WO1998056772A1 - Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3--triazole-4-carboxamide and its use as antiepileptic - Google Patents
Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3--triazole-4-carboxamide and its use as antiepileptic Download PDFInfo
- Publication number
- WO1998056772A1 WO1998056772A1 PCT/EP1998/003427 EP9803427W WO9856772A1 WO 1998056772 A1 WO1998056772 A1 WO 1998056772A1 EP 9803427 W EP9803427 W EP 9803427W WO 9856772 A1 WO9856772 A1 WO 9856772A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- modification
- weak
- triazole
- carboxamide
- medium
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
Definitions
- Example 4 Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as an antiepileptic.
- the compound 1- (2,6-dif luorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide is obtained according to EP 199262, starting from 2,6-difluorobenzyl azide via the formation of 1-(2,6-difluorobenzyl)-1 H-1 ,2,3- triazoie-4-carboxylic acid, the procedure being analogous to Example 2.
- EP 199262 provides no information at all about possible crystal modifications obtained. If the method according to the Example 4 is used in conjunction with Example 2, the crude 1 - (2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide product obtained is finally crystallized from ethanol. However, EP 199262 gives no indication that such recrystallization is specifically to be applied, or on particular conditions that might be adopted. It has now surprisingly been found that the different crystal modifications (polymorphism) characterized below can be prepared by choice of specially selected process conditions, for example through the choice of an appropriate solvent for the recrystallization or the duration of the recrystallization. Description of the invention
- the invention relates to the novel crystal modifications A and A', their preparation and use in pharmaceutical preparations comprising this crystal modification.
- the modification A' compared with A, has defects in the crystal lattice. These are detectable, for example, by X-ray analysis, e.g. by smaller line spacings with otherwise predominantly identical lines or bands.
- novel crystal modification A of 1-(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide melts at 242 °C (239-245 °C).
- modification A or A' differs from modifications B and C predominantly in the shape and in the relative intensity of many bands. Particularly characteristic are the bands at 3412 cm '1 and 3092 cm '1 [cf. Figure 1], which are not present in the FT-IR spectra of the modifications B and C. In the range 4000-600 cm '1 , inter alia the following bands are obtained for modification A: 3412, 3189, 3092, 1634, 1560, 1473, 1397, 1325, 1300, 1284, 1235, 1125, 1053, 1036, 1014, 885, 840, 799, 781 , 723, 688 and 640 cm '1 .
- the apparatus IFS 88 (Bruker) can be used for the recording of each of the FT-IR spectra.
- the modification A or A' differs from modifications B and C predominantly in the shape and in the relative intensity of many bands. Particularly characteristic are the band at 1080 cm "1 [cf. Figure 2], which is not present in the Raman spectra of the modifications B and C. In the range 3400-300 cm '1 , inter alia the following bands are obtained for the modification A: 3093, 2972, 1628, 1614, 1558, 1465, 1446, 1393, 1279, 1245, 1147, 1080, 1061 , 1036, 1014, 840, 724, 691 , 667, 550, 499, 437 and 368 cm "1 .
- the apparatus RFS 100 (Bruker) can be used for the recording of each of the FT Raman spectra.
- the novel modification A has an X-ray powder pattern with characteristic lines with interplanar spacings (d values) of 10.5 A, 5.14 A, 4.84 A, 4.55 A, 4.34 A, 4.07 A, 3.51 A, 3.48 A, 3.25 A, 3.19 A, 3.15 A, 3.07 A, 2.81 A [cf. Table 1].
- the patterns recorded on X-ray film were measured using an LS-18 line scanner from Johannsson, Taby (Sweden) and evaluated using the Scanpi software (P.E.Werner, University of Sweden).
- Characteristic for the novel modification A is the thermogram in differential scanning calorimetry. It has an endothermic peak in the range from 230 °C to 260 °C. The peak temperature is 239-245 °C, and the endothermic signal is 209 J/g +/- 10 J/g. The measurement was carried out on a Perkin Elmer DSC 7 in a closed pan with a heating rate of 20 K/minute. The typical sample quantity is about 4 mg. As a typical distinguishing feature compared with the modifications B and C, the thermogram of the modification A has no further thermal signal.
- Crystals fo the modification A' have the same crystal structure as modification A. They differ from the modification A in the X-ray powder pattern in that they have slightly smaller line spacings between specific pairs of lines. These are the pairs of lines with the following interplanar spacings: 3.68 A and 3.64 A, 3.51 A and 3.48 A, 3.19 A and 3.15 A.
- the modification B differs from the modification A or A' and C predominantly in the shape and in the relative intensity of many bands. Particularly characteristic is a band at 1678 cm '1 [cf . Figure 1 ], which is not to be observed in the corresponding spectra of the modifications A and C.
- the following bands are obtained for the modification B: 3404, 3199, 3125, 1678, 1635, 1560, 1475, 1393, 1357, 1322, 1286, 1237, 1051 , 1036, 1028, 889, 837, 800, 719, 667 and 645 cm "1 .
- the apparatus IFS 85 (Bruker) can be used for recording of each of the FT-IR spectra.
- the modification B differs from the modifications A or A' and C predominantly in the shape and in the relative intensity of many bands. Particularly characteristic are the bands at 3166 cm “1 and 1086 cm '1 [cf. Figure 2], which are not present in the Raman spectra of the modifications A and C.
- the following bands are obtained for the modification B: 3166, 3089, 2970, 1678, 1628, 1614, 1559, 1464, 1441 , 1391 , 1275, 1244, 1147, 1086, 1062, 1036, 1014, 839, 773, 724, 690, 668, 595, 549, 500, 493, 430 and 365 cm '1 .
- the apparatus RFS 100 (Bruker) can be used for recording of each of the FT Raman spectra.
- the modification B has an X-ray powder pattern with characteristic lines with interplanar spacings (d values) of 11.0 A, 8.3 A, 5.18 A, 4.88 A, 4.80 A, 4.42 A, 4.33 A, 4.19 A, 4.12 A, 3.81 A, 3.50 A, 3.41 A, 3.36 A, 3.32 A, 3.28 A, 3.24 A, 3.05 A, 2.83 A [cf. Table 1].
- the modification B has, in addition to an endothermic signal in the range from 230 °C to 260 °C (peak temperature 239-245 °C), a weak thermal signal at 205 °C (180° - 220°C) as a typical distinguishing feature compared with the modifications A or A' and C.
- the modification C differs from the modifications A or A' and B predominantly in the shape and in the relative intensity of many bands. Particularly characteristic is a band at 3137 cm “1 [cf. Figure 1], which is not to be observed in the corresponding spectra of the modifications A and B.
- the following bands are obtained for the modification C: 3396, 3287, 3137, 1657, 1631 , 1602, 1559, 1475, 1392, 1323, 1287, 1237, 1122, 1104, 1047, 1035, 1012, 876, 839, 797, 773, 729 and 653 cm '1 .
- the apparatus IFS 85 (Bruker) can be used for recording of each of the FT-IR spectra.
- the modification C differs from the modifications A or A' and B predominantly in the shape and in the relative intensity of many bands. Particularly characteristic are the bands at 3137 cm “1 and 1602 cm '1 [cf. Figure 2], which are not present in the Raman spectra of the modifications A and B.
- the following bands are obtained for the modification C: 3137, 3080, 3012, 2971 , 1673, 1629, 1602, 1561 , 1436, 1271 , 1248, 1105, 1065, 1035, 1013, 839, 800, 767, 726, 690, 672, 593, 549, 500, 492, 435 and 370 cm '1 .
- the apparatus RFS 100 (Bruker) can be used for recording of each of the FT Raman spectra.
- the modification C has an X-ray powder pattern with characteristic lines with interplanar spacings (d values) of 9.0 A, 4.73 A, 4.65 A, 3.75 A, 3.54 A, 3.42 A, 3.25 A [cf. Table 1].
- the modification C has, in addition to an endothermic signal in the range of 230 °C to 260 °C (peak temperature 239-245 °C), a very broad, weak, exothermic signal in the region of 180 °C compared with the modifications A or A' and B.
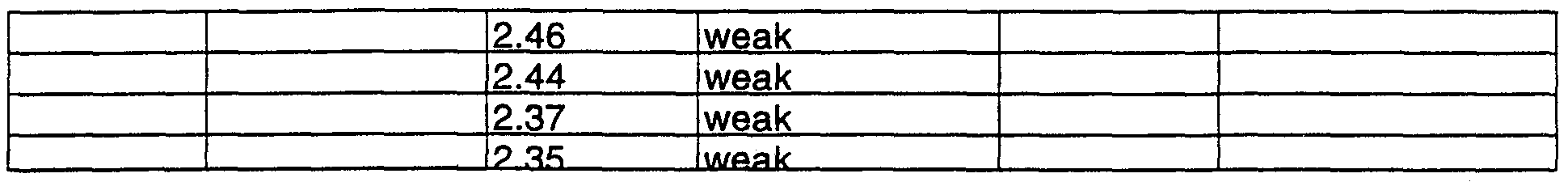
- Table 1 Characterization of the modifications A. B and C (X-rav powder patterns):
- Modifications A, A', B and C have valuable pharmacological properties; in particular, they can be used for the treatment of epilepsy.
- the modification A or A' has significant advantages compared with the modification B and compared with the modification C.
- comprehensive thermodynamic investigations such as thermomicroscopy, X-ray powder diffractometry, DSC, solubility tests and other experiments, have shown that the modification A or A' surprisingly has substantially better thermodynamic stability than the modifications B and C.
- Modification C which can be obtained only under specific conditions, is the least stable of the three modifications.
- the crystals of the modification C are converted into modification B at as low as room temperature within a few weeks.
- the modification C is converted either into the modification A or A' or into the modification B, depending on experimental conditions. It is particularly important for drug that its pharmaceutical formulation ensures high and reproducible stability over a long period.
- a constant stability also permits reproducible bioavailability of an active ingredient. If an active ingredient is subjected to a conversion process, this may readily also cause the bioavailability to fluctuate, which is undesirable. Accordingly, pharmaceutical active ingredients or polymorphic forms thereof which are of primary interest for pharmaceutical developments are those which exhibit high stability and do not have the above-mentioned disadvantages.
- the crystal modification A or A' fulfils these preconditions.
- the modification A or A' has, for example, a slower dissolution rate in water or in gastric fluid (so-called “slow-release effect”). This effect can be utilized primarily for long- term therapy where a slow or delayed release is desired.
- the invention relates to the modification A of 1 -(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4- carboxamide, characterized by the following absorptions in the infrared spectrum (KBr pellet - transmission method): bands at 3092 cm '1 and 3412 cm "1 .
- the invention relates to the modification A of 1 -(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4- carboxamide, characterized by the characteristic lines with interplanar spacings (d values) as shown in Table 1.
- the invention relates to the modification A of 1 -(2,6-dif luorobenzyl)-1 H-1 ,2,3-triazole-4- carboxamide, characterized by an endothermic peak in the range from 230 °C to 260 °C, the peak temperature being 239-245 °C and the endothermic signal being 209 J/g +/- 10 J/g.
- the invention relates to the crystal modification A' which, compared with modification A, has defects in the crystal lattice.
- the invention relates to the modification A' which, compared with modification A, has smaller line spacings between the pairs of lines with interplanar spacings 3.68 A and 3.64 A, 3.51 A and 3.48 A, and 3.19 A and 3.15 A.
- the invention relates to the essentially pure form of the modification A or A' of 1 -(2,6- difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide.
- the term "essentially pure form” means purity of >95%, in particular >98%, primarily >99%, based on the modification A or A'.
- the invention relates to pharmaceutical preparations comprising the modification A or A' of 1-(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide.
- the invention relates in particular to corresponding pharmaceutical preparations for the treatment of epilepsy and subindications thereof.
- the invention relates to the use of the modification A or A' of 1 -(2,6-difluorobenzyl)- 1 H-1 ,2,3-triazole-4-carboxamide for the preparation of pharmaceutical preparations, in particular for the treatment of epilepsy and subindications thereof.
- novel modification A or A' of 1 -(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide can be used, for example, in the form of pharmaceutical preparations which comprise a therapeutically effective amount of the active ingredient, if desired together with inorganic or organic, solid or liquid, pharmaceutically usable carriers, which are suitable for enteral, for example oral, or parenteral administration.
- novel modification A or A' of 1- (2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide can be used in the form of preparations which can be administered parenterally or of infusion solutions.
- the pharmaceutical preparations may be sterilized and/or may comprise excipients, for example preservatives, stabilizers, wetting agents and/or emulsifiers, solubilizers, salts for regulating the osmotic pressure and/or buffers.
- excipients for example preservatives, stabilizers, wetting agents and/or emulsifiers, solubilizers, salts for regulating the osmotic pressure and/or buffers.
- the present pharmaceutical preparations comprise from about 0.1% to 100%, in particular from about 1% to about 50%, of lyophilisates to about 100% of the active ingredient.
- the invention also relates to the use of modification A or A' of 1-(2,6-difluorobenzyl)-1 H- 1 ,2,3-triazole-4-carboxamide as a drug, preferably in the form of pharmaceutical preparations.
- the dosage may depend on various factors, such as method of administration, species, age and/or individual condition.
- the doses to be administered daily are between about 0.25 and about 10 mg/kg in the case of oral administration, and preferably between about 20 mg and about 500 mg for warm-blooded species having a body weight of about 70 kg.
- modification A or A' is carried out, for example, as described in the embodiments below.
- a suspension of methyl 1-(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxylate (about 62 parts by weight), methanol (475.2 parts by weight) and anhydrous ammonia (29.4 parts by weight) is stirred for about 24 hours at 50-55°C in a closed vessel.
- the suspension is cooled to about 20°C and stirred for about a further 2 hours.
- the product is isolated by filtration, washed with methanol (240 parts by weight) and dried at 40-60°C in vacuo. Yield:
- the starting compounds can be prepared, for example, as follows: A mixture of 1 -(2,6-dif luorobenzyl)-1 H-1 ,2,3-triazole-4-carboxylic acid (167.1 parts by weight), methanol (552 parts by weight) and 96% sulfuric acid (35.7 parts by weight) is stirred for about 5 hours at 60-66°C. The suspension is cooled to about 20°C and stirred for about a further 2 hours. The product is isolated by filtration and washed with methanol (198 parts by weight). A yield of about 160 parts by weight is obtained by drying at 40-60°C in vacuo.
- the starting material can be prepared, for example, as follows: 4-Cyano-1 -(2,6-dif luorobenzyl)-1 H-1 ,2,3-triazole
- 1 -(2,6-dif luorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide (pure active ingredient; 4.0 g) is dissolved in 96% ethanol (500 ml, without denaturing agent) at about 80°C while stirring.
- the solution is filtered into a suction bottle (1 litre) at about 20°C (glass suction filter, pore size 10-20 ⁇ m), a suspension forming. After stirring has been continued for 5 minutes at about 20°C and for 15 minutes at about 0°C, the product is isolated by filtration (about 0° to about 20°C).
- the solvent-moist product (9.6 g) is investigated without subsequent drying. Modification A'.
- Film-coated tablets each containing, for example, 100, 200 or 400 mg of modification A or A' of 1 -(2,6-difluorobenzyl)-1 H-1 ,2,3-triazole-4-carboxamide with the following composition per dosage unit:
- Titanium dioxide 0.83 1.66 3.32
- the active ingredient is granulated with demineralised water. Milled lactose, maize starch, Avicel PH 102, cellulose-HP-M-603 and sodium laurylsulfate are added to the above mixture and granulated with demineralised water.
- the moist material is dried and milled. After the addition of the remaining ingredients, the homogeneous mixture is compressed to give tablet cores having the stated active ingredient content.
- the tablet cores are coated with the film coat which is formed from the appropriate ingredients, the latter being dissolved or being suspended in water or in small amounts of ethanol with 5% of isopropanol.
- Figure 1 shows the FT-IR spectra of the KBr pellets of modifications A, B and C.
- Figure 2 shows the FT-Raman spectra of the powder of modification A, B and C.
- the modification A is denoted by the symbol *
- the modification B by the symbol **
- the modification C by the symbol * * *.
Abstract
Description


Claims

Priority Applications (24)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR9804947A BR9804947A (en) | 1997-06-10 | 1998-06-08 | Crystal modification of a pharmaceutical agent |
| SK1093-98A SK283685B6 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole- 4-carboxamide, pharmaceutical compositions containing it and use thereof |
| DE69811500T DE69811500T2 (en) | 1997-06-10 | 1998-06-08 | CRYSTAL MODIFICATION OF 1- (2,6-DIFLUORBENZYL) -1H-1,2,3-TRIAZOL-4-CARBONIC ACID AMIDE AND ITS USE AS AN ANTI-PILEPTIC |
| NZ331371A NZ331371A (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxamide and use thereof |
| AT98934929T ATE232852T1 (en) | 1997-06-10 | 1998-06-08 | CRYSTAL MODIFICATION OF 1-(2,6-DIFLUORBENZYL)-1H-1,2,3-TRIAZOLE-4-CARBON AUREAMIDE AND ITS USE AS AN ANTI-EPILEPTIC |
| US09/125,329 US6740669B1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| PL330764A PL191943B1 (en) | 1997-06-10 | 1998-06-08 | Crystalline for of a pharmaceutic agent |
| IL12573398A IL125733A (en) | 1997-06-10 | 1998-06-08 | Crystal modification of the compound 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and pharmaceutical compositions for treating epilepsy comprising the same |
| EP98934929A EP0994863B1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| HU0000798A HU226107B1 (en) | 1997-06-10 | 1998-06-08 | New crystal modifications of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and pharmaceutical compositions containing them |
| SI9830405T SI0994863T1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| AU84371/98A AU725528B2 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1H-1,2,3- triazole-4-carboxamide and its use as antiepileptic |
| JP53628898A JP3672574B2 (en) | 1997-06-10 | 1998-06-08 | Drug crystal deformation |
| CA002256013A CA2256013C (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| DE122007000051C DE122007000051I2 (en) | 1997-06-10 | 1998-06-08 | CRYSTAL MODIFICATION OF 1- (2,6-DIFLUORBENZYL) -1H-1,2,3-TRIAZOL-4-CARBOXYL ACID AND ITS USE AS ANTIEPILEPTICUM |
| DK98934929T DK0994863T3 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1- (2,6-difluorobenzyl) -1H-1,2,3-triazole-4-carboxamide and its use as an antiepileptic agent |
| NO19983667A NO329315B1 (en) | 1997-06-10 | 1998-08-11 | Crystal modification of compound, pharmaceutical compositions containing it and use of the crystal modification. |
| HK00106804A HK1028241A1 (en) | 1997-06-10 | 2000-10-25 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| US11/329,945 US7750028B2 (en) | 1997-06-10 | 2006-01-11 | Crystal modifications of 1-(2,6-difluorobenzyl)-1H-1, 2,3-triazole-4-carboxamide |
| NL300284C NL300284I2 (en) | 1997-06-10 | 2007-07-03 | Crystal modification of |
| LU91345C LU91345I2 (en) | 1997-06-10 | 2007-07-04 | Rufinamide and its pharmaceutically acceptable salts (inovelon) |
| FR07C0037C FR07C0037I2 (en) | 1997-06-10 | 2007-07-06 | |
| CY200700014C CY2007014I2 (en) | 1997-06-10 | 2007-07-13 | CONVERSION IN CRYSTALLINE FORM OF 1-(2,6-DIFLUOROBENZYL)-1H-1,2,3-TRIAZOLE-4-CARBOXAMIDE AND THEIR USE AS AN ANTI-SEPILLANT |
| US12/767,003 US8076362B2 (en) | 1997-06-10 | 2010-04-26 | Crystal modification A of 1-(2,6-difluorobenzyI)-1 H-1,2,3-triazole-4-carboxamide and dosage forms and formulations thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CH1404/97 | 1997-06-10 | ||
| CH140497 | 1997-06-10 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/125,329 A-371-Of-International US6740669B1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| US10/294,408 Continuation US20030125568A1 (en) | 1997-06-10 | 2002-11-14 | Crystal modifications of 1-(2,6-difluoro benzyl)-1H-1,2,3-triazole-4-carboxamide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998056772A1 true WO1998056772A1 (en) | 1998-12-17 |
Family
ID=4209650
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/003428 WO1998056773A1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
| PCT/EP1998/003427 WO1998056772A1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3--triazole-4-carboxamide and its use as antiepileptic |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/003428 WO1998056773A1 (en) | 1997-06-10 | 1998-06-08 | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
Country Status (37)
| Country | Link |
|---|---|
| US (6) | US6740669B1 (en) |
| EP (2) | EP0994864B1 (en) |
| JP (2) | JP3672574B2 (en) |
| KR (2) | KR100425656B1 (en) |
| CN (3) | CN1298708C (en) |
| AR (4) | AR012945A1 (en) |
| AT (2) | ATE232852T1 (en) |
| AU (2) | AU725528B2 (en) |
| BR (2) | BR9804946A (en) |
| CA (3) | CA2256013C (en) |
| CO (2) | CO4940452A1 (en) |
| CY (1) | CY2007014I2 (en) |
| CZ (2) | CZ292260B6 (en) |
| DE (3) | DE69811500T2 (en) |
| DK (2) | DK0994864T3 (en) |
| ES (2) | ES2192779T3 (en) |
| FR (1) | FR07C0037I2 (en) |
| HK (3) | HK1028242A1 (en) |
| HU (2) | HU225153B1 (en) |
| ID (2) | ID27660A (en) |
| IL (2) | IL125733A (en) |
| LU (1) | LU91345I2 (en) |
| MY (2) | MY125854A (en) |
| NL (1) | NL300284I2 (en) |
| NO (2) | NO329315B1 (en) |
| NZ (2) | NZ331370A (en) |
| PE (2) | PE80999A1 (en) |
| PL (2) | PL192114B1 (en) |
| PT (1) | PT994864E (en) |
| RU (2) | RU2198167C2 (en) |
| SI (2) | SI0994864T1 (en) |
| SK (2) | SK283685B6 (en) |
| TR (2) | TR199801630T1 (en) |
| TW (2) | TW526195B (en) |
| UY (1) | UY25844A1 (en) |
| WO (2) | WO1998056773A1 (en) |
| ZA (2) | ZA984967B (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010043849A1 (en) * | 2008-10-13 | 2010-04-22 | Cipla Limited | Process for the preparation of rufinamide |
| ITMI20091412A1 (en) * | 2009-08-04 | 2011-02-05 | Dipharma Francis Srl | CRYSTALLINE FORMS OF RUFINAMIDE |
| WO2011135105A1 (en) * | 2010-04-30 | 2011-11-03 | Laboratorios Lesvi, S.L. | Improved process for preparing rufinamide intermediate |
| EP2465853A1 (en) * | 2010-12-14 | 2012-06-20 | Laboratorios Lesvi, S.L. | Polymorph of rufinamide and process for obtaining it |
| ITMI20110718A1 (en) * | 2011-04-29 | 2012-10-30 | Dipharma Francis Srl | PROCEDURE FOR PURIFICATION OF RUFINAMIDE |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW526195B (en) | 1997-06-10 | 2003-04-01 | Novartis Ag | Crystal modifications of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxamide and their use |
| JP2005529933A (en) * | 2002-05-31 | 2005-10-06 | シェーリング コーポレイション | Xanthine phosphodiesterase V inhibitor polymorph |
| GB0507298D0 (en) | 2005-04-11 | 2005-05-18 | Novartis Ag | Organic compounds |
| US7875284B2 (en) * | 2006-03-10 | 2011-01-25 | Cook Incorporated | Methods of manufacturing and modifying taxane coatings for implantable medical devices |
| US20090069390A1 (en) * | 2007-09-12 | 2009-03-12 | Protia, Llc | Deuterium-enriched rufinamide |
| CN101768124B (en) * | 2008-12-30 | 2012-01-04 | 北京本草天源药物研究院 | Medicine crystal, preparation method and purpose thereof |
| EP2473051B1 (en) * | 2009-09-04 | 2016-02-10 | Tactical Therapeutics, Inc. | Novel compositions and processes for preparing 5-amino or substituted amino 1,2,3-triazoles and triazole orotate formulations |
| WO2014013511A2 (en) * | 2012-07-20 | 2014-01-23 | Hetero Research Foundation | Rufinamide solid dispersion |
| WO2021099481A1 (en) | 2019-11-20 | 2021-05-27 | Medichem, S.A. | Solid composition containing rufinamide |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0199262A2 (en) * | 1985-04-18 | 1986-10-29 | Ciba-Geigy Ag | Fluorinated benzyl triazole compounds |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3992378A (en) | 1973-12-26 | 1976-11-16 | Eli Lilly And Company | Fluoralkyl quinoxadinediones |
| US4156734A (en) | 1976-02-13 | 1979-05-29 | Merck & Co., Inc. | Antihypertensive compositions containing an aryl-substituted alanine azo and an arylhydrazino-propionic acid |
| GB1511195A (en) | 1976-10-18 | 1978-05-17 | Ici America Inc | Triazole derivatives |
| US4536518A (en) | 1979-11-01 | 1985-08-20 | Pfizer Inc. | Antidepressant derivatives of cis-4-phenyl-1,2,3,4-tetrahydro-1-naphthalenamine |
| US4346097A (en) * | 1980-09-30 | 1982-08-24 | Warner-Lambert Company | Method for treating convulsions with pyrazole-4-carboxamide derivatives |
| US4789680A (en) * | 1982-12-23 | 1988-12-06 | Ciba-Geigy Corporation | Aralkyltriazole compounds |
| FI834666A (en) | 1982-12-23 | 1984-06-24 | Ciba Geigy Ag | FOERFARANDE FOER FRAMSTAELLNING AV NYA ARALKYLTRIAZOLFOERENINGAR. |
| US4511572A (en) * | 1983-03-18 | 1985-04-16 | The University Of Kentucky Research Foundation | Triazoline anticonvulsant drugs |
| CA2002864C (en) | 1988-11-29 | 1999-11-16 | Eddy J. E. Freyne | (1h-azol-1-ylmethyl) substituted quinoline, quinazoline or quinoxaline derivatives |
| JPH02214504A (en) | 1989-02-15 | 1990-08-27 | Nissan Chem Ind Ltd | Method for depositing crystal of compound having plural crystal forms |
| JPH04506967A (en) | 1989-07-27 | 1992-12-03 | ジー.ディー.サール アンド カンパニー | Renal selective products for the treatment of hypertension |
| IL96507A0 (en) | 1989-12-08 | 1991-08-16 | Merck & Co Inc | Nitrogen-containing spirocycles and pharmaceutical compositions containing them |
| AU656154B2 (en) | 1990-11-06 | 1995-01-27 | Astellas Pharma Inc. | Fused pyrazine derivative |
| JP2753911B2 (en) | 1991-12-06 | 1998-05-20 | キッセイ薬品工業株式会社 | Crystal polymorph of N-tert-butyl-1-methyl-3,3-diphenylpropylamine hydrochloride and method for producing the same |
| DE4217952A1 (en) | 1992-05-30 | 1993-12-02 | Basf Ag | Quinoxaline-2,3 (1H, 4H) diones |
| US5248699A (en) | 1992-08-13 | 1993-09-28 | Pfizer Inc. | Sertraline polymorph |
| US5631373A (en) | 1993-11-05 | 1997-05-20 | State Of Oregon, Acting By And Through The Oregon State Board Of Higher Education, Acting For And On Behalf Of The Oregon Health Sciences University And The University Of Oregon, Eugene Oregon | Alkyl, azido, alkoxy, and fluoro-substituted and fused quinoxalinediones |
| GB9418443D0 (en) | 1994-09-13 | 1994-11-02 | Pfizer Ltd | Therapeutic agents |
| GB9419318D0 (en) | 1994-09-24 | 1994-11-09 | Pfizer Ltd | Therapeutic agents |
| US6096779A (en) | 1995-02-22 | 2000-08-01 | Hoechst Pharmaceuticals & Chemicals K.K. | Amorphous piretanide, piretanide polymorphs, process for their preparation and their use |
| NZ333505A (en) * | 1996-07-11 | 2000-06-23 | Novartis Ag | Preparing 1-substituted 4-cyano-1,2,3-triazoles using 4-cyano-1-(2,6-difuorobenzyl)-1H-1,2,3-triazole |
| TW526195B (en) | 1997-06-10 | 2003-04-01 | Novartis Ag | Crystal modifications of 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxamide and their use |
-
1998
- 1998-06-04 TW TW087108858A patent/TW526195B/en not_active IP Right Cessation
- 1998-06-04 TW TW087108859A patent/TW403740B/en not_active IP Right Cessation
- 1998-06-08 NZ NZ331370A patent/NZ331370A/en not_active IP Right Cessation
- 1998-06-08 CA CA002256013A patent/CA2256013C/en not_active Expired - Lifetime
- 1998-06-08 WO PCT/EP1998/003428 patent/WO1998056773A1/en active IP Right Grant
- 1998-06-08 TR TR1998/01630T patent/TR199801630T1/en unknown
- 1998-06-08 HU HU0002113A patent/HU225153B1/en unknown
- 1998-06-08 AU AU84371/98A patent/AU725528B2/en not_active Expired
- 1998-06-08 ID IDW980065D patent/ID27660A/en unknown
- 1998-06-08 DK DK98934930T patent/DK0994864T3/en active
- 1998-06-08 ID IDW980066D patent/ID21014A/en unknown
- 1998-06-08 RU RU99104300/04A patent/RU2198167C2/en active
- 1998-06-08 EP EP98934930A patent/EP0994864B1/en not_active Expired - Lifetime
- 1998-06-08 CZ CZ19982534A patent/CZ292260B6/en not_active IP Right Cessation
- 1998-06-08 DE DE69811500T patent/DE69811500T2/en not_active Expired - Lifetime
- 1998-06-08 WO PCT/EP1998/003427 patent/WO1998056772A1/en active IP Right Grant
- 1998-06-08 EP EP98934929A patent/EP0994863B1/en not_active Expired - Lifetime
- 1998-06-08 KR KR10-1998-0708150A patent/KR100425656B1/en not_active IP Right Cessation
- 1998-06-08 CA CA002256015A patent/CA2256015C/en not_active Expired - Lifetime
- 1998-06-08 CN CNB2004100473675A patent/CN1298708C/en not_active Expired - Lifetime
- 1998-06-08 CN CN988000113A patent/CN1132820C/en not_active Expired - Lifetime
- 1998-06-08 KR KR10-1998-0708149A patent/KR100409168B1/en not_active IP Right Cessation
- 1998-06-08 CN CNB988056755A patent/CN1159300C/en not_active Expired - Lifetime
- 1998-06-08 SI SI9830450T patent/SI0994864T1/en unknown
- 1998-06-08 IL IL12573398A patent/IL125733A/en active Protection Beyond IP Right Term
- 1998-06-08 BR BR9804946A patent/BR9804946A/en not_active Application Discontinuation
- 1998-06-08 JP JP53628898A patent/JP3672574B2/en not_active Expired - Lifetime
- 1998-06-08 AT AT98934929T patent/ATE232852T1/en active
- 1998-06-08 BR BR9804947A patent/BR9804947A/en not_active Application Discontinuation
- 1998-06-08 PE PE1998000479A patent/PE80999A1/en not_active IP Right Cessation
- 1998-06-08 PL PL330798A patent/PL192114B1/en unknown
- 1998-06-08 SK SK1093-98A patent/SK283685B6/en not_active IP Right Cessation
- 1998-06-08 PE PE1998000478A patent/PE79799A1/en not_active IP Right Cessation
- 1998-06-08 TR TR1998/01631T patent/TR199801631T1/en unknown
- 1998-06-08 CZ CZ19982533A patent/CZ292481B6/en not_active IP Right Cessation
- 1998-06-08 DE DE69813560T patent/DE69813560T2/en not_active Expired - Lifetime
- 1998-06-08 HU HU0000798A patent/HU226107B1/en unknown
- 1998-06-08 ES ES98934929T patent/ES2192779T3/en not_active Expired - Lifetime
- 1998-06-08 US US09/125,329 patent/US6740669B1/en not_active Expired - Lifetime
- 1998-06-08 NZ NZ331371A patent/NZ331371A/en not_active IP Right Cessation
- 1998-06-08 PL PL330764A patent/PL191943B1/en unknown
- 1998-06-08 IL IL12573298A patent/IL125732A/en not_active IP Right Cessation
- 1998-06-08 JP JP53628998A patent/JP3672575B2/en not_active Expired - Lifetime
- 1998-06-08 ES ES98934930T patent/ES2197485T3/en not_active Expired - Lifetime
- 1998-06-08 AT AT98934930T patent/ATE237599T1/en active
- 1998-06-08 RU RU99104299/04A patent/RU2194041C2/en active
- 1998-06-08 SI SI9830405T patent/SI0994863T1/en unknown
- 1998-06-08 DE DE122007000051C patent/DE122007000051I2/en active Active
- 1998-06-08 AU AU84372/98A patent/AU725517B2/en not_active Expired
- 1998-06-08 SK SK1094-98A patent/SK283734B6/en not_active IP Right Cessation
- 1998-06-08 CA CA2614926A patent/CA2614926C/en not_active Expired - Lifetime
- 1998-06-08 DK DK98934929T patent/DK0994863T3/en active
- 1998-06-08 PT PT98934930T patent/PT994864E/en unknown
- 1998-06-09 CO CO98032879A patent/CO4940452A1/en unknown
- 1998-06-09 MY MYPI98002572A patent/MY125854A/en unknown
- 1998-06-09 ZA ZA984967A patent/ZA984967B/en unknown
- 1998-06-09 CO CO98032885A patent/CO4940448A1/en unknown
- 1998-06-09 ZA ZA984966A patent/ZA984966B/en unknown
- 1998-06-09 AR ARP980102707A patent/AR012945A1/en not_active Application Discontinuation
- 1998-06-09 MY MYPI98002570A patent/MY120156A/en unknown
- 1998-06-09 AR ARP980102708A patent/AR012946A1/en not_active Application Discontinuation
- 1998-08-11 NO NO19983667A patent/NO329315B1/en not_active IP Right Cessation
- 1998-08-11 NO NO19983666A patent/NO329314B1/en not_active IP Right Cessation
-
1999
- 1999-12-10 UY UY25844A patent/UY25844A1/en not_active Application Discontinuation
-
2000
- 2000-10-25 HK HK00106807A patent/HK1028242A1/en not_active IP Right Cessation
- 2000-10-25 HK HK00106804A patent/HK1028241A1/en not_active IP Right Cessation
- 2000-11-17 HK HK00107340A patent/HK1028031A1/en not_active IP Right Cessation
-
2001
- 2001-05-31 US US09/871,366 patent/US6455556B2/en not_active Expired - Lifetime
-
2002
- 2002-11-14 US US10/294,408 patent/US20030125568A1/en not_active Abandoned
-
2004
- 2004-02-26 US US10/787,528 patent/US20040167186A1/en not_active Abandoned
-
2006
- 2006-01-11 US US11/329,945 patent/US7750028B2/en not_active Expired - Fee Related
-
2007
- 2007-05-17 AR ARP070102131A patent/AR061004A2/en unknown
- 2007-05-17 AR ARP070102132A patent/AR061005A2/en not_active Application Discontinuation
- 2007-07-03 NL NL300284C patent/NL300284I2/en unknown
- 2007-07-04 LU LU91345C patent/LU91345I2/en unknown
- 2007-07-06 FR FR07C0037C patent/FR07C0037I2/fr active Active
- 2007-07-13 CY CY200700014C patent/CY2007014I2/en unknown
-
2010
- 2010-04-26 US US12/767,003 patent/US8076362B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0199262A2 (en) * | 1985-04-18 | 1986-10-29 | Ciba-Geigy Ag | Fluorinated benzyl triazole compounds |
Non-Patent Citations (2)
| Title |
|---|
| MÜNZEL K: "Der Einfluss der Formgebung auf die Wirkung eines Arzneimittels", FORTSCHRITTE DER ARZNEIMITTELFORSCHUNG - PROGRESS IN DRUG RESEARCH - PROGRÈS DES RECHERCHES PHARMACEUTIQUES, vol. 10, 1966, Basel, CH, pages 227 - 30, XP002078506 * |
| MÜNZEL K: "Galenische Formgebung und Arzneimittelwirkung / Neue Erkenntnisse und Feststellungen", PROGRESS IN DRUG RESEARCH - FORTSCHRITTE DER ARZNEIMITTELFORSCHUNG - PROGRÈS DES RECHERCHES PHARMACEUTIQUES, vol. 14, 1970, Basel, CH, pages 309 - 21, XP002078507 * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8183269B2 (en) | 2008-10-13 | 2012-05-22 | Cipla Limited | Process for the preparation of rufinamide |
| EP2334653A1 (en) | 2008-10-13 | 2011-06-22 | Cilpa Limited | Process for the preparation of rufinamide |
| JP2012505191A (en) * | 2008-10-13 | 2012-03-01 | シプラ・リミテッド | Method for producing rufinamide |
| WO2010043849A1 (en) * | 2008-10-13 | 2010-04-22 | Cipla Limited | Process for the preparation of rufinamide |
| ITMI20091412A1 (en) * | 2009-08-04 | 2011-02-05 | Dipharma Francis Srl | CRYSTALLINE FORMS OF RUFINAMIDE |
| EP2292609A1 (en) | 2009-08-04 | 2011-03-09 | Dipharma Francis S.r.l. | Crystalline forms of rufinamide |
| WO2011135105A1 (en) * | 2010-04-30 | 2011-11-03 | Laboratorios Lesvi, S.L. | Improved process for preparing rufinamide intermediate |
| EP2727914A1 (en) * | 2010-04-30 | 2014-05-07 | Laboratorios Lesvi, S.L. | Polymorph of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-carboxamide |
| US8884026B2 (en) | 2010-04-30 | 2014-11-11 | Laboratorios Lesvi, S.L. | Process for preparing rufinamide intermediate |
| EP2465853A1 (en) * | 2010-12-14 | 2012-06-20 | Laboratorios Lesvi, S.L. | Polymorph of rufinamide and process for obtaining it |
| ITMI20110718A1 (en) * | 2011-04-29 | 2012-10-30 | Dipharma Francis Srl | PROCEDURE FOR PURIFICATION OF RUFINAMIDE |
| EP2518057A1 (en) | 2011-04-29 | 2012-10-31 | Dipharma Francis S.r.l. | Process for the purification of rufinamide |
| US8461190B2 (en) | 2011-04-29 | 2013-06-11 | Dipharma Francis S.R.L. | Process for the purification of rufinamide |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7750028B2 (en) | Crystal modifications of 1-(2,6-difluorobenzyl)-1H-1, 2,3-triazole-4-carboxamide | |
| MXPA98006951A (en) | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3--triazole-4-carboxamide and its use as antiepileptic | |
| MXPA98006950A (en) | Crystal modification of 1-(2,6-difluorobenzyl)-1h-1,2,3-triazole-4-carboxamide and its use as antiepileptic |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 125733 Country of ref document: IL Ref document number: 98800011.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998934929 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1998-2533 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 109398 Country of ref document: SK Ref document number: 84371/98 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2256013 Country of ref document: CA Ref document number: 2256013 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 331371 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1199800713 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998/01630 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1998/006951 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 1998 536288 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09125329 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019980708149 Country of ref document: KR |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWP | Wipo information: published in national office |
Ref document number: PV1998-2533 Country of ref document: CZ |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 1019980708149 Country of ref document: KR |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998934929 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 84371/98 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998934929 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1019980708149 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1998-2533 Country of ref document: CZ |