL-IB, and IL-6) induced by TNF-α- or IL-IB. These results may explain, at least in part, the therapeutic effect of IV Ig in vascular and inflammatory disorders. Additionally, these results suggest that inhibition of M-CSF activity at the level of interaction with Fms is an efficacious treatment strategy.
[0408] The role of M-CSF and Fms in emphysema appears to involve the regulation of elastin metabolism through control of matrix metalloproteins. M-CSF has a role in the modulation of the accumulation and function of alveolar macrophages (AMs) in vivo (Shibata et al, Blood 2001, 98: pp. 2845-2852). Osteopetrotic (Op/Op) mice have no detectable M-CSF and show variable tissue- specific reductions in macrophage numbers. Accordingly, it was hypothesized that AMs would be decreased in number and have altered function in Op/Op mice because of the absence of M-CSF. Shibata et al found that lung macrophages identified in lung sections were decreased in number in 20- day-old Op/Op mice but not Op/Op mice older than 4 months compared with findings in age-matched littermate controls. The numbers of AMs recovered by bronchoalveolar lavage (BAL) were also reduced in young but not adult Op/Op mice compared with controls. Importantly, AMs of Op/Op mice spontaneously release higher levels of matrix metalloproteinases (MMPs) than AMs of controls. Consistent with an increased release of MMP, Op/Op mice have abnormal elastin deposition and spontaneously develop emphysema in the absence of molecular or cellular evidence of lung inflammation. Accordingly, the modulation of metalloelastase activity in macrophages by M-CSF may control the degradation of elastin fibers in lungs or blood vessels.
[0409] Metastatic cancer cells cause bone destruction, with associated fracture, pain, deformation, and hypercalcaemia, due to production of osteoclasticogenic factors including M-CSF by tumor cells (Clohisy et al, Clin. Orthop. 2000, 373: 104-14). Binding of M-CSF to the Fms product stimulates formation of osteoclasts and osteolytic activity (Kodama et al, J. Exp. Med. 1991, 173: 269-72; Feng
et al, Endocrinology 2002, 143: 4868-74). Accordingly, inhibition of osteoclast activity at the level of Fms offers a compelling target for amelioration of bone metastasis.
[0410] Nephritis is inflammation of the kidneys. It may be caused for example by a bacterial infection of the kidneys or exposure to a toxin. However, nephritis more commonly develops from an abnormal immune reaction, which can occur, for example, when an antibody attacks either the kidney itself or an antigen attached to kidney cells, or when an antigen-antibody complex formed elsewhere in the body attachs to cells in the kidney. Some types of nephritis involve infiltration of kidney tissues by white blood cells and deposits of antibodies. In other types of nephritis, inflammation may consist of tissue swelling or scarring without white blood cells or antibodies. Furthermore, nephritis can occur anywhere in the kidneys. With respect to the glomeruli, progressive damage to glomeruli causes urine production to fall and metabolic waste products to build up in the blood. When damage to glomeruli is severe, inflammatory cells and injured glomerular cells accumulate, compressing the capillaries within the glomerulus and interfering with filtration. Scarring may develop, impairing kidney function and reducing urine production. In some cases, microthrombi may form in the small blood vessels, further decreasing kidney function. Less commonly, nephritis involves the tubulointerstitial tissues; such inflammation is called tubulointerstitial nephritis. When inflammation damages the tubules and the tubulointerstitial tissues, the kidneys may become unable to concentrate urine, eliminate (excrete) metabolic waste products from the body, or balance the excretion of sodium and other electrolytes, such as potassium. When the tubules and tubulointerstitial tissues are damaged, kidney failure often develops. Accordingly, inhibition of Fms offers a target for therapeutic intervention in nephritis due to the modulation of the inflammatory response comprising the etiology of the disease.
[0411] Lupus nephritis, i.e., renal involvement in systemic lupus erythematosus (SLE), is a common disease manifestation with a poor prognosis. At least three potentially overlapping, immuno- pathogenic mechanisms for lupus nephritis are supported by experimental data. First, circulating immune complexes consisting chiefly of DNA and anti-DNA are deposited in the kidney. Resulting complement activation and chemotaxis of neutrophils leads to a local inflammatory process. Second, in situ formation of antigen and antibody complexes may similarly lead to complement activation and leucocyte mediated injury. Third, antibodies against specific cellular targets may produce renal injury. An additional mechanism is observed in SLE patients with the antiphospholipid antibody syndrome. Glomerular thrombosis can result from the hypercoagulability that accompanies antibodies directed against negatively charged phospholipid-protein complexes (e.g. biologic false positive VDRL, anticardiolipin antibodies, and lupus anticoagulant). Mesangial lupus nephritis is accompanied by normal diagnostic findings or with a mild degree of proteinuria but typically absence of hypertension or abnormal urinary sediment. Focal and diffuse proliferative lupus
glomerulonephritis are often associated with the worst prognosis for renal survival and can be accompanied by nephrotic syndrome, significant hypertension and abnormal urine sediment. Membranous lupus nephritis often presents with proteinuria, moderate to high grade, but usually normal urinary sediment in the absence of hypertension. Mesangial lupus nephropathy is generally associated with an excellent prognosis, whereas proliferative lupus nephropathy, especially diffuse variant, is often characterized by hypertension, red cell casts and significant deterioration of renal function. Nephrotic syndrome in the absence of hypertension, active urinary sediment or significant hypocomplementemia suggest the membranous variant of lupus nephropathy. Membranous nephropathy generally is associated with a good prognosis and relative preservation of renal function. However, in the presence of persistent nephrotic range proteinuria, membranous lupus nephropathy can, in fact, lead to loss of renal function and end stage renal disease (ESRD). Accordingly, inhibition of Fms offers a target for therapeutic intervention in lupus due to the modulation of the inflammatory response comprising the etiology of the disease.
[0412] Macrophage accumulation is a prominent feature in many forms of glomerulonephritis. Local proliferation of macrophages within the kidney has been described in human and experimental glomerulonephritis and may have an important role in augmenting the inflammatory response. Isbel et al (Nephrol Dial Transplant 2001, 16: 1638-1647) examined the relationship between local macrophage proliferation and renal expression of M-CSF. Glomerular and tubulointerstitial M-CSF expression was found to be up-regulated in human glomerulonephritis, being most prominent in proliferative forms of disease. Because this correlates with local macrophage proliferation, it suggests that increased renal M-CSF production plays an important role in regulating local macrophage proliferation in human glomerulonephritis. In a model of renal inflammation (UUO- unilateral ureteric obstruction) anti-Fms antibody treatment reduced macrophage accumulation (Le Meur et.al., J Leukocyte Biology, 2002, 72: 530-537). Accordingly, inhibition of Fms offers a target for therapeutic intervention in glomerulonephritis.
[0413] Insulin resistance and obesity are hallmark of type II diabetes and there is a strong correlation exists between insulin resistance and abdominal visceral fact accumulation (Bjorntrop, Diabetes Metab. Res. Rev., 1999, 15: 427-441). Current evidence indicates that macrophages accumulating in adipose tissue release TNF-a and other factors that cause adipocyte changes (hypertrophy, lipolysis, reduced insulin sensitivity) and also promote insulin resistance in surrounding tissues. Therefore, macrophage accumulation in type 2 diabetes is important for disease progression. Accordingly, inhibition of Fms has potential in preventing the development of insulin resistance and hyperglycemia.
[0414] Similarly, the observation that production of M-CSF, the major macrophage growth factor, is increased in tissues during inflammation points out a role for Fms in diseases, such as for example
inflammatory diseases. More particularly, because elevated levels of M-CSF are found in the disease state, modulation of the activity of Fms can ameliorate disease associated with increased levels of M- CSF.
[0415] Fms inhibitors may be useful in treating to immune disorders, including rheumatoid arthritis, systemic lupus erythematosis (SLE), Wegener's granulomatosis, and transplant rejection, inflammatory diseases including Chronic Obstructive Pulmonary Disease (COPD), emphysema, and atherosclerosis, metabolic disorders, including insulin resistance, hyperglycemia, and lipolysis, disorders of bone structure or mineralization, including osteoporosis, increased risk of fracture, hypercalcemia, and bone metastases, kidney diseases, including nephritis (e.g. glomerulonephritis, interstitial nephritis, Lupus nephritis), tubular necrosis, diabetes-associated renal complications, and hypertrophy and cancers, including multiple myeloma, acute myeloid leukemia, chronic myeloid leukemia (CML), breast cancer, and ovarian cancer.
[0416] Frk: Target kinase Frk (Fyn-related kinase) is a 58.5 kDa tyrosine kinase encoded by chromosome 6q21-q22.3 (symbol: FRK). The structure comprises an SH2, an SH3 and a tyrosine kinase domain. Hosoya et al., report the the identification of a SRC-like tyrosine kinase gene, FRK (Fyn-related kinase), fused with ETV6 in a patient with acute myelogenous leukemia carrying t(6; 12)(q21 ;p 13). The ETV6/FRK protein was shown to be constitutively autophosphorylated on its tyrosine residues. ETV6/FRK phosphorylated histones H2B and H4 in vitro to a greater extent than did FRK, suggesting it had elevated kinase activity. ETV6/FRK could transform both Ba/F3 cells and NDH3T3 cells, which depended on its kinase activity (Hosoya et al., Genes Chromosomes Cancer 2005, 42(3):269-79). Welsh et al. concluded that FRK/RAK contributes to cytokine-induced beta-cell death, and inhibition of this kinase could provide means to suppress beta-cell destruction in Type I diabetes (Welsh et al., Biochem J 2004, 382(l):261-8). Frk inhibitors may be useful in treating acute myeloid leukemia and type I diabetes.
[0417] Fyn: Target kinase Fyn (i.e., Fyn oncogene related to Src, Fgr, Yes) is a 60.6 kDa non-receptor tyrosine kinase encoded by chromosome 6q21 (symbol: FYN). Fyn is involved in regulation of mast cell degranulation in a synergistic confluence of Fyn and Lyn (i.e., v-Yes-1 Yamaguchi sarcoma viral related oncogene homolog) pathways at the level of protein kinase C and calcium regulation. Fyn inhibitors may be useful in treating Alzheimer's disease, schizophrenia and in prevention of metastases, e.g. in melanoma and squamous cell carcinoma.
[0418] Gsk3«, Gsk33: Target kinase Gsk3J3 (i.e., Glycogen synthase kinase 3 beta) is a 46.8 kDa STK encoded by chromosome 3ql3.3 (symbol: GSK3B). Target kinase Gsk3α (i.e., Glycogen synthase kinase 3 alpha) is a 51.0 kDa STK encoded by chromosome 19ql3.2 (symbol: GSK3A).Gsk3 is a proline-directed serine-threonine kinase that was initially identified as a
phosphorylating and inactivating glycogen synthase. Two isoforms, alpha and beta, show a high degree of amino acid homology (Stambolic & Woodgett. Biochem. J. 1994, 303: 701-704,). GSK3B is involved in energy metabolism, neuronal cell development, and body pattern formation (Plyte et al., Biochim. Biophys. Acta 1992, 1114: 147-162,). The X-ray crystallographic structure of Gsk3 has been reported by Dajani et al. (CeK 2001, 105: 721-732). Klein & Melton (Proc. Nat. Acad. ScL 1996, 93: 8455-8459) proposed that Gsk3 is the endogenous target of lithium in diverse systems. For example, lithium potently and specifically inhibits Gsk3 activity in vitro. This suggests a mechanism whereby lithium can mimic insulin action, and lithium inhibition of the Gsk3 pathway in the brain could explain the actions of lithium action in manic-depressive illness in addition to its effects on development and its insulinlike activity.
[0419] Phiel et al., show that therapeutic concentrations of lithium, a GSK-3 inhibitor, block the production of Abeta peptides by interfering with APP cleavage at the gamma-secretase step, but do not inhibit Notch processing. Importantly, lithium also blocks the accumulation of Abeta peptides in the brains of mice that overproduce APP. The target of lithium in this setting is GSK-3alpha, which is required for maximal processing of APP. Since GSK-3 also phosphorylates tau protein, the principal component of neurofibrillary tangles, inhibition of GSK-3alpha offers a new approach to reduce the formation of both amyloid plaques and neurofibrillary tangles, two pathological hallmarks of Alzheimer's disease (Phiel et al., Nature 2003, 423:435-439). Eldar-Finkelman states that GSK-3 inhibitors might prove useful as therapeutic compounds in the treatment of conditions associated with elevated levels of enzyme activity, such as type 2 diabetes and Alzheimer's disease. The pro-apoptotic feature of GSK-3 activity suggests a potential role for its inhibitors in protection against neuronal cell death, and in the treatment of traumatic head injury and stroke. Finally, selective inhibitors of GSK-3 could mimic the action of mood stabilizers such as lithium and valproic acid and be used in the treatment of bipolar mood disorders (Eldar-Finkelman, Trends MoI Med 2002, 8:126-132). Martinez et al. state that glycogen synthase kinase 3 (GSK-3) was initially described as a key enzyme involved in glycogen metabolism, but is now known to regulate a diverse array of cell functions. Two forms of the enzyme, GSK-3alpha and GSK-3beta, have been previously identified. Small molecules inhibitors of GSK-3 may, therefore, have several therapeutic uses, including the treatment of neurodegenerative diseases, diabetes type II, bipolar disorders, stroke, cancer, and chronic inflammatory disease (Martinez et al., Med Res Rev 2002, 22(4):373-84). GSK inhibitors may be useful in treating CNS disorders such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, diabetes type II, bipolar disorders, stroke, cancer, chronic inflammatory disease, leucopenia, schizophrenia, chronic pain, neuropathic pain, and traumatic head injury.
[0420] HCK: Target kinase HCK (hemopoietic cell kinase) is a 59.5 kDa tyrosine kinase encoded by chromosome 20ql 1.21 (symbol: HCK). The protein structure comprises an SH3, and SH2 and a
bipartite kinase domain. HCK inhibitors may be useful in treating chronic myelogenous leukemia and acute lymphocytic leukemia.
[0421] Her2/Erbb2; Target kinase Her2/Erbb2 (i.e., Human EGF receptor 2) is a 137.9 kDa transmembrane tyrosine kinase encoded by chromosome 17ql 1.2-ql2/l 7q21.2 (symbol: ERBB2). According to OMM, the ERBB2 locus 17q21 is the chromosome 17 breakpoint in acute promyelocytic leukemia (APL). Amplification of ERBB2 is observed in human salivary gland adenocarcinoma (Semba et al., Proc. Nat. Acad. Sci. 1985, 82:6497-6501) and in a gastric cancer cell line (Fukushige et al., Molec. Cell. Biol. 1986, 6:955-958). Overexpression of ERBB2 has been implicated in the neoplastic transformation of prostate cancer. Interleukin-6 (IL6) is a cytokine that was initially recognized as a regulator of immune and inflammatory responses, but also regulates the growth of many tumor cells, including prostate cancer. Qui et al. showed that treatment of a prostate cancer cell line with IL6 induces tyrosine phosphorylation of ERBB2 and ERBB3, but not ERBB1/EGFR. ERBB2 also forms a complex with the gpl30 subunit of the TL6 receptor (IL6R) in an IL6-dependent manner. This association is important because the inhibition of ERBB2 activity results in abrogation of IL6-induced MAPK activation (Qui et al., Nature 1998, 393:83-85). Thus, ERBB2 is a critical component of IL6 signaling through the MAP kinase pathway. Additionally, overexpression of ERBB2 confers Taxol resistance in breast cancers by inhibiting p34 (CDC2) activation (Yu et al., Molec. Cell 1998, 2:581-91) (OMIM MIM Number: 164870: 01/30/2006). Her2/Erbb2 inhibitors may be useful in treating prostate and breast cancer.
[0422] Her4/Erbb4: Target kinase Her4/Erbb4 (i.e., Human EGF receptor 4) is a 146.8 kDa transmembrane tyrosine kinase encoded by chromosome 2q33.3-q34 (symbol: ERBB4). According to OMIM, the HER4/ERBB4 gene is a member of the type I receptor tyrosine kinase subfamily that includes EGFR, ERBB2, and ERBB3. The gene product of ERBB4 is a receptor for NDF/heregulin, which are essential for neuronal development. Her4/Erbb4 -/- mouse embryos exhibit axonal misprojections which correlate with aberrant migration of a subpopulation of hindbrain-derived cranial neural crest cells. Accordingly, Her4/Erbb4 signaling provides patterning information essential for the proper migration of neural crest cells (OMM MM Number: 600543: 07/27/2005). Her4/Erbb4 inhibitors may be useful in treating childhood medulloblastoma.
[0423] IGFlR: Target kinase IGFlR (insulin-like growth factor 1 receptor) is a 154.8 kDa receptor tyrosine kinase encoded by chromosome 15q26.1 (symbol: IGFlR). Overexpressed in breast and prostate cancer, acting to enhance tumor cell survival. IGFlR inhibitors may be useful in treating prostate cancer and hepatocellular carcinoma.
[0424] IKK beta: Target kinase IKK beta (i.e., inhibitor of nuclear factor kappa B kinase beta) is a 86.6 kDa STK encoded by chromosome 8pl 1.2 (symbol: IKBKB). According to OMM, IKK beta
phosphorylates serine residues of I-kappa-B proteins which marks them for destruction via the ubiquitination pathway, thereby allowing activation of the NF-kappa-B complex. Activated NF-κB complex translocates into the nucleus and binds DNA at kappa-B-binding motifs. Yin et al (Nature 396: 77-80, 1998) have shown that the antiinflammatory properties of aspirin and salicylate are mediated in part by their specific inhibition of IKK-beta, thereby preventing activation by NF-kappa- B of genes involved in the pathogenesis of the inflammatory response. Rossi et al (403: 103-108, 2000) demonstrated a novel mechanism of antiinflammatory activity that was based on the direct inhibition and modification of the IKK-beta subunit of IKK. Since IKK-beta is responsible for the activation of NF-kappa-B by proinflammatory stimuli, Rossi et al. (ibid.) suggested that their findings explained how cyclopentenone prostaglandins function and can be used to improve the utility of COX2 inhibitors (OMEVI MIM Number: 603258: 11/16/2005).
[0425] IKKbeta inhibitors may be useful in treating leukemia of T-cells, necrosis, neoplasms, insulin resistance, and malignant neoplasms.
[0426] Irak4; Target kinase Irak4 (i.e., Interleukin 1 receptor associated kinase 4) is a 51.5 kDa serine/threonine kinase encoded by chromosome 12ql2 (symbol: IRAK4). Interleukin-1 receptor associated kinases (e.g., IRAKI) are important mediators in the signal transduction of Toll-like receptor (TLR, e.g., TLR4) and ILlR family members are collectively referred to as TIRs. Irak4 functions in this signal transduction pathway. The structure of Irak4 comprises a DEATH domain adjacent a STK domain. The DEATH domain is a protein-protein interaction motif found in certain proteins of the apoptotic pathway.
[0427] Irak4 was originally identified as NY-REN-64, one of 65 human tumor antigens recognized by autologous antibodies from patients with renal cell carcinoma using serological analysis of recombinant cDNA expression libraries (SEREX). Sequence analysis of the NY-REN-64 cDNA clone identified in the SEREX screen revealed a novel gene encoding a transcript of 2.8 kilobases and a predicted protein of 460 amino acids (Genbank Accession AF 155118) noted to bear a protein kinase motif (Seaman et al., Int. J. Cancer, 1999, 83, 456-464). Based on its homology to the other IL-I receptor-associated kinases, this gene has more recently been placed in the IRAK family and given the name IL-I receptor-associated kinase-4.
[0428] Irak4 is required for the efficient recruitment of IRAKI to the IL-I receptor complex following IL-I engagement, triggering intracellular signaling cascades leading to transcriptional up- regulation and mRNA stabilization. Irak4 Phosphorylates Iraki . Effective Irak4 functioning is crucial for protective immunity against specific bacteria, including pyogenic bacterial, but is redundant against other microorganism.
[0429] Lrak4 inhibitors may be useful in treating immunodeficiency syndrome, Crohn's disease, ulcerative colitis, asthma, chronic bronchitis, cardio hypertrophy, and kidney hypertension.
[0430] Mc: Target kinase Itk (i.e., IL-2 inducible T-cell kinase) is a tyrosine kinase of 71.8 kDa encoded by chromosome 5q31-q32 (symbol: ITK). Itk is a T-cell specific homology of kinase Btk. The EMT Tec family kinases are non-receptor type protein-tyrosine kinase that are highly expressed in many hematopoietic cell lines. The TEC-family protein tyrosine kinases ITK, RLK(TXK) and TEC have been identified as key components of T-cell-receptor signalling that contribute to the regulation of phospholipase C-gamma, the mobilization of Ca2+ and the activation of mitogen- activated protein kinases. Recent data also show that TEC kinases contribute to T-cell-receptor-driven actin reorganization and cell polarization, which are required for productive T-cell activation. Functional studies have implicated TEC kinases as important mediators of pathways that control the differentiation of CD4+ T helper cells (Schwartzberg et al., 2005, Nature immunology, 5:284).
[0431] T cells express three TEC kinases, ITK, RLK and TEC, all of which are activated downstream of the T-cell receptor (TCR) ( Berg, L. J et al., Annu. Rev. Immunol., 2005, 23:549) and have been shown to be involved in signaling through the TCR (Schaeffer, E. M. et al., 1999, Science, 284,:638). Although ITK, RLK and TEC are all found in T cells, they are expressed at different levels and by different subpopulations. (Lucas, J. A et al., 2003, Immunol. Rev., 191 ;119. Colgan, J. et al. 2004, Immunity, 21:189). High expression of TEC was seen in each of 3 patients examined with myelodysplastic syndrome(Sato K et al., 1994, Leukemia, 8:1663). Although no human disease has been associated with mutations of the TEC kinases that are expressed by T cells, ITK-deficient mice have specific defects in T Helper 2 (TH2)-cell responses and reduced pathology in models of allergic asthma (Fowell et al. 1999, Immunity, 11 :399). Specific ITK inhibitors reduce disease in a mouse model of allergic asthmal9, TEC kinases are activated through phosphorylation by SRC-family kinases, such as LCK, and recruitment to the plasma membrane through binding of PtdIns(3,4,5)P3, where they are brought into TCRsignalling complexes through interactions with SLP76, LAT and other molecules (Bunnell, S. C. et al. 2000, J. Biol. Chem., 275:2219.).
[0432] Consistent with a role for ITK in allergic responses, increased ITK expression has been seen in peripheralblood T cells from humans with atopic dermatitis(Matsumoto, Y. et al., 2002, Int. Arch. Allergy Immunol. 129:327). Importantly, Itk-/- mice cannot mount effective TH2-cell responses to infection with many pathogens that are used to evaluate TH2-cell differentiation, including Nippostrongylus brasiliensis, Schistosoma niansoni and Leishmania major (Fowell, D. J. et al. 1999, Immunity, 11 :399. Schaeffer, E. et al. 2001, Nature Immunol., 2:1183).
[0433] TH2-cell responses have been implicated in the pathology of allergic asthma, which is characterized by an increased number of TH2 cells in the lungs, increased TH2-cytokine production,
increased mucus production in the lungs and inflammation of the airways (Cohn, L et al. 2004, Annu. Rev.Immunol., 22:789). For several reasons, ITK, however, might be an ideal therapeutic target for TH2-cell-mediated diseases, provided that the inhibitor has a high degree of specificity. Itk inhibitors may be useful in treating allergic asthma.
[0434] Jakl; Target kinase Jakl (i.e., Janus kinase 1) is a 132 kDa tyrosine kinase encoded by chromosome Ip31.3 (symbol: JAKl). Jakl inhibitors may be useful in treating Hepatitis C virus infection.
[0435] Jak2: Target kinase Jak2 (i.e., Janus kinase 2) is a 130.7 kDa tyrosine kinase encoded by chromosome 9p24 (symbol: JAK2). Jak2 inhibitors may be useful in treating myeloproliferative disorders such as polycvthaemia vera, myelofibrosis, essential thrombocythemia, myeloid metaplasia and leukemias, including acute lymphoblastic leukemia, chronic neutrophilic leukemia, juvenile myelomonocytic leukemia, CMML, Philadelphia chromosome-negative CML, megakaryocyte leukemia, and acute erythroid leukemia
[0436] Jak3: Target kinase Jak3 (i.e., Janus kinase 3) is a 125.1 kDa tyrosine kinase encoded by chromosome 19pl3.1 (symbol: JAK3). According to OMHVI, JAK3 is a member of the Janus kinase (JAK) family of tyrosine kinases involved in cytokine receptor-mediated intracellular signal transduction. Interleukin-2 (IL2) signaling requires the dimerization of JJL2 receptor-beta (JX2RB) with the common gamma chain (gamma-c; IL2RG). Mutations in the JX2RG gene cause X-linked severe combined immunodeficiency. Interleukins JX2, JX4, IL7, JX9, and ILl 5, whose receptors are known to contain the common gamma chain, induce the tyrosine phosphorylation and activation of Jak3. Truncations of gamma-c and a point mutation of gamma-c, causing moderate X-linked combined immunodeficiency, decrease the association between the common gamma chain and Jak3. Since mutations in the IL2RG gene in at least some XSCJX) and XCID patients prevent normal Jak3 activation, mutations in Jak3 may result in an XSCJD-like phenotype (OMM MIM Number: 600173: 04/04/2006). A related kinase, Jak2, is activated through mutation in patients with a variety of myeloproliferative disorders (Kralovics R. et al. N Engl J Med. 2005 352:1779-90). The role of Jak3 in B and T lymphocyte maturation and T cell function makes Jak3 a target for treating transplant rejection and autoimmune diseases. Jak3 inhibitors may be useful in treating X-linked severe combined immunodeficiency, myeloproliferative disorders, transplant rejection and autoimmune diseases such as rheumatoid arthritis, inflammatory bowel syndrome, Crohn's disease, systemic lupus erythematosis, ulcerative colitis, psoriasis and multiple sclerosis.
[0437] Jnkl: Target Jnkl (i.e., c-Jun kinase 1) is a 48.3 kDa serine/threonine kinase encoded by chromosome 1 OqI 1.22 (symbol: MAPK8), also known as mitogen-activated protein kinase 8. Jnkl is a mitogen-activated protein kinase (i.e., MAPK) which form a family of serine-threonine protein
kinases that participate in a major signaling system by which cells transduce extracellular stimuli into intracellular responses. MAPKs comprise the extracellular regulated kinases, or ERKs, for example Erk2, and the stress-activated protein kinases (SAPKs). MAPKs respond to activation by environmental stress and pro-inflammatory cytokines by phosphorylating a number of transcription factors, primarily components of AP-I such as c-Jun and ATF2 and thus regulates AP-I transcriptional activity. In T- cells, Jnkl and Jnk2 are required for polarized differentiation of T- helper cells into ThI cells. Jnkl is activated by threonine and tyrosine phosphorylation by either of two dual specificity kinases, MAP2K4 and MAP2K7 and inhibited by dual specificity phosphatases, such as DUSPl. Jnkl inhibitors may be useful in treating type 1 diabetes, type 2 diabetes, metabolic syndrome, obesity and hepatic steatosis.
[0438] Jnk2: Target kinase Jnk2 (i.e., c-Jun kinase 2) is 48.1 kDa serine/threonine kinase encoded by chromosome 5q35 (symbol: MAPK9). According to OMIM, the transcriptional activity of the c- Jun protooncoprotein is augmented through phosphorylation at two sites by c-Jun kinases (JNKs). Using in-gel kinase assays, Hibi et al. (1993) identified 2 JNKs, 46 and 55 kD in size. The 46-kD protein Jnkl was shown to be a member of the mitogen-activated protein kinase (MAPK) family. Using a JNKl cDNA as a probe, Kallunki et al. (1994) and Sluss et al. (1994) isolated cDNAs encoding the 55-kD protein, which both designated Jnk2. Kallunki et al. (1994) reported that the sequence of the predicted 424-amino acid JNK2 protein is 83% identical to that of JNKl . Both JNKs contain a thr-pro-tyr phosphorylation motif. Northern blot analysis revealed that JNK2 is expressed as multiple transcripts in many cell types (OMJJvI MIM Number: 602896: 07/07/2005).
[0439] Jnk2 responds to activation by environmental stress and pro- inflammatory cytokines by phosphorylating a number of transcription factors, primarily components of AP-I such as c-Jun and ATF2 and thus regulates AP-I transcriptional activity. In T- cells, JNKl and JNK2 are required for polarized differentiation of T-helper cells into ThI cells. Jnk2 isoforms display different binding patterns: alpha- 1 and alpha-2 preferentially bind to c-Jun, whereas beta-1 and beta-2 bind to ATF2. However, there is no correlation between binding and phosphorylation, which is achieved at about the same efficiency by all isoforms. Jnk2 is activated by threonine and tyrosine phosphorylation by either of two dual specificity kinases, MAP2K4 and MAP2K7. Further, Jnk2 is inhibited by dual specificity phosphatases, such as DUSPl . Jnk2 inhibitors may be useful in treating atherosclerosis.
[0440] Jnk3: Target kinase Jnk3 (i.e., c-Jun kinase 3) is 52.6 kDa serine/threonine kinase encoded by chromosome 4q21-q22 (symbol: MAPKlO). According to OMJJVI, the c-Jun kinases (JNKs) are members of the mitogen-activated protein kinase (MAPK) family that activate the Jun transcription factor. Gupta et al. (EMBO J. 1996, 15: 2760-2770) isolated brain cDNAs encoding 10 different JNK isoforms, 8 of which were derived from either JNKl or JNK2. The other 2 cDNAs were from a gene that the authors designated JNK3. JNK3 contains an extended N-terminal region not found in JNKl or
JNK2. The 2 JNK3 isoforms, called JNK3-alpha-l and JNK3-alpha-2, have different C termini. By SDS-PAGE of in vitro transcription/translation products, Gupta et al. (ibid.) determined that JNK3- alpha-1 migrates as a 45-to-48-kD doublet and XNK3-alpha-2 migrates as a 54-to-57-kD doublet. They stated that the lower band probably represents translation from a second in-frame start codon that corresponds to the first codon in JNKl and JNK2. All the JNECs were activated by treatment of cells with the inflammatory cytokine ILl . Multiple JNK isoforms were shown to be inactivated by MKPl . Comparison of the binding activity of the JNK isoforms demonstrated that they differ in their interactions with the ATF2 (CREB2), ELKl, and Jun transcription factors. Gupta et al. (ibid.) suggested that individual JNKs selectively target specific transcription factors in vivo, providing a mechanism for the generation of tissue-specific responses to the activation of the JNK signal transduction pathway. Mohit et al. (Neuron 1995, 14: 67-78) identified JNK3, or p49-3F12 kinase, as the gene encoding a 49-kD antigen found in the hippocampus and neocortex. The distribution of JNK3 -expressing neurons closely matches that of Alzheimer disease targeted neurons in those areas of the brain. Northern blot analysis revealed that JNK3 is expressed as a 2.7-kb mRNA exclusively in the nervous system. Mice defective for Jnk3 are resistant to excitotoxicity induced apoptosis (Yang D.D. et al., Nature 1997, 389:865) (OMIM MIM Number: 602897: 03/13/2006).
[0441] Jnk3 responds to activation by environmental stress and pro- inflammatory cytokines by phosphorylating a number of transcription factors, primarily components of AP-I such as c-Jun and ATF2 and thus regulates AP-I transcriptional activity. Jnk3 is required for stress-induced neuronal apoptosis and the pathogenesis of glutamate excitotoxicity. Jnk3 is activated by threonine and tyrosine phosphorylation by two dual specificity kinases, MAP2K4 and MAP2K7. MAP2K7 phosphorylates MAPKlO on Thr-221 causing a conformational change and a large increase in Vmax. MAP2K4 then phosphorylates Tyr-223 resulting in a further increase in Vmax. Jnk3 is inhibited by dual specificity phosphatases, such as DUSPl .
[0442] Jnk3 inhibitors may be useful in treating inflammatory diseases including autoimmune diseases such as rheumatoid arthritis, inflammatory bowel syndrome, Crohn's disease, systemic lupus erythematosis, Sjogren's Syndrome, psoriasis and multiple sclerosis, airway inflammatory diseases such as asthma, allergy, pulmonary fibrosis, and chronic obstructive pulmonary disease, and inflammation in other organs, such as CNS inflammation, pancreatitis, nephritis, and hepatitis; neurologic diseases such as stroke, cerebrovascular ischemia, and neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, and Huntington's disease; and neoplastic diseases such as prostate tumors and myeloid leukemia.
[0443] Kdr: Target kinase Kdr (i.e., Kinase Insert Domain Receptor) is a transmembrane tyrosine kinase of 151.5 kDa encoded by chromosome 4ql2 (symbol: KDR). Kdr has a complex secondary structure comprising three Ig-like (i.e., immunoglobulin-like) domains, one IGC2 (i.e.,
iπrtnunoglobulin-like C2-type) domain, two additional Ig-like domains, one additional IGC2 domain, one TM (i.e., transmembrane) domain, and a split TK domain. Kdr, also known as VEGFR2 (i.e., Vascular Endothelial Growth Factor Receptor 2), Flt2, and Flkl (i.e., fetal liver kinase 1), is the receptor for Vegf and VegfC (i.e., Vascular endothelial growth factor C) and plays a key role in vascular development and regulation of vascular permeability. Walter et al. (Genes Chromosomes Cancer, 2002, 33:295-303) has proposed based on an observed mutation in the kinase domain of KDR that a potential mechanism involved in hemangioma formation is the alteration of the VEGF signaling pathway in endothelial and/or pericytic cells.
[0444] Due to the role of angiogenesis, and aberrant control thereof in pathologic states, Kdr is a target for therapeutic intervention. Angiogenesis is the process by which new blood vessel growth occurs from pre-existing vasculature and is mediated through multiple pro-angiogenic factors, including for example Kdr. Under normal adult physiological conditions, angiogenesis occurs during wound healing, organ regeneration, and in some aspects of female reproductive function. Angiogenesis is also important for the progression of many pathological disorders such as solid tumor growth (ovarian, lung, breast, prancreatic, prostate, colon, gastrointestinal stromal tumor, non small cell lung cancer, and epidermoid cancer), metastasis, psoriasis, rheumatoid arthritis, diabetic retinopathy and age related macular degeneration. (Hoeben et al., 2004, Pharmacol. Rev. 56:549-580).
[0445] When the dimeric cytokine VEGF binds to the receptor tyrosine kinases FIt-I and/or KDR, receptor dimerization occurs, followed by autophosphorylation which leads to kinase activation and phosphorylation of intracellular substrates. This receptor tyrosine kinase activity initiates a cellular signaling pathway which leads to endothelial cell proliferation and migration that is necessary for the process of angiogenesis.
[0446] Tumors that grow beyond 1 -2 mm in size require the process of angiogenesis in order to receive the appropriate nutrients and oxygen that is required for tumor progression. Accordingly, inhibition of this process by small molecules that bind to the surface of receptor tyrosine kinases such as, for example, Kdr, inhibits tumor growth in both animal and human models. Direct evidence of the role of VEGF as a tumor angiogenesis factor in vivo is shown in studies in which VEGF expression or VEGF activity was inhibited. This has been achieved with anti-VEGF antibodies, with dominant- negative VEGFR-2 mutants which inhibited signal transduction, and with anti-sense VEGF RNA techniques. All approaches led to the reduction in tumor cell lines in vivo as a result of inhibited tumor angiogenesis. (Scappaticci., 2002, J. Clin. Oncology 20(18) :3906-3927 and references within).
[0447] Kdr inhibitors may be useful in treating solid tumor growth (e.g. ovarian, lung, breast, prancreatic, prostate, colon, gastrointestinal stromal tumor, non small cell lung cancer, and
epidermoid cancer), metastasis, psoriasis, rheumatoid arthritis, diabetic retinopathy and age related macular degeneration.
[0448] Kit: Target kinase Kit (i.e., feline Hardy-Zuckerman 4 sarcoma viral oncogene) is a 109.9 kDa transmembrane tyrosine kinase encoded by chromosome 4ql2 (symbol: KIT). Receptor protein tyrosine kinases (RPTKs) regulate key signal transduction cascades that control cellular growth and proliferation. The Stem Cell Factor (SCF) receptor Kit is a type III transmembrane RPTK that includes five extracellular immunoglobulin (IG) domains, a single transmembrane domain, and a split cytoplasmic kinase domain separated by a kinase insert segment. Kit plays an important role in the development of melanocytes, mast, germ, and hematopoietic cells.
[0449] Stem Cell Factor (SCF) is a protein encoded by the S 1 locus, and has also been called kit ligand (KL) and mast cell growth factor (MGF), based on the biological properties used to identify it (reviewed in Tsujimura, Pathol Int 1996, 46:933-938; Loveland, et al., J. Endocrinol 1997, 153:337- 344; Vliagoftis, et al., Clin Immunol 1997, 100:435-440; Broudy, Blood 1997, 90:1345-1364; Pignon, Hermatol Cell Ther 1997, 39:114-116; and Lyman, et al., Blood 1998, 91 :1101-1134.). Herein the abbreviation SCF refers to the ligand for Kit.
[0450] SCF is synthesized as a transmembrane protein with a molecular weight of 220 or 248 Dalton, depending on alternative splicing of the mRNA to encode exon 6. The larger protein can be proteolytically cleaved to form a soluble, glycosylated protein which noncovalently dimerizes. Both the soluble and membrane-bound forms of SCF can bind to and activate Kit. For example, in the skin, SCF is predominantly expressed by fibroblasts, keratinocytes, and endothelial cells, which modulate the activity of melanocytes and mast cells expressing Kit. In bone, marrow stromal cells express SCF and regulate hematopoiesis of Kit expressing stem cells. In the gastrointestinal tract, intestinal epithelial cells express SCF and affect the interstitial cells of Cajal and intraepithelial lymphocytes. In the testis, Sertoli cells and granulosa cells express SCF which regulates spermatogenesis by interaction with Kit on germ cells.
[0451] According to OMIM, Signaling from Kit is essential for primordial germ cell growth both in vivo and in vitro. Many downstream effectors of the KIT signaling pathway have been identified in other cell types, but how these molecules control primordial germ cell survival and proliferation are unknown. Determination of the KIT effectors acting in primordial germ cells has been hampered by the lack of effective methods to manipulate easily gene expression in these cells. De Miguel et al. (2002) overcame this problem by testing the efficacy of retroviral-mediated gene transfer for manipulating gene expression in mammalian germ cells. They found that primordial germ cells can successfully be infected with a variety of types of retroviruses. They used this method to demonstrate
an important role of the AKTl in regulating primordial germ cell growth (OMIM MIM Number: 164920: 04/17/2006).
[0452] Aberrant expression and/or activation of Kit has been implicated in a variety of pathologic states. For example, evidence for a contribution of Kit to neoplastic pathology includes its association with leukemias and mast cell tumors, small cell lung cancer, testicular cancer, and some cancers of the gastrointestinal tract and central nervous system. In addition, Kit has been implicated in playing a role in carcinogenesis of the female genital tract sarcomas of neuroectodermal origin, and Schwann cell neoplasia associated with neurofibromatosis. It was found that mast cells are involved in modifying the tumor microenvironment and enhancing tumor growth (Yang et al., J Clin Invest. 2003, 112:1851-1861; Viskochil, J Clin Invest. 2003, 112:1791-1793).
[0453] Kit inhibitors may be useful in treating malignancies, including mast cell tumors, small cell lung cancer, testicular cancer, gastrointestinal stromal tumors (GISTs), glioblastoma, astrocytoma, neuroblastoma, carcinomas of the female genital tract, sarcomas of neuroectodermal origin, colorectal carcinoma, carcinoma in situ, Schwann cell neoplasia associated with neurofibromatosis, acute myelocytic leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, mastocytosis, melanoma, and canine mast cell tumors, and inflammatory diseases, including asthma, rheumatoid arthritis, allergic rhinitis, multiple sclerosis, inflammatory bowel syndrome, transplant rejection, and hypereosinophilia.
[0454] LCK: Target kinase MCK (i.e., lymphocyte-specific protein tyrosine kinase) is a 57.9 kDa membrane associated non receptor tyrosine kinase encoded by chromosome Ip34.3 (symbol: LCK). The protein structure comprises an SH3 and SH2 domain. LCK inhibitors may be useful in treating acute lymphoblastic leukemia, T-cell lymphoma, lymphopenia, renal carcinoma, colon carcinoma, severe combined immunodeficiency, multiple sclerosis, inflammatory bowel and type I diabetes.
[0455] MAP2K1: Target kinase MAP2K1 (i.e., Mitogen-activated protein kinase kinase 1) is a threonine/tyrosine kinase of 43.3 kDa encoded by chromosome 15q22.1-q22.33 (symbol: MAP2K1). According to OMM, MAP2K1 is also known as MEKl (i.e., MAPK/ERK Kinase 1). Mitogen- activated protein (MAP) kinases, also known as extracellular signal-regulated kinases (ERKs) are thought to act as an integration point for multiple biochemical signals because they are activated by a wide variety of extracellular signals, are rapidly phosphorylated on threonine and tyrosine residues, and are highly conserved in evolution (Crews et al., Science 1992 258: 478-480). MAP2K1 is a critical protein kinase lying upstream of MAP kinase which stimulates the enzymatic activity of MAP kinase Crews et al. (ibid.) found that Mekl (i.e., MAP2K1) expressed in bacteria phosphorylates the Erk gene product in vitro. They showed that the Mekl gene is highly expressed in murine brain. Seger et al. (J. Biol. Chem., 1992, 267: 25628-25631) cloned a cDNA encoding the human homolog
of Mekl, symbolized MKKl by them, from a human T-cell cDNA library. When overexpressed in COS cells, the predicted 43,439-Da protein led to increased phorbol ester-stimulated MAP kinase kinase activity. They also isolated a related cDNA, called MKKIb, that appears to be an alternatively spliced form of MKKl . Seger et al. (ibid.) detected a 2.6-kb MKKl transcript by Northern blot analysis in all tissues examined. Zheng and Guan (J. Biol. Chem., 1993, 268: 11435-11439) also cloned a human cDNA corresponding to MEKl . They noted that the 393-amino acid protein shares 99% amino acid identity with murine Mekl and 80% homology with human MEK2. The authors characterized biochemically the human MEKl and MEK2 gene products. The gene is also symbolized MAP2K1 , or PRKMKl . MAP2K1 catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases and activates ERKl and ERK2 MAP kinases. Certain inhibitors of MEKl are potent anti-cancer agents (Sebolt- Leopold, J.S., et al., Nat. Med. 1999, 5:810) (OMM MM Number: 176872: 06/06/2005). MAP2K1 inhibitors may be useful in treating acute myeloid leukemia, breast, ovarian and liver cancer.
[0456] MAP2K2: Target kinase MAP2K2 (i.e., Mitogen-activated protein kinase kinase 2) is a threonine/tyrosine kinase of 44.4 kDa encoded by chromosome 7q32 (symbol: MAP2K2); MAP2K2 is also known as Mek2; see MAP2K1 above. According to OMM, Zheng and Guan (ibid.) isolated and sequenced 2 human cDNAs encoding members of the MAP kinase kinase (MAP2K) family, designated MEKl and MEK2 by them. The MEK2 cDNA encodes a predicted 400-amino acid protein that shares 80% sequence identity with human MEKl. Zheng and Guan (ibid.) showed that recombinant MEK2 and MEKl both could activate human Erkl in vitro. They further characterized biochemically both MAP2Ks (OMM MM Number: 601263: 10/23/2003).
[0457] The mitogen-activated protein kinase (MAPK) pathway is a major pathway in the cellular signal transduction cascade from growth factors to the cell nucleus. The pathway involves kinases at two levels: MAP kinase kinases (MAPKK), and their substrates MAP (mitogen activated protein) kinases (MAPK). There are different isoforms in the MAP kinase family. [For review, see Seger, R.; Krebs, E. G. FASEB, 9, 726, (1995)]. The compounds of this invention can inhibit the action of one or both of these kinases: MEK, a MAP kinase kinase, and its substrate ERK, a MAP kinase. ERK (extracellular regulated kinases), a p42 MAPK, is found to be essential for cell proliferation and differentiation. Overexpression and/or over activation of MEK or ERK has been found to be associated with various human cancers [For example, Sivaraman, V. S. et al., C. C. J. Clin. Invest., 99, 1478 (1997)]. It has been demonstrated that inhibition of MEK prevents activation of ERK and subsequent activation of ERK substrates in cells, resulting in inhibition of cell growth stimulation and reversal of the phenotype of ras-transformed cells [Dudley, D. T. et al., Proc. Nat. Acad. Sci., 92, 7686 (1995)]. MAP2K2 inhibitors may be useful in treating cancer and inflammation.
[0458] MAP4K4: Target kinase MAP4K4 (i.e., Mitogen-activated protein kinase kinase 4) is a serine threonine kinase of 152.1 kDa encoded by chromosome 2ql 1.2 (symbol: MAP4K4) and is also known as HGK. MAP4K4 inhibitors may be useful in treating cancer, tumor metastasis, diabetes and metabolic syndrome.
[0459] MAPKAPK2: Target kinase MAPKAPK2 (i.e., Mitogen activated protein kinase activated protein kinase 2) is 45.6 kDa serine/threonine kinase encoded by chromosome Iq32 (symbol: MAPKAPK2). According to OMIM, Stokoe et al. (Biochem. J. 1993, 296:843-849) described a protein kinase, which they designated MAPK-AP kinase-2, that was active only after phosphorylation by mitogen-activated protein kinase (MAP kinase). They identified several features that distinguish MAPKAP kinase-2 from the MAPKAP kinase-1 family. Stokoe et al. (ibid.) stated that MAPKAP kinase-2 was identified based on its in vitro phosphorylation of glycogen synthase; however, its phosphorylation of glycogen synthase had not been shown in vivo. Stokoe et al. (ibid.) cloned a partial human MAPKAP kinase-2 cDNA from a teratocarcinoma cell line cDNA library. The cDNA sequence revealed the following features (in 5-prime to 3-prime order): a proline-rich region containing 2 putative SH3-binding sites, a kinase catalytic domain, a threonine residue phosphorylated by MAP kinase, and a nuclear localization signal. By Northern analysis, Stokoe et al. (ibid.) demonstrated that the gene is expressed as a 3.3-kb transcript in all of the 6 human tissues tested. The physiological substrate of MAPKAP kinase 2 appears to be the small heat shock protein (HSP27/HSP25) (OMIM MIM Number: 602006: 03/03/2005).
[0460] In vitro, MAPKAP kinase 2 can phosphorylate glycogen synthase at Ser-7 and tyrosine hydroxylase (on Ser-19 and Ser-40). This kinase phosphorylates Ser in the peptide sequence, Hyd-X- R- X(2)-S, where Hyd is a large hydrophobic residue. MAPKAP kinase 2 is activated by two distinct pathways: the first involves the stimulation of p42/p44 MAPK by growth factors, and the second, triggered by stress and heat shock, depends on the activation of MPK2 and upstream MAPKKMAPKKK. MAPKAPK2 inhibitors may be useful in treating cancer (e.g. prostate, breast), stroke, menengitis, and inflammatory disorders.
[0461] Met: Target kinase Met (i.e., Hepatocyte growth factor receptor) is 155.5 kDa transmembrane tyrosine kinase encoded by chromosome 7q31 (symbol: MET). According to OMIM, Cooper et al. (Nature 1984, 311 : 29-33) cloned a transforming gene from a chemically transformed human osteosarcoma-derived cell line and mapped it to 7pl 1.4-qter. Identity to all previously known oncogenes except ERBB was ruled out by the fact that they are encoded by other chromosomes; identity to ERBB is probably excluded by failure of direct hybridizations of the 2 probes. MET was the designation suggested by Cooper et al. (ibid.). Dean et al. (Nature 1985, 318: 385-388) showed that MET is in the tyrosine kinase family of oncogenes. It appeared to be most closely related in sequence to the human insulin receptor and ABL oncogene. From the sequence of MET cDNA, Park
et al. (Proc. Nat. Acad. Sci. 1987, 84: 6379-6383) concluded that this oncogene is a cell-surface receptor for a then unknown ligand. The cellular MET protooncogene product is a receptor-like tyrosine kinase comprised of disulfϊde-linked subunits of 50 kD (alpha) and 145 kD (beta). In the fully processed Met product, the alpha subunit is extracellular, and the beta subunit has extracellular, transmembrane, and tyrosine kinase domains as well as sites of tyrosine phosphorylation (OMIM MIM Number: 164860: 10/18/2005). Met inhibitors may be useful in treating a variety of neoplasms including kidney, breast, bladder, non-small-cell lung, colorectal, and bladder cancers, and in hepatocellular carcinoma.
[0462] MLKl: Target kinase MLKl (i.e., mixed-lineage kinase 1, aka mitogen-activated protein kinase kinase kinase 9) is a 121.9 kDa serine/threonine kinase encoded by chromosome 14q24.3-q31 (symbol: MAP3K9). MLKl is expressed in epithelial tumor cell lines of colon, breast and esophageal origin. Silva et al. review the mixed lineage kinase (MLK) -c-jun N-terminal kinase (JNK) signaling cascade, which leads to the phosphorylation and activation of the transcription factor c-jun. There is much evidence, from in vitro and in vivo studies, that this cascade can mediate cell death. In addition, there is evidence that it is operative upstream in the death process. It is possible that abrogation of this pathway may forestall death before irreversible cellular injury. They review the evidence that inhibition of the MLKs can prevent dopamine neuron cell death and the degeneration of their axons (Silva et al., Mov Disord 2005, 20(6):653-64). Lund et al. state that MLK inhibitor CEP-1347 blocks the activation of the c-Jun/JNK apoptotic pathway in neurons exposed to various stressors and attenuates neurodegeneration in animal models of Parkinson's disease (PD). Microglial activation may involve kinase pathways controlled by MLKs and might contribute to the pathology of neurodegenerative diseases. They explored the possibility that CEP-1347 modulates the microglial inflammatory response [tumour necrosis factor-alpha (TNF-alpha), interleukin-6 (IL-6), and monocyte chemotactic protein-1 (MCP-I)] and report that the MLK inhibitor CEP-1347 reduced cytokine production in primary cultures of human and murine microglia, and in monocyte/macrophage-derived cell lines, stimulated with various endotoxins or the plaque forming peptide Abetal -40. Moreover, CEP-1347 inhibited brain TNF production induced by intracerebroventricular injection of lipopolysaccharide in mice. As expected from a MLK inhibitor, CEP-1347 acted upstream of p38 and c-Jun activation in microglia by dampening the activity of both pathways. These data imply MLKs as important, yet unrecognized, modulators of microglial inflammation, and demonstrate a novel anti-inflammatory potential of CEP-1347 (Lund et al., J Neurochem 2005, 92(6): 1439-51). MLKl inhibitors may be useful in treating neurodegenerative disorders such as Alzheimer's and Parkinson's disease and inflammatory disorders.
[0463] Mnkl: Target kinase Mnkl (i.e., MAP kinase interacting serine/threonine kinase 1) is a 51.3 kDa STK encoded by chromosome Ip34.1 (symbol: MKNKl). According to OMIM, Fukunaga
and Hunter (EMBO J. 1997, 16:1921-1933) observed that the C-terminal region of Mnkl was phosphorylated and activated in vivo and in vitro by Erkl and p38 MAP kinases, but not by JNK/SAPK. Waskiewicz et al., (EMBO J. 1997, 16:1909-1920) reported that in vitro, Mnkl rapidly phosphorylates eIF4E at the physiologically relevant site, ser209. In cells, they observed that Mnkl is posttranslationally modified and enzymatically activated in response to mitogenic and stress stimuli. This activation could be blocked by inhibitors of MAP kinase kinase-1 and p38, and Waskiewicz et al. (ibid.) concluded that Mnkl is downstream of multiple MAP kinases (OMIM MEVI Number: 606724: 02/27/2002).
[0464] Accordingly, dephosphorylation of eIF4E strongly correlates with inhibition or impairment of cap-dependent mRNA translation under certain stress conditions such as heat shock, nutrient deprivation, oxidative or osmotic stress, and infection of mammalian cells with certain viruses such as adenovirus (Ad) or influenza virus, among others. Mnkl inhibitors may be useful in treating conditions associated with heat shock, nutrient deprivation, oxidative or osmotic stress, infection of mammalian cells (e.g. with viruses such as adenovirus (Ad) or influenza virus), and autoimmune diseases.
[0465] p38: Target kinase p38 (i.e., Mitogen-activated Protein Kinase 14) is a 41.5 kDa STK encoded by chromosome 6p21.3-p21.2 (symbol: MAPKl 4). According to OMBVI, production of interleukin-1 and tumor necrosis factor (TNF) from stimulated human monocytes is inhibited by a series of pyridinyl-imidazole compounds called CSAIDs (cytokine-suppressive antiinflammatory drugs). These agents have shown activity in a variety of animal models of acute and chronic inflammation. Using radiolabeled chemical probes for radioligand binding assays and photoaffmity labeling experiments, Lee et al. (Nature 1994, 372:739-746) identified, purified, cDNA-cloned, and biochemically characterized 2 CSBPs (CSAID-binding proteins) as molecular targets of pyridinyl- imidazole cytokine inhibitors. They designated the 2 closely related mitogen-activated protein kinases (MAPKs) CSBPl and CSBP2. Binding of pyridinyl-imidazole compounds inhibited CSBP kinase activity and was directly correlated with their ability to inhibit cytokine production, suggesting that the CSBPs are critical for cytokine production. Lee et al. (ibid.) considered the 2 to be products of alternative splicing. The 4.2-kb CSBP mRNA encodes a predicted 360-amino acid protein and was expressed in all tissues tested. CSBPl and CSBP2 are identical except for a 75-nucleotide stretch within the coding region. Han et al. (Science 1994, 265: 808-811) cloned the mouse homolog as a protein that is tyrosine phosphorylated as part of the protein kinase cascades induced by endotoxic lipopolysaccharide. They named this 38-kD protein p38. As p38 is a member of the stress-activated protein kinase (SAPK) class of MAPKs, Goedert et al. (Genomics 1997, 41:501-502) referred to this protein as SAPK2A. Zervos et al. (Proc. Nat. Acad. Sci. 1995, 92:10531-10534) identified p38 as a human protein that interacts with MAX protein and designated it MXI2. The MXI2 gene encodes a
297-residue protein whose sequence indicates that it is related to the extracellular signal-regulated kinases (ERK protein kinases). MXI2 in yeast interacts with Max and with the C terminus of c-Myc. MXI2 phosphorylates MAX both in vitro and in vivo. The authors speculated that phosphorylation by MXI2 may effect the ability of MAX to oligomerize with itself and its partners, bind DNA, or regulate gene expression (OMIM MM Number: 600289: 02/13/2006).
[0466] There are four known isoforms of p38, i.e., p38-ct!, p38/3, p38γ, and p38δ. The a and β isoforms are expressed in inflammatory cells and are key mediators of TNF-α production. Inhibiting the p38α and β enzymes in cells results in reduced levels of TNF-o; expression. Also, administering p38α and β inhibitors in animal models of inflammatory disease has proven that such inhibitors are effective in treating those diseases. Accordingly, the p38 enzymes serve an important role in inflammatory processes mediated by IL-I and TNF-α. Compounds that reportedly inhibit p38 kinase and cytokines such as IL-I and TNF-α for use in treating inflammatory diseases are disclosed in the following published international patent applications: WO 00/12497 (quinazoline derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors).
[0467] p38 responds to activation by environmental stress, pro- inflammatory cytokines and lipopolysaccharide (LPS) by phosphorylating a number of transcription factors, such as ELKl and ATF2 and several downstream kinases, such as MAPKAPK2 and MAPKAPK5. Additionally, p38 plays a critical role in the production of some cytokines, for example IL-6. p38 phosphorylates ELKl and ATF2.
[0468] p38 is activated by threonine and tyrosine phosphorylation by either of two dual specificity kinases, MAP2K3 or MAP2K6, and potentially also MAP2K4 and it is inhibited by dual specificity phosphatases, such as DUSPl . p38 is specifically inhibited by the binding of pyridinyl-imidazole compounds, which are cytokine- suppressive anti-inflammatory drugs (CSAID).
[0469] p38 inhibitors have the potential to treat a number of diseases, including, but not limited to acute coronary syndrome, stroke, atherosclerosis, and inflammatory diseases such as rheumatoid arthritis, inflammatory bowel disease, and Crohn's disease.
[0470] PDGFR kinase family: Related to Fms and Kit are two platelet-derived growth factor receptors, alpha (i.e., PDGFRA) and beta (PDGFRB). The gene coding for PDGFRA is located on chromosome 4ql2 in the same region of chromosome 4 as the oncogene coding for Kit. Most gastrointestinal stromal tumors (GIST) have activating mutations in Kit, and most patients with GISTs respond well to Gleevec, which inhibits Kit. Heinrich et al. (Science 2003, 299:708-10.) have shown
that approximately 35% of GISTs lacking Kit mutations have intragenic activation mutations in the gene encoding pdgfra, and that tumors expressing Kit or PDGFRA were indistinguishable with respect to activation of downstream signaling intermediates and cytogenetic changes associated with tumor progression. Thus, Kit and PDGFRA mutations appear to be alternative and mutually exclusive oncogenic mechanisms in GISTs. PDGF is a potent growth factor and chemoattractant for smooth muscle cells (SMCs), and the renarrowing of coronary arteries following angioplasty is due in part to the enhanced proliferation of SMCs in response to increased levels of PDGF. Therefore, compounds that inhibit the kinase activity of PDGFr may be useful in the treatment of restenosis. In addition, since PDGF and PDGFr are overexpressed in several types of human gliomas, small molecules capable of suppressing PDGFr activity have potential utility as anticancer therapeutics [Nister, M., J. Biol. Chem., 266, 16755 (1991); Strawn, L. M., J. Biol. Chem. 269, 21215 (1994)].
[0471] PDGFRo;: Target PDGFRα (i.e., Plate Derived Growth Factor Receptor, alpha) is a 122.7 kDa transmembrane tyrosine kinate encoded by chromosome 4ql2 (symbol: PDGFRA). According to OMlM, The KIT oncogene, another member of the PDGF growth factor receptor subfamily, is located in the same region of chromosome 4 (Stenman et al., Genes Chromosomes Cancer 1989, 1:155-158). PDGFRl (i.e.PDGFRB) and CSFlR (i.e. FMS) are also membrane-spanning growth factor receptors with tyrosine kinase activity. The PDGFRl and CSFlR genes appear to have evolved from a common ancestral gene by gene duplication, inasmuch as these 2 genes are tandemly linked on chromosome 5 (Roberts et al., Cell 1988, 55: 655-661). They are oriented head-to-tail with the 5- prime exon of FMS located only 500 bp from the last 3-prime exon of PDGFRB. An analogous situation may exist for the PDGFR2 (i.e. PDGFRA) and KIT genes on chromosome 4. From an evolutionary point of view, it is possible that the distribution of these 4 loci, PDGFR2, KIT, PDGFRl, and FMS, on chromosomes 4 and 5 is a result of gene duplication and chromosome doubling (tetraploidization) (OMM MM Number: 173490: 03/21/2005). PDGFRA inhibitors may be useful in treating idiopathic hypereosinophilic syndrome, chronic eosinophilic leukemia, glioma, gastrointestinal stromal tumors (GISTs), juvenile myelomonocytic leukemia, metastatic medulloblastoma, atherogenesis, and restenosis.
[0472] PDGFRβ: Target PDGFRβ (i.e., Plate Derived Growth Factor Receptor, beta) is a 124.0 kDa transmembrane tyrosine kinate encoded by chromosome 5q31-q32 (symbol: PDGFRB). According to OMM, stimulation of cell proliferation of the receptor for PDGF has been implicated in atherogenesis and in cell transformation by the SIS oncogene. Escobedo et al. (1986) sequenced the receptor and cloned its gene. Gronwald et al. (Proc. Nat. Acad. Sci. 1988, 85:3435-3439) cloned a cDNA coding for human PDGFR and studied its expression. The cDNA contained an open reading frame that coded for a protein of 1,106 amino acids. In transfectants, Gronwald et al. (ibid.) found that the PDGFR clone expressed a high affinity receptor specific for the BB isoform of PDGF, i.e.,
PDGF dimers composed of 2 B chains. There may be a separate class of PDGF receptor that binds both the homodimers and the heterodimer. Claesson-Welsh et al. (Molec. Cell. Biol. 1988, 8:3476- 3486) determined the structure of the human PDGF receptor as deduced from a full-length cDNA clone. The receptor expressed in Chinese hamster ovary cells was found to bind specifically to B- chain-containing PDGF molecules. With the description of a second PDGF receptor, it is necessary to use the symbol PDGFRl . Matsui et al. (1989) designated the second type of PDGFR as type alpha because PDGF binding was blocked by AA as well as BB isoforms of the ligand; the product of the earlier cloned PDGF receptor was termed type beta (OMIM MM Number: 173410: 11/19/2003).
[0473] PDGFRβ has been implicated in hematological cancers, such as chronic myelomonocytic leukemia (CMML), in which a significant number of patients have a t(5;12)(q33;pl3) translocation resulting in TEL-PDGFRβ fusion protein. Golub et al. report that the consequence of the t(5;12) translocation is expression of a fusion transcript in which the tyrosine kinase domain of the platelet- derived growth factor receptor beta (PDGFRβ) on chromosome 5 is coupled to a novel ets-like gene, tel, on chromosome 12. The tel-PDGFR beta fusion demonstrates the oncogenic potential of PDGFRβ and may provide a paradigm for early events in the pathogenesis of AML (Golub et al., Cell 1994, 77:307-316). PDGFRB inhibitors may be useful in treating idiopathic hypereosinophilic syndrome, chronic eosinophilic leukemia, juvenile myelomonocytic leukemia, and metastatic medulloblastoma.
[0474] PDPKl; Target PDPKl (3-phosphoinositide dependent protein kinase-1) is a 63.2 kDa serine/threonine kinase encoded by chromosome 16pl3.3 (symbol: PDPKl). PDPKl inhibitors may be useful in treating cancers and diabetes.
[0475] Piml: Target Piml (i.e., Proviral Integration Site 1) is a 35.7 kDa serine/threonine kinase encoded by chromosome 6p21.2 (symbol: PIMl) found in the cytoplasm and nucleus. The structure of Piml comprises a STK (i.e., serine/threonine kinase) domain. X-ray crystal structures of Piml bound to various small molecules have recently been solved (Jacobs, et al., J Biol Chem 2005 280: 13728-34; Qian, et al., J Biol Chem 2005 280: 6130-7; Kumar, et al., J MoI Biol 2005 348: 183-93).
[0476] Piml is the first described member of a unique family of serine/threonine kinases, which includes at least two other kinases (PIM2 and P1M3) with significant sequence homology to Piml (van der Lugt, et al., Embo J 1995 14: 2536-44; Feldman, et al., J Biol Chem 1998 273: 16535-43). The PIMl protooncogene was originally identified as a genetic locus frequently activated by the proviral insertion of Moloney murine leukemia virus into mouse T cell lymphomas (Cuypers et al., Cell 1984, 37:141-150). Several substrates of Piml phosphorylation have been identified, including c-Myb (Winn, et al., Cell Cycle 2003 2: 258-62), BAD (Yan, et al., J Biol Chem 2003 278: 45358-67; Aho, et al., FEBS Lett 2004 571: 43-9), SOCS-I (Chen, et al., Proc Natl Acad Sci U S A 2002 99: 2175-
80), Cdc25A (Mochizuki, et al., J Biol Chem 1999 274: 18659-66), HPl (Koike, et al., FEBS Lett 2000 467: 17-21), PAP-I (Maita, et al., Eur J Biochem 2000 267: 5168-78), p2loiPI/wafl (Wang, et al., Biochim Biophys Acta 2002 1593: 45-55), PTP-U2S (Wang, et al., Arch Biochem Biophys 2001 390: 9-18), and NFATcI (Rainio, et al., J Immunol 2002 168: 1524-7). Piml has been shown to have diverse biological roles in cell survival, proliferation, differentiation, and immune response (Wang, et al., J Vet Sci 2001 2: 167-79; Bachmann, et al., M J Biochem Cell Biol 2005 37: 726-30). However, mice lacking all three Pirn genes have recently been shown to be viable and demonstrate that the PlM kinases are important for growth factor signaling, but are not essential for development (Mikkers, et al., MoI Cell Biol 2004 24: 6104-15). During embryonal development PIM genes are expressed in partially overlapping fashion in cells in both immune and central nervous system as well as in epithelia (Eichmann A, Yuan L, Breant C, Alitalo K, and Koskinen PJ. (2000) Developmental expression of PIM kinases suggests functions also outside of the hematopoietic system. Oncogene 19: 1215-1224). PIM-I, the prototypical member of the PIM family is located both in the cytoplasm and nucleus, but its precise role in these two locations has not been fully elucidated.
[0477] Dysfunction of Piml has been implicated in the progression of multiple cancers, including several hematopoietic and prostate cancers. Although the exact mechanisms by which Piml participates in cell transformation have not been completely elucidated, several reports point to the ability of Piml to prolong cell survival (Lilly, et al., Cancer Res 1997 57: 5348-55; Lilly, et al., Oncogene 1999 18: 4022-31; Moroy, et al., Proc Natl Acad Sci U S A 1993 90: 10734-8). Overexpression of Piml has been observed in myeloid and lymphoid acute leukemia and Piml is constitutively expressed in some myeloid leukemia cell lines (Lilly, et al., Oncogene 1992 7: 727-32; Amson, et al., Proc Natl Acad Sci U S A 1989 86: 8857-61). Increased Piml expression has also been identified in neoplastic prostate cancer specimens from patients by cDNA microarray analysis and by anti-Piml antibody staining (Dhanasekaran, et al., Nature 2001 412: 822-6). In a transgenic murine model of prostate cancer in which human c-myc is expressed, the gene expression profile is consistent with that seen in human prostate cancer, including upregulation of Piml (Ellwood-Yen, et al., Cancer Cell 2003 4: 223-38). In addition, Piml may participate in deregulated cell growth in prostate cancer through the hormone independent activation of the androgen receptor, a typical characteristic of advanced prostate cancer that offers poor patient prognosis (Kim, et al., Oncogene 2004 23: 1838-44). The PIM-I proto-oncogene has also been implicated in human hematopoietic malignancies with its overexpression frequently detected in human hematopoietic cell lines as well as in fresh tumor cells from patients with leukemia (Nagarajan et al. Proc. Natl. Acad. Sci. USA, 1986, 83:2556-2560; Meeker et al., Oncogene Res. 1987, 1: 87-101; Amson et al., Proc. Natl. Acad. Sci. USA, 1989, 86: 8857-8861).
[0478] In diffuse large cell lymphoma (DLCL), the most common form of non-Hodgkin's lymphoma, Piml has been shown to undergo chromosomal translocations, resulting in its overexpression (Akasaka, et al.; Cancer Res 2000 60: 2335-41). A recent study showed that Piml was also the target of an aberrant somatic hypermutation in DLCL (Pasqualucci, et al., Nature 2001 412: 341-6). Hypermutation sites are distributed in both 5' UTR and coding sequence, and independent of the chromosomal translocations. Notably, there are seven missense mutations introduced into the coding exons of the gene. These missense mutations may affect the three- dimensional structure and, in some cases, the kinase activity of the Piml protein. Hypermutations are also detected in Piml found in primary central nervous system lymphomas (Montesinos-Rongen, et al., Blood 2004 103: 1869-75) and multiple subtypes of AIDS-induced non-Hodgkin's lymphomas (Gaidano, et al., Blood 2003 102: 1833-41). Inhibition of Piml kinase activity by small molecules has the potential to offer a therapeutic benefit in these diseases.
[0479] Transgenic mice with PIM-I driven by Emu enhancer sequences demonstrated that PIM-I function as a weak oncogene because by itself it does not lead to tumor formation but does so after a second oncogenic gene become overexpressed. In 75% of the tumors over-expressing PIM-I, the second gene found to be over-expressed is c-myc (van der Houven van Oordt CW, Schouten TG, van Krieken JH, van Dierendonck JH, van der Eb AJ, Breuer ML.(1998) X-ray-induced lymphomagenesis in E mu-PIM-1 transgenic mice: an investigation of the co-operating molecular events. Carcinogenesis 19:847-853). In fact when crosses were made between Emu-PIM transgenic mice and Emu-myc transgenic mice, the combination of genes is so oncogenic that the offsprings die in utero due to pre B cell lymphomas (Verbeek S, van Lohuizen M, van der VaIk M, Domen J, Kraal G, and Berns A. (1991) Mice bearing the Emu-myc and Emu-PIM-1 transgenes develop pre-B-cell leukemia prenatally. MoI. Cell. Biol., 11: 1176-1179).
[0480] Mice deficient for PIM- 1 show normal synaptic transmission and short-term plasticity but failed to consolidate enduring LTP (i.e., long term potentiation) even though PIM-2 and PIM-3 are expressed in the hippocampus (Konietzko U, Kauselmann G, Scafidi J, Staubli U, Mikkers H, Berns A, Schweizer M, Waltereit R, and Kuhl D.(1999) PIM kinase expression is induced by LTP stimulation and required for the consolidation of enduring LTP. EMBO J. 18: 3359-3369).
[0481] Various factors are known to enhance the transcription of PIM-I kinase in mouse and human. PIM-I closely cooperates with another oncoprotein, c-myc, in triggering intracellular signals leading to both transformation and apoptosis and the selective inhibition of apoptotic signaling pathways leading to Bcl-2 (van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, and Berns A. (1989) Predisposition to lymphomagenesis in PIM-I transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 56:673-682; Breuer ML, Cuypers HT, Berns A. (1989). Evidence for the involvement of PIM-2, a new common
proviral insertion site, in progression of lymphomas. EMBO J. 8:743-748.; Verbeek S, van Lohuizen M, van der VaIk M, Domen J, Kraal G, and Berns A. (1991) Mice bearing the E mu-myc and E mu- PIM-I transgenes develop pre-B-cell leukemia prenatally. MoI. Cell. Biol. 11: 1176-1179; Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, and Hirano T. (1999) Synergistic roles for PIM-I and c- Myc in STAT3 -mediated cell cycle progression and antiapoptosis. Immunity, 11: 709-719). PIM-I kinase is induced by T cell antigen receptor cross linking by cytokines and growth factors and by mitogens including EL2, IL3, TL6, EL9, IL12, IL15, GM-CSF, G-CSF, IFNa, INFg, prolactin, ConA, PMA and anti-CD3 antibodies (Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, and Gold MR.(2002) CD40 signaling in B cells regulates the expression of the PIM-I kinase via the NF-kappa B pathway. J Immunol. 168: 744-754). PIM-I expression is rapidly induced after cytokine stimulation and the proliferative response to cytokines is impaired in cells from PIM-I deficient mice (Domen J, van der Lugt NM, Acton D, Laird PW, Linders K, Berns A.(1993) PIM-I levels determine the size of early B lymphoid compartments in bone marrow. J. Exp. Med. 178: 1665-1673).
[0482] Members of the PIM family of kinases interact with Socs-1 protein, a potent inhibitor of JAK activation thereby playing a major role in signaling down stream of cytokine receptors. The phosphorylation of Socs-1 by PDvI family of kinases prolongs the half-life of Socs-1 protein, thus potentiating the inhibitory effect of Socs-1 on JAK-STAT activation (Chen XP, Losman JA, Cowan S, Donahue E, Fay S, Vuong BQ, Nawijn MC, Capece D, Cohan VL, Rothman P. (2002) PIM serine/threonine kinases regulate the stability of Socs-1 protein. Proc. Natl. Acad. Sci. USA 99:2175- 2180.). PJJVI-I is expressed during Gl/S phase of the cell cycle suggesting that it is involved in cell cycle regulation (Liang H, Hittelman W, Nagarajan L., Ubiquitous expression and cell cycle regulation of the protein kinase PJM-I. (1996) Arch Biochem Biophys. 330:259-265). ). PM-I kinase activity and the protein level is increased in CD 40 mediated B cell signaling and this increase in PM-I level is mediated through the activation of NF-kB (Zhu et al. 2002. supra). PM-I can physically interact with NFATc transcription factors enhancing NFATc dependant transactivation and IL2 production in Jurkat cells (Rainio EM, Sandholm J, Koskinen PJ. (2002) Cutting edge: Transcriptional activity of NFATcI is enhanced by the PM-I kinase. J. Immunol. 168:1524-1527). This indicates a novel phosphorylation dependant regulatory mechanism targeting NFATcI through which PM-I acts as down stream effector of ras to facilitate JJL2 dependant proliferation and survival of lymphoid cells (ibid.).
[0483] Piml is shown to interact with many other targets. Phosphorylation of Cdc25A phosphatase, a direct transcriptional target of c-myc, increase its phosphatase activity both in-vivo and in-vitro indicating that Cdc25A link PM-I and c-myc in cell transformation and apoptosis (Mochizuki T, Kitanaka C, Noguchi K, Muramatsu T, Asai A, and Kuchino Y. (1999) Physical and functional interactions between PM-I kinase and Cdc25A phosphatase. Implications for the PM-I -mediated
activation of the c-Myc signaling pathway; J. Biol. Chem. 274:18659-18666). PIM-I also phosphorylate PTP-U2S, a tyrosine phosphatase associated with differentiation and apoptosis in myeloid cells, decreasing its phosphatase activity and hence preventing premature onset of apoptosis following PMA-induced differentiation (Wang et al. (2001) Pim-1 negatively regulates the activity of PTP-U2S phosphatase and influences terminal differentiation and apoptosis of monoblastoid leukemia cells. Arch. Biochem. Biophys. 390:9-18). The phosphorylation of another PIM-I target, heterochromatin protein 1(HPl) has been shown to be involved in transcription repression (Koike et al., FEBS Lett. 2000, 467: 17-21).
[0484] Piml inhibitors may be useful in treating cancers such as hematopoietic (e.g. acute myeloid and acute lymphoid leukemias) and prostate cancers, and non-Hodgkin's lymphomas.
[0485] Pim2: Target kinase Pim2 (i.e., Serine/threomne-protein kinase Proviral Integration Site 2) is a 34.2 kDa STK encoded by chromosome XpI 1.23 (symbol: PM2), Pim2 has also been shown to play a role in cell survival and the control of apoptosis and its value as an inhibitor target has been considered (Yan, et al., J Biol Chem 2003 278: 45358-67; Giles, Blood 2005 105: 4158-4159; Fox, et al., Genes Dev 2003 17: 1841-54). Pim2 has been found to be overexpressed in some lymphomas (Cohen, et al., Leuk Lymphoma 2004 45: 951-5). Additionally, Pim2 is required for the rapamycin resistant T-cell survival and the rapamycin-resistant growth of nontransformed hematopoietic cells (Hammerman, et al., Blood 2005 105: 4477-83; Fox, et al., J Exp Med 2005 201: 259-66). Pim2 inhibitors may be useful in treating lymphomas.
[0486] Pim3; Target kinase Pim3 (i.e., Serine/threonine-protem kinase Proviral Integration Site 3) is a 35.8 kDa STK encoded by chromosome 22ql3 (symbol: PIM3). Pim3 has recently been shown to be overexpressed in human hepatocellular carcinoma cells and its ablation resulted in attenuated cell proliferation and enhanced apoptosis, suggesting that Pim3 can also participate in abnormal cell growth and inhibition of apoptosis (Fujii, et al., Int J Cancer 2005 114: 209-18). Pim3 inhibitors may be useful in treating hepatocellular carcinoma.
[0487] PKC alpha: Target kinase PKC alpha (i.e., Protein kinase C alpha) is a 76.8 kDa STK encoded by chromosome 17q22-q23.2 (symbol: PRKCA). Protein kinase C (PKC) is the major phorbol ester receptor. Nine mammalian members of the PKC family have been identified and designated alpha, beta, gamma, delta, epsilon, zeta, eta, theta, and lambda. According to OMIM, Parker et al..(Science 1986, 233:853-859) purified PKC from bovine brain and through the use of oligonucleotide probes based on partial amino acid sequence, derived cDNA clones from bovine cDNA libraries. Activation of PKC by calcium ions and the second messenger diacylglycerol is thought to play a central role in the induction of cellular responses to a variety of ligand-receptor systems and in the regulation of cellular responsiveness to external stimuli. Birnbaum et al. (Science
2004, 306:882-884) showed that high levels of PKC activity in prefrontal cortex, as seen for example during stress exposure, markedly impaired behavioral and electrophysiologic measures of working memory. Birnbaum (ibid.) concluded that excessive PKC activation can disrupt prefrontal cortical regulation of behavior and thought, possibly contributing to signs of prefrontal cortical dysfunction such as distractibility, impaired judgment, impulsivity, and thought disorder (OMM MIM Number: 176960: 04/17/2006). A mutation in PKC-alpha (D294G) correlates with pituitary tumor. PKC alpha inhibitors may be useful in treating pituitary tumors and prefrontal cortical dysfunction such as distractibility, impaired judgment, impulsivity, and thought disorder, also may be used to sensitize chemotherapy in breast, colon, and non small cell lung cancers.
[0488] PKC beta: Target kinase PKC beta (i.e., Protein kinase C, beta 1) is a 76.7 kDa STK encoded by chromosome 16pl 1.2 (symbol: PRKCBl). According to OMIM, Leitges et al. (Science 1996, 273:788-791) found that mice homozygous for a targeted disruption of the PRKCBl gene develop an immunodeficiency characterized by impaired humoral immune responses and reduced cellular responses of B cells similar to X-linked immunodeficiency (Xid) in mice. Thus, they concluded that the 2 isoforms, PKC-beta-I (PRKCBl) and PKC-beta-II (PRKCB2), play an important role in B-cell activation and may be functionally linked to Bruton tyrosine kinase in antigen receptor- mediated signal transduction (OMM MM Number: 176970: 03/03/2006). In general, inhibitors PKC beta and PKC isoforms may be effective in treating disorders characterized by dysregulated NFKB survival signaling. PKC beta inhibitors may be useful in treating diabetic retinopathy.
[0489] PKC theta: Target kinase PKC-theta (i.e., Protein kinase, theta) is a 81.9 kDa STK encoded by chromosome 10pl5 (symbol: PRKCQ). According to OMM, in an attempt to find PKC isoforms that are involved in growth control and/or activation of T lymphocytes, Baier et al., (J. Biol. Chem. 1993, 268:4997-5004) used a human peripheral blood lymphocyte-derived cDNA library was employed to identify a novel PKC isoform, termed PKC-theta. The gene encodes a protein of approximately 80 kD, expressed predominantly in lymphoid tissues and hematopoietic cell lines, particularly T cells. The alpha form (PRKCA) has been mapped to chromosome 17, the beta form (PRKCBl) to chromosome 16, the gamma form (PRKCG) to chromosome 19, and the delta form (PRKCD) to chromosome 3. By fluorescence in situ hybridization, Erdel et al. (Genomics 1995, 25:595-597) assigned the PRKCQ gene to 10ρl5. Blanco and Brown (Mammalian Genome 1997, 8:70-71) mapped the homolog Pkcq to mouse chromosome 2 by analysis of an interspecific backcross. Sun et al. (Nature 2000, 404:402-407) demonstrated that PKC-theta is essential for T-cell antigen receptor (TCR)-mediated T-cell activation but dispensable during TCR-dependent thymocyte development. They generated mice deficient in Pkc-theta by homologous recombination. Mutant mice were normal and fertile. TCR-initiated NF-kappa-B activation was absent from PKC-theta -/- mature T lymphocytes, but was intact in thymocytes. Activation of NF-kappa-B by tumor necrosis
factor-alpha and interleukin-1 was unaffected in the mutant mice. Induction of JNK was normal in T cells from mutant mice. Sun et al. (ibid.) concluded that PKC-theta functions in a unique pathway that links the TCR signaling complex to the activation of NF-kappa-B in mature T lymphocytes. Using hyperinsulinemic-euglycemic clamps, Kim et al. (J. Clin. Invest. 2004, 114:823-827) demonstrated that skeletal muscle and hepatic insulin action did not differ between wildtype and Pkc- theta null mice. A 5-hour lipid infusion decreased insulin-stimulated skeletal muscle glucose uptake in the wildtype mice that was associated with 40 to 50% decreases in insulin-stimulated tyrosine phosphorylation of insulin receptor substrate-1 (ERSl) and IRSl -associated PDK activity. In contrast, Pkc-theta inactivation prevented fat-induced defects in insulin signaling and glucose transport in skeletal muscle. Kim et al. (ibid.) concluded that PKC-theta is a crucial component mediating fat- induced insulin resistance in skeletal muscle (OMIM MIM Number: 600448: 10/15/2004).
[0490] Li et al. report that PKC plays a critical role in competitive activity-dependent synapse modification at the neuromuscular synapse in vitro and in vivo. This action involves a reduction of the strength of inactive inputs to muscle cells that are activated by other inputs. A decrease of postsynaptic responsiveness and a loss of postsynaptic acetyl choline receptors account for the heterosynaptic loss in vitro. The loss is not seen in preparations in which PKC has been blocked pharmacologically. Here, they show that the loss does not occur in in vitro preparations made from animals genetically modified to lack the theta isoform of PKC. Synapse elimination in the newborn period in vivo is delayed but is eventually expressed in knock-out animals. PKC-dependent synapse reduction is suppressed in heterologous cultures combining normal nerve and PKC-theta-deficient muscle, as might be expected from the postsynaptic locus of the changes that underlie the activity- dependent plasticity. Preparations in which PKC-theta-deficient neurons innervated normal muscle also exhibited a marked deficit in PKC-deficient synapse reduction. The presynaptic action of PKC- theta implied by this observation is blocked by TTX, and the authors propose that the activity-related synapse strengthening is decreased by presynaptic PKC-theta. Thus, PKC-theta in both presynaptic and postsynaptic elements plays a critical role in activity-dependent synapse modulation and loss (Li et al., Journal of Neuroscience 2004, 24(15):3762-3769). PKC theta inhibitors may be useful in treating insulin resistance, T-cell lymphoma.
[0491] PIkI: Target kinase Plkl (i.e., Polo like kinase 1) is a 68.3 kDa STK encoded by chromosome 16pl2.3 (symbol: PLKl). Plkl is a regulator required for multiple mitotic processes, including bipolar mitotic spindle formation, actin ring formation, and chromosomal segregation. According to OMIM, Holtrich et al. (Proc. Nat. Acad. Sci. 1994, 91:1736-1740) observed that PLKl transcripts are present at high level in tumors of various origins. In vertebrate cells, the nuclear entry of mitosis-promoting factor (MPF) during prophase is thought to be essential for the induction and coordination of M-phase events. Phosphorylation of cyclin Bl is central to its nuclear translocation.
Toyoshima-Morimoto et al. (Nature 2001, 410:215-220) purified a protein kinase from Xenopus M- phase extracts that phosphorylates a crucial serine residue (S 147) in the middle of the nuclear export signal sequence of cyclin Bl. They identified this kinase as Plxl, a Xenopus homolog of PLKl. During cell cycle progression in HeLa cells, a change in the kinase activity of endogenous PIs 1 toward S147 and/or S133 correlates with a kinase activity in the cell extracts. Aa anti-PLKl antibody depleted the M-phase extracts of the kinase activity toward S 147 and/or S 133. An anti-phospho-S147 antibody reacted specifically with cyclin Bl only during G2/M phase. A mutant cyclin Bl in which S 133 and S 147 were replaced by alanines remained in the cytoplasm, whereas wildtype cyclin Bl accumulated in the nucleus during prophase. Further, coexpression of constitutively active Plkl stimulates nuclear entry of cyclin Bl . Tovoshima-Morimoto et al. (ibid.") concluded that Plkl may be involved in targeting MPF to the nucleus during prophase (OMM MM Number: 602098: 08/05/2005). Plkl inhibitors may be useful in treating cancers (e.g. lymphoma of the thyroid, non-Hodgkin's lymphomas, colorectal cancers, leukemias and melanoma), also useful as sensitizer in chemotherapy.
[0492] Pyk2: Target kinase Pyk2 (i.e., Protein-Tyrosine Kinase 2) is a 115.9 kDa tyrosine kinase of the FAK family (see e.g., target Fak) encoded by chromosome 8p21.1 (symbol: PTK2B). As with target kinase Fak, Pyk2 comprises B41 and TK domains, and Pyk2 is also known as Fak2 (i.e., Focal Adhesion Kinase 2). Because Pyk2 is calcium dependent, it is also known as CADAK (i.e., calcium- dependent tyrosine kinase).
[0493] As mentioned, another member of the FAK subfamily is kinase Fak. Pyk2 and Fak shares 65% sequence identity in the kinase domain and have similar domain structure: an N-terminus domain for integrin binding, and a C-terminus domain for Paxillin binding. Fak is ubiquitously expressed while Pyk2 exhibits a more restricted tissue expression pattern primarily in neuronal and hematopoetic tissues.
[0494] According to OMM, Focal adhesion kinases (i.e., FAKs) are cytoplasmic protein-tyrosine kinases associated with focal adhesions and whose activity is induced by ligand binding to various receptors including those of, for example, integrin and growth factors. FAKs are known to target paxillin and are substrates for Src family kinases (Calalb et al., Molec. Cell. Biol. 1995, 15:954-963). Herzog et al. (Genomics 1996, 32:484-486) identified a gene for another focal adhesion kinase by low-stringency screening of a hippocampus cDNA library. They symbolized the gene FAK2. The FAK2 cDNA encodes a predicted 1 ,009-amino acid protein with 42% identity to FAKl . Northern blot analysis detected a 4.5-kb mRNA in brain, kidney, spleen, and lymphocytes. Protein-tyrosine kinases in the central nervous system are activated in response to a variety of neurotrophic factors that control neuronal differentiation and survival via cell surface receptors. Also, protein phosphorylation
is involved in membrane excitability and the function of ion channels. Lev et al. (Nature 1995, 376:737-745) discovered a nonreceptor type protein kinase that is highly expressed in adult rat brain. The kinase, which they symbolized PYK2 (proline-rich tyrosine kinase-2), was cloned from a rat spinal cord cDNA library using degenerate PCR primers corresponding to conserved tyrosine kinase motifs of PYKl (see Manser et al., Nature 1993, 363:364-367). Lev et al. (ibid.) cloned the human homolog from a human fetal brain cDNA library using the rat sequence as a probe. The predicted protein of 1,009 amino acids has 61% sequence identity to the FAKl protein (Ptk2) (OMM MM Number: 601212: 01/19/2005). PKC-theta may represent an important signaling intermediate between neuropeptide activated receptors or neurotransmitters that increase calcium flux and the downstream signals that regulate neuronal activity. PKC-theta interacts with the SH2 domain of Grb2, and may phosphorylate the voltage-gated potassium channel protein KvI .2. Its activation is highly correlated with the stimulation of c-Jun N-terminal kinase activity.
[0495] Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. It mediates the Jak-dependent activation of MAPK and Statl . By rapidly translocating to the vicinity of the immune synapse after T cell receptor stimulation, Pyk2 plays an essential role in T cell activation and polarized secretion of cytokines. The morphology and behavior of macrophages in Pyk2-/- mice were impaired. Macrophages isolated from mutant mice failed to become polarized, to undergo membrane ruffling, and to migrate in response to chemokine stimulation. Moreover, the contractile activity in the lamellipodia of Pyk2-/- macrophages was impaired, as revealed by measuring the rearward movement toward the nucleus of fibronectin-coated beads on the lamellipodia in opposition to an immobilizing force generated by optical tweezers.
[0496] Pyk2 is implicated in several therapeutic areas including inflammation (e.g. osteoporosis, Polycystic Kidney Disease, rheumatoid arthiritis and some bowel diseases) and CNS disease like Parkinson's disease and Alzheimer's disease). Pyk2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of v3 integrin, and phosphorylated by Src kinase. Methods for preventing cell death in a subject and their application in the treatment of neurodegenerative diseases and conditions, such as Alzheimer's disease, stroke, Parkinson's disease have been pattened by Griswold-Prenner Irene and Powell Kyle.
[0497] Pyk2 is also a potential therapeutic target for tumors. Pyk2 is a novel effector of fibroblast growth factor receptor 3 activation. Pyk2 facilitates EGFR- and c-Src-mediated Stat3 activation and has a role in triggering Stat3-induced oncogenesis. HER3, but not HER2, mediates the phosphorylation of the C-terminal region of PYK2 to promote a mitogenic response through activation of the MAPK pathway. Furthermore, PYK2 phosphorylation by HER3 induces tumor invasion. A central role of PYK2 in signaling downstream of HER3 is substantiated by the
demonstration that expression of a dominant-negative PYK2-KM construct abrogates the Heregulin- induced MAPK activity and inhibits the invasive potential of glioma cells.
[0498] Overexpression of wild-type RAFTK significantly enhanced breast cancer cell invasion, while overexpression of the mutants Tyr402 or Tyr881 of RAFTK inhibited this migration. Therefore, Pyk2 may serve as a mediator and an integration point between focal adhesion molecules in HRG- mediated signaling in T47D breast cancer cells.
[0499] A murine pancreatic cancer cell line overexpressing Pyk2, mPancO2, was treated with a Pyk2-dominant negative adenovirus (Ad-Pyk2DN), or GFP (ad-GFP) adenovirus. The dominant negative Pyk2 adenovirus is able to decrease tumor growth and increase survival in several in vivo tumor models.
[0500] Although no point mutation of Pyk2 has been reported to be significant in any disease, human umbilical vein endothelial cells express mRNA transcripts for both the full length isoform Pyk2 and the truncated isoform Pyk2-H containing the C-terminal deletion.
[0501] Pyk 2 inhibitors may be useful in treating inflammation (e.g. osteoporosis, polycystic kidney disease, rheumatoid arthiritis and inflammatory bowel disease), CNS disease (e.g. Parkinson's disease and Alzheimer's disease), stroke and cancers (e.g. gliomas, breast cancer, and pancreatic cancer).
[0502] Ret: Target Ret (i.e., Rearranged during Transfection) is a 124.3 kDa tyrosine kinase encoded by chromosome 1OqI 1.2 (symbol: RET). Ret is also known as c-ret (i.e., cellular ret). The domain structure of Ret comprises cadherin, transmembrane, and TK domains. Cadherins are glycoproteins involved in Ca2+-mediated cell-cell adhesion; see e.g., Yap et al., Annu Rev Cell Dev Biol. 1997; 13:119-46).
[0503] According to OMDvI, the RET protooncogene is one of the receptor tyrosine kinases, cell- surface molecules that transduce signals for cell growth and differentiation. The RET gene was defined as an oncogene by a classical transfection assay. RET can undergo oncogenic activation in vivo and in vitro by cytogenetic rearrangement (Grieco et al., Cell 1990, 60:557-563). Mutations in the RET gene are associated with multiple endocrine neoplasia, type IIA (MEN2A), multiple endocrine neoplasia, type IB (MEN2B), Hirschsprung disease (HSCR; aganglionic megacolon), and medullary thyroid carcinoma (MTC) (OMIM MIM Number: 164761: 01/27/2006).
[0504] Ret (Rearranged during Transformation) was identified as a rearranged human oncogene in the classic NIH3T3 transformation assay (Takahashi et al., 1985, Cell 42(2):581-8) and subsequently characterized as a Receptor Tyrosine kinase (Takahashi et al., 1988, Oncogene 3(5):571-8).
[0505] Ret and NTRKl (i.e., Neurotrophic tyrosine receptor kinase 1) are receptor tyrosine kinase (RTK) proteins which play a role in the development and maturation of specific components of the nervous system. Their alterations have been associated to several human diseases, including some forms of cancer and developmental abnormalities. These features have contributed to the concept that one gene can be responsible for more than one disease. Moreover, both genes encoding for the two RTKs show genetic alterations that belong to either "gain of function" or "loss of function" class of mutations. In fact, receptor rearrangements or point mutations convert Ret and NTRKl into dominantly acting transforming genes leading to thyroid tumors, whereas inactivating mutations, associated with Hirschsprung's disease (HSCR) and congenital insensitivity to pain with anhidrosis (CIPA), impair Ret and NTRKl functions, respectively.
[0506] Implication of Ret in human tumorigenesis was indicated by the frequent identification of rearranged Ret sequences that transformed NIH3T3 cells in the DNA isolated from Papillary Thyroid Carcinoma DNAs. In these cases, the Ret gene was fused to as yet unknown PTC DNA sequences in the tumor DNA but not the normal patient DNA (Grieco et al, 1990, Cell 60(4):557-63). In addition, the chromosomal mapping of Ret to chromosome 1OqI 1.2 co-localized with genetic mapping data that implicated a gene involved in patients with MEN2A (Multiple Endocrine Neoplasia 2A) (Ishizaka et al. 1989 Oncogene 4(12):1519-21). Expression analysis of the RET oncogene in a number of human tumors consistently detected expression of normal-sized transcripts of the RET proto- oncogene in human pheochromocytomas and in human medullary thyroid carcinomas, both of familial and sporadic type (Santoro et al., 1990, Oncogene 5:1595-8).
[0507] Further analysis of the tumor DNA of patients with Multiple endocrine neoplasia type 2 A (MEN 2A) and familial medullary thyroid carcinoma (FMTC) identified mutations in the RET sequence resulting in amino acid changes in the encoded Ret protein (Donis-Keller 1993, Hum MoI Genet. 2(7):851-6). Likewise, mutations in the RET gene were correlated with Hirschprung disease, a developmental disorder with genetic deletions and mutations in the chromosomal location of the RET gene (Luo et al., 1993, Hum MoI Genet. 2(11): 1803-8).
[0508] By early 1994, multiple papers describe the inactivation of the RET gene in patients with Hirschsprung disease and similar phenotype in knock out mice. In addition, activating mutations in Ret are now identified in patients with MEN2A, MEN2B, and FMTC (reviewed by van Heyningen V., 1994, Nature 367(6461):319-20).
[0509] It was determined that Ret regulates cell survival. Signal transduction molecules that form a complex with Ret as a result of these phosphoryl moieties, such as GRB2, SOS, ras, and raf, propagate a signal in the cell that promotes neural survival. Thus, compounds that promote the interactions of the se stimulatory molecules of Ret would enhance the activity of c-Ret. Alternatively,
protein phosphatases can remove the phosphoryl moieties placed on the intracellular region of Ret in response to GDNF, and thus inhibit the signaling capability c-Ret. Thus, compounds that inhibit, phosphatases of Ret will probably enhance the signaling capacity of c-Ret.
[0510] Ret is implicated in the development and survival of enteric, synaptic, and sensory neurons and neurons of the renal system upon stimulation by GDNF (Jing, et al., 1996, Cell 85:1113-1124; Trupp, et al., 1996, Nature 381:785-789; Durbec, et al., 1996, Nature 381:789-793). Lack of function mutations in Ret can lead to Hirschsprung's disease, for example, which manifests itself as a decrease in intestinal tract innervation in mammals. Thus, compounds that activate Ret are potential therapeutic agents for the treatment of neurodegenerative disorders, including, but not limited to, Hirschsprung's disease, Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis. Compounds that inhibit Ret function can also be anti-cancer agents as over-expression of Ret in cells is implicated in cancers, such as cancer of the thyroid.
[0511] Modulation of Ret activity may also be useful in treating cancers of the nerve tissue, such as neuroblastoma, even if an abnormality is not found the signaling pathway.
[0512] As stated above, RET gene is responsible for MEN2 syndromes, which are inherited in an autosomal dominant fashion with high penetrance and diverse clinical manifestations. The predominant RET mutation is missense mutation which is restricted to 9 codons (codons 609, 611, 618, 620, 630, 634, 768, 804 and 918). The MEN2 syndromes have 3 subtypes: multiple endocrine neoplasia type 2A (MEN2A), MEN2B, and familial medullary thyroid carcinoma (FMTC). Missense mutations at exon 10 (codons 609, 611, 618, and 620) and exon 11 (codons 630 and 634) have been identified in 98% of MEN2A families and in 85% of FMTC families. Missense mutations at codons 768 and 804 have been known to be responsible for 5.about.l0% of FMTC cases. In addition, missense mutations at exon 16 (codon 918) have been found in 95% of MEN2B cases.
[0513] Ret inhibitors may be useful in treating cancer of the thyroid, neuroblastoma, familial medullary thyroid carcinoma (FMTC), multiple endocrine neoplasia type HA and HB (MEN2A, MEN2B), and neurodegenerative disorders (e.g. Hirschsprung's disease, Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis)
[0514] Ron: Target kinase Ron (i.e., Ron protein tyrosine kinase) is a 152.2 kDa transmembrane tyrosine kinase encoded by chromosome 3p21.3 (symbol: MSTlR), also known as macrophage stimulating protein receptor (i.e., MSP receptor). Ron, a member of the Met hepatocyte growth factor receptor family, was originally cloned from a human foreskin keratinocyte cDNA library by Ronsin et al {Oncogene 1993, 8: 1195-1202). RON is expressed in various cell types including macrophages, epithelial and hematopoietic cells. Ron activation results in a variety of cellular responses in vitro,
such as activation of macrophages, proliferation, migration, and invasion, which suggest a broad biologic role in vivo. Hemizygous mice (Ron +/-) grow to adulthood; however, these mice are highly susceptible to endotoxic shock and appeared to be compromised in their ability to downregulate nitric oxide production. Accordingly, Ron plays a role in early mouse development and may play a limited role in the inflammatory response. Further, Ron may be involved in cancer development and progression (OMIM MIM Number: 600168: 01/20/2006). Ron inhibitors may be useful in treating cancer and inflammation.
[0515] ROCK CROCK! and ROCK2): Target kinase ROCKl (i.e., Rho-associated, coiled-coil containing protein kinase 1) is a 158.2 kDa serine/threonine kinase encoded by chromosome 18ql 1.1 (symbol: ROCKl). Target kinase ROCK2 (i.e., Rho-associated, coiled-coil containing protein kinase 2) is a 160.9 kDa serine/threonine kinase encoded by chromosome 2p24 (symbol: ROCK2). ROCK inhibitors may be useful in treating related to cancers (e.g. ovarian cancer, hepatocellular carcinoma, pancreatic cancer), ocular disease (e.g. glaucoma), cardiac hypertrophy, improved renal perfusion, transplant rejection, and acute respiratory distress syndrome.
[0516] Src: Target kinase Src (i.e., v-Src Avian Sarcoma Schmidt-Ruppin A-2 viral oncogene) is a 59.8 kDa non-receptor tyrosine kinase encoded by chromosome 20ql2-ql3 (symbol: SRC). The structure of Src comprises SH3 and SH2 domains adjacent to the TK domain. According to OMIM, Azarnia et al. (Science 1988, 239:398-401) found that overexpression of the SRC gene in MH 3T3 cells caused reduction of cell-to-cell transmission of molecules in the 400- to 700-dalton range. Downregulation was enhanced by point mutation of tyrosine-527, whereas mutation of tyrosine-416 suppressed both the downregulation of communication by the tyr-527 mutation and that by gene overexpression. The regulation of communication by SRC may be important in the control of embryonic development and cellular growth. Luttrell et al. (Science 1999, 283:655-661) demonstrated that c-src binds to the amino terminus of beta-arrestin-1 in a complex resulting from the stimulation of beta-2 adrenergic receptors. Activated beta-2-adrenergic receptor bound beta-arrestin- 1, which then bound c-src. This interaction targeted the complex to clathrin-coated pits and allowed for beta-2-adrenergic activation of the MAP kinases Erkl and Erk2. TRANCE, a TNF family member, and its receptor, RANK, are critical regulators of dendritic cell and osteoclast function. Wong et al. (Molec. Cell 1999, 4:1041-1049) demonstrated that TRANCE activates the antiapoptotic serine/threonine kinase PKB (AKTl) through a signaling complex involving SRC and TRAF6. A deficiency in SRC or addition of SRC family kinase inhibitors blocked TRANCE-mediated PKB activation in osteoclasts. SRC and TRAF6 interacted with each other and with RANK upon receptor engagement. TRAF6, in turn, enhanced the kinase activity of SRC, leading to tyrosine phosphorylation of downstream signaling molecules such as CBL. These results defined a mechanism by which TRANCE activates SRC family kinases and PKB, and provided evidence of
cross-talk between TRAF proteins and SRC family kinases. Using a colon cancer cell line, Avizienyte et al. (Nature Cell Biol. 2002, 4:632-638) studied the role of SRC in cell adhesion and metastasis. Transfection and overexpression of a constitutively active SRC mutant reduced cell-cell contacts and caused redistribution of adherens junction components to discrete adhesion-like structures at the tips of membrane protrusions. Expression of active SRC also impaired the movement of E-cadherin from the cell interior to the plasma membrane following exposure to high calcium. Avizienyte et al. (ibid.) provided evidence that the alpha-V and beta-1 integrins and FAK were required for the adhesion changes induced by SRC. Sandilands et al. (Dev. Cell 2004, 7:855-869) found that RhoB colocalized with active Src in the cytoplasm of mouse embryonic fibroblasts, and they presented evidence that RhoB is a component of 'outside-in' signaling pathways that coordinate Src activation with translocation to transmembrane receptors (OMIM MIM Number: 190090: 01/07/2005).
[0517] The Src family of cytoplasmic protein tyrosine kinases consists of at least eight members (Src, Fyn, Lyn, Yes, Lck, Fgr, Hck and BIk) that participate in a variety of signaling pathways [Schwartzberg, P. L., Oncogene, 17, 1463 (1998)]. The prototypical member of this tyrosine kinase family is p60src (Src). Src is involved in proliferation and migration responses in many cell types. In limited studies, Src activity has been shown to be elevated in breast, colon (~90%), pancreatic (>90%) and liver (>90%) tumors. Greatly increased Src activity is also associated with metastasis (>90%) and poor prognosis. Antisense Src message impedes growth of colon tumor cells in nude mice [Staley et al., Cell Growth & Differentation, 8, 269 (1997)], suggesting that Src inhibitors should slow tumor growth. In addition to its role in cell proliferation, Src also acts in stress response pathways, including the hypoxia response. Previous studies have shown that colonic tumor cells genetically engineered to express antisense Src message form tumors demonstrating reduced vascularization in nude mouse models [Ellis, et al., J. Biol. Chem., 273, 1052 (1998)], suggesting that Src inhibitors would be anti- angiogenic as well as anti-proliferative.
[0518] Apart from its role in cancer, Src also appears to play a role in osteoporosis. Mice genetically engineered to be deficient in src production were found to exhibit osteopetrosis, the failure to resorb bone [Soriano, P., Cell, 64, 693 (1991); Boyce, B. Fv J. Clin. Invest., 90, 1622 (1992)]. This defect was characterized by a lack of osteoclast activity. Since osteoclasts normally express high levels of Src, inhibition of Src kinase activity may be useful in the treatment of osteoporosis [Missbach, M., Bone, 24, 437 (1999)]. Src inhibitors may be useful in treating cancer and osteoporosis.
[0519] Stk6: Target Stk6 (i.e., Serine/Threonine protein kinase 6) is a 45.8 kDa serine/threonine kinase encoded by chromosome 20ql3.2-ql3.3 (symbol: STK6). According to OMIM, Kimura et al. (J. Biol. Chem. 1997, 272:13766-13771) cloned a cDNA encoding a novel human serine/threonine kinase, STK6, that has high homology with the Aurora and IpIl kinases. Mutations in these yeast
kinases are known to cause abnormal spindle formation and nαissegregation of chromosomes. Northern and Western blot analyses revealed a high level of STK6 expression product in testis and proliferating culture cells such as HeLa cells. The endogenous levels of STK6 protein and protein kinase activity were tightly regulated during cell cycle progression in HeLa cells. T he protein was upregulated during G2/M and rapidly reduced after mitosis. Immunofluorescence studies revealed specific localization of STK6 protein to the spindle pole region during mitosis. The results suggested that STK6, like Aurora and IpIl, is involved in cell growth and/or chromosome segregation (OMIM MlM Number: 602687: 04/01/2003).
[0520] Aurora A belongs to the family of STKs that are involved in mitotic events such as centrosome separation and chromosome segregation and are therefore essential for cell proliferation (Bischoff & Plowman, Trends Cell Biol. 1999, 9:454-459); Giet & Prigent, 1999, Cell Science 112: 3591-3601). Inhibitors of the Aurora kinase family therefore have the potential to block growth of all tumors.
[0521] The three identified Aurora kinases of this family are known under various names: Aurora-A (Aurora 1), Aurora B (Aurora 2) and Aurora C (Aurora 3). Alternate names for Aurora A, described here further are serine/threonine protein kinase 15 (STK 15), BTAK Aurora-related kinase 1 (ARKl). The mouse homolog of Aurora A, STK6 also referred to as AIK, is highly homologous to STKl 5. All Aurora kinase family members are highly homologous proteins responsible for mitotic events such as centrosome maturation and seggregation, chromosome segregation, mitotic spindle function and cytokinesis. Peak expression of Aurora occurs during the G2 and mitotic phase in cycling cells and then decreases and remains low or undetectable in resting cells (Shindo et al., 1998, Biochem. Biophys. Res. Commun. 244: 285-292). In mammalian cells proposed substrates for Aurora include histone H3, a protein involved in chromosome condensation, and CENP-A, myosin II regulatory light chain, protein phosphatase 1, TPX2, all of which are required for cell devision.
[0522] Zhou et al., (Zhou et al., 1998, Nature Genet. 20: 189-193) found that Aurora is involved in the induction of centrosome duplication-distribution abnormalities and aneuploidy in mammalian cells. Centrosomes appear to maintain genomic stability through the establishment of bipolar spindles during cell division, ensuring equal segregation of replicated chromosomes to 2 daughter cells. Deregulated duplication and distribution of centrosomes are implicated in chromosome segregation abnormalities, leading to aneuploidy seen in many cancer cell types. Zhou et al., (Zhou et al., 1998, Nature Genet. 20: 189-193) found amplification of Aurora A in approximately 12% of primary breast tumors, as well as in breast, ovarian, colon, prostate, neuroblastoma, and cervical cancer cell lines. Additionally, high expression of Aurora A mRNA was detected in tumor cell lines without evidence of gene amplification. Ectopic expression of Aurora A in mouse NBH 3T3 cells led to the appearance of abnormal centrosome number (amplification) and transformation in vitro. Finally, over-expression
of Aurora A in near-diploid human breast epithelial cells revealed similar centrosome abnormality, as well as induction of aneuploidy. These findings suggested that Aurora A is a critical kinase-encoding gene, whose over-expression leads to centrosome amplification, chromosomal instability, and transformation in mammalian cells.
[0523] The AURE-A gene is over expressed in many human cancers. Ectopic over-expression of aurora kinase A in mammalian cells induces centrosome amplification, chromosome instability, and oncogenic transformation, a phenotype characteristic of loss-of-function mutations of p53. Katayama et al. (Katayama et al., 2004, Nature Genet. 36: 55-62) showed that Aurora A kinase phosphorylates p53 at ser315, leading to its ubiquitination by MDM2 and proteolysis. P53 is not degraded in the presence of inactive Aurora A or ubiquitination-defective MDM2. Silencing of Aurora kinase A results in less phosphorylation of p53 at ser315, greater stability of p53, and cell-cycle arrest at G2-M. Cells depleted of Aurora kinase A are more sensitive to cisplatin-induced apoptosis, and elevated expression of aurora kinase A abolishes this response. In a sample of bladder tumors with wildtype p53, (Katayama et al., 2004, Nature Genet. 36: 55-62) found a correlation between elevated expression of aurora kinase A and low p53 concentration. They concluded that Aurora A kinase is a key regulatory component of the p53 pathway and that over-expression of Aurora A leads to increased degradation of p53, causing down-regulation of checkpoint-response pathways and facilitating oncogenic transformation of cells.
[0524] By immunoprecipitation of epitope-tagged proteins from transfected HEK293 cells, Kunitoku et al. (Kunitoku et al., 2003, Dev. Cell 5: 853-854) demonstrated direct interaction between the genes CENPA and AURKA. In vitro, AURKA phosphorylated CENPA on ser7, a residue that is also phosphorylated by AURKB. Examination of the role of both kinases in the phosphorylation of CENPA revealed that the reaction is mediated sequentially by AURKA and AURKB in early mitosis. Mitotic cells in which phosphorylation of CENPA on ser7 was prevented exhibited a substantial proportion of misaligned chromosomes resulting from a defect in the ability of kinetochores to attach to microtubules.
[0525] By yeast 2-hybrid analysis of HeLa cells, Hirota et al. (Hirota et al., 2003, Cell 114: 585- 598) determined that AURKA interacts with Ajuba (JUB). The two proteins interacted in mitotic cells and became phosphorylated as they did so. In vitro analysis revealed that Ajuba induced the autophosphorylation and consequent activation of AURKA. Depletion of Ajuba prevented activation of AURKA at centrosomes in late G2 phase and inhibited mitotic entry. Hirota et al. (Hirota et al., 2003, Cell 114: 585-598) concluded that Ajuba is an essential activator of AURKA in mitotic commitment.
[0526] The mammalian Aurora kinase family has been implicated in tumorigenesis of a variety of different cancers. The main role of Aurora A in tumor development is in controlling chromosome segregation during mitosis (Bischoff & Plowman, Trends Cell Biol. 1999, 9:454-459). Over- expression of Aurora A transforms rodent fibroblasts (Bischoff et al., 1998, EMBO J. 17:3052-3065). The elevated levels of Aurora A induce misregulation of chromosome segregation that results in cells containing multiple centrosomes and multipolar spindles leading to aneuploidy, a distinctive feature of most cancers. (Zhou et al., 1998, Nature Genet. 20: 189-193). Ewart-Toland et al. (Ewart-Toland et al., 2003, Nature Genet. 34: 403-412) found that tumors from individuals carrying the 91A allele showed more evidence of aneuploidy than those from individuals who were homozygous for the common 91T allele. They concluded that individuals with even one copy of the Aurora A 91A allele develop tumors that have on average a higher degree of aneuploidy than those from individuals homozygous for 91T. The oncogenic activity of Aurora kinases is likely to be linked to the generation of such genetic instability. Miyoshi et al. (Miyoshi et al., 2001, Int. J. Cancer 92: 370-373) and Sakakura et al. (Sakakura, et al., 2001, Br. J. Cancer 84: 824-831) report a correlation between amplification of the Aurora A locus and chromosomal instability in mammary and gastric tumors.
[0527] Over-expression of Aurora kinases have been reported in a wide range of human tumors. Aurora A expression is elevated in tumor cell lines derived from colon, breast, lung, melanoma, kidney, ovary, pancreas, CNS, gastric tract and leukemia cells (Tatsuka et al., 1998, Cancer Res. 58(21): 4811-6.) Elevated expression of Aurora A has been detected in over 50% of colorectal, ovarian and gastric tumors and in 94% of invasive duct adenocarcinomas of the breast (Colorectal tumors: Bischoff et al., 1998, EMBO J. 17:3052-3065, Takahashi et al., 2000, Jpn. J. Cancer Res. 91.-1007-1014, Ovarian tumors: Gritsko et al., 2003, Clin. Cancer Res. 9:1420-1426; gastric tumors: Sakakura, et al., 2001, Br. J. Cancer 84: 824-831; breast tumors: Tanaka et al., 1999, Cancer Res. 59:2041-2044). High levels of Aurora A have also been reported in renal, cervical, neuroblastoma, melanoma, lymphoma, pancreatic and prostate tumor cell lines (Bischoff et al., 1998, EMBO J. 17:3052-3065; Zhou et al., 1998, Nature Genetics 20:189-193; Li et al., 2003, Clin. Cancer Res. 9(3): 991-997). Amplification and over-expression of Aurora A is further observed in human bladder cancers and where it is associated with aneuploidy and aggressive clinical behavior (Sen et al., 2002, J. Natl. Cancer Inst. 94(17): 1320-1329). Isola et al. (Isola et al., 1995, Am. J. Pathology 147: 905- 911) further found that amplification of the Aurora A locus correlates with poor prognosis for patients with node-negative breast cancer.
[0528] Based on the known function of the Aurora kinases, inhibition of their activity should disrupt mitosis leading to cell cycle arrest. In vivo, an Aurora inhibitor therefore slows tumor growth and induces regression. Stk6 inhibitors may be useful in treating gastric, bladder, breast, lung, CNS,
ovarian, kidney, colon, prostate, pancreas, and cervical cancers, melanoma, leukemia, and neuroblastoma.
[0529] Syk: Target kinase Syk (i.e., spleen tyrosine kinase) is a 72.1 kDa tyrosine kinase encoded by chromosome 9q22.2 (symbol: SYK). Syk inhibitors may be useful in treating lymphomas, such as mantle cell lymphoma.
[0530] TEC: Target kinase TEC (i.e., tec protein tyrosine kinase) is a 73.6 kDa non receptor tyrosine kinase encoded by chromosome 4pl2 (symbol: TEC). TEC inhibitors may be useful in treating sepsis, septic shock, inflammation, rheumatoid arthritis, Crohn's disease, irritable bowel disease (IBD), and ulcerative colitis.
[0531] Tie2: Target kinase Tie2 (i.e., tyrosine kinase, endothelial) is a 125.8 kDa receptor tyrosine kinase encoded by chromosome 9p21 (symbol: TEK). Tie2 inhibitors may be useful in treating cancer, arthritis (e.g. rheumatoid arthritis), and atherosclerosis.
[0532] TrkA; Target kinase TrkA (i.e., neurotrophic tyrosine kinase, receptor, type 1) is a 87.5 kDa tyrosine kinase encoded by chromosome Iq21 -q22 (symbol: NTRKl). TrkA inhibitors may be useful in treating pain (e.g. chronic pain, neuropathic pain), cancer, arthritis, diabetic retinopathy, macular degeneration and psoriasis.
[0533] Yes: Target kinase Yes (i.e., Yamaguchi Sarcoma Oncogene homolog 1) is a 60.8 kDa tyrosine kinase encoded by chromosome 18pl 1.31-pl 1.21 (symbol: YESl). The structure of Yes comprises SH3 and SH2 domains followed by a TK domain. The YES oncogene is homologous to the Yamaguchi sarcoma virus gene, and the amino acid sequence of Yes shows a high degree of homology with that of the SRC gene product of Rous sarcoma virus. The Yes kinase is highly expressed in multiple mammalian cell types, including neurons, spermatozoa, platelets, and epithelial cells. The target kinase Yes is amplified and overexpressed in various cancers including esophageal squamous cell carcinoma. Yes inhibitors may be useful in treating cancers including esophageal squamous cell carcinoma.
[0534] Zap70: Target kinase Zap70 (i.e., Zeta-chain associated protein kinase, 70 kDa) is a 69.9 kDa tyrosine kinase encoded by chromosome 2ql 1-13 (symbol: ZAP70). Zap70 was first reported by Chan et al. (Cell 1992, 71 : 649-662). The mature protein comprises two SH2 domains and a TK domain.
[0535] Zap70 is crucial to the transduction of the T-cell receptor (TCR) signalling pathway, which leads ultimately to cellular differentiation and proliferation. (Weiss & Imboden 1987, Adv. Immunol. 41 : 1-38). On stimulation of the T-cell antigen receptor tyrosine phosphorylation takes place in a
number of intracellular substrates, mediated by sequential activation of two distinct families of cytoplasmic PTKs. One substrate is the TCR-zeta chain, which can mediate the transduction of extracellular stimuli into cellular effector functions. The Src kinases Lck and Fyn phosphorylate tyrosine residues on the TCR zeta-chain, contained within conserved sequences known as immunoreceptor tyrosine-based activation motifs (ITAMs). The Zap70 protein associates with the phosphorylated ITAMs in the zeta chain of the activated TCR complex (Chan et al., 1991, PNAS 88: 9166-9170. Recruitment of Zaρ70 to the TCR and its subsequent phosphorylation and activation triggers all downstream signaling events (Irving & Weiss, 1991, Cell 64: 891-902). This interaction is believed to be critical for TCR signaling, since zeta-phosphopeptides that block the interaction of Zap70 with the zeta-chain also inhibit TCR signaling events (Wange et al., 1995, J. Biol. Chem. 270:944-948).
[0536] The essential role of Zap70 in T-cell function has been demonstrated in human patients, human T-cell lines and mice. Elder et al. (Elder et al., 1997, J. of Pedriatic Hematology/Oncology 19(6): 546-550) reported studies of human patient suffering from a rare form of severe combined immune deficiency syndrome (SCID). The patient was found to be homozygous for a 13-bp deletion involving nucleotides 1719-1731 of the ZAP70 gene, resulting in premature termination 35 codons downstream and yielding a mutant protein 82 residues shorter than wildtype Zap70 . This kind of patients have profound immunodeficiency, lack CD8+ T-cells and have CD4+ T-CeIIs that are unresponsive to T-cell receptor (TCR)-mediated stimulation. Following TCR activation the CD4+ cells show severe defects in Ca2+ mobilization, tyrosine phosphorylation of down-stream substrates, proliferation and IL-2 production (Elder et al., Pedriatric Research 39: 743-748).
[0537] ZAP70 deficient human Jurkat cells also demonstrate the important function of ZAP70 in T- cell receptor signaling. A Jurkat clone (pi 16) lacking the Zap70 protein was shown to have defects in TCR signaling which was correctable by re-introduction of wild type ZAP70 (Williams et al., 1998, Molecular and Cellular Biology 18 (3): 1388-1399.)
[0538] Studies of ZAPlQ deficient mice also underline the crucial role of the PTK in T-cell signal transduction. ZAP70 deficient mice had neither CD4 nor CD8 single-positive T cells, but human Zap70 reconstitutes both CD4 and CD8 single-positive populations. Besides the defects in T-cell development the TCR signalling in thymocytes was found to be profoundly impaired, suggesting that Zap70 is a central signalling molecule during thymic selection for CD4 and CD8 lineage. (Negishi et al.,1995, Nature 376: 435-438, Sakaguchi et al. (Sakaguchi et al. 2003, Nature 426: 454-460) reported that the mouse strain SKG, which is derived from a closed breeding colony of BALB/c mice, spontaneously develops chronic arthritis. This autosomal recessive trait was found to be caused by a mutation (Wl 63C) in the second SH2 domain of Zap70. The phenotype showed altered signal
transduction from TCRs and a change in the threshold of T-cells to thymic selection, leading to the positive selection of otherwise negatively selected autoimmune T-cells.
[0539] The importance of the Zaρ70 kinase domain function has been demonstrated by Elder et al. (Elder et al., 2001, J. Immunology 166(1): 656-661). In studies of humans patients and mice showed that missense mutations within the highly conserved DLAARN motif within the kinase domain of Zap70 result in SCBD. This mutation caused the loss of catalytic function of Zap70, resulting in defective T-cell receptor signaling. The requirement of the catalytic function of Zap70 was further illustrated by Williams et al., who found an inactive Zap70 mutant (Lys369Arg) was unable to restore TCR signaling in a ZAP70 deficient Jurkat cell clone (pi 16) (Williams et al., 1998, MoI. Cell Biology 18 (3): 1388-1399).
[0540] Zap70 further participates in early B-cell differentiation and is a prognostic factor in chronic lymphocytic leukaemia (CLL). The course of CLL is variable. Crespo and coworkers (Crespo et al., 2003, New Eng. J. Med. 348: 1764-1775) found that Zap70 expression by cells in CLL is a simple and reliable surrogate for the identification of immunoglobulin heavy chain variable region mutations. In the aggressive progression of the disease, Zap70 is associated with CLL cells expressing an unmutated configuration of the immunoglobulin heavy chain variable region gene (IgVH) (Carreras et al., 2005, Journal of Pathology 205 (4): 507-513). Whereas in indolent disease, the CLL cells usually express a mutated immunoglobulin heavy chain variable region gene but lack expression of ZAP70. Rassenti et al. (Rassenti et al., 2004, New Eng. J. Med. 351: 893-901) found that although the presence of an unmutated immunoglobulin heavy chain variable region gene in CLL patients was strongly associated with expression of Zap70, Zap70 was a stronger predictor of the need for treatment in B-cell CLL. T-cell Zap70 overexpression in CLL patients was found to not only correlate with Zap70 levels in CLL cells, but also with clinical stage and disease progression. (Herishanu et al., 2005, Leukemia advance online publication).
[0541] The protein tyrosine kinase Zap70 functions in the signaling pathway that plays an essential role in T-cell activation and development. After TCR stimulation, Zap70 is associated with the receptor complex through the interaction of its two SH2 domains with the doubly phosphorylated ITAMs. The association of Zap70 with the TCR ITAMs facilitates its autophosphorylation and the tyrosine phosphorylation of Zap70 mediated by Src family PTKs, e.g. Lck and Fyn. (Iwashima et al., 1994, Science 263:1136-1139). The recruitment of adaptor molecules like Lat and Slp76 to Zap70 in turn couple to more distal signaling pathways including Ras and PLC-gamma (Chu et al., 2003, Journal of Biology 2 (3): 2-21).
[0542] Mutations in the ZAP70 gene are associated with the selective T-cell defect (STD) and severe combined irnmunodeficiency (SCID) in human patients. The variety of point mutations, missense mutations, deletions and chromosomal rearrangements are identified in Zaρ70 all result in the same phenotype (OMIM database with genetic mutations).
[0543] Zap70 inhibitors may be useful in treating autoimmune, inflammatory, proliferative and hyperproliferative diseases, immunologically mediated diseases, AIDS, systemic lupus erythematosus, myasthenia gravis, atherosclerosis, rejection of transplanted organs or tissues, allograft rejection including acute and chronic allograft rejection, graft versus host disease, rheumathoid arthritis, psoriasis, systemic sclerosis, atopic dermatitis, eczematous dermatitis, alopecia, and inflammation of the nasal mucus membrane, including all forms of rhinitis.
III. Binding Assays
[0544] The methods of the present invention can involve assays that are able to detect the binding of compounds to a target molecule. Such binding is at a statistically significant level, preferably with a confidence level of at least 90%, more preferably at least 95, 97, 98, 99% or greater confidence level that the assay signal represents binding to the target molecule, i.e., is distinguished from background. Preferably controls are used to distinguish target binding from non-specific binding. A large variety of binding assays are known for different target types and can be used for this invention.
[0545] Binding compounds can also be characterized by their effect on the activity of the target molecule. Compounds of the present invention (i.e., compounds of Formula I, including Formulae Ia - Iz, and all sub-embodiments disclosed herein, or Formula II, including Formulae IIa - Ho, and all sub-embodiments disclosed herein) may be assayed with respect to a particular kinase to assess inhibitory concentration (IC50) or excitation concentration (EC50) of the compound with respect to that kinase. The ICs0 (or EC50) is defined as the concentration of compound at which 50% of the activity of the target kinase activity being measured is lost (or gained) relative to activity when no compound is present. Activity can be measured using methods known to those of ordinary skill in the art, e.g., by measuring any detectable product or signal produced by occurrence of an enzymatic reaction, or other activity by a protein being measured. Compounds will have an IC50 or EC50 of less than 10 μM, also less than 1 μM, also less than 100 nM, also less than 10 nM or less than 1 nM.
[0546] By "background signal" in reference to a binding assay is meant the signal that is recorded under standard conditions for the particular assay in the absence of a test compound, molecular scaffold, or ligand that binds to the target molecule. Persons of ordinary skill in the art will realize that accepted methods exist and are widely available for determining background signal.
[0547] By "standard deviation" is meant the square root of the variance. The variance is a measure of how spread out a distribution is. It is computed as the average squared deviation of each number from its mean. For example, for the numbers 1, 2, and 3, the mean is 2 and the variance is: σ2 = (l-2)2 + f2-2)2 + f3-2) 2 = 0.667 . 3
Surface PIasmon Resonance
[0548] Binding parameters can be measured using surface plasmon resonance, for example, with a BIAcore® chip (Biacore, Japan) coated with immobilized binding components. Surface plasmon resonance is used to characterize the microscopic association and dissociation constants of reaction between an sFv or other ligand directed against target molecules. Such methods are generally described in the following references which are incorporated herein by reference. VeIy F. et al., (2000) BIAcore® analysis to test phosphopeptide-SH2 domain interactions, Methods in Molecular Biology. 121 :313-21; Liparoto et al., (1999) Biosensor analysis of the interleukin-2 receptor complex, Journal of Molecular Recognition. 12:316-21; Lipschultz et al., (2000) Experimental design for analysis of complex kinetics using surface plasmon resonance, Methods. 20(3):310-8; Malmqvist, (1999) BIACORE: an affinity biosensor system for characterization of biomolecular interactions, Biochemical Society Transactions 27:335-40; Alfthan, (1998) Surface plasmon resonance biosensors as a tool in antibody engineering, Biosensors & Bioeiectronics. 13:653-63; Fivash et al., (1998) BIAcore for macromolecular interaction, Current Opinion in Biotechnology. 9:97-101; Price et al.; (1998) Summary report on the ISOBM TD-4 Workshop: analysis of 56 monoclonal antibodies against the MUCl mucin. Tumour Biology 19 Suppl 1:1-20; Malmqvist et al, (1997) Biomolecular interaction analysis: affinity biosensor technologies for functional analysis of proteins, Current Opinion in Chemical Biology. 1:378-83; O'Shannessy et al., (1996) Interpretation of deviations from pseudo-fϊrst-order kinetic behavior in the characterization of ligand binding by biosensor technology, Analytical Biochemistry. 236:275-83; Malmborg et al., (1995) BIAcore as a tool in antibody engineering, Journal of Immunological Methods. 183:7-13; Van Regenmortel, (1994) Use of biosensors to characterize recombinant proteins, Developments in Biological Standardization, 83:143-51; and O'Shannessy, (1994) Determination of kinetic rate and equilibrium binding constants for macromolecular interactions: a critique of the surface plasmon resonance literature, Current Opinions in Biotechnology. 5:65-71.
[0549] BIAcore® uses the optical properties of surface plasmon resonance (SPR) to detect alterations in protein concentration bound to a dextran matrix lying on the surface of a gold/glass sensor chip interface, a dextran biosensor matrix. In brief, proteins are covalently bound to the dextran matrix at a known concentration and a ligand for the protein is injected through the dextran matrix. Near infrared light, directed onto the opposite side of the sensor chip surface is reflected and also induces an evanescent wave in the gold film, which in turn, causes an intensity dip in the
reflected light at a particular angle known as the resonance angle. If the refractive index of the sensor chip surface is altered (e.g., by ligand binding to the bound protein) a shift occurs in the resonance angle. This angle shift can be measured and is expressed as resonance units (RUs) such that 1000 RUs is equivalent to a change in surface protein concentration of 1 ng/mm2. These changes are displayed with respect to time along the y-axis of a sensorgram, which depicts the association and dissociation of any biological reaction.
High Throughput Screening (HTS) Assays
[0550] HTS typically uses automated assays to search through large numbers of compounds for a desired activity. Typically HTS assays are used to find new drugs by screening for chemicals that act on a particular enzyme or molecule. For example, if a chemical inactivates an enzyme it might prove to be effective in preventing a process in a cell which causes a disease. High throughput methods enable researchers to assay thousands of different chemicals against each target molecule very quickly using robotic handling systems and automated analysis of results.
[0551] As used herein, "high throughput screening" or "HTS" refers to the rapid in vitro screening of large numbers of compounds (libraries); generally tens to hundreds of thousands of compounds, using robotic screening assays. Ultra high-throughput Screening (uHTS) generally refers to the high- throughput screening accelerated to greater than 100,000 tests per day.
[0552] To achieve high-throughput screening, it is advantageous to house samples on a multicontainer carrier or platform. A multicontainer carrier facilitates measuring reactions of a plurality of candidate compounds simultaneously. Multi-well microplates may be used as the carrier. Such multi-well microplates, and methods for their use in numerous assays, are both known in the art and commercially available.
[0553] Screening assays may include controls for purposes of calibration and confirmation of proper manipulation of the components of the assay. Blank wells that contain all of the reactants but no member of the chemical library are usually included. As another example, a known inhibitor (or activator) of an enzyme for which modulators are sought, can be incubated with one sample of the assay, and the resulting decrease (or increase) in the enzyme activity used as a comparator or control. It will be appreciated that modulators can also be combined with the enzyme activators or inhibitors to find modulators which inhibit the enzyme activation or repression that is otherwise caused by the presence of the known the enzyme modulator. Similarly, when ligands to a sphingolipid target are sought, known ligands of the target can be present in control/calibration assay wells.
Measuring Enzymatic and Binding Reactions During Screening Assays
[0554] Techniques for measuring the progression of enzymatic and binding reactions, e.g., in multicontainer carriers, are known in the art and include, but are not limited to, the following.
[0555] Spectrophotometric and spectrofluorometric assays are well known in the art. Examples of such assays include the use of colorimetric assays for the detection of peroxides, as described in Gordon, A. J. and Ford, R. A., (1972) The Chemist's Companion: A Handbook Of Practical Data, Techniques, And References, John Wiley and Sons, N. Y., Page 437.
[0556] Fluorescence spectrometry may be used to monitor the generation of reaction products. Fluorescence methodology is generally more sensitive than the absorption methodology. The use of fluorescent probes is well known to those skilled in the art. For reviews, see Bashford et al., (1987) Spectrophotometry and Spectrofluorometry: A Practical Approach, pp. 91-114, JRL Press Ltd.; and Bell, (1981) Spectroscopy In Biochemistry, Vol. I, pp. 155-194, CRC Press.
[0557] In spectrofluorometric methods, enzymes are exposed to substrates that change their intrinsic fluorescence when processed by the target enzyme. Typically, the substrate is nonfiuorescent and is converted to a fluorophore through one or more reactions. As a non-limiting example, SMase activity can be detected using the Amplex® Red reagent (Molecular Probes, Eugene, OR). In order to measure sphingomyelinase activity using Amplex® Red, the following reactions occur. First, SMase hydrolyzes sphingomyelin to yield ceramide and phosphorylcholine. Second, alkaline phosphatase hydrolyzes phosphorylcholine to yield choline. Third, choline is oxidized by choline oxidase to betaine. Finally, H2O2, in the presence of horseradish peroxidase, reacts with Amplex® Red to produce the fluorescent product, Resorufin, and the signal therefrom is detected using spectrofluorometry.
[0558] Fluorescence polarization (FP) is based on a decrease in the speed of molecular rotation of a fluorophore that occurs upon binding to a larger molecule, such as a receptor protein, allowing for polarized fluorescent emission by the bound ligand. FP is empirically determined by measuring the vertical and horizontal components of fluorophore emission following excitation with plane polarized light. Polarized emission is increased when the molecular rotation of a fluorophore is reduced. A fluorophore produces a larger polarized signal when it is bound to a larger molecule (i.e. a receptor), slowing molecular rotation of the fluorophore. The magnitude of the polarized signal relates quantitatively to the extent of fluorescent ligand binding. Accordingly, polarization of the "bound" signal depends on maintenance of high affinity binding.
[0559] FP is a homogeneous technology and reactions are very rapid, taking seconds to minutes to reach equilibrium. The reagents are stable, and large batches may be prepared, resulting in high reproducibility. Because of these properties, FP has proven to be highly automatable, often performed
with a single incubation with a single, premixed, tracer-receptor reagent. For a review, see Owickiet al., (1997), Application of Fluorescence Polarization Assays in High-Throughput Screening, Genetic Engineering News, 17:27.
[0560] FP is particularly desirable since its readout is independent of the emission intensity (Checovich, W. J., et al., (1995) Nature 375:254-256; Dandliker, W. B., et al., (1981) Methods in Enzymology 74:3-28) and is thus insensitive to the presence of colored compounds that quench fluorescence emission. FP and FRET (see below) are well-suited for identifying compounds that block interactions between sphingolipid receptors and their ligands. See, for example, Parker et al., (2000) Development of high throughput screening assays using fluorescence polarization: nuclear receptor-ligand-binding and kinase/phosphatase assays, J Biomol Screen 5:77-88.
[0561] Fluorophores derived from sphingolipids that may be used in FP assays are commercially available. For example, Molecular Probes (Eugene, OR) currently sells sphingomyelin and one ceramide flurophores. These are, respectively, N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s- indacene- 3-pentanoyl)sphingosyl phosphocholine (BODIPY® FL C5-sphingomyelin); N-(4,4- difiuoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene- 3-dodecanoyl)sphingosyl phosphocholine (BODIPY® FL C12-sphingomyelin); and N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s- indacene- 3-pentanoyl)sρhingosine (BODIPY® FL C5-ceramide). U.S. Patent No. 4,150,949, (Immunoassay for gentamicin), discloses fluorescein-labelled gentamicins, including fluoresceinthiocarbanyl gentamicin. Additional fluorophores may be prepared using methods well known to the skilled artisan.
[0562] Exemplary normal-and-polarized fluorescence readers include the POLARION® fluorescence polarization system (Tecan AG, Hombrechtikon, Switzerland). General multiwell plate readers for other assays are available, such as the VERSAMAX® reader and the SPECTRAMAX® multiwell plate spectrophotometer (both from Molecular Devices).
[0563] Fluorescence resonance energy transfer (FRET) is another useful assay for detecting interaction and has been described. See, e.g., Heim et al., (1996) Curr. Biol. 6:178-182; Mitra et al., (1996) Gene 173:13-17; and Selvin et al., (1995) Meth. Enzymol. 246:300-345. FRET detects the transfer of energy between two fluorescent substances in close proximity, having known excitation and emission wavelengths. As an example, a protein can be expressed as a fusion protein with green fluorescent protein (GFP). When two fluorescent proteins are in proximity, such as when a protein specifically interacts with a target molecule, the resonance energy can be transferred from one excited molecule to the other. As a result, the emission spectrum of the sample shifts, which can be measured by a fluorometer, such as a fMAX multiwell fluorometer (Molecular Devices, Sunnyvale Calif.).
[0564] Scintillation proximity assay (SPA) is a particularly useful assay for detecting an interaction with the target molecule. SPA is widely used in the pharmaceutical industry and has been described (Hanselman et al., (1997) J. Lipid Res. 38:2365-2373; Kahl et al., (1996) Anal. Biochem. 243:282- 283; Undenfriend et al., (1987) Anal. Biochem. 161:494-500). See also U.S. Patent Nos. 4,626,513 and 4,568,649, and European Patent No. 0,154,734. One commercially available system uses FLASHPLATE® scintillant-coated plates (NEN Life Science Products, Boston, MA).
[0565] The target molecule can be bound to the scintillator plates by a variety of well known means. Scintillant plates are available that are derivatized to bind to fusion proteins such as GST, His6 or Flag fusion proteins. Where the target molecule is a protein complex or a multrmer, one protein or subunit can be attached to the plate first, then the other components of the complex added later under binding conditions, resulting in a bound complex.
[0566] In a typical SPA assay, the gene products in the expression pool will have been radiolabeled and added to the wells, and allowed to interact with the solid phase, which is the immobilized target molecule and scintillant coating in the wells. The assay can be measured immediately or allowed to reach equilibrium. Either way, when a radiolabel becomes sufficiently close to the scintillant coating, it produces a signal detectable by a device such as a TOPCOUNT NXT® microplate scintillation counter (Packard BioScience Co., Meriden Conn.). If a radiolabeled expression product binds to the target molecule, the radiolabel remains in proximity to the scintillant long enough to produce a detectable signal.
[0567J In contrast, the labeled proteins that do not bind to the target molecule, or bind only briefly, will not remain near the scintillant long enough to produce a signal above background. Any time spent near the scintillant caused by random Brownian motion will also not result in a significant amount of signal. Likewise, residual unincorporated radiolabel used during the expression step may be present, but will not generate significant signal because it will be in solution rather than interacting with the target molecule. These non-binding interactions will therefore cause a certain level of background signal that can be mathematically removed. If too many signals are obtained, salt or other modifiers can be added directly to the assay plates until the desired specificity is obtained (Nichols et al., (1998) Anal. Biochem. 257:112-119).
IV. Kinase Activity Assays
[0568] A number of different assays for kinase activity can be utilized for assaying for active modulators and/or determining specificity of a modulator for a particular kinase or group or kinases. In addition to the assay mentioned in the Examples below, one of ordinary skill in the art will know of
other assays that can be utilized and can modify an assay for a particular application. For example, numerous papers concerning kinases describe assays that can be used.
[0569] Additional alternative assays can employ binding determinations. For example, this sort of assay can be formatted either in a fluorescence resonance energy transfer (FRET) format, or using an AlphaScreen (amplified /uminescent proximity homogeneous assay) format by varying the donor and acceptor reagents that are attached to streptavidin or the phosphor-specific antibody.
V. Organic Synthetic Techniques
[0570] The versatility of computer-based modulator design and identification lies in the diversity of structures screened by the computer programs. The computer programs can search databases that contain very large numbers of molecules and can modify modulators already complexed with the enzyme with a wide variety of chemical functional groups. A consequence of this chemical diversity is that a potential modulator of kinase function may take a chemical form that is not predictable. A wide array of organic synthetic techniques exist in the art to meet the challenge of constructing these potential modulators. Many of these organic synthetic methods are described in detail in standard reference sources utilized by those skilled in the art. One example of suh a reference is March, 1994, Advanced Organic Chemistry; Reactions, Mechanisms and Structure, New York, McGraw Hill. Thus, the techniques useful to synthesize a potential modulator of kinase function identified by computer-based methods are readily available to those skilled in the art of organic chemical synthesis.
[0571] Regarding the synthetic examples described herein, solvents include polar and non- polar solvents known to those of skill in the art, including polar aprotic and polar protic solvents. Polar solvents include, without limitation, protic solvents such as methanol, ethanol, isopropyl alcohol, t-butanol, n-butanol, acetic acid, formic acid or water, or aprotic solvents such as tetrahydrofuran (THF), acetonitrile, dioxane, methylene chloride, dimethylsulfoxide (DMSO), acetone, N,N-dimethylformamide (DMF), N5N- dimethylacetamide (DMA), ethyl acetate, 1,2-dimethoxyethane, 1,2-dichloroethane, chloroform, 1 ,2-dichloroethane, or pyridine. Polar solvents include a mixture of water with any of the above, or a mixture of any two or more of the above. Apolar solvents include, without limitation, toluene, benzene, chlorobenzene, xylenes and hexanes.
[0572] Regarding the synthetic examples described herein, reducing agent includes, without limitation, a reducing agent such as catalytic reducing agents using hydrogen and transition metal catalysts such as palladium, platinum, rhodium, etc.(e.g. Pt/acetic acid/H2); a mixture of trifluoroacetic acid and triethylsilane, borane tetrahydrofuran complex, diborane, borane
dimethylsulfide complex, and a combination of sodium borohydride and boron trifluoride; metals such as reduced iron, zinc powder, magnesium etc.; metal hydrogen complex compounds such as alkali metal borohydrides (for example, potassium borohydride, sodium borohydride, lithium borohydride, zinc borohydride, sodium triacetoxyborohydride, etc.), aluminum lithium hydride, etc.; metal hydrides such as sodium hydride, etc.; organic tin compounds (triphenyltin hydride, etc.); and metal salts such as nickel compounds, zinc compounds, tin compounds (for example tin(II) chloride), and samarium iodide/pivalic acid/hexamethylphorphoric triamide.
[0573] Regarding the synthetic examples described herein, oxidizing agent includes, without limitation, an oxidizing agent such as Dess-Martin reagent, TEMPO (2,2,6,6- tetramethylpiperidine-N-oxide), DDQ (2,3-Dichloro~5,6-dicyano-l,4-benzoquinone), PDC (pyridinium dichromate), PCC (pyridinium chlorochromate), Pyridine. SO3, Chromium trioxide, p-nitroperbenzoic acid, magnesium monoperoxyphthalate, sodium periodate, potassium periodate, hydrogen peroxide, urea peroxide, alkali metal bromates, cumene hydroperoxide, tert-butyl peroxide, peracids such as performic acid, peracetic acid, pertrifluoroacetic acid, perbenzoic acid, m-chloroperbenzoic acid, o-carboxyperbenzoic acid and the like; sodium metaperiodate, bichromic acid; bichromates such as sodium bichromate, potassium bichromate; permanganic acid; permanganates such as potassium permanganate, sodium permanganate; and lead salts such as lead tetraacetate.
VI. Alternative Compound Forms or Derivatives
[0574] Compounds contemplated herein are described with reference to both generic formulae and specific compounds. In addition, invention compounds may exist in a number of different forms or derivatives, all within the scope of the present invention. These include, for example, tautomers, stereoisomers, racemic mixtures, regioisomers, salts, prodrugs (e.g., carboxylic acid esters), solvated forms, different crystal forms or polymorphs, and active metabolites.
(a) Tautomers, Stereoisomers, Regioisomers, and Solvated Forms
[0575] It is understood that some compounds may exhibit tautomerism. In such cases, the formulae provided herein expressly depict only one of the possible tautomeric forms. It is therefore to be understood that the formulae provided herein are intended to represent any tautomeric form of the depicted compounds and are not to be limited merely to the specific tautomeric form depicted by the drawings of the formulae.
[0576] Likewise, some of the compounds according to the present invention may exist as stereoisomers, i.e. having the same atomic connectivity of covalently bonded atoms yet differing in the spatial orientation of the atoms. For example, compounds may be optical stereoisomers, which contain one or more chiral centers, and therefore, may exist in two or more stereoisomeric forms (e.g. enantiomers or diastereomers). Thus, such compounds may be present as single stereoisomers (i.e., essentially free of other stereoisomers), racemates, and/or mixtures of enantiomers and/or diastereomers. As another example, stereoisomers include geometric isomers, such as cis- or trans- orientation of substituents on adjacent carbons of a double bond. All such single stereoisomers, racemates and mixtures thereof are intended to be within the scope of the present invention. Unless specified to the contrary, all such steroisomeric forms are included within the formulae provided herein.
[0577] In some embodiments, a chiral compound of the present invention is in a form that contains at least 80% of a single isomer (60% enantiomeric excess ("e.e.") or diastereomeric excess ("d.e.")), or at least 85% (70% e.e. or d.e.), 90% (80% e.e. or d.e.), 95% (90% e.e. or d.e.), 97.5% (95% e.e. or d.e.), or 99% (98% e.e. or d.e.). As generally understood by those skilled in the art, an optically pure compound having one chiral center is one that consists essentially of one of the two possible enantiomers (i.e., is enantiomerically pure), and an optically pure compound having more than one chiral center is one that is both diastereomerically pure and enantiomerically pure. In some embodiments, the compound is present in optically pure form.
[0578] For compounds in which synthesis involves addition of a single group at a double bond, particularly a carbon-carbon double bond, the addition may occur at either of the double bond-linked atoms. For such compounds, the present invention includes both such regioisomers.
[0579] Additionally, the formulae are intended to cover solvated as well as unsolvated forms of the identified structures. For example, the indicated structures include both hydrated and non-hydrated forms. Other examples of solvates include the structures in combination with a suitable solvent such as isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, or ethanolamine.
(b) Prodrugs and Metabolites
[0580] In addition to the present formulae and compounds described herein, the invention also includes prodrugs (generally pharmaceutically acceptable prodrugs), active metabolic derivatives (active metabolites), and their pharmaceutically acceptable salts.
[0581] Prodrugs are compounds or pharmaceutically acceptable salts thereof which, when metabolized under physiological conditions or when converted by solvolysis, yield the desired active compound. Prodrugs include, without limitation, esters, amides, carbamates, carbonates, ureides,
solvates, or hydrates of the active compound. Typically, the prodrug is inactive, or less active than the active compound, but may provide one or more advantageous handling, administration, and/or metabolic properties. For example, some prodrugs are esters of the active compound; during metabolysis, the ester group is cleaved to yield the active drug. Also, some prodrugs are activated enzymatically to yield the active compound, or a compound which, upon further chemical reaction, yields the active compound.
[0582] In this context, a common example or a prodrug is an alkyl ester of a carboxylic acid. Relative to compounds of Formula I, further examples include, without limitation, an amide or carbamate derivative at the 1 -position nitrogen of the azaindole core.
[0583] As described in The Practice of Medicinal Chemistry, Ch. 31-32 (Ed. Wermuth, Academic Press, San Diego, CA, 2001), prodrugs can be conceptually divided into two non-exclusive categories, bioprecursor prodrugs and carrier prodrugs. Generally, bioprecursor prodrugs are compounds that are inactive or have low activity compared to the corresponding active drug compound, that contain one or more protective groups and are converted to an active form by metabolism or solvolysis. Both the active drug form and any released metabolic products should have acceptably low toxicity. Typically, the formation of active drug compound involves a metabolic process or reaction that is one of the follow types:
[0584] Oxidative reactions: Oxidative reactions are exemplified without limitation to reactions such as oxidation of alcohol, carbonyl, and acid functionalities, hydroxylation of aliphatic carbons, hydroxylation of alicyclic carbon atoms, oxidation of aromatic carbon atoms, oxidation of carbon- carbon double bonds, oxidation of nitrogen-containing functional groups, oxidation of silicon, phosphorus, arsenic, and sulfur, oxidative N-dealkylation, oxidative O- and S-dealkylation, oxidative deamination, as well as other oxidative reactions.
[0585] Reductive reactions: Reductive reactions are exemplified without limitation to reactions such as reduction of carbonyl functionalities, reduction of alcholo functionalities and carbon-carbon double bonds, reduction of nitrogen-containing function groups, and other reduction reactions.
[0586] Reactions without change in the oxidation state: Reactions without change in the state of oxidation are exemplified without limitation to reactions such as hydrolysis of esters and ethers, hydrolytic cleavage of carbon-nitrogen single bonds, hydrolytic cleavage of non-aromatic heterocycles, hydration and dehydration at multiple bonds, new atomic linkages resulting from dehydration reactions, hydrolytic dehalogenation, removal of hydrogen halide molecule, and other such reactions.
[0587] Carrier prodrugs are drug compounds that contain a transport moiety, e.g., that improves uptake and/or localized delivery to a site(s) of action. Desirably for such a carrier prodrug, the linkage between the drug moiety and the transport moiety is a covalent bond, the prodrug is inactive or less active than the drug compound, the prodrug and any release transport moiety are acceptably non-toxic. For prodrugs where the transport moiety is intended to enhance uptake, typically the release of the transport moiety should be rapid. In other cases, it is desirable to utilize a moiety that provides slow release, e.g., certain polymers or other moieties, such as cyclodextrins. (See, e.g., Cheng et al., U.S. Patent Publ. No. 20040077595, App. No. 10/656,838, incorporated herein by reference). Such carrier prodrugs are often advantageous for orally administered drugs. Carrier prodrugs can, for example, be used to improve one or more of the following properties: increased lipophilicity, increased duration of pharmacological effects, increased site-specificity, decreased toxicity and adverse reactions, and/or improvement in drug formulation (e.g., stability, water solubility, suppression of an undesirable organoleptic or physiochemical property). For example, lipophilicity can be increased by esterifϊcation of hydroxyl groups with lipophilic carboxylic acids, or of carboxylic acid groups with alcohols, e.g., aliphatic alcohols. Wermuth, supra.
[0588] Prodrugs may proceed from prodrug form to active form in a single step or may have one or more intermediate forms which may themselves have activity or may be inactive.
[0589] Metabolites, e.g., active metabolites overlap with prodrugs as described above, e.g., bioprecursor prodrugs. Thus, such metabolites are pharmacologically active compounds or compounds that further metabolize to pharmacologically active compounds that are derivatives resulting from metabolic process in the body of a subject. Of these, active metabolites are such pharmacologically active derivative compounds. For prodrugs, the prodrug compound is generally inactive or of lower activity than the metabolic product. For active metabolites, the parent compound may be either an active compound or may be an inactive prodrug.
[0590] Prodrugs and active metabolites may be identified using routine techniques know in the art. See, e.g., Bertolini et al, 1997, J Med Chem 40:2011-2016; Shan et al., J Pharm Sci 86:756-757; Bagshawe, 1995, DrugDev Res 34:220-230; Wermuth, supra.
(c) Pharmaceutically acceptable salts
[0591] Compounds can be formulated as or be in the form of pharmaceutically acceptable salts. Contemplated pharmaceutically acceptable salt forms include, without limitation, mono, bis, tris, tetrakis, and so on, Pharmaceutically acceptable salts are non-toxic in the amounts and concentrations at which they are administered. The preparation of such salts can facilitate the pharmacological use by altering the physical characteristics of a compound without preventing it from
exerting its physiological effect. Useful alterations in physical properties include lowering the melting point to facilitate transmucosal administration and increasing the solubility to facilitate administering higher concentrations of the drug.
[0592] Pharmaceutically acceptable salts include acid addition salts such as those containing sulfate, chloride, hydrochloride, fumarate, maleate, phosphate, sulfamate, acetate, citrate, lactate, tartrate, methanesulfonate, ethanesulfonate, benzenesulfonate,/>-toluenesulfonate, cyclohexylsulfamate and quinate. Pharmaceutically acceptable salts can be obtained from acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid,j9-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
[0593] Pharmaceutically acceptable salts also include basic addition salts such as those containing benzathine, chloroprocaine, choline, diethanolamine, ethanolamine, t-butylamine, ethylenediamine, meglumine, procaine, aluminum, calcium, lithium, magnesium, potassium, sodium, ammonium, alkylamine, and zinc, when acidic functional groups, such as carboxylic acid or phenol are present. For example, see Remington's Pharmaceutical Sciences, 19th ed., Mack Publishing Co., Easton, PA, Vol. 2, p. 1457, 1995. Such salts can be prepared using the appropriate corresponding bases.
[0594] Pharmaceutically acceptable salts can be prepared by standard techniques. For example, the free-base form of a compound can be dissolved in a suitable solvent, such as an aqueous or aqueous- alcohol solution containing the appropriate acid and then isolated by evaporating the solution. In another example, a salt can be prepared by reacting the free base and acid in an organic solvent.
[0595] Thus, for example, if the particular compound is a base, the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
[0596] Similarly, if the particular compound is an acid, the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like. Illustrative examples of suitable salts include organic salts derived
from amino acids, such as L-glycine, L-lysine, and L-arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as hydroxyethylpyrrolidine, piperidine, morpholine or piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium. Further examples of pharmaceutically acceptable salts of compounds of Formulae I-iπ include, without limitation, the mono-sodium and bis-potassium salts thereof.
[0597] The pharmaceutically acceptable salt of the different compounds may be present as a complex. Examples of complexes include 8-chlorotheophylline complex (analogous to, e.g., dimenhydrinate: diphenhydramine 8-chlorotheophylline (1:1) complex; Dramamine) and various cyclodextrin inclusion complexes.
[0598] Unless specified to the contrary, specification of a compound herein includes pharmaceutically acceptable salts of such compound.
(d) Polymorphic forms
[0599] In the case of agents that are solids, it is understood by those skilled in the art that the compounds and salts may exist in different crystal or polymorphic forms, all of which are intended to be within the scope of the present invention and specified formulae.
VIL Administration
[0600] The methods and compounds will typically be used in therapy for human subjects. However, they may also be used to treat similar or identical indications in other animal subjects. In this context, the terms "subject," "animal subject," and the like refer to human and non-human vertebrates, e.g. mammals, such as non-human primates, sports and commercial animals, e.g., equines, bovines, porcines, ovines, rodents, and pets, e.g., canines and felines.
[0601] Suitable dosage forms, in part, depend upon the use or the route of administration, for example, oral, transdermal, transmucosal, inhalant or by injection (parenteral). Such dosage forms should allow the compound to reach target cells. Other factors are well known in the art, and include considerations such as toxicity and dosage forms that retard the compound or composition from exerting its effects. Techniques and formulations generally may be found in The Science and Practice of Pharmacy, 21st edition, Lippincott, Williams and Wilkins, Philadelphia, PA, 2005 (hereby incorporated by reference herein).
[0602] Compounds of the present invention, i.e. Formula I, can be formulated as pharmaceutically acceptable salts.
[0603] Carriers or excipients can be used to produce compositions. The carriers or excipients can be chosen to facilitate administration of the compound. Examples of carriers include calcium carbonate, calcium phosphate, various sugars such as lactose, glucose, or sucrose, or types of starch, cellulose derivatives, gelatin, vegetable oils, polyethylene glycols and physiologically compatible solvents. Examples of physiologically compatible solvents include sterile solutions of water for injection (WFI), saline solution, and dextrose.
[0604] The compounds can be administered by different routes including intravenous, intraperitoneal, subcutaneous, intramuscular, oral, transmucosal, rectal, transdermal, or inhalant. Oral administration is preferred. For oral administration, for example, the compounds can be formulated into conventional oral dosage forms such as capsules, tablets, and liquid preparations such as syrups, elixirs, and concentrated drops.
[0605] For inhalants, compounds of the invention may be formulated as dry powder or a suitable solution, suspension, or aerosol. Powders and solutions may be formulated with suitable additives known in the art. For example, powders may include a suitable powder base such as lactose or starch, and solutions may comprise propylene glycol, sterile water, ethanol, sodium chloride and other additives, such as acid, alkali and buffer salts. Such solutions or suspensions may be administered by inhaling via spray, pump, atomizer, or nebulizer, and the like. The compounds of the invention may also be used in combination with other inhaled therapies, for example corticosteroids such as fluticasone proprionate, beclomethasone dipropionate, triamcinolone acetonide, budesonide, and mometasone furoate; beta agonists such as albuterol, salmeterol, and formoterol; anticholinergic agents such as ipratroprium bromide or tiotropium; vasodilators such as treprostinal and iloprost; enzymes such as DNAase; therapeutic proteins; immunoglobulin antibodies; an oligonucleotide, such as single or double stranded DNA or RNA, siRNA; antibiotics such as tobramycin; muscarinic receptor antagonists; leukotriene antagonists; cytokine antagonists; protease inhibitors; cromolyn sodium; nedocril sodium; and sodium cromoglycate.
[0606] Pharmaceutical preparations for oral use can be obtained, for example, by combining the active compounds with solid excipients, optionally grinding a resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores. Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose (CMC), and/or polyvinylpyrrolidone (PVP: povidone). If desired, disintegrating agents may be added, such as the cross — linked polyvinylpyrrolidone, agar, or alginic acid, or a salt thereof such as sodium alginate.
[0607J Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain, for example, gum arabic, talc, polyvinylpyrrolidone, carbopol gel, polyethylene glycol (PEG), and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dye-stuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
[0608] Pharmaceutical preparations that can be used orally include push-fit capsules made of gelatin ("gelcaps"), as well as soft, sealed capsules made of gelatin, and a plasticizer, such as glycerol or sorbitol. The push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. Ia soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols (PEGs). In addition, stabilizers may be added.
[0609] Alternatively, injection (parenteral administration) may be used, e.g., intramuscular, intravenous, intraperitoneal, and/or subcutaneous. For injection, the compounds of the invention are formulated in sterile liquid solutions, preferably in physiologically compatible buffers or solutions, such as saline solution, Hank's solution, or Ringer's solution. In addition, the compounds may be formulated in solid form and redissolved or suspended immediately prior to use. Lyophilized forms can also be produced.
[0610] Administration can also be by transmucosal, topical, or transdermal means. For transmucosaϊ, topical or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art, and include, for example, for transmucosal administration, bile salts and fusidic acid derivatives. In addition, detergents may be used to facilitate permeation. Transmucosal administration, for example, may be through nasal sprays or suppositories (rectal or vaginal). The topical compositions of this invention are formulated preferably as oils, creams, lotions, ointments and the like by choice of appropriate carriers known in the art. Suitable carriers include vegetable or mineral oils, white petrolatum (white soft paraffin), branched chain fats or oils, animal fats and high molecular weight alcohol (greater than C12). The preferred carriers are those in which the active ingredient is soluble. Emulsifiers, stabilizers, humectants and antioxidants may also be included as well as agents imparting color or fragrance, if desired. Creams for topical application are preferably formulated from a mixture of mineral oil, self-emulsifying beeswax and water in which mixture the active ingredient, dissolved in a small amount solvent (e.g., an oil), is admixed. Additionally, administration by transdermal means may comprise a transdermal patch or dressing such as a bandage impregnated with an active ingredient and optionally one or more carriers or diluents known in the art. To be administered in the
form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen.
[0611] The amounts of various compounds to be administered can be determined by standard procedures taking into account factors such as the compound IC50, the biological half-life of the compound, the age, size, and weight of the subject, and the disorder associated with the subject. The importance of these and other factors are well known to those of ordinary skill in the art. Generally, a dose will be between about 0.01 and 50 mg/kg, preferably 0.1 and 20 mg/kg of the subject being treated. Multiple doses may be used.
[0612] The compounds of the invention may also be used in combination with other therapies for treating the same disease. Such combination use includes administration of the compounds and one or more other therapeutics at different times, or co-administration of the compound and one or more other therapies. In some embodiments, dosage may be modified for one or more of the compounds of the invention or other therapeutics used in combination, e.g., reduction in the amount dosed relative to a compound or therapy used alone, by methods well known to those of ordinary skill in the art.
[0613] It is understood that use in combination includes use with other therapies, drugs, medical procedures etc., where the other therapy or procedure may be administered at different times (e.g. within a short time, such as within hours (e.g. 1, 2, 3, 4-24 hours), or within a longer time (e.g. 1-2 days, 2-4 days, 4-7 days, 1 -4 weeks)) than a compound of the present invention, or at the same time as a compound of the invention. Use in combination also includes use with a therapy or medical procedure that is administered once or infrequently, such as surgery, along with a compound of the invention administered within a short time or longer time before or after the other therapy or procedure. In some embodiments, the present invention provides for delivery of compounds of the invention and one or more other drug therapeutics delivered by a different route of administration or by the same route of administration. The use in combination for any route of administration includes delivery of compounds of the invention and one or more other drug therapeutics delivered by the same route of administration together in any formulation, including formulations where the two compounds are chemically linked in such a way that they maintain their therapeutic activity when administered. In one aspect, the other drug therapy may be co-administered with one or more compounds of the invention. Use in combination by co-administration includes administration of co- formulations or formulations of chemically joined compounds, or administration of two or more compounds in separate formulations within a short time of each other (e.g. within an hour, 2 hours, 3 hours, up to 24 hours), administered by the same or different routes. Co-administration of separate formulations includes co-administration by delivery via one device, for example the same inhalant device, the same syringe, etc., or administration from separate devices within a short time of each other. Co-formulations of compounds of the invention and one or more additional drug therapies
delivered by the same route includes preparation of the materials together such that they can be administered by one device, including the separate compounds combined in one formulation, or compounds that are modified such that they are chemically joined, yet still maintain their biological activity. Such chemically joined compounds may have a linkage that is substantially maintained in vivo, or the linkage may break down in vivo, separating the two active components.
EXAMPLES
[0614] Examples related to the present invention are described below. In most cases, alternative techniques can be used. The examples are intended to be illustrative and are not limiting or restrictive to the scope of the invention. In some examples, the mass spectrometry result indicated for a compound may have more than one value due to the isotope distribution of an atom in the molecule, such as a compound having a bromo or chloro substituent.
Example 1: Synthesis of propane-1-sulfonic acid [3-(5-bromo-lH-pyrroIo[2,3-b]pyridine-3- carbonyl)-2,4-difluoro-phenyI]-amide P-0773 and related compounds.
[0615] Compound P-0773 was synthesized in five steps from 2,4-difluoro-phenylamine 42 as shown in Scheme 13.
Scheme 13
Step 1 -Preparation of 3-amino-2,6-difluoro-benzoic acid benzyl ester (43): [0616] To 2,4-difluoro-phenylamine (42, 5.11 mL, 50.7 mmol) in tetrahydrofuran (250 niL), cooled with dry ice/acetone bath under an atmosphere of nitrogen, was added n-butyllithium (1.60 M in hexane, 34.0 mL, 54.4 mmol) slowly. After 30 minutes, l,2-Bis-(chloro-dimethyl-silanyl)-ethane (11.5 g, 53.4 mmol) dissolved in tetrahydrofuran (40.0 mL) was added to the reaction slowly. After 1 hour, n-butyllithium (1.60 M in hexane, 31.9 mL, 51.0 mmol) was added slowly to the reaction. The reaction was stirred at -78 0C for 30 minutes and then allowed to warm to room temperature over 40 minutes. The reaction was cooled to -78 0C, followed by addition of n-butyllithium (1.60 M in
hexane, 35.1 mL, 56.2 mmol) slowly. After 70 minutes, benzyl chlorofoπnate (7.97 mL, 55.8 mmol) was added to the reaction. The reaction mixture was stirred at -78 0C overnight followed by addition of 2 N HCl (120 mL). The reaction was allowed to warm to room temperature for 2 hours. The organic layer was separated. The aqueous layer was basified with potassium carbonate and extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate/hexane 20%) to give a colorless oil (43, 10.6 g, 79.7%). MS(ESI) PVRH+I+ = 264.1.
Step 2 ~ Preparation of2,6-difluoro-3~(propane-l-sulfonylamino)-benzoic acid benzyl ester (44): [0617] To 3-amino-2,6-difluoro-benzoic acid benzyl ester (43, 6.00 g, 22.8 mmol) in methylene chloride (150 mL) was added pyridine (2.76 mL, 34.2 mmol) and propane- 1-sulfonyl chloride (3.80 mL, 33.8 mmol). The reaction was stirred at room temperature overnight. Then the reaction was poured into water, and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated with silica gel column chromatography to give a colorless oil (44, 7.0 g, 83.1%). MS(ESI) [M+H"1"]"*" = 370.1.
Step 3 — Preparation of2,6-difluoro-3-(propane-l-sulfonylamino)-benzoic acid (45): [0618] To 2,6-difluoro-3 -(propane- 1 -sulfonylamino)-benzoic acid ben∑yl ester (44, 2.0 g, mmol) in methanol (30 mL) was added 20% palladium hydroxide on carbon (100 mg). The reaction was stirred under hydrogen at 1 atm for 15 minutes. The reaction was filtrated and concentrated to give white solid 45 that was used in the next step.
Step 4 — Preparation of2,6-difluoro-3-(propane-l-sulfonylamino)-benzoyl chloride (46): [0619] To 2,6-difluoro-3-(propane-l -sulfonylamino)-benzoic acid (45, 1.50 g, 5.4 mmol) was added toluene (7.0 mL) and thionyl chloride (15.0 mL, 0.21 mmol). The reaction was heated to reflux for 3 hours. The reaction was concentrated to give crude compound that was used in the next step.
Step 5- Preparation of propane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- 2, 4-difluoro-phenyl] -amide (P-0773):
[0620] To aluminum trichloride (8.89 g, 66.7 mmol) was added methylene chloride (150 mL) under an atmosphere of nitrogen below 5 0C. Into this, 5-bromo-7-azaindole (67, 1.64 g, 8.34 mmol) in methylene chloride (20 mL) was added. The reaction was stirred for 60.0 minutes and 2,6-difluoro-3- (propane-l-sulfonylamino)-benzoyl chloride (46, 3.50 g, 11.8 mmol) in methylene chloride (20 mL) was added. The reaction was stirred for 6 hours and warmed to room temperature overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated by
silica gel column chromatography (methylene chloride/methanol 5%) to give a white solid (P-0773, 1.2 g, 31.4%). MS(ESI) [M+H+]+ = 460.0, 462.0.
[0621] N-[3-(5-Bromo-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-4-chloro-2-fluoro-phenyl]- methanesulfonamide P-0868
was prepared following the protocol of Scheme 13, substituting propane- 1-sulfonyl chloride with methanesulfonyl chloride in Step 2 and 2,4-difluoro-ρhenylamine with 4-chloro-2-fluoro-phenylamine in Step 1. MS (ESI) [M - H
+]
" = 443.9, 445.9.
[0622] N-[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]- methanesulfonamide P-0162
was prepared following the protocol of Scheme 13, substituting propane-1-sulfonyl chloride with methanesulfonyl chloride hi Step 2. This was isolated and reacted further (50 mg, 0.10 mrnoi) in methanol (20 mL) by adding 20% palladium hydroxide on carbon (53 mg). The reaction was stirred under hydrogen at 40 psi for 12 hours. The reaction mixture was filtered and concentrated to give product methanesulfonic acid [2,4-difluoro-3-lH-pyrrolo-[2,3-b]pyridrne-3-carbonyl)-phenyl]-amide P-0811 (40 mg, 95%).
MS(ESI)[M +H+] " = 352.3.
[0623] N-[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]- benzenesulfonamide P-0798 and N-[3-(5-bromo-lH-pyrrolo[2,3-b]pyridnie-3-carbonyl)-2,4-difluoro- phenyl] -3 -fluoro-benzenesulf onamide
were prepared following the protocol of Scheme 13, substituting propane- 1-sulfonyl chloride with benzenesulfonyl chloride and 3-fluoro-benzenesulfonyl chloride, respectively, in Step 2. P-0798 MS(ESI) [M - H
+]
" = 489.9, 491.9.
[0624] Propane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-4-chloro-2- fluoro-phenyl] -amide P-0805
was prepared following the protocol of Scheme 13, substituting 2,4-difluoro-phenylamine with A- chlQro-2-fiuoro-phenylamine in Step 1. MS(ESI) [M - H
+]
" = 471.9, 473.9.
[0625] Propane- 1 -sulfonic acid [2,4-difluoro-3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- amide P-0007
was prepared following the protocol of Scheme 13, substituting 5-bromo-7-azaindole with 7- azaindole in Step 5. MS(ESI) [M + H
+J
+ = 380.1.
[0626] Propane- 1 -sulfonic acid [2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide T-0806
was prepared following the protocol of Scheme 13, substituting 5-bromo-7-azaindole with 5- methoxy-7-azaindole 104 (prepared as described in Example 16) in Step 5. MS(ESI) [M - H
+]
' = 410.1.
[0627] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-methoxy-phenyl)-methanone P-0265
was prepared using the protocol of steps 4 and 5 of Scheme 13, substituting 2,6-difluoro-3-(propane- l-sulfonylamino)-benzoic acid with 2-fluoro-3-methoxy-benzoic acid in Step 4. MS (ESI) [M - H
+]
" = 347, 349.
[0628] N-[3-(5-Bromo-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-4-fluoro-phenyl]- methanesulfonamide P-0170
was prepared using the protocol of steps 2, 4, and 5 of Scheme 13, substituting 3-amino-2,6-difluoro- benzoic acid benzyl ester with 5-amino-2-fluoro-benzoic acid and ρropaπe-1 -sulfonyl chloride with methanesulfonyl chloride in Step 2, to provide the acid that is carried through in Step 4. MS (ESI) [M - H
+]
" = 410.0, 412.0.
[0629] N-[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2-methyl-phenyl]- methanesulfonamide P-0180
was prepared using the protocol of steps 2, 4, and 5 of Scheme 13, substituting 3-amino-2,6-difluoro- benzoic acid benzyl ester with 3-amino-2-methyl-benzoic acid and propane- 1 -sulfonyl chloride with methanesulfonyl chloride in Step 2, to provide the acid that is carried through in Step 4. MS (ESI) [M - H
+]
" = 405.9, 407.9.
[0630] (5-Bromo-lH-pvrrolo[2,3-b]pyridin-3-yl)-(3-chloro-2-fluoro-phenyl)-methanone P-0299
was prepared using the protocol of Step 5 of Scheme 13, substituting 2, 6-difluoro-3 -(propane- 1 - sulfonylamino)-benzoyl chloride with 3-chloro-2-fluoro-benzoyl chloride. MS (ESI) [M - H
+]
" = 350.9, 352.9.
Example 2: Synthesis of Propane-l-sulfonic acid [3-(5-bronio-lH-pyrrolo[2,3-b]pyridine-3- carbonyI)-2-fluoro-phenyl] -amide P-0955 and related compounds.
[0631] Compound P-0955 was synthesized in six steps from 4-chloro-2-fluoro-phenylamine 47 as shown in Scheme 14.
Scheme 14
Step 1 - Preparation ofS-Amino-δ-chloro-l-fluoro-benzoic acid benzyl ester (48): [0632] To 4-chloro-2-fluoro-phenylamine (47, 6.30 niL, 57.0 mmol) in tetrahydrofuran (300 niL), cooled with dry ice/acetone bath under an atmosphere of nitrogen, was added n-butyllithium (2.500 M in hexane, 24.4 niL) slowly. After 20 minutes, l,2-Bis-(chloro-dimethyl-silanyl)-ethane (12.9 g, 60.0 mmol) dissolved in tetrahydrofuran (40.0 mL) was added to the reaction slowly. After 1 hour, n- butyllithium (2.50 M in hexane, 25.0 mL) was added slowly to the reaction. The reaction was stirred at -78 0C for 20 minutes and allowed to warm to room temperature over 60 minutes. The reaction was cooled to -78 0C, followed by addition of n-butyllithium (2.50 M in hexane, 26.0 mL) slowly. After 80 minutes, benzyl chloroformate (10.0 mL, 70.0 mmol) was added to the reaction. The reaction mixture was stirred at -78 0C overnight followed by addition of water (80 mL) and concentrated hydrochloric acid (25 mL). The reaction was allowed to warm to room temperature for 2 hours. The organic layer was separated and the aqueous layer was basified with potassium carbonate and extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate/hexane 20%) to give a colorless oil (48, 12.5 g, 78.3%). MS(ESI) [M+Iff = 280.0.
Step 2 — Preparation of6-chloro-2~fluoro-3-(propane-l-sulfonylamino)-benzoic acid benzyl ester
(49):
[0633] To 3-amino-6-chloro-2-fluoro-benzoic acid benzyl ester (48, 1.20 g, 4.3 mmol) in methylene chloride (28 mL) was added pyridine (0.52 mL, 6.4 mmol) and propane-1-sulfonyl chloride (0.685 g,
4.8 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated with silica gel column chromatography to give a colorless oil (49, 960 mg, 58.0%). MS(ESI) [M-H+]" = 384.1.
Step 3 - Preparation of6-chloro-2-fluoro-3-(propane-l-sulfonylanιino)-be?ιzoic acid (115): [0634] To 6-chloro-2-fluoro-3 -(propane- 1 -sulfonylamino)-benzoic acid benzyl ester (49, 6.00 g, 15.6 mmol) in tetrahydrofuran (100 mL) was added 1.0 M aqueous potassium hydroxide (100 mL). The reaction was heated to reflux overnight. The reaction was poured into water, acidified to pH 2 with 1 N hydrochloric acid and extracted with ethyl acetate. The organic portion was dried over anhydrous sodium sulfate, filtrated and concentrated to give a white solid 115 (3.95 g, 85.8%).
Step 4 - Preparation of2-fluoro-3~(propane-l-sulfonylamino)-benzoic acid (50): [0635] To 6-chloro-2-fluoro-3-(propane-l-sulfonylamino)-benzoic acid (115, 0.69 g, 2.3 mmol) in methanol (10 mL) was added 20% palladium hydroxide on carbon (200 mg). The reaction was stirred under hydrogen at 50 psi for 2 hours. The reaction was filtrated and concentrated to give white solid 50 that was used in the next step. MS(ESI) [M-H+]"=260.1.
Step 5 - Preparation of2-fluoro-3-(propane-l-sulfonylamino)-benzoyl chloride (51): [0636] To 2-fluoro -3-(propane-l -sulfonylamino)-benzoic acid (50, 1.17 g, 4.5 mmol) was added thionyl chloride (10.0 mL). The reaction was heated to reflux for 3 hours. The reaction was concentrated to give crude compound 51 that was used in the next step.
Step 6- Preparation of Propane- 1 -sulfonic acid [3-(5~bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- 2-fluoro-phenylj '-amide (P-0955):
[0637] Aluminum trichloride (2.52 g, 18.9 mmol) and methylene chloride (60.0 mL) were combined under an atmosphere of nitrogen. Into the reaction mixture, was added 5-bromo-7-azaindole (67, 630.0 mg, 3.2 mmol) in methylene dichloride (20.0 mL). The reaction was stirred at room temperature for 70 minutes, then 2-fluoro-3-(propane-l-sulfonylamino)-benzoyl chloride (51, 0.749 g, 2.68 mmol) in methylene chloride (20 mL) was added. The reaction was stirred at room temperature for 2 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give compound P-0955 (65 mg, 5.5%). MS(ESI) [M+HT = 440.2, 442.2.
[0638] Using the protocol of Scheme 14, substituting 5-bromo-azaindole with either 5-chloro-7- azaindole (80, prepared as described in Example 9), 5-fluoro-7-azaindole (81, prepared as described in Example 9) or 7-azaindole in Step 6, propane-1 -sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-
3-carbonyl)-2-fluoro-ρhenyl]-amide P-1013 (MS(ESI) [M - H+]' = 394.1), propane-1 -sulfonic acid [2- fluoro-3-(5-fluoro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-amide P-1028 (MS(ESI) [M - H+]" = 378.1), and propane- 1 -sulfonic acid [2-fluoro-3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-plienyl]- amide P-1056 (MS(ESI) [M + H+]+ = 362.2) were prepared, respectively;
Example 3: Propane-l-sulfonic acid [2,4-difihioro-3-(5-pyridin-3-yI-lH-pym>lo[2,3-b]pyridine- 3-carbonyl)-phenyI]-amide P-0088 and related compounds.
[0639] Compound P-0088 was synthesized in one step from propane-l-sulfonic acid [3-(5-bromo- lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-amide P-0773 by Suzuki coupling ((Muyaura and Suzuki, Chem. Rev. 1995, 95:2457) as shown in Scheme 15.
Scheme 15
Step 1 -Preparation of propane-l-sulfonic acid [2,4-difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3- b]ρyridine-3-carbonyl)-phenyl] -amide (P-0088) :
[0640] To propane-1 -sulfonic acid [3-(5-bromo-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenyl] -amide (P-0773, prepared as described in Example 1, 65.0 mg, 0.14 mmoϊ) in acetonitrile (4.0 mL) was added pyridine-3-boronic acid (609, 25.0 mg, 0.20 mmol), tetrakis (triphenylρhosphine)palladium(O) (11 mg, 1.0% mmol) and aqueous potassium carbonate (1.0 M, 2.0 mL). The reaction was heated to 160 0C for 10 minutes in a CEM Discover microwave instrument. The reaction was poured into water, and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtrated and concentrated. The desired compound was isolated by silica gel column chromatography (methylene chloride/methanol 5%) to give a white solid (P-0088, 30 mg, 46.9%). MS(ESI) [M+H+]+= 457. 2.
[0641] Additional compounds were prepared following the protocol of Scheme 15, optionally replacing proρane-1-sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide P-0773 with an appropriate 5-bromo azaindole and/or ρyridine-3-boronic acid 609
with an appropriate boronic acid or boronic acid ester. The 5-bromo azaindole used was synthesized as described in either Example 1, 2, 5, 10, or 73. The following compounds were made following this procedure:
N-[2-Methyl-3-(5-phenyl- 1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-methanesulfonamide (P-0082),
N-[2-Methyl-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- methanesulfonamide (P-0121),
N-[4-Fluoro-3-(5-pyridin-3-yl- 1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- methanesulfonamide (P-0079),
N-[4-Fluoro-3-(5-phenyl-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-phenyl]-methanesulfonamide
(P-0308),
N-{4-Fluoro-3-[5-(3-methanesulfonylamino-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- phenyl }-methanesulfonamide (P-0156),
N-{4-Fluoro-3-[5-(3-methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridine-
3 -carbonyl] -phenyl} -methanesulfonamide (P-0297),
3-[3-(2-Fluoro-5-methanesulfonylamino-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-benzamide (P-
0228),
3-[3-(2-Fluoro-5-hydroxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-benzamide (P-0037),
(2-Fluoro-5-hydroxy-phenyl)-[5-(3-methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]- methanone (P-0008),
N-{3-[3-(2-Fluoro-5-hydroxy-benzoyl)-lH-ρyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-0716),
N-[2,4-Difiuoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- methanesulfonamide (P-0700),
N-{4-Chloro-2-fluoro-3-[5-(3-methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b] pyridine-3-carbonyl]- phenyl} -methanesulfonamide (P-0841),
N-[4-Chloro-2-fluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- methanesulfonamide (P-0734),
N-{4-Chloro-2-fluoro-3-[5-(3-methanesulfonylamino-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl} -methanesulfonamide (P-0745),
N-{2,4-Difluoro-3-[5-(3-methanesulfonylamino-phenyl)-lH-pyrrolo[2,3-b3 pyridine-3-carbonyl]- phenyl}-methanesulfonamide (P-0746),
3-[3-(2,6-Difluoro-3-methanesulfonylamino-benzoyl)-lH-pyrrolo[2,3-b]pyridm-5-yl]-benzamide
(P-0721),
N-[2,4-Difluoro-3-(5-ρhenyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]- methanesulfonamide (P-0184),
N-[2,4-Difluoro-3-(5-pyridm-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-
benzenesulfonamide (P-0685),
Propane- 1 -sulfonic acid [4-chloro-2-fhioro-3 -(5 -ρyridin-3 -yl-1 H-pyrrolo[2,3 -b]pyridine-3 - carbonyl)-ρhenyl] -amide (P-0753),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-phenyl-lH-pyrrolo[253-b]pyridine-3-carbonyl)- phenylj-amide (P-0636),
Propane-1 -sulfonic acid [4-chloro-2-fluoro-3-(5-phenyl-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)- phenyl]-amide (P-0776)
Propane-1 -sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4- difluoro-phenyl} -amide (P-0956),
Propane-1 -sulfonic acid {3-[5-(4-dimethylamino-ρhenyl)-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl]-
2,4-difluoro-phenyl} -amide (P-0889),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(4-methoxy-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl} -amide (P-0877),
Propane-1 -sulfonic acid {2,4-difl-uoro-3-[5-(4-trifluoromethyl-ρhenyl)-lH-pyrrolo[2,3-b]pyridine-
3-carbonyl]-phenyl} -amide (P-0912),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(3-metb.oxy-ρhenyl)-lH-pyrrolot2,3-b]ρyridine-3- carbonyl] -phenyl} -amide (P-0874),
Propane-1 -sulfonic acid {3-[5-(3-dimethylamino-phenyl)-lH-pyrrolo]2,3-b]pyridine-3-carbonyl)-
2,4-difluoro-phenyl} -amide (P-0876),
Propane-l-sulfonic acid [2-fluoro-3-(5-pb.enyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- amide (P-0897),
Propane-1 -sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2J3-b]pyridine-3-carbonyl]-2-fluoro- phenyl} -amide (P-1009),
(2-Fluoro-3 -hydroxy-phenyl)-(5-pyridin-3 -yl- 1 H-pyrrolo [2,3-b]pyridin-3 -yl)-methanone
(P-0027),
(3-Chloro-2-fluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-0165),
5-ρhenyl- 1 H-pyrrolo[2,3-b]pyridine,
Proρane-1 -sulfonic acid [2-fluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-1251),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(3-fluoro-phenyl)-1H-ρyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl} -amide (P-1259),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(4-fluoro-phenyl)-lH-ρyrrolo[2,3-b]pyridine-3- carbonylj-phenyl} -amide (P-1260),
Propane-l-sulfonic acid [2-fluoro-3-(5-ρyridin-3-yl-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)- phenyl]-amide (P-1261),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-ρyridin-4-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenylj-amide (P-1262),
3-{3-[2,6-Difluoro-3-(jpropane-l-sulfonylamino)-benzoyl]-lH-pyrrolo[2,3-b]pyridm-5-yl}- benzoic acid (P-1266),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(3-morpholin-4-yl-plienyl)-lH-pyrrolo[2,3-b]pyridine-
3-carbonyl]-phenyl} -amide (P-1873),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(3-morpholin-4-ylmethyl-ρhenyl)-lH-pyrrolo[2,3- b]pyridine-3-carbonyl]-ρhenyl} -amide (P-1878),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(6-methoxy-pyridin-3-yl)-lH-pyrrolo[2,3-b]pyridine-
3-carbonyl]-phenyl}-amide (P-1879),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(6-morpholin-4-yl-pyridin-3-yl)-lH-pyrrolo[2,3- b]pyridine-3-carbonyl] -phenyl} -amide (P-1881),
Propane-1 -sulfonic acid (2,4-difluoro-3-{5-[6-(4-metliyl-piperazin-l -yl)-pyridin-3-yl]-lH- pyrrolo[2,3-b]pyridine-3-carbonyl} -phenyl)-amide (P-1882),
Propane-1 -sulfonic acid {3-[5-(4-cyano-3,5-dimethyl-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-2,4-difiuoro-phenyl} -amide (P-1980),
N-{2,4-Difluoro-3-[5-(l-methyl-lH-pyrazol-4-yl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- phenyl} -4-trifluoromethyl-benzenesulfonamide (P-1996),
N-{2,4-Difluoro-3-[5-(l-methyl-lH-pyrazol-4-yl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- phenyl} -3 -fluoro-benzenesulfonamide (P-1997),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(l -methyl-lH-pyrazol-4-yl)-lH-pyrrolo[2,3- b]pyridine-3-carbonyl]-phenyl} -amide (P-1864),
Propane-1 -sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-4-fluoro- phenyl} -amide (P-1432),
4-{3-[2,6-Difluoro-3-(propane-l-sulfonylamino)-benzoyl]-lH-pyrrolo[2,3-b]pyridin-5-yl}- benzamide (P-1546),
4-{3-[2,6-Difluoro-3-(proρane-l-sulfonylamino)-benzoyl]-lH-pyrrolo[2,3-b]pyridin-5-yl}-N,N- dimethyl-benzamide (P-1547),
Propane-1 -sulfonic acid (2,4-difluoro-3-{5-[4-(morpholine-4-carbonyl)-phenyl]-lH-pyrrolo[2,3- b]pyridine-3 -carbonyl} -phenyl)-amide (P-1548),
3- {3-[2,6-Difluoro-3-(proρane-l -sulfonylamino)-benzoyl]-lH-pyrrolo[2,3-b]pyridm-5-yl} - benzamide (P-1549),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(4-methyl-lH-imidazol-2-yl)-lH-pyrrolo[2,3- b]pyridine-3-carbonyl]-phenyl}-amide (P-2006),
N-{2,4-Difluoro-3-[5-(6-methoxy-pyridm-3-yl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-ρhenyl}-
3-fluoro-benzenesulfonamide (P-2012),
(3-Difluoromethoxy-2,6-difluoro-phenyl)-(5-pyridhi-3-yl-lH-ρyrrolo[2,3-b]pyridin-3-yl)- methanone (P-1528),
N-{3-[3<3-Difluoromethoxy-2,6-difluoro-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-
methanesulfonamide (P-1527),
(2,2-Difluoro-beiizo[l,3]dioxol-5-yl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-inethanone (P-1558),
N-{3-[3-(2,2-Difluoro-benzo[l,3]dioxole-5-carbonyl)-lH-ρyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1564),
3-(2,3-Dihydro-benzo[l,4]dioxin-6-ylmethyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]ρyridme (P-1529),
N-{3-[3-(2,3-Dihydro-benzo[l,4]dioxin-6-ylmethyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1530),
(3,5-Bis-difluoromethoxy-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1520),
N-{3-[3-(3,5-Bis-difluoromethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridm-5-yl]-phenyl}- methanesulfonamide (P-1522),
(2,2-Difluoro-benzo[l,3]dioxol-4-yl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1515),
N-{3-[3-(2,2-Difluoro-benzo[l,3]dioxole-4-carbonyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1516),
(2,6-Difluoro-3-methoxy-phenyl)-(5-phenyl-lH-pyiτolo[2,3-b]pyridin-3-yl)-methanone (P-1387),
(2,6-Difluoro-3 -methoxy-phenyl)-(5 -pyridin-3 -yl- 1 H-pyrrolo[2,3 -b]pyridin-3 -yl)-methanone
(P-1388),
(2,6-Difluoro-3-methoxy-phenyl)-(5-pyridin-4-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1389),
N-{3-[3-(2,6-Difiuoro-3-methoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acetamide
(P-1390),
N-{3-[3-(2,6-Difluoro-3-methoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1391),
3-(2-Fluoro-3-methoxy-benzyl)-5-phenyl-lH-pyrrolo[2,3-b]ρyridine (P-1323),
3-(2-Fluoro-3-methoxy-benzyl)-5-(4-trifluoromethyl-phenyl)-lH-ρyrrolo[2,3-b]ρyridine
(P-1324),
3-(2-Fluoro-3 -methoxy-benzyl)-5 -(3 -trifluoromethyl-phenyl)- 1 H-ρyrrolo [2,3 -b]ρyridine
(P-1325),
5-(3-Chloro-phenyl)-3-(2-fluoro-3-methoxy-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1326),
3-(2-Fluoro-3-methoxy-benzyl)-5-(3-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine (P-1327),
3 -(2-Fluoro-3 -methoxy-benzyl)-5-(3 -trifluoromethoxy-phenyl)- 1 H-pyrrolo [2,3 -b]pyridine
(P-1328),
N-{3-[3-(2-Fluoro-3-methoxy-benzyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1329),
(2-Fluoro-3-methoxy-phenyl)-[5-(4-trifluoromethyl-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]- methanone (P-1330),
(2-Fluoro-3-methoxy-phenyl)-[5-(3-trifluoromethyl-phenyl)-lH-pyrrolo[253-b]pyridin-3-yl]- methanone (P-1331),
[5-(3-Chloro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-(2-fluoro-S-methoxy-plienyO-methanone
(P-1332),
[5-(4-Chloro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-(2-fluoro-3-methoxy-phenyl)-methanone
(P-1333),
[5-(3,5-Difluoro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-(2-fluoro-3-methoxy-phenyl)-niethanone
(P-1334),
4-[3-(2-Fluoro-3-methoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-benzonitrile (P-1335),
N-{3-[3-(2-Fluoro-3-methoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acetamide
(P-1336),
(2-Fluoro-3-methoxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1338),
(2-Fluoro-3-methoxy-phenyl)-[5-(4-trifluoromethoxy-phenyl)-lH-pyrrolot2,3-b]ρyridin-3-yl]- methanone (P-1339),
(2-Fluoro-3-methoxy-ρhenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1343),
(2-Fluoro-3-methoxy-ρhenyl)-[5-(3-trifluoromethoxy-phenyl)-lH-pyrrolo[2,3-b]ρyridin-3-yl]- methanone (P-1344),
(5-Phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-trifluoromethoxy-phenyl)-methanone (P-1408),
(5 -Pyridin-3 -yl- 1 H-ρyrrolo[2,3 -b]pyridin-3 -yl)-(3 -trifluoromethoxy-phenyl)-methanone (P-1413),
N-{3-[3-(3-Trifluoromethoxy-benzoyl)-lH-ρyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1428),
N-{3-[3-(3-Trifluoromethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acetamide
(P-1422),
(E)-3-{3-[3-(3-Trifluoromethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acrylic acid
(P-1417),
(3-Difluoromethoxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1409),
(3-Difluoromethoxy-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1415),
N-{3-[3-(3-Difluoromethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P- 1430),
N-{3-[3-(3-Difluoromethoxy-benzoyl)-1H-pyrrolo[2,3-b]pyridin-5-yl]-phenyl }-acetamide
(P-1424),
(E)-3-{3-[3-(3-Difluoromethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acrylic acid
(P-1418),
(3,5-Dimethoxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1406), (315-Dimethoxy-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1411), N-{3-[3-(3,5-Dimethoxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-methanesulfonamide (P-1426),
N-{3-[3-(3,5-Dimetb.oxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acetamide (P-1420),
(3-Fluoro-5 -trifluoromethyl-phenyl)-(5-phenyl- 1 H-pyirolo [2,3 -b]pyridin-3 -yl)-methanone
(P-1407),
(3-Fluoro-5-trifluoromethyl-phenyl)-(5-pyridm-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1412),
N-{3-[3-(3-Fluoro-5-trifluoromethyl-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P- 1427),
N- { 3 -[3 -(3 -Fluoro-5 -trifluoromethyl-benzoyl)-l H-pyrrolo[2,3 -b]ρyridin-5-yl]-phenyl} -acetamide
(P-1421),
(3,5-Dichloro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1414),
N--{3-[3-(3,5-Dichloro-benzoyl)-1H-pyrrolo[2,3-b]pyridin-5-y -phenyl}-methanesulfonamide
(P-1429),
N-{3-[3-(3,5-Dichloro-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}-acetamide (P-1423),
(5-Phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-trifluoromethyl-phenyl)-methanone (P-1405),
(5-Pyridin-3-yl-lH-pyπ-olo[2,3-b]pyridin-3-yl)-(3-trifluoroinethyl-ph.enyl)-inethanone (P-1410),
N-{3-[3-(3-Trifluoromethyl-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-1425),
N-{3-[3-(3-Trifluoromethyl-benzoyl)-1H-pyrrolo[2,3-b]pyridin-5-yl}-phenyl}-acetamide
(P-1419), and
(E)-3-{3-[3-(3-Trifluoromethyl-benzoyl)-lH-pyπOlo[2,3-b]pyridin-5-yl]-phenyl}-acrylic acid
(P-1416).
The following table indicates the 5-bromo azaindole (column 2) and boronic acid (column 3) used to afford the compound (column 4). Column 1 provides the compound number and column 5 the observed mass.
Example 4: Synthesis of N-[2,4-difluoro-3-(5-pyridin-3-yl-lH-pyrroIo[2,3-b]pyridine-3- carbonyl)-phenyl]-ethanesulfonamide P-0728.
[0642] Compound P-0728 was synthesized in eight steps from 2,4-difmorophenylamine 42 as shown in Scheme 16.
Step 1 ~ Preparation ofdibenzyl-(2,4-difluoro-phenyl)-amine (52):
[0643] To 2,4-difluoro-phenylamine (42, 10.0 g, 77.4 mmol) in N,N-dimethylformamide (130 mL) were added potassium carbonate (32.1 g, 0.23 mol) and benayl bromide (21.2 mL, 0.18 mol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtrated. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% ethyl acetate in hexane. The appropriate fractions were combined and concentrated to provide the compound (52, 12.0 g, 50%). MS(ESI) [M+H4]4" = 310.2.
Step 2 - Preparation of 3-dibenzylamino-2, 6-difluoro-benzaldehyde (53): [0644] To dibenzyl-(2,4-difluoro-ρhenyl)-amine (52, 4.30 g, 13.9 mmol) in tetrahydrofuran (60 mL), under an atmosphere of nitrogen, cooled in a -78 0C acetone/dry ice bath, was added n- butyllithium (2.50 M in hexane, 6.1 mL, 15.3 mmol) slowly. The reaction was stirred for 1 hour, N,N-dimethylformamide (1.2 mL, 15.3 mmol) was added and the reaction was allowed to warm to room temperature for 1 hour. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% ethyl acetate in hexane to provide the compound (53, 4.0 g, 85%). MS(ESI) [M+HT = 33,7."2.
Step 3 - Preparation of(3-dibenzylamino-2,6-difluoro-phenyl)-(5-pyridin~3-yl-lH-pyrrolo[2,3- b]pyridin-3-yl)~methanol (54):
[0645] To 3-dibenzylamino-2,6-difluoro-benzaldehyde (53, 0.76 g, 2.3 mmol) in methanol (50 mL) were added 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (89, 0.40 g, 2.1 mmol, prepared as described in Example 17) and potassium hydroxide (0.50 g, 8.9 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% methanol in methylene chloride to provide the compound (54, 0.60 g, 50%). MS(ESI) PVRH+J+ = 533.2.
Step 4 - Preparation of (3-dibenzylamino-2, 6-difluoro-phenyl)-(5-pyridin-3-yl-lH'pyrrolo[2, 3- b]pyridin-3~yl)-methanone (55):
[0646] To (3-dibenzylamino-2,6-difluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3--b]pyridm-3-yl)- methanol (54, 0.9Og, 1.7 mmol) in methylene chloride (20 mL) under an atmosphere of nitrogen was added Dess-Martin periodane (0.97 g, 2.3 mmol). The reaction was stirred at room temperature for 15 minutes. The reaction was poured into a solution of sodium bicarbonate and sodium thiosulfate and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography
eluting with 5% methanol in methylene chloride to provide the compound (55, 0.70 g, 78%). MS(ESI) [M^-H+J+ = 531.2.
Step 5 ~ Preparation of (3-dibenzylamino~2, 6-difluoro-phenyl)-(5-pyridin-3-yl-l-triisopropylsilanyl- lH-pyrrolo[2, 3-bJpyridin-3-yl)-methanone (56):
[0647] To (3 -dibenzylamino-2,6-difluoro-phenyl)-(5 -pyridin-3 -yl- 1 H-pyrrolo[2,3-b]pyridm-3 -yl)- methanone (55, 0.84 g, 1.6 mmol) in tetrahydrofuran (150 mL) was added sodium hydride (210.0 mg, 60% in mineral oil, 5.3 mmol) under an atmosphere of nitrogen. The reaction was stirred for 5 minutes. Triisopropylsilyl chloride (0.80 mL, 3.8 mmol) was added and the reaction was stirred at room temperature for 3 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% ethyl acetate in hexane to provide the compound (56, 420 mg, 39%). MS(ESl) [M+H*]+ = 687.4.
Step 6 - Preparation ofβ-amino-2, 6-difluoro-phenyl)-(5-pyridin-3-yl-l~triisopropylsilanyl-lH- pyrrolo[2, 3-b]pyridin-3-yϊ)-methanone (57): f[0648] To (3-dibenzylamino-2,6-difluoro-phenyl)-(5-pyridin-3-yl-l-triisopropylsilanyl-lH- pyrrolo[2,3-b]ρyridin-3-yl)-methanone (56, 55.0 mg, 0.080 mmol) in methanol (15 mL) was added 20% palladium hydroxide on carbon (20 mg). The reaction was stirred under an atmosphere of hydrogen overnight. The reaction was filtered to remove the catalyst, and then concentrated to give the crude compound that was used in the next step.
Step 7 - Preparation ofN-[2,4-difluoro-3-(5-pyridin-3-yl~l-triisopropylsilanyl-lH-pyrrolo[2,3- b]pyridine-3-carbonyl)-phenyl]-ethanesulfonamide (58):
[0649] To (3-amino-2,6-difluoro-phenyl)-(5-pyridin-3-yl-l -triisopropylsilanyl-lH-ρyrrolo[2,3- b]pyridin-3-yi)-methanone (57, 35.0 mg, 0.069 mmol) in methylene chloride (6 mL) was added methanesulfonyl chloride (0.30 mL, 3.9 mmol) and triethylamine (0.40 mL, 2.9 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated to give the crude compound that was used in the next step.
Step 8 - Preparation ofN-[2, 4-difluoro-3-(5-pyridin-3-yl-lH~pyrrolo[2, 3-b]pyridine-3-carbonyl)- phenylj-ethanesulfonamide (P-0728):
[0650] To N-[2,4-difluoro-3-(5-ρyridin-3-yl-l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-ethanesulfonamide (58, 35.0 mg, 0.060 mmol) in tetrahydrofuran (10 mL) was added tetra-n-butylammonium fluoride (19 mg, 0.072 mmol). The reaction was stirred at room temperature for 5 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate
was concentrated and purified by silica gel column chromatography eluting with 5% methanol in methylene chloride to provide the compound (P-0728, 5.6 mg, 22%). MS(ESI) [M+H1]"1" = 443.1 Λ
[0651] Also, (3-Ben2ylammo-2,6-difluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone P-0807 and (3-amino-2,6-difluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone P-0763
were synthesized by reacting (3-dibenzylamino-2,6-difluoro-phenyl)-(5-pyridin-3-yl-l- triisoproρylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (56, 20.0 mg, 0.033 mtnol) and (3- Amino-2,6-difluoro-phenyl)-(5 -pyridin-3 -yl- 1 -triisopropylsilanyl- 1 H-pyrrolo [2,3 -b]pyridin-3 -yl)- methanone (57, 20.0 mg, 0.039 mmol), respectively, in tetrahydrofuran (5.0 mL) with tetra-n- butylammonium fluoride (13 mg, 0.050 mmol). The reaction mixtures were stirred at room temperature for 10 minutes, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The products were isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give P-0807 (2.0 mg, MS(ESI) [M+H
4]
"1" = 441.1) and P-0763 (1.7 mg, MS(ESI)[M-H
+]
' = 351.1) as white solids.
Example 5: Preparation of propane-2-sulfonic acid [2,4-difluoro-3-(5-pyridin-3-yl-lH- pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-amide P-0850.
[0652] Compound P-0850 was synthesized in four steps from 2,4-difluorophenylamine 42 as shown in Scheme 17.
Scheme 17
Step 1 - Preparation ofpropane-2-sulfonic acid (2,4-difluoro-phenyl)-amide (59): [0653] To 2,4-difluoro-phenylamine (42, 4.0 niL, 40.0 mmol) in methylene chloride (50 mL) were added pyridine (3.37 mL, 42.3 mmol), propane-2-sulfonyl chloride (6.00 g, 42.3 mmol) and dimethylaminopyridine (0.20 g, 1.64 mmol) under an atmosphere of nitrogen. The reaction was stirred at 45
0C overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography 3% methanol in methylene chloride to give a white solid (59, 8.0 g, 85%). MS(ESI) [M-H
1]
' = 234.0.
Step 2 - Preparation ofpropane-2-sulfonic acid (2,4-difluoro-3~fortnyl-phenyl)-amide (60): [0654] To ρropane-2-sulfonic acid (2,4-difluoro-phenyl)-amide (59, 2.35 g, 9.95 mmol) in tetrahydrofuran (70 mL) under an atmosphere of nitrogen cooled with a dry ice/acetone bath was added 1.60 M of n-butyllithium (1.60 M in hexane, 6.53 mL, 10.45 mmol). The reaction was stirred for 40 minutes, and then another portion of n-butyllithium (1.60 M in hexane, 6.84 mL, 10.94 mmol). The reaction was stirred for 1 hour and N,N-dimethylformamide (0.92 mL, 11.9 mmol) was added. The reaction was allowed to warm to room temperature overnight. The reaction was poured into water extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtrated. The filtrate was concentrated and purified by silica gel column chromatography (dichloromethane/methanol 5%) to give the compound (60, 1.4 g, 53.4%). MS(ESI) [M - If]-= 263.4.
Step 3 — Preparation ofpropane-2-sulfonic acid {2,4-difluoro-3-[hydroxy-(5-pyridin-3~yl-lH- pyrrolo[2, 3-b]pyridin-3-yl)-methyl]-phenyl}-amide (61):
[0655] To propane-2-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide (60, 220.0 mg, 0.83 mmol) in methanol (15 mL) was added 5-ρyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (89, 150.0 mg, 0.77 mmol, prepared as described in Example 17) and potassium hydroxide (537.0 mg, 9.6 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and filtrated. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% methanol in dichloromethane to give the compound (61, 160 mg, 45.3%). In this step, minor compound Proρane-2-sulfonic acid {2,4-difluoro-3-[methoxy-(5-pyridin- 3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-ρhenyl} -amide was also formed and isolated. MS(ESI)
Step 4 - Preparation ofpropane-2-sulfonic acid [2,4-difluoro~3-(5-pyridin-3-yl-lH~pyrrolo[2,3- b]pyridine-3-carbonyl)-phenyl] -amide (P-0850) :
[0656] To propane-2-sulfonic acid {2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3- b]ρyridin-3-yl)-methyl]-phenyl}-amide (61, 40.0 mg, 0.087 mmol) in tetrahydrofuran (10 mL) was
added Dess-Martin periodane (48.0 mg, 0.11 mmol). The reaction was stirred at room temperature for 5 minutes. The reaction was poured into sodium thiosulfate and potassium carbonate solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtrated. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% methanol in methylene chloride to give the compound (P-0850, 13.4 mg, 33.5%). MS(ESI) [M + H+]+ - 458.1.
[0657] N-(2,4-Difluoro-3-formyl-ρhenyl)-3-trifluoromethyl-benzenesulfonamide 579, N-(2,4- difluoro-3-formyl-phenyl)-4-trifluoromethyl-benzenesulfonamide 580, and N-(2,4-difluoro-3-formyI- phenyl)-4-fϊuoro-benzenesulfonamide 581, and (2,4-difluoro-3-formyl-phenyl)-carbamic acid benzyl ester
were prepared following Steps 1 and 2 of Scheme 17, substituting proρane-2-sulfonyl chloride with 3- trifluoromethyl-benzenesulfonyl chloride, 4-trifluoromethyl-benzenesulfonyl chloride, 4-fluoro- benzenesulfonyl chloride, and benzyl chloroformate, respectively, in Step 1.
[0658] Propane-1 -sulfonic acid [4-chloro-3-(5-chloro-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-2- fluoro-phenyl]-amide P-1004
was prepared using the protocol of Scheme 17, substituting 2,4-difluoro-phenylamine with 4-chloro- 2-fluoro-phenylamine and propane-2-sulfonyl chloride with propane- 1-sulfonyl chloride in Step 1, and 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 with 5-chloro-7-azaindole 80 (see Example 9) in Step 3. MS(ESI) [M + H*]
+ = 430.1.
[0659] Propane-1 -sulfonic acid [4-chloro-2-fluoro-3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide P-0904
was prepared using the protocol of Scheme 17, substituting 2,4-difluoro-phenylamine with 4-chloro- 2-fluoro-phenylamine and propane-2-sulfonyl chloride with propane-1-sulfonyl chloride in Step 1, and 5-ρyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 with 7-azaindole 94 in Step 3. MS(ESI) [M + H
+]
"*
" = 396.2.
[0660] [2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-carbamic acid methyl ester P-0974 and [2,4-Difluoro-3-(lH-pyrrolo[2,3-bjpyridine-3-carbonyl)-phenyl]-carbamic acid benzyl ester P-0894
were prepared using the protocol of Scheme 17, substituting propane-2-sulfonyl chloride with benzyl chloroformate in Step 1 and 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 with 7-azaindole in Step 3. The products of Step 3 were a mixture of {2,4-Difluoro-3-[hydroxy-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methyl]-phenyl}-carbamic acid methyl ester and {2,4-Difluoro-3-[hydroxy-(lH-pyrrolo[2,3- b]ρyridin-3 -yl)-methyl] -phenyl} -carbamic acid benzyl ester that were separated by silica gel column chromatography and carried through Step 4 separately to provide P-0974 and P-0894, respectively. P-0974 MS (ESI) [M - H
+]
" = 330.1. P-0894 MS (EST) [M - H
+]
" = 406.1.
[0661] [2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-carbamic acid methyl ester P-0486 and [2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-carbamic acid benzyl ester P-0302
were prepared using the protocol of Scheme 17, substituting proρane-2-sulfonyl chloride with benzyl chloroformate in Step 1. The products of Step 3 were a mixture of {2,4-Difluoro-3-[hydroxy-(5- pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-phenyl}-carbamic acid methyl ester and {2,4- Difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-phenyl}-carbamic acid benzyl ester that were separated by silica gel column chromatography and carried through Step 4 separately to provide P-0486 and P-0302, respectively. P-0486 MS (ESI) [M + H
+J
+ = 409.1. P-0302 MS (ESI) [M + H
+J
+ = 485.1.
[0662] [5-(3-Methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-(2,3,6-trifluoro-phenyl)- methanoneP-0102
was prepared using the protocol of Steps 3 and 4 of Scheme 17, substituting propane-2-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide with 2,3,6-Trifluoro-benzaldehyde and 5-pyridin-3-yl-lH- pyrrolo[2,3-b]pyridine 89 with 5-(3-methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridine in Step 3. MS (ESI) [M - H
+]
" = 429.0.
[0663] Thiophene-2-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide P-1267
was prepared using the protocol of Steps 3 and 4 of Scheme 17, substituting 5-pyridin-3-yl-lH- ρyrrolo[2,3-b]pyridine 89 with 5-chloro-7-azaindole 80 (see Example 9) and propane-2-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 60 with thiophene-2-sulfonic acid (2,4-difluoro-3-formyl- phenyl)-amide 512 (see Example 21) in Step 3. MS(ESl) [M + H
"]
" = 451.9.
[0664] Thiophene-3 -sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide P-1268
was prepared using the protocol of Steps 3 and 4 of Scheme 17, substituting 5-pyridin-3-yl-lH- pyrrolo[2,3-b]ρyridme 89 with 5-chloro-7-azaindole 80 (see Example 9) and propane-2-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 60 with thiophene-3 -sulfonic acid (2,4-difluoro-3-formyl- phenyl)-amide 513 (see Example 21) in Step 3. MS(ESI) [M + ϊt]
+ = 454.1.
[0665] Additional compounds were prepared following the protocol of Scheme 17, optionally replacing propane-2=sulfonyl chloride with an appropriate acid chloride in Step 1 and/or 5-pyridin-3- yl-lH-pyrrolo[2,3-b]pyridine with an appropriate azaindole in Step 3. Azaindoles were purchased or synthesized as described in Examples 6, 13, 14, 16 and 17. Some compounds were isolated after Step 3, as either hydroxy or methoxy derivatives. The following compounds were made following this procedure:
Dimethylamine-1 -sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- 2,4-difluoro-plienyl} -amide (P-1257),
N-[3-(5-Bromo-lH-pyrτolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-benzenesulfonamide (P-0798),
Propane-1 -sulfonic acid [3-(5-bromo-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-0773),
Dimethylamine-1 -sulfonic acid [3-(5-bromo-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-0898),
N-[3-(5-Chloro-1H-pyrrolo [2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyy-benzenesulfonamide
(P-0885),
N-[2,4-Difluoro-3-(5-fluoro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-benzenesulfonamide
(P-0902),
Propane-1 -sulfonic acid {2,4-difluoro-3-fliydroxy-(5-isopropenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methylj-phenyl} -amide (P-1239),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-isopropenyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-0991),
Propane-1 -sulfonic acid {2,4-difluoro-3 -[hydroxy-(5 -methyl- 1 H-pyrrolo[2,3-b]pyridin-3 -yl)- methyl] -phenyl} -amide (P-1240),
Propane-1 -sulfonic acid {2,4-difluoro-3-[methoxy-(5-methyl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methyl]-phenyl} -amide (P-1241),
Propane-1 -sulfonic acid {2,4-difluoro-3-[hydroxy-(5-isopropyl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methyl]-phenyl} -amide (P-1242),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-isopropyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-0997),
Propane-1 -sulfonic acid {2,4-difluoro-3-[hydroxy-(5-methoxy-lH-ρyrrolo[2,3-b]ρyridin-3-yl)- methylj-phenyl} -amide (P-1243),
Propane-1 -sulfonic acid {2,4-difluoro-3-[methoxy-(5-methoxy-lH-pyrrolo[2,3-b]pyridin-3-yl)- methyl]-phenyl} -amide (P-1244),
Propane-1 -sulfonic acid (2J4-difluoro-3-{hydroxy-[5-(4-methyl-piperazin-l-yl)-lH-pyrrolo[2,3- b]pyridin-3-yl]-methyl} -phenyl)-amide (P-1245),
Propane-l-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-0933),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-fluoro-1H- pyrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- amide (P-0907),
Piperidine-1 -sulfonic acid [3-(5-chloro-1H-pyr olo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-1020),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-prienyl]-4-metrioxy-
benzenesulfonamide (P-0983),
N-[3-(4-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-plienyl]-benzenesulfonamide (P-09S4),
N-[2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-acetamide (P-
1002),
Dimethylamine-1 -sulfonic acid [3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-ρhenyl]- amide (P-0950),
Dimethylamine-1 -sulfonic acid [3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenyl]-amide (P-0837),
Dimethylamine-1 -sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenylj-amide (P-1258),
Butane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2!4-difluoro-ρhenyl]- amide (P-1263),
Butane- 1 -sulfonic acid [2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-1264),
Butane- 1 -sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2J4-difluoro-ρhenyl]- amide (P-1265),
Propane-1 -sulfonic acid [3-(5-ethoxy-lH-pyrrolo[2,3-b]pyridiαe-3-carbonyl)-2,4-difIuoro- phenyl]-amide (P-1252),
Propane-1 -sulfonic acid {2,4-difluoro-3-[5-(2-methoxy-ethoxy)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl} -amide (P-1253),
Propane-1 -sulfonic acid {3-[5-(2-diethylamino-ethoxy)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-
2,4-difluoro-phenyl} -amide (P-1254),
N-[3-(5-Chloro-1H-pyrrolo [2,3-b]pyridine-3-carbonyl) 2,4-difluoro-ρhenyl]-4-ethyl- benzenesulfonamide (P-1700),
N-[3-(5-Ethyl-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-2,4-difluoro-phenyl]-4-trifluoromethyl- benzenesulfonamide (P-1783),
Thiophene-3 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-1798),
Benzo[b]thiophene-2-sulfonic acid p-CS-chloro-lH-pyrrol[2,3-b]pyridine-3-carbonyl) -2,4- difluoro-phenyl]-amide (P-1799),
5-Pyridin-2-yl-thiophene-2-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenylj-amide (P-1800),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-ρhenyl]-4-cyano- benzenesulfonamide (P-1822),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3-fluoro-4-methyl- benzenesulfonamide (P-1823),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-isopropyl- benzenesulfonamide (P-1839),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-2,4-difluoro-phenyl]-4-fluoro- benzenesulfonamide (P-1840),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3,5-difluoro- benzenesulfonamide (P-1841),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2;4-difluoro-phenyl]-4-methyl- benzenesulfonamide (P-1842),
N-[3-(5-Chloro-1H-pyrrolo [2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-oxazol-5-yl- benzenesulfonamide (P-1843),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-fluoro-benzenesulfonamide
(P-1865),
N-{2,4-Difluoro-3-[5-(2-methoxy-ethoxy)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-3- fluoro-benzenesulfonamide (P-1871),
N-{2,4-Difluoro-3-[5-(2-methoxy-ethoxy)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-4- fluoro-benzenesulfonamide (P-1872),
Propane-l-sulfonie acid (2,4-difluoro-3-{5-[4-(2-methoxy-ethoxy)-phenyl]-lH-pyrrolo[2,3- b]pyridine-3-carbonyl} -phenyl)-amide (P-1998),
N-{2,4-Difluoro-3-[5-(2-methoxy-ethoxy)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-4- trifluoromethyl-benzenesulfonamide (P-2005), and
N-(2,4-Difluoro-3-{5-[4-(2-metb.oxy-ethoxy)-phenyl]-lH-pyrrolo[2,3-b]pyridine-3-carbonyl}- phenyl)-4-trifluoromethyl-benzenesulfonamide (P-2013).
The following table indicates the acid chloride (column 2) and the azaindole (column 3) used to afford the target compounds (column 4). Column 1 provides the compound number and column 5 the observed mass. Compounds isolated after Step 3 of Scheme 17 are so noted in column 1.
Example 6: Synthesis of propane-l-sulfonic acid [2,4-difluoro-3-(5-phenylamino-lH- pyrroIo[2,3-b]pyridine-3-carbonyl)-phenyl]-amide P-0848 and related compounds.
[0666] Propane-l-sulfonic acid [2,4-difluoro-3-(5-phenylamino-lH-ρyrrolo[2,3-b]ρyridine-3- carbonyl)-phenyl] -amide P-0848 was synthesized in five steps from 5-bromo-7 -azaindole 67 as shown in Scheme 18.
Scheme 18
Step 1 - Preparation of5-bromo-l-triisopropylsilanyl-lH-pyrrolo[2,3~b]pyridine (68): [0667] To 5-bromo-7-azaindole (67, 1.5 g, 7.6 mmol) in N,N-dimethylformamide (20 mL) were added sodium hydride (60% in mineral oil, 0.27 g, 11.0 mmol) and trriisopropylsilyl chloride ( 2.6 mL, 12.0 mmol), under an atmosphere of nitrogen. The reaction was stirred for 2 hours at room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10 % ethyl acetate in hexane to give the compound (68, 1.6 g, 59%). MS(ESI)[M+lf]+ = 352.3.
Step 2 - Preparation of5-phenyl-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine-5-yl)-amine (69): [0668] To 5-bromo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (68, 0.10 g, 0.3 mmol) in toluene (5 mL) were added aniline (0.04 mL, 0.42 mmol), sodium tert-butoxide (0.15 g, 1.56 mmol), tris(dibenzyllideneacetone)dipalladium(0) (9.2 mg, 0.01 mmol) and (S) ~(-)-2,2'- bis(diphenylphosphino)-l,l '-binaphthyl (6.3 mg, 0.01 mmol). The reaction was heated to 160 0C for 10 minutes in a CEM Discover microwave instrument. The reaction was concentrated and purified by silica gel column chromatography eluting with 3 % ethyl acetate in hexane to give the compound (69, 40 mg, 40%). MS(ESI)PVH-H+J+ = 366.6.
Step 3 - Preparation ofphenyl-(-lH-pyrrolo[2,3-b]pyridine-5-yl)-amine (70): [0669] To 5-phenyl-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine-5-yl)-amine (69, 0.14 g, mmol) in tetrahydrofuran (3.0 mL) was added tetra-n-butylammonium fluoride (0.197 g, 0.76 mmol). The reaction was stirred for 1 hour at room temperature. The reaction was concentrated and purified by silica gel column chromatography eluting with 3 % ethyl acetate in hexane to give the compound (70, 60 mg, 76 %). MS(ESI)[M+BTT = 210.3.
Step 4 - Preparation of propane-1 -sulfonic acid {2,4-difluoro-3~[hydroxy-(5-phenyIamino-lH- pyrrolo[2, 3-b]pyridin-3-yl)-methyl]-phenyl}-amide (71) :
[0670] To phenyl-(-lH-ρyrrolo[2,3-b]pyridme-5-yl)-amine (70, 17.0 mg, 0.09 mmol) in methanol (5.0 mL) were added potassium hydroxide (92.0 mg, 1.6 mmol) and propane-1 -sulfonic acid (2,4- difluoro-3-formyl-phenyl)-amide (73, 19.0 mg, 0.072 mmol, prepared as described in Example 7) under an atmosphere of nitrogen. The reaction was stirred for 12 hours at room temperature. The reaction was concentrated and purified by silica gel column chromatography eluting with 1 % methanol in dichloromethane to give the compound (71, 17 mg, 50%). MS(ESI)[M+H+]+ = 473.5.
Step 5 - Preparation of propane-1 -sulfonic acid [2,4-difluoro-3-(5-phenylamno-lH-pyrrolo[2,3~ b]pyridine-3-carbonyl)-phenyl] -amide (P-0848):
[0671] To propane-1 -sulfonic acid {2,4-difluoro-3-[hydroxy-(5-ρhenylamino-lH-pyrrolo[2,3- b]pyridin-3-yl)-methyl]-phenyl}-amide (71, 7.5 mg, 0.016 mmol) in tetrahydrofuran (3 mL) was added Dess-Martin periodane (6.70 mg, 0.0158 mmol) under an atmosphere of nitrogen. The reaction was stirred for 20 minutes. The reaction was concentrated and purified by silica gel column chromatography eluting with 1 % methanol in dichloromethane to give the compound (P-0848, 6.2 mg, 84%). MS(ESI)[M+^ = 471.2.
[0672] Propane-1 -sulfonic acid [2,4-difluoro-3-(5-morpholin-4-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl] -amide P-08S3, propane-1 -sulfonic acid {2,4-difluoro-3-[5-(4-methyl-piperidin-l- yl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-amide P-0860, and propane-1 -sulfonic acid {2,4- difluoro-3-[5-(4-methyl-piperazin-l-yl)-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-amide
P-1246,
, , were prepared using the protocol of Scheme 18, substituting aniline with morpholine, 4-methyl- piperidine, and 4-methyl-piperazine, respectively, in Step 2. P-0853 MS(ESI) [M+H
+]
+ = 465.2. P-0860 MS(ESI) [M + H
+J
+ = 477.3. P-1246 MS(ESI) [M - H
+]
' = 478.4.
[0673] 4-[5-(3-Chloro-4-methoxy-phenylamino)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-indole-l- carboxylic acid butylamide P-1859
was prepared using the protocol of Scheme 18, substituting aniline with 3-chloro-4-methoxy- phenylamine in Step 2 and propane- 1 -sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 73 with 4- Formyl-indole-1 -carboxylic acid butylamide 519 (see Example 22) in Step 4. MS(ESI) [M+H
+]
+ = 516.2.
[0674] (lH-Indol-4-yl)-[5-(4-morpholin-4-yl-phenylamino)-lH-pyrrolo[2,3-b]pvridin-3-yl]- methanone P-1792, [5-(4-Chloro-phenylamino)-lH-pyrrolo[2,3-b]pyridin-3-yl]-(lH-indol-4-yl)- methanone P-1793, (lH-Indol-4-yl)-[5-(4-ρiperidin-l-yl-phenylamino)-lH-pyrrolo[2,3-b]pyridin-3- yl]-methanone P-1794, and [5-(3-Chloro-4-methoxy-phenylammo)-lH-pyrrolo[2,3-b]pyridin-3-yl]-
were prepared using the protocol of Steps 1-3 of Scheme 18, substituting 5-bromo-7-azaindole with 4-
(S-Bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l-carboxylic acid butylamide (P-1687, prepared as described in Example 22, Scheme 41) in Step 1 and replacing aniline with 4-morpholin-4- yl-ρhenylamine, 4-chloro-phenylamine, 4-piperidin-l-yl-ρhenylamine, and 3-chloro-4-methoxy- phenylamine, respectively, in Step 2. P-1792 MS (ESl) [M + H
+J
+ = 438.3. P-1793 MS (ESI) [M + H
+J
+ = 387.1. P-1794 MS (ESl) [M + H
+J
+ = 436.3. P-1795 MS (ESI) [M + H
4J
1- = 417.1.
Example 7: Synthesis of propane-1-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 73.
[0675] Compound 73 was synthesized in two steps from 2,4-difluorophenylamine 42 as shown in Scheme 19.
Scheme 19
Step 1 - Preparation of Propane-1-sulfonic acid (2,4-difluoro-phenyl)-amide (72): [0676] To 2,4-difluoro-phenylamine (42, 3.0 mL, 29.8 mmol) in tetrahydrofuran (50 mL) were added triethylamine (9.13 mL, 65,5 mmol) and propane- 1-sulfonyl chloride (2.90 mL, 25.8 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into 1 M HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to give the compound (72, 2.0 g, 28%) that was used in the next step.
Step 2 - Preparation of propane-1-sulfonic acid (2,4~difluoro-3-formyl-phenyl)-amide (73): [0677] To proρane-1-sulfonic acid (2,4-difluoro-phenyl)-amide (72, 1.5 g, 6.38 mmol) in tetrahydrofuran (10 mL) under an atmosphere of nitrogen, cooled in a -78 0C acetone/dry ice bath was added lithium diisopropylamide (0.80 M in tetrahydrofuran, 24 mL, freshly prepared from n- butyllithium and diisopropylamine). After 30 minutes, N,N-dimethyl-formamide (542 μL, 7.018 mmol) was added dropwise to the reaction. The reaction was stirred for 30 minutes at -78 0C and then allowed to warm to room temperature for 40 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5 % ethyl acetate in hexane to give a light yellow solid (73, 300 mg, 18 %). MS(ESI)[M- If]- = 262.3.
Example 8: Synthesis of propane-1-sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-amide P-1116.
[0678] Compound P-1116 was synthesized in four steps from 3 -amino-benzoic acid ethyl ester 74 as shown in Scheme 20.
Scheme 20
Step 1 - Preparation of3-(propane-l-sulfonylamino)-benzoic acid ethyl ester (75): [0679] To 3-amino-benzoic acid ethyl ester (74, 5.0 g, 0.030 mol) in methylene chloride (30.0 niL) were added pyridine (3.67 mL, 0.045 mol) and propane- 1-sulfonyl chloride (3.75 mL, 33.0 mmol). The reaction was stirred at room temperature for 2 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a white solid (75, 6.0 g, 74.1%).
Step 2 - Preparation of3-(propane-l-sulfonylamino)-benzoic acid (76):
[0680] To 3-(propane-l -sulfonylamino)-benzoic acid ethyl ester (75, 1.60 g, 5.90 mmol) in water was added lithium hydroxide (1.0 g, 4.2 mmol) and tetrahydrofuran (20 mL). The reaction was stirred at room temperature overnight. The reaction mixture was acidified with 1 N HCl, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and washed with ethyl acetate to give the compound (76, 1.2 g, 84.5%) as white solid. MS(ESI)[M-H+J+= 242.1.
Step 3 - Preparation of 3-(propane-l-sulfonylamino)-benzoyl chloride (77): [0681] A solution of 3 -(propane- l-sulfonylamino)-benzoic acid (76, 1.20 g, 4,93 mmol) in thionyl chloride was heated to reflux for 3.0 hours. Evaporation of the solvent gave compound 77 as white solid that was used for the next step.
Step 4 - Preparation of propane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl] -amide (P-1116):
[0682] To aluminum trichloride (4.8 g, 36.0 mmol) in methylene chloride (70.0 mL), under an atmosphere of nitrogen, was added 5-bromo-lH-pyrrolo[2,3-b]pyridine (67, 797 mg, 4.04 mmol) dissolved in methylene chloride (5.0 mL). The reaction was stirred at room temperature for 30 minutes, followed by addition of 3 -(propane- l-sulfonylamino)-benzoyl chloride (77, 1.10 g, 4.20 mmol) dissolved in methylene dichloride (4.0 mL). The reaction was stirred at room temperature for 2 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give a white solid (P-1116, 300.0 mg). MS(ESI)[M-H+]- = 420.1, 422.1.
Example 9: Synthesis of 5-chloro-lH-pyrrolo[2,3-b]pyridine 80.
[0683] Compound 80 was synthesized in two steps from 5-bromo-l-triisopropylsilyl-7-azaindole 68 as shown in Scheme 21.
Scheme 21
Step 1 - Preparation 5-chloro-l-triisopropylsilanyl-lH-pyrrolo[2,S~b]pyridine (79): [0684] To 5-bromo-l-triisopropylsilyl-7-azaindole (68, 1.60 g, 4.53 mmol, prepared as described in Example 6) in tetrahydroiuran (50.0 mL), under an atmosphere of nitrogen at -78 0C, was added tert- butyllithium (1.70 M in hexane, 6.12 mL). The reaction was stirred for 1 hour, followed by addition of hexachloroethane (1.29 g, 5.43 mmol). The reaction was stirred for 3 hours, poured into water, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to give the crude compound (79, 1.60 g). MS(ESI)[M+H+]+ = 309.3.
Step 2 - Preparation 5-chloro-lH-pyrrolo[2,3-b]pyridine (80):
[0685] To 5-chloro-l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (79, 1.40 g, 4.53 mmol) in tetrahydrofuran (15 mL) was added fefrα-n-butylammonium fluoride (1.42 g, 5.43 mmol). The reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was concentrated and isolated by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound (80, 0.40 g. 58% over 2 steps). MS(ESI)[M-H+]" = 153.1.
[0686] 5-Fluoro-lH-pyrrolo[2,3-b]pyridine 81
was prepared using the protocol of Scheme 21 , substituting hexachloroethane with N-fluoro-N- (phenylsulfonyl) benzenesulfonamide in Step 1. MS(ESI) [M + H
+]* = 137.1.
Example 10: Synthesis of 2,4-difluoro-3-(5-pyridin-3-yl-lH-pyrroIo[2,3-b]pyridin-3-ylmethyl)- phenol P-0078 and related compounds.
[0687] Compound P-0078 was synthesized in two steps from 5-pyridin-3-yl-lH-pyrrolo[2,3- b]pyridine 89 as shown in Scheme 24.
Scheme 24
Step 1 — Preparation of2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH~pyrrolo[2,3-b]pyridin-3-yl)~ methyl] -phenol (P-0009):
[0688] To carbonic acid tert-bυtyl ester 2,4-difluoro-3-formyl-phenyl ester (39, 0.405 g, 15.7 mmol) in methanol (36 mL), under an atmosphere of nitrogen, was added 5-pyridin-3-yl-lH-pyrrolo[2,3- b]pyridine (89, 288.0 mg, 14.8 mmol, prepared as described in Example 17) and potassium hydroxide (145.0 mg, 25.9 mmol). The reaction was stirred at room temperature overnight. Then, the reaction was poured into water, and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The mixture was purified by silica gel column chromatography eluting with 4% methanol in methylene chloride to provide two separate compounds, a colorless oil (P-0009, 0.23 g, 44.1%, MS(ESl) [M+H+]+ = 354.1), and a colorless oil (P-0042, 0.050 g, 9.2%, MS(ESI) [M+HT = 367.1).
Step 2 - Preparation of2,4-difluoro-3~(5-pyridin~3-yl-lH-pyrrolo[2,3-b]pyridin--3-ylmethyl)-phenol
(P-0078):
[0689] To 2,4-difluoro-3-[hydroxy-(5-p)'ridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-ρhenol
(P-0009, 34.0 mg, 0.096 mmol) in acetonitrile (15 mL), were added trifluoroacetic acid (1.0 mL, 13.0 mmol) and triethylsilane (2.0 mL, 12.0 mmol). The reaction was stirred at room temperature for 48 hours. The reaction mixture was poured sodium bicarbonate solution, and extracted with ethyl acetate.
The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound (P-0078, 6.0 mg, 19%). MS(ESI)[M + H+J+ = 338.1.
[0690] Additional compounds were prepared following the protocol of Scheme 24, replacing either or both of carbonic acid tert-buty\ ester 2,4-difluoro-3-formyl-phenyl ester 39 with an appropriate aldehyde and/or 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 with an appropriate azaindole in Step 1. Azaindoles were purchased or synthesized as described in Examples 6 and 17. Aldehydes were prepared as described in Example 5 (through Step 2). Some compounds were isolated after Step 1, as either hydroxy or methoxy derivatives. The following compounds were made following this procedure:
3-[(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-methoxy-methyl]-254-difluoro-phenol (P-0126),
3-[(5-Bromo-lH-pyrrolo[2,3-b]ρyridin-3-yl)-hydroxy-methyl]-2}4-difluoro-phenol (P-1180),
3-(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenol (P-0122),
{2,4-Difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3b]pyridin-3-yl)-methyl]-phenyl}- carbamic acid benzyl ester (P-0095),
[2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridm-3-ylmethyl)-phenyl]-carbamic acid benzyl ester (P-0396),
{2,4-Difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-ρyrrolo[2,3-b]pyridin-3-yl)-methyl]-phenyl}- carbamic acid methyl ester (P-0065),
[2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-carbamic acid methyl ester (P-0257),
Propane-2-sulfonic acid {3- [(5-bromo- 1 H-pyrrolo[2,3 -b]pyridin-3 -yl)-methoxy-methyl] -2,4- difluoro-phenyl} -amide (P-0356),
Propane-2-sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]- amide (P-0867),
Propane-2-sulfonic acid {2,4-difluoro-3-[methoxy-(5-ρyridin-3-yl-lH-pyrrolo[2,3-b]ρyridin-3- yl)-methyl]-phenyl}-amide (P~0947),
Propane- 1 -sulfonic acid {3-[(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-hydroxy-methyl]-2,4- difluoro-phenyl} -amide (P-0188),
Propane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-plienyl]- amide (P-0910),
Dimethylamino-1 -sulfonic acid {2,4-diiluoro-3-[hydroxy-(5-pyridm-3-yl-lH-pyrrolo[2,3- b]pyridin-3-yl)-methyl]-phenyl} -amide (P-0944),
3-(2-Fluoro-3-methoxy-benzyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (P-0269),
Propane- 1 -sulfonic acid [2,4-difluoro-3-(5-phenylamino-lH-pyrrolo[2,3-b]pyridm-3-ylmethyl)- phenyl]-amide (P-0818),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]- amide (P-0911),
Propane-1 -sulfonic acid [2,4-difluoro-3-(5-moipholin-4-yl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenyl]-amide (P-0964),
Propane-l-sulfonic acid {2,4-difluoro-3-[5-(4-methyl-piperidin-l-yl)-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl]-phenyl} -amide (P-0984),
{3-[(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-hydroxy-methyl]-2,4-difluoro-phenyl}-carbamic acid tert-butyl ester (P-0318),
{3-[(5-Chloro-1H-pyrrolo[2,3 -b] pyridin-3- yl) -hydroxy-methyl]-2,4-difluoro-phenyl}-carbamic acid methyl ester,
{2,4-Difluoro-3-[hydroxy-(5-methoxy-lH-ρyrrolo[2,3-b]pyridin-3-yl)-metliyl]-phenyl}-carbamic acid methyl ester, and
{2,4-Difluoro-3-[hydroxy-(lH-pyrrolo[2,3-b]pyridin-3-yl)-meth.yl]-phenyl}-carbamic acid methyl ester,
5-Bromo-3-(2,3-dihydro-benzo[l ,4]dioxin-6-ylmethyl)-lH-pyrrolo[2,3-b]pyridine (P-1474), and
5-Bromo-3-(2-fluoro-3-methoxy-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-0535). The following table indicates the aldehyde (column 2) and azaindole (column 3) used to afford the target compound (column 4). Column 1 provides the compound number and column 5 the observed mass. Compounds isolated after Step 1 of Scheme 24 are so noted in column 1.
* P-0095 and P-0065 were both produced in Step 1 and isolated from the mixture. These were carried through to step 2 to provide P-0396 and P-0257, respectively.
Example 11: Synthesis of N-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl) phenyl]-3- methoxy-benzenesulf onamide P-0971.
[0691] Compound P-0971 was synthesized in four steps from 2,4-difluorophenylamine 42 as shown in Scheme 26.
Scheme 26
Step 1 - Preparation ofN-(2,4-difluoro~phenyl)-3-methoxy-benzenesulfonamide (91): [0692] To 2,4-difluoro-phenylamine(42, 0.44 mL, 4.4 mmol) in methylene chloride (10.0 mL), under an atmosphere of nitrogen, were added pyridine (1.00 mL, 12.4 mmol) and 3-methoxy- benzenesulfonyl chloride (1.00 g, 4.84 mmol). After 12 hours, the reaction was poured into cold 1 M HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20 % ethyl acetate in hexane to give light yellow solid (91, 0.90 g, 69%). MS(ESl)[M+!!+]"1" = 300.
Step 2 — Preparation ofN-{2, 4-difluoro-3-[hydroxy-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin- 3-yl)-methyl]-phenyl}-3-methoxy-benzenesulfonamide (92):
[0693] To N-(2,4-difluoro-phenyl)-3-methoxy-benzenesulfonamide (91, 0.148 g, 0.494 mmol) in tetrahydrofuran (10.0 mL) cooled in a -78 0C acetone/dry ice bath, under an atmosphere of nitrogen, was added lithium diisopropylamide (0.85 M in tetrahydrofuran, 1.45 mL, 1.23 mmol) dropwise. After 30 minutes, l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine-3~carbaldehyde (96, 0.15 g, 0.500 mmol, prepared as described in Example 12) was added in tetrahydrofuran (2.0 mL) into the reaction dropwise. Then the reaction was stirred for 1 hour at -78 0C and allowed to reach room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 40 % ethyl acetate in hexane to give light yellow solid (92, 0.080 g, 26.8%). MS(ESI)[M+H+]+ = 602.
Step 3 — Preparation ofN-{2,4-difluoro-3~[hydroxy-(lH-pyrrolo[2,3-b]pyridin-3-yl)methylJ-phenyl}- 3-methoxy-benzenesulfonamide (93):
[0694] To N-{2,4-difiuoro-3-[hydroxy-(l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridm-3-yl)- methyl] -phenyl} -3 -methoxy-benzenesulfonamide (92, 0.075 g, 0.12 mmol) in tetrahydrofuran (3.0
mL), was added tetra-n-butylammonium fluoride (0.039 g, 0.15 mmol). The reaction was stirred at room temperature for 20 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 3 % methanol in dichloromethane to give light yellow solid (93, 0.030 g, 55%). MS(ESI) [M+H+]+ = 446.
Step 4 - Preparation ofN-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)phenyl]-3-methoxy- benzenesulfonamide (P-0971):
[0695] To N-{2,4-difluoro-3-[hydroxy-(lH-pyrrolo[2,3-b]pyridin-3-yl)methyl]-ρhenyl}-3-methoxy- benzenesulfonamide (93, 0.02 g, 0.05 mmol) in tetrahydrofuran (3.0 mL) was added Dess-Martin periodane ( 0.02 g, 0.015 mmol) under an atmosphere of nitrogen. The reaction was stirred for 10 minutes at room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 3 % methanol in dichloromethane to give light yellow solid (P-0971, 0.010 g, 50%). MS(ESI) [M+H+]+ = 444.
[0696] Additional compounds were prepared following the protocol of Scheme 26, replacing 3- methoxy-benzene sulfonyl chloride with the appropriate sulfonyl chloride in Step 1. The following compounds were made following this procedure:
N-[2,4-Difluoro-3-( 1H-pyrrolo-[2,3-b]pyridine-3-carbonyl)-phenyl]-2,5-dmiethoxy- benzenesulfonamide (P-1131),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-methoxy- benzenesulfonamide (P-0958),
Piρeridine-1 -sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-amide
(P-0952),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-trifluoromethyl- benzenesulfonamide (P-0931),
4-Butoxy-N-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]-benzenesulfonamide
(P-1006),
4-Chloro-N-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-benzenesulfonamide
(P-0937),
N- [2,4-Difluoro-3 -(I H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-phenyl] -3 ,4-dimethoxy- benzenesulfonamide, (P-1090), and
3,4-Dichloro-N-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- benzenesulfonamide (P-1015).
The following table indicates the sulfonyl chloride (column 2) used to afford the target compound (column 3). Column 1 provides the compound number and column 4 gives the observed mass.
* Piperidine-1 -sulfonyl chloride prepared from sulfuryl chloπde and piperidine in acetonitrile, refluxed for 8 hours, concentrated, and used without further purification.
Example 12: Synthesis of l-triisopropyIsiIanyl-lH-pyrrolo[2,3-b]pyridine-3-carbaldehyde 96. [0697J Compound 96 was synthesized in two steps from 7-azaindole 94 as described in Scheme 27.
Scheme 27
Step 1 -Preparation oflH-pyrrolo[2,3-b]pyridine-3~carbaldehyde (95): [0698] To lH-Pyrrolo[2,3-b]pyridine (94, 16.0 g, 135 mmol) in water (110 mL), were added hexamethylenetetramine (26.0 g, 185 mmol), and acetic acid (55.0 mL, 967 mmol). The reaction was refluxed for 12 hours. Water (329 mL) was added and the reaction was cooled to room temperature. The reaction was filtrated and washed with water to give the compound (95, 15.0 g, 76%).
Step 2 - Preparation ofl-triisopropylsilanyl~lH-pyrrolo[2,3-b]pyridine-3-carbaldehyde (96): [0699] To lH-Pyrrolo[2,3-b]pyridine-3-carbaldehyde (95, 4.05 g, 27.71 mmol) in tetrahydrofuran (30.0 mL) were added sodium hydride (60% in mineral oil, 1.5 g, 38 mmol) and trriisopropylsilyl chloride (8.0 mL, 38 mmol) under an atmosphere of nitrogen. The reaction was stirred for 2 hours at room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10 % ethyl acetate in hexane to give the compound (96, 3.0 g, 36%). MS(ESI)[M+H+]+ = 303.
Example 13: Synthesis of 5-isopropyl-lH-pyrroIo[2,3-b]pyridine 99.
[0700] Compound 98 was synthesized in three steps from 5-bromo-l -triisopropylsilanyl-lH- pyrrolo[2,3-b]ρyridine 68 described in Scheme 28.
Scheme 28
Step 1 - Preparation of2-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-5-yl)-propan-2-ol (97): [0701] To 5-bromo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (68, 2.0 g, 5.66 mmol, prepared as described in Example 6) in tetrahydrofuran (20.0 mL), cooled in a -78 0C acetone/dry ice bath, under an atmosphere of nitrogen, was added tert-butyllithium (1.7 M in tetrahydrofuran, 7.3 mL, 12
mmol) dropwise. After 20 minutes, acetone (0.830 mL, 11 mmol) was added dropwise to the reaction. The reaction was stirred for 30 minutes at -78 0C and then allowed to reach room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10 % ethyl acetate in hexane to give the compound (97, 1.30 g, 69%). MS(ESI)[M^-H+J+ = 333.
Step 2 - Preparation of 5-isopropenyl-lH-pyrrolo [2, 3-b] pyridine (98):
[0702] To 2-(l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-5-yl)-propan-2-ol (97, 0.500 g, 1.5 mmol) in acetonitrile (10.0 mL) were added triethylsilane (1.00 mL, 6.3 mmol) and trifluoroacetic acid (0.50 mL, 6.5 mmol) under an atmosphere of nitrogen. The reaction was refluxed for 3 hours, then cooled down to room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 50 % ethyl acetate in hexane to give the compound (98, 0.200 g, 84%). MS(ESI)[M+H+]+ = 159.
Step 3- Preparation of5-isopropyl-lH-pyrrolo[2,3-b]pyridine (99):
[0703] To 5-isopropenyl-lH-pyrrolo[2,3-b]ρyridine (98, 0.080 g, 0.501 mmol) in tetrahydrofuran (5.0 mL) was added 20 % palladium hydroxide on carbon (5.0 mg). The reaction was stirred under hydrogen at 40 psi for 30 minutes. The reaction mixture was filtered and concentrated to give the compound (99, 0.078 g, 96%). MS(ESI)[M+H+]+ = 161.
Example 14: Synthesis of 5-Methyl-lH-pyrrolo[2,3-b]pyridine 101.
[0704] Compound 101 was synthesized in two steps from 5-bromo-l -triisopropylsilanyl-lH- pyrrolo[2J3-b]pyridine 68 described in Scheme 29.
Scheme 29
Step 1 - Preparation of5~Methyl-l-trnsopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (100): [0705] To PdCl2(dppf) (0.04 g, 0.05 mmol) in toluene (10.0 mL) under an atmosphere of nitrogen were added 5-bromo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (68, 0.3 g, 0.8 mmol, prepared as described in Example 6, 1.0 mL in toluene) and methylmagnesium bromide (1.0 M in tetrahydrofuran, 3.0 mL, 3.0 mmol). The reaction was stirred 90 °C for 2 hours and then allowed to reach to room temperature. The reaction was poured into citric acid (0.1 M in water) and extracted
with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 50 % ethyl acetate in hexane to give the compound (100, 0.16 g, 60.0 %). MS(ESI)[M+H+j+ = 289.4.
Step 2 -Preparation of5-Methyl-lH-pyrrolo[2,3-b]pyridine (101):
[0706] To 5-Methyl-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (10O5 0.160 g, 0.55 mmol) in tetrahydrofuran (3.0 mL) was added tetra-n-butylammonium fluoride (0.145 g, 0.55 mmol). The reaction was stirred for 1 hour at room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 3% methanol in dichloromethane to provide light yellow solid (101, 0.07 g, 95%). MS(ESI) [M+Hy = 133.2.
[0707] 5-Methyl-lH-pyrrolo[2,3-b]pyridine
was prepared following the protocol of Scheme 29, substituting methylmagnesium bromide with ethylmagnesium bromide in Step 1.
Example 15: Synthesis of l-[2,4-difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-3-propyl-urea P-0774 and related compounds.
[0708] Compound P-0774 was synthesized in two steps from {2,4-difluoro-3-[hydroxy-(5-pyridin- 3-yl-lH-pyrrolo[2,3b]pyridin-3-yl)-methyl]-phenyl}-carbamic acid methyl ester P-0065 described in Scheme 30.
Scheme 30
Step 1 - Preparation ofl-{2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3b]pyridin-3-yl)- methyl]-phenyl}-3-propyl-urea (103) :
[0709] To {2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3b]pyτidin-3-yl)-methyl]- phenyl} -carbamic acid methyl ester (P-0065, 30.0 mg, 0.07 mmol, prepared as described in Example 10) was added 1-propanamine (2.0 mL, 20 mmol) and the reaction was heated to 120 0C for 20 minutes in a CEM Discover microwave instrument. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% methanol in methylene chloride to give the compound (103, 20.0 mg, 60%). MS(ESI)[MMf]+ = 438.51.
Step 2 - Preparation ofl-[2,4-diflιιoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-3-propyl-urea (P-0774):
[0710] To l-{2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3b]ρyridin-3-yl)-methyl]- phenyl} -3-propyl-urea (103, 20.0 mg, 0.04 mmol) in tetrahydrofuran (3 mL) was added Dess-Martin periodane (23.0 mg, 0.055 mmol) under an atmosphere of nitrogen. The reaction was stirred for 10 minutes. The reaction was concentrated and purified by silica gel column chromatography eluting with 1 % methanol in dichloromethane to give the compound (P-0774, 6.0 mg, 30 %). MS(ESI)[M+H*]+ = 436.5.
[0711] Additional compounds were prepared following the protocol of Scheme 30, replacing 1- propanamine with an appropriate amine and optionally replacing {2,4-difluoro-3-[hydroxy-(5-pyridin- 3-yl-lH-pyrrolo[2,3b]pyridin-3-yl)-methyl]-ρhenyl} -carbamic acid methyl ester P-0065 with an appropriate carbamic acid methyl ester (see Example 10) in Step 1. The following compounds were made following this procedure:
1 -sec-Butyl-3 -[2,4-difluoro-3 -(5-pyridin-3 -yl- 1 H-pyrrolo [2, 3 -b]pyridine-3 -carbonyl)-phenyl] - urea (P-1289),
1 -Cyclopentyl-3 -[2,4-difluoro-3 -(5-pyridin-3 -yl- 1 H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-ρhenyl] - urea (P-1317), l-Butyl-3-[2,4-difluoro-3-(5-ρyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1318), l-Butyl-3-[2,4-difluoro-3-(5-methoxy-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]-urea
(P-1S67), l-[3-(5-Chloro-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3-(2-morpholin-4-yl- ethyl)-urea (P-1580),
1 -Butyl-3 -[3 -(5 -chloro- 1 H-pyrrolo[2,3 -b]pyridine-3 -carbonyl)-2,4-difluoro-phenyl] -urea
(P-1586),
Moφholme-4-carboxylic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-1606), l-Butyl-3-[3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-l-methyl- urea (P-1612), l-Butyl-3-[2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-plienyl]-l-methyl- urea (P-1884),
Mθφholine-4-carboxylic acid [2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridme-3- carbonyl)-phenyl]-amide (P-1894), l-Butyl-3-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-l -ethyl-urea (P-1983), l-Butyl-3-[2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-l-etliyl- urea (P-1994), and
3-Diethylamino-pyrrolidme-l-carboxylic acid [2,4-difluoro-3-(5-methoxy-1H-pyrrolo[2,3-- b]pyridine-3 -carbonyl)-phenyl]-amide (P-2015).
The following table indicates the amine (column 2) and the carbamic acid methyl ester (column 3) used to afford the target compounds (column 4). Column 1 provides the compound number and column 5 the observed mass.
[0712] The intermediate product of Step 1 of Scheme 30 can alternatively be reacted under reducing conditions to provide analogous compounds in which the carbonyl linker of the 3 -position of azaindole to the phenyl ring is methylene. The product of Step 1 is a mixture of the hydroxyl and methoxy methyl linker of the 3 -position, which may be carried through the reduction Step as the mixture or may be isolated as either the hydroxyl or methoxy for use in this reaction. This reduction is exemplified as follows using the product of Step 1 in the preparation of P-1567 to prepare 1-Butyl- 3-[2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridin-3-ylmethyI)-phenyl]-urea P-1571.
Step 1 - Preparation ofl-ButylS-[2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenylj-urea (P-1571):
[0713] A mixture of l-Butyl-S-2,4-difluoro-3-[hydroxy-(5-methoxy-lH- 1H-pyrrolo[2,3-b]pyridin-3 yl)-methyl]-phenyl-urea (41 mg, 0.000081 mol, isolated from Step 1 of Scheme 30), triethylsilane (2 mL, 0.01 mol), and trifiuoroacetic acid (1 mL, 0.01 mol) in 20 ml of acetonitrile was refluxed for 3 hrs. The mixture was concentrated and the residue was redissolved in ethyl acetate and sodium bicarbonate solution. The organic layer was collected and dried over MgSC^. An off-white solid compound was obtained after chromatography (P-1571, 18 mg, 57%). MS(ESI) [M-H+]" = 389.2.
[0714] Additional compounds were prepared following the protocol of Step 1 of Scheme 30 followed by the reduction step above as Step 2, where the product of Scheme 30 Step 1 may be isolated as either the hydroxyl or methoxy derivative, or used as the mixture, replacing 1-proρanamine with an appropriate amine and {2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-ρyrrolo[2,3b]pyridm-3- yl)-methyl]-phenyl}-carbamic acid methyl ester P-0065 with an appropriate carbamic acid methyl ester (see Example 10) in Step 1. The following compounds were made following this procedure:
1 -Cyclopentyl-3 -[2,4-difluoro-3 -(5 -methoxy- 1 H-pyrrolo[2,3 -b]pyridin-3 -ylmethyl)-phenyl] -urea
(P-1572), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]-3-cyclopentyl-urea
(P-1575), l-Butyl-3-[3-(5-chloro-lH-ρyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]-urea (P-1587), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]-3-(2-morpholin-4-yl- ethyl)-urea (P-1594),
Morpholine-4-carboxylic acid [3-(5-chloiO-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-
phenyl]-amide (P-1595), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-plienyl]-3-(2,2,2-trifluoro- ethyl)-urea (P-1601), l-Cyclopentyl-3-{2,4-difluoro-3-[5-(3-methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl]-phenyl}-urea (P-1615), l-Butyl-3-[3-(5-chloro-1H-pyrrolo[2,3-b]pyridin-3- ylmethyl)-2,4-difluoro-phenyl]-1-methyl-urea (P-1625), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]-3~cyclopropylmethyl- urea (P-1652), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmetliyl)-2,4-difluoro-phenyl]-3-(4-fluoro-phenyl)- urea (P-1657), l-[3-(5-Cb.loro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenyl]-3-(4-fluoro-benzyl)- urea (P-1654), and
S-Diethylammo-pyrrolidine-l-carboxylic acid [2,4-difluoro-3-(5-methoxy-lH-pyrrolo[2,3- b]pyridin-3-ylmethyl)-phenyl]-amide (P-2014).
The following table indicates the amine (column 2) and the carbamic acid methyl ester (column 3) used to afford the target compound (column 4). For the carbamic acid methyl ester, R can be H or CH3, where the compound was either a mixture of the two, or either of the isolated compounds wherein R is H or R is CH3. Column 1 provides the compound number and column 5 the observed mass.
Example 16: Synthesis of 5-Methoxy-lH-pyrrolo[2,3-b]pyridine 104 and related compounds.
[0715] Compound 104 was synthesized in one step from 5-bromo-lH-pyrrolo[2,3-b]pyridine 67 as described in Scheme 31.
Scheme 31
Step 1 -Preparation of 5-Methoxy-lH-pyrrolo [2, 3-b] pyridine (104):
[0716] To 5-bromo-7-azaindole (67, 500.0 mg, 2.53 mmol) in N,N-dimethylformamide (8 niL) were added copper(I) iodide (966 mg, 5.08 mmol) and sodium methoxide in methanol (3 M, 5 mL). The reaction was stirred overnight at 120 0C under an atmosphere of Argon. The reaction was poured into water, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, filtered. The filtrate was concentrated and purified with silica gel column chromatograph eluting with 20% ethyl acetate in hexane to give white solid (104, 140 mg, 28%). MS(ESI)[M+H+]+ = 149.1. In an alternative method, 2.3 g (11.7 mmol) 5-bromo-7-azaindole (67, 2.3 g, 11.7 mmol) was dissolved in 75 mL N,N-dimethylforrnamide and 50 mL methanol (50 mL), adding sodium methoxide (32 g, 0.6 mol) and copper-(I) bromide (3.2 g, 22.4 mmol) at room temperature. The reaction was stirred for three hours at 100 0C under an atmosphere of argon. The mixture was diluted with ethyl acetate and poured into a solution of ammonium chloride:ammonium hydroxide (4:1). The organic layer was extracted with ammonium chloride:ammonium hydroxide (4:1), washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 30% to 70% ethyl acetate in hexanes to give a yellow solid (104, 0.27 g, 15.6%). MS(ESI) [M + H+J+= 149.2.
[0717] 5-Ethoxy-lH-pyrrolo[2,3-b]pyridine 506
was prepared using the protocol of Scheme 31, substituting methanol with ethanol and sodium methoxide with sodium ethoxide.
[0718] 5-(2-Methoxy-ethoxy)-lH-ρyrrolo[2,3-b]pyridine 507
was prepared using the protocol of Scheme 31, substituting methanol with 2-Methoxy-ethanoI and
sodium methoxide with sodium 2-Methoxy-ethoxide (prepared from 2-Methoxy-ethanol and sodium hydride). MS(ESI) [M + H
+J
+ = 193.3.
[07191 Diethyl-[2-(lH-pyrrolo[2,3-b]pyridin-5-yloxy)-etliyl]-amme 508
was prepared using the protocol of Scheme 31, substituting methanol with 2-diethylamino-ethanol and sodium methoxide with sodium 2-diethylamino-ethoxide (prepared from 2 2-diethylamino- ethanol and sodium hydride). MS(ESI) [M + H
+J
+ = 234.5.
Example 17: Synthesis of 5-Pyridin-3-yl-lH-pyrrolo[2,3-b] pyridine 89.
[0720] 5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 was synthesized in one step from 5-bromo-lH- pyrrolo[2,3-b]pyridine 67 as described in Scheme 32.
Scheme 32
Step 1 - Preparation of5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (89):
[0721] To 5-bromo-7-azaindole (67, 1.00 g, 5.08 mmol) in water (13.0 mL) and acetonitrile (36 mL) were added pyridine-3-boronic acid (609, 1.0 g, 8.1 mmol), potassium carbonate (1.79 g, 0.0130 mol) and Tetrakis(triphenylphosphine)palladium(0) (50.0 mg, 0.043 mmol) under an atmosphere of nitrogen. The reaction mixture was heated to 170
0C overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residue was purified with silica gel column chromatography eluting with 25% ethyl acetate in hexane to provide a light yellow solid (89, 820 mg, 82%).
[0722] Additional compounds were prepared following the protocol of Scheme 32, either by substituting pyridine-3-boronic acid with an appropriate boronic acid or by substituting the 5-bromo- 7-azaindole with 5-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyrrolo[2,3-b]pyridine and reacting with a suitable aryl or heteroaryl halide (i.e. coupling with the boronic acid ester on the azaindole, and the halide on the group to be coupled to the 5-position of the azaiαdole). The following compounds were prepared by this procedure:
5-(4-Chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine (514),
5-(4-Fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine (605),
5-Phenyl-lH-pyrrolo[2,3-b]pyridine,
5-(6-Methoxy-pyridin-3-yl)-lH-pyrrolo[2,3-b]pyridine,
5-(2-Methoxy-pyrimidin-5-yl)-lH-ρyrrolo[2,3-b]pyridine,
5-Pyridin-4-yl- 1 H-pyrrolo [2,3 -b]pyridine,
4-(lH-Pyrrolo[2,3-b]pyridin-5-yl)-benzenesulfonamide,
3-(lH-Pyrrolo[2,3-b]pyridin-5-yl)-benzenesulfonamide,
5-Pyrimidin-5-yl-lH-pyrrolo[2,3~b]pyridine,
5-(3-Methanesulfonyl-phenyl)-lH-pyrrolo[2,3-b]pyridine (P-0173), and
3-(lH-Pyrrolo[2,3-b]ρyridin-5-yl)-benzamide (P-1622).
The following table indicates either 5-bromo-7-azaindole or 5-(4,4,5,5-Tetramethyl-[l,3,2] dioxaborolan-2-yl)-lH-pyrrolo[2,3-b]pyridine starting material (column 1) and the appropriate reagent to be coupled to the 5 position of the azaindole (column 2) to afford the resulting compound (column 3), with the observed mass given in column 4.
Example 18: Synthesis of 3-(4-(4-chlorobenzyloxy)-3-methoxybeiizyl)-lH-pyrrolo[2,3- b]pyridine P-1247.
[0723] Compound P-1247 was synthesized in three steps from 4-hydroxy-3-methoxybenzaldehyde 105 as shown in Scheme 33.
Scheme 33
Step 1 - Preparation of4-(4-chlorobenzyloxy)-3-methoxybenzaldehyde (106): [0724] To 4-hydroxy-3-methoxybenzaldehyde (105, 600.0 mg, 3.94 mmol) and 4-chloroben2yl bromide (557, 1.20 g, 5.84 mmol) in acetonitrile (6 niL) was added potassium carbonate (0.390 g, 2.82 mmol). The reaction was microwaved on 300 watts, 120
0C for 10 minutes. The reaction was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over magnesium sulfate, filtered and the volatiles removed by evaporation. The desired compound was purified by recrystallization from hexanes to provide 106 (1.01 g, 93%). MS(ESI) [M-H
+]
" = 275.1.
Step 2 - Preparation of3-((4-(4-chlorobenzyloxy)-3-methoxyphenyl)(meihoxy)meihyl)-lH- pyrrolo[2,3-b]pyridine (107):
[0725] To lH-Pyrrolo[2,3-b]pyridine (94, 0.235 g, 1.99 mmol) and 4-(4-chlorobenzyloxy)-3- methoxybenzaldehyde (106, 0.500 g, 1.81 mmol) was added 5 mL of methanol followed by the addition of solid potassium hydroxide (0.203 g, 3,61 mmol). The reaction was allowed to stir at ambient temperature for 18 days. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was separated and volatiles removed to give a solid which was suspended in hot ethyl acetate. The suspension was allowed to cool and the solid collected by vacuum filtration to provide 107 (548 mg, 74%). MS(ESI) [MH-H+J+ = 409.4.
Step 3 - Preparation of 3-(4~(4-chlorobenzyloxy)-3-methoxybenzyl)-lH-pyrrolo[2,3-b] pyridine (P- 1247):
[0726] To 3-((4-(4-chlorobenzyloxy)-3-methoxyphenyl)(methoxy)methyl)-lH-pyrrolo[2,3- b]ρyridine (107, 0.548 g, 1.34 mmol) in acetonitrile (20 mL) was added trifmoroacetic acid (1.7 mL, 2.21 mmol) and triethylsilane (3.47 mL, 2.17 mmol). The reaction was stirred at 60
0C for 15 hours. The volatiles were removed and the desired compound was purified by silica gel chromatography, eluting with a gradient from 0% to 60% ethyl acetate in hexanes to provide a white solid (P-1247, 505 mg, 99%). MS(ESI)
379.4.
[0727] Additional compounds were prepared using the protocol of Scheme 33, Steps 2 and 3, replacing 4-(4-chlorobenzyloxy)-3-methoxybenzaldehyde 106 with a suitable aldehyde (prepared as described in Example 34), and optionally replacing lH-Pyrrolo[2,3-b]pyridine 94 with an appropriate substituted 7-azaindole (see Example 9 or Example 16) in Step 2. The following compounds were made following this procedure:
3-[3-Methoxy-4-(4-trifluoromethyl-benzyloxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1721),
3-[3-Trifluoromethyl-4-(4-trifluoromethyl-benzyloxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-1797),
3-{3-Methoxy-4-[4-(4-methyl-piperazin-l-ylmethyl)-benzyloxy]-benzyl}-lH- ρyrrolo[2,3-b]pyridme (P-1821),
3-[4-(4-Chloro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]ρyridine (P-1844);,
3-[4-(3-Fluoro-4-trifluoromethyl-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolot2,3-b]ρyridine (P-1849),
3-[4-(4-Chloro-3-trifluoromethyl-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1851),
2-[2-Methoxy-4-(lH-pyπrolo[2,3-b]pyridin-3-ylmethyl)-phenoxyrQethyl]-lH-benzoimidazole
(P-1870),
3-t4-(4-Chloro-2-fluoro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-5-methoxy-lH- pyrrolo[2,3-b]pyridine (P-1885),
3-[4-(3,4-Dichloro-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1886),
3-[4-(4-Chloro-benzyloxy)-3-fluoro-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1896),
2-[2-Fluoro-4-( 1 H-pyrrolo [2,3 -b]pyridin-3 -ylmetliyl)-ρhenoxymethyl] - 1 H-benzoimidazole
(P-1899),
3-(4-Ben2yloxy-2,5-difluoro-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1901),
5-Chloro-3 - [4-(4-chloro-benzyloxy)-2-fluoro-5-methoxy-benzyI] - 1 H-pyrrolo[2,3 -b]pyridine
(P-1970),
5-Chloro-3-[4-(4-chloro-2-fluoro-ben2yloxy)-2-fluoro-5-methoxy-benzyl]-lH- pyrrolo[2,3-b]pyridine (P-1972),
3-[4-(4-Chloro-2-fluoro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-1973),
2-[4-(5-Chloro-lH-pyπ-olo[2,3-b]pyridin-3-ylmetliyl)-5-fluoro-2-methoxy-plienoxymetb.yl]-lH- benzoimidazole (P-1976),
2-[5-Fluoro-2-methoxy-4-(5-methoxy-lH-pyrrolo[2,3-b]pyridin--3-ylmetliyl)-ρhenoxym6thyl]- lH-benzoimidazole (P-1977),
2-[5-Fluoro-2-methoxy-4-(lH-ρyrrolo[2,3-b]pyridin-3-ylmethyl)-phenoxymetliyl]-lH- benzoimidazole (P-1978),
3-{4-[2-(2-Bromo-ethoxy)-ethoxy]-2-fluoro-5-methoxy-benzyl}-5-chloro-lH-pyrrolo[2,3- bjpyridine (P-1984),
5-Chloro-3-[2,5-difluoro-4-(2-methoxy-ethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1986),
5-Chloro-3-[2-fluoro-5-methoxy-4-(2-methoxy-ethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-1990),
{3-[4-(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-5-fluoro-2-methoxy-phenoxy]-propyl}- diethyl-amine (P-2004),
5-Chloro-3 - {2-fluoro-5-methoxy-4-[2-(2-methoxy-etlioxy)-ethoxy]-benzyl} -lH-ρyrrolo[2,3- b]pyridine (P-2002),
3-(4-Benzyloxy-2,6-difluoro-benzyl)-lH-ρyrrolo[2,3-b]pyridine (P-2022),
3-{2-Fluoro-5-methoxy-4-[2-(2-methoxy-ethoxy)-etlioxy]-benzyl}-5-methoxy-lH-pyrrolo[2,3- b]pyridine (P-2025), and
3-{2-Fluoro-5-metlioxy-4-[2-(2-methoxy-ethoxy)-ethoxy]-benzyl}-lH-pyrrolo[2,3-b]pyridine
(P-2026).
The following table indicates the aldehyde (column 2) and the azaindole (column 3) used to afford the target compound (column 4). Column 1 indicates the compound number and column 5 the observed mass.
Example 19: Synthesis of Propane-l-sulfonic acid [3-(5-bromo-lH-pyrroIo[2,3-b]pyridine-3- carbonyl)-2-fluoro-phenyl] -amide P-0955 and related compounds.
[0728] As an alternative method to that of Example 2, compound P-0955 was synthesized in nine steps from 4-chloro-2-fluoro-phenylamine 47 as shown in Scheme 37.
Scheme 37
Step 1 - Preparation of3-Amino'6-chloro~2-fluoro'benzoic acid benzyl ester (48): [0729] To 4-chloro-2-fluoro-phenylamine (47, 6.30 mL, 57.0 mmol) in tetrahydrofuran (300 mL), cooled with dry ice/acetone bath under an atmosphere of nitrogen, n-butyllithium (2.50 M in hexane, 24.4 mL) was added slowly. After 20 minutes, l,2-Bis-(chloro-dimethyl-silanyl)-ethane (12.9 g, 60.0 mmol) dissolved in tetrahydrofuran (40.0 mL) was added slowly to the reaction. After 1 hour, n- butyllithium (2.50 M in hexane, 25.0 mL) was added slowly to the reaction. The reaction was stirred at -78 0C for 20 minutes and then allowed to warm to room temperature over 60 minutes. The
reaction was cooled to -78 0C, followed by addition of n-butyllithium (2.50 M in hexane, 26.0 mL) slowly. After 80 minutes, benzyl chloroformate (10.0 mL, 70.0 mmol) was added to the reaction. The reaction mixture was stirred at -78 0C overnight followed by addition of water (80 mL) and concentrated hydrochloric acid (25 mL). The reaction was allowed to warm to room temperature for 2 hours. The organic layer was separated. The aqueous layer was basified with potassium carbonate and extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate/hexane 20%) to give a colorless oil (48, 12.5 g, 78.3%). MS(ESl) [M+H+f = 280.0.
Step 2 - Preparation of6-chloro-2-fluoro-3-(propane-l-sulfonylamino)~benzoic acid benzyl ester (49):
[0730] To S-amino-β-chloro^-fluoro-benzoic acid benzyl ester (48, 1.20 g, 4.3 mmol) in methylene chloride (28 mL) was added pyridine (0.52 mL, 6.4 mmol) and propanesulfonyl chloride (0.685 g, 4.8 mmol). The reaction was stirred at room temperature overnight, then poured into water, and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated with silica gel column chromatography to give a colorless oil (49, 960 mg, 58.0%). MS(ESI) [M-H"]" = 384.1.
Step 3 - Preparation of6-chloro-2-fluoro-3~(propane-l-sulfonylamino)-benzoic acid (115): [0731] To 6-chloro-2-fluoro-3-(propane-l -sulfonylamino)-benzoic acid benzyl ester (49, 6.00 g, 15.6 mmol) in tetrahydrofuran (100 mL) was added 1.0 M aqueous potassium hydroxide (100 mL). The reaction was heated to reflux overnight. The reaction was poured into water, acidified to pH 2 with 1 N hydrochloric acid and extracted with ethyl acetate. The organic portion was dried over anhydrous sodium sulfate, filtered and concentrated to give a white solid 115 (3.95 g, 85.8%).
Step 4 - Preparation of2-fluoro-3-(propane-l-sulfonylamino)~benzoic acid (50): [0732] To 6-chloro-2-fluoro-3-(propane-l-sulfonylamino)-benzoic acid (115, 0.69 g, 2.3 mmol) in methanol (10 mL) was added 20% palladium hydroxide on carbon (200 mg). The reaction was stirred under hydrogen at 50 psi for 2 hours. The reaction was filtered and concentrated to give white solid 50 that was used in the next step. MS(ESI) [M-H+] =260.1.
Step 5 - Preparation of2-fluoro-3-(propane-l-sulfonylamino)-benzoic acid methyl ester (501): [0733] To a 2-fluoro-3-(proρane-l-sulfonylamino)-benzoic acid (50, 5.05 g, 0.0193 mol) in methylene chloride (100 mL) was added N,N-dimethylfoπnaraide (0.075 mL, 0.97 mmol) under an atmosphere of nitrogen. The reaction was cooled with ice/water, followed by slow addition of Oxalyl chloride (2.00 M of in methylene chloride, 10.8 mL, 21.6 mmol). The reaction mixture was stirred at room temperature for 3.0 hours. The reaction was cooled with ice/water, followed by addition of
methanol (36.0 mL, 0.89 mol) slowly. The reaction was stirred at room temperature overnight. The reaction was concentrate and purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give a crude white solid 4.Og.
Step 6 '- Preparation of Propane- 1 -sulfonic acid (2-fluoro-3-hydroxymethyl~phenyl)-aιnide (502): [0734] To 2-fluoro-3-(propane-l-sulfonylamino)-benzoic acid methyl ester (501, 3.80 g, 13.8 mmol) in tetrahydrofuran (133 mL) was added lithium tetrahydroaluminate (1.00 M in tetrahydrofuran, 20.0 mL, 20.0 mmol) under an atmosphere of nitrogen at room temperature. The reaction was stirred at room temperature for 8 hours, followed by addition of 10 g OfNaSO4-IOH2O. After 12 hours, the reaction was filtered, concentrated and purified with silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (502, 3.0 g, 87.9%).
Step 7 — Preparation of propane- 1 -sulfonic acid (2-fluoro-3-formyl-phenyl)-amide (503): [0735] To propane-1 -sulfonic acid (2-fluoro-3-hydroxymethyl-phenyl)-amide (502, 0.20 g, 0.81 mmol) in tetrahydrofuran (5.0 mL) was added Dess-Martin periodinane (0.377 g, 0.89 mmol). The reaction was stirred at room temperature for 10 minutes, then poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a white solid (503, 100 mg, 50.0%). MS(ESI) [M-H+J+ = 244.1.
Step 8 - Preparation of Propane-1 -sulfonic acid {3-[(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)- hydroxy-methyl]-2-fluoro-phenyl}-amide (504) :
[0736] To 5-bromo-7-azaindole 67 (312 mg, 1.58 mmol) in methanol (28 mL) were added propane- 1 -sulfonic acid (2-fluoro-3-formyl-phenyl)-amide (503, 370 mg, 1.5 mmol) and potassium hydroxide (422.8 mg, 7.5 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight, then poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the desired compound as white solid (504, 300 mg, 45.0%).
Step 9 - Preparation of propane-1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- 2, 4-difluoro-phenyl] -amide (P-0955) :
[0737] To Propane-1 -sulfonic acid {3-[(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-hydroxy-methyl]- 2-fluoro-phenyl} -amide (504, 0.650 g, 1.47 mmol) in tetrahydrofuran (25.0 mL) cooled with ice/water was added Dess-Martin periodinane (0.748 g, 1.76 mmol). The reaction was stirred at room temperature for 15 minutes. The reaction was poured into water containing sodium thiosulfate and potassium carbonate and extracted with ethyl acetate. The organic layer was dried over anhydrous
sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane and washed with ethyl acetate to give white solid. (P-0955, 0.35 g, 54.1%). MS(ES]) [JVH-H+J1" = 460.0, 462.0.
[0738] Butane-1 -sulfonic acid [3-(5-chloro-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-2-fluoro- phenyl] -amide P-1250
was prepared following the protocol of Scheme 37, substituting propane-2-sulfonyl chloride with butane- 1-sulfonyl chloride in Step 1 and 5-bromo-lH-ρyrrolo[2,3-b]pyridine 67 with 5-chloro-lH- pyrrolo[2,3-b]pyridine 80 (see Example 9) in step 8. MS(ESI) [M - H
+]
" = 408.1.
[0739] Propane- 1 -sulfonic acid [2-fIuoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl] -amide P-1256
was prepared following the protocol of Scheme 37, substituting 5-bromo-lH-pyrrolo[2,3-b]pyridine 67 with 5-methoxy-lH-pyrrolo[2,3-b]pyridine 104 (see Example 16) in step 8. MS(ES]) [M - H
+]
" = 390.1.
[0740] N-[3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2-fluoro-phenyl] benzenesulfonamide P-1255
was prepared following the protocol of Scheme 37, substituting propane-2-sulfonyl chloride with benzenesulfonyl chloride in Step 1 and 5-bromo-lH-pyrrolo[2,3-b]pyridine 67 with 5-chloro-lH- pyrrolo[2,3-b]ρyridine 80 (see Example 9) in step 8. MS(ESI) [M - H
+]
" = 428.0.
Example 20: Synthesis of 3-3-[2,6-difluoro-3-(propane-l-sulfonylamino)~benzoyl]-lH- pyrrolo[2,3 -b]pyridin-5-yl-propionic acid P-1270.
[0741] Compound P-1270 was synthesized in three steps from propane- 1 -sulfonic acid [3-(5- bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-amide P-0773 as shown in Scheme 38.
Scheme 38
Step 1 - Preparation of(E)-3-3-[2,6-difluoro-3-(propane-l -sulfonylamino)~benzoyl]-lH-pyrrolo [2,3-b]pyridin-5-yl-acrylic acidm ethyl ester (505):
[0742] To propane- 1 -sulfonic acid [3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difiuoro-phenyl] -amide (P-0773, 125.0 mg, 0.27 mmol, prepared as described in Example 4) in N,N- dimethylformamide (4.0 mL) were added palladium acetate (15 mg, 0.068 mmol), triphenylphosphine (36 mg, 0.14 mmol), methyl acrylate (0.098 mL, 1.1 mmol) and triethylamine (0.114 mL, 0.82 mmol) under an atmosphere of nitrogen. The reaction was stirred at 140 0C overnight, then poured into water, acidified with water and extracted with ethyl acetate. To the filtrate in methylene chloride (5.0 mL) was added l,8-diazabicyclo[5.4.0]undec-7-ene (0.50 mL, 3.3 mmol). The reaction was stirred at room temperature for 3 hours. The reaction was concentrated and purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give a light yellow oil that was used directly in the next step.
Step 2 - Preparation of3-3-[2,6-difluoro-3-(propane-l-sulfonylamino)-henzoyl]-lH-pyrrolo[2,3 - bJpyridin-5-yl-acrylic acid (P-1269):
[0743] To (E)-3-3-[2,6-difluoro-3-(propane-l -sulfonylamino)-benzoyl]-lH-pyrrolo [2,3-b]pyridin- 5-yl-acrylic acid methyl ester (100.0 mg, 0.22 mmol) in tetrahydrofuran (5.0 mL) and water (1.50 mL) was added lithium hydroxide (21 mg, 0.86 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water, acidified with IN HCl to pH around 1, and then extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a white solid (P-1269, 30 mg). MS(ESI) [M-H+]' = 448.0.
Step 3 ~ 3-3-[2,6-difluoro-3-(propane-l-sulfonylmnino)-benzoyl]-lH-pyrrolo[2,3 -bJpyridin-5-yl- propionic acid (P-1270):
[0744] To 3-3-[2,6-difluoro-3-(propane-l-sulfonylamino)-benzoyl]-lH-ρyrrolo[253 -b]ρyridin-5-yl- acrylic acid (P-1269, 20.0 mg, 0.045 mmol) in methanol (5.0 niL) was added 20% Pd(OH)2/C (10 mg) under an atmosphere of hydrogen. The reaction was stirred at room temperature for 2 hours. The reaction mixture was filtered, concentrated and purified with silica gel column chromatography eluting with 10% methanol in methylene chloride to give a white solid (P-1270, 8.8 mg). MS(ESI) [M-H+]- = 450.1.
Example 21: Synthesis of thiophene-2-suIfonic acid (2,4-difluoro-3-formy]-phenyl)-amide 508.
[0745] Compound 512 was synthesized in four steps from 2,4-difluorophenylamme 42 as shown in Scheme 39.
Scheme 39
Step 1 -Preparation of 3-amino-4,2-difluoro-henzoic acid ethyl ester (509): [0746] To 4,2-difluoro-phenylamine (42, 6.30 mL, 57.0 mmol) in tetrahydrofuran (300 mL), cooled with dry ice/acetone bath under an atmosphere of nitrogen, n-butyllithium (2.50 M in hexane, 24.4 mL) was added slowly. After 20 minutes, l,2-Bis-(chloro-dimethyl-silanyl)-ethane (12.9 g, 60.0 mmol) dissolved in tetrahydrofuran (40.0 mL) was slowly added to the reaction. After 1 hour, n- butyllithium (2.50 M in hexane, 25.0 mL) was slowly added to the reaction. The reaction was stirred at -78 0C for 20 minutes and then allowed to warm to room temperature over 60 minutes. The reaction was cooled to -78 0C, followed by addition of n-butyllithium (2.50 M in hexane, 26.0 mL) slowly. After 80 minutes, ethyl chloroformate (6.69 mL, 70.0 mmol) was added to the reaction. The reaction mixture was stirred at -78 0C overnight followed by addition of water (80 mL) and concentrated hydrochloric acid (25 mL). The reaction was allowed to warm to room temperature for 2 hours. The organic layer was separated. The aqueous layer was basified with potassium carbonate and extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate/hexane 20%) to give a colorless oil (509, 4.6 g, 39%). MS(ESI) [M + H+J+ = 218.1.
Step 2 - Preparation of 2, 6-difluoro-3-(thiophene-2-sulfonylamino)-benzoic acid ethyl ester (510): [0747] To 3-amino-2,4-difluoro-benzoic acid ethyl ester (509, 1.20 g, 5.93 mmol) in methylene chloride (28 mL) was added pyridine (0.52 mL, 6.4 mmol) and thiophene-2-sulfonyl chloride (0.97 g, 5.38 mmol). The reaction was stirred at room temperature overnight, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated with silica gel column chromatography (ethyl acetate/hexane 20%) to give a colorless oil (510, 1.2 g, 65.0%). MS(ESI) [M + HT= 348.2.
Step 3 — Preparation ofthiophene-2-sulfonic acid (2,4-dtfluoro-3-hydroxymethyl-phenyl)-amide (511):
[0748] To 2,6-difluoro-3-(thiophene-2-sulfonylamino)-benzoic acid ethyl ester (510, 1.6 g, 3.5 mmol) in tetrahydrofuran (25.0 mL) was added lithium tetrahydroaluminate (1.00 M in tetrahydrofuran, 8.08 mL, 8.08 mmol) under an atmosphere of nitrogen at room temperature. The reaction was stirred at room temperature for 8 hours, followed by addition of 10 g OfNaSO4-IOH2O. After 12 hours, the reaction was filtered, concentrated and purified with silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (511, 300.0 mg, 21.0%).
Step 4 - Preparation ofthiophene-2-sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide (512): [0749] To thioρhene-2-sulfonic acid (2,4-difluoro-3-hydroxymethyl-phenyl)-amide (511, 0.46 g, 1.52 mmol) in tetrahydrofuran (5.0 mL) was added Dess-Martin periodinane (0.71g, 1.67 mmol). The reaction was stirred at room temperature for 10 minutes, then poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a white solid (512, 100 mg, 21%). MS(ESI) [M + H+J+ = 304.2.
[0750] Thiophene-3 -sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 513
was prepared following the protocol of Scheme 39, substituting thiophene-2-sulfonyl chloride with thiophene-3-sulfonyl chloride in Step 2. MS(ESI) [M + iff = 304.2.
[0751] N-(2,4-Difluoro-3-formyl-phenyl)-methanesulfonamide 577
was prepared following the protocol of Scheme 39, substituting thiophene-2-sulfonyl chloride with methanesulfonyl chloride in Step 2.
[0752] N-(2,4-Difluoro-3-formyl-phenyl)-3-fluoro-benzenesulfonamide 578
was prepared following the protocol of Scheme 39, substituting thiophene-2-sulfonyl chloride with 3- fluoro-benzenesulfonyl chloride in Step 2.
Example 22: Synthesis of 4-(5-Pyridm-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l- carboxylic acid butylamide P-1486 and related compounds
[0753] Compound P-1486 was synthesized in three steps from lH-indole-4-carbaldehyde 518 as shown in Scheme 41.
Scheme 41
Step 1 - Preparation of 4-Formyl-indole-l-carboxylic acid butylamide (519): [0754] To lH-indole-4-carbaldehyde (518, 1.57 g, 10.8 mmol) in acetonitrile (20 niL) was added 1- isocyanatobutane (1.81 mL, 16.2 mmol), followed by 4-dimethylaminopyridine (130 mg, 1.1 mmol). The reaction was refluxed for 48 hours. The reaction solution was quenched with 1 M HCl (aq.) and extracted with ethyl acetate. The organic layer was washed with sodium bicarbonate and brine, dried
over anhydrous magnesium sulfate, filtrated and concentrated to give a light yellow solid (519, 2.62 g, 45%). MS(ESI) [M+lf]+= 245.2.
Step 2 - Preparation of4~[Hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-indole-l- carboxylic acid butylamide (520):
[07S5] To 5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (89, 51 mg, 0.26 mmol, prepared as in Example 17) in methanol (2 mL), was added 4-Formyl-indole-l -carboxylic acid butylamide (519, 84 mg, 0.34 mmol) and potassium hydroxide (44 mg, 0.78 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous magnesium sulfate, filtrated and concentrated. The desired compound was isolated with silica gel column chromatography to give an off-white solid (520, 7 mg, 6 %). MS(ESI) [M+Εtf = 440.3.
Step 3 — Preparation of 4-(5-Pyridin-3-yl-lH-pyrrolo [2,3 ~b]pyridine-3-carbonyl)-indole-l -carboxylic acid butylamide (P-1486):
[0756] To 4-[Hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-indole-l -carboxylic acid butylamide (520, 7 mg, 0.016 mmol) in tetrahydrofuran (1 mL) was added Dess-Martin periodinane (7.4 mg, 0.017 mmol). The reaction was stirred at room temperature for 30 minutes, then poured into water and extracted with ethyl acetate. The organic layer was washed with saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, filtrated and concentrated. The desired compound was purified by Prep HPLC using a gradient of buffer A (5% acetonitrile, 95% water, 0.1% formic acid) and buffer B (95% acetonitrile, 5% water, 0.1% formic acid). P-1486 was isolated as a fluffy white solid (2.8 mg, 40%). MS(ESI) [M+HT = 438.3.
[0757] Additional compounds were prepared following the protocol of Scheme 41 , optionally substituting 1-isocyanatobutane with an appropriate isocyanate in Step 1 under suitable base/solvent conditions (dimethylaminopyridine and acetonitrile per Scheme 41 Step 1 or appropriate conditions) and optionally substituting 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 with a suitable 7-azaindole in Step 2. The azaindole was purchased or synthesized as described in Examples 16 and 17. The following compounds were made following this procedure:
( 1 -Benzyloxymethyl- 1 H-indol-4-yl)-(5 -pyridin-3 -yl- 1 H-pyrrolo[2,3 -b]pyridin-3 -yl)-methanone
(P-1523),
(l-Benzenesulfonyl-lH-indol-4-yl)-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1524),
4-(5-Phenyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-mdole-I-carboxylic acid butylamide
(P-1576),
4-[5-(4-Chloro-phenyl)-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl]-indole-l -carboxylic acid butylamide (P-1602),
4-(5-Phenyl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l-carboxylic acid ben2ylamide (P-1611),
4-[5-(4-Chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-indole-l-carboxylic acid benzylamide (P-1618),
4-(5-Bromo-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-indole-l -carboxylic acid butylamide
(P-1687),
4-(5 -Pyridin-3 -yl- 1 H-pyrrolo [2,3 -b]ρyridine-3 -carbonyl)-indole-l -sulfonic acid dimethylamide
(P-1744),
(l-But-2-ynyl-lH-mdol-4-yl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-metlianone
(P-1820),
4-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-indole-l -sulfonic acid butyl-methyl- amide (P-1830),
4-[5-(4-Sulfamoyl-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-indole-l-carboxylic acid butylamide (P-1854),
4-[5-(3-Sulfamoyl-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-indole-l-carboxylic acid butylamide (P-1858),
4-[5-(2-Methoxy-pyrimidin-5-yl)-lH-pyrrolo[2,3-b]pyridme-3-carbonyl]-indole-l -carboxylic acid butylamide (P-1860),
4-(5-Pyrimidin-5-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l-carboxylic acid butylamide
(P-1862),
4-(5-Methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l -carboxylic acid butylamide
(P-1875), and
4-[5-(6-Methoxy-pyridin-3-yl)-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl]-indole-l -carboxylic acid
(pyridin-3-ylmethyl)-amide (P-1887).
The following table indicates the base/solvent (Column 2) and isocyanate used in Step 1 (Column 3), and the 7-azaindole used in Step 2 (column 4) to afford the target compound (Column 5). Column 1 provides the compound number and Column 6 the observed mass.
Step l MS(ESI)
Step 1 Base / Azaindole Compound [M+HY reagent Solvent observed
* per Scheme 41 (i.e. dimethylaminopyridine and acetonitrile)
[0758] The product of Step 2 of Scheme 41 can alternatively be reacted to form the corresponding compounds with methylene linker at the 3 position of the azaindole. For example, 4-(5-ρyridin-3-yl- lH-ρyrrolo[2,3-b]pyridin-3-ylmethyl)-indole-l-carboxylic acid butylamide P-1656 was prepared from 4-[Hydroxy-(5 -pyridin-3 -yl- 1 H-pyrrolo[2,3 -b]pyridin-3 -yl)-methyl] -indole- 1 -carboxylic acid butylamide 520 as shown in Scheme 41a.
Scheme 41a
Step 1 - Preparation of 4-(5-pyridin-3-yl-lH-pyrrolo [2, 3-b]pyridin-3-ylmethyϊ)--indole-l -carboxylic acid butylamide (P-1656):
[0759] A mixture of 4-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-indole-l- carboxylic acid butylamide (520, 18 mg, 0.041 mmol), trifluoroacetic acid (0.5 mL), triethylsilane (1 mL), and acetonitrile (8 mL) was refluxed for 4 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, filtrated and concentrated. The desired compound was purified by Prep HPLC using a gradient of buffer A (5% acetonitrile, 95% water, 0.1% formic acid) and buffer B (95% acetonitrile, 5% water, 0.1% formic acid). P-1656 was isolated as an off-
white solid (4.8 mg, 28%). MS(ESI) [M+H*]* = 424.2.
[0760] The corresponding hydroxy-methyl derivative of Scheme 41 Step 2 was reacted following the protocol of Scheme 41a to prepare 4-[5-(2-methoxy-pyrimidin-5-yl)-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl]-indole-l-carboxylic acid butylamide P-1861, 4-(5-methoxy-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl)-indole-l-carboxylic acid butylamide P-1876, and 4-(5-Pyridin-4-yl-lH-pyrrolo[2,3- b]pyridin-3-ylmethyl)-indole-l-carboxylic acid butylamide P-1877, with structures shown below.
Example 23: Synthesis of (3-Ben2yIoxy-2,6-difluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone P-1467 and related compounds
[0761] Compound P-1467 was synthesized in four steps from 2,4-difluorophenol 35 as shown in Scheme 43. Scheme 43
Step 1 —Preparation ofl-Benzyloxy-2,4-difluoro-benzene (525):
[0762] To 2,4-difluoro-phenol (35, 7.60 g, 0.0584 moϊ) in N,N-dimethylformamide (50.0 mL) were added benzyl bromide (8.0 mL, 0.067 mol) and potassium carbonate (9.00 g, 0.0651 rnol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as white solid (525, 3.20 g, 25%).
Step 2 - Preparation of3-Benzyloxy-2,6-difluoro-benzaldehyde (526):
[0763] To l-Ben2yloxy-2,4-difluoro-benzene (525, 3.00 g, 13.6 mmol) in Tetrahydrofuran (48 mL) under an atmosphere of nitrogen and cooled with dry ice/acetone was added n-Butyllithium (1.60 M in hexane, 8.94 mL). After 20 minutes, N,N-dimethylformamide (1.46 mL, 0.0189 mol) was added to the reaction. After another 20 minutes, the flask was stirred at room temperature for 30 minutes. The reaction mixture was poured into water, acidified to pH = 1, and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound as a yellow solid (526, 2.5g, 74%).
Step 3 - Preparation of(3~Benzyloxy-2,6-difluoro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3~b]pyridin- 3-yl)-methanol (527):
[0764] To 5-Pyridm-3-yl-lH-pyrrolo[2,3-b]pyridine (89, 750.0 mg, 0.003842 mol, prepared as in Example 17) in methanol (20.0 mL) were added 3-Benzyloxy-2,6-difluoro-benzaldehyde (526, 1.12 g, 4.5 mmol) and potassium hydroxide (1.50 g, 0.0267 mol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight and then poured into water, acidified with IN HCl to pH around 2 and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound (527, 700 mg, 35%).
Step 4 - Preparation of(3-Benzyloxy-2, 6-difluoro-phenyl)-( 5-pyridin-3-yl-lH-pyrrolo[2, 3-bJpyridin- 3-yl)-methanone (P-1467):
[0765] To (3-Benzyloxy-2,6-difluoro-phenyl)-( 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanol (527, 300.0 mg, 0.68 mmol) in tetrahydrofuran (10.0 mL) was added Dess-Martin periodinane (344 mg, 0.81 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction mixture was concentrated with silica and purified with silica gel column chromatography eluting with 10% methanol in dichloromethane to give the compound (P-1467, 240 mg, 80%).
[0766] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-[2,6-difluoro-3-(2-methoxy-ethoxy)-ρhenyl]- methanone P-1453
was prepared following the protocol of Scheme 43, substituting benzyl bromide with l-Bromo-2- methoxy-ethane in Step 1 and 5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine with 5-Bromo-lH- pyrrolo[2,3-b]pyridine (67) in Step 3. MS (ESI) [M + H
+I
+ = 410.1, 412.1.
[0767] [2,6-Difluoro-3-(2-methoxy-ethoxy)-phenyl]<5-methoxy-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone P-1584
was prepared following the protocol of Scheme 43, substituting benzyl bromide with l-Bromo-2- methoxy-ethane in Step 1 and 5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine with 5-methoxy-lH- pyrrolo[2,3-b]pyridine (104, prepared as in Example 22) in Step 3. MS (ESI) [M + H
+]
"1" = 363.2.
[0768] (3-Benzyloxy-2,6-difluoro-phenyl)-(5-methoxy-lH-pyrroloL2,3-b]ρyridin-3-yl)-methanone P-1597
was prepared following the protocol of Scheme 43, substituting 5-Pyridin-3-yl-lH-pyrrolo[2,3- b]ρyridine with 5-methoxy-lH-pyrrolo[2,3-b]ρyridine (104, prepared as in Example 16) in Step 3. MS(ESI) [M + H
+J
+ = 395.2.
[0769] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2,6-difluoro-3-methoxy-phenyl)-methanone P- 1386
was prepared following the protocol of Steps 2, 3 and 4 of Scheme 43, substituting l-Benzyloxy-2,4- difluoro-benzene 525 with 2,4-Difluoro-l-methoxy-benzene in Step 2 and 5-Pyridin-3-yl-lH- pyrrolo[2,3-b]ρyridine with 5-bromo-lH-pyrrolo[2,3-b]pyridine (67) in Step 3. MS (ESI) [M + H
+]* = 367.0, 369.0.
[0770] (3-Benzyloxy-2,6-difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone P-1802
was prepared following the protocol of Scheme 43, substituting 5-pyridin-3-yl-lH-pyrrolo[2,3- b]pyridine with 7-azaindole. To a solution of (3-benzyloxy-2,6-difluoro-ρhenyl)-(lH-pyrrolo[2,3- Z>]ρyridin-3-yl)-methanone (P-1802, 0.5 g, 1.37 mol) in methanol (70 niL) and tetrahydrofuran (30 mL) was added palladium on carbon (120 mg, 10% wt, 0.58 mol). The mixture was stirred under hydrogenation (60 psi) for six hours. After removal of solvent, the residue was dried under vacuum, which provided (2,6-Difluoro-3-hydroxy-phenyl)-(lH-pyrrolo[2,3-b]pyridine-3-yl)-methanone 651
as a white solid (363 mg, 96%). MS (ESI) [M+ϊtγ = 275.36.
[0771] Additional compounds were prepared following steps 3 and 4 of Scheme 43, replacing 3- benzyloxy-2,6-difluoro-benzaldehyde 526 with an appropriate aldehyde and/or pyridin-3-yl-lH- pyrrolo[2,3-b]pyridine 89 with an appropriate azaindole in Step 3. The azaindoles used were synthesized as described in Examples 9 or 16. The aldehydes used were synthesized as described in Example 5 or 21. The following compounds were made following this procedure:
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3,5-dimethoxy-phenyl)-methanone (P-1463),
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-fluoro-5-trifluoromethyl-phenyl)-methanone,
(5-Bromo-lH-pyrrolo[2,3-b]pyridm-3-yl)-(3-trifluoromethoxy-phenyl)-methanone,
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-trifluoromethyl-phenyl)-methanone,
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3,5-dichloro-phenyl)-methanone,
(2)6-Dichloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone,
(2,6-Difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone,
(2,6-Dimethyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone,
(2,6-Dichloro-phenyl)-(5-ρyridin-3-yl-lH-pyπ"olo[2,3-b]pyridin-3-yl)-methanone,
(2,6-Difluoro-ρhenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone,
(2,6-Dimethyl-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone,
(5-Bromo-lH-pyrrolo[2,3-b]ρyridin-3-yl)-(2,2-difluoro-benzo[l,3]dioxol-4-yl)-methanone (P-
1513),
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-difluoromethoxy-phenyl)-methanone (P-1514)J
(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2,2-difluoro-benzo[l,3]dioxol-5-yl)-methanone (P-
1551),
N-[3 -(5-Chloro- 1 H-pyrrolo [2, 3 -b]pyridine-3 -carbonyl)-2,4-difluoro-phenyl]-4-trifluoromethyl- benzenesulfonamide (P-1541),
N-[2,4-Difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-trifluoromethyl- benzenesulfonamide (P-1542),
N-[2,4-Difluoro-3-(5-methoxy-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-fluoro- benzenesulfonamide (P-1581),
N-[2,4-Difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-fluoro- benzenesulfonamide (P-1582),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-ρhenyl]-3-fluoro- benzenesulfonamide (P-1583),
N-[2,4-Difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-trifluoromethyl- benzenesulfonamide (P-1598),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3-trifluoromethyl- benzenesulfonamide (P-1599),
(5-Chloro-lH-pyrrolot2,3-b]pyridin-3-yl)-{2-fluoro-5-methoxy-4-[2-(2-methoxy-ethoxy)- ethoxy] -phenyl} -methanone (P-2003),
(4-Benzyloxy-2,6-difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-2020), and
[4-(4-Chloro-benzyloxy)-3-methoxy-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1698).
The following table indicates the aldehyde (column 2) and the azaindole (column 3) used to afford the target compound (column 4). Column 1 provides the compound number and column 5 the observed mass.
Example 24: Synthesis of 3-(3-BenzyIoxy-2,6-difluoro-benzyI)-5-pyridin-3-yl-lH-pyrrolo[2,3- bjpyridine P-1455:
[0772] Compound P-1455 was synthesized in four steps from 2,4-difluorophenol 35 as shown in Scheme 43a.
Scheme 43a
[0773] Steps 1 -3 are identical to Steps 1-3 of Scheme 43.
Step 4 - Preparation of3-(3-Benzyloxy-2,6-difluoro-benzyl) -5-pyridin-3-yl-lH-pyrrolo[2,3- b] pyridine (P-1455):
[0774] To (3-Benzyloxy-2,6-difluoro-phenyl)-( 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridm-3-yl)- methanol (527, 580.0 mg, 1.3 mmol) in acetonitrile (29.0 mL) were added trifluoroacetic acid (1.9 mL, 0.025 mol) and triethylsilane (3.9 mL, 0.024 mol). The reaction was stirred at 80 0C for 1 hour. The reaction was poured into water, basified with 1 M potassium carbonate to pH = 4, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 50% ethyl acetate in hexane to give as a yellow solid (P-1455, 530 mg). MS(ESI) [M+Εtf = 428.3.
[0775] 5-Bromo-3-[2,6-difluoro-3-(2-methoxy-ethoxy)-benzyl]-lH-ρyrrolo[2,3-b] pyridineP-1454
was prepared following the protocol of Scheme 43a by substituting benzyl bromide with l-Bromo-2- methoxy-ethane in Step 1 and 5-Pyridm-3-yl-lH-pyrrolo[2,3-b]pyridine with 5-Bromo-lH- ρyrrolo[2,3-b]pyridine (67) in Step 3. MS (ESI) [M + ϊtf = 410.1, 412.1.
[0776] Additional compounds were prepared following steps 3 and 4 of Scheme 43a, replacing 3- benzyloxy-2,6-difluoro-benzaldehyde 526 with an appropriate aldehyde and/or pyridin-3-yl-lH- pyrrolo[2,3-b]pyridine 89 with an appropriate azaindole (see Example 9 or Example 16) in Step 3. The following compounds were made following this procedure:
5-Bromo-3-(3,5-dimethoxy-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1464),
3-(3,5-Bis-difluoromethoxy-benzyl)-5-bromo-lH-ρyrrolo[2,3-b]pyridine (P-1538),
3-(2,6-Dichloro-benzyl)-lH-pyrrolo[2,3-b]pyridbe (P-1483),
3-(2,6-Difluoro-benzyl)-lH-pyrrolo[2,3-b]pyridme (P-1482),
3-(2,6-Dimethyl-benzyl)-l H-pyrrolo[2,3-b]pyridine (P-0333),
3-(2,6-Dichloro-benzyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (P-1478),
3-(2,6-Difluoro-benzyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridme (P-1477),
3-(2,6-Dimethyl-benzyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (P-1481),
N-[2,4-Difluoro-3-(5-methoxy-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-3-trifluoromethyl- benzenesulfonamide (P-1590),
N-[2,4-Difluoro-3-(5-chloro-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-3-trifluoromethyl- benzenesulfonamide (P-1600),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-ρhenyl]-methanesulfonamide (P-1603), and
N-[2,4-Difluoro-3-(5-pyridm-3-yl-lH-ρyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]- methanesulfonamide (P-1605).
The following table indicates the aldehyde (column 2) and the azaindole (column 3) used to afford the target compound (column 4). Column 1 indicates the compound number and column 5 the observed mass.
Example 25: Synthesis of 3-[2,6-difluoro-3-(propane-l-sulfonylamino)-benzoy]]-lH-pyrrolo[2,3- b]pyridine-5-carboxylic acid ethylamide P-1630
[0777] Compound P-1630 was synthesized in six steps from 5-bromo-l -triisoρropylsilyl-7- azaindole 68 as shown in Scheme 45.
Scheme 45
Step 1 -Preparation ofl-Triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine-5-carboxylic acid methyl ester (531):
[0778] To 5-bromo-l -triisopropylsilyl-7-azaindole (68, 1.50 g, 4.2 mmol, prepared as described in Example 6) in tetrahydrofuran (20.0 niL) under an atmosphere of nitrogen, cooled with dry ice/acetone, was slowly added n-Butyllithium (10.0 M in hexane, 0.467 mL). After 60 minutes,
methyl chloroformate (0.394 mL, 5.1 mmol) was added to the reaction. After another hour, the reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to give the crude compound as a light yellow solid that was used directly in the next step.
Step 2 - Preparation oflH-Pyrrolo[2,3-b]pyridine-5-carboxylic acid methyl ester (532):
[0779] To l-Triisopropylsilanyl-lH-ρyrrolo[2,3-b]pyridine-5-carboxylic acid methyl ester (531,
0.950 g, 2.9 mmol) in tetrahydrofuran (20.0 mL) was added tetrabutyl-ammonium fluoride, trihydrate
(1.20 g, 3.8 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction was concentrated and purified with silica gel column chromatography eluting with 4% methanol in methylene chloride to give the compound as a white solid (532, 300 mg, 60%). MS(ESI) [MH-H+I+ =
177.2.
Step 3 — Preparation of 3- [2, 6-difluoro-3~(propane-l-sulfonylamino)-phenyl]-hydroxy-methyl-lH- pyrrolo[2,3-b]pyridine-5-carboxylic acid methyl ester (P-1545):
[0780] To lH-Pyrrolo[2,3-b]pyridine-5-carboxylic acid methyl ester (532, 155.0 mg, 0.88 mmol) in methanol (15.0 mL) were added propane- 1 -sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide (73, 260.0 mg, 0.99 mmol, prepared as described in Example 7) and potassium hydroxide (859 mg, 15.3 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as white solid (P- 1545, 110 mg, 28%). MS(ESI) [M+HY = 440.2.
Step 4 — Preparation of3-[2, 6~difluoro-3-(propane-lsulfonylamino)-benzoyl]-lH-pyrrolo[2, 3- bJpyridine-S-carboxylic acid methyl ester (P-1552):
[0781] To 3-[2,6-difmoro-3-(propane-l -sulfonylamino)-phenyl]-hydroxy-methyl-lH-pyrrolo[2,3- b]ρyridine-5-carboxylic acid methyl ester (P-1545, 100.0 mg, 0.23 mmol) in tetrahydrofuran (10 mL) was added Dess-Martin periodinane (107 mg, 2.5 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction mixture was concentrated with silica gel and then purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound as white solid (P-1552, 80 mg, 80%). MS(ESI) [MH-H+J+ = 438.2.
Step 5 - Preparation of3-[2, 6~difluoro-3-(propane~l-sulfonylamino)-benzoyl]-lH-pyrrolo[2, 3- bJpyridine-5-carboχylic acid (P-1559):
[0782] To 3-[2,6-difluoro-3-(propane-l -sulfonylamino)-benzoyl]-lH-pyrrolo[2,3~b]ρyridine-5- carboxylic acid methyl ester (P-1552, 80.0 mg, 0.18 mmol) in tetrahydrofuran (10.0 mL) were added water (3.0 mL) and lithium hydroxide (82 mg, 3.4 mmol). The reaction was stirred at room
temperature overnight. The reaction was poured into water, acidified with 1 N HCl to pH around 1, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, filtered, concentrated, and washed with ethyl acetate to give an off-white solid (P-1559, 60 mg, 77%) MS(ESI) [M+HT = 424.2.
Step 6: Preparation of3-[2, 6-difluoro-3-(propane-l-sulfonylamino)-benzoyl]-lH-pyrrolo[2,3- bJpyridine-5-carboxylic acid ethylamide (P-1630):
[0783] To 3-[2,6-difluoro-3-(ρroρane-l -sulfonylamino)-benzoyl]-lH-pyrrolo[2,3-b]pyridine-5- carboxylic acid (P-1559, 38.0 mg, 0.090 mmol) in tetrahydrofuran (2.3 mL) was added a solution of ethylamine (2.0 M in tetrahydrofuran, 0.20 mL), bromo-tris-pyrrolidino-phosphonium hexafluorophosphate (80.0 mg, 0.17 mmol) and triethylamine (0.30 mL, 2.2 mmol) under an atmosphere of nitrogen. The reaction mixture was stirred overnight at room temperature. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 40% ethyl acetate in hexane to give the compound as a white solid (P- 1630, 13.2 mg, 33%). MS(ESI) [M-H+]" = 449.0.
Example 26: Synthesis of l-butyl-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yImethyl)- pheπyl]-urea P-1445.
[0784] Compound P-1445 was synthesized in six steps from 5-pyridm-3-yl-lH-ρyrrolo[2,3- b]pyridine 89 as shown in Scheme 49.
Scheme 49
Step 1 - Preparation of(3-nitro-phenyl)~(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (P- 1399):
[0785] To 3-nitrobenzaldehyde (534, 1.08 g, 7.17 mmol) in methanol (34 niL) was added 5- pyridm-3-yl-lH-ρyrrolo[2,3-b]pyridine (89, 1.08 g, 5.52 mmol, prepared as described in Example 17) and potassium hydroxide (1.55 g, 27.6 mmol). The reaction was stirred at room temperature for four hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The mixture was purified by silica gel column chromatography eluting with 4% methanol in dichloromethane to provide two different compounds, a white solid (P-1399, R = H, 1.20 g, 63%) MS(ESI) 01+H+J+ = 347.2, and a light yellow solid (535, R = Me, 0.434 g, 22%).
Step 2 - Preparation of(3-nitro-phenyl)~(5-pyridin-3-yl-lH-pyrrolo[2, 3-b]pyridin-3-yl)-methanone
(536):
[0786] To (3-nitro-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (P-1399,
R=H, 500 mg, 1.44 mmol) in dimethylformamide (26 mL) was added Dess-Martin periodane (674 mg, 1.59 mmol). The reaction was stirred for one hour and the reaction was poured into water. All solids were filtered and purified by silica gel column chromatography eluting with 3 % methanol in dichloromethane to give the compound (536, 295 mg, 59 %). MS(ESI)[M^-H+J+ = 345.2.
Step 3 - Preparation of(3-nitro-phenyl)-[5~pyridin-3-yl-l-(toluene-4~sulfonyl)-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone (537) :
[0787] To (3-nitro-phenyl)-(5-ρyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (536, 291 mg, 0.85 mmol) in tetrahydrofuran (7 mL) was added 1.5 M of lithium diisopropylamide in cyclohexane (676 μl, 1.59 mmol) at -78 0C under an atmosphere of nitrogen. After 30 minutes, p- toluenesulfonyl chloride (209 mg, 1.10 mmol) was added in tetrahydrofuran and the reaction was stirred for three hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. All solids were filtered and purified by silica gel column chromatography eluting with 60 % ethyl acetate in hexane to give the compound (537, 182 mg, 43 %). MS(ESI)[IyB-H+]"1" = 499.2.
Step 4 - Preparation of(3-Amino-phenyl)-[5-pyridin-3-yl-l-(toluene-4-sulfonyl)-lH-pyrrolo[2,3- bJpyridin-3-ylJ-methanone (538):
[0788] To (3-nitro-phenyl)-[5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (537, 180 mg, 0.361 mmol) in methanol (4 mL) was added 10% palladium on carbon (20 mg) and few drops of concentrated aqueous hydrochloric acid. The resulting mixture was stirred under an atmosphere of hydrogen overnight and the catalyst was filtered out through a bed of celite. The filtrate was concentrated and purified by silica gel column chromatography eluting with 90 % ethyl acetate in hexane to give the compound (538, 58 mg, 34 %). MS(ESI)[M+Hf]+ = 469.3.
Step 5 - Preparation ofl-Butyl-3-3-[5-pyridin-3-yl-l-(toluene-4-sulfonyl)-lH-pyri'olo[2,3- bJpyridine-3-carbonylJ-phenyl-urea (539):
[0789] To (3-Amino-phenyl)-[5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridin-3- yl]-methanone (538, 53 mg, 0.11 mmol) in tetrahydrofuran (1.6 mL) was added 1 -isocyanatobutane (12 mg, 0.12 mmol). The reaction was heated at 90 0C overnight and it was concentrated and purified by silica gel column chromatography eluting with 2 % methanol in dichloromethane to give the compound (539, 39 mg, 61%). MS(ESI)[M+^]4" = 568.4.
Step 6 - Preparation ofl-Butyl-3-[3-(5-pyridin-3~yl-lH-pyrrolo[2,3~b]pyridine-3-carbonyl)-phenyl]- urea (P-1445):
[0790] To 1 -Butyl-3-3-[5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl-urea (539, 33 mg, 0.058 mmol) in tetrahydrofuran (2 mL) was added 1.0 M tetra-n- butylammonium fluoride in tetrahydrofuran (192 μl) under an atmosphere of nitrogen and the reaction was stirred for three hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. All solids were filtered and purified by silica gel column chromatography eluting with 4 % methanol in dichloromethane to give the compound (P-1445, 8 mg, 30 %). MS(ESI)PVB-IrT = 414.3.
Example 27: Synthesis of l-butyI-3-[3-(5-pyridin-3-yHH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenyl]-urea P-1447
[0791] Compound P-1447 was synthesized in five steps from 3-[(3-nitro-phenyl)-methoxy-methyl]- 5-ρyridm-3-yl-lH-pyrrolo[2,3-b]pyridme 535 as shown in Scheme 50.
Scheme 50
Step 1 - Preparation of3-(3-nitro-benzyl)-5-pyridin-3-yl~lH-pyrrolo[2,3-b]pyridine (P-1402): [0792] To 3-[(3-nitro-phenyl)-methoxy-methyl]-5-pyridin-3-yl-lH-pyrrolo[2J3-b]pyridine (535, 431 mg, 1.20 mmol, per Example 26, Scheme 49 Step 1) in acetonitrile (130 mL), were added trifluoroacetic acid (18 mL, 230 mmol) and triethylsilane (36 mL, 230 mmol). The reaction was refluxed for three hours. The reaction mixture was poured into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 80% ethyl acetate in hexane to give the compound (P-1402, 323 mg, 82%). MS(ESI)[MH-H+J+ = 331.2.
Step 2 - Preparation of3-(3-nitro-benzyl)~5-pyridin-3-yl-l-(toluene-4-sulfonyl)~lH-pyrrolo[2,3- bjpyridine (552):
[0793] To 3-(3-nitro-benzyl)-5-pyridm-3-yl-lH-ρyrrolo[2,3-b]pyridine (P-1402, 141 mg, 0.43 mmol) in N,N-dimethylformamide (3 mL) was added sodium hydride (60% dispersion in mineral oil, 21 mg, 0.512 mmol) under an atmosphere of nitrogen. After thirty minutes, p-toluenesulfonyl chloride (114 mg, 0.60 mmol) in N,N-dimethyl-formamide was added and the reaction was stirred for three hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. All solids were filtered and purified by silica gel column chromatography eluting with 40 % ethyl acetate in hexane to give the compound (552, 120 mg, 58 %). MS(ESI)[M+H+]+ = 485.25.
Step 3 ~ Preparation of3-[5-Pyridin-3-yl-l-(toluene~4-sulfonyl)-lH-pyrrolo[2,3-b]pyridin~3- ylmethy I] -phenyl amine (553) :
[0794] To 3-(3-nitro-benzyl)-5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridine (552, 230 mg, 0.14 mmol) in methanol (5 mL) was added 10% palladium on carbon (10 mg) and few drops of concentrated aqueous hydrochloric acid. The resulting mixture was stirred under an atmosphere of hydrogen overnight, and the catalyst was filtered out through a bed of celite. The filtrate was concentrated and purified by silica gel column chromatography eluting with 90 % ethyl acetate in hexane to give the compound (553, 88 mg, 41 %). MS(ESl)[M+H+]+ = 455.3.
Step 4 ~ Preparation ofl~butyl-3-3-[5-pyridin-3-yl-l-(toluene-4-sulfonyl)-lH-pyrrolo[2, 3-bJpyridin- 3-ylmethyl] -phenyl-urea (554) :
[0795] To 3-[5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridm-3-ylmethyl]- phenylamine (553, 14 mg, 0.031 mmol) in tetrahydrofuran (0.5 mL) was added 1-isocyanatobutane (3.4 mg, 0.03 mmol). The reaction was heated at 90 0C overnight and was concentrated and purified by silica gel column chromatography eluting with 2 % methanol in dichloromethane to give the compound (554, 7.2 mg, 42 %). MS(ESI)[M+H+]+ = 554.4.
Step 5 - Preparation ofl-Butyl-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2, 3-b]pyridin-3-ylmethyl)-phenyl]- urea (P-1447):
[0796] To 1 -butyl-3-3-[5-pyridin-3-yl-l -(toluene-4-sulfonyl)-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl]-phenyl-urea (554, 11 mg, 0.02 mmol) in tetrahydrofuran (0.7 mL) was added 1.0 M tetra-n- butylammonium fluoride in tetrahydrofuran (66 μl) under an atmosphere of nitrogen and the reaction was stirred for three hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. All solids were filtered and purified by silica gel column chromatography eluting with 4 % methanol in dichloromethane to give the compound (P-1447, 2.5 mg, 31 %). MS(ESI)[M+ϊf ]+ = 400.3.
[0797] 1 -Cyclopentyl-3-[3-(5-pyridm-3-yl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-urea P- 1446
was prepared following the protocol of Scheme 50, substituting 1-isocyanatobutane with isocyanato- cyclopentane in Step 4. MS(ESI) [M + H+]"1" = 412.4.
Example 28: Synthesis of 3-[3-chIoro-4-(4-chIoro-benzyloxy)-benzyI]-lH-pyrrolo[2,3-b]pyridine P-1449.
[0798] Compound P-1449 was synthesized in three steps from 3-chloro-4-hydroxy-benzaldehyde 556 as shown in Scheme 51.
Scheme 51
Step 1 -Preparation of3-chloro-4-(4-chloro-ben∑yloxy)-benzaldehyde (558): [0799] To acetonitrile (15.0 mL) were added 3-chloro-4-hydroxy-benzaldehyde (556, 0.6 g, 4 mmol), 4-chlorobenzyl bromide (557, 1.2 g, 6 mmol), and potassium carbonate (0.9 g, 7 mmol). The reaction was heated to 150 0C for 10 minutes in a CEM Discover microwave instrument. The reaction was poured into water, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate: hexanes) (558, 0.85 g, 76%).
Step 2 — Preparation of3-[3-chloro-4-(4-chloro-ben∑yloxy)-phenyl]-methoxy-methyl-lH~pyrrolo[2,3- bjpyridine (559):
[0800] lH-Pyrrolo[2,3-b]pyridine (94, 0.3 g, 2.8 mmol) was mixed with 3-chloro-4-(4-chloro- ben2yloxy)-benzaldehyde (558, 0.8 g, 3 mmol), potassium hydroxide (0.9 g, 17 mmol) and methanol (90.0 mL). The reaction was heated to 50 0C under an atmosphere of nitrogen for six days. After neutralization with 6N hydrochloric acid the reaction was poured into water, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate: hexanes) to give a yellow solid (559, 0.6 g, 41%). MS(ESI) [M + H+J+= 413.2, 415.2 [M- H+]"= 411.1, 413.1.
Step 3 - Preparation of3-[3-chlorO'4-(4~chloro-benzyloxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P- 1449):
[0801] 3-[3-Chloro-4-(4-chloro-beiizyloxy)-phenyl]-methoxy-methyl-lH-pyrrolo[2,3-b]pyridine (559, 0.2 g, 0.6 mmol) was mixed with trifluoroacetic acid (0.226 mL, 3 mmol), triethylsilane (0.4 mL, 3 mmol) and acetonitrile (5 rαL). The reaction was heated at 50 0C and stirred for two days. The reaction was concentrated. The residue was diluted with ethyl acetate and neutralized with 2M aqueous sodium hydroxide. The reaction was poured into water, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography (ethyl acetate: hexanes) to give a yellow solid (P-1449, 0.0744 g, 33%). MS(ESI) [M + ET1I+= 383.2, 385.2.
[0802] Additional compounds were prepared following the protocol of Scheme 51, replacing 3- chloro-4-hydroxy-benzaldehyde 556 with an appropriate aldehyde and optionally replacing 4- chlorobenzyl bromide 557 with an appropriate benzyl halide in Step 1, and optionally replacing IH- pyrrolo[2,3-b]pyridine 94 with an appropriate azaindole in Step 2. The following compounds were made following this procedure:
3-[4-(4-chloro-benzyloxy)-2-methoxy-benzyl]-lH-ρyrrolo[2,3-b]ρyridine (P-1450),
3-[4-(4-Chloro-benzyloxy)-benzyl]-lH-ρyrrolo[2,3-b]pyridine (P-1462), 3-[4-(4-Chloro-benzyloxy)-3-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]ρyridine (P-1466), 3-[4-(4-Chloro-benzyloxy)-3-ethoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1470), 3-[2-Chloro-4-(4-chloro-benzyloxy)-benzyl]-lH-ρyrrolo[2,3-b]pyridine (P-1471), 3-[4-(4-Chloro-benzyloxy)-3-trifluoromethoxy-benzyl]-lH-pyrrolot2,3-b]pyridine (P-1487), 3-[4-(4-Chloro-benzyloxy)-3-methoxy-benzyl]-5-methoxy-lH-pyrrolo[2,3-b]ρyridine (P-1531), 5-Chloro-3-[4-(4-chloro-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridme (P-1532), 3-[4-(4-Chloro-2-fluoro-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]ρyridine (P-1544), 3-[4-(2,4-Dichloro-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1568), 3-[3-Methoxy-4-(4-methoxy-benzyloxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1569), 3-[3-Methoxy-4-(2,4,6-trifluoro-benzyloxy)-ben2yl]-lH-pyrrolo[2,3-b]pyridine (P-1578), 3-[4-(2,6-Dichloro-benzyloxy)-3-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1579), and 3-[3- Chloro-4-(4-chloro-benzyloxy)-5-methoxy-benzyl]-lH-ρyrrolo[2,3-b]pyridine (P-1616). The following table indicates the aldehyde (Column 2), the benzyl halide (Column 3), and the azaindole (Column 4) used to afford the target compound (Column 5). Column 1 indicates the compound number and column 6 the observed mass.
Example 29: Synthesis of 3-(4-benzyloxy-3-methoxy-benzyI)-lH-pyrrolo[2,3-bJpyridine P-1613.
[0803] Compound P-1613 was synthesized in two steps from 4-benzyloxy-3-methoxy- benzaldehyde 564 as shown in Scheme 53.
Scheme 53
Step 1 - Preparation of3~[(4-benzyloxy-3-methoxy-phenyl)-methoxy-methyl]-lH-pyrrolo[2,3- b] pyridine (565):
[0804] Methanol (125 mL) and potassium hydroxide (4.4 g, 79 mmol) were mixed with IH- pyrrolo[2,3-b]pyridine (94, 3.1 g, 26.6 mmol) and 4-benzyloxy-3-methoxy-benzaldehyde (564, 12.9 g, 53.2 mmol). The reaction was stirred at room temperature for 2 days. The resulting white solid was filtered and washed with water. Crude material was carried forward without further purification.
Step 2 ~ Preparation of 3-(4-benzyloxy-3-methoxy-benzyl)-lH-pyrrolo[2,3-b] pyridine (P-1613): [0805] 3 -[(4-Benzyloxy-3 -methoxy-phenyl)-methoxy-methyl] - 1 H-pyrrolo [2,3-b]pyridine (565, 0.9g, 2.4 mmol) and acetonitrile (50 mL) were mixed with trifluoroacetic acid (0.360 mL, 4.7 mmol) and triethylsilane (0.746 mL, 4.7 mmol). The reaction was heated at 80 0C and stirred overnight. The reaction was concentrated. The mixture was extracted with ethyl acetate and saturated sodium bicarbonate. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The desired compound was isolated by silica gel column chromatography to give the compound (P- 1613, 0.454 g 54.8%). MS(ESI) [M + H^]+= 345.3.
Example 30: Synthesis of l-[3-(5-bromo-lH-pyrroIo[2,3-b]pyridine-3-carbonyl)-phenyl]-3- butyl-urea P-1596.
[0806] Compound P-1596 was synthesized in one step from 5-bromo-lH-pyrrolof2,3-b]pyridine 67 as shown in Scheme 55.
Scheme 55
Step 1 - Preparation ofl-[3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]-3-butyl-urea (P-1596):
[0807] To aluminum trichloride (3.67 g, 0.0275 mol) in dichloromethane (100 mL, 2 mol) under an atmosphere of nitrogen was added 5-bromo-7-azaindole (67, 1.08 g, 0.00548 mol) at room temperature. After one hour, 3-isocyanato-benzoyl chloride (584, 5.00 g, 0.0275 mol) was added under an atmosphere of nitrogen at room temperature. The resulting mixture was stirred over night at room temperature. 1-Butanamine (585, 54 mL, 0.54 mol) was added carefully. All solvents were removed. The residue was purified by silica gel column chromatography to give the compound (P- 1596, 172 tag, 8%). MS(ESI) [M-H+]- = 413.1, 415.0.
[0808] Additional compounds were prepared following the protocol of Scheme 55, replacing 1 - butanamine 585 with an appropriate amine and optionally replacing 5-bromo-7-azaindole 67 with 5- pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine 89 (prepared as described in Example 17). The following compounds were made following this procedure: l-Benzyl-3-[3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1553), 1 -Benzyl-3 -[3 -(5 -pyridin-3 -yl- 1 H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl] -urea (P-I 554), l-(2-Methoxy-ethyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1566), and l-Phenyl-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1591). The following table indicates the amine (column 2) and the azaindole (column 3) used to afford the target compound (column 4). The compound number is provided in column 1 and the observed mass in column 5.
Example 31: Synthesis of l-Butyl-3-3-[5-(l-methyl-lH-pyrazol-4-yl)-lH-pyrrolo[2,3-b]pyridine- 3-carbonyl]-phenyl-urea P-1880
[0809] Compound P-1880 was synthesized in one step from l-[3-(5-bromo-lH-pyrrolo[2,3- b]pyridine-3-carbonyl)-phenyl] -3 -butyl-urea P-1596 as shown in Scheme 56.
Scheme 56
Step 1 - Preparation ofl-Butyl-3-3-[5-(l-methyl-lH-pyrazol-4-yl)-lH-pyrrolo[2,3-b]pyridine-3- carbonylj-phenyl-urea (P-1880):
[0810] In a microwave tube, l-[3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]-3-bιιtyl- urea (P-1596, 0.077 g, 0.00018 mol, prepared as described in Example 30, Scheme 55), l-Methyl-4- (4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyτazole (0.0964 g, 0.000464 mol), and Tetrakis(triphenylphosphine)palladium(0) (0.011 g, 0.0000093 mol) were mixed in 1.00 M of potassium carbonate in water (1.2 mL), acetonitrile (2.0 mL, 0.037 mol), and tetrahydrofuran (1.0 mL, 0.012 mol). The resulting mixture was heated at 100 0C in the microwave for 20 minutes, then at 120 0C for 10 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography to give the compound (P-1880, 52 mg, 67%). MS(ESI)PvMf]+ = 417.4.
Example 32: Synthesis of l-Butyl-3-[2-chloro-3-(5-pyridin-3-yHH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-urea P-1828
[0811] Compound P-1828 was synthesized in two steps from 3-amino-2-chlorobenzoic acid 586 as shown in Scheme 57.
Scheme 57
Step 1 -Preparation of-(3~Butyl-ureido)-2~chloro-benzoic acid (587):
[0812] To N,N-diisoproρylamine (1.72 mL, 0.0122 mol) in tetrahydrofuran (12 mL, 0.14 mol), was added 1.6 M n-butyllithium in hexane (7.6 mL) at -78 0C under an atmosphere of nitrogen. After 30 minutes, 3-amino-2-chlorobenzoic acid (586, 1.00 g, 0.00583 mol) was added. After another 30 minutes, 1-isocyanatobutane (2.60 mL, 0.0233 mol) was added at -78 °C under an atmosphere of nitrogen and allowed to stir for two hours. The reaction mixture was warmed up to room temperature
and stirred at room temperature for 30 minutes. The reaction was quenched with IM HCl (aqueous) solution and extracted with ethyl acetate twice. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with dichloromethane:methanol:acetic acid 40:2:1 to give the compound as an off-white solid (587, 147 mg, 9%).
Step 2 ~ Preparation ofl-Butyl~3-[2-chloro-3-(5~pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl] -urea (P-1828):
[0813] To 3-(3-Butyl-ureido)-2-chloro-benzoic acid (587, 103 mg, 0.000380 mol) was added dichloromethane (10 mL, 0.2 mol) followed by thionyl chloride (110 μL, 0.0015 mol) and 1 drop of dimethylformamide to give a suspension. The reaction was stirred at room temperature for 2 hours. Solid material was still present in the reaction mixture, so tetrahydrofuran (0.5 mL, 0.006 mol) was added and continued stirring at room temperature. The reaction became a clear solution after 2 hours, then stirred for another hour. All volatiles were removed under vacuum and the residue stripped from toluene, twice. The solid was then dried under high vacuum for 60 minutes and dissolved in dichloromethane (5 mL). This was added to 5-(pyridin-3-yl)-lH-pyrrolo[2,3-b]pyridine (89, 0.074 g, 0.00038 mol, prepared as described in Example 17) which had been treated with aluminum trichloride (0.25 g, 0.0019 mol) in dichloromethane (10 mL) for 1 hour. The reaction was stirred at room temperature overnight, then quenched with methanol (5 mL). The resulting solution was extracted with ethyl acetate and water with added saturated sodium bicarbonate to adjust pH ~ 8. The organic layer was washed with sodium bicarbonate and brine and dried over magnesium sulfate and filtered. The organic layer was concentrated and purified by silica gel column chromatography eluting with 2% methanol in dichloromethane followed by 5% methanol in dichloromethane to give the compound as a white solid (P-1828, 45 mg, 26%). MS(ESI) [M+lf"]+= 448.3.
[0814] Additional compounds were prepared following the protocol of Scheme 57, replacing 3- amino-2-chlorobenzoic acid 586 with an appropriate carboxylic acid and optionally replacing 1- isocyanatobutane with an appropriate isocyanate in Step 1 and optionally replacing 5-(ρyridin-3-yl)- lH-pyrrolo[2,3-b]pyridine 89 with an appropriate substituted 7-azaindole (see Example 17) in Step 2. The following compounds were made following this procedure: l-Butyl-3-[2-methyl-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-
1742),
3-Butyl-l -methyl-1 -[2-methyl-3-(5-pyridin-3-yl-lH- pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- urea (P-1855),
[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-4-fluoro-phenyl]-urea (I>-1570),
[4-Fluoro-3-(5-phenyl-lH-pyrrolof2, 3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1589),
3-{3-[5-(3-Butyl-ureido)-2-fluoro-benzoyl]-lH-pyrrolo[2,3-b]pyridin-5-yl}-benzamide (P-1621), l-Butyl-3-{4-fluoro-3-[5-(3-methane sulfonyl-phenyl)-lH-pyrrolo[2,3~b]pyridine-3-carbonyl]- phenyl} -urea (P-1627), and
1 -[3-(5-Bromo-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-4-fluoro-phenyl]-3 -butyl-urea (P-1637). The following table indicates the carboxylic acid (column 2), the isocyanate (column 3), and the azaindole (column 4) used to afford the target compound (column 5). Column 1 indicates the
Example 33: Synthesis of l-Butyl-3-[4-fluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-urea (P-1534):
[0815] Compound P-1534 was synthesized in two steps from 5-(3-butylureido)-2-fluorobenzoic acid 588 (prepared from 3-fluoro-5-aminobenzoic acid and 1-isocyanatobutane following the protocol described in Step 1 of Scheme 57, Example 32) and 5-bromo-7-azaindole 67 as shown in Scheme 58.
Scheme 58
Step 1 — Preparation ofl-[3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-4-fluoro-pheτιyl]-3 - butyl-urea (P-1637):
[0816] To aluminum trichloride (0.524 g, 0.00393 mol) and dichloromethane (20 mL, 0.3 mol) under an atmosphere of nitrogen was added 5-bromo-7-azaindole (67, 0.155 g, 0.000787 mol) in dichloromethane. To 5-(3-butylureido)-2-fluorobenzoic acid (588, 0.200 g, 0.000787 mol) was added 4 mL of dichloromethane (4 mL) followed by thionyl chloride (69 μL, 0.00094 mol) and a drop of N,N-dimethylformamide. After 1 hour, the reaction remained a suspension so additional thionyl chloride was added along with tetrahydroiuran. The reaction remained a suspension, so was placed in a 50 0C oil bath. After another hour, the reaction was still a suspension, and so was left to react at 50 0C overnight. The reaction had become a clear solution. All volatiles were removed under vacuum, then the residue dissolved in dichloromethane and added to the 5-bromo-7-azaindole and aluminum trichloride suspension. The reaction was allowed to stir at room temperature for 4.5 hours, followed by the addition of water and extraction with ethyl acetate. The organic layer was dried over magnesium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with a gradient of methanol (0 to 10%) in dichloromethane to give the compound (P-1637, 14 mg, 4%).
Step 2 - Preparation ofl-Butyl-3-[4-fluoro-3-(5-pyridin-3~ yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl] -urea (P-1534):
[0817] To l-[3-(5-bromo-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-4-fluoro-phenyl]-3 -butyl-urea (P- 1637, 14.0 mg, 0.0000323 mol), 3-pyridylboronic acid (5.96 mg, 0.0000485 mol), and
Tetrakis(triphenylphosphine)palladium(0) (0.820 mg, 7.09E-7 mol) were mixed in 1.00 M potassium carbonate in water (1.00 niL) and acetonitrile (2.00 niL, 0.0383 mol). The resulting mixture was heated at 1200C in the microwave for 40 minutes. The reaction was extracted with ethyl acetate and water twice, and the combined organic layers were washed with IM sodium bicarbonate followed by brine and the organic layer was dried over magnesium sulfate and filtered. The organic layer was concentrated and purified by reverse phase HPLC (acetonitrile and water with 0.1% formic acid) to give the compound as a white solid (P-1637, 8.5 mg, 61%). MS(ESI) [M+H*]+= 432.3.
[0818] l-Butyl-3-{4-fluoro-3-[5-(3-trifluoromethoxy-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-ρhenyl}-urea P-1660
was prepared following the protocol of Scheme 58, replacing 3-Pyridylboronic acid with 3- trifiuoromethoxy-phenylboronic acid in Step 2. MS(ESI) [M^-H
+J
+= 515.2.
Example 34: Synthesis of aldehyde reagents for coupling to 7-azaindoles
[0819] Aldehyde compounds for coupling to the 3-position of a 7-azaindole are shown in the following Schemes. 3-Methoxy-4-[4-(4-methyl-piperazin-l-ylmethyl)-benzyloxy]-benzaldehyde 591 was prepared in one Step as shown in Scheme 59.
Step 1- Synthesis of3~Methoxy~4-[4-(4-rnethyl-piperazin-l-ylmethyl)-benzyloxy]-benzaldehyde (591): [0820] To 4-Hydroxy-3-methoxybenzaldehyde (105, 2.1 g, 0.014 mol) in N,N-dimethylfoπnamide (40.0 TΏL) were added l,4-bis(bromomethyl)-benzene (589, 4.00 g, 0.0152 mol) and potassium carbonate (5.0 g, 0.036 mol) under an atmosphere of nitrogen. After 12 hours 1-methyl-piρerazine (590, 3.8 mL, 0.034 mol) was added to the reaction. After 2 hours, the reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with
20% methanol in dicliloromethane to give the compound (589, 1.2 g, 25.0%). MS(ESI) [M+H1"]* = 355.3.
[0821] 2-Fluoro-4-hydroxy-5-methoxy-benzaldehyde 593 was synthesized in one step from 2-fluoro-4,5-dimethoxy-benzaldehyde 592 as shown in Scheme 60.
Scheme 60
Step 1 - Synthesis of2-fluoro-4~hydroxy-5-methoxy-benzaldehyde (593):
[0822] To 2-fluoro-4,5-dimethoxy-benzaldehyde (592, 1.00 g, 5.43 mol) in dichloromethane (50.0 mL) was added aluminum trichloride (4.34 g, 32.6 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and washed with ethyl acetate and hexane to give a white solid (593, 0.7Og,
76.0%).
[0823] 2,5-Difluoro-4-hydroxy-benzaldehyde 597 was synthesized in three steps from 2,5- difluorophenol 594 as shown in Scheme 61.
Step J — Synthesis of 4-bromo-2,5-difluoro-phenol (595):
[0824] To 2,5-difluoroρhenol (594, 5.50 g, 0.0423 mol) in chloroform (110.0 mL), bromine (2.18 mL, 0.0423 mol) was added slowly. After 3 hours, the reaction was poured into a solution of sodium thiosulfate and extracted with ethyl acetate. The organic layer was dried over sodium sulfate, concentrated and purified with silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a colorless oil (595, 6.20 g, 70.2%).
Step 2 - (4-Bromo-2,5-difluoro-phenoxy)-tert-butyl-dimethyl-silane (596):
[0825] To 4-bromo-2,5-difluoro-phenol (595, 3.50 g, 0.0167 mol) in N,N-dimethylformamide (50.0 mL) were added tert-butyldimethylsilyl chloride (3.83 g, 0.0254 mol) and lH-imidazole (6.00 g,
0.0529 mol). The reaction was stirred at room temperature overnight, then poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound (596, 3.0 g, 55.4%).
Step 3 - 2,5-Difluoro-4-hydroxy~benzaldehyde (597):
[0826] To (4-bromo-2,5-difluoro-phenoxy)-tert-butyl-dimethyl-silane (596, 3.00 g, 9.28 mmol) in tetrahydrofuran (37.5 tnL), under an atmosphere of nitrogen at -78 0C, n-butyllithium (3.90 mL, 2.50 M in hexane) was added slowly. After 30 minutes, N,N-dimethylformamide (0.825 mL, 0.0106 mol) was added to the reaction. One hour later, the reaction was allowed to come to room temperature. The reaction was poured into water and 1 N HCl, then extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as an off-white solid (597, 0.86 g, 59.0%).
[0827] 4-(4-Chloro-benzyloxy)-3-fluoro-benzaldehyde 599 was synthesized in one step from 3- fluoro-4-hydroxy-benzaldehyde 598 as shown in Scheme 62.
Scheme 62
Step 1 - Synthesis of4-(4-chloro-benzyloxy)-3-fluoro-benzaldehyde (599):
[0828] To 3-fluoro-4-hydroxy-benzaldehyde (598, 0.800 g, 5.71 mmol) in N,N-dimethylformamide (50.0 mL) was added sodium hydride (260.0 mg, 60% in mineral oil, 6.50 mmol). After 15 minutes, 4-chlorobenzyl bromide (557, 1.29 g, 6.28 mmol) was added to the reaction mixture. The reaction was stirred at 80 0C for 5 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound (599, 1.3 g, 86.0%).
[0829] Additional aldehydes were prepared using the protocol of Scheme 62, replacing either 4- chlorobenzyl bromide 557 with a suitable alkylating agent, and/or 3-fluoro-4-hydroxy-benzaldehyde 598 with a suitable aldehyde. The following table indicates the alkylating agent (column 1) and the
starting aldehyde (column 2) used to afford the aldehyde (column 3) synthesized following this protocol.
Example 35: Synthesis of [4-(4-chloro-benzyloxy)-3-fluoro-phenyI]-(IH-pyrroIo[2,3-b]pyridin-3- yl)-methanone P-1897 and related compounds
[0830] Compound P-1897 was synthesized in two steps from 4-(4-chloro-benzyloxy)-3-fluoro- benzaldehyde 599 as shown in Scheme 63.
e e
Step 1 - Synthesis of[4-(4-chloro-benzyloxy)-3-fluoro-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)' methanol (P-1895):
[0831] To lH-Pyrrolo[2,3-b]pyridine (94, 100.0 mg, 0.85 mmol) in methanol (50.0 niL) were added 4-(4-chloro-benzyloxy)-3-fluoro-benzaldehyde (599, 250.0 mg, 0.94 mmol, prepared as described in Example 34) and potassium hydroxide (1.0Og, 17.82 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound (P-1895, 55 mg, 17.0%). MS(ESI) [M+H+]+ = 383.3.
Step 2 — Synthesis of[4-(4-chloro-benzyloxy)-3-fluoro-phenyl]-(lH-pyrrolo[2,3~b]pyridin-3-yl)- methanone (P-1897):
[0832] To [4-(4-chloro-benzyloxy)-3-fluoro-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (P- 1895, 17.7 mg, 0.046 mmol) in tetrahydrofuran (10.0 niL) was added Dess-Martin periodinane (23.5 mg, 0.056 mmol). The reaction was stirred at room temperature for 15 minutes. The reaction was concentrated, then purified with silica gel column chromatography eluting with 50% ethyl acetate in hexane to give a white solid (P-1897, 6.4 mg, 36.3%). MS(ESI) [M+HT = 381.3.
[0833] Additional compounds were prepared using the protocol of Scheme 63, replacing 4-4-(4- chloro-benzyloxy)-3-fluoro-benzaldehyde 599 with a suitable aldehyde (prepared as described in Example 34), and optionally replacing lH-Pyrrolo[2,3-b]pyridine 94 with an appropriate substituted 7-azaindole (see Example 9 or Example 16) in Step 1. The following compounds were made following this procedure:
[4-(4-Chloro-benzyloxy)-2-fluoro-5-methoxy-ρhenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1845),
[4-(4-Chloro-3-trifluoromethyl-benzyloxy)-3-methoxy-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-1850),
[4-(lH-Benzoimidazol-2-ylmethoxy)-3-fluoro-ρhenyl]-(lH-pyrrolo[2,3-b]ρyridin-3-yl)- methanone (P-1900),
(4-Benzyloxy-2,5-difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1903),
[4-(lH-Benzoimidazol-2-ylmethoxy)-2-fluoro-5-methoxy-phenyl]-(5-methoxy-lH-pyrrolo[2,3- b]pyridin-3-yl)~methanone (P-1979),
[4-(lH-Benzoimidazol-2-ylmethoxy)-2-fluoro-5-methoxy-phenyI]-(lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone (P-1982),
[4-(lH-Benzoimidazol-2-ylmethoxy)-2,5-difluoro-phenyl]-(lH-pyrrolo[2,3-b]pyridm-3-yl)- methanone (P-1987),
{4-[2-(2-Bromo-ethoxy)-ethoxy]-2-fluoro-5-methoxy-phenyl}-(5-chloro-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone (P-1988),
(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-yl)-[2,5-difluoro-4-(2-methoxy-ethoxy)-phenyl]- methanone (P-1989), and
(5-Chloro-lH-pyrrolo[2,3-b]pyridin-3-yl)-[2-fluoro-5-methoxy-4-(2-methoxy-ethoxy)-phenyl]- methanone (P-1991).
The following table indicates the aldehyde (column 2) and the azaindole (column 3) used to afford the target compound (column 4). Column 1 indicates the compound number and column 5 the observed mass.
Example 36: Synthesis of 3-(4-Benzyloxy-2,5-difluoro-benzyl)-lH-pyrrolo[2,3-b]pyridine P-1901
[0834] Compound P-1901 was synthesized in four steps from 4-bromo-2,5-difluoro-phenol 595 as shown in Scheme 64.
Scheme 64
Step 1 -Synthesis ofl-Benzyloxy-4-bromo-2,5-difluoro-benzene (600): [0835] To 4-bromo-2,5-difluoro-ρhenol (595, 0.90 g, 0.0043 mol, prepared as described in Example 34, Scheme 61) in N,N-dimethylfbrmamide (30.0 mL) were added sodium hydride (0.21 g, 60% in mineral oil, 0.0052 mol) and benzyl bromide (0.563 mL, 0.00474 mol). The reaction was
stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% ethyl acetate in hexane to give a white solid (600, 0.84 g, 65.0%).
Step 2 - (4-Benzyloxy-2,5-difluoro-phenyl)-(l-triisopropylsilanyl-lH~pyrrolo[2,3-b]pyridin-3-yl)- methanol (601):
[0836] To l-Benzyloxy-4-bromo-2J5-difluoro-benzene (600, 0.84 g, 2.80 mmol) in tetrahydrofiiran (15.0 mL) and ether (15.0 mL), under an atmosphere of nitrogen at -78 0C, n-butyllithium (1.20 mL, 2.50 M in hexane) was added slowly. After 20 minutes, l-Triisoρropylsilanyl-lH-pyrrolo[2,3- b]ρyridine-3-carbaldehyde (96, 0.82 g, 0.0027 mol, prepared as described in Example 18) was added to the reaction. After 20 minutes, the reaction was allowed to warm to room temperature for 10 minutes, then poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified with silica gel column chromatography eluting with 20% ethyl acetate in hexane to a white solid (601, 1.Og, 70.0%). MS(ESI) [M+H+]+= 523.4.
Step 3 — Synthesis of(4-Benzyloxy~2,5-difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (P- 1902):
[0837] To (4-Benzyloxy-2,5-difluoro-phenyl)-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3- yl)-methanol (601, 1.00 g, 1.91 mmol) in tetrahydrofuran (15.0 mL) was added tetrabutylammonium fluoride, trihydrate (0.63 g, 2.04 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction was roto-evaporated and purified with silica gel column chromatography eluting with 50% ethyl acetate in hexane to give the compound as a white solid (P-1902, 0.59 g, 84.0%). MS(ESI)
Step 4 — Synthesis of3-(4-Benzyloxy-2,5-difluoro-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1901): [0838] To (4-Benzyloxy-2,5-difluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (P-1902, 500.0 mg, 1.37 mmol) in acetonitrile (25.0 mL) were added triethylsilane (2.00 mL, 0.0125 mol) and trifluoroacetic acid (1.00 mL, 0.0130 mol). The reaction was heated to reflux for 2 hours. The reaction was concentrated and purified with silica gel column chromatography eluting with 50% ethyl acetate in hexane to give a white solid (P-1901, 60.0 mg, 94.1 %). MS(ESI) [M+H4]"1" = 351.4.
[0839] 3-[3-Trifluoromethyl-4-(4-trifluoromethyl-benzyloxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine P-1797
was prepared using the protocol of Scheme 64, substituting 4-bromo-2,5-difluoro-phenol 595 with 4- bromo-2-trifluoromethyl-phenol (prepared as described in Example 34, Scheme 61, Step 1, substituting 2,5-difluoro-phenol 594 with 2-trifluoromethyl-phenol) and benzyl bromide with 1- bromomethyl-4-trifluoromethyl-benzene in Step 1. MS(ESI) [M+HY = 451.
Example 37: Synthesis of 3-[4-(4-chloro-benzyIoxy)-2,5-difluoro-benzyl]-lH-pyrroIo[2,3- b]pyridine P-1974
[0840] Compound P-1974 was synthesized in four steps from 3-(4-benzyloxy-2,5-difluoro-benzyi)- lH-ρyrrolo[2,3-b]pyridine P-1901 as shown in Scheme 65.
Scheme 65
Step 1 — Synthesis of3-(4-Benzyloxy-2,5-difluoro-benzyl)-l-triisopropylsilanyl-lH-pyrrolo[2,3~ bjpyridine (602):
[0841] To 3-(4-Benzyloxy-2,5-difluoro-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1901, 560.0 mg, 1.60 mmol, prepared as described in Example 18, Scheme 33) in tetrahydrofuran (28.0 mL) was added sodium hydride (100.0 mg, 60% in mineral oil, 2.50 mmol). After 10 minutes, triisopropylsilyl chloride (0.500 mL, 2.36 mmol) was added to the reaction. After 4 hours, the reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound (602, 0.70 g, 86.1%).
Step 2 - Synthesis of2,5-difluoro-4-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenol (603):
[0842] To 3-(4-Benzyloxy-2,5-difluoro-benzyl)-l-triisoρropylsilanyl-lH-pyrrolo[2,3-b]pyridine
(602, 0.70 g, 0.0014 mol) in methanol (30.0 niL) was added 50% palladium hydroxide on carbon (0.1 g) under an atmosphere of hydrogen. The reaction was stirred at room temperature overnight. The reaction was filtered and concentrated to give a colorless oil (603, 0.47 g, 82.0%).
Step 3 - 3-[4-(4-Chloro-benzyloxy)-2, 5-difluoro-benzyl]-l-triisopropylsilanyl-lH~pyrrolo[2, 3- bjpyridine (604):
[0843] To 2,5-difluoro-4-(l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenol (603, 120.0 mg, 0.29 mmol) in N,N-dimethylformamide (15.0 mL) was added sodium hydride (18.0 mg, 60% in mineral oil, 0.45 mol) under an atmosphere of nitrogen. After 10 minutes, 4-chlorobenzyl bromide (65.1 mg, 0.32 mol) was added to the reaction. The reaction was stirred at 40 0C overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to give the crude compound (604, 0.15 g) that was used directly in the next step.
Step 4 - Synthesis of 3-[4-(4-chloro-benzyloxy)-2,5-difluoro-benzyl] ~lH-pyrrolo[2,3-b] pyridine (P-
1974):
[0844] To 3-[4-(4-chloro-benzyloxy)-2,5-difluoro-benzyl]-l -triisopropylsilanyl-1 H-pyrrolo[2,3- b]pyridine (604, 0.150 g, 0.28 mmol) in tetrahydrofuran (10.0 mL) was added tetra-n-butylammonium fluoride (80.0 mg, 0.31 mmol). After 10 minutes, the reaction was concentrated and purified by silica gel column chromatography eluting with 50% ethyl acetate in hexane to give the compound as a white solid (P-1974, 30.8 mg, 28.9%). MS(ESI) |M+H*]+= 385.3.
[0845] 2-[2,5-Difluoro-4-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenoxymethyl]-lH- benzoimidazole P-1975
was prepared using the protocol of Scheme 65, substituting 4-chlorobenzyl bromide with 2- chloromethyl-lH-benzoimidazole in step 3. MS(ESI) [M+H
4]
4 = 391.3.
Example 38: Synthesis of l-(4-Butoxy-phenyI)-3-{3-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3- b]pyridine-3-carbonyI]-phenyl}-urea P-1754
[0846] Compound P-1754 was synthesized in three steps from 5-(4-fluorophenyl)-lH-pyrrolo[2,3- bjpyridine 605 as shown in Scheme 66.
Scheme 66
Step 1 - Preparation of(3-chloro-phenyl)-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]- methanone (606):
[0847] To 5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine (605, 530 mg, 2.5 mmol, prepared as described in Example 23) dissolved in 20 mL of dioxane was added 3-nitrobenzaldehyde (S34, 758 mg, 5 mmol) and potassium hydroxide (4 mL of 2.5M aqueous). The vial was shaken on an orbital shaker for 16 hrs and the dioxane was removed under reduced pressure. The residue was partitioned between ethyl acetate and water. The aqueous layer was neutralized with the addition of IM HCl. The organic layer was washed with brine, dried over magnesium sulfate and concentrated under reduced pressure to give yellow-orange oil (1.5g). The crude material was dissolved in dichloromethane (150 mL) and chilled to 0° C. With vigorous stirring, pyridinium chlorochromate (3.O g, 14 mmol) was added slowly, maintaining the solution temperature at 0° C. After complete addition the solution was allowed to stir at ambient temperature for 1 hour. The resulting dark brown/black solution was diluted with chloroform and passed through a plug of silica. Elution with methanol gave 1.5 g of crude 606 that was carried on to the next step without further purification.
Step 2 - Preparation of(3-Amino-phenyl)-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-bJpyridin-3-ylJ- methanone (607):
[0848] Crude (3-chloro-phenyl)-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-methanone (606, 1.5 g) was dissolved in a minimal amount of methanol (~5 mL) and Pd/C (5%, ~10 mg) was added. The reaction mixture was shaken on a Parr shaker under 70 psi H2 overnight. The reaction mixture was filtered through Celite® and concentrated under reduced pressure to give 1.4 g of crude 607 that was carried on to the next step without further purification.
Step 3 - Preparation ofl-(4-Butoxy-phenyl)-3-{3~[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl}-urea (P-1754):
[0849] To a solution of- (3-Amino-phenyl)-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]- methanone (607, 7.5 mg) in anhydrous pyridine (200 μL) was added neat l-butoxy-4-isocyanato-
benzene (608, 1.6 mg) and the reaction mixture was stirred at ambient temperature for 2 hours. The reaction mixture was concentrated under reduced pressure and the residue was dissolved in DMSO (200 μL) and purified using reverse phase HPLC with an acetonitrile/water gradient. MS(ESl) [M+!!1"]4= 523.5.
[08S0] Additional compounds were prepared following the protocol of Scheme 66, optionally substituting 5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine 605 with a suitable azaindole in Step 1 and/or optionally substituting l-butoxy-4-isocyanato-benzene 608 with a suitable isocyanate in Step 3. Azaindoles were purchased or prepared as described in Examples 9 or 17. The following compounds were prepared by this procedure: l-(2-Methoxy-ethyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2!3-b]pyridine-3-carbonyl)-ρhenyl]-urea
(P-1566), l-Phenyl-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1591), l-Phenyl-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1703), l-(4-Fluoro-phenyl)-3-[3-(lH-pyrrolo[253-b]pyridine-3-carbonyl)-phenyl]-urea (P-1704), l-(4-Methoxy-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1705), l-(3,4-Difluoro-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1706), l-(3-Methoxy-phenyl)-3-[3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1707), l-(3,4-Dimethoxy-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1708), l-(4-Chloro-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1709), l-(3-Chloro-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1710),
1 -(4-Chloro-3 -trifluoromethyl-phenyl)-3 -[3 -( 1 H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-phenyl]-urea
(P-1711), l-(2-Chloro-5-trifluoromethyl-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1712), l-(2-Chloro-4-trifluoromethyl-phenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1713), l-(2-Fluoro-3-trifluoromethyl-phenyl)-3-[3-(lH-pyrrolo[2)3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1714), l-(4-Butoxy-ρhenyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1715), l-(3-Fluoro-phenyl)-3-[3-(lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-phenyl]-urea (P-1716), l-[3-(lH-Pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(4-trifluoromethyl-phenyl)-urea (P-1717), l-[3-(lH-Pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-p-tolyl-urea (P-1718), l-[3-(lH-Pyrrolo[2,3-b]pyridine-3-carbonyl)-pheny]]-3-m-tolyl-urea (P-1719), l-[3-(lH-Pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-o-tolyl-urea (P-1720), l-(4-Methoxy-phenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2)3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1723),
1-(3,4-Difluoro-ρhenyl)-3-[3-(5-pyridin-3-yl-lH-pyrτolo[253-b]pyridine-3-carbonyl)-plienyl]-urea (P-1724),
1-(3,4-Dimethoxy-phenyl)-3-[3-(5-pyridin-3-yl- 1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- urea (P-1725),
1-(3-Chloro-phenyl)-343-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1726),
1-(2-Chloro-4-trifluoromethyl-ρhenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-urea (P-1727), l-(2-Fluoro-3-trifluoromethyl-phenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-urea (P-1728),
1 -(4-Butoxy-ρhenyl)-3 -[3 -(5-pyridin-3 -yl-1 H-pyrrolo[2,3 -b]ρyridine-3 -carbonyl)-phenyl] -urea
(P-1729),
1 -(3 -Fluoro-phenyl)-3 -[3 -(5-pyridin-3 -yl- 1 H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-phenyl]-urea
(P-1730),
1 -[3 -(5 -Pyridin-3 -yl- 1 H-pyrrolo [2,3-b]pyridine-3 -carbonyl)-phenyl]-3 -(4-trifluoromethyl- phenyl)-urea (P-1731),
1-(2-Chloro-5-trifluoromethyl-phenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-urea (P-1732),
1-[3-(5-Chloro-lH-pyirolo[253-b]pyridine-3-carbonyl)-phenyl]-3-phenyl-urea (P-1733),
1-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-phenyl]-3-(4-metlioxy-phenyl)-urea
(P-1734),
1-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(3,4-difluoro-phenyl)-urea
(P-1735),
1-(3-Chloro-ρhenyl)-3-[3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1736),
1 -[3 -(5 -Chloro- 1 H-pyrrolo[2,3 -b]pyridine-3 -carbonyl)-phenyl]-3 -(4-chloro-3 -trifluoromethyl- plienyl)-urea (P-1737), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(2-chloro-5-trifluorometliyl- phenyl)-urea (P-1738),
1-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(2-chloro-4-trifluoromethyl- phenyl)-urea (P-1739),
1-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(2-fluoro-3-trifluoromethyl- phenyl)-urea (P-1740),
1-(4-Butoxy-phenyl)-3-[3-(5-chloro-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1741),
1-[3-(lH-Pyrrolo[2,3-b]ρyridine-3-carbonyl)-phenyl]-3-(3-trifluoromethyl-ρhenyl)-urea (P-1746),
l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(3-trifluoromethyl-ρhenyl)-urea (P-1747), l-(4-Fluoro-phenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-plienyl]-urea (P-1748),
1-(3-Methoxy-phenyl)-3-[3-(5-pyridin-3-yl- 1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea
(P-1749),
1 -(4-Chloro-phenyl)-3 - [3 -(5-pyridin-3 -yl- 1 H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-phenyl]-urea
(P-1750),
1 -(4-Chloro-3 -trifluoromethyl-phenyl)-3 -[3 -(5-pyridin-3 -yl- 1 H-pyrrolo f 2,3 -b]pyridine-3 - carbonyl)-ρhenyl]-urea (P-1751), l-(2-Chloro-5-trifluoromethyl-phenyl)-3-{3-[5-(4-fluoro-ρhenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl}-urea (P-1752), l-(2-Chloro-4-trifluoromethyl-phenyl)-3-{3-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carbonyl]-phenyl}-urea (P-1753), l-(4-Butoxy-phenyl)-3-{3-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}- urea (P-1754),
1 -[3 -(5-Pyridin-3 -yl- 1 H-pyrrolo [2,3-b]pyridine-3 -carbonyl)-phenyl] -3 -(3 -trifluoromethyl- phenyl)-urea (P-1755), l-[3-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-phenyl]-3-p-tolyl-urea (P-1756), l-[3-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-plienyl]-3-m-tolyl-urea (P-1757), l-{3-[5-(4-Fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}-3-m-tolyl-urea
(P-1758), l-[3-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-o-tolyl-urea (P-1759)5 l-Pyridin-4-yl-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1760), l-(2-Methoxy-ethyl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1761), l-(3-Methoxy-5-trifluoromethyl-phenyl)-3-[3-(lH-pyrroIo[2,3-b]pyridine-3-carbonyl)-phenyl]- urea (P-1762), l-(6-Methoxy-pyridin-3-yl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1763), l-Isoxazol-3-yl-3-[3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1764), l-(3-Methyl-isoxazol-5-yl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1765),
1 -(3 -Chloro-4-trifluoromethyl-phenyl)-3 -[3-(I H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-ρhenyl] -urea
(P-1766), l-(3,4-Dimethyl-isoxazol-5-yl)-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-pb.enyl]-urea
(P-1767), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]ρyridine-3-carbonyl)-phenyl]-3-pyridin-4-yl-urea (P-1770), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]ρyridme-3-carbonyl)-phenyl]-3-pyridin-3-yl-urea (P-1771),
l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(2-methoxy-ethyl)-urea (P-1772), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(3-methoxy-5-trifluoromethyl- phenyl)-urea (P-1773),
1 -[3 -(5-Chloro- 1 H-pyrrolo[2,3 -b]pyridine-3 -carbonyl)-phenyl] -3 -(6-methoxy-pyridin-3 -yl)-urea
(P-1774), l-t3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-isoxazol-3-yl-urea (P-1775), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(3,4-dimethyl-isoxazol-5-yl)- urea (P-1776),
1 -Pyridin-4-yl-3 -[3 -(5 -pyridin-3 -yl-1 H-pyrrolo [2,3 -b]ρyridine-3-cafbonyl)-phenyl]-urea
(P-1777), l-(3-Methoxy-5-trifluoromethyl-phenyl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridme-3- carbonyl)-phenyl]-urea (P-1778), l-(6-Methoxy-pyridm-3-yl)-3-[3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- urea (P-1779),
1 -(4-Dimethylamino-phenyl)-3 -[3 -( 1 H-pyrrolo[2,3 -b]pyridine-3 -carbonyl)-phenyl]-urea
(P-1780), l-Pyridm-3-yl-3-[3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-urea (P-1781), l-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-(4-dimethylamino-phenyl)-urea
(P-1782), l-(4-Fluoro-phenyl)-3-{3-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}- urea (P-1816), l-(3,4-Difluoro-phenyl)-3-{3-t5-(4-fluoro-ρhenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- phenyl}-urea (P-1817), l-(3,4-Dimethoxy-phenyl)-3-{3-[5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]- phenyl}-urea (P-1818), and l-(3-Fluoro-plienyl)-3-{3-t5-(4-fluoro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3-carbonyl]-phenyl}- urea (P-1819).
The following table indicates the azaindole (column 2) and isocyanate (column 3) used to afford the target compound (column 4). Column 1 provides the compound number and the observed mass is given in column 5.
Example 39: Synthesis of N-[3-(5-chloro-lH-pyrroIo[2,3-b ]pyridiiie-3-carbonyl)-2,4-difluoro- phenyl]-3,5-difluorobenzenesuIfonamide P-1841
[0851] Compound P-1841 was synthesized in six steps from 2,4-difluoroaniline 42 as shown in
Scheme 67.
Scheme 67
Step 1 - Preparation of(2,4-difluoro-phenyl)-carbamic acid benzyl ester (613): [0852] To 2,4-difluoroaniline (42, 7.0 mL, 0.070 mol) in 1 OO mL of dichloromethane was added pyridine (11 mL, 0.14 mol) and benzyl chloroformate (11.9 mL, 0.0834 mol). The reaction mixture was stirred at ambient temperature for 1.5 hours. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate and KHSO4 solution. The organic layer was dried (MgSO4), concentrated and crystallized from hexanes to give compound 613 (15.6 g, 85%).
Step 2 - Preparation of(2,4~difluoro-3-formyl-phenyl)-carbamic acid benzyl ester (614): [0853] Into a round bottom flask was added (2,4-difluoro-phenyl)-carbamic acid benzyl ester (613, 3.83 g, 14.5 mmol) in tetrahydrofuran (148 mL, 1.82 mol). The solution was chilled to - 78 0C and n-butyllithium (1.60 M in hexane, 19.1 mL, 30.0 mmol) was added over 30 minutes followed by the addition of, N,N-dimethylformamide (1.12 mL, 14.5 mol). The reaction mixture was allowed to warm to ambient temperature and was stirred overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and crystallized from ether to give compound 614 (3.0 g, 71%).
Step 3 - Preparation of {2,4-difluoro-3-[hydroxy-(lH-pyrrolo[2,3-h]pyridin-3--yl)-methyl] -phenyl}- carbamic acid benzyl ester (615):
[0854] Into a round bottom flask was added 5-chloro-lH-pyrrolo[2,3-b]pyridine (80, 0.524 g, 3.43 mmol, prepared as described in Example 9) in methanol (5.00 mL, 0.123 mol). Potassium hydroxide (0.800 g, 14.2 mmol) and (2,4-difluoro-3-formyl-phenyl)-carbamic acid benzyl ester (614, 1.02 g, 3.5 mmol) were added and the reaction mixture was stirred overnight. The reaction mixture was poured into IN HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and crystallized from ethyl acetate to give compound 615 (710 mg, 46%). MS(ESI)[M+HT = 444.
Step 4 - Preparation off2,4-diβuoro-3-(lH-pyrrolof2,3-bJpyridine-3-carbonyl)-phenylJ-carbamic acid benzyl ester (616):
[0855] Into a round bottom flask was added {2,4-difluoro-3-[hydroxy-(lH-pyrrolo[2,3-b]ρyridin-3- yl)-methyl]-phenyl}-carbamic acid benzyl ester (615, 1.01 g, 2.28 mmol) in tetrahydrofuran (5.00 mL, 0.0616 mol). Dess-Martin periodinane (1.20 g, 2.89 mmol) was added in portions. The reaction mixture was stirred at ambient temperature for 10 minutes, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography to give compound 616 (914 mg, 91%). MS(ESI)[M+H+]+ = 442.
Step 5 - Preparation of(3-Amino-2,6~difluoro-phenyl)-(S-chloro-lH-pyrrolo[2,3-b]pyridin-3-yl) - methanone (P-1801):
[0856] [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-carbamic acid benzyl ester (616, 800 mg, 1.81 mmol) was added to 10 M NaOH (15.00 mL) and warmed to reflux overnight. The reaction mixture was diluted with 30 mL of water and was extracted with ethyl acetate to give compound P-1801 (450 mg, 81% ).
Step 6 -Preparation ofN-[3-(5-chloro-lH-pyrrolo[2, 3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]- 3, 5-difluorobenzenesulfonamide (P-1841)
[0857] Into a microwave reaction vessel were combined (3-amino-2,6-difluoro-ρhenyl)-(5-chloro- lH-pyrrolo[2,3-b]pyridin-3-yl) -methanone (P-1801, 50 mg, 0.16 mmol), 3,5-difluorobenzenesulfonyl chloride (610, 103 mg, 0.49 mmol), pyridine (0.5 mL, 6.1820 mol) and tetrahydrofuran (3.0 mL,). The reaction was warmed in the CEM microwave at 300 watts, 130 0C for 10 minutes. The reaction mixture was partitioned between ethyl acetate and brine. The organic layer was collected, dried over Na2SO4, filtered and concentrated. The compound (P-1841) was isolated using column chromatography (silica, hexane:ethyl acetate 70:30) to obtain 36 mg (46%) compound. MS = 482.0.
[0858] Additional compounds were prepared following the protocol of Scheme 67 Step 6, optionally substituting (3-Amino-2,6-difluoro-phenyl)-(5-chloro-lH-pyrrolo[2,3-b]pyridin-3-yl) - methanone P-1801 with (3-Amino-2,6-difluoro-ρhenyl)-(lH-pyrrolo[2,3-b]pyridm-3-yl) -methanone P-2021 (prepared per Scheme 67 Steps 1-5, substituting 5-chloro-lH-pyrrolo[2,3-b]pyridine 80 with lH-pyrrolo[2,3-b]pyridine 94 in Step 3) and/or 3,5-difluorobenzenesulfonyl chloride 610 with an appropriate sulfonyl chloride. The following compounds were prepared by this procedure:
N-[3-(5-chloro-lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-isoproρyl- benzenesulfonamide (P-1839),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-benzenesulfonamide
(P-0913),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-nitro-benzenesulfonamide
(P-1937),
N-{4-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenylsulfamoyl]-phenyl}- acetamide (P-1938),
N-[2,4-Difluoro-3 -(I H-pyrrolo [2,3 -b]pyridine-3 -carbonyl)-phenyl] -4-methoxy- benzenesulfonamide (P-0958),
5-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenylsulfamoyl]-furan-2-carboxylic acid methyl ester (P-1941),
5-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenylsulfamoyl]-2-methyl-furan-3- carboxylic acid methyl ester (P-1942),
5 -Oxazol-5 -yl-thioρhene-2-sulfonic acid [2,4-difluoro-3 -(I H-pyrrolo [2,3-b]ρyridine-3 -carbonyl)- phenyl]-amide (P-1943),
5-Isoxazol-5-yl-thiophene-2-sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-amide (P-1948),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-phenyl]-2,4-dimethoxy- benzenesulfonamide (P-1951),
2,5-Dimethyl-thiophene-3-sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-1952),
2,5-Dimethyl-f\iran-3-sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridme-3-carbonyl)- phenyl]-amide (P-1953),
N-[2,4-Difluoro-3-(lH"pyrrolo[2,3-b]pyridme-3-carbonyl)-phenylJ-2-methyl-benzenesulfonamide
(P-1954),
2,3-Dihydro-benzo[l ,4]dioxine-6-sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-phenyl]-amide (P-1955),
2,4-Dimethyl-thiazole-5-sulfonic acid [2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)- phenyl]-amide (P-1956),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-ρhenyl]-4-trifluoromethyl- benzenesulfonamide (P-0931),
N-[2,4-Difluoro-3-(1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-5-fluoro-2-methyl- benzenesulfonamide (P-1961),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-3-methyl-benzenesulfonamide
(P-1962),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-oxazol-5-yl- benzenesulfonamide (P-1963),
N-[2,4-Difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-2,5-dimethoxy- benzenesulfonamide (P-1131),
2-Cyano-N-[2,4-difluoro-3-(lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-benzenesulfonamide
(P-1965),
3-Cyano-N-[2,4-difluoro-3-(1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-benzenesulfonamide (P-1966),
N-[2,4-Difluoro-3-(1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]-4-isopropyl- benzenesulfonamide (P-1968),
Benzothiazole-6-sulfonic acid [2,4-difluoro-3-(lH-ρyrrolo[2,3-b]pyridine-3-carbonyl)-phenyl]- amide (P-1969),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3-methoxy- benzenesulfonamide (P-2011),
N-[3-(5-Chloro-lH-pyiτolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-benzenesulfonamide
(P-0885),
Thiophene-2-sulfonic acid [3-(5-chloro-lH-pyiτolo[2,3-b]pyridme-3-carbonyl)-2,4-difluoro- phenyl]-amide (P-1267),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-metliyl- benzenesulfonamide (P-1842),
N-{4-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2;4-difluoiO-phenylsulfamoyl]- phenyl} -acetamide (P-1905),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-methoxy- benzenesulfonamide (P-0983),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-pb.enyl]-3-trifluoromethyl- benzenesulfonamide (P-1599),
5-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-2,4-difluoro-phenylsulfamoyl]-flιran-2- carboxylic acid methyl ester (P-1907),
5-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenylsulfamoyl]-2 -methyl- furan-3 -carboxylic acid methyl ester (P-1908), l,2-Dimethyl-lH-imidazole-4-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-
2,4-difluoro-phenyl]-amide (P-1911),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-2-fluoro- benzenesulfonamide (P-1912),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-4-difluoromethoxy- benzenesulfonamide (P-1916),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-2,4-dimethoxy- benzenesulfonamide (P-1918),
2,5-Dimethyl-thiophene-3-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenyl]-amide (P-1919),
2,5-Dimethyl-furan-3-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4- difluoro-phenyl]-amide (P-1920),
N-[3-(5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-2-methyl-
benzenesulfonamide (P-1921),
2,3-Dihydro-benzo[l ,4]dioxine-6-sulfonic acid [3-(5-chloro-lH-ρyrrolo[2,3-b]pyridine-3- carbonyl)-2,4-difluoro-phenyl]-amide (P-1922),
2,4-Dimethyl-thiazole-5-sulfonic acid [3-(5-chloro-lH-pyrrolo[2,3-b]pyridme-3-carbonyl)-2,4- difluoro-phenylj-amide (P-1923),
N-[3-(5-Chloro-1H-pyrrol[2,3-b]pyridine-3-carbonyl) -2,4-difluoro-phenylJ-2,4-difluoro- benzenesulfonamide (P-1926),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-5-flxioro-2-methyl- benzenesulfonamide (P-1927),
N-[3 -(5 -Chloro- 1 H-pyrrolo[2,3 -b]pyridine-3-carbonyl)-2,4-difluoro-phenyl] -3 -methyl- benzenesulfonamide (P-1928),
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-2,5-dimethoxy- benzenesulfonamide (P-1929),
N-[3-(5-Chloro-lH-ρyrrolo[2,3-b]pyridme-3-carbonyl)-2,4-difluoro-phenyl]-2-cyano- benzenesulfonamide (P-1931), and
N-[3-(5-Chloro-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluoro-phenyl]-3-cyano- benzenesulfonamide (P-1932).
The following table indicates the azaindole (column 2) and the sulfonyl chloride (column 3) used to afford the target compound (column 4). The compound number is provided in column 1 , with the observed mass given in column 5.
Example 40: Synthesis of 4-(5-pyridm-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l- carboxylic acid dibutylamide P-1636
[0859] Compound P-1636 was synthesized in two steps from lH-indole-4-carboxylic acid 611 as shown in Scheme 68.
Scheme 68
Step 1 — Preparation ofl-dibutylcarbamoyl-lH-indole-^carboxylic acid (612): [0860] To lH-indole-4-carboxylic acid (611, 251 mg, 1.56 mmol) in tetrahydrofuran (3 mL), was added 2.5M n-butyllithium in hexane (1.28 mL, 3.19 mmol) at -78 0C. After 30 minutes, dibutyl carbamyl chloride (657 mg, 3.43 mmol) was added and stirred for two hours. The reaction solution was quenched with IM HCl (aq.) and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous magnesium sulfate, filtrated and concentrated. The desired compound was isolated with silica gel column chromatography using 10% ethyl acetate in hexane to give a white solid (612, 88 mg, 18%). MS(ESI) [M-H+]"= 315.1.
Step 2 - Preparation of4-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l-carboxylic acid dibutylamide (P-1636):
[0861] To l-dibutylcarbamoyl-lH-indole-4-carboxylic acid (612, 78 mg, 0.25 mmol) in dichloromethane (2 mL), tbionyl chloride (25 μL, 0.34 mmol) was added and stirred for one hour, followed by rotary evaporation to remove solvents to provide the dried acid chloride, which was dissolved in dichloromethane for later use. Meanwhile, 5-ρyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (89, 55 mg, 0.28 mmol, prepared as described in Example 17) in dichloromethane (8 mL), was mixed with aluminum trichloride (215 mg, 1.6 mmol) and stirred for one hour, followed by addition of the dried acid chloride in dichloromethane (3 mL). The reaction was stirred at room temperature overnight, then quenched with methanol and all volatiles were removed. The desired compound was isolated with silica gel column chromatography using 10% methanol in dichloromethane to give a solid (P- 1636, 11 mg, 9%). MS(ESI) [M+HY = 494.3.
[0862] Additional compounds were prepared following the protocol of Scheme 68, substituting dibutyl carbamyl chloride with a suitable reagent in Step 1 and optionally substituting 5-pyridin-3-yl-
lH-pyrrolo[2,3-b]pyridme 89 with 5-(6-Methoxy-pyridin-3-yl)-lH-pyrrolo[2,3-b]pyridine (see Example 17) in Step 2. The following compounds were prepared following this procedure:
[1-(Butane-l-sulfonyl)-lH-indol-4-yl]-(5-pyridin-3-yl-1H-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-1661),
4-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-1-carboxylic acid ρentylamide
(P-1702),
4-(5-Pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-indole-l-carboxylic acid dipropylamide
(P-1722), and
4-[5-(6-Methoxy-pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-indole-1-carboxylic acid butylamide (P-1827).
The following table indicates the reagent used in place of dibutyl carbamyl chloride (column 2) and the azaindole (column 3) used to afford the target compound (column 3). The compound number is provided in column 1, and the observed mass is given in column 5.
Example 41: Synthesis of 3-(3-Benzyloxy-2-chloro-6-fluoro-benzyl)-lH-pyrrolo[2,3-A]pyridine P-1852, (3-Benzyloxy-2-chloro-6-fluoro-phenyl)-(lH-pyrroio[2,3-Zι]pyridin-3-yl)-inethanone P- 1853 and related compounds
[0863] Compounds P-1852 and P-18S3 were synthesized in four steps from 2-chloro-4- fluorophenol 617 and lH-pyrrolo[2,3-έ]pyridine 94 as shown in Scheme 69.
Scheme 69
Step 1 —Preparation ofl-Benzyloxy-2-chloro-4-fluoro-benzene (618):
[0864] To a solution of 2-chloro-4-fluorophenol (617, 7 g, 0.05 mol) in tetrahydrofuran (100 mL) was added sodium hydride (1.8 g, 95% dry powder, 0.071 mol) at room temperature over 15 minutes under an atmosphere of nitrogen. The reaction mixture was stirred at room temperature for 30 minutes. Benzyl bromide (10 g, 0.060 mol) was added slowly to the reaction mixture, then stirred at room temperature overnight. The reaction mixture was poured into ice water, extracted with ethyl acetate, washed with hydrochloric acid (10%), water, brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide compound as a white solid (618, 7.6 g, 60%).
Step 2 - Preparation ofS-Benzyloxy-l-chloro-ό-fluoro-benzaldehyde (619): [0865] To a solution of l-benzyloxy-2-chloro-4-fluoro-benzene (618, 5.8 g, 0.024 mol) in tetrahydrofuran (100 mL) was added 2.50 M of n-butyllithium (2.7 mL, 2.50 M in hexane, 0.029 mol) slowly at -78 0C over 15 minutes under nitrogen. The reaction mixture was stirred at -78 °C for 30 minutes. To the reaction mixture was then added N,N-dimethylformamide (4.2 mL, 0.054 mol). The reaction was allowed to warm to room temperature and was continued at room temperature overnight. The reaction mixture was poured into ice water, extracted with ethyl acetate, washed with hydrochloric acid (10%), water, brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to
provide the compound as a white solid (619, 2.1 g, 32%). MS(ESI) [M+H+]+ = 265.08.
Step 3 — Preparation of3-[(3-Benzyloxy-2-chloro-6-fluoro-phenyl)-methoxy-methyl]-lH-pyrrolo[2,3- b] pyridine (P-1867) and (3-Benzyloxy-2-chloro-6-fluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl) - methanol (P-1868):
[0866] A mixture of lH-pyrrolo[2,3-6]pyridine (94, 0.5 g, 4 mmol), 3-benzyloxy-2-chloro-6- fluoro-benzaldehyde (619, 1.3 g, 4.9 mmol), and potassium hydroxide (0.99 g, 18 mmol) in methanol (30 mL) was stirred at room temperature overnight. The reaction mixture was concentrated and the residue was dissolved in ethyl acetate and water. The organic layer was collected and washed with brine. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide compound P-1867 as a white solid (1.3 g, 70%, MS(ESI) [M+HY = 397.16), and compound P-1868 as an off-white solid (0.2 g, 10, MS(ESI) [M+HT = 383.14).
Step 4a - Preparation of3-(3~Benzyloxy-2-chloro-6-fluoro-benzyl)-lH-pyrrolo[2,3-b]pyridine (P- 1852):
[0867] A mixture of 3-[(3-Benzyloxy-2-chloro-6-fluoro-phenyl)-methoxy-methyl]-lH-ρyrrolo[2,3- &]pyridine (P-1867, 0.1 g, 0.2 mmol), trifluoroacetic acid (0.6 mL, 8 mmol), and triethylsilane (0.3 mL, 2 mmol) in acetonitrile (10 mL) was refluxed for 2 hours. The mixture was concentrated and the residue was dissolved in ethyl acetate. The solution was washed with saturated sodium bicarbonate, brine, and dried over sodium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with methanol in dichloromethane to provide compound as an off- white solid (P-1852, 62 mg, 70%). MS(ESl) [M+H+f = 367.16.
Step 4b - Preparation of(3-Benzyloxy-2-chloro-6-fluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl) - methanone (P-1853):
[0868] To a solution of (3-Benzyloxy-2-chloro-6-fluoro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanol (P-1868, 65 mg, 0.17 mmol) in tetrahydrofuran (10 mL) was added Dess-Martin periodinane (79 mg, 0.19 mmol) at 0 0C. The reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched with a saturated solution of sodium thiosulfate, extracted with ethyl acetate, washed with sodium bicarbonate, brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with methanol in dichloromethane to provide the compound as a light yellow solid (P-1853, 32 mg, 50%). MS(ESI) [M+ϊrT = 381.13.
[0869] Additional compounds were prepared following the protocol of Scheme 69, optionally replacing 2-chloro-4-fluorophenol 617 with 2, 6-difluorophenol or 2, 6-dichloroρhenol, optionally replacing benzyl bromide with an appropriate substituted benzyl bromide, and optionally replacing
1H-pyrrolo[2,3-b]pyridine 94 with an appropriate substituted lH-pyrrolo[2,3-b]pyridine. Azaindoles were purchased or prepared as described in Examples 9 or 16. The following compounds were made following this procedure:
3-[2,6-Dichloro-3-(4-chloro-benzyloxy)-benzyl]- 1H-pyrrolo[2,3-b]pyridine (P-1768), [2,6-Dichloro-3-(4-chloro-benzyloxy)-phenyl]-( 1H-pyrrolo[2,3-b]pyridine-3-yl) methanone (P-1769),
(3-Benzyloxy-2,6-difluoro-phenyl)-( 1H-pyrrolo[2,3-b]pyridine-3-yl)-methanone (P-1802), 3-(3-Benzyloxy-2,4-difluoro-benzyl)- 1H-pyrrolo[2,3-b]pyridine (P-1803), 3-(3-Ben2yloxy-2,6-difluoro-benzyl)-5-methoxy-lH-pyrrolo[2,3- b]pyridine (P-1804), 3-(3-Benzyloxy-2,6-difluoro-benzyl)-5-chloro-lH-pyrrolo[2,3- b]pyridine (P-1824),
(3-Benzyloxy-2,6-difluoro-phenyl)-5-chl oro-1H-pyrrolo[2,3-b]pyridin-3-yl) -methanone (P-1825),
3-[(3-Benzyloxy-2-chloro-6-fluoro-phenyl)-methoxy-methyl]-lH-pyrrolo[2,3- έ]pyridine (P-1867),
(3-Benzyloxy-2-chloro-6-fluoro-phenyl)-(lH-pyrrolo[2,3-b]ρyridin-3-yl)-methanol (P-1868), [2-Chloro-3-(3-chloro-benzyloxy)-6-fluoro-phenyl]-(lH-pyrrolo[2,3- έ]pyridin-3-yl)-methanone (P-1869),
[2-Chloro-3-(4-chloro-benzyloxy)-6-fluoro-ρhenyl]-(lH-pyrrolo[2,3- έ]pyridin-3-yl)-methanone (P-1874),
3-[2,6-Difluoro-3-(pyridin-4-ylmethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1993), and 3-[3-(4-Chloro-2-fiuoro-benzyloxy)-2,6-difluoro-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-1992). The phenol, benzyl bromide and azaindole used in Steps 1, 2, and 3, respectively, are indicated in columns 2, 3, and 4 of the following table, respectively, to afford the target compound (column 5). The compound number is provided in column 1, and the observed mass is given in column 6.
Example 42: Synthesis of (3-Benzyloxy-2-methyI-phenyl)-(lH-pyrrolo [2,3-6] pyridin-3-yl)- methanone P-1848 and 3-(3-Benzyloxy-2-methyl-benzyl)-lH-pyrrolo[2,3-6]pyridine P-1857
[0870] Compounds P-1848 and P-1857 were synthesized in five steps from compounds 620 and lH-pyrrolo[2,3-6]pyridine 94 as shown in Scheme 70.
Scheme 70
Step 1 — Preparation of 3-Benzyloxy-2-methyl-benzoic acid (621):
[0871] To a solution of 3-hydroxy-2-methyl-benzoic acid (620, 5.0 g, 0.033 mol) in tetrahydrofuran (100 niL) and N,N-dimethylformamide (50 mL), sodium hydride (4.4 g as 60% dispersion in mineral oil, 0.11 mol) was added slowly over 30 minutes and the reaction was stirred at 0 0C under an atmosphere of nitrogen. The reaction mixture was allowed to warm to room temperature, then stirred at room temperature for 1 hour. Benzyl bromide (9.0 mL, 0.076 mol) was added slowly into the reaction mixture, and the reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into water, extracted with ethyl acetate, washed with a solution of ammonium chloride and ammonium hydroxide (4: 1), brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide the compound as a white solid (621, 5.8 g, 73%).
Step 2 - Preparation of(3-Benzyloxy-2-methyl-phenyl)-methanol (622): [0872] To a solution of 3-benzyloxy-2-methyl-benzoic acid (621, 3.0 g, 0.012 mol) in tetrahydrofuran (100 mL), lithium aluminum hydride (25 mL, IM solution in tetrahydrofuran, 0.025 mol) was added dropwise at 0 °C for 5 minutes. The reaction mixture was then stirred at room
temperature overnight under an atmosphere of nitrogen. After sodium sulfate decahydrate (20.0 g, 0.062 mol) was added, the reaction mixture was stirred at room temperature for 10 minutes. A white solid was collected by filtration. The solid compound was further washed with a mixture of hexane and dichloromethane (9:1) and dried under high-vacuum (622, 2.8 g, 91%).
Step 3 - Preparation of3-Benzyloxy-2-methyl-benzaldehyde (623):
[0873] To a solution of (3-benzyloxy-2-methyl-phenyl)-methanol (622, 627 mg, 2.75 mmol) in tetrahydrofuran (60 mL) was added Dess-Martin periodinane (2.9 g, 6.87 mmol) at 0 0C. The resulting mixture was stirred at 0 °C for 50 minutes. The reaction mixture was quenched with a solution of saturated sodium thiosulfate, extracted with ethyl acetate, washed with sodium bicarbonate, brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide the compound as a white solid (623, 0.55 g, 84%).
Step 4 — Preparation of(3-Benzyloxy~2-methyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (624) and 3-[(3-Benzyloxy-2-methyl-phenyl)-methoxy-methyl]-lH-pyrrolo[2,3-b]pyridine (625): [0874] A mixture of lH-pyrrolo[2,3-%yridine (94, 0.33 g, 2.8 mmol), 3-benzyloxy-2-methyl- benzaldehyde (623, 0.55 g, 2.4 mmol), and potassium hydroxide (0.39 g, 6.1 mmol) in methanol (40 mL) was stirred at room temperature for 17 hours. The reaction mixture was poured into water and then extracted with ethyl acetate. The organic layer was collected, washed with brine, and dried over sodium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide compound 624 as an off-white solid (330 mg, 39%, MS(ESI) PS/MIT = 345.29, and compound 625 as a white solid (24 mg, 3%, MS(ESI) [M+Εff = 359.30).
Step 5a - Preparation of(3-Benzyloxy-2-methyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1848):
[0875] To a solution of (3-Benzyloxy-2-methyl-ρhenyl)-(lH-pyrrolo[2,3-£)]ρyridm-3-yl)-methanol (624, 0.12g, 0.35 mmol) in tetrahydrofuran (15 mL) was added Dess-Martin periodinane (0.37 g, 0.S9 mmol) at 0 0C . The reaction mixture was stirred at 0 0C for 50 minutes, then quenched with a saturated solution of sodium thiosulfate, extracted with ethyl acetate, washed with sodium bicarbonate, brine, and dried over magnesium sulfate. After removal of solvent, the residue was washed with a mixture of ethyl ether and hexanes (1 :1) to provide the compound as a yellow solid (P- 1848, 108 mg, 90%). MS(ESI) [M+rff = 343.22.
Step 5b - Preparation of3-(3-Benzyloxy-2-methyl-benzyl)-lH-pyrrolo[2,3-b]pyridine (P-1857): [0876] A mixture of 3-[(3-benzyloxy-2-methyl-phenyl)-methoxy-methyl]-lH-pyrrolo[2,3- b]ρyridine (625, 24 mg, 0.067 mmol), trifluoroacetic acid (1 mL, 13 mmol), and triethylsilane (2 mL,
12.5 mmol) in acetonitrile (10 mL) was refluxed for 4 hours. The mixture was concentrated and the residue was dissolved in ethyl acetate. The solution was washed with saturated sodium bicarbonate, brine, and dried over sodium sulfate. After removal of solvent, the residue was washed with a mixture of ethyl ether and hexanes (1:1) to provide the compound as a yellow solid (P-1857, 17 mg, 75%). MS(ESI) [M+H+]+ = 329.24.
Example 43: Synthesis of [3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-(lH-pyrrolo [2,3-b] pyridin- 3-yl)-methanone P-1892 and 3-[3-(4-chloro-benzyIoxy)-2-ethoxy-benzyl]-lH-pyrroIo[2,3- b]pyridine P-1893
[0877] Compounds P-1892 and P-1893 were synthesized in five steps from compounds 626, 557 and lH-pyrrolo[2,3-b]pyridine 94 as shown in Scheme 71.
Scheme 71
Step 1 -Preparation of2,3-Bis-(4-chloro-benzyloxy)-benzaldehyde (627): [0878] To a solution of 2,3-dihydroxybenzaldehyde (626, 2.0 g, 14.5 mmol) in tetrahydrofuran (100 mL) was added sodium hydride (0.52 g, 13.0 mmol) at 0 0C under an atmosphere of nitrogen. The reaction mixture was allowed to warm to room temperature and was stirred at room temperature for 30 minutes. To the reaction mixture was then added 4-chlorobenzyl bromide (557, 2.7 g, 13.0 mmol). The reaction mixture was stirred at room temperature under an atmosphere of nitrogen overnight. N, N-dimethylformamide (50 mL) was added into the reaction mixture and it was stirred at room temperature for 24 hours. The reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was collected, washed with brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with
ethyl acetate in hexane to provide the compound as an off-white solid (627, 2.3 gm, 46%).
Step 2 - Preparation of3-(4-chloro-benzyloxy)-2-hydroxy-benzaldehyde (628): [0879] To magnesium (0.098 g, turnings, 4.0 mmol) in a mixture of anhydrous ether (20 mL) and benzene (20 mL) at 0 0C, bromine (0.10 mL, 2.0 mmol) was added dropwise. When the reaction had started, stirring was commenced and the addition of bromine continued until complete. The ice bath was removed and the reaction mixture was heated until the solution was almost colorless. After cooling down, the reaction mixture was slowly added to a solution of 2, 3-bis-(4-chloro-benzyloxy)- benzaldehyde (627, 0.78 g, 2.0 mmol) in benzene (60 mL) at room temperature while stirring vigorously. Upon completion of the addition, the reaction mixture was stirred at room temperature overnight, then refiuxed for 36 hours. After the reaction mixture was cooled down to room temperature, a solid was collected by filtration and washed with benzene, then boiled in hydrochloric acid (100 mL, 1.0 M) for 30 minutes. After cool down, the solution was extracted with dichloromethane. The organic layer was washed with brine and dried over magnesium sulfate. An off-white solid was obtained after removal of the solvent (628, 0.32 mg, 60%). MS(ESI) [M-H-] =261.25.
Step 3 — Preparation of3-(4-chloro-benzyloxy)-2-ethoxy-benzaldehyde (629): [0880] To a mixture of 3-(4-chloro-benzyloxy)-2-hydroxy-benzaldehyde (110 mg, 0.42 mmol), potassium carbonate (150 mg, 1.1 mmol) in acetonitrile (8 mL) was added iodoethane (0.2 mL, 2.5 mmol) at room temperature. The mixture was stirred at 98 0C for 18 hours. The reaction mixture was poured into a solution of saturated ammonium chloride and was extracted with ethyl acetate. The organic layer was collected, washed with brine, and dried over magnesium sulfate. After removal of solvent, a light yellow solid was obtained (629, 116 mg, 95%).
Step 4 - Preparation of[3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-(lH-pyrrolo[2,3-b]pyήdin-3-yl)- methanol (630) and 3-{[3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-methoxy-methyl}-lH-pyrrolo[2, 3- b] pyridine (631):
[0881] A mixture of lH-Pyrrolo[2,3-6]pyridine (94, 26 mg, 0.22 mmol), 3-(4-chloro-benzyloxy)-2- ethoxy-benzaldehyde (629, 54 mg, 0.19 mmol), and potassium hydroxide (30 mg, 0.4.6 mmol) in methanol (5 mL) was stirred at room temperature for 4 days. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was collected, washed with brine, and dried over sodium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide compound 630 as an off-white solid (20 mg, 26%, MS(ESI) [MH-H+J+ = 409.32) and compound 631 as an off-white solid (44 mg, 56%, MS(ESI) [MH-H+I+ = 423.33.
Step 5a - Preparation of[3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-(lH~pyrrolo[2,3-b]pyridin-3-yl)-
methanone (P-1892):
[0882] To a solution of [3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanol (630, 20 mg, 0.05 mmol) in tetrahydrofuran (8 mL) was added Dess-Martin periodinane (52 mg, 0.12 mmol) at 0 0C. The reaction mixture was stirred at 0 0C for 50 minutes. The reaction was quenched with a saturated solution of sodium thiosulfate, extracted with ethyl acetate, washed with sodium bicarbonate, brine, and dried over magnesium sulfate. After removal of solvent, the residue was washed with a mixture of ethyl ether and hexanes (1:1) to provide the compound as a yellow solid (P-1892, 15 mg, 75%). MS(ESI) [M+Hf]+ = 407.38.
Step 5b - Preparation of3-[3-(4-chloro-benzyloxy)-2-ethoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P- 1893):
[0883] A mixture of 3-{[3-(4-chloro-benzyloxy)-2-ethoxy-phenyl]-methoxy-methyl}-lH- pyrrolo[2,3-6]pyridine (631, 44 mg, 0.1 mmol), trifluoroacetic acid (1 mL, 13 mmol), and triethylsilane (2 mL, 12.5 mmol) in acetonitrile (10 mL) was refluxed for 4 hours. The mixture was concentrated and the residue was dissolved in ethyl acetate. The solution was washed with saturated sodium bicarbonate, brine, and dried over sodium sulfate. After removal of solvent, the residue was washed with a mixture of ethyl ether and hexanes (1 : 1) to provide the compound as a yellow solid (P-1893, 40 mg, 98%). MS(ESI) [M+H*]+ = 393.39.
[0884] [3 -(4-Chloro-benzyloxy)-2-methoxy-phenyl] -(I H-pyrrolo [2,3 -b]pyridin-3 -yl)-methanone (P-1891), [3-(4-Chloro-benzyloxy)-2-(2,2,2-trifluoroethoxy)-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone (P-2076), and [3-(4-chloro-2-fluoro-benzyloxy)-2-ethoxy-phenyl]-(5-methoxy-lH- pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-2016)
were prepared following the protocol of Scheme 71, substituting iodoethane with iodomethane in Step 3 to provide P-1891, or substituting iodoethane with 2-iodo-l,l,l-trifluoroethane in Step 3 to provide P-2076, or substituting 4-chlorobenzyl bromide 557 with 4-chloro-2-fluoro-benzyl bromide in Step 1 and 7-azaindole 94 with 5-methoxy-7-azaindole in Step 4 to provide P-2016. MS(ESI) [M+H
4']
+ = 393.4 (P-1891), 461.08 (P-2076), and 455.2 (P-2016).
Example 44. Synthesis of Propane-l-sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2,3- b]pyridme-3-carboiiyl]-2,4-difluoro-phenyl}-amide P-0956
[0885] Compound P-0956 was synthesized in three steps from 5-(4-chlorophenyl)-lH-pyrrolo[2,3- b]pyridine 514 and propane- 1 -sulfonic acid (2,4-difluoro-3-formyl-phenyl)-amide 73 as shown in Scheme 72.
Scheme 72
632 R = H 633 R = CH3
Step 1-Preparation of Propane-l-sulfonic acid (3-{[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3- yl]-hydroxy-methyl}-2,4-difluoro-phenyl)-anιide (632) and Propane-l-sulfonic acid (3-{[5-(4-chloro- phenyl)-lH-pyrrolo[2, 3-b]pyridin-3-yl]-methoxy-methyl}-2, 4-difluoro-phenyl)-amide (633) : [0886] To a suspension of 5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine (514, 64.9 g, 158 mM, prepared as described in Example 17) and propane-l-sulfonic acid (2,4-difluoro-3-formyl-phenyl)- amide (73, 90.4 g, 191 mM, prepared as described in Example 7) in methanol in a water bath was added potassium hydroxide (128.8 g, 1.28 M). The reaction was stirred 72 hours at room temperature and then adjusted to pH 7 with 4N hydrochloric acid. The resulting mixture was evaporated in vacuo to remove methanol and extracted 3x with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated in vacuo to give a crude oil. The crude oil was triturated with 3:1 MTBE/heptane to give a 1 :3 solid mixture of 632 and 633 that was used directly for the next step.
Step 2-Preparation of Propane-l-sulfonic acid (3-{[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridin-3- yl]-hydroxy-meihyl}-2,4-difluoro-phenyl)-amide (632):
[0887] To a solution of 632 and 633 (ca. 315 mM) in acetic acid was added 48% hydrobromic acid (final 8%). The resulting mixture was stirred overnight at room temperature and then evaporated in vacuo. The crude residue was taken up with equal volumes of ethyl acetate and water, and adjusted to pH 7 with solid potassium carbonate. The layers were split and the aqueous layer was extracted 2x with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated in vacuo to give 632 as a viscous oil that was used directly for the next step.
Step 3-Preparation of Propane- 1-sulfonic acid {3-[5-(4-chloro-phenyl)-lH-pyrrolo[2,3-b]pyridine-3- carhonylJ-2, 4-difluoro-phenylj-amide (P-0956):
[0888] To a solution of 632 (ca. 386 mM) in 1 ,4-dioxane was added 2,3-dichloro-5,6- dicyanobenzoquinone (83.8 g, 502 mM) followed by water (final 4.8%). The resulting mixture was stirred 2 hours at room temperature and then quenched with one volume of saturated sodium bicarbonate. The mixture was evaporated in vacuo to remove 1 ,4-dioxane and extracted 3x with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated in vacuo to give a crude solid that was purified on a silica-gel column with 94:5:1 dichloromethane/methanol/ammonium hydroxide as eluent to give P-0956 (approximately 50% yield for 3 steps) as a white solid.
Example 45: Synthesis of 3-Iodo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine 635
[0889] 3-Iodo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine 635 was synthesized in one step from 3-Iodo-lH-pyrrolo[2,3-b]pyridine 634 as shown in Scheme 73.
Scheme 73
635
Step 1 — Preparation of 3 -Iodo-1-triisopropylsilanyl-lH-pyrrolo [2,3 -b] pyridine (635): [0890] 3-Iodo-lH-pyrrolo[2,3-b]pyridine 634 (2.00 g, 8.20 mmol) was dissolved in N,N- dimethylformamide (50 mL). Sodium hydride (60% dispersion in mineral oil, 390 mg, 9.8 mmol) was added. After 20 minutes, triisopropylsilyl chloride (1.74 mL, 8.20 mmol) was added dropwise. After 1.5 hours, the reaction was poured into water and extracted with ethyl acetate, washed with saturated sodium bicarbonate and brine. The organic portions were dried over anhydrous sodium sulfate and concentrated. Purification by silica gel chromatography, 0-25% gradient ethyl acetate/ hexane gave compound 635 as a white solid (3.224 g, 98.2%). 1H-NMR was consistent with the desired compound.
Example 46: Synthesis of l-(tert-Butyl-dimethyl-silanyl)-3-iodo-lH-pyrroIo[2,3-b]pyridine 636
[0891] 1 -(tert-Butyl-dimethyl-silanyl)-3 -iodo-1 H-pyrrolo[2,3-b]pyridine 636 was synthesized in one step from 3-Iodo-lH-pyrrolo[2,3-b]pyridine 634 as shown in Scheme 74.
Step 1 -Preparation ofl-(tert-Butyl-dimethyl-silanyl)-3-iodo-lH-pyrrolo[2,3-b]pyridine (636): [0892] 3-Iodo-lH-ρyrrolo[2,3-b]pyridine 634 (1.11 g, 4.6 mmol) was dissolved in tetrahydrofuran (120 mL). Sodium hydride (60% dispersion in mineral oil, 0.13 g, 5.5 mmol) was added, followed by tert-butyldimethylsilyl chloride (0.85 g, 5.5 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic portion was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound as a white solid (636, 100 mg, 15%).
Example 47: Synthesis of [5-(4-chloro-benzyloxy)-4-methoxy-pyridin-2-yl]-(lH-pyrrolo[2,3- b]pyridin-3~yl)-methanone P-2024
[0893] [5-(4-Chloro-benzyloxy)-4-methoxy-pyridin-2-yl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone P-2024 was synthesized in six steps from Kojic acid, 3, and 3-iodo-l-triisopropylsilanyl- lH-pyrrolo[2,3-b]pyridineJ 2, as shown in Scheme 75.
Scheme 75
Step 1 -Preparation of5-(4-chloro-benzyloxy)-2~hydroxymethyl-pyran~4-one (638): [0894] Kojic acid (637, 5.00 g, 35.2 mmol) and 4-chlorobenzyl bromide (557, 7.95 g, 38.7 mmol) were suspended in methanol (40 mL) in an 80 mL sealed tube. Sodium hydroxide in water (12 M, 2.93 mL) was added. The reaction was heated at 80 0C overnight. The resulting suspension was concentrated. Water was added and the mixture was filtered and washed with water to provide a brown solid. Washing with minimal methanol on the filter removed the brown color. A white solid (638, 7.58 g, 80%) was isolated. 1H-NMR was consistent with the desired compound.
Step 2 - Preparation of5-(4-chloro-benzyloxy)~2-hydroxymethyl-lH-pyridin-4-one (639): [0895] 5-(4-Chloro-benzyloxy)-2-hydroxymethyl-pyran-4-one (638, 8.00 g, 3.00 mmol) was suspended in ammonium hydroxide (200 mL) in an 80 mL sealed tube. The reaction was heated at 90 0C overnight. Upon cooling, the reaction was lowered to pH 10 with 6N HCl to provide a beige solid that was collected by filtration (639, 7.8 g, 98%).
Step 3 - Preparation of [5-(4-chloro-benzyloxy)-4-methoxy-pyridin-2-yl] -methanol (640):
[0896] 5-(4-Chloro-benzyloxy)-2-hydroxymethyl-lH-pyridin-4-one (639, 1.06 g, 3.99 mmol) was dissolved in methanol (8.5 mL) and N,N-dimethylformamide (46 mL). Trimethylsilyldiazomethane in
hexane (2.00 M, 3.99 mL) was added. The reaction was stirred at room temperature overnight, then additional trimethylsilyldiazomethane in hexane (2.00 M, 3.99 mL) was added. The reaction was stirred at room temperature for 2 days. The mixture was adsorbed onto silica and purified by silica gel chromatography, methanobdichloromethane to provide the compound (640, 798 mg, 72 %). MS(ESl) [M+H*]+ a= 280.4, 282.4.
Step 4 - Preparation of5-(4-chloro-benzyloxy)-4-methoxy-pyridine-2-carbaldehyde (641): [0897] [5-(4-Chloro-benzyloxy)-4-methoxy-pyridin-2-yl]-methanol (640, 480 mg, 1.7 mmol) was dissolved in dimethyl sulfoxide (26 mL) and Dess-Martin periodinane (909 mg, 2.1 mmol) was added. The reaction was allowed to stir at room temperature for 2 hours. The reaction was concentrated under high vacuum and then poured into a solution OfNaHCO3 and Na2S2O3. The mixture was extracted with ethyl acetate. The organic portions were dried with anhydrous sodium sulfate and filtered. The filtrate was adsorbed onto silica and purified by silica gel chromatography, ethyl acetate:hexanes, to provide the desired compound as a white powder (641, 343 mg, 72 %).
Step 5 - Preparation of[5-(4-chloro-benzyloxy)-4-methoxy-pyridin-2-yl]-(l-triisopropylsilanyl~lH-- pyrrolo[2, 3-bJpyridin-3-yl)-methanol (642):
[0898] 3-Iodo-l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (635, 180 mg, 0.450 mmol) was dissolved in tetrahydrofuran (2.5 mL) and the reaction was cooled to -20
0C under an atmosphere of nitrogen. Isopropylmagnesium chloride in tetrahydrofuran (2.00 M, 0.243 mL) was added. The reaction was stirred for 1 hour, during which the temperature rose to 0
0C. The reaction was cooled to -20
0C and 5-(4-chloro-benzyloxy)-4-methoxy-pyridine-2-carbaldehyde (641, 80.0 mg, 0.288 mmol) in tetrahydrofuran (0.75 mL) was added. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was quenched with methanol and adsorbed onto silica, then purified by silica gel chromatography, methanol: dichloromethane, to provide the desired product, (642, 94 mg, 59%).
1H-NMR was consistent with the desired compound. MS(ESI)
552.4, 554.4, 555.4.
Step 6- Preparation of(5-(4-chloro~benzyloxy)-4-methoxy-pyridin-2-yl]-(lH-pyrrolo[2,3-b]pyridin- 3-yl)-methanone (P-2024):
[0899] [5-(4-Chloro-benzyloxy)-4-methoxy-pyridin-2-yl]-(l-triisopropylsilanyl-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanol (642, 60.0 mg, 0.11 mmol) was dissolved in tetrahydrofuran (2.00 mL). Dess-Martin periodinane (55.3 mg, 0.13 mmol) was added to the reaction and it was stirred at room temperature overnight. The mixture was extracted with ethyl acetate and saturated sodium bicarbonate. The organic portions were dried with anhydrous sodium sulfate, filtered and the filtrate was adsorbed onto silica and purified by silica gel chromatography, methanol:dichloromethane, to provide the desired compound (P-2024, 10.7 mg, 25%). 1H-NMR was consistent with the desired compound. MS(ESI) [M+HY = 394.1, 396.1.
Example 48: Synthesis of 3-4-[l-(4-chloro-pheuyl)-etho3sy]-3-methoxy-benzyl-lH-pyrrolo[2,3- b]pyridine P-2000
[0900] 3-4-[l-(4-Chloro-plienyl)-ethoxy]-3-methoxy-benzyl-lH-pyrrolo[2,3-b]pyridme P-2000 was synthesized in three steps from vanillin 105, 4-chloroρhenylmethylcarbinol 643, and l-(tert- butyl-dimethyl-silanyl)-3-iodo-lH-ρyrrolo[2,3-b]pyridine 636, as shown in Scheme 76.
Scheme 76
Step 1 - Preparation of4-[l-(4-chloro-phenyl)-ethoxy]-3-methoxy-benzaldehyde (644): [0901] 4-Chlorophenylmethylcarbinol (643, 0.668 mL, 6.57 mmol) was dissolved in tetrahydroiuran (60.0 mL) at 0 0C under an atmosphere of nitrogen. 4-Hydroxy-3- methoxybenzaldehyde (105, 1.00 g, 6.57 mmol) and triphenylphosphine (2.07 g, 7.89 mmol) were added to the reaction, followed by diisopropyl azodicarboxylate (1.55 mL, 7.89 mmol) over 10 minutes. The reaction was stirred for 2 hours. The mixture was adsorbed onto silica and purified by silica gel chromatography, ethyl acetate:hexanes, to provide the desired compound, (644, 1.14 g, 60 %). 1H-NMR was consistent with the desired compound.
Step 2 - Preparation of[l-(tert-Butyl-dimethyl'Silanyl)-lH-pyrrolo[2,3-b]pyridin-3-yl]-4-[l-(4~ chloro-phenyl)-ethoxy] ' -3-methoxy-phenyl-methanol (645):
[0902] 1 -(tert-Butyl-dimethyl-silanyl)-3-iodo-lH-pyrrolo[2,3-b]pyridine (636, 647.0 mg, 1.81 mmol) was dissolved in tetrahydrofuran (10.0 mL) at -20 0C under an atmosphere of nitrogen. Isopropylmagnesium chloride in tetrahydrofuran (2.0 M, 0.98 mL) was added to the reaction. The reaction was stirred for 1 hour, during which the temperature rose to 0 0C. The reaction was cooled to -20 0C and 4-[l-(4-chloro-phenyl)-ethoxy]-3-methoxy-benzaldehyde (644, 420 mg, 1.4 mmol) in
tetrahydrofuran (3.00 niL) was added. The reaction was stirred for 2 hours during which time the temperature rose to 10 0C. The reaction was quenched with methanol and adsorbed onto silica, then purified by silica gel chromatography, ethyl acetate:hexanes, to provide the desired compound, (645, 463 mg, 61%). 1H-NMR was consistent with the desired compound.
Step 2 — Preparation of3-4-[l-(4-chloro-phenyl)-ethoxy]-3-methoxy-benzyl-lH-pyrrolo[2,3- bjpyήdine (P-2000):
[0903] [l-(tert-Butyl-dimethyl-silanyl)-lH -pyrrolo[2,3-b]pyridm-3-yl]-4-[l-(4-chloro-pheiiyl)- ethoxy]-3-methoxy-phenyl-methanol (645, 0.200 g, 0.382 mmol) was dissolved in acetonitrile (5.00 mL). Trifluoroacetic acid (0.138 niL) was added and the reaction was stirred for five minutes. Triethylsilane (0.285 mL) was added and the reaction was heated at 80 0C for 2 hours. The reaction was concentrated, then redissolved in ethyl acetate and adsorbed onto silica and purified by silica gel chromatography, ethyl acetate±exanes, to provide the desired compound (P-2000, 57 mg, 38 %). 1H- NMR was consistent with the desired compound. MS(ESI): [M^-H+J+= 393.3, 395.3.
Example 49: Synthesis of 5-[4-(2-methoxyethoxy)-phenyl]-lH-pyrrolo[2,3-b]pyridine 648
[0904] 5-[4-(2-Methoxyethoxy)-phenyl]-lH-pyrrolo[2,3-b]pyridine 648 was synthesized in two steps from 4-bromophenol 646 as shown in Scheme 77.
Scheme 77
Step 1 - Preparation l-Bromo-4-(2-methoxy-ethoxy)-benzene (647):
[0905] To a solution of 4-bromophenol (646, 5.0 g, 28.9 mmol) in dimethylformamide (15 mL) were added potassium carbonate (4.40 g, 31.8 mmol) and l-bromo-2-methoxyethane (5.00 g, 36.0 mmol) under an atmosphere of nitrogen. The reaction mixture was stirred at ambient temperature overnight and concentrated under reduced pressure. The residue was slurried in ethyl acetate (50 mL) and filtered. The filtrate was washed with saturated sodium bicarbonate solution, dried over magnesium sulfate and filtered. Silica gel column chromatography (0-10% ethyl acetate in hexanes) gave the desired compound as a colorless oil (647, 3.2 g, 48%).
Step 2 - Preparation of5-[4-(2-Methoxy-ethoxy)-phenyl]~lH-pyrrolo[2,3~b]pyridine (648): [0906] To a solution of 5-(4,4,5,5,-tetramethyl-[l ,3,2]dioxaborolan-2-yl)-lH-pyrrolol[2,3- b]pyridine (1.1 g, 4.3 mmol) in tetrahydrofuran (40 mL) was added l-bromo-4-(2-methoxy-ethoxy)- benzene (647, 1.50 g, 6.49 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.25 g, 0.21 mmol).
The reaction mixture was stirred with potassium carbonate solution (10 mL, 1.0 M) and warmed to reflux overnight. The biphasic reaction mixture was diluted with ethyl acetate (50 mL) and saturated sodium carbonate solution (20 mL). The organic layer was separated, washed with brine, dried over magnesium sulfate and purified by silica gel column chromatography (50-100% ethyl acetate in hexanes) to give the desired compound as a colorless solid (648, 782 mg, 67%). MS (ESI) PVM-H4I+= 267.4.
Example 50: Synthesis of 3-[2-fluoro-5-methoxy-4-φyridin-4-ylmethoxy)-beiizyl]-lH- pyrrolo[2,3-b]pyridine P-2040 and related compounds.
[0907] Compound P-2040 was synthesized in one step from 5-fluoro-2-methoxy-4-(l- triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenol 649 and pyridin-4-yl-methanol 650 as shown in Scheme 78.
Sche
Step 1 - Preparation of3-[2-fluoro-5-methoxy-4-(pytϊdin-4-ylmethoxy)~benzyl]-lH~pyrrolo[2,3- b] pyridine (P-2040):
[0908] 5-Fluoro-2-methoxy-4-(l-triiisopropylsilanyl-lH-pyrrolo[2,3-b]pyridrne-3-ylmethyl)- phenol (649, 10 mg, 0.024 mmol, prepared as described in Example 52) was combined with pyridin- 4-yl-methanol (650, 3.2mg, 0.029 mmol) in a 4 mL vial and dissolved in dry tetrahydrofuran (200 μl). Triphenylphosphine (7.7 mg) was added and the solution was shaken until homogenous. The mixture was cooled to below 00C in a liquid nitrogen bath and diisopropyl azodicarboxylate solution (50μl of 20mg/50μl in THF) was added. The reaction mixture was allowed to warm to room temperature. After 2 hours, the solvent was removed under reduced atmosphere. The crude material was dissolved in dimethyl sulfoxide (300μl) and potassium fluoride (10 mg, 0.1 Smmol) was added. The mixture was heated gently and allowed to react overnight at room temperature. The vial was centrifuged and the DMSO solution was purified by reverse phase HPLC using a YMC-Pack ODS-A C-18 column (50mm x 10mm ID), and eluting with water with 0.1% TFA and a gradient of 15%-80% acetonitrile with 0.1% TFA over 8 minutes and a flow rate of 6 mL/minute to provide the compound (P-2040, 4.4 mg, 50%). MS (ESI) [M-HT]+= 364.3.
[0909] Additional compounds were prepared following the protocol of Scheme 78, replacing pyridin-4-yl-methanol 650 with an appropriate alcohol. The following compounds were made following this procedure:
3-[2-Fluoro-5 -methoxy-4-(2-morpholin-4-yl-ethoxy)-benzyl] - 1 H-pyrrolo [2,3 -b]pyridine
(P-2037),
3-[2-Fluoro-5-methoxy-4-(pyridin-3-ylmethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2038),
3-[2-Fluoro-5-methoxy-4-(6-methyl-pyridm-2-ylmethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-2039),
3-[2-Fluoro-5-methoxy-4-(pyridin-2-ylmethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2041),
3-[2-Fluoro-4-(2-fluoro-4-trifluoromethyl-benzyloxy)-5-methoxy-benzyl]-lH-pyrrolo[2,3- b]pyridine (P-2042),
3-[4-(4-Chloro-2-fluoro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-1973),
3-[4-(2,4-Dimethyl-thiazol-5-ylmethoxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridme
(P-2043),
3-[4-(2,5-Dimethyl-2H-pyrazol-3-ylmethoxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3- b]pyridine (P-2044),
3 -[2-Fluoro-5 -methoxy-4-(3 -morpholin-4-yl-propoxy)-benzyl] - 1 H-pyrrolo [2,3 -b]pyridine
(P-2045), l-{2-[5-Fluoro-2-methoxy-4-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenoxy]-ethyl}-pyrrolidin-
2-one (P-2046),
3-[2-Fluoro-4-(2-fluoro-benzyloxy)-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2047),
3-[2-Fluoro-5-methoxy-4-(3-methyl-pyridin-4-ylmethoxy)-benzyl]-lH-pyrrolo[2,3-b]pyridine
(P-2048),
3-[2-Fluoro-5-methoxy-4-(6-trifluoromethyl-pyridin-3-ylmethoxy)-benzyl]-lH-pyrrolo[2,3- bjpyridine (P-2049),
3-[4-(2,4-Dichloro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2050),
3 -[2-Fluoro-4-(4-imidazol- 1 -yl-benzyloxy)-5 -methoxy-benzyl] - 1 H-pyrrolo [2,3 -b]pyridine
(P-2051),
3-[4-(2,4-Difluoro-benzyloxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2052),
3-{2-Fluoro-4-[l-(2-fluoro-phenyl)-ethoxy]-5-methoxy-benzyl}-lH-pyrrolo[2,3-b]pyridine
(P-2053),
3-[4-(3-Cyclopentyl-propoxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2054),
3-[4-(l,5-Dimethyl-lH-pyrazol-3-ylmethoxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3- b]pyridine (P-2055), and
3-[4-(2-Cyclopentyl-ethoxy)-2-fluoro-5-methoxy-benzyl]-lH-pyrrolo[2,3-b]pyridine (P-2056)
The following table indicates the alcohol (column 2) used in Scheme 78 to provide the compounds (column 4). Column 1 provides the compound number and column 4 the observed mass.
Example 51: Synthesis of [2,6-difluoro-3-(pyridin-3-ylmethoxy)-phenyl]-(lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone P-2058 and related compounds.
[0910] Compound P-2058 was synthesized in 1 step from (2,6-Difluoro-3-hydroxy-plienyi)-(lH- pyrrolo[2,3-b]pyridin-3-yl)-methanone 651 and Pyridin-3-yl-methanol 652 as shown in Scheme 79.
Scheme 79
651
P-2058
Step 1 - Preparation of [2, 6-Difluoro-3-(ρyridin-3-ylmethoxy)-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone (P-2058):
[0911] In a 4mL vial, (2,6-Difluoro-3-hydroxy-phenyl)-(lH-ρyrrolo[2,3-b]pyridine-3-yl)- methanone (651, 10 mg, 0.037 mmol, prepared as described in Example 23) was combined with pyridin-3-yl-methanol (652, 4.9 mg 0.044 mmol). The solids were dissolved in dry tetrahydrofuran (200μl) and triphenylphosphine (11.5 mg, 0.044 mmol) was added. Once the solution was homogenous, the mixture was cooled to below 0 0C in liquid nitrogen bath and diisopropyl azodicarboxylate solution (50μl of 20mg/50μl TBDF) was added. The reaction mixture was allowed to warm to room temperature and the reaction was continued for 2 hours. The solvents were removed under reduced atmosphere. The resultant residue was diluted with 200μl DMSO and the mixture purified by reverse phase HPLC using a YMC-Pack ODS-A C- 18 column (50mm x 10mm ID), and eluting with water with 0.1% TFA and a gradient of 15%-80% acetonitrile with 0.1% TFA over 8 minutes and a flow rate of 6 mL/minute to provide P-2058 (5.9 mg, 44 %). MS (ESI) PVH-HT = 365.9.
[0912] Additional compounds were prepared following the protocol of Scheme 79, replacing pyridin-3-yl-methanol 652 with an appropriate alcohol. The following compounds were made following this procedure:
[2,6-Difluoro-3-(l-methyl-lH-imidazol-2-ylmethoxy)-phenyl]-(lH-pyrrolo[2,3-b]ρyridin-3-yl)- methanone (P-2033),
[2,6-Difluoro-3-(6-morpholin-4-yl-pyridin-3-ylmethoxy)-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone (P-2034),
{2,6-Difluoro-3-[4-(5-methyl-[l,2,4]oxadiazol-3-yl)-benzyloxy]-phenyl}-(lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone (P-2035),
[3-(6-Diethylamino-ρyridin-3-ylmethoxy)-2,6-difluoro-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-2036),
[3-(2-Chloro-4-fluoro-benzyloxy)-2,6-difluoro-ρhenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-20S7),
[2)6-Difluoro-3-(6-methyl-pyridin-2-ylmethoxy)-phenyl]-(lH-ρyrrolo[2,3-b]pyridin-3-yl)- methanone (P-20S9),
[2,6-Difluoro-3-(pyridin-4-ylmethoxy)-phenyl]-(lH~pyiτolo[2,3-b]pyridin-3-yl)-methanone
(P-2060),
[3-(4-Chloro-2-fluoro-benzyloxy)-2,6-difluoro-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-2061),
[3-(2,4-Dimethyl-thiazol-5-ylmethoxy)-2,6-difluoro-ρhenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-2062),
[3-(2,5-Dimethyl-2H-pyrazol-3-ylmethoxy)-2,6-difluoro-phenyl]-(lH-pyrrolo[253-b]pyridin-3- yl)-methanone (P-2063), and
[2,6-Difluoro-3-(3-moφholin-4-yl-propoxy)-ρhenyl]-(lH-ρyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-2064).
The following table indicates the alcohol (column 2) used to afford the compound (column 3). Column 1 provides the compound number and column 4 the observed mass.
Example 52: Synthesis of 5-fluoro-2-methoxy-4-(l-triisopropylsiIanyl-lH-pyrrolo[2,3-b]pyridin- 3-ylmethyl)-phenol 649.
[0913] 5-Fluoro-2-methoxy-4-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenol 649 was synthesized in five steps from 2-fluoro-4-hydroxy-5-methoxy-benzaldehyde 653 and benzyl bromide as shown in Scheme 80.
Scheme 80
Step 1 —Preparation of4-Benzyloxy-2-fluoro-5-methoxy-benzaldehyde (654): [0914] 2-Fluoro-4-hydroxy-5-methoxy-benzaldehyde (653, 1.62 g, 9.52 mmol) was dissolved in N,N-dimethylformamide (50 mL) and sodium hydride (60% dispersion in mineral oil, 530 mg, 13 mmol) was added. After 20 minutes, benzyl bromide (1.5 mL, 12 mmol) was added to the reaction mixture. The reaction was stirred at room temperature under an atmosphere of nitrogen for 5.5 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 0-50 % ethyl acetate in hexane to provide compound as a white solid, consistent with the desired structure by 1H-NMR (654, 2.0 g, 81 %).
Step 2 — Preparation of(4-Benzyloxy-2-fluoro-5-methoxy-phenyl)-(l-triisopropylsilanyl-lH- pyrrolo[2,3-b]pyridin-3-yl)-methanol (65S)\
[0915] 3-Iodo-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (635, 620 mg, 1.5 mmol, prepared as described in Example 45) was dissolved in tetrahydrofuran (15 mL) at -20 0C under an atmosphere of nitrogen. Isopropylmagnesium chloride (2.0 M in tetrahydrofuran, 840 μL) was added to the reaction.
The reaction was stirred for 1.5 hours, during which the temperature rose to 5
0C. The reaction was cooled to -20
0C. 4-Benzyloxy-2-fluoro-5-methoxy-benzaldehyde (654, 250 mg, 0.9606 mmol) in
tetrahydrofuran (5.0 mL) was added to the reaction. The reaction was stirred for 2.5 hours during which time the temperature rose to 5
0C. The reaction was poured into water. The mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 2-25 % ethyl acetate in hexane to provide compound as a white solid (655, 501 mg, 63 %). MS (ESI)
Step 3 - Preparation of3-(4-Benzyloxy-2-fluoro-5-methoxy-benzyl)-lH-pyrrolo[2,3-bJpyridine (656): [0916] (4-Benzyloxy-2-fluoro-5-methoxy-phenyl)-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin- 3-yl)-methanol (655, 1.49 g, 2.79 mmol) was dissolved in acetonitrile (50 mL) and trifluoroacetic Acid (1.1 mL) was added. The reaction was stirred for 5 minutes. Triethylsilane (2.2 mL) was added to the reaction. The reaction was heated at 80 0C for 6 hours. The reaction was concentrated and the crude material was dissolved into ethyl acetate and washed with 1 N HCl, saturated sodium bicarbonate, and brine. The organic portion was dried over anhydrous sodium sulfate and concentrated. The solid obtained was used in the next reaction without further purification (656, 833 mg, 83%). MS (ESI) [M+HY = 363.4.
Step 4 - Preparation of3-(4-Benzyloxy-2-fluoro-5-methoxy-benzyl)-l-triisopropylsilanyl-lH- pyrrolo [2, 3-b] pyridine (657):
[0917] 3-(4-Benzyloxy-2-fluoro-5-methoxy-benzyl)-lH-pyrrolo[2,3-b]pyridine (656, 0.877 g, 2.42 mmol) was dissolved in N,N-dimethylformamide (30 mL). Sodium hydride (60% dispersion in mineral oil, 140 mg, 3.6 mmol) was added at room temperature. After 20 minutes, triisopropylsilyl chloride (513 μL, 2.42 mmol) was added dropwise. The reaction was stirred for four hours. The reaction was poured into water and extracted with ethyl acetate. The organic portion was washed with saturated sodium bicarbonate and brine. The organic portion was dried over anhydrous sodium sulfate and filtered. The filtrate was adsorbed onto silica gel and purified by silica gel chromatography using 20-80% ethyl acetate/ hexane. The resulting material was purified a second time with 5-30% gradient ethyl acetate/ hexane to provide the desired compound (657, 831 mg, 66%). MS (ESI) [M-Hf]+ = S^A
Step 5 - Preparation of5-Fluoro-2-methoxy-4-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl)-phenol (649):
[0918] 3-(4-Benzyloxy-2-fluoro-5-methoxy-benzyl)-l-triisopropylsilanyl-lH-pyrrolo[2,3- b]ρyridine (657, 0.831 g, 1.60 mmol) was dissolved in methanol (40 mL) and tetrahydrofuran (40 mL). 10% Palladium on carbon (3.41 g) was added. The reaction was shaken at 50 psi for 1 hour. The reaction was filtered through Celite and washed with methanol. The organic portion was passed through celite several times until a clear solution was obtained. The organic portion was concentrated
under reduced pressure to provide the desired compound as an off-white solid (649, 587 mg, 86%). MS (ESI) [M+HT = 429.5.
Example 53: Synthesis of (5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fiuoro-3-hydroxy- phenyD-methanone P-0067 and related compounds.
[0919] Compound P-0067 was synthesized in two steps from 5-bromo-lH-pyrrolo[2,3-b]pyridine 67 as shown in Scheme 10.
Scheme 10
Step 1 — Preparation of(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-methoxy-phenyl)- methanone (P-0265):
[0920] To 5-bromo-lH-pyrrolo[2,3-b]pyridine (67, 2.00 g, 0.0102 mol) in methylene chloride (120 rnL), under an atmosphere of nitrogen, was added aluminum trichloride (8.20 g, 0.0615 mol). The reaction was stirred at room temperature for 60 minutes, then 2-fluoro-3-methoxy-benzoyl chloride (34, 2.12 g, 0.0112 mol, prepared from the corresponding carboxylic acid using the protocol of Example 1, Scheme 13, Step 4), dissolved in methylene chloride (20.0 mL), was added. After 2 hours, the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography, eluting with 25% ethyl acetate in hexane to provide P-0265 (approx 2.6 g, 73%). MS (ESI) [M-BT]" = 347, 349.
Step 2 - Preparation of(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-hydroxy-phenyl)- methanone (P-0067):
[0921] To (5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-methoxy-phenyl)-methanone (P-0265, 430.0 mg, 1.23 mmol) in methylene chloride (60 mL) was added boron tribromide (1.0 M in methylene chloride, 10 mL). The reaction mixture was stirred at room temperature overnight. The reaction was quenched with methanol (3.0 mL), poured into water and extracted with ethyl acetate. The organic layer was dried, concentrated and purified by silica gel chromatography eluting with 25% ethyl acetate in hexane to provide the compound (P-0067, 180 mg, 44%). MS (ESI) [M-H+]" = 333.0, 335.0.
[0922] 2-Fluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenol P-0778
was prepared using the protocol of Scheme 10, Step 2, replacing (5-Bromo-lH-pyrrolo[2,3-b]pyridin- 3-yl)-(2-fluoro-3-methoxy-phenyl)-methanone P-0265 with 3-(2-Fluoro-3-methoxy-benzyl)-5- pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (P-0269, prepared as described in Example 10, Scheme 24). MS(ESI) |M+H*]
+ = 320.2.
[0923] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-5-hydroxy-phenyl)-methanone P-0068
was prepared using the protocol of Scheme 10, replacing 2-Fluoro-3-methoxy-benzoyl chloride 34 with 2-Fluoro-5-methoxy-benzoyl chloride (prepared from the corresponding carboxylic acid using the protocol of Example 1, Scheme 13, Step 4) in Step 1. MS(ESI) [M-H
+]
" = 347, 349.
Example 54: Synthesis of (2-fluoro-3-hydroxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b] pyridin-3- yl)-methanone P-0036.
[0924] Compound P-0036 was synthesized in three steps from (5-Bromo-lH-pyrrolo[2,3-b]pyridin- 3-yl)-(2-fluoro-3-hydroxy-phenyl)-methanone P-0067 as shown in Scheme 11.
Step 1 — Preparation of5-bromo-3-(3-tert-butoxycarboιιyloxy~2-fluoro-benzoyl)-pyrrolo[2,3- b]pyridine-l -carboxylic acid tert-butyl ester (32):
[0925] To (5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-hydroxy-phenyl)-methanone (P-0067, 310.0 mg, 0.93 mmol, prepared as described in Example 53, Scheme 10) in tetrahydrofuran (40 mL) were added sodium hydride (60% dispersion, 81.4 mg, 2.04 mmol) and di-tert-
butyldicarbonate (0.50 g, 2.3 mmol). The reaction was stirred at room temperature for 1 hour. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography, eluting with 20% ethyl acetate in hexane to provide the compound (32, 0.45 g, 91%).
Step 2 - Preparation of3-(3-tert~butoxycarbonyloxy-2-fluoro-benzoyl)-5-phenyl~pyrrolo [2,3- bjpyridine-l -carboxylic acid tert-butyl ester (33):
[0926] To 5-bromo-3-(3-tert-butoxycarbonyloxy-2-fluoro-benzoyl)-pyrrolo[2,3-b]pyridine-l- carboxylic acid tert-butyl ester (32, 55.0 mg, 0.10 mmol) in tetrahydrofuran (10 mL) were added 1.0 M potassium carbonate in water (3.5 mL), phenylboronic acid (18.8 mg, 0.15 mmol) and tetrakis(triphenylphosphine)palladium(0) (3.9 mg, 0.0034 mmol) under an atmosphere of nitrogen. The reaction was stirred at 65 0C for 3 hours, and then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, and concentrated to give crude compound 33 that was used in the next step.
Step 3 — Preparation of(2-fluoro-3-hydroxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-0036):
[0927] To 3-(3-tert-butoxycarbonyloxy-2-fluoro-benzoyl)-5-phenyl-pyrrolo[2,3-b]pyridine-l - carboxylic acid tert-butyl ester (33, 30.0 mg, 0.056 mmol) in methylene chloride (6.0 mL) was added 4 N HCl in dioxane (2.0 mL). The reaction was stirred at room temperature for 36 hours. Solvents were removed by evaporation. The residue was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography, eluting with 25% ethyl acetate in hexane to give a white solid (P-0036, 4.8 mg, 24%). MS(ESI) [M+HT = 333.2.
[0928] Additional compounds were prepared following the protocol of Scheme 11 , optionally replacing (5-bromo-lH-ρyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-hydroxy-phenyl)-methanone P-0067 with (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-hydroxy-2-methyl-phenyl)-methanone P-0055 (prepared as described in Example 56, Scheme 22) in Step 1 and optionally replacing phenylboronic acid with an appropriate boronic acid in Step 2. The following compounds were made following this procedure:
N- {3-[3 -(2-Fluoro-3 -hydroxy-benzoyl)- 1 H-pyrrolo[2,3-b]pyridin-5-yl] -phenyl} -acetamide
(P-0035),
N-{3-[3-(2-Fluoro-3-hydroxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]-phenyl}- methanesulfonamide (P-0038),
3-[3-(2-Fluoro-3-hydroxy-benzoyl)-lH-pyrrolo[2,3-b]pyridin-5-yl]benzamide (P-0034),
(3-Hydroxy-2-methyl-phenyl)-(5-ρyridin-3-yl-lH-pyrrolo[2,3b]pyridin-3-yl)-methanone
(P-0006),
(3-Hydroxy-2-methyl-phenyl)-(5-thiophen-2-yl-lH-pyrrolo[2,3b]pyridm-3-yl)-methanone
(P-0114),
(3-Hydroxy-2-methyl-phenyl)-(5-phenyl-lH-pyrrolo[2,3b]pyridin-3-yl)-methanone (P-0016),
(2-Fluoro-3-hydroxy-phenyl)-[5-(3-methanesulfonylphenyl)lH-pyrrolo[2,3-b]pyridin-3-yl]- methanone (P-0026),
(2-fluoro-3-hydroxy-phenyl)-(5-pyrimidin-5-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone
(P-0744), and
[5-(3 -amino-phenyl)- 1 H-pyrrolo[2,3 -b]pyridin-3 -yl] -(2-fmoro-3 -hydroxy-phenyl)methanone
(P-0004).
The following table indicates the boronic acid (Column2) and the 5-Br azaindole (Column 3) used to afford the compound (Column 4). Column 1 provides the compound number and Column 5 the observed mass.
Example 55: Synthesis of (2,6-Difluoro-3-hydroxy-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2,3- b] pyridin-3-yl)-methanone P-0066.
[0929] Compound P-0066 was synthesized in six steps from 2,4-difluoro-phenol 35 as shown in Scheme 12.
Scheme 12
Step 1 - Preparation oftert~butyl-(2,4~difluoro-phenoxy)-dimethyl-silane (36): [0930] To 2,4-difluoro-phenol (35, 5.00 g, 38.4 mmol) in N, N-dimethylformamide (100 mL) were added tert-butyldimethylsilyl chloride (7.00 g, 46.4 mmol) and imidazole (6.28 g, 92.2 mmol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtrated. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% ethyl acetate in hexane. The appropriate fractions were combined and concentrated to provide the compound (36, 7.50 g, 80.0%). MS(ESI) [M+H*]+ = 245.1.
Step 2 - Preparation of3-(tert-butyl-dimethyl-silanyloxy)-2,6-difluoro-benzaldehyde (37): [0931] To ter^butyl-(2,4-difluoro-phenoxy)-dimethyl-silane (36, 5.90 g, 24.1 mmol) in tetrahydrofuran (100 mL), under an atmosphere of nitrogen, cooled in a -78 0C acetone/dry ice bath, was added n-butyllithium (2.50 M in hexane, 10.6 mL, 26.5 mmol) slowly. The reaction was allowed to stir for 1 hour, then N,N-dimethylformamide (2.24 mL, 29.0 mmol) was added and the reaction was allowed to warm to room temperature overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The compound was isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give a colorless oil (37, 5.0 g, 76.0%). MS(ESI) [M+H+f = 272.2.
Step 3 — Preparation of2,6-difluoro-3-hydroxy-benzaldehyde (38):
[0932] To 3-(ter/-butyl-dimethyl-silanyloxy)-2,6-difluoro-benzaldehyde (37, 3.50 g, 12.9 mmol) in N,N-dimethylformamide (50 mL) was added tetrabutylammonium fluoride trihydrate (4.40 g, 14.0 mmol). The reaction was stirred at room temperature for 1 hour. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated to give crude compound 38 that was used in the next step.
Step 4 — Preparation of carbonic acid tert-butyl ester 2,4-difluoro-3-formyl-phenyl ester (39): [0933] To 2,6-difluoro-3-hydroxy-benzaldehyde (38, 1.90 g, 12.0 mmol) in tetrahydrofuran (60 mL) was added sodium hydride (60% in mineral oil, 0.58 g, 14.5 mmol) and di-tø^-butyldicarbonate (3.90 g, 17.9 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give a colorless oil (39, 2.50 g, 80.8%). MS(ESI) [M+!^]+ = 258.1.
Step 5 - Preparation of2,4-difluoro-3-[hydroxy-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methyl] -phenol (P-0009):
[0934] To carbonic acid tert-butyl ester 2,4-difluoro-3-formyl-phenyl ester (39, 0.405 g, 15.7 mmol) in methanol (36 mL), under an atmosphere of nitrogen, was added 5-pyridin-3-yl-lH-pyrrolo[2,3- b]pyridine (89, 288.0 mg, 14.8 mmol, prepared as in Example 17, Scheme 32) and potassium hydroxide (145.0 mg, 25.9 mol). The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 4% methanol in methylene chloride to give a colorless oil (P-0009, 0.23 g, 44.1%). MS(ESI) [M+HT = 354.1.
Step 6 - Preparation of (2, 6-difluoro-3-hydroxy-phenyl)-(5-pyridin-3-yl-lH-pyrrolo[2, 3-b]pyridin-3- yl)-methanone (P-0066):
[0935] To 2,4-difluoro-34hydroxy-(5-pyridm-3-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methyl]-phenol (P-0009, 45.0 mg, 1.27 mmol) in tetrahydrofuran (15 niL) and N,N-dimethylformamide (5 mL) was added dichlorodicyanoquinone (87 mg, 3.8 mmol). The reaction was stirred at room temperature for 50 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (P-0066, 7 mg, 15.6%). MS(ESI) [M+H+]+ = 352.1.
[0936] (5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2,6-difluoro-3-hydroxy-phenyl)-methanone P-1271
was prepared following the protocol of Scheme 12, substituting 5-pyridin-3-yl-lH-pyrrolo[2,3- bjpyridine 89 with 5-bromo-lH-pyrrolo[2,3-b]pyridine 67 in Step 5. This was reacted further (70.0 mg, 0.20 mmol) in methylene chloride (15 mL) by adding pyridine (0.50 mL, 6.2 mmol), propane-1- sulfonyl chloride (0.50 g, 3.5 mmol) and 4-dimethylaminopyridine (0.10 g, 0.82 mmol). The reaction was,.stirred at room temperature overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The compond, propane- 1 -sulfonic acid 3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-2,4-difluoro-phenyl ester P-0919
was isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give a white solid (15 mg, 16%). MS(ESI) [M - H
+]
' = 456.9, 458.9.
Example 56: Synthesis of (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-hydroxy-2-methyl- phenyl)-methanone P-0055.
[0937] Compound P-0055 was synthesized in two steps from 5-Bromo-7-azaindole 67 as shown in Scheme 22.
Scheme 22
Step 1 - Preparation of acetic acid 3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-caτ-bonyl)-2-methyl- phenyl ester (84):
[0938] To aluminum trichloride (6.14 g, 0.046 mol) in methylene chloride (26 mL) under an atmosphere of nitrogen was added 5-Bromo-7-azaindole (67, 1.05 g, 5.31 mmol) dissolved in methylene dichloride (20.0 mL). The reaction was stirred at room temperature for 65.0 minutes, then acetic acid 3-chlorocarbonyl-2-methyl-phenyl ester (1.25 g, 5.88 mmol) in methylene dichloride (7.0 mL) was added. The reaction was stirred for 2 hours at room temperature, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated to give crude compound 84 that was used directly in the next step.
Step 2 - Preparation of(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-hydroxy-2-methyl-phenyl)- methanone (P-0055):
[0939] A solution of acetic acid 3-(5-bromo-lH-pyrrolo[2,3-b]pyridine-3-carbonyl)-2-methyl- phenyl ester (84, 25.0 mg, 0.067 mmol) in 6 N HCl (4.0 mL) was heated with a CEM Discover microwave instrument at 110 0C for 10 min. The reaction mixture was poured into water, neutralized with 1 M aqueous potassium carbonate, and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give a colorless oil (P-0055, 4.7 mg, 21%). MS(ESI) [M - ϊtj = 329.0, 331.0.
Example 57: Synthesis of 2-Fluoro-3-(5-thiophen-2-yI-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenol P-0052.
[0940] Compound P-0052 was synthesized in five steps from (5-Bromo-lH-pyrrolo[2,3-b]pyridin- 3-yl)-(2-fluoro-3-methoxy-phenyl)-methanone P-0265 as shown in Scheme 23.
Scheme 23
Step 1 - Preparation of5-bromo-3-(2-fluoro-3-methoxy-benzoyl)-pyrrolo[2,3-bJpyridine-l-carboxylic acid tert-butyl ester (86):
[0941] To (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(2-fluoro-3-methoxy-phenyl)-metlianone (P-0265, 600.0 mg, 1.72 mmol, prepared as described in Example 1 or 53) in tetrahydrofuran (20 mL) was added sodium hydride (60% dispersion, 103 mg, 2.58 mmol) under an atmosphere of nitrogen. After 15 minutes, di-tert-butyldicarbonate (648 mg, 2.97 mmol) was added. The reaction mixture was stirred at room temperature for 1 hour, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 10% ethyl acetate in hexane to give a colorless oil (86, 0.64 g, 82.3%).
Step 2 - Preparation of3-(2-fluoro-3-methoxy-benzoyl)-5-thiophen-2-yl-pyrrolo[2,3-b]pyridine-l- carboxylic acid tert-butyl ester (87):
[0942] To 5-bromo-3-(2-fluoro-3-methoxy-benzoyl)-pyrrolo[2,3-b]pyridine-l-carboxylic acid tert- butyl ester (86, 350.0 mg, 0.78 mmol) in tetrahydrofuran (30 mL) and water (10 mL) were added 2- thiophene boronic acid (190 mg, 1.49 mmol), tetrakis(triphenyl-phosphine)palladium(0) (40.0 mg, 0.0346 mmol) and potassium carbonate (780 mg, 5.64 mmol) under an atmosphere of nitrogen. The reaction mixture was refluxed for 2 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated to give crude compound (87, 0.30 g, 85%).
Step 3 - Preparation of(2-fluoro-3-hydroxy-phenyl)-(5-thiophen-2-yl-lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone (P-0521):
[0943] To 3-(2-fluoro-3-methoxy-benzoyl)-5-thiophen-2-yl-pyrrolo[2,3-b]pyridine-l-carboxylic acid tert-hxxtyl ester (87, 200 mg, 0.44 mmol) in methylene chloride (10 mL), under an atmosphere of nitrogen, cooled in a -78 0C acetone/dry ice bath, was added boron tribromide (1.0 M in hexane, 2.0 mL). The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched with methanol (3.0 mL), poured into water and extracted with ethyl acetate.
The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (P-0521, 119 mg, 82.6%). MS(ESI)[M-H+]" = 337.
Step 4 - Preparation of2-Fluoro-3-(5-thiophen-2-yl-lH-pyrrolo[2,3-b]pyήdin-3-ylmethyl)-phenol (P-0052):
[0944] To (2-fluoro-3-hydroxy-phenyl)-(5-thiophen-2-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-0521, 50.0 mg, 0.15 mmol) in diethylene glycol (5.0 mL) were added hydrazine hydrate (1.0 mL, 0.020 mol) and potassium hydroxide (200.0 mg, 3.56 mmol). The reaction mixture was heated in a CEM Discovery microwave instrument at 180 0C for 20 minutes, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated. The desired compound was isolated by silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (P-0052, 10 mg, 20%). MS(ESI) [M + Yt)+ = 325.2.
Example 58: Synthesis of (2,4-Difluoro-3-formyl-phenyl)-carbamic acid tert-butyl ester 124.
[0945] (2,4-Difluoro-3-formyl-phenyl)-carbamic acid tert-butyl ester 124 was synthesized in two steps from 2,4-Difluoro-phenylamine 42 as shown in Scheme 25.
Scheme 25
Step 1 - Preparation of (2,4-Difluoro-phenyl)-carbamic acid tert-butyl ester (123): [0946] To 2,4-difluoro-phenylamine (42, 5.0 g, 0.039 mol) in methylene chloride (100 mL) were added di-tert-butyldicarbonate (21.1 g, 0.0968 mol), triethylamine (16 mL, 0.12 mol) and 4- dimethylaminopyridine (0.2 g, 0.002 mol). The reaction was stirred at room temperature overnight, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography, eluting with 20% ethyl acetate in hexane to provide compound 123.
Step 2 - Preparation of(2,4-Difluoro-3-formyl-phenyl)-carbamic acid tert-butyl ester (124): [0947] To 2,4-difluoro-phenyl)-carbamic acid tert-butyl ester (123, 1.41 g, 6.17 mmol) in tetrahydrofuran (60.0 mL) under an atmosphere of nitrogen at 78 0C were added n-butyllithium (2.50 M in hexane, 2.59 mL) for 30 min. Lithium diisopropylamide (2.0 M in hexane, 3.4 mL) was added
to the reaction. After 35 min, N,N-dimethylformamide (1.05 mL, 0.0136 mol) was added to the reaction mixture. The reaction was allowed to cool to room temperature overnight. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography, eluting with 20% ethyl acetate in hexane to provide compound 124.
Example 59: Synthesis of l-[4-(lH-Pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-3-(3- trifluoromethyl-phenyl)-urea P-0262.
[0948] Compound P-0262 was synthesized in seven steps from lH-Pyrrolo[2,3-b]pyridine 94 as shown in Scheme 34.
Scheme 34
Step 1 - Preparation ofDimethyl-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-amine (109): [0949] lH-Pyrrolo[2,3-b]pyridine (94, 5.0 g, 42 mmol), dimethylamine hydrochloride (3.8 g, 46 mmol), formaldehyde (1.4 g, 46 mmol) and isopropyl alcohol (220.0 mL) were combined in a pressure tube. The reaction mixture was stirred at 20 0C for 12 hours and then refluxed for 2 hours. The clear solution was evaporated to dryness in vacuo. Water (40 mL) and concentrated hydrochloric acid (4 mL) were added to the residue. The mixture was extracted with ether and then made strongly basic with potassium carbonate. The basified aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified by silica gel chromatography to give the compound (109, 5.Og, 68%).
Step 2 - Preparation ofDimethyl-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-amine (110):
[0950] Dimethyl-(lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-amine 109 (5.0 g, 28 mmol), was dissolved in N,N-dimethylformamide (80.0 mL) and sodium hydride (60% dispersion in mineral oil, 1.25 g, 31.4 mmol) was added, followed by triisopropylsilyl chloride (6.3 mL, 30.0 mmol). The reaction was stirred at room temperature for 12 hours, then poured into water and extracted with ethyl acetate. The organic portion was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel chromatography to give the compound (110, 6.5 g, 70%).
Step 3 — Preparation of3-(4-Bromo-benzyl)-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (111): [0951] l-bromo-4-iodobenzene (2.1 g, 7.4 mmol) was dissolved in tetrahydrofuran (15.0 mL) under an atmosphere of nitrogen at -30 0C. Isopropylmagnesium chloride in tetrahydrofuran (2.0 M, 4.0 mL) was added. The reaction mixture was stirred below -20 0C for 1 hour to provide a solution of 1- bromo-4-benzene magnesium chloride.
[0952] Dimethyl-(1 -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-amine 110 (1.25 g, 3.77 mmol) was added to toluene (20.0 mL) under an atmosphere of nitrogen at room temperature. Isopropyl chloroformate in toluene (1.0 M, 4.0 mL) was added. The reaction mixture was stirred at room temperature for 3 hours to provide a solution of 3-chloromethyl-l-triisopropylsilanyl-lH- pyrrolo[2,3-b]pyridine.
[0953] CuCN.2LiCl in tetrahydrofuran (0.7 M, 10 mL) was added to the solution of l-bromo-4- benzene magnesium chloride at -20 0C. The reaction mixture was allowed to warm to room temperature for 10 minutes. Trimethyl phosphite (1.8 mL, 0.015 mol) was added to the reaction. After 5 minutes, the solution of 3-chloromethyl-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine was added to the reaction at room temperature. The reaction was stirred for 12 hours at room temperature. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel chromatography to give the compound (111, 850 mg, 51%).
Step 4 - Preparation ofBenτyl-[4-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)- phenyl] -amine (112):
[0954] 3-(4-Bromo-benzyl)-l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridine (111, 330.0 mg, 0.74 mmol), benzylamine (160 mg, 1.5 mmol), 2-[di(tert-butyl)phosphino]-l,l'-biphenyl (15.0 mg, 0.05 mmol), tris(dibeπzylideneacetone)dipalladium(0) (15.0 mg, 0.016 mmol), potassium tert-butoxide (100 mg, 0.89 mmol) and toluene (10.0 mL) were combined under an atmosphere of nitrogen. The reaction mixture was refluxed overnight, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel chromatography to give the compound (112, 260 mg, 75%).
Step 5 — Preparation of4-(l-Triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenylamine
(113):
[0955] Benzyl-[4-(l -triisoρropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenyl]-amine (112,
210.0 mg, 0.447 mmol), methanol (15.0 niL) and palladium hydroxide (30.0 mg, 0.214 mmol) were combined. The reaction mixture was hydrogenated under under an atmosphere of hydrogen (1 arm) at room temperature for 2 hours. Filtration and concentration gave the compound (113, 154 mg, 91%).
Step 6 - Preparation ofl-(3-Trifluoromethyl-phenyl)-3-[4-(l-triisopropylsilanyl-lH-pyrrolo[2, 3- b]pyridin-3-ylmethyl)-phenyl]-urea (114):
[0956] 4-(l -Triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenylamine (113, 70.0 mg, 0.184 mmol) was dissolved in tetrahydrofuran (12 mL) and 3-trifluoromethyl-phenyl isocyanate (41 mg, 0.22 mmol) and triethylamine (28 mg, 0.28 mmol) were added under an atmosphere of nitrogen. The reaction mixture was stirred at room temperature overnight, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated to give crude compound 114 used in the next step.
Step 7 - Preparation of l-[4-(lH-Pyrrolo[2, 3-b]pyridin-3-ylmethyl)-phenyl]-3-(3-trifluoromethyl- phenyl)-urea (P-0262):
[0957] 1 -(3-Trifluoromethyl-phenyl)-3-[4-(l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl)-phenyl]-urea (114, 85.0 mg, 0.150 mmol) was dissolved in tetrahydrofuran (10.0 mL) and tetra-n-butylammonium fluoride (52.2 mg, 0.200 mmol) was added. After 10 minutes, the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with by silica gel chromatography to give the compound (P-0262, 23 mg, 38% over steps 6 & 7). MS(ESI)[M+!^]4 = 411.2
Example 60: Synthesis of (4-Hydroxy-phenyl)-(5-thiophen-2-yI-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone P-0024.
[0958] Compound P-0024 was synthesized in four steps from 5-bromo-7-azaindole 67 as shown in Scheme 35.
Scheme 35
Step 1 —Preparation of(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(4-methoxy-phenyl)-methanone (116):
[0959] Aluminum trichloride (2.7 g, 20.0 mmol) and methylene chloride (30 mL) were combined under an atmosphere of nitrogen and 5-bromo-7-azaindole (67, 400.0 mg, 2.30 mmol) in methylene dichloride (20 mL) was added. The reaction was stirred at room temperature for 70 minutes. 4-Methoxybenzoic acid chloride (0.38 g, 2.2 mmol) (prepared from the corresponding carboxylic acid using the protocol described in Example 1, Scheme 13, Step 4) in methylene dichloride (7.0 mL) was added. The reaction was stirred at room temperature for 2 hours, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, and concentrated to give the compound (116, 250 mg, 34%).
Step 2 — Preparation of5-Bromo-3-(4-methoxy-benzoyl)-pyrrolo[2,3-b]pyridine-l-carboxylic acid tert-butyl ester (117):
[0960] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(4-methoxy-phenyl)-methanone (116, 220.0 mg, 0.66 mmol) was dissolved in tetrahydrofuran (15.0 mL) and sodium hydride (60% dispersion, 29 mg, 0.73 mmol) was added under an atmosphere of nitrogen. The reaction was stirred for 15 minutes. Di-tert-butyldicarbonate (220 mg, 1.0 mmol) was added. The reaction was stirred for one hour, then poured into water and extracted with ethyl acetate. The organic portion was washed with brine, dried over sodium sulfate and concentrated to give compound 117.
Step 3 - Preparation of3-(4-Methoxy-benzoyl)-5-thiophen-2-yl-pyrrolo[2,3-b]pyridine-l-carboxylic acid tert-butyl ester (118):
[0961] 5-Bromo-3-(4-methoxy-benzoyl)-pyrrolo[2,3-b]pyridine-l -carboxylic acid tert-butyl ester (117, 100.0 mg, 0.23 mmol), thiophene-2-boronic acid (59 mg, 0.46 mmol), tetrakis(triphenylphosphine)palladium(0) (20.0 mg, 0.017 mmol), potassium carbonate (80 mg, 0.58 mmol), tetrahydrofuran (15.0 mL) and water (5.0 mL) were combined under an atmosphere of nitrogen. The reaction mixture was refluxed overnight, then poured into water and extracted with
ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel chromatography to give compound 118 (100 mg).
Step 4 - Preparation of(4-Hydroxy-phenyl)-(5-thiophen-2-yl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-0024):
[0962] 3-(4-Methoxy-benzoyl)-5-thiophen-2-yl-pyrrolo[2,3-b]pyridine-l-carboxylic acid tert-butyl ester (118, 100.0 mg, 0.23 mmol) and methylene chloride (15.0 mL) were combined under an atmosphere of nitrogen. Boron tribromide (1.0 M in methylene chloride, 3.0 mL) was added. The reaction was stirred at room temperature overnight. Methanol (2.0 mL) was added into the the reaction mixture. The reaction was concentrated to remove solvents. The residue was extracted with water and ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated and purified with silica gel chromatography. The product was washed with methanol to provide compound (P-0024, 25.0 mg, 34%). MS(ESI) [M+H*]^ 321.
Example 61: Synthesis of 4-Benzofuran-2-yl-lH-pyrroIo[2,3-b]pyridine-2-carboxylic acid ethyl ester P-1248.
[0963] Compound P-1248 was synthesized in four steps from 4-chloro-7-azaindole 119 as shown in Scheme 36.
Scheme 36
Step 1 - Preparation of4-Benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine (120): [0964] 4-Chloro-7-azaindole (119, 100 mg, 0.66 mmol, prepared as described in Example 70, Scheme 81), 2-Benzofuranboronic acid (212 mg, 1.31 mmol) and Potassium fluoride (127 mg, 2.18 mmol) were stirred in 3 mL of anhydrous dioxane. Tris(dibenzylideneacetone)dipalladium(0) chloroform adduct (10.2 mg, 9.83E-3 mmol) and tri J-butylphosphine (6.6 mg, 0.033 mol) were added and the suspension was stirred at 100 0C for 4 hours in a pressure tube under inert atmosphere. The reaction mixture was cooled to room temperature and was adsorbed onto silica gel and purified by chromatography to provide the compound (120, 56 mg, 36%). MS(ESI) [M+H1"]"1" = 235.20.
Step 2 — Preparation ofl-Benzenesulfonyl-4-benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine (121): [0965] Into a Round bottom flask 4-benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine (120, 100 mg, 0.43 mmol) was suspended in methylene chloride (10 mL) and 50% KOH solution (5mL) was added. Tetrabutylammonium hydrogen sulfate (10 mg, 0.03 mmol) and benzenesulfonyl chloride (81.4 mg, 0.46 mmol) were added. The resulting yellow solution was stirred at ambient temperature for 2 hours. The reaction mixture was partitioned between ethyl acetate and water and the organic layer was separated and washed with water and brine then dried over MgSO4, filtered and concentrated under reduced pressure to provide compound 121. MS(ESI) [M+HY = 375.29.
Step 3 - l-Benzenesulfonyl-4-benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine-2-carboxylic acid ethyl ester (122):
[0966] To a chilled (-78 0C acetone/dry ice bath) solution of l-benzenesulfonyl-4-benzofuran-2-yl- lH-pyrrolo[2,3-b]pyridine (121, 66.0 mg, 0.18 mmol) in tetrahydrofuran (5.0 mL) under an atmosphere of nitrogen, rc-butylithium (1.6 M in tetrahydrofuran, 0.14 mL, 0.23 mmol) was added dropwise. The reaction mixture was stirred for 30 minutes and ethyl chloroformate (0.02 mL, 0.19 mmol) was added. The reaction mixture was stirred for 1 hour while allowing it to reach room temperature, poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and purified by silica gel column chromatography, eluting with 20 % ethyl acetate in hexane, to give light yellow solid (122, 0.015 g, 19 %). MS(ESI)[M+H+]+ = 447.3.
Step 4 - 4-Benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine-2-carboocylic acid ethyl ester (P-1248): [0967] To a solution of l-benzenesulfonyl-4-benzofuran-2-yl-lH-pyrrolo[2,3-b]pyridine-2- carboxylic acid ethyl ester (122, 14.0 mg, 0.03 mmol) in tetrahydrofuran (3.0 mL), was added tetra N-butylammonium fluoride (26.0 mg, 0.10 mmol). The reaction was stirred for 48 hours at room temperature, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and purified by silica gel column chromatography, eluting with 50 % Ethyl acetate in hexane, to give light yellow solid (P-1248, 3.0 mg, 36%). MS(ESI)
307.2.
Example 62: Synthesis of Ν-phenyl-lH-pyrrolo[2,3-b]pyridin-6-amine P-1350 and related compounds
[0968] Compound P-1350 was synthesized in five steps from lH-pyrrolo[2,3-b]pyridine 94 as shown in Scheme 40.
Scheme 40
Step 1 — Preparation oflH-pyrrolo[2,3-b]pyridine-N-oxide (585):
[0969] To lH-pyrrolo[2,3-b]pyridine (94, 3.0 g, 25.3mmol) dissolved in 175 mL of diethyl ether was added m-CPBA (1.5 equiv) in portions over 30 minutes with vigorous stirring. The solution turned yellow, and precipitates formed. After two hours the solid was collected, washed with 2 x 50 mL of ether, and recrystallized from acetone:ether. Yield was approximately 125% due to contaminating acid. This crude material was carried through to the next step.
Step 2 - (6-bromo-lH-pyrrolo[2,3-b]pyridin-l-yl)(phenyl)methanone (586):
[0970] lH-pyrrolo[2,3-b]pyridine-N-oxide (585, 500 mg, 3.72mmol) was dissolved in 40 mL of dry benzene. In a separate dry flask, benzoyl bromide (2.5 equiv) and 1,1,1,3,3,3-hexamethyldisilazane (1.0 equiv) were combined in 20 mL of dry benzene. The bromide solution was added in 5 mL aliquots over 30 minutes to the reaction flask. The reaction was stirred at ambient temperature for two hours. It was then washed with 3 x 30 mL NaHCO3 (aq., satd.) and 1 x 30 mL brine. The organic layer was dried over sodium sulfate and evaporated. The crude material was taken directly to the next step without further purification.
Step 3 - Preparation of 6-bromo-l H-pyrrolo [2, 3-b] pyridine (587):
[0971] (6-bromo-lH-pyrrolo[2,3-b]pyridin-l-yl)(phenyl)methanone, 586 was dissolved in 20 mL of dioxane and 20 mL of 2M KOH (aq). This was stirred at ambient temperature until analysis indicated all of the starting material had been consumed (2 to 4 hours). The reaction was diluted with 50 mL of ethyl acetate and washed with 2 x 25 mL OfNaHCO3 (aq. satd.) and 25 mL of brine. The organic layer was dried with sodium sulfate, evaporated and purified by column chromatography. Combined steps 2 and 3 gave approximately 65% overall yield.
Step 4 - Preparation of 6-bromo-l -(triisopropylsilyl)-lH-pyrrolo[2,3-b]pyridine (588): [0972] 6-bromo-lH-pyrrolo[2,3-b]pyridine (587, 275 mg, 1.39mmol) was dissolved in 4 mL of dry dioxane. DIEA (3 equiv) and TIPS-OTf (2.5 equiv) were added and the reaction was stirred at 50 0C overnight. The reaction was then diluted with 20 mL of ethyl acetate and washed 2 times with 10 mL NaHCO3 (aq., 5%) and once with 10 mL brine. The organic fraction was dried over MgSO4,
evaporated and diluted with 5.5 mL of dry toluene (~ 10 mg bromide per 0.2 mL of solution) to use directly in the next reaction step.
Step 5- Preparation ofN-phenyl-lH-pyrrolo[2,3-b]pyridin-6-amine (P-1350): [0973] A 1 dram vial was charged with aniline (2 -3 equiv), and 0.200 mL of the 588 stock solution of bromide in toluene was added. A catalyst stock solution containing 3 mmol Pd(OAc)2, 3 mmol biphenyl-2-yl-di-tert-butyl-phosphane and 15 mL of toluene was prepared and 0.050 mL of the catalyst solution was added to the reaction. An excess of NaOtBu was added as a solid to the reaction. The vial was placed in an 80 0C oven for 60 minutes (shaken several times over the hour). After cooling, the reaction was neutralized with 0.100 mL of TFA. After 30 minutes the sample was evaporated and resolvated in 0.300 mL of DMSO. The desired compound was isolated by preparative HPLC/MS. MS(ESI) [MHhH+I+ = 210.4.
[0974] The following compounds were prepared following the protocol of scheme 40, substituting aniline with a suitable amine is Step 5:
Cyclohexyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1290),
Benzyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1291),
Cyclopropylmethyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1292),
(3-Methoxy-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1293),
(lH-Pyrrolo[2,3-b]pyridin-6-yl)-(3-trifluoromethyl-benzyl)-amine (P-1294),
(lH-Pyπ-olo[2,3-b]pyridin-6-yl)-(2-trifluoromethyl-benzyl)-amine (P-1295),
Cyclohexylmethyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1296),
(lH-Pyrrolo[2,3-b]pyridin-6-yl)-(4-trifluoromethoxy-benzyl)-amine (P-1297),
(lH-Pyrrolo[2,3-b]pyridin-6-yl)-(3-trifluoromethoxy-benzyl)-amine (P-1298),
(lH-Pyrrolo[2,3-b]pyridin-6-yl)-(2-trifluoromethoxy-benzyl)-amine (P-1299),
Pyridin-2-ylmethyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1300),
(4-Methanesulfonyl-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1301),
(4-Methoxy-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1302),
Ethyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1303),
(3-Chloro-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1304),
(4-Methyl-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1305),
(l-Methyl-piperidin-4-yl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1306),
Pyridin-3-ylmethyl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1307),
[4-(Moφholine-4-sulfonyl)-phenyl]<lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1308),
(4-Methanesulfonyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1309),
(2-Chloro-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1310),
(lH-Pyrrolo[2,3-b]pyridin-6-yl)-(tetrahydro-pyran-4-yl)-amine (P-1311),
(4-Chloro-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1312), (3-Methyl-benzyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amme (P-1313), [3-(Morpholine-4-sulfonyl)-phenyl]-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1314), (3-Methanesulfonyl-phenyl)-(lH-ρyrrolo[2,3-b]pyridin-6-yl)-amine (P-1315), Pyridm-3-yl-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1351), (2-Methoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1352), (lH-Pyrrolo[2,3-b]pyridin-6-yl)-(3-trifluoromethyl-phenyl)-amine (P-1353), (lH-Pyrrolo[2,3-b]pyridin-6-yl)-(4-trifluoromethoxy-phenyl)-amine (P-1354), (4-Methoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1355), N,N-Dimethyl-Nl-(lH-pyrrolo[2J3-b]pyridin-6-yl)-benzene-l,4-diamine (P-1356), (3-Methoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amme (P-13S7), (4-Morpholin-4-yl-phenyl)-(lH-pyrrolo[2,3-b]pyridm-6-yl)-amine (P-1358), (4-Piperidin-l-yl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1359), (lH-Pyrrolo[2,3-b]pyridin-6-yl)-(3-trifluoromethoxy-phenyl)-amine (P-1360), (lH-Pyrrolo[2,3-b]pyridin-6-yl)-p-tolyl-amine (P-1361), (3-tert-Butyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amme (P-1362), (3-Dimethylamino-benzyl)-(lH-pyrrolo[2,3-b]pyridm-6-yl')-amme (P-1363), (3,5-Dichloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1364), (lH-Pyrrolo[2,3-b]pyridin-6-yl)-(4-trifluoromethyl-benzyl)-amine (P-1371), N,N-Dimethyl-N'-(lH-pyrrolo[2,3-b]pyridin-6-yl)-benzene-l,3-diamine (P-1372), (3-Chloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1373), (4-Chloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1374), (2-Chloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1375), (5-Methyl-isoxazol-3-yl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1376), (2-Mθφholin-4-yl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1377), and (2-Methanesulfonyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-6-yl)-amine (P-1378). The following table indicates the amine (Column 2) that is substituted in place of the aniline in Step 5 to afford the compound (Column 3). Column 1 provides the compound number and Column 4 the observed mass.
Example 63: Synthesis of 2-[2,4-Difluoro-3-(5-pyridin-3-yl-l H-pyrroIo[2,3-b]pyridin-3- ylmethyl) -phenoxy]-ethanol P-1395 and related compounds
[0975] Compound P-1395 was synthesized in four steps from 4-Chloro-2-fiuoro-phenol 521 as shown in Scheme 42.
Scheme 42
Step 1 —Preparation of4-Chloro-2-fluoro-l-(2,2,2-trifluoro-ethoxy)-benzene (522): [0976] To 4-Chloro-2-fluoro-phenol (521, 5.0 g, 0.034 mol) in methanol (50.0 mL) was added potassium fluoride (2.2 g, 0.038 mol). The solvent was removed. The resulting salt was added to N,N-dimethylformaldehyde (25 mL), followed by adding l,l,l-trifluoro-2-iodo-ethane (8.60 g, 40.9 mmol). The reaction was stirred at 50 0C overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give colorless oil (522, 2.0 g, 26%).
Step 2 — Preparation of6-Chloro-2-fluoro-3-(2,2,2-trifluoro-ethoxy)-benzaldehyde (523): [0977] To 4-Chloro-2-fluoro-l-(2,2,2-trifluoro-ethoxy)-benzene (522, 0.80 g, 3.5 mmol) in THF (20 mL), cooled in dry ice/acetone bath and under an atmosphere of nitrogen, was slowly added n- butyllithium (1.60 M in Hexane, 2.30 mL). After an hour, N,N-dimethylformamide (0.298 mL, 3.85 mmol) was added to the reaction. After 30 minutes, the reaction was allowed to reach room temperature and was stirred for 10 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound (523, 450 mg, 50% ).
Step 3 - Preparation of(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-[6-chloro-2-fluoro-3-(2,2,2- trifluoro-ethoxy) -phenyl] -methanol (524):
[0978] To 5-bromo-7-azaindole (67, 291 mg, 1.48 mmol) in methanol (22 mL) were added 6-Chloro- 2-fluoro-3-(2,2,2-trifluoro-ethoxy)-benzaldehyde (523, 400.0 mg, 1.6 mmol) and potassium hydroxide (1.49 g, 26.6 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 48 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 25% ethyl acetate in hexane to give the compound
(524, 300 mg, 42%). MS (ESI) [M+HT = 453.1, 455.1.
Step 4 - Preparation of(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3 -yl)-[6-chloro-2-fluoro-3-(2,2,2-tr ifluoro-ethoxy)-phenyl] -methanol (P-1393):
[0979] To (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-[6-chloro-2-fluoro-3-(2,2,2-trifluoro-ethoxy)- phenyl] -methanol (524, 140.0 mg, 0.31 mmol) in trahydrofuran (6.0 mL) was added Dess-Martin periodinane (157 mg, 0.37 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound (P-1393, 100 mg, 72%). MS (ESI) [M-H+]- = 448.9, 450.9.
Step 5 - Preparation of[6-Chloro-2-fluoro-3-(2,2,2-trifluoro-ethoxy)-phenyl]-(5-pyridin-3-yl- IH- pyrrolo[2, 3-b]pyridin-3-yl)-methanone (P-1395):
[0980] To (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-[6-chloro-2-fluoro-3-(2,2,2-trifluoro-ethoxy)- phenyl] -methanol (P-1393, 53.0 mg, 0.12 mmol) in acetonitrile (4.0 mL) was added Tetrakis(triphenylphospliine)palladium(0) (5.0 mg, 0.0043 mmol), 3-pyridylboronic acid (15.1 mg, 0.12 mmol) and 1 M potassium carbonate solution (1.5 mL). The reaction was microwaved (300 watts) at 160 0C for 7 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 60% ethyl acetate in hexane to give the compound (P-1395, 3.8 mg, 53%) as light yellow solid. MS (ESI) [M+HT = 450.2.
[0981] [2,6-Difluoro-3-(2-methoxy-ethoxy)-phenyl]-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3- yl)-methanone P-1456
was prepared following the protocol of Scheme 42, substituting 4-Chloro-2-fluoro-phenol 521 with 2,4-difluoro-phenol and 1,1, l-Trifluoro-2-iodo-ethane with 1 -Bromo-2-methoxy-ethane in Step 1. MS (ESI) [M + H
+J
+ = 410.2.
[0982] N-(3-{3-[2,6-Difluoro-3-(2-methoxy-ethoxy)-benzoyl]-lH-yrrolo[2,3-b]pyridrn-5-yl}- phenyl)-methanesulfonamide P-1472
was prepared following the protocol of Scheme 42, substituting 4-Chloro-2-fluoro-phenol 521 with 2,4-difluoro-phenol and l,l,l-Trifluoro-2-iodo-ethane with l-Bromo-2-methoxy-ethane in Step 1, and substituting pyridine-3-boronic acid with [(3-methylsulfonyl)aminophenyl]-boronic acid in step 4. MS (ESI) [M + H
+]
"1" = 502.2.
Example 64: Synthesis of (3,5-Bis-difluoromethoxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanone P-1521
[0983] Compound P-1521 was synthesized in four steps from 3, 5-Dihydroxy-benzaldehyde 528 as shown in Scheme 44.
Scheme 44
Step 1 —Preparation of3,5-Bis-difluoromethoxy-benzaldehyde (529):
[0984] To 3, 5-Dihydroxy-benzaldehyde (528, 1.50 g, 10.9 mmol) in N,N-Dimethylformamide (100 mL) were added sodium chlorodifluoroacetate (5.90 g, 0.0387 mol), potassium carbonate (6.20 g, 0.0448 mol) and water (10 mL) under an atmosphere of nitrogen. The reaction was stirred at 100 0C overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as colorless oil (529, 50 mg, 2%).
Step 2 - Preparation of(3,5-Bis-difluoromethoxy-phenyl)-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanol (530):
[0985] To 5-bromo-7-azaindole (67, 86.4 mg, 0.000438 mol) in methanol (10.0 mL) were added 3,5-
bis-difluoromethoxy-benzaldehyde (529, 110.0 mg, 0.46 mmol) and potassium hydroxide (200.0 mg, 3.6 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as a white solid (530, 70 mg, 35%). MS (ESI) {M+ϊtγ = 435.1, 437.1.
Step 3 - Preparation of(3,5-Bis-difluoronιethoxy-phenyl)-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-1519):
[0986] To (3,5-Bis-difluoromethoxy-phenyl)-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanol (530, 70.0 mg, 0.16 mmol) in tetrahydrofuran (6.0 niL) was added Dess-Martin periodinane (75.0 mg, 0.18 mmol). The reaction was stirred at room temperature for 10 minutes. The reaction mixture was concentrated with silica gel and purified with silica gel column chromatography eluting with 30% ethyl acetate in hexane to give the compound as a white solid (P-1519, 65.0 mg, 93%.). MS (ESI) [M-H+]"= 431.0. 433.0.
Step 4 - Preparation of(3,5-Bis-difluoromethoxy-phenyl)-(5-phenyl-lH-pyrrolo[2,3-b]pyridin-3-yl)- methanone (P-1521):
[0987] To (3,5-Bis-difluoromethoxy-phenyl)-(5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-methanone (P-1519, 29.0 mg, 0.067 mmol) in acetonitrile (4.0 niL) were added phenylboronic acid (12.2 mg, 0.10 mmol), Tetrakis(triphenylphosphine)palladium(0) (10.0 mg, 8.65E-6 mol) and 1.0 M potassium carbonate in water (1.5 mL). The reaction was microwaved (300 watts) at 160 0C for 7 minutes. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, concentrated, and purified with silica gel column chromatography to give the compound as a white solid (P-1521, 9.4 mg, 33%). MS (ESI) [M+H1"]* = 431.2.
[0988] (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)-(3-difluoromethoxy-2,6-difluoro-phenyl)- methanone P-1526
was prepared using Steps 1-3 of Scheme 44 by substituting 3,5-Dihydroxy-benzaldehyde 528 with N- 2,4-Difluoro-phenol in Step 1. MS (ESI) [M + H
+]
"1" = 401.0, 403.0.
Example 65: Synthesis of N-Benzyl-2-[2,4-difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-
3- ylmethyl)-phenoxy]-acetamide P-1469
[0989] Compound P-1469 was synthesized in four steps from 3-(3-Ben2yloxy-2,6-difluoro-benzyl) 5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridme P-1455 as shown in Scheme 46.
Scheme 46
Step 1 — Preparation of 3-(3-Benzyloxy-2, 6-difluoro-benzyl) -5-pyridin-3-yl-l~triisopropylsilanyl-lH- pyrrolo [2, 3 -h] pyridine (548):
[0990] To 3-(3-Benzyloxy-2,6-difluoro-benzyl)-5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridine (P-1455, 700.0 mg, 1.64 mmol, prepared as described in Example 24, Scheme 43a) in tetrahydrofuran (40.0 mL) was added sodium hydride (60% in mineral oil, 100.0 mg, 2.5 mmol). After 10 minutes, triisopropylsilyl chloride (0.60 mL, 0.0028 mol) was added to the reaction. The reaction was stirred at room temperature for 2 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as colorless oil (548, 0.85g, 89%).
Step 2 — Preparation of2,4-Difluoro-3-(5-pyridin-3-yl-l-triisopropylsilanyl-lH-pyrrolo[2,3- b]pyridin-3-ylmethyl)-phenol (549) :
[0991] To 3-(3-Benzyloxy-2,6-difluoro-benzyl)-5-pyridin-3-yl-l -triisopropylsilanyl-lH-pyrrolo[2,3- b]pyridine (548, 510.0 mg, 0.87 mmol) in methanol (20.0 mL) was added 20% Pd(OH)2/C (50 mg). The reaction was stirred under an atmosphere of hydrogen for 2 hours. Filtration and concentration gave the compound as white solid (549, 427 mg, 99%).
Step 3 - Preparation of[2,4-Difluoro-3-(5-pyridin-3-yl-l-triisopropylsilanyl-lH-pyrrolo[2,3-b ] pyridin-3-ylnιethyl)-phenoxy] -acetic acid methyl ester (550):
[0992] To 2,4-Difluoro-3-(5-pyridin-3-yl-l-triisopropylsilanyl-lH-pyrrolo[2,3-b] pyridin-3- ylmethyl)-phenol (549, 427.0 mg, 0.87 mmol) in tetrahydrofuran (20.0 mL) was added sodium hydride (60% in mineral oil, 42.0 mg, 1.1 mmol). 30 minutes later, methyl bromoacetate (146 mg,
0.95 nimol) was added to the reaction. The reaction was stirred at room temperature for 4 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to give the crude compound that was used directly in the next step.
Step 4 - Preparation ofN-Benzyl-2-[2,4-difluoro-3-(5-pyrid in-3-yl-lH-pyrrolo[2,3-b]pyridin-3- ylmeihyl)-phenoxy]-acetamide (P-I 469):
[0993] To [2,4-Difluoro-3-(5-pyridm-3-yl-l-triisopropylsilanyl-lH-pyrrolo[2,3-b ]pyridin-3- ylmethyl)-phenoxy]-acetic acid methyl ester (550, 210.0 mg, 0.37 mmol) in methanol (6.0 niL) was added benzylamine (0.45 mL, 4.1 mmol). The reaction was stirred at room temperature for 80 hours. To the reaction was added TBAF (0.31 g, 1.0 mmol). The reaction was concentrated and purified with silical gel column chromatography eluting with 10% methanol in methylene chloride to give the compound as a white solid (P-1469, 60 mg, 33%). MS (ESI) [M+H+f = 423.3.
[0994] 2-[2,4-Difluoro-3-(5-pyridin-3-yl-l H-pyrrolo[2,3-b]pyridin-3-yhnethyl) -phenoxy]-N-ethyl- acetamide P-1475
was prepared using the same protocol as Scheme 46 by substituting benzylamine with ethylamine in Step 4. MS (ESI) [M + If]
+ = 423.3.
Example 66: Synthesis of [2,4-difluoro-3-(5-pyridin-3-yl-lH- pyrrolo[2,3-b]pyridin-3-ylmethyI)- phenoxy] -acetic acid P-1468
[0995] Compound P-1468 was synthesized in two steps from [2,4-Difluoro-3-(5-pyridin-3-yl-l- triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenoxy]-acetic acid methyl ester 550 as shown in Scheme 47.
Scheme 47
Stepl - Preparation of [2, 4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2, 3-b]pyridin-3-ylmethyl)- phenoxy] -acetic acid methyl ester (P-1465):
[0996] To [2,4-Difluoro-3-(5-pyridin-3-yl-l -triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin-3- ylmethyl)-phenoxy] -acetic acid methyl ester (550, 40.0 mg, 0.071 mmol, prepared as described in Example 65, Scheme 46) in tetrahydrofuran (5.0 mL) was added tetra-n-butylammonium fluoride (22.2 mg, 0.085 mmol). The reaction was stirred at room temperature for 10 minutes. The crude compound was concentrated with silica gel and purified by silica gel column chromatography eluting with 3% methanol in methylene chloride to give the compound as white solid (P-1465, 20 mg, 69%).
Step 2 - Preparation of[2,4-Difluoro-3-(5-pyridin-3-yl-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-p henoxy] -acetic acid (P-1468):
[0997] To [2,4-Difluoro-3-(5-pyridin-3-yl-lH- pyrrolo[2,3-b]pyridin-3-ylmethyl)-phenoxy]-acetic acid methyl ester (P-1465, 20.0 mg, 0.049 mmol) in tetrahydrofuran (8.0 mL) were added potassium hydroxide (100.0 mg, 1.8 mmol) and water (3.0 mL). The reaction was stirred at room temperature overnight and then poured into water, acidified to pH=5 with 1 N HCl, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% methanol in methylene chloride to give the compound as light yellow solid (P-1468, 9.1 mg, 47%). MS (ESI) [M+^ = 396.2.
Example 67: Synthesis of 2-[2,4-Difluoro-3-(5-pyridin-3-yHH-pyrroIo[2,3-b]pyridin-3- ylmethyl) -phenoxy]-ethanol P-1394
[0998] Compound P-1394 was synthesized in four steps from 2,6-Difluoro-3-hydroxy-benzaldehyde 540 as shown in Scheme 48.
Scheme 48
Step 1 —Preparation of2,6-Difluoro-3-[2-(tetrahydro-pyran-2-yloxy)-ethoocy]-benzaldehyde (541): [0999] To 2,6-difluoro-3-hydroxy-benzaldehyde (540, 0.150 g, 0.95 mmol) in N,N- dimethylformamide (8.0 mL) were added 2-(2-bromo-ethoxy)-tetrahydro-pyran (0.218 g, 1.04 mmol) and potassium carbonate (0.52 g, 3.8 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 72 hours. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give the compound as colorless oil (541, 180 mg, 66%).
Step 2 - Preparation of(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3 -yl)-2,6-difluoro-3-[2-(tetrahydro - pyran-2-yloxy)-ethoxy]-pheιtyl-methanol (542) :
[1000] To 5-bromo-7-azaindole (67, 118 mg, 0.000597 mol) in methanol (9.0 mL) were added 2,6- difluoro-3-[2-(tetrahydro-pyran-2-yloxy)-ethoxy]-benzaldehyde (541, 180.0 mg, 0.63 mmol) and potassium hydroxide (601.9 mg, 10.7 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to give crude compound that was used directly in the next step.
Step 3 - Preparation of2-[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenoxy]- ethanol (P-1392):
[1001] To (5-Bromo-lH-pyrrolo[2,3-b]pyridin-3 -yl)-2,6-difluoro-3-[2-(tetrahydro -pyran-2-yloxy)- ethoxy]-phenyl-methanol (542, 0.22 g, 0.46 mmol) in acetonitrile (6.0 mL) were added trifluoroacetic acid (0.14 mL, 1.8mmol) and triethylsilane (0.29 mL, 1.8 mmol). The reaction was stirred at room temperature overnight. The filtrate was concentrated and purified by silica gel column chromatography eluting with 35% ethyl acetate in hexane to give the compound as a white solid (P-1392, 62 mg, 35%). MS (M+lf)+ = 383.1, 385.1.
Step 4 - Preparation of2-[2,4-Difluoro-3-(5-pyridin-3-yl-l H-pyrrolo[2,3-bJpyridin-3-ylmethyl) - phenoxyj-ethanol (P-1394):
[1002] To 2-[3-(5-Bromo-lH-pyrrolo[2,3-b]pyridin-3-ylmethyl)-2,4-difluoro-phenoxy]-ethanol (P-1392, 35.0 mg, 0.091 mmol) in acetonitrile (4.0 mL) were added 3-pyridylboronic acid (14.6 mg, 0.12 mmol), tetrakis(triphenylphosphine)palladium(0) (3.0 mg, 0.0026 mmol) and 1 M potassium carbonate solution (1.5 mL). The reaction was microwaved (300 watts) at 1600C for 7 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was collected and dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting 4% methanol in methylene chloride to give the compound as white solid (P-1394, 11.0 mg, 32%). MS (ESI) [M+lff = 382.2.
Example 68: Synthesis of 3-[4-methoxy-3-(2,2,2-trifluoro-ethoxy)-benzyl]-lH-pyrrolo[2,3- bjpyridine P-1626.
[0633] Compound P-1626 was synthesized in three steps from 3-hydroxy-4-methoxy-benzaldehyde 560 as shown in Scheme 52.
Scheme 52
Step 1 - Preparation of4-Methoxy-3-(2,2,2-trifluoro-ethoxy)-benzaldehyde (562): [0633] 3-Hydroxy-4-methoxy- benzaldehyde (560, 2.9 g, 19.5 mmol), 2-iodo-l,l,l-trifluoroethane (561, 2.2 mL, 23 mmol), and potassium carbonate (3.7 g, 27 mmol) were dissolved inN,N- dimethylformamide (100 mL). The solution was stirred at 90 0C overnight under an atmosphere of nitrogen. The reaction was poured into water, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The compound was isolated by silica gel column chromatography eluting with 25% to 50% ethyl acetate in hexanes to give white powder (562, 1.6 g, 40%).
Step 2 - Preparation of3-Methoxy~[4-methoxy~3-(2,2,2-trifluoro-ethoxy)-phenyl]-methyl-lH- pyrrolo[2, 3-b] pyridine (563) :
[0633] lH-Pyrrolo[2,3-b]pyridine (0.2 g, 1.8 mmol), 4-methoxy-3-(2,2,2-trifluoro-ethoxy)- benzaldehyde (562, 0.4 g, 1.9 mmol) and potassium hydroxide (0.5 g, 9.6 mmol) were combined in methanol (20 mL). The reaction was heated to 50 0C under an atmosphere of nitrogen and stirred for two days. The reaction was poured into water, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The compound was isolated by silica gel column chromatography eluting with 40% to 75% ethyl acetate in hexanes to give orange oil (563, 0.2 g, 31%). MS (ESI) [M + H4I+ = 367.2 [M - H1]'= 365.1.
Step 3 — Preparation of3-[4-Methoxy-3-(2,2,2-trifluoro-ethoxy)-benzylJ-lH-pyrrolo[2,3-bJpyridine
(P-1626):
[0633] 3-methoxy-[4-methoxy-3-(2,2,2-trifluoro-ethoxy)-phenyl]-methyl-lH-pyrrolo[2,3-b]pyridine
(563) was reacted using the same protocol described in Example 28, Scheme 51, Step 3. The compound P-1626 was isolated by silica gel column chromatography eluting with 50% to 75% ethyl acetate in hexanes. MS (ESI) [M + If]+= 337.1.
Example 69: Synthesis ofN-phenyl-lH-pyrrolo[2,3-b]pyridin-3-amine P-0221.
[1003] Compound P-0221 was synthesized in two or three steps from lH-pyrrolo[2,3-b]pyridine 94 as shown in Scheme 54.
Scheme 54
583 P-0221
Step 1 -Preparation of 3-bromo-l H-pyrrolo [2,3 -b] pyridine (582):
[1004] To lH-pyrrolo[2,3-b]pyridine (94, 600 mg) dissolved in 10 mL of 100% acetic acid was added the brominating agent DMAP-Br3 (1.001 equiv.). The reaction was heated at 75° C until the orange color was gone and a precipitate formed (about 60 min). It was dissolved with 50 mL of water (ppt dissolved) and adjusted to pH 8 with NaHCO3 (aq. sat'd). The compound precipitated from the solution and was collected. The aqueous fraction was extracted with 3 x 30 mL with ethyl acetate. The combined organics were dried over Na2SC^, evaporated and the solids combined with the previously collected solid (582, 87%).
Route A: Step 2 — 3-bromo-l-(triisopropylsilyl)-lH-pyrrolo [2, 3-b] pyridine (583):
[1005] To 3-bromo-lH-pyrτolo[2,3-b]pyridine (582, 0.5 mmol) dissolved in 5 mL of dry dioxane was added BEMP (2.5 equiv). The TJPS-OTf (2.0 equiv) was then added and the reaction was stirred overnight (15 hours). The solution was diluted with 20 mL of ethyl acetate and washed with 5% acetic acid (2x10 mL), water (2x10 mL), and brine (10 mL). The organic layer was dried over
Na2SO4, filtered, evaporated and dried under vacuum. This material was used without further purification.
Route A: Step 3 — Preparation ofN-phenyl-lH-pyrrolo[2,3-b]pyridin-3-amine (P-0221): [1006] A 1 dram vial was charged with aniline (2 -3 equiv), and 10 mg of 3-bromo-l- (triisopropylsilyl)-lH-pyrrolo[2,3-b]pyridine 583 in 0.4 mL toluene (dry, degassed). A catalyst stock solution containing 3 mmol Pd(OAc)2, 3 mmol biphenyl-2-yl-di-tert-butyl-phosphane and 15 mL of toluene was prepared. To the reaction mixture, 0.050 mL of the catalyst solution was added. Excess NaOtBu was added as a solid to the reaction. The vial was then placed in a 75 0C oven for 60 minutes (shaken several times over an hour). After cooling, the reaction was neutralized with 0.100 mL of TFA. After 30 minutes the sample was evaporated and re-solvated in 0.300 mL of DMSO. The desired compound was isolated by preparative HPLC/MS.
Rout B: Step 2 - Preparation o/N-phenyl-lH-pyrrolo[2,3-b]pyridin-3-amine (P-0221): [1007] To 10 mg of 3-bromo-lH-pyrrolo[2,3-b]pyridine 582 was added 0.4 mL of sulfolane and aniline (2 -3 equiv). The vial was sealed and heated at 180 0C for 10 minutes. After cooling, the desired compound was isolated by preparative HPLC/MS.
[1008] The following compounds were prepared by one of the procedures described in scheme 54, substituting aniline with a suitable amine in Step 5:
Methyl-phenyl-(lH-ρyrrolo[2,3-b]pyridin-3-yl)-amine (P-1434),
4-[Methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amino]-benzoic acid (P-1435),
(4-Chloro-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1436),
Methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-m-tolyl-amme (P-1437),
(4-Methoxy-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1438),
(3-Chloro-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1439),
(4-Fluoro-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1440),
Methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-(4-trifluoromethoxy-phenyl)-amine (P-1441),
(2,4-Difluoro-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridrn-3-yl)-amine (P-1442),
(3-Fluoro-ρhenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1443),
(4-Methoxy-phenyl)-(m-pyrrolo[2,3-b]pyridm-3-yl)-amiαe (P-1488),
(3-Chloro-ρhenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1489),
(3-Methoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1490),
(4-Chloro-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1491), (lH-Pyrrolo[2,3-b]pyridin-3-yl)-(4-trifluoromethoxy-phenyl)-amine (P-1492), (lH-Pyrrolo[2,3-b]pyridin-3-yl)-(3-trifluoromethoxy-phenyl)-amine (P-1493), (3,4-Dichloro-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1494), (4-Phenoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1495), (3-Phenoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1496), (3-Benzyl-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1497), N-[3 -( 1 H-Pyrrolo [2,3 -b]pyridin-3 -ylamino)-phenyl] -methanesulfonamide (P-1498), 3-Fluoro-4-(lH-pyrrolo[2,3-b]pyridin-3-ylamino)-plienol (P-1499), 3-(lH-Pyrrolo[2,3-b]pyridin-3-ylamino)-benzenesulfonamide (P-1500), (2-Phenoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1501), (S^-Dichloro-phenyO-ClH-pyrroloP^-blpyridin-S-y^-amine CP-lSOl), (3,5-Dimethoxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1503), [3-(4-Methyl-4H-[l,2,4]triazol-3-yl)-phenyl]-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1504), (2-Benzyloxy-phenyl)-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1505), and (3-Methoxy-phenyl)-methyl-(lH-pyrrolo[2,3-b]pyridin-3-yl)-amine (P-1506). The following table indicates the amine (Column 2) that is substituted in place of the aniline in Route B, Step 2, or Route A, Step 3, to afford the compounds (Column 3). Column 1 provides the
Example 70: Synthesis of 4-chloro-7-azaindole 119
[1009] 4-chloro-7-azaindole 119 was synthesized in two steps from 7-azaindole according to the protocol of Scheme 81.
Scheme 81
-Step -7 — Synthesis of lH-Pyrrolo[2,3-b] pyridine 7-oxide (658):
[1010] lH-Pyrrolo[2,3-b]pyridine 7-oxide 658 was synthesized by reacting 7-azaindole 94 with an oxidizing agent (e.g. m-CPBA) in a non-reactive solvent (e.g. dimethoxyethane) as described by Schneller, S. W.; Luo, Jiann-Kuan. J. Org. Chem. 1980, 45:4045-4048. The compound was isolated by filtration of the resulting solid that forms upon standing at 5 0C for typically 1-3 h.
Step - 2 — Synthesis of 4-chloro-7-azaindole (119):
[1011] 4-chloro-7-azaindole 119 was synthesized by reacting lH-Pyrrolo[2,3-b]pyridine 7-oxide 658 with a chlorinating agent (e.g. POCl3) neat as described by Schneller, S. W.; Luo, Jiann-Kuan. J. Org. Chem. 1980, 45:4045-4048. The resulting solution after heating for 3-5 h at elevated temperatures (100-150 0C) was neutralized with a base (e.g. NH4OH) until a solid precipitated. The solid was isolated by filtration.
Example 71: Synthesis of [3-(4-Chloro-2-fluoro-benzyloxy)-2- (2-fluoro-ethoxy)-phenyl]-(lH- pyrro Io[2,3-b]pyridin-3-yl)-methanone P-2086 and 3-[3-(4-Chloro-2-fluoro-benzyIoxy)- 2-(2- fluoro-ethoxy)-benzyl]-lH-pyrr olo[2,3-b] pyridine P-2085
[1012] Compounds P-2086 and P-2085 were synthesized in three steps from compounds 659 and lH-pyrrolo[2,3-ό]pyridine 94 as shown in Scheme 82.
660
Step 1 — Preparation of3-(4-Chloro-2-fluoro-benzyloxy)-2-(2-fluoro-ethoxy)-benzaldehyde (660): [1013] To a solution of 3-(4-Chloro-2-fluoro-ben2yloxy)-2-hydroxy-benzaldehyde (659, 140 mg, 0.5 mmol, prepared by protocol of Example 43, Steps 1 and 2 of Scheme 71, using 4-chloro-2-fluoro- benzyl bromide in place of 4-chloro-benzyl bromide in Step 1) in tetrahydrofuran (8 mL) was added dropwise a mixture of 2-fluoro-ethanol (64 mg, 1.0 mmol), triphenylphosphine (180 mg, 0.7 mmol), and diisopropyl azodicarboxylate (120 mg, 0.6 mol) in tetrahydrofuran (5 ml) at 0 0C. The reaction mixture was stirred at 0 0C for 10 minutes and then at 40 0C for 3 days. The reaction mixture was dissolved in water and ethyl acetate. The organic layers were collected, washed with brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide the compound as a white solid (660, 88 mg, 54%). MS (ESI) [M+lff =327.12.
Step 2 - Preparation of[3-(4-Chloro-2-fluoro-benzyloxy)-2- (2-fluoro-ethoxy)-phenyl]-(lH-pyrro lo[2,3-b]pyridin-3-yl)-methanol (661) and 3-{[3-(4-Chloro-2-fluoro-benzyloxy) -2-(2-fluoro-ethoxy)- phenyl] -methox y-methyl}-lH-pyrrolo[2,3-b]ρyridine (662):
[1014] A solution of 3-(4-chloro-2-fluoro-benzyloxy)-2-( 2-fluoro-ethoxy)-benzaldehyde (660, 88 mg, 0.27 mmol), lH-pyrrolo[2,3-έ]pyridine (94, 38 mg, 0.32 mmol), and potassium hydroxide (45 mg, 0.81 mol) in methanol (5 mL) was stirred at room temperature for 24 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was collected, washed with brine, and dried over sodium sulfate. After removal of solvent, the residue was purified by silica gel
column chromatography eluting with ethyl acetate in hexane to provide compound 661 as a white solid (67 mg, 56%), MS(ESI) [M+H+]+ = 445.13 and compound 662 as a white solid (36 mg, 29%), MS(ESI) [MH-H+J+ = 459.15.
Step Sa — Preparation of[3-(4-Chloro-2-fluoro-benzyloxy)-2- (2-fluoro-ethoxy)-phenyl]-(lH-pyrro Io [2, 3-b]pyridin-3-yl)-rnethanone (P-2086):
[1015] To a solution of [3-(4-chloro-2-fiuoro-benzyloxy)-2- (2-fluoro-ethoxy)-phenyl]-(lH- ρyrrolo[2,3-6]pyridin-3-yl)-methanol (661, 60 mg, 0.1 mmol) in tetrahydrofuran (10 mL) was added Dess-Martin periodinane (69 mg, 0.16 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 3 hours. The reaction was quenched with a saturated solution of sodium thiosulfate, extracted with ethyl acetate, washed with sodium bicarbonate, brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide the compound as a white solid (P-2086, 15 mg, 20%). MS(ESI) [M+H+]+ = 441.06.
Step 3b - Preparation of3-[3-(4-Chloro-2-fluoro-benzyloxy)- 2-(2-fluoro-ethoxy)-benzyl]-lH-pyrr olo[2,3-b]pyridine (P-2085):
[1016] A mixture of 3-{[3-(4-chloro-2-fluoro-benzyloxy) -2-(2-fluoro-ethoxy)-phenyl]-methoxy- methyl}-lH-pyrrolo[2,3-6]pyridine (662, 36 mg, 0.078 mmol) , triethylsilane (0.5 mL, 3 mmol), and trifluoroacetic acid (0.2 mL, 2 mmol) in acetonitrile (20 mL) was stirred at 80 0C for 2 hours. The mixture was concentrated and the residue was dissolved in ethyl acetate. The solution was washed with saturated sodium bicarbonate, brine, and dried over sodium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide the compound as a yellow solid (P-2085, 24 mg, 71%). MS(ESI) [M+HY = 429.15. [1017] 3-(4-Chloro-benzyloxy)-2-(2,2-difluoro-ethoxy)-phenyl]-(lH-pyrrolo[2, 3-έ]pyridin-3-yl)- methanone (P-2075)
was prepared following the protocol of Scheme 82, substituting 2-fiuoro-ethanol with 2,2-difluoro- ethanol and substituting 3-(4-Chloro-2-fluoro-benzyloxy)-2-hydroxy-benzaldehyde with 3-(4-chloro- benzyloxy)-2-hydroxy-benzaldehyde (628 of Example 43) in Step 1 to provide P-2075. MS(ESI) PdH-H
+J
1" = 443.1.
Example 72: Synthesis of [3-(2-Chloro-4-methanesulfonyI-benzyloxy)-2-ethoxy-phenyl]-(lH- pyrrolo[2,3-b]pyridin-3-yl)-methanone P-2094
[1018] Compound P-2094 was synthesized in four steps from compounds 635 and 663 as shown in Scheme 83.
Scheme 83
Step 1 — Preparation of(3-Benzyloxy-2-ethoxy-phenyl)-(l-triisopropylsilaιιyl-lH-pyrrolo[2,3- b]pyridin-3-yl)-methanol (664):
[1019] To a solution of 3-iodo-l -triisopropylsilanyl-lH-pyrrolo[2,3-Z>]pyridine (635,1.306 g, 3.26 mmol) in tetrahydrofuran (42 mL) at -20 0C under nitrogen was added isopropylmagnesium chloride (1.70 mL, 2.0 M solution in tetrahydrofuran, 3.40 mmol). The reaction mixture was stirred at -20 0C for 1.5 hours. It was allowed to warm to 5 0C and then kept at 5 0C for 1 hour. The reaction mixture was then cooled down to -20 0C. To this solution was slowly added a solution of 2-ethoxy-3- benzyloxybenzaldehyde (663, 0.698 g, 2.72 mmol, prepared by protocol of Example 43, Steps 1-3 of Scheme 71, using benzyl bromide in place of 4-chloro-benzyl bromide in Step 1) in tetrahydrofuran (42 mL). The reaction mixture was stirred at -20 0C for 2.5 hrs, and was allowed to warm to 5 0C for 2.5 hours. The reaction mixture was poured into iced water, extracted with ethyl acetate, washed with saturated ammonium chloride and brine, and dried over magnesium sulfate. After removal of solvent, the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane to provide the compound as light-yellow oil (664, 200 mg, 13.9%).
Step 2 - Preparation of(2-ethoxy-3-hydroxy-phenyl)-(l-triisopropylsilanyl-lH-pyrrolo[2,3-b]pyridin- 3-yl)-methanone (665):
[1020] To a solution of (3-benzyloxy-2-ethoxy-phenyl)-(l -triisopropylsilanyl-lH-pyrrolo[2,3- έ]ρyridin-3-yl)-methanol (664,195 mg, 0.37 mmol) in a mixture of methanol (20 mL) and tetrahydrofuran (50 mL) was added palladium on carbon (50 mg, 10% wt., 0.2 mmol). The mixture was stirred under hydrogenation for seventeen hours. After removal of solvent, the residue was
washed with a mixture of ethyl ether and hexanes to provide the compound as a white solid (665, 63 mg, 95%). MS(ESI) [M+rf]+ = 439.37.
Step 3 - Preparation of[3-(2-Chloro-4~methanesulfonyl-benzyloocy)-2-ethoxy-phenyl]-(l- triisopropylsilanyl-lH-pyrrolo[2, 3-b]pyridin-3-yl)-methanone (666) :
[1021] To a solution of (2-ethoxy-3-hydroxy-phenyl)-(l -triisopropylsilanyl-lH-pyrrolo[2,3- έ]pyridin-3-yi)-methanone (665, 40 mg, 0.064 mmol) in tetrahydrofuran (15 mL) was added sodium hydride (3.32 mg, 0.083 mmol) at room temperature under an atmosphere of nitrogen. The mixture was stirred at room temperature for 40 minutes, thenl-bromomethyl-2-chloro-4-methanesulfonyl- benzene (21.72 mg, 0.077 mmol) was added to the reaction mixture. It was stirred at room temperature overnight. The mixture was then poured into water and was extracted with ethyl acetate. The organic layer was collected and washed with brine, dried over magnesium sulfate. After removal of the solvent, a crude compound as light yellow oil was obtained (666, 84 mg).
Step 4 - Preparation of[3-(2-Chloro-4-methanesulfonyl-benzyloxy)-2-ethoxy-phenyl]-(lH- pyrrolo[2, 3-b]pyridin-3-yl)-methanone (P-2094):
[1022] To a solution of (2-ethoxy-3-hydroxy-phenyl)-(l -triisopropylsilanyl-lH-ρyrrolo[2,3- b]pyridin-3-yl)-methanone (666, 84 mg, 0.054 mmol) in methanol (10 mL) was added potassium hydroxide (6 N solution) until pH of the solution turned to over 10. Potassiun fluoride (30 mg, 0.5 mmol) was then added to the reaction mixture and the mixture was stirred at room temperature for 6 hours. The reaction mixture was then poured into saturated sodium carbonate and was extracted with ethyl acetate. The organic layer was collected and washed with brine, dried over magnesium sulfate. After removal of the solvent, the residue was purified by preparative HPLC to provide as a white solid (P-2094, 5 mg, 19%). MS(ESI) [M+H+]+ = 485.17.
Example 73: Synthesis of Propane-1-sulfonic acid [3-(5-bromo -lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-4-fluoro-phenyl]-amide P-1403
[1023] Compound P-1403 was synthesized in seven steps from 3-fluoro-5-nitrobenzoic acid 667 as shown in Scheme 84.
Scheme 84
Step 1 -Preparation of 3-fluoro-5-aminobenzoic acid (668):
[1024] Into a Parr pressure reactor were added 3-fluoro-5-nitrobenzoic acid (667, 5.0 g, 0.027 mol), methanol (50.0 rnL), 20% Pd(OH)2 on carbon (300 mg). The reaction was shaken under an atmosphere of hydrogen at 50 psi overnight. The reaction was filtered through celite and was concentrated to dryness to provide a white solid (668, 4.0 g, 95.0%).
Step 2 - Preparation of2-Fluoro-5-(propane-l-sulfonylamino)-benzoic acid (669): [1025] To 3-fluoro-5-aminobenzoic acid (668, 3.00 g, 0.0180 mol) in methylene chloride (204 mL) were added pyridine (41 mL, 0.50 mol) and propane- 1-sulfonyl chloride (2.23 mL, 0.0198 mol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 5 days. The reaction was poured into water, adjusted pH to 1 with IN HCl, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 5% methanol in methylene chloride to give a white solid (669, 2.0 g, 42%). MS (ESI) [M-H+]" = 260.1.
Step 3 - Preparation of 2-Fluoro-5-(propane-l-sulfonylamino )-benzoic acid methyl ester (670): [1026] To 2-Fluoro-5 -(propane- 1 -sulfonylamino)-benzoic acid (669, 2.0 g, 0.0076 mol) in methanol (20.0 mL) was added sulfuric acid (0.90 mL, 0.017 mol). The reaction was stirred at room temperature overnight. The reaction was concentrated and purified with silica gel column chromatograph eluting with 20% ethyl acetate in hexane to give a white solid (670, 1.27 g, 62%). MS (ESI) [M-H+]- = 274.1.
Step 4 - Preparation of Propane- 1 -sulfonic acid (4-fluoro-3 -hydroxymethyl-phenylj-amide (671): [1027] To 2-Fluoro-5-(propane-l -sulfonylamino )-benzoic acid methyl ester (670, 1.20 g, 0.00436 mol) in tetrahydrofuran (100.0 mL) was added lithium tetrahydroaluminate (1.00 M in tetrahydrofuran, 10.0 mL) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. To the reaction was added Na2SO4JOH2O (5 g), and then stirred at room temperature for 1 hour. The reaction was filtered, concentrated and purified with silica gel column
chromatography eluting with 50% ethyl acetate in hexane to give the compound (671, 0.90 g, 83%). MS (ESI) [M-H+]" = 246.1.
Step 5 - Preparation of Propane- 1 -sulfonic acid (4-fluoro-S -formyl-phenyl) -amide (672): [1028] To propane-1 -sulfonic acid (4-fluoro-3 -hydroxymethyl-phenyl)-amide (671, 0.483 g, 0.00195 mol) in tetrahydrofuran (10.0 mL), cooled with ice/water, was added Dess-Martin periodinane (1.00 g, 0.00236 mol). The reaction was stirred at room temperature for 10 minutes. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 30% ethyl acetate in hexane to give a white solid (672, 360 mg, 75%). MS (ESI)[M-H+]" = 244.1.
Step 6 — Preparation of Propane-1 -sulfonic acid 3-[(5-brom o-lH-pyrrolo[2,3-b]pyridin-3-yl)~ hydroxy-methylJ-4-fluoro-phenyl-amide (673):
[1029] To 5-bromo-7-azaindole (67, 170.0 mg, 0.86 mmol) in methanol (7.0 mL) were added propane-1 -sulfonic acid (4-fluoro-3 -formyl-phenyl)-amide (672, 220.0 mg, 0.90 mmol) and potassium hydroxide (0.50 g, 0.0089 mol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water, acidified with IN HCl to pH = 5, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 10% methanol in methylene chloride to give the compound (673, 55.0 mg, 14.0%). MS (ESI) PVH-H+]"1" = 442.1, 444.1.
Step 7 — Preparation of Propane-1 -sulfonic acid [3-(5-bromo -lH-pyrrolo[2,3-b]pyridine-3- carbonyl)-4-fluoro-phenyl] -amide (P-1403):
[1030] To propane-1 -sulfonic acid 3-[(5-brom o-lH-pyrrolo[2,3-b]pyridin-3-yl)-hydroxy-methyl]-4- fluoro-phenyl-amide (673, 55.0 mg, 0.12 mmol) in tetrahydrofuran (8.0 mL) was added Dess-Martin periodinane (70.0 mg, 0.17 mmol). The reaction was stirred at room temperature for 5 minutes. The reaction was concentrated with silica gel and purified with silica gel column chromatography eluting with 25% ethyl acetate in hexane to give an off-white solid (P-1403, 26.2 mg, 47%). MS (ESI) [M+H+]+ = 437.9, 439.9.
Example 74: Additional compounds
[1031] Additional compounds of the invention were synthesized following the methods of the Examples above or similar methods, or by methods known to those of skill in the art, and are shown in the following Table 1.
Table 1. Additional compounds of the invention
Example 75: Kinase Activity Assays
[1032] The effect of potential modulators of kinase activity can be measured in a variety of different assays known in the art. For example, direct radiometric, indirect FRET or AlphaScreen assays may be used to assess the level of phosphorylation of a substrate in the presence of test compounds in order to determine the inhibitory affect of the compound on the kinase. Invitrogen (Carlsbad, CA) uses a FRET based assay for Btk, EGFR, EphB2, Flt3, Irak4, Kdr, MAP2K1, MAPKAPK2, PDGFRB, PKC theta, Stk6 and Yes. For these assays, compounds of the invention were screened by Invitrogen using Z'-Lyte™ kinase assay.
[1033] Briefly, the Invitrogen kinase assay involves use of a specific peptide substrate optimized for each kinase, containing a fluorophore at each end that make up the FRET pair. The peptide sensitivity to proteolytic cleavage depends on phosphorylation of the peptide. Non-phosphorylated peptide is cleaved by a protease while peptide phosphorylated by the kinase is not cleaved. Cleavage of the peptide disrupts the FRET between the donor (coumarin) and acceptor (fluorescein) fluorophores, resulting in an increase in the ratio of donor emission to acceptor emission. The emission ratio of coumarin to fluorescein is used to assess the reaction progress. The extent of phosphorylation is determined from the emission ratio, which is low when the kinase is active (phosphorylated peptide is not cleaved, FRET pair connected) or higher for inhibited kinase (non-phosphorylated peptide is cleaved, FRET pair separated). Thus, the assay involves a kinase reaction in the presence of varying concentrations of a given compound, a development reaction with site specific protease, and detection of the fluorescent emission ratio of coumarin and fluorscein. The emission ratio as a function of compound concentration was used to determine the IC50 value. Reaction conditions for each kinase are determined to provide optimal reaction times, incubation temperature, kinase and ATP concentrations. Test samples are made up in Ix kinase buffer (50 mM HEPES pH 7.5, 50 mM MgCl2, 5 mM EGTA, 0.05% BRIJ-35) and compound at desired concentration prepared in DMSO such that the final DMSO is 1%. Assay controls include 0% phosphorylation, which contains no ATP, 100% phosphorylation control, which consists of a synthetically phosphorylated peptide, and 0% inhibition control, which contains active kinase conditions without compound. Typically, assay plates are prepared to 10 μl final volume per sample, adding 2.5 μl of compound at 4x the desired concentration in 4% DMSO (serial dilution of compound provides concentration curve), 5 μl of the desired kinase mixed with Z'-LYTE™ peptide substrate (2x in 2x kinase buffer, final peptide at 2 μM), and 2.5 μl of 4x ATP solution. All kinase except for MAP2K1 were at 10 μM ATP in the kinase reaction, while MAP2K1 was at 100 μM. Samples are mixed and the kinase reaction is incubated 1 hour at room temperature, after which 5 μl of development solution is added and mixed. After incubation for
another 1 hour at room temperature, 5 μl of stop reagent is added to each sample and mixed. The fluorescence signals are measured to determine the emission ratio.
[1034] AlphaScreen assay was used to screen additional kinases. The assay is similarly dependent on the phosphorylation of a peptide substrate. In this assay, an antibody that recognizes phosphorylated substrate is bound to an acceptor bead. The peptide substrate is attached to biotin, which binds to a donor bead that contains streptavidin. Thus phosphorylated substrate is bound by antibody and streptavidin, bringing the donor and acceptor beads into close proximity when kinase is not inhibited. The donor produces singlet oxygen which results in emission from the acceptor when they are in close proximity. Conversely, when kinase is inhibited, the donor and acceptor beads are not associated and the emission from the acceptor is reduced. The fluorescence signal vs. compound concentration was used to determine the IC50 values. Genetic Engineering
[1035] For some of the kinase screens, preparation of the kinase was required. Plasmids encoding a selection of kinase enzymes were engineered using common polymerase chain reaction (PCR) methods. The relevant DNA sequences and encoded protein sequences used in the assay are shown for each (see below). Complementary DNA cloned from various human tissues were purchased from Invitrogen, and these were used as substrates in the PCR reactions. Specific custom synthetic oligonucleotide primers (Invitrogen see below) were designed to initiate the PCR product, and also to provide the appropriate restriction enzyme cleavage sites for ligation with the plasmids. In selected cases additional pairs of oligonucleotides (see below) were used to introduce mutations into the coding sequence to alter the sequence for enzyme activation (BRAF), for removing problematic surface Cys residues (FGFRl), or for disabling the capacity to bind ATP (MEKl substrate).
[1036] In the case of KIT, the entire sequence encoding the enzyme was made through a gene synthesis procedure, using custom synthetic oligonucleotides covering the entire coding sequence (Invitrogen).
[1037] The plasmids used for ligation with the kinase-encoding inserts were derivatives of either pET (Novagen) for expression using E. coli, or pFastBac (Invitrogen) for expression using baculovirus infection of insect cell cultures. In each of these cases the kinase was engineered to include a Histidine tag for purification using metal affinity chromatography.
[1038] In some cases the kinase-encoding plasmids were engineered as bicistronic mRNA to co- express a second protein that modifies the kinase protein during its expression in the host cell. In the cases of AbI, FGFRl, Fltl, Kdr, Kit, Met, Ret, and Src protein tyrosine phosphatase IB (PTP), was co-expressed for dephosphorylation of the phospho-Tyrosines. In the case of ERK2, an activated
form of MEKl (MEKlDD), was co-expressed for phosphorylation and activation of the ERK2. In the case of p38a, an activated form of MEK6 (MKK6) was co-expressed for phosphorylation and activation of the p38a. In the case of BRAF, the chaperone CDC37, was co-expressed for more efficient protein folding of the BRAF.
[1039] Plasmids encoding phosphorylation substrate proteins were expressed as N-terminal GST fusions and C-terminal biotinylation fusions, using pGEX vectors (Amersham) modified to include sequences encoding a C-terminal biotinylation tag. These substrates include MEKl, a substrate for BRAF, and BAD, a substrate for Piml .
Protein Expression in E. coli and Purification.
[1040] For protein expression, plasmids containing genes of interest were transformed into E.coli strains BL21(DE3)RIL or pLyS (Invitrogen) and transformants selected for growth on LB agar plates containing appropriate antibiotics. Single colonies were grown overnight at 370C in 200ml TB (Terrific broth) media. 16x1 L of fresh TB media in 2.8L flasks were inoculated with 10ml of overnight culture and grown with constant shaking at 37°C. Once cultures reached an absorbance of 1.0 at 600nm, IPTG was added and cultures were allowed to grow for a further 12 to 18hrs at temperatures ranging from 12-3O0C. Cells were harvested by centrifugation and pellets frozen at — 800C until ready for lysis.
[1041] For protein Purification; frozen E.coli cell pellets were resuspended in lysis buffer and lysed using standard mechanical methods. Soluble proteins were purified via poly-Histidine tags using immobilized metal affinity purification MAC. In the case of all kinases described here all have been purified using a 3 step purification process utilizing; IMAC, size exclusion chromatography and ion exchange chromatography. In most cases the poly-Histidine tag was removed using Thrombin (Calbiochem).
[1042] In some cases the purification protocol required modifications in order to stabilize soluble protein during purification and concentration. In the case of BRaf 5mM MgCl2 was required throughout purification. In the case of Ret a combination of ImM ATP and 5mM MgCl2 was required during cell lysis and throughout purification. In the case of 2ap70 a 5M excess of AMP-PCP over protein was required as well as 5mM MgCl2.
Baculovirus Expression Vector System
Virus production:
[1043] The transfection of a monolayer of Spodopterafrugiperda (Sf9) cells was performed utilizing a bacmid containing the gene of interest and Cellfectin (Invitrogen) transfection reagent in antibiotic-free, serum-free Grace's complete media (Invitrogen). After a five hour incubation, the
transfection media was removed, and the monolayer was fed with Grace's media containing 10% FBS and antibiotics. After a 72 to 96hr incubation, the cell supernatant containing the virus was harvested. The titer of the virus stock was then determined using a baculo virus titer kit (BD). The virus stock was then expanded using low Multiplicity of Infection (MOI of 0.1) cultures and harvested 48hrs post-infection. The titer of the expanded virus stock was then determined for use in recombinant protein production.
Protein Production:
[1044] The protein expression level was optimized by varying the MOI (1-10) and time of harvest (48-72hrs). Sf9 cells were adapted to SF-900 II serum-free media and grown in suspension in spinner flasks. The cell suspension was then used to inoculate a Wave bioreactor for 25L production scale. Cells were harvested 48-72hrs post-infection and stored at -8O0C until ready for lysis. Proteins were purified similarly to those expressed in E. coli.
Kinase assay
[1045] AlphaScreen assays were done using compounds dissolved in DMSO to a concentration of 20 mM. The compounds were diluted accordingly to the desired final concentrations in each sample well, using 1 :3 serial dilution for a total of 8 concentration points. Plates were prepared such that each kinase reaction is 20 μl in Ix kinase buffer, 5% DMSO and 10 μM ATP. Kinase and assay conditions (defined below) used for each kinase are shown in Table 3. After incubation of the kinase reaction for 1 hour at room temperature, 5 μl of donor beads in stop buffer (5OmM EDTA in Ix kinase buffer) was added, the sample was mixed and incubated for 20 minutes at room temperature before adding 5 μl of acceptor beads in stop buffer. The samples were incubated for 60 minutes at room temperature and the signal per well was read on AlphaQuest reader. Phosphorylated substrate results in binding of the antibody and association of the donor and acceptor beads such that signal correlates with kinase activity. The signal vs. compound concentration was used to determine the IC50.
[1046] Assay Condition A:
Kinase buffer HEPES 5OmM, pH7.2, 5mM MgCl2, 5mM MnCl2, 0.2% BSA, 0.01% NP-40
Substrate 10OnM biotin-(E4Y)3 (Open Source Biotech, Inc.)
Donor bead 1 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 1 μg/ml PY20 coated bead (Perkin Elmer Life Science)
[1047] Assay Condition B :
Kinase buffer 50 mM HEPES pH 7.0, 50 mM NaCl, 2 mM MgCl2, 1 mM MnCl2, 1 mM
DTT, 0.01% Tween-20.
Substrate 100 nM biotin-MEKl (prepared as described above).
Donor bead 10 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 10 μg/ml Protein A coated, bound to anti phospho MEK1/2 antibody
(CellSignal)
[1048] Assay Condition C:
Kinase buffer 8 niM HEPES pH 7.0, 4 niM MgCl2, 1 niM DTT, 0.01% Tween-20.
Substrate 100 nM biotin-MBP (Upstate Biotechnology, Waltham, MA).
Donor bead 10 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 10 μg/ml Protein A coated, bound to anti phospho MBP antibody
(CellSignal)
[1049] Assay Condition D:
Kinase buffer 8 rnM MOPS pH 7.4, 2 mM MgCl2, 8 mM MnCl2, 2 mM DTT, 0.01%
Tween-20.
Substrate 30 nM biotin-(E4Y)10 (Upstate Biotechnology).
Donor bead 20 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 20 μg/ml PY20 antibody coated (Perkin Elmer Life Science)
[1050] Assay Condition E:
Kinase buffer 20 mM HEPES pH 7.0, 10 mM MgCl2, 1 mM DTT, 0.01% Tween-20.
Substrate 30 nM biotin-ATF2 (Upstate Biotechnology).
Donor bead 10 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 10 μg/ml Protein A coated, bound to anti phospho ATF2 antibody
(CellSignal)
[1051] Assay Condition F:
Kinase buffer 8 mM HEPES pH 7.0, 4 mM MgCl2, 1 mM DTT, 0.01 % Tween-20.
Substrate 100 nM biotin-BAD (prepared as described above).
Donor bead 10 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 10 μg/ml Protein A coated, bound to anti phospho BAD (Serl 12) antibody
(CellSignal)
[1052] Assay Condition G:
Kinase buffer MOPS 25mM, pH7.1, O.lmM MgCl2, 5mM MnCl2, 0.2% BSA, ImM DTT,
0.01% Tween-20.
Substrate 10OnM biotin-(E4Y)3 (Open Source Biotech, Inc.)
Donor bead 1 μg/ml Streptavidin coated bead (Perkin Elmer Life Science).
Acceptor bead 1 μg/ml PY20 coated bead (Perkin Elmer Life Science)
Table 3 Kinase reaction conditions (20 μl reaction volume) for kinase screens.
Kinase Vendor* Plasmid Number Expression Host Assay Condition
AbI P1121 E. coli A (1 ng kinase)
B-Raf Upstate B (0.1 ng kinase)
B-RafV600E P4254 Sf9 B (0.1 ng kinase) c-Raf-1 Upstate B (0.1 ng kinase)
Erk2 P4227 E. coli C (4 ng kinase)
Fak P1358 Sf9 A (0.1 ng kinase)
FGFRl P1351 E. coli A (0.1 ng kinase)
Fltl Pl 826 E. coli A (0.1 ng kinase)
Flt4 ProQinase A (0.1 ng kinase)
Fms Upstate D (0.5 ng kinase)
Jnkl Upstate E (0.1 ng kinase)
Jnk2 Roche E (0.05 ng kinase)
Jnk3 Upstate E (0.1 ng kinase)
Kit P1332 E. coli A (0.1 ng kinase)
Met P1818 E. coli A (0.1 ng kinase) p38 P4292 E. coli E (6 ng kinase)
Piml P1215 E. coli F (0.01 ng kinase)
Pyk2 Upstate D (1 ng kinase)
Ret P1378 E. coli A (0.01 ng kinase)
Src Pl 144 E. coli A (0.01 ng kinase)
Zap70 P1868 Sf9 G (0.1 ng kinase)
* Upstate = Upstate Biotechnology Roche = Roche Protein Expression Group (Indianapolis, IN) ProQinase = ProQinase GmbH (Freiburg, Germany)
[1053] Kit was alternatively purchased from Cell Signalling Technology. Kit and Fms assays were also run at 100 μM ATP, where for Fms the Ix buffer was 8 mM MOPS pH 7.0, 2 mM MgCl2, 8 mM MnCl2, 2 mM DTT, 50 mM NaCl, 0.01% BSA and 0.01% Tween-20 and stop buffer was 8 mM MOPS, pH 7.0, 100 mM EDTA, 0.01% BSA and for kit, 1 ng kinase was used and the Ix buffer was 8 mM MOPS pH 7.0, 1 mM MgCl2, 2 mM MnCl2, 1 mM DTT, 0.001% BSA and 0.01% Tween-20, with substrate of 30 nM biotin-(E4Y)i0 (Upstate Biotechnology) and beads were at 10 μg/ml in stop buffer of 8 mM MOPS, pH 7.0, 100 mM EDTA, 0.3% BSA.
[1054] Compounds screened by at least one of the methods described above, or by similar methods, having IC50 of less than 10 μM are shown in tables 2a (AbI), 2b (B-Raf), 2c (B-Raf V600E), 2d (Btk), 2e (c-Raf-1), 2f (EGFR), 2g (EphB2), 2h (Erk2), 2i (Fak), 2j (FGFRl), 2k (Fltl), 21 (Flt3), 2m (Flt4), 2n (Fms), 2o (Irak4), 2p (Jnkl), 2q (Jnk2), 2r (Jnk3), 2s (Kdr), 2t (Kit), 2u (MAP2K1), 2v (MAPKAPK2), 2w (Met), 2x (p38), 2y (PDGFRB), 2z (Piml), 2aa (PKC theta), 2bb (Pyk2), 2cc (Ret), 2dd (Src), 2ee (Stk6), and 2ff (Yes), 2gg (Zap70), 2hh (Akt3), 2ii (ALK), 2jj (Cdk2), 2kk (Csk), 211 (EphA2), 2mm (EphB4), 2nn (Frk), 2oo (Gsk/3), 2pp (Hck), 2qq (MAP4K4), 2rr (IGFlR), 2ss (IKK beta), 2tt (Itk), 2uu (Jak3), 2w (MLKl), 2ww (TrkA), 2xx (PDGFRA), 2yy (Plkl), 2zz (Brk), 2ab (ROCKl), 2ac (Syk), 2ad (TEC), and 2ae (Tie2).
Table 2a. Compounds with activity toward kinase AbI with IC
50 < 10 μM. P-0025, P-0054, P-0142, P-0453, P-0604, P-0708, P-1012,
Table 2b. Compounds with activity toward kinase B-Raf with IC50 < 10 μM.
P-0001, P-0002, P-0003, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0010, P-0016, P-0019, P-0021, P-0024, P-0025, P-0026, P-0027, P-0032, P-0034, P-0035, P-0036, P-0037, P-0038, P-0041, P-0042, P-0045, P-0050, P-0052, P-0054, P-0055, P-0056, P-0059, P-0060, P-0065, P-0066, P-0067, P-0068, P-0078, P-0079, P-0082, P-0088, P-0090, P-0093, P-0095, P-0102, P-Ol 12, P-Ol 14, P-0122, P-Ol 26, P-0140, P-0143, P-0162, P-0165, P-0166, P-0171, P-0178, P-Ol 80, P-O 184, P-0188, P-Ol 92, P-0196, P-0200, P-0210, P-0228, P-0257, P-0262, P-0265, P-0269, P-0271, P-0284, P-0293, P-0297, P-0302, P-0307, P-0310, P-0351, P-0356, P-0369, P-0382, P-0396, P-0414, P-0448, P-0486, P-0521, P-0550, P-0556, P-0559, P-0579, P-0594, P-0599, P-0604, P-0613, P-0636, P-0683, P-0685, P-0693, P-0697, P-0700, P-0716, P-0721, P-0728, P-0734, P-0744, P-0745, P-0746, P-0753, P-0763, P-0773, P-0774, P-0776, P-0778, P-0779, P-0794, P-0798, P-0805, P-0806, P-0807, P-0818, P-0837, P-0841, P-0842, P-0848, P-0850, P-0851, P-0853, P-0857, P-0860, P-0861, P-0863, P-0866, P-0867, P-0868, P-0874, P-0876, P-0877, P-0883, P-0885, P-0889, P-0894, P-0896, P-0897, P-0898, P-0902, P-0904, P-0907, P-0909, P-0910, P-0911, P-0912, P-0913, P-0919, P-0924, P-0928, P-0931, P-0932, P-0933, P-0937, P-0939, P-0941, P-0944, P-0946, P-0947, P-0952, P-0954, P-0955, P-0956, P-0958, P-0959, P-0973, P-0975, P-0978, P-0980, P-0983, P-0984, P-0987, P-0991, P-0997, P-0998, P-1003, P-1004, P-1006, P-1009, P-1013, P-1014, P-1020, P-1027, P-1028, P-1056, P-1076, P-1080, P-1110, P-1116, P-1243, P-1244, P-1246, P-1247, P-1249, P-1250, P-1251, P-I 252, P-1253, P-1254, P-1255, P-1256, P-1257, P-1258, P-1259, P-1260, P-1261, P-1262, P-1263, P-1264, P-1265, P-1266, P-1267, P-1268, P-1269, P-1270, P-1279, P-1280, P-1281, P-1282, P-1288, P-1289, P-1317, P-1318, P-1336, P-1338, P-1341, P-1343, P-1346, P-1347, P-1348, P-1349, P-1365, P-1383, P-1384, P-1385, P-1386, P-1387, P-1388, P-1389, P-1390, P-1391, P-1395, P-1397, P-1419, P-1420, P-1429, P-1430, P-1431, P-1432, P-1433, P-1445, P-1446, P-1447, P-1451, P-1452, P-1453, P-1454, P-1455, P-1456, P-1457, P-1458, P-1459, P-1467, P-1469, P-1472, P-1473, P-1475,
P-1477, P-1479, P-1480, P-1481, P-1485, P-1486, P ■1526, P-1527, P-1528, P-■1529, P-1532,P-1534, P-1539, P-1541, P-1542, P-1544, P ■1546, P-1547, P-1548, P-•1549, P-1552,P-1553, P-1554, P-1555, P-1556, P-1559, P ■1566, P-1567, P-1568, P-•1569, P-1570,P-1576, P-1577, P-1580, P-1581, P-1582, P •1583, P-1584, P-1585, P-•1586, P-1589,P-1590, P-1591, P-1592, P-1593, P-1596, P ■1597, P-1598, P-1599, P-•1600, P-1602,P-1605, P-1608, P-1609, P-1610, P-1612, P 1613, P-1616, P-1621, P-•1627, P-1630,P-1631, P-1636, P-1637, P-1638, P- 1639, P•1656, P-1660, P-1663, P-•1664, P-1665,P-1670, P-1671, P-1687, P- 1700, P- 1701, P■1702, P-1703, P- 1704, P.1705, P-1706,P-1707, P-1708, P-1709, P- 1710, P- 1711 1712, P-1713, P-1714, P-•1715, P-1716,P-1717, P-1718, P-1719, P-1720, P- 1721 ■1722, P-1723, P- 1724, P-•1725, P-1726,P-1727, P-1728, P-1729, P-1730, P- 1731 ■1732, P-1733, P- 1734, P-•1735, P-1736,P-1737, P-1738, P-1739, P-1740, P- 1741 ■1742, P-1746, P- 1747, P-■1748, P-1749,P-1750, P-1751, P-1752, P- 1753, P- 1755, P.■1756, P-1757, P-1758, P-1759, P-1760,P-1762, P-1763, P-1764, P-1765, P- 1766, P•1767, P-1768, P- 1769, P-1770, P-1771,P-1772, P-1773, P-1774, P- 1775, P- 1776, P.■1777, P-1778, P- 1779, P-•1780, P-1781,P-1782, P-■1783, P-1784, P- 1798, P-1799, P■1800, P- 1802, P-1804, P■1816, P-1817,P- 1818, P-■1819, P-1822, P- 1823, P- 1825, P■1827, P-1828, P-1839, P-1840, P-1841,P- 1842, P-1864, P-1865, P-1871, P- 1872, P■1873, P-1878, P-1879, P-1881, P-1882, P- 1907, P- 1912, P-1916, P- 1980, P- 1996, P-1997, P-1998, P-■2005, P-2006, P-2007, P-2012, P-2013
TableJZc. Compounds with activitytoward kinase B-RafV600E with IC50< 10 μM.
P-0001, P-0002, P-0003, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0010, P-0011, P-0012, P-0013, P-0014, P-OOl5, P-0016, P-0017, P-OO19, P-0021, P-0022, P-0024, P-0025, P-0026, P-0027, P-0028, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-0038, P-0039, P-0040, P-0041, P-0042, P-0043, P-0044, P-0045, P-0046, P-0047, P-0048, P-0049, P-0050, P-0051, P-0052, P-0053, P-0054, P-0055, P-0056, P-0059, P-0060, P-0062, P-0063, P-0066, P-0067, P-0068, P-0070, P-0071, P-0072, P-0073, P-0074, P-0075, P-0078, P-0079, P-0082, P-0085, P-0088, P-0089, P-0090, P-0092, P-0093, P-0095, P-0097, P-0099, P-0100, P-O102, P-O107, P-0108, P-0109, P-0110, P-Ol12, P-Ol14, P-Ol18, P-Ol19, P-0121, P-O122, P-O124, P-O126, P-O129, P-0134, P-0135, P-Ol37, P-0139, P-0140, P-0141, P-0146, P-0147, P-0148, P-0152, P-0153, P-0156, P-Ol60, P-0162, P-Ol65, P-0166, P-0170, P-0171, P-0174, P-0175, P-0178, P-Ol80, P-0181, P-O184, P-Ol85, P-Ol86, P-Ol87, P-Ol88, P-0191, P-Ol92, P-0196, P-Ol99, P-0200, P-0205, P-0208, P-0210, P-0211, P-0215, P-0228, P-0231, P-0237, P-0242, P-0243, P-0244, P-0246, P-0248, P-0252, P-0255, P-0257, P-0261, P-0262, P-0265, P-0269, P-0271, P-0274, P-0284, P-0287, P-0293, P-0295, P-0297, P-0302, P-0307, P-0308, P-0311, P-0318, P-0320, P-0325, P-0333, P-0344, P-0347, P-0351, P-0352, P-0355, P-0356, P-0358, P-0369, P-0373, P-0381, P-0382, P-0386, P-0388, P-0396, P-0409, P-0414, P-0418, P-0420, P-0421, P-0434, P-0441, P-0447, P-0448, P-0453, P-0472, P-0483, P-0486, P-0493, P-0515, P-0521, P-0535, P-0550, P-0552, P-0559, P-0573, P-0579, P-0592, P-0594, P-0599, P-0600, P-0603, P-0604, P-0613, P-0623, P-0624, P-0636, P-0638, P-0645, P-0646, P-0647, P-0651, P-0656, P-0668, P-0671, P-0679, P-0683, P-0685, P-0691, P-0693, P-0696, P-0698, P-0700, P-0710, P-0713, P-0716, P-0721, P-0728, P-0730, P-0734, P-0744, P-0745, P-0746, P-0751, P-0753, P-0762, P-0763, P-0771, P-0773, P-0774, P-0776, P-0777, P-0778, P-0779, P-0794, P-0798, P-0803, P-0805, P-0806, P-0807, P-0810, P-0811, P-0816, P-0818, P-0819, P-0833, P-0837, P-0841, P-0842, P-0848, P-0850, P-0851, P-0853, P-0857, P-0860, P-0861, P-0863, P-0866, P-0867, P-0868, P-0874, P-0876, P-0877, P-0885, P-0886, P-0889, P-0893, P-0894, P-0896, P-0897, P-0898, P-0902, P-0904, P-0905, P-0907, P-0910, P-0911, P-0912, P-0913, P-0919, P-0927, P-0928, P-0931, P-0933, P-0937, P-0939, P-0944, P-0946, P-0947, P-0950, P-0951, P-0952, P-0954, P-0955, P-0956, P-0957, P-0958, P-0959, P-0962, P-0964, P-0971, P-0976, P-0980,
P-0981, P-0983, P-•0984, P-■0991, P-•0997, P-■0998, P-■1000, P-1002, P-1003, F 1004, P-1006, P-1007, P-•1008, P-■1009, P-•1010, P-■1013, P-•1014, P-1015, P-1016, P-1018, P-1020,P-1021, P-■1022, P-•1024, P-■1025, P-■1026, P-•1028, P-1029, P-1030, P- 1031, P-1035,P-1043, P-■1049, P-•1056, P-■1061, P-•1063, P-•1064, P-1065, P-1067, P- 1069, P-1070, P-■1071, P-•1074, P-•1082, P-■1085, P-■1090, P-■1091, F1112, P-1113, P- 1116, P-1117,P-■1120, P--1123, P-■1128, F■1131, P--1138, P-■1139, P- 1140, P-1160, F■1177, P-1179, P-•1180, P--1181, P-■1183, F•1187, F■1194, P-■1195, P- 1199, P-1243, P-■1244, P-1246, P-■1247, P-■1249, F■1250, P-■1251, F■1252, P-■1253, P-1254, P-1255, P- 1256, P-1257, P-■1258, P-■1259, P--1260, P-■1261, F■1262, P-■1263, F 1264, P-1265, P-•1266, P-1267, P-■1269, P-■1270, P-■1279, P-■1280, P-■1281, P-■1282, P- 1283, P-1288, P- 1289, P-1316, P-•1317, P-•1318, F■1323, F■1329, F-1336, F■1341, P- 1343, P-1345, P-■1346, P-1347, P-■1348, P-•1349, P-•1365, F■1366, F-1368, F•1369, P- 1370, P-■1381, P-•1383, P-1384, P-•1385, P-■1386, F■1387, P--1388, F■1389, P-■1390, F 1391, P-1392, P-■1394, P-1395, P-■1396, P-■1397, F■1398, F-1399, F-1402, P-■1403, P- 1409, P-1411, F•1413, P-1414,P-■1415, P-■1416, F■1417, F■1418, P--1419, P-■1420, P- 1423, P-1425, P-■1426, P-1429, P-■1430, P-■1431, F■1432, F■1433, P--1444, P-■1445, F 1446, P-1447, P-■1448, P-1449, p.•1450, P-■1451, F■1452, P-■1453, P--1454, P-■1455, F 1456, P-1457, P-■1458, P-1459, P-•1462, P--1465, F-1466, P-■1467, P--1469, P-■1470, P- 1471, P-1472, P-■1473, P-1474, P--1475, P--1477, F■1478, P-■1479, P--1480, F■1481, P- 1485, P-1486, P-•1495, P-1505, P-•1506, P.■1516, F-1526, F-1527, P--1528, F■1529, F 1530, P-1531, P■1532, P-1534,P-■1539, P-■1540, F-1541, F-1542, P--1544, P-■1545, F 1546, P-1547, P-■1548, P-1549, F•1550, P-■1552, F-1553, F■1554, F-1556, F■1558, F 1559, P-1561, P-■1564, P-1566, P-■1567, F-1568, F-1569, F-1570, P--1572, P--1575, F 1576, P-•1577, F•1578, P-1579,F■1581, F-1582, F-1583, F-1584, P--1585, F■1586, P- 1589, P-•1590, F•1591, P-1592,F•1594, F-1596, F-1597, F-1598, F-1599, F-1600, F 1601, F•1602, P-■1605, P-1606,F-1607, F-1608, F-1609, F-1610, F-1611, F-1612, F 1613, F■1614, F•1621, P-1627,F-1630, P-1631, F-1636, P--1637, F-1638, F■1639, P- 1656, P-■1660, P-1663, P-1664, F■1665, F-1666, F-1670, F-1671, F-1687, F-1698, F 1700, P-•1701, F•1702, P-1703, F■1704, F-1705, F-1706, P--1707, P--1708, P-1709, F■1710, P-■1711 ■1712, P-1713,F■1714, F-1715, F-1716, F-1717, F-1718, P--1719, F■1720, P-•1721 •1722, P-1723, P-■1724, F-1725, P-1726, F-1727, P--1728, P--1729, P-■1730, P-■1731 ■1732, P-1733, P-1734, F-1735, P'-1736, F-1737, F-1738, F-1739, P•1740, P-•1741 ■1742, P-1746, P--1747, F-1748, F'-1749,P-1750, F-1751, F-1752, P•1753, P--1755, P-■1756, P-1757, P--1758, F-1759, P-1760, P-1762, F-1763, P-1764, P ■1765, P-■1766, P■1767, P-1768, P-1769, P-1770, P'-1771,P'-1772, P-1773, P-1774, P■1775, P-■1776, F■1777, P-1778, P-1779, F-1780, P'-1781, P'-1782, F-1783, P-1784, F•1797, P-■1798, P-■1799, P-I800, P 1802, F-1804, P 1816, F•1817, F-1818, F-1819, P■1822, P-•1823, F■1828, P-1839, P-1840, F'-1841,P'-1842,P'-1843,P-1864, P-1865, P■1871, F-1872, P-1873, P-1878, P-1879, P-1881, P'-1882,P'-1907,F-1912, F-1916, P■1980, F■1996, P■1997, P-1998,P-2005, P-2006, P'-2007, P'-2012, P-2013
Table 2d. Compounds with activitytowardkinaseBtkwith IC50< 10 μM.
P-1389, P-1390, P-1420, P-1426, P-1459, P-1473, P-1475, P-1479, P-1480, P-1481, P-1485, P-1486
Table 2e. Compounds with activity toward kinase c-Raf-1 with IC50 < 10 μM c-Raf-1 : P-0001, P-0002, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0010, P-OOl 5, P-OOl 6, P-0021, P-0024, P-0025, P-0026, P-0027, P-0032, P-0034, P-0035, P-0036, P-0037, P-0038, P-0042, P-0045, P-0052, P-0055, P-0066, P-0078, P-0079, P-0082, P-0088, P-0090, P-0102, P-Ol 12, P-Ol 14, P-0121, P-0122, P-0137, P-0156, P-Ol 62, P-0165, P-0166, P-0170, P-0178, P-Ol 80, P-O 184, P-0188, P-0210, P-0228, P-0257, P-0262, P-0265, P-0269, P-0297, P-0302, P-0307, P-0356, P-0369, P-0382, P-0396, P-0418, P-0486, P-0521, P-0535, P-0542, P-0559, P-0604, P-0613, P-0636, P-0656, P-0685, P-0700, P-0716, P-0721, P-0728, P-0734, P-0744, P-0745, P-0746, P-0753, P-0763, P-0773, P-0774, P-0776, P-0778, P-0779, P-0794, P-0798, P-0805, P-0806, P-0807, P-0811, P-0818, P-0837, P-0841, P-0842, P-0848, P-0850, P-0851, P-0853, P-0857, P-0860, P-0861, P-0863, P-0866, P-0867, P-0868, P-0874, P-0876, P-0877, P-0883, P-0885, P-0889, P-0890, P-0894, P-0896, P-0897, P-0898, P-0902, P-0904, P-0907, P-0909, P-0910, P-0911, P-0912, P-0913, P-0919, P-0924, P-0928, P-0931, P-0933, P-0937, P-0939, P-0941, P-0944, P-0946, P-0947, P-0950, P-0952, P-0954, P-0955, P-0956, P-0957, P-0958, P-0959, P-0964, P-0971, P-0973, P-0974, P-0975, P-0978, P-0983, P-0987, P-0991, P-0997, P-0998, P-1002, P-1003, P-1004, P-1006, P-1009, P-1013, P-1014, P-1015, P-1017, P-1020, P-I 027, P-1028, P-1047, P-1056, P-1061, P-1063, P-I 064, P-1065, P-1070, P-1071, P-1076, P-1077, P-1078, P-1079, P-1118, P-1122, P-1145, P-1243, P-1244, P-1246, P-1247, P-1249, P-1250, P-1251, P-1253, P-1254, P-1255, P-1256, P-1257, P-1258, P-1260, P-1261, P- 1262, P-1265, P-1279, P-1283, P-1288, P-1289, P-1316, P-1317, P-1318, P-1336, P-1338, P-1365, P-1386, P- 1387, P-1388, P-1389, P-1390, P-1391, P-1395, P-1396, P-1397, P-1398, P-1403, P-1413, P-1419, P-1431, P- 1432, P-1433, P-1448, P-1451, P-1452, P-1453, P-1454, P-1455, P-1456, P-1458, P-1541, P-1542, P-1546, P-1547, P-1581, P-1583, P-1630, P-1671, P-1712, P-1713, P-1714, P-1733, P-1737, P-1738, P-1739, P-1740, P-1783, P-1839, P-1864, P-1871, P-I 873, P-1878, P-1879, P-1881, P-I 882
Table 2f. Compounds with activity toward kinase EGFR with IC50 < 10 μM
EGFR: P-0001, P-0002, P-0003, P-0004, P-0025, P-0095, P-0153, P-0877
Table 2g. Compounds with activity toward kinase EphB2 with ICso≤ 10 μM P-0013, P-0025, P-0027, P-0041, P-0056, P-0083, P-0126,
Table 2h. Compounds with activity toward kinase Erk2 with IC50 < 10 μM
Erk2: P-0031, P-0041, P-0058, P-0154, P-0550, P-0611, P-1336
Table 2i. Compounds with activity toward kinase Fak with IC5o< 10 μM
P-OO 18,
P-1256, P-1288, P-1347, P-1389, P-1403, P-1451, P-1475, P- 1495,
Table 2j. Compounds with activity toward kinase FGFR with IC50 < 10 μM
P-0001, P-0002, P-0003, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0010, P-0011, P-OO 12, P-0013, P-0015, P-0016, P-0017, P-0019, P-0020, P-0021, P-0024, P-0025, P-0026, P-0027, P-0028, P-0029, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-0038, P-0039, P-0040, P-0041, P-0042, P-0043, P-0045, P-0050, P-0052, P-0054, P-0055, P-0056, P-0058, P-0059, P-0066, P-0067, P-0068, P-0072, P-0073, P-0078, P-0079, P-0082, P-0088, P-0089, P-0090, P-0093, P-0096, P-0097, P-0099, P-0101, P-0102, P-O 103, P-Ol 12, P-Ol 14, P-Ol 19, P-0121, P-0129, P-0132, P-0134, P-0137, P-0140, P-0142, P-0149, P-Ol 50, P-O 152, P-O 156, P-0158, P-0165, P-Ol 66, P-Ol 67, P-0168, P-0170, P-0171, P-0175, P-0178, P-O 180, P-Ol 84, P-O 189, P-O 192, P-0196, P-0198, P-0204, P-0205, P-0206, P-0210, P-0211, P-0215, P-0217, P-0218, P-0219, P-0220, P-0227, P-0228, P-0232, P-0233, P-0239, P-0242, P-0244, P-0246, P-0248, P-0257, P-0262, P-0265, P-0267, P-0269, P-0271, P-0274, P-0284, P-0287, P-0289, P-0293, P-0294, P-0297, P-0298, P-0302, P-0304, P-0308, P-0309, P-0316, P-0320, P-0321, P-0325, P-0326, P-0329, P-0335, P-0339, P-0341, P-0344, P-0346, P-0351, P-0363, P-0368, P-0369, P-0371, P-0373, P-0374, P-0379, P-0383, P-0385, P-0391, P-0392, P-0396, P-0400, P-0404, P-0409, P-0411, P-0418, P-0420, P-0421, P-0424, P-0426, P-0442, P-0447, P-0448, P-0452, P-0453, P-0459, P-0473, P-0474, P-0479, P-0483, P-0486, P-0489, P-0493, P-0495, P-0501, P-0507, P-0510, P-0515, P-0520, P-0521, P-0535, P-0550, P-0556, P-0559, P-0561, P-0562, P-0563, P-0575, P-0579, P-0594, P-0599, P-0611, P-0613, P-0615, P-0623, P-0624, P-0632, P-0636, P-0640, P-0645, P-0647, P-0656, P-0658, P-0668, P-0671, P-0679, P-0683, P-0685, P-0691, P-0693, P-0697, P-0698, P-0699, P-0700, P-0708, P-0710, P-0721, P-0728, P-0730, P-0734, P-0737, P-0744, P-0745, P-0746, P-0749, P-0751, P-0753, P-0763, P-0771, P-0773, P-0774, P-0776, P-0798, P-0805, P-0806, P-0807, P-0811, P-0818, P-0819, P-0826, P-0828, P-0835, P-0837, P-0841, P-0848, P-0850, P-0851, P-0853, P-0854, P-0857, P-0860, P-0865, P-0867, P-0868, P-0874, P-0876, P-0877, P-0885, P-0889, P-0894, P-0896, P-0897, P-0898, P-0902, P-0904, P-0907, P-0910, P-0911, P-0912, P-0913, P-0919, P-0927, P-0933, P-0935, P-0937, P-0944, P-0947, P-0950, P-0951, P-0952, P-0954, P-0955, P-0956, P-0958, P-0964, P-0974, P-0976, P-0977, P-0979, P-0983, P-0991, P-0997, P-1002, P-1004, P-1008, P-1009, P-1015, P-1017, P-1018, P-1020, P-1021, P-1027, P-1066, P-1074, P-1078, P-I 110, P-Il H, P-1116, P-1120, P-1123, P-1125, P-1142, P-1181, P-I 182, P-1188, P-1194, P-1246, P-1249, P-1250, P-1251, P-1252, P-1253, P-1254, P-1255, P-1256, P-1257, P-1258, P-1259,
P-1260, P-1261, P-1262, P- 1263, P-1264, P-•1265, P-■1266, P-1267, P-1269, P-1270, P-1272, P-1273, P-1274, P-1279, P-1280, P-■1281, P-■1282, P-1283, P-1287, P-1288, P-1289, P-1316, P-1317, P-1318, P-1321, P-•1322, P-■1323, P-1325, P-1326, P-1327, P-1328,P-1329, P-1330, P-1331, P-1332, P-•1333, P-■1334, P-1335, P-1336, P-1337, P-1338,P-1339, P-1340, P-1341, P-1342, P-■1343, P-•1344, P-1345, P-1346, P-1347, P-1348,P-1349, P-1365, P-1366, P-1367, P-■1369, P-■1377, P-1380, P-1381, P-1382, P-1383,P-1384, P-1385, P- 1386, P-1387, P-•1388, P-■1389, P-1390, P-1391, P- 1392, P-1393,P-1394, P-1395, P- 1396, P-1397, P-■1398, P-•1399, P- 1402, P- 1403, P- 1404, P-1406, P-1407, P-1409, P- 1411, P-1415, P-•1416, P-■1418, P- 1419, P- 1420, P- 1423, P-1424, P-1426, P- 1428, P- 1429, P-1430, P.■1431, P-■1433, P- 1445, P- 1446, P- 1447, p-1448, p.1451, P- 1452, P- 1453, P-1454, P■1455, P■1456, P- 1458, P- 1459, P- 1460, P-1461,P- 1463, P- 1464, P- 1465, P- 1467, P■1468, P■1469, P- 1472, P- 1473, P- 1474, P-1475, P- 1476, P- 1477, P- 1478, P- 1479, P-1480, P■1481, P- 1482, P- 1485, P- 1486, P-1512,P- 1516, P- 1522, P- 1524, P- 1525, P-1526, P-1527, P- 1528, P- 1529, P- 1530, P-1534, P- 1538, P- 1539, P- 1542, P-•1545, P-1546, P-1547, P- 1548, P- 1549, P- 1550, P-1554, P- 1555, P- 1556, P- 1561, P-1564, P■1577, P■1581, P- 1582, P- 1583, P- 1584, P-1585, P-1589, P- 1591, P- 1592, P-1593, P■1595, P■1597, P- 1603, P- 1605, P- 1608, P-1609, P- 1610, P- 1614, P- 1621,P- 1622, P■1624, P■1625, P- 1685
Table 2k. Compounds with activity toward kinase Fltl with IC50 < 10 μM
P-0001, P-0002, P-0003, P-0004, P-0005, P-0008, P-0009, P-OOl 1, P-0012, P-0013, P-0016, P-0017, P-0018, P-0019, P-0020, P-0021, P-0024, P-0026, P-0027, P-0032, P-0033, P-0034, P-0036, P-0037, P-0038, P-0039, P-0041, P-0054, P-0055, P-0056, P-0067, P-0068, P-0072, P-0078, P-0082, P-0088, P-0090, P-0091, P-OlOl, P-Ol 03, P-0112, P-0114, P-0127, P-0134, P-0150, P-0154, P-0166, P-0177, P-0180, P-0184, P-0194, P-0196, P-0206, P-0211, P-0214, P-0224, P-0244, P-0269, P-0274, P-0278, P-0287, P-0298, P-0302, P-0315, P-0320, P-0325, P-0326, P-0337, P-0371, P-0373, P-0383, P-0404, P-0409, P-0421, P-0448, P-0455, P-0461, P-0470, P-0477, P-0483, P-0486, P-0491, P-0514, P-0515, P-0521, P-0550, P-0559, P-0579, P-0603, P-0624, P-0629, P-0632, P-0636, P-0640, P-0656, P-0668, P-0679, P-0683, P-0685, P-0691, P-0700, P-0708, P-0721, P-0733, P-0737, P-0749, P-0751, P-0757, P-0768, P-0771, P-0773, P-0774, P-0777, P-0805, P-0866, P-0868, P-0951, P-0958, P-0962, P-1002, P-1008, P-1010, P-1018, P-1021, P-1027, P-1082, P-1110, P-1112, P-1147, P-1160, P-1181, P-1194, P-1246, P-1247, P-1250, P-1251, P-1255, P-1256, P-1259, P-1260, P-1261, P-1262, P-1266, P-1269, P-1279, P-1289, P-1317, P-1318, P-1365, P-1366, P-1370, P-1372, P-1373, P-1383, P-1393, P-1395, P-1403, P-1404, P-1406, P-1411, P-1415, P-1416, P-1417, P-1418, P-1420, P-1422, P-1423, P-1424, P-1425, P-1426, P-1427, P-1428, P-1429, P-1430, P-1431, P-1432, P-1433, P-1445, P-1446, P-1447, P-1457, P-1460, P-1461, P-1462, P-1463, P-1464, P-1465, P-1467, P-1468, P-1469, P-1472, P-1475, P-1486, P-1491, P-1492, P-1495, P-1497, P-1499, P-1502, P-1505, P-1523, P-1526, P-1527, P-1528, P-1529, P-1530, P-1531, P-1532, P-1533, P-1534, P-1541, P-1542, P-1544, P-1546, P-1547, P-1548, P-1549, P-1552, P-1553, P-1554, P-1556, P-1557, P-1559, P-1564, P-1566, P-1567, P-1569, P-1570, P-1571, P-1572, P-1575, P-1576, P-1577, P-1580, P-1581, P-1582, P-1583, P-1584, P-1585, P-1586, P-1587, P-1589, P-1590, P-1591, P-1592, P-1593, P-1594, P-1595, P-1596, P-1597, P-1598, P-1599, P-1600, P-1601, P-1602, P-1603, P-1605, P-1606, P-1608, P-1609, P-1610, P-1612, P-1613, P-1614, P-1615, P-1618, P-1619, P-1621, P-1622, P-1624, P-1625, P-1686
Table 21. Compounds with activity toward kinase Flt3 with IC50 < 10 μM
Flt3: P-0088, P-0262, P-0409, P-0636, P-0806, P-1244, P-1280, P-1318, P-1336, P-1394, P-1426
Table 2m. Compounds with activity toward kinase Flt4 with IC
50 < 10 μM P-0039, P-0196, P-0325, P-0550, P-0668,
P-0813
Table 2n. Compounds with activity toward kinase Fms with IC50 < 10 μM
Fms: P-0001, P-0002, P-0003, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0010, P-OO 12, P-0016, P-0019, P-0020, P-0021, P-0025, P-0026, P-0027, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-0038, P-0042, P-0045, P-0048, P-0054, P-0067, P-0068, P-0072, P-0078, P-0079, P-0082, P-0088, P-0090, P-0093, P-0101, P-Ol 02, P-0103, P-Ol 12, P-0114, P-0121, P-O 129, P-0134, P-O 145, P-O 152, P-0166, P-0170, P-0175, P-Ol 80, P-Ol 84, P-Ol 89, P-0196, P-0210, P-0211, P-0228, P-0233, P-0237, P-0239, P-0244, P-0257, P-0262, P-0269, P-0274, P-0284, P-0287, P-0291, P-0293, P-0297, P-0298, P-0302, P-0317, P-0325, P-0327, P-0333, P-0351, P-0369, P-0373, P-0383, P-0391, P-0396, P-0409, P-0418, P-0420, P-0421, P-0461, P-0483, P-0486, P-0491, P-0501, P-0515, P-0535, P-0559, P-0563, P-0613, P-0636, P-0656, P-0685, P-0721, P-0733, P-0753, P-0763, P-0771, P-0773, P-0774, P-0778, P-0798, P-0805, P-0806, P-0807, P-0811, P-0818, P-0835, P-0837, P-0848, P-0850, P-0851, P-0853, P-0854, P-0857, P-0867, P-0874, P-0876, P-0883, P-0885, P-0898, P-0911, P-0913, P-0919, P-0927, P-0931, P-0933, P-0952, P-0954, P-0955, P-0956, P-0958, P-I 002, P-1008, P-1009, P-1013, P-I 110, P-I 112, P-I 124, P-1194, P-1246, P-1247, P-1249, P-1250, P-1251, P-1252, P-1253, P-1255, P-1259, P-1260, P-1262, P-1263, P-1264, P-1265, P-1266, P-1267, P-1269, P-1279, P-1280, P-1281, P-1282, P-1289, P-1316, P-1317, P-1318, P-1321, P-1323, P-I 324, P-I 325, P-1326, P-1327, P-1328, P-1329, P-1330, P-1331, P-1332, P-1333, P-1334, P-1335, P-1336, P-1337, P-1338, P-1339, P-1340, P-1341, P-1342, P-1343, P-1344, P-1346, P-1365, P-1366, P-1368, P-1369, P-1370, P-1372, P-1376, P-1380, P-1381, P-1384, P-1385, P-1386, P-1387, P-1388, P-1389, P-1390, P-1391, P-1392, P-1393, P-1394, P-1395, P-1397, P-1399, P-1400, P-1402, P-1403, P-I 404, P-1405, P-1406, P-1409, P-1410, P-1411, P-1415, P-1416, P-1419, P-1420, P-1421, P-1423, P-1425, P-1426, P-1427, P-1428, P-1429, P-1430, P-1431, P-1432, P-1433, P-1445, P-1447, P-1448, P-1449, P-1450, P-1451, P-1452, P-1453, P-1454, P-1455, P-1456, P-1457, P-1458, P-1459, P-1460, P-1461, P-1462, P-1463, P- 1464, P-1465, P-1466, P-1467, P-1468, P-1469, P-1470, P-1471, P-1472, P-1473, P-1474, P-1475, P-1476, P-1477, P-1478, P-1479, P-1480, P-1481, P-1482, P-1483, P-1485, P-1486, P-1488, P-1489, P-1490, P-1491, P-1492, P-1493, P-1494, P-1495, P-1496, P-I 497, P-1498, P-1499, P-1500, P-1501, P-I 502, P-1503, P-1504, P-1505, P-1506, P-1511, P-1522, P-I 525, P-I 526, P-1527, P-1528, P-1529, P-1530, P-1532, P-1534, P-1541, P-I 542, P-1544, P-I 545, P-1546, P-I 547, P-1548, P-1549, P-1552, P-1553, P-1554, P-1555, P-1556, P-1558, P-1559, P-1564, P-1566, P-1567, P-1568, P-1569, P-1570, P-1571, P-I 572, P-1575, P-1576, P-1580, P-1581, P-1583, P-1586, P-I 587, P-1589, P-1591, P-1594, P-1595, P-1596, P-1597, P-1598, P-1599, P-1602, P-1606, P-I 608, P-I 609, P-1610, P-1611, P-1612, P-1613, P-1615, P-1616, P-1618, P-1621, P-I 625, P-1627, P-1630, P-I 631, P-1636, P-1637, P-1638, P-1639, P-1652, P-1653, P-I 654, P-1656, P-1657, P-I 660, P-1663, P-I 664, P-1665, P-1670, P-1671, P-1685, P-1687, P-1700, P-1701, P-I 702, P-1703, P-1704, P-1705, P-1706, P-1707, P-1708, P-1709, P-1710, P-1711, P-1712, P-1713, P-1714, P-1715, P-1716, P-1717, P-1718, P-1719, P-1720, P-1721, P-1722, P-I 723, P-1724, P-1725, P-1726, P-1727, P-1728, P-1729, P-1730, P-I 731, P-1732, P-I 733, P-1734, P-1735, P-1736,
P-1737, P■1738, P-1739, P-1740, P■1741, P-•1742, P■1746, P-■1747, P- 1748, P-1749, P-1750, P•1751, P-1753, P-1754, P■1755, P-■1756, P■1757, P-■1758, P- 1759, P-1760, P-1761,P■1762, P-1763, P-1764, P-■1765, P-■1766, P•1767, P-■1768, P- 1769, P-1771, P-1772, P■1773, P-1774, P-1775, P-■1776, P-■1778, P■1779, P-■1780, P- 1781, P-1782, P-1783, P■1784, P-1796, P- 1798, P■1799, P-•1800, P■1802, P-■1803, P- 1804, P-1816, P-1817, P■1818, P-1819, P- 1821, P-•1822, P-•1827, P■1828, P-■1839, P- 1840, P-1864, P-1871, P■1872, P-1873, P- 1878, P•1879, P-•1881, P■1882, P-■1907, P- 1912, P-1916, P-1980, P■1996, P-1997,P- 1998, P■2005, P■2006, P■2007, P-■2012, P-2013
Table 2o. Compounds with activity toward kinase Irak4 with IC50 < 10 μM
Irak4: P-0002, P-0020, P-0076, P-0087, P-0091, P-0130
Table 2p Compounds with activity toward kinase Jnkl with IC50 < 10 μM
P-OO 16, P-0037, P-0079, P-0156, P-0302, P-0636, P-0763, P-0956, P-1256, P-1318, P-1365, P-1433, P-1475, P-1556, P-1596, P-1638, P-I 722, P-1750, P-I 770, P-I 864,
Table 2q. Compounds with activity toward kinase Jnk2 with IC50 < 10 μM
Table 2r. Compounds with activity toward kinase Jnk3 with IC
50 < 10 μM
P-OO 16, P-0036, P-0058, P-Ol 02, P-Ol 66, P-0213, P-0300, P-0559, P-0640, P-0697, P-0763, P-I 005, P-1087, P-1348, P-1400, P-1486, P-1561, P-1610, P-1707, P-1753, P-1777,
Table 2s. Compounds with activity toward kinase Kdr with IC50 < 10 μM
P-0001, P-0003, P-0005, P-0006, P-0007, P-0009, P-OOI l5 P-OO 12, P-0013, P-OO 14, P-0015, P-0016, P-0017, P-OO 18, P-0019, P-0020, P-0021, P-0022, P-0023, P-0024, P-0025, P-0027, P-0028, P-0029, P-0030, P-0031, P-0032, P-0033, P-0039, P-0040, P-0041, P-0042, P-0043, P-0044, P-0045, P-0046, P-0048, P-0049, P-0050, P-0051, P-0052, P-0053, P-0054, P-0055, P-0056, P-0057, P-0059, P-0060, P-0061, P-0063, P-0064, P-0065, P-0067, P-0068, P-0069, P-0070, P-0071, P-0073, P-0074, P-0075, P-0076, P-0077, P-0078, P-0080, P-0081, P-0082, P-0084, P-0085, P-0086, P-0087, P-0088, P-0089, P-0090, P-0091, P-0092, P-0093, P-0096, P-0097, P-0098, P-0100, P-0101, P-Ol 02, P-0103, P-0105, P-O 106, P-0107, P-0108, P-0110, P-0111, P-Ol 12, P-Ol 13, P-Ol 14, P-Ol 15, P-Ol 17, P-0120, P-0122, P-0125, P-0126, P-O 127, P-0128, P-0129, P-0130, P-0131, P-0133, P-0134, P-0135, P-0136, P-Ol 37, P-0139, P-0140, P-0141, P-O 142, P-O 143, P-0144, P-0145, P-0147, P-0149, P-0152, P-0157, P-0158, P-0161, P-0162, P-Ol 63, P-0164, P-0165, P-0167, P-0168, P-O 169, P-0170, P-0172, P-0173, P-0174, P-0175, P-0176, P-0177, P-0179, P-O 180, P-Ol 82, P-Ol 84, P-0189, P-0190, P-Ol 92, P-0194, P-0195, P-0196, P-0197, P-Ol 98, P-0199, P-0202, P-0203, P-0204, P-0206, P-0212, P-0213, P-0216, P-0218, P-0224, P-0225, P-0226, P-0230, P-0234, P-0242, P-0243, P-0260, P-0261, P-0262, P-0267, P-0268, P-0269, P-0270, P-0274, P-0279, P-0287, P-0288, P-0289, P-0293, P-0295, P-0296, P-0299, P-0308, P-0316, P-0319, P-0320, P-0321, P-0322, P-0326, P-0337, P-0339, P-0369, P-0376, P-0379, P-0391, P-0409, P-0418, P-0427, P-0448, P-0455, P-0458, P-0473, P-0482, P-0495, P-0521, P-0550, P-0559, P-0562, P-0579, P-0611, P-0623, P-0624, P-0632, P-0640, P-0645, P-0668, P-0679, P-0683, P-0691, P-0703, P-0708, P-0730, P-0737, P-0738, P-0751, P-0762, P-0771, P-0777, P-0796, P-0806, P-0813, P-0933, P-0951, P-0956, P-0962, P-0981, P-1023, P-1244, P-1280, P-1318, P-1394, P-1426
Table 2t. Compounds with activity toward kinase Kit with IC50 < 10 μM
Kit: P-OOOl, P-0002, P-0003, P-0004, P-0005, P-0006, P-0007, P-0008, P-0009, P-0011, P-0012, P-0013, P-0016, P-0017, P-OOl 8, P-0019, P-0020, P-0021, P-0024, P-0026, P-0027, P-0028, P-0029, P-0030, P-0031, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-0038, P-0039, P-0040, P-0041, P-0042, P-0050, P-0054, P-0056, P-0059, P-0064, P-0067, P-0069, P-0072, P-0078, P-0079, P-0080, P-0082, P-0086, P-0088, P-0090, P-0093, P-0097, P-OlOl, P-0102, P-0103, P-Ol 12, P-Ol 14, P-Ol 15, P-Ol 17, P-0120, P-0121, P-0122, P-0123, P-0127, P-0129, P-0132, P-0133, P-0134, P-Ol 36, P-0140, P-0142, P-0152, P-Ol 54, P-0157, P-0166, P-0167, P-0168, P-0171, P-Ol 75, P-0176, P-0179, P-0180, P-0184, P-0189, P-0190, P-0195, P-0196, P-0204, P-0205, P-0206, P-0210, P-0211, P-0213, P-0216, P-0217, P-0218, P-0224, P-0233, P-0235, P-0237, P-0244, P-0248, P-0253, P-0257, P-0262, P-0263, P-0265, P-0269, P-0274, P-0279, P-0284, P-0287, P-0289, P-0293, P-0298, P-0300, P-0302, P-0304, P-0315, P-0316, P-0321, P-0323, P-0325, P-0326, P-0333, P-0341, P-0346, P-0352, P-0367, P-0369, P-0371, P-0373, P-0378, P-0384, P-0385, P-0391, P-0392, P-0396, P-0402, P-0404, P-0409, P-0411, P-0418, P-0420, P-0421, P-0427, P-0447, P-0448, P-0450, P-0453, P-0459, P-0461, P-0473, P-0483, P-0491, P-0495, P-0499, P-0501, P-0511, P-0519, P-0521, P-0535, P-0550, P-0554, P-0556, P-0559, P-0561, P-0562, P-0563, P-0579, P-0594, P-0599, P-0604, P-0611, P-0613, P-0617, P-0623, P-0624, P-0625, P-0626, P-0632, P-0636, P-0640, P-0644, P-0645, P-0647, P-0656, P-0658, P-0659, P-0668, P-0671, P-0679, P-0682, P-0683, P-0685, P-0690, P-0691, P-0693, P-0697, P-0699, P-0700, P-0703, P-0708, P-0721, P-0733, P-0736, P-0737, P-0738, P-0749, P-0751, P-0753, P-0755, P-0757, P-0762, P-0763, P-0771, P-0773, P-0774, P-0776, P-0778, P-0794, P-0796, P-0798, P-0800, P-0806, P-0810, P-O813, P-O815, P-0818, P-0825, P-0835, P-0837, P-0848, P-0850, P-0851, P-0853, P-0854, P-0857, P-0860, P-0861, P-0865, P-0866, P-0867, P-0874, P-0876, P-0877, P-0885, P-0889, P-0898, P-0905, P-0907, P-0910, P-0911, P-0913, P-0919, P-0924, P-0927, P-0931, P-0933, P-0935, P-0937, P-0951, P-0952, P-0954, P-0955, P-0956, P-0958, P-0962, P-0964, P-0978, P-0983, P-1002, P-1008, P-1009, P-1010, P-1013, P-1016, P-1018, P-1021, P-1033, P-1082, P-1084, P-1096, P-1110, P-1112, P-1160, P-1181, P-1194, P-1246, P-1247, P-1249, P-1250, P-1251, P-1252, P-1253, P-1254, P-1255, P-1256, P-1257, P-1259, P-1260, P-1261, P-1262, P-1263, P-1264, P-1266, P-1267, P-1268, P-1269, P-1275, P-1279, P-1280, P-1281, P-1289, P-1316, P-1317, P-1318, P-1320, P-1321, P-1323, P-1329, P-1336, P-1338, P-1341, P-1343, P-1346, P-1347, P-1348, P-1349, P-1365, P-1366, P-1367, P-1368, P-1369, P-1370, P-1372, P-1376, P-1380, P-1382, P-1383, P-1384, P-1385, P-1386, P-1387, P-1388, P-1389, P-1390, P-1391, P-1392, P-1393, P-1394, P-1395, P-1396, P-1397, P-1399, P-1400, P-1402, P-1403, P-1404, P-1406, P-1407, P-1408, P-1409, P-1410, P-1411, P-1413, P-1414, P-1415, P-1416, P-1417, P-1419, P-1420, P-1422, P-1423, P-1424, P-1425, P-1426, P-1427, P-1428, P-1429, P-1430, P-1431, P-1432, P-1433, P-1445, P-1446, P-1447, P-1449, P-1450, P-1451, P-1452, P-1453, P-1454, P-1455, P-1456, P-1457, P-1458, P-1460, P-1461, P-1462, P-1463, P-1464, P-1465, P-1466, P-1467, P-1468, P-1469, P-1470, P-1471, P-1472, P-1474, P-1475, P-1476, P-1478, P-1479, P-1480, P-1481, P-1482, P-1483, P-1484, P-1486, P-1488, P-1489, P-1490, P-1493, P-1495, P-1497, P-1498, P-1499, P-1500, P-1501, P-1502, P-1503, P-1505, P-1506, P-1514, P-1521, P-1522, P-1525, P-1526, P-1527, P-1528, P-1529, P-1530, P-1531, P-1532, P-1534, P-1538, P-1541, P-1542, P-1543, P-1544, P-1545, P-1546, P-1547, P-1548, P-1549, P-1550, P-1551, P-1552, P-1553, P-1554, P-1557, P-1559, P-1562, P-1564, P-1565, P-1566, P-1567, P-1568, P-1569, P-1574, P-1575, P-1576, P-1578, P-1580, P-1581, P-1582, P-1583, P-1590, P-1591, P-1593, P-1598, P-1599, P-1605, P-1630, P-1671, P-1685, P-1700, P-1703, P-1704, P-1705, P-1706, P-1707, P-1708, P-1709, P-1711, P-1712, P-1713, P-1714, P-1718, P-1719, P-1720, P-1733, P-1737, P-1739, P-1740, P-1767, P-1776, P-1783, P-1798, P-1822, P-1839, P-1840, P-1864, P-1865, P-1871, P-1872, P-I 873, P-1878, P-1879, P-1881, P-1882, P-1980, P-1996, P-1997, P-1998
Table 2u. Compounds with activity toward kinase MAP2K1 with IC50 < 10 μM
MAP2K1: P-OOOl, P-0002, P-0003, P-0005, P-0006, P-0008, P-OOlO, P-0011, P-0012, P-0014, P-0015, P-0017, P-0018, P-0022, P-0023, P-0029, P-0031, P-0041, P-0046, P-0051, P-0057, P-0058, P-0061, P-0076, P-0079, P-0081, P-0087, P-0098, P-0099, P-0105, P-0108, P-Ol I l, P-0120, P-0149, P-0152, P-0158, P-0167, P-0170, P-0177, P-0194, P-0198, P-0331, P-0337, P-0568, P-0806
Table 2v. Compounds with activity toward kinase MAPKAPK2 with IC50 < 10 μM
MAPKAPK P-0007, P-0041, P-0057, P-0058, P-0077, P-0086, P-0104, P-0106, P-0151, P-0226
2:
Table 2w. Compounds with activity toward kinase Met with IC
50 < 10 μM P-0026, P-0058, P-0149, P-0419, P-0763, P-1012,
P-1527
Table 2x. Compounds with activity toward kinase p38 with IC5Q < 10 μM
P-0019, P-0038, P-0082, P-0158, P-0228, P-0448, P-0611, P-0700, P-0746, P-0807,
Table 2y. Compounds with activity toward kinase PDGFRB with IC50 < 10 μM
PDGFRB: P-0088, P-0262
Table 2z. Compounds with activity toward kinase Piml with IC50 < 10 μM
Piml: P-0024, P-0090
Table 2aa. Compounds with activity toward kinase PKC theta with IC50 < 10 μM
PKC theta: P-0001, P-0002, P-0003, P-0004, P-0006, P-0008, P-0011, P-0012, P-0013, P-0014, P-0017, P-OO 19, P-0020, P-0021, P-0022, P-0024, P-0025, P-0026, P-0030, P-0031,
P-0069,
Table 2bb. Compounds with activity toward kinase Pyk2 with IC50 < 10 μM
P-0056, P-Ol 37, P-0320, P-0562, P-0837, P-0897, P-0947, P-1280, P-1372,
Table 2cc. Compounds with activity toward kinase Ret with IC50 < 10 μM
Ret: P-0001, P-0001, P-0002, P-0003, P-0003, P-0004, P-0005, P-0005, P-0006, P-0008, P-0009, P-0010, P-0011, P-0012, P-0013, P-0014, P-0015, P-OO 16, P-0017, P-OO 19, P-0020, P-0022, P-0024, P-0025, P-0026, P-0027, P-0028, P-0028, P-0029, P-0032, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-0038, P-0039, P-0040, P-0040, P-0041, P-0041, P-0042, P-0043, P-0044, P-0045, P-0046, P-0047, P-0048, P-0049, P-0050, P-0051, P-0052, P-0053, P-0054, P-0055, P-0056, P-0058, P-0059, P-0060, P-0061, P-0062, P-0063, P-0064, P-0065, P-0066, P-0069, P-0070, P-0071, P-0072, P-0073, P-0075, P-0078, P-0079, P-0082, P-0083, P-0085, P-0088, P-0089, P-0090, P-0094, P-0095, P-0096, P-0097, P-0099, P-0100, P-0101, P-0102, P-0103, P-Ol 07, P-0108, P-O 109, P-0110, P-0113, P-Ol 14, P-Ol 15, P-0116, P-Ol 17, P-Ol 19, P-0121, P-0122, P-0123, P-0124, P-0134, P-0135, P-0137, P-0138, P-Ol 39, P-0140, P-0141, P-0142, P-0148, P-0152, P-0156, P-0158, P-Ol 59, P-0165, P-0167, P-0171, P-0175, P-O 179, P-0181, P-Ol 84, P-0186, P-O 190, P-0196, P-0196, P-0204, P-0205, P-0206, P-0210, P-0211, P-0215, P-0218, P-0224, P-0228, P-0231, P-0232, P-0236, P-0244, P-0244, P-0245, P-0246, P-0248, P-0250, P-0257, P-0262, P-0265, P-0269, P-0280, P-0286, P-0289, P-0293, P-0297, P-0302, P-0304, P-0307, P-0308, P-0314, P-0316, P-0316, P-0320, P-0320, P-0321, P-0325, P-0329, P-0339, P-0341, P-0344, P-0347, P-0351, P-0352, P-0363, P-0367, P-0369, P-0371, P-0378, P-0385, P-0392, P-0396, P-0412, P-0418, P-0434, P-0448, P-0448, P-0452, P-0453, P-0453, P-0469, P-0472, P-0486, P-0495, P-0501, P-0501, P-0517, P-0520, P-0521, P-0521, P-0533, P-0536, P-0542, P-0550, P-0550, P-0559, P-0559, P-0561, P-0579, P-0594, P-0596, P-0599, P-0599, P-0604, P-0608, P-0611, P-0623, P-0623, P-0624, P-0624, P-0632, P-0636, P-0638, P-0640, P-0644, P-0645, P-0645, P-0647, P-0656, P-0659, P-0668, P-0668, P-0671, P-0675, P-0678, P-0679, P-0682, P-0683, P-0691, P-0693, P-0697, P-0698, P-0699, P-0700, P-0703, P-0708, P-0710, P-0716, P-0721, P-0726, P-0728, P-0730, P-0734, P-0735, P-0736, P-0737, P-0738, P-0744, P-0745, P-0746, P-0749, P-0751, P-0753, P-0757, P-0761, P-0762, P-0763, P-0771, P-0778, P-0794, P-0795, P-0796, P-0807, P-0810, P-0811, P-0813, P-0822, P-0825, P-0826, P-0835, P-0841, P-0863, P-0865, P-0881, P-0939, P-0976, P-0977, P-0985, P-0998, P-1000, P-1005, P-1007, P-1011, P-1019, P-1024, P-1025, P-1026, P-1029, P-I 031, P-1033, P-1036, P-1066, P-1072, P-1073, P-1075, P-1081, P-1085, P-1089, P-1097, P-I 111, P-I 113, P-I 115, P-1117, P-I 119, P-1121, P-I 125, P-1126, P-1129, P-1130, P-I 133, P-1134, P-I 135, P-I 137, P-I 178, P-1181, P-1185, P-1188, P-1189, P-1191, P-I 192, P-I 193, P-1196, P-1198, P-I 200, P-1201
Table 2dd. Compounds with activity toward kinase Src with IC50 < 10 μM
P-0010, P-0035, P-0072, P-O 152, P-0316, P-0550, P-0700, P-0837, P-0885, P-0958, P-1250, P-1262, P-1289, P-1348, P-1394, P-1433, P-1473, P-1486,
Table 2ee. Compounds with activity toward kinase Stk6 with IC50 < 10 μM
P-OO 14, P-0033, P-0051, P-0065, P-0087, P-O 104, P-0123, P-0146, 63, P-Ol 64, 85, P-Ol 87, P-0207, P-0250, P-0283, P-0348, P-0441, P-1013,
Table 2ff. Compounds with activity toward kinase Yes with IC50 < 10 μM
P-0017, P-0033, P-0049, P-0062, P-0082, P-Ol 09, P-0145, P-0209,
Table 2gg. Compounds with activity toward kinase Zap70 with IC
50 < 10 μM
Table 2hh. Compounds with activity toward kinase Akt3 with IC50 < 10 μM
P-0024, P-0048, P-0127, P-0861, P-0962, P-1034, P-1145,
Table 2ii. Compounds with activity toward kinase ALK with IC50 < 10 μM
ALK: P-0806, P-1280, P-1244, P-1336, P-1394, P-1426
Table 2jj. Compounds with activity toward kinase Cdk2 with IC50 < 10 μM
Cdk2: P-0806, P-1280, P-1244, P-1394
Table 2kk. Compounds with activity toward kinase Csk with IC50 < 10 μM
Csk: P-0007, P-0805, P-0806, P-0885, P-0933, P-0955, P-0956, P-1013, P-1020, P-1336, P-1394, P-1426
Table 211. Compounds with activity toward kinase EphA2 with IC50 < 10 μM
EphA2: P-1336, P-1394
Table 2mm. Compounds with activity toward kinase EphB4 with IC50 < 10 μM
EρhB4: P-0806, P-1336
Table 2nn. Compounds with activity toward kinase Frk with IC50 < 10 μM
Frk: P-0007, P-0805, P-0885, P-0933, P-0955, P-0956, P-1013, P-1020, P-1244, P-1318, P-1336, P-1394
Table 2oo. Compounds with activity toward kinase Gsk3/3 with IC
50 < 10 μM P-0094, P-0180, P-0850, P-1372,
Table 2pp. Compounds with activity toward kinase Hck with IC50 < 10 μM
Hck: P-0007, P-0806, P-0885, P-0933, P-1318, P-1336, P-1394, P-1426
Table 2qq. Compounds with activity toward kinase MAP4K4 with IC50 < 10 μM
MAP4K4: P-0007, P-0057, P-0069, P-0079, P-0082, P-0088, P-0130, P-0131, P-0152, P-0174, P-0176, P-0198, P-0202, P-0214, P-0220, P-0256, P-0269, P-0287, P-0300, P-0317, P-0357, P-0367, P-0369, P-0391, P-0402, P-0442, P-0449, P-0477, P-0488, P-0495, P-0518, P-0527, P-0537, P-0573, P-0601, P-0685, P-0695, P-0700, P-0728, P-0734, P-0753, P-0800, P-0806, P-0811, P-0850, P-0851, P-0853, P-0862, P-0885, P-0896, P-0902, P-0904, P-0909, P-0913, P-0931, P-0933, P-0937, P-0954, P-0958, P-0971, P-0986, P-1017, P-1042, P-1056, P-1252, P-1253, P-1279, P-1280, P-1289, P-1317, P-1318, P-1336, P-1372, P-1383, P-1394, P-1406, P-1411, P-1414, P-1415, P-1417, P-1418, P-1426, P-1429, P-1685
Table 2rr. Compounds with activity toward kinase IGFlR with IC50 < 10 μM
Table 2ss. Compounds with activity toward kinase IKK beta with IC50 < 10 μM
DCK beta: P-0007, P-0013, P-0014, P-0029, P-0057, P-0073, P-0084, P-0085, P-0086, P-0087, P-0096, P-0098, P-0106, P-Ol I l, P-Ol 15, P-0120, P-0127, P-0128, P-0133, P-0135, P-0163, P-0164, P-0172, P-0177, P-0179, P-0216, P-0270, P-0272, P-0315, P-0376, P-0404, P-0410, P-0436, P-0629, P-0682, P-0690, P-0790, P-0896, P-0920, P-0962, P-1223
Table 2tt. Compounds with activity toward kinase Itk with IC
50 < 10 μM P-0020, P-0173, P-1337,
Table 2uu. Compounds with activity toward kinase Jak3 with IC50 < 10 μM P-0034, P-0084, P-0146,
Table 2w. Compounds with activity toward kinase MLKl with IC50 < 10 μM
Table 2ww. Compounds with activity toward kinase TrkA with IC50 ≤ 10 μM
TrkA: P-0409, P-0806, P-1244, P-1426
Table 2xx. Compounds with activity toward kinase PDGFRA with IC50 < 10 μM
PDGFRA: P-0007, P-0409, P-0806, P-0885, P-0933, P-1280, P-1336, P-1394, P-1426
Table 2yy. Compounds with activity toward kinase Plkl with IC50 < 10 μM
Plkl: P-0018, P-0022, P-0031, P-0044, P-0046, P-0067, P-0075, P-0083, P-0085, P-0099, P-Ol 13, P-0123, P-0128, P-0135, P-0146, P-0148, P-0154, P-0178, P-0286, P-0332, P-0345, P-0366, P-0480, P-0490, P-0581, P-0863, P-0954, P-1138
Table 2zz. Compounds with activity toward kinase Brk with IC50 < 10 μM
Brk: P-0007, P-0805, P-0806, P-0885, P-0933, P-0955, P-0956, P-1013, P-1020, P-1244, P-1318, P-1336, P-1394
Table 2ab. Compounds with activity toward kinase ROCKl with IC50 < 10 μM
ROCKl: P-0057
Table 2ac. Compounds with activity toward kinase Syk with IC50 < 10 μM
Syk: P-0002, P-OOlO, P-0033, P-0054, P-0056, P-0057, P-0089, P-0196, P-0448, P-0521, P-0599, P-1336
Table 2ad. Compounds with activity toward kinase TEC with IC50 <10 μM
TEC: P-0017, P-0033, P-0044, P-0088, P-0156, P-0166, P-0228, P-0257, P-0297, P-0429, P-0897, P-0954, P-0983, P-0991, P-0997, P-1020, P-1317
Table 2ae. Compounds with activity toward kinase Tie2 with IC50 < 10 μM
Tie2: P-0806, P-1280, P-1336, P-1394, P-1426
Plasmid sequence and PCR primer information:
AbI
PCR primers
P1121. pET-SPEC BI-PTP AbI G227-V515-X taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccaccacggtgtgtcc
M G H H H H H H G V S cccaactacgacaagtgggagatggaacgcacggacatcaccatgaagcacaagctgggc
P N Y D K W E M E R T D I T M K H K L G gggggccagtacggggaggtgtacgagggcgtgtggaagaaatacagcctgacggtggcc
G G Q Y G E V Y E G V W K K Y S L T V A gtgaagaccttgaaggaggacaccatggaggtggaagagttcttgaaagaagctgcagtc
V K T L K E D T M E V E E F L K E A A V atgaaagagatcaaacaccctaacctggtgcagctccttggggtctgcacccgggagccc
M K E I K H P N L V Q L L G V C T R E P ccgttctatatcatcactgagttcatgacctacgggaacctcctggactacctgagggag
P F Y I I T E F M T Y G N L L D Y L R E tgcaaccggcaggaggtgaacgccgtggtgctgctgtacatggccactcagatctcgtca
C N R Q E V N A V V L L Y M A T Q I S S gccatggagtacctggagaagaaaaacttcatccacagagatcttgctgcccgaaactgc
A M E Y L E K K N F I H R D L A A R N C ctggtaggggagaaccacttggtgaaggtagctgattttggcctgagcaggttgatgaca
L V G E N H L V K V A D F G L S R L M T ggggacacctacacagcccatgctggagccaagttccccatcaaatggactgcacccgag
G D T Y T A H A G A K F P I K W T A P E agcctggcctacaacaagttctccatcaagtccgacgtctgggcatttggagtattgctt
S L A Y N K F S I K S D V W A F G V L L tgggaaattgctacctatggcatgtccccttacccgggaattgacctgtcccaggtgtat
W E I A T Y G M S P Y P G I D L S Q V Y gagctgctagagaaggactaccgcatggagcgcccagaaggctgcccagagaaggtctat
E L L E K D Y R M E R P E G C P E K V Y gaactcatgcgagcatgttggcagtggaatccctctgaccggccctcctttgctgaaatc
E L M R A C W Q W N P S D R P S F A E I caccaagcctttgaaacaatgttccaggaatccagtatctcagacgaagtggaaaaggag
H Q A F E T M F Q E S S I S D Ξ V E K E ctggggaaacaaggcgtctgagtcgac (SEQ ID NO: )
L G K Q G V - (SEQ ID NO: )
B-RafV600E PCRprimers
P4254. pFastBacBD-CDC37 BRAF D437-K722-X, V600E tattccggattattcataccgtcccaccatcgggcgcggatctcggtccgaaacc atgtcgtactaccatcaccatcaccatcacgattacgatatcccaacgaccgaaaacctg
M S Y Y H H H H H H D Y D I P T T E N L tattttcagggccatatggatgattgggagattcctgatgggcagattacagtgggacaa
Y F Q G H M D D W E I P D G Q I T V G Q agaattggatctggatcatttggaacagtctacaagggaaagtggcatggtgatgtggca
R I G S G S F G T V Y K G K W H G D V A gtgaaaatgttgaatgtgacagcacctacacctcagcagttacaagccttcaaaaatgaa
V K M L N V T A P T P Q Q L Q A F K N E gtaggagtactcaggaaaacacgacatgtgaatatcctactcttcatgggctattccaca
V G V L R K T R H V N I L L F M G Y S T aagccacaactggctattgttacccagtggtgtgagggctccagcttgtatcaccatctc
K P Q L A I V T Q W C E G S S L Y H H L catatcattgagaccaaatttgagatgatcaaacttatagatattgcacgacagactgca
H I I E T K F E M I K L I D I A R Q T A cagggcatggattacttacacgccaagtcaatcatccacagagacctcaagagtaataat
Q G M D Y L H A K S I I H R D L K S N N atatttcttcatgaagacctcacagtaaaaataggtgattttggtctagctacagaaaaa
I F L H E D L T V K I G D F G L A T E K tctcgatggagtgggtcccatcagtttgaacagttgtctggatccattttgtggatggca
S R W S G S H Q F E Q L S G S I L W M A ccagaagtcatcagaatgcaagataaaaatccatacagctttcagtcagatgtatatgca
P E V I R M Q D K N P Y S F Q S D V Y A tttggaattgttctgtatgaattgatgactggacagttaccttattcaaacatcaacaac
F G I V L Y E L M T G Q L P Y S N I N N agggaccagataatttttatggtgggacgaggatacctgtctccagatctcagtaaggta
R D Q I I F M V G R G Y L S P D L S K V cggagtaactgtccaaaagccatgaagagattaatggcagagtgcctcaaaaagaaaaga
R S N C P K A M K R L M A E C L K K K R gatgagagaccactctttccccaaattctcgcctctattgagctgctggcccgctcattg
D E R P L F P Q I L A S I E L L A R S L ccaaaatagtcgactagagcctgcagtctcgaggcatgcggtaccaagctt (SEQ ID NO:
P K - (SEQ ID NO:
Erk2
PCRprimers
P4227.pET15S ERK2/MEK1DD taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattt tgtttaactttaagaaggagatataccatgggcagcagccatcatcatcatcatcacagc
M G S S H H H H H H S agcggcctggtgccgcgcggcagccatatggcggcggcggcgggcgcgggcccggagatg
S G L V P R G S H M A A A A G A G P E M gtccgcgggcaggtgttcgacgtggggccgcgctacaccaacctctcgtacatcggcgag
V R G Q V F D V G P R Y T N L S Y I G E ggcgcctacggcatggtgtgctctgcttatgataatgtcaacaaagttcgagtagctatc
G A Y G M V C S A Y D N V N K V R V A I aagaaaatcagcccctttgagcaccagacctactgccagagaaccctgagggagataaaa
K K I S P F E H Q T Y C Q R T L R E I K atcttactgcgcttcagacatgagaacatcattggaatcaatgacattattcgagcacca
I L L R F R H E N I I G I N D I I R A P accatcgagcaaatgaaagatgtatatatagtacaggacctcatggaaacagatctttac
T I E Q M K D V Y I V Q D L M E T D L Y aagctcttgaagacacaacacctcagcaatgaccatatctgctattttctctaccagatc
K L L K T Q H L S N D H I C Y F L Y Q I ctcagagggttaaaatatatccattcagctaacgttctgcaccgtgacctcaagccttcc
L R G L K Y I H S A N V L H R D L K P S aacctgctgctcaacaccacctgtgatctcaagatctgtgactttggcctggcccgtgtt
N L L L N T T C D L K I C D F G L A R V gcagatccagaccatgatcacacagggttcctgacagaatatgtggccacacgttggtac
A D P D H D H T G F L T E Y V A T R W Y agggctccagaaattatgttgaattccaagggctacaccaagtccattgatatttggtct
R A P E I M L N S K G Y T K S I D I W S gtaggctgcattctggcagaaatgctttctaacaggcccatctttccagggaagcattat
V G C I L A E M L S N R P I F P G K H Y cttgaccagctgaaccacattttgggtattcttggatccccatcacaagaagacctgaat
L D Q L N H I L G I L G S P S Q E D L N tgtataataaatttaaaagctaggaactatttgctttctcttccacacaaaaataaggtg
C I I N L K A R N Y L L S L P H K N K V ccatggaacaggctgttcccaaatgctgactccaaagctctggacttattggacaaaatg
P W N R L F P N A D S K A L D L L D K M ttgacattcaacccacacaagaggattgaagtagaacaggctctggcccacccatatctg
L T F N P H K R I E V E Q A L A H P Y L gagcagtattacgacccgagtgacgagcccatcgccgaagcaccattcaagttcgacatg
E Q Y Y D P S D E P I A E A P F K F D M gaattggatgacttgcctaaggaaaagctcaaagaactaatttttgaagagactgctaga
E L D D L P K E K L K E L I F E E T A R ttccagccaggatacagatcttaaatgtcgac (SEQ ID NO: )
F Q P G Y R S - (SEQ ID NO: )
Fak
PCR primers
P1358.pFastBacHtb FAK S411-Q686-X tattccggattattcataccgtcccaccatcgggcgcggatctcggtccgaaacc atgtσgtactaccatcaccatcaccatcacgattacgatatcccaacgaccgaaaacctg
M S Y Y H H H H H H D Y D I P T T E N L tattttcagggcgccatgggatccaccagggattatgagattcaaagagaaagaatagaa
Y F Q G A M G S T R D Y E I Q R E R I E cttggacgatgtattggagaaggccaatttggagatgtacatcaaggcatttatatgagt
L G R C I G E G Q F G D V H Q G I Y M S ccagagaatccagctttggcggttgcaattaaaacatgtaaaaactgtacttcggacagc
P E N P A L A V A I K T C K N C T S D S gtgagagagaaatttcttcaagaagccttaacaatgcgtcagtttgaccatcctcatatt
V R E K F L Q E A L T M R Q F D H P H I gtgaagctgattggagtcatcacagagaatcctgtctggataatcatggagσtgtgcaca
V K L I G V I T E N P V W I I M E L C T cttggagagctgaggtcatttttgcaagtaaggaaatacagtttggatctagcatctttg
L G E L R S F L Q V R K Y S L D L A S L atcctgtatgcctatcagcttagtacagctcttgcatatctagagagcaaaagatttgta
I L Y A Y Q L S T A L A Y L E S K R F V cacagggacattgctgctcggaatgttctggtgtcctcaaatgattgtgtaaaattagga
H R D I A A R N V L V S S N D C V K L G gactttggattatcccgatatatggaagatagtacttactacaaagcttccaaaggaaaa
D F G L S R Y M E D S T Y Y K A S K G K ttgcctattaaatggatggctccagagtcaatcaattttcgacgttttacctcagctagt
L P I K W M A P E S I N F R R F T S A S gacgtatggatgtttggtgtgtgtatgtgggagatactgatgcatggtgtgaagcctttt
D V W M F G V C M W E I L M H G V K P F caaggagtgaagaacaatgatgtaatcggtcgaattgaaaatggggaaagattaccaatg
Q G V K N N D V I G R I E N G E R L P M cctccaaattgtcctcctaccctctacagccttatgacgaaatgctgggcctatgacccc
P P N C P P T L Y S L M T K C W A Y D P agcaggcggcccaggtttactgaacttaaagctcagctcagcacaatcctggaggaagag
S R R P R F T E L K A Q L S T I L E E E aaggctcagtagtcgacgagctcactagtcgcggccgctttcgaatctagagcctgcagt
K A Q - S T S S L V A A A F E S R A C S ctcgaggcatgσggtaccaagcttgtcgagaagtactagaggatcataatc (SEQ ID NO.-
L E A C G T K L V E K Y - (SEQ ID NO:
FGFRl PCR primers
P1351.pET N6 BI-PTP FGFR A458-E765-X C488A, C584S taatacgactcactataggggaattgtgagcggataacaatt cccct ct agaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccatcatatggcaggg
M G H H H H H H M A G gtctctgagtatgagcttcccgaagaccctcgctgggagctgcctcgggacagactggtc
V S E Y E L P E D P R W E L P R D R L V ttaggcaaacccctgggagagggcgcgtttgggcaggtggtgttggcagaggctatcggg
L G K P L G E G A F G Q V V L A E A I G ctggacaaggacaaacccaaccgtgtgaccaaagtggctgtgaagatgttgaagtcggac
L D K D K P N R V T K V A V K M L K S D gcaacagagaaagacttgtcagacctgatctcagaaatggagatgatgaagatgatcggg
A T E K D L S D L I S E M E M M K M I G aagcataagaatatcatcaacctgctgggggcctgcacgcaggatggtcccttgtatgtc
K H K N I I N L L G A C T Q D G P L Y V atcgtggagtatgcctccaagggcaacctgcgggagtacctgcaggcccggaggccccca
I V E Y A S K G N L R E Y L Q A R R P P gggctggaatacagctacaaccccagccacaacccagaggagcagctctcctccaaggac
G L E Y S Y N P S H N P E E Q L S S K D ctggtgtcctgcgcctaccaggtggcccgaggcatggagtatctggcctccaagaagtgc
L V S C A Y Q V A R G M E Y L A S K K C atacaccgagacctggcagccaggaatgtcctggtgacagaggacaatgtgatgaagata
I H R D L A A R N V L V T E D N V M K I gcagactttggcctcgcacgggacattcaccacatcgactactataaaaagacaaccaac
A D F G L A R D I H H I D Y Y K K T T N ggccgactgcctgtgaagtggatggcacccgaggcattatttgaccggatctacacccac
G R L P V K W M A P E A L F D R I Y T H cagagtgatgtgtggtctttcggggtgctcctgtgggagatcttcactctgggcggctcc
Q S D V W S F G V L L W E I F T L G G S ccataccccggtgtgcctgtggaggaacttttcaagctgctgaaggagggtcaccgcatg
P Y P G V P V E E L F K L L K E G H R M gacaagcccagtaactgcaccaacgagctgtacatgatgatgcgggactgctggcatgca
D K P S N C T N E L Y M M M R D C W H A gtgccctcacagagacccaccttcaagcagctggtggaagacctggaccgcatcgtggcc
V P S Q R P T F K Q L V E D L D R I V A ttgacctccaaccaggagtagtcgacgaaggagatatatcc (SEQ ID NO: )
L T S N Q E - (SEQ ID NO: )
Fltl
PCR primers
P1826.pETN6 BI-PTP FLTl M799-D1165-X WT taatacgactcactataggggaattgtgagcggataacaatt cccct ct agaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccatcatatggaccca
M G H H H H H H M D P gat gaagtt cct ttggatgagcagtgtgagcggct ccct tat gat gccagcaagtgggag
D E V P L D E Q C E R L P Y D A S K W E tttgcccgggagagacttaaactgggcaaatcacttggaagaggggcttttggaaaagtg
F A R E R L K L G K S L G R G A F G K V gttcaagcatcagcatttggcattaagaaatcacctacgtgccggactgtggctgtgaaa
V Q A S A F G I K K S P T C R T V A V K atgctgaaagagggggccacggccagcgagtacaaagctctgatgactgagctaaaaatc
M L K E G A T A S E Y K A L M T E L K I ttgacccacattggccaccatctgaacgtggttaacctgctgggagcctgcaccaagcaa
L T H I G H H L N V V N L L G A C T K Q ggagggcctctgatggtgattgttgaatactgcaaatatggaaatctctccaactacctc
G G P L M V I V E Y C K Y G N L S N Y L aagagcaaacgtgacttattttttctcaacaaggatgcagcactacacatggagcctaag
K S K R D L F F L N K D A A L H M E P K aaagaaaaaatggagccaggcctggaacaaggcaagaaaccaagactagatagcgtcacc
K E K M E P G L E Q G K K P R L D S V T agcagcgaaagctttgcgagctccggctttcaggaagataaaagtctgagtgatgttgag
S S E S F A S S G F Q E D K S L S D V E gaagaggaggattctgacggtttctacaaggagcccatcactatggaagatctgatttct
E E E D S D G F Y K E P I T M E D L I S tacagttttcaagtggccagaggcatggagttcctgtcttccagaaagtgcattcatcgg
Y S F Q V A R G M E F L S S R K C I H R gacctggcagcgagaaacattcttttatctgagaacaacgtggtgaagatttgtgatttt
D L A A R N I L L S E N N V V K I C D F ggccttgcccgggatatttataagaaccccgattatgtgagaaaaggagatactcgactt
G L A R D I Y K N P D Y V R K G D T R L cctctgaaatggatggctcccgaatctatctttgacaaaatctacagcaccaagagcgac
P L K W M A P E S I F D K I Y S T K S D gtgtggtcttacggagtattgctgtgggaaatcttctccttaggtgggtctccataccca
V W S Y G V L L W E I F S L G G S P Y P ggagtacaaatggatgaggacttttgcagtcgcctgagggaaggcatgaggatgagagct
G V Q M D E D F C S R L R E G M R M R A cctgagtactctactcctgaaatctatcagatcatgctggactgctggcacagagaccca
P E Y S T P E I Y Q I M L D C W H R D P aaagaaaggccaagatttgcagaacttgtggaaaaactaggtgatttgcttcaagcaaat
K E R P R F A E L V E K L G D L L Q A N gtacaacaggattaggtcgaccaccaccaccaccaccactgagatccggctggccctact
V Q Q D - (SEQ ID NO: ) ggccgaaaggaattcgaggccagcagggccaccgctgagcaataactagcataacccctt ggggcctctaaacgggtcttgaggggttttttg (SEQ ID NO: )
Kit
PCR primers
P1332.N6 BIPTP KITM552-K948-X COD taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccatcatatgtacgaa
M G H H H H H H M Y E gttcagtggaaagttgttgaagaaatcaacggtaacaactacgtttacatcgacccgacc
V Q W K V V E E I N G N N Y V Y I D P T cagctgccgtacgaccacaaatgggagttcccgcgtaaccgtctgtctttcggtaaaacc
Q L P Y D H K W E F P R N R L S F G K T ctgggtgcgggtgcgttcggtaaagttgttgaagcgaccgcgtacggtctgatcaaatct
L G A G A F G K V V E A T A Y G L I K S gacgcggcgatgaccgttgcggttaaaatgctgaaaccgtctgcgcacctgaccgaacgt
D A A M T V A V K M L K P S A H L T E R gaagcgctgatgtctgaactgaaagttctgtcttacctgggtaaccacatgaacatcgtt
E A L M S E L K V L S Y L G N H M N I V aacctgctgggtgcgtgcaccatcggtggtccgacσctggttatcaccgaatactgctgc
N L L G A C T I G G P T L V I T E Y C C tacggtgacctgctgaacttcctgcgtcgtaaacgtgactctttcatctgctctaaacag
Y G D L L N F L R R K R D S F I C S K Q gaagaccacgcggaagcggcgctgtacaaaaacctgctgcactctaaagaatcttcttgc
E D H A E A A L Y K N L L H S K E S S C tctgactctaccaacgaatacatggacatgaaaccgggtgtttcttacgttgttccgacc
S D S T N E Y M D M K P G V S Y V V P T aaagcggacaaacgtcgttctgttcgtatcggttcttacatcgaacgtgacgttaccccg
K A D K R R S V R I G S Y I E R D V T P gcgatcatggaagacgacgaactggcgctggacctggaagacctgctgtctttctcttac
A I M E D D E L A L D L E D L L S F S Y caggttgcgaaaggtatggcgttcctggcgtctaaaaactgcatccaccgtgacctggcg
Q V A K G M A F L A S K N C I H R D L A gcgcgtaacatcctgctgacccacggtcgtatcaccaaaatctgcgacttcggtctggcg
A R N I L L T H G R I T K I C D F G L A cgtgacatcaaaaacgactctaactacgttgttaaaggtaacgcgcgtctgccggttaaa
R D I K N D S N Y V V K G N A R L P V K tggatggcgccggaatctatcttcaactgcgtttacaccttcgaatctgacgtttggtct
W M A P E S I F N C V Y T F E S D V W S tacggtatcttcctgtgggaactgttctctctgggttcttctccgtacccgggtatgccg
Y G I F L W E L F S L G S S P Y P G M P gttgactctaaattctacaaaatgatcaaagaaggtttccgtatgctgtctccggaacac
V D S K F Y K M I K E G F R M L S P E H gcgccggcggaaatgtacgacatcatgaaaacctgctgggacgcggacccgctgaaacgt
A P A E M Y D I M K T C W D A D P L K R ccgaccttcaaacagatcgttcagctgatcgaaaaacagatctctgaatctaccaaccac
P T F K Q I V Q L I E K Q I S E S T N H atctactctaacctggcgaactgctctccgaaccgtcagaaatagtcgactgaaaaagga
I Y S N L A N C S P N R Q K - (SEQ ID NO: ) agagt (SΞQ ID NO: )
Met
PCR primers
P1818.pETN6 BI-PTP MET G1056-G1364-X WT taatacgactcactataggggaattgtgagcggataacaatt cccct ct agaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccatcatatgggggac
M G H H H H H H M G D tctgatatatccagtccattactgcaaaatactgtccacattgacctcagtgctctaaat
S D I S S P L L Q N T V H I D L S A L N ccagagctggtccaggcagtgcagcatgtagtgattgggcccagtagcctgattgtgcat
P E L V Q A V Q H V V I G P S S L I V H ttcaatgaagtcataggaagagggcattttggttgtgtatatcatgggactttgttggac
F N E V I G R G H F G C V Y H G T L L D aatgatggcaagaaaattcactgtgctgtgaaatccttgaacagaatcactgacatagga
N D G K K I H C A V K S L N R I T D I G gaagtttcccaatttctgaccgagggaatcatcatgaaagattttagtcatcccaatgtc
E V S Q F L T E G I I M K D F S H P N V ctctcgctcctgggaatctgcctgcgaagtgaagggtctccgctggtggtcctaccatac
L S L L G I C L R S E G S P L V V L P Y atgaaacatggagatcttcgaaatttcattcgaaatgagactcataatccaactgtaaaa
M K H G D L R N F I R N E T H N P T V K gatcttattggctttggtcttcaagtagccaaaggcatgaaatatcttgcaagcaaaaag
D L I G F G L Q V A K G M K Y L A S K K tttgtccacagagacttggctgcaagaaactgtatgctggatgaaaaattcacagtcaag
F V H R D L A A R N C M L D E K F T V K gttgctgattttggtcttgccagagacatgtatgataaagaatactatagtgtacacaac
V A D F G L A R D M Y D K E Y Y S V H N aaaacaggtgcaaagctgccagtgaagtggatggctttggaaagtctgcaaactcaaaag
K T G A K L P V K W M A L E S L Q T Q K tttaccaccaagtcagatgtgtggtcctttggcgtgctcctctgggagctgatgacaaga
F T T K S D V W S F G V L L W E L M T R ggagccccaccttatcctgatgtaaacaccttt gat ataactgtt tact tgttgcaaggg
G A P P Y P D V N T F D I T V Y L L Q G agaagactcctacaacccgaatactgcccagaccccttatatgaagtaatgctaaaatgc
R R L L Q P E Y C P D P L Y E V M L K C tggcaccctaaagccgaaatgcgcccatccttttctgaactggtgtcccggatatcagcg
W H P K A E M R P S F S E L V S R I S A atcttctctactttcattgggtagtcgac (SEQ ID NO: )
I F S T F I G - (SEQ ID NO: )
p38
PCRprimers
P4292.pET15S P38A M1-S360-X MKK6DD taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattt tgtttaactttaagaaggagatataccatgggcagcagccatcatcatcatcatcacagc
M G S S H H H H H H S agcggcctggtgccgcgcggcagccatatgtctcaggagaggcccacgttctaccggcag
S G L V P R G S H M S Q E R P T F Y R Q gagctgaacaagacaatctgggaggtgcccgagcgttaccagaacctgtctccagtgggc
E L N K T I W E V P E R Y Q N L S P V G tctggcgcctatggctctgtgtgtgctgcttttgacacaaaaacggggttacgtgtggca
S G A Y G S V C A A F D T K T G L R V A gtgaagaagctctccagaccatttcagtccatcattcatgcgaaaagaacctacagagaa
V K K L S R P F Q S I I H A K R T Y R E ctgcggttacttaaacacatgaaacatgaaaatgtgattggtctgttggacgtttttaca
L R L L K H M K H E N V I G L L D V F T cctgcaaggtctctggaggaattcaatgatgtgtatctggtgacccatctcatgggggca
P A R S L E E F N D V Y L V T H L M G A gatctgaacaacattgtgaaatgtcagaagcttacagatgaccatgttcagttccttatc
D L N N I V K C Q K L T D D H V Q F L I taccaaattctccgaggtctaaagtatatacattcagctgacataattcacagggaccta
Y Q I L R G L K Y I H S A D I I H R D L aaacctagtaatctagctgtgaatgaagactgtgagctgaagattctggattttggactg
K P S N L A V N E D C E L K I L D F G L gctcggcacacagatgatgaaatgacaggcta'cgtggccactaggtggtacagggctcct
A R H T D D E M T G Y V A T R W Y R A P gagatcatgctgaactggatgcattacaaccagacagttgatatttggtcagtgggatgc
E I M L N W M H Y N Q T V D I W S V G C ataatggccgagctgttgactggaagaacattgtttcctggtacagaccatattgatcag
I M A E L L T G R T L F P G T D H I D Q ttgaagctcattttaagactcgttggaaccccaggggctgagcttttgaagaaaatctcc
L K L I L R L V G T P G A E L L K K I S tcagagtctgcaagaaactatattcagtctttgactcagatgccgaagatgaactttgcg
S E S A R N Y I Q S L T Q M P K M N F A aatgtatttattggtgccaatcccctggctgtcgacttgctggagaagatgcttgtattg
N V F I G A N P L A V D L L E K M L V L gactcagataagagaattacagcggcccaagcccttgcacatgcctactttgctcagtac
D S D K R I T A A Q A L A H A Y F A Q Y cacgatcctgatgatgaaccagtggccgatccttatgatcagtcctttgaaagcagggac
H D P D D E P V A D P Y D Q S F E S R D ctccttatagatgagtggaaaagcctgacctatgatgaagtcatcagctttgtgccacca
L L I D E W K S L T Y D E V I S F V P P ccccttgaccaagaagagatggagtcctgactcgac (SEQ ID NO: )
P L D Q E E M E S - (SEQ ID NO: )
Piml
PCR primers
P1215.pET29SRI PIMl E29-K313 HIS WT agatcgatctcgatcGcgcgaaattaatacgactcactataggggaattgtgagcggataa caatt cccct ctagaaataattttgtttaactttaagaaggagatatacatatgaaggag
M K E aaggagcccctggagtcgcagtaccaggtgggcccgctactgggcagcggcggcttcggc
K E P L E S Q Y Q V G P L L G S G G F G tcggtctactcaggcatccgcgtctccgacaacttgccggtggccatcaaacacgtggag
S V Y S G I R V S D N L P V A I K H V E aaggaccggatttccgactggggagagctgcctaatggcactcgagtgcccatggaagtg
K D R I S D W G E L P N G T R V P M E V gtcctgctgaagaaggtgagctcgggtttctccggcgtcattaggctcctggactggttc
V L L K K V S S G F S G V I R L L D W F gagaggcccgacagtttcgtcctgatcctggagaggcccgagccggtgcaagatctcttc
E R P D S F V L I L E R P E P V Q D L F gacttcatcacggaaaggggagccctgcaagaggagctggcccgcagcttcttctggcag
D F I T E R G A L Q E E L A R S F F W Q gtgctggaggccgtgcggcactgccacaactgcggggtgctccaccgcgacatcaaggac
V L E A V R H C H N C G V L H R D I K D gaaaacatccttatcgacctcaatcgcggcgagctcaagctcatcgacttcgggtcgggg
E N I L I D L N R G E L K L I D F G S G gcgctgctcaaggacaccgtctacacggacttcgatgggacccgagtgtatagccctcca
A L L K D T V Y T D F D G T R V Y S P P gagtggatccgctaccatcgctaccatggcaggtcggcggcagtctggtccctggggatc
E W I R Y H R Y H G R S A A V W S L G I ctgctgtatgatatggtgtgtggagatattcctttcgagcatgacgaagagatcatcagg
L L Y D M V C G D I P F E H D E E I I R ggccaggttttcttcaggcagagggtctcttcagaatgtcagcatctcattagatggtgc
G Q V F F R Q R V S S E C Q H L I R W C ttggccctgagaccatcagataggccaaccttcgaagaaatccagaaccatccatggatg
L A L R P S D R P T F E E I Q N H P W M caagatgttctcctgccccaggaaactgctgagatccacctccacagcctgtcgccgggg
Q D V L L P Q E T A E I H L H S L S P G cccagcaaagtcgaccaccaccaccaccaccactgagatccggctgctaacaaagcccga
P S K V D H H H H H H - (SEQ ID NO: ) aaggaattcgagttggctgctgccaccgctgagcaataactagcataaccccttggggcc tctaaacgggtcttgaggggttttttg (SEQ ID NO: )
Ret
PCR primers
P1378.pET-SF BI-PTP RET H661-R1012 HIS taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataatttt gtttaactttaagaaggagatatacatatgcacaagtttgcccacaagccacccatctcc
M H K F A H K P P I S tcagctgagatgaccttccggaggcccgcccaggccttcccggtcagctactcctcttcc
S A E M T F R R P A Q A F P V S Y S S S ggtgcccgccggccctcgctggactccatggagaaccaggtctccgtggatgccttcaag
G A R R P S L D S M E N Q V S V D A F K atcctggaggatccaaagtgggaattccctcggaagaacttggttcttggaaaaactcta
I L E D P K W E F P R K N L V L G K T L ggagaaggcgaatttggaaaagtggtcaaggcaacggccttccatctgaaaggcagagca
G E G E F G K V V K A T A F H L K G R A gggtacaccacggtggccgtgaagatgctgaaagagaacgcctccccgagtgagcttcga
G Y T T V A V K M L K E N A S P S E L R gacctgctgtcagagttcaacgtcctgaagcaggtcaaccacccacatgtcatcaaattg
D L L S E F N V L K Q V N H P H V I K L tatggggcctgcagccaggatggcccgctcctcctcatcgtggagtacgccaaatacggc
Y G A C S Q D G P L L L I V E Y A K Y G tccctgcggggcttcctccgcgagagccgcaaagtggggcctggctacctgggcagtgga
S L R G F L R E S R K V G P G Y L G S G ggcagccgcaactccagctccctggaccacccggatgagcgggccctcaccatgggcgac
G S R N S S S L D H P D E R A L T M G D ctcatctcatttgcctggcagatctcacaggggatgcagtatctggccgagatgaagctc
L I S F A W Q I S Q G M Q Y L A E M K L gttcatcgggacttggcagccagaaacatcctggtagctgaggggcggaagatgaagatt
V H R D L A A R N I L V A E G R K M K I tcggatttcggcttgtcccgagatgtttatgaagaggattcctacgtgaagaggagccag
S D F G L S R D V Y E E D S Y V K R S Q ggtcggattccagttaaatggatggcaattgaatccctttttgatcatatctacaccacg
G R I P V K W M A I E S L F D H I Y T T caaagtgatgtatggtcttttggtgtcctgctgtgggagatcgtgacGctagggggaaac
Q S D V W S F G V L L W E I V T L G G N ccctatcctgggattcctcctgagcggctcttcaaccttctgaagaccggccaccggatg
P Y P G I P P E R L F N L L K T G H R M gagaggccagacaactgcagcgaggagatgtaccgcctgatgctgcaatgctggaagcag
E R P D N C S E E M Y R L M L Q C W K Q gagccggacaaaaggccggtgtttgcggacatcagcaaagacctggagaagatgatggtt
E P D K R P V F A D I S K D L E K M M V aagagggtcgaccaccaccaccaccaccactgagatccggctggccctactggccgaaag
K R V D H H H H H H - (SEQ ID NO: ) gaattcgaggccagcagggccaccgctgagcaataactagcataaccccttggggcctct aaacgggtcttgaggggttttttg (SEQ ID NO: )
Src
PCR primers
P1144. pET-N6 BI-PTP SRC V86-L536-X taatacgactcactataggggaattgtgagcggataacaatt cccct ctagaaataattt tgtttaactttaagaaggagatataccatgggtcaccaccatcaccatcatatggtgacc
M G H H H H H H M V T acctttgtggccctctatgactatgagtctaggacggagacagacctgtccttcaagaaa
T F V A L Y D Y E S R T E T D L S F K K ggcgagcggctccagattgtcaacaacacagagggagactggtggctggcccactcgctc
G E R L Q I V N N T E G D W W L A H S L agcacaggacagacaggctacatccccagcaactacgtggcgccctccgactccatccag
S T G Q T G Y I P S N Y V A P S D S I Q get gaggagtggtattttggcaagatcaccagacgggagtcagagcggt tact get caat
A E E W Y F G K I T R R E S E R L L L N gcagagaacccgagagggaccttcctcgtgcgagaaagtgagaccacgaaaggtgcctac
A E N P R G T F L V R E S E T T K G A Y tgcctctcagtgtctgacttcgacaacgccaagggcctcaacgtgaagcactacaagatc
C L S V S D F D N A K G L N V K H Y K I cgcaagctggacagcggcggcttctacatcacctcccgcacccagttcaacagcctgcag
R K L D S G G F Y I T S R T Q F N S L Q cagctggtggcctactactccaaacacgccgatggcctgtgccaccgcctcaccaccgtg
Q L V A Y Y S K H A D G L C H R L T T V tgccccacgtccaagccgcagactcagggcctggccaaggatgcctgggagatccctcgg
C P T S K P Q T Q G L A K D A W E I P R gagtcgctgcggctggaggtcaagctgggccagggctgctttggcgaggtgtggatgggg
E S L R L E V K L G Q G C F G E V W M G acctggaacggtaccaccagggtggccatcaaaaccctgaagcctggcacgatgtctcca
T W N G T T R V A I K T L K P G T M S P gaggccttcctgcaggaggcccaggtcatgaagaagctgaggcatgagaagctggtgcag
E A F L Q E A Q V M K K L R H E K L V Q ttgtatgctgtggtttcagaggagcccatttacatcgtcacggagtacatgagcaagggg
L Y A V V S E E P I Y I V T E Y M S K G agtttgctggactttctcaagggggagacaggcaagtacctgcggctgcctcagctggtg
S L L D F L K G E T G K Y L R L P Q L V gacatggctgctcagatcgcctcaggcatggcgtacgtggagcggatgaactacgtccac
D M A A Q I A S G M A Y V E R M N Y V H cgggaccttcgtgcagccaacatcctggtgggagagaacctggtgtgcaaagtggccgac
R D L R A A N I L V G E N L V C K V A D tttgggctggctcggctcattgaagacaatgagtacacggcgcggcaaggtgccaaattc
F G L A R L I E D N E Y T A R Q G A K F cccatcaagtggacggctccagaagctgccctctatggccgcttcaccatcaagtcggac
P I K W T A P E A A L Y G R F T I K S D gtgtggtccttcgggatcctgctgactgagctcaccacaaagggacgggtgccctaccct
V W S F G I L L T E L T T K G R V P Y P gggatggtgaaccgcgaggtgctggaccaggtggagcggggctaccggatgccctgcccg
G M V N R E V L D Q V E R G Y R M P C P ccggagtgtcccgagt ccct gcacgacct cat gtgccagtgctggcggaaggagcctgag
P E C P E S L H D L M C Q C W R K E P E
gagcggcccaccttcgagtacctgcaggccttcctggaggactacttcacgtccaccgag E R P T F E Y L Q A F L E D Y F T S T E ccccagtaccagcccggggagaacctctaggtcgacgaaggagatatatcc (SEQ ID NO: )
P Q Y Q P G E N L - (SEQ ID NO: )
Zap70 PCRprimers
P1868.pFastBacl ZAP70 D327-G606 HIS, WT agatcatggagataattaaaatgataaccatctcgcaaataaataagtattttactgtttt cgtaacagttttgtaataaaaaaacctataaatattccggattattcataccgtcccacc atcgggcgcggatccgccaccatggacaagaagctcttcctgaagcgcgataacctcctc
M D K K L F L K R D N L L atagctgacattgaacttggctgcggcaactttggctcagtgcgccagggcgtgtaccgc
I A D I E L G C G N F G S V R Q G V Y R atgcgcaagaagcagatcgacgtggccatcaaggtgctgaagcagggcacggagaaggca
M R K K Q I D V A I K V L K Q G T E K A gacacggaagagatgatgcgcgaggcgcagatcatgcaccagctggacaacccctacatc
D T E E M M R E A Q I M H Q L D N P Y I gtgcggctcattggcgtctgccaggccgaggccctcatgctggtcatggagatggctggg
V R L I G V C Q A E A L M L V M E M A G ggcgggccgctgcacaagttcctggtcggcaagagggaggagatccctgtgagcaatgtg
G G P L H K F L V G K R E E I P V S N V gccgagctgctgcaccaggtgtccatggggatgaagtacctggaggagaagaactttgtg
A E L L H Q V S M G M K Y L E E K N F V caccgtgacctggcggcccgcaacgtcctgetggttaaccggcactacgccaagatcage
H R D L A A R N V L L V N R H Y A K I S gactttggcctctccaaagcactgggtgccgacgacagctactacactgcccgctcagca
D F G L S K A L G A D D S Y Y T A R S A gggaagtggccgctcaagtggtacgcacccgaatgcatcaacttccgcaagttctccagc
G K W P L K W Y A P E C I N F R K F S S cgcagcgatgtctggagctatggggtcaccatgtgggaggccttgtcctacggccagaag
R S D V W S Y G V T M W E A L S Y G Q K ccctacaagaagatgaaagggccggaggtcatggccttcatcgagcagggcaagcggatg
P Y K K M K G P E V M A F I E Q G K R M gaatgcccaccagagtgtccacccgaactgtacgcactcatgagtgactgctggatctac
E C P P E C P P E L Y A L M S D C W I Y aagtgggaggatcgccccgacttcctgaccgtggagcagcgcatgcgagcctgttactac
K W E D R P D F L T V E Q R M R A C Y Y agcctggccagcaaggtggaagggcaccaccaccaccaccaccactagaattc (SEQ ID NO: )
S L A S K V E G H H H H H H H - (SEQ ID NO: )
BAD substrate PCR primers
P963.pET-BH BAD M1-Q168-X tacgactcactataggggaattgtgagcggataacaattcccctctagaaataattttgt ttaactttaagaaggagatataccatggctggttgcctgaacgacatcttcgaagctcag
M A G C L N D I F E A Q aaaatcgaatggcaccatcaccatcaccatatgttccagatcccagagtttgagccgagt
K I E W H H H H H H M F Q I P E F E P S gagcaggaagactccagctctgcagagaggggcctgggccccagccccgcaggggacggg
E Q E D S S S A E R G L G P S P A G D G ccctcaggctccggcaagcatcatcgccaggccccaggcctcctgtgggacgccagtcac
P S G S G K H H R Q A P G L L W D A S H cagcaggagcagccaaccagcagcagccatcatggaggcgctggggctgtggagatccgg
Q Q E Q P T S S S H H G G A G A V E I R agtcgccacagctcctaccccgcggggacggaggacgacgaagggatgggggaggagccc
S R H S S Y P A G T E D D E G M G E E P agcccctttcggggccgctcgcgctcggcgccccccaacctctgggcagcacagcgctat
S P F R G R S R S A P P N L W A A Q R Y ggccgcgagctccggaggatgagtgacgagtttgtggactcctttaagaagggacttcct
G R E L R R M S D E F V D S F K K G L P cgcccgaagagcgcgggcacagcaacgcagatgcggcaaagctccagctggacgcgagtc
R P K S A G T A T Q M R Q S S S W T R V ttccagtcctggtgggatcggaacttgggcaggggaagctccgccccctcccagtgagtc
F Q S W W D R N L G R G S S A P S Q - gaccaccaccaccaccaccactgagatccggctggccctactggccgaaaggaattcgag gccagcagggccaccgctgagcaataactagcataaccccttggggcctctaaacgggtc ttgaggggttttttg (SEQ ID NO: )
(polypeptide SEQ ID NO: )
MEKl substrate PCR primers
P1277.pGEX-BIO MEKl K97A atgtcccctatactaggttattggaaaattaagggccttgtgcaacccactcgacttctt
M S P I L G Y W K I K G L V Q P T R L L ttggaatatcttgaagaaaaatatgaagagcatttgtatgagcgcgatgaaggtgataaa
L E Y L E E K Y E E H L Y E R D E G D K tggcgaaacaaaaagtttgaattgggtttggagtttcccaatcttccttattatattgat
W R N K K F E L G L E F P N L P Y Y I D ggtgatgttaaattaacacagtctatggccatcatacgttatatagctgacaagcacaac
G D V K L T Q S M A I I R Y I A D K H N atgttgggtggttgtccaaaagagcgtgcagagatttcaatgcttgaaggagcggttttg
M L G G C P K E R A E I S M L E G A V L gatattagatacggtgtttcgagaattgcatatagtaaagactttgaaactctcaaagtt
D I R Y G V S R I A Y S K D F E T L K V gattttcttagcaagctacctgaaatgctgaaaatgttcgaagatcgtttatgtcataaa
D F L S K L P E M L K M F E D R L C H K acatatttaaatggtgatcatgtaacccatcctgacttcatgttgtatgacgctcttgat
T Y L N G D H V T H P D F M L Y D A L D gttgttttatacatggacccaatgtgcctggatgcgttcccaaaattagtttgttttaaa
V V L Y M D P M C L D A F P K L V C F K aaacgtattgaagctatcccacaaattgataagtacttgaaatccagcaagtatatagca
K R I E A I P Q I D K Y L K S S K Y I A tggcctttgcagggctggcaagccacgtttggtggtggcgaccatcctccaaaatcggat
W P L Q G W Q A T F G G G D H P P K S D ctggttccgcgtggatctcatatgcccaagaagaagccgacgcccatccagctgaacccg
L V P R G S H M P K K K P T P I Q L N P gcccccgacggctctgcagttaacgggaccagctctgcggagaccaacttggaggccttg
A P D G S A V N G T S S A E T N L E A L cagaagaagctggaggagctagagcttgatgagcagcagcgaaagcgccttgaggccttt
Q K K L E E L E L D E Q Q R K R L E A F cttacccagaagcagaaggtgggagaactgaaggatgacgactttgagaagatcagtgag
L T Q K Q K V G E L K D D D F E K I S E ctgggggctggcaatggcggtgtggtgttcaaggtctcccacaagccttctggcctggtc
L G A G N G G V V F K V S H K P S G L V atggccagagcgctaattcatctggagatcaaacccgcaatccggaaccagatcataagg
M A R A L I H L E I K P A I R N Q I I R gagctgcaggttctgcatgagtgcaactctccgtacatcgtgggcttctatggtgcgttc
E L Q V L H E C N S P Y I V G F Y G A F tacagcgatggcgagatcagtatctgcatggagcacatggatggaggttctctggatcaa
Y S D G E I S I C M E H M D G G S L D Q gtcctgaagaaagctggaagaattcctgaacaaattttaggaaaagttagcattgctgta
V L K K A G R I P E Q I L G K V S I A V
ataaaaggcctgacatatctgagggagaagcacaagatcatgcacagagatgtcaagccc
I K G L T Y L R E K H K I M H R D V K P tccaacatcctagtcaactcccgtggggagatcaagctctgtgactttggggtcagcggg
S N I L V N S R G E I K L C D F G V S G cagctcatcgactccatggccaactccttcgtgggcacaaggtcctacatgtcgccagaa
Q L I D S M A N S F V G T R S Y M S P E agactccaggggactcattactctgtgcagtcagacatctggagcatgggactgtctctg
R L Q G T H Y S V Q S D I W S M G L S L gtagagatggcggttgggaggtatcccatccctcctccagatgccaaggagctggagctg
V E M A V G R Y P I P P P D A K E L E L atgtttgggtgccaggtggaaggagatgcggctgagaccccacccaggccaaggaccccc
M F G C Q V E G D A A E T P P R P R T P gggaggccccttagctcatacggaatggacagccgacctcccatggcaatttttgagttg
G R P L S S Y G M D S R P P M A I F E L ttggattacatagtcaacgagcctcctccaaaactgcccagtggagtgttcagtctggaa
L D Y I V N E P P P K L P S G V F S L E tttcaagattttgtgaataaatgcttaataaaaaaccccgcagagagagcagatttgaag
F Q D F V N K C L I K N P A E R A D L K caactcatggttcatgcttttatcaagagatctgatgctgaggaagtggattttgcaggt
Q L M V H A F I K R S D A E E V D F A G tggctctgctccaccatcggccttaaccagcccagcacaccaacccatgctgctggcgtc
W L C S T I G L N Q P S T P T H A A G V gtcgacctgaacgacatcttcgaagctcagaaaatcgaatggcaccgttagaattc
V D L N D I F E A Q K I E W H R - (SEQ ID NO: )
(nucleic acid SEQ ID NO: )
Example 76: Efficacy of Compounds in Combination with Standard-of-Care Chemotherapeutic agents in four human cancer cell lines.
[1055] Compounds of the invention, such as compounds of Formula IE, in combination with a standard chemotherapeutic agent, such as 5-fluorouracil, carboplatin, dacarbazine, gefϊtinib, oxaliplatin, paclitaxel, SN-38, temozolomide, or vinblastine, can be assessed for their effectiveness in killing human tumor cells. Human tumor cell lines, such as A-375 (malignant melanoma), SK-MEL- 2 (malignant melanoma, skin metastasis), COLO 205 (colorectal adenocarcinoma, ascites metastasis) or SW-620 (colorectal adenocarcinoma, lymph node metastasis) can be treated with a compound of Formula III alone, or in combination with one of the above-mentioned chemotherapeutic agents.
[1056] Tumor cells are grown as a monolayer at 370C in a humidified atmosphere (5% CO2, 95% air). Cells are grown in a suitable culture medium, e.g. RPMI 1640 (Ref BE12-702F, Cambrex, Venders, Belgium) containing 2 mM L-glutamine and supplemented with 10% fetal bovine serum (Ref DE14-801E, Cambrex). For experimental use, the tumor cells are detached from the culture flask by a 5-minute treatment with trypsin-versene (Ref 02-007E, Cambrex), diluted in Hanks' medium without calcium or magnesium (Ref BE10-543F, Cambrex). Trypsin treatment is neutralized by culture medium addition. The cells are counted in a hemocytometer and their viability assessed by 0.25% trypan blue exclusion.
The cell lines are checked for mycoplasma contamination with the Mycotect assay kit (Ref 15672-017, Invitrogen, Cergy-Pontoise, France) in accordance with the manufacturer's instructions. The mycoplasma test is assayed from the culture supernatants of the cell lines and compared to negative and positive controls.
[1057] The tumor cells (10,000 per well) are plated in 96-well flat-bottom microtitration plates (Ref 055260, Nunc, Dutscher, Brumath, France) and incubated at 37 0C for 24 hours before treatment in 100 μl of drug-free culture medium supplemented with 10% FBS. In order to assess the IC50 of each compound to be used for each cell line, the tumor cells are incubated in a 200 μl final volume of RPMI 1640 supplemented with 10% FBS and containing either a compound of Formula III or one of 5-fluorouracil, carboplatin, dacarbazine, gefϊtinib, oxaliplatin, paclitaxel, SN-38, temozolomide, or vinblastine. The compounds are tested in a suitable concentration range, such as 10"8 to 10"3 M for a compound of Formula DI, 5-fluorouracil, dacarbazine or gefϊtinib, 10"9 to 10"4 M for carboplatin, oxaliplatin, or temozolomide, 10"11 to 10"6 M for paclitaxel or SN-38, and 10'15 to 10"10 M for vinblastine. Compounds of Formula III are dissolved in DMSO and diluted with culture medium to the desired concentrations. 5-fluorouracil (50 mg/ml, Dakota Pharm, LePlessis Robinson, France), carboplatin (10 mg/ml, Aguettant, Lyon, France), and paclitaxel (6 mg/ml, Bristol-Myers Squibb SpA, Rueil Malmaison, France), are diluted with culture medium to the desired concentrations. Dacarbazine (Sigma, Saint Quentin Fallavier, France) and vinblastine (Lilly France S.A., Saint Cloud, France) are dissolved in NaCl 0.9% and diluted with culture medium to the desired concentrations. Gefitinib is dissolved in a mixed solution of RPMI 1640 and DMSO and diluted with culture medium to the desired concentrations (maximum final DMSO of 0.1% v/v). SN-38 (LKT Laboratories, Inc., St. Paul, Minnesota) is dissolved in DMSO and diluted with culture medium to the desired concentrations (maximum final DMSO of 0.1% v/v). Temozolomide (LKT Laboratories, Inc., St. Paul, Minnesota) is dissolved in water for injection and diluted with culture medium to the desired concentrations. Cells are incubated for 96 hours in the presence of test substances at 37 0C under 5% CO2. At the end of treatments, the cytotoxic activity is evaluated by an MTT assay.
[1058] For the MTT assay, at the end of the cells treatment, 20 μl of a 5 mg/ml solution 0.22 μm filtered tetrazolium reagent (MTT, Ref M2128, Sigma) in Phosphate Buffered Saline (PBS, Ref BE17-517Q, Cambrex), is added in each well. Culture plates are incubated for 2 h at 37 °C. The resulting supernatant is removed and formazan crystals dissolved with 200 μl of OMSO per well. Absorbency (OD) is measured at 570 nm in each well using VICTOR3™ 1420 multilabeled counter (Wallac, PerkinElmer, Courtaboeuf, France).
[1059] The IC50 for each compound on each cell line is determined from the OD measurements of each sample. The dose response inhibition of cell proliferation is expressed as: IC = (OD of drug exposed cells / OD of drug free wells) x 100.
The mean of multiple measurements for each concentration is plotted vs. the drug concentration. The dose-response curves are plotted using XLFit 3 (IDBS, United Kingdom). The IC50 (drug concentration to obtain 50% inhibition of cell proliferation) determination values are calculated using the XLFit 3 from semi-log curves. The IC50 value determined for each compound in each cell line is used to determine the concentration of a compound of Formula III and of the standard chemotherapeutic to be used in combination.
[1060] The cells are treated with a combination of five concentrations of a compound of Formula III and five concentrations of one of 5-fluorouracil, carboplatin, dacarbazine, gefϊtinib, oxaliplatin, paclitaxel, SN-38, temozolomide, or vinblastine, based on the IC50 results. The compounds and cells are treated per the IC50 determination described above and assayed by the MTT assay.
[1061] The results are assessed to determine whether the combination is synergistic or antagonistic. The compound interactions are calculated by multiple drug effect analysis and are performed by the median equation principle according to the methodology described by Chou and Talalay (Adv. Enzyme Regul. 1984, 22: 27-55).
[1062] The combination index (CI) will be calculated by the Chou et al. equation (Adv. Enzyme Regul. 1984, 22: 27-55 ; Encyclopaedia of human biology, Academic Press, 1991, 2: 371-9; Synergism and Antagonism in Chemotherapy, Academic Press, 1991, 61-102) which takes into account both the potency (Dm or IC50) and the shape of the dose-effect curve (the m value). The general equation for the CI of the two compounds is given by:
(D)1 (D)2 i (P)1(D)2 (Dx\ (Dx)2 (DX\(DX)2 where:
(Dx)i and (Dx)2 in the denominators are the doses (or concentrations) for compound 1 and compound 2 alone which demonstrate x% of inhibition, whereas (D) i and (D)2 in the numerators are doses of both compounds (1 and 2) in combination that also inhibit x% (iso-effective). CI<1, =1, and >1 indicate synergism, additive effect and antagonism, respectively.
[1063] The (D
x)] and (D
x)
2 can be calculated from the median-effect equation of Chou et al. (J. Natl. Cancer Inst. 1994, 86: 1517-24):
where:
Dn, is the median-effect dose that is obtained from the anti-log of x-intercept of the median-effect plot, x = log(D) versus y = log{fj(l-fa)}, or Dm = IO^^P^ and m is the slope of the median-effect plot and/, is the fraction of cells affected by the treatment.
Each CI will be calculated with CalcuSyn software (Biosoft, UK) from the mean affected fraction at each drug ratio concentration.
[1064] Additional examples. Unless specifically indicated otherwise, the Formula enumeration and R group enumeration used in the following examples is not related to such enumeration in other sections of this application. The reagents and solvents used in these examples can be readily substituted with appropriate alternatives as are known in the art and isolation of products is readily achieved by methods known in the art, including, but not limited to, extraction, crystallization, and chromatographic methods. For the following examples, the following formulae are defined:
Formula I Formula Il Formula III an(j Formula XV wherein:
R4 and R5 are as defined in paragraph [0008]; R12, R13 and R16 are as defined in paragraph [0103];
R28 is -SO2R25, -C(=O)R25, -C(=O)NR25, -C(=O)OR25, -C(=S)R25, -C(=S)NR25, -C(=S)OR25, or -SO2NR25R26; wherein R25 and R26 are hydrogen, optionally substituted lower alkyl, optionally substituted lower alkenyl, optionally substituted lower alkynyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl, or
R and R ,26 together with the nitrogen form optionally substituted 5-7 membered heterocycloalkyl or optionally substituted 5 or 7 membered nitrogen containing heteroaryl.
R29 is hydrogen or optionally substituted lower alkyl; and
R30 is optionally substituted lower alkyl or optionally substituted ben2yl, wherein optionally substituted benzyl refers to an optionally substituted aralkyl with unsubstituted structure - CH2-phenyl.
Example 77. Synthesis of compound of Formula π, where R and R are independently fluoro or chloro
III Il where R12 where R12 and R16 and R16 are are F or Cl F or Cl
Step 1— Synthesis of compound of Formula II
[1065] Compound of Formula II, where R12 and R16 are fluoro or chloro, may be synthesized by reacting a compound of Formula III with an organolithium reagent (e.g. n-butyllithium, lithium diisopropylamine) in an inert solvent (e.g. THF), followed by the addition of a formylating reagent (e.g. DMF). The reaction is allowed to proceed, typically at -78 0C, for 1-2 hours and the desired product is isolated by standard procedures (e.g. extraction, silica gel chromatography).
Example 78. Synthesis of compound of Formula HI, where R13 is NR28 R29
IV where R13 is NR28R29
Step - 1 - Synthesis of compound of Formula III where R13 is NR28 R29 [1066] Compound of Formula HI, where R13 is NR28 R29, may be synthesized by reacting a compound of Formula IV with a base (e.g. pyridine, sodium hydride) in an inert solvent (e.g. DMF, CH2Cl2), followed by an appropriate reagent (R25SO2Cl, e.g. propane- 1-sulfonyl chloride; R25C(=O)C1, e.g. acetyl chloride; R25NCO, e.g. propyl isocyanate; R25OC(=O)C1, e.g. benzyl chloroformate; R26R25NSO2Cl, e.g. dimethylsulfamoyl chloride.) The reaction is allowed to proceed, typically at room temperature, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction and silica gel chromatography).
Example 79. Synthesis of compound of Formula m, where R13 is CONR25R26:
III where R13 is C(=O)NR25R26
Step - 1 - Synthesis of compound of Formula III, where R13 is C(=O)NR25R26 [1067] Compound of Formula III, where R13 is C(=O)NR25R26, may be synthesized by reacting a compound of Formula V with a base (e.g. N,N-diisopropylethylamine) and condensing reagents (e.g. N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride and 1-hydroxybenzotriazole) in an inert solvent (e.g. tetrahydrofuran), followed by an amine (R25R26NH, e.g. 1-propanamine). The reaction is allowed to proceed, typically at room temperature, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction, silica gel chromatography).
Example 80. Synthesis of compound of Formula m, where R13 is SO2NR25R26:
Vl III where R13 is SO2NR25R26
Step - 1 - Synthesis of compound of Formula III
[1068] Compound of Formula III, where R13 is -SO2NR25R26, is synthesized by reacting a compound of Formula VI with a base (e.g. triethylamine) in an inert solvent (e.g. CH2Cl2), followed by an appropriate reagent (R25R25NH2, e.g. 1-ρropanamine). The reaction is allowed to proceed, typically at room temperature, for 2-5 hours and the desired product is isolated by standard procedures (e.g. extraction, silica gel chromatography).
Example 81. Synthesis of compound of Formula HI, where R13 is OP where P is a protecting group
VII where R13 is OP
Step - I - Synthesis of compound of Formula III, where R13 is OP and P is a protecting group [1069] Compound of Formula III, where R13 is -OP and P is a protecting group, is synthesized by reacting a compound of Formula VII with a base (e.g. imidazole) in a polar solvent (e.g. DMF), followed by an appropriate reagent (e.g. ter^butyl-chloro-dimethyl-silane) to introduce the protecting group. The reaction is allowed to proceed, typically at room temperature, for 8-12 hours, and the desired product is isolated by standard procedures (e.g. extraction, silica gel chromatography).
Example 82. Synthesis of compound of Formula VΗI
R25
Step i O R25 Cl-S-N
R26
O R26 IX VIII
Step - 1 - Synthesis of compound of Formula VIII
[1070] Compound of Formula Vm is synthesized by reacting a compound of Formula IX with a base (e.g. pyridine) in an inert solvent (e.g. CH2Cl2), followed by sulfuryl chloride. The reaction is allowed to proceed, typically under reflux, for 8-12 hours and the desired product is isolated by standard procedures (e.g. evaporation).
Example 83. Synthesis of compounds of Formula X
XIIb X = OCH3
Step 1 - Preparation of compounds of Formula XIIa and XIIb
[1071] To a compound of Formula XVIII and a compound of Formula II is added an appropriate solvent (e.g. methanol) followed by an appropriate base (e.g. potassium hydroxide, sodium methoxide). The reaction is typically allowed to stir at room temperature overnight. Isolation by convention means (e.g. extraction, washing and filtering) affords a mixture of compounds of Formula XIIa and XIIb which may be separated by silica gel chromatography if desired.
Step 2 - Preparation of compounds of Formula X
[1072] To a compound of Formula XIIa or XIIb in an appropriate solvent (e.g. acetonitrile) is added a reducing agent (e.g. trifluoroacetic acid and triethylsilane). Typically, the reaction is allowed to stir at room temperature overnight. Isolation by conventional means (e.g. extraction and silica gel column chromatography) affords compounds of Formula X.
Example 84. Synthesis of compounds of Formula I
XIIa I
Step 1 - Preparation of compounds of Formula I:
[1073] To a compound of Formula XIIa in an appropriate solvent (e.g. THF) is added an oxidizing agent (e.g.Dess-Martin periodane, TEMPO, DDQ). Typically, the reaction is allowed to stir at room temperature for 20 minutes. Isolation by conventional means (e.g. extraction and silica gel column chromatography) affords compounds of Formula I.
Example 85. Synthesis of Compounds of Formula XIII where R12 and R16 are independently chloro or fluoro
F or Cl F or Cl
Step -1- Synthesis of compound of Formula XIII, where R12 and R are chloro orfluoro [1074] Compound of Formula XHI, where R12 and R16 are chloro or fluoro, is synthesized by reacting a compound of Formula XIV with an organolithium reagent (e.g. n-butyllithium) and a temporary protecting group (e.g. l,2-Bis-(chloro-dimethyl-silanyl)-ethane) and DMF in a inert solvent (e.g.THF) under inert atmosphere (e.g. argon) at -78 0C for 2-4 hours, followed by removal of the temporary protecting group using an acid (e.g. IN HCl). The product is isolated by extraction and silica gel column chromatography.
Example 86. Synthesis of Compounds of Formula XV, where R13 is NHR28
XV where R13 is NHR28
Step 1- Synthesis of compound of Formula XVI
[1075] Compound of Formula XVI may be synthesized by reacting a compound of Formula XIII with a base (e.g. pyridine, sodium hydride) in an inert solvent (e.g. DMF, CH2Cl2), followed by an appropriate reagent (R25SO2Cl, e.g., propane-1 -sulfonyl chloride; R25C(=O)C1, e.g., acetyl chloride; R25NCO, e.g., propyl isocyanate; R25OC(=O)C1, e.g., benzyl chloroformate; R26R25NSO2Cl, e.g., dimethylsulfamoyl chloride). The reaction is allowed to proceed, typically at room temperature, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction and silica gel chromatography) .
Step 2- Synthesis of compound of Formula XVII
[1076] Compound of Formula XVII, may be synthesized by hydrolyzing a compound of Formula
XVI with an aqueous base solution (e.g. sodium hydroxide). The reaction is allowed to proceed, typically under reflux, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction).
Step 3- Synthesis of compound of Formula XV, where R13 is NHR28
[1077] Compound of Formula XV, where R13 is NHR28, may be synthesized by reacting a compound of Formula XVII with thionyl chloride. The reaction is allowed to proceed, typically under reflux, for 3 hours and the desired product is isolated by standard procedures (e.g. evaporation).
Example 87. Synthesis of Compounds of Formula XV
XIX XV
Step 1- Synthesis of compound of Formula XV
[1078] Compound of Formula XV may be synthesized by reacting a compound of Formula XIX with thionyl chloride. The reaction is allowed to proceed, typically under reflux, for 3 hours and the desired product is isolated by standard procedures (e.g. evaporation).
Example 88. Synthesis of Compounds of Formula I
Step -1- Synthesis of compound of Formula I
[1079] Compound of Formula I is synthesized by reacting a compound of Formula XVIII with a compound of Formula XV (Example 83, e.g. benzoyl chloride) in the presence of a Lewis acid (e.g. aluminum trichloride) in an inert solvent (e.g. methylene chloride) under an inert atmosphere (e.g. argon) at room temperature or with heating up to reflux for 1-18 hours. The product is isolated by extraction and silica gel column chromatography.
Example 89. Synthesis of Compounds of Formula I where R5 is aryl or heteroaryl
I I where where R5 R5 is Br is aryl or heteroaryl
Step 1: Synthesis of compound of Formula I, where R5 is aryl or heteraryl [1080] Compound of the Formula I, where R5 is aryl or heteroaryl, is prepared by reacting a compound of Formula I, where R5 is bromo, under Suzuki coupling conditions, with boronic acid (e.g. phenyl boronic acid) in the presence of a base (e.g., potassium carbonate) and catalyst (e.g., Pd(Ph3P)4) in aqueous/THF solvent system. After 4-12 hours with heating to 80 0C or heating in a microwave instrument at 120 0C for 15 minutes, the product is isolated by standard workup procedures (e.g. silica gel column chromatography).
Example 90. Synthesis of Compounds of Formula I
Step - 1 — Synthesis of compound XX
[1081] Compound of Formula XX can be synthesized by reacting a compound of Formula XVIII with hexamethyltetramine and acetic acid in water with heating to reflux for two hours. After cooling, the desired product precipitates and may be collected by filtration.
Step - 2 - Synthesis of compound of Formula XXI
[1082] Compound of Formula XXI, where P is a protecting group, is synthesized by reacting a compound XX with an appropriate reagent to introduce a protecting group (P-X, e.g. triisopropylsilylchloride) and a base (e.g. sodium hydride) in a solvent (e.g. THP) typically at room temperature for 8-12 hours. The product is isolated by conventional means (e.g. extraction).
Step - 3 — Synthesis of compound of Formula XXII
[1083] Compound of Formula XXII is synthesized by reacting a compound of Formula XXI in a solvent (e.g. THF) with an organolithium reagent (e.g. phenyl lithium) in a solvent (e.g. TEDF) under an inert atmosphere, cooled to -78 0C. An appropriate organolithium reagent can also be prepared by reacting compounds of Formula III, where R12 and R16 are independently fluoro or chloro, with an organolithium reagent (e.g. butyllithium) in a solvent (e.g. THF) under an inert atmosphere, cooled to -78 0C. The reaction is typically allowed to warm to room temperature and stirred for 30 minutes. The product is isolated by conventional means (e.g. extraction).
Step - 4 — Synthesis of an intermediate of compound of Formula I
[1084] An intermediate of compound of Formula I is synthesized by reacting a compound of Formula XXII with an appropriate reagent to remove the protecting group, P, (e.g. tetra-n-bxityl ammonium fluoride) in an appropriate solvent (e.g. THF). The final product is isolated by standard procedures (e.g. extraction).
Step - 5 — Synthesis of compound of Formula I
[1085] Compound of Formula I is synthesized by reacting the intermediate from Step 4 with an oxidizing agent (e.g. Dess-Martin periodane, TEMPO) in an aprotic solvent (e.g. THF) typically at room temperature for 20 minutes. The product is isolated by conventional means (e.g. extraction and silica gel chromatography).
Example 91. Synthesis of compounds of Formula I, where R13 is NHC(O)NR25R26.
Il NHC(=O)OR28 where R13 is NHC(=O)OR28
Step 1 - Preparation of compounds of Formula XIIa where R13 is NHC(=O)OR2S [1086] To a compound of Formula XVHI and a compound of Formula II, where R13 is NHC(=O)OR28, is added an appropriate solvent (e.g. methanol) followed by an appropriate base (e.g. potassium hydroxide). The reaction is typically allowed to stir at room temperature overnight. Isolation by convention means (e.g. extraction, washing and filtering) affords compound of Formula XIIa where R13 is NHC(=O)OR28.
Step 2 - Preparation of compounds of Formula XIIa, where R13 is NHC(=O)NR25R26: [1087] Compound of Formula XIIa, where R13 is NHC(=O)NR25R26, is synthesized by reacting a compound of Formula XIIa, where R13 is NHC(=O)OR28, with an amine of the formula NHR25R26 (e.g. propylamine) in an appropriate solvent (e.g. dioxane) followed by an appropriate base (e.g. triethylamine). The reaction is typically heated at 140 0C for 15 minutes in a CEM Discover microwave instrument. Isolation by convention means (e.g. extraction, washing and filtering) affords compounds of Formula XIIa, where R13 is NHC(=O)NR25R26.
Step 3 - Preparation of compounds of Formula I, where R13 is NHC(=O)NR2SR26: [1088] Compound of Formula I, where R13 is NHC(=O)NR25R26, is synthesized by reacting a compound of Formula XIIa, where R13 is NHC(^O)NR25R26, with an oxidizing agent (e.g. Dess- Martin periodane, TEMPO) in an appropriate solvent (e.g. THF). The reaction is typically stirred at
room temperature for 15 minutes. Isolation by convention means (e.g. extraction, washing and filtering) affords compounds of Formula I, where R13 is NHC(=O)NR25R26.
Example 92. Synthesis of compounds of Formula I, where R13 is NHC(=O)OR25.
XlIa XIIa where R13 is where R13 is
I where R13 is NHC(=O)OR25
Step 1 - Preparation of compounds of Formula XIIa, where R13 is NHC(=O)OR25 : [1089] Compound of Formula XIIa, where R13 is NHC(=O)OR25, is synthesized by reacting a compound of Formula XIIa, where R13 is NHC(=O)OR28, with an alcohol of the formula R25OH (e.g. methanol) in an appropriate solvent (e.g. dioxane) followed by an appropriate base (e.g. triethylamine). The reaction is typically heated at 140 0C for 15 minutes in a CEM Discover microwave instrument. Isolation by convention means (e.g. extraction, washing and filtering) affords compounds of Formula XIIa, where R13 is NHC(=O)OR25.
Step 2 - Preparation of compounds of Formula I, where R13 is NHC(=O)OR25: [1090] Compound of Formula I, where R13 is NHC(=O)OR25, is synthesized by reacting a compound of Formula XIIa, where R13 is NHC(=O)OR25, with an oxidizing agent (e.g. Dess-Martin periodane, TEMPO) in an appropriate solvent (e.g. THF). The reaction is typically stirred at room temperature for 15 minutes. Isolation by convention means (e.g. extraction, washing and filtering) affords compounds of Formula I, where R13 is NHC(=O)OR25.
Example 93. Synthesis of compounds of Formula I, where R4 and R5 are hydrogen.
I I where R4 is H where R4 and R5 is Br and R5 are H
Step 1 — Preparation of compounds of Formula I where R4 and R5 are hydrogen
[1091] Compound of Formula I, where R4 and R5 are hydrogen, is synthesized by hydrogenating compound of Formula I, where R4 is hydrogen and R5 is bromo, in the presence of an appropriate solvent (e.g methanol) and catalyst (e.g. 10% Pd/C), under an atmosphere of hydrogen gas. The reaction is typically allowed to stir at room temperature for 8-12 hours. Isolation by convention means (e.g. extraction, washing and filtering) affords compounds of Formula I, where R4 and R5 are hydrogen.
Synthesis of compound of Formula Ia:
[1092] Compounds of Formula Ia are compounds of Formula XVIH in which R4 is hydrogen and R5 is the only substituent on the core structure. Exemplary synthetic schemes for groups of compounds within Formula Ia are shown in Examples 91 to 99 for different selections of R5.
Example 94. Synthesis of Compounds of Formula Ia where R5 is aryl or heteroaryl
Formula Ia Where R5 is aryl or heteroaryl
[1093] Compound of Formula Ia, where R5 is aryl or heteroaryl, is synthesized from compound 1 under Suzuki reaction conditions using aryl or heteroaryl bornonic acids (e.g. phenyl bornonic acid) in the presence of a base (e.g. potassium carbonate) and a catalyst (e.g. Pd(PPh3^) in aqueous/THF
system with thermal heating (e.g. 80 0C for 12 hours) or microwave heating (e.g. 120 0C for 15 minutes). The product is isolated by conventional means (e.g. silica gel column chromatography).
Example 95. Synthesis of Compounds of Formula Ia where R5 is alkyl or cycloalkyl
Step - 1 - Synthesis of compound of Formula XXIII
[1094] Compound of Formula XXIH, where P is a protecting group, is synthesized by reacting compound 1 with a base (e.g. sodium hydride) in an inert solvent (e.g. THF), followed by an appropriate reagent (P-X, e.g., triisopropylsilylchloride) for introduction of a protecting group. The reaction is allowed to proceed, typically at room temperature, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction) (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis I, 3rd ed.; John Wiley & Sons: New York, 1981).
Step - 2 - Synthesis of an intermediate of compound of Formula Ia1 where R5 is alkyl and cycloalkyl [1095] An intermediate of compound of Formula Ia, where R5 is alkyl or cycloalkyl, is synthesized by reacting a compound of Formula XXIII with an alkyl or cycloalkyl Grignard reagent (e.g. ethyl magnesium bromide) in the presence of catalyst (e.g. [1,1 '- bis(diphenylphosphino)ferrocene]dichloropalladium(II)) in an inert solvent (e.g. toluene ) at low temperature (e.g. -78 °C) or under reflux for 2-8 hours. The product is isolated by standard procedures (e.g. extraction and silica gel column chromatography) as described by literature. (T. Hayashi, M.Konishi, Y. Kobori, M. Kumada, T. Higuchi, K. Hirotsu; J. Am .Chem. Soc. 1984, 106, 158-163).
Step - 3 - Synthesis of compound of Formula Ia, where R5 is alkyl and cycloalkyl [1096] Compound of Formula Ia, where R5 is alkyl or cycloalkyl, is synthesized by reacting an intermediate of compound Formula Ia from Step 2 with an appropriate reagent to remove the protecting group (e.g. tetrabutylammonium fluoride) in an appropriate solvent (e.g. tetrahydrofuran). The product is isolated by standard procedures (e.g. extraction and silica gel column chromatography) .
Example 96. Synthesis of Compounds of Formula Ia where R5 is NR22R23
XXIII XXIV Formula Ia
Where R5 is NR22R23
Step - 1 - Synthesis of an intermediate of compound of Formula XXIV
[1097] An intermediate of compound of Formula XXIV, is synthesized by reacting a compound of Formula XXIII, with an amine of the formula NHR
22R
23 (e.g. aniline) in a solvent (e.g. toluene), in the presence of a base (e.g. sodium tert-butoxide) and a catalyst composed of a metal (e.g., Tris(dibenzylideneacetone)dipalladium(0)) and a ligand (e.g.,
with heating, typically to 95
0C, for 8-12 hours as described (Thomas, et. al., J. Am. Chem. Soc, 2001, 123, 9404) by substituting a compound of Formula XXIII for the N-substituted-3,6-dibromocarbazole. The desired compound is purified by silica gel column chromatography. This intermediate is used directly in Step 3 to provide compound of Formula Ia where R
5 is NR
22R
23 and R
22 and R
23 are not -C(X)R
20, -C(X)NR
17R
18, -S(O)
2R
21, or S(O)
2NR
17R
18 or alternatively, it can be additionally substituted as described in Step 2.
Step - 2 - Synthesis of compound of Formula XXIV
[1098] The intermediate from Step 1 can be further modified when R22or R23 is hydrogen. In this case, the intermediate from Step 1 can be reacted with a base (e.g. sodium hydride) in a solvent (e.g. N,N-dimethylformamide), followed by reaction with an alkylating reagent (e.g. benzyl bromide) or an acylating reagent (e.g. benzoyl chloride, phenyl isocyanate, phenyl isothiocyanate, phenylsulfonyl chloride) typically at room temperature or with heating up to 80 0C for 1-12 hours. The desired product can be purified by conventional means (e.g. silica gel column chromatography). Alternatively, when R22 or R23 is a suitable protecting group (e.g. benzyl), it may be removed by appropriate treatment (e.g. hydrogenation) to provide a compound where R22 and/or R23 are hydrogen, which is suitable for further modification with an alkylating reagent or acylating reagent as described herein.
Step - 3 - Synthesis of compound of Formula Ia, where R5 is - NR22R23
[1099] Compound of Formula Ia, where R5 is -NR22R23, is synthesized by reacting a compound of Formula XXTV with an appropriate reagent to remove the protecting group (e.g. tetra-n- butylammonium fluoride) in an appropriate solvent (e.g. methanol). The final product can be isolated by standard procedures (e.g. extraction).
Example 97. Synthesis of Compounds of Formula Ia where R5 is C(O)NR25R ,26
XXIII XXVI XXVII
XXViIi Formula Ia
Where R5 is CONR25R26
Step -1 - Synthesis of compound of Formula XXIII
[1100] Compound of Formula XIII, where P is a protecting group, is synthesized by reacting compound 1 with a base (e.g. sodium hydride) in a solvent (e.g. THF), followed by an appropriate reagent (P-X, e.g. triisopropylsilylchloride) for introduction of a protecting group. The reaction is allowed to proceed, typically at room temperature for 8-12 hours, and the desired product is isolated by standard procedures (e.g. extraction and silica gel column chromatography) (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis I, 3rd ed.; John Wiley & Sons: New York, 1981).
Step -2- Synthesis of compound of Formula XXVI
[1101] Compound of Formula XXVI may be synthesized by reacting a compound of Formula XXIII with sodium cyanide in a polar aprotic solvent (e.g. DMF) in an inert atmosphere (e.g. argon), in the presence of a catalyst (e.g. cuprous iodide or Tris(dibenzylideneacetone)dipalladium(0)) following the procedure described by Buchwald et. al., J. Am. Chem. Soc, 2003, 125, 2890-2891, by substituting 5-bromo-7-azaindole for 5-bromo-indole.
Step -3 - Synthesis of compound of Formula XXVII
[1102] Compound of Formula XXVII may be synthesized by heating a compound of Formula XXVI with aqueous base (e.g. aq. KOH) in the presence of an alcohol (e.g. ethanol) at higher temperatures (e.g. 90 °C) for required time, typically 24 h, as described in Org. Syn. Collective Volume 2, 292 (1943). Alternatively, compounds of Formula XXVII may be synthesized directly from a compound of Formula XXIII by reacting a compound of Formula XXIII with a strong base (e.g. n-butyllithium) and benzyl chloroformate in an inert solvent (e.g. THF), and further debenzylation by hydrogenating the obtained benzyl ester with hydrogen, in the presence of a catalyst (e.g. 20% Pd(OH)2/C) at room temperature. The product may be isolated by filtration and evaporation.
Step -4 - Synthesis of compound of Formula XXVIII
[1103] Compound of Formula XXVIII may be synthesized by reacting a compound of Formula
XXVII with an amine (e.g. benzylamine) in a polar aprotic solvent (e.g. DMF) in an inert atmosphere, in presence of an activating agent (e.g. PyBroP (Bromotri(pyrrolidino)phosphonium hexafluorophosphate) following the procedure described by Coste et. al., J. Org. Chem., 1994, 59,
2437.
Step -5 - Synthesis of compound of Formula Ia
[1104] Compound of Formula Ia, where R5 is C(O)NR25R26, may be synthesized by cleaving the protective group (e.g. TIPS) of a compound of Formula XXVDQ with appropriate reagents (e.g. TBAF) and isolating the product (e.g. extraction and silica gel column chromatography).
Example 98. Synthesis of Compounds of Formula Ia where R5 is CH2NHR25R26
1 XXlIl
Where R5 is CH2NR25R26
Step -1 — Synthesis of compound of Formula XXIII
[1105] Compound of Formula XXIII, where P is a protecting group, is synthesized by reacting compound 1 with a base (e.g. sodium hydride) in a solvent (e.g. THF), followed by an appropriate reagent (P-X, e.g. triisopropylsilylchloride) for introduction of a protecting group. The reaction is allowed to proceed, typically at room temperature, for 8-12 hours and the desired product is isolated by standard procedures (e.g. extraction and silica gel column chromatography) (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis I, 3rd ed.; John Wiley & Sons: New York, 1981).
Step -2- Synthesis of compound of Formula XXVI
[1106] Compound of Formula XXVI may be synthesized by reacting a compound of Formula XXπi with sodium cyanide in a polar aprotic solvent (e.g. DMF) in an inert atmosphere, in presence of a catalyst (e.g. Tris(dibenzylideneacetone)dipalladium(0) or cuprous iodide) following the procedure described by Buchwald et. al., J Am. Chem. Soc, 2003, 125, 2890-2891, by substituting 5- bromo-7-azaindole for 5-bromo-indole.
Step -3 - Synthesis of compound of Formula XXIX
[1107] Compound of Formula XXEX can be synthesized from a compound of Formula XXVI under hydrogenation conditions using a catalyst (e.g. PtO2) in an atmosphere of H2 as described by Secrist HI et. al., J Org. Chem., 1972, 37, 335-336.
Step -4 — Synthesis of compound of Formula XXX
[1108] Compound of Formula XXX can be synthesized from a compound of Formula XXIX with an electrophilic reagent (e.g. benzyl bromide, benzenesulfonyl chloride, benzoyl chloride, phenyl isocyanate, phenyl isothiocyanate) in a polar aprotic solvent (e.g. DMF) in an inert atmosphere, in the presence of abase (e.g. K2CO3, Et3N). The product can be isolated by standard methods (e.g. aqueous workup and silica gel column chromatography).
Step -5 — Synthesis of compound of Formula Ia
[1109] Compound of Formula Ia, where R5 is CH2NHR25R26, can be synthesized from a compound of Formula XXX with an electrophilic reagent (e.g. benzyl bromide, benzenesulfonyl chloride, benzoyl chloride, phenyl isocyanate, phenyl isothiocyanate) in a polar aprotic solvent (e.g. DMF) in an inert atmosphere, in the presence of a base (e.g. K2CO3, Et3N), followed by deprotection of the protecting group with appropriate conditions (e.g. tetra-n-butylammonium fluoride) and purification by conventional meand (e.g. silica gel chromatography).
Example 99. Synthesis of Compounds of Formula Ia where R5 is OR25
1 Formula Ia
Where R5 is OR25
Step - 1 - Synthesis of compound of Formula Ia, where Rs is OR25
[1110] A compound of Formula Ia, where R5 is OR25, is synthesized by reacting compound 1 with a reagent of formula R25OH (e.g. methanol) in the presence of base (e.g. sodium methoxide) and copper (I) bromide in a solvent (e.g. N,N-dimethylformamide) typically with heating to reflux for 2-8 hours as described by Mazeas, et. al. in Heterocycles, 1999, 50:1065. The desired intermediate is purified by conventional means (e.g. silica gel column chromatography).
Example 100. Synthesis of Compounds of Formula Ia where R5 is SR25
1 Formula Ia
Where R5 is SR25
[1111] Compound of Formula Ia, where R5 is SR25, can be prepared by reacting compound 1 with a strong base (e.g. potassium hydride or ^-butyl lithium) and dialkyldisulfides (e.g. dimethyldisulfane) or thiophenols (e.g. 4-methoxythiophenol) in a polar aprotic solvent (e.g. N,N-dimethylformamide) in an inert atmosphere following the procedure described by Yang et. al., Heterocycles, 1992, 34, 1169, by substituting 5-bromo-7-azaindole for 5-bromo-indole.
Example 101. Synthesis of Compounds of Formula Ia where R5 is S(O)R25 or S(O)2R »'25
Formula Ia Formula Ia Formula Ia Where R5 is SR25 Where R5 is S(O)R25 Where R5 is S(O)2R25
[1112] Compounds of Formula Ia, where R5 is S(O)R25, or S(O)2R25 can be prepared by reacting compound of Formula Ia, where R5 is SR25, with 1 or 2 equivaluents of oxidizing agent (e.g. Oxone), respectively, in a polar solvent (e.g. DMF), using standard procedures.
Example 102. Synthesis of compound of Formula Ic, where Y3 is C(O) or CH2; Y6 is NH; and R27 is as defined in paragraph [0020]
Formula XXXI Formula Ic
Step - 1 Synthesis of compound of Formula XXXI
[1113] Compound of Formula XXXI in turn can be prepared by reacting 6-bromo-7-azaindole (Minakata, S., etal, Synthesis, 1992, p661-663) with an appropriate nucleophile (e.g. acid chlorides, alkyl halides, and the like) in polar aprotic solvents (e.g. THF, DMF) in the presence of a Lewis acid (e.g. AlCl3, InCl3) etc.). The product may be isolated by following standard procedures (work up and silica gel column chromatography).
Step — 2 Synthesis of compound of Formula Ic
[1114] Compound of Formula Ic can be prepared by reacting compound of Formula XXXI with R27NH2 under Buckwald reaction conditions (Palladium tetrakis trialkylphosphine and a base). The product may be isolated by following standard procedures (work up and silica gel column chromatography) .
Example 103. Synthesis of compound of Formula Id, where Y3 is C(O) or CH2 ; Y2 is CH2; and
R27 is as defined in paragraph [0024]
Step — 1 Synthesis of compound of Formula XXXII
[1115] Compound of Formula XXXII can be prepared from 2-methyl-7-azaindole (Clemo, S;
J. Chem. Soc. , 1945, p 603-607) by reacting with N-bromosuccinimide in carbon tetrachloride followed by the reaction of the di bromo compound with a nucleophile (e.g. amine, alkoxy, phenoxy, etc). The product may be isolated by following standard procedures (work up and silica gel column chromatography) .
Step — 2 Synthesis of compound of Formula Id
[1116] Compound of Formula XXXII can be reacted with a lithium reagent (e.g. butyl lithium) followed by the reaction with an aldehyde to provide the key intermediate to compound of Formula Id. This intermediate can be reacted with a reducing agent (e.g. trifluoroacetic acid and triethylsilane) at room temperature overnight to provide a compound of Formula Id where Y3 is CH2 as described in Example 79 step-2, or with an oxidizing agent (e.g.Dess-Martin periodane, TEMPO) at room temperature for 20 minutes to provide a compound of Formula Id where Y3 is C(O) as described in Example 80.
Example 104. Synthesis of compound of Formula Ie, where Y4 is NH ; Y2 is CH2; and R27 is as defined in paragraph [0027]
Step - 1 Synthesis of compound of Formula XXXIII
[1117] Compound of Formula XXXIII can be prepared by reacting 4-chloro-l-phenylsulfonyl-7- azaindole (Mendiola, J., etal.; J.Org. Chem., 2004, p4974-4983) with lithium reagent (e.g. LDA) at low temperature (e.g. -40 0C) followed by the addition of an alkyl halide (e.g. benzyl bromide). The product may be isolated by following standard procedures (work up and silica gel column chromatography) .
Step — 2 Synthesis of compound of Formula Ie
[1118] Compound of Formula Ie can be prepared by reacting a compound of Formula XXXIII with an amine of formula R27NH2 under Buckwald reaction conditions followed by deprotection and isolating the product by following standard procedures (work up and silica gel column chromatography) .
Example 105. Synthesis of compound of Formula If, where Y5 is NH ; Y2 is CH2; R27 is as defined in paragraph [0030]
Formula XXXV Formula XXXIV Formula If
Step - 1 Synthesis of compound of Formula XXXIV
[1119] Compound of Formula XXXIV can be prepared by reacting a compound of Formula XXXV with a strong base (e.g. KH, potassium tert-butoxide) in a suitable solvent (e.g. NMP) at ambient temperature (Koradin, C, et.al.; Tetrahedron, 2003, pl571-1588). The product may be isolated by following standard procedures (work up and silica gel column chromatography).
Step — 2 Synthesis of compound of Formula If
[1120] Compound of Formula If can be prepared by reacting a compound of Formula XXXIV with an amine of formula R27NH2 under Buckwald reaction conditions followed by deprotection and isolating the product by following standard procedures (work up and silica gel column chromatography) .
Example 106. Synthesis of compound of Formula Ij, where Y4 is NH ; Y5 is O; and R27 is as defined in paragraph [0042]
Formula XXXVI Formula Ij
Step — 1 Synthesis of compound of Formula XXXVI
[1121] Compound of Formula XXXVI can be synthesized by reacting N-triisopopylsilyl-4-chloro- 5-hydroxy-7-azaindole (L'Heureux, A., Thibault, C, and Ruel, R.; Tetrahedron Letters, 2004, p2317- 2319) with an alcohol (e.g. benzyl alcohol) under Mitsunobu reaction conditions and isolating the product by following standard procedures (work up and silica gel column chromatography).
Step - 2 Synthesis of compound of Formula Ij
[1122] Compound of Formula Ij can be prepared by reacting a compound of Formula XXXVI with an amine of formula R27NH2 under Buckwald reaction conditions followed by deprotection and isolating the product by following standard procedures (work up and silica gel column chromatography).
[1123] Additional examples of certain methods contemplated by the present invention may be found in the following applications: U.S. Prov. App. No. 60/580,898, filed June 17, 2004; U.S. Prov. App. No. 60/682,076, filed May 17, 2005; U.S. Prov. App. No. 60/682,058, filed May 17, 2005; U.S. Prov. App. No. 60/682,063, filed May 17, 2005; U.S. Prov. App. No. 60/682,051, filed May 17, 2005; U.S. Prov. App. No. 60/682,042, filed May 17, 2005; U.S. Prov. App. No. 60/692,750, filed June 22, 2005; and U.S. Prov. App. No. 60/692,960, filed June 22, 2005; each of which are hereby incorporated by reference herein in their entireties including all specifications, figures, and tables, and for all purposes.
[1124] All patents and other references cited in the specification are indicative of the level of skill of those skilled in the art to which the invention pertains, and are incorporated by reference in their entireties, including any tables and figures, to the same extent as if each reference had been incorporated by reference in its entirety individually.
[1125] One skilled in the art would readily appreciate that the present invention is well adapted to obtain the ends and advantages mentioned, as well as those inherent therein. The methods, variances, and compositions described herein as presently representative of preferred embodiments are
exemplary and are not intended as limitations on the scope of the invention. Changes therein and other uses will occur to those skilled in the art, which are encompassed within the spirit of the invention, are defined by the scope of the claims.
[1126] It will be readily apparent to one skilled in the art that varying substitutions and modifications may be made to the invention disclosed herein without departing from the scope and spirit of the invention. For example, variations can be made to crystallization or co-crystallization conditions for Ret and Ret surrogate proteins and/or various kinase domain sequences can be used. Thus, such additional embodiments are within the scope of the present invention and the following claims.
[1127] The invention illustratively described herein suitably may be practiced in the absence of any element or elements, limitation or limitations which is not specifically disclosed herein. Thus, for example, in each instance herein any of the terms "comprising", "consisting essentially of and "consisting of may be replaced with either of the other two terms. The terms and expressions which have been employed are used as terms of description and not of limitation, and there is no intention that in the use of such terms and expressions of excluding any equivalents of the features shown and described or portions thereof, but it is recognized that various modifications are possible within the scope of the invention claimed. Thus, it should be understood that although the present invention has been specifically disclosed by preferred embodiments and optional features, modification and variation of the concepts herein disclosed may be resorted to by those skilled in the art, and that such modifications and variations are considered to be within the scope of this invention as defined by the appended claims.
[1128] In addition, where features or aspects of the invention are described in terms of Markush groups or other grouping of alternatives, those skilled in the art will recognize that the invention is also thereby described in terms of any individual member or subgroup of members of the Markush group or other group.
[1129] Also, unless indicated to the contrary, where various numerical values are provided for embodiments, additional embodiments are described by taking any 2 different values as the endpoints of a range. Such ranges are also within the scope of the described invention.
[1130] Thus, additional embodiments are within the scope of the invention and within the following claims.