WO1988005661A1 - Vasopressin-based pharmaceutical compositions - Google Patents
Vasopressin-based pharmaceutical compositions Download PDFInfo
- Publication number
- WO1988005661A1 WO1988005661A1 PCT/US1988/000349 US8800349W WO8805661A1 WO 1988005661 A1 WO1988005661 A1 WO 1988005661A1 US 8800349 W US8800349 W US 8800349W WO 8805661 A1 WO8805661 A1 WO 8805661A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- administration
- polypeptide
- vasopressin
- composition according
- composition
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/095—Oxytocins; Vasopressins; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
Definitions
- the present invention relates to novel pharmaceutical compositions containing biologically-active vasopressin-based. polypeptides in combination with carboxymethylcellulose.
- the invention is concerned with vasopressin-based polypeptides which have been mixed with carboxymethylcellulose to produce non-orally administered pharmaceutical compositions, and which exhibit prolonged activity and increased bioavailability of the polypeptide.
- vasopressin has biological effects, such as antidiuretic activity and vasoconstriction of visceral blood flow. Its utility, however, is limited by its relatively short half-life in the blood stream, as well as its well known general circulatory systemic pressor effect, and cardiac toxicity.
- vasopressin A variety of analogs of vasopressin have been synthesized in an effort to modify the properties of vasopressin and provide products having increased pharmaceutical utility.
- vasopressin has been modified by deamination of cysteine at the 1 position and replacement of arginine by its D-isomer at the 8 position to yield desmopressin ("dDAVP").
- dDAVP desmopressin
- Desmopressin which is immune to enzymatic cleavage of the 1-2 and 8-9 carbon-nitrogen bonds, exhibits prolonged antidiuretic activity with low pressor activity. See U.S. Patent No. 3,497,491.
- the disulfide bridge has been replaced with a "monocarba” linkage (-CH-S- or -SCH 2 ⁇ ) or a dicarba (-CH 2 -CH_-) linkage.
- vasopressin analogs having a chain of cleavable peptide residues attached to the active molecule.
- a short peptide chain is attached to the N-terminal amino group.
- N(glycyl-glycyl-glycyl)-8-lysine-vasopressin, or tGLVP also referred to as terlipressin or glypressin
- tGLVP also referred to as terlipressin or glypressin
- a short peptide side chain may be attached to the N position of 8-lysine. In any event, the additional amino acids of the side chain are slowly cleaved enzymatically so that prolonged release of active nonapeptide 8-lysine-vasopressin is achieved.
- vasopressin-based polypeptides have been administered by a variety of routes and in combination with a variety of pharmaceutical carriers and additives.
- terlipressin has been administered by i.v. injection in the form of a physiological saline solution at pH 4
- desmopressin has been administered intranasally, subcutaneously, intravenously, and intramuscularly in physiological saline at pH 4.
- vasopressin-based polypeptides such as desmopressin. It has traditionally been accepted that such peptides are decomposed in the gastrointestinal tract with little nonapeptide absorption taking place. Nonetheless, desmopressin has been incorporated in gelatin-based sub-lingual lozenges. (A. Grossman et al. "Two new modes of desmopressin (dDAVP) administration", British Medical Journal, May 17, 1980, 1215.) Additionally, desmopressin has been formulated into an orally administered composition in the form of a tablet with various fillers and inert constituents, including crosslinked carboxymethylcellulose. (International Patent Application No. PCT/US84/01860, H. Hagsta et al. ) , but the effective unit dosage appears to be ten times the effective intranasal dosage conventionally required, and no difference in duration of antidiuretic action was reported or claimed.
- U.S. Patent No. 3,454,549 discloses still another vasopressin analog, l-desamino-[8-L-Arg]-vasopressin, and states that it can be used in the form of its free base, or as a salt of an organic or inorganic acid, on an acid-radical containing polymer, with carboxymethylcellulose being mentioned as an illustrative acid polymer. There is no recognition, however, that any acid salt, much less the CMC salt, has any effect on the activity of the vasopressin analog.
- Still another object of this invention is the provision of a method for administering a vasopressin-based polypeptide to a patient in which the activity of the polypeptide is prolonged.
- the present invention provides a pharmaceutical composition which comprises a vasopressin-based polypeptide combined with carboxymethylcellulose in an amount sufficient to provide improved activity of the polypeptide.
- the inventive composition has enhanced- physiological activity and a long effective lifetime as compared with the polypeptide alone.
- vasopressin-based polypeptides employed in the invention may be represented by the following formula: B-Tyr-X 1 -Gln-Asn-CH-CO-X 2 -D-X 3 -NH. t I ⁇
- A is disulfide or a monocarba (-CH-S- or -SCH--) linkage;
- B is -C IH Z_, -CIHNH Z, or -CjHENH Z_;
- E is a peptide chain of from 1 to 3 amino acid residues;
- D is a basic amino acid residue, e.g. D-Arg or Lys, or a lysine peptide residue which is substituted at the W ⁇ position with a peptide chain of from 1 to 3 peptide residues; and each of X 1, X2 and X3 is an amino acid residue.
- an "amino acid residue” is the divalent moiety obtained by removing a hydrogen from the alpha amino group and the hydroxyl group from the carboxyl group of an alpha-amino acid.
- Especially preferred polypeptides which can be used according to the invention, and which are encompassed by the above formula, are desmopressin ("dDAVP") and terlipressin ("tGLVP").
- dDAVP desmopressin
- tGLVP terlipressin
- X is phenylalanine (Phe)
- X 2 is prolm. e (Pro)
- X3 is glycine (Gly).
- the invention is also directed to a method for treating a patient with a vasopressin-based polypeptide, especially a patient suffering from diabetes insipidus, incontinence, enuresis, or sickle cell anemia. Additionally, the invention pertains to a method for minimizing or preventing gastrointestinal or uterine bleeding in a patient suffering such distress, and for controlling the blood pressure changes associated with hemorrage and the management of burns. According to the inventive method, a pharmaceutical composition which comprises a vasopressin-based polypeptide having the above formula and carboxymethylcellulose is administered to a patient in an amount effective to produce and prolong the intended physiological response.
- vasopressin-based pharmaceutical composition which has enhanced pharmacological activity and increased duration of action by combining a vasopressin-based polypeptide with carboxmethylcellulose.
- vasopressin-based polypeptide includes vasopressin itself, as well as its homologs, analogs, hormonogens and other variants possessing similar activity.
- polypeptides of the invention may be represented by the following formula:
- A is a disulfide (-S-S-) or monocarba (-CH-S- or -SCH--) linkage?
- B is a divalent moeity selected from the group consisting of -CHNH 2 , _ CH 2 , and -CHENH 2 ;
- E is a peptide chain of from 1 to 3 amino acid residues;
- D is a basic amino acid residue, such as arginine (Arg), lysine (Lys), or ornithine (Orn) , or a lysine peptide residue substituted at the N position with a chain of 1 to 3 amino acid residues; and each of X 1, X2 and X3 is an amino acid residue.
- the polypeptide may be vasopressin. If so, B is -CHNH 2 ; A is disulfide; D is Arg; and the 1 and 6 positions are cysteine (Cys).
- the peptide preferably is an analog or hormonogen which exhibits increased resistance to enzymatic cleavage so that the biovailability of the peptide is enhanced.
- the peptide is desmopressin or terlipressin.
- desmopressin A is disulfide
- B is -CH (the 1 position is mercaptopropionic acid)
- D is -[-D-Arg]-.
- terlipressin A is disulfide
- B is -CH-Gly-Gly-Gly-NH 2
- E is a tripeptide chain of three Gly residues
- D is Lys.
- CMC carboxymethylcellulose
- CMC has been used as an inert and safe "filler" in tablet formulations, while the sodium salt of CMC has been given orally as a weak antacid. CMC has also been safely used in injectable formulations in connection with other molecules than the peptides encompassed by the formula given.
- the present invention is premised on the previously unrecognized fact that CMC can be noncovalently complexed or otherwise combined with a vasopressin-based polypeptide to provide a pharmaceutical composition showing increased bioavailability and prolonged effect of the polypeptide.
- CMC is not critical, provided it is water soluble.
- the amount of CMC should be sufficient to impart improved polypeptide activity, as reflected by increased activity and/or increased half-life.
- the weight ratio of CMC to polypeptide should be at least 100:1. Ratios of from about 100:1 to 200:1 are preferred, with a ratio of about 100:1 being especially preferred.
- higher amounts of CMC should be avoided. Otherwise/ the viscosity of the resulting solution will be too high.
- a minimum amount of CMC must be complexed with the peptide in order to achieve the advantages of the invention.
- the degree to which the peptide is complexed at any pH will depend to some extent on the isoelectric point of the peptide selected, and the amount and type of other ionic and polar molecules which are present to compete with the peptide and CMC.
- compositions of the invention dosage and route of administration will ultimately depend upon the'patient's unique conditions, and the judgment of his attending physician, they can generally be administered to the patient being treated at dosages and by routes calculated to deliver effective amounts of polypeptide to the site of action.
- they may be administered by intravenous, subcutaneous, or intramuscular injection, or they may be administered intranasally in the form of nose drops or spray.
- the composition of this invention can be administered as a dispersion or in solution in a suitable liquid medium, such as water, saline, isotonic aqueous solution and alcohol.
- a suitable liquid medium such as water, saline, isotonic aqueous solution and alcohol.
- the medium may contain various pharmaceutically acceptable fillers and additives, such as anticlotting agents, dispersing agents, acidifying agents and the like.
- a preferred medium is physiological saline solution.
- concentration of the polypeptide in the solution is not narrowly critical and and will depend upon the polypeptide, the presence or absence of a disulfide linkage, the pH, the intended mode of administration, and dosage.
- solutions intended for intranasal applications may contain higher concentrations than solutions intended to be administered by injection, but dimerisation and peptide stability are usually the decisive factors here.
- the dosage of the composition which is administered will depend greatly upon the specific action(s) of the polypeptide, the level(s) of such action(s), the effect intended, and the mode of administration. For example, when administered in the form of nose drops, the applied dosage will be about 10 times the applied dose administered by intravenous routes.
- the effective dosage may be significantly decreased for all modes of administration pursuant to the invention.
- Aii acid physiological saline solution (pH 4) containing 10 ug/ml of Desmopressin (dDAVP) (manufactured by Ferring AB) and chlorobutanol was prepared and divided into two aliquots. One of the aliquots was set aside, while the other aliquot was mixed with sufficient Na-CMC (Tylopur C30, manufactured by Kalle) to obtain a CMC concentration of 2 mg/ml.
- the CMC:dDAVP weight ratio of the CMC-containing aliquot was 200:1, at a pH of 4.
- mice having a body weight of 180-200 g Male rats having a body weight of 180-200 g were anesthetised with Methohexital-Na (Brevimytal, manufactured by Lilly), and then surgically prepared by inserting a tracheal cannula into a carotid artery in order to draw blood samples (0.5 ml/sample) into heparinised tuberculin syringes, and a polyethylene cannula into a jugular vein to rehydrate and replace blood volumes lost during sampling, by injecting equal volumes of iso-oncotic dextran in saline. Both cannulas were inserted in a central direction. Zero-time samples were withdrawn to serve as a baseline.
- Methohexital-Na (Brevimytal, manufactured by Lilly)
- dDAVP and dDAVP-CMC solutions were then subcutaneously injected into the rats in a volume dose of 0.1 ml/lOOg body weight. Blood samples were withdrawn at 5, 10, 20, and 40 minutes after injection. These times were chosen because the half-life ( ⁇ -,/ 2 ) °f dDVAP in man after i.v. injection has already been determined to be 17-18 min. (K.E. Andersson et al., Acta Endocrinol, 69:640, 1972), as opposed to about 3 min. for the natural vasopressins and tGLVP. (V. Pliska et al., Exoerientia, 29:767, 1973).
- the heparinised samples were stirred and centrifuged. Then, the supernatant plasma was pipetted off and stored in separate plastic tubes at -70°C until analyzed.
- RIA radio-immune assay
- a commercial goat-anti-rabbit plasma was used to precipitate An-Ab complexes, while 125I was used to label the 2-Tyr of the dDAVP. Even though the Ab used was Arg-vasopressin specific, it showed sufficient cross-reactivity with other vasopressins, including dDAVP, to allow detection of plasma levels of dDAVP down to 7 pg/ml.
- Terlipressin (manufactured by the Chemical Works of Gedeon Richter, Ltd., Budapest) was prepared in two aliquots of tGLVP and tGLVP-CMC at the same concentrations and under the same conditions as was dDAVP in Example 1 above.
- mice having weights of 180-200 g, were anaesthetised with urethane i.p. and s.c, but otherwise were surgically prepared exactly as in Example 1 above. This time, however, after the withdrawal of the 0-time blood samples, tGLVP and tGLVP-CMC were given i.v. into the jugular vein cannula. Because tGLVP is a long-acting polypeptide, the times of sampling were longer than in Example 1: 10, 20, 40, 60 and 180 min. sampling times were used.
Abstract
Pharmaceutical compositions containing biologically active vasopressin-based polypeptides in combination with carboxymethyl cellulose. The compositions exhibit prolonged activity and increased bioavailability of the polypeptide.
Description
VASOPRESSIN-BASED PHARMACEUTICAL COMPOSITIONS
BACKGROUND OF THE INVENTION
The present invention relates to novel pharmaceutical compositions containing biologically-active vasopressin-based. polypeptides in combination with carboxymethylcellulose.
More particularly, the invention is concerned with vasopressin-based polypeptides which have been mixed with carboxymethylcellulose to produce non-orally administered pharmaceutical compositions, and which exhibit prolonged activity and increased bioavailability of the polypeptide.
It has long been recognized that natural vasopressin has biological effects, such as antidiuretic activity and vasoconstriction of visceral blood flow. Its utility, however, is limited by its relatively short half-life in the blood stream, as well as its well known general circulatory systemic pressor effect, and cardiac toxicity.
A variety of analogs of vasopressin have been synthesized in an effort to modify the properties of vasopressin and provide products having increased
pharmaceutical utility. For example, vasopressin has been modified by deamination of cysteine at the 1 position and replacement of arginine by its D-isomer at the 8 position to yield desmopressin ("dDAVP"). Desmopressin, which is immune to enzymatic cleavage of the 1-2 and 8-9 carbon-nitrogen bonds, exhibits prolonged antidiuretic activity with low pressor activity. See U.S. Patent No. 3,497,491. In further modifications, the disulfide bridge has been replaced with a "monocarba" linkage (-CH-S- or -SCH2~) or a dicarba (-CH2-CH_-) linkage.
A different approach has been used in the preparation of hormonogens, i.e., vasopressin analogs having a chain of cleavable peptide residues attached to the active molecule. Typically, a short peptide chain is attached to the N-terminal amino group. Of these analogs, N(glycyl-glycyl-glycyl)-8-lysine-vasopressin, or tGLVP (also referred to as terlipressin or glypressin) is of particular interest. See, FRG Patent No. 1,493,561. Alternatively, a short peptide side chain may be attached to the N position of 8-lysine. In any event, the additional amino acids of the side chain are slowly cleaved enzymatically so that prolonged release of active nonapeptide 8-lysine-vasopressin is achieved.
These vasopressin-based polypeptides have been administered by a variety of routes and in combination with a variety of pharmaceutical carriers and additives. For example, terlipressin has been administered by i.v. injection in the form of a physiological saline solution at pH 4, while desmopressin has been administered intranasally, subcutaneously, intravenously, and intramuscularly in physiological saline at pH 4.
Various attempts have also been made to effectively orally administer vasopressin-based polypeptides, such
as desmopressin. It has traditionally been accepted that such peptides are decomposed in the gastrointestinal tract with little nonapeptide absorption taking place. Nonetheless, desmopressin has been incorporated in gelatin-based sub-lingual lozenges. (A. Grossman et al. "Two new modes of desmopressin (dDAVP) administration", British Medical Journal, May 17, 1980, 1215.) Additionally, desmopressin has been formulated into an orally administered composition in the form of a tablet with various fillers and inert constituents, including crosslinked carboxymethylcellulose. (International Patent Application No. PCT/US84/01860, H. Hagsta et al. ) , but the effective unit dosage appears to be ten times the effective intranasal dosage conventionally required, and no difference in duration of antidiuretic action was reported or claimed.
Hagstam et al. disclose that such compositions are capable of dissolving and being absorbed in the gastrointestinal tract. However, this absorption apparently results from the selective use of the cleavage-resistant analog dDAVP; there is no indication that carboxymethylcellulose functions as other than an inert constituent. Indeed, in the example given, the crosslinked carboxymethylcellulose was only a minor component of a mixture of several excipients, and was present at a rate of only 2 to 4 parts per 100 parts of desmopressin.
U.S. Patent No. 3,454,549 discloses still another vasopressin analog, l-desamino-[8-L-Arg]-vasopressin, and states that it can be used in the form of its free base, or as a salt of an organic or inorganic acid, on an acid-radical containing polymer, with carboxymethylcellulose being mentioned as an illustrative acid polymer. There is no recognition,
however, that any acid salt, much less the CMC salt, has any effect on the activity of the vasopressin analog.
It is an object of the present invention to provide a novel pharmaceutical composition containing a vasopressin-based polypeptide which is combined with carboxymethylcellulose in an amount sufficient to provide prolonged and increased activity of the polypeptide.
It is a further object of the invention to provide a non-orally administered vasopressin-based pharmaceutical composition which is longer acting, and which has greater activity than the known vasopressin-based pharmaceutical compositions.
Still another object of this invention is the provision of a method for administering a vasopressin-based polypeptide to a patient in which the activity of the polypeptide is prolonged.
These objects and other advantages 'of the invention will be apparent from the detailed description which follows.
SUMMARY OF THE INVENTION
The present invention provides a pharmaceutical composition which comprises a vasopressin-based polypeptide combined with carboxymethylcellulose in an amount sufficient to provide improved activity of the polypeptide. The inventive composition has enhanced- physiological activity and a long effective lifetime as compared with the polypeptide alone.
The vasopressin-based polypeptides employed in the invention may be represented by the following formula:
B-Tyr-X1-Gln-Asn-CH-CO-X2-D-X3-NH. t I ^
CH^-A-CH
wherein A is disulfide or a monocarba (-CH-S- or -SCH--) linkage; B is -C IH Z_, -CIHNH Z, or -CjHENH Z_; E is a peptide chain of from 1 to 3 amino acid residues; D is a basic amino acid residue, e.g. D-Arg or Lys, or a lysine peptide residue which is substituted at the W~position with a peptide chain of from 1 to 3 peptide residues; and each of X 1, X2 and X3 is an amino acid residue.
In the context of the present invention, an "amino acid residue" is the divalent moiety obtained by removing a hydrogen from the alpha amino group and the hydroxyl group from the carboxyl group of an alpha-amino acid. Especially preferred polypeptides which can be used according to the invention, and which are encompassed by the above formula, are desmopressin ("dDAVP") and terlipressin ("tGLVP"). In these polypeptides, X is phenylalanine (Phe), X 2 is prolm. e (Pro), and X3 is glycine (Gly).
The invention is also directed to a method for treating a patient with a vasopressin-based polypeptide, especially a patient suffering from diabetes insipidus, incontinence, enuresis, or sickle cell anemia. Additionally, the invention pertains to a method for minimizing or preventing gastrointestinal or uterine bleeding in a patient suffering such distress, and for controlling the blood pressure changes associated with hemorrage and the management of burns. According to the inventive method, a pharmaceutical composition which comprises a vasopressin-based polypeptide having the above formula and carboxymethylcellulose is administered to a patient in an amount effective to produce and prolong the intended physiological response.
DETAILED DESCRIPTION OF THE INVENTION
While the invention is susceptible to various modifications and alternative forms, the preferred embodiments are described herein in detail. It is to be understood, however, that it is not intended to limit the invention to the specific forms disclosed. On the contrary, it is intended to cover all modifications and alternative forms falling within the spirit and scope of the invention.
'In general, the present invention is predicated on the discovery that a vasopressin-based pharmaceutical composition can be provided which has enhanced pharmacological activity and increased duration of action by combining a vasopressin-based polypeptide with carboxmethylcellulose. As used herein, the phrase "vasopressin-based polypeptide" includes vasopressin itself, as well as its homologs, analogs, hormonogens and other variants possessing similar activity.
The polypeptides of the invention may be represented by the following formula:
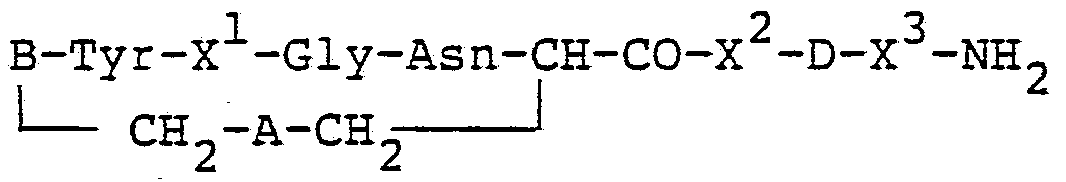
wherein A is a disulfide (-S-S-) or monocarba (-CH-S- or -SCH--) linkage? B is a divalent moeity selected from the group consisting of -CHNH2, _CH2, and -CHENH2; E is a peptide chain of from 1 to 3 amino acid residues; D is a basic amino acid residue, such as arginine (Arg), lysine (Lys), or ornithine (Orn) , or a lysine peptide residue substituted at the N position with a chain of 1 to 3 amino acid residues; and each of X 1, X2 and X3 is an amino acid residue.
The polypeptide may be vasopressin. If so, B is -CHNH2; A is disulfide; D is Arg; and the 1 and 6 positions are cysteine (Cys).
The peptide preferably is an analog or hormonogen which exhibits increased resistance to enzymatic cleavage so that the biovailability of the peptide is enhanced. Most preferably, the peptide is desmopressin or terlipressin. For desmopressin, A is disulfide, B is -CH (the 1 position is mercaptopropionic acid), and D is -[-D-Arg]-. For terlipressin, A is disulfide, B is -CH-Gly-Gly-Gly-NH2, E is a tripeptide chain of three Gly residues, and D is Lys.
It is believed that CMC is able to prolong and enhance the activity of vasopressin and its analog or hormonogen forms because it is able to form a complex with the vasopressin-based polypeptide. The polypeptides encompassed by the formula given above have an unusually high density of amino groups which are available for complexing via non-covalent association with the carboxyl groups of carboxymethylcellulose ("CMC"), i.e. the N at position 1, and the positions of 4, 5, and 8 (if Arg occupies position 8, the omega side-chain group is guanidino, which is also capable of associating with the carboxyl group of CMC). Vasopressin has five such amino groups available in nine amino acid residues, dDAVP has four such amino groups in nine residues, while tGLVP has 5 amino groups in 12 residues.
Previously, CMC has been used as an inert and safe "filler" in tablet formulations, while the sodium salt of CMC has been given orally as a weak antacid. CMC has also been safely used in injectable formulations in connection with other molecules than the peptides encompassed by the formula given. However, the present invention is premised on the previously unrecognized
fact that CMC can be noncovalently complexed or otherwise combined with a vasopressin-based polypeptide to provide a pharmaceutical composition showing increased bioavailability and prolonged effect of the polypeptide.
The specific form of CMC is not critical, provided it is water soluble. The amount of CMC should be sufficient to impart improved polypeptide activity, as reflected by increased activity and/or increased half-life. As a general rule, however, the weight ratio of CMC to polypeptide should be at least 100:1. Ratios of from about 100:1 to 200:1 are preferred, with a ratio of about 100:1 being especially preferred. In particular, if the composition is used in the form of an injectable solution, higher amounts of CMC should be avoided. Otherwise/ the viscosity of the resulting solution will be too high.
On the other hand, a minimum amount of CMC must be complexed with the peptide in order to achieve the advantages of the invention. The degree to which the peptide is complexed at any pH will depend to some extent on the isoelectric point of the peptide selected, and the amount and type of other ionic and polar molecules which are present to compete with the peptide and CMC.
Although the use of the pharmaceutical compositions of the invention, dosage and route of administration will ultimately depend upon the'patient's unique conditions, and the judgment of his attending physician, they can generally be administered to the patient being treated at dosages and by routes calculated to deliver effective amounts of polypeptide to the site of action. For example, they may be administered by intravenous, subcutaneous, or intramuscular injection, or they may be
administered intranasally in the form of nose drops or spray.
With these routes of administration, the composition of this invention can be administered as a dispersion or in solution in a suitable liquid medium, such as water, saline, isotonic aqueous solution and alcohol. The medium may contain various pharmaceutically acceptable fillers and additives, such as anticlotting agents, dispersing agents, acidifying agents and the like. A preferred medium is physiological saline solution. If the formulation is for multiple use, it is also desirable to include in the dispersion or solution a small amount of a physiologically acceptable bacteriostat, e.g., chlorobutanol, to minimize bacterial contamination. This is especially useful in the intranasal preparation.
The concentration of the polypeptide in the solution is not narrowly critical and and will depend upon the polypeptide, the presence or absence of a disulfide linkage, the pH, the intended mode of administration, and dosage. In general, solutions intended for intranasal applications may contain higher concentrations than solutions intended to be administered by injection, but dimerisation and peptide stability are usually the decisive factors here.
The dosage of the composition which is administered will depend greatly upon the specific action(s) of the polypeptide, the level(s) of such action(s), the effect intended, and the mode of administration. For example, when administered in the form of nose drops, the applied dosage will be about 10 times the applied dose administered by intravenous routes.
However, as shown in the Examples, because the
pharmaceutical compositions of the invention have enhanced bioavailability and prolonged effect, the effective dosage may be significantly decreased for all modes of administration pursuant to the invention.
The invention will be further described by reference to the following detailed examples, which are not intended to be limiting, but rather, illustrative of some approaches taken. These examples may, of course, be varied in accordance with the spirit and scope of this description.
EXAMPLE 1
Subcutaneous Administration of dDAVP to Rats
Aii acid physiological saline solution (pH 4) containing 10 ug/ml of Desmopressin (dDAVP) (manufactured by Ferring AB) and chlorobutanol was prepared and divided into two aliquots. One of the aliquots was set aside, while the other aliquot was mixed with sufficient Na-CMC (Tylopur C30, manufactured by Kalle) to obtain a CMC concentration of 2 mg/ml. Thus, the CMC:dDAVP weight ratio of the CMC-containing aliquot was 200:1, at a pH of 4.
Male rats having a body weight of 180-200 g were anesthetised with Methohexital-Na (Brevimytal, manufactured by Lilly), and then surgically prepared by inserting a tracheal cannula into a carotid artery in order to draw blood samples (0.5 ml/sample) into heparinised tuberculin syringes, and a polyethylene cannula into a jugular vein to rehydrate and replace blood volumes lost during sampling, by injecting equal volumes of iso-oncotic dextran in saline. Both cannulas were inserted in a central direction. Zero-time samples were withdrawn to serve as a baseline.
dDAVP and dDAVP-CMC solutions were then subcutaneously injected into the rats in a volume dose of 0.1 ml/lOOg body weight. Blood samples were withdrawn at 5, 10, 20, and 40 minutes after injection. These times were chosen because the half-life (τ-,/2) °f dDVAP in man after i.v. injection has already been determined to be 17-18 min. (K.E. Andersson et al., Acta Endocrinol, 69:640, 1972), as opposed to about 3 min. for the natural vasopressins and tGLVP. (V. Pliska et al., Exoerientia, 29:767, 1973).
The heparinised samples were stirred and centrifuged. Then, the supernatant plasma was pipetted off and stored in separate plastic tubes at -70°C until analyzed.
The plasma levels of dDAVP in the withdrawn samples were detected by radio-immune assay ( "RIA") , using a high affinity rabbit antibody prepared by Dr. M. Sofroniew of the Department of Anatomy at the University of Munchen.
A commercial goat-anti-rabbit plasma was used to precipitate An-Ab complexes, while 125I was used to label the 2-Tyr of the dDAVP. Even though the Ab used was Arg-vasopressin specific, it showed sufficient cross-reactivity with other vasopressins, including dDAVP, to allow detection of plasma levels of dDAVP down to 7 pg/ml.
Because of the small plasma samples involved, no extraction methods were applied to the plasma samples. For this reason, higher than usual 0-time levels were recorded and in all calculations the 0-time level was subtracted from all levels recorded after drug injection.
(Table 1 )
dDAVP dDAVP-CMC
Plasma Peak Time (min)
Means 8.8* 21.5*
±S.D. 1.3 6.6
±SEM 0.9 2.7
+95CL 1.7 5.3
Plasma (T, ,-) (min) Means 20.5* 58.8*
±S.D. 0.5 10.9
±SEM 0.35 4.9
+95CL 0.7 9.6
Plasma peak concentrations (pg/ml)
Means 27.0* 82.0*
+S.D. 1.5 38.7
+SEM 1.1 17.3
+95CL 2.1 34.0
All means pairs followed by * indicate a statistically significant difference P < 0.05.
The reported data clearly show that increased plasma concentration half-lives were obtained using the dDAVP-CMC compositons of the invention. In connection with the above, it is noted that the reported T,/2 values for dDAVP alone were generally in agreement with the previously reported values obtained with i.v. injection in man. The larger mean value reported here undoubtedly resulted because absorption is relatively slower after s.c. injection.
Additionally, the reported plasma T. ,_ values should not be confused with a biological response T 1/2 which would be much longer in all cases (c.f. Example 2)
However, the increased 1/2 value obtained using a dDAVP/CMC combination indicates that dDAVP activity would be even more prolonged in vivo.
The above data also show that a peak concentration was reached after a significantly longer time when the dDAVP/CMC combination was used. Again, both mean peak values were greater than would be obtained after i.v. injection.
Perhaps the most striking difference associated with the use of CMC was the significantly higher peak plasma concentrations achieved with dDAVP-CMC vs. dDAVP alone. Although no definitive explanation is offered at this time, it is possible that slower release of dDAVP from the dDAVP-CMC complex decreases the rate of either enzymatic cleavage or disappearance of dDAVP into other receptor fields than the target organ. In other words, combining dDAVP with CMC appears to increase the bioavailability of the active peptide.
EXAMPLE 2
Antidiuretic Response to Intranasal Administration of dDAVP in Man
Normal test subjects were loaded with 1.5% body weight of water under constantly maintained conditions, with urine being collected every 30 minutes, as described by J.H. Cort et al., Kidney International, 8:292, 1975, and then they were administered dDAVP intranasally, using a standard 0.1 g/ml formulation obtained from Ferring AB of Malmo. A portion of the dDAVP was set aside, while Na-CMC was added to another portion to form a solution having a CMC:dDAVP weight ratio of 100:1.
The dDAVP and dDAVP-CMC solutions were intranasally administered to each subject in doses of 2.5 ug/nostril or 5 ug/patient. The results are given in Table 2.
(Table 2)
dDAVP dDAVP-CMC
Peak % Inhibition of Diuretic Urine Flow
Means 76.7* -97.4* +S.D. 3.7 1.9 +SEM 1.5 1.1 +95 CL 3.0 2.2
τι/2 °^ Antidiuretic Response (h.)
Means 4.92* 11.0* +S.D. 1.30 4.08 +SEM 0.53 2.36 +95 CL 1.04 4.65
There were no side-effects or discomfort associated with the intranasal use of dDAVP-CMC but there was a 15-20 min delay in the onset of the antidiuretic response. Increased bioavailability induced by the CMC complexing was evidenced both by the significant increased in peak antidiuretic response and the approximate doubling of the biological (antidiuretic) T, ,~ . This doubling of the duration half-time (total time would be expected to show an even greater increase in prolongation) would indicate that: a) the effective average dose of peptide in dDAVP-CMC could be halved from 1 ug to 5 ug/adult Diabetes insipidus patient and b) this halved dose could be given once daily instead of b.i.d.
EXAMPLE 3
Intravenous Administration of Terlipressin
Terlipressin (tGLVP) (manufactured by the Chemical Works of Gedeon Richter, Ltd., Budapest) was prepared in two aliquots of tGLVP and tGLVP-CMC at the same concentrations and under the same conditions as was dDAVP in Example 1 above.
Female rats, having weights of 180-200 g, were anaesthetised with urethane i.p. and s.c, but otherwise were surgically prepared exactly as in Example 1 above. This time, however, after the withdrawal of the 0-time blood samples, tGLVP and tGLVP-CMC were given i.v. into the jugular vein cannula. Because tGLVP is a long-acting polypeptide, the times of sampling were longer than in Example 1: 10, 20, 40, 60 and 180 min. sampling times were used.
Peptide plasma levels were again determined by radio- immune assay (RIA) methods. As shown by M.L. Forsling et al., J. Endocr. , 85:237, 1980, immunoreactive vasopressin concentrations in plasma following administration of tGLVP show an initial exponential decay curve with a T. /2 of about 24 min, which is considered to represent the tGLVP dodecapeptide, followed by a second peak representing lysine- vasopressin released from tGLVP. Since the Ab reacts with the Pro-Lys-Gly-NH2 portion of the peptide hormone, it detects both LVP and tGLVP, and may also detect tGLVP-CMC.
Because single RIA measurements obtained from the complex three component system involved in the present experiment would have little meaning, if indeed such measurements could be obtained at all, the only data
considered to be of significance was the following tGLVP vs. tGLVP-CMC comparison of bioavailability and the time of appearance of the second (released LVP) peak in the curve. These latter parameters are presented in Table 3:
Table 3
tGLVP tGLVP-CMC Initial plasma peak cone. (10 min)(pg.ml)
Means 125* 2820* S . D . 39 784 +SEM 17 351 +95CL 33 691
Time of .second peak (min)
Means 44* 120* +S.D. 15 60 +SEM 7 27 +95CL 14 53
The highly significant increased peak concentration cannot be analysed or explained only on the basis of RIA measurements, but does present a distinct improvement in bioavailability.
Either due to the increased bioavailability or to the slow release of LVP from tGLVP, or to a combination of both, it would appear that the total biological T. ,„ would be significantly prolonged by combining the peptide with CMC.
Claims
1. A pharmaceutical composition which comprises carboxymethylcellulose and a vasopressin-based polypeptide represented by the formula:

wherein A is disulfide or a monocarba linkage;
B is -CIH_2, -CiHNH2-, or -CiHENH2-;
D is a basic amino acid residue or a lysine peptide residue which is substituted at the N position with a peptide chain of from 1 to 3 amino acid residues;
E is a peptide chain of from 1 to 3 amino acid residues; and
each of X 1, X2 and X3 is a ammo acid residue;
and wherein the polypeptide is is combined with carboxymethylcellulose in an amount sufficient to provide improved activity of the polypeptide.
2. The composition according to claim 1, wherein the weight ratio of CMC to polypeptide is from about 100:1 to about 200:1.
3. The composition according to claim 2, wherein A is disulfide; B is -CH_; and D is [8-D-Arg]-.
I
4. The composition according to claim 2, wherein A is disulfide; B is -CH-Gly-Gly-Gly-NH_; and D is Lys.
5. The composition according to claim 2, wherein D is a basic peptide residue selected from the group consisting of Arg, Lys, and Orn.
6. The composition according to claim 1, wherein the weight ratio of carboxymethylcellulose to polypeptide is about 100:1.
7. The composition according to claim 1, wherein the composition is in the form of a freeze dried powder.
8. The composition according to claim 1, wherein the composition is in the form of an aqueous solution.
9. The composition according to claim 8, further comprising a bacterial growth inhibitor.
10. The composition according to claim 1, further comprising an inert and pharmaceutically acceptable filler.
11. A method for administering a vasopressin-based polypeptide to a patient, comprising administering to said patient an amount of the composition according to claim 1 effective to produce and prolong the intended pharmacological response.
12. The method according to claim 11, wherein the patient suffers from diabetes insipidus, and the route of administration is selected from the group consisting of intravenous, subcutaneous, intramuscular, and intranasal administration.
13. The method according to claim 11, wherein the patient suffers from enuresis or incontinence, and the route of administration is selected from the group consisting of intravenous, subcutanous, intra uscular, and intranasal administration.
14. The method according to claim 1, wherein the patient suffers from sickle cell anemia, and the route of administration is selected from the group consisting of intravenous, subcutanous, intramuscular and intranasal administration.
15. The method according to claims 12, 13, or 14 wherein the polypeptide is desmopressin.
16. The method according to claim 11, wherein the patient has gastrointestinal bleeding, and the route of administration is selected from the group consisting of intravenous, subcutanous, intramuscular, and intranasal administration.
17. The method according to claim 11, wherein the patient suffers from uterine bleeding, and the route of administration is selected from the group consisting of intravenous, subcutanous, intramuscular, and intranasal administration.
18. The method according to claim 11 for the management of the blood pressure of a patient suffering from burns, and the route of administration is selected from the group consisting of intravenous, subcutanous, intramuscular, and intranasal administration.
19. The method accordi...: co claims 16, 17 or 18 wherein the polypeptide is terlipressin.
20. The method according to claims 11, 12, 13, 15, 16 17 or 18, wherein the pharmaceutical composition is administered in the form of a solution.
21. The method according to claims 11, 12, 13, 15, 16, 17 or 18, wherein the pharmaceutical composition is in the form of a freeze-dried powder and the method further comprises dissolving the powder to form a solution of the composition prior to administration.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US884187A | 1987-01-30 | 1987-01-30 | |
| US008,841 | 1987-01-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1988005661A1 true WO1988005661A1 (en) | 1988-08-11 |
Family
ID=21733999
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1988/000349 WO1988005661A1 (en) | 1987-01-30 | 1988-01-27 | Vasopressin-based pharmaceutical compositions |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP0300036A4 (en) |
| WO (1) | WO1988005661A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0381345A1 (en) * | 1989-01-30 | 1990-08-08 | Corint, Ltd. | Aqueous liquid desmopressin-CMC pharmaceutical composition |
| DE4229880A1 (en) * | 1992-09-04 | 1994-03-31 | Knauf Siegfried | Medicaments contg. oxytocin, vasopressin and/or endorphins - for treating sex crime, bed-wetting, autism, homosexulaity and bulimia |
| WO1995001183A1 (en) * | 1993-06-29 | 1995-01-12 | Ferring B.V. | Compositions for nasal administration of desmopressin |
| WO1998043661A1 (en) * | 1997-03-27 | 1998-10-08 | Thomas Lundeberg | Use of substances having oxytocin activity for preparation of medicaments for wound healing |
| US5968895A (en) * | 1996-12-11 | 1999-10-19 | Praecis Pharmaceuticals, Inc. | Pharmaceutical formulations for sustained drug delivery |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB155919A (en) * | 1919-09-29 | 1920-12-29 | Eugene Bregeon Temple | An improved hydraulic variable speed transmitter |
| US3502545A (en) * | 1968-09-27 | 1970-03-24 | Monsanto Co | Process for the separation of water-soluble polymeric materials from unbound protein or peptide using semiporous gel |
| US3679653A (en) * | 1968-09-27 | 1972-07-25 | Monsanto Co | Hormonally-active reaction product of a polymer with a hormone |
| US3980631A (en) * | 1973-11-09 | 1976-09-14 | Ceskoslovenska Akademie Ved | Novel analogs of deamino-vasopressin with a modified disulfide bridge and manufacturing process thereof |
| US4093610A (en) * | 1977-01-31 | 1978-06-06 | American Home Products Corporation | Process for producing triglycyl-lysine vasopressin and intermediates therefor |
| US4261980A (en) * | 1980-05-16 | 1981-04-14 | Vega Laboratories, Inc. | Method for prophylaxis and/or treatment of sickle cell disease |
| US4263283A (en) * | 1980-05-16 | 1981-04-21 | Ferring Pharmaceuticals, Inc. | Method for prophylaxis and/or treatment of sickle cell disease |
| US4399125A (en) * | 1981-03-24 | 1983-08-16 | Maurice Manning | Novel antagonists of the antidiuretic action of arginine vasopressin |
| US4613500A (en) * | 1983-03-09 | 1986-09-23 | Teijin Limited | Powdery pharmaceutical composition for nasal administration |
| US4649130A (en) * | 1984-01-26 | 1987-03-10 | Medical College Of Ohio | Novel derivatives of arginine vasopressin antagonists |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE8306367L (en) * | 1983-11-18 | 1985-05-19 | Ferring Ab | ANTIDIURETICALLY EFFECTIVE PHARMACEUTICAL PREPARATION |
| SE457055B (en) * | 1986-08-18 | 1988-11-28 | Ferring Ab | TOPIC RADIATION GEL FOR MUKOSA CONTAINING VARIETY CONDUCTIVE SUBSTANCES |
-
1988
- 1988-01-27 WO PCT/US1988/000349 patent/WO1988005661A1/en not_active Application Discontinuation
- 1988-01-27 EP EP19880903003 patent/EP0300036A4/en not_active Withdrawn
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB155919A (en) * | 1919-09-29 | 1920-12-29 | Eugene Bregeon Temple | An improved hydraulic variable speed transmitter |
| US3502545A (en) * | 1968-09-27 | 1970-03-24 | Monsanto Co | Process for the separation of water-soluble polymeric materials from unbound protein or peptide using semiporous gel |
| US3679653A (en) * | 1968-09-27 | 1972-07-25 | Monsanto Co | Hormonally-active reaction product of a polymer with a hormone |
| US3980631A (en) * | 1973-11-09 | 1976-09-14 | Ceskoslovenska Akademie Ved | Novel analogs of deamino-vasopressin with a modified disulfide bridge and manufacturing process thereof |
| US4093610A (en) * | 1977-01-31 | 1978-06-06 | American Home Products Corporation | Process for producing triglycyl-lysine vasopressin and intermediates therefor |
| US4261980A (en) * | 1980-05-16 | 1981-04-14 | Vega Laboratories, Inc. | Method for prophylaxis and/or treatment of sickle cell disease |
| US4263283A (en) * | 1980-05-16 | 1981-04-21 | Ferring Pharmaceuticals, Inc. | Method for prophylaxis and/or treatment of sickle cell disease |
| US4399125A (en) * | 1981-03-24 | 1983-08-16 | Maurice Manning | Novel antagonists of the antidiuretic action of arginine vasopressin |
| US4613500A (en) * | 1983-03-09 | 1986-09-23 | Teijin Limited | Powdery pharmaceutical composition for nasal administration |
| US4649130A (en) * | 1984-01-26 | 1987-03-10 | Medical College Of Ohio | Novel derivatives of arginine vasopressin antagonists |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0300036A4 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0381345A1 (en) * | 1989-01-30 | 1990-08-08 | Corint, Ltd. | Aqueous liquid desmopressin-CMC pharmaceutical composition |
| DE4229880A1 (en) * | 1992-09-04 | 1994-03-31 | Knauf Siegfried | Medicaments contg. oxytocin, vasopressin and/or endorphins - for treating sex crime, bed-wetting, autism, homosexulaity and bulimia |
| WO1995001183A1 (en) * | 1993-06-29 | 1995-01-12 | Ferring B.V. | Compositions for nasal administration of desmopressin |
| US5498598A (en) * | 1993-06-29 | 1996-03-12 | Ferring Ab | Composition for nasal administration of desmopressin |
| CN1101701C (en) * | 1993-06-29 | 2003-02-19 | 凡林有限公司 | Compositions for nasal administration of desmopressin |
| US5968895A (en) * | 1996-12-11 | 1999-10-19 | Praecis Pharmaceuticals, Inc. | Pharmaceutical formulations for sustained drug delivery |
| US6180608B1 (en) | 1996-12-11 | 2001-01-30 | Praecis Pharmaceuticals, Inc. | Pharmaceutical formulations for sustained drug delivery |
| US6699833B1 (en) | 1996-12-11 | 2004-03-02 | Praecis Pharmaceuticals, Inc. | Pharmaceutical formulations for sustained drug delivery |
| WO1998043661A1 (en) * | 1997-03-27 | 1998-10-08 | Thomas Lundeberg | Use of substances having oxytocin activity for preparation of medicaments for wound healing |
| US6262021B1 (en) | 1997-03-27 | 2001-07-17 | Entretech Medical Ab | Use of substances having oxytocin activity for preparation of medicaments for wound healing |
| AU749479B2 (en) * | 1997-03-27 | 2002-06-27 | Entretech Medical Ab | Use of substances having oxytocin activity for preparation of medicaments for wound healing |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0300036A1 (en) | 1989-01-25 |
| EP0300036A4 (en) | 1989-06-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100256715B1 (en) | Composition for the sublingual or buccal administration of therapeutic agents | |
| JP4033498B2 (en) | Pharmaceutical formulations containing water-insoluble complexes for sustained drug delivery | |
| US5487898A (en) | Compositions and method for the sublingual or buccal administration therapeutic agents | |
| US5725852A (en) | Transmucosal therapeutic composition | |
| KR930003332B1 (en) | Process for preparing composition contained antidiuretically effective amount of 1-deamino-8-d-arginine | |
| JP2004504406A (en) | Ghrelin antagonist | |
| JPH0430926B2 (en) | ||
| ZA200500585B (en) | Aqueous preparation containing oligopeptides and etherified cyclodextrin | |
| US6150331A (en) | Human growth hormone-containing aqueous pharmaceutical composition | |
| US7718599B2 (en) | Pharmaceutical administration form for peptides, process for its preparation, and use | |
| JP2000504696A (en) | Stabilized growth hormone formulation and method of making same | |
| GB2193891A (en) | Nasal pharmaceutical compositions containing octreotide | |
| CA2062659A1 (en) | Composition for sustained-release of erythropoietin | |
| WO1988005661A1 (en) | Vasopressin-based pharmaceutical compositions | |
| EP0381345B1 (en) | Aqueous liquid desmopressin-CMC pharmaceutical composition | |
| US4816443A (en) | Piptides having ANF activity | |
| AU768882B2 (en) | Sustained release salts of pharmaceutically active peptides and their production | |
| AU2004283526A1 (en) | Therapeutic applications for C-peptide | |
| IE60761B1 (en) | Novel galenical formulations | |
| WO1983002271A1 (en) | Use of peptides as a medicament |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE FR GB IT LU NL SE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1988903003 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1988903003 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1988903003 Country of ref document: EP |