WO1998018498A2 - Improvements in or relating to diagnostic/therapeutic agents - Google Patents
Improvements in or relating to diagnostic/therapeutic agents Download PDFInfo
- Publication number
- WO1998018498A2 WO1998018498A2 PCT/GB1997/002958 GB9702958W WO9818498A2 WO 1998018498 A2 WO1998018498 A2 WO 1998018498A2 GB 9702958 W GB9702958 W GB 9702958W WO 9818498 A2 WO9818498 A2 WO 9818498A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- agent
- vector
- microbubbles
- gas
- reporter
- Prior art date
Links
- 239000003814 drug Substances 0.000 title claims description 48
- 239000000032 diagnostic agent Substances 0.000 title description 6
- 229940039227 diagnostic agent Drugs 0.000 title description 6
- 229940124597 therapeutic agent Drugs 0.000 title description 6
- 230000006872 improvement Effects 0.000 title description 2
- 239000013598 vector Substances 0.000 claims abstract description 163
- 230000000975 bioactive effect Effects 0.000 claims abstract description 42
- 239000000463 material Substances 0.000 claims abstract description 40
- 239000000725 suspension Substances 0.000 claims abstract description 21
- 239000002961 echo contrast media Substances 0.000 claims abstract description 19
- 239000007788 liquid Substances 0.000 claims abstract description 8
- 239000013543 active substance Substances 0.000 claims abstract description 7
- 239000008365 aqueous carrier Substances 0.000 claims abstract description 4
- 239000007789 gas Substances 0.000 claims description 113
- 239000003795 chemical substances by application Substances 0.000 claims description 81
- 239000000203 mixture Substances 0.000 claims description 79
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 76
- 230000027455 binding Effects 0.000 claims description 71
- 210000004027 cell Anatomy 0.000 claims description 57
- 238000002360 preparation method Methods 0.000 claims description 57
- 230000015572 biosynthetic process Effects 0.000 claims description 45
- 238000000034 method Methods 0.000 claims description 44
- 229940079593 drug Drugs 0.000 claims description 39
- 108090000623 proteins and genes Proteins 0.000 claims description 38
- 230000001225 therapeutic effect Effects 0.000 claims description 37
- 102000004169 proteins and genes Human genes 0.000 claims description 36
- KAVGMUDTWQVPDF-UHFFFAOYSA-N perflubutane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)F KAVGMUDTWQVPDF-UHFFFAOYSA-N 0.000 claims description 35
- 230000008685 targeting Effects 0.000 claims description 35
- 210000002889 endothelial cell Anatomy 0.000 claims description 34
- 238000010168 coupling process Methods 0.000 claims description 32
- 229950003332 perflubutane Drugs 0.000 claims description 32
- 230000008878 coupling Effects 0.000 claims description 30
- 238000005859 coupling reaction Methods 0.000 claims description 30
- 239000000523 sample Substances 0.000 claims description 30
- 239000002872 contrast media Substances 0.000 claims description 26
- 150000001875 compounds Chemical class 0.000 claims description 25
- 108091034117 Oligonucleotide Proteins 0.000 claims description 21
- 238000002604 ultrasonography Methods 0.000 claims description 21
- 238000003384 imaging method Methods 0.000 claims description 17
- 239000012528 membrane Substances 0.000 claims description 16
- 150000003904 phospholipids Chemical class 0.000 claims description 16
- 239000004094 surface-active agent Substances 0.000 claims description 15
- 239000000126 substance Substances 0.000 claims description 14
- 229960002685 biotin Drugs 0.000 claims description 13
- 239000011616 biotin Substances 0.000 claims description 13
- 239000003112 inhibitor Substances 0.000 claims description 13
- 230000008569 process Effects 0.000 claims description 13
- 230000003993 interaction Effects 0.000 claims description 12
- 102000004190 Enzymes Human genes 0.000 claims description 11
- 108090000790 Enzymes Proteins 0.000 claims description 11
- 230000008512 biological response Effects 0.000 claims description 11
- 238000001727 in vivo Methods 0.000 claims description 11
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 10
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 10
- 150000002148 esters Chemical class 0.000 claims description 9
- 238000009472 formulation Methods 0.000 claims description 9
- 108010067225 Cell Adhesion Molecules Proteins 0.000 claims description 8
- 230000004071 biological effect Effects 0.000 claims description 7
- 150000002576 ketones Chemical class 0.000 claims description 7
- 239000003446 ligand Substances 0.000 claims description 7
- 102000016289 Cell Adhesion Molecules Human genes 0.000 claims description 6
- 108091005804 Peptidases Proteins 0.000 claims description 6
- SFZCNBIFKDRMGX-UHFFFAOYSA-N sulfur hexafluoride Chemical compound FS(F)(F)(F)(F)F SFZCNBIFKDRMGX-UHFFFAOYSA-N 0.000 claims description 6
- 239000004365 Protease Substances 0.000 claims description 5
- 108010000499 Thromboplastin Proteins 0.000 claims description 5
- 102000002262 Thromboplastin Human genes 0.000 claims description 5
- 239000003102 growth factor Substances 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 claims description 5
- 239000000813 peptide hormone Substances 0.000 claims description 5
- 239000004215 Carbon black (E152) Substances 0.000 claims description 4
- 102000004127 Cytokines Human genes 0.000 claims description 4
- 108090000695 Cytokines Proteins 0.000 claims description 4
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 claims description 4
- 239000011230 binding agent Substances 0.000 claims description 4
- 230000021164 cell adhesion Effects 0.000 claims description 4
- 239000003638 chemical reducing agent Substances 0.000 claims description 4
- 229950003499 fibrin Drugs 0.000 claims description 4
- 229940088597 hormone Drugs 0.000 claims description 4
- 239000005556 hormone Substances 0.000 claims description 4
- 229930195733 hydrocarbon Natural products 0.000 claims description 4
- 150000002430 hydrocarbons Chemical class 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 230000002209 hydrophobic effect Effects 0.000 claims description 4
- 229960004065 perflutren Drugs 0.000 claims description 4
- 150000008106 phosphatidylserines Chemical class 0.000 claims description 4
- 239000000758 substrate Substances 0.000 claims description 4
- 230000004568 DNA-binding Effects 0.000 claims description 3
- 108010073385 Fibrin Proteins 0.000 claims description 3
- 102000009123 Fibrin Human genes 0.000 claims description 3
- 230000003444 anaesthetic effect Effects 0.000 claims description 3
- 230000001772 anti-angiogenic effect Effects 0.000 claims description 3
- 150000002321 glycerophosphoglycerophosphoglycerols Chemical class 0.000 claims description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 3
- 239000011261 inert gas Substances 0.000 claims description 3
- NJCBUSHGCBERSK-UHFFFAOYSA-N perfluoropentane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F NJCBUSHGCBERSK-UHFFFAOYSA-N 0.000 claims description 3
- 150000008103 phosphatidic acids Chemical class 0.000 claims description 3
- 229940067626 phosphatidylinositols Drugs 0.000 claims description 3
- 150000003905 phosphatidylinositols Chemical class 0.000 claims description 3
- 229960000909 sulfur hexafluoride Drugs 0.000 claims description 3
- 229940088594 vitamin Drugs 0.000 claims description 3
- 229930003231 vitamin Natural products 0.000 claims description 3
- 235000013343 vitamin Nutrition 0.000 claims description 3
- 239000011782 vitamin Substances 0.000 claims description 3
- ABQLAMJAQZFPJI-UHFFFAOYSA-N 3-heptyloxolan-2-one Chemical compound CCCCCCCC1CCOC1=O ABQLAMJAQZFPJI-UHFFFAOYSA-N 0.000 claims description 2
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 2
- 230000000884 anti-protozoa Effects 0.000 claims description 2
- 230000003356 anti-rheumatic effect Effects 0.000 claims description 2
- 230000002365 anti-tubercular Effects 0.000 claims description 2
- 239000000043 antiallergic agent Substances 0.000 claims description 2
- 229940121375 antifungal agent Drugs 0.000 claims description 2
- 239000003429 antifungal agent Substances 0.000 claims description 2
- 229940034982 antineoplastic agent Drugs 0.000 claims description 2
- 239000002246 antineoplastic agent Substances 0.000 claims description 2
- 239000003435 antirheumatic agent Substances 0.000 claims description 2
- 239000010836 blood and blood product Substances 0.000 claims description 2
- 229940125691 blood product Drugs 0.000 claims description 2
- 229940097217 cardiac glycoside Drugs 0.000 claims description 2
- 239000002368 cardiac glycoside Substances 0.000 claims description 2
- 102000008395 cell adhesion mediator activity proteins Human genes 0.000 claims description 2
- 238000005345 coagulation Methods 0.000 claims description 2
- 230000015271 coagulation Effects 0.000 claims description 2
- 150000002170 ethers Chemical class 0.000 claims description 2
- 230000005291 magnetic effect Effects 0.000 claims description 2
- 230000002503 metabolic effect Effects 0.000 claims description 2
- 239000000842 neuromuscular blocking agent Substances 0.000 claims description 2
- 229940127240 opiate Drugs 0.000 claims description 2
- 239000002861 polymer material Substances 0.000 claims description 2
- 239000000932 sedative agent Substances 0.000 claims description 2
- 229930002534 steroid glycoside Natural products 0.000 claims description 2
- QTJXVIKNLHZIKL-UHFFFAOYSA-N sulfur difluoride Chemical compound FSF QTJXVIKNLHZIKL-UHFFFAOYSA-N 0.000 claims description 2
- TXEYQDLBPFQVAA-UHFFFAOYSA-N tetrafluoromethane Chemical compound FC(F)(F)F TXEYQDLBPFQVAA-UHFFFAOYSA-N 0.000 claims description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 1
- 239000003570 air Substances 0.000 claims 1
- 230000000840 anti-viral effect Effects 0.000 claims 1
- 230000003115 biocidal effect Effects 0.000 claims 1
- UBAZGMLMVVQSCD-UHFFFAOYSA-N carbon dioxide;molecular oxygen Chemical compound O=O.O=C=O UBAZGMLMVVQSCD-UHFFFAOYSA-N 0.000 claims 1
- 230000020764 fibrinolysis Effects 0.000 claims 1
- 238000011835 investigation Methods 0.000 claims 1
- 239000003607 modifier Substances 0.000 claims 1
- 230000003533 narcotic effect Effects 0.000 claims 1
- LPMXVESGRSUGHW-HBYQJFLCSA-N ouabain Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H](C=5COC(=O)C=5)[C@@]4(C)C[C@@H](O)[C@@H]3[C@@]2(CO)[C@H](O)C1 LPMXVESGRSUGHW-HBYQJFLCSA-N 0.000 claims 1
- 229960004692 perflenapent Drugs 0.000 claims 1
- 230000001624 sedative effect Effects 0.000 claims 1
- 229940124549 vasodilator Drugs 0.000 claims 1
- 239000003071 vasodilator agent Substances 0.000 claims 1
- 150000003722 vitamin derivatives Chemical class 0.000 claims 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 87
- 108010028921 Lipopeptides Proteins 0.000 description 84
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- 239000000243 solution Substances 0.000 description 73
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 69
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 66
- 208000033921 delayed sleep phase type circadian rhythm sleep disease Diseases 0.000 description 56
- 239000000047 product Substances 0.000 description 52
- -1 perfluoro-n-butane Chemical compound 0.000 description 39
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- 238000003786 synthesis reaction Methods 0.000 description 34
- 238000006243 chemical reaction Methods 0.000 description 29
- 235000018102 proteins Nutrition 0.000 description 29
- 125000003396 thiol group Chemical group [H]S* 0.000 description 29
- 230000033115 angiogenesis Effects 0.000 description 27
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 26
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 25
- 229920001223 polyethylene glycol Polymers 0.000 description 25
- 238000005406 washing Methods 0.000 description 25
- 150000001413 amino acids Chemical class 0.000 description 24
- 229920000642 polymer Polymers 0.000 description 24
- 235000001014 amino acid Nutrition 0.000 description 23
- 229940024606 amino acid Drugs 0.000 description 23
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 22
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 22
- 239000004005 microsphere Substances 0.000 description 22
- 229960004063 propylene glycol Drugs 0.000 description 22
- 235000013772 propylene glycol Nutrition 0.000 description 22
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 21
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 21
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 21
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 20
- 125000006850 spacer group Chemical group 0.000 description 20
- 238000004949 mass spectrometry Methods 0.000 description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 239000003153 chemical reaction reagent Substances 0.000 description 18
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 18
- 102000005962 receptors Human genes 0.000 description 17
- 108020003175 receptors Proteins 0.000 description 17
- 206010028980 Neoplasm Diseases 0.000 description 16
- 235000012000 cholesterol Nutrition 0.000 description 16
- 238000012512 characterization method Methods 0.000 description 15
- 239000013058 crude material Substances 0.000 description 15
- 239000012043 crude product Substances 0.000 description 15
- 239000011541 reaction mixture Substances 0.000 description 15
- 239000011347 resin Substances 0.000 description 15
- 229920005989 resin Polymers 0.000 description 15
- 210000001519 tissue Anatomy 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 14
- 238000005119 centrifugation Methods 0.000 description 14
- 230000021615 conjugation Effects 0.000 description 14
- 239000006185 dispersion Substances 0.000 description 14
- 238000004128 high performance liquid chromatography Methods 0.000 description 14
- 230000014759 maintenance of location Effects 0.000 description 14
- 210000004379 membrane Anatomy 0.000 description 14
- 125000006239 protecting group Chemical group 0.000 description 14
- 238000000746 purification Methods 0.000 description 14
- 102100037362 Fibronectin Human genes 0.000 description 13
- 108010067306 Fibronectins Proteins 0.000 description 13
- 238000004108 freeze drying Methods 0.000 description 13
- 150000002632 lipids Chemical class 0.000 description 13
- 108090001008 Avidin Proteins 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 12
- 238000001514 detection method Methods 0.000 description 12
- 125000005647 linker group Chemical group 0.000 description 12
- 239000002953 phosphate buffered saline Substances 0.000 description 12
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 12
- 229920000656 polylysine Polymers 0.000 description 12
- 239000011734 sodium Substances 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- 235000021314 Palmitic acid Nutrition 0.000 description 11
- 108010039918 Polylysine Proteins 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 125000003277 amino group Chemical group 0.000 description 11
- 235000020958 biotin Nutrition 0.000 description 11
- 201000011510 cancer Diseases 0.000 description 11
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 11
- 238000002953 preparative HPLC Methods 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 10
- 239000008367 deionised water Substances 0.000 description 10
- 229940088598 enzyme Drugs 0.000 description 10
- 125000000524 functional group Chemical group 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 239000011859 microparticle Substances 0.000 description 10
- 239000002245 particle Substances 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 238000012285 ultrasound imaging Methods 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 9
- 108010090804 Streptavidin Proteins 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 229920000669 heparin Polymers 0.000 description 9
- 229960002897 heparin Drugs 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- 238000000386 microscopy Methods 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- 239000012581 transferrin Substances 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 8
- 238000012377 drug delivery Methods 0.000 description 8
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 8
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 8
- 238000010792 warming Methods 0.000 description 8
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 7
- 239000000306 component Substances 0.000 description 7
- 229920001577 copolymer Polymers 0.000 description 7
- 239000002502 liposome Substances 0.000 description 7
- 239000011159 matrix material Substances 0.000 description 7
- 229960000485 methotrexate Drugs 0.000 description 7
- MILWSGRFEGYSGM-UHFFFAOYSA-N propane-1,2-diol;propane-1,2,3-triol Chemical compound CC(O)CO.OCC(O)CO MILWSGRFEGYSGM-UHFFFAOYSA-N 0.000 description 7
- 239000012460 protein solution Substances 0.000 description 7
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 6
- 102000009027 Albumins Human genes 0.000 description 6
- 108010088751 Albumins Proteins 0.000 description 6
- 241000183024 Populus tremula Species 0.000 description 6
- 101001000212 Rattus norvegicus Decorin Proteins 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 6
- 102000004338 Transferrin Human genes 0.000 description 6
- 108090000901 Transferrin Proteins 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 238000009825 accumulation Methods 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 210000004204 blood vessel Anatomy 0.000 description 6
- FVJZSBGHRPJMMA-UHFFFAOYSA-N distearoyl phosphatidylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCCCCCCCCCCCC FVJZSBGHRPJMMA-UHFFFAOYSA-N 0.000 description 6
- 239000012634 fragment Substances 0.000 description 6
- 230000004927 fusion Effects 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 229910021653 sulphate ion Inorganic materials 0.000 description 6
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- FZTIWOBQQYPTCJ-UHFFFAOYSA-N 4-[4-(4-carboxyphenyl)phenyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(C=2C=CC(=CC=2)C(O)=O)C=C1 FZTIWOBQQYPTCJ-UHFFFAOYSA-N 0.000 description 5
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 102000035195 Peptidases Human genes 0.000 description 5
- 108010067787 Proteoglycans Proteins 0.000 description 5
- 102000016611 Proteoglycans Human genes 0.000 description 5
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 5
- 238000010976 amide bond formation reaction Methods 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 108091007433 antigens Proteins 0.000 description 5
- 102000036639 antigens Human genes 0.000 description 5
- 229960002274 atenolol Drugs 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 5
- 229960000830 captopril Drugs 0.000 description 5
- 150000001718 carbodiimides Chemical class 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 210000002216 heart Anatomy 0.000 description 5
- 238000000099 in vitro assay Methods 0.000 description 5
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 5
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 230000002285 radioactive effect Effects 0.000 description 5
- 238000002390 rotary evaporation Methods 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- ADFXKUOMJKEIND-UHFFFAOYSA-N 1,3-dicyclohexylurea Chemical compound C1CCCCC1NC(=O)NC1CCCCC1 ADFXKUOMJKEIND-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 229920002307 Dextran Polymers 0.000 description 4
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 4
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 4
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 4
- SBKRTALNRRAOJP-BWSIXKJUSA-N N-[(2S)-4-amino-1-[[(2S,3R)-1-[[(2S)-4-amino-1-oxo-1-[[(3S,6S,9S,12S,15R,18R,21S)-6,9,18-tris(2-aminoethyl)-15-benzyl-3-[(1R)-1-hydroxyethyl]-12-(2-methylpropyl)-2,5,8,11,14,17,20-heptaoxo-1,4,7,10,13,16,19-heptazacyclotricos-21-yl]amino]butan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1-oxobutan-2-yl]-6-methylheptanamide (6S)-N-[(2S)-4-amino-1-[[(2S,3R)-1-[[(2S)-4-amino-1-oxo-1-[[(3S,6S,9S,12S,15R,18R,21S)-6,9,18-tris(2-aminoethyl)-15-benzyl-3-[(1R)-1-hydroxyethyl]-12-(2-methylpropyl)-2,5,8,11,14,17,20-heptaoxo-1,4,7,10,13,16,19-heptazacyclotricos-21-yl]amino]butan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1-oxobutan-2-yl]-6-methyloctanamide sulfuric acid Polymers OS(O)(=O)=O.CC(C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@@H](NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](Cc2ccccc2)NC(=O)[C@@H](CCN)NC1=O)[C@@H](C)O.CC[C@H](C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@@H](NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](Cc2ccccc2)NC(=O)[C@@H](CCN)NC1=O)[C@@H](C)O SBKRTALNRRAOJP-BWSIXKJUSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 150000001350 alkyl halides Chemical class 0.000 description 4
- 239000002870 angiogenesis inducing agent Substances 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 229920001400 block copolymer Polymers 0.000 description 4
- 239000011575 calcium Substances 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 4
- 229960004630 chlorambucil Drugs 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 238000000684 flow cytometry Methods 0.000 description 4
- 229960000304 folic acid Drugs 0.000 description 4
- 235000019152 folic acid Nutrition 0.000 description 4
- 239000011724 folic acid Substances 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 150000004676 glycans Chemical class 0.000 description 4
- 239000000787 lecithin Substances 0.000 description 4
- 235000010445 lecithin Nutrition 0.000 description 4
- 210000005240 left ventricle Anatomy 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000002107 myocardial effect Effects 0.000 description 4
- 210000004165 myocardium Anatomy 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229920001184 polypeptide Polymers 0.000 description 4
- 229920001282 polysaccharide Polymers 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 230000005747 tumor angiogenesis Effects 0.000 description 4
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- KIUMMUBSPKGMOY-UHFFFAOYSA-N 3,3'-Dithiobis(6-nitrobenzoic acid) Chemical compound C1=C([N+]([O-])=O)C(C(=O)O)=CC(SSC=2C=C(C(=CC=2)[N+]([O-])=O)C(O)=O)=C1 KIUMMUBSPKGMOY-UHFFFAOYSA-N 0.000 description 3
- JDDWRLPTKIOUOF-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl n-[[4-[2-[bis(4-methylphenyl)methylamino]-2-oxoethoxy]phenyl]-(2,4-dimethoxyphenyl)methyl]carbamate Chemical compound COC1=CC(OC)=CC=C1C(C=1C=CC(OCC(=O)NC(C=2C=CC(C)=CC=2)C=2C=CC(C)=CC=2)=CC=1)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 JDDWRLPTKIOUOF-UHFFFAOYSA-N 0.000 description 3
- 102000014914 Carrier Proteins Human genes 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 3
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 3
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 3
- 229920002971 Heparan sulfate Polymers 0.000 description 3
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 3
- 108091006905 Human Serum Albumin Proteins 0.000 description 3
- 102000008100 Human Serum Albumin Human genes 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 3
- 239000012979 RPMI medium Substances 0.000 description 3
- 239000012317 TBTU Substances 0.000 description 3
- 208000007536 Thrombosis Diseases 0.000 description 3
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 3
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 3
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 3
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 3
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 3
- 239000003875 Wang resin Substances 0.000 description 3
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 230000001656 angiogenetic effect Effects 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 150000001720 carbohydrates Chemical class 0.000 description 3
- 235000014633 carbohydrates Nutrition 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical class C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 238000001212 derivatisation Methods 0.000 description 3
- VJVMIIAKISKTDK-UHFFFAOYSA-N dichloro-[4-[dichloro(methyl)silyl]butyl]-methylsilane Chemical compound C[Si](Cl)(Cl)CCCC[Si](C)(Cl)Cl VJVMIIAKISKTDK-UHFFFAOYSA-N 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 238000005188 flotation Methods 0.000 description 3
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 3
- 238000000799 fluorescence microscopy Methods 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 102000035118 modified proteins Human genes 0.000 description 3
- 108091005573 modified proteins Proteins 0.000 description 3
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 125000000962 organic group Chemical group 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 229920002635 polyurethane Polymers 0.000 description 3
- 239000004814 polyurethane Substances 0.000 description 3
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 3
- 239000001120 potassium sulphate Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 3
- 230000009145 protein modification Effects 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 229960003766 thrombin (human) Drugs 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- UVGHPGOONBRLCX-NJSLBKSFSA-N (2,5-dioxopyrrolidin-1-yl) 6-[5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]hexanoate Chemical compound C([C@H]1[C@H]2NC(=O)N[C@H]2CS1)CCCC(=O)NCCCCCC(=O)ON1C(=O)CCC1=O UVGHPGOONBRLCX-NJSLBKSFSA-N 0.000 description 2
- KLBPUVPNPAJWHZ-UMSFTDKQSA-N (2r)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-tritylsulfanylpropanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)SC(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 KLBPUVPNPAJWHZ-UMSFTDKQSA-N 0.000 description 2
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical class C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 2
- CROLNSXOPSNSFL-UHFFFAOYSA-N 1-(16-hydroxyhexadecanoyloxy)ethyl 16-hydroxyhexadecanoate Chemical compound OCCCCCCCCCCCCCCCC(=O)OC(C)OC(=O)CCCCCCCCCCCCCCCO CROLNSXOPSNSFL-UHFFFAOYSA-N 0.000 description 2
- MEZJQXVOMGUAMP-UHFFFAOYSA-N 1-(2-methylnaphthalen-1-yl)pyrrole-2,5-dione Chemical class CC1=CC=C2C=CC=CC2=C1N1C(=O)C=CC1=O MEZJQXVOMGUAMP-UHFFFAOYSA-N 0.000 description 2
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 2
- GEULZAPFKVWTHK-UHFFFAOYSA-N 16-hexadecanoyloxyhexadecanoic acid;methoxymethane Chemical compound COC.CCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC(O)=O GEULZAPFKVWTHK-UHFFFAOYSA-N 0.000 description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 2
- XXBVXLIDXFUGNT-UHFFFAOYSA-N 2-[4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]acetic acid;hydrochloride Chemical compound Cl.CC(C)NCC(O)COC1=CC=C(CC(O)=O)C=C1 XXBVXLIDXFUGNT-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- LJGFXAKKZLFEDR-LBPRGKRZSA-N 9h-fluoren-9-ylmethyl n-[(2s)-1-oxopropan-2-yl]carbamate Chemical compound C1=CC=C2C(COC(=O)N[C@@H](C)C=O)C3=CC=CC=C3C2=C1 LJGFXAKKZLFEDR-LBPRGKRZSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 102000000412 Annexin Human genes 0.000 description 2
- 108050008874 Annexin Proteins 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 108010024976 Asparaginase Proteins 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- WDJUZGPOPHTGOT-OAXVISGBSA-N Digitoxin Natural products O([C@H]1[C@@H](C)O[C@@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@@](C)([C@H](C6=CC(=O)OC6)CC5)CC4)CC3)CC2)C[C@H]1O)[C@H]1O[C@@H](C)[C@H](O[C@H]2O[C@@H](C)[C@@H](O)[C@@H](O)C2)[C@@H](O)C1 WDJUZGPOPHTGOT-OAXVISGBSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 2
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 244000068988 Glycine max Species 0.000 description 2
- 235000010469 Glycine max Nutrition 0.000 description 2
- 229920002527 Glycogen Polymers 0.000 description 2
- 229920002683 Glycosaminoglycan Polymers 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 108010031792 IGF Type 2 Receptor Proteins 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- 229930182816 L-glutamine Natural products 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- 102100032131 Lymphocyte antigen 6E Human genes 0.000 description 2
- 101710157879 Lymphocyte antigen 6E Proteins 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- GTUQCKXJFBMPTI-UHFFFAOYSA-N O.P.P Chemical compound O.P.P GTUQCKXJFBMPTI-UHFFFAOYSA-N 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical group OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- 102000003790 Thrombin receptors Human genes 0.000 description 2
- 108090000166 Thrombin receptors Proteins 0.000 description 2
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 2
- 108010033576 Transferrin Receptors Proteins 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- PWAXUOGZOSVGBO-UHFFFAOYSA-N adipoyl chloride Chemical compound ClC(=O)CCCCC(Cl)=O PWAXUOGZOSVGBO-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- VIROVYVQCGLCII-UHFFFAOYSA-N amobarbital Chemical compound CC(C)CCC1(CC)C(=O)NC(=O)NC1=O VIROVYVQCGLCII-UHFFFAOYSA-N 0.000 description 2
- 238000004873 anchoring Methods 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- 210000002469 basement membrane Anatomy 0.000 description 2
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 239000012620 biological material Substances 0.000 description 2
- 230000006287 biotinylation Effects 0.000 description 2
- 238000007413 biotinylation Methods 0.000 description 2
- 239000012503 blood component Substances 0.000 description 2
- 230000036760 body temperature Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 2
- 239000000084 colloidal system Substances 0.000 description 2
- 238000012790 confirmation Methods 0.000 description 2
- 229940039231 contrast media Drugs 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- RMRCNWBMXRMIRW-BYFNXCQMSA-M cyanocobalamin Chemical compound N#C[Co+]N([C@]1([H])[C@H](CC(N)=O)[C@]\2(CCC(=O)NC[C@H](C)OP(O)(=O)OC3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)C)C/2=C(C)\C([C@H](C/2(C)C)CCC(N)=O)=N\C\2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O RMRCNWBMXRMIRW-BYFNXCQMSA-M 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical class P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 229960000394 droperidol Drugs 0.000 description 2
- RMEDXOLNCUSCGS-UHFFFAOYSA-N droperidol Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC=C(N2C(NC3=CC=CC=C32)=O)CC1 RMEDXOLNCUSCGS-UHFFFAOYSA-N 0.000 description 2
- 229940000406 drug candidate Drugs 0.000 description 2
- 230000009881 electrostatic interaction Effects 0.000 description 2
- 239000006274 endogenous ligand Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000007667 floating Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 238000005227 gel permeation chromatography Methods 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 229940096919 glycogen Drugs 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- WXEHBUMAEPOYKP-UHFFFAOYSA-N methylsulfanylethane Chemical compound CCSC WXEHBUMAEPOYKP-UHFFFAOYSA-N 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 230000001338 necrotic effect Effects 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 229960001412 pentobarbital Drugs 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 230000010363 phase shift Effects 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 102000036213 phospholipid binding proteins Human genes 0.000 description 2
- 108091011000 phospholipid binding proteins Proteins 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229940127126 plasminogen activator Drugs 0.000 description 2
- 238000004262 preparative liquid chromatography Methods 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 235000021251 pulses Nutrition 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 238000010187 selection method Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000012064 sodium phosphate buffer Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000010183 spectrum analysis Methods 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 150000005846 sugar alcohols Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 229920001059 synthetic polymer Polymers 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 229940126585 therapeutic drug Drugs 0.000 description 2
- 150000007970 thio esters Chemical class 0.000 description 2
- 150000003568 thioethers Chemical group 0.000 description 2
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 2
- ATGUDZODTABURZ-UHFFFAOYSA-N thiolan-2-ylideneazanium;chloride Chemical group Cl.N=C1CCCS1 ATGUDZODTABURZ-UHFFFAOYSA-N 0.000 description 2
- 238000006177 thiolation reaction Methods 0.000 description 2
- 229960000187 tissue plasminogen activator Drugs 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 102000003390 tumor necrosis factor Human genes 0.000 description 2
- 210000003954 umbilical cord Anatomy 0.000 description 2
- 210000005167 vascular cell Anatomy 0.000 description 2
- 210000005166 vasculature Anatomy 0.000 description 2
- 229960003636 vidarabine Drugs 0.000 description 2
- 229910052724 xenon Inorganic materials 0.000 description 2
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 2
- GXFZCDMWGMFGFL-KKXMJGKMSA-N (+)-Tubocurarine chloride hydrochloride Chemical compound [Cl-].[Cl-].C([C@H]1[N+](C)(C)CCC=2C=C(C(=C(OC3=CC=C(C=C3)C[C@H]3C=4C=C(C(=CC=4CC[NH+]3C)OC)O3)C=21)O)OC)C1=CC=C(O)C3=C1 GXFZCDMWGMFGFL-KKXMJGKMSA-N 0.000 description 1
- CRPUJAZIXJMDBK-BDAKNGLRSA-N (-)-camphene Chemical compound C1C[C@H]2C(=C)C(C)(C)[C@@H]1C2 CRPUJAZIXJMDBK-BDAKNGLRSA-N 0.000 description 1
- 229930006714 (-)-camphene Natural products 0.000 description 1
- YMXHPSHLTSZXKH-RVBZMBCESA-N (2,5-dioxopyrrolidin-1-yl) 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoate Chemical compound C([C@H]1[C@H]2NC(=O)N[C@H]2CS1)CCCC(=O)ON1C(=O)CCC1=O YMXHPSHLTSZXKH-RVBZMBCESA-N 0.000 description 1
- JQWAHKMIYCERGA-UHFFFAOYSA-N (2-nonanoyloxy-3-octadeca-9,12-dienoyloxypropoxy)-[2-(trimethylazaniumyl)ethyl]phosphinate Chemical compound CCCCCCCCC(=O)OC(COP([O-])(=O)CC[N+](C)(C)C)COC(=O)CCCCCCCC=CCC=CCCCCC JQWAHKMIYCERGA-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- VKCGSTZAZMNTRV-DHUJRADRSA-N (2s)-2,6-bis(hexadecanoylamino)hexanoic acid Chemical compound CCCCCCCCCCCCCCCC(=O)NCCCC[C@@H](C(O)=O)NC(=O)CCCCCCCCCCCCCCC VKCGSTZAZMNTRV-DHUJRADRSA-N 0.000 description 1
- NTUPOKHATNSWCY-PMPSAXMXSA-N (2s)-2-[[(2s)-1-[(2r)-2-amino-3-phenylpropanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound C([C@@H](N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)C1=CC=CC=C1 NTUPOKHATNSWCY-PMPSAXMXSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- WSJULBMCKQTTIG-OWOJBTEDSA-N (e)-1,1,1,2,3,4,4,4-octafluorobut-2-ene Chemical compound FC(F)(F)C(/F)=C(\F)C(F)(F)F WSJULBMCKQTTIG-OWOJBTEDSA-N 0.000 description 1
- ZEHYJZXQEQOSON-AATRIKPKSA-N (e)-1-chloro-3-ethylpent-1-en-4-yn-3-ol Chemical compound CCC(O)(C#C)\C=C\Cl ZEHYJZXQEQOSON-AATRIKPKSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- COQIQRBKEGPRSG-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoro-2-(trifluoromethyl)propane Chemical compound FC(F)(F)C(F)(C(F)(F)F)C(F)(F)F COQIQRBKEGPRSG-UHFFFAOYSA-N 0.000 description 1
- FNVLGCVAWPSVSK-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7-tetradecafluorocycloheptane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F FNVLGCVAWPSVSK-UHFFFAOYSA-N 0.000 description 1
- RKIMETXDACNTIE-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6-dodecafluorocyclohexane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F RKIMETXDACNTIE-UHFFFAOYSA-N 0.000 description 1
- QIROQPWSJUXOJC-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6-undecafluoro-6-(trifluoromethyl)cyclohexane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F QIROQPWSJUXOJC-UHFFFAOYSA-N 0.000 description 1
- PWMJXZJISGDARB-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5-decafluorocyclopentane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F PWMJXZJISGDARB-UHFFFAOYSA-N 0.000 description 1
- BCNXQFASJTYKDJ-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5-nonafluoro-5-(trifluoromethyl)cyclopentane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F BCNXQFASJTYKDJ-UHFFFAOYSA-N 0.000 description 1
- CIWUYWQUYMZILR-UHFFFAOYSA-N 1,1,2,2,3,3,4,4-octafluoro-5,5-bis(trifluoromethyl)cyclopentane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C(F)(F)C1(F)F CIWUYWQUYMZILR-UHFFFAOYSA-N 0.000 description 1
- ZVXOHSHODRJTCP-UHFFFAOYSA-N 1,1,2,2,3,3,4-heptafluoro-4-(trifluoromethyl)cyclobutane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(F)F ZVXOHSHODRJTCP-UHFFFAOYSA-N 0.000 description 1
- TXGPGHBYAPBDAG-UHFFFAOYSA-N 1,1,2,2,3,3-hexafluoro-4,4-bis(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C1(F)F TXGPGHBYAPBDAG-UHFFFAOYSA-N 0.000 description 1
- YUFJLVUCHXMKKM-UHFFFAOYSA-N 1,1,2,2,3-pentafluoro-3,4,4-tris(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(C(F)(F)F)C(F)(F)F YUFJLVUCHXMKKM-UHFFFAOYSA-N 0.000 description 1
- ZVJOQYFQSQJDDX-UHFFFAOYSA-N 1,1,2,3,3,4,4,4-octafluorobut-1-ene Chemical class FC(F)=C(F)C(F)(F)C(F)(F)F ZVJOQYFQSQJDDX-UHFFFAOYSA-N 0.000 description 1
- LGPPATCNSOSOQH-UHFFFAOYSA-N 1,1,2,3,4,4-hexafluorobuta-1,3-diene Chemical compound FC(F)=C(F)C(F)=C(F)F LGPPATCNSOSOQH-UHFFFAOYSA-N 0.000 description 1
- NPNPZTNLOVBDOC-UHFFFAOYSA-N 1,1-difluoroethane Chemical compound CC(F)F NPNPZTNLOVBDOC-UHFFFAOYSA-N 0.000 description 1
- 229940051271 1,1-difluoroethane Drugs 0.000 description 1
- CITHEXJVPOWHKC-UUWRZZSWSA-N 1,2-di-O-myristoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCC CITHEXJVPOWHKC-UUWRZZSWSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 1
- SLKDGVPOSSLUAI-PGUFJCEWSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCCCCCCCCCC SLKDGVPOSSLUAI-PGUFJCEWSA-N 0.000 description 1
- MTZUIIAIAKMWLI-UHFFFAOYSA-N 1,2-diisocyanatobenzene Chemical compound O=C=NC1=CC=CC=C1N=C=O MTZUIIAIAKMWLI-UHFFFAOYSA-N 0.000 description 1
- VPVXHAANQNHFSF-UHFFFAOYSA-N 1,4-dioxan-2-one Chemical class O=C1COCCO1 VPVXHAANQNHFSF-UHFFFAOYSA-N 0.000 description 1
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical group O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- RFCAUADVODFSLZ-UHFFFAOYSA-N 1-Chloro-1,1,2,2,2-pentafluoroethane Chemical compound FC(F)(F)C(F)(F)Cl RFCAUADVODFSLZ-UHFFFAOYSA-N 0.000 description 1
- XQFRJNBWHJMXHO-UHFFFAOYSA-N 1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodopyrimidine-2,4-dione Chemical compound C1C(O)C(CO)OC1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-UHFFFAOYSA-N 0.000 description 1
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 1
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- TXLHNFOLHRXMAU-UHFFFAOYSA-N 2-(4-benzylphenoxy)-n,n-diethylethanamine;hydron;chloride Chemical compound Cl.C1=CC(OCCN(CC)CC)=CC=C1CC1=CC=CC=C1 TXLHNFOLHRXMAU-UHFFFAOYSA-N 0.000 description 1
- DTCYNRIBBFRFNN-UHFFFAOYSA-N 2-(4-oxaldehydoylphenyl)-2-oxoacetaldehyde Chemical compound O=CC(=O)C1=CC=C(C(=O)C=O)C=C1 DTCYNRIBBFRFNN-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- BHANCCMWYDZQOR-UHFFFAOYSA-N 2-(methyldisulfanyl)pyridine Chemical compound CSSC1=CC=CC=N1 BHANCCMWYDZQOR-UHFFFAOYSA-N 0.000 description 1
- KLDZYURQCUYZBL-UHFFFAOYSA-N 2-[3-[(2-hydroxyphenyl)methylideneamino]propyliminomethyl]phenol Chemical compound OC1=CC=CC=C1C=NCCCN=CC1=CC=CC=C1O KLDZYURQCUYZBL-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- LRYZPFWEZHSTHD-HEFFAWAOSA-O 2-[[(e,2s,3r)-2-formamido-3-hydroxyoctadec-4-enoxy]-hydroxyphosphoryl]oxyethyl-trimethylazanium Chemical class CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](NC=O)COP(O)(=O)OCC[N+](C)(C)C LRYZPFWEZHSTHD-HEFFAWAOSA-O 0.000 description 1
- IZQAUUVBKYXMET-UHFFFAOYSA-N 2-bromoethanamine Chemical compound NCCBr IZQAUUVBKYXMET-UHFFFAOYSA-N 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- IJVRPNIWWODHHA-UHFFFAOYSA-N 2-cyanoprop-2-enoic acid Chemical compound OC(=O)C(=C)C#N IJVRPNIWWODHHA-UHFFFAOYSA-N 0.000 description 1
- GTQFGAJYMBBUSO-UHFFFAOYSA-N 2-diazoacetamide Chemical class NC(=O)C=[N+]=[N-] GTQFGAJYMBBUSO-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- GHCZTIFQWKKGSB-UHFFFAOYSA-N 2-hydroxypropane-1,2,3-tricarboxylic acid;phosphoric acid Chemical compound OP(O)(O)=O.OC(=O)CC(O)(C(O)=O)CC(O)=O GHCZTIFQWKKGSB-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- TUATYNXRYJTQTQ-BVRBKCERSA-N 3,6-diamino-n-[[(2s,5s,8z,11s,15s)-15-amino-11-(2-amino-1,4,5,6-tetrahydropyrimidin-6-yl)-8-[(carbamoylamino)methylidene]-2-(hydroxymethyl)-3,6,9,12,16-pentaoxo-1,4,7,10,13-pentazacyclohexadec-5-yl]methyl]hexanamide;3,6-diamino-n-[[(2s,5s,8z,11s,15s)-15-a Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CNC(=O)CC(N)CCCN)NC(=O)[C@H](C)NC(=O)[C@@H](N)CNC(=O)[C@@H]1C1NC(=N)NCC1.N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CNC(=O)CC(N)CCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CNC(=O)[C@@H]1C1NC(=N)NCC1 TUATYNXRYJTQTQ-BVRBKCERSA-N 0.000 description 1
- WCFJUSRQHZPVKY-UHFFFAOYSA-N 3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NCCC(O)=O WCFJUSRQHZPVKY-UHFFFAOYSA-N 0.000 description 1
- KFKRXESVMDBTNQ-UHFFFAOYSA-N 3-[18-(2-carboxylatoethyl)-8,13-bis(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-21,24-diium-2-yl]propanoate Chemical class N1C2=C(C)C(C(C)O)=C1C=C(N1)C(C)=C(C(O)C)C1=CC(C(C)=C1CCC(O)=O)=NC1=CC(C(CCC(O)=O)=C1C)=NC1=C2 KFKRXESVMDBTNQ-UHFFFAOYSA-N 0.000 description 1
- CUNJTOHTJOOFJQ-WZTVWXICSA-N 3-acetamido-2,4,6-triiodobenzoic acid;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CC(=O)NC1=C(I)C=C(I)C(C(O)=O)=C1I CUNJTOHTJOOFJQ-WZTVWXICSA-N 0.000 description 1
- 101150066375 35 gene Proteins 0.000 description 1
- UHBAPGWWRFVTFS-UHFFFAOYSA-N 4,4'-dipyridyl disulfide Chemical compound C=1C=NC=CC=1SSC1=CC=NC=C1 UHBAPGWWRFVTFS-UHFFFAOYSA-N 0.000 description 1
- UPXRTVAIJMUAQR-UHFFFAOYSA-N 4-(9h-fluoren-9-ylmethoxycarbonylamino)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound C1C(C(O)=O)N(C(=O)OC(C)(C)C)CC1NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 UPXRTVAIJMUAQR-UHFFFAOYSA-N 0.000 description 1
- FZPBXNLCWHEGFO-UHFFFAOYSA-N 4-[2-(4-carboxyphenoxy)propan-2-yloxy]benzoic acid Chemical compound C(=O)(O)C1=CC=C(OC(C)(C)OC2=CC=C(C=C2)C(=O)O)C=C1 FZPBXNLCWHEGFO-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- JBVJKCHENYIOLW-UHFFFAOYSA-N 4-chloro-1,1,1,2,2,6-hexafluorohexane Chemical compound FCCC(Cl)CC(F)(F)C(F)(F)F JBVJKCHENYIOLW-UHFFFAOYSA-N 0.000 description 1
- FLDROJIECSYZST-UHFFFAOYSA-N 4-methyl-2-[(4-methyl-3-nitropyridin-2-yl)disulfanyl]-3-nitropyridine Chemical compound CC1=CC=NC(SSC=2C(=C(C)C=CN=2)[N+]([O-])=O)=C1[N+]([O-])=O FLDROJIECSYZST-UHFFFAOYSA-N 0.000 description 1
- PJJGZPJJTHBVMX-UHFFFAOYSA-N 5,7-Dihydroxyisoflavone Chemical compound C=1C(O)=CC(O)=C(C2=O)C=1OC=C2C1=CC=CC=C1 PJJGZPJJTHBVMX-UHFFFAOYSA-N 0.000 description 1
- CIVGYTYIDWRBQU-UFLZEWODSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoic acid;pyrrole-2,5-dione Chemical group O=C1NC(=O)C=C1.N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 CIVGYTYIDWRBQU-UFLZEWODSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 1
- 229920000945 Amylopectin Polymers 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 108090000672 Annexin A5 Proteins 0.000 description 1
- 102000004121 Annexin A5 Human genes 0.000 description 1
- 102000006410 Apoproteins Human genes 0.000 description 1
- 108010083590 Apoproteins Proteins 0.000 description 1
- 244000186140 Asperula odorata Species 0.000 description 1
- YXSLJKQTIDHPOT-UHFFFAOYSA-N Atracurium Dibesylate Chemical compound C1=C(OC)C(OC)=CC=C1CC1[N+](CCC(=O)OCCCCCOC(=O)CC[N+]2(C)C(C3=CC(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=CC=2)(C)CCC2=CC(OC)=C(OC)C=C21 YXSLJKQTIDHPOT-UHFFFAOYSA-N 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 1
- 102100030802 Beta-2-glycoprotein 1 Human genes 0.000 description 1
- 102000004954 Biglycan Human genes 0.000 description 1
- 108090001138 Biglycan Proteins 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 108010058905 CD44v6 antigen Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 108010065839 Capreomycin Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 102100032231 Caveolae-associated protein 2 Human genes 0.000 description 1
- 108050005259 Caveolae-associated protein 2 Proteins 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 description 1
- 239000004340 Chloropentafluoroethane Substances 0.000 description 1
- 235000001258 Cinchona calisaya Nutrition 0.000 description 1
- 108010048623 Collagen Receptors Proteins 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- ITRJWOMZKQRYTA-RFZYENFJSA-N Cortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)CC2=O ITRJWOMZKQRYTA-RFZYENFJSA-N 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine group Chemical group N[C@H](CCCCN)C(=O)O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 229920004934 Dacron® Polymers 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- CKNOLMVLQUPVMU-XOMFLMSUSA-N Digitalin Natural products O(C)[C@H]1[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@@H](O)[C@H](CO)O2)[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@@](C)([C@@H]([C@H](O)C5)C5=CC(=O)OC5)CC4)CC3)CC2)[C@@H]1O CKNOLMVLQUPVMU-XOMFLMSUSA-N 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- IIUZTXTZRGLYTI-UHFFFAOYSA-N Dihydrogriseofulvin Natural products COC1CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 IIUZTXTZRGLYTI-UHFFFAOYSA-N 0.000 description 1
- 241000551547 Dione <red algae> Species 0.000 description 1
- 229940123907 Disease modifying antirheumatic drug Drugs 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010041308 Endothelial Growth Factors Proteins 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- VTUSIVBDOCDNHS-UHFFFAOYSA-N Etidocaine Chemical compound CCCN(CC)C(CC)C(=O)NC1=C(C)C=CC=C1C VTUSIVBDOCDNHS-UHFFFAOYSA-N 0.000 description 1
- 108050001049 Extracellular proteins Proteins 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 1
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 1
- 102000003971 Fibroblast Growth Factor 1 Human genes 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 108090000368 Fibroblast growth factor 8 Proteins 0.000 description 1
- ZIIJJOPLRSCQNX-UHFFFAOYSA-N Flurazepam hydrochloride Chemical compound Cl.Cl.N=1CC(=O)N(CCN(CC)CC)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F ZIIJJOPLRSCQNX-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 235000008526 Galium odoratum Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- JMBQKKAJIKAWKF-UHFFFAOYSA-N Glutethimide Chemical compound C=1C=CC=CC=1C1(CC)CCC(=O)NC1=O JMBQKKAJIKAWKF-UHFFFAOYSA-N 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- UXWOXTQWVMFRSE-UHFFFAOYSA-N Griseoviridin Natural products O=C1OC(C)CC=C(C(NCC=CC=CC(O)CC(O)C2)=O)SCC1NC(=O)C1=COC2=N1 UXWOXTQWVMFRSE-UHFFFAOYSA-N 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229920001499 Heparinoid Polymers 0.000 description 1
- 108010007267 Hirudins Proteins 0.000 description 1
- 102000007625 Hirudins Human genes 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101001024703 Homo sapiens Nck-associated protein 5 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 229920001612 Hydroxyethyl starch Polymers 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102100025947 Insulin-like growth factor II Human genes 0.000 description 1
- 102100025305 Integrin alpha-2 Human genes 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 102100026236 Interleukin-8 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- UXIGWFXRQKWHHA-UHFFFAOYSA-N Iotalamic acid Chemical compound CNC(=O)C1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I UXIGWFXRQKWHHA-UHFFFAOYSA-N 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- 125000003412 L-alanyl group Chemical group [H]N([H])[C@@](C([H])([H])[H])(C(=O)[*])[H] 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- ODLGFPIWRAEFAN-PFEQFJNWSA-N Levomepromazine hydrochloride Chemical compound Cl.C1=CC=C2N(C[C@H](C)CN(C)C)C3=CC(OC)=CC=C3SC2=C1 ODLGFPIWRAEFAN-PFEQFJNWSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000019218 Mannose-6-phosphate receptors Human genes 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- SIDLZWOQUZRBRU-UHFFFAOYSA-N Methyprylon Chemical compound CCC1(CC)C(=O)NCC(C)C1=O SIDLZWOQUZRBRU-UHFFFAOYSA-N 0.000 description 1
- DIGFQJFCDPKEPF-OIUSMDOTSA-L Metocurine iodide Chemical compound [I-].[I-].C1([C@@H]([N+](CCC1=CC=1OC)(C)C)CC2=CC=C(C=C2)O2)=CC=1OC(=C1)C(OC)=CC=C1C[C@H]1[N+](C)(C)CCC3=C1C2=C(OC)C(OC)=C3 DIGFQJFCDPKEPF-OIUSMDOTSA-L 0.000 description 1
- BAQCROVBDNBEEB-UBYUBLNFSA-N Metrizamide Chemical compound CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C(=O)N[C@@H]2[C@H]([C@H](O)[C@@H](CO)OC2O)O)=C1I BAQCROVBDNBEEB-UBYUBLNFSA-N 0.000 description 1
- BYBLEWFAAKGYCD-UHFFFAOYSA-N Miconazole Chemical compound ClC1=CC(Cl)=CC=C1COC(C=1C(=CC(Cl)=CC=1)Cl)CN1C=NC=C1 BYBLEWFAAKGYCD-UHFFFAOYSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- NRFJZTXWLKPZAV-UHFFFAOYSA-N N-(2-oxo-3-thiolanyl)acetamide Chemical compound CC(=O)NC1CCSC1=O NRFJZTXWLKPZAV-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 125000005118 N-alkylcarbamoyl group Chemical group 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 102100036946 Nck-associated protein 5 Human genes 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- DDUHZTYCFQRHIY-UHFFFAOYSA-N Negwer: 6874 Natural products COC1=CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 102000005348 Neuraminidase Human genes 0.000 description 1
- 108010006232 Neuraminidase Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- BXEFQPCKQSTMKA-UHFFFAOYSA-N OC(=O)C=[N+]=[N-] Chemical class OC(=O)C=[N+]=[N-] BXEFQPCKQSTMKA-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 239000008896 Opium Substances 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- HYRKAAMZBDSJFJ-LFDBJOOHSA-N Paramethasone acetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]2(C)C[C@@H]1O HYRKAAMZBDSJFJ-LFDBJOOHSA-N 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- TZRXHJWUDPFEEY-UHFFFAOYSA-N Pentaerythritol Tetranitrate Chemical compound [O-][N+](=O)OCC(CO[N+]([O-])=O)(CO[N+]([O-])=O)CO[N+]([O-])=O TZRXHJWUDPFEEY-UHFFFAOYSA-N 0.000 description 1
- 239000000026 Pentaerythritol tetranitrate Substances 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- IGVPBCZDHMIOJH-UHFFFAOYSA-N Phenyl butyrate Chemical compound CCCC(=O)OC1=CC=CC=C1 IGVPBCZDHMIOJH-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 102000003666 Placenta Growth Factor Human genes 0.000 description 1
- 108010082093 Placenta Growth Factor Proteins 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 102000004211 Platelet factor 4 Human genes 0.000 description 1
- 108090000778 Platelet factor 4 Proteins 0.000 description 1
- 102100039277 Pleiotrophin Human genes 0.000 description 1
- 229920001744 Polyaldehyde Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000362 Polyethylene-block-poly(ethylene glycol) Polymers 0.000 description 1
- 108010040201 Polymyxins Proteins 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 241001415846 Procellariidae Species 0.000 description 1
- 101710149951 Protein Tat Proteins 0.000 description 1
- 108010085249 Purinergic P2 Receptors Proteins 0.000 description 1
- 102000007466 Purinergic P2 Receptors Human genes 0.000 description 1
- 102000044126 RNA-Binding Proteins Human genes 0.000 description 1
- 108700020471 RNA-Binding Proteins Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 108020001027 Ribosomal DNA Proteins 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010023197 Streptokinase Proteins 0.000 description 1
- CPFNIKYEDJFRAT-UHFFFAOYSA-N Strospasid Natural products OC1C(OC)C(O)C(C)OC1OC1CC(CCC2C3(CC(O)C(C3(C)CCC32)C=2COC(=O)C=2)O)C3(C)CC1 CPFNIKYEDJFRAT-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical group O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 229920001963 Synthetic biodegradable polymer Polymers 0.000 description 1
- SEQDDYPDSLOBDC-UHFFFAOYSA-N Temazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 SEQDDYPDSLOBDC-UHFFFAOYSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- IUJDSEJGGMCXSG-UHFFFAOYSA-N Thiopental Chemical compound CCCC(C)C1(CC)C(=O)NC(=S)NC1=O IUJDSEJGGMCXSG-UHFFFAOYSA-N 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 102100031372 Thymidine phosphorylase Human genes 0.000 description 1
- 108700023160 Thymidine phosphorylases Proteins 0.000 description 1
- CRPUJAZIXJMDBK-UHFFFAOYSA-N Toxaphene Natural products C1CC2C(=C)C(C)(C)C1C2 CRPUJAZIXJMDBK-UHFFFAOYSA-N 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108060008539 Transglutaminase Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 208000035896 Twin-reversed arterial perfusion sequence Diseases 0.000 description 1
- CKNOLMVLQUPVMU-UHFFFAOYSA-N UNPD183315 Natural products O1C(C)C(OC2C(C(O)C(O)C(CO)O2)O)C(OC)C(O)C1OC(C1)CCC2(C)C1CCC(C1(CC3O)O)C2CCC1(C)C3C1=CC(=O)OC1 CKNOLMVLQUPVMU-UHFFFAOYSA-N 0.000 description 1
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 1
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 238000007329 Woodward reaction Methods 0.000 description 1
- 108010093894 Xanthine oxidase Proteins 0.000 description 1
- 102100033220 Xanthine oxidase Human genes 0.000 description 1
- VRGWBRLULZUWAJ-XFFXIZSCSA-N [(2s)-2-[(1r,3z,5s,8z,12z,15s)-5,17-dihydroxy-4,8,12,15-tetramethyl-16-oxo-18-bicyclo[13.3.0]octadeca-3,8,12,17-tetraenyl]propyl] acetate Chemical compound C1\C=C(C)/CC\C=C(C)/CC[C@H](O)\C(C)=C/C[C@@H]2C([C@@H](COC(C)=O)C)=C(O)C(=O)[C@]21C VRGWBRLULZUWAJ-XFFXIZSCSA-N 0.000 description 1
- KMLCRELJHYKIIL-UHFFFAOYSA-N [1-(azanidylmethyl)cyclohexyl]methylazanide;platinum(2+);sulfuric acid Chemical compound [Pt+2].OS(O)(=O)=O.[NH-]CC1(C[NH-])CCCCC1 KMLCRELJHYKIIL-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- FFINMCNLQNTKLU-UHFFFAOYSA-N adipiodone Chemical compound OC(=O)C1=C(I)C=C(I)C(NC(=O)CCCCC(=O)NC=2C(=C(C(O)=O)C(I)=CC=2I)I)=C1I FFINMCNLQNTKLU-UHFFFAOYSA-N 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical compound C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- YVPYQUNUQOZFHG-UHFFFAOYSA-N amidotrizoic acid Chemical compound CC(=O)NC1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I YVPYQUNUQOZFHG-UHFFFAOYSA-N 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960001301 amobarbital Drugs 0.000 description 1
- 229960005143 amobarbital sodium Drugs 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 1
- 229960003942 amphotericin b Drugs 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 229940124326 anaesthetic agent Drugs 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 238000011122 anti-angiogenic therapy Methods 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000001188 anti-phage Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 210000002403 aortic endothelial cell Anatomy 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- KXNPVXPOPUZYGB-XYVMCAHJSA-N argatroban Chemical compound OC(=O)[C@H]1C[C@H](C)CCN1C(=O)[C@H](CCCN=C(N)N)NS(=O)(=O)C1=CC=CC2=C1NC[C@H](C)C2 KXNPVXPOPUZYGB-XYVMCAHJSA-N 0.000 description 1
- 229960003856 argatroban Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000000712 assembly Effects 0.000 description 1
- 238000000429 assembly Methods 0.000 description 1
- PUQIRTNPJRFRCZ-UHFFFAOYSA-N atenolol acid Chemical compound CC(C)NCC(O)COC1=CC=C(CC(O)=O)C=C1 PUQIRTNPJRFRCZ-UHFFFAOYSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229960001862 atracurium Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- BOFZOTMTKBQRAB-UHFFFAOYSA-N azanium;2-carboxyphenolate Chemical compound N.OC(=O)C1=CC=CC=C1O BOFZOTMTKBQRAB-UHFFFAOYSA-N 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- PFOLLRNADZZWEX-FFGRCDKISA-N bacampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)[C@H](C(S3)(C)C)C(=O)OC(C)OC(=O)OCC)=CC=CC=C1 PFOLLRNADZZWEX-FFGRCDKISA-N 0.000 description 1
- 229960002699 bacampicillin Drugs 0.000 description 1
- 229950000210 beclometasone dipropionate Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- IYXMNTLBLQNMLM-UHFFFAOYSA-N benzene-1,4-diamine;hydron;dichloride Chemical compound Cl.Cl.NC1=CC=C(N)C=C1 IYXMNTLBLQNMLM-UHFFFAOYSA-N 0.000 description 1
- 108010023562 beta 2-Glycoprotein I Proteins 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 229920000229 biodegradable polyester Polymers 0.000 description 1
- 239000004622 biodegradable polyester Substances 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000008081 blood perfusion Effects 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- MEXUFEQDCXZEON-UHFFFAOYSA-N bromochlorodifluoromethane Chemical compound FC(F)(Cl)Br MEXUFEQDCXZEON-UHFFFAOYSA-N 0.000 description 1
- RJCQBQGAPKAMLL-UHFFFAOYSA-N bromotrifluoromethane Chemical compound FC(F)(F)Br RJCQBQGAPKAMLL-UHFFFAOYSA-N 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960003150 bupivacaine Drugs 0.000 description 1
- 230000009172 bursting Effects 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 229940068366 butabarbital sodium Drugs 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229960002968 capreomycin sulfate Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- FPPNZSSZRUTDAP-UWFZAAFLSA-N carbenicillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C(C(O)=O)C1=CC=CC=C1 FPPNZSSZRUTDAP-UWFZAAFLSA-N 0.000 description 1
- 229960003669 carbenicillin Drugs 0.000 description 1
- 125000000837 carbohydrate group Chemical group 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 238000013155 cardiography Methods 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- QYIYFLOTGYLRGG-GPCCPHFNSA-N cefaclor Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CS[C@@H]32)C(O)=O)=O)N)=CC=CC=C1 QYIYFLOTGYLRGG-GPCCPHFNSA-N 0.000 description 1
- 229960005361 cefaclor Drugs 0.000 description 1
- 229960004841 cefadroxil Drugs 0.000 description 1
- NBFNMSULHIODTC-CYJZLJNKSA-N cefadroxil monohydrate Chemical compound O.C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=C(O)C=C1 NBFNMSULHIODTC-CYJZLJNKSA-N 0.000 description 1
- 229960002588 cefradine Drugs 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 210000002390 cell membrane structure Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229940106164 cephalexin Drugs 0.000 description 1
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 description 1
- RDLPVSKMFDYCOR-UEKVPHQBSA-N cephradine Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CCC=CC1 RDLPVSKMFDYCOR-UEKVPHQBSA-N 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 229940045110 chitosan Drugs 0.000 description 1
- RNFNDJAIBTYOQL-UHFFFAOYSA-N chloral hydrate Chemical compound OC(O)C(Cl)(Cl)Cl RNFNDJAIBTYOQL-UHFFFAOYSA-N 0.000 description 1
- 229960002327 chloral hydrate Drugs 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- NEHMKBQYUWJMIP-NJFSPNSNSA-N chloro(114C)methane Chemical compound [14CH3]Cl NEHMKBQYUWJMIP-NJFSPNSNSA-N 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical class OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 1
- 235000019406 chloropentafluoroethane Nutrition 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- AFYPFACVUDMOHA-UHFFFAOYSA-N chlorotrifluoromethane Chemical compound FC(F)(F)Cl AFYPFACVUDMOHA-UHFFFAOYSA-N 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229960004753 citiolone Drugs 0.000 description 1
- 229960002227 clindamycin Drugs 0.000 description 1
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 1
- 239000012568 clinical material Substances 0.000 description 1
- 238000004581 coalescence Methods 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000002607 contrast-enhanced ultrasound Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229960003290 cortisone acetate Drugs 0.000 description 1
- 239000006059 cover glass Substances 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- NLCKLZIHJQEMCU-UHFFFAOYSA-N cyano prop-2-enoate Chemical class C=CC(=O)OC#N NLCKLZIHJQEMCU-UHFFFAOYSA-N 0.000 description 1
- 229960002104 cyanocobalamin Drugs 0.000 description 1
- 235000000639 cyanocobalamin Nutrition 0.000 description 1
- 239000011666 cyanocobalamin Substances 0.000 description 1
- HGBLNBBNRORJKI-WCABBAIRSA-N cyclacillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C1(N)CCCCC1 HGBLNBBNRORJKI-WCABBAIRSA-N 0.000 description 1
- 229960004244 cyclacillin Drugs 0.000 description 1
- 150000001923 cyclic compounds Chemical group 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 108010010449 dansyl-glutamyl-glycyl-arginyl-factor VIIa Proteins 0.000 description 1
- 229960000860 dapsone Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- BELUJIRQWUXEKA-UHFFFAOYSA-N decanedioic acid;octanedioic acid Chemical compound OC(=O)CCCCCCC(O)=O.OC(=O)CCCCCCCCC(O)=O BELUJIRQWUXEKA-UHFFFAOYSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 201000001098 delayed sleep phase syndrome Diseases 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 125000004420 diamide group Chemical group 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 229960005223 diatrizoic acid Drugs 0.000 description 1
- 150000008049 diazo compounds Chemical group 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 229960001585 dicloxacillin Drugs 0.000 description 1
- YFAGHNZHGGCZAX-JKIFEVAISA-N dicloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(Cl)C=CC=C1Cl YFAGHNZHGGCZAX-JKIFEVAISA-N 0.000 description 1
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 1
- 229960000616 diflunisal Drugs 0.000 description 1
- CKNOLMVLQUPVMU-YMMLYESFSA-N digitalin Chemical compound C1([C@@H]2[C@@]3(C)CC[C@H]4[C@H]([C@]3(C[C@@H]2O)O)CC[C@H]2[C@]4(C)CC[C@@H](C2)O[C@H]2[C@H](O)[C@H]([C@H]([C@@H](C)O2)O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)OC)=CC(=O)OC1 CKNOLMVLQUPVMU-YMMLYESFSA-N 0.000 description 1
- 229950004590 digitalin Drugs 0.000 description 1
- WDJUZGPOPHTGOT-XUDUSOBPSA-N digitoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)CC5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O WDJUZGPOPHTGOT-XUDUSOBPSA-N 0.000 description 1
- 229960000648 digitoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- 229960005156 digoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 230000000916 dilatatory effect Effects 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- UBHZUDXTHNMNLD-UHFFFAOYSA-N dimethylsilane Chemical compound C[SiH2]C UBHZUDXTHNMNLD-UHFFFAOYSA-N 0.000 description 1
- 229960003724 dimyristoylphosphatidylcholine Drugs 0.000 description 1
- 150000002009 diols Chemical group 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 235000013345 egg yolk Nutrition 0.000 description 1
- 210000002969 egg yolk Anatomy 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- 108010048367 enhanced green fluorescent protein Proteins 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- SNFOERUNNSHUGP-ZXZARUISSA-N erythrityl tetranitrate Chemical compound [O-][N+](=O)OC[C@@H](O[N+]([O-])=O)[C@@H](O[N+]([O-])=O)CO[N+]([O-])=O SNFOERUNNSHUGP-ZXZARUISSA-N 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 229960003276 erythromycin Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- 229960000285 ethambutol Drugs 0.000 description 1
- 229960004447 ethchlorvynol Drugs 0.000 description 1
- GXRZIMHKGDIBEW-UHFFFAOYSA-N ethinamate Chemical compound NC(=O)OC1(C#C)CCCCC1 GXRZIMHKGDIBEW-UHFFFAOYSA-N 0.000 description 1
- 229960002209 ethinamate Drugs 0.000 description 1
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 1
- 229960002001 ethionamide Drugs 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N ethyl formate Chemical compound CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960003976 etidocaine Drugs 0.000 description 1
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 1
- 229960001690 etomidate Drugs 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 229960004207 fentanyl citrate Drugs 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 229940126864 fibroblast growth factor Drugs 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- SYWHXTATXSMDSB-GSLJADNHSA-N fludrocortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O SYWHXTATXSMDSB-GSLJADNHSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960003336 fluorocortisol acetate Drugs 0.000 description 1
- 229960003628 flurazepam hydrochloride Drugs 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- VRGWBRLULZUWAJ-UHFFFAOYSA-N fusaproliferin Natural products C1C=C(C)CCC=C(C)CCC(O)C(C)=CCC2C(C(COC(C)=O)C)=C(O)C(=O)C21C VRGWBRLULZUWAJ-UHFFFAOYSA-N 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 229960005271 gallamine triethiodide Drugs 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 229960002972 glutethimide Drugs 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 229960002867 griseofulvin Drugs 0.000 description 1
- DDUHZTYCFQRHIY-RBHXEPJQSA-N griseofulvin Chemical compound COC1=CC(=O)C[C@@H](C)[C@@]11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-RBHXEPJQSA-N 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 239000002554 heparinoid Substances 0.000 description 1
- 229940025770 heparinoids Drugs 0.000 description 1
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 1
- DXVUYOAEDJXBPY-NFFDBFGFSA-N hetacillin Chemical compound C1([C@@H]2C(=O)N(C(N2)(C)C)[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 DXVUYOAEDJXBPY-NFFDBFGFSA-N 0.000 description 1
- 229960003884 hetacillin Drugs 0.000 description 1
- 229940084108 hexafluorenium bromide Drugs 0.000 description 1
- WBCLXFIDEDJGCC-UHFFFAOYSA-N hexafluoro-2-butyne Chemical compound FC(F)(F)C#CC(F)(F)F WBCLXFIDEDJGCC-UHFFFAOYSA-N 0.000 description 1
- VBZWSGALLODQNC-UHFFFAOYSA-N hexafluoroacetone Chemical compound FC(F)(F)C(=O)C(F)(F)F VBZWSGALLODQNC-UHFFFAOYSA-N 0.000 description 1
- WMIYKQLTONQJES-UHFFFAOYSA-N hexafluoroethane Chemical compound FC(F)(F)C(F)(F)F WMIYKQLTONQJES-UHFFFAOYSA-N 0.000 description 1
- HCDGVLDPFQMKDK-UHFFFAOYSA-N hexafluoropropylene Chemical compound FC(F)=C(F)C(F)(F)F HCDGVLDPFQMKDK-UHFFFAOYSA-N 0.000 description 1
- WDEFPRUEZRUYNW-UHFFFAOYSA-L hexafluronium bromide Chemical compound [Br-].[Br-].C12=CC=CC=C2C2=CC=CC=C2C1[N+](C)(C)CCCCCC[N+](C)(C)C1C2=CC=CC=C2C2=CC=CC=C21 WDEFPRUEZRUYNW-UHFFFAOYSA-L 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 229940006607 hirudin Drugs 0.000 description 1
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 229940050526 hydroxyethylstarch Drugs 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 150000002463 imidates Chemical class 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229920000592 inorganic polymer Polymers 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- XKTZWUACRZHVAN-VADRZIEHSA-N interleukin-8 Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@@H](NC(C)=O)CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N1[C@H](CCC1)C(=O)N1[C@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC(O)=CC=1)C(=O)N[C@H](CO)C(=O)N1[C@H](CCC1)C(N)=O)C1=CC=CC=C1 XKTZWUACRZHVAN-VADRZIEHSA-N 0.000 description 1
- 229940096397 interleukin-8 Drugs 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 229940029355 iodipamide Drugs 0.000 description 1
- DGIAUNUPXILTJW-VRWDCWMNSA-N iodipamide dimeglumine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC(=O)C1=C(I)C=C(I)C(NC(=O)CCCCC(=O)NC=2C(=C(C(O)=O)C(I)=CC=2I)I)=C1I DGIAUNUPXILTJW-VRWDCWMNSA-N 0.000 description 1
- 229960004359 iodixanol Drugs 0.000 description 1
- NBQNWMBBSKPBAY-UHFFFAOYSA-N iodixanol Chemical compound IC=1C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C(I)C=1N(C(=O)C)CC(O)CN(C(C)=O)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I NBQNWMBBSKPBAY-UHFFFAOYSA-N 0.000 description 1
- 229960001025 iohexol Drugs 0.000 description 1
- NTHXOOBQLCIOLC-UHFFFAOYSA-N iohexol Chemical compound OCC(O)CN(C(=O)C)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I NTHXOOBQLCIOLC-UHFFFAOYSA-N 0.000 description 1
- 229960004647 iopamidol Drugs 0.000 description 1
- XQZXYNRDCRIARQ-LURJTMIESA-N iopamidol Chemical compound C[C@H](O)C(=O)NC1=C(I)C(C(=O)NC(CO)CO)=C(I)C(C(=O)NC(CO)CO)=C1I XQZXYNRDCRIARQ-LURJTMIESA-N 0.000 description 1
- 229960000824 iopentol Drugs 0.000 description 1
- IUNJANQVIJDFTQ-UHFFFAOYSA-N iopentol Chemical compound COCC(O)CN(C(C)=O)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I IUNJANQVIJDFTQ-UHFFFAOYSA-N 0.000 description 1
- 229960002603 iopromide Drugs 0.000 description 1
- DGAIEPBNLOQYER-UHFFFAOYSA-N iopromide Chemical compound COCC(=O)NC1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)N(C)CC(O)CO)=C1I DGAIEPBNLOQYER-UHFFFAOYSA-N 0.000 description 1
- 229960000929 iotalamic acid Drugs 0.000 description 1
- 229960001707 ioxaglic acid Drugs 0.000 description 1
- TYYBFXNZMFNZJT-UHFFFAOYSA-N ioxaglic acid Chemical compound CNC(=O)C1=C(I)C(N(C)C(C)=O)=C(I)C(C(=O)NCC(=O)NC=2C(=C(C(=O)NCCO)C(I)=C(C(O)=O)C=2I)I)=C1I TYYBFXNZMFNZJT-UHFFFAOYSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical class C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 229960004184 ketamine hydrochloride Drugs 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- FRQMUZJSZHZSGN-HBNHAYAOSA-N medroxyprogesterone Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FRQMUZJSZHZSGN-HBNHAYAOSA-N 0.000 description 1
- 229960003464 mefenamic acid Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- MIKKOBKEXMRYFQ-WZTVWXICSA-N meglumine amidotrizoate Chemical compound C[NH2+]C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CC(=O)NC1=C(I)C(NC(C)=O)=C(I)C(C([O-])=O)=C1I MIKKOBKEXMRYFQ-WZTVWXICSA-N 0.000 description 1
- 229940053382 meglumine antimonate Drugs 0.000 description 1
- XOGYVDXPYVPAAQ-SESJOKTNSA-M meglumine antimoniate Chemical compound O[Sb](=O)=O.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO XOGYVDXPYVPAAQ-SESJOKTNSA-M 0.000 description 1
- 229940100477 meglumine iodipamide Drugs 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229960002409 mepivacaine Drugs 0.000 description 1
- INWLQCZOYSRPNW-UHFFFAOYSA-N mepivacaine Chemical compound CN1CCCCC1C(=O)NC1=C(C)C=CC=C1C INWLQCZOYSRPNW-UHFFFAOYSA-N 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- KDXZREBVGAGZHS-UHFFFAOYSA-M methohexital sodium Chemical compound [Na+].CCC#CC(C)C1(CC=C)C(=O)N=C([O-])N(C)C1=O KDXZREBVGAGZHS-UHFFFAOYSA-M 0.000 description 1
- 229960001620 methohexital sodium Drugs 0.000 description 1
- 229940069038 methotrimeprazine hydrochloride Drugs 0.000 description 1
- XGDZEDRBLVIUMX-UHFFFAOYSA-N methyl 2-(4-hydroxyphenyl)acetate Chemical compound COC(=O)CC1=CC=C(O)C=C1 XGDZEDRBLVIUMX-UHFFFAOYSA-N 0.000 description 1
- UGCLEKVSHFQKOD-UHFFFAOYSA-N methyl 2-[4-(oxiran-2-ylmethoxy)phenyl]acetate Chemical compound C1=CC(CC(=O)OC)=CC=C1OCC1OC1 UGCLEKVSHFQKOD-UHFFFAOYSA-N 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 229960000316 methyprylon Drugs 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 229940091062 metocurine iodide Drugs 0.000 description 1
- 229960000554 metrizamide Drugs 0.000 description 1
- GGGDNPWHMNJRFN-UHFFFAOYSA-N metrizoic acid Chemical compound CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I GGGDNPWHMNJRFN-UHFFFAOYSA-N 0.000 description 1
- 229960004712 metrizoic acid Drugs 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960002509 miconazole Drugs 0.000 description 1
- PLYSCVSCYOQVRP-UHFFFAOYSA-N midazolam hydrochloride Chemical compound Cl.C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F PLYSCVSCYOQVRP-UHFFFAOYSA-N 0.000 description 1
- 229960002853 midazolam hydrochloride Drugs 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- GPXLMGHLHQJAGZ-JTDSTZFVSA-N nafcillin Chemical compound C1=CC=CC2=C(C(=O)N[C@@H]3C(N4[C@H](C(C)(C)S[C@@H]43)C(O)=O)=O)C(OCC)=CC=C21 GPXLMGHLHQJAGZ-JTDSTZFVSA-N 0.000 description 1
- 229960000515 nafcillin Drugs 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-M naproxen(1-) Chemical compound C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-M 0.000 description 1
- 239000004081 narcotic agent Substances 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 229940127264 non-peptide agonist Drugs 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000004305 normal phase HPLC Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229960000988 nystatin Drugs 0.000 description 1
- VQOXZBDYSJBXMA-NQTDYLQESA-N nystatin A1 Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/CC/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 VQOXZBDYSJBXMA-NQTDYLQESA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- BCCOBQSFUDVTJQ-UHFFFAOYSA-N octafluorocyclobutane Chemical compound FC1(F)C(F)(F)C(F)(F)C1(F)F BCCOBQSFUDVTJQ-UHFFFAOYSA-N 0.000 description 1
- 235000019407 octafluorocyclobutane Nutrition 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 229960001027 opium Drugs 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 125000002092 orthoester group Chemical group 0.000 description 1
- 230000010355 oscillation Effects 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 150000002924 oxiranes Chemical class 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- NPIJXCQZLFKBMV-YTGGZNJNSA-L pancuronium bromide Chemical compound [Br-].[Br-].C[N+]1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 NPIJXCQZLFKBMV-YTGGZNJNSA-L 0.000 description 1
- 229960003379 pancuronium bromide Drugs 0.000 description 1
- SQYNKIJPMDEDEG-UHFFFAOYSA-N paraldehyde Chemical compound CC1OC(C)OC(C)O1 SQYNKIJPMDEDEG-UHFFFAOYSA-N 0.000 description 1
- 229960003868 paraldehyde Drugs 0.000 description 1
- 230000005298 paramagnetic effect Effects 0.000 description 1
- 229960000865 paramethasone acetate Drugs 0.000 description 1
- 239000008414 paregoric Substances 0.000 description 1
- 229940069533 paregoric Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 229960004321 pentaerithrityl tetranitrate Drugs 0.000 description 1
- QIYZKVMAFMDRTP-UHFFFAOYSA-N pentafluoro(trifluoromethyl)-$l^{6}-sulfane Chemical compound FC(F)(F)S(F)(F)(F)(F)F QIYZKVMAFMDRTP-UHFFFAOYSA-N 0.000 description 1
- 150000002972 pentoses Chemical class 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- LGUZHRODIJCVOC-UHFFFAOYSA-N perfluoroheptane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F LGUZHRODIJCVOC-UHFFFAOYSA-N 0.000 description 1
- ZJIJAJXFLBMLCK-UHFFFAOYSA-N perfluorohexane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F ZJIJAJXFLBMLCK-UHFFFAOYSA-N 0.000 description 1
- 210000003516 pericardium Anatomy 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 108010089198 phenylalanyl-prolyl-arginine Proteins 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 1
- 229940067605 phosphatidylethanolamines Drugs 0.000 description 1
- 102000023884 phosphatidylserine binding proteins Human genes 0.000 description 1
- 108091008423 phosphatidylserine binding proteins Proteins 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920001308 poly(aminoacid) Polymers 0.000 description 1
- 229920002627 poly(phosphazenes) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 239000003910 polypeptide antibiotic agent Substances 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 229930185346 proliferin Natural products 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- MWWATHDPGQKSAR-UHFFFAOYSA-N propyne Chemical compound CC#C MWWATHDPGQKSAR-UHFFFAOYSA-N 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 229960005206 pyrazinamide Drugs 0.000 description 1
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- 229960000948 quinine Drugs 0.000 description 1
- 239000012857 radioactive material Substances 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 229920013730 reactive polymer Polymers 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 229940107685 reopro Drugs 0.000 description 1
- 125000006853 reporter group Chemical group 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 210000005241 right ventricle Anatomy 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- AHTFMWCHTGEJHA-UHFFFAOYSA-N s-(2,5-dioxooxolan-3-yl) ethanethioate Chemical compound CC(=O)SC1CC(=O)OC1=O AHTFMWCHTGEJHA-UHFFFAOYSA-N 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 229960003141 secobarbital sodium Drugs 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 229960001153 serine Drugs 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- QORQZMBCPRBCAB-UHFFFAOYSA-N sodium;5-butan-2-yl-5-ethyl-1,3-diazinane-2,4,6-trione Chemical compound [Na+].CCC(C)C1(CC)C(=O)NC(=O)NC1=O QORQZMBCPRBCAB-UHFFFAOYSA-N 0.000 description 1
- BNHGKKNINBGEQL-UHFFFAOYSA-M sodium;5-ethyl-5-(3-methylbutyl)pyrimidin-3-ide-2,4,6-trione Chemical compound [Na+].CC(C)CCC1(CC)C(=O)NC(=O)[N-]C1=O BNHGKKNINBGEQL-UHFFFAOYSA-M 0.000 description 1
- AXXJTNXVUHVOJW-UHFFFAOYSA-M sodium;5-pentan-2-yl-5-prop-2-enylpyrimidin-3-ide-2,4,6-trione Chemical compound [Na+].CCCC(C)C1(CC=C)C(=O)NC(=O)[N-]C1=O AXXJTNXVUHVOJW-UHFFFAOYSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 229950004330 spiroplatin Drugs 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 150000008143 steroidal glycosides Chemical class 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 229960005202 streptokinase Drugs 0.000 description 1
- QTENRWWVYAAPBI-YCRXJPFRSA-N streptomycin sulfate Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O QTENRWWVYAAPBI-YCRXJPFRSA-N 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 229940120904 succinylcholine chloride Drugs 0.000 description 1
- YOEWQQVKRJEPAE-UHFFFAOYSA-L succinylcholine chloride (anhydrous) Chemical compound [Cl-].[Cl-].C[N+](C)(C)CCOC(=O)CCC(=O)OCC[N+](C)(C)C YOEWQQVKRJEPAE-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000003774 sulfhydryl reagent Substances 0.000 description 1
- 150000003457 sulfones Chemical group 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical class ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 229960005314 suramin Drugs 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 229960004000 talbutal Drugs 0.000 description 1
- BJVVMKUXKQHWJK-UHFFFAOYSA-N talbutal Chemical compound CCC(C)C1(CC=C)C(=O)NC(=O)NC1=O BJVVMKUXKQHWJK-UHFFFAOYSA-N 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229960003188 temazepam Drugs 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-N tert-butylcarbamic acid Chemical compound CC(C)(C)NC(O)=O XBXCNNQPRYLIDE-UHFFFAOYSA-N 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- 229960002372 tetracaine Drugs 0.000 description 1
- GKCBAIGFKIBETG-UHFFFAOYSA-N tetracaine Chemical compound CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 GKCBAIGFKIBETG-UHFFFAOYSA-N 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 238000009210 therapy by ultrasound Methods 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 230000034005 thiol-disulfide exchange Effects 0.000 description 1
- 229960003279 thiopental Drugs 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 102000003601 transglutaminase Human genes 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 229940126307 triamcinolone acetate Drugs 0.000 description 1
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 description 1
- 229960003386 triazolam Drugs 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- 229960002655 tubocurarine chloride Drugs 0.000 description 1
- 229960005356 urokinase Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- VEPSYABRBFXYIB-PWXDFCLTSA-M vecuronium bromide Chemical compound [Br-].N1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 VEPSYABRBFXYIB-PWXDFCLTSA-M 0.000 description 1
- 229960004298 vecuronium bromide Drugs 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- 108010047303 von Willebrand Factor Proteins 0.000 description 1
- 102100036537 von Willebrand factor Human genes 0.000 description 1
- 229960001134 von willebrand factor Drugs 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 238000000733 zeta-potential measurement Methods 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0058—Antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/542—Carboxylic acids, e.g. a fatty acid or an amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
- A61K47/6925—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a microcapsule, nanocapsule, microbubble or nanobubble
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0021—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent group being a small organic molecule
- A61K49/0041—Xanthene dyes, used in vivo, e.g. administered to a mice, e.g. rhodamines, rose Bengal
- A61K49/0043—Fluorescein, used in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0063—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres
- A61K49/0069—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres the agent being in a particular physical galenical form
- A61K49/0089—Particulate, powder, adsorbate, bead, sphere
- A61K49/0091—Microparticle, microcapsule, microbubble, microsphere, microbead, i.e. having a size or diameter higher or equal to 1 micrometer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/22—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations
- A61K49/222—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations characterised by a special physical form, e.g. emulsions, liposomes
- A61K49/223—Microbubbles, hollow microspheres, free gas bubbles, gas microspheres
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0497—Organic compounds conjugates with a carrier being an organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/088—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins conjugates with carriers being peptides, polyamino acids or proteins
Definitions
- This invention relates to diagnostic and/or therapeutically active agents, more particularly to diagnostic and/or therapeutically active agents incorporating moieties which interact with or have affinity for sites and/or structures within the body so that diagnostic imaging and/or therapy of particular locations within the body may be enhanced.
- diagnostic agents for use in ultrasound imaging which are hereinafter referred to as targeted ultrasound contrast agents.
- ultrasound imaging comprises a potentially valuable diagnostic tool, for example in studies of the vascular system, particularly in cardiography, and of tissue microvasculature .
- contrast agents has been proposed to enhance the acoustic images so obtained, including suspensions of solid particles, emulsified liquid droplets, gas bubbles and encapsulated gases or liquids. It is generally accepted that low density contrast agents which are easily compressible are particularly efficient in terms of the acoustic backscatter they generate, and considerable interest has therefore been shown in the preparation of gas-containing and gas-generating systems .
- Gas-containing contrast media are also known to be effective in magnetic resonance (MR) imaging, e.g. as susceptibility contrast agents which will act to reduce MR signal intensity.
- Oxygen-containing contrast media also represent potentially useful paramagnetic MR contrast agents .
- gases such as carbon dioxide may be used as negative oral contrast agents or intravascular contrast agents.
- radioactive gases e.g. radioactive isotopes of inert gases such as xenon
- scintigraphy for example for blood pool imaging .
- Targeted ultrasound contrast agents may be regarded as comprising (i) a reporter moiety capable of interacting with ultrasound irradiation to generate a detectable signal; (ii) one or more vectors having affinity for particular target sites and/or structures within the body, e.g. for specific cells or areas of pathology; and (iii) one or more linkers connecting said reporter and vector (s), in the event that these are not directly joined.
- the molecules and/or structure to which the agent is intended to bind will hereinafter be referred to as the target .
- the target In order to obtain specific imaging of or a therapeutic effect at a selected region/structure in the body the target must be present and available in this region/structure.
- the target may either be a defined molecular species (i.e. a target molecule) or an unknown molecule or more complex structure (i.e. a target structure) which is present in the area to be imaged and/or treated, and is able to bind specifically or selectively to a given vector molecule.
- the vector is attached or linked to the reporter moiety in order to bind these moieties to the region/structure to be imaged and/or treated.
- the vector may bind specifically to a chosen target, or it may bind only selectively, having affinty also for a limited number of other molecules/structures, again creating possible background problems.
- US-A-5531980 is directed to systems in which the reporter comprises an aqueous suspension of air or gas microbubbles stabilised by one or more film- forming surfactants present at least partially in lamellar or laminar form, said surfactant (s) being bound to one or more vectors comprising "bioactive species designed for specific targeting purposes".
- the microbubbles are not directly encapsulated by surfactant material but rather that this is incorporated in liquid- filled liposomes which stabilise the microbubbles.
- lamellar or laminar surfactant material such as phospholipids present in such liposomes will inevitably be present in the form of one or more lipid bilayers with the lipophilic tails "back-to-back” and the hydrophilic heads both inside and outside (see e.g. Schneider, M. on “Liposomes as drug carriers: 10 years of research” in Drug targeting, Nyon, Swi tzerland, 3 -5 October 1984 , Buri, P. and Gumma, A. (Ed) , Elsevier, Amsterdam 1984) .
- EP-A-0727225 describes targeted ultrasound contrast agents in which the reporter comprises a chemical having a sufficient vapour pressure such that a proportion of it is a gas at the body temperature of the subject.
- This chemical is associated with a surfactant or albumin carrier which includes a protein-, peptide- or carbohydrate-based cell adhesion molecule ligand as vector.
- the reporter moieties in such contrast agents correspond to the phase shift colloid systems described in WO-A-9416739 ; it is now recognised that administration of such phase shift colloids may lead to generation of microbubbles which grow uncontrollably, possibly to the extent where they cause potentially dangerous embolisation of, for example, the myocardial vasculature and brain (see e.g.
- tissue-specific ultrasonic image enhancement may be achieved using acoustically reflective oligolamellar liposomes conjugated to tissue-specific ligands such as antibodies, peptides, lectins etc.
- the liposomes are deliberately chosen to be devoid of gas and so will not have the advantageous echogenic properties of gas-based ultrasound contrast agents .
- Further references to this technology e.g. in targeting to fibrin, thrombi and atherosclerotic areas are found in publications by Alkanonyuksel , H. et al . in J “ . Pharm . Sci . (1996) 85(5), 486-490; J “ . Am. Coll . Cardiol . (1996) 27(2) Suppl A,
- the present invention is based on the finding that gas-containing and gas-generating diagnostic and/or therapeutic agents coupled to non-bioactive vectors are particularly useful targeting agents by virtue of their enhanced safety relative to conventional targeting agents, which may elicit undesirable and unwanted biological effects in the subject.
- non- bioactive vectors may exhibit improved targeting efficacy as compared to bioactive vectors.
- bioactive vectors usually will have to compete with endogenous ligands for the same binding site on the target molecule.
- non- bioactive vectors will often bind to target molecules for which endogenous ligands do not exist, or alternatively non-bioactive vectors may bind at sites in a target molecule that is not involved in the biological function of the target molecule and for which natural ligands are not present in the body.
- non-bioactive denotes two kinds of substances. First, it denotes a substance capable of interacting with or binding to a target molecule or target structure that is not normally involved in generating biological responses. Second, it denotes a substance capable of interacting with or binding to a given target without either giving or inhibiting the biological response normally elicited upon binding of a bioactive substance to the target, a bioactive substance being one which interacts with another molecule (i.e. a target) and generates a defined and measurable biological response.
- receptors for transport proteins such as receptors for transferrin or lipoproteins
- a moiety carried in the protein may be bioactive, but the apoprotein itself is not.
- many cofactors, vitamins etc. are bioactive only after they have been carried into the cell, that is to say, they do not elicit a biological response simply upon binding to the carrier protein.
- Vectors useful in accordance with the invention may, for example, be non-bioactive per se or may be non- bioactive at doses useful for diagnostic and/or therapeutic purposes.
- a combination of otherwise bioactive vectors may be employed in such a way that a biological activity elicited by one type of vector is exactly counterbalanced by another kind or other kinds of vector molecules coupled to the same agent or otherwise present in the same diagnostic and/or therapeutic composition.
- One advantageous embodiment of the invention is based on the additional finding that limited adhesion to targets is a highly useful property of diagnostic and/or therapeutically active agents, which property may be achieved using non-bioactive vectors giving temporary retention rather than fixed adhesion to a target.
- agents rather than being fixedly retained at specific sites, may for example effectively exhibit a form of retarded flow along the vascular endothelium by virtue of their transient interactions with endothelial cells.
- Such agents may thus become concentrated on the walls of blood vessels, in the case of ultrasound contrast agents providing enhanced echogenicity thereof relative to the bulk of the bloodstream, which is devoid of permanent structural features. They therefore may permit enhanced imaging of the capillary system, including the microvasculature, and so may facilitate distinction between normal and inadequately perfused tissue, e.g. in the heart, and may also be useful in visualising structures such as Kupffer cells, thrombi and atherosclerotic lesions or for visualising neo- vascularized and inflamed tissue areas.
- the present invention is also well suited to image changes occurring to normal blood vessels which are situated in areas of tissue necrosis.
- a targetable diagnostic and/or therapeutic agent e.g. an ultrasound contrast agent
- a suspension in an aqueous carrier liquid e.g. an injectable carrier liquid
- a reporter comprising gas-containing or gas-generating material and coupled to one or more vectors, characterised in that said vector or vectors are non- bioactive .
- Vectors useful in accordance with the invention include proteins which bind to cell-surface proteoglycans , e.g. so that the contrast agent becomes concentrated on cell surfaces.
- proteoglycans are large glycoproteins containing glucosaminoglycan side chains which, in many cases, are heparan sulphate. Binding to glucosoaminoglycans has not been shown to elicit a biological response.
- one may isolate or synthesise the proteoglycan-binding (or, where appropriate, heparan sulphate-binding) part of the molecule for use as a vector, while avoiding any biological activity associated with other parts of the molecule.
- non-bioactive monomeric or oligomeric vectors and the use of non-bioactive peptide vectors represent preferred aspects of this invention.
- a further aspect of the present invention is for example where a vector or vectors is attached to the reporter or included non-covalently into the reporter in a manner where the said vector or vectors is not readily exposed to the targets or receptors.
- Increased tissue specificity may therefore be achieved by applying an additional process to expose the vectors, e.g. the agent is exposed to external ultrasound to change the diffusibility of the moieties containing the vectors.
- Any biocompatible gas may be present in the reporter of contrast agents according to the invention, the term "gas” as used herein including any substances (including mixtures) substantially or completely in gaseous (including vapour) form at the normal human body temperature of 37°C.
- the gas may thus, for example, comprise air; nitrogen; oxygen; carbon dioxide; hydrogen; an inert gas such as helium, argon, xenon or krypton; a sulphur fluoride such as sulphur hexafluoride, disulphur decafluoride or trifluoromethylsulphur pentafluoride ; selenium hexafluoride; an optionally halogenated silane such as methylsilane or dimethylsilane; a low molecular weight hydrocarbon (e.g.
- an alkane such as methane, ethane, a propane, a butane or a pentane, a cycloalkane such as cyclopropane, cyclobutane or cyclopentane, an alkene such as ethylene, propene, propadiene or a butene, or an alkyne such as acetylene or propyne; an ether such as dimethyl ether; a ketone; an ester; a halogenated low molecular weight hydrocarbon (e.g. containing up to 7 carbon atoms); or a mixture of any of the foregoing.
- an alkane such as methane, ethane, a propane, a butane or a pentane
- a cycloalkane such as cyclopropane, cyclobutane or cyclopentane
- an alkene such as ethylene, propene, propadiene or a butene
- biocompatible halogenated hydrocarbon gases may, for example, be selected from bromochlorodifluoromethane , chlorodifluoromethane , dichlorodifluoromethane , bromotrifluoromethane , chlorotrifluoromethane, chloropentafluoroethane, dichlorotetrafluoroethane , chlorotrifluoroethylene , fluoroethylene, ethylfluoride, 1, 1-difluoroethane and perfluorocarbons , e.g.
- perfluoroalkanes such as perfluoromethane , perfluoroethane, perfluoropropanes , perfluorobutanes (e.g. perfluoro-n-butane, optionally in admixture with other isomers such as perfluoro-iso- butane) , perfluoropentanes, perfluorohexanes and perfluoroheptanes ; perfluoroalkenes such as perfluoropropene, perfluorobutenes (e.g.
- perfluorobut-2- ene and perfluorobutadiene; perfluoroalkynes such as perfluorobut-2-yne; and perfluorocycloalkanes such as perfluorocyclobutane , perfluoromethylcyclobutane , perfluorodimethylcyclobutanes, perfluorotrimethyl- cyclobutanes, perfluorocyclopentane, perfluoromethyl- cyclopentane, perfluorodimethylcyclopentanes , perfluorocyclohexane, perfluoromethylcyclohexane and perfluorocycloheptane .
- halogenated gases include methyl chloride, fluorinated (e.g. perfluorinated) ketones such as perfluoroacetone and fluorinated (e.g. perfluorinated) ethers such as perfluorodiethyl ether.
- perfluorinated gases for example sulphur hexafluoride and perfluorocarbons such as perfluoropropane , perfluorobutanes and perfluoropentanes, may be particularly advantageous in view of the recognised high stability in the bloodstream of microbubbles containing such gases.
- the reporter may be in any convenient form, for example being any appropriate gas-containing or gas- generating ultrasound contrast agent formulation.
- Representative examples of such formulations include microbubbles of gas stabilised (e.g. at least partially encapsulated) by a coalescence-resistant surface membrane (for example gelatin, e.g. as described in 0- A-8002365) , a filmogenic protein (for example an albumin such as human serum albumin, e.g.
- a polymer material for example a synthetic biodegradable polymer as described in EP-A- 0398935 , an elastic interfacial synthetic polymer membrane as described in EP-A-0458745 , a microparticulate biodegradable polyaldehyde as described in EP-A-0441468 , a microparticulate N-dicarboxylic acid derivative of a polyamino acid - polycyclic imide as described in EP-A- 0458079, or a biodegradable polymer as described in O- A-9317718 or O-A-9607434 ) , a non-polymeric and non- polymerisable wall-forming material
- gas-containing contrast agent formulations include gas-containing solid systems, for example microparticles (especially aggregates of microparticles) having gas contained therewithin or otherwise associated therewith (for example being adsorbed on the surface thereof and/or contained within voids, cavities or pores therein, e.g. as described in EP-A-0122624, EP-A- 0123235 , EP-A-0365467 , WO-A-9221382 , WO-A-9300930, WO-A-9313802 , WO-A-9313808 or WO-A- 9313809) .
- the echogenicity of such microparticulate contrast agents may derive directly from the contained/associated gas and/or from gas (e.g. microbubbles) liberated from the solid material (e.g. upon dissolution of the microparticulate structure) .
- Gas microbubbles and other gas-containing materials such as microparticles preferably have an initial average size not exceeding 10 ⁇ m (e.g. of 7 ⁇ m or less) in order to permit their free passage through the pulmonary system following administration, e.g. by intravenous injection.
- phospholipid-containing compositions are employed in accordance with the invention, e.g. in the form of phospholipid-stabilised gas microbubbles
- useful phospholipids include lecithins (i.e. phosphatidylcholines) , for example natural lecithins such as egg yolk lecithin or soya bean lecithin and synthetic or semisynthetic lecithins such as dimyristoylphosphatidylcholine , dipalmitoylphosphatidylcholine or distearoylphosphatidylcholine; phosphatidic acids; phosphatidylethanolamines ; phosphatidylserines ; phosphatidylglycerols ; phosphatidylinositols ; cardiolipins; sphingomyelins ; fluorinated analogues of any of the foregoing; mixtures of any of the foregoing and mixtures with other lipids such as
- phospholipids predominantly (e.g. at least 75%) comprising molecules individually bearing net overall charge, e.g. negative charge, for example as in naturally occurring (e.g. soya bean or egg yolk derived), semisynthetic (e.g. partially or fully hydrogenated) and synthetic phosphatidylserines, phosphatidylglycerols, phosphatidylinositols, phosphatidic acids and/or cardiolipins, may be particularly advantageous.
- naturally occurring e.g. soya bean or egg yolk derived
- semisynthetic e.g. partially or fully hydrogenated
- synthetic phosphatidylserines e.g. partially or fully hydrogenated
- phosphatidylglycerols phosphatidylglycerols
- phosphatidylinositols phosphatidic acids and/or cardiolipins
- exemplary lipids which may be used to prepare gas-containing contrast agents include fatty acids, stearic acid, palmitic acid, 2-n-hexadecylstearic acid, oleic acid and other acid containing lipid structures. These lipid structures are considered particularly interesting when coupled by amide bond formation to amino acids containing one or more amino groups.
- the resulting lipid modified amino acids e.g. dipalmitoyllysine, distearoyl-2 , 3 -diaminopropionic acid
- a further extension of this invention relates to the synthesis of lipopeptide structures comprising a lipid reporter attached to a linker portion (e.g. PEG, polyamino acid, alkylhalide etc) the said linker being suitably functionalised for coupling to one or more vector molecules.
- a linker portion e.g. PEG, polyamino acid, alkylhalide etc
- the said linker being suitably functionalised for coupling to one or more vector molecules.
- a positively charged linker element eg. two or more lysine residues
- microbubbles carrying one or more reactive groups for non-specific modification of a multiplicity of receptor molecules located on cell surfaces.
- Microbubbles comprising a thiol moiety can bind to cell surface receptors via disulphide exchange reactions. The reversible nature of this covalent bond means that bubble flow can be controlled by altering the redox environment.
- activated' microbubbles of membranes comprising active esters such as N-hydroxysuccinimide esters can be used to modify amino groups found on a multiplicity of cell surface molecules.
- gas-containing microparticulate materials which may be useful in accordance with the invention include carbohydrates (for example hexoses such as glucose, fructose or galactose; disaccharides such as sucrose, lactose or maltose; pentoses such as arabinose, xylose or ribose; - , ⁇ - and ⁇ -cyclodextrins ; polysaccharides such as starch, hydroxyethyl starch, amylose, amylopectin, glycogen, inulin, pulullan, dextran, carboxymethyl dextran, dextran phosphate, ketodextran, aminoethyldextran, alginates, chitin, chitosan, hyaluronic acid or heparin; and sugar alcohols, including alditols such as mannitol or sorbitol), inorganic salts (e.g.
- X-ray contrast agents e.g. any of the commercially available carboxylic acid and non-ionic amide contrast agents typically containing at least one 2, 4 , 6-triiodophenyl group having substituents such as carboxyl, carbamoyl, N-alkylcarbamoyl , N- hydroxyalkylcarbamoyl , acylamino, N-alkylacylamino or acylaminomethyl at the 3- and/or 5 -positions, as in metrizoic acid, diatrizoic acid, iothalamic acid, ioxaglic acid, iohexol , iopentol, iopamidol , iodixanol, iopromide, metrizamide, iodipamide, meglumine iodipamide, meglumine acetrizo
- the reporter may be made by any convenient process, for example by making gas-containing or gas- generating formulations.
- Representative examples include the preparation of a suspension of gas microbubbles by contacting a surfactant with gas and mixing them in the presence of an aqueous carrier, as described in WO
- a suitable process for attachment of the desired vector to the reporter comprises a surface modification of the preformed reporter with a suitable linker employing reactive groups on the surface of both the reporter and vector. It may be particularly advantageous physically to mix the reporter material with the vector- containing substance at any step of the process. Such a process will result in incorporation or an attachment of the vector to the reporter. An optional process step may remove the excess of vector not bound to the reporter by washing the gas-containing particles following separation, by for example, floatation.
- a preferred aspect is the use of lipopeptide structures incorporating functional groups such as thiol, maleimide biotin etc. which can be premixed if desired with other reporter molecules before formation of gas-containing agents .
- the attachment of vector molecules may be carried out using the linker reagents listed below.
- Coupling of a reporter unit to the desired vectors may be achieved by covalent or non-covalent means, usually involving interaction with one or more functional groups located on the reporter and/or vectors.
- functional groups located on the reporter and/or vectors.
- chemically reactive functional groups include amino, hydroxyl, sulfhydryl, carboxyl, and carbonyl groups, as well as carbohydrate groups, vicinal diols, thioethers, 2-aminoalcohols , 2-aminothiols , guanidinyl, imidazolyl and phenolic groups.
- Covalent coupling of reporter and vectors may therefore be effected using linking agents containing reactive moities capable of reaction with such functional groups .
- N-Maleimide derivatives are also considered selective towards sulfhydryl groups, but may additionaly be useful in coupling to amino groups under certain conditions.
- N-maleimides may be incorporated into linking systems for reporter-vector conjugation as described by Kitagawa, T. et al . in Chem . Pharm. Bull . (1981) 29, 1130 or used as polymer crosslinkers for bubble stabilisation as described by Kovacic, P. et al . in J. Am . Chem . Soc . (1959) 81, 1887.
- Reagents such as 2-iminothiolane, e.g. as described by Traut, R. et al. in Biochemistry (1973) 12, 3266, which introduce a thiol group through conversion of an amino group, may be considered as sulfhydryl reagents if linking occurs through the formation of disulphide bridges .
- reagents which introduce reactive disulphide bonds into either the reporter or the vector may be useful, since linking may be brought about by disulphide exchange between the vector and reporter;
- examples of such reagents include Ellman's reagent (DTNB), 4,4'- dithiodipyridine, methyl-3-nitro-2-pyridyl disulphide and methyl -2 -pyridyl disulphide (described by Kimura, T. et al . in Analyt . Biochem . (1982) 122, 271) .
- Examples of reactive moieties capable of reaction with amino groups include alkylating and acylating agents.
- Representative amino-reactive acylating agents include : i) isocyanates and isothiocyanates, particularly aromatic derivatives, Which form stable urea and thiourea derivatives respectively and have been used for protein crosslinking as described by Schick, A.F. et al. in J " . Biol . Chem . (1961) 236, 2477; ii) sulfonyl chlorides, which have been described by Herzig, D.J. et al .
- Carbonyl groups such as aldehyde functions may be reacted with weak protein bases at a pH such that nucleophilic protein side-chain functions are protonated.
- Weak bases include 1 , 2-aminothiols such as those found in N-terminal cysteine residues, which selectively form stable 5-membered thiazolidine rings with aldehyde groups, e.g. as described by Ratner, S. et al . in J. Am. Chem . Soc . (1937) 59, 200.
- Other weak bases such as phenyl hydrazones may be used, e.g. as described by Heitzman, H. et al . in Proc . Na tl . Acad . Sci . USA (1974) 71, 3537.
- Aldehydes and ketones may also be reacted with amines to form Schiff's bases, which may advantageously be stabilised through reductive amination.
- Alkoxylamino moieties readily react with ketones and aldehydes to produce stable alkoxamines, e.g. as described by Webb, R. et al . in Bioconjuga te Chem . (1990) 1, 96.
- reactive moieties capable of reaction with carboxyl groups include diazo compounds such as diazoacetate esters and diazoacetamides, which react with high specificity to generate ester groups, e.g. as described by Herriot R.M. in Adv. Protein Chem . (1947) 3, 169.
- Carboxylic acid modifying reagents such as carbodiimides , which react through O-acylurea formation followed by amide bond formation, may also usefully be employed; linking may be facilitated through addition of an amine or may result in direct vector-receptor coupling.
- Useful water soluble carbodiimides include 1- cyclohexyl-3- (2-morpholinyl-4-ethyl) carbodiimide (CMC) and l-ethyl-3 - (3 -dimethylaminopropyl) carbodiimide (EDC) , e.g. as described by Zot, H.G. and Puett, D. in J " . Biol . Chem . (1989) 264, 15552.
- CMC 1- cyclohexyl-3- (2-morpholinyl-4-ethyl) carbodiimide
- EDC l-ethyl-3 - (3 -dimethylaminopropyl) carbodiimide
- carboxylic acid modifying reagents include isoxazolium derivatives such as Woodwards reagent K; chloroformates such as p- nitrophenylchloroformate; carbonyldiimidazoles such as 1 , 1 ' -carbonyldiimidazole ; and N- carbalkoxydihydroquinolines such as N- (ethoxycarbonyl) - 2-ethoxy-l, 2-dihydroquinoline .
- isoxazolium derivatives such as Woodwards reagent K
- chloroformates such as p- nitrophenylchloroformate
- carbonyldiimidazoles such as 1 , 1 ' -carbonyldiimidazole
- N- carbalkoxydihydroquinolines such as N- (ethoxycarbonyl) - 2-ethoxy-l, 2-dihydroquinoline .
- vicinal diones such as p-phenylenediglyoxal , which may be used to react with guanidinyl groups, e.g. as described by Wagner e ⁇ al . in Nucleic acid Res . (1978) 5, 4065; and diazonium salts, which may undergo electrophilic substitution reactions, e.g. as described by Ishizaka, K. and Ishizaka T. in J " . Immunol . (1960) 85, 163.
- Bis-diazonium compounds are readily prepared by treatment of aryl diamines with sodium nitrite in acidic solutions.
- functional groups in the reporter and/or vector may if desired be converted to other functional groups prior to reaction, e.g. to confer additional reactivity or selectivity.
- methods useful for this purpose include conversion of amines to carboxylic acids using reagents such as dicarboxylic anhydrides; conversion of amines to thiols using reagents such as N-acetylhomocysteine thiolactone, S-acetylmercaptosuccinic anhydride, 2- iminothiolane or thiol -containing succinimidyl derivatives; conversion of thiols to carboxylic acids using reagents such as ⁇ -haloacetates; conversion of thiols to amines using reagents such as ethylenimine or 2-bromoethylamine ; conversion of carboxylic acids to amines using reagents such as carbodiimides followed by diamines ; and conversion of alcohols to thio
- Vector-reporter coupling may also be effected using enzymes as zero-length crosslinking agents; thus, for example, transglutaminase, peroxidase and xanthine oxidase have been used to produce crosslinked products.
- Reverse proteolysis may also be used for crosslinking through amide bond formation.
- Non-covalent vector-reporter coupling may, for example, be effected by electrostatic charge interactions e.g. between a polylysinyl-functionalised reporter and a polyglutamyl-functionalised vector, through chelation in the form of stable metal complexes or through high affinity binding interaction such as avidin/biotin binding.
- Polylysine, coated non- covalently to the negatively charged membrane surface can also increase non-specifically the affinity of a microbubble for a cell through charge interactions.
- a vector may be coupled to a protein known to bind phospholipids.
- a single molecule of phospholipid may attach to a protein such as a translocase, while other proteins may attach to surfaces consisting mainly of phospholipid head groups and so may be used to attach vectors to phospholipid microspheres; one example of such a protein is ⁇ 2-glycoprotein I (Chonn, A., Semple, S.C. and
- a conjugate of a vector with such a phosphatidylserine-binding protein may therefore be used to attach the vector to phosphatidylserine-encapsulated microbubbles .
- the amino acid sequence of a binding protein is known, the phospholipid-binding portion may be synthesised or isolated and used for conjugation with a vector, thus avoiding the biological activity which may be located elsewhere in the molecule.
- phage libraries displaying small peptides could be used for such selection.
- the selection may be made by simply mixing the microspheres and the phage display library and eluting the phages binding to the floating microspheres. If desired, the selection can be done under "physiological conditions" (e.g. in blood) to eliminate peptides which cross-react with blood components.
- physiological conditions e.g. in blood
- An advantage of this type of selection procedure is that only binding molecules that do not destabilize the microspheres should be selected, since only binding molecules attached to intact floating microspheres will rise to the top. It may also be possible to introduce some kind of "stress" during the selection procedure (e.g. pressure) to ensure that destabilizing binding moieties are not selected.
- binding moieties identified in this way may be coupled (by chemical conjugation or via peptide synthesis, or at the DNA-level for recombinant vectors) to a vector molecule, constituting a general tool for attaching any vector molecule to the microspheres .
- a vector which comprises or is coupled to a peptide, lipo-oligosaccharide or lipopeptide linker which contains a element capable of mediating membrane insertion may also be useful.
- a peptide, lipo-oligosaccharide or lipopeptide linker which contains a element capable of mediating membrane insertion
- Non-bioactive molecules consisting of known membrane insertion anchor/signal groups may also be used as vectors for certain applications, an example being the HI hydrophobic segment from the Na,K-ATPase - subunit described by Xie, Y. and Morimoto, T. in J " . Biol . Chem. (1995) 270(20), 11985-11991.
- the anchor group may also be fatty acid(s) or cholesterol.
- Coupling may also be effected using avidin or streptavidin, which have four high affinity binding sites for biotin. Avidin may therefore be used to conjugate vector to reporter if both vector and reporter are biotinylated. Examples are described by Bayer, E.A. and Wilchek, M. in Methods Biochem . Anal . (1980) 26, 1. This method may also be extended to include linking of reporter to reporter, a process which may encourage bubble association and consequent potentially increased echogenicity . Alternatively, avidin or streptavidin may be attached directly to the surface of reporter microparticles. Non-covalent coupling may also utilise the bifunctional nature of bispecific immunoglobulins .
- bispecific IgG or chemically engineered bispecific F(ab)'2 fragments may be used as linking agents.
- Heterobifunctional bispecific antibodies have also been reported for linking two different antigens, e.g. as described by Bode, C. et al . in J “ . Biol . Chem . (1989) 264, 944 and by Staerz, U.D. et al. in Proc . Na tl . Acad . Sci . USA (1986) 83, 1453.
- any reporter and/or vector containing two or more antigenic determinants e.g. as described by Chen, Aa et al . in Am. J “ . Pathol . (1988) 130, 216) may be crosslinked by antibody molecules and lead to formation of multi-bubble cross-linked assemblies of potentially increased echogenicity.
- Linking agents used in accordance with the invention will in general bring about linking of vector to reporter or reporter to reporter with some degree of specificity, and may also be used to attach one or more therapeutically active agents.
- a PEG component as a stabiliser in conjunction with a vector or vectors or directly to the reporter in the same molecule where the PEG does not serve as a spacer.
- the reporter unit will usually remain attached to the vectors.
- the vector (often, a monoclonal antibody) is administered alone; subsequently, the reporter is administered, coupled to a moiety which is capable of specifically binding the vector molecule (when the vector is an antibody, the reporter may be coupled to an immunoglobulin-binding molecule, such as protein A or an anti-immunoglobulin antibody) .
- an immunoglobulin-binding molecule such as protein A or an anti-immunoglobulin antibody
- Ultrasound imaging modalities which may be used in accordance with the invention include two- and three- dimensional imaging techniques such as B-mode imaging (for example using the time-varying amplitude of the signal envelope generated from the fundamental frequency of the emitted ultrasound pulse, from sub-harmonics or higher harmonics thereof or from sum or difference frequencies derived from the emitted pulse and such harmonics, images generated from the fundamental frequency or the second harmonic thereof being preferred) , colour Doppler imaging and Doppler amplitude imaging, and combinations of the two latter with any of the modalities (techniques) above.
- B-mode imaging for example using the time-varying amplitude of the signal envelope generated from the fundamental frequency of the emitted ultrasound pulse, from sub-harmonics or higher harmonics thereof or from sum or difference frequencies derived from the emitted pulse and such harmonics, images generated from the fundamental frequency or the second harmonic thereof being preferred
- colour Doppler imaging and Doppler amplitude imaging and combinations of the two latter with any of the modalities (techniques) above.
- the present invention accordingly provides a tool for therapeutic drug delivery in combination with vector-mediated direction of the product to the desired site.
- therapeutic or “drug” is meant an agent having a beneficial effect on a specific disease in a living human or non-human animal.
- combinations of drugs and ultrasound contrast agents have been proposed in, for example, WO-A-9428873 and WO-A-9507072 , these products lack vectors having affinity for particular sites and thereby show comparitively poor specific retention at desired sites prior to or during drug release .
- Therapeutic compounds used in accordance with the present invention may be encapsulated in the interior of the microbubbles/microparticles or attached to or incorporated into the structure thereof.
- the therapeutic compound may be linked to a part of the wall or matrix, for example through covalent or ionic bonds, or may be physically mixed into the encapsulating or matrix material, particularly if the drug has similar polarity or solubility to this material, so as to prevent it from leaking out of the product before it is intended to act in the body.
- the release of the drug may be initiated merely by wetting contact with blood following administration or as a consequence of other internal or external influences, e.g. dissolution processes catalyzed by enzymes or the use of ultrasound.
- the destruction of gas-containing microparticles using external ultrasound is a well known phenomenon in respect of ultrasound contrast agents, e.g. as described in WO-A-9325241; the rate of drug release may be varied depending on the type of therapeutic application, using a specific amount of ultrasound energy from the transducer.
- the therapeutic may be covalently linked to the membrane or matrix surface using a suitable linking agent, e.g. as described herein.
- a suitable linking agent e.g. as described herein.
- one may initially prepare a phospholipid or lipopeptide or derivative thereof to which the drug is bonded through a biodegradable bond or linker, and then incorporate this derivative into the material used to prepare the reporter, as described above.
- the product may initially be prepared without the therapeutic, which may then be coupled to or coated on the microbubbles or microparticles prior to use.
- a therapeutic could be added to a suspension of microbubbles or microparticles in aqueous media and shaken in order to attach or adhere the therapeutic thereto.
- Exemplary drug delivery systems suitable for use in the present compositions include any known therapeutic drugs or active analogues thereof containing thiol groups which are coupled to thiol containing microbubbles under oxidative conditions yielding disulphide bridges.
- the drug/vector modified microbubbles are allowed to accumulate in the target tissue.
- Administration of a reducing agent such as reduced glutathione then liberates the drug molecule from the targeted microbubble in the vicinity of the target cell increasing the local concentration of the drug and enhancing therapeutic effect.
- the product may also be prepared without the therapeutic if desired.
- the drug may then be coupled to or coated on the microbubbles prior to use.
- a therapeutic could be added to a suspension of microbubbles in aqueous media and shaken in order to attach or adhere the therapeutic to the microbubbles.
- Other drug delivery systems include vector modified phospholipid membranes doped with lipopeptide structures comprising a poly-L-lysine or poly-D-lysine chain in combination with a targeting vector.
- the microbubble carrier is condensed with DNA or RNA via elecrostatic interaction with the polycation. This method has the advantage that the vector or vectors used for targeted delivery are not directly attached to the polysine carrier moiety.
- the polylysine chain is also anchored more tightly in the microbubble membrane due to the presence of the lipid chains.
- the use of ultrasound to increase the effectiveness of delivery is also considered useful.
- free polylysine chains are firstly modified with drug or vector molecules then condensed onto the negative surface of targeted microbubbles.
- drugs useful in accordance with the invention include antineoplastic agents such as vincristine, vinblastine, vindesine, busulfan, chlorambucil , spiroplatin, cisplatin, carboplatin, methotrexate, adriamycin, mitomycin, bleomycin, cytosine arabinoside, arabinosyl adenine, mercaptopurine, mitotane, procarbazine, dactinomycin (antinomycin D) , daunorubicin, doxorubicin hydrochloride, taxol, plicamycin, aminoglutethimide, estramustine, flutamide, leuprolide, megestrol acetate, tamoxifen, testolactone, trilostane, amsacrine (m-AMSA) , asparaginase (L-asparaginase) , etoposide, interferon a- 2
- RNA RNA
- DNA of natural or synthetic origin, including recombinant RNA and DNA. DNA encoding certain proteins may be used in the treatment of many different types of diseases .
- tumor necrosis factor or interleukin-2 genes may be provided to treat advanced cancers; thymidine kinase genes may be provided to treat ovarian cancer or brain tumors; interleukin-2 genes may be provided to treat neuroblastoma, malignant melanoma or kidney cancer; and interleukin-4 genes may be provided to treat cancer.
- Lipophilic derivatives of drugs linked to the microbubble wall through hydrophobic interactions may exhibit therapeutic effects as part of the microbubble or after release from the microbubble, e.g. by use of ultrasound.
- a lipophilic group may be introduced for anchoring the drug to the membrane .
- the lipophilic group should be introduced in a way that does not influence the in vivo potency of the molecule, or the lipophilic group may be cleaved releasing the active drug.
- Lipophilic groups may be introduced by various chemical means depending on functional groups available in the drug molecule. Covalent coupling may be effected using functional groups in the drug molecule capable of reacting with appropriately functionalised lipophilic compounds.
- lipophilic moieties include branched and unbranched alkyl chains, cyclic compounds, aromatic residues and fused aromatic and non-aromatic cyclic systems. In some instances the lipophilic moiety will consist of a suitably functionalised steroid, like cholesterol and related compounds .
- functional groups particularly suitable for derivatisation include nucleophilic groups like amino, hydroxy and sulfhydryl groups.
- Suitable processes for lipophilic derivatisation of any drug containing a sulfhydryl group, like captopril may include direct alkylation, e.g. reaction with an alkyl halide under basic conditions and thiol ester formation by reaction with an activated carboxylic acid.
- derivatisation of any drug having carboxylic functions like atenolol and chlorambucil, include amide and ester formation by coupling of amines and alcohols, respectively, possesing requested physical properties.
- a preferred aspect is attachment of cholesterol to a therapeutic compound by forming a degradable ester bond.
- a preferred application of the present invention relates to angiogenesis, which is the formation of new blood vessels by branching from existing vessels.
- the primary stimulus for this process may be inadequate supply of nutrients and oxygen (hypoxia) to cells in a tissue.
- the cells may respond by secreting angiogenetic factors, of which there are many; one example is vascular endothelial growth factor. These factors initiate the secretion of proteolytic enzymes which break down the proteins of the basement membrane, as well as inhibitors which limit the action of these potentially harmful enzymes.
- the combined effect of loss of attachment and signals from the receptors for angiogenetic factors is to cause the endothelial cells to move, multiply, and rearrange themselves, and finally to synthetise a basement membrane around the new vessels .
- Tumors must initiate angiogenesis when they reach millimeter size in order to keep up their rate of growth.
- angiogenesis is accompanied by characteristic changes in the endothelial cells and their environment, this process is a promising target for therapeutic intervention.
- the transformations accompanying angiogenesis are also very promising for diagnosis, a preferred example being malignant disease, but the concept also shows great promise in inllammation and a variety of inflammation-related diseases. These factors are also involved in re-vascularisation of infarcted parts of the myocardium, which occurs if the stenosis is released within a short time.
- angiogenesis mav be detected by the majority of the imaging modalities in use in medicine. Contrast -enhanced ultrasound may possess additional advantages, the contrast medium being microspheres which are restricted to the interior of blood vessels. Even if the target antigens are found on many cell types, the microspheres will attach exclusively to endothelial cells.
- prodrugs may also be used in agents according to the invention.
- drugs may be derivatised to alter their physicochemical properties and to adapt them for inclusion into the reporter; such derivatised drugs may be regarded as prodrugs and are usually inactive until cleavage of the derivatising group regenerates the active form of the drug.
- Another alternative is to incorporate the prodrug, the prodrug-activating enzyme and the vector in the same microbubble in a system where the prodrug will only be activated after some external stimulus.
- a stimulus may, for example, be a tumour-specific protease as described above, or bursting of the bubbles by external ultrasound after the desired targeting has been achieved.
- Therapeutics may easily be delivered in accordance with the invention to diseased and necrotic areas including the heart and vasculature in general, and to the liver, spleen and kidneys and other regions such as the lymph system, body cavities or gastrointestinal system.
- Products according to the present invention may be used for targeted therapeutic delivery either in vivo or in vi tro .
- the products may be useful in in vi tro systems such as kits for diagnosis of different diseases or characterisation of different components in blood or tissue samples.
- Similar techniques to those used to attach certain blood components or cells to polymer particles (e . g. monodisperse magnetic particles) in vi tro to separate them from a sample may be used in the present invention, using the low density of the reporter units in agents of the present invention to effect separation of the gas-containing material by floatation and repeated washing.
- So-called zero-length linking agents which induce direct covalent joining of two reactive chemical groups without introducing additional linking material (e.g. as in amide bond formation induced using carbodiimides or enzymatically) may, if desired, be used in accordance with the invention, as may agents such as biotin/avidin systems which induce non-covalent reporter-vector linking and agents which induce hydrophobic or electrostatic interactions.
- the linking agent will comprise two or more reactive moieties, e.g. as described above, connected by a spacer element.
- a spacer element permits bifunctional linkers to react with specific functional groups within a molecule or between two different molecules, resulting in a bond between these two components and introducing extrinsic linker-derived material into the reporter- vector conjugate.
- the reactive moieties in a linking agent may be the same (homobifunctional agents) or different (heterobifunctional agents or, where several dissimilar reactive moieties are present, heteromultifunctional agents) , providing a diversity of potential reagents that may bring about covalent bonding between any chemical species, either intramolecularly or intermolecularly .
- extrinsic material introduced by the linking agent may have a critical bearing on the targeting ability and general stability of the ultimate product .
- labile linkages e.g. containing spacer arms which are biodegradable or chemically sensitive or which incorporate enzymatic cleavage sites.
- the spacer may include polymeric components, e.g. to act as surfactants and enhance bubble stability.
- the spacer may also contain reactive moieties, e.g. as described above to enhance surface crosslinking, or it may contain a tracer element such as a fluorescent probe, spin label or radioactive material.
- Contrast agents according to the present invention are therefore useful in all imaging modalities since contrast elements such as X-ray contrast agents, light imaging probes, spin labels or radioactive units may readily be incorporated in or attached to the reporter units .
- Spacer elements may typically consist of aliphatic chains which effectively separate the reactive moieties of the linker by distances of between 5 and 30 A. They may also comprise macromolecular structures such as poly (ethylene glycols). Such polymeric structures, hereinafter referred to as PEGs, are simple, neutral polyethers which have been given much attention in biotechnical and biomedical applications (see e.g. Milton Harris, J.
- PEGs Poly (ethylene glycol ) chemistry, biotechnical and biomedical applica tions" Plenum Press, New York, 1992
- PEGs are soluble in most solvents, including water, and are highly hydrated in aqueous environments, with two or three water molecules bound to each ethylene glycol segment; this has the effect of preventing adsorption either of other polymers or of proteins onto PEG-modified surfaces.
- PEGs are known to be nontoxic and not to harm active proteins or cells, whilst covalently linked PEGs are known to be non- immunogenic and non-antigenic .
- PEGs may readily be modified and bound to other molecules with only little effect on their chemistry. Their advantageous solubility and biological properties are apparent from the many possible uses of PEGs and copolymers thereof, including block copolymers such as PEG-polyurethanes and PEG-polypropylenes .
- Appropriate molecular weights for PEG spacers used in accordance with the invention may, for example, be between 120 Daltons and 20 kDaltons.
- the major mechanism for uptake of particles by the cells of the reticuloendothelial system (RES) is opsonisation by plasma proteins in blood; these mark foreign particles which are then taken up by the RES.
- the biological properties of PEG spacer elements used in accordance with the invention may serve to increase contrast agent circulation time in a similar manner to that observed for PEGylated liposomes (see e.g. Klibanov, A.L. et al . in FEBS Let ters (1990) 268, 235- 237 and Blume, G. and Cevc, G. in Biochim . Biophys . Acta (1990) 1029, 91-97).
- Increased coupling efficiency to areas of interest may also be achieved using antibodies bound to the terminii of PEG spacers (see e.g. Maruyama, K. et al . in Biochim . Biophys . Acta (1995) 1234, 74-80 and Hansen, C.B. et al . in Biochim . Biophys . Acta (1995) 1239, 133-144) .
- a PEG component as a stabiliser in conjunction with a vector or vectors or directly to the reporter in the same molecule where the PEG does not serve as a spacer .
- Other representative spacer elements include structural-type polysaccharides such as polygalacturonic acid, glycosaminoglycans, heparinoids, cellulose and marine polysaccharides such as alginates, chitosans and carrageenans ; storage-type polysaccharides such as starch, glycogen, dextran and aminodextrans ; polyamino acids and methyl and ethyl esters thereof, as in homo- and co-polymers of lysine, glutamic acid and aspartic acid; and polypeptides, oligonucleotides and oligosaccharides, which may or may not contain enzyme cleavage sites.
- structural-type polysaccharides such as polygalacturonic acid, glycosaminoglycans, heparinoids, cellulose and marine polysaccharides such as alginates, chitosans and carrageenans ; storage-type polysaccharides such as starch, glycogen, dextran and
- spacer elements may contain cleavable groups such as vicinal glycol, azo, sulfone, ester, thioester or disulphide groups .
- X and Z are selected from -0-, -S-, and -NR- (where R is hydrogen or an organic group) ; each Y is a carbonyl, thiocarbonyl, sulphonyl, phosphoryl or similar acid-forming group: m and n are each zero or 1; and R 1 and R 2 are each hydrogen, an organic group or a group -X.Y.(Z) m -, or together form a divalent organic group] may also be useful; as discussed in, for example, WO-A- 9217436 such groups are readily biodegraded in the presence of esterases, e.g. in vivo, but are stable in the absence of such enzymes. They may therefore advantageously be linked to therapeutic agents to permit slow release thereof .
- Poly[N- (2-hydroxyethyl)methacrylamides] are potentially useful spacer materials by virtue of their low degree of interaction with cells and tissues (see e.g. Volfova, I., Rihova, B. and V.R. and Vetvicka, P. in J. Bioact . Comp . Polymers (1992) 7, 175-190) .
- Work on a similar polymer consisting mainly of the closely related 2-hydroxypropyl derivative showed that it was endocytosed by the mononuclear phagocyte system only to a rather low extent (see Goddard, P., Williamson, I., Bron, J. , Hutchkinson, L.E., Nicholls, J. and Petrak, K. in J " . Bioct . Compa t . Polym . (1991) 6, 4-24.).
- poymeric spacer materials include : i) copolymers of methyl methacrylate with methacrylic acid; these may be erodible (see Lee, P.I. in Pharm. Res . (1993) 10, 980) and the carboxylate substituents may cause a higher degree of swelling than with neutral polymers ; ii) block copolymers of polymethacrylates with biodegradable polyesters (see e.g. San Roman, J. and Guillen-Garcia, P. in Bioma terials (1991) 12, 236-241); iii) cyanoacrylates , i.e.
- polymers of esters of 2- cyanoacrylic acid - these are biodegradable and have been used in the form of nanoparticles for selective drug delivery (see Forestier, F., Gerrier, P., Chaumard, C, Quero, A.M., Couvreur, P. and Labarre, C. in J " . Antimicrob . Chemoter. (1992) 30, 173-179) ; iv) polyvinyl alcohols, which are water-soluble and generally regarded as biocompatible (see e.g. Langer, R. in J. Control . Release (1991) 16, 53-60); v) copolymers of vinyl methyl ether with maleic anhydride, which have been stated to be bioerodible (see Finne, U.
- Dacron R which are non-degradable but highly biocompatible; ix) block copolymers comprising biodegradable segments of aliphatic hydroxyacid polymers (see e.g. Younes, H., Nataf, P.R., Cohn, D., Appelbaum, Y.J., Pizov, G. and Uretzky, G. in Bioma ter. Artif . Cells Artif . Organs (1988) 16, 705-719) , for instance in conjunction with polyurethanes (see Kobayashi , H., Hyon, S.H. and Ikada, Y. in "Water-curable and biodegradable prepolymers" - J". Biomed . Mater. Res .
- polyurethanes which are known to be well- tolerated in implants, and which may be combined with flexible "soft" segments, e.g. comprising poly(tetra methylene glycol), poly (propylene glycol) or poly (ethylene glycol)) and aromatic "hard” segments, e.g. comprising 4 , 4 ' -methylenebis (phenylene isocyanate) (see e.g. Ratner, B.D., Johnston, A.B. and Lenk, T.J. in J " . Biomed. Ma ter. Res : Applied Bioma terials (1987) 21, 59-90; Sa Da Costa, V. et al . in J.
- DPPE dipalmitoylphosphatidylethanolamine
- LC long chain Agents for protein modification
- Targetable agents include the following:
- Antibodies which can be used as vectors for a very wide range of targets, and which have advantageous properties such as very high specificity, high affinity (if desired) , the possiblity of modifying affinity according to need etc. Whether or not antibodies will be bioactive will depend on the specific vector/target combination. Both conventional and genetically engineered antibodies may be employed, the latter permitting engineering of antibodies to particular needs, e.g. as regards affinity and specificity. The use of human antibodies may be preferred to avoid possible immune reactions against the vector molecule.
- a further useful class of antibodies comprises so-called bi- and multi-specific antibodies, i.e. antibodies having specificity for two Or more different antigens in one antibody molecule.
- Such antibodies may, for example, be useful in promoting formation of bubble clusters and may also be used for various therapeutic purposes, e.g. for carrying toxic moieties to the target.
- Various aspects of bispecific antibodies are described by McGuinness, B.T. et al . in Na t . Biotechnol . (1996) 14, 1149-1154; by George, A.J. et al . in J " . Immunol . (1994) 152, 1802-1811; by Bonardi et al . in Cancer Res . (1993) 53, 3015-3021; and by French, R.R. et al . in Cancer Res . (1991) 51, 2353-2361.
- Non-bioactive binders of receptors for cell adhesion molecules, cytokines, growth factors or peptide hormones This category may include peptidic or non- peptidic non-bioactive vectors which will be neither agonists nor antagonists.
- Oligonucleotides and modified oligonucleotides which bind DNA or RNA through Watson-Crick or other types of base-pairing DNA is usually only present in extracelluar space as a consequence of cell damage, so that such oligonucleotides, which will usually be non- bioactive, may be useful in, for example, targeting of necrotic regions, which are associated with many different pathological conditions.
- Oligonucleotides may also be designed to bind to specific DNA- or RNA-binding proteins, for example transcription factors which are very often highly overexpressed or activated in tumour cells or in activated immune or endothelial cells.
- Combinatorial libraries may be used to select oligonucleotides which bind specifically to any possible target molecules (from the examples of proteins to caffeine) and which therefore may be employed as vectors for targeting.
- DNA-binding drugs may behave similarly to oligonuclotides , but may exhibit biological acitvity and/or toxic effects if taken up by cells.
- Protease substrates/inhibitors are involved in many pathological conditions. Many substrates/inhibitors are non-peptidic but, at least in the case of inhibitors, are often bioactive.
- Vector molecules may be generated from combinatorial libraries without necessarily knowing the exact molecular target, by functionally selecting (in vitro, ex vivo or in vivo) for molecules binding to the region/structure to be imaged.
- Various small molecules including bioactive compounds known to bind to biological receptors of various kinds. Such vectors or their targets may be used for generate non-bioactive compounds binding to the same targets.
- proteins or peptides which bind to glucosaminoglycan side chains eg heparan sulphate, including glucosoaminoglycan-binding portions of larger molecules, as binding to glucosoaminoglycans does not result in a biological response.
- Proteoglycans are not found on red blood cells, which eliminates undesirable adsorption to these cells.
- the following tables identify various vectors which may be targeted to particular types of targets and indicated areas of use for targetable diagnostic and/or therapeutic agents according to the invention which contain such vectors .
- the vectors stated are bioactive it is understood that either: (1) non-bioactive analogs are used; (2) the vectors are used in doses too low to generate a biological response; or (3) the vectors are used in combinations in such a way that the resulting diagnostic and/or therapeutic composition gives no biological response.
- Protein and peptide vectors - cell adhesion molecules etc Protein and peptide vectors - cell adhesion molecules etc .
- Vectors comprising cytokines/growth factors/peptide hormones and fracrments thereof
- Vectors comprising non-peptide agonists/antagonists or non-bioactive binders of receptors for cytokines/growth factors/peptide hormones/cell adhesion molecules
- Receptors comprising DNA-binding drugs
- Receptors comprising protease substrates
- Receptors comprising protease inhibitors
- targeted agents to bind specifically to cells expressing a target may be studied by microscopy and/or using a flow chamber containing immobilised cells, for example employing a population of cells expressing the target structure and a further population of cells not expressing the target.
- Radioactive or fluorescent enzyme- labelled streptavidin/avidin may used to analyse biotin attachment .
- Example 1 Gas-filled microbubbles encapsulated with phosphatidylserine , phosphatidylcholine and biotin- amidocaproate-PEG 3 ⁇ nn-Ala-cholesterol
- Ala-cholesterol is added to a solution of Boc-NH-PEG 3400 - SC (t-butyl carbamate poly (ethylene glycol) - succinimidyl carbonate) (Shearwater) in chloroform, followed by triethylamine .
- the suspension is stirred at 41 °C for 10 minutes.
- the crude product is purified by chromatography .
- Boc-NH-PEG 3400 -Ala-cholesterol is stirred in 4 M hydrochloric acid in dioxane for 2.5 hours at ambient temperature.
- the solvent is removed by rotary evaporation and the residue is taken up in chloroform and washed with water.
- the organic phase is rotary evaporated to dryness .
- the crude product may be purified by chromatography.
- Biotinamidocaproate-PEG 3400 -Ala- cholesterol dissolved in water is added the washed microbubbles, which are placed on a roller table for several hours. The washing procedure is repeated following incorporation of the biotinamidocaproate- PEG 3400 -Ala-cholesterol into the microbubble membranes.
- Example 2 Gas-containing microparticles comprising phosphatidylserine. phosphatidylcholine. biotin- amidocaproate-PEG 3 , 100 -Ala-Cholesterol and drug-cholesterol
- Cholesterol (4mmol) a drug having an acid group (see Example 2(b) for a list of cholesterol-derivatised drugs) and dimethylaminopyridine (4 mmol) are dissolved in dimethylformamide/tetrahydrofuran (20 ml + 5 ml) and dicyclohexylcarbodiimide is added. The reaction mixture is stirred at ambient temperature overnight. Dicyclohexylurea is filtered off and the solvent is rotary evaporated. The title compound is purified by chromatography .
- Biotin may be attached to microbubbles in many different ways, e.g. in a similar way to that described by Corley, P. and Loughrey, H.C. in (1994) Biochim . Biophys . Acta 1195, 149-156.
- the resulting bubbles are analysed by flow cytometry, e.g. by employing fluorescent streptavidin to detect attachment of biotin to the bubbles.
- radioactive or enzyme-labelled streptavidin/avidin is used to analyse biotin attachment .
- Example 4 -Gas- filled microbubbles encapsulated with 1.2-distearoyl-sn-Glycero-3- fPhospo-L-Serinel and biotin-DPPE
- Example 5 Gas-filled microbubbles encapsulated with phosphatidylserine and biotinylated oligonucleotide non- covalently bound to streptavidin-Succ-PEG-DSPE
- NH 2 -PEG 3400 -DSPE (prepared as in Preparation 1) is carboxylated using succinic anhydride, e.g. by a similar method to that described by Nayar, R. and Schroit, A.J. in Biochemis try (1985) 24, 5967-5971.
- Streptavidin is covalently bound to succ-PEG 3400 -DSPE in the microbubbles by standard coupling methods using a water-soluble carbodiimide.
- the sample is placed on a roller table during the reaction. After centrifugation the infranatant is exchanged with water and the washing is repeated.
- the functionality of the attached streptavidin is analysed by binding, e.g. to fluorescently labeled biotin, biotinylated antibodies (detected with a fluorescently labeled secondary antibody) or biotinylated and fluorescence- or radioactively-labeled oligonucleotides. Analysis is performed by fluorescence microscopy or scintillation counting.
- Microbubbles from (c) above are incubated in a solution containing a biotinylated oligonucleotide.
- the oligonucleotide-coated bubbles are washed as described above. Binding of the oligonucleotide to the bubbles is detected e.g. by using fluorescent-labeled oligonucleotides for attachment to the bubbles, or by hybridising the attached oligonucleotide to a labeled (fluorescence or radioactivity) complementary oligonucleotide.
- the functionality of the oligonucleotide-carrying microbubbles is analysed, e.g.
- an oligonucleotide complementary to ribosomal DNA (of which there are many copies per haploid genome) and an oligonucleotide complementary to an oncogene (e.g. ras of which there is one copy per haploid genome) are used.
- Example 6 The peptide FNFRLKAGOKIRFGAAAWEPPRARI attached to gas-filled microbubbles encapsulated with phosphatidylserine
- the peptide FNFRLKAGOKIRFGAAAWEPPRARI comprising phosphatidylserine-binding and heparin-binding sections, is synthesised.
- the peptide is added to preformed phosphatidylserine-encapsulated perfluorobutane microbubbles and thoroughly mixed.
- Example 7 Gas-filled microbubbles encapsulated with phosphatidylserine and inactivated human thrombin-Succ- PEG 400 ⁇ DSPE
- Human thrombin is inactivated by incubation with a 20 % molar excess of D-Phe-L-Pro-L-Arg-chloromethyl ketone in 0.05 M HEPES buffer, pH 8.0, at 37 °C for 30 minutes.
- Example 5(a) To a mixture (5 mg) of phosphatidylserine (90-99.9 mol%) and Succ-PEG 3400 -DSPE (10 - 0.1 mol%, prepared as in Example 5(a)) is added 5% propyleneglycol -glycerol in water (1 ml) . The dispersion is heated to not more than 80 °C for 5 minutes and is then cooled to ambient temperature. The dispersion (0.8 ml) is transferred to a vial (1 ml) and the head space is flushed with perfluorobutane . The vial is shaken in a cap-mixer for 45 seconds, whereafter the sample is put on a roller table. After centrifugation the infranatant is exchanged with water and the washing is repeated. Alternatively the microbubbles may be prepared as described in Preparation 1(f).
- Inactivated human thrombin is covalently bound to succ- PEG 3400 -DSPE in the microbubbles from (b) above by standard coupling methods using a water-soluble carbodiimide.
- the sample is placed on a roller table during the reaction. After centrifugation the infranatant is exchanged with water and the washing is repeated.
- Example 8 Gas-containing microparticles comprising polymer from ethylidene bis (16-hydroxyhexadecanoate) and adipoyl chloride and biotin-amidocaproate-Ala covalently attached to the polymer a) Synthesis of Z-Ala-polymer (3 -0- (carbobenzyloxy-L- alanyl) -polymer)
- the polymer is prepared from ethylidene bis (16- hydroxyhexadecanoate) and adipoyl chloride as described in WO-A- 9607434 , and a polymer fraction with molecular weight 10000 is purified using gel permeation chromatography (GPC) .
- GPC gel permeation chromatography
- 10 g of the material (corresponding to 1 mmol OH groups), Z-alanine (5 mmol) and dimethylaminopyridine (4 mmol) are dissolved in dry dimethylformamide/tetrahydrofuran and dicyclohexylcarbodiimide is then added.
- the reaction mixture is stirred at ambient temperature overnight.
- Dicyclohexylurea is filtered off and the solvent is removed using rotary evaporation.
- the product is purified by chromatography, fractions containing the ti tle compound are combined and the solvent is removed using rotary evaporation.
- the structure of the product is
- Z-Ala-polymer (0.1 mmol) is stirred in toluene/tetrahydrofuran and glacial acetic acid (15% of the total volume) and hydrogenated in the presence of 5 % palladium on charcoal for 2 hours. The reaction mixture is filtered and concentrated in vacuo .
- a solution of biotinamidocaproate N-hydroxysuccinimide ester in tetrahydrofuran is added to H 2 N-Ala-polymer dissolved in a mixture of tetrahydrofuran and dimethylformamide and 0.1 M sodium phosphate buffer having a pH of 7.5.
- the reaction mixture is heated to 30 °C and stirred vigorously; the reaction is followed by TLC to completion. The solvent is evaporated and the crude product is used without further purification.
- streptavidin conjugated to horseradish peroxidase was added to both suspensions and the tubes were rotated for 1 hour to allow for reaction.
- the microspheres were then washed three times, resuspended in 100 mM citrate-phosphate buffer (pH 5) containing 0.1 mg/ml phenylenediamine dihydrochloride and 0.01% hydrogen peroxide, and rotated for 10 minutes. Development of a yellow-green colour was indicative of the presence of enzyme. The following results were obtained:
- the agent from Example 7, comprising phosphatidylserine- encapsulated microbubbles having inactivated human thrombin-Succ-PEG 3400 -DSPE incorporated into the encapsulating membrane is lyophilised from 0.01 M phosphate buffer, pH 7.4.
- the product is redispersed in sterile water and injected intravenously into a patient with suspected venous thrombosis in a leg vein.
- the leg is examined by standard ultrasound techniques.
- the thrombus is located by increased contrast as compared with surrounding tissue.
- Example 11 Adhesion of poly-L-lysine-coated phosphatidylserine-encapsulated microbubbles to endothelial cells
- Poly-L-lysine (8 mg, Sigma P-1274 lot no. 14H5546) having a molecular weight of 115 kDa was dissolved in water (400 ⁇ l) .
- a cell adhesion study using human endothelial cells grown in culture dishes (Type CRL 1730) was performed with the above-described microbubbles, the uncoated microbubbles being used as a control. Microscopy of the endothelial cells after incubation showed a much increased number of poly-L-lysine-coated microbubbles adhering to endothelial cells in comparison to the uncoated microbubbles .
- Example 12 Preparation and biological evaluation of gas-containing microbubbles of DSPS 'doped' with a lipopeptide consisting of a heparin sulphate binding peptide (KRKR) and a fibronectin peptide fWOPPRARI) .
- KRKR heparin sulphate binding peptide
- fWOPPRARI fibronectin peptide
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- DSPS vanti, 4.5 mg
- lipopeptide from a 0.5 mg
- DSPS 0.8 mL
- a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial.
- the mixture was warmed to 80°C for 5 minutes (vials shaken during warming) .
- the samples were cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles rolled overnight.
- Bubbles were washed several times with deionised water and analysed by Coulter counter (Size: 1-3 micron (87 %), 3-5 micron (11.5%)) and acoustic attenuation (frequency max att . : 3.5 MHz) .
- the microbubbles were stable at 120 mm Hg .
- MALDI mass spectral analysis was used to confirm incorporation into DSPS microbubbles as follows; ca . 0.05-0.1 mL of microbubble suspension was transferred to a clean vial and 0.05-0.1 mL methanol added. The suspension was sonicated for 30 s and the solution analysed by MALDI MS. Positive mode gave M+H at 2200, expected for lipopeptide, 2198.
- the human endothelial cell line ECV 304 derived from a normal umbilical cord (ATCC CRL-1998) was cultured in 260 mL Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whittaker) to which L-Glutamine 200 mM,
- Penicillin/ Streptomycin (10.000 U/mL and 10.000 mcg/mL) and 10% Fetal Bovine Serum (Hyclone Lot no. AFE 5183) were added.
- the cells were subcultured with a split ratio of 1:5 to 1:7 when reaching confluence.
- Cover-glasses, 22mm in diameter were sterilised and placed on the bottom of 12 well culture plates (Costar) before cells in 0,5 mL complete medium with serum was added on top.
- the chamber consists of a groove carved into a glass plate upon which the cover slip with cells was placed with the cells facing the groove forming a flow channel.
- Ultrasound microbubbles from section b) were passed from a reservoir held at 37 degree Celsius through the flow chamber and back to the reservoir using a peristaltic pump. The flow rate was adjusted to simulate physiological relevant shear rates .
- the flow chamber was placed under a microscope and the interaction between the microspheres and cells viewed directly. A camera mounted on the microscope was connected to a colour video printer and a monitor.
- a 22 kg mongrel dog was anaesthetized with pentobarbital and mechanically ventilated.
- the chest was opened by a midline sternotomy, the anterior pericardium was removed, and a 30 mm gelled silicone rubber spacer was inserted between the heart and a P5-3 transducer of an ATL HDI-3000 ultrasound scanner.
- the scanner was set for intermittent short axis imaging once in each end-systole by delayed EGC triggering.
- a net volume of 2 mL of microbubbles from b) were injected as a rapid intravenous bolus. 3 seconds later, the imaged right ventricle was seen to contain contrast material, another 3 seconds later, the left ventricle was also filled, and a transient attenuation shadow that obscured the view of the posterior parts of the left ventricle was observed. A substantial increase in brightness of the myocardium was seen, also in the portions of the heart distal to the left ventricle when the attenuation shadow subsided. After passage of the inital bolus, the ultrasound scanner was set to continuous, high frame rate high output power imaging, a procedure known to cause destruction of utrasound contrast agent bubbles in the imaged tissue regions.
- the scanner was adjusted back to its initial setting.
- the myocardium was then darker, and closer to the baseline value. Moving the imaged slice to a new position resulted in re-appearance of contrast effects, moving the slice back to the initial position again resulted in a tissue brightness again close to baseline.
- a net volume of 2 mL microbubbles prepared in an identical manner to b) above with the exception that no lipopeptide was included in the preparation was injected, using the same imaging procedure as above.
- the myocardial echo enhancement was far less intense and of shorter duration than observed in case 1.
- At the completion of the left ventricular attenuation phase there was also almost complete loss of myocardial contrast effects, and a myocardial echo increases in the posterior part of the left ventricle as in case 1 was not observed.
- Example 13 Targeted gas-containing microbubbles of DSPS coated non-covalently with polylysine and a fusion peptide comprising a PS binding component and a Fibronectin peptide sequence
- the peptide was synthesised on an ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling. The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol , 5% EDT and 5% H 2 0 for 2 hours giving a crude product yield of 302 mg .
- DSPS (5 mg, Avanti) was weighed into a clean vial along with poly-L-lysine (Sigma, 0.2 mg) and peptide from a) above (0.2 mg) .
- To the vial was added 1.0 mL of a solution of 1.4% propylene glycol/ 2.4% glycerol. The mixture was warmed to 80°C for 5 minutes. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes . Following extensive washing with water, PBS and water the final solution was examined for polylysine and peptide content using MALDI MS.
- the spacer element contained within the PS binding/Fibronectin fusion peptide can also be replaced with other spacers such as PEG 200o or P°ly alanine (-AAA-) . It is also envisaged that a form of pre-targeting may be employed, whereby the DSPS binding/Fibronectin fragment fusion peptide is firstly allowed to associate with cells via the fibronectin peptide binding. This is followed by administration of PS microbubbles which then bind to the PS binding peptide .
- Example 14 Gas containing microbubbles of DSPS covalently modified with CD71 FITC-labelled anti- transferrin receptor antibody and 'doped' with a lipopeptide with affinity for endothelial cells.
- This example is directed at the preparation of multiple vector targeted ultrasound agents .
- the lipopeptide shown below was synthesised on a ABI 433A automatic peptide synthesiser starting with a Rink amide resin on a 0.1 mmol scale using 1 mmol ammo acid cartridges .
- DSPS vanti, 4.5 mg
- lipopeptide from a) 0.5 mg
- PE-PEG 2000 -Maleimide from example 2 0.5 mg
- the mixture was warmed to 80°C for 5 minutes then filtered through a 4.5 micron filter.
- the sample was cooled to room temperature and the head space flushed with perfluorobutane gas .
- the vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with distilled water.
- FITC labelled CD71 anti-transferrin receptor Ab 100 ⁇ g/ mL, Becton Dickinson
- 0.7 mL in PBS was modified with Traut's reagent (0.9 mg, Pierce) at room temperature for 1 h.
- Excess reagent was separated from modified protein on a NAP-5 column (Pharmacia) .
- Fig. 1 of the accompanying drawing represents the flow cytometric comparison of negative control microbubbles of DSPS (left curve) with bubbles conjugated with CD71 FITC- labelled anti-transferrin antibody (filled curve, right), showing that 92% of the population fluoresce. Incorporation into the microbubbles of lipopeptide was confirmed by MALDI mass spectrometry as described in example 12 b) .
- Example 15 Preparation of Transferrin/Avidin coated ⁇ a.- filled microbubbles for targeted ultrasound imaging.
- This example is directed to the preparation of microbubbles containing multiple protein vectors for targeted ultrasound/therapy.
- the lipid structure shown above was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc- Cys (Trt) -Wang resin (Novabiochem) on a 0.25 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU coupling chemistry .
- DSPS (Avanti, 4.5 mg) and the lipid structure from a) above (0.5 mg) were weighed into a clean vial and 0.8 mL of a solution containing 1.4% propylene glycol/ 2.4% glycerol in water added. The mixture was warmed to 80°C for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter. The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans Reagent.
- Example 16 Preparation of functionalised gas-filled microbubbles for targeted ultrasound imaging.
- This example is directed to the preparation of microbubbles having a reactive group on the surface for non-specific targeting, principally utilising disulphide exchange reactions to effect binding to a multiplicity of cellular targets.
- DSPS vanti, 5.0 mg
- thiol containing lipid structure from example 15 a 1.0 mg
- the mixture was warmed to 80°C for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter.
- the samples were cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans
- Example 17 Gas-containing microbubbles of DSPS comprising a lipopeptide for endothelial cell targeting and a captopril containing molecule. This example is directed to the preparation of ultrasound agents for combined targeting and therapeutic applications .
- the structure shown above was synthesised using a manual nitrogen bubbler apparatus starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale. All amino acids were purchased from Novabiochem and palmitic acid from Fluka. Coupling was carried out using standard TBTU/HOBt/DIEA protocols. Bromoacetic acid was coupled through the side-chain of Lys as a symmetrical anhydride using DIC preactivation . Captopril (Sigma) dissolved in DMF was introduced on the solid- phase using DBU as base.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Rink amide resin
- DSPS (Avanti, 4.5 mg) , product from a) (0.5 mg) and product from b) (0.5 mg) were weighed into a vial and 1.0 mL of a solution of 1.4% propylene glycol/ 2.4% glycerol was added to each vial. The mixture was warmed to 80°C for 5 minutes (vials shaken during warming) . The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were firstly shaken in a cap mixer for 45 s then rolled for 1 h followed by extensive washing with deionised water. No detectable levels of starting material was found in the final wash solution as evidenced by MALDI MS . MALDI mass spectral analysis was used to confirm incorporation of the products from section a) and b) into the microbubbles as described in example 12 b) .
- Example 18 Preparation of gas-containing microbubbles of DSPS loaded with a lipopeptide comprising a helical peptide with affinity for cell membranes and the peptide antibiotic polymixin B sulphate.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors having a combined targeting and a therapeutic application.
- DSPS (Avanti, 5.0 mg) , lipopeptide from a) (0.3 mg)and polymixin B sulphate (Sigma, 0.5 mg) was weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/ 2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80°C for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes.
- microbubbles were washed in water until no polymixin B sulphate or lipopeptide could be detected in the infranatant by MALDI-MS. Microscopy showed that the size distribution of the bubble population was between 1-8 micron as desired. To the washed bubbles (ca. 0.2 mL) was added methanol (0.5 mL) and the mixture placed in a sonic bath for 2 min. The resulting clear solution, following analysis by MALDI-MS, was found to contain both lipopeptide and polymixin B sulphate (expected 1203, found 1207).
- Example 19 Preparation of multiple-specific gas- containing microbubbles of DSPS 'doped' with a lipopeptide comprising a IL-1 receptor binding sequence and modified with a branched structure containing the drug methotrexate .
- This example is directed at the preparation of targeted microbubbles comprising a non-bioactive vector for targeting and a component for drug delivery.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ala-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- the methotrexate structure was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc- Cys(Trt) Tentagel resin on a 0.1 mmol scale.
- the simultaneous removal of product from the resin and deprotection of protecting groups was carried out in TFA containing 5% EDT and 5% H 2 0 for 2 hours giving a crude product yield of 160 mg .
- DSPS (Avanti, 4.5 mg) and thiol containing lipopeptide from example 15 a) (0.5 mg) and lipopeptide from a) (0.2 mg) above were weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/ 2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80°C for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes following which the infranatant was discarded.
- methotrexate structure from b) above (0.5 mg) was dissolved in PBS pH 8.0. The solution was then added to the thiol containing bubbles from c) and disulphide bond formation allowed to proceed for 16 h. Following extensive washing with PBS and water the bubbles were analysed by microscopy and MALDI MS.
- the disulphide bond linking the methotrexate structure to the microbubble may be reduced in vivo liberating the free drug molecule.
- This in combination with a tumour specific vector is a drug delivery system.
- a physiologically relevant reducing agent such as glutathione may be used to bring about drug release.
- Example 20 Preparation of microbubbles encapsulated with DSPS and unctionalised with a thrombi-tar ⁇ eting lipopeptide and the thrombolytic enzyme tissue plasminogen activator.
- This example is directed at the preparation of thrombus targeted US agents comprising a therapeutic thromolytic agent .
- the lipopeptide was synthesised on a ABI 433 A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All ammo acids and palmitic acid were preactivated using HBTU before coupling. The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT, 5% anisole and 5% H 2 0 for 2 h giving a crude product yield of 80 mg . Purification by preparative HPLC (Vydac 218TP1022 column) of a 20 mg aliquot of the crude material was carried out. After lyophilization 6 mg of pure material was obtained. The product was characterized by MALDI mass spectrometry and analytical HPLC.
- a solution of 0.1 mL of ammonim carbonate buffer containing 0.1 mg of t-PA (Sigma) was made up to 0.2 mL by the addition of water.
- To this solution was added 0.4 mg of Sulpho-SMPB (Pierce) dissolved in 0.05 mL DMSO.
- the protein solution was left standing at room temperature for 45 min then purification carried out on a Superdex 200 column. The product was eluted in PBS and the modified protein fraction collected.
- DSPS (Avanti, 5.0 mg) was weighed into a clean vial along with 0.5 mg of the lipopeptide from a) and 0.5 mg of the thiol containing lipopeptide from example 15 a) . To this was added 1.0 mL of a solution of 1.4% propylene glycol/ 2.4% glycerol and the mixture sonicated for 2 min then warmed to 80°C for 5 minutes. Immediately following warming the solution was filtered through a 4 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas . The vials were shaken in a cap mixer for 45 s and the microbubbles washed 2 times with deionised water.
- Example 21 Preparation of gas-containing microbubbles encapsuled with DSPS comprising thiolated anti-CD34-MAL-
- DSPS Advanti, 4,5 mg
- PE-PEG 2000 -maleimide from example x (0,5 mg) were weighed into a clean vial and 1 mL of a solution of 1.4% propylene glyco/2.4% glycerol added. The mixture was warmed to 80°C for 5 minutes then filtered through a 4.5 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorbutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with destilled water.
- 0.5mL of the thiolated antibody praparation from b) was added to an aliqout of microbubbles from a) and the conjugation reaction allowed to proceed for 30 min on a roller table. Following centifugation at 2000 rpm for 5 min the infranatant was removed. The microbubbles were washed a further three times with water.
- Example 22 Preparation of gas-containing microbubbles of DSPS loaded with a lipopeptide comprising a helical peptide with affinity for cell membranes
- This example is directed at the preparation of targeted microbubbles comprising a non-bioactive peptidic vector for targeting of cell membrane structures. a) Synthesis of a lipopeptide comprising a helical peptide with affinity for cell membranes
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.2 mmol scale using lmmol amino acid cartridges. All amino acids and 2-n-hexadecylstearic acid were preactivated using HBTU before coupling.
- the simultaneous removal of lipopeptide from the resin and side-chain protecting groups was carried out in TFA containing 5% H 2 0 for 2 hours giving a crude product yield of 520 mg.
- Purification by preparative HPLC (Vydac 218TP1022 column) of a 30 mg aliqout of crude material was carried out using a gradient of 90 to 100 % B over
- DSPS vanti, 4.5 mg and lipopeptide from a) (0.5 mg) was weighed into a clean vial and 1.0 ml of a solution of 1.4% propylene glycol/ 2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80°C for 5 minutes then filtered through a 4.5 mm filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes. The bubbles were washed in water until no lipopeptide could be detected by MALDI-MS.
- Example 23 Preparation of gas-containing microbubbles of DSPS loaded with a lipopeptide comprising a non- bioactive interleukin 1 receptor binding peptide
- This example is directed at the preparation of targeted microbubbles comprising a non-bioactive peptidic vector for targeting at the IL-1 recptor which does not induce signal tranduction or prevent IL-1 binding.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ala-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- the simultaneous removal of lipopeptide from the resin and side-chain protecting groups was carried out in TFA containing 5% H 2 0, 5% anisole, 5 % phenol and 5% EDT for 2 hours giving a crude product yield of 150 mg.
- DSPS vanti, 4.5 mg
- lipopeptide from a 0.5 mg
- the mixture was sonicated for 3-5 mins , warmed to 80°C for 5 minutes then filtered through a 4.5 micron filter.
- the mixture was cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes. The bubbles were washed in water until no lipopeptide could be detected by MALDI-MS. To the washed bubbles (ca.
- Example 24 Preparation of PFP containing microbuhhl p of DSPC. DSPS and endothelial cell binding lipopeptide for targeted ultrasound imaging.
- the headspace was again flushed with perfluorobutane and the sample was kept on a table roller until a homogenous appearance was obtained. The washing procedure was repeated. The resulting ultrasound contrast agent was confirmed by Coulter counter, acoustic attenuation measurements and resistance to external pressure. The microbubbles were tested in the in vitro assay as detailed in example 12. A gradual accumulation of bubbles binding to the cells was observed.
- Example 25 Preparation of SF f containing microbubbles of DSPC. DSPS and endothelial cell binding lipopeptide for targeted ultrasound imaging.
- DSPC DSPS (3:1) (5mg/ ml) in propyleneglycol/glycerol (4% in water) was added 0.5 mg of the lipopeptide from example 17 b) .
- the mixture was heated to 80°C for 5 minutes and shaken.
- the solution was then cooled to ambient temperature and the headspace was flushed with SF 6 gas.
- the vial was shaken on a Capmix (Espe Capmix) for 45 seconds and placed on a roller table for 5 min.
- the sample was centrifuged (Juan MR 14.11) at 2000 rpm for 5 minutes and the infranatant removed and replaced with destilled water.
- the headspace was again flushed with perfluorobutane and the sample was kept on a table roller until a homogenous appearance was obtained. The washing procedure was repeated. The resulting ultrasound contrast agent was confirmed by Coulter counter, acoustic attenuation measurements and resistance to external pressure.
- Example 26 Preparation of PFB containing microbubbles of DSPG and endothelial cell binding lipopeptide for targeted ultrasound imaging.
- Example 27 Preparation of PFP containing microbubbles of DSPG and endothelial cell binding lipopeptide for targeted ultrasound imaging.
- the headspace was again flushed with perfluorobutane and the sample was kept on a table roller until a homogenous appearance was obtained. The washing procedure was repeated. The resulting ultrasound contrast agent was confirmed by Coulter counter, acoustic attenuation measurements and resistance to external pressure. The microbubbles were tested in the in vitro assay as detailed in example 12. A gradual accumulation of bubbles binding to the cells was observed.
- Example 28 Preparation of SF 6 containing microbubbles of DSPG and endothelial cell binding lipopeptide for targeted ultrasound imaging.
- the headspace was again flushed with perfluorobutane and the sample was kept on a table roller until a homogenous appearance was obtained. The washing procedure was repeated. The resulting ultrasound contrast agent was confirmed by Coulter counter, acoustic attenuation measurements and resistance to external pressure.
- Example 29 Targeted gas -containing microbubbles of DSPS coated non-covalently with polylysine
- DSPS (5 mg, Avanti) was weighed into a clean vial along with poly-L-lysine (Sigma, 0.2 mg) .
- poly-L-lysine (Sigma, 0.2 mg) was added to the vial.
- To the vial was added 1.0 ml of a solution of 1.4% propylene glycol/ 2.4% glycerol.
- the mixture was warmed to 80°C for 5 minutes .
- the sample was cooled to room temperature and the head space flushed with perfluorobutane gas .
- the vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes.
- the lipopeptide was synthesised on a ABI 433 A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling. The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT, 5% anisole and 5% H 2 0 for 2 h giving a crude product yield of 80 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 20 mg aliquot of the crude material was carried out. After lyophilization 6 mg of pure material was obtained. The product was characterized by MALDI mass spectrometry and analytical HPLC.
- DSPS vanti, 4.5 mg
- lipopeptide from a 1.0 mg
- the mixture was warmed to 80°C for 5 minutes then the sample filtered through a 4 micron filter. After cooling to room temperature the head space was flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles washed extensively with deionised water. The bubbles were analysed by microscopy Coulter counter. MALDI-MS was used to confirm the presence of lipopeptide as previously described.
- Example 31 Gas-filled microbubbles of DSPS comprising a lipopeptide consisting of a heparin sulphate binding peptide (KRKR) and a fibronectin peptide (WOPPRARI) for targeting and a lipopeptide containing atenolol for therapeutic applications
- KRKR heparin sulphate binding peptide
- WOPPRARI fibronectin peptide
- the reaction mixture was poured onto water saturated with potassium hydrogensulphate and organic material was extracted into ethyl acetate. The organic phase was washed with water and brine, dried (Na 2 S0 4 ) and filtered to give 0.6 g of crude material.
- the product was purified by chromatography (silica, CHCl 3 /MeOH/AcOH 85:10:5). The solution was concentrated and the residue was taken up in glacial acetic acid and lyophilised. Yield 415 mg (56%) , white solid. The structure was confirmed by : H and 13 C NMR analysis.
- the structure shown above was synthesised using a manual nitrogen bubbler apparatus starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale, using amino acids from Novabiochem, palmitic acid from Fluka and the compound from a) . Coupling was carried out using standard TBTU/HOBt/DIEA protocols. Simultaneous removal of the peptide from the resin and deprotection of side-chain protecting groups was carried out in TFA containing 5% EDT and 5% water for 2h.
- a solution of 1.4% propylene glycol / 2.4% glycerol (1.0 ml) was added to a mixture of DSPD (Avanti, 5.0 mg) , product from Example 12 a) (0.5 mg) and product from b) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 °C for 5 min (vial was shaken during warming) .
- the solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45s followed by extensive washing with deionised water.
- Example 32 Gas-filled microbubbles encapsulated with DSPS and a compound containing folic acid for diagnostic applications
- a solution of 1.4% propylene glycol / 2.4% glycerol (1.0 ml) was added t a mixture of DSPS (Avanti, 4.5 mg) and product from a) (0.5 mg) in a vial.
- Dilute ammonia (to pH 8) and DMSO (40 ⁇ l) were added and the mixture was sonicated for 5 min and then heated at 80 °C for 5 min (vial was shaken during warming) .
- the solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45s followed by extensive washing with deionised water.
- microbubbles were tested in the in vitro assay as detailed in example 12. A gradual accumulation of bubbles binding to the cells was observed.
- Example 33 Gas-filled microbubbles of phosphatidylserine comprising biotinamide-PEG- ⁇ -Ala- Cholesterol and a cholesteryl ester of chlorambucil for diagnostic and therapeutic applications
- a solution of 1.4% propylene glycol / 2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5 mg) and product from c) (0.5 mg) and d) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 °C for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45s followed by extensive washing with deionised water.
- MALDI mass spectrometry showed no detectable level of compound from c and d) in the final wash solution.
- Example 34 Flotation of endothelial cells by micro bubbles with vectors that specifically bind to the endothelial cells
- the human endothelial cell line ECV 304 derived from a normal umbilical cord (ATCC CRL-1998) was cultures in Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whitaker) to which L-Glutamine 200 mM, Penicillin/Streptomycin (10.000 U/ml and 10.00 mcg/ml) and 10% Fetal Calf Serum (Hyclone Lot no AFE 5183) were added. The cells were subcultured following trypsination with a split ratio of 1:5 to 1:7 when reaching confluence. 2 mill . cells from trypsinated confluent cultures were added to each set of 5 centrifuge tubes .
- control microbubbles or bubbles carrying the vector including WQPPARI (example 12) , or the endothelial cell binding peptide vector (example 14) were added at 2, 4, 6 ,8 or 10 mill bubbles per tube.
- the cells at the bottom of the tubes after centrifugation at 400 g for 5 minutes were counted with a Coulter counter. It was found the 4 or more microbubbles binding to a cell did bring the cells to top of the fluid in the centrifugation tube. All cells were floated by the endothelial cell binding peptide vector and about 50 % with the WQPPARI vector.
- This example is directed at the preparation of targeted microbubbles for gene transfer.
- DSPS 4,5 mg
- lipopeptide from 17 b 0.5 mg
- DSPS lipopeptide from 17 b
- 0.8 ml propyleneglycol/glycerol 4%) in water was added.
- the solution was heated at 80°C for 5 minutes and shaken.
- the solution was then cooled to ambient temperature and the headspace flushed with perfluorobutane .
- the vials were shaken on a Capmix (Espe Capmix, 4450 oscillations/min) for 45 seconds and put on a roller table for 5 minutes.
- the content of the vials were mixed and the sample washed by centrifugation at 2000 rpm for 5 minutes.
- the infranatant was removed and the same volume of distilled water added.
- the washing procedure was repeated once.
- Poly-L-lysine HBr (Sigma, 20.6 mg) was dissolved in 2 mL water then an aliquot (0.4 mL) made up to 2 mL water. To 1.2 mL of the diluted poly-L-lysine solution was added 0.12 mL of the DSPS-lipopeptide bubble suspension. Following incubation excess polylysine was removed by extensive washing with water.
- Endothelial cells (ECV 304) were cultured in 6 well plates to a uniform subconfluent layer.
- a transfection mixture consisting of 5 ⁇ g DNA (an Enhanced Green Fluorescent Protein vector from CLONTECH) and 50 ⁇ l of microbubble suspension from a) in RPMI medium at a final volume of 250 ⁇ l was prepared. The mixture was left standing for 15 min at room temperature then 1 mL of complete RPMI medium added. The medium was removed from the cell culture dish, and the DNA-microbubble mixture added to the cells. The cells were incubated in a cell culture incubator (37 °C) .
Abstract
Description













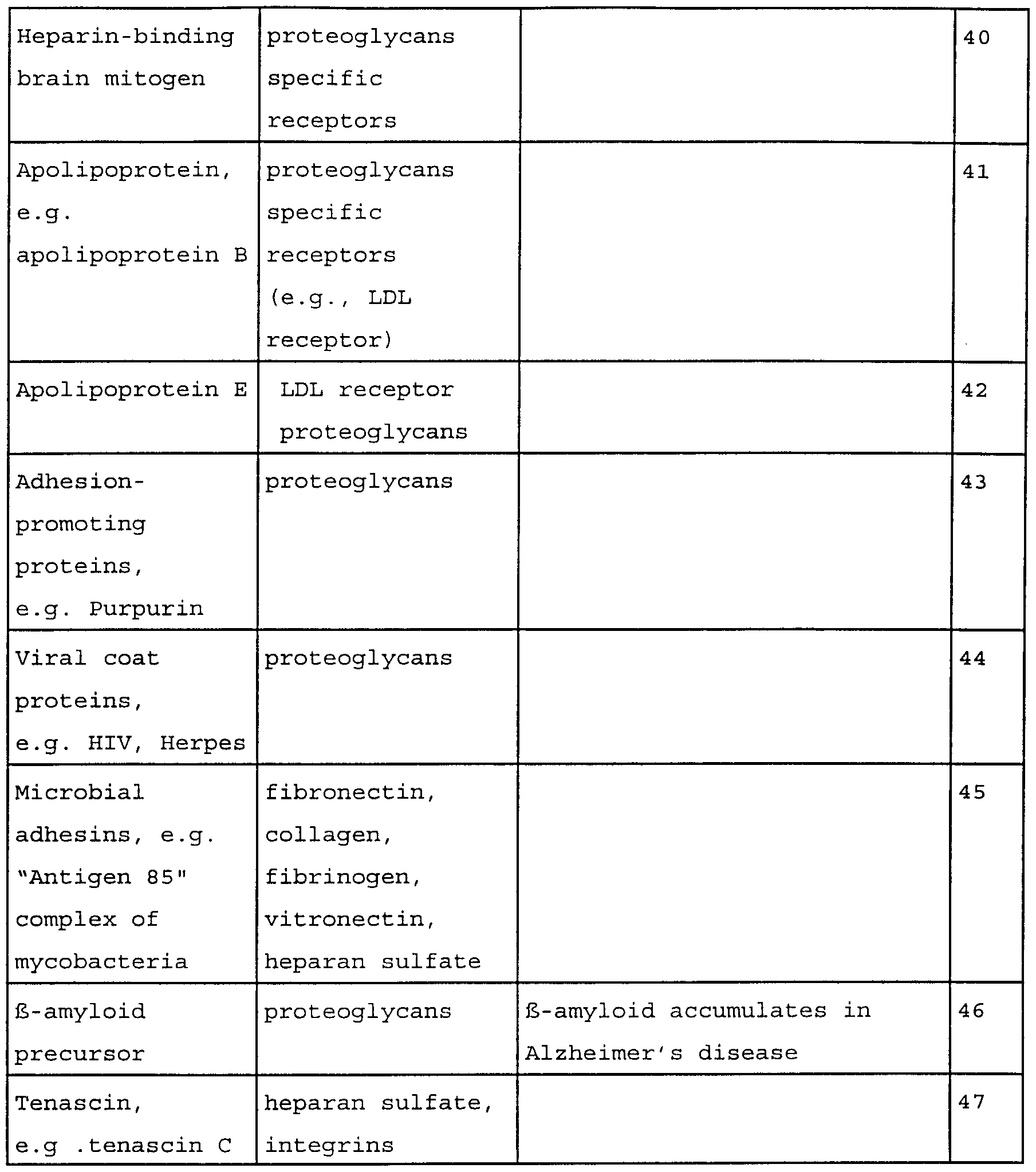



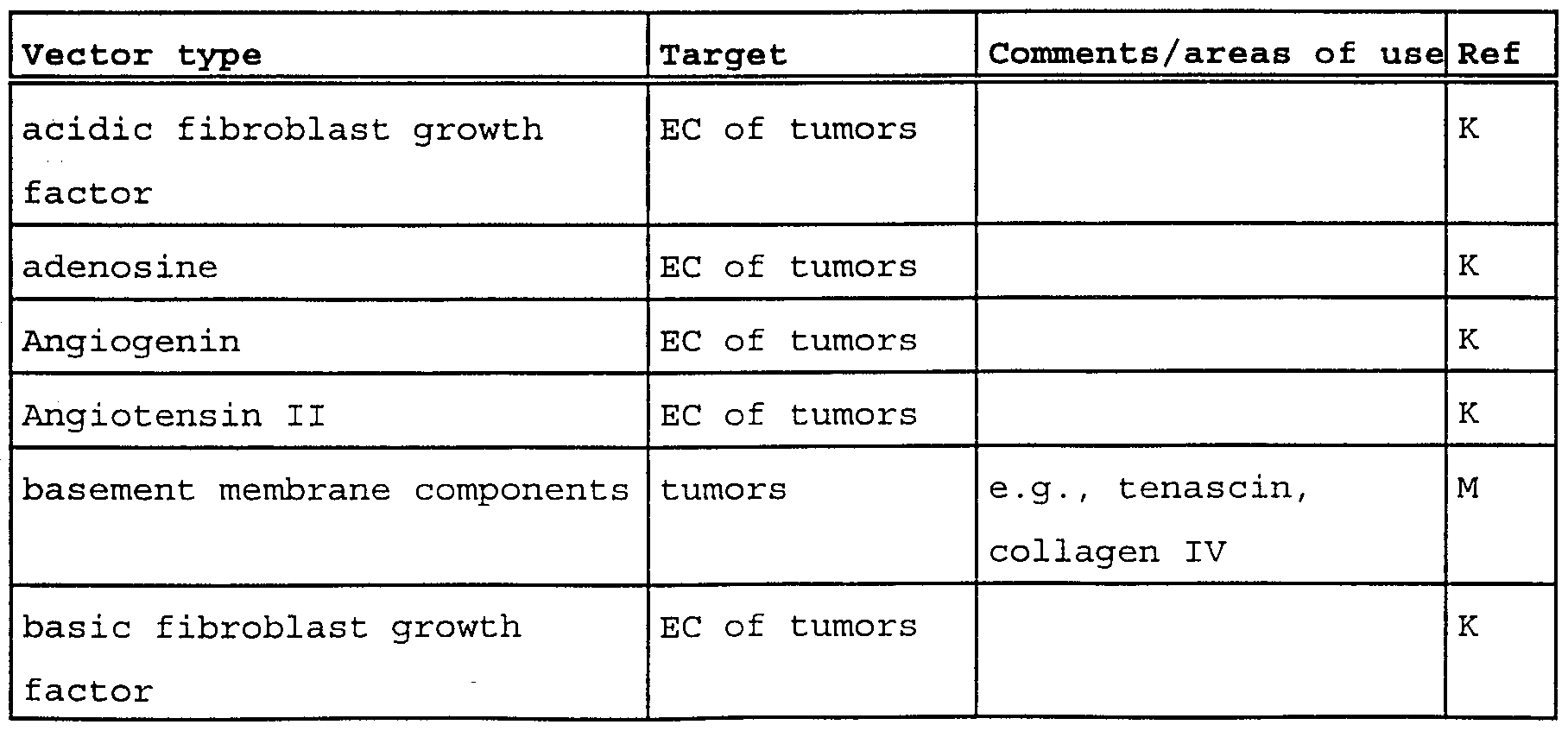



















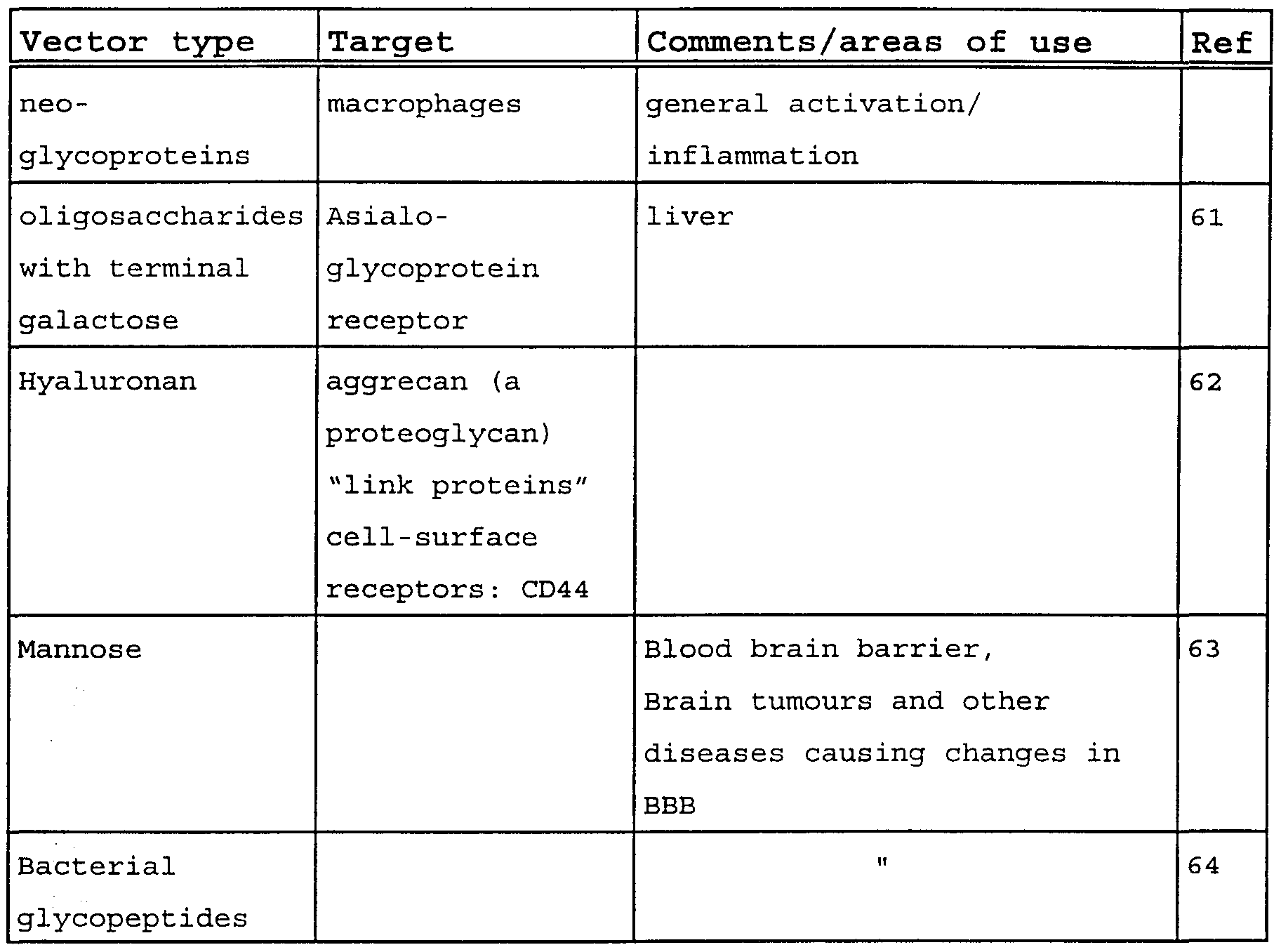












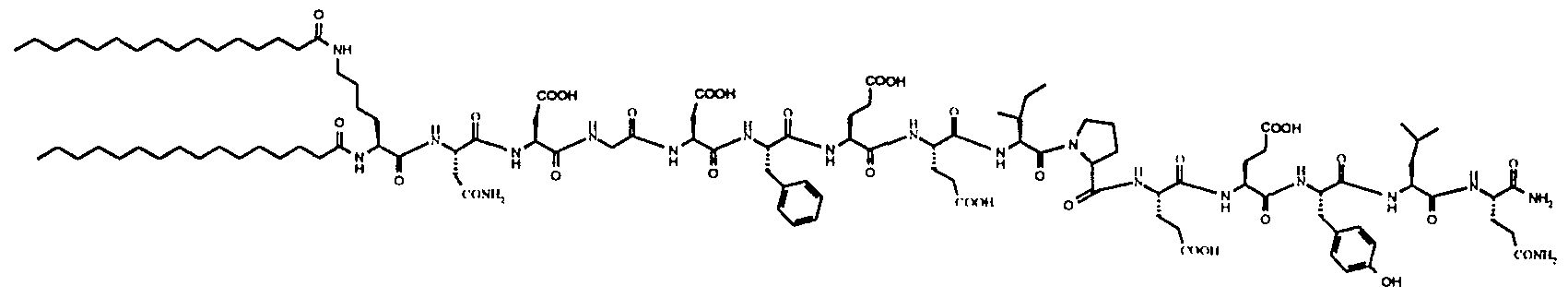


Claims
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU47870/97A AU4787097A (en) | 1996-10-28 | 1997-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| JP10520191A JP2001502719A (en) | 1996-10-28 | 1997-10-28 | Improved diagnostic / therapeutic agents |
| EP97910518A EP0963209A2 (en) | 1996-10-28 | 1997-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
Applications Claiming Priority (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9622364.9A GB9622364D0 (en) | 1996-10-28 | 1996-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| GBGB9622367.2A GB9622367D0 (en) | 1996-10-28 | 1996-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9622367.2 | 1996-10-28 | ||
| GBGB9622366.4A GB9622366D0 (en) | 1996-10-28 | 1996-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9622366.4 | 1996-10-28 | ||
| GB9622364.9 | 1996-10-28 | ||
| GB9700699.3 | 1997-01-15 | ||
| GBGB9700699.3A GB9700699D0 (en) | 1997-01-15 | 1997-01-15 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9700698.5 | 1997-01-15 | ||
| GBGB9700698.5A GB9700698D0 (en) | 1997-01-15 | 1997-01-15 | Improvements in or relating to diagnostic/therapeutics agents |
| GB9708265.5 | 1997-04-24 | ||
| GBGB9708265.5A GB9708265D0 (en) | 1997-04-24 | 1997-04-24 | Contrast agents |
| US4926597P | 1997-06-06 | 1997-06-06 | |
| US4926797P | 1997-06-06 | 1997-06-06 | |
| US4926497P | 1997-06-06 | 1997-06-06 | |
| GB9711844.2 | 1997-06-06 | ||
| GB9711842.6 | 1997-06-06 | ||
| GBGB9711844.2A GB9711844D0 (en) | 1997-06-06 | 1997-06-06 | Improvements in or relating to diagnostic/therapeutic agents |
| GBGB9711842.6A GB9711842D0 (en) | 1997-06-06 | 1997-06-06 | Improvements in or relating to diagnostic/therapeutic agents |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1998018498A2 true WO1998018498A2 (en) | 1998-05-07 |
| WO1998018498A3 WO1998018498A3 (en) | 1998-07-16 |
Family
ID=27581684
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB1997/002958 WO1998018498A2 (en) | 1996-10-28 | 1997-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP0963209A2 (en) |
| WO (1) | WO1998018498A2 (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999020312A1 (en) * | 1997-10-21 | 1999-04-29 | Nycomed Imaging As | Ultrasound imaging with contrast agent targeted to microvasculature and a vasodilator drug |
| WO1999053963A1 (en) * | 1998-04-22 | 1999-10-28 | Marsden, John, Christopher | Improvements in or relating to contrast agents |
| WO1999055383A2 (en) * | 1998-04-28 | 1999-11-04 | Nycomed Imaging As | Improvements in or relating to diagnostic/therapeutic agents |
| WO2000002588A1 (en) * | 1998-07-13 | 2000-01-20 | The Board Of Regents Of The University Of Nebraska | Targeted site specific drug delivery compositions and method of use |
| EP0977597A4 (en) * | 1996-09-11 | 2000-02-09 | Imarx Pharmaceutical Corp | Improved methods for diagnostic imaging using a contrast agent and a vasodilator |
| EP1017425A1 (en) * | 1996-09-11 | 2000-07-12 | IMARx PHARMACEUTICAL CORP. | Improved methods for diagnostic imaging involving the use of a contrast agent and a coronary vasodilator |
| WO2001017566A2 (en) * | 1999-09-08 | 2001-03-15 | Institut für Diagnostikforschung GmbH an der Freien Universität Berlin | L-selectin contrast agents |
| US6245747B1 (en) | 1996-03-12 | 2001-06-12 | The Board Of Regents Of The University Of Nebraska | Targeted site specific antisense oligodeoxynucleotide delivery method |
| US6548048B1 (en) | 1998-04-28 | 2003-04-15 | Amersham Health As | Lipopeptide stabilized microbubbles as diagnostic/therapeutic agents |
| WO2005044313A2 (en) * | 2003-11-06 | 2005-05-19 | Amersham Health As | Conjugates of angiotensin ii and an imaging moiety |
| US6984373B2 (en) | 2000-12-23 | 2006-01-10 | Dyax Corp. | Fibrin binding moieties useful as imaging agents |
| US7109167B2 (en) | 2000-06-02 | 2006-09-19 | Bracco International B.V. | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| EP1944312A1 (en) | 2003-03-03 | 2008-07-16 | Dyax Corporation | Peptides that specifically bind HGF receptor (CMET) and uses thereof |
| EP2014310A2 (en) | 2002-03-01 | 2009-01-14 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| EP2147684A1 (en) | 2008-07-22 | 2010-01-27 | Bracco Imaging S.p.A | Diagnostic Agents Selective Against Metalloproteases |
| EP2281580A2 (en) | 2003-01-13 | 2011-02-09 | Bracco Imaging S.p.A | Labeled gastrin releasing peptides (GRP) |
| EP2359864A2 (en) | 2005-12-09 | 2011-08-24 | Bracco Suisse SA | Targeting Vector-Phospholipid Conjugates |
| US8263739B2 (en) | 2000-06-02 | 2012-09-11 | Bracco Suisse Sa | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| US8642010B2 (en) | 2002-03-01 | 2014-02-04 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9056138B2 (en) | 2002-03-01 | 2015-06-16 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US9545457B2 (en) | 1998-01-14 | 2017-01-17 | Lantheus Medical Imaging, Inc. | Preparation of a lipid blend and a phospholipid suspension containing the lipid blend |
| US9789210B1 (en) | 2016-07-06 | 2017-10-17 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US10022460B2 (en) | 2014-12-31 | 2018-07-17 | Lantheus Medical Imaging, Inc. | Lipid-encapsulated gas microsphere compositions and related methods |
| US10471163B2 (en) | 2013-09-13 | 2019-11-12 | The General Hospital Corporation | Activatable fibrin-binding probes |
| US10588988B2 (en) | 2016-05-04 | 2020-03-17 | Lantheus Medical Imaging, Inc. | Methods and devices for preparation of ultrasound contrast agents |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8623822B2 (en) | 2002-03-01 | 2014-01-07 | Bracco Suisse Sa | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US7794693B2 (en) | 2002-03-01 | 2010-09-14 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4232755A1 (en) * | 1992-09-26 | 1994-03-31 | Schering Ag | Microparticle preparations made from biodegradable copolymers |
| WO1994028873A1 (en) * | 1993-06-11 | 1994-12-22 | Unger Evan C | Novel therapeutic drug delivery systems |
| WO1995003357A1 (en) * | 1993-07-23 | 1995-02-02 | Massachusetts Institute Of Technology | Biodegradable particles |
| WO1995003356A1 (en) * | 1993-07-23 | 1995-02-02 | Massachusetts Institute Of Technology | Nanoparticles and microparticles of non-linear hydrophilic-hydrophobic multiblock copolymers |
| WO1995007072A2 (en) * | 1993-09-09 | 1995-03-16 | Schering Aktiengesellschaft | Active principles and gas containing microparticles |
| WO1995015118A1 (en) * | 1993-11-30 | 1995-06-08 | Imarx Pharmaceutical Corp. | Gas microspheres for topical and subcutaneous application |
| US5534241A (en) * | 1993-07-23 | 1996-07-09 | Torchilin; Vladimir P. | Amphipathic polychelating compounds and methods of use |
| EP0727225A2 (en) * | 1995-02-14 | 1996-08-21 | Sonus Pharmaceuticals, Inc. | Compositions and methods for directed ultrasound imaging |
| WO1996040277A2 (en) * | 1995-06-07 | 1996-12-19 | Brown University Research Foundation | Spray dried polymeric microparticles containing imaging agents |
| WO1996041647A1 (en) * | 1995-06-08 | 1996-12-27 | Barnes-Jewish Hospital D/B/A | Site specific binding system, imaging compositions and methods |
| WO1998000172A2 (en) * | 1996-06-28 | 1998-01-08 | Board Of Regents Of The University Of Nebraska | Compositions and methods for altering the biodistribution of biological agents |
-
1997
- 1997-10-28 WO PCT/GB1997/002958 patent/WO1998018498A2/en not_active Application Discontinuation
- 1997-10-28 EP EP97910518A patent/EP0963209A2/en not_active Withdrawn
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4232755A1 (en) * | 1992-09-26 | 1994-03-31 | Schering Ag | Microparticle preparations made from biodegradable copolymers |
| WO1994028873A1 (en) * | 1993-06-11 | 1994-12-22 | Unger Evan C | Novel therapeutic drug delivery systems |
| WO1995003357A1 (en) * | 1993-07-23 | 1995-02-02 | Massachusetts Institute Of Technology | Biodegradable particles |
| WO1995003356A1 (en) * | 1993-07-23 | 1995-02-02 | Massachusetts Institute Of Technology | Nanoparticles and microparticles of non-linear hydrophilic-hydrophobic multiblock copolymers |
| US5534241A (en) * | 1993-07-23 | 1996-07-09 | Torchilin; Vladimir P. | Amphipathic polychelating compounds and methods of use |
| WO1995007072A2 (en) * | 1993-09-09 | 1995-03-16 | Schering Aktiengesellschaft | Active principles and gas containing microparticles |
| WO1995015118A1 (en) * | 1993-11-30 | 1995-06-08 | Imarx Pharmaceutical Corp. | Gas microspheres for topical and subcutaneous application |
| EP0727225A2 (en) * | 1995-02-14 | 1996-08-21 | Sonus Pharmaceuticals, Inc. | Compositions and methods for directed ultrasound imaging |
| WO1996040277A2 (en) * | 1995-06-07 | 1996-12-19 | Brown University Research Foundation | Spray dried polymeric microparticles containing imaging agents |
| WO1996041647A1 (en) * | 1995-06-08 | 1996-12-27 | Barnes-Jewish Hospital D/B/A | Site specific binding system, imaging compositions and methods |
| WO1998000172A2 (en) * | 1996-06-28 | 1998-01-08 | Board Of Regents Of The University Of Nebraska | Compositions and methods for altering the biodistribution of biological agents |
Non-Patent Citations (2)
| Title |
|---|
| A.L. KLIBANOV ET AL.: "TARGETING OF ULTRASOUND CONTRAST MATERIAL. AN IN VITOR FEASIBILTY STUDY." ACTA RADIOLOGICA, vol. 38, no. 412, 1997, page 113-120 XP002063479 * |
| V.R. MUZYKANTOV: "IMMUNOTARGETING OF STREPTAVIDIN TO THE PULMONARY ENDOTHELIUM" JOURNAL OF NUCLEAR MEDICINE, vol. 35, no. 8, August 1994, NEW YORK US, pages 1358-1365, XP002063478 * |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6245747B1 (en) | 1996-03-12 | 2001-06-12 | The Board Of Regents Of The University Of Nebraska | Targeted site specific antisense oligodeoxynucleotide delivery method |
| EP1323434A3 (en) * | 1996-09-11 | 2004-03-24 | Imarx Pharmaceutical Corp. | Improved methods for diagnostic imaging using a contrast agent and a vasodilator |
| EP1323434A2 (en) * | 1996-09-11 | 2003-07-02 | Imarx Pharmaceutical Corp. | Improved methods for diagnostic imaging using a contrast agent and a vasodilator |
| EP1017425A4 (en) * | 1996-09-11 | 2002-03-06 | Imarx Pharmaceutical Corp | Improved methods for diagnostic imaging involving the use of a contrast agent and a coronary vasodilator |
| EP0977597A4 (en) * | 1996-09-11 | 2000-02-09 | Imarx Pharmaceutical Corp | Improved methods for diagnostic imaging using a contrast agent and a vasodilator |
| EP0977597A1 (en) * | 1996-09-11 | 2000-02-09 | Imarx Pharmaceutical Corp. | Improved methods for diagnostic imaging using a contrast agent and a vasodilator |
| EP1017425A1 (en) * | 1996-09-11 | 2000-07-12 | IMARx PHARMACEUTICAL CORP. | Improved methods for diagnostic imaging involving the use of a contrast agent and a coronary vasodilator |
| WO1999020312A1 (en) * | 1997-10-21 | 1999-04-29 | Nycomed Imaging As | Ultrasound imaging with contrast agent targeted to microvasculature and a vasodilator drug |
| US6811766B1 (en) | 1997-10-21 | 2004-11-02 | Amersham Health As | Ultrasound imaging with contrast agent targeted to microvasculature and a vasodilator drug |
| US9545457B2 (en) | 1998-01-14 | 2017-01-17 | Lantheus Medical Imaging, Inc. | Preparation of a lipid blend and a phospholipid suspension containing the lipid blend |
| WO1999053963A1 (en) * | 1998-04-22 | 1999-10-28 | Marsden, John, Christopher | Improvements in or relating to contrast agents |
| WO1999055383A2 (en) * | 1998-04-28 | 1999-11-04 | Nycomed Imaging As | Improvements in or relating to diagnostic/therapeutic agents |
| US6548048B1 (en) | 1998-04-28 | 2003-04-15 | Amersham Health As | Lipopeptide stabilized microbubbles as diagnostic/therapeutic agents |
| AU763191B2 (en) * | 1998-04-28 | 2003-07-17 | Amersham Health As | Improvements in or relating to diagnostic/therapeutic agents |
| WO1999055383A3 (en) * | 1998-04-28 | 2000-07-06 | Nycomed Imaging As | Improvements in or relating to diagnostic/therapeutic agents |
| WO2000002588A1 (en) * | 1998-07-13 | 2000-01-20 | The Board Of Regents Of The University Of Nebraska | Targeted site specific drug delivery compositions and method of use |
| WO2001017566A2 (en) * | 1999-09-08 | 2001-03-15 | Institut für Diagnostikforschung GmbH an der Freien Universität Berlin | L-selectin contrast agents |
| WO2001017566A3 (en) * | 1999-09-08 | 2001-12-20 | Diagnostikforschung Inst | L-selectin contrast agents |
| US8263739B2 (en) | 2000-06-02 | 2012-09-11 | Bracco Suisse Sa | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| US7109167B2 (en) | 2000-06-02 | 2006-09-19 | Bracco International B.V. | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| US7820621B2 (en) | 2000-06-02 | 2010-10-26 | Bracco International B.V. | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| US7884183B2 (en) | 2000-06-02 | 2011-02-08 | Bracco Suisse Sa | Compounds for targeting endothelial cells, compositions containing the same and methods for their use |
| US6984373B2 (en) | 2000-12-23 | 2006-01-10 | Dyax Corp. | Fibrin binding moieties useful as imaging agents |
| EP2301587A2 (en) | 2002-03-01 | 2011-03-30 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9629934B2 (en) | 2002-03-01 | 2017-04-25 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9056138B2 (en) | 2002-03-01 | 2015-06-16 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| EP2014310A2 (en) | 2002-03-01 | 2009-01-14 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US8642010B2 (en) | 2002-03-01 | 2014-02-04 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| EP2281580A2 (en) | 2003-01-13 | 2011-02-09 | Bracco Imaging S.p.A | Labeled gastrin releasing peptides (GRP) |
| EP2500040A1 (en) | 2003-01-13 | 2012-09-19 | Bracco Imaging S.p.A | Improved Gastrin Releasing Peptide Receptor-Binding Compounds |
| US9000124B2 (en) | 2003-03-03 | 2015-04-07 | Dyax Corp. | Peptides that specifically bind HGF receptor (cMet) and uses thereof |
| US9845340B2 (en) | 2003-03-03 | 2017-12-19 | Dyax Corp. | Peptides that specifically bind HGF receptor (cMet) and uses thereof |
| EP2949658A2 (en) | 2003-03-03 | 2015-12-02 | Dyax Corp. | Peptides that specifically bind HGF receptor (cMet) and uses thereof |
| US8044175B2 (en) | 2003-03-03 | 2011-10-25 | Dyax Corp. | Peptides that specifically bind HGF receptor (CMET) and uses thereof |
| EP1944312A1 (en) | 2003-03-03 | 2008-07-16 | Dyax Corporation | Peptides that specifically bind HGF receptor (CMET) and uses thereof |
| US7785566B2 (en) | 2003-11-06 | 2010-08-31 | Ge Healthcare, Inc. | Pharmaceutical compounds |
| WO2005044313A3 (en) * | 2003-11-06 | 2006-05-11 | Amersham Health As | Conjugates of angiotensin ii and an imaging moiety |
| WO2005044313A2 (en) * | 2003-11-06 | 2005-05-19 | Amersham Health As | Conjugates of angiotensin ii and an imaging moiety |
| JP2007510643A (en) * | 2003-11-06 | 2007-04-26 | ジーイー・ヘルスケア・アクスイェ・セルスカプ | Pharmaceutical compounds |
| EP2359864A2 (en) | 2005-12-09 | 2011-08-24 | Bracco Suisse SA | Targeting Vector-Phospholipid Conjugates |
| EP2147684A1 (en) | 2008-07-22 | 2010-01-27 | Bracco Imaging S.p.A | Diagnostic Agents Selective Against Metalloproteases |
| US10471163B2 (en) | 2013-09-13 | 2019-11-12 | The General Hospital Corporation | Activatable fibrin-binding probes |
| US11395856B2 (en) | 2014-12-31 | 2022-07-26 | Lantheus Medical Imaging, Inc. | Lipid-encapsulated gas microsphere compositions and related methods |
| US10022460B2 (en) | 2014-12-31 | 2018-07-17 | Lantheus Medical Imaging, Inc. | Lipid-encapsulated gas microsphere compositions and related methods |
| US10583207B2 (en) | 2014-12-31 | 2020-03-10 | Lantheus Medical Imaging, Inc. | Lipid-encapsulated gas microsphere compositions and related methods |
| US10588988B2 (en) | 2016-05-04 | 2020-03-17 | Lantheus Medical Imaging, Inc. | Methods and devices for preparation of ultrasound contrast agents |
| US10220104B2 (en) | 2016-07-06 | 2019-03-05 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US10583208B2 (en) | 2016-07-06 | 2020-03-10 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US9913919B2 (en) | 2016-07-06 | 2018-03-13 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11266750B2 (en) | 2016-07-06 | 2022-03-08 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11266749B2 (en) | 2016-07-06 | 2022-03-08 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11344636B2 (en) | 2016-07-06 | 2022-05-31 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US9789210B1 (en) | 2016-07-06 | 2017-10-17 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11529431B2 (en) | 2016-07-06 | 2022-12-20 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11857646B2 (en) | 2016-07-06 | 2024-01-02 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
| US11925695B2 (en) | 2016-07-06 | 2024-03-12 | Lantheus Medical Imaging, Inc. | Methods for making ultrasound contrast agents |
Also Published As
| Publication number | Publication date |
|---|---|
| WO1998018498A3 (en) | 1998-07-16 |
| EP0963209A2 (en) | 1999-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU733477B2 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| AU733495B2 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| EP0973552B1 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| EP0963209A2 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| WO1998018495A2 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| US7109167B2 (en) | Compounds for targeting endothelial cells, compositions containing the same and methods for their use | |
| AU777304B2 (en) | Novel methods of imaging and treatment with targeted compositions | |
| US8257683B2 (en) | Multicomponent assemblies having enhanced binding properties for diagnosis and therapy | |
| JP4215820B2 (en) | Novel targeted compositions for diagnostic and therapeutic uses | |
| US8263739B2 (en) | Compounds for targeting endothelial cells, compositions containing the same and methods for their use | |
| US20090162293A1 (en) | Methods and compositions for ultrasound imaging of apoptosis | |
| MXPA99003867A (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| JP2001502719A (en) | Improved diagnostic / therapeutic agents | |
| JP2002515889A (en) | Improved diagnostic / therapeutic agents | |
| CZ149599A3 (en) | Diagnostic and/or therapeutical preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US US US UZ VN YU ZW AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| CFP | Corrected version of a pamphlet front page |
Free format text: ADD INID NUMBER (63) "RELATED BY CONTINUATION (CON) OR CONTINUATION-IN-PART (CIP) TO EARLIER APPLICATION" WHICH WAS INADVERTENTLY OMITTED FROM THE FRONT PAGE |
|
| ENP | Entry into the national phase in: |
Ref country code: JP Ref document number: 1998 520191 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997910518 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997910518 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997910518 Country of ref document: EP |
|
| NENP | Non-entry into the national phase in: |
Ref country code: CA |