WO2007002238A2 - Drug/polymer composite materials and methods of making the same - Google Patents
Drug/polymer composite materials and methods of making the same Download PDFInfo
- Publication number
- WO2007002238A2 WO2007002238A2 PCT/US2006/024221 US2006024221W WO2007002238A2 WO 2007002238 A2 WO2007002238 A2 WO 2007002238A2 US 2006024221 W US2006024221 W US 2006024221W WO 2007002238 A2 WO2007002238 A2 WO 2007002238A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- drug
- composite
- derivatives
- particles
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
- A61K9/1694—Processes resulting in granules or microspheres of the matrix type containing more than 5% of excipient
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/54—Improvements relating to the production of bulk chemicals using solvents, e.g. supercritical solvents or ionic liquids
Definitions
- the present invention concerns methods of making drug/polymer composite materials, the materials so made, and shaped articles fo ⁇ ned from such drug/polymer composite materials.
- Drug/polymer composite materials are traditionally formed either by solvent-based processing where a solvent or combination of solvents is used to facilitate intimate mixing of the drug with polymer(s) by a combination of reducing the polymer viscosity and by dispersing/dissolving the drug into a fluid-like phase or by processing a mixture of drug(s) - polymer(s) at an elevated temperature sufficient to cause flow of the polymer into a desired shape.
- the solvents commonly utilized include all common organic solvents, halogenated solvents and aqueous solvent compositions.
- Solvent-based processing can adversely affect the drug by reacting, bonding or binding with the chemical functionality of many drugs.
- Solvent-based processing can also adversely affect the secondary structure of sophisticated therapeutics such as proteins, enzymes, hormones, which changes the drug's efficacy and may denature the drug compound rendering it useless or toxic or change its effective shelf-life
- Solvent-based processing can also adversely affect the polymorph of the drug; changing crystalline structure or providing amorphous materials that have different bioavailability profiles and adversely affecting shelf-life.
- I ' OQMii ⁇ llilt.e ⁇ lItWi;,tSitS,pna process uses elevated tempera ures to provide a lower viscosity polymer(s) for mixing with the drug.
- U.S. Patent 6,190,699 (Luzzi) describes compositions of protein and peptide infused polymer particles and methods for production using densified solvents including supercritical fluids. Luzzi claims that the proteins and peptides are partially adsorbed into (infused) the polymer particles. Since proteins and peptides are not soluble in supercritical carbon dioxide, it can be reasonable assumed that dense carbon dioxide is not a suitable densified solvent to practice this art as sorption would be disfavored due to a lack of solubility of the protein in the densified solvent. Additionally, Luzzi discloses methods for making particles and does not address shaped or formed articles or semi-porous or porous articles.
- a first aspect of the invention provides composite comprising at least one polymer; at least one pharmaceutical agent in a therapeutically desirable morphology; and optionally at least one excipient; wherein the pharmaceutical agent is sequestered in interstices formed by fusing particles of the polymer.
- the pharmaceutical agent is a drug or a biological agent having a secondary, tertiary and/or quaternary structure required for biological activity, wherein the structure is substantially retained when the particles of the biological agent are sequestered within interstices formed by the fused polymer particles.
- the pharmaceutical agent is a peptide, protein, enzyme, nucleic acid, antibody, therapeutic vaccine, antisense nucleic acid, antimicrobial, vitamin, hormone, steroid, lipid, polysaccharide or carbohydrate.
- the pharmaceutical agent may be a therapeutic protein, such as erythropoietin, interferon, insulin, blood factor, colony stimulating factor, growth hormone, interleuldn and growth factor.
- the pharmaceutical agent is a drug in substantially crystalline form.
- the composite provides a rate of elution that remains within a 15% variation range from a selected rate of elution between day 2 and day 30 after the composite is implanted in a subject under physiological conditions.
- the composite provides an elution profile wherein about 10% to about 50% of the pharmaceutical agent is eluted at week 1 after the composite is implanted in a subject under physiological conditions, about 25% to about 75% of the pharmaceutical agent is eluted at week 2 and about 50% to about 75% of the pharmaceutical agent is eluted at week 4. .
- the polymer and the pharmaceutical agent form two substantially separate phases.
- Another aspect of the invention provides a method of preparing a composite comprising [0014] (a) forming a mixture by combining particles of at least one polymer with particles of at least one pharmaceutical agent in a therapeutically desirable morphology, and optionally with particles of at least one excipient; and (b) plasticizing said mixture under conditions that do not substantially modify the morphology of said pharmaceutical agent wherein the pharmaceutical agent is sequestered in interstices formed by fused particles of the polymer.
- the method further comprises forming a shaped article from said composite.
- the shaped article is formed by placing the mixture of step (a) in a mold prior to performing said plasticizing of step (b).
- the shaped article is a biomedical implant., for example, a drug depot or a stent.
- present invention is a metho o orm ng a drug/polymer compos te material by combining a drug material with a polymer material under pressure in the presence of a densified gas solvent to form the drug/polymer composite material.
- a further aspect of the present invention is a drug/polymer composite material (in some embodiments a "medicament” herein), which may be produced by a process as described above.
- a further aspect of the present invention is a shaped article (in some embodiments also referred to as a "medicament” herein) comprising, consisting of or consisting essentially of a drug/polymer composite material as described above.
- a further aspect of the present invention is a method of treating a subject with a drug, comprising administering a drug/polymer composite material as described herein to said subject in an amount effective to treat said subject with said drug.
- a further aspect of the present invention is the use of a drug for the preparation of a medicament for carrying out a method of treatment as described herein.
- FIGURE 1 shows supercritical CO 2 molding process and formation of an implant according to the invention.
- FIGURE 2 shows supercritical CO 2 molding process "plasticization" leading to particle fusion according to the invention.
- FIGURE 3 shows final implant, cross section SEM according to the invention.
- FIGURE 4 shows In-vitro esculetin elution from PGLA implant according to the invention.
- FIGURE 5 shows In-vitro esculetin mass elution from PGLA implant according to the invention.
- Subjects that may be treated by the present invention include both human subjects for medical purposes and animal subjects for veterinary and drug screening and development purposes.
- Other suitable animal subjects are, in general, mammalian subjects such as primates, bovines, ovines, caprines, porcines, equines, felines, canines, rodents (e.g., rats and mice), etc.
- Human subjects are the most preferred. Human subjects include fetal, neonatal, infant, juvenile and adult subjects.
- Shaped articles as used herein include, but are not limited to, pills, tablets, drug depots or drug delivery devices ⁇ e.g., subcutaneous implants), biomedical implants, etc.
- Biomedical implant includes but is not limited to drug depots and drug delivery devices, stents (e.g., vascular stents), electrodes, catheters, leads, implantable pacemaker or cardioverter housings, joints, screws, rods, ophthalmic implants (including, but not limited to, intraocular lens implants, glaucoma implants or drainage implants, and punctal implants or plugs), etc.
- the implants may be of any suitable material, including but not limited to organic polymers (including stable or inert polymers and polymers), metals such as stainless steel and titanium, inorganic materials such as silicon, and composites thereof.
- drug depot or “drug delivery device” include those be configured for any route of administration, including those that may be implanted (luminal, venous, subcutaneous, muscular, ocular), inserted (oral, rectal, vaginal, ocular) or topically applied (transdermal, transmucual, sublingual).
- Treatment refers to any type of treatment or prevention that imparts a benefit to a subject afflicted with a disease or at risk of developing the disease, including improvement in the condition of the subject (e.g., in one or more symptoms), delay in the progression of the disease, delay the onset of symptoms or slow the progression of symptoms, etc.
- treatment also includes prophylactic treatment of the subject to prevent the onset of symptoms.
- treatment and “prevention” are not necessarily meant to imply cure or complete abolition of symptoms.” to any type of treatment that imparts a benefit to a patient afflicted with a disease, including improvement in the condition of the patient (e.g., in one or more symptoms), delay in the progression of the disease, etc.
- IPO ⁇ BIIIiitndqiSii ⁇ aifcJljient 11 as used herein includes refers to any pharmaceutically acceptable material that is included in a drug composition to enhance the pharmaceutical (including manufacturing and shelf-stability) and/or pharmacological properties thereof.
- compositions include, but are not limited to, adjuvants, surfactants, stabilizers, morphology modifiers, porogens, diluents, carriers, solubilizers, antioxidants, lubricants (or glidants), binders, disintigrants, and mixtures thereof. Definitions
- Substrate refers to any surface upon which it is desirable to deposit a coating comprising a polymer and a pharmaceutical or biological agent, wherein the coating process does not substantially modify the morphology of the pharmaceutical agent or the activity of the biological agent.
- Biomedical implants are of particular interest for the present invention; however the present invention is not intended to be restricted to this class of substrates.
- substrates that could benefit from the coating process described herein, such as pharmaceutical tablet cores, as part of an assay apparatus or as components in a diagnostic kit (e.g. a test strip).
- the implants may be formed from any suitable material, including but not limited to organic polymers (including stable or inert polymers and biodegradable polymers), metals, inorganic materials such as silicon, and composites thereof, including layered structures with a core of one material and one or more coatings of a different material.
- organic polymers including stable or inert polymers and biodegradable polymers
- metals including stable or inert polymers and biodegradable polymers
- inorganic materials such as silicon
- composites thereof including layered structures with a core of one material and one or more coatings of a different material.
- the biomedical implant is an expandable intraluminal vascular graft or stent (e.g., comprising a wire mesh tube) that can be expanded within a blood vessel by an angioplasty balloon associated with a catheter to dilate and expand the lumen of a blood vessel, such as described in US Patent No. 4,733,665 to Palmaz Shaz.
- "Pharmaceutical agent” as used herein refers to any of a variety of drugs or pharmaceutical compounds that can be used as active agents to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e.
- the pharmaceutical agents of the invention may also comprise two or more drugs or pharmaceutical compounds.
- Pharmaceutical agents include but are not limited to antirestenotic agents, antidiabetics, analgesics, antiinflammatory agents, antirheumatics, antihypotensive agents, antihypertensive agents, psychoactive drugs, ,tfan(
- Suitable active ingredients are acarbose, antigens, beta-receptor blockers, non-steroidal antiinflammatory drugs (NSAIDs], cardiac glycosides, acetylsalicylic acid, virustatics, aclarubicin, acyclovir, cisplatin, actinomycin, alpha- and beta- sympatornimetics, (dmeprazole, allopurinol, alprostadil, prostaglandins, amantadine, ambroxol, amlodipine, methotrexate, S-aminosalicylic acid, amitriptyline, amoxicillin, anastrozole, atenolol, azathioprine, balsalazide, beclomethasone, betahistine, bezaf ⁇ brate, bicalutamide, diazepam and diazepam derivatives, budesonide, bufexamac, buprenorphine, methadone, calcium
- ketoconazole ketoprofen, ketotifen, lacidipine, lansoprazole, levodopa, levomethadone, thyroid hormones, lipoic acid and lipoic acid derivatives, lisinopril, lisuride, lofepramine, lomustine, loperamide, loratadine, maprotiline, mebendazole, mebeverine, meclozine, mefenamic acid, mefloquine, meloxicam, mepindolol, meprobamate, meropenem, mesalazine, mesuximide, metamizole, metformin, methotrexate, methylphenidate, methylprednisolone, metixene, metoclopramide, metoprolol, metronidazole, mianserin, miconazole, minocycline, minoxidil, misoprostol, mitomycin, mizola
- the active ingredients may, if desired, also be used in the form of their pharmaceutically acceptable salts or derivatives (meaning salts which retain the biological effectiveness and properties of the compounds of this invention and which are not biologically or otherwise undesirable), and in the case of chiral active ingredients it is possible to employ both optically active isomers and racemates or mixtures of diastereoisomers.
- active biological agent refers to a substance, originally produced by living organisms, that can be used to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e. arresting the development of clinical symptoms; and/or relieving the disease, i.e. causing the regression of clinical symptoms). It is possible that the active biological agents of the invention may also comprise two or more active biological agents or an active biological agent combined with a pharmaceutical agent, a stabilizing agent or chemical or biological entity.
- the active biological agent may have been originally produced by living organisms, those of the present invention may also have been synthetically prepared, or by methods combining biological isolation and synthetic modification.
- a nucleic acid could be isolated form from a biological source, or prepared by traditional techniques, known to those skilled in the art of nucleic acid synthesis.
- the nucleic acid may be further modified to contain non-naturally occurring moieties.
- Non-limiting examples of active biological agents include peptides, proteins, enzymes, glycoproteins, nucleic acids (including deoxyribonucleotide or ribonucleotide polymers in either single or double stranded form, and unless otherwise limited, encompasses known analogues of natural nucleotides that hybridize to nucleic acids in a manner similar to naturally occurring nucleotides), antisense nucleic acids, fatty acids, antimicrobials, vitamins, hormones, steroids, lipids, polysaccharides, carbohydrates and the like.
- antirestenotic agents antidiabetics
- analgesics antiinflammatory agents, antirheumatics, antihypotensive agents, antihypertensive agents, psychoactive drugs, tranquillizers, antiemetics, muscle relaxants, glucocorticoids, agents for treating ulcerative colitis or Crohn's disease, antiallergics, antibiotics, antiepileptics, anticoagulants, antimycotics, antitussives, arteriosclerosis remedies, diuretics, proteins, peptides, enzymes, enzyme inhibitors, gout remedies, hormones and inhibitors thereof, cardiac glycosides, immunotherapeutic agents aMifiifal ⁇ Mssj-'laii ⁇ lvieSjiillpld-lowering agents, migraine remedies, mineral products, otologicals, anti parkinson agents, thyroid therapeutic agents, spasmolytics, platelet aggregation inhibitors, vitamins, cytostatics and metastasis inhibitors, phytopharmaceutic
- Activity refers to the ability of a pharmaceutical or active biological agent to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e. arresting the development of clinical symptoms; and/or relieving the disease, i.e. causing the regression of clinical symptoms).
- a pharmaceutical or active biological agent should be of therapeutic or prophylactic value.
- the active biological agents of the present invention will typically possess some degree of secondary, tertiary and/or quaternary structure, upon which the activity of the agent depends.
- proteins possess secondary, tertiary and quaternary structure.
- Secondary structure refers to the spatial arrangement of amino acid residues that are near one another in the linear sequence.
- the ⁇ -helix and the /3-strand are elements of secondary structure.
- Tertiary structure refers to the spatial arrangement of amino acid residues that are far apart in the linear sequence and to the pattern of disulfide bonds.
- Proteins containing more than one polypeptide chain exhibit an additional level of structural organization. Each polypeptide chain in such a protein is called a subunit.
- Quaternary structure refers to the spatial arrangement of subunits and the nature of their contacts.
- hemoglobin consists of two a. and two ⁇ chains.
- protein function arises from its conformation or three dimensional arrangement of atoms (a stretched out polypeptide chain is devoid of activity).
- one aspect of the present invention is to manipulate active biological agents, while being careful to maintain their conformation, so as not to lose their therapeutic activity.
- Polymer refers to a series of repeating monomelic units that have been cross-linked or polymerized. Any suitable polymer can be used to carry out the present invention. It is possible that the polymers of the invention may also comprise two, three, four or more different polymers. In some embodiments, of the invention only one polymer is used. In some preferred embodiments a combination of two polymers are used. Combinations of polymers can be in varying ratios, to provide the polymer part of the composite materials of the invention including plasticized polymers that form a matrix with interstices wherein the drugs are sequestered. In addition to their role in forming the matrices of the invention, the polymers can be used as coating for composite materials of the invention.
- Polymer refers to organic polymers, and includes copolymers of a named polymer with other constituents.
- the polymer is preferably an absorbable and/or resorbable polymer.
- the polymer is preferably non-resorbable and biocompatible.
- ploymers that may be used in the present invention include, but are not limited to polycarboxylic acids, cellulosic polymers, proteins, polypeptides, polyvinylpyrrolidone, maleic anhydride polymers, polyamides, polyvinyl alcohols, polyethylene oxides, glycosaminoglycans, polysaccharides, polyesters, polyurethanes, polystyrenes, copolymers, silicones, polyorthoesters, polyanhydrides, copolymers of vinyl monomers, polycarbonates, polyethylenes, polypropylenes, polylactic acids, polyglycolic acids, polycaprolactones, polyhydroxybutyrate valerates, polyacrylamides, polyethers, polyurethane dispersions, polyacrylates, acrylic latex dispersions, polyacrylic acid, mixtures and copolymers thereof.
- the polymers of the present invention may be natural or synthetic in origin, including gelatin, chitosan, dextrin, cyclo
- Poly(urethanes), Poly(siloxanes) or silicones Poly(acrylates) such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate), Poly(vinyl alcohol) Poly(olefms) such as poly(ethylene), poly(isoprene), halogenated polymers such as Poly(tetrafluoroethylene) - and derivatives and copolymers such as those commonly sold as Teflon® products, Poly(vinylidine fluoride), Polyvinyl acetate), Polyvinyl pyrrolidone),.
- Poly(acrylates) such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate)
- Poly(vinyl alcohol) Poly(olefms) such as poly(ethylene), poly(isoprene), halogenated polymers such as Poly(tetrafluoroethylene) - and derivatives
- Suitable polymers also include absorbable and/or resorbable polymers including the following, combinations, copolymers and derivatives of the following: Polylactides (PLA), Polyglycolides (PGA), Poly ⁇ actide-co-glycolides) (PLGA), Polyanhydrides, Polyorthoesters, Poly(N-(2-hydroxypropyl) methacrylamide), PoIy(I- aspartamide), etc.
- Any suitable polymer can preferably be used to carry out the present invention, including but not limited to: natural and synthetic polymers, gelatin, chitosan, dextrin, cyclodextrin, Poly(urethanes), Poly(siloxanes) or silicones , Poly(acrylates) such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate), Poly(vinyl alcohol) Poly(olefinds) such as poly(ethylene), poly(isoprene), halogenated polymers such as Poly(tetrafluoroethylene) - and derivatives and copolymers such as those commonly sold as Teflon® products, Poly(vinylidine fluoride), Poly(vinyl acetate), Polyvinyl pyrrolidone),.
- natural and synthetic polymers such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate)
- Suitable polymers also include absorbable ⁇ ' ' '" ""” • ""”' "- ⁇ - "” • •" ⁇ " • **-" - 1 ⁇ 1 - -; the following, combinations, copolymers and e ⁇ vau of the following: Polylactides (PLA), Polyglycolides (PGA), Poly(lactide-co-glycolides) (PLGA), Polyanhydrides, Polyorthoesters, Poly(N-(2-hydroxypropyl) methacrylamide), PoIy(I- aspartamide), etc.
- “Therapeutically desirable morphology” refers to the gross form and structure of the pharmaceutical agent, once deposited on the substrate, so as to provide for optimal conditions of ex vivo storage, in vivo preservation and/or in vivo release. Such optimal conditions may include, but are not limited to increased shelf life, increased in vivo stability, good biocompatibility, good bioavailability or modified release rates.
- the desired morphology of a pharmaceutical agent would be crystalline or semi- crystalline, although this may vary widely depending on many factors including, but not limited to, the nature of the pharmaceutical agent, the disease to be treated/prevented, the intended storage conditions for the substrate prior to use or the location within the body of any biomedical implant.
- Preferably at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% or 100% of the pharmaceutical agent is in crystalline or semi-crystalline form.
- Stabilizing agent refers to any substance that maintains or enhances the stability of the biological agent. Ideally these stabilizing agents are classified as Generally Regarded As Safe (GRAS) materials by the US Food and Drug Administration (FDA). Examples of stabilizing agents include, but are not limited to carrier proteins, such as albumin, gelatin, metals or inorganic salts.
- Supercritical fluid refers to a substance under pressure greater than ambient conditions, where it demonstrates a density greater than 0.4 g/cc but the mobility of a gas, i.e. a gas with liquid-like densities in which the pressure and temperature are above the critical point (the temperature and pressure at which the density of the liquid and vapor phases become identical).
- Examples of near critical fluids include fluids which are in gaseous state at standard temperature and pressure (STP) conditions and have a critical density above 0.2 g/cc. See, e.g., US Patent ⁇ os.
- substances that demonstrate supercritical or near critical behavior suitable for the present invention include, but are not limited to carbon dioxide, ammonia, water, methanol, ethanol, ethane, propane, butane, pentane, dimethyl ether, xenon, sulfur hexafluoride, halogenated and partially halogenated materials such as chlorofluorocarbons, hydrochlorofluorocarbons, hydrofluorocarbons, perfluorocarbons (such as perfluoromethane and perfuoropropane, chloroform, trichloro-fluoromethane, dichloro- difluoromethane, dichloro-tetrafluoroethane) and mixtures thereof.
- chlorofluorocarbons such as chlorofluorocarbons, hydrochlorofluorocarbons, hydrofluorocarbons, perfluorocarbons (such as perfluoromethane and perfuoropropane, chloroform, trichloro-fluo
- Carbon dioxide is preferred.
- ⁇ .Fblfcjaa ⁇ flia.c'ai'irl ⁇ fliQ ⁇ dfe conditions including a temperature etween 0° C and 100° C and a pressure between 30 psig and 10,000 psig are preferred.
- “Sintering” or “plasticizing” as used herein refers to the process by which the co- deposited pharmaceutical agent or biological agent-polymer matrix, as described above, becomes fused by treatment of the polymer matrix or substrate with a compressed gas, compressed liquid, or densified fluid that is a non-solvent for both the polymer and the pharmaceutical agent and biological agents, but a plasticizing agent for the polymer.
- carbon dioxide is used to treat a matrix of polymer or a substrate that has been coated with a polymer and a drug, using dry powder and RESS electrostatic coating processes.
- "Bulk properties" properties of a coating including a pharmaceutical or a biological agent that can be enhanced through the methods of the invention include for example: adhesion, smoothness, conformality, thickness, and compositional mixing.
- Rapid Expansion of Supercritical Solutions involves the dissolution of a polymer into a compressed fluid, typically a densified fluid, followed by rapid expansion into a chamber at atmospheric pressure. The rapid expansion of the densified fluid solution through a small opening, with its accompanying decrease in density, reduces the dissolution capacity of the fluid and results in the nucleation and growth of polymer particles.
- Solution Enhanced Dispersion of Supercritical Solutions or “SEDS” as used herein involves a spray process for the generation of polymer particles, which, are formed when a compressed fluid (e.g.
- densified fluid preferably densified CO2
- a diluent to a vehicle in which a polymer or the drug is dissolved, (one that can dissolve both the polymer or the drug and the densified gas).
- the mixing of the densified fluid diluent with the polymer-containing solution may be achieved by encounter of a first stream containing the polymer solution and a second stream containing the diluent densified fluid, for example, within one spray nozzle or by the use of multiple spray nozzles.
- the solvent in the polymer solution may be one compound or a mixture of two or more ingredients and may be or comprise an alcohol (including diols, triols, etc.), ether, amine, ketone, carbonate, or alkanes, or hydrocarbon (aliphatic or aromatic) or may be a mixture of compounds, such as mixtures of alkanes, or mixtures of one or more alkanes in combination with additional compounds such as one or more alcohols, (e.g., from 0 or 0.1 to 5% of a Cl to C15 alcohol, including diols, triols, etc.). See for example US Patent No. 6,669,785.
- the solvent may optionally contain a surfactant, as also described in (for example) US Patent No. 6,669,785.
- a first stream of fluid comprising a polymer dissolved in a common solvent is co-sprayed with a second stream of densified fluid.
- Polymer particles are produced as the second stream acts as a diluent that weakens the solvent in the p ⁇ y H r l idh ⁇ BW €il ⁇ iSCfetream.
- the now combined streams of fluid, along with the polymer particles, flow out into a collection vessel which may or may not be at elevated pressure.
- a first stream of fluid comprising a drug dissolved in a common solvent is co-sprayed with a second stream of densified fluid.
- Drug particles are produced as the second stream acts as a diluent that weakens the solvent in the drug solution of the first stream.
- the now combined streams of fluid, along with the drug particles, flow out into a collection vessel which may or may not be at elevated pressure.
- Control of particle size, particle size distribution, and morphology is achieved by tailoring the following process variables: temperature, pressure, solvent composition of the first stream, flow-rate of the first stream, flow-rate of the second stream, composition of the second stream (where soluble additives may be added to the densified gas), and conditions of the capture vessel.
- the capture vessel contains a fluid phase that is at least five to ten times (5-1Ox) atmospheric pressure.
- Electrostatic capture refers to the collection of the spray-produced particles upon a substrate that has a different electrostatic potential than the sprayed particles.
- the substrate is at an attractive electronic potential with respect to the particles exiting, which results in the capture of the particles upon the substrate, i.e. the substrate and particles are oppositely charged, and the particles transport through the fluid medium of the capture vessel onto the surface of the substrate is enhanced via electrostatic attraction. This may be achieved by charging the particles and grounding the substrate or conversely charging the substrate and grounding the particles, or by some other process, which would be easily envisaged by one of skill in the art of electrostatic capture.
- Open vessel refers to a vessel open to the outside atmosphere, and thus at substantially the same temperature and pressure as the outside atmosphere.
- “Closed vessel” as used herein refers to a vessel sealed from the outside atmosphere, and thus may be at significantly different temperatures and pressures to the outside atmosphere.
- the drug or active ingredient may be in any physical form, such as crystalline (including semicrystalline) and amorphous.
- Solvents that may be used to carry out the present invention are, in some embodiments, gases (that is, compounds that are in the form of a gas at atmospheric pressure and 25 0 C).
- solvents include but are not limited to carbon dioxide, ammonia, water, methanol, ethanol, ethane, propane, butane, pentane, dimethyl ether, xenon, sulfur hexafluoride, , , _ , .
- hanogemate ⁇ aB'ciiiplaraanyii a Dgenate mate ⁇ als suc as c oro uorocar ons hydrochlorofluorocarbons, hydrofluorocarbons, perfluorocarbons (such as perfluoromethane and perfuoropropane, chloroform, trichloro-fluoromethane, dichloro-difluoromethane, dichloro- tetrafluoroethane) and mixtures thereof. Carbon dioxide is preferred.
- the solvent may be utilized per se or a cosolvent may be included therewith (e.g., in an amount of from 0.01 or 0.1 to 20 or 30 percent by weight or more).
- cosolvents include, but are not limited to, water and organic co-solvents.
- the organic co-solvent may be one compound or a mixture of two or more ingredients.
- the organic co-solvent may be or comprise an alcohol (including diols, triols, etc.), ether, amine, ketone, carbonate, or alkanes, or hydrocarbon (aliphatic or aromatic)
- the organic co-solvent may be a mixture of compounds, such as mixtures of alkanes as given above, or mixtures of one or more alkanes in combination with additional compounds such as one or more alcohols as described above, (e.g., from 0 or 0.1 to 5% of a Cl to C15 alcohol (including diols, triols, etc.)). See, e.g., US Patent No. 6,669,785.
- the solvent may optionally contain a surfactant, as also described in (for example) US Patent No. 6,669,785.
- the solvent is preferably provided in densified form.
- This densified form can be a gas at densities greater than 1.1 times the gas density at STP, a liquid (including near-supercritical fluids) or as a densified fluid, these three forms together sometimes being referred to as a "densified” fluid or "densified” gas. See, e.g., US Patent Nos. 6,860,123; 6,837,611; and 6,755,871.
- the drug- polymer composition may contain pharmaceutical excipients materials for: 1) enhancing the stability of the drug, 2) modifying the ultimate morphology of the drug or polymer, or drug polymer composite 3) inserting a porogen into the composite for subsequent removal in or during dense fluid processing, 4) improving the solubility characteristics of the drug in- vitro and in-vivo.
- these excipients are classified as Generally Regarded As Safe (GRAS) materials by the US Food and Drug Administration (FDA).
- the excipient serves to stabilize the drug material.
- a primary example is represented by the use of sugars and other carbohydrates to stabilize proteins and peptides in pharmaceutical formulations.
- one particularly useful sugar .deiMWI ⁇ S ⁇ ®fefrEfc «BUS etate (SOA) which can serve to sta l ze prote ns n solution or n the solid state during compounding of the drug with the polymer.
- the SOA may also serve to benefit the composite in downstream processing described below.
- the excipient serves to modify the morphology of the drug or polymer or the composite during and after processing with a dense gas fluid.
- dense gas fluids for processing drug-polymer composites relates to the "plasticizing" effect of the fluid (such as densified carbon dioxide) on the polymer.
- the fluid essentially permeates the free-volume of the polymer micro-structure lowering the glass transition temperature of the amorphous polymer and enhancing particle fusion at temperature much lower than those needed for heat bonding or fusion. This enhanced flow allows for suitable cohesion or adhesion of the formulated drug-polymer composite creating a semi-rigid composite product.
- excipients such as SOA may also serve to further plasticize the polymer thus enhancing the particle fusion and the overall solid-state integrity of the final composite.
- the excipient serves as a removable material (porogen) during the dense fluid processing step.
- the excipient material for non-absorbable polymers it may be desirable to create increased surface area to affect drug removal in- vivo.
- a porous or semi-porous structure is created upon exposure to the dense fluid. In this case, the excipient is extracted from the formed composite leaving a micro- or nano-porous internal structure after completed dense fluid processing.
- SOA One particular excipient of interest is SOA.
- Sucrose octaacetate is know to be soluble in dense carbon dioxide and in this case may serve as a stabilizer, a plasticizer, and a porogen. Other partially or fully acetylated sugars and carbohydrates may also be employed for these same purposes.
- the excipient increases the solubility of the drug as measure in- vitro and as applied in- vivo by preventing drug aggregation/agglomeration and by increasing the hydration capacity of the drug particle in-situ. Many drugs have poor aqueous solubility and therefore limited efficacy based on there ability to reach sufficient levels in the blood.
- excipients are used to prevent particle agglomeration and to enhance dissolution characteristics by increasing hydration in and around the particle.
- Noteworthy examples useful in the current invention include dextrin and its derivatives, other carbohydrates and simple sugars, and partially or fully acetylated sugars such as SOA.
- the excipient may serve one or several of the purposes described.
- vom11 ⁇ l;;:i( ,/QtnewsBimLlsxcipients include surfactants. Ideally, these suriac ⁇ ants are classified as GRAS materials by the FDA.
- Suitable examples include but are not limited to sorbitan monooleate, Twean® trademarked surfactants, soy derived surfactants, and fatty acid derived GRAS surfactants. These surfactants may serve one or multiple roles as described above in this section.
- HDCs hydrophobically derivatized carbohydrates
- SOA and other such hydrophobically derivatized carbohydrates can be utilized as the pharmaceutical excipient.
- HDCs are a wide variety of hydrophobically derivatized carbohydrates where at least one hydroxyl group is substituted with a hydrophobic moiety including, but not limited to, esters and ethers.
- suitable HDCs and their syntheses are described in Developments in Food Carbohydrate, C. K. Lee, Applied Science Publishers, London (2d Ed. 1980) and PCT publication No. 96/03978.
- Other syntheses are described in, for example, Akoh et al. (1987) J. Food Sci. 52:1570; Khan et al. (1933) Tetra.
- HDCs include, but are not limited to, sorbitol hexaacetate (SHAC), alpha-glucose pentaacetate (alpha-GPAC), beta-glucose pentaacetate (beta-GPAC), l-O-Octyl-.beta.-D-glucose tetraacetate (OGTA), trehalose octaacetate (TOAC), trehalose octapropionate (TOP), trehalose octa-3,3,dimethylbutyrate
- HAC sorbitol hexaacetate
- alpha-GPAC alpha-glucose pentaacetate
- beta-GPAC beta-glucose pentaacetate
- OGTA l-O-Octyl-.beta.-D-glucose tetraacetate
- TOAC trehalose octacetate
- TOP trehalose
- TO33DMB trehalose diisobutyrate hexaacetate, trehalose octaisobutyrate, lactose octaacetate, sucrose octaacetate (SOAC), cellobiose octaacetate (COAC), raffmose undecaacetate (RUDA), sucrose octapropanoate, cellobiose octapropanoate, raffmose undecapropanoate, tetra-O-methyl trehalose, trehalose octapivalate, trehalose hexaacetate dipivalate and di-O-methyl-hexa-O-actyl sucrose and mixtures thereof. See, e.g., US Patent No. 6,517,860.
- the method of the invention may be carried out by first, combining the drug with the polymer and optionally an excipient(s) to form a mixture.
- This mixing step may be carried out by any suitable technique or in any suitable apparatus, such as in a blender, extruder, etc.
- both the drug and the polymer are provided in solid particulate form, and hence the mixture so formed will also be in the form of a solid.
- the polymer and the drug particles range between 0.02 and 50 microns in size. In some embodiments the particle size is in a larger size range than the drug. In this case the polymer may range from 0.2 micron and 50 microns and the drug from 0.02 to 20 microns.
- the mixture is contacted under pressure with a densified gas solvent as described above to form the composite material.
- a densified gas solvent is at a pressure sufficient to reduce the yiiidbl ⁇ ifWiih ⁇ ipbljhKfeuMterial, trapping the fluid insoluble drug material in the polymer matrix as polymer particles fuse with adjacent polymer particles and hence form the drug/polymer composite article.
- dense fluid gases such as carbon dioxide at high pressures
- many drugs, particularly protein-based drugs are not soluble in the dense fluid and therefore are not efficiently infused into polymer matrices.
- the drug such as a protein-based therapeutic may remain largely unchanged as the polymer particle fuse around the drug particles.
- the drug/polymer composite can be in the form of discrete particles (which may for example be the same size but likely larger than the polymer particles previously provided) or may be in the form of a shaped article.
- the composite mixture is used in conjunction with a mechanical article such as a mold or a template and the final composite article takes on the shape or general shape of that mold or template. So in working practice the mixture of the drug, polymer and excipients is added to a three-dimensional article, mechanically constrained such that the particles of both the drug and the polymer are immobilized.
- this contacting step is carried out at a pressure between 300 and 15,000 psig and a temperature of between 2OC and 175 0 C. Most preferably the contacting step is carried out at between300 and 3000 psig at a temperature between 2OC and 100 0 C
- the step of combining the mixture with the solvent can be carried out by any suitable technique or in any suitable apparatus, such as in an extruder (which may be the same or different from the extruder noted above), mold (e.g., injection mold, blow mold, compression mold, etc.), reaction vessel, etc.
- a shaped article as described herein may, in some embodiments, be formed concurrently with this combining step, for example when the combining is carried out in a mold, or when the combining is carried out in an extruder and the composite formed therein then extruded through a die. In other embodiments, however, the shaped article will be formed in a subsequent step.
- the drug/composite material may comprise, consist of, or consist essentially of: U-BKiSi 0'02 ⁇ r ⁇ !J:;:!b,$rcent to 40,.50, or 60 percent y weight of drug (which may be a single compound or a combination of different active agents); and from 40 or 50 percent to 99.9 or 99.99 percent by weight of polymer; optionally, from 0.01 or 0.1 percent to 20 or 30 percent pharmaceutical excipient.
- the physical form of the drug in the composite is substantially the same as the physical form of the drug before the combining step (b).
- a drug initially provided in crystalline form remains in crystalline form in the composite;
- a drug initially provided in amorphous form remains in amorphous form in the composite; etc.
- the composite is porous (this term including "semiporous"), with porous composites being made by inclusion of a porogen as a pharmaceutical excipient and subsequent removal of at least a portion thereof by an appropriate solvent (e.g., organic solvents; densified carbon dioxide solvent compositions as described herein) thereof after formation of the composite, in accordance with known techniques.
- an appropriate solvent e.g., organic solvents; densified carbon dioxide solvent compositions as described herein
- the porogen is an SOA or other such hydrophobically derivatized carbohydrate as described above.
- Secondary coatings Drug/polymer composites prepared as described above may optionally be coated (e.g., by spraying, dipping, or any suitable technique) with a second material to aid in the subsequent binding, forming, dispersion, structure or drug-elution profile of the drug/ polymer composite.
- This second material can be any of several different chemical functionalities and several different functions in the resulting drug/polymer composite material.
- the second material can be a pharmaceutical excipient, providing a means to alter the pharmacological effect of the drug or providing a means to alter the release profile of the drug-delivery.
- the second material can be a CO 2 -philic material.
- a second condition of densified fluid can be used to remove the CO 2 -philic material, thereby forming pores in and rendering porosity to the formed part.
- a cylindrical composite article consisting of 3 parts ⁇ oly(butyl methacrylate), 2 parts recombinant Human Growth hormone (rHGh), and 1 part sucrose octaacetate is created in the following manner.
- the resulting formulation is then added to a cylindrical hollow mold constructed from sintered metal creating a fluid permeable three-dimensional article with an average pore size of 0.2 microns.
- the cylinder is open on both ends. Prior to the addition of the drug-polymer composition to the mold, one end is closed off using a matching cap designed to lock in place at the end of the cylinder.
- the composition is then mechanically compressed using a metal plunger matching the approximate inner diameter of the cylinder minus 0.001 -inch to remove the majority of the free-volume. The other end of the cylinder is then closed using an end cap that locks in place constraining the composition in three dimensions.
- the mold containing the polymer drug composition is then placed in a sterile pressure vessel to which 99.99% pure carbon dioxide is added to a pressure of 4000 psig at a temperature of 80C.
- the article is maintained in the CO2 environment at this temperature for 20 minutes after which the vessel is vented to atmospheric conditions.
- the mold is then removed from the vessel and the end caps are removed.
- the drug-polymer composite is then removed from the mold using a metal plunger fed from the open top of the mold thus pushing the composite out the bottom as the cylinder is mechanically restrained. Upon inspection the sample is a semi-rigid solid article in the shape of the mold.
- the solid article consists of a porous network of fused polymer particles with protein residing largely between adjacent fused particles and in void spaces created by the partial extraction of the sucrose octaacetate.
- SEM Scanning Electron Microscopy
- the porous structure is largely inter-connected and partially opened to the outer surface of the article and the ratio of polymer to drug to sucrose octaacetate was 3:2:0.2 indicating substantial removal of the sucrose derivative during fluid processing.
- a cylindrical composite article consisting of 4 parts poly(butyl methacrylate), 2 parts recombinant Human Growth hormone (rHGh), and 2 part sucrose octaacetate is created in the following manner.
- Dry sucrose octaacetate powder in the appropriate ratio is then added under constant mixing.
- the resulting formulation is then added to a cylindrical hollow mold constructed from sintered metal creating a fluid permeable three-dimensional article with an average pore size of 0.2 microns.
- the cylinder is open on both ends.
- one end Prior to the addition of the drug-polymer composition to the mold, one end is closed off using a matching cap designed to lock in place at the end of the cylinder.
- the mold containing the polymer-drug- excipient mixture is then added to a pressure vessel equipped with a mechanical device designed with a piston actuator to exert pressure on the open end of the mold.
- the sealed pressure vessel is then filled with densified CO2 to a pressure of 3000 psi at a temperature of 80C.
- the piston is actuated to apply mechanical pressure through the open end of the mold compressing the composition with 25 IbS-(Ui 2 ) "1 of mechanical force.
- the CO2 is vented from the chamber and the piston is removed from the open end of the cylindrical mold.
- the drug-polymer composite is then removed from the mold using a metal plunger fed from the open top of the mold thus pushing the composite out the bottom as the cylinder is mechanically restrained. Upon inspection the sample is a semi-rigid solid article in the shape of the mold.
- FIG. 1-5 illustrate embodiments of the invention including SEM images of fused
Abstract
A method of forming a drug/polymer composite material is carried out by combining a drug material with a polymer material under pressure in the presence of a densified gas solvent (e.g., carbon dioxide) to form the drug/polymer composite material. Drug/polymer composite materials and shaped articles (e.g., subcutaneous drug depots) which may be produced by a process are also described, along with methods of use thereof.
Description
IMG/POLYMER COMPOSITE MATERIALS AND
METHODS OF MAKING THE SAME
RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. patent application number 11/158,724, entitled "Drug.Polymer Composite Materials and Methods of Making the Same," filed June 22,
2005, which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
[0002] The present invention concerns methods of making drug/polymer composite materials, the materials so made, and shaped articles foπned from such drug/polymer composite materials.
BACKGROUND OF THE INVENTION
[0003] Drug/polymer composite materials are traditionally formed either by solvent-based processing where a solvent or combination of solvents is used to facilitate intimate mixing of the drug with polymer(s) by a combination of reducing the polymer viscosity and by dispersing/dissolving the drug into a fluid-like phase or by processing a mixture of drug(s) - polymer(s) at an elevated temperature sufficient to cause flow of the polymer into a desired shape. The solvents commonly utilized include all common organic solvents, halogenated solvents and aqueous solvent compositions. However, Solvent-based processing can adversely affect the drug by reacting, bonding or binding with the chemical functionality of many drugs. In addition, removal of solvent and solvent residues from the composite material is problematic and requires extensive processing with heat, vacuum, etc. Further, these processes can be process/cost intensive, lack precise material control and can adversely affect the drug. For example: (i) Trace solvent residues are unavoidable and are often toxic or can negatively interact with the drug or polymer molecules altering the therapeutic effect. (H) Solvent-based processing can also adversely affect the primary structure of the drug in the polymer matrix. For example, making very difficult the production of small particles/domains of drug in the polymer matrix. (Hi) Solvent-based processing can also adversely affect the secondary structure of sophisticated therapeutics such as proteins, enzymes, hormones, which changes the drug's efficacy and may denature the drug compound rendering it useless or toxic or change its effective shelf-life, (in) Solvent-based processing can also adversely affect the polymorph of the drug; changing crystalline structure or providing amorphous materials that have different bioavailability profiles and adversely affecting shelf-life.
I'OQMiiΛllilt.eϊlItWi;,tSitS,pna process uses elevated tempera ures to provide a lower viscosity polymer(s) for mixing with the drug. Again, however, high temperature processing can adversely affect many thermally sensitive drugs, rendering them ineffective or toxic, and elevated temperature processing is often used in conjunction with solvent-based methods (one still has to dissolve/disperse the drug molecule(s)), resulting in combined challenges of high temperature and solvents.
[0005] Densifϊed gases, liquid and supercritical fluids have been described in the art as processing media for the incorporation of active materials including drugs into polymeric matrices. U.S. patent 5,340,614 (Perman) describes impregnating materials into polymeric matrices by using a carrier liquid that carries the active ingredient(s) where the carrier fluid is substantially insoluble in the supercritical fluid as is the active ingredient(s). A polymeric material is added to a pressure vessel after which the carrier liquid containing the active material(s) is( are) added, and then the system is exposed to supercritical carbon dioxide. After removal of the supercritical fluid and the carrier fluid, the polymer is found to have absorbed a portion of the active and presumably the carrier fluid.
[0006] U.S. Patent 6,190,699 (Luzzi) describes compositions of protein and peptide infused polymer particles and methods for production using densified solvents including supercritical fluids. Luzzi claims that the proteins and peptides are partially adsorbed into (infused) the polymer particles. Since proteins and peptides are not soluble in supercritical carbon dioxide, it can be reasonable assumed that dense carbon dioxide is not a suitable densified solvent to practice this art as sorption would be disfavored due to a lack of solubility of the protein in the densified solvent. Additionally, Luzzi discloses methods for making particles and does not address shaped or formed articles or semi-porous or porous articles. [0007] What is needed in the art is a method that allows for the formation of polymer-drug composites that does not require the use of a carrier liquid or emulsions to make soluble or make mobile the drug for addition to the polymer. What is needed in the art is a method that allows for production of a polymer-drug composite that does not physically or chemically change the state of the drug during processing (solid to liquid). What is needed in the art is a method that allows for the creation of formed articles of a desired and controllable geometry. What is needed in the art is a method that allows for a low temperature forming of a semi-porous or porous solid article that does not physically or chemically change the state of the drug during processing. [0008] Accordingly, there is a need for new approaches to the production of drug/polymer composite materials, and for new materials produced by such methods.
SUMMARY OF THE INVENTION
[0009] A first aspect of the invention provides composite comprising at least one polymer; at least one pharmaceutical agent in a therapeutically desirable morphology; and optionally at least one excipient; wherein the pharmaceutical agent is sequestered in interstices formed by fusing particles of the polymer. In one embodiment, the pharmaceutical agent is a drug or a biological agent having a secondary, tertiary and/or quaternary structure required for biological activity, wherein the structure is substantially retained when the particles of the biological agent are sequestered within interstices formed by the fused polymer particles. In one embodiment The pharmaceutical agent is a peptide, protein, enzyme, nucleic acid, antibody, therapeutic vaccine, antisense nucleic acid, antimicrobial, vitamin, hormone, steroid, lipid, polysaccharide or carbohydrate. The pharmaceutical agent may be a therapeutic protein, such as erythropoietin, interferon, insulin, blood factor, colony stimulating factor, growth hormone, interleuldn and growth factor. In another embodiment, the pharmaceutical agent is a drug in substantially crystalline form. [0010] In yet another embodiment, the composite provides a rate of elution that remains within a 15% variation range from a selected rate of elution between day 2 and day 30 after the composite is implanted in a subject under physiological conditions.
[0011] In yet another embodiment, the composite provides an elution profile wherein about 10% to about 50% of the pharmaceutical agent is eluted at week 1 after the composite is implanted in a subject under physiological conditions, about 25% to about 75% of the pharmaceutical agent is eluted at week 2 and about 50% to about 75% of the pharmaceutical agent is eluted at week 4. . [0012] In still another embodiment, the polymer and the pharmaceutical agent form two substantially separate phases. [0013] Another aspect of the invention provides a method of preparing a composite comprising [0014] (a) forming a mixture by combining particles of at least one polymer with particles of at least one pharmaceutical agent in a therapeutically desirable morphology, and optionally with particles of at least one excipient; and (b) plasticizing said mixture under conditions that do not substantially modify the morphology of said pharmaceutical agent wherein the pharmaceutical agent is sequestered in interstices formed by fused particles of the polymer. In one embodiment, the method further comprises forming a shaped article from said composite. In yet another embodiment, the shaped article is formed by placing the mixture of step (a) in a mold prior to performing said plasticizing of step (b).
[0015] hi one embodiment, the shaped article is a biomedical implant., for example, a drug depot or a stent.
Jrøβ
 present invention is a metho o orm ng a drug/polymer compos te material by combining a drug material with a polymer material under pressure in the presence of a densified gas solvent to form the drug/polymer composite material.
present invention is a metho o orm ng a drug/polymer compos te material by combining a drug material with a polymer material under pressure in the presence of a densified gas solvent to form the drug/polymer composite material.
[0017] A further aspect of the present invention is a drug/polymer composite material (in some embodiments a "medicament" herein), which may be produced by a process as described above.
[0018] A further aspect of the present invention is a shaped article (in some embodiments also referred to as a "medicament" herein) comprising, consisting of or consisting essentially of a drug/polymer composite material as described above.
[0019] A further aspect of the present invention is a method of treating a subject with a drug, comprising administering a drug/polymer composite material as described herein to said subject in an amount effective to treat said subject with said drug.
[0020] A further aspect of the present invention is the use of a drug for the preparation of a medicament for carrying out a method of treatment as described herein.
[0021] The foregoing and other objects and aspects of the present invention are explained in greater detail in the drawings herein and the specification set forth below.
BRIEF DESCRIPTION OF THE DRAWINGS
[0022] FIGURE 1 shows supercritical CO2 molding process and formation of an implant according to the invention.
[0023] FIGURE 2 shows supercritical CO2 molding process "plasticization" leading to particle fusion according to the invention.
[0024] FIGURE 3 shows final implant, cross section SEM according to the invention.
[0025] FIGURE 4 shows In-vitro esculetin elution from PGLA implant according to the invention.
[0026] FIGURE 5 shows In-vitro esculetin mass elution from PGLA implant according to the invention.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
[0027] The present invention is explained in greater detail below. This description is not intended to be a detailed catalog of all the different ways in which the invention may be implemented, or all the features that may be added to the instant invention. For example, features illustrated with respect to one embodiment may be incorporated into other embodiments, and features illustrated with respect to a particular embodiment may be deleted from that embodiment. In addition, numerous variations and additions to the various embodiments suggested herein will be apparent to those skilled in the art in light of the instant disclosure
 instant invention. Hence, the following specification is intended to illustrate some particular embodiments of the invention, and not to exhaustively specify all permutations, combinations and variations thereof.
instant invention. Hence, the following specification is intended to illustrate some particular embodiments of the invention, and not to exhaustively specify all permutations, combinations and variations thereof.
[0028] The disclosures of all United States patents cited herein are to be incorporated herein by reference in their entirety.
A. Definitions.
[0029] Subjects that may be treated by the present invention include both human subjects for medical purposes and animal subjects for veterinary and drug screening and development purposes. Other suitable animal subjects are, in general, mammalian subjects such as primates, bovines, ovines, caprines, porcines, equines, felines, canines, rodents (e.g., rats and mice), etc. Human subjects are the most preferred. Human subjects include fetal, neonatal, infant, juvenile and adult subjects. [0030] Shaped articles as used herein include, but are not limited to, pills, tablets, drug depots or drug delivery devices {e.g., subcutaneous implants), biomedical implants, etc.
[0031] "Biomedical implant" as used herein includes but is not limited to drug depots and drug delivery devices, stents (e.g., vascular stents), electrodes, catheters, leads, implantable pacemaker or cardioverter housings, joints, screws, rods, ophthalmic implants (including, but not limited to, intraocular lens implants, glaucoma implants or drainage implants, and punctal implants or plugs), etc. The implants may be of any suitable material, including but not limited to organic polymers (including stable or inert polymers and polymers), metals such as stainless steel and titanium, inorganic materials such as silicon, and composites thereof. [0032] "Drug depot" or "drug delivery device" include those be configured for any route of administration, including those that may be implanted (luminal, venous, subcutaneous, muscular, ocular), inserted (oral, rectal, vaginal, ocular) or topically applied (transdermal, transmucual, sublingual).
[0033] "Treat" as used herein refers to any type of treatment or prevention that imparts a benefit to a subject afflicted with a disease or at risk of developing the disease, including improvement in the condition of the subject (e.g., in one or more symptoms), delay in the progression of the disease, delay the onset of symptoms or slow the progression of symptoms, etc. As such, the term "treatment" also includes prophylactic treatment of the subject to prevent the onset of symptoms. As used herein, "treatment" and "prevention" are not necessarily meant to imply cure or complete abolition of symptoms." to any type of treatment that imparts a benefit to a patient afflicted with a disease, including improvement in the condition of the patient (e.g., in one or more symptoms), delay in the progression of the disease, etc.
IPO^BIIIiitndqiSiiφaifcJljient11 as used herein includes refers to any pharmaceutically acceptable material that is included in a drug composition to enhance the pharmaceutical (including manufacturing and shelf-stability) and/or pharmacological properties thereof. Pharmaceutical excipients include, but are not limited to, adjuvants, surfactants, stabilizers, morphology modifiers, porogens, diluents, carriers, solubilizers, antioxidants, lubricants (or glidants), binders, disintigrants, and mixtures thereof. Definitions
[0035] As used in the present specification, the following words and phrases are generally intended to have the meanings as set forth below, except to the extent that the context in which they are used indicates otherwise.
[0036] "Substrate" as used herein, refers to any surface upon which it is desirable to deposit a coating comprising a polymer and a pharmaceutical or biological agent, wherein the coating process does not substantially modify the morphology of the pharmaceutical agent or the activity of the biological agent. Biomedical implants are of particular interest for the present invention; however the present invention is not intended to be restricted to this class of substrates. Those of skill in the art will appreciate alternate substrates that could benefit from the coating process described herein, such as pharmaceutical tablet cores, as part of an assay apparatus or as components in a diagnostic kit (e.g. a test strip). [0037] The implants may be formed from any suitable material, including but not limited to organic polymers (including stable or inert polymers and biodegradable polymers), metals, inorganic materials such as silicon, and composites thereof, including layered structures with a core of one material and one or more coatings of a different material.
[0038] In a preferred embodiment the biomedical implant is an expandable intraluminal vascular graft or stent (e.g., comprising a wire mesh tube) that can be expanded within a blood vessel by an angioplasty balloon associated with a catheter to dilate and expand the lumen of a blood vessel, such as described in US Patent No. 4,733,665 to Palmaz Shaz. [0039] "Pharmaceutical agent" as used herein refers to any of a variety of drugs or pharmaceutical compounds that can be used as active agents to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e. arresting the development of clinical symptoms; and/or relieving the disease, i.e. causing the regression of clinical symptoms). It is possible that the pharmaceutical agents of the invention may also comprise two or more drugs or pharmaceutical compounds. Pharmaceutical agents, include but are not limited to antirestenotic agents, antidiabetics, analgesics, antiinflammatory agents, antirheumatics, antihypotensive agents, antihypertensive agents, psychoactive drugs,
,tfan(|€'.lllJ^(-is,/≤ϋ;!diϊiEΪ!idi .muscle relaxants, glucocorticoids, agents for treating ulcerative colitis or Crohn's disease, antiallergics, antibiotics, antiepileptics, anticoagulants, antimycotics, antitussives, arteriosclerosis remedies, diuretics, proteins, peptides, enzymes, enzyme inhibitors, gout remedies, hormones and inhibitors thereof, cardiac glycosides, immunotherapeutic agents and cytokines, laxatives, lipid-lowering agents, migraine remedies, mineral products, otologicals, anti parkinson agents, thyroid therapeutic agents, spasmolytics, platelet aggregation inhibitors, vitamins, cytostatics and metastasis inhibitors, phytopharmaceuticals, chemotherapeutic agents and amino acids. Examples of suitable active ingredients are acarbose, antigens, beta-receptor blockers, non-steroidal antiinflammatory drugs (NSAIDs], cardiac glycosides, acetylsalicylic acid, virustatics, aclarubicin, acyclovir, cisplatin, actinomycin, alpha- and beta- sympatornimetics, (dmeprazole, allopurinol, alprostadil, prostaglandins, amantadine, ambroxol, amlodipine, methotrexate, S-aminosalicylic acid, amitriptyline, amoxicillin, anastrozole, atenolol, azathioprine, balsalazide, beclomethasone, betahistine, bezafϊbrate, bicalutamide, diazepam and diazepam derivatives, budesonide, bufexamac, buprenorphine, methadone, calcium salts, potassium salts, magnesium salts, candesartan, carbamazepine, captopril, cefalosporins, cetirizine, chenodeoxycholic acid, ursodeoxycholic acid, theophylline and theophylline derivatives, trypsins, cimetidine, clarithromycin, clavulanic acid, clindamycin, clobutinol, clonidine, cotrimoxazole, codeine, caffeine, vitamin D and derivatives of vitamin D, colestyramine, cromoglicic acid, coumarin and coumarin derivatives, cysteine, cytarabine, cyclophosphamide, ciclosporin, cyproterone, cytabarine, dapiprazole, desogestrel, desonide, dihydralazine, diltiazem, ergot alkaloids, dimenhydrinate, dimethyl sulphoxide, dimeticone, domperidone and domperidan derivatives, dopamine, doxazosin, doxorubizin, doxylamine, dapiprazole, benzodiazepines, diclofenac, glycoside antibiotics, desipramine, econazole, ACE inhibitors, enalapril, ephedrine, epinephrine, epoetin and epoetin derivatives, morphinans, calcium antagonists, irinotecan, modafinil, orlistat, peptide antibiotics, phenytoin, riluzoles, risedronate, sildenafil, topiramate, macrolide antibiotics, oestrogen and oestrogen derivatives, progestogen and progestogen derivatives, testosterone and testosterone derivatives, androgen and androgen derivatives, ethenzamide, etofenamate, etofibrate, fenoflbrate, etofylline, etoposide, famciclovir, famotidine, felodipine, fenofϊbrate, fentanyl, fenticonazole, gyrase inhibitors, fluconazole, fludarabine, fluarizine, fluorouracil, fluoxetine, flurbiprofen, ibuprofen, flutamide, fluvastatin, follitropin, formoterol, fosfomicin, furosemide, fusidic acid, gallopamil, ganciclovir, gemfibrozil, gentamicin, ginkgo, Saint John's wort, glibenclamide, urea derivatives as oral antidiabetics, glucagon, glucosamine and glucosamine derivatives, glutathione, glycerol and glycerol derivatives, hypothalamus hormones, goserelin, gyrase inhibitors, guanethidine, halofantrine, haloperidol, heparin and heparin derivatives, hyaluronic acid, hydralazine,
'hylldW^ilfeiiaiWfflffil^lirochlorothiaz de derivatives, salicylates, hydroxyzine, idarubicin, ifosfamide, imipramine, indometacin, indoramine, insulin, interferons, iodine and iodine derivatives, isoconazole, isoprenaline, glucitol and glucitol derivatives, itraconazole, . ketoconazole, ketoprofen, ketotifen, lacidipine, lansoprazole, levodopa, levomethadone, thyroid hormones, lipoic acid and lipoic acid derivatives, lisinopril, lisuride, lofepramine, lomustine, loperamide, loratadine, maprotiline, mebendazole, mebeverine, meclozine, mefenamic acid, mefloquine, meloxicam, mepindolol, meprobamate, meropenem, mesalazine, mesuximide, metamizole, metformin, methotrexate, methylphenidate, methylprednisolone, metixene, metoclopramide, metoprolol, metronidazole, mianserin, miconazole, minocycline, minoxidil, misoprostol, mitomycin, mizolastine, moexipril, morphine and morphine derivatives, evening primrose, nalbuphine, naloxone, tilidine, naproxen, narcotine, natamycin, neostigmine, nicergoline, nicethamide, nifedipine, nifhimic acid, nimodipine, nimorazole, nimustine, nisoldipine, adrenaline and adrenaline derivatives, norfloxacin, novamine sulfone, noscapine, nystatin, ofloxacin, olanzapine, olsalazine, omeprazole, omoconazole, ondansetron, oxaceprol, oxacillin, oxiconazole, oxymetazoline, pantoprazole, paracetamol, paroxetine, penciclovir, oral penicillins, pentazocine, pentifylline, pentoxifylline, perphenazine, pethidine, plant extracts, phenazone, pheniramine, barbituric acid derivatives, phenylbutazone, phenytoin, pimozide, pindolol, piperazine, piracetam, pirenzepine, piribedil, piroxicam, pramipexole, pravastatin, prazosin, procaine, promazine, propiverine, propranolol, propyphenazone, prostaglandins, protionamide, proxyphylline, quetiapine, quinapril, quinaprilat, ramipril, ranitidine, reproterol, reserpine, ribavirin, rifampicin, risperidone, ritonavir, ropinirole, roxatidine, roxithromycin, ruscogenin, rutoside and rutoside derivatives, sabadilla, salbutamol, salmeterol, scopolamine, selegiline, sertaconazole, sertindole, sertralion, silicates, sildenafil, simvastatin, sitosterol, sotalol, spaglumic acid, sparfloxacin, spectinomycin, spiramycin, spirapril, spironolactone, stavudine, streptomycin, sucralfate, sufentanil, sulbactam, sulphonamides, sulfasalazine, sulpiride, sultamicillin, sultiam, sumatriptan, suxamethonium chloride, tacrine, tacrolimus, taliolol, tamoxifen, taurolidine, tazarotene, temazepam, teniposide, tenoxicam, terazosin, terbinafme, terbutaline, terfenadine, terlipressin, tertatolol, tetracyclins, teryzoline, theobromine, theophylline, butizine, thiamazole, phenothiazines, thiotepa, tiagabine, tiapride, propionic acid derivatives, ticlopidine, timolol, tinidazole, tioconazole, tioguanine, tioxolone, tiropramide, tizanidine, tolazoline, tolbutamide, tolcapone, tolnaftate, tolperisone, topotecan, torasemide, antioestrogens, tramadol, tramazoline, trandolapril, tranylcypromine, trapidil, trazodone, triamcinolone and triamcinolone derivatives, triamterene, trifluperidol, trifluridine, trimethoprim, trimipramine, tripelennamine, triprolidine, trifosfamide, tromantadine, trometamol, tropalpin, troxerutine, tulobuterol, tyramine, tyrothricin, urapidil, ursodeoxycholic acid, chenodeoxycholic
ai:c iϊbiα, vancomycin, vecuronium chloride, Viagra, venlafaxme, verapamil, vidarabine, vigabatrin, viloazine, vinblastine, vincamine, vincristine, vindesine, vinorelbine, vinpocetine, viquidil, warfarin, xantinol nicotinate, xipamide, zafirlukast, zalcitabine, zidovudine, zolmitriptan, Zolpidem, zoplicone, zotipine and the like. See, e.g., US Patent No. 6,897,205; see also US Patent No. 6,838,528; US Patent No. 6,497,729.
[0040] The active ingredients may, if desired, also be used in the form of their pharmaceutically acceptable salts or derivatives (meaning salts which retain the biological effectiveness and properties of the compounds of this invention and which are not biologically or otherwise undesirable), and in the case of chiral active ingredients it is possible to employ both optically active isomers and racemates or mixtures of diastereoisomers.
[0041] "Active biological agent" as used herein refers to a substance, originally produced by living organisms, that can be used to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e. arresting the development of clinical symptoms; and/or relieving the disease, i.e. causing the regression of clinical symptoms). It is possible that the active biological agents of the invention may also comprise two or more active biological agents or an active biological agent combined with a pharmaceutical agent, a stabilizing agent or chemical or biological entity. Although the active biological agent may have been originally produced by living organisms, those of the present invention may also have been synthetically prepared, or by methods combining biological isolation and synthetic modification. By way of a non-limiting example, a nucleic acid could be isolated form from a biological source, or prepared by traditional techniques, known to those skilled in the art of nucleic acid synthesis. Furthermore, the nucleic acid may be further modified to contain non-naturally occurring moieties. Non-limiting examples of active biological agents include peptides, proteins, enzymes, glycoproteins, nucleic acids (including deoxyribonucleotide or ribonucleotide polymers in either single or double stranded form, and unless otherwise limited, encompasses known analogues of natural nucleotides that hybridize to nucleic acids in a manner similar to naturally occurring nucleotides), antisense nucleic acids, fatty acids, antimicrobials, vitamins, hormones, steroids, lipids, polysaccharides, carbohydrates and the like. They further include, but are not limited to, antirestenotic agents, antidiabetics, analgesics, antiinflammatory agents, antirheumatics, antihypotensive agents, antihypertensive agents, psychoactive drugs, tranquillizers, antiemetics, muscle relaxants, glucocorticoids, agents for treating ulcerative colitis or Crohn's disease, antiallergics, antibiotics, antiepileptics, anticoagulants, antimycotics, antitussives, arteriosclerosis remedies, diuretics, proteins, peptides, enzymes, enzyme inhibitors, gout remedies, hormones and inhibitors thereof, cardiac glycosides, immunotherapeutic agents
aMifiifalϊMssj-'laii^lvieSjiillpld-lowering agents, migraine remedies, mineral products, otologicals, anti parkinson agents, thyroid therapeutic agents, spasmolytics, platelet aggregation inhibitors, vitamins, cytostatics and metastasis inhibitors, phytopharmaceuticals and chemotherapeutic agents. Preferably, the active biological agent is a peptide, protein or enzyme, including derivatives and analogs of natural peptides, proteins and enzymes.
[0042] "Activity" as used herein refers to the ability of a pharmaceutical or active biological agent to prevent or treat a disease (meaning any treatment of a disease in a mammal, including preventing the disease, i.e. causing the clinical symptoms of the disease not to develop; inhibiting the disease, i.e. arresting the development of clinical symptoms; and/or relieving the disease, i.e. causing the regression of clinical symptoms). Thus the activity of a pharmaceutical or active biological agent should be of therapeutic or prophylactic value. [0043] "Secondary, tertiary and quaternary structure " as used herein are defined as follows. The active biological agents of the present invention will typically possess some degree of secondary, tertiary and/or quaternary structure, upon which the activity of the agent depends. As an illustrative, non-limiting example, proteins possess secondary, tertiary and quaternary structure. Secondary structure refers to the spatial arrangement of amino acid residues that are near one another in the linear sequence. The α-helix and the /3-strand are elements of secondary structure. Tertiary structure refers to the spatial arrangement of amino acid residues that are far apart in the linear sequence and to the pattern of disulfide bonds. Proteins containing more than one polypeptide chain exhibit an additional level of structural organization. Each polypeptide chain in such a protein is called a subunit. Quaternary structure refers to the spatial arrangement of subunits and the nature of their contacts. For example hemoglobin consists of two a. and two β chains. It is well known that protein function arises from its conformation or three dimensional arrangement of atoms (a stretched out polypeptide chain is devoid of activity). Thus one aspect of the present invention is to manipulate active biological agents, while being careful to maintain their conformation, so as not to lose their therapeutic activity.
[0044] "Polymer" as used herein, refers to a series of repeating monomelic units that have been cross-linked or polymerized. Any suitable polymer can be used to carry out the present invention. It is possible that the polymers of the invention may also comprise two, three, four or more different polymers. In some embodiments, of the invention only one polymer is used. In some preferred embodiments a combination of two polymers are used. Combinations of polymers can be in varying ratios, to provide the polymer part of the composite materials of the invention including plasticized polymers that form a matrix with interstices wherein the drugs are sequestered. In addition to their role in forming the matrices of the invention, the polymers can be used as coating for composite materials of the invention. Selection of appropriate
 composite materials w th differing properties. Those of skill in the art of polymer chemistry will be familiar with the different properties of polymeric compounds. "Polymer" as used herein refers to organic polymers, and includes copolymers of a named polymer with other constituents. In some embodiments, such as in the preparation of drug depots or drug delivery devices, the polymer is preferably an absorbable and/or resorbable polymer. In other embodiments the polymer is preferably non-resorbable and biocompatible. Examples of ploymers that may be used in the present invention include, but are not limited to polycarboxylic acids, cellulosic polymers, proteins, polypeptides, polyvinylpyrrolidone, maleic anhydride polymers, polyamides, polyvinyl alcohols, polyethylene oxides, glycosaminoglycans, polysaccharides, polyesters, polyurethanes, polystyrenes, copolymers, silicones, polyorthoesters, polyanhydrides, copolymers of vinyl monomers, polycarbonates, polyethylenes, polypropylenes, polylactic acids, polyglycolic acids, polycaprolactones, polyhydroxybutyrate valerates, polyacrylamides, polyethers, polyurethane dispersions, polyacrylates, acrylic latex dispersions, polyacrylic acid, mixtures and copolymers thereof. The polymers of the present invention may be natural or synthetic in origin, including gelatin, chitosan, dextrin, cyclodextrin,
composite materials w th differing properties. Those of skill in the art of polymer chemistry will be familiar with the different properties of polymeric compounds. "Polymer" as used herein refers to organic polymers, and includes copolymers of a named polymer with other constituents. In some embodiments, such as in the preparation of drug depots or drug delivery devices, the polymer is preferably an absorbable and/or resorbable polymer. In other embodiments the polymer is preferably non-resorbable and biocompatible. Examples of ploymers that may be used in the present invention include, but are not limited to polycarboxylic acids, cellulosic polymers, proteins, polypeptides, polyvinylpyrrolidone, maleic anhydride polymers, polyamides, polyvinyl alcohols, polyethylene oxides, glycosaminoglycans, polysaccharides, polyesters, polyurethanes, polystyrenes, copolymers, silicones, polyorthoesters, polyanhydrides, copolymers of vinyl monomers, polycarbonates, polyethylenes, polypropylenes, polylactic acids, polyglycolic acids, polycaprolactones, polyhydroxybutyrate valerates, polyacrylamides, polyethers, polyurethane dispersions, polyacrylates, acrylic latex dispersions, polyacrylic acid, mixtures and copolymers thereof. The polymers of the present invention may be natural or synthetic in origin, including gelatin, chitosan, dextrin, cyclodextrin,
Poly(urethanes), Poly(siloxanes) or silicones, Poly(acrylates) such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate), Poly(vinyl alcohol) Poly(olefms) such as poly(ethylene), poly(isoprene), halogenated polymers such as Poly(tetrafluoroethylene) - and derivatives and copolymers such as those commonly sold as Teflon® products, Poly(vinylidine fluoride), Polyvinyl acetate), Polyvinyl pyrrolidone),. Poly(acrylic acid), Polyacrylamide, Poly(ethylene-co-vinyl acetate), Poly(ethylene glycol), Poly(ρropylene glycol), Poly(methacrylic acid); etc. Suitable polymers also include absorbable and/or resorbable polymers including the following, combinations, copolymers and derivatives of the following: Polylactides (PLA), Polyglycolides (PGA), Polyøactide-co-glycolides) (PLGA), Polyanhydrides, Polyorthoesters, Poly(N-(2-hydroxypropyl) methacrylamide), PoIy(I- aspartamide), etc.
[0045] Any suitable polymer can preferably be used to carry out the present invention, including but not limited to: natural and synthetic polymers, gelatin, chitosan, dextrin, cyclodextrin, Poly(urethanes), Poly(siloxanes) or silicones , Poly(acrylates) such as poly(methyl methacrylate), poly(butyl methacrylate), and Poly(2-hydroxy ethyl methacrylate), Poly(vinyl alcohol) Poly(olefinds) such as poly(ethylene), poly(isoprene), halogenated polymers such as Poly(tetrafluoroethylene) - and derivatives and copolymers such as those commonly sold as Teflon® products, Poly(vinylidine fluoride), Poly(vinyl acetate), Polyvinyl pyrrolidone),. Poly(acrylic acid), Polyacrylamide, Poly(ethylene-co-vinyl acetate), Poly(ethylene glycol), Poly(ρroρylene glycol), Poly(methacrylic acid); etc. Suitable polymers also include absorbable
■' ' '" """• """' "- τ- ""• •"■"• **-" -1 Δ1 - -; the following, combinations, copolymers and eπvau of the following: Polylactides (PLA), Polyglycolides (PGA), Poly(lactide-co-glycolides) (PLGA), Polyanhydrides, Polyorthoesters, Poly(N-(2-hydroxypropyl) methacrylamide), PoIy(I- aspartamide), etc. [0046] "Therapeutically desirable morphology" as used herein refers to the gross form and structure of the pharmaceutical agent, once deposited on the substrate, so as to provide for optimal conditions of ex vivo storage, in vivo preservation and/or in vivo release. Such optimal conditions may include, but are not limited to increased shelf life, increased in vivo stability, good biocompatibility, good bioavailability or modified release rates. Typically, for the present invention, the desired morphology of a pharmaceutical agent would be crystalline or semi- crystalline, although this may vary widely depending on many factors including, but not limited to, the nature of the pharmaceutical agent, the disease to be treated/prevented, the intended storage conditions for the substrate prior to use or the location within the body of any biomedical implant. Preferably at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% or 100% of the pharmaceutical agent is in crystalline or semi-crystalline form.
[0047] "Stabilizing agent" as used herein refers to any substance that maintains or enhances the stability of the biological agent. Ideally these stabilizing agents are classified as Generally Regarded As Safe (GRAS) materials by the US Food and Drug Administration (FDA). Examples of stabilizing agents include, but are not limited to carrier proteins, such as albumin, gelatin, metals or inorganic salts.
[0048] "Supercritical fluid", "near-critical fluid", "critical fluid", "densifϊed fluid" or "densified gas" as used herein refers to a substance under pressure greater than ambient conditions, where it demonstrates a density greater than 0.4 g/cc but the mobility of a gas, i.e. a gas with liquid-like densities in which the pressure and temperature are above the critical point (the temperature and pressure at which the density of the liquid and vapor phases become identical). Examples of near critical fluids include fluids which are in gaseous state at standard temperature and pressure (STP) conditions and have a critical density above 0.2 g/cc. See, e.g., US Patent Νos. 6,860,123; 6,837,611; and 6,755,871. Examples of substances that demonstrate supercritical or near critical behavior suitable for the present invention include, but are not limited to carbon dioxide, ammonia, water, methanol, ethanol, ethane, propane, butane, pentane, dimethyl ether, xenon, sulfur hexafluoride, halogenated and partially halogenated materials such as chlorofluorocarbons, hydrochlorofluorocarbons, hydrofluorocarbons, perfluorocarbons (such as perfluoromethane and perfuoropropane, chloroform, trichloro-fluoromethane, dichloro- difluoromethane, dichloro-tetrafluoroethane) and mixtures thereof. Carbon dioxide is preferred.
■.FblfcjaaΘflia.c'ai'irlδfliQϋdfe, conditions including a temperature etween 0° C and 100° C and a pressure between 30 psig and 10,000 psig are preferred.
[0049] "Sintering" or "plasticizing" as used herein refers to the process by which the co- deposited pharmaceutical agent or biological agent-polymer matrix, as described above, becomes fused by treatment of the polymer matrix or substrate with a compressed gas, compressed liquid, or densified fluid that is a non-solvent for both the polymer and the pharmaceutical agent and biological agents, but a plasticizing agent for the polymer. In one example, carbon dioxide is used to treat a matrix of polymer or a substrate that has been coated with a polymer and a drug, using dry powder and RESS electrostatic coating processes. [0050] "Bulk properties" properties of a coating including a pharmaceutical or a biological agent that can be enhanced through the methods of the invention include for example: adhesion, smoothness, conformality, thickness, and compositional mixing.
[0051] "Rapid Expansion of Supercritical Solutions" or "RESS" as used herein involves the dissolution of a polymer into a compressed fluid, typically a densified fluid, followed by rapid expansion into a chamber at atmospheric pressure. The rapid expansion of the densified fluid solution through a small opening, with its accompanying decrease in density, reduces the dissolution capacity of the fluid and results in the nucleation and growth of polymer particles. [0052] "Solution Enhanced Dispersion of Supercritical Solutions" or "SEDS" as used herein involves a spray process for the generation of polymer particles, which, are formed when a compressed fluid (e.g. densified fluid, preferably densified CO2) is used as a diluent to a vehicle in which a polymer or the drug is dissolved, (one that can dissolve both the polymer or the drug and the densified gas). The mixing of the densified fluid diluent with the polymer-containing solution may be achieved by encounter of a first stream containing the polymer solution and a second stream containing the diluent densified fluid, for example, within one spray nozzle or by the use of multiple spray nozzles. The solvent in the polymer solution may be one compound or a mixture of two or more ingredients and may be or comprise an alcohol (including diols, triols, etc.), ether, amine, ketone, carbonate, or alkanes, or hydrocarbon (aliphatic or aromatic) or may be a mixture of compounds, such as mixtures of alkanes, or mixtures of one or more alkanes in combination with additional compounds such as one or more alcohols, (e.g., from 0 or 0.1 to 5% of a Cl to C15 alcohol, including diols, triols, etc.). See for example US Patent No. 6,669,785. The solvent may optionally contain a surfactant, as also described in (for example) US Patent No. 6,669,785.
[0053] In one embodiment of the SEDS process, a first stream of fluid comprising a polymer dissolved in a common solvent is co-sprayed with a second stream of densified fluid. Polymer particles are produced as the second stream acts as a diluent that weakens the solvent in the
pό y H r l idhϊBW€ilϊiSCfetream. The now combined streams of fluid, along with the polymer particles, flow out into a collection vessel which may or may not be at elevated pressure. In another embodiment of the SEDS process, a first stream of fluid comprising a drug dissolved in a common solvent is co-sprayed with a second stream of densified fluid. Drug particles are produced as the second stream acts as a diluent that weakens the solvent in the drug solution of the first stream. The now combined streams of fluid, along with the drug particles, flow out into a collection vessel which may or may not be at elevated pressure. [0054] Control of particle size, particle size distribution, and morphology is achieved by tailoring the following process variables: temperature, pressure, solvent composition of the first stream, flow-rate of the first stream, flow-rate of the second stream, composition of the second stream (where soluble additives may be added to the densified gas), and conditions of the capture vessel. Typically the capture vessel contains a fluid phase that is at least five to ten times (5-1Ox) atmospheric pressure. [0055] "Electrostatically charged" or "electrical potential" or "electrostatic capture" as used herein refers to the collection of the spray-produced particles upon a substrate that has a different electrostatic potential than the sprayed particles. Thus, the substrate is at an attractive electronic potential with respect to the particles exiting, which results in the capture of the particles upon the substrate, i.e. the substrate and particles are oppositely charged, and the particles transport through the fluid medium of the capture vessel onto the surface of the substrate is enhanced via electrostatic attraction. This may be achieved by charging the particles and grounding the substrate or conversely charging the substrate and grounding the particles, or by some other process, which would be easily envisaged by one of skill in the art of electrostatic capture. [0056] "Open vessel" as used herein refers to a vessel open to the outside atmosphere, and thus at substantially the same temperature and pressure as the outside atmosphere. [0057] "Closed vessel" as used herein refers to a vessel sealed from the outside atmosphere, and thus may be at significantly different temperatures and pressures to the outside atmosphere. [0058] The drug or active ingredient may be in any physical form, such as crystalline (including semicrystalline) and amorphous.
Solvents.
[0059] Solvents that may be used to carry out the present invention are, in some embodiments, gases (that is, compounds that are in the form of a gas at atmospheric pressure and 25 0C).
Examples of such solvents include but are not limited to carbon dioxide, ammonia, water, methanol, ethanol, ethane, propane, butane, pentane, dimethyl ether, xenon, sulfur hexafluoride,
, , _ , . hanogemateβaB'ciiiplaraanyii a Dgenate mateπals suc as c oro uorocar ons, hydrochlorofluorocarbons, hydrofluorocarbons, perfluorocarbons (such as perfluoromethane and perfuoropropane, chloroform, trichloro-fluoromethane, dichloro-difluoromethane, dichloro- tetrafluoroethane) and mixtures thereof. Carbon dioxide is preferred. [0060] The solvent may be utilized per se or a cosolvent may be included therewith (e.g., in an amount of from 0.01 or 0.1 to 20 or 30 percent by weight or more). Examples of cosolvents include, but are not limited to, water and organic co-solvents. The organic co-solvent may be one compound or a mixture of two or more ingredients. The organic co-solvent may be or comprise an alcohol (including diols, triols, etc.), ether, amine, ketone, carbonate, or alkanes, or hydrocarbon (aliphatic or aromatic) The organic co-solvent may be a mixture of compounds, such as mixtures of alkanes as given above, or mixtures of one or more alkanes in combination with additional compounds such as one or more alcohols as described above, (e.g., from 0 or 0.1 to 5% of a Cl to C15 alcohol (including diols, triols, etc.)). See, e.g., US Patent No. 6,669,785. The solvent may optionally contain a surfactant, as also described in (for example) US Patent No. 6,669,785.
[0061] The solvent is preferably provided in densified form. This densified form can be a gas at densities greater than 1.1 times the gas density at STP, a liquid (including near-supercritical fluids) or as a densified fluid, these three forms together sometimes being referred to as a "densified" fluid or "densified" gas. See, e.g., US Patent Nos. 6,860,123; 6,837,611; and 6,755,871.
Excipients.
[0062] Numerous pharmaceutical excipients that may be used to carry out the present invention are known. See, e.g., US Patent Nos. 6,767,558; 6,720,003; 6,710,059; and 6,649,627. Comprehensive examples are included in the Handbook of Pharmaceutical Excipients, edited by Raymond Rowe, Paul Sheskey and Paul Weller (4th Ed. 2003). Among other things, the drug- polymer composition may contain pharmaceutical excipients materials for: 1) enhancing the stability of the drug, 2) modifying the ultimate morphology of the drug or polymer, or drug polymer composite 3) inserting a porogen into the composite for subsequent removal in or during dense fluid processing, 4) improving the solubility characteristics of the drug in- vitro and in-vivo. Ideally these excipients are classified as Generally Regarded As Safe (GRAS) materials by the US Food and Drug Administration (FDA).
[0063] In category ' 1 ' above the excipient serves to stabilize the drug material. A primary example is represented by the use of sugars and other carbohydrates to stabilize proteins and peptides in pharmaceutical formulations. In the current invention one particularly useful sugar
.deiMWI ©Sύ®fefrEfc«BUS etate (SOA) which can serve to sta l ze prote ns n solution or n the solid state during compounding of the drug with the polymer. The SOA may also serve to benefit the composite in downstream processing described below.
[0064] In category '2' above the excipient serves to modify the morphology of the drug or polymer or the composite during and after processing with a dense gas fluid. One highlighted advantage to using dense gas fluids for processing drug-polymer composites relates to the "plasticizing" effect of the fluid (such as densified carbon dioxide) on the polymer. The fluid essentially permeates the free-volume of the polymer micro-structure lowering the glass transition temperature of the amorphous polymer and enhancing particle fusion at temperature much lower than those needed for heat bonding or fusion. This enhanced flow allows for suitable cohesion or adhesion of the formulated drug-polymer composite creating a semi-rigid composite product. The inclusion of excipients such as SOA may also serve to further plasticize the polymer thus enhancing the particle fusion and the overall solid-state integrity of the final composite. [0065] In category '3' above the excipient serves as a removable material (porogen) during the dense fluid processing step. For non-absorbable polymers it may be desirable to create increased surface area to affect drug removal in- vivo. By inclusion of the excipient material during the compounding step, a porous or semi-porous structure is created upon exposure to the dense fluid. In this case, the excipient is extracted from the formed composite leaving a micro- or nano-porous internal structure after completed dense fluid processing. One particular excipient of interest is SOA. Sucrose octaacetate is know to be soluble in dense carbon dioxide and in this case may serve as a stabilizer, a plasticizer, and a porogen. Other partially or fully acetylated sugars and carbohydrates may also be employed for these same purposes. [0066] In category '4' above the excipient increases the solubility of the drug as measure in- vitro and as applied in- vivo by preventing drug aggregation/agglomeration and by increasing the hydration capacity of the drug particle in-situ. Many drugs have poor aqueous solubility and therefore limited efficacy based on there ability to reach sufficient levels in the blood. Aside from particle size control (smaller particle size equals better dissolution profiles) excipients are used to prevent particle agglomeration and to enhance dissolution characteristics by increasing hydration in and around the particle. Noteworthy examples useful in the current invention include dextrin and its derivatives, other carbohydrates and simple sugars, and partially or fully acetylated sugars such as SOA. [0067] As outlined above the excipient may serve one or several of the purposes described.
„11 ιl;;:i( ,/QtnewsBimLlsxcipients include surfactants. Ideally, these suriacτants are classified as GRAS materials by the FDA. Suitable examples include but are not limited to sorbitan monooleate, Twean® trademarked surfactants, soy derived surfactants, and fatty acid derived GRAS surfactants. These surfactants may serve one or multiple roles as described above in this section.
[0069] As indicated above, SOA and other such hydrophobically derivatized carbohydrates (HDCs) can be utilized as the pharmaceutical excipient. HDCs are a wide variety of hydrophobically derivatized carbohydrates where at least one hydroxyl group is substituted with a hydrophobic moiety including, but not limited to, esters and ethers. Numerous examples of suitable HDCs and their syntheses are described in Developments in Food Carbohydrate, C. K. Lee, Applied Science Publishers, London (2d Ed. 1980) and PCT publication No. 96/03978. Other syntheses are described in, for example, Akoh et al. (1987) J. Food Sci. 52:1570; Khan et al. (1933) Tetra. Letts 14:1161; Khan (1984) Pure & Appl Chem. 56:833-844; and Khan et al. (1990) Carb. Res. 198:275-283. Specific examples of HDCs include, but are not limited to, sorbitol hexaacetate (SHAC), alpha-glucose pentaacetate (alpha-GPAC), beta-glucose pentaacetate (beta-GPAC), l-O-Octyl-.beta.-D-glucose tetraacetate (OGTA), trehalose octaacetate (TOAC), trehalose octapropionate (TOP), trehalose octa-3,3,dimethylbutyrate
(TO33DMB), trehalose diisobutyrate hexaacetate, trehalose octaisobutyrate, lactose octaacetate, sucrose octaacetate (SOAC), cellobiose octaacetate (COAC), raffmose undecaacetate (RUDA), sucrose octapropanoate, cellobiose octapropanoate, raffmose undecapropanoate, tetra-O-methyl trehalose, trehalose octapivalate, trehalose hexaacetate dipivalate and di-O-methyl-hexa-O-actyl sucrose and mixtures thereof. See, e.g., US Patent No. 6,517,860.
Methods of making and using. [0070] The method of the invention may be carried out by first, combining the drug with the polymer and optionally an excipient(s) to form a mixture. This mixing step may be carried out by any suitable technique or in any suitable apparatus, such as in a blender, extruder, etc. Typically both the drug and the polymer are provided in solid particulate form, and hence the mixture so formed will also be in the form of a solid. [0071] Typically the polymer and the drug particles range between 0.02 and 50 microns in size. In some embodiments the particle size is in a larger size range than the drug. In this case the polymer may range from 0.2 micron and 50 microns and the drug from 0.02 to 20 microns. [0072] Next, the mixture is contacted under pressure with a densified gas solvent as described above to form the composite material. Without wishing to be bound to any particular theory of the invention, it is believed that the densified gas solvent is at a pressure sufficient to reduce the
yiiidbl^ifWiihέipbljhKfeuMterial, trapping the fluid insoluble drug material in the polymer matrix as polymer particles fuse with adjacent polymer particles and hence form the drug/polymer composite article. As contrasted with other art utilizing dense fluid gases such as carbon dioxide at high pressures, many drugs, particularly protein-based drugs, are not soluble in the dense fluid and therefore are not efficiently infused into polymer matrices. In the current invention the drug, such as a protein-based therapeutic may remain largely unchanged as the polymer particle fuse around the drug particles. Depending upon the specific manner in which this step is carried out the drug/polymer composite can be in the form of discrete particles (which may for example be the same size but likely larger than the polymer particles previously provided) or may be in the form of a shaped article. Ideally, the composite mixture is used in conjunction with a mechanical article such as a mold or a template and the final composite article takes on the shape or general shape of that mold or template. So in working practice the mixture of the drug, polymer and excipients is added to a three-dimensional article, mechanically constrained such that the particles of both the drug and the polymer are immobilized. The densified fluid at the desired pressure and temperature is then allowed to permeate the three- dimensional article such to effect the fusion of the polymer particles without extraction or removal of either the drug or the polymer from the mechanical article. Finally the fluid is removed from the mechanical article by reducing the pressure to ambient levels and the final composite is then removed from the template as a semi-rigid solid composite. In general, this contacting step is carried out at a pressure between 300 and 15,000 psig and a temperature of between 2OC and 175 0C. Most preferably the contacting step is carried out at between300 and 3000 psig at a temperature between 2OC and 1000C
[0073] The step of combining the mixture with the solvent can be carried out by any suitable technique or in any suitable apparatus, such as in an extruder (which may be the same or different from the extruder noted above), mold (e.g., injection mold, blow mold, compression mold, etc.), reaction vessel, etc. A shaped article as described herein may, in some embodiments, be formed concurrently with this combining step, for example when the combining is carried out in a mold, or when the combining is carried out in an extruder and the composite formed therein then extruded through a die. In other embodiments, however, the shaped article will be formed in a subsequent step. Such subsequent forming may likewise be carried out by any suitable technique such as by spraying or dipping a pre-formed substrate with the composite material (e.g., to form a stent or biomedical implant). By use of a subsequent extruder or mold, etc. [0074] The drug/composite material may comprise, consist of, or consist essentially of:
U-BKiSi 0'02 βrø!J:;:!b,$rcent to 40,.50, or 60 percent y weight of drug (which may be a single compound or a combination of different active agents); and from 40 or 50 percent to 99.9 or 99.99 percent by weight of polymer; optionally, from 0.01 or 0.1 percent to 20 or 30 percent pharmaceutical excipient. [0075] In some embodiments, the physical form of the drug in the composite is substantially the same as the physical form of the drug before the combining step (b). For example, a drug initially provided in crystalline form remains in crystalline form in the composite; a drug initially provided in amorphous form remains in amorphous form in the composite; etc. [0076] In some embodiments, the composite is porous (this term including "semiporous"), with porous composites being made by inclusion of a porogen as a pharmaceutical excipient and subsequent removal of at least a portion thereof by an appropriate solvent (e.g., organic solvents; densified carbon dioxide solvent compositions as described herein) thereof after formation of the composite, in accordance with known techniques. In some embodiments the porogen is an SOA or other such hydrophobically derivatized carbohydrate as described above. [0077] Secondary coatings. Drug/polymer composites prepared as described above may optionally be coated (e.g., by spraying, dipping, or any suitable technique) with a second material to aid in the subsequent binding, forming, dispersion, structure or drug-elution profile of the drug/ polymer composite. This second material can be any of several different chemical functionalities and several different functions in the resulting drug/polymer composite material. For example, the second material can be a pharmaceutical excipient, providing a means to alter the pharmacological effect of the drug or providing a means to alter the release profile of the drug-delivery. In some embodiments the second material can be a CO2-philic material. In this case an additional process step can be utilized where after compressive forming of the part, a second condition of densified fluid can be used to remove the CO2-philic material, thereby forming pores in and rendering porosity to the formed part.
[0078] The present invention is explained in greater detail in the following non-limiting Examples.
Example 1
Preparation of a Drug Polymer Composite Article using Densified Fluid Processing
[0079] A cylindrical composite article consisting of 3 parts ρoly(butyl methacrylate), 2 parts recombinant Human Growth hormone (rHGh), and 1 part sucrose octaacetate is created in the following manner. Spherical emulsion prepared poly(butyl methacrylate) of an average size
ra S 'SlDliikiύTOhfeiisi'b αled with lyophilized HGh with an average particle size of 1.0 microns using an ultrasonic mixer. Dry sucrose octaacetate powder in the appropriate ratio is then added under constant mixing. The resulting formulation is then added to a cylindrical hollow mold constructed from sintered metal creating a fluid permeable three-dimensional article with an average pore size of 0.2 microns. The cylinder is open on both ends. Prior to the addition of the drug-polymer composition to the mold, one end is closed off using a matching cap designed to lock in place at the end of the cylinder. Once added to the mold, the composition is then mechanically compressed using a metal plunger matching the approximate inner diameter of the cylinder minus 0.001 -inch to remove the majority of the free-volume. The other end of the cylinder is then closed using an end cap that locks in place constraining the composition in three dimensions. The mold containing the polymer drug composition is then placed in a sterile pressure vessel to which 99.99% pure carbon dioxide is added to a pressure of 4000 psig at a temperature of 80C. The article is maintained in the CO2 environment at this temperature for 20 minutes after which the vessel is vented to atmospheric conditions. The mold is then removed from the vessel and the end caps are removed. The drug-polymer composite is then removed from the mold using a metal plunger fed from the open top of the mold thus pushing the composite out the bottom as the cylinder is mechanically restrained. Upon inspection the sample is a semi-rigid solid article in the shape of the mold. Upon thorough analysis of the polymer drug composite using Scanning Electron Microscopy (SEM) and routine chemical analysis it is determined that the solid article consists of a porous network of fused polymer particles with protein residing largely between adjacent fused particles and in void spaces created by the partial extraction of the sucrose octaacetate. Upon detailed morphological and chemical examination of the composite it is determined that the porous structure is largely inter-connected and partially opened to the outer surface of the article and the ratio of polymer to drug to sucrose octaacetate was 3:2:0.2 indicating substantial removal of the sucrose derivative during fluid processing.
Example 2
Preparation of a Drug Polymer Composite
Article using Densified Fluid Processing
[0080] A cylindrical composite article consisting of 4 parts poly(butyl methacrylate), 2 parts recombinant Human Growth hormone (rHGh), and 2 part sucrose octaacetate is created in the following manner. Spherical emulsion prepared poly(butyl methacrylate) of an average size
 with lyophilized HGh with an average particle size of LU microns using an ultrasonic mixer. Dry sucrose octaacetate powder in the appropriate ratio is then added under constant mixing. The resulting formulation is then added to a cylindrical hollow mold constructed from sintered metal creating a fluid permeable three-dimensional article with an average pore size of 0.2 microns. The cylinder is open on both ends. Prior to the addition of the drug-polymer composition to the mold, one end is closed off using a matching cap designed to lock in place at the end of the cylinder. The mold containing the polymer-drug- excipient mixture is then added to a pressure vessel equipped with a mechanical device designed with a piston actuator to exert pressure on the open end of the mold. The sealed pressure vessel is then filled with densified CO2 to a pressure of 3000 psi at a temperature of 80C. After 5 minutes at static pressure and temperature, the piston is actuated to apply mechanical pressure through the open end of the mold compressing the composition with 25 IbS-(Ui2)"1 of mechanical force. After 5 minutes of mechanical compression and exposure to CO2 at a pressure of 3000 psi (80C) the CO2 is vented from the chamber and the piston is removed from the open end of the cylindrical mold. The drug-polymer composite is then removed from the mold using a metal plunger fed from the open top of the mold thus pushing the composite out the bottom as the cylinder is mechanically restrained. Upon inspection the sample is a semi-rigid solid article in the shape of the mold.
with lyophilized HGh with an average particle size of LU microns using an ultrasonic mixer. Dry sucrose octaacetate powder in the appropriate ratio is then added under constant mixing. The resulting formulation is then added to a cylindrical hollow mold constructed from sintered metal creating a fluid permeable three-dimensional article with an average pore size of 0.2 microns. The cylinder is open on both ends. Prior to the addition of the drug-polymer composition to the mold, one end is closed off using a matching cap designed to lock in place at the end of the cylinder. The mold containing the polymer-drug- excipient mixture is then added to a pressure vessel equipped with a mechanical device designed with a piston actuator to exert pressure on the open end of the mold. The sealed pressure vessel is then filled with densified CO2 to a pressure of 3000 psi at a temperature of 80C. After 5 minutes at static pressure and temperature, the piston is actuated to apply mechanical pressure through the open end of the mold compressing the composition with 25 IbS-(Ui2)"1 of mechanical force. After 5 minutes of mechanical compression and exposure to CO2 at a pressure of 3000 psi (80C) the CO2 is vented from the chamber and the piston is removed from the open end of the cylindrical mold. The drug-polymer composite is then removed from the mold using a metal plunger fed from the open top of the mold thus pushing the composite out the bottom as the cylinder is mechanically restrained. Upon inspection the sample is a semi-rigid solid article in the shape of the mold.
Example 3 [0081] Figures 1-5 illustrate embodiments of the invention including SEM images of fused
PGLA matrix formed according to the invention with and without drug present in the interstices formed by fusing the PGLA polymer. Elution profiles for an implant according to the invention formed with PGLA as the polymer and esculetin as the drug are also shown. [0082] The foregoing is illustrative of the present invention, and is not to be construed as limiting thereof. The invention is defined by the following claims, with equivalents of the claims to be included therein.
Claims
WHAT IS CLAIMED IS:
1. A composite comprising at least one polymer; at least one pharmaceutical agent in a therapeutically desirable morphology; and optionally at least one excipient; wherein the pharmaceutical agent is sequestered in interstices formed by fusing particles of the polymer.
2. The composite of Claim 1 wherein the pharmaceutical agent is a biological agent having a secondary, tertiary and/or quaternary structure required for biological activity, wherein said structure is substantially retained when the particles of the biological agent are sequestered within interstices formed by the fused polymer particles.
3. The composite of Claim 1 wherein the pharmaceutical agent is a peptide, protein, enzyme, nucleic acid, antibody, therapeutic vaccine, antisense nucleic acid, antimicrobial, vitamin, hormone, steroid, lipid, polysaccharide or carbohydrate. • 4. The composite of Claim 1, wherein the pharmaceutical agent is a therapeutic protein.
5. The composite of Claim 4, wherein the therapeutic protein is one or more of erythropoietin, interferon, insulin, blood factor, colony stimulating factor, growth hormone, interleukin and growth factor.
6. The composite of Claim 1, wherein the pharmaceutical agent is one or more of antirestenotic agents, antidiabetics, analgesics, antiinflammatory agents, antirheumatics, antihypotensive agents, antihypertensive agents, psychoactive drugs, tranquillizers, antiemetics, muscle relaxants, glucocorticoids, agents for treating ulcerative colitis or Crohn's disease, antiallergics, antibiotics, antiepileptics, anticoagulants, antimycotics, antitussives, arteriosclerosis remedies, diuretics, proteins, peptides, enzymes, enzyme inhibitors, gout remedies, hormones and inhibitors thereof, cardiac glycosides, immunotherapeutic agents and cytokines, laxatives, lipid-lowering agents, migraine remedies, mineral products, otologicals, anti parkinson agents, thyroid therapeutic agents, spasmolytics, platelet aggregation inhibitors, vitamins, cytostatics and metastasis inhibitors, phytopharmaceuticals, chemotherapeutic agents and amino acids. Examples of suitable active ingredients are acarbose, antigens, beta-receptor blockers, non-steroidal antiinflammatory drugs {NSAIDs], cardiac glycosides, acetylsalicylic acid, virustatics, aclarubicin, acyclovir, cisplatin, actinomycin, alpha- and beta-sympatomimetics, (dmeprazole, allopurinol, alprostadil, prostaglandins, amantadine, ambroxol, amlodipine, methotrexate, S-aminosalicylic acid amitriptyline, amoxicillin, anastrozole, atenolol, azathioprine, balsalazide, beclomethasone, betahistine, bezafibrate, bicalutamide,
 u examac, uprenoφ ne, met a one, calcium salts, potassium salts, magnesium salts, candesartan, carbamazepine, captopril, cefalosporins, cetirizine, chenodeoxycholic acid, ursodeoxycholic acid, theophylline and theophylline derivatives, trypsins, cimetidine, clarithromycin, clavulanic acid, clindamycin, clobutinol, clonidine, cotrimoxazole, codeine, caffeine, vitamin D and derivatives of vitamin D, colestyramine, cromoglicic acid, coumarin and coumarin derivatives, cysteine, cytarabine, cyclophosphamide, ciclosporin, cyproterone, cytabarine, dapiprazole, desogestrel, desonide, dihydralazine, diltiazem, ergot alkaloids, dimenhydrinate, dimethyl sulphoxide, dimeticone, domperidone and domperidan derivatives, dopamine, doxazosin, doxorubizin, doxylamine, dapiprazole, benzodiazepines, diclofenac, glycoside antibiotics, desipramine, econazole, ACE inhibitors, enalapril, ephedrine, epinephrine, epoetin and epoetin derivatives, morphinans, calcium antagonists, irinotecan, modafmil, orlistat, peptide antibiotics, phenytoin, riluzoles, risedronate, sildenafil, topiramate, macrolide antibiotics, oestrogen and oestrogen derivatives, progestogen and progestogen derivatives, testosterone and testosterone derivatives, androgen and androgen derivatives, ethenzamide, etofenamate, etofibrate, fenofibrate, etofylline, etoposide, famciclovir, famotidine, felodipine, fenofibrate, fentanyl, fenticonazole, gyrase inhibitors, fluconazole, fludarabine, fluarizine, fluorouracil, fluoxetine, flurbiprofen, ibuprofen, flutamide, fluvastatin, follitropin, formoterol, fosfomicin, furosemide, fusidic acid, gallopamil, ganciclovir, gemfibrozil, gentamicin, ginkgo, Saint John's wort, glibenclamide, urea derivatives as oral antidiabetics, glucagon, glucosamine and glucosamine derivatives, glutathione, glycerol and glycerol derivatives, hypothalamus hormones, goserelin, gyrase inhibitors, guanethidine, halofantrine, haloperidol, heparin and heparin derivatives, hyaluronic acid, hydralazine, hydrochlorothiazide and hydrochlorothiazide derivatives, salicylates, hydroxyzine, idarubicin, ifosfamide, imipramine, indometacin, indoramine, insulin, interferons, iodine and iodine derivatives, isoconazole, isoprenaline, glucitol and glucitol derivatives, itraconazole, ketoconazole, ketoprofen, ketotifen, lacidipine, lansoprazole, levodopa, levomethadone, thyroid hormones, lipoic acid and lipoic acid derivatives, lisinopril, lisuride, lofepramine, lomustine, loperamide, loratadine, maprotiline, mebendazole, mebeverine, meclozine, mefenamic acid, mefloquine, meloxicam, mepindolol, meprobamate, meropenem, mesalazine, mesuximide, metamizole, metformin, methotrexate, methylphenidate, methylprednisolone, metixene, metoclopramide, metoprolol, metronidazole, mianserin, miconazole, minocycline, minoxidil, misoprostol, mitomycin, mizolastine, moexipril, morphine and morphine
gs|.|4SeJi|n^ primrose, nalbuphine, naloxone, tilidine, naproxen, narcotine, natamycin, neostigmine, nicergoline, nicethamide, nifedipine, niflumic acid, nimodipine, nimorazole, nimustine, nisoldipine, adrenaline and adrenaline derivatives, norfloxacin, novamine sulfone, noscapine, nystatin, ofloxacin, olanzapine, olsalazine, omeprazole, omoconazole, ondansetron, oxaceprol, oxacillin, oxiconazole, oxymetazoline, pantoprazole, paracetamol, paroxetine, penciclovir, oral penicillins, pentazocine, pentifylline, pentoxifylline, perphenazine, pethidine, plant extracts, phenazone, pheniramine, barbituric acid derivatives, phenylbutazone, phenytoin, pimozide, pindolol, piperazine, piracetam, pirenzepine, piribedil, piroxicam, pramipexole, pravastatin, prazosin, procaine, promazine, propiverine, propranolol, propyphenazone, prostaglandins, protionamide, proxyphylline, quetiapine, quinapril, quinaprilat, ramipril, ranitidine, reproterol, reserpine, ribavirin, rifampicin, risperidone, ritonavir, ropinirole, roxatidine, roxithromycin, ruscogenin, rutoside and rutoside derivatives, sabadilla, salbutamol, salmetefol, scopolamine, selegiline, sertaconazole, sertindole, sertralion, silicates, sildenafil, simvastatin, sitosterol, sotalol, spaglumic acid, sparfloxacin, spectinomycin, spiramycin, spirapril, spironolactone, stavudine, streptomycin, sucralfate, sufentanil, sulbactam, sulphonamides, sulfasalazine, sulpiride, sultamicillin, sultiam, sumatriptan, suxamethonium chloride, tacrine, tacrolimus, taliolol, tamoxifen, taurolidine, tazarotene, temazepam, teniposide, tenoxicam, terazosin, terbinafine, terbutaline, terfenadine, terlipressin, tertatolol, tetracyclins, teryzoline, theobromine, theophylline, butizine, thiamazole, phenothiazines, thiotepa, tiagabine, tiapride, propionic acid derivatives, ticlopidine, timolol, tinidazole, tioconazole, tioguanine, tioxolone, tiropramide, tizanidine, tolazoline, tolbutamide, tolcapone, tolnaftate, tolperisone, topotecan, torasemide, antioestrogens, tramadol, tramazoline, trandolapril, tranylcypromine, trapidil, trazodone, triamcinolone and triamcinolone derivatives, triamterene, trifluperidol, trifluridine, trimethoprim, trimipramine, tripelennamine, triprolidine, trifosfamide, tromantadine, trometamol, tropalpin, troxerutine, tulobuterol, tyramine, tyrothricin, urapidil, ursodeoxycholic acid, chenodeoxycholic acid, valaciclovir, valproic acid, vancomycin, vecuronium chloride, Viagra, venlafaxine, verapamil, vidarabine, vigabatrin, viloazine, vinblastine, vincamine, vincristine, vindesine, vinorelbine, vinpocetine, viquidil, warfarin, xantinol nicotinate, xipamide, zafirlukast, zalcitabine, zidovudine, zolmitriptan, Zolpidem, zoplicone, and zotipine. 7. The composite of Claim 1 wherein the pharmaceutical agent is a drug in substantially crystalline form.
!l$im 1, wherein the composite provides a rate of elution that remains within a 15% variation from a selected rate of elution between day 2 and day 30 after the composite is implanted in a subject under physiological conditions.
u examac, uprenoφ ne, met a one, calcium salts, potassium salts, magnesium salts, candesartan, carbamazepine, captopril, cefalosporins, cetirizine, chenodeoxycholic acid, ursodeoxycholic acid, theophylline and theophylline derivatives, trypsins, cimetidine, clarithromycin, clavulanic acid, clindamycin, clobutinol, clonidine, cotrimoxazole, codeine, caffeine, vitamin D and derivatives of vitamin D, colestyramine, cromoglicic acid, coumarin and coumarin derivatives, cysteine, cytarabine, cyclophosphamide, ciclosporin, cyproterone, cytabarine, dapiprazole, desogestrel, desonide, dihydralazine, diltiazem, ergot alkaloids, dimenhydrinate, dimethyl sulphoxide, dimeticone, domperidone and domperidan derivatives, dopamine, doxazosin, doxorubizin, doxylamine, dapiprazole, benzodiazepines, diclofenac, glycoside antibiotics, desipramine, econazole, ACE inhibitors, enalapril, ephedrine, epinephrine, epoetin and epoetin derivatives, morphinans, calcium antagonists, irinotecan, modafmil, orlistat, peptide antibiotics, phenytoin, riluzoles, risedronate, sildenafil, topiramate, macrolide antibiotics, oestrogen and oestrogen derivatives, progestogen and progestogen derivatives, testosterone and testosterone derivatives, androgen and androgen derivatives, ethenzamide, etofenamate, etofibrate, fenofibrate, etofylline, etoposide, famciclovir, famotidine, felodipine, fenofibrate, fentanyl, fenticonazole, gyrase inhibitors, fluconazole, fludarabine, fluarizine, fluorouracil, fluoxetine, flurbiprofen, ibuprofen, flutamide, fluvastatin, follitropin, formoterol, fosfomicin, furosemide, fusidic acid, gallopamil, ganciclovir, gemfibrozil, gentamicin, ginkgo, Saint John's wort, glibenclamide, urea derivatives as oral antidiabetics, glucagon, glucosamine and glucosamine derivatives, glutathione, glycerol and glycerol derivatives, hypothalamus hormones, goserelin, gyrase inhibitors, guanethidine, halofantrine, haloperidol, heparin and heparin derivatives, hyaluronic acid, hydralazine, hydrochlorothiazide and hydrochlorothiazide derivatives, salicylates, hydroxyzine, idarubicin, ifosfamide, imipramine, indometacin, indoramine, insulin, interferons, iodine and iodine derivatives, isoconazole, isoprenaline, glucitol and glucitol derivatives, itraconazole, ketoconazole, ketoprofen, ketotifen, lacidipine, lansoprazole, levodopa, levomethadone, thyroid hormones, lipoic acid and lipoic acid derivatives, lisinopril, lisuride, lofepramine, lomustine, loperamide, loratadine, maprotiline, mebendazole, mebeverine, meclozine, mefenamic acid, mefloquine, meloxicam, mepindolol, meprobamate, meropenem, mesalazine, mesuximide, metamizole, metformin, methotrexate, methylphenidate, methylprednisolone, metixene, metoclopramide, metoprolol, metronidazole, mianserin, miconazole, minocycline, minoxidil, misoprostol, mitomycin, mizolastine, moexipril, morphine and morphine
gs|.|4SeJi|n^ primrose, nalbuphine, naloxone, tilidine, naproxen, narcotine, natamycin, neostigmine, nicergoline, nicethamide, nifedipine, niflumic acid, nimodipine, nimorazole, nimustine, nisoldipine, adrenaline and adrenaline derivatives, norfloxacin, novamine sulfone, noscapine, nystatin, ofloxacin, olanzapine, olsalazine, omeprazole, omoconazole, ondansetron, oxaceprol, oxacillin, oxiconazole, oxymetazoline, pantoprazole, paracetamol, paroxetine, penciclovir, oral penicillins, pentazocine, pentifylline, pentoxifylline, perphenazine, pethidine, plant extracts, phenazone, pheniramine, barbituric acid derivatives, phenylbutazone, phenytoin, pimozide, pindolol, piperazine, piracetam, pirenzepine, piribedil, piroxicam, pramipexole, pravastatin, prazosin, procaine, promazine, propiverine, propranolol, propyphenazone, prostaglandins, protionamide, proxyphylline, quetiapine, quinapril, quinaprilat, ramipril, ranitidine, reproterol, reserpine, ribavirin, rifampicin, risperidone, ritonavir, ropinirole, roxatidine, roxithromycin, ruscogenin, rutoside and rutoside derivatives, sabadilla, salbutamol, salmetefol, scopolamine, selegiline, sertaconazole, sertindole, sertralion, silicates, sildenafil, simvastatin, sitosterol, sotalol, spaglumic acid, sparfloxacin, spectinomycin, spiramycin, spirapril, spironolactone, stavudine, streptomycin, sucralfate, sufentanil, sulbactam, sulphonamides, sulfasalazine, sulpiride, sultamicillin, sultiam, sumatriptan, suxamethonium chloride, tacrine, tacrolimus, taliolol, tamoxifen, taurolidine, tazarotene, temazepam, teniposide, tenoxicam, terazosin, terbinafine, terbutaline, terfenadine, terlipressin, tertatolol, tetracyclins, teryzoline, theobromine, theophylline, butizine, thiamazole, phenothiazines, thiotepa, tiagabine, tiapride, propionic acid derivatives, ticlopidine, timolol, tinidazole, tioconazole, tioguanine, tioxolone, tiropramide, tizanidine, tolazoline, tolbutamide, tolcapone, tolnaftate, tolperisone, topotecan, torasemide, antioestrogens, tramadol, tramazoline, trandolapril, tranylcypromine, trapidil, trazodone, triamcinolone and triamcinolone derivatives, triamterene, trifluperidol, trifluridine, trimethoprim, trimipramine, tripelennamine, triprolidine, trifosfamide, tromantadine, trometamol, tropalpin, troxerutine, tulobuterol, tyramine, tyrothricin, urapidil, ursodeoxycholic acid, chenodeoxycholic acid, valaciclovir, valproic acid, vancomycin, vecuronium chloride, Viagra, venlafaxine, verapamil, vidarabine, vigabatrin, viloazine, vinblastine, vincamine, vincristine, vindesine, vinorelbine, vinpocetine, viquidil, warfarin, xantinol nicotinate, xipamide, zafirlukast, zalcitabine, zidovudine, zolmitriptan, Zolpidem, zoplicone, and zotipine. 7. The composite of Claim 1 wherein the pharmaceutical agent is a drug in substantially crystalline form.
!l$im 1, wherein the composite provides a rate of elution that remains within a 15% variation from a selected rate of elution between day 2 and day 30 after the composite is implanted in a subject under physiological conditions.
9. The composite of Claim 1 wherein the composite provides an elution profile wherein about 10% to about 50% of the pharmaceutical agent is eluted at week 1 after the composite is implanted in a subject under physiological conditions, about 25% to about 75% of the pharmaceutical agent is eluted at week 2 and about 50% to about 75% of the phaπnaceutical agent is eluted at week 4. .
10. The composite of Claim 1 wherein said polymer and said pharmaceutical agent form two substantially separate phases.
11. A method of preparing a composite comprising
(a) forming a mixture by combining particles of at least one polymer with particles of at least one pharmaceutical agent in a therapeutically desirable morphology, and optionally with particles of at least one excipient; and (b) plasticizing said mixture under conditions that do not substantially modify the morphology of said pharmaceutical agent wherein the pharmaceutical agent is sequestered in interstices formed by fused particles of the polymer.
12. The method of claim 11, further comprising forming a shaped article from said composite. 13. The method of claim 12 wherein said shaped article is formed by placing the mixture of step (a) in a mold prior to performing said plasticizing of step (b).
14. The method of claim 12 wherein said shaped article is a biomedical implant.
15. The method of claim 12 wherein said biomedical implant is a drug depot or a stent.
16. The method of claim 15, wherein said shaped article is a porous subcutaneous drug depot.
17. The method of claim 11, further comprising the step of coating said composite with a polymer film to further control elution of said pharmaceutical agent.
18. The method of claim 11 wherein said mixing step (a) is performed in an extruder.
19. The method of claim 11 wherein said plasticizing step (b) is carried out at a temperature from about O0C to about 1000C.
20. The method of claim 11 wherein said plasticizing step (b) is carried out at a pressure of from about 30 psig to about 10,000 psig.
21. The method of claim 11 wherein said plasticizing step (b) comprises treating said mixture with a densified gas, densified liquid, or densified fluid that is a non-solvent for said at
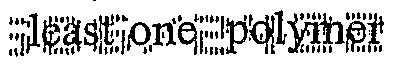 ,&d said at least one pharmaceut ca agent or active biological agent, but a plasticizing agent for the polymer. 22. The method of claim 11 wherein said densified gas, densifϊed liquid, or densified fluid comprises carbon dioxide, isobutylene or a mixture thereof. 23. The method of claim 11 wherein the ratio of pharmaceutical agent or active biological agent to polymer is from about 1 : 1 to about 1 : 10,000.
,&d said at least one pharmaceut ca agent or active biological agent, but a plasticizing agent for the polymer. 22. The method of claim 11 wherein said densified gas, densifϊed liquid, or densified fluid comprises carbon dioxide, isobutylene or a mixture thereof. 23. The method of claim 11 wherein the ratio of pharmaceutical agent or active biological agent to polymer is from about 1 : 1 to about 1 : 10,000.
24. The method of claim 11 wherein said polymer is selected from the group og PGLA, (DL)-PLA, and (L)-PLA.
25. The method of claim 11 wherein said therapeutically desirable morphology is crystalline or semi-crystalline.
26. The method of claim 11 wherein at least 50% of said pharmaceutical agent in the plasticized mixture is crystalline or semicrystalline.
27. The method of claim 11 wherein said therapeutically desirable morphology is not substantially changed after said plasticizing step (b). 28. The method of claim 11 wherein the pharmaceutical agent is an active biological agent having a secondary, tertiary and/or quaternary structure.
29. The method of claim 11 wherein said secondary, tertiary and/or quaternary structure is not substantially changed after said plasticizing step (b).
30. The method of claim 11 wherein said excipient is selected from the group consisting of adjuvants, surfactants, stabilizers, morphology modifiers, porogens, diluents, carriers, solubilizers, antioxidants, lubricants, binders!, disintigrants, and mixtures thereof.
31. The method of claim 11 wherein said excipient is a porogen.
32. The method of claim 31 further comprising contacting said composition of matter with a solvent to at least partially solubilize said porogen and form pores in said composite. 33. The method of claim 11 wherein said excipient is a hydrophobically derivatized carbohydrate.
34. The method of claim 33 wherein said hydrophobically derivatized carbohydrateis selected from the group consisting of sorbitol hexaacetate, alpha-glucose pentaacetate, beta-glucose pentaacetate, 1-0-Octyl-beta-D-glucose tetraacetate, trehalose octaacetate, tetralose octapropionate, trehalose octa-3,3,dimethylbutyrate, trehalose diisobutyrate hexaacetate, trehalose octaisobutyrate, lactose octaacetate, sucrose octaacetate, cellobiose octaacetate, raffϊnose undecaacetate, sucrose octapropanoate, cellobiose octapropanoate, raffmose undecapropanoate, tetra-0-methyl trehalose, trehalose octapivalate, trehalose hexaacetate dipivalate and di-0-methyl-hexa-O-actyl sucrose and mixtures thereof.
tMiϋm^U'WI-β itøj||n 11 wherein said composite comprises a substantially continuous polymer phase and a substantially discontinuous, discrete pharmaceutical agent or active biological agent particulate phase.
36. The method of claim 11 further comprising preparing said particles of polymer and/or said particles of pharmaceutical agent using a RESS process or a SEDS process.
37. The method of claim 11 further comprising electrostatic capture to deposit said particles of polymer and/or said particles of pharmaceutical agent in a mold for forming said composite, wherein said mold and said particles of polymer and/or said particles of pharmaceutical agent are maintained at an electrical potential. 38. The method of claim 11 wherein step (b) comprises forming a polymer microstructure.
39. The method of Claim 38 wherein the pharmaceutical particles are sequestered or encapsulated within said microstructure.
40. The method of claim 39 wherein said microstructure comprises microchannels, micropores and/or microcavities. 41. A method of forming a drug/polymer composite material, comprising the steps of:
(a) mixing a solid particulate drug material with a solid particulate polymer material, and optionally with a pharmaceutical excipient, to form a particle mixture of polymer particles and interspersed drug particles; and then
(b) combining said particle mixture with a densified gas solvent at a pressure sufficient to reduce the viscosity of said polymer material, fuse said polymer particles to one another and capture said drug particles therebetween and form a drug/polymer composite material from said particulate mixture.
42. The method of claim 41, wherein said combining step (b) is carried out in a mold so that a shaped article of said drug/polymer composite material is thereby produced. 43. The method of claim 42, wherein said shaped article is a stent, drug depot, or biomedical implant.
44. The method of claim 1, further comprising the step:
(a) forming a shaped article from said drug/polymer composite material.
45. The method of claim 44, wherein said combining step (b) is carried out in an extruder. 46. The method of claim 44, wherein said forming step (c) is carried out by molding.
47. The method of claim 44, wherein said forming step (c) is carried out by coating a preformed substrate with said drug/polymer composite material.
48. The method of claim 44, wherein said shaped article is a stent, drug depot, or biomedical implant. 49. The method of claim 41, wherein said drug is in crystalline or amorphous form.
I, where n sa d drug is a prote n or peptide.
51. The method of claim 1, wherein said composite material comprises: from 0.01 percent to 50 percent by weight of drug; from 50 to 99.99 percent by weight of polymer; and optionally, from 0.01 to 30 percent by weight of pharmaceutical excipient.
52. The method of claim 41, wherein said pharmaceutical excipient is absent.
53. The method of claim 41, wherein said pharmaceutical excipient is present.
54. The method of claim 53, wherein said pharmaceutical excipient is selected from the group consisting of adjuvants, surfactants, stabilizers, morphology modifiers, porogens, diluents, carriers, solubilizers, antioxidants, lubricants, binders, disintigrants, and mixtures thereof.
55. The method of claim 53, wherein said pharmaceutical excipient is a hydrophobically derivatized carbohydrate.
56. The method of claim 55, wherein said hydrophobically derivatized carbohydrate is selected from the group consisting of sorbitol hexaacetate, alpha-glucose pentaacetate, beta-glucose pentaacetate, 1-0-Octyl-beta-D-glucose tetraacetate, trehalose octaacetate, tetralose octapropionate, trehalose octa-3,3,dimethylbutyrate, trehalose diisobutyrate hexaacetate, trehalose octaisobutyrate, lactose octaacetate, sucrose octaacetate, cellobiose octaacetate, raffinose undecaacetate, sucrose octapropanoate, cellobiose octapropanoate, raffinose undecapropanoate, tetra-0-methyl trehalose, trehalose octapivalate, trehalose hexaacetate dipivalate and di-0-methyl-hexa-O-actyl sucrose and mixtures thereof.
57. The method of claim 41, further comprising the step of coating said composite material with a secondary material.
58. The method of claim 41, wherein said excipient is a porogen, said method further comprising the step of contacting said composite material to a solvent to at least partially solubilize said porogen and form pores in said composite material.
59. A drug/polymer composite material produced by the process of claim 41
60. The composite of claim 59 wherein said composite is porous.
61. A method of treating a subject with a drug, comprising administering a drug/polymer composite material of claim 59 to said subject in an amount effective to treat said subject with said drug.
62. A shaped article comprising a drug/polymer composite material of claim 59.
63. shaped article of claim 62, wherein said shaped article is a stent, drug depot, or biomedical implant.
i Q .cam 62, wherein said shaped article is a porous subcutaneous drug depot.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2613280A CA2613280C (en) | 2005-06-22 | 2006-06-21 | Drug/polymer composite materials and methods of making the same |
| EP06773731.2A EP1898878B1 (en) | 2005-06-22 | 2006-06-21 | Drug/polymer composite materials and methods of making the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/158,724 | 2005-06-22 | ||
| US11/158,724 US20070009564A1 (en) | 2005-06-22 | 2005-06-22 | Drug/polymer composite materials and methods of making the same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007002238A2 true WO2007002238A2 (en) | 2007-01-04 |
| WO2007002238A3 WO2007002238A3 (en) | 2007-05-10 |
Family
ID=37595812
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/024221 WO2007002238A2 (en) | 2005-06-22 | 2006-06-21 | Drug/polymer composite materials and methods of making the same |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US20070009564A1 (en) |
| EP (1) | EP1898878B1 (en) |
| CA (2) | CA2613280C (en) |
| WO (1) | WO2007002238A2 (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009013552A1 (en) | 2007-07-23 | 2009-01-29 | Richter Gedeon Nyrt. | Controlled release pharmaceutical composition of tolperison hydrochloride |
| DE102007043883A1 (en) * | 2007-09-14 | 2009-03-26 | Biotronik Vi Patent Ag | Stent with a coating |
| WO2012083594A1 (en) * | 2010-12-24 | 2012-06-28 | Dongguan Tiantianxiangshang Medical Technology Co., Ltd | Biodegradable drug eluting stent and methodsof making the same. |
| US8642631B2 (en) | 2008-05-27 | 2014-02-04 | University Of Melbourne | Methods of treating mammals with eustachian tube dysfunctions |
| CN103877049A (en) * | 2014-04-04 | 2014-06-25 | 白玲强 | Tablet containing fenofibrate and preparation technology thereof |
| US8921365B2 (en) | 2007-07-23 | 2014-12-30 | Biomet Deutschland Gmbh | Pharmaceutical composition, substrate comprising a pharmaceutical composition, and use of a pharmaceutical composition |
| US9486338B2 (en) | 2007-04-17 | 2016-11-08 | Micell Technologies, Inc. | Stents having controlled elution |
| AU2015200571B2 (en) * | 2007-04-17 | 2016-11-17 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US9539593B2 (en) | 2006-10-23 | 2017-01-10 | Micell Technologies, Inc. | Holder for electrically charging a substrate during coating |
| US9636309B2 (en) | 2010-09-09 | 2017-05-02 | Micell Technologies, Inc. | Macrolide dosage forms |
| CN106619532A (en) * | 2016-11-16 | 2017-05-10 | 山东裕欣药业有限公司 | Roxithromycin ambroxol hydrochloride dry suspension and preparation method thereof |
| US9687864B2 (en) | 2010-03-26 | 2017-06-27 | Battelle Memorial Institute | System and method for enhanced electrostatic deposition and surface coatings |
| US9737645B2 (en) | 2006-04-26 | 2017-08-22 | Micell Technologies, Inc. | Coatings containing multiple drugs |
| US9737642B2 (en) | 2007-01-08 | 2017-08-22 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US9789233B2 (en) | 2008-04-17 | 2017-10-17 | Micell Technologies, Inc. | Stents having bioabsorbable layers |
| US9827117B2 (en) | 2005-07-15 | 2017-11-28 | Micell Technologies, Inc. | Polymer coatings containing drug powder of controlled morphology |
| US9981072B2 (en) | 2009-04-01 | 2018-05-29 | Micell Technologies, Inc. | Coated stents |
| US9981071B2 (en) | 2008-07-17 | 2018-05-29 | Micell Technologies, Inc. | Drug delivery medical device |
| CN108456714A (en) * | 2013-03-14 | 2018-08-28 | 简·探针公司 | Diagnostic system and method |
| US10117972B2 (en) | 2011-07-15 | 2018-11-06 | Micell Technologies, Inc. | Drug delivery medical device |
| US10188772B2 (en) | 2011-10-18 | 2019-01-29 | Micell Technologies, Inc. | Drug delivery medical device |
| US10232092B2 (en) | 2010-04-22 | 2019-03-19 | Micell Technologies, Inc. | Stents and other devices having extracellular matrix coating |
| US10272606B2 (en) | 2013-05-15 | 2019-04-30 | Micell Technologies, Inc. | Bioabsorbable biomedical implants |
| US10350391B2 (en) | 2008-07-17 | 2019-07-16 | Micell Technologies, Inc. | Drug delivery medical device |
| US10464100B2 (en) | 2011-05-31 | 2019-11-05 | Micell Technologies, Inc. | System and process for formation of a time-released, drug-eluting transferable coating |
| US10835396B2 (en) | 2005-07-15 | 2020-11-17 | Micell Technologies, Inc. | Stent with polymer coating containing amorphous rapamycin |
| US11039943B2 (en) | 2013-03-12 | 2021-06-22 | Micell Technologies, Inc. | Bioabsorbable biomedical implants |
| US11369498B2 (en) | 2010-02-02 | 2022-06-28 | MT Acquisition Holdings LLC | Stent and stent delivery system with improved deliverability |
| US11426494B2 (en) | 2007-01-08 | 2022-08-30 | MT Acquisition Holdings LLC | Stents having biodegradable layers |
| US11904118B2 (en) | 2010-07-16 | 2024-02-20 | Micell Medtech Inc. | Drug delivery medical device |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2432556T3 (en) | 2004-08-04 | 2013-12-04 | Evonik Corporation | Methods for manufacturing supply devices and their devices |
| US20080181958A1 (en) * | 2006-06-19 | 2008-07-31 | Rothrock Ginger D | Nanoparticle fabrication methods, systems, and materials |
| US8636767B2 (en) | 2006-10-02 | 2014-01-28 | Micell Technologies, Inc. | Surgical sutures having increased strength |
| EA020509B1 (en) * | 2007-04-17 | 2014-11-28 | Миселл Текнолоджиз, Инк. | Stents having biodegradable layers |
| AU2008256684B2 (en) * | 2007-05-25 | 2012-06-14 | Micell Technologies, Inc. | Polymer films for medical device coating |
| US20100298928A1 (en) * | 2007-10-19 | 2010-11-25 | Micell Technologies, Inc. | Drug Coated Stents |
| US8124601B2 (en) * | 2007-11-21 | 2012-02-28 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| EP2222281B1 (en) | 2007-12-20 | 2018-12-05 | Evonik Corporation | Process for preparing microparticles having a low residual solvent volume |
| US8298466B1 (en) * | 2008-06-27 | 2012-10-30 | Abbott Cardiovascular Systems Inc. | Method for fabricating medical devices with porous polymeric structures |
| US8834913B2 (en) * | 2008-12-26 | 2014-09-16 | Battelle Memorial Institute | Medical implants and methods of making medical implants |
| CA2756307C (en) * | 2009-03-23 | 2017-08-08 | Micell Technologies, Inc. | Peripheral stents having layers and reinforcement fibers |
| US20100239635A1 (en) * | 2009-03-23 | 2010-09-23 | Micell Technologies, Inc. | Drug delivery medical device |
| EP2411440B1 (en) * | 2009-03-23 | 2018-01-17 | Micell Technologies, Inc. | Improved biodegradable polymers |
| EP2480200A2 (en) * | 2009-09-22 | 2012-08-01 | Evonik Degussa Corporation | Implant devices having varying bioactive agent loading configurations |
| WO2011146483A1 (en) | 2010-05-17 | 2011-11-24 | Aerie Pharmaceuticals, Inc. | Drug delivery devices for delivery of ocular therapeutic agents |
| US10028858B2 (en) | 2011-07-11 | 2018-07-24 | Medicines360 | Intrauterine systems, IUD insertion devices, and related methods and kits therefor |
| PT106063B (en) * | 2011-12-19 | 2017-05-30 | Inst Superior Técnico | THERMORRESISTENT CRYSTALLINE MINOCYCLINE PRODUCED BY RECRYSTALLATION WITH CARBON DIOXIDE |
| ITTO20130284A1 (en) * | 2013-04-09 | 2014-10-10 | Fond Istituto Italiano Di Tecnologia | PROCEDURE FOR THE PRODUCTION OF SHAPED POLYMERIC MICROPARTELS |
| US11696902B2 (en) | 2018-08-14 | 2023-07-11 | AltaThera Pharmaceuticals, LLC | Method of initiating and escalating sotalol hydrochloride dosing |
| US11610660B1 (en) | 2021-08-20 | 2023-03-21 | AltaThera Pharmaceuticals LLC | Antiarrhythmic drug dosing methods, medical devices, and systems |
| US11344518B2 (en) | 2018-08-14 | 2022-05-31 | AltaThera Pharmaceuticals LLC | Method of converting atrial fibrillation to normal sinus rhythm and loading oral sotalol in a shortened time frame |
| CN109298105B (en) * | 2018-12-06 | 2019-09-13 | 广东省生物资源应用研究所 | A kind of method that ultra high efficiency closes L-pantoprazole and dextral-pantoprazole in phase chromatograph-mass spectrometer coupling quantitative detection biological sample |
| US11338119B2 (en) * | 2020-03-20 | 2022-05-24 | The Regents Of The University Of California | Implantable drug delivery devices for localized drug delivery |
| US11344526B2 (en) | 2020-03-20 | 2022-05-31 | The Regents Of The University Of California | Implantable drug delivery devices for localized drug delivery |
| US11173291B2 (en) * | 2020-03-20 | 2021-11-16 | The Regents Of The University Of California | Implantable drug delivery devices for localized drug delivery |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996003978A1 (en) | 1994-08-04 | 1996-02-15 | Quadrant Holdings Cambridge Limited | Solid delivery systems for controlled release of molecules incorporated therein and methods of making same |
| US6517860B1 (en) | 1996-12-31 | 2003-02-11 | Quadrant Holdings Cambridge, Ltd. | Methods and compositions for improved bioavailability of bioactive agents for mucosal delivery |
Family Cites Families (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3123077A (en) * | 1964-03-03 | Surgical suture | ||
| US3087860A (en) * | 1958-12-19 | 1963-04-30 | Abbott Lab | Method of prolonging release of drug from a precompressed solid carrier |
| US3087660A (en) * | 1962-07-24 | 1963-04-30 | Yankee Plasties Inc | Two-step garment hanger |
| US4326532A (en) * | 1980-10-06 | 1982-04-27 | Minnesota Mining And Manufacturing Company | Antithrombogenic articles |
| SE445884B (en) * | 1982-04-30 | 1986-07-28 | Medinvent Sa | DEVICE FOR IMPLANTATION OF A RODFORM PROTECTION |
| US4734451A (en) * | 1983-09-01 | 1988-03-29 | Battelle Memorial Institute | Supercritical fluid molecular spray thin films and fine powders |
| US4734227A (en) * | 1983-09-01 | 1988-03-29 | Battelle Memorial Institute | Method of making supercritical fluid molecular spray films, powder and fibers |
| US4582731A (en) * | 1983-09-01 | 1986-04-15 | Battelle Memorial Institute | Supercritical fluid molecular spray film deposition and powder formation |
| US4733665C2 (en) * | 1985-11-07 | 2002-01-29 | Expandable Grafts Partnership | Expandable intraluminal graft and method and apparatus for implanting an expandable intraluminal graft |
| US4985625A (en) * | 1986-03-06 | 1991-01-15 | Finnigan Corporation | Transfer line for mass spectrometer apparatus |
| US5106650A (en) * | 1988-07-14 | 1992-04-21 | Union Carbide Chemicals & Plastics Technology Corporation | Electrostatic liquid spray application of coating with supercritical fluids as diluents and spraying from an orifice |
| US5000519A (en) * | 1989-11-24 | 1991-03-19 | John Moore | Towed vehicle emergency brake control system |
| JP2641781B2 (en) * | 1990-02-23 | 1997-08-20 | シャープ株式会社 | Method of forming semiconductor element isolation region |
| US5090419A (en) * | 1990-08-23 | 1992-02-25 | Aubrey Palestrant | Apparatus for acquiring soft tissue biopsy specimens |
| US6524698B1 (en) * | 1990-09-27 | 2003-02-25 | Helmuth Schmoock | Fluid impermeable foil |
| US5195969A (en) * | 1991-04-26 | 1993-03-23 | Boston Scientific Corporation | Co-extruded medical balloons and catheter using such balloons |
| JPH07505316A (en) * | 1992-03-31 | 1995-06-15 | ボストン サイエンティフィック コーポレーション | medical wire |
| US5288711A (en) * | 1992-04-28 | 1994-02-22 | American Home Products Corporation | Method of treating hyperproliferative vascular disease |
| US5500180A (en) * | 1992-09-30 | 1996-03-19 | C. R. Bard, Inc. | Method of making a distensible dilatation balloon using a block copolymer |
| US5385776A (en) * | 1992-11-16 | 1995-01-31 | Alliedsignal Inc. | Nanocomposites of gamma phase polymers containing inorganic particulate material |
| US5494620A (en) * | 1993-11-24 | 1996-02-27 | United States Surgical Corporation | Method of manufacturing a monofilament suture |
| US5837313A (en) * | 1995-04-19 | 1998-11-17 | Schneider (Usa) Inc | Drug release stent coating process |
| US5609629A (en) * | 1995-06-07 | 1997-03-11 | Med Institute, Inc. | Coated implantable medical device |
| AU716005B2 (en) * | 1995-06-07 | 2000-02-17 | Cook Medical Technologies Llc | Implantable medical device |
| US6256529B1 (en) * | 1995-07-26 | 2001-07-03 | Burdette Medical Systems, Inc. | Virtual reality 3D visualization for surgical procedures |
| JP3249357B2 (en) * | 1995-11-01 | 2002-01-21 | 三菱重工業株式会社 | Magnetic separation device and pulverized coal combustion device using the magnetic separation device |
| US5876426A (en) * | 1996-06-13 | 1999-03-02 | Scimed Life Systems, Inc. | System and method of providing a blood-free interface for intravascular light delivery |
| GB9623634D0 (en) * | 1996-11-13 | 1997-01-08 | Bpsi Holdings Inc | Method and apparatus for the coating of substrates for pharmaceutical use |
| GB9800936D0 (en) * | 1997-05-10 | 1998-03-11 | Univ Nottingham | Biofunctional polymers |
| EP1752112B1 (en) * | 1997-11-07 | 2009-12-23 | Salviac Limited | An embolic protection device |
| SE9801288D0 (en) * | 1998-04-14 | 1998-04-14 | Astra Ab | Vaccine delivery system and method of production |
| GB9808052D0 (en) * | 1998-04-17 | 1998-06-17 | Secr Defence | Implants for administering substances and methods of producing implants |
| US6206914B1 (en) * | 1998-04-30 | 2001-03-27 | Medtronic, Inc. | Implantable system with drug-eluting cells for on-demand local drug delivery |
| US6190699B1 (en) * | 1998-05-08 | 2001-02-20 | Nzl Corporation | Method of incorporating proteins or peptides into a matrix and administration thereof through mucosa |
| US8070796B2 (en) * | 1998-07-27 | 2011-12-06 | Icon Interventional Systems, Inc. | Thrombosis inhibiting graft |
| US6248127B1 (en) * | 1998-08-21 | 2001-06-19 | Medtronic Ave, Inc. | Thromboresistant coated medical device |
| US6342062B1 (en) * | 1998-09-24 | 2002-01-29 | Scimed Life Systems, Inc. | Retrieval devices for vena cava filter |
| US6355691B1 (en) * | 1998-11-12 | 2002-03-12 | Tobias M. Goodman | Urushiol therapy of transitional cell carcinoma of the bladder |
| US6858598B1 (en) * | 1998-12-23 | 2005-02-22 | G. D. Searle & Co. | Method of using a matrix metalloproteinase inhibitor and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| US6706283B1 (en) * | 1999-02-10 | 2004-03-16 | Pfizer Inc | Controlled release by extrusion of solid amorphous dispersions of drugs |
| US6171327B1 (en) * | 1999-02-24 | 2001-01-09 | Scimed Life Systems, Inc. | Intravascular filter and method |
| SE9901002D0 (en) * | 1999-03-19 | 1999-03-19 | Electrolux Ab | Apparatus for cleaning textile articles with a densified liquid processing gas |
| US8016873B1 (en) * | 1999-05-03 | 2011-09-13 | Drasler William J | Intravascular hinge stent |
| WO2001001969A2 (en) * | 1999-07-06 | 2001-01-11 | Endorecherche, Inc. | Methods of treating and/or suppressing weight gain |
| US6537310B1 (en) * | 1999-11-19 | 2003-03-25 | Advanced Bio Prosthetic Surfaces, Ltd. | Endoluminal implantable devices and method of making same |
| TWI284048B (en) * | 2000-01-27 | 2007-07-21 | Zentaris Ag | Compressed microparticles for dry injection |
| EP1132058A1 (en) * | 2000-03-06 | 2001-09-12 | Advanced Laser Applications Holding S.A. | Intravascular prothesis |
| AU2001287349B2 (en) * | 2000-09-01 | 2006-03-02 | Palmaya Pty Ltd | Slow release pharmaceutical preparation and method of administering same |
| US6521258B1 (en) * | 2000-09-08 | 2003-02-18 | Ferro Corporation | Polymer matrices prepared by supercritical fluid processing techniques |
| US6506213B1 (en) * | 2000-09-08 | 2003-01-14 | Ferro Corporation | Manufacturing orthopedic parts using supercritical fluid processing techniques |
| US20040018228A1 (en) * | 2000-11-06 | 2004-01-29 | Afmedica, Inc. | Compositions and methods for reducing scar tissue formation |
| US6682757B1 (en) * | 2000-11-16 | 2004-01-27 | Euro-Celtique, S.A. | Titratable dosage transdermal delivery system |
| GB0100760D0 (en) * | 2001-01-11 | 2001-02-21 | Biocompatibles Ltd | Drug delivery from stents |
| TWI246524B (en) * | 2001-01-19 | 2006-01-01 | Shearwater Corp | Multi-arm block copolymers as drug delivery vehicles |
| US20040022853A1 (en) * | 2001-04-26 | 2004-02-05 | Control Delivery Systems, Inc. | Polymer-based, sustained release drug delivery system |
| US7201940B1 (en) * | 2001-06-12 | 2007-04-10 | Advanced Cardiovascular Systems, Inc. | Method and apparatus for thermal spray processing of medical devices |
| US7485113B2 (en) * | 2001-06-22 | 2009-02-03 | Johns Hopkins University | Method for drug delivery through the vitreous humor |
| US7015875B2 (en) * | 2001-06-29 | 2006-03-21 | Novus Partners Llc | Dynamic device for billboard advertising |
| TW497494U (en) * | 2001-12-28 | 2002-08-01 | Metal Ind Redearch & Amp Dev C | Fluid driven stirring device for compressing gas cleaning system |
| KR20040076278A (en) * | 2002-01-10 | 2004-08-31 | 노파르티스 아게 | Drug delivery systems for the prevention and treatment of vascular diseases comprising rapamycin and derivatives thereof |
| ES2276084T3 (en) * | 2002-02-15 | 2007-06-16 | Cv Therapeutics, Inc. | POLYMER COATING FOR MEDICAL DEVICES. |
| GB0205868D0 (en) * | 2002-03-13 | 2002-04-24 | Univ Nottingham | Polymer composite with internally distributed deposition matter |
| US20040013792A1 (en) * | 2002-07-19 | 2004-01-22 | Samuel Epstein | Stent coating holders |
| US20050019747A1 (en) * | 2002-08-07 | 2005-01-27 | Anderson Daniel G. | Nanoliter-scale synthesis of arrayed biomaterials and screening thereof |
| US7029495B2 (en) * | 2002-08-28 | 2006-04-18 | Scimed Life Systems, Inc. | Medical devices and methods of making the same |
| US7060051B2 (en) * | 2002-09-24 | 2006-06-13 | Scimed Life Systems, Inc. | Multi-balloon catheter with hydrogel coating |
| US6770729B2 (en) * | 2002-09-30 | 2004-08-03 | Medtronic Minimed, Inc. | Polymer compositions containing bioactive agents and methods for their use |
| AU2003277332B2 (en) * | 2002-10-11 | 2009-03-12 | University Of Connecticut | Shape memory polymers based on semicrystalline thermoplastic polyurethanes bearing nanostructured hard segments |
| US20050079199A1 (en) * | 2003-02-18 | 2005-04-14 | Medtronic, Inc. | Porous coatings for drug release from medical devices |
| US20080051866A1 (en) * | 2003-02-26 | 2008-02-28 | Chao Chin Chen | Drug delivery devices and methods |
| US7326734B2 (en) * | 2003-04-01 | 2008-02-05 | The Regents Of The University Of California | Treatment of bladder and urinary tract cancers |
| US20050038498A1 (en) * | 2003-04-17 | 2005-02-17 | Nanosys, Inc. | Medical device applications of nanostructured surfaces |
| GB0310300D0 (en) * | 2003-05-06 | 2003-06-11 | Univ Belfast | Nanocomposite drug delivery composition |
| US7662864B2 (en) * | 2003-06-04 | 2010-02-16 | Rutgers, The State University Of New Jersey | Solution polymerization processes to prepare a polymer that degrades to release a physiologically active agent |
| US7318945B2 (en) * | 2003-07-09 | 2008-01-15 | Medtronic Vascular, Inc. | Laminated drug-polymer coated stent having dipped layers |
| US8025637B2 (en) * | 2003-07-18 | 2011-09-27 | Boston Scientific Scimed, Inc. | Medical balloons and processes for preparing same |
| US7169404B2 (en) * | 2003-07-30 | 2007-01-30 | Advanced Cardiovasular Systems, Inc. | Biologically absorbable coatings for implantable devices and methods for fabricating the same |
| US20050033417A1 (en) * | 2003-07-31 | 2005-02-10 | John Borges | Coating for controlled release of a therapeutic agent |
| US7318944B2 (en) * | 2003-08-07 | 2008-01-15 | Medtronic Vascular, Inc. | Extrusion process for coating stents |
| US20050070990A1 (en) * | 2003-09-26 | 2005-03-31 | Stinson Jonathan S. | Medical devices and methods of making same |
| US7198675B2 (en) * | 2003-09-30 | 2007-04-03 | Advanced Cardiovascular Systems | Stent mandrel fixture and method for selectively coating surfaces of a stent |
| US20070254035A1 (en) * | 2003-10-23 | 2007-11-01 | Jianyuan Hao | Preparing Active Polymer Extrudates |
| US20060001011A1 (en) * | 2004-07-02 | 2006-01-05 | Wilson Neil R | Surface conditioner for powder coating systems |
| US20060020325A1 (en) * | 2004-07-26 | 2006-01-26 | Robert Burgermeister | Material for high strength, controlled recoil stent |
| US8541078B2 (en) * | 2004-08-06 | 2013-09-24 | Societe Bic | Fuel supplies for fuel cells |
| US8119153B2 (en) * | 2004-08-26 | 2012-02-21 | Boston Scientific Scimed, Inc. | Stents with drug eluting coatings |
| US20080077232A1 (en) * | 2004-09-08 | 2008-03-27 | Kaneka Corporation | Stent for Placement in Body |
| US20070059350A1 (en) * | 2004-12-13 | 2007-03-15 | Kennedy John P | Agents for controlling biological fluids and methods of use thereof |
| AU2006270221B2 (en) * | 2005-07-15 | 2012-01-19 | Micell Technologies, Inc. | Polymer coatings containing drug powder of controlled morphology |
| US8343170B2 (en) * | 2005-08-12 | 2013-01-01 | Massicotte J Mathieu | Method and device for extracting objects from the body |
| US20080075753A1 (en) * | 2006-09-25 | 2008-03-27 | Chappa Ralph A | Multi-layered coatings and methods for controlling elution of active agents |
| US8636767B2 (en) * | 2006-10-02 | 2014-01-28 | Micell Technologies, Inc. | Surgical sutures having increased strength |
| US8430055B2 (en) * | 2008-08-29 | 2013-04-30 | Lutonix, Inc. | Methods and apparatuses for coating balloon catheters |
| WO2008070996A1 (en) * | 2006-12-13 | 2008-06-19 | Angiotech Pharmaceuticals Inc. | Medical implants with a combination of compounds |
| US9737642B2 (en) * | 2007-01-08 | 2017-08-22 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US20090068266A1 (en) * | 2007-09-11 | 2009-03-12 | Raheja Praveen | Sirolimus having specific particle size and pharmaceutical compositions thereof |
| US20090076446A1 (en) * | 2007-09-14 | 2009-03-19 | Quest Medical, Inc. | Adjustable catheter for dilation in the ear, nose or throat |
| HUE055815T2 (en) * | 2007-10-05 | 2021-12-28 | Univ Wayne State | Dendrimers for sustained release of compounds |
| US20100042206A1 (en) * | 2008-03-04 | 2010-02-18 | Icon Medical Corp. | Bioabsorbable coatings for medical devices |
| JP2011528275A (en) * | 2008-07-17 | 2011-11-17 | ミセル テクノロジーズ,インク. | Drug delivery medical device |
| US20100055145A1 (en) * | 2008-08-29 | 2010-03-04 | Biosensors International Group | Stent coatings for reducing late stent thrombosis |
| US8367090B2 (en) * | 2008-09-05 | 2013-02-05 | Abbott Cardiovascular Systems Inc. | Coating on a balloon comprising a polymer and a drug |
| WO2010056754A2 (en) * | 2008-11-11 | 2010-05-20 | The Board Regents Of The University Of Texas System | Inhibition of mammalian target of rapamycin |
| US9327060B2 (en) * | 2009-07-09 | 2016-05-03 | CARDINAL HEALTH SWITZERLAND 515 GmbH | Rapamycin reservoir eluting stent |
| CA2810842C (en) * | 2010-09-09 | 2018-06-26 | Micell Technologies, Inc. | Macrolide dosage forms |
-
2005
- 2005-06-22 US US11/158,724 patent/US20070009564A1/en not_active Abandoned
-
2006
- 2006-06-21 CA CA2613280A patent/CA2613280C/en not_active Expired - Fee Related
- 2006-06-21 CA CA2922492A patent/CA2922492C/en not_active Expired - Fee Related
- 2006-06-21 EP EP06773731.2A patent/EP1898878B1/en active Active
- 2006-06-21 WO PCT/US2006/024221 patent/WO2007002238A2/en active Application Filing
-
2019
- 2019-01-28 US US16/259,541 patent/US20190151453A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996003978A1 (en) | 1994-08-04 | 1996-02-15 | Quadrant Holdings Cambridge Limited | Solid delivery systems for controlled release of molecules incorporated therein and methods of making same |
| US6517860B1 (en) | 1996-12-31 | 2003-02-11 | Quadrant Holdings Cambridge, Ltd. | Methods and compositions for improved bioavailability of bioactive agents for mucosal delivery |
Non-Patent Citations (6)
| Title |
|---|
| AKOH ET AL., J. FOOD SCI., vol. 52, 1987, pages 1570 |
| C. K. LEE: "Developments in Food Carbohydrate (2d Ed.)", 1980, APPLIED SCIENCE PUBLISHERS |
| KHAN ET AL., CARBO RES, vol. 198, 1990, pages 275 - 283 |
| KHAN ET AL., TETRA. LETTS, vol. 34, 1933, pages 7767 |
| KHAN, PURE & APPL. CHEM., vol. 56, 1984, pages 833 - 844 |
| See also references of EP1898878A4 |
Cited By (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11911301B2 (en) | 2005-07-15 | 2024-02-27 | Micell Medtech Inc. | Polymer coatings containing drug powder of controlled morphology |
| US9827117B2 (en) | 2005-07-15 | 2017-11-28 | Micell Technologies, Inc. | Polymer coatings containing drug powder of controlled morphology |
| US10898353B2 (en) | 2005-07-15 | 2021-01-26 | Micell Technologies, Inc. | Polymer coatings containing drug powder of controlled morphology |
| US10835396B2 (en) | 2005-07-15 | 2020-11-17 | Micell Technologies, Inc. | Stent with polymer coating containing amorphous rapamycin |
| US11850333B2 (en) | 2006-04-26 | 2023-12-26 | Micell Medtech Inc. | Coatings containing multiple drugs |
| US11007307B2 (en) | 2006-04-26 | 2021-05-18 | Micell Technologies, Inc. | Coatings containing multiple drugs |
| US9737645B2 (en) | 2006-04-26 | 2017-08-22 | Micell Technologies, Inc. | Coatings containing multiple drugs |
| US9539593B2 (en) | 2006-10-23 | 2017-01-10 | Micell Technologies, Inc. | Holder for electrically charging a substrate during coating |
| US10617795B2 (en) | 2007-01-08 | 2020-04-14 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US11426494B2 (en) | 2007-01-08 | 2022-08-30 | MT Acquisition Holdings LLC | Stents having biodegradable layers |
| US9737642B2 (en) | 2007-01-08 | 2017-08-22 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US9775729B2 (en) | 2007-04-17 | 2017-10-03 | Micell Technologies, Inc. | Stents having controlled elution |
| US9486338B2 (en) | 2007-04-17 | 2016-11-08 | Micell Technologies, Inc. | Stents having controlled elution |
| AU2015200571B2 (en) * | 2007-04-17 | 2016-11-17 | Micell Technologies, Inc. | Stents having biodegradable layers |
| AU2017200794B2 (en) * | 2007-04-17 | 2018-07-26 | Micell Technologies, Inc. | Stents having biodegradable layers |
| US9968710B2 (en) | 2007-07-23 | 2018-05-15 | Biomet Deutschland Gmbh | Pharmaceutical composition, substrate comprising a pharmaceutical composition, and use of a pharmaceutical composition |
| WO2009013552A1 (en) | 2007-07-23 | 2009-01-29 | Richter Gedeon Nyrt. | Controlled release pharmaceutical composition of tolperison hydrochloride |
| EA018885B1 (en) * | 2007-07-23 | 2013-11-29 | Рихтер Гедеон Нирт. | Controlled release pharmaceutical composition of tolperison hydrochloride |
| US8921365B2 (en) | 2007-07-23 | 2014-12-30 | Biomet Deutschland Gmbh | Pharmaceutical composition, substrate comprising a pharmaceutical composition, and use of a pharmaceutical composition |
| DE102007043883A1 (en) * | 2007-09-14 | 2009-03-26 | Biotronik Vi Patent Ag | Stent with a coating |
| US9789233B2 (en) | 2008-04-17 | 2017-10-17 | Micell Technologies, Inc. | Stents having bioabsorbable layers |
| US10350333B2 (en) | 2008-04-17 | 2019-07-16 | Micell Technologies, Inc. | Stents having bioabsorable layers |
| US8642631B2 (en) | 2008-05-27 | 2014-02-04 | University Of Melbourne | Methods of treating mammals with eustachian tube dysfunctions |
| US9981071B2 (en) | 2008-07-17 | 2018-05-29 | Micell Technologies, Inc. | Drug delivery medical device |
| US10350391B2 (en) | 2008-07-17 | 2019-07-16 | Micell Technologies, Inc. | Drug delivery medical device |
| US9981072B2 (en) | 2009-04-01 | 2018-05-29 | Micell Technologies, Inc. | Coated stents |
| US10653820B2 (en) | 2009-04-01 | 2020-05-19 | Micell Technologies, Inc. | Coated stents |
| US11369498B2 (en) | 2010-02-02 | 2022-06-28 | MT Acquisition Holdings LLC | Stent and stent delivery system with improved deliverability |
| US9687864B2 (en) | 2010-03-26 | 2017-06-27 | Battelle Memorial Institute | System and method for enhanced electrostatic deposition and surface coatings |
| US10232092B2 (en) | 2010-04-22 | 2019-03-19 | Micell Technologies, Inc. | Stents and other devices having extracellular matrix coating |
| US11904118B2 (en) | 2010-07-16 | 2024-02-20 | Micell Medtech Inc. | Drug delivery medical device |
| US10293050B2 (en) | 2010-09-09 | 2019-05-21 | Micell Technologies, Inc. | Macrolide dosage forms |
| US9636309B2 (en) | 2010-09-09 | 2017-05-02 | Micell Technologies, Inc. | Macrolide dosage forms |
| WO2012083594A1 (en) * | 2010-12-24 | 2012-06-28 | Dongguan Tiantianxiangshang Medical Technology Co., Ltd | Biodegradable drug eluting stent and methodsof making the same. |
| US10464100B2 (en) | 2011-05-31 | 2019-11-05 | Micell Technologies, Inc. | System and process for formation of a time-released, drug-eluting transferable coating |
| US10729819B2 (en) | 2011-07-15 | 2020-08-04 | Micell Technologies, Inc. | Drug delivery medical device |
| US10117972B2 (en) | 2011-07-15 | 2018-11-06 | Micell Technologies, Inc. | Drug delivery medical device |
| US10188772B2 (en) | 2011-10-18 | 2019-01-29 | Micell Technologies, Inc. | Drug delivery medical device |
| US11039943B2 (en) | 2013-03-12 | 2021-06-22 | Micell Technologies, Inc. | Bioabsorbable biomedical implants |
| CN108456714A (en) * | 2013-03-14 | 2018-08-28 | 简·探针公司 | Diagnostic system and method |
| CN108456714B (en) * | 2013-03-14 | 2022-07-19 | 简·探针公司 | Diagnostic system and method |
| US11761026B2 (en) | 2013-03-14 | 2023-09-19 | Gen-Probe Incorporated | Diagnostic system and method |
| US10272606B2 (en) | 2013-05-15 | 2019-04-30 | Micell Technologies, Inc. | Bioabsorbable biomedical implants |
| CN103877049A (en) * | 2014-04-04 | 2014-06-25 | 白玲强 | Tablet containing fenofibrate and preparation technology thereof |
| CN106619532A (en) * | 2016-11-16 | 2017-05-10 | 山东裕欣药业有限公司 | Roxithromycin ambroxol hydrochloride dry suspension and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1898878A4 (en) | 2012-10-31 |
| CA2613280A1 (en) | 2007-01-04 |
| CA2613280C (en) | 2016-05-10 |
| EP1898878B1 (en) | 2020-01-08 |
| EP1898878A2 (en) | 2008-03-19 |
| CA2922492A1 (en) | 2007-01-04 |
| WO2007002238A3 (en) | 2007-05-10 |
| CA2922492C (en) | 2018-02-20 |
| US20190151453A1 (en) | 2019-05-23 |
| US20070009564A1 (en) | 2007-01-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1898878B1 (en) | Drug/polymer composite materials and methods of making the same | |
| US20230181802A1 (en) | Stents Having Biodegradable Layers | |
| US11911301B2 (en) | Polymer coatings containing drug powder of controlled morphology | |
| US11007307B2 (en) | Coatings containing multiple drugs | |
| US10617795B2 (en) | Stents having biodegradable layers | |
| AU2017200794B2 (en) | Stents having biodegradable layers | |
| Liu | Update on polymers for oral drug delivery | |
| US11426494B2 (en) | Stents having biodegradable layers | |
| AU2011256902B2 (en) | Polymer coatings containing drug powder of controlled morphology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2613280 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006773731 Country of ref document: EP |