WO1992020808A1 - Genomic modifications with homologous dna targeting - Google Patents
Genomic modifications with homologous dna targeting Download PDFInfo
- Publication number
- WO1992020808A1 WO1992020808A1 PCT/US1992/004054 US9204054W WO9220808A1 WO 1992020808 A1 WO1992020808 A1 WO 1992020808A1 US 9204054 W US9204054 W US 9204054W WO 9220808 A1 WO9220808 A1 WO 9220808A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- cells
- marker
- sequence
- indigenous
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/795—Porphyrin- or corrin-ring-containing peptides
- C07K14/805—Haemoglobins; Myoglobins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/15—Vector systems having a special element relevant for transcription chimeric enhancer/promoter combination
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/60—Vector systems having a special element relevant for transcription from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/80—Vector systems having a special element relevant for transcription from vertebrates
- C12N2830/85—Vector systems having a special element relevant for transcription from vertebrates mammalian
Definitions
- the field of this invention is genomic modification using homologous DNA for targeting.
- genetic disorders involving hematopoietic cells are such defects as sickle cell anemia, 3-thalassemia, various hemoglobinopathies, and disorders of erythrocyte metabolism including hereditary spherocytosis, pyruvate kinase deficiency, G6PD deficiency, etc.
- genetic disorders involving circulating plasma proteins and enzymes are inherited disorders of the complement system such as hereditary angioneurotic edema, agammaglobulinemia syndromes, ⁇ .-antitrypsin deficiency and disorders of hemostasis, such as hemophilia A, hemophilia B and Von Willebrand's disease.
- genetic disorders involving connective tissue, bone and muscle are the muscular dystrophies, the mucopolysaccharidosis syndromes, amyloidosis and various disorders of calcium and phosphate metabolisms including hypophospha asia, rickets and pseudo- hypoparathyroidism.
- disorders of metabolism are inherited disorders of amino acid metabolism, such as phenylketonuria, homocystinuria, albinism and tyrosinosis; inherited disorders of carbohydrate metabolism such as the glycogen storage diseases, diabetic syndromes and galactosemia; inherited disorders of lipid metabolism such as the hyperlipoproteinemias, the hyperlipidemias, the lipoprotein deficiency syndromes, the gangliosidoses, including Tay-Sach's disease, the lipidoses including Fabry's disease, Gaucher's disease, Refsum's disease, and Neimann-Pick disease; inherited disorders of steroid metabolism, such as adrenal hyperplasia; inherited disorders of purine and pyrimidine metabolism such as gout, Lesch- Nyhan Syndrome, and xanthinuria; and other metabolic disorders such as Wilson's disease, the porphyria syndromes, and hemochromatosis.
- amino acid metabolism such as phenylketonuria, homo
- disorders involving membrane transport of substances in the kidney, lung and other organs are the malabsorption syndromes, cystinuria, the renal tubular acidoses, cystinosis, Fanconi's syndrome and cystic fibrosis.
- disorders involving a genetic predisposition based on MHC antigen haplotype which may be potentially addressable by gene therapy are multiple sclerosis, ankylosing spondylitis, juvenile diabetes, rheumatoid arthritis and other autoimmune disorders.
- target cells for targeting for gene therapy or gene modification.
- embryonal stem cells somatic cells, hematopoietic stem cells, other stem cells, cells of connective tissue origin, including myoblasts, osteoblasts, or chondroblasts; hepatic cells, endothelial cells, neural cells, epitithelial cells, and cells of endocrine origin, including islet cells, or the like.
- the techniques and methodology used for modifying the genotype of the target cells require that the modification provide the desired function. While one may rely upon random integration and selection of the clones of the integrants, selecting for function may not be sufficient.
- Random integration may result in a variety of situations, where the integrated DNA may be subject to regulation, depending upon the site at which it is integrated, where the functioning gene may become inactivated upon differentiation or proliferation of the cells, where the site of integration may result in a change in the functioning of one or more indigenous genes, or where the site of integration may lead to neoplasia.
- Valancius and Smithies Mol. Cell. Biol. (1991) 11:1402-1408 describe an in-out targeting procedure for making genomic modifications in mouse embryonic stem cells.
- Hasty et al., Nature (1991) 350:243-246 describe the introduction of a mutation into the HOX-2.6 locus in embryonic stem cells by in-out targeting.
- Rothstein, Targeting, Disruption, Replacement and Allele Rescue; Integrative DNA Transformation in Yeast, Methods in Enzymology (1991) ; 281-301 describes in-out targeting in yeast.
- Smithies et aJL. Nature (1985) 317:230-234 describe targeting the human ⁇ -globin locus in a mouse erythroleukemia hybrid cell line containing a single human chromosome eleven.
- Methods for performing homologous recombination employing one or more selectable markers and a homologous region for introducing a modification in an indigenous chromosomal gene in a mammalian host cell.
- Two methods are employed for diminishing interference of the marker with the functioning of the target locus: (1) use of an ⁇ - (replacement) targeting vector, which allows retention of the selectable marker(s) in such a manner that the marker does not interfere with the desired function or expression of the indigenous gene; and (2) use of an O- (insertional) targeting vector, which allows for excision of the selectable marker(s) .
- the resulting cells have the modified target locus without the indigenous sequence at the target site.
- Methods are provided for targeted modification of indigenous genes using vectors comprising extended homology with the target gene, but differing at at least one site in conjunction with a marker gene.
- a linear DNA construct is employed with two regions of homology to the target locus. Frequently, these regions will be proximal to the ends of the linear DNA molecule.
- the linear DNA molecule is transformed into the target cell by any convenient means and selection for integrants is performed using the selection provided by the marker gene.
- the selected clones are then further screened for target genes having the desired modification. If desired, one may further screen for loss of the selection gene and other foreign DNA with retention of the target gene with the introduced modification.
- the first strategy an ⁇ -vector is employed, where hybridization results in a loop or D-loop of the hybrid.
- the resulting integrant retains the marker and provides a functioning target locus.
- the selectable marker is situated in such a manner in which at does not significantly interfere with the function or expression of the target locus.
- the marker may be located 5' of the known local transcription regulatory sequences, within an intron or 3' of the coding region of the gene, etc. Usually the marker will be within 10 kbp, more usually within 5 kbp, of the gene.
- an 0- vector is employed where insertion results in two copies of the target locus homologous sequence: the indigenous sequence and the modified sequence.
- an excision step the indigenous sequence and marker(s) are excised leaving only the modified sequence.
- the subject method can be used in a variety of ways for treating a variety of genetic diseases, mapping chromosomes, identifying loci, and the like.
- the modification of dysfunctional genes is the modification of dysfunctional genes, where the dysfunctional gene may be substituted with a functional gene.
- gene therapy may be carried out on a variety of types of cells, resulting in functional or modified genes from dysfunctional or undesired allelic genes.
- the DNA employed for targeting will have a region of homology with the target locus differing from the locus by a modification, which may be a substitution, deletion, insertion, or combinations thereof. Also included will be a marker gene which allows for selection. One or more unique primer sites may be present for subsequent PCR analysis.
- the homologous DNA will usually be not more than about 100 kbp, usually not more than about 20 kbp and usually more than about 0.5 kbp.
- the target cells may be any of a variety of vertebrate cells, particularly animals cells, more particularly mammalian cells, which may include any of the cells previously described.
- the constructs will comprise a region of homology associated with the target gene, where the region of homology may be noncoding, coding, or combinations thereof.
- a noncoding region may comprise the 5' non-coding region, introns, and in some instances the 3• noncoding region.
- the homologous region will normally encompass the modification, which may be a single site or a polynucleotide, usually not greater than about ten percent of the homologous region, e.g. 500 bp, usually not more than about five percent of the homologous region, e.g. 250 bp.
- the modification will normally be bordered by a total of at least about 50 bp of homology, usually 100 bp and less than about 100 kbp usually less than about 50 kbp.
- the restriction site which provides for the site of linearity of the DNA for the O-type vector employed for integration will be desirably between the site(s) of modification and the shorter stretch of homology. Desirably, one may have a short gap in the homology at the termini.
- the missing sequence which is filled in during the targeting can provide a primer site, so that targeted integration may be readily detected.
- the areas of homology will include regions flanking the marker(s) and the modified homologous region, where the flanking regions may or may not be immediately adjacent in the target locus.
- a double cross ⁇ over event is targeted resulting in replacement of the chromosomal region lying between the flanking homologous sequences.
- the regions of homology to the chromosome are usually adjacent in the chromosomal target locus.
- the homologous region includes the modified region. Cross-over events resulting in the integration of the vector are selected, since the termini of the linear construct, when joined, define a sequence of substantially continuous homology.
- the ⁇ -vector has an internal loop while the O-vector has an external loop of non-homologous sequence.
- the termini are distant at the target locus, while in the O- vector the termini are proximal at the target locus.
- markers may be employed for selection. These markers include the HPRT minigene (Reid et a . (1990) Proc. Natl. Acad. Sci. USA 87.:4299-4303, the neo gene for resistance to G418, the HSV thymidine kinase (tk) gene for sensitivity to gancyclovir, the hygromycin resistance gene, etc. As indicated above for the O-vector, by linearizing within the region of homology, the marker gene(s) , with accompanying foreign DNA (by foreign is intended foreign to the target host) will be situated between the duplicated genes, where one of the genes will have the introduced modification, while the other will be the indigenous sequence.
- HPRT minigene Reid et a . (1990) Proc. Natl. Acad. Sci. USA 87.:4299-4303
- the neo gene for resistance to G418, the HSV thymidine kinase (tk) gene for sensitivity to
- a marker that can be employed for both positive selection and negative selection of thee out step
- one may use separate markers for positive selection e.g., neo, hygromycin resistance, etc.
- negative selection e.g. , HSV-tk gene, cytosine deaminase, etc.
- the positive selection marker allows one to choose integrants lacking antibiotic resistance.
- the negative selection markers allows one to select against cells which retain the negative selection markers.
- a negative selection marker situated outside of the flanking homologous regions to enrich for double cross ⁇ over events.
- Other aspects of the construct may include sequences which allow for specific primer regions for polymerase chain reaction (PCR) identification of homologous recombinants, one or more restriction sites, which allow for identification by gel electrophoresis, removal of a restriction site at the target locus, or other modification which allows for identification of target cells which have undergone the desired modification.
- PCR polymerase chain reaction
- restriction sites which allow for identification by gel electrophoresis, removal of a restriction site at the target locus, or other modification which allows for identification of target cells which have undergone the desired modification.
- the changes outside of the coding region should allow for retention of the transcriptional regulation region, unless some change in the transcriptional regulation region is desired. Therefore, the gene for selection, restriction sites, primer sites, etc.
- the integrated DNA sequence will usually be at least about 0.5 kbp, more usually at least about 1 kbp and usually less than about 100 kbp, more usually less than about 50 kbp.
- the linear DNA may be introduced into the target cell.
- Techniques include electroporation, calcium precipitated DNA, fusion, transfection, and the like.
- the particular manner by which the DNA is introduced is not critical to this invention, although electroporation is preferred.
- the cells may then be selected by means of the marker gene.
- the cells may be plated in a selective medium or grown in selective culture and clones identified for further investigation.
- the clones may be analyzed using PCR, employing primers which will provide for different sized fragments, depending upon whether homologous recombination has occurred and whether the modi ied gene or the wild type gene is retained or other event has occurred to modify the target gene.
- target cells which have undergone the desired modification may be identified.
- the gene expresses a surface membrane protein
- Genes which may be targeted for gene therapy include jS-globin, enzymes of erythrocyte metabolism, the complement system, coagulation factors, dystrophin, enzymes of carbohydrate, lipid, amino acid, steroid and purine and pyrimidine metabolism, transport proteins, e.g., cystic fibrosis transmembrane regulator, and the like.
- the targeting construct /34.7NEO is a 4.7 kb BamHI/Xba I fragment that includes the ⁇ globin gene and surrounding sequences. It also contains a 20 bp oligomer inserted at the Sph I site 614 bp upstream of the start of the normal ⁇ globin transcript, and a 1.2 kb Xho I/Sal I fragment from pMClneo Poly A (Thomas and Capecchi (1987) Cell 51:503-512) (the neo ycin-resistance gene in this particular version of pMCl neo Poly A, from Stratagene, contains a point mutation that reduces its ability to confer resistance to G418) inserted into a Bgl II site in the oligomer by blunt ending both oligomer and the insert DNA with Klenow polymerase.
- the targeting sequences were excised from the vector plasmid Bluescript ⁇ by a Sma I/Xba I double digest. This leaves one base of nonhomology at the 5' end of the targeting construct.
- the DNA was precipitated with EtOH after digestion and resuspended in phosphate buffered saline.
- the cell line BMS is a hybrid murine-human cell line derived from the fusion of murine erythroleuke ia cells (MEL 179, APRT-, available from Dr. A. Deisseroth) and human EBV-transfor ed lymphoblasts derived from an individual heterozygous for HPFH-2 and for the ⁇ * globin gene. Fusion between the two cell lines was carried out in the presence of PEG (polyethylene glycol) 15 (50% PEG 15 in 75 mM Hepes) , and hybrid selection was achieved in AA media (50 ⁇ M Adenine, 40 ⁇ M Alanosine) in which only hybrid cells are expected to survive.
- PEG polyethylene glycol
- hybrids After 2-3 weeks of selection, these hybrids appeared as clonal outgrowths and were tested for the presence of human chromosome 11 using a monoclonal antibody against a chromosome 11-encoded antigen (Papayannopolou et al. , (1986) Cell 46:469-476). Hybrids were maintained in non-selective media and were occasionally enriched for the presence of human chromosome 11 by immunoadherence ("panning") to the monoclonal antibody.
- Cell line BSM carries only the copy of human chromosome 11 with the ⁇ s allele, as judged by gene-specific polymerase chain reaction (PCR) amplification, and by determining the pattern of human globin expression after induction using previously reported methods (Papayannopolou et al. , (1988) Science 242:1056-58)
- PCR polymerase chain reaction
- Cell line PC4 was constructed as a positive control for PCR amplification with primers 1 and 2 (SEQ ID NO:
- the cell line contains a portion of the sequences in the targeting construct /34.7NEO (from the 3' end of oligomer used as a primer binding site for primer 2 (SEQ ID NO:2) through the 5' BamHI site), plus further upstream sequences from the globin region.
- PC4 therefore contains the binding sites for primers 1 and 2 (SEQ ID NOS: 1 and 2, respectively) and probes A and B, but lacks the neomycin gene and the ⁇ globin gene and therefore lacks binding sites for primers 3, 4, and 6 (SEQ ID NOS: 3, 4 and 6, respectively) and probe C.
- Clone PC4 was obtained by co-electroporating the construct into BSM cells with pMClneo Poly A, followed by selection for G418 resistance and screening for PCR amplification of with primers 1 and 2 (SEQ ID NOS: 1 and 2, respectively). All cells were grown at 37°C and 5% C0 2 in RPMI- 1640 (Gibco) with 13% heat-inactivated fetal calf serum, supplemented with 2 mM L-Glutamine. G418 selection was carried out with 300 ⁇ g/ml G418 sulfate (Gibco) .
- Electroporation Cells were fed with fresh medium the day prior to electroporation, and were harvested when at a density of approximately 10 6 cells/ml. For electroporation, they were resuspended at 2xl0 7 cells/ml in warm growth medium, and digested. p/54.7NEO was added to a final concentration of 5nM. Electroporation was with 10 7 cells in a chamber of 5 mm length and 100 mm 2 cross sectioned as described (Boggs et al., (1986) Exp. He atol. 14:988-994). The electric pulse, from a 400 ⁇ F capacitor charged to 400V (800V/cm) , was for one second.
- the cells were diluted into warm growth medium, and 10 ml was immediately plated into each of sixteen 100 mm diameter dishes at either 5xl0 5 or 1.5xl0 5 cells/dish. Electroporated cells were also plated into microtiter plates at 100 ⁇ l/well, at the same concentration and diluted threefold and tenfold. The next day an equal volume of medium containing 600 ⁇ g/ml G418 was added to the cultures. The number of G418-resistant clones in the 100 mm dishes was estimated by the Poisson distribution from the frequency of microtiter wells having no G 18-resistant cells.
- Probes are a 218 bp Sty I/BamHI fragment from the genome just 5' of the targeting construct sequences.
- Probe B is a 627 bp Hpa I fragment from the 5• region of the targeting construct.
- Probe C is a 920 bp BamHI/EcoRI fragment covering the human ⁇ globin IVS2 region.
- An RNA probe assaying human globin gene transcription was transcribed in vitro from a genomic 0.77 kb EcoRI/Pst I fragment cloned in the antisense orientation into a T7 vector; it contains the 3' end of the human ⁇ globin gene and has 212 bases of homology to the transcript.
- the mouse probe was transcribed from a 0.65 kb BamEl/Hinfl cDNA-derived fragment cloned into an Sp6 vector; it has 298 bases of homology to the mouse ⁇ TM* gene transcript.
- the ⁇ globin replacement (or ⁇ ) type targeting construct has 4.7 kb of sequences homologous to the human ⁇ globin region; it also contains a unique oligomer for use as a PCR primer binding site (primer 2) (SEQ ID NO:2), and a neomycin-resistance gene, both placed 5' of any known local ⁇ globin transcriptional regulation sites.
- the neomycin gene is a 1.2 kb fragment derived from pMClneo PolyA and is driven by the herpes simplex thymidine kinase (tk) promoter plus the duplicated mutant polyoma virus enhancer originally designed for use in mouse embryonic stem cells (Thomas and Capecchi (1987) , supra) .
- the targeting construct was introduced into BSM cells in a series of eight electroporations. After electroporation, the cells were diluted, and a total of 4.1xl0 7 were immediately plated into tissue culture dishes. The following day they were placed under G418 selection which yielded 126 pools of between 10 and 1000 G418- resistant clones per dish (average about 200) , each clone having incorporated the targeting construct somewhere in the genome.
- primer 1 SE ID N0:1
- primer 2 SEQ ID NO:2
- the PCR analysis included positive control lysates made from mixtures of PC4 cells with untreated BSM cells at ratios of 1 to 10 and 1 to 100.
- PC4 is a pseudo-recombinant cell line which contains integrated copies of foreign DNA having primers 1 and 2 (SEQ ID NOS: 1 and 2, respectively) already juxtaposed.
- This positive control proved essential for working out appropriate PCR conditions, but introduced the risk that false positives might arise from contamination of nontargeted cells with the diagnostic 1.2 kb fragment from amplified controls.
- the possibility that the positives were artifactual contaminants of this type was excluded by carrying out a second set of PCR amplifications using a neomycin gene-derived primer (primer 3) (SEQ ID NO:3) in place of primer 2 (SEQ ID NO:2). All three pools that the test PCR assay had indicated contained a targeted clone yielded the expected 1.6 kb band that hybridized to probe A during this second PCR reaction; as expected, the positive control PC4 cells did not.
- Sib-selection was used to isolate a clone of targeted cells from one of the PCR- positive pools that contained about 200 independent G418- resistant clones. Cells from this pool were diluted into 96 smaller pools with approximately 10 cells in each. After expansion, these smaller pools were rescreened by PCR for the presence of the diagnostic 1.2 kb band. Three of the smaller pools gave a positive PCR result, and the signal level corresponded to that expected for mixtures having one targeted cell for every ten nontargetted. One of the enriched pools was then diluted into microtiter dishes so that only 10 to 20% of the wells received a cell.
- a BamEI site that begins 1 bp from the 5' end of the targeting construct is still intact after targeting, as shown by the presence of a predicted 2 kb fragment that hybridizes to probe B following BamEI digestion of DNA from the targeted clone; the parental BSM DNA gives a 1.9 kb band.
- the 3' Xba I site at the 3' end of the targeting construct was shown to be intact by the presence of a 4.5 kb band that hybridized to probe C following an Xba I/Pvu II double digest of the targeted clone, compared to the 10 kb band from parental BSM DNA.
- a pool of G418- resistant, but nontargetted cells yielded an amplified band when either primer set was used, although a more intense band was observed with the /3 A -specific primer, than with the / ⁇ -specific primer.
- This result is expected because the human chromosome 11 is not present in all of the hybrid cells, whereas a randomly integrated targeting construct must be present in all cells (in order to obtain G418 resistance) , and often occurs in multiple copies.
- the targeted clone again as expected, amplifies a band when the 0 A -specific primer 6 (SEQ ID NO:6) is used, but not with the (8 s -specific primer 5 (SEQ ID NO:5).
- allele-specific antibodies were used to investigate the globin polypeptides synthesized after the parental BSM cells and the targeted clone have been induced with HMBA (hexamethylenebisacetamide) .
- a human 0 s -specific antibody binds to the globins produced by the induced parental BSM cells, but not by the targeted clone.
- Human globin genes introduced on their native human chromosomes introduced into the BSM cells by somatic cell fusion are regulated and expressed after induction in a manner comparable to that shown by the endogenous mouse globin genes (Marks and Rifkind (1978) in Ann. Rev. Bioche . 42:419-448; Willing et al. (1979) Nature 277:534-538; Deisseroth and Hendrick f!979) Proc. Natl. Acad. Sci. USA 76:2185-2189.
- the results demonstrate that gene targeting can correct a human ⁇ s globin gene to ⁇ A , an essential requirement before gene targeting can be considered for human gene therapy.
- the induction ratio of the corrected gene was not significantly altered by the introduction of a neomycin- resistance helper gene into the target locus to facilitate identification and isolation of the targeted clone.
- Example 2 The Use of In-Out for Making Subtle Genomic Modifications in Mouse Embryonic Stem Cells.
- the mouse ES cell line E-14TG2a was isolated as described previously (Hooper et al. (1987) Nature 326:292-295; Thomas and Capecchi (1987), supra) . Cells were grown in Dulbecco's modified Eagle's medium (GIBCO) supplemented with 15% heat-inactivated fetal calf serum (Flow) and 10 ⁇ M 2-mercaptoethanol (Sigma) . The pluripotential nature of the ES cells was retained by supplementing each liter of growth medium with 10 6 U of recombinant human leukemia inhibitory factor (available from N. Gough, Walter and Eliza Hall Institute, Melbourne, Victoria, Australia) .
- HAT medium was standard culture medium supplemented with 120 ⁇ M hypoxanthine, 0.4 ⁇ M aminopterin, and 20 ⁇ M thy idine.
- 6-TG (thioguanosine) selection was carried out in standard medium containing 10 ⁇ M 6-TG. Cultures were incubated at 37°C in an atmosphere of 5% C0 2 . They were checked periodically for mycoplasma contamination.
- Plasmid pNMR133 has already been described (Doetschman et al. , (1987) J. Embryo1. Exp. Morphol. 330:576-578) . It contains 5 kb of DNA identical to the exon 3 target region of the mouse HPRT gene, except for a 4-bp insertion that destroys a unique Hindlll site and consequently generates a new Nhel site in intron 2. It also carries the human HPRT promoter and exon 1 sequences (which have been shown to function in mouse cells) and the mouse exon 2 region.
- Plasmid pNMR133D200 was derived from pNMR133 by removing a 200-bp Bglll fragment from intron 2. DNA preparation. Targeting vector DNAs were prepared by standard methods, omitting the CsCl purification, which was found unnecessary. All targeting DNAs were linearized by restriction enzyme digestion, using the manufacturers' recommended conditions, prior to electroporations. Digested DNAs were ethanol precipitated and resuspended in sterile TE buffer (0.05 M Tris, 0.001 M EDTA) .
- the vectors were introduced into the ES cells by electroporation (Boggs et al. (1986) Exp. Hematol. .14 . :988-994) .
- the cells were grown in 100-mm culture dishes (as described above) to a density of 1 x 10 7 to 2 x 10 7 cells per dish in nonselective medium. Cultures were trypsinized, centrifuged, and then resuspended in nonselective medium to a density of 4 x 10 7 to 10 x 10 7 cells per ml. A 0.5-ml sample of the cell suspension was added to each microfuge tube, and prepared DNA was then added to a final concentration of 5 nM.
- the cell-DNA mixtures were incubated on ice for 20 min, loaded into an electroporation chamber precooled on ice (length, 5 mm; cross section, 100 mm 2 ) , and exposed to a 1-s electrical pulse from a 250- ⁇ F capacitor charged to 300 V. Cells were immediately removed from the chamber and plated into five 100-mm cultures dishes. The plates had been prepared by gelatinization and contained 7 ml of nonselective medium. The cells were allowed to recover overnight. The next day, the number of colonies in each dish was determined by counting, and HAT selection was then applied.
- Cultures to be assayed for the loss of HPRT function by selection in 6-TG were maintained under HAT selection for at least 1 month prior to the start of the assay in order to kill any accumulated hprt " cells. These cultures were trypsinized, counted, and then replated at a density of 0.5 x 10 7 to 1 x 10 7 cells per plate in nonselective medium. They were without selection for 3 or 4 days to allow spontaneous revertants time to purge residual HPRT transcripts or protein. Selection was then started by applying 6-TG medium. All selections were maintained for 16 days, with feeding as necessary. Targeting and reversion frequencies were determined by counting the number of resistant colonies obtained for each experiment. Individual colonies were picked by using cloning rings into 24-well (1 ml per well) dishes and maintained under selection. Cultures were transferred to 60-mm culture dishes and then either harvested for genomic DNA preparation or transferred to 100- mm dishes for further expansion.
- Genomic DNA preparation and characterization DNA was prepared from expanded clones by using conventional procedures. Restriction enzyme digestions were done according to manufacturers* specifications, incubating overnight. After electrophoresis on 0.8% agarose gels, Southern blotting was done by standard techniques.
- Probes Two probes were used, a 250-bp Rsal fragment from intron 3 and a 300-bp Hindlll-Xhol fragment from the human cDNA which includes exons 3 to 6 but is specific for the mouse exon 3 element (Doetschman et al. , (1987) Nature 330:576-578) . Both probes hybridize to sequences present in the endogenous locus as well as on the targeting vectors. For each blot, 25 to 50 ng of purified fragment was radiolabeled with 32 P-dCTP by the random-primed oligonucleotide method, using a Boehringer Mannheim kit. Four-hour prehybridizations and overnight hybridizations were done in 50% formamide solutions at 42°C.
- Blots were washed to a stringency of 1 x SSC (0.15M NaCl plus 0.015 M sodium citrate) at 68°C. Washed blots were exposed to preflashed XAR-5 film at -70°C.
- the first step in the two- step targeting procedure is a homologous integration event that incorporates vector DNA carrying the desired modification into the genome.
- the method of (Doetschman et al. (1987) supra. , was used to introduce into mouse ES cells an integrating targeting vector that carries a 4-bp insertion in the second intron of the HPRT gene.
- Either plasmid pNMR133 or plasmid pNMRl33D200 (which has a 200-bp gap in the region homologous to the target locus) was electroporated into the male mouse-derived ES cell line E- 14TG2a. This cell line, isolated by Hooper et al.
- Both targeting vectors contain approximately 5kb of DNA identical in sequence to the exon 3 target region of the hprt"gene except for the intended modification: a 4-bp insertion in intron 2 that destroys a unique Hindlll site. They also carry the human HPRT promoter and exon 1 sequences and the mouse exon 2 region.
- the homologous integration event generates a duplication of the 5-kb target region separated by the remainder of the vector sequences.
- the duplicated regions are identifiable with the exception of the 4-bp insertion, identified by a missing Hindlll site, that is located on the downstream repeat.
- This event restores the promoter and first two exons deleted from the locus, generating HPRT+ targeted recombinants that can be directly selected with HAT-containing medium.
- Three independent HPRT+ cell lines were isolated by selection in HAT medium at an average frequency of 2.8 x 10" 6 per electroporated cell and it was then confirmed that these cell lines were targeted by genomic Southern blot hybridization.
- the blots were probed either with a 250-bp Rsal fragment from intron 3 or with a 300-bp Hindlll-Xhol fragment from the human cDNA that specifically hybridizes to the mouse exon 3. Both probes hybridize to sequences found in the genome as well as on the targeting vectors. All of the cell lines examined contained the expected recombinant locus, indicating that a single copy of the targeting vectors had integrated into the E-14TG2a hprt"gene.
- the second in the two-step targeting procedure is a spontaneous event that excises from the genome the vector sequences that integrated in the first step.
- a homologous recombination event between the regions duplicated during the in reaction can occur by either intrachromatid recombination (Doetschman et aJL. , (1987) supra) or unequal sister chromatid exchange.
- a crossover event in the 2-kb region comprising the 5'-terminal portion and the Hindlll site will leave the 4-bp insertion in the genome; crossing over in the 3 kb region comprising the 3'- terminal portion will excise the 4-bp modification along with the vector sequences or move it to the (HPRT+) triplicated chromosome.
- the excision event removes the vector-derived promoter and first two exons, causing a reversion to the hprt " phenotype.
- Such revertants can be selected with the nucleoside analog 6-TG.
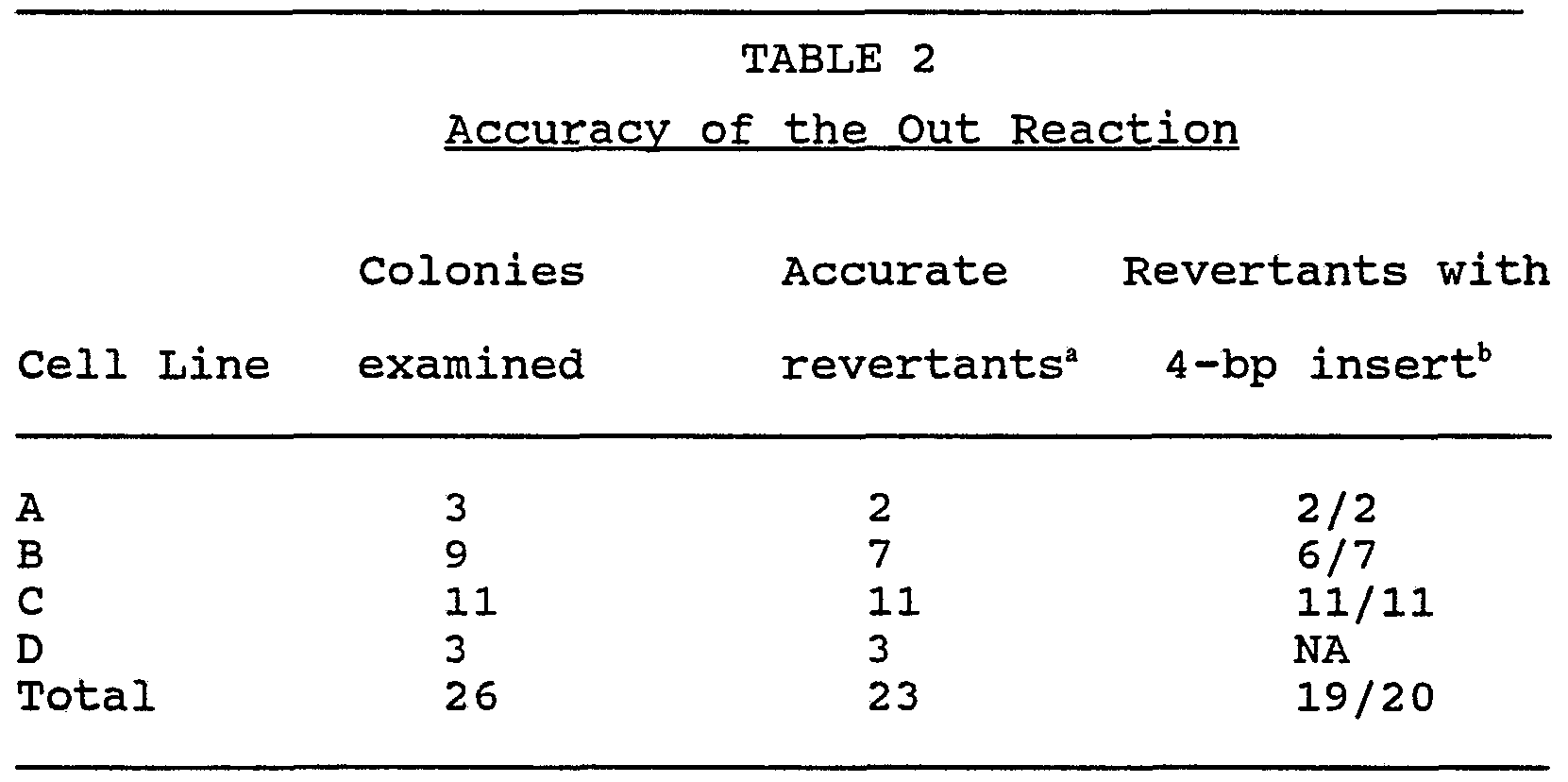
- the four HAT cell lines described above were used to study the excision (out) reaction. They all carry essentially the same HPRT locus: a duplication of 5 kb separated by 7 kb of plasmid-derived unique sequence.
- ES cell line D-3 Doetschman (1985) J. Embrvol. Exp. Morphol.
- ES cells form one colony for every 5 to 10 cells plated (Table 1) .
- each colony found the day after replating is composed of 5 to 10 individual cells. Although this aggregation may interfere with the 6-TG selections as a result of metabolic cross-feeding (Hooper et al. , (1981) Int. Rev. Cyto. jj :45-104) , it cannot be avoided.
- the number of 6-TG r colonies obtained for each line examined and calculated reversion frequencies are listed in Table 1. As shown, all four HAT lines initially generated by gene targeting reverted to the hprt " phenotype at similar frequencies, averaging 8 x 10 "7 6-TG r colonies isolated for every HAT r cell plated.
- Control cell line D-3 which carries the wild-type HPRT locus, failed to produce any 6-TG r colonies from 5.6 x 10 7 cells plated.
- the spontaneous mutation frequency at the HPRT locus, for cells preselected with HAT is less than 1.8 x 10 "8 . This result is consistent with the rate of 1.5 x 10 "8 per cell generation reported for the locus by Caskey and Kruh, (1979) Cell 16:1- 19.
- this cell line does not carry the 4-bp insertion.
- Hindlll digestion of the revertant DNAs is expected to reveal one of two bands upon hybridization: either an 11-kb fragment, if the crossover occurs in the 5' region and the 4-bp insertion introduced by the in event is retained, or a 7-kb fragment, if the crossover occurs in the 3' region and the modification is removed from the hprt"genome.
- the accurately modified revertant does contain a 2.7-kb Nhel fragment which hybridizes to the probe, confirming the presence of the 4-bp insertion, and the revertant which has lost the 4-bp insertion reveals a 4.9-kb fragment upon hybridization.
- the other three colonies examined were found to contain aberrant hprt " loci that did not arise by the predicted homologous excision reaction. They contained a single 14-kb Hindlll fragment and a 16-kb BamHI fragment that hybridized to the probes. These fragments failed to hybridize to a plasmid-specific probe, indicating that the target vector sequences have been excised from the genome. Since the bands are not the expected sizes, these colonies were probably generated by an alternate excision reaction. Because they account for only 12% of the 6-TG' colonies obtained, they were not examined further.
- the above data demonstrate the successful in-out targeting in modifying the genome of a mouse ES cell line by introducing a 4-bp insertion, in a two step procedure, the second step being automatic.
- the average frequency of the in reaction was found to be 2.8 x 10 "6 .
- the frequency of the out reaction, 8 x 10 "7 per HAT r cell plated is approximately 30% that of the in reaction. This frequency is 40-fold higher than the spontaneous mutation rate at the normal HPRT locus. Of the 6-TG r colonies isolated, 88% had accurately excised the target vector sequences from the genome.
- the targeted cell lines can then be identified by the polymerase chain reaction, for example, or other means, depending upon the nature of the targeting gene and the modification. Including a small gap in the region of homology on the insertional vector provides a convenient primer binding site, since all gaps are repaired during the homologous insertion event. Excision-derived hprt" revertants are likely to be found after the outstep at a frequency of nearly 1 in 10 6 per targeted cell line.
- the above techniques allow for modification of genomes of viable cells, particularly embryonic cells, where a stable modification can be achieved, which can be inherited by progeny cells.
- the modifications can be subtle and functional target genes achieved even when a marker is allowed to remain in the genome.
- the subject invention demonstrates the feasibility of gene therapy with stem cells or other cells, which can be used for the treatment of a variety of genetic or other diseases.
- ADDRESSEE Bertram I. Rowland
- B STREET: 4 Embarcadero Center, Suite 3400
- MOLECULE TYPE cDNA
- xi SEQUENCE DESCRIPTION: SEQ ID N ⁇ :2:
Abstract
Description

Claims
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU21402/92A AU664847B2 (en) | 1991-05-15 | 1992-05-13 | Genomic modifications with homologous DNA targeting |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US70050191A | 1991-05-15 | 1991-05-15 | |
| US700,501 | 1991-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992020808A1 true WO1992020808A1 (en) | 1992-11-26 |
Family
ID=24813739
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1992/004054 WO1992020808A1 (en) | 1991-05-15 | 1992-05-13 | Genomic modifications with homologous dna targeting |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP0539573A4 (en) |
| AU (1) | AU664847B2 (en) |
| CA (1) | CA2092695A1 (en) |
| WO (1) | WO1992020808A1 (en) |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993009222A2 (en) * | 1991-11-05 | 1993-05-13 | Transkaryotic Therapies, Inc. | Transfection of vertebrate cells e.g. by homologous recombination |
| WO1993022443A1 (en) * | 1992-04-24 | 1993-11-11 | Sri International | In vivo homologous sequence targeting in eukaryotic cells |
| WO1994004667A1 (en) * | 1992-08-25 | 1994-03-03 | Kölner Verein Zur Förderung Der Immunologie | Targeted replacement of a gene without endogenous and selectable residual sequences |
| WO1994012650A2 (en) | 1992-12-03 | 1994-06-09 | Transkaryotic Therapies, Inc. | Activating expression of an amplifying endogenous gene by homologous recombination |
| WO1994024301A1 (en) * | 1993-04-21 | 1994-10-27 | The University Of Edinburgh | Expression of heterologous genes according to a targeted expression profile |
| EP0656747A1 (en) * | 1992-08-21 | 1995-06-14 | The Regents Of The University Of California | Composition and method for altering dna sequences by homologous recombination |
| EP0702720A1 (en) * | 1993-03-15 | 1996-03-27 | Cell Genesys, Inc. | Method for defined deletions of dna |
| US5589362A (en) * | 1993-06-14 | 1996-12-31 | Basf Aktiengesellschaft | Tetracycline regulated transcriptional modulators with altered DNA binding specificities |
| US5641670A (en) * | 1991-11-05 | 1997-06-24 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US5650298A (en) * | 1993-06-14 | 1997-07-22 | Basf Aktiengesellschaft | Tight control of gene expression in eucaryotic cells by tetracycline-responsive promoters |
| US5654168A (en) * | 1994-07-01 | 1997-08-05 | Basf Aktiengesellschaft | Tetracycline-inducible transcriptional activator and tetracycline-regulated transcription units |
| US5733761A (en) * | 1991-11-05 | 1998-03-31 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US5789156A (en) * | 1993-06-14 | 1998-08-04 | Basf Ag | Tetracycline-regulated transcriptional inhibitors |
| US5814618A (en) * | 1993-06-14 | 1998-09-29 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US5851796A (en) * | 1995-06-07 | 1998-12-22 | Yale University | Autoregulatory tetracycline-regulated system for inducible gene expression in eucaryotes |
| US5859310A (en) * | 1993-06-14 | 1999-01-12 | Basf Aktiengesellschaft | Mice transgenic for a tetracycline-controlled transcriptional activator |
| US5866755A (en) * | 1993-06-14 | 1999-02-02 | Basf Aktiengellschaft | Animals transgenic for a tetracycline-regulated transcriptional inhibitor |
| WO1999005264A2 (en) * | 1997-07-23 | 1999-02-04 | Roche Diagnostics Gmbh | Production of human mutant proteins in human cells by homologous recombination |
| US5888981A (en) * | 1993-06-14 | 1999-03-30 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US5888809A (en) * | 1997-05-01 | 1999-03-30 | Icos Corporation | Hamster EF-1α transcriptional regulatory DNA |
| US5912411A (en) * | 1993-06-14 | 1999-06-15 | University Of Heidelberg | Mice transgenic for a tetracycline-inducible transcriptional activator |
| US5932465A (en) * | 1997-10-16 | 1999-08-03 | Icos Corporation | Phosphodiesterase 8A |
| US6004941A (en) * | 1993-06-14 | 1999-12-21 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US6048729A (en) * | 1987-05-01 | 2000-04-11 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6054288A (en) * | 1991-11-05 | 2000-04-25 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6063630A (en) * | 1991-11-05 | 2000-05-16 | Transkaryotic Therapies, Inc. | Targeted introduction of DNA into primary or secondary cells and their use for gene therapy |
| US6071691A (en) * | 1998-04-27 | 2000-06-06 | Oregon Health Science University | Materials and methods for modulating differentiation |
| US6100445A (en) * | 1994-10-14 | 2000-08-08 | Basf Aktiengesellschaft | Transgenic knockout mouse having functionally disrupted interleukin-1β converting enzyme gene |
| US6130071A (en) * | 1997-02-05 | 2000-10-10 | Helsinki University Licensing, Ltd. | Vascular endothelial growth factor C (VEGF-C) ΔCys156 protein and gene, and uses thereof |
| US6194633B1 (en) | 1998-01-26 | 2001-02-27 | University Of Iowa Research Foundation | Non-human animal having a functionally disrupted SLP-76 gene |
| US6200812B1 (en) | 1997-03-21 | 2001-03-13 | Sri International | Sequence alterations using homologous recombination |
| US6200951B1 (en) | 1998-03-12 | 2001-03-13 | Icos Corporation | Chitinase chitin-binding fragments |
| US6265218B1 (en) * | 1994-08-11 | 2001-07-24 | Roche Diagnostics Gmbh | Plasmids without a selection marker gene |
| US6294655B1 (en) | 1998-04-03 | 2001-09-25 | Hyseq, Inc. | Anti-interleukin-1 receptor antagonist antibodies and uses thereof |
| US6335013B1 (en) | 1999-03-19 | 2002-01-01 | Hyseq, Inc. | Methods and materials relating to CD39-like polypeptides |
| US6337072B1 (en) | 1998-04-03 | 2002-01-08 | Hyseq, Inc. | Interleukin-1 receptor antagonist and recombinant production thereof |
| US6344549B1 (en) | 1999-10-14 | 2002-02-05 | Icos Corporation | ATR-2 cell cycle checkpoint |
| US6350447B1 (en) | 1999-01-29 | 2002-02-26 | Hyseq, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6350603B1 (en) | 1998-02-23 | 2002-02-26 | Icos Corporation | Phosphodiesterase 10 |
| US6361946B1 (en) | 1997-02-05 | 2002-03-26 | Licentia Ltd | Vascular endothelial growth factor C (VEGF-C) protein and gene, mutants thereof, and uses thereof |
| US6387645B1 (en) | 1998-07-16 | 2002-05-14 | Hyseq, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6399747B1 (en) | 1998-05-21 | 2002-06-04 | Amgen Canada, Inc. | Shc-binding protein |
| US6426191B1 (en) | 1998-04-03 | 2002-07-30 | Hyseq, Inc. | Assays involving an IL-1 receptor antagonist |
| US6447771B1 (en) | 1999-03-19 | 2002-09-10 | Hyseq, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6465620B1 (en) | 2000-01-21 | 2002-10-15 | Hyseq, Inc. | Methods and materials relating to novel von Willebrand/Thrombospondin-like polypeptides and polynucleotides |
| US6476211B1 (en) | 1998-07-16 | 2002-11-05 | Hyseq, Inc. | Methods and materials relating to CD39-like polypeptides |
| US6479256B1 (en) | 1998-03-04 | 2002-11-12 | Icos Corporation | Lectomedin materials and methods |
| US6498015B1 (en) | 1995-06-07 | 2002-12-24 | Icos Corporation | Methods of identifying agents that modulate the binding between MDC and an MDC receptor |
| US6524818B1 (en) | 1997-09-26 | 2003-02-25 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US6531124B1 (en) | 1992-07-10 | 2003-03-11 | Transkaryotic Therapies, Inc. | In vivo production and delivery of insulinotropin for gene therapy |
| US6541623B1 (en) | 1998-04-03 | 2003-04-01 | Hyseq, Inc. | Interleukin—1 receptor antagonist and uses thereof |
| US6599727B1 (en) | 1999-06-16 | 2003-07-29 | Icos Corporation | Human poly (ADP-ribose) polymerase 2 materials and methods |
| US6632665B1 (en) | 1999-08-09 | 2003-10-14 | Wake Forest University | Human gene encoding 3′-5′ exonuclease |
| US6635742B1 (en) | 2000-01-25 | 2003-10-21 | Nuvelo, Inc. | Antibodies specific for semaphorin-like polypeptides |
| US6645933B1 (en) | 1995-08-01 | 2003-11-11 | Helsinki University Licensing Ltd. Oy | Receptor ligand VEGF-C |
| US6670178B1 (en) | 1992-07-10 | 2003-12-30 | Transkaryotic Therapies, Inc. | In Vivo production and delivery of insulinotropin for gene therapy |
| US6692737B1 (en) | 1991-11-05 | 2004-02-17 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6734005B2 (en) | 2000-05-22 | 2004-05-11 | Pharmacia & Upjohn Company | Matrix metalloproteinases |
| US6737513B1 (en) | 1996-06-07 | 2004-05-18 | Icos Corporation | Macrophage derived chemokine (MDC) and chemokine analogs and assay to identify modulators of MDC activity, and therapeutic uses for same |
| US6740503B1 (en) | 1997-09-26 | 2004-05-25 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US6780977B1 (en) | 1999-01-29 | 2004-08-24 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6787684B2 (en) | 2000-10-16 | 2004-09-07 | E. & J. Gallo Winery | Lipoxygenase genes from Vitis vinifera |
| US6818220B1 (en) | 1994-11-14 | 2004-11-16 | Licentia Ltd. | Vascular endothelial growth factor C (VEGF-C) protein and gene mutants thereof, and uses thereof |
| US6835546B1 (en) | 1999-10-22 | 2004-12-28 | Pharmacia & Upjohn Company | Drosophila G protein coupled receptors, nucleic acids, and methods related to the same |
| US6858207B2 (en) | 1998-07-16 | 2005-02-22 | Nuvelo, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6899875B1 (en) | 1999-01-29 | 2005-05-31 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| WO2005107796A2 (en) | 2004-04-23 | 2005-11-17 | Pharmacia & Upjohn Company, Llc | Cellular permissivity factor for viruses, and uses thereof |
| WO2006028967A2 (en) | 2004-09-02 | 2006-03-16 | Yale University | Regulation of oncogenes by micrornas |
| US7018627B1 (en) | 1995-06-07 | 2006-03-28 | Icos Corporation | Macrophage derived chemokine (MDC), MDC analogs, MDC inhibitor substances, and uses thereof |
| US7067626B2 (en) | 2000-07-05 | 2006-06-27 | Pharmacia & Upjohn Company | Human ion channel proteins |
| US7125714B2 (en) | 1997-02-05 | 2006-10-24 | Licentia Ltd. | Progenitor cell materials and methods |
| US7314716B2 (en) | 1999-11-19 | 2008-01-01 | Mount Sinai School Of Medicine | Gustducin γ subunit materials and methods |
| US7423125B2 (en) | 1995-08-01 | 2008-09-09 | Vegenics Limited | Antibodies to VEGF-C |
| WO2008140449A1 (en) | 2007-05-11 | 2008-11-20 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| USRE40594E1 (en) | 1998-12-23 | 2008-12-02 | Mount Sinai School Of Medicine Of New York University | Inhibitors of the bitter taste response |
| US7527944B2 (en) | 2003-06-27 | 2009-05-05 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic cat |
| US7531523B2 (en) | 2005-02-17 | 2009-05-12 | Vertex Pharmaceuticals Incorporated | Sodium channel protein type III alpha-subunit splice variant |
| US7541158B2 (en) | 2004-04-14 | 2009-06-02 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic dog |
| US7785807B2 (en) | 2004-12-13 | 2010-08-31 | Monell Chemical Senses Center | Voltage-gated, pH-sensitive anion channel and its novel splice variant involved in taste sensation |
| US7795494B2 (en) | 2001-03-22 | 2010-09-14 | Abbott Laboratories | Transgenic mice expressing antibodies specific for genes of interest and uses thereof |
| US7803982B2 (en) | 2001-04-20 | 2010-09-28 | The Mount Sinai School Of Medicine Of New York University | T1R3 transgenic animals, cells and related methods |
| US7972813B2 (en) | 2005-09-30 | 2011-07-05 | Vertex Pharmaceuticals Incorporated | Tetrodotoxin-resistant sodium channel alpha subunit |
| WO2013003697A1 (en) | 2011-06-30 | 2013-01-03 | Trustees Of Boston University | Method for controlling tumor growth, angiogenesis and metastasis using immunoglobulin containing and proline rich receptor-1 (igpr-1) |
| US8354384B2 (en) | 2005-06-23 | 2013-01-15 | Yale University | Anti-aging micrornas |
| WO2013009971A1 (en) | 2011-07-12 | 2013-01-17 | E. I. Du Pont De Nemours And Company | Detection and screening method and materials useful in performance thereof |
| WO2013020044A1 (en) | 2011-08-03 | 2013-02-07 | The Charlotte-Mecklenburg Hospital Authority D/B/A Carolinas Medical Center | Treatment of fibrosis using microrna-19b |
| US8420373B2 (en) | 2007-07-27 | 2013-04-16 | Universiteit Gent | Permissive cells and uses thereof |
| US9029383B2 (en) | 2007-05-11 | 2015-05-12 | The Trustees Of The University Of Pennsylvania | Methods of treatment of skin ulcers |
| US9052317B2 (en) | 2008-02-20 | 2015-06-09 | Universiteit Gent | Mucosal membrane receptor and uses thereof |
| WO2016044271A2 (en) | 2014-09-15 | 2016-03-24 | Children's Medical Center Corporation | Methods and compositions to increase somatic cell nuclear transfer (scnt) efficiency by removing histone h3-lysine trimethylation |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2646438B1 (en) * | 1989-03-20 | 2007-11-02 | Pasteur Institut | A METHOD FOR SPECIFIC REPLACEMENT OF A COPY OF A GENE PRESENT IN THE RECEIVER GENOME BY INTEGRATION OF A GENE DIFFERENT FROM THAT OR INTEGRATION |
| ATE244763T1 (en) * | 1992-02-11 | 2003-07-15 | Cell Genesys Inc | ACHIEVEMENT OF HOMOZYGOTE THROUGH TARGETED GENETIC EVENTS |
-
1992
- 1992-05-13 WO PCT/US1992/004054 patent/WO1992020808A1/en not_active Application Discontinuation
- 1992-05-13 CA CA 2092695 patent/CA2092695A1/en not_active Abandoned
- 1992-05-13 EP EP19920913238 patent/EP0539573A4/en not_active Withdrawn
- 1992-05-13 AU AU21402/92A patent/AU664847B2/en not_active Ceased
Non-Patent Citations (10)
| Title |
|---|
| Cell, Vol. 51, No. 6. issued 6 November 1987, THOMAS et al., "Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells", pages 503-512, see entire article. * |
| Molecular and Cellular Biology, Vol. 11, No. 3, issued March 1991, VALANCIUS et al., "Testing an 'in-out' targeting procedure for making subtle genomic modifications in mouse embryonic stem cells", pages 1402-1408. see entire article. * |
| Nature, Vol. 317, issued 19 September 1985, SMITHIES et al., "Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination", pages 230-234, see entire article. * |
| Nature, Vol. 336, issued 24 November 1988, MANSOUR et al., "Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes", pages 348-352. see entire article. * |
| Nature, Vol. 345, issued 3 May 1990, DECHIARA et al., "A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth fctor II gene disrupted by targeting", pages 78-80. see entire article. * |
| Nucleic Acids Research, Vol. 16, No. 18, issued 1988, KIM et al., "Recombinant fragment assay for gene targetting based on the polymerase chain reaction", pages 8887-8903. see entire aricle. * |
| Proceedings of the National Academy of Sciences USA, Vol. 76, No. 10, issued October 1979, SCHERER et al., "Replacement of chromosome ssegments with altered DNA sequences constructed in vitro", pages 4951-4955, see entire article. * |
| Proceedings of the National Academy of Sciences USA, Vol. 78, No. 10, issued October 1981, ORR-WEAVER et al., "Yeast transformation: A model system for the study of recombination", pages 6354, 6358. see entire article. * |
| Science, Vol. 242, issued 18 November 1988, PAPAYANNOPOULOU et al., "Activation of developmentally mutated human globin genes by cell fusion", pages 1056-1058. see entire article. * |
| See also references of EP0539573A4 * |
Cited By (169)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6048729A (en) * | 1987-05-01 | 2000-04-11 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6303379B1 (en) | 1987-05-01 | 2001-10-16 | Transkaryotic Therapies, Inc. | Vivo protein production and delivery system for gene therapy |
| US5968502A (en) * | 1991-11-05 | 1999-10-19 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US5641670A (en) * | 1991-11-05 | 1997-06-24 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US6692737B1 (en) | 1991-11-05 | 2004-02-17 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6048724A (en) * | 1991-11-05 | 2000-04-11 | Transkaryotic Therapies Inc. | Method of producing clonal cell strains which express exogenous DNA encoding glucagon-like peptide 1 |
| US5733761A (en) * | 1991-11-05 | 1998-03-31 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US6214622B1 (en) | 1991-11-05 | 2001-04-10 | Transkaryotic Therapies, Inc. | Targeted introduction of DNA into primary or secondary cells and their use for gene therapy |
| US6048524A (en) * | 1991-11-05 | 2000-04-11 | Transkaryotic Therapies, Inc. | In vivo production and delivery of erythropoietin for gene therapy |
| US6063630A (en) * | 1991-11-05 | 2000-05-16 | Transkaryotic Therapies, Inc. | Targeted introduction of DNA into primary or secondary cells and their use for gene therapy |
| WO1993009222A3 (en) * | 1991-11-05 | 1993-10-28 | Transkaryotic Therapies Inc | Transfection of vertebrate cells e.g. by homologous recombination |
| US6537542B1 (en) | 1991-11-05 | 2003-03-25 | Transkaryotic Therapies, Inc. | Targeted introduction of DNA into primary or secondary cells and their use for gene therapy and protein production |
| US6187305B1 (en) | 1991-11-05 | 2001-02-13 | Transkaryotic Therapies, Inc. | Targeted introduction of DNA into primary or secondary cells and their use for gene therapy and protein production |
| US6846676B2 (en) | 1991-11-05 | 2005-01-25 | Transkaryotic Therapies, Inc. | In Vivo production and delivery of erythropoietin or insulinotropin for gene therapy |
| US6054288A (en) * | 1991-11-05 | 2000-04-25 | Transkaryotic Therapies, Inc. | In vivo protein production and delivery system for gene therapy |
| US6355241B1 (en) | 1991-11-05 | 2002-03-12 | Transkaryotic Therapies, Inc. | In vivo production and delivery of erythropoietin |
| US7410799B2 (en) | 1991-11-05 | 2008-08-12 | Shire Human Genetic Therapies, Inc. | In vivo production and delivery of erythropoietin or insulinotropin for gene therapy |
| EP1221477A2 (en) * | 1991-11-05 | 2002-07-10 | Transkaryotic Therapies, Inc. | Transfection of vertebrate cells e.g. by homologous recombination |
| WO1993009222A2 (en) * | 1991-11-05 | 1993-05-13 | Transkaryotic Therapies, Inc. | Transfection of vertebrate cells e.g. by homologous recombination |
| EP1221477A3 (en) * | 1991-11-05 | 2002-07-24 | Transkaryotic Therapies, Inc. | Transfection of vertebrate cells e.g. by homologous recombination |
| WO1993022443A1 (en) * | 1992-04-24 | 1993-11-11 | Sri International | In vivo homologous sequence targeting in eukaryotic cells |
| US6531124B1 (en) | 1992-07-10 | 2003-03-11 | Transkaryotic Therapies, Inc. | In vivo production and delivery of insulinotropin for gene therapy |
| US6670178B1 (en) | 1992-07-10 | 2003-12-30 | Transkaryotic Therapies, Inc. | In Vivo production and delivery of insulinotropin for gene therapy |
| EP0656747A4 (en) * | 1992-08-21 | 1997-05-07 | Univ California | Composition and method for altering dna sequences by homologous recombination. |
| EP0656747A1 (en) * | 1992-08-21 | 1995-06-14 | The Regents Of The University Of California | Composition and method for altering dna sequences by homologous recombination |
| US6570061B1 (en) | 1992-08-25 | 2003-05-27 | Klaus Rajewsky | Targeted replacement of an immunoglobulin gene without endogenous and selectable residual sequences in mice |
| WO1994004667A1 (en) * | 1992-08-25 | 1994-03-03 | Kölner Verein Zur Förderung Der Immunologie | Targeted replacement of a gene without endogenous and selectable residual sequences |
| AU689455B2 (en) * | 1992-12-03 | 1998-04-02 | Transkaryotic Therapies, Inc. | Activating expression of an amplifying endogenous gene by homologous recombination |
| WO1994012650A3 (en) * | 1992-12-03 | 1994-08-04 | Transkaryotic Therapies Inc | Activating expression of an amplifying endogenous gene by homologous recombination |
| WO1994012650A2 (en) | 1992-12-03 | 1994-06-09 | Transkaryotic Therapies, Inc. | Activating expression of an amplifying endogenous gene by homologous recombination |
| EP0702720A4 (en) * | 1993-03-15 | 1997-07-02 | Cell Genesys Inc | Method for defined deletions of dna |
| EP0702720A1 (en) * | 1993-03-15 | 1996-03-27 | Cell Genesys, Inc. | Method for defined deletions of dna |
| US6150169A (en) * | 1993-04-21 | 2000-11-21 | The University Of Edinburgh | Expression of the heterologous genes according to a targeted expression profile |
| US7005299B1 (en) | 1993-04-21 | 2006-02-28 | The University Of Edinburgh | Expression of heterologous genes according to a targeted expression profile |
| WO1994024301A1 (en) * | 1993-04-21 | 1994-10-27 | The University Of Edinburgh | Expression of heterologous genes according to a targeted expression profile |
| US6004941A (en) * | 1993-06-14 | 1999-12-21 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US5650298A (en) * | 1993-06-14 | 1997-07-22 | Basf Aktiengesellschaft | Tight control of gene expression in eucaryotic cells by tetracycline-responsive promoters |
| US5789156A (en) * | 1993-06-14 | 1998-08-04 | Basf Ag | Tetracycline-regulated transcriptional inhibitors |
| US5814618A (en) * | 1993-06-14 | 1998-09-29 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US5859310A (en) * | 1993-06-14 | 1999-01-12 | Basf Aktiengesellschaft | Mice transgenic for a tetracycline-controlled transcriptional activator |
| US6136954A (en) * | 1993-06-14 | 2000-10-24 | Basf Aktiengesellschaft | Tetracycline-inducible transcriptional activator fusion proteins |
| US5866755A (en) * | 1993-06-14 | 1999-02-02 | Basf Aktiengellschaft | Animals transgenic for a tetracycline-regulated transcriptional inhibitor |
| US5922927A (en) * | 1993-06-14 | 1999-07-13 | Basf Aktiengesellschaft | Methods for producing tetracycline-regulated transgenic mice |
| US6783756B2 (en) | 1993-06-14 | 2004-08-31 | Abbott Gmbh & Co., Kg | Methods for regulating gene expression |
| US5888981A (en) * | 1993-06-14 | 1999-03-30 | Basf Aktiengesellschaft | Methods for regulating gene expression |
| US6914124B2 (en) | 1993-06-14 | 2005-07-05 | Tet Systems Holding Gmbh & Co. Kg | Tetracycline-regulated transcriptional activator fusion proteins |
| US5912411A (en) * | 1993-06-14 | 1999-06-15 | University Of Heidelberg | Mice transgenic for a tetracycline-inducible transcriptional activator |
| US6242667B1 (en) | 1993-06-14 | 2001-06-05 | Basf Aktiengesellschaft | Transgenic organisms having tetracycline-regulated transcriptional regulatory systems |
| US6252136B1 (en) | 1993-06-14 | 2001-06-26 | Basf Aktiengesellschaft | Transgenic organisms having tetracycline-regulated transcriptional regulatory systems |
| US5589362A (en) * | 1993-06-14 | 1996-12-31 | Basf Aktiengesellschaft | Tetracycline regulated transcriptional modulators with altered DNA binding specificities |
| US6271348B1 (en) * | 1993-06-14 | 2001-08-07 | Basf Aktiengesellschaft | Tetracycline-inducible transcriptional inhibitor fusion proteins |
| US5654168A (en) * | 1994-07-01 | 1997-08-05 | Basf Aktiengesellschaft | Tetracycline-inducible transcriptional activator and tetracycline-regulated transcription units |
| US6265218B1 (en) * | 1994-08-11 | 2001-07-24 | Roche Diagnostics Gmbh | Plasmids without a selection marker gene |
| US6100445A (en) * | 1994-10-14 | 2000-08-08 | Basf Aktiengesellschaft | Transgenic knockout mouse having functionally disrupted interleukin-1β converting enzyme gene |
| US6818220B1 (en) | 1994-11-14 | 2004-11-16 | Licentia Ltd. | Vascular endothelial growth factor C (VEGF-C) protein and gene mutants thereof, and uses thereof |
| US6498015B1 (en) | 1995-06-07 | 2002-12-24 | Icos Corporation | Methods of identifying agents that modulate the binding between MDC and an MDC receptor |
| US7018627B1 (en) | 1995-06-07 | 2006-03-28 | Icos Corporation | Macrophage derived chemokine (MDC), MDC analogs, MDC inhibitor substances, and uses thereof |
| US5851796A (en) * | 1995-06-07 | 1998-12-22 | Yale University | Autoregulatory tetracycline-regulated system for inducible gene expression in eucaryotes |
| US7423125B2 (en) | 1995-08-01 | 2008-09-09 | Vegenics Limited | Antibodies to VEGF-C |
| US6730658B1 (en) | 1995-08-01 | 2004-05-04 | Helsinki University Licensing, Ltd. | Stimulation of lymphatic growth with an FLT4 ligand |
| US7709270B2 (en) | 1995-08-01 | 2010-05-04 | Vegenics Ltd. | Vascular endothelial growth factor C (VEGF-C) protein diagnostic |
| US7807412B2 (en) | 1995-08-01 | 2010-10-05 | Vegenies Limited | VEGF-C ΔR226ΔR227 mutants and uses thereof |
| US7727761B2 (en) | 1995-08-01 | 2010-06-01 | Vegenics Limited | Vascular endothelial growth factor C (VEGF-C) protein and gene, mutants thereof, and uses thereof |
| US6645933B1 (en) | 1995-08-01 | 2003-11-11 | Helsinki University Licensing Ltd. Oy | Receptor ligand VEGF-C |
| US8282931B2 (en) | 1995-08-01 | 2012-10-09 | Vegenics Pty Limited | Vascular endothelial growth factor C (VEGF-C) protein and gene, mutants thereof, and uses thereof |
| US8637262B2 (en) | 1995-08-01 | 2014-01-28 | Vegenics Pty Limited | Vascular endothelial growth factor C (VEGF-C) protein and gene, mutants thereof, and uses thereof |
| US7794756B1 (en) | 1995-08-01 | 2010-09-14 | Vegenics Limited | Methods of using antibodies to VEGF-C |
| US6737513B1 (en) | 1996-06-07 | 2004-05-18 | Icos Corporation | Macrophage derived chemokine (MDC) and chemokine analogs and assay to identify modulators of MDC activity, and therapeutic uses for same |
| US7125714B2 (en) | 1997-02-05 | 2006-10-24 | Licentia Ltd. | Progenitor cell materials and methods |
| US6361946B1 (en) | 1997-02-05 | 2002-03-26 | Licentia Ltd | Vascular endothelial growth factor C (VEGF-C) protein and gene, mutants thereof, and uses thereof |
| US6130071A (en) * | 1997-02-05 | 2000-10-10 | Helsinki University Licensing, Ltd. | Vascular endothelial growth factor C (VEGF-C) ΔCys156 protein and gene, and uses thereof |
| US6200812B1 (en) | 1997-03-21 | 2001-03-13 | Sri International | Sequence alterations using homologous recombination |
| US5888809A (en) * | 1997-05-01 | 1999-03-30 | Icos Corporation | Hamster EF-1α transcriptional regulatory DNA |
| WO1999005264A3 (en) * | 1997-07-23 | 1999-04-22 | Roche Diagnostics Gmbh | Production of human mutant proteins in human cells by homologous recombination |
| US6444441B1 (en) | 1997-07-23 | 2002-09-03 | Roche Diagnostics Gmbh | Production of human mutated proteins in human cells by means of homologous recombination |
| WO1999005264A2 (en) * | 1997-07-23 | 1999-02-04 | Roche Diagnostics Gmbh | Production of human mutant proteins in human cells by homologous recombination |
| US7785831B2 (en) | 1997-09-26 | 2010-08-31 | Abt Holding Company | Compositions and methods for non-targeted activation of endogenous genes |
| US6740503B1 (en) | 1997-09-26 | 2004-05-25 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US6541221B1 (en) | 1997-09-26 | 2003-04-01 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US6602686B1 (en) | 1997-09-26 | 2003-08-05 | Athersys, Inc. | Compositions and method for non-targeted activation of endogenous genes |
| US6623958B1 (en) | 1997-09-26 | 2003-09-23 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US7842792B2 (en) | 1997-09-26 | 2010-11-30 | Abt Holding Company | Compositions and methods for non-targeted activation of endogenous genes |
| US6524818B1 (en) | 1997-09-26 | 2003-02-25 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US7569220B2 (en) | 1997-09-26 | 2009-08-04 | Abt Holding Company | Compositions and methods for non-targeted activation of endogenous genes |
| US7033782B2 (en) | 1997-09-26 | 2006-04-25 | Athersys, Inc. | Compositions and methods for non-targeted activation of endogenous genes |
| US6566087B1 (en) | 1997-10-16 | 2003-05-20 | Icos Corporation | Phosphodiesterase 8A |
| US6133007A (en) * | 1997-10-16 | 2000-10-17 | Icos Corporation | Phosphodiesterase 8A |
| US5932465A (en) * | 1997-10-16 | 1999-08-03 | Icos Corporation | Phosphodiesterase 8A |
| US6194633B1 (en) | 1998-01-26 | 2001-02-27 | University Of Iowa Research Foundation | Non-human animal having a functionally disrupted SLP-76 gene |
| US6350603B1 (en) | 1998-02-23 | 2002-02-26 | Icos Corporation | Phosphodiesterase 10 |
| US6864070B2 (en) | 1998-02-23 | 2005-03-08 | Icos Corporation | Phosphodiesterase 10 |
| US6734003B2 (en) | 1998-02-23 | 2004-05-11 | Icos Corporation | Phosphodiesterase 10 |
| US6479256B1 (en) | 1998-03-04 | 2002-11-12 | Icos Corporation | Lectomedin materials and methods |
| US6200951B1 (en) | 1998-03-12 | 2001-03-13 | Icos Corporation | Chitinase chitin-binding fragments |
| US6399571B1 (en) | 1998-03-12 | 2002-06-04 | Icos Corporation | Chitinase chitin-binding fragments |
| US6426191B1 (en) | 1998-04-03 | 2002-07-30 | Hyseq, Inc. | Assays involving an IL-1 receptor antagonist |
| US6541623B1 (en) | 1998-04-03 | 2003-04-01 | Hyseq, Inc. | Interleukin—1 receptor antagonist and uses thereof |
| US6294655B1 (en) | 1998-04-03 | 2001-09-25 | Hyseq, Inc. | Anti-interleukin-1 receptor antagonist antibodies and uses thereof |
| US6337072B1 (en) | 1998-04-03 | 2002-01-08 | Hyseq, Inc. | Interleukin-1 receptor antagonist and recombinant production thereof |
| US6071691A (en) * | 1998-04-27 | 2000-06-06 | Oregon Health Science University | Materials and methods for modulating differentiation |
| US6492138B1 (en) | 1998-05-21 | 2002-12-10 | Amgen Canada Inc. | Polynucleotides encoding a novel SHC-binding protein |
| US6399747B1 (en) | 1998-05-21 | 2002-06-04 | Amgen Canada, Inc. | Shc-binding protein |
| US6858207B2 (en) | 1998-07-16 | 2005-02-22 | Nuvelo, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6387645B1 (en) | 1998-07-16 | 2002-05-14 | Hyseq, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6476211B1 (en) | 1998-07-16 | 2002-11-05 | Hyseq, Inc. | Methods and materials relating to CD39-like polypeptides |
| USRE40594E1 (en) | 1998-12-23 | 2008-12-02 | Mount Sinai School Of Medicine Of New York University | Inhibitors of the bitter taste response |
| US6884872B1 (en) | 1999-01-29 | 2005-04-26 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6899875B1 (en) | 1999-01-29 | 2005-05-31 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6787328B1 (en) | 1999-01-29 | 2004-09-07 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6828423B1 (en) | 1999-01-29 | 2004-12-07 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6780977B1 (en) | 1999-01-29 | 2004-08-24 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6350447B1 (en) | 1999-01-29 | 2002-02-26 | Hyseq, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6780410B1 (en) | 1999-01-29 | 2004-08-24 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6759214B1 (en) | 1999-01-29 | 2004-07-06 | Nuvelo, Inc. | Methods and compositions relating to CD39-like polypeptides and nucleic acids |
| US6335013B1 (en) | 1999-03-19 | 2002-01-01 | Hyseq, Inc. | Methods and materials relating to CD39-like polypeptides |
| US6447771B1 (en) | 1999-03-19 | 2002-09-10 | Hyseq, Inc. | Methods and materials relating to novel CD39-like polypeptides |
| US6989260B2 (en) | 1999-06-16 | 2006-01-24 | Icos Corporation | Human poly(ADP-ribose) polymerase 2 materials and methods |
| US6599727B1 (en) | 1999-06-16 | 2003-07-29 | Icos Corporation | Human poly (ADP-ribose) polymerase 2 materials and methods |
| US6632665B1 (en) | 1999-08-09 | 2003-10-14 | Wake Forest University | Human gene encoding 3′-5′ exonuclease |
| US6815532B2 (en) | 1999-10-14 | 2004-11-09 | Icos Corporation | ATR-2 cell cycle checkpoint |
| US6344549B1 (en) | 1999-10-14 | 2002-02-05 | Icos Corporation | ATR-2 cell cycle checkpoint |
| US6835546B1 (en) | 1999-10-22 | 2004-12-28 | Pharmacia & Upjohn Company | Drosophila G protein coupled receptors, nucleic acids, and methods related to the same |
| US7354724B2 (en) | 1999-10-22 | 2008-04-08 | Pharmacia & Upjohn Company | Drosophila G protein coupled receptors, nucleic acids, and methods related to the same |
| US7314716B2 (en) | 1999-11-19 | 2008-01-01 | Mount Sinai School Of Medicine | Gustducin γ subunit materials and methods |
| US6465620B1 (en) | 2000-01-21 | 2002-10-15 | Hyseq, Inc. | Methods and materials relating to novel von Willebrand/Thrombospondin-like polypeptides and polynucleotides |
| US6635742B1 (en) | 2000-01-25 | 2003-10-21 | Nuvelo, Inc. | Antibodies specific for semaphorin-like polypeptides |
| US7070976B2 (en) | 2000-05-22 | 2006-07-04 | Pfizer Inc. | Matrix metalloproteinase polypeptide |
| US6734005B2 (en) | 2000-05-22 | 2004-05-11 | Pharmacia & Upjohn Company | Matrix metalloproteinases |
| US7067626B2 (en) | 2000-07-05 | 2006-06-27 | Pharmacia & Upjohn Company | Human ion channel proteins |
| US6787684B2 (en) | 2000-10-16 | 2004-09-07 | E. & J. Gallo Winery | Lipoxygenase genes from Vitis vinifera |
| EP2294917A1 (en) | 2001-03-22 | 2011-03-16 | Abbott GmbH & Co. KG | Transgenic animals expressing antibodies specific for genes of interest and uses thereof |
| US7795494B2 (en) | 2001-03-22 | 2010-09-14 | Abbott Laboratories | Transgenic mice expressing antibodies specific for genes of interest and uses thereof |
| US7803982B2 (en) | 2001-04-20 | 2010-09-28 | The Mount Sinai School Of Medicine Of New York University | T1R3 transgenic animals, cells and related methods |
| US8710186B2 (en) | 2003-06-27 | 2014-04-29 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic cat |
| US8173769B2 (en) | 2003-06-27 | 2012-05-08 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic cat |
| US7527944B2 (en) | 2003-06-27 | 2009-05-05 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic cat |
| US8765912B2 (en) | 2004-04-14 | 2014-07-01 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic dog |
| US7541158B2 (en) | 2004-04-14 | 2009-06-02 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic dog |
| US8168395B2 (en) | 2004-04-14 | 2012-05-01 | Monell Chemical Senses Center | Taste receptors of the T1R family from domestic dog |
| US8486685B2 (en) | 2004-04-23 | 2013-07-16 | Zoetis P&U Llc | Cellular permissivity factor for viruses and uses thereof |
| US7754464B2 (en) | 2004-04-23 | 2010-07-13 | Pharmacia And Upjohn Company, Llc | Cellular permissivity factor for viruses and uses thereof |
| EP2520310A1 (en) | 2004-04-23 | 2012-11-07 | Pharmacia & Upjohn Company LLC | Cellular permissivity factor for viruses, and uses thereof |
| WO2005107796A2 (en) | 2004-04-23 | 2005-11-17 | Pharmacia & Upjohn Company, Llc | Cellular permissivity factor for viruses, and uses thereof |
| US8058050B2 (en) | 2004-04-23 | 2011-11-15 | Pharmacia And Upjohn Company Llc | Cellular permissivity factor for viruses and uses thereof |
| US9102912B2 (en) | 2004-04-23 | 2015-08-11 | Zoetis Services Llc | Cellular permissivity factor for viruses and uses thereof |
| EP2338994A1 (en) | 2004-09-02 | 2011-06-29 | Yale University | Regulation of oncogenes by microRNAs |
| EP2338993A1 (en) | 2004-09-02 | 2011-06-29 | Yale University | Regulation of oncogenes by microRNAs |
| WO2006028967A2 (en) | 2004-09-02 | 2006-03-16 | Yale University | Regulation of oncogenes by micrornas |
| US7741306B2 (en) | 2004-09-02 | 2010-06-22 | Yale University | Regulation of oncogenes by microRNAs |
| EP2298897A1 (en) | 2004-09-02 | 2011-03-23 | Yale University | Regulation of oncogenes by microRNAs |
| US7893034B2 (en) | 2004-09-02 | 2011-02-22 | Yale University | Regulation of oncogenes by microRNAs |
| US7785807B2 (en) | 2004-12-13 | 2010-08-31 | Monell Chemical Senses Center | Voltage-gated, pH-sensitive anion channel and its novel splice variant involved in taste sensation |
| US7531523B2 (en) | 2005-02-17 | 2009-05-12 | Vertex Pharmaceuticals Incorporated | Sodium channel protein type III alpha-subunit splice variant |
| US8252541B2 (en) | 2005-02-17 | 2012-08-28 | Vertex Pharmaceuticals Incorporated | Sodium channel protein type III α-subunit splice variant |
| US7915385B2 (en) | 2005-02-17 | 2011-03-29 | Vertex Pharmaceuticals Incorporated | Sodium channel protein type III α-subunit splice variant |
| US8663936B2 (en) | 2005-02-17 | 2014-03-04 | Vertex Pharmaceuticals Incorporated | Sodium channel protein type III α-subunit splice variant |
| US8354384B2 (en) | 2005-06-23 | 2013-01-15 | Yale University | Anti-aging micrornas |
| US7972813B2 (en) | 2005-09-30 | 2011-07-05 | Vertex Pharmaceuticals Incorporated | Tetrodotoxin-resistant sodium channel alpha subunit |
| EP2977452A2 (en) | 2007-05-11 | 2016-01-27 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| US9029383B2 (en) | 2007-05-11 | 2015-05-12 | The Trustees Of The University Of Pennsylvania | Methods of treatment of skin ulcers |
| WO2008140449A1 (en) | 2007-05-11 | 2008-11-20 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| US8420373B2 (en) | 2007-07-27 | 2013-04-16 | Universiteit Gent | Permissive cells and uses thereof |
| US9052317B2 (en) | 2008-02-20 | 2015-06-09 | Universiteit Gent | Mucosal membrane receptor and uses thereof |
| US9636417B2 (en) | 2008-02-20 | 2017-05-02 | Universiteit Gent | Mucosal membrane receptor and uses thereof |
| WO2013003697A1 (en) | 2011-06-30 | 2013-01-03 | Trustees Of Boston University | Method for controlling tumor growth, angiogenesis and metastasis using immunoglobulin containing and proline rich receptor-1 (igpr-1) |
| WO2013009971A1 (en) | 2011-07-12 | 2013-01-17 | E. I. Du Pont De Nemours And Company | Detection and screening method and materials useful in performance thereof |
| WO2013020044A1 (en) | 2011-08-03 | 2013-02-07 | The Charlotte-Mecklenburg Hospital Authority D/B/A Carolinas Medical Center | Treatment of fibrosis using microrna-19b |
| WO2016044271A2 (en) | 2014-09-15 | 2016-03-24 | Children's Medical Center Corporation | Methods and compositions to increase somatic cell nuclear transfer (scnt) efficiency by removing histone h3-lysine trimethylation |
| US11390885B2 (en) | 2014-09-15 | 2022-07-19 | Children's Medical Center Corporation | Methods and compositions to increase somatic cell nuclear transfer (SCNT) efficiency by removing histone H3-lysine trimethylation |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0539573A4 (en) | 1993-12-29 |
| AU2140292A (en) | 1992-12-30 |
| CA2092695A1 (en) | 1992-11-16 |
| AU664847B2 (en) | 1995-12-07 |
| EP0539573A1 (en) | 1993-05-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU664847B2 (en) | Genomic modifications with homologous DNA targeting | |
| Valancius et al. | Testing an “in-out” targeting procedure for making subtle genomic modifications in mouse embryonic stem cells | |
| AU2019250224B2 (en) | Enhanced transgene expression and processing | |
| Fiering et al. | An" in-out" strategy using gene targeting and FLP recombinase for the functional dissection of complex DNA regulatory elements: analysis of the beta-globin locus control region. | |
| Hanson et al. | Analysis of biological selections for high-efficiency gene targeting | |
| Koller et al. | Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. | |
| US5830698A (en) | Method for integrating genes at specific sites in mammalian cells via homologous recombination and vectors for accomplishing the same | |
| US6689610B1 (en) | Cells and non-human organisms containing predetermined genomic modifications and positive-negative selection methods and vectors for making same | |
| Thomas et al. | Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells | |
| US5631153A (en) | Cells and non-human organisms containing predetermined genomic modifications and positive-negative selection methods and vectors for making same | |
| KR100233781B1 (en) | Production of proteins using homologous recombination | |
| US5922601A (en) | High efficiency gene trap selection of regulated genetic loci | |
| JP3059481B2 (en) | Method for specifically replacing a gene present in a receptor by incorporating the gene | |
| JP5377806B2 (en) | Expression of endogenous genes by non-homologous recombination of vector constructs with cellular DNA | |
| JP3844656B2 (en) | Methods for transformation of animal cells | |
| JPH06507550A (en) | Gene manipulation and expression using genomic elements | |
| US5543319A (en) | Recombination-proficient avian/mammalian microcell hybrids | |
| WO1997026327A1 (en) | Compositions and methods for mediating cell cycle progression | |
| AU684275B2 (en) | Method for defined deletions of DNA | |
| CN112218945A (en) | Method for producing homozygous cells | |
| JPH06508745A (en) | Increased expression by targeting genes into endogenous retrovirus-like sequences | |
| US8361711B2 (en) | Tools and methods useful in characterising the immunotoxic activity of xenobiotic substances | |
| WO1993005149A1 (en) | Subtractive hybridization cloning for probes for gene isolation and mapping | |
| CA2265492A1 (en) | Mammalian and human rec2 | |
| JP2002505858A (en) | Non-human transgenic mammal, method of production and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP KR NO |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LU MC NL SE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1992913238 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2092695 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1992913238 Country of ref document: EP |
|
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication |
Free format text: UA |
|
| COP | Corrected version of pamphlet |
Free format text: PAGES 36-38,AMENDED CLAIMS,REPLACED BY CORRECT PAGES 36-39 |
|
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication |
Free format text: BY |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1992913238 Country of ref document: EP |