WO2000077012A1 - Bisphenol-a bis(diphenyl phosphate)-based flame retardant - Google Patents
Bisphenol-a bis(diphenyl phosphate)-based flame retardant Download PDFInfo
- Publication number
- WO2000077012A1 WO2000077012A1 PCT/US2000/015598 US0015598W WO0077012A1 WO 2000077012 A1 WO2000077012 A1 WO 2000077012A1 US 0015598 W US0015598 W US 0015598W WO 0077012 A1 WO0077012 A1 WO 0077012A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- bisphenol
- flame retardant
- area
- bis
- diphenyl phosphate
- Prior art date
Links
- 239000003063 flame retardant Substances 0.000 title claims abstract description 69
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 title claims abstract description 47
- BQPNUOYXSVUVMY-UHFFFAOYSA-N [4-[2-(4-diphenoxyphosphoryloxyphenyl)propan-2-yl]phenyl] diphenyl phosphate Chemical compound C=1C=C(OP(=O)(OC=2C=CC=CC=2)OC=2C=CC=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OP(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 BQPNUOYXSVUVMY-UHFFFAOYSA-N 0.000 title claims abstract description 45
- 239000000539 dimer Substances 0.000 claims abstract description 37
- 238000004128 high performance liquid chromatography Methods 0.000 claims abstract description 25
- XZZNDPSIHUTMOC-UHFFFAOYSA-N triphenyl phosphate Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)(=O)OC1=CC=CC=C1 XZZNDPSIHUTMOC-UHFFFAOYSA-N 0.000 claims abstract description 12
- WLXXENAUDPZXIB-UHFFFAOYSA-N diphenyl (2-prop-1-en-2-ylphenyl) phosphate Chemical compound CC(=C)C1=CC=CC=C1OP(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 WLXXENAUDPZXIB-UHFFFAOYSA-N 0.000 claims abstract description 6
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 claims description 73
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 56
- 229940106691 bisphenol a Drugs 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 29
- 239000000203 mixture Substances 0.000 claims description 28
- 229910019213 POCl3 Inorganic materials 0.000 claims description 27
- 238000000034 method Methods 0.000 claims description 20
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 17
- 229920005989 resin Polymers 0.000 claims description 15
- 239000011347 resin Substances 0.000 claims description 15
- 239000000178 monomer Substances 0.000 claims description 13
- 239000003054 catalyst Substances 0.000 claims description 11
- 239000004721 Polyphenylene oxide Substances 0.000 claims description 6
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 239000004417 polycarbonate Substances 0.000 claims description 5
- 229920000515 polycarbonate Polymers 0.000 claims description 5
- 229920006380 polyphenylene oxide Polymers 0.000 claims description 5
- 230000002829 reductive effect Effects 0.000 claims description 5
- 229910019142 PO4 Inorganic materials 0.000 claims description 4
- XECAHXYUAAWDEL-UHFFFAOYSA-N acrylonitrile butadiene styrene Chemical compound C=CC=C.C=CC#N.C=CC1=CC=CC=C1 XECAHXYUAAWDEL-UHFFFAOYSA-N 0.000 claims description 4
- 229920000122 acrylonitrile butadiene styrene Polymers 0.000 claims description 4
- 239000004676 acrylonitrile butadiene styrene Substances 0.000 claims description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 4
- 239000010452 phosphate Substances 0.000 claims description 4
- 235000010290 biphenyl Nutrition 0.000 claims description 3
- 239000004305 biphenyl Substances 0.000 claims description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 3
- 230000003197 catalytic effect Effects 0.000 claims description 2
- 239000004793 Polystyrene Substances 0.000 claims 2
- 229920005992 thermoplastic resin Polymers 0.000 claims 2
- 125000006267 biphenyl group Chemical group 0.000 claims 1
- 229920002223 polystyrene Polymers 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- 239000012535 impurity Substances 0.000 description 17
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000013638 trimer Substances 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 9
- 238000004821 distillation Methods 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 8
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- JREYOWJEWZVAOR-UHFFFAOYSA-N triazanium;[3-methylbut-3-enoxy(oxido)phosphoryl] phosphate Chemical compound [NH4+].[NH4+].[NH4+].CC(=C)CCOP([O-])(=O)OP([O-])([O-])=O JREYOWJEWZVAOR-UHFFFAOYSA-N 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- -1 isopropenylphenyl diphenylphosphate Chemical compound 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000470 constituent Substances 0.000 description 4
- 229910001629 magnesium chloride Inorganic materials 0.000 description 4
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 239000011574 phosphorus Substances 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 239000008399 tap water Substances 0.000 description 4
- 235000020679 tap water Nutrition 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 3
- 239000003518 caustics Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- ASMQGLCHMVWBQR-UHFFFAOYSA-M diphenyl phosphate Chemical compound C=1C=CC=CC=1OP(=O)([O-])OC1=CC=CC=C1 ASMQGLCHMVWBQR-UHFFFAOYSA-M 0.000 description 3
- 229920005669 high impact polystyrene Polymers 0.000 description 3
- 239000004797 high-impact polystyrene Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 3
- 238000004679 31P NMR spectroscopy Methods 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 229920007019 PC/ABS Polymers 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229910001507 metal halide Inorganic materials 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- 239000012925 reference material Substances 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- WUQYBSRMWWRFQH-UHFFFAOYSA-N 2-prop-1-en-2-ylphenol Chemical compound CC(=C)C1=CC=CC=C1O WUQYBSRMWWRFQH-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000004609 Impact Modifier Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- SISZSEGHLTVIJW-UHFFFAOYSA-N OC1=CC=C(C=C1)C(C)(C)C1=CC=C(C=C1)O.[P] Chemical compound OC1=CC=C(C=C1)C(C)(C)C1=CC=C(C=C1)O.[P] SISZSEGHLTVIJW-UHFFFAOYSA-N 0.000 description 1
- 239000012963 UV stabilizer Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 230000002528 anti-freeze Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- YHRRLJVICROTOJ-UHFFFAOYSA-N diphenyl phosphono phosphate Chemical compound C=1C=CC=CC=1OP(=O)(OP(O)(=O)O)OC1=CC=CC=C1 YHRRLJVICROTOJ-UHFFFAOYSA-N 0.000 description 1
- MQHNKCZKNAJROC-UHFFFAOYSA-N dipropyl phthalate Chemical compound CCCOC(=O)C1=CC=CC=C1C(=O)OCCC MQHNKCZKNAJROC-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 239000012035 limiting reagent Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 150000005309 metal halides Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001707 polybutylene terephthalate Polymers 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/52—Phosphorus bound to oxygen only
- C08K5/521—Esters of phosphoric acids, e.g. of H3PO4
- C08K5/523—Esters of phosphoric acids, e.g. of H3PO4 with hydroxyaryl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/12—Esters of phosphoric acids with hydroxyaryl compounds
Definitions
- This invention relates to a novel liquid flame retardant having a high bisphenol-A bis(diphenyl phosphate) content.
- Bisphenol-A bis(diphenyl phosphate) is a well known flame retardant for use in normally flammable resins and is especially useful in flame retarding polycarbonate/acrylonitrile-butadiene- styrene (PC/ABS) compositions. It also finds use as a flame retardant in polyphenylene oxide/styrene compositions.
- this invention provides a bisphenol-A bis(diphenyl phosphate) monomer-based flame retardant which is a liquid at room temperature, i.e., 20 to 25°C, and which is resistant to the formation of crystals over time.
- this invention provides a flame retardant comprising bisphenol-A bis(diphenyl phosphate) and its dimer, the former having from 78 to 87 HPLC area % and from about 85 to less than 90 normalized area %, the normalized area % being based on the total HPLC area % of the bisphenol-A bis(diphenyl phosphate) and the dimer.
- this invention provides a bisphenol-A bis(diphenyl phosphate)-based flame retardant having a low isopropenylphenyl diphenyl phosphate content.
- this invention provides a bisphenol-A bis(diphenyl phosphate)-based flame retardant having a low triphenyl phosphate content. Still further, this invention provides a bisphenol-A bis(diphenyl phosphate)-based flame retardant which is a liquid and is resistant to the formation of crystals and which has a high phosphorus content, a low isopropenylphenyl diphenylphosphate content and a low triphenyl phosphate content.
- the dimer not only attenuates the formation of a solid flame retardant or crystallization in the liquid form, it is also a contributor to the total phosphorus content of the flame retardant. (It is pointed out that the trimer of bisphenol-A bis(diphenyl phosphate) is also an impurity which contributes to the same functions as the dimer. However, the amount of trimer present is usually quite small and thus, focus is kept on the dimer in defining the flame retardants of this invention.) The dimer is easily obtained in situ as it is a product of the process for producing the bisphenol-A bis(diphenyl phosphate) from the reaction of POCl 3 and bisphenol-A followed by the reaction of phenol with the first reaction product. Not any amount of dimer, however, is suitable for these purposes.
- the amount of dimer needed is tied to the amount of bisphenol-A bis(diphenyl phosphate) in the flame retardant.
- the amount of bisphenol-A bis(diphenyl phosphate) lies within the range of from 78 to 87 area %, preferably 80 to 85 area %, and most preferably 82 to 85 area %.
- the amount of dimer needed is that amount which will give a normalized area % for the bisphenol-A bis(diphenyl phosphate) which is within the range of from 83 to less than 90 %, preferably from 85 to 89 %, and most preferably from 85 to 88%.
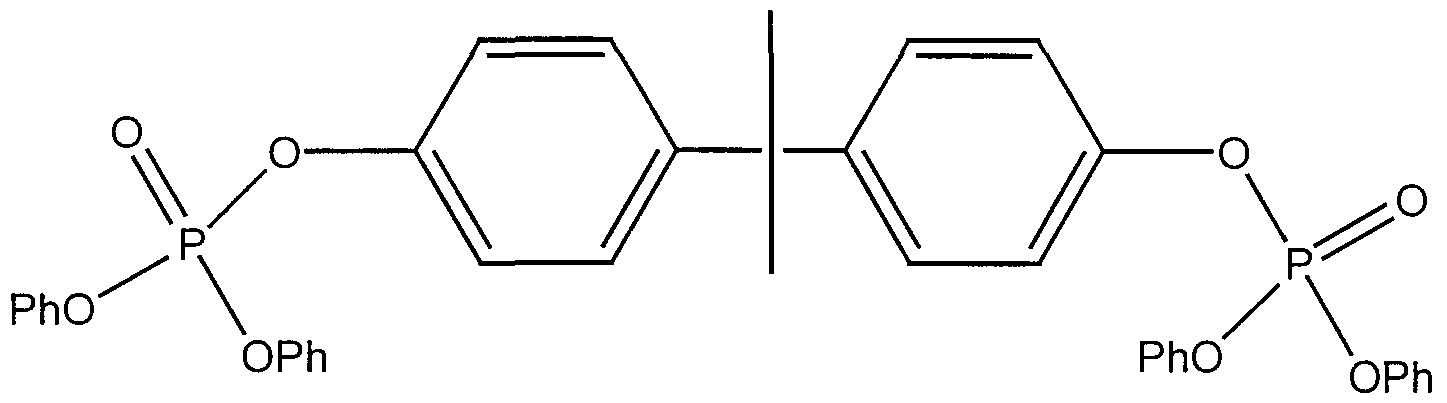
- the normalized area % is based on the total of the area %'s for the bisphenol-A bis(diphenyl phosphate) and its dimer. If less dimer than recited above is used, its useful effects are diminished, if more dimer is used, then the character of the flame retardant and its use in a resin formulation is affected. For each bisphenol-A molecule used to produce bisphenol-A bis(diphenyl phosphate), there are two phosphorus substituents, whereas for each molecule of bisphenol-A used to produce the dimer, there are only one and one-half phosphorus substituents. The structural formulas of the two make that clear.
- the bisphenol-A bis(diphenyl phosphate) molecule which can be referred to as a monomer, has the structure:
- the dimer has the structure:
- the amount of dimer in the flame retardants of this invention is within the range of from 10 to 13 area %, preferably from 11 to 13 area %, and most preferably from 12 to 13 area %.
- the flame retardants of this invention will also have a trimer content, the trimer having the structure:
- the flame retardants of this invention also feature a very low isopropenylphenyl diphenyl phosphate content. This compound is considered by some in the resin formulation industry as a deleterious impurity whose presence must be minimized.
- the flame retardants of this invention preferably contain no more than 0.01 area % of this impurity.
- the structure for this impurity is:
- a widely recognized and particularly troublesome impurity found in bisphenol-A bis(diphenyl phosphate)-based flame retardants is triphenylphosphate.
- This impurity tends to "juice" in the resin formulation and adversely affect the formulation's physical characteristics.
- the flame retardants of this invention are advantaged as they contain less than about 2.5 area % triphenyl- phosphate and preferably less than about 2.0 area %. Most preferred are flame retardants containing less than about 1.5 area % of this impurity.
- the flame retardants of this invention, after washing and neutralization, will have an acid number less than about 2.0 mg KOH/g and preferably less than about 0.15 mg KOH/g .
- the melting point range of the flame retardants of this invention is found to be below room temperature, say 20-25°C.
- the flame retardants of this invention are best described as viscous oils at room temperature.
- Their viscosity is within the range of from 16,000 to 18,500 cP at 25°C, 2200 to 2400 cP at 40°C and 40-60 cP at 100°C.
- the flame retardants of this invention are produced in a two step process.
- the first step entails producing the intermediate, diphosphorotetrachloridate of bisphenol- A, and, to a specified extent, its dimer (and trimer), by gradually adding, over time, bisphenol-A to a reactor containing an excess of phosphorus oxyhalide, the halide being bromine or chlorine, and a catalytic amount of a metal halide, e.g., magnesium chloride.
- the ratio of POCl 3 to bisphenol-A be within the range of from 3.5:1 to 4.5: 1. These ratios define a large excess of POCl 3 above the stoichiometric amounts. Such excesses serve an additional purpose, that is, the excess POCl 3 acts as a solvent for the process. Thus, no other process solvents, organic or otherwise, are needed.
- the preferred catalyst is MgCl 2 for use in both process steps.
- the amount of catalyst used in the first step is generally in an amount of from 0.01 to 4.0 wt% based on the weight of the bisphenol-A fed.
- the second step if the catalyst is the same as that used in the first step, the original catalyst provided remains in the reaction mass and is sufficient for the second step. If the second step catalyst is different than the first step catalyst, then there is provided about 0.8 wt% catalyst, based on the weight of the bisphenol-A fed to the reaction mass.
- Other suitable catalyst include, metal halide salts such as aluminum chloride, calcium chloride, zinc chloride and titanium tetrachloride.
- the bisphenol-A be added to the POCl 3 in increments or on a continuous basis.
- the monomer/dimer relationship i.e., the normalized area % of the bisphenol-A bis(diphenyl phosphate)
- the rate of feed of the bisphenol-A to the POCl 3 can be affected by adjusting the rate of feed of the bisphenol-A to the POCl 3 .
- the trend suggests that high rates of bisphenol-A addition favor the production of the monomer. It is believed that for a particular set of process parameters and for a particular reactor size and configuration, the determination of the best bisphenol-A feed rates is performed by trial and error.
- the reaction mass temperature during the bisphenol-A addition is kept within the range of from 85 to 106°C to insure that the reaction between the POCl 3 and bisphenol-A proceeds expeditiously. After the bisphenol-A addition is complete, the reaction mass is maintained until the reaction is deemed complete.
- the bisphenol-A feed and temperature maintenance periods together can range from 3 to 6 hours, and more usually from 4 to 5 hours. If the temperature is too low during the bisphenol-A addition and temperature maintenance periods, say below 70°C, it is believed that the final bisphenol-A bis(diphenyl phosphate)-based product will contain a high amount of isopropenylphenyl diphenylphosphate.
- the reaction mass is heated under a reduced pressure to distill off the excess POCl 3 . Pressures of about 50 torr can be used. Distillation pot temperatures beginning at 50°C and ending at 150 to 160°C are suitable. Alternatively, the POCl 3 can be removed by stripping the heated reaction mass with an inert gas such as nitrogen. The distillation continues until typically ⁇ 3.5 mole % phosphorus as POCl 3 is detected in the reaction mass by 31 P-NMR.
- the resultant intermediate product (monomer, dimer and trimer) from the POCl 3 distillation is reacted with phenol in the presence of any one of the previously discussed catalysts.
- the phenol in the molten state, is fed to the intermediate reaction mass which is at a temperature of from 130 to 160°C.
- the amount of phenol fed provides from 3.8 to 4, and preferably about 3.9 moles of phenol per mole of bisphenol-A fed in the first step.
- the reaction mass is kept at a temperature of from 130 to 180°C until no further evolution of HCl is detected. In some cases, it may be advantageous to add a small amount of phenol after the last HCl is detected to insure that the reaction is indeed complete.
- reaction time (at the above temperatures) for the second step is within 24 hours and preferably occurs in about 6 hours which includes addition times of less than three hours.
- the reaction mass is dissolved in an organic solvent and washed with caustic, which can be sodium or potassium hydroxide. Multiple water washes are also used. After each washing, there is a phase separation. After the washing, the organic solvent is removed by heating under a reduced pressure.
- Typical organic solvents are methyl- cyclohexane, toluene, xylene, cyclohexane, heptane, and mixtures of any two or more of the foregoing. Most preferred is a 50 wt% mix of methylcyclohexane and toluene.
- BPDAP normalized area % was calculated in accordance with, BPDAP area %
- Step l A 4 necked 2000 ml round-bottom flask was equipped with a mechanical stirrer, a Friedrich condenser stacked on top of an Allihn condenser (tap water used for coolant), and a thermocouple well. The glassware was dried and flushed with nitrogen. A nitrogen blanket was maintained on the contents of the flask by having a nitrogen flow (0.5-1.0 SCFH) T'eed into a line connecting the condenser and a scrubber solution (water or caustic will work) to absorb the HCl that is evolved. The flask containing the scrubber solution was placed on a balance to determine the mass of the HCl evolved.
- a nitrogen flow 0.5-1.0 SCFH
- the magnetic stirrer was replaced with a mechanical stirrer and the stack of condensers returned in place of the distillation takeoff/condenser.
- a 250 ml jacketed addition funnel on an offset adapter was mounted on the 2000 ml flask.
- a nitrogen blanket was maintained on the contents of the flask by having a nitrogen flow (0.2-1.0 SCFH) T'eed into a line connecting the condenser and a scrubber solution (water or caustic will work) to absorb the HCl that is evolved.
- the flask containing the scrubber solution was placed on a balance to determine the mass of the HCl evolved.
- the reaction mixture was stirred while it was warmed to about 145°C and the molten phenol (752 g, 7.99 mol) was added as shown in the table below.
- reaction mixture (1362 g) was transferred to a jacketed wash kettle (5 liter 4 necked flask with a bottom drain and a mechanical stirrer) with a mixture of 1000 g of toluene and 1004 g of methylcyclohexane.
- the reaction mixture was washed at 60-72°C with 300 g of 10 wt % aqueous potassium hydroxide (all of the other experiments used 10 wt % aq.
- sodium hydroxide obtained 434 g of aqueous phase, pH ⁇ 14), 300 g of 5 wt% aqueous potassium hydroxide (obtained 334 g of aqueous phase, pH ⁇ 14), 301 g of tap water (obtained 304 g of aqueous phase, pH ⁇ l 1). 302 g of tap water (obtained 304 g of aqueous phase, pH ⁇ 8). and then 302 g of tap water (obtained 307 g of aqueous phase, pH ⁇ 7). There was obtained 3157 g of organic phase which was gravity filtered (Whatman 2 V paper). The volatiles were removed on a rotavap (2 torr/90°C).
- the residual solvent was removed in a vacuum oven at 150°C/2 torr to give 1189 g of slight cloudy colorless product as a viscous oil.
- the product by HPLC analysis contained 0.07 area % DPP, 0.11 area % phenol, 0.49 area % half-ester, 0.002 area % IPP, 84.17 area % BPADP, 12.35 area % dimer, and 1.53 area % trimer. 87.2 normalized area % for the BPADP was caculated.
- Example II was run in a manner similar to Example I except that it was run in 1/4 scale and used laboratory glassware.
- Example III was run in the manner of Example I.
- the process parameters of Examples I and II are recited in Table III.
- Examples IN-IX were run in the manner of Example I.
- the process parameters of Examples IV-IX are recited in Table III.
- Phosphorus oxychloride (POCl 3 , 306.7 g, 2.0 mol) and MgCl 2 (0.95 g, 0.01 mol) were added into a 1 L round-bottomed flask equipped with a Friedrich's condenser (8.5°C). The reaction mass was stirred at 90-95°C under a pad of nitrogen. BPA was added in six increments 114.9 g (total, 0.50 mol) over 1.5 hours. After an additional 3 hours at 105°C, reaction completion was shown by the weight of HCl evolved into a water scrubber (35.8 g HCl trapped, 98% of theoretical). Excess POCl 3 was then removed by distillation from the reaction mass until no POCl 3 was detected using 31 P-NMR. Step 2
- the reactor was reconfigured with a jacketed addition funnel and nitrogen pad inlet through the addition funnel.
- the evolved HCl passed through an expansion piece in the reactor neck into an aqueous trap, its weight also used for monitoring the reaction progress.
- Molten phenol (174.1 g , 1.9 moles at 72°C) was added dropwise over 1.5 hours into the reactor held at 128-134°C and, after addition, reaction proceeded at 148°C for an additional 3 hours, followed by NMR and HPLC analysis of the crude product.
- the total mass yield of crude product was 322.1 g (92.3% yield, with BPA as limiting reagent).
- HPLC showed 69.9 % BPADP, 9.07 % oligomerics, 11.0 % unknowns, 2.35 % PhOH, no TPP was detected, and the IPP level was ⁇ 0.01 %.
- An 88.5 normalized area % for BPADP was calculated.
- the crude BPADP was purified by dissolving 286.4 g into 483.5 g of mixed solvent (50 % wt methylcyclohexane : toluene), then washing with two 150 g portions 10 % wt. NaOH. There was a phase separation at 70°C after each wash. The organic phase was then washed with three portions of water, which was separated from the aqueous phase after each wash.
- Solvent removal and drying of the organic phase were accomplished by distillation and nitrogen stripping.
- the organic phase recovery in the purification step was 91.3 wt %.
- the residual solvent was removed in a vacuum oven (12 hours, 30 mm Hg; 140°C).
- the product analysis by HPLC showed 84J area % BPADP, 12.0 area % dimer, 1.46 area % trimer, ⁇ 0.01 area % IPP, 0.11 area % half ester, 0.27 area % DPP.
- Example X (Of the Invention) The procedure of Example X was followed except that 413.1 g POCl 3 , 176.6 g of bisphenol-
- HPLC Analysis Method The HPLC method used to obtain the area % values reported herein is described below. The method uses UV detection at 254 nm with and acetonitrile/water gradient on a reverse phase C18 column.
- the sample is dissolved in acetonitrile at a concentration of approximately 2500 ppm. An aliquot of the solution is then transferred to an autosampler vial. A small portion (10 ⁇ L) is injected into the HPLC and analyzed under gradient conditions at a wavelength of 254 nm. Area % values are calculated for all peaks in the chromatogram.
- External standard reference materials are available for the following impurities: DPP, Phenol, BPA, and TPP. Solutions of these reference materials are made up at concentrations of 100 ppm. Each is injected and analyzed following the conditions listed below. Response factors are calculated for each of these reference peaks to allow weight % values for these impurities to be calculated from the sample chromatograms.
- IPP impurity
- HPLC any suitable HPLC system equipped with a multisolvent delivery system capable of binary gradient elution, UV detection at 254 nm, automatic sample injector capable of 10 ⁇ L sample injection.
- the HPLC instrument used to obtain the area % values reported was a Hewlett-Packard Model 1090.
- the flame retardants of this invention can be used in a wide variety of polymer resins. As before noted, they are useful in polycarbonate and acrylonitrile-butadiene-styrene blends (PC/ABS) and in polyphenylene oxide containing blends, especially blends with high impact polystyrene (PPO/HIPS). Other resins in which the flame retardants of this invention are useful are poly- phenylene oxide, high-impact polystyrene, polycarbonate, polyurethane, polyvinyl chloride, acrylonitirle-butadiene-styrene and polybutylene terephthalate.
- PC/ABS polycarbonate and acrylonitrile-butadiene-styrene blends
- PPO/HIPS high impact polystyrene
- Other resins in which the flame retardants of this invention are useful are poly- phenylene oxide, high-impact polystyrene, polycarbonate, polyurethane, poly
- the flame retardants of this invention will generally be use in amounts ranging from 7 to 20 wt% in the resin, based upon the total weight of the entire resin formulation.
- the flame retardants of this invention are also suitable for use in combination with other formulation constituents.
- plasticizers, impact modifiers, antioxidants, UV stabilizers, pigments, fillers may be used.
- Reference to the prior art will identify further constituents which are suitable additives in for resin formulations.
Abstract
Description







Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001503869A JP2003502451A (en) | 1999-06-11 | 2000-06-06 | Flame retardant based on bisphenol-A bis (diphenyl phosphate) |
| EP00939617A EP1194435A1 (en) | 1999-06-11 | 2000-06-06 | Bisphenol-a bis(diphenyl phosphate)-based flame retardant |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/330,688 US6319432B1 (en) | 1999-06-11 | 1999-06-11 | Bisphenol-A bis(diphenyl phosphate)-based flame retardant |
| US09/330,688 | 1999-06-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000077012A1 true WO2000077012A1 (en) | 2000-12-21 |
Family
ID=23290873
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/015598 WO2000077012A1 (en) | 1999-06-11 | 2000-06-06 | Bisphenol-a bis(diphenyl phosphate)-based flame retardant |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US6319432B1 (en) |
| EP (1) | EP1194435A1 (en) |
| JP (1) | JP2003502451A (en) |
| KR (1) | KR100720873B1 (en) |
| CN (1) | CN1210284C (en) |
| WO (1) | WO2000077012A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002008329A1 (en) * | 2000-07-25 | 2002-01-31 | Bayer Aktiengesellschaft | Flame-resistant polycarbonate compositions |
| WO2002055526A1 (en) * | 2001-01-09 | 2002-07-18 | Bayer Aktiengesellschaft | Phosphoric flame-retardant and flame-retarding thermoplastic molding compounds |
| WO2011054862A1 (en) | 2009-11-05 | 2011-05-12 | Bayer Materialscience Ag | Polycarbonate plates with improved flame resistance |
| EP3312239A4 (en) * | 2015-06-18 | 2019-01-02 | Kingfa Sci&Tech Co. Ltd. | Polycarbonate composition and preparation method thereof |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3926938B2 (en) * | 1998-12-03 | 2007-06-06 | 三菱エンジニアリングプラスチックス株式会社 | Flame retardant polycarbonate resin composition |
| US6399685B1 (en) * | 2000-12-11 | 2002-06-04 | Albemarle Corporation | Purification of arylene polyphosphate esters |
| US7303810B2 (en) * | 2001-03-05 | 2007-12-04 | 3Form, Inc. | Fire-resistant architectural resin materials |
| US7691470B2 (en) * | 2001-03-05 | 2010-04-06 | 3Form | Laminate structure with polycarbonate sheets |
| US7008700B1 (en) | 2001-03-05 | 2006-03-07 | 3-Form | Architectural laminate panel with embedded compressible objects and methods for making the same |
| US6667355B2 (en) * | 2001-08-30 | 2003-12-23 | Pabu Services, Inc. | Higher alkylated triaryl phosphate ester flame retardants |
| DE10216736A1 (en) * | 2002-04-16 | 2004-02-05 | Bayer Ag | Flame retardants for polymers containing a mixture of two different aryl phosphates, their preparation and their use |
| CN1708503A (en) * | 2002-10-26 | 2005-12-14 | 阿克苏诺贝尔公司 | Retardation of crystallization in oligomeric phosphate compositions |
| US20060046017A1 (en) | 2004-09-01 | 2006-03-02 | 3Form | Architectural glass panels with embedded objects and methods for making the same |
| US20060068317A1 (en) * | 2004-09-30 | 2006-03-30 | Klei Steven R | Poly(arylene ether) composition |
| EP1945711A1 (en) * | 2005-11-08 | 2008-07-23 | Supresta LLC | Flame retardant composition and hydrolysis-susceptible resin containing same |
| US20070254991A1 (en) * | 2006-04-28 | 2007-11-01 | Saadat Hussain | Phosphorus-containing tetrabromobisphenol A |
| WO2008082690A2 (en) * | 2006-07-07 | 2008-07-10 | Albemarle Corporation | Phosphorous-based flame retardant recovery method |
| US8182903B2 (en) | 2007-05-08 | 2012-05-22 | 3Form, Inc. | Multivariate color system with texture application |
| US8968611B2 (en) * | 2008-06-09 | 2015-03-03 | Adeka Corporation | Process for solidification of phosphoric ester-based flame retardants |
| CN101348500B (en) * | 2008-08-19 | 2012-02-22 | 山东默锐化学有限公司 | Preparation of bisphenol A bis(diphenyl phosphoester ) |
| KR101013666B1 (en) * | 2008-09-18 | 2011-02-10 | 한밭대학교 산학협력단 | Radiant heat device of computer |
| EP2358816B1 (en) * | 2008-12-08 | 2021-03-24 | SABIC Global Technologies B.V. | Flame retardant polycarbonate compositions, method of manufacture thereof, and articles therefrom |
| TWI382988B (en) * | 2009-12-25 | 2013-01-21 | Grand Tek Advance Material Science Co Ltd | Organic silicon phosphate and fabrication method thereof |
| CN101831308A (en) * | 2010-04-19 | 2010-09-15 | 江苏大明科技有限公司 | Solid phosphate ester flame retardant with high molecular weight and preparation method and applications thereof |
| CN101993586B (en) * | 2010-11-30 | 2012-10-10 | 金发科技股份有限公司 | Flame-resistant polycarbonate/ABS (Acrylonitrile Butadiene Styrene) material |
| TWI410446B (en) | 2011-01-05 | 2013-10-01 | Grand Tek Advance Material Science Co Ltd | Phosphorus containing novalac phenol resins, methods for manufacturing the same, and formulae containing the same |
| SE1100784A1 (en) * | 2011-10-21 | 2013-01-08 | Perstorp Ab | New phosphate compound |
| USD691289S1 (en) | 2012-09-05 | 2013-10-08 | 3Form, Inc. | Panel with cut and aligned thatch interlayer |
| CN104829647B (en) * | 2014-04-10 | 2016-08-24 | 北京理工阻燃科技有限公司 | A kind of preparation method and application of phosphorous polyether ester |
| US10240030B2 (en) * | 2014-12-02 | 2019-03-26 | Sabic Global Technologies B.V. | Article comprising a high flow polyetherimide composition |
| JP6532276B2 (en) * | 2015-04-23 | 2019-06-19 | 株式会社Adeka | Flame-retardant water-based polyurethane resin composition, and flame-retardant polyester fiber coated with the same |
| CN106699805B (en) * | 2016-12-15 | 2019-02-01 | 衢州普信新材料有限公司 | A kind of preparation method of bisphenol-A bis- (diphenyl phosphoesters) |
| CN108676282A (en) * | 2018-05-18 | 2018-10-19 | 武汉理工大学 | A kind of environment friendly flame-retardant PVC/ABS alloys and preparation method thereof |
| CN111040457B (en) * | 2019-12-27 | 2022-05-06 | 广东精一塑胶实业有限公司 | Preparation method of curing coated flame-retardant waterproof agent for plastics |
| CN112142779B (en) * | 2020-10-13 | 2023-01-13 | 中国科学院宁波材料技术与工程研究所 | Rapid preparation method of bisphenol A bis (diaryl phosphate) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997047631A1 (en) * | 1996-06-13 | 1997-12-18 | Great Lakes Chemical Corporation | Process to prepare aryldiphosphoric esters |
| WO1998004566A1 (en) * | 1996-07-29 | 1998-02-05 | Akzo Nobel N.V. | Process for the formation of hydrocarbyl bis(dihydrocarbyl phosphate) |
| WO1998035970A1 (en) * | 1997-02-14 | 1998-08-20 | Great Lakes Chemical Corporation | Process for making and using bisaryl diphosphates |
| WO1999055771A1 (en) * | 1998-04-29 | 1999-11-04 | Great Lakes Chemical Corporation | Continuous process for the manufacture of phosphoric acid esters |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2520090A (en) | 1947-12-30 | 1950-08-22 | Monsanto Chemicals | Polyphosphates of divalent aryl hydrocarbons |
| GB734767A (en) | 1952-01-10 | 1955-08-10 | Lankro Chem Ltd | New neutral orthophosphate esters |
| US3174931A (en) | 1961-12-05 | 1965-03-23 | Sinclair Research Inc | Grease compositions |
| US3360591A (en) | 1962-07-31 | 1967-12-26 | Mobil Oil Corp | Diphosphates of divalent aryl hydrocarbons |
| US3254973A (en) | 1962-07-31 | 1966-06-07 | Socony Mobil Oil Co Inc | Gasolines and phosphorus-containing additives therefor |
| US3317636A (en) | 1964-01-10 | 1967-05-02 | American Cyanamid Co | O, o, o', o'-tetramethyl o, o'-thiodi-p-phenylene phosphorothioate |
| JPS5051154A (en) | 1973-09-06 | 1975-05-07 | ||
| AU8671075A (en) | 1974-12-06 | 1977-05-26 | Gen Electric | Flame retardant, non-dripping composition of polyphenylene ether and acrylonitrile - butadiene-styrene |
| NL7901769A (en) | 1979-03-06 | 1980-09-09 | Gen Electric | FLAME RETARDANT, POLYPHENYLENE ETHER CONTAINING COMPOSITIONS. |
| US4223100A (en) | 1979-04-18 | 1980-09-16 | Mobay Chemical Corporation | Flame retardant aromatic polycarbonate with good mechanical properties and melt stability |
| EP0074112B2 (en) | 1981-09-09 | 1993-06-23 | Sumitomo Naugatuck Co., Ltd. | Thermoplastic composition |
| JPS5924736A (en) | 1982-08-02 | 1984-02-08 | Adeka Argus Chem Co Ltd | Flame-retarding polystyrene resin composition |
| JPS5945351A (en) | 1982-09-08 | 1984-03-14 | Adeka Argus Chem Co Ltd | Flame-retardant composition |
| EP0103230B1 (en) | 1982-09-10 | 1988-06-22 | Bayer Ag | Flame-retardant polymer mixtures |
| JPS59202240A (en) | 1983-05-02 | 1984-11-16 | Daihachi Kagaku Kogyosho:Kk | Flame-retardant thermoplastic resin composition |
| DE3523314A1 (en) | 1985-06-29 | 1987-01-02 | Bayer Ag | THERMOPLASTIC POLYCARBONATE MOLDINGS |
| JPS63117057A (en) | 1986-10-31 | 1988-05-21 | Daihachi Kagaku Kogyosho:Kk | Flame-retardant, heat-resistant aromatic vinyl resin composition |
| DE3700208A1 (en) | 1987-01-07 | 1988-07-21 | Bayer Ag | OLIGOMERS AND POLYMERS CONTAINING PHOSPHORUS WITH POLYPHENYLENE ETHERLOCKS AND MIXTURES OF OTHER PLASTICS WITH YOU |
| DE3819081A1 (en) | 1988-06-04 | 1989-12-07 | Bayer Ag | FLAME-RESISTANT, IMPACT TOOL POLYCARBONATE MOLDS |
| CA1334695C (en) | 1988-09-07 | 1995-03-07 | Jurou Ohzeki | Electroless plating-susceptive, fire retardant polyphenylene ether resin |
| US5204394A (en) | 1988-09-22 | 1993-04-20 | General Electric Company | Polymer mixture having aromatic polycarbonate, styrene I containing copolymer and/or graft polymer and a flame-retardant, articles formed therefrom |
| USRE36188E (en) | 1989-09-20 | 1999-04-06 | General Electric Company | Polymer mixture having aromatic polycarbonate styrene I containing copolymer and/or graft polymer and a flame-retardant, articles formed therefrom |
| US5281741A (en) | 1990-11-13 | 1994-01-25 | Fmc Corporation | Process for preparing aryldiphosphate esters |
| JPH0598118A (en) | 1991-10-14 | 1993-04-20 | Asahi Chem Ind Co Ltd | Styrene-based flame-retardant and thermal shockresistant resin composition |
| JP3137400B2 (en) | 1992-01-16 | 2001-02-19 | 旭化成工業株式会社 | Polyphenylene ether flame retardant resin composition |
| US5278279A (en) | 1992-03-11 | 1994-01-11 | Daicel Chemical Industries, Ltd. | Process for producing (co)polycarbonate with transesterification catalyst and compound |
| KR0163445B1 (en) | 1992-08-06 | 1999-01-15 | 유미꾸라 레이이찌 | Resin composition |
| US5420327A (en) | 1992-09-14 | 1995-05-30 | Akzo Nobel N.V. | Process for forming hydrocarbyl bisphosphate compound |
| DE4328656A1 (en) | 1993-08-26 | 1995-03-02 | Bayer Ag | Flame retardant, stress crack resistant polycarbonate ABS molding compounds |
| US5952408A (en) | 1994-12-01 | 1999-09-14 | Cheil Industries, Inc. | Flameproof thermoplastic resin compositions |
| JPH0995610A (en) | 1995-09-30 | 1997-04-08 | Nippon G Ii Plast Kk | Fire resisting resin composition |
| WO1997047361A1 (en) * | 1996-06-12 | 1997-12-18 | Southwest Research Institute | Fire extinguishing trash can liner |
| JP3558458B2 (en) | 1996-07-10 | 2004-08-25 | 旭化成ケミカルズ株式会社 | Method for producing aryl phosphate |
| ES2176676T3 (en) * | 1997-02-17 | 2002-12-01 | Bessey & Sohn | HOLDING DEVICE. |
| MY121010A (en) | 1997-08-29 | 2005-12-30 | Sabic Innovative Plastics Ip | Polycarbonate resin composition |
| TW455605B (en) | 1998-02-13 | 2001-09-21 | Gen Electric | Flame retardant carbonate polymer composition with improved hydrolytic stability |
-
1999
- 1999-06-11 US US09/330,688 patent/US6319432B1/en not_active Expired - Fee Related
-
2000
- 2000-06-06 KR KR1020017015969A patent/KR100720873B1/en not_active IP Right Cessation
- 2000-06-06 CN CNB008087938A patent/CN1210284C/en not_active Expired - Fee Related
- 2000-06-06 WO PCT/US2000/015598 patent/WO2000077012A1/en active Search and Examination
- 2000-06-06 JP JP2001503869A patent/JP2003502451A/en not_active Withdrawn
- 2000-06-06 EP EP00939617A patent/EP1194435A1/en not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997047631A1 (en) * | 1996-06-13 | 1997-12-18 | Great Lakes Chemical Corporation | Process to prepare aryldiphosphoric esters |
| WO1998004566A1 (en) * | 1996-07-29 | 1998-02-05 | Akzo Nobel N.V. | Process for the formation of hydrocarbyl bis(dihydrocarbyl phosphate) |
| WO1998035970A1 (en) * | 1997-02-14 | 1998-08-20 | Great Lakes Chemical Corporation | Process for making and using bisaryl diphosphates |
| WO1999055771A1 (en) * | 1998-04-29 | 1999-11-04 | Great Lakes Chemical Corporation | Continuous process for the manufacture of phosphoric acid esters |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002008329A1 (en) * | 2000-07-25 | 2002-01-31 | Bayer Aktiengesellschaft | Flame-resistant polycarbonate compositions |
| WO2002055526A1 (en) * | 2001-01-09 | 2002-07-18 | Bayer Aktiengesellschaft | Phosphoric flame-retardant and flame-retarding thermoplastic molding compounds |
| US7019056B2 (en) | 2001-01-09 | 2006-03-28 | Bayer Aktiengesellschaft | Flame retardants which contain phosphorus, and flame-retardant thermoplastic molding compositions |
| WO2011054862A1 (en) | 2009-11-05 | 2011-05-12 | Bayer Materialscience Ag | Polycarbonate plates with improved flame resistance |
| US9371437B2 (en) | 2009-11-05 | 2016-06-21 | Covestro Deutschland Ag | Polycarbonate plates with improved flame resistance |
| EP3312239A4 (en) * | 2015-06-18 | 2019-01-02 | Kingfa Sci&Tech Co. Ltd. | Polycarbonate composition and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2003502451A (en) | 2003-01-21 |
| KR100720873B1 (en) | 2007-05-22 |
| CN1355808A (en) | 2002-06-26 |
| KR20020010927A (en) | 2002-02-06 |
| CN1210284C (en) | 2005-07-13 |
| US6319432B1 (en) | 2001-11-20 |
| EP1194435A1 (en) | 2002-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6319432B1 (en) | Bisphenol-A bis(diphenyl phosphate)-based flame retardant | |
| EP1327635B1 (en) | Preparation of phenylphosphate esters of 4,4'-biphenol | |
| CN102264747B (en) | Water miscible solvent based process for purifying bisphosphate | |
| KR940001061B1 (en) | Process for the preparation of hydrolysis-stable organic phosphits | |
| JP2004511564A (en) | Manufacturing method of phosphate ester | |
| WO1998055486A1 (en) | Process for producing phosphoric ester compounds | |
| WO1998037046A1 (en) | PROCESS FOR THE MANUFACTURE OF TETRABROMOBISPHENOL-A WITH CO-PRODUCTION OF n-PROPYL BROMIDE | |
| US4897502A (en) | Process for making solid polyhalotriaryl phosphate esters | |
| EP0172633A1 (en) | Preparation of 2,2,4-trimethyl-1,2-dihydroquinoline | |
| US4241034A (en) | Process for purifying cyclic phosphonitrilic chlorides | |
| US6504065B1 (en) | Method of making metal salts of 2,4,6-tri-t-butylphenol | |
| KR100240175B1 (en) | Method for preparing the flame retardants of phosphoric ester with high flame-retardation and low fogging properties | |
| JP4084096B2 (en) | Method for producing pentaerythritol diphosphonate | |
| US3087960A (en) | Alkali metal salts of diglycol borates and methods for preparing the same | |
| KR20010021646A (en) | Method for Producing Tetrabutyl Ammonium Phenolate Diphenol Adduct | |
| JPH05247067A (en) | Method for preparation of polybrominated triarylphosphate | |
| RU2188826C2 (en) | Method of synthesis of ferrocene | |
| US9024054B2 (en) | Process for production of purified O-(2,6-dichloro-4-methyl-phenyl) O,O-dimethyl phosphorothioate | |
| Conant | THE PREPARATION OF SODIUM p-HYDROXYPHENYL-ARSONATE. | |
| JP2004018387A (en) | Method for producing pentaerythritol diphosphonate | |
| CA2216329A1 (en) | Process for the preparation of dimethylamine-borane | |
| JP2004035481A (en) | Method for producing spirocyclic diphosphonate compound | |
| WO1998050390A1 (en) | Formation of 1-phenylvinyl-1-phosphonic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 00808793.8 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CN JP KR |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2001 503869 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020017015969 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000939617 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017015969 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000939617 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000939617 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017015969 Country of ref document: KR |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) |