SUSTAINED RELEASE PHARMACEUTICAL COMPOSITION
Field of the invention
The present invention relates to a sustained release pharmaceutical composition, having a beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters in a polymer mixture such that the beta lactam antibiotic is released over an extended period, suitable for once or twice daily oral administration. The invention preferably relates to a sustained release pharmaceutical composition of the beta lactam antibiotic having gelling tendency when in contact with the gastrointestinal fluids. The invention more preferably relates to sustained release pharmaceutical compositions of prodrugs of cefpodoxime and cefuroxime such as cefpodoxime proxetil and cefuroxime axetil.
Background of the Invention While many compounds are known to be useful as pharmacologically active substances, some of them have relatively short biological half life and needs to be administered several times a day in order to achieve desired therapeutic effects. Especially, the drugs used in the treatment of microbial infections are required to be given more than once during a dosage regimen. In an anti-microbial therapy the main requirement is to maximize the blood concentration, preferably several folds higher than the minimum inhibitory concentration (MIC) for the active agent, yet to minimize both the risk of toxicity to the patient and of promoting microbial resistance to the active agent. Although oral administration will be the preferred route, in the case of antibiotics this route is frequently unattractive because of their low or variable oral bioavailability. In addition extremely high plasma concentrations of antibiotics are frequently required to achieve the MIC values towards certain gram-negative bacteria (Antibiotics and Chemotherapy; Anti infective
agents and their use in therapy, 7th edition Ed. By O'grady F, Finch RG, Lambert HP; Greenwood D; Churchill Livingstone 1997).
Sustained release preparation of drugs are advantageous as the administration frequency can be reduced by maintaining a constant plasma concentration of drug over an extended period of time to ensure sustained effect of active ingredient well above the MIC levels. In addition, these preparations are also expected to decrease side effects by suppressing the rapid rise in the blood levels of the drugs.
This has been primarily achieved by development of a novel drug delivery system utilizing diverse techniques and principles. Amongst these, known in the art is one such delivery system, which employs hydrophihc polymers to sustained or modified release pharmaceutical composition. For modified release solid dosage forms comprising a drug dispersed uniformly in mixture of polymers, release of the drug is controlled primarily by diffusion of the drug, or by surface erosion of the hydrophihc polymers into the surrounding medium or by a combination of both processes. Control of the rate of release can produce constant blood levels of the active ingredient that may result in reducing the frequency of administration, thereby improving patient compliance to the dosage regimen. The relevant prior art references, which disclose diverse delivery systems for the sustained release of the active ingredient, are summarized below.
United States Patent No.6, 120,803 discloses an active agent dosage form, which is adapted for retention in the stomach and useful for the prolonged delivery of an active agent to a fluid environment of use. The active agent dosage form is a polymer matrix that swells upon contact with the fluid of the stomach. A portion of the polymer matrix is surrounded by a band of insoluble material that prevents the covered portion of the polymer matrix from swelling and provides a segment of the dosage form that is of sufficient
rigidity to withstand the contraction of the stomach and delay expulsion of the dosage form from the stomach until substantially all the active agent has been dispensed.
United States Patent No.5,128,142 discloses a controlled release formulation comprising an adsorbate of a mixture of a pharmaceutically useful active ingredient and an inactive substance adsorbed on a cross linked polymer. The inactive substance is selected to modify the dissolution of active ingredients from the cross-linked polymer in vivo. The inactive substance is preferably present in the adsorbate in an amount of 0.5 - 3 parts by weight relative to 1 part by weight of the active ingredients.
United States Patent No.4,968,508 discloses a sustained release matrix tablet comprising from about 0.1% to about 90% by weight of cefaclor, about 5% to about 29% by weight by hydrophihc polymer and about 0.5% to about 25% by weight of an acrylic polymer which dissolve at a pH in the range of about 5.0 to about 7.4, the total weight of polymers being less than 30% by weight of the formulation. Although a specific cefaclor formulation is claimed, the text suggests that the matrix formulation is suitable for weakly basic drugs and particularly suitable for cephalexin and cefaclor.
United States Patent No.5,948,440 discloses a controlled release tablet of an active ingredient comprising of cefaclor, cephalexin or their pharmaceutically acceptable hydrates, salts or esters as active ingredients and a mixture of hydrophihc polymers selected from the group consisting of at least one hydroxypropyl methylcellulose and at least one hydroxypropyl cellulose. The composition optionally also contains one or more of a water soluble or water dispersible diluent, the quantities are such that the therapeutically effective active ingredient is released at a rate suitable for twice daily administration of the pharmaceutical composition.
International Publication number WOOO/15198 teaches controlled delivery pharmaceutical composition having temporal and spatial control, comprising a drag, a gas generating component, a swelling agent, a viscolyzing agent and optionally a gel forming polymer. The viscolyzing agent initially and the gel forming polymer thereafter form a hydrated gel matrix which entraps the gas, causing the tablet to float so that it is retained in the stomach thereby providing spatial control and at the same time resulting in sustained release of the drag providing temporal control. The combination of gas generating component, swelling agent and viscolyzing agent results in the controlled drag delivery systems. Thus all these components are essential for achieving the temporal and spatial control. A preferred once daily ciprofloxacin formulation comprising 69.9% ciprofloxacin base, 0.34% sodium alginate, 1.03% xanthan gum, 13.7% sodium bicarbonate, 12.1% cross linked polyvinyl pyrrolidone and optionally other excipients is disclosed. International Publication number WO 02/41876 discloses a pharmaceutical composition in the form of a tablet for controlled release of an active ingredient comprising a beta lactam antibiotic such as cephalexin, cefaclor or their pharmaceutically acceptable hydrates, salts or esters as active ingredients, and a mixture of hydrophihc polymers selected from the group consisting of at least one sodium alginate and at least one xanthan gum as controlled release matrix and optionally probenecid as an antibiotic adjuvant as either immediate release or controlled release part. The composition may contain one or more of a water-soluble and/or water dispersible diluent. The quantity of the hydrophihc polymer matrix still provides the desired once a day drug release profile.
United States Patent No.6,399.086 discloses a controlled release beta lactam antibiotic agent preferably amoxicillin trihydrate in a hydrophihc and/or hydrophobic polymer matrix such that at least 50% but not more than 67.61 + 5.78 % of the active agent is released within 3-4 hrs from oral
administration and remainder is released at a controlled rate from the said composition. The composition teaches the use of commonly used hydrophihc polymers such as hydrophihc cellulose derivatives, hydrophihc methacrylic acid derivatives, chitosan, and alginates. Preferably, the use of hydrophihc cellulose derivative such as methylcellulose, hydroxypropyl methylcellulose, and hydroxyethyl cellulose, is disclosed. The hydrophobic polymers used in the invention are acrylamides and polyamido derivatives and hydrophobic methacrylic acid derivatives. The preferred hydrophobic polymer is ethyl cellulose. However, with this drag release profile wherein 67.61 + 5.78 of the drag is already out of the matrix in 3-4 hrs and considering a very short half- life of both cefpodoxime proxetil and cefuroxime axetil of 2-3 hrs and about lhr respectively, the composition may not be suitable for once daily administration. Hence, it is necessary that the matrix formulation should release the drag over extended period of time. International Publication number WO 02/36126 discloses a fast disintegrating controlled release oral composition comprising a core material containing cefuroxime axetil present as controlled release form, the cefuroxime axetil being provided with an outer coating of a copolymer selected from aqueous dispersions of enteric methacrylic acid and methacrylic acid esters anionic copolymers having carboxyl group as the functional group or mixture thereof and an inner coating of a sustained release copolymer selected from aqueous dispersions of acrylate or methacrylate pH independent copolymers having quaternary ammonium group as a functional group or mixture thereof. Additionally the coating composition may contain plasticizers. The composition is suitable for once daily administration.
In multi-particulate controlled release dosage form the particles are less sensitive to the effect of gastric/stomach emptying time. As the particles are smaller, a portion of the particles gradually reaches the small intestine and passes small intestine relatively faster than the tablet dosage form even in the
presence of food. Hence, there is a potential chance of drag dissolution/delivery in the non-absorbable gastro-intestinal tract site. Therefore, there are chances for failure of the mul ti-particula te control release system especially if the drag has narrow absorption window in the gastro- intestinal tract. Cefuroxime axetil and cefpodoxime proxetil are reported to be absorbed from the upper part of the gastrointestinal tract. Hence, developing and achieving once daily administration through the multi-particulate controlled release system for the said drags having absorption in upper part of GIT with short biological half-life, and the cumbersome process involved in developing such type of dosage form, makes it undesirable.
Cefpodoxime proxetil and cefuroxime axetil, are broad spectrum antibiotics active against both gram positive and gram negative microorganisms. Cefpodoxime proxetil and cefuroxime axetil oral bioavailability is reported to be 50%, with a slight increase in the bioavailability in the presence of food, and they are orally administered twice daily. Cefpodoxime proxetil and cefuroxime axetil molecules tend to form gels, upon exposure to the environmental fluids. These drags form gel in contact with acidic media of the stomach, leading to reduction in dissolution and that, in turn, leads to poor bioavailability. Both cefuroxime axetil and cefpodoxime proxetil have narrow absorption window, i.e., absorbed less from the upper part of gastrointestinal tract, and at the same time, have plasma half- lives of 1.2 and 2.1 to 2.9 hrs, respectively.
The relevant prior arts, which describe the formulations to overcome the gelling of cefuroxime axetil, are as follows: United States Patent No. 4,897,270 discloses a film coated cefuroxime axetil tablet to mask the bitter taste of the drag upon oral administration. The patent teaches that conventional film coated tablets result in low levels of absorption of cefuroxime axetil from gastrointestinal tract (GIT) and this is overcome by control of the film coat rapture time and use of a tablet core,
which disintegrates immediately following rapture of the film coat. The patent further teaches that cefuroxime axetil, once in contact with aqueous media, can form a gelatinous mass. This gelling effect is temperature dependent but does occur at temperature of about 37 °C, i.e., at the physiological temperature at which the disintegration of orally administered tablets takes place. The relatively slow permeation of moisture from the film coat to the core, which occurs upon administration of the tablets provided with conventional film coats, leads to gelling of cefuroxime axetil present in the core. The gel formation leads to poor disintegration of the tablet core and hence poor dissolution of cefuroxime axetil, thus the absorption from the GIT is greatly reduced. This occurs with both the crystalline and amorphous forms of cefuroxime axetil.
International Publication No. WQ99/44614. however, comments that such thin film coating with the water soluble film-forming material cannot completely block the absoφtion of moisture into the core upon long term storage and thus rather may cause gelation of the active ingredient within the core of the tablet upon storage. The patent further claims a pharmaceutical composition comprising amorphous cefuroxime axetil and silicon dioxide or its hydrate as a micro environmental pH adjuster and an anti-gelling agent, which is not gelled by moisture absoφtion and is thus stable during storage period for cefuroxime axetil.
International Publication No. WO00/30647 discloses cefuroxime axetil in non-gelatinous form on contact with an aqueous liquid comprising cefuroxime axetil in the form of a solid solution in a polymer or in the form of a solid dispersion on an adsorbent, useful for pharmaceutical composition. The composition disclosed is a conventional formulation and it does not relate to a method to prevent the gelation of cefuroxime axetil if exposed to aqueous liquid for extended period of more than 45 minutes.
United States Patent No. 6,323,193 discloses that addition of sodium citrate to the formulation containing amoφhous cefuroxime axetil, inhibits the tendency of amoφhous cefuroxime axetil to form a gel. This may be due to the presence of citrate ions, which prevents cefuroxime axetil molecules from bridging with each other to form a gel, thereby helping in the tablet dissolution.
United States Patent No. 4,865,851 discloses cefuroxime axetil composition comprising particulate cefuroxime axetil coated with an integral coating of a lipid or mixture of lipids which are insoluble in water and which serves to mask the bitter taste of cefuroxime axetil but disperses or dissolves on contact with gastro-intestinal fluid. The resulting particles can be incoφorated into pharmaceutical compositions for oral administration, for example aqueous suspension, dry product for reconstitution with water or granules. However, the drawback of this composition is that, coated particles having diameter of less than 250 microns are preferred and, the coating procedure employed is rather elaborate, resulting in the process which is inconvenient for preparing solid dosage forms of cefuroxime axetil for oral administration.
International Publication No. WO99/62559 discloses a pharmaceutical tablet comprising cefuroxime axetil and a carbonate to enhance the rate of disintegration of the tablets in gastric fluid.
International Publication No. WO99/08683 discloses a pharmaceutical composition comprising of co-precipitate of cefuroxime axetil and a water soluble excipient selected from the group consisting of povidone, hydroxypropyl cellulose, methyl cellulose, lactose, mannitol and sorbitol. Tablets made from the said co-precipitate exhibit high bioavailability without requiring that the tablets disintegrate immediately in gastro-intestinal fluid.
In the above prior art references, attempts have been made to overcome the gelling tendency of cefuroxime axetil for rapid release of the drag in the gastro-intestinal tract. However, the compositions described above cannot be used for controlled release of the drag to achieve and sustain therapeutic blood levels for a prolonged period of time.
Most of the polymers used to prepare sustained release polymeric matrix dosage form, has a tendency to swell in the presence of water and form gel like consistency. When this happens, the gel provides a natural barrier for drag diffusion through the swollen, hydrated gelled layer, in addition to the erosion from the surface of the tablet. Since the gel like material is quite viscous and may not disperse for hours, this provides a means of sustaining the drag release for extended hours until all the drag has been dissolved and diffused out in to the intestinal fluid. Similarly, the plastic matrix provides a rapid geometric surface for drag diffusion so that a relatively constant rate of drag release is obtained.
Ideally, a sustained release dosage form should deliver the medicament at a constant rate throughout the GIT. However this cannot be helpful for beta lactam antibiotics, as they have a narrow absoφtion window, instability in higher pH and some of the beta lactam antibiotics also forms gel upon exposure to the GIT fluids. A decline in the solubility due to gel formation in response to pH fluctuations within the body may result in a decreased release rate and thus poor bioavailability. Hence it is a very difficult task to develop a sustained release dosage form in the form of matrix tablets for drugs like cefuroxime axetil and ce odoxime proxetil, which form gelatinous mass upon contact with the environmental fluids.
The sustained release matrix form may be retained in the stomach for a long time due to the size of the dosage form and delayed stomach emptying time in the presence of food. Stomach emptying is particularly important in
the preparation of sustained release formulation for drags having narrow absoφtion window.
Sustained release preparations of drags are advantageous since the frequency of dosing can be reduced by maintaining a constant plasma concentration of drug over an extended period of time to ensure sustained effect of active ingredient.
Polymer blends in sustained release compositions are known and used in the pharmaceutical industry because of the blend versatility of being able to create different release properties. Polysaccharides such as carrageenans are desirable biopolymers for use in sustained release compositions because they are derived from naturally occurring seaweeds.
None of the prior art references teach about sustained release polymeric matrix tablets of drugs having gelling tendency when in contact with gastrointestinal fluids such as cefpodoxime proxetil or cefuroxime axetil, thus a need exists for such a composition wherein the active ingredient is released in a sustained manner maintaining above the effective plasma levels such that the dosage form is suitable for once or twice daily administration and thereby improving the patient compliance to the dosage regimen.
Although the antibiotics often require high dose and/or higher frequency of administration, extended release drag delivery systems have not been very successful in reducing the frequency. The present invention is based on the observation that the release of active ingredient from the delivery system is controlled by the specific polymers present in the matrix and in specific concentrations, thus allowing blood levels above MIC over extended period of time such that the frequency of the dosage form can be reduced to twice daily or once daily dosing. We have found suφrisingly that when we combined the copolymers and a release enhancer along with a biopolymer, the gelling characteristics of the active ingredient was eliminated, and we could
achieve a constant release of the drag in- vitro over a extended period of time both in acidic and alkaline pH.
Objective of the Invention The main objective of the present invention is to provide an oral sustained release formulation of a beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters.
Further objective of the present invention is to provide an oral sustained release formulation for beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters, having gelling tendency when in contact with gastro-intestinal fluids.
Further objective of the present invention is to avoid gelling of beta lactam antibiotic on contact with the gastro-intestinal fluid, yet controlling the release of the beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters in a non disintegrating matrix such that it provides therapeutically effective blood levels of the medicaments for prolonged period suitable for twice or once daily administration.
Summary of the Invention
The present invention provides a sustained release oral pharmaceutical composition comprising a beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters; mixture of polymers comprising of a water soluble N-vinyl-2-pyrrolidone/vinyl acetate copolymer and polysaccharide(s); a release enhancer(s); and other pharmaceutically acceptable excipients.
In an embodiment of the present invention the polysaccharide is selected from different grades of carrageenan, used either alone or in combination thereof.
In an embodiment of the present invention the pharmaceutical composition is a solid dosage form, preferably in the form of a tablet.
In an embodiment of the present invention the pharmaceutical composition comprises from about 25 to 90% by weight of beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters.
In an embodiment of the present invention the pharmaceutical composition comprises of a mixture of polymers from about 1 to 35% by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises from about 1 to 30% of N-vinyl-2-pyrrolidone/vinyl acetate copolymer by weight of the composition. In an embodiment of the present invention the pharmaceutical composition comprises from about 0.5 to 20% of carrageenan(s) by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises drug release enhancers from about 0.1 to 25% by weight of the total weight of the composition, used either alone or in combination.
In an embodiment of the present invention the pharmaceutically acceptable excipients are selected from integrity enhancers, disintegrants, solubilizers, lubricants, diluents, coating agents, and optionally binders. In an embodiment of the present invention the pharmaceutical composition comprises from about 0.1 to 10% of integrity enhancers by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises from about 0.1 to 15% of disintegrant by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises from about 0.1 to 10% of solubilizer by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises of lubricant from about 0.2 % to about 5% by weight of the total weight of the composition, used either alone or in combination .
In an embodiment of the present invention the pharmaceutical composition comprises one or more pharmaceutically acceptable diluents in an amount from about 1 to about 40% by weight of the composition.
In an embodiment of the present invention the pharmaceutical composition comprises from about 0.1 to 20% of binder by weight of the composition. In an embodiment of the present invention the pharmaceutical composition in the form of tablets are coated using conventional coating agents.
According to the present invention, the pharmaceutical composition may be prepared by dry granulation using the following procedure. (i) blending the beta lactam antibiotic or its pharmaceutically acceptable salts, hydrates or esters, diluent(s) and copolymer; polysaccharide(s), disintegrant(s) and lubricant(s) either whole or in part; release enhancer(s), optionally integrity enhancer(s) and solubilizer(s) together, (ii) passing the blended mixture through a suitable sieve, (iii) compacting the blend using a roller compactor,
(iv) sizing the compacted material using a suitable mill, (v) blending the resultant granules with lubricant(s) in whole or in part; optionally with polysaccharide(s), release enhancer(s), and integrity enhancer(s), in whole or in part, (vi) compressing the lubricated granules using a tableting machine and, (vii) coating the tablets.
In another embodiment of the present invention, the process for the preparation of the sustained release pharmaceutical composition, comprises the steps of:
(i) blending the beta lactam antibiotic or their pharmaceutically acceptable salts, hydrates or esters, diluent(s), copolymer and polysaccharide(s); disintegrant(s) and lubricant(s) either whole or in part; release enhancer(s), optionally integrity enhancer(s) and solubilizer(s) together, (ii) passing the blended mixture through a suitable sieve, (iii) compacting the blend using a roller compactor, (iv) sizing the compacted material using an suitable mill, (v) resultant granules coated with ethyl cellulose in a suitable fluid bed processor, (vi) optionally sizing the coated granules by passing through a suitable sieve,
(vii) blending the resultant granules with lubricant(s) and disintegrant(s), (viii) compressing the lubricated granules using a tableting machine and, (ix) coating the tablets. Detailed Description of the Invention
According to the present invention the beta lactam antibiotic include amoxycillin, ampicillin, cephalexin, cefprozil, cefadroxil, cefaclor, cefamandole, cefoxitin, cephalothin, cephapirin, ceftizoxime, cefonicid, cefpodoxime proxetil, cefuroxime axetil, cefotiam, hexetil, cefteram pivoxil, cefditoren pivoxil, cefcapene pivoxil, cefetamet pivoxil or their pharmaceutically acceptable salts, hydrates or esters.
Preferably, the beta lactam antibiotic is in the form of a prodrag ester such as cefpodoxime proxetil, cefuroxime axetil, cefotiam hexetil, cefteram pivoxil, cefditoren pivoxil, cefcapene pivoxil or cefetamet pivoxil. More preferably, the beta lactam antibiotic is selected from cefpodoxime proxetil or cefuroxime axetil.
According to another embodiment of the present invention the beta lactam antibiotic can be optionally combined with a beta lactamase inhibitor in a sustained release composition. The examples of such beta lactamase
inhibitors include clavulonic acid, sulbactam, tazobactam or their pharmaceutically acceptable salts, hydrates or esters.
Carrageenan is a naturally occurring family of polysaccharides extracted from red seaweed. It is a high molecular weight polysaccharide made up of repeating galactose and 3,6 anhydrogalactose (3,6-AG) units, both sulfated and nonsulfated. The units are joined by alternating alpha 1-3 and beta 1-4 glycosidic linkages. Three types of carrageenan are commercially available. They are iota, kappa and lambda. The primary differences which influence the properties of kappa, iota and lambda carrageenan are the number and position of the ester sulfate groups on the repeating galactose units. Higher levels of ester sulfate, lower the solubility and produce lower strength gels, or contribute to gel inhibition. All three types are soluble in hot water. Lambda is soluble in cold water whereas iota and kappa are insoluble, but their sodium salt is soluble in cold water. According to the present invention carrageenan used is selected from Viscarin GP328, Niscarin GP 209, and Gelcarin GP911 (FMC).
Ν-vinyl-2-pyrrolidone and vinyl acetate is a synthetic water-soluble copolymer in a random 60:40 ratio (copolyvidonum). Copolyvidonum is a highly effective film forming adhesive, the k-value for which is specified between 25.4 and 34.2. The copolymer that may be used in the present invention include Plasdone S-630 (ISP) or Kollidon VA64 (BASF).
According to our observation, when N-vinyl-2-pyrrolidone and vinyl acetate copolymer alone was used as matrix forming agent, the initial release of the cefpodoxime proxetil or cefuroxime axetil was higher and almost 100% of drag is released in first hour in acidic media. This is due to faster erosion of tablet because of water-soluble N-vinyl-2-pyrrolidone and vinyl acetate copolymer, which in turn dissolves the drag and gives higher drag release in the initial hour itself. Hence, incoφoration of a polymer for controlling the release was desired. The cephalosporins have pH dependent solubility; they
have high solubility in acidic pH, which decrease with increase in pH. Hence, the polymer must have low solubility in acidic environment but dissolves relatively faster in the pH 6.8. Due to the inherent gelling tendency of some cephalosporins such as cefuroxime axetil or cefpodoxime proxetil, the polymer should not form strong gel, but sufficient to keep the integrity of the tablet throughout the gastro-intestinal tract. A polysaccharide such as carrageenan was incoφorated along with N-vinyl-2-pyrrolidone and vinyl acetate copolymer, the initial release was controlled, but the release retarded in the later stages for both cefuroxime axetil and cefpodoxime proxetil. Suφrisingly, we found that, when we incoφorated a release enhancing agent such as sodium chloride or lactose to the above composition the release was controlled initially and uniformly over extended period. It was also found out that there was no gelling of cefuroxime axetil or cefpodoxime proxetil in such composition. Hence, in the present invention, the N-vinyl-2-pyrrolidone/vinyl acetate copolymer, carrageenan and sodium chloride or lactose when used in appropriate concentrations achieves a sustained release of the active ingredient over a extended period thereby achieving desired blood levels suitable for twice or once daily dosage regimen. In addition, the gelling tendency of beta lactam antibiotic was also eliminated.
In an embodiment of the present invention the pharmaceutical composition comprises release enhancer such as sodium chloride, potassium chloride; sulfates of sodium, potassium, calcium, magnesium; lactose or the like, used either alone or in combinations thereof. In an embodiment of the present invention the pharmaceutical composition comprises integrity enhancerss such as polyacrylic acid derivatives e.g. Carbopol 971P, Carbopol 974P, Novenon AA1, or the like, used either alone or in combinations thereof.
In an embodiment of the present invention the pharmaceutical composition comprises of water soluble or water dispersible diluents.
In an embodiment of the present invention the water soluble diluents are selected from mannitol, glucose, sorbitol, maltose, dextrates, dextrans, dextrins and the like, used either alone or in combinations thereof.
In an embodiment of the present invention the water dispersible diluents are selected from carboxymethyl cellulose, calcium carboxymethyl cellulose, microcrystalline cellulose, pregelatinized starch, and the like, used either alone or in combinations thereof. In an embodiment of the present invention the pharmaceutical composition comprises of the lubricant such as talc, stearic acid, magnesium stearate, colloidal silicon dioxide, calcium stearate, zinc stearate, hydrogenated vegetable oil, and the like, used either alone or in combinations thereof.
In an embodiment of the present invention the pharmaceutical composition comprises of binders such as cellulosic ether like methyl cellulose, hydroxy propyl cellulose, hydroxy ethyl cellulose, hydroxy propyl methyl cellulose or ethyl cellulose, polyvinyl pyrrolidone and its derivatives or copolymers; used either alone or in combinations thereof.
In an embodiment of the present invention the pharmaceutical composition comprises of disintegrant such as croscarmellose sodium (Acdisol), crospovidone, calcium carboxymethyl cellulose, sodium starch glycolate, and the like, used either alone or in combinations thereof.
In an embodiment of the present invention the pharmaceutical composition comprises of solubilizer such as cationic surfactants like cetrimide, benzalkonium chloride, benzethonium chloride; anionic surfactants like sodium lauryl sulphate, docusate sodium; and non-ionic surfactants like glyceryl monooleate, polyoxyethylene sorbitan fatty acid, polyoxyethylene sorbitan fatty esters, polyvinyl alcohol or sorbitan esters, used either alone or in combinations thereof.
In an embodiment of the present invention the pharmaceutical composition in the form of tablets are coated with coating agents such as Opadry (Colorcon).
In an embodiment of the present invention the coating agent may comprise of film forming agents such as hydroxypropyl methylcellulose, hydroxyethyl cellulose, hydroxypropyl cellulose; plasticizers such as propylene glycol, polyethylene glycol; color pigments; opacifϊers such as titanium dioxide; and optionally talc. Examples The present invention is illustrated by the following examples, which are not intended to limit the scope of the invention, but are provided by way of illustration only.
In all the examples mentioned, the coating agent comprises of an aqueous dispersion of hydroxypropyl methylcellulose, propylene glycol, and color pigment.
The procedure for carrying out the dissolution study for Cefpodoxime proxetil and Cefuroxime axetil is given below.
Procedure for the dissolution study for formulations of Cefpodoxime proxetil The prepared cefpodoxime proxetil sustained release tablets were tested for cefpodoxime proxetil in 900 ml of pH 3.0 glycine buffer for 1 hr, after which the dissolution media was changed to pH 6.8 phosphate buffer 900ml every hour up to 6 hrs. The tablets were placed in the dissolution vessel (USP type 2) and rotated at 75φm.
Procedure for the dissolution study for formulations of Cefuroxime axetil The prepared cefuroxime axetil sustained release tablets were tested for cefuroxime axetil in 900 ml of 0.07N hydrochloric acid in water for 1 hr, after which the dissolution media was changed to pH 6.8 phosphate buffer 900 ml every hour up to 7 hrs. The tablets were placed in the dissolution vessel (USP type 2) and rotated at 75φm.
Example 1
Composition
Procedure for making the formulation
Cefpodoxime proxetil, calcium carboxymethyl cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose anhydrous, Plasdone S-630, and Viscarin GP328, were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The granules were blended with magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Example 2 Composition
Example 3
Composition
Example 4
Composition
Procedure for making the formulations of examples 2-4
Cefpodoxime proxetil, calcium carboxymethyl cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose anhydrous, Plasdone S-630, Viscarin
GP328, and Carbopol 97 IP, were screened through 40 mesh sieve and roll compacted. The compacts were crushed and passed through 20 mesh to get granules. The granules were blended with magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated.
Example 5
Composition
Procedure for making the formulation
Cefpodoxime proxetil, calcium carboxymethyl cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose anhydrous, Plasdone S-630, Viscarin GP209, Carbopol 97 IP, and Gelcarin GP 911, were screened through 40 mesh sieve and roll compacted. The compacts were crushed and passed through 20 mesh to get granules. The granules were blended with magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Dissolution profile
Example 6
Composition
Procedure for making the formulation
Cefpodoxime proxetil, calcium carboxymethyl cellulose, croscarmellose sodium, sodium lauryl sulfate, lactose anhydrous, Plasdone S-630, and
Viscarin GP209, were screened through 40 mesh sieve and roll compacted.
The compacts were crashed and passed through 20 mesh to get granules. The granules were blended with Gelcarin GP911, Carbopol 97 IP, and magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated.
Example 7
Composition
Procedure for making the formulation
Cefpodoxime proxetil, lactose anhydrous, Plasdone S-630, Gelcarin GP911, and, magnesium stearate, were screened through 40 mesh sieve and roll
compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Dissolution profile
Example 8
Composition
Procedure for making the formulation
Cefpodoxime proxetil, lactose anhydrous, Plasdone S-630, Gelcarin GP911, and, magnesium stearate, were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with croscarmellose sodium, and magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press.
The tablets were coated.
Example 9
Composition
Procedure for making the formulation
Cefpodoxime proxetil, lactose anhydrous, croscarmellose sodium, Plasdone S- 630, Gelcarin GP911, and, magnesium stearate, were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed
through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Example 10 Composition
Procedure for making the formulation Cefpodoxime proxetil, lactose anhydrous, calcium carboxymethyl cellulose, Plasdone S-630, Gelcarin GP911, and, magnesium stearate, were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and magnesium stearate. The lubricated
granules were then compressed into the tablets using a tablet press. The tablets were coated. Example 11 Composition
Procedure for making the formulation
Cefpodoxime proxetil, lactose anhydrous, Plasdone S-630, Gelcarin GP911, and, magnesium stearate, were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and magnesium stearate. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated.
Examples 12
Composition
Examples 13
Composition
Examples 14
Composition
Example 15
Composition
Dissolution profile
Procedure for making the formulations of examples 12-15
Cefuroxime axetil, calcium carboxymethyl cellulose, sodium lauryl sulfate, sodium chloride, Plasdone S-630, and Viscarin GP328, were screened through
40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The granules were blended with hydrogenated vegetable oil. The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated.
Example 16
Composition
Example 17
Composition
Dissolution profile
Procedure for making the formulations of examples 16-17 Cefuroxime axetil, lactose anhydrous, Plasdone S-630, Gelcarin GP911, and a portion of hydrogenated vegetable oil (0.48% w/w), were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and remaining hydrogenated vegetable oil (0.95% w/w). The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated.
Example 18
Composition
Procedure for making the formulation
Cefuroxime axetil, lactose anhydrous, Plasdone S-630, Gelcarin GP911, and a portion of hydrogenated vegetable oil (0.45% w/w), were screened through 40 mesh sieve and roll compacted. The compacts were crashed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with croscarmellose sodium, and remaining hydrogenated vegetable oil (0.91% w/w). The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Dissolution profile
Example 19
Composition
Procedure for making the formulation
Cefuroxime axetil, lactose anhydrous, croscarmellose sodium, Plasdone S- 630, Gelcarin GP911, and a portion of hydrogenated vegetable oil (0.50% w/w), were screened through 40 mesh sieve and roll compacted. The compacts were crushed and passed through 20 mesh to get granules. The resultant granules were coated with ethyl cellulose aqueous dispersion in a fluid bed processor. The resultant dried granules were screened and blended with calcium carboxymethyl cellulose, croscarmellose sodium, and remaining
hydrogenated vegetable oil (1.00% w/w). The lubricated granules were then compressed into the tablets using a tablet press. The tablets were coated. Bioavailability study
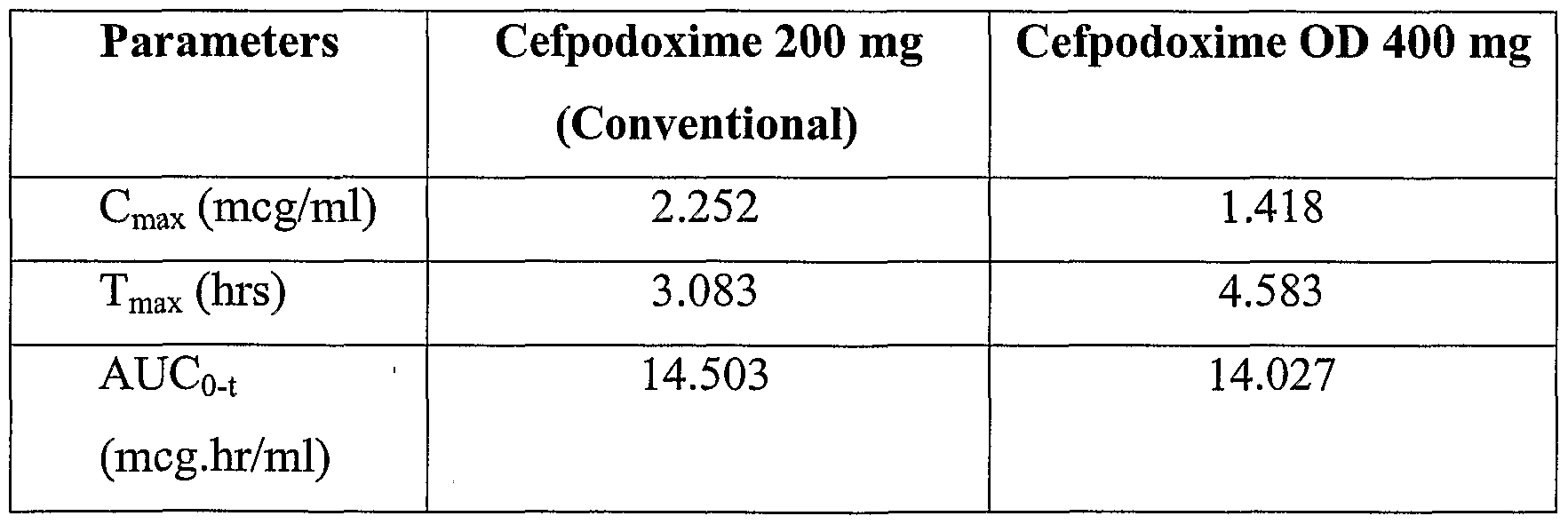
Bioavailability study was done using the composition of Example 1 in normal healthy human volunteers. The study was conducted for comparison between conventional Cefpodoxime tablets 200mg and sustained release Cefpodoxime tablets 400mg.
Six healthy male volunteers were selected for the study in which each volunteer was administered a dose of the drag with 180 ml of water. The volunteers fasted overnight and had a US FDA recommended breakfast before taking the drag. The desired blood levels up to 17 hrs were achieved with the composition prepared according the present invention. The data of the comparative pharmacokinetic parameters are summarized in table below (Figure 1). Comparative pharmacokinetic parameters:
Description of figure
Figure 1: Plot of comparative plasma profile of Cefpodoxime OD tablets 400mg (Test) v/s Cefpodoxime conventional tablets 200mg (Ref)