USRE41571E1 - Method of providing sustained analgesia with buprenorphine - Google Patents
Method of providing sustained analgesia with buprenorphine Download PDFInfo
- Publication number
- USRE41571E1 USRE41571E1 US11/799,610 US79961007A USRE41571E US RE41571 E1 USRE41571 E1 US RE41571E1 US 79961007 A US79961007 A US 79961007A US RE41571 E USRE41571 E US RE41571E
- Authority
- US
- United States
- Prior art keywords
- dosing interval
- hours
- initiation
- plasma concentration
- mean plasma
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 title claims abstract description 328
- 229960001736 buprenorphine Drugs 0.000 title claims abstract description 325
- 238000000034 method Methods 0.000 title claims abstract description 107
- 230000036592 analgesia Effects 0.000 title abstract description 35
- 230000002459 sustained effect Effects 0.000 title description 4
- 230000036470 plasma concentration Effects 0.000 claims abstract description 358
- 208000002193 Pain Diseases 0.000 claims abstract description 50
- 230000036407 pain Effects 0.000 claims abstract description 48
- 230000000977 initiatory effect Effects 0.000 claims description 335
- 230000037317 transdermal delivery Effects 0.000 claims description 185
- 241000282414 Homo sapiens Species 0.000 claims description 69
- 230000001225 therapeutic effect Effects 0.000 claims description 23
- 239000004480 active ingredient Substances 0.000 claims description 17
- 229920000058 polyacrylate Polymers 0.000 claims description 10
- 239000002253 acid Substances 0.000 claims description 5
- 230000007423 decrease Effects 0.000 claims description 5
- 230000006872 improvement Effects 0.000 claims description 4
- 230000000052 comparative effect Effects 0.000 description 79
- 239000003814 drug Substances 0.000 description 64
- 229940079593 drug Drugs 0.000 description 62
- 210000002381 plasma Anatomy 0.000 description 37
- 239000010410 layer Substances 0.000 description 28
- 230000000694 effects Effects 0.000 description 24
- 239000000203 mixture Substances 0.000 description 23
- 238000001990 intravenous administration Methods 0.000 description 21
- 230000003285 pharmacodynamic effect Effects 0.000 description 20
- 239000013543 active substance Substances 0.000 description 18
- 239000000463 material Substances 0.000 description 18
- 238000011282 treatment Methods 0.000 description 18
- 229920000642 polymer Polymers 0.000 description 17
- 238000001802 infusion Methods 0.000 description 16
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 16
- 230000000202 analgesic effect Effects 0.000 description 15
- -1 discussed above) Chemical compound 0.000 description 15
- 239000011159 matrix material Substances 0.000 description 15
- 230000002035 prolonged effect Effects 0.000 description 14
- 238000009472 formulation Methods 0.000 description 13
- 239000006211 transdermal dosage form Substances 0.000 description 13
- 239000000014 opioid analgesic Substances 0.000 description 12
- 239000012790 adhesive layer Substances 0.000 description 11
- 229920001577 copolymer Polymers 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- JOOXCMJARBKPKM-UHFFFAOYSA-N 4-oxopentanoic acid Chemical compound CC(=O)CCC(O)=O JOOXCMJARBKPKM-UHFFFAOYSA-N 0.000 description 10
- 210000004369 blood Anatomy 0.000 description 10
- 239000008280 blood Substances 0.000 description 10
- 239000002552 dosage form Substances 0.000 description 10
- 230000004907 flux Effects 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- 238000007726 management method Methods 0.000 description 9
- 239000003961 penetration enhancing agent Substances 0.000 description 9
- 229920000728 polyester Polymers 0.000 description 9
- 238000010521 absorption reaction Methods 0.000 description 8
- 239000000853 adhesive Substances 0.000 description 8
- 230000001070 adhesive effect Effects 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 238000010241 blood sampling Methods 0.000 description 8
- 229960005181 morphine Drugs 0.000 description 8
- 229940005483 opioid analgesics Drugs 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 230000036765 blood level Effects 0.000 description 7
- 230000008030 elimination Effects 0.000 description 7
- 238000003379 elimination reaction Methods 0.000 description 7
- 239000011888 foil Substances 0.000 description 7
- 239000004031 partial agonist Substances 0.000 description 7
- 206010015150 Erythema Diseases 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 238000007918 intramuscular administration Methods 0.000 description 6
- 239000011241 protective layer Substances 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 206010028813 Nausea Diseases 0.000 description 5
- YOYLLRBMGQRFTN-IOMBULRVSA-N Norbuprenorphine Chemical compound C([C@@H](NCC1)[C@]23CC[C@]4([C@H](C3)[C@](C)(O)C(C)(C)C)OC)C3=CC=C(O)C5=C3[C@@]21[C@H]4O5 YOYLLRBMGQRFTN-IOMBULRVSA-N 0.000 description 5
- 206010030113 Oedema Diseases 0.000 description 5
- 229940127450 Opioid Agonists Drugs 0.000 description 5
- 229920002367 Polyisobutene Polymers 0.000 description 5
- 206010041349 Somnolence Diseases 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- 239000002131 composite material Substances 0.000 description 5
- 230000008693 nausea Effects 0.000 description 5
- 229920001296 polysiloxane Polymers 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 238000005070 sampling Methods 0.000 description 5
- 229920005573 silicon-containing polymer Polymers 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 208000032140 Sleepiness Diseases 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- 229940035676 analgesics Drugs 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 239000000730 antalgic agent Substances 0.000 description 4
- 210000003169 central nervous system Anatomy 0.000 description 4
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 208000002173 dizziness Diseases 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 102000051367 mu Opioid Receptors Human genes 0.000 description 4
- 229920006267 polyester film Polymers 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 108020001612 μ-opioid receptors Proteins 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000004820 Pressure-sensitive adhesive Substances 0.000 description 3
- 206010047700 Vomiting Diseases 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 229960001889 buprenorphine hydrochloride Drugs 0.000 description 3
- UAIXRPCCYXNJMQ-RZIPZOSSSA-N buprenorphine hydrochlorie Chemical compound [Cl-].C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)C[NH+]2CC1CC1 UAIXRPCCYXNJMQ-RZIPZOSSSA-N 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 230000001186 cumulative effect Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000009792 diffusion process Methods 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 235000019439 ethyl acetate Nutrition 0.000 description 3
- 229940093499 ethyl acetate Drugs 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 210000003608 fece Anatomy 0.000 description 3
- 229960002428 fentanyl Drugs 0.000 description 3
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 3
- 230000003601 intercostal effect Effects 0.000 description 3
- 102000048260 kappa Opioid Receptors Human genes 0.000 description 3
- 229940040102 levulinic acid Drugs 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 229920001684 low density polyethylene Polymers 0.000 description 3
- 239000004702 low-density polyethylene Substances 0.000 description 3
- 210000004379 membrane Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 description 3
- 229960005301 pentazocine Drugs 0.000 description 3
- 229920002635 polyurethane Polymers 0.000 description 3
- 239000004814 polyurethane Substances 0.000 description 3
- 229920000915 polyvinyl chloride Polymers 0.000 description 3
- 239000004800 polyvinyl chloride Substances 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 230000002385 psychotomimetic effect Effects 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000037321 sleepiness Effects 0.000 description 3
- 238000013268 sustained release Methods 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 230000008673 vomiting Effects 0.000 description 3
- 108020001588 κ-opioid receptors Proteins 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 2
- 208000000003 Breakthrough pain Diseases 0.000 description 2
- 208000000094 Chronic Pain Diseases 0.000 description 2
- 206010010774 Constipation Diseases 0.000 description 2
- 206010012335 Dependence Diseases 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- 206010019233 Headaches Diseases 0.000 description 2
- 208000026251 Opioid-Related disease Diseases 0.000 description 2
- 101100244562 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) oprD gene Proteins 0.000 description 2
- 239000004902 Softening Agent Substances 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 2
- 239000002998 adhesive polymer Substances 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- 230000037058 blood plasma level Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 210000000038 chest Anatomy 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 108700023159 delta Opioid Receptors Proteins 0.000 description 2
- 102000048124 delta Opioid Receptors Human genes 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 150000001991 dicarboxylic acids Chemical class 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 230000000857 drug effect Effects 0.000 description 2
- 229940099191 duragesic Drugs 0.000 description 2
- 231100000321 erythema Toxicity 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 239000004744 fabric Substances 0.000 description 2
- PJMPHNIQZUBGLI-UHFFFAOYSA-N fentanyl Chemical compound C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 PJMPHNIQZUBGLI-UHFFFAOYSA-N 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 231100000869 headache Toxicity 0.000 description 2
- 230000000774 hypoallergenic effect Effects 0.000 description 2
- 230000000642 iatrogenic effect Effects 0.000 description 2
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000003978 infusion fluid Substances 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 239000003402 opiate agonist Substances 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 230000000079 pharmacotherapeutic effect Effects 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920006254 polymer film Polymers 0.000 description 2
- 229920000193 polymethacrylate Polymers 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- VQJMAIZOEPPELO-KYGIZGOZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-(2-hydroxy-5-methylhexan-2-yl)-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol hydrochloride Chemical compound Cl.CO[C@]12CC[C@@]3(C[C@@H]1C(C)(O)CCC(C)C)[C@H]1Cc4ccc(O)c5O[C@@H]2[C@]3(CCN1CC1CC1)c45 VQJMAIZOEPPELO-KYGIZGOZSA-N 0.000 description 1
- WJTCHBVEUFDSIK-NWDGAFQWSA-N (2r,5s)-1-benzyl-2,5-dimethylpiperazine Chemical compound C[C@@H]1CN[C@@H](C)CN1CC1=CC=CC=C1 WJTCHBVEUFDSIK-NWDGAFQWSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- AXTGDCSMTYGJND-UHFFFAOYSA-N 1-dodecylazepan-2-one Chemical compound CCCCCCCCCCCCN1CCCCCC1=O AXTGDCSMTYGJND-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- GOXQRTZXKQZDDN-UHFFFAOYSA-N 2-Ethylhexyl acrylate Chemical class CCCCC(CC)COC(=O)C=C GOXQRTZXKQZDDN-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- BHIZVZJETFVJMJ-UHFFFAOYSA-N 2-hydroxypropyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCC(C)O BHIZVZJETFVJMJ-UHFFFAOYSA-N 0.000 description 1
- WLAMNBDJUVNPJU-UHFFFAOYSA-N 2-methylbutyric acid Chemical compound CCC(C)C(O)=O WLAMNBDJUVNPJU-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000006734 Beta-Globulins Human genes 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- 239000004709 Chlorinated polyethylene Substances 0.000 description 1
- 239000004821 Contact adhesive Substances 0.000 description 1
- 229920001634 Copolyester Polymers 0.000 description 1
- 206010013654 Drug abuse Diseases 0.000 description 1
- 229920000219 Ethylene vinyl alcohol Polymers 0.000 description 1
- 241001539473 Euphoria Species 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- XADCESSVHJOZHK-UHFFFAOYSA-N Meperidine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(C)CC1 XADCESSVHJOZHK-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108090000137 Opioid Receptors Proteins 0.000 description 1
- 102000003840 Opioid Receptors Human genes 0.000 description 1
- 208000000114 Pain Threshold Diseases 0.000 description 1
- 229920002614 Polyether block amide Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038678 Respiratory depression Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical class CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 229920002433 Vinyl chloride-vinyl acetate copolymer Polymers 0.000 description 1
- RMRJXGBAOAMLHD-CTAPUXPBSA-N [H][C@]12Cc3ccc(O)c4c3[C@]3(CCN1CC1CC1)[C@@]21CC[C@](OC)([C@@]([H])([C@](C)(O)C(C)(C)C)C1)[C@]3([H])O4 Chemical compound [H][C@]12Cc3ccc(O)c4c3[C@]3(CCN1CC1CC1)[C@@]21CC[C@](OC)([C@@]([H])([C@](C)(O)C(C)(C)C)C1)[C@]3([H])O4 RMRJXGBAOAMLHD-CTAPUXPBSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000002390 adhesive tape Substances 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 229910001583 allophane Inorganic materials 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 238000012466 analgesic assay Methods 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229940087828 buprenex Drugs 0.000 description 1
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 description 1
- 229960001113 butorphanol Drugs 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 238000006900 dealkylation reaction Methods 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000002951 depilatory effect Effects 0.000 description 1
- 238000001784 detoxification Methods 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000001544 dysphoric effect Effects 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000010235 enterohepatic circulation Effects 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- HIHIPCDUFKZOSL-UHFFFAOYSA-N ethenyl(methyl)silicon Chemical compound C[Si]C=C HIHIPCDUFKZOSL-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 229920001903 high density polyethylene Polymers 0.000 description 1
- 239000004700 high-density polyethylene Substances 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229940057917 medium chain triglycerides Drugs 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- UOBSVARXACCLLH-UHFFFAOYSA-N monomethyl adipate Chemical compound COC(=O)CCCCC(O)=O UOBSVARXACCLLH-UHFFFAOYSA-N 0.000 description 1
- IBMRTYCHDPMBFN-UHFFFAOYSA-N monomethyl glutaric acid Chemical compound COC(=O)CCCC(O)=O IBMRTYCHDPMBFN-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- INAXVFBXDYWQFN-XHSDSOJGSA-N morphinan Chemical compound C1C2=CC=CC=C2[C@]23CCCC[C@H]3[C@@H]1NCC2 INAXVFBXDYWQFN-XHSDSOJGSA-N 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 239000002756 mu opiate receptor agonist Substances 0.000 description 1
- NETZHAKZCGBWSS-CEDHKZHLSA-N nalbuphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]1(O)CC[C@@H]3O)CN2CC1CCC1 NETZHAKZCGBWSS-CEDHKZHLSA-N 0.000 description 1
- 229960000805 nalbuphine Drugs 0.000 description 1
- 239000004081 narcotic agent Substances 0.000 description 1
- 239000004084 narcotic analgesic agent Substances 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 230000003533 narcotic effect Effects 0.000 description 1
- 210000001640 nerve ending Anatomy 0.000 description 1
- 230000001473 noxious effect Effects 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical class CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- YYZUSRORWSJGET-UHFFFAOYSA-N octanoic acid ethyl ester Natural products CCCCCCCC(=O)OCC YYZUSRORWSJGET-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- BARWIPMJPCRCTP-UHFFFAOYSA-N oleic acid oleyl ester Natural products CCCCCCCCC=CCCCCCCCCOC(=O)CCCCCCCC=CCCCCCCCC BARWIPMJPCRCTP-UHFFFAOYSA-N 0.000 description 1
- BARWIPMJPCRCTP-CLFAGFIQSA-N oleyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCCOC(=O)CCCCCCC\C=C/CCCCCCCC BARWIPMJPCRCTP-CLFAGFIQSA-N 0.000 description 1
- 229940127240 opiate Drugs 0.000 description 1
- 239000003401 opiate antagonist Substances 0.000 description 1
- 230000037040 pain threshold Effects 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 229960000482 pethidine Drugs 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229920003168 pharmaceutical polymer Polymers 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 239000011505 plaster Substances 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920001195 polyisoprene Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000346 polystyrene-polyisoprene block-polystyrene Polymers 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 239000005033 polyvinylidene chloride Substances 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 229940026235 propylene glycol monolaurate Drugs 0.000 description 1
- 230000003236 psychic effect Effects 0.000 description 1
- 210000001747 pupil Anatomy 0.000 description 1
- 210000002321 radial artery Anatomy 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 229920000260 silastic Polymers 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000013464 silicone adhesive Substances 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 229920003048 styrene butadiene rubber Polymers 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- ZQZCOBSUOFHDEE-UHFFFAOYSA-N tetrapropyl silicate Chemical compound CCCO[Si](OCCC)(OCCC)OCCC ZQZCOBSUOFHDEE-UHFFFAOYSA-N 0.000 description 1
- 239000004753 textile Substances 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 229940100640 transdermal system Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 150000004072 triols Chemical class 0.000 description 1
- KJIOQYGWTQBHNH-UHFFFAOYSA-N undecanol Chemical compound CCCCCCCCCCCO KJIOQYGWTQBHNH-UHFFFAOYSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4748—Quinolines; Isoquinolines forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
- A61K9/703—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms characterised by shape or structure; Details concerning release liner or backing; Refillable patches; User-activated patches
- A61K9/7038—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer
- A61K9/7046—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer the adhesive comprising macromolecular compounds
- A61K9/7053—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer the adhesive comprising macromolecular compounds obtained by reactions only involving carbon to carbon unsaturated bonds, e.g. polyvinyl, polyisobutylene, polystyrene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
- A61K9/703—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms characterised by shape or structure; Details concerning release liner or backing; Refillable patches; User-activated patches
- A61K9/7038—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer
- A61K9/7046—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer the adhesive comprising macromolecular compounds
- A61K9/7053—Transdermal patches of the drug-in-adhesive type, i.e. comprising drug in the skin-adhesive layer the adhesive comprising macromolecular compounds obtained by reactions only involving carbon to carbon unsaturated bonds, e.g. polyvinyl, polyisobutylene, polystyrene
- A61K9/7061—Polyacrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Definitions
- sustained-release pharmaceutical preparations It is the intent of all sustained-release pharmaceutical preparations to provide a longer period of pharmacologic effect after the administration of a drug than is ordinarily experienced after the administration of immediate release preparations of the same drug. Such longer periods of efficacy can provide many inherent therapeutic benefits that are not achieved with corresponding immediate release preparations.
- the benefits of prolonged analgesia afforded by sustained release oral analgesic preparations have become universally recognized and oral opioid analgesic sustained-release preparations are commercially available.
- Prolonged analgesia is particularly desirable in patients suffering from moderate to severe pain, such as cancer patients.
- Available oral preparations provide a duration of effect lasting e.g., about twelve hours (and sometimes 24 hours) such that a drug may only have to be administered to a patient one to three times a day.
- morphine which has been considered to be the prototypic opioid analgesic, has been formulated into twice-daily, oral controlled release formulations (e.g., MS Contin® tablets, commercially available from The Purdue Frederick Company).
- transdermal delivery systems such as transdermal patches.
- transdermal patches contain a therapeutically active agent (e.g., an opioid analgesic), a reservoir or matrix containing the opioid or other active ingredient(s) and an adhesive which allows the transdermal device to adhere to the skin, allowing for the passage of the active agent from the device through the skin of the patient.
- a therapeutically active agent e.g., an opioid analgesic
- a reservoir or matrix containing the opioid or other active ingredient(s) e.g., an opioid analgesic
- an adhesive which allows the transdermal device to adhere to the skin, allowing for the passage of the active agent from the device through the skin of the patient.
- Transdermal delivery systems in which an opioid analgesic is the active ingredient have been contemplated.
- an opioid analgesic transdermal formulation is Duragesic® (commercially available from Janssen Pharmaceutical; active ingredient is fentanyl).
- Duragesic® patch is said to provide adequate analgesia for up to 48 to 72 hours (2 to 3 days).
- Buprenorphine a partially synthetic opiate
- Buprenorphine has also been contemplated for prolonged analgesia.
- other types of opioid analgesic transdermal formulations have been reported in the literature (such as fentanyl, discussed above)
- buprenorphine transdermal delivery systems are of particular interest because buprenorphine is a potent, partial agonist opioid analgesic with desirable therapeutic properties.
- buprenorphine is 50 to 100 times more potent than morphine, but has a much safer therapeutic index than morphine (see Wallenstein S L, et al., Crossover Trials in Clinical Analgesic Assays: Studies of Buprenorphine and Morphine, Pharmacotherapy, G(5): 225-235, 1986 hereby incorporated by reference). Further, the partial agonist properties of buprenorphine are useful in the treatment of opioid addiction.
- Buprenorphine has a low oral bioavailability and has been considered by certain of those skilled in the art to be like other narcotics which are habit-forming (see, e.g., U.S. Pat. No. 5,240,711 to Hille, et. al.) and induce tolerance (see, e.g., U.S. Pat. No. 5,613,958 to Kochinke, et. al.).
- transdermal delivery system which includes buprenorphine or one of its pharmaceutically compatible salts and which releases the drug over a period of at least 24 hours in a controlled manner, and ensures that the buprenorphine does not notably decompose when the transdermal delivery system is stored, and which further ensures that the buprenorphine in-vivo penetrates through the skin at the required amount.
- Kochinke et al. describe a transdermal system for the modulated administration of tolerance-inducing drugs.
- Buprenorphine is identified therein as such a drug.
- the system is designed to deliver the drug through the patient's skin via a three-phase drug delivery profile.
- first phase which begins with patch application and ends at 2-10 hours after patch application, plasma levels of the drug are obtained.
- second phase in which therapeutic plasma levels of the drug are maintained.
- the second phase begins at about two to ten hours after patch application and ends at about 8-18 hours after patch application.
- sub-therapeutic levels of the drug are maintained, via inherent patch design and/or patch removal.
- initial high blood levels may be more effective when followed by a period of decreasing dosage (down to sub-therapeutic levels), than if the blood levels are maintained either at the higher or lower level (i.e., sub-therapeutic levels) throughout the entire administration period.
- this modulated profile it is said that the onset of tolerance to the drug being administered can be prevented or greatly reduced.
- a further object of the invention is to provide a method for treating opioid-addicted patients in a manner which gradually reduces the plasma concentration of opioid in the patients' plasma while at the same time providing effective plasma concentrations for those patients to be detoxified.
- the invention is directed in part to the surprising result that effective pain management is provided by providing a substantially first order rate of increase of blood plasma concentrations of buprenorphine over about a three day (e.g., 72 hours) time interval, followed by a prolonged time period of at least about two days (e.g., 48 hours) during which the plasma concentrations of buprenorphine are maintained according to substantially zero order pharmacokinetics.
- the invention relates in part to a method of effectively treating pain in humans, comprising administering buprenorphine to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 0.3 to about 113 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 3 to about 296 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 11 to about 644 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 13 to about 630 pg/ml at about 30 hours after initiation of the dosing interval; a mean plasma concentration from about 15 to about 715 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 20 to about 984 pg/ml at about 48 hours after initiation of the dosing
- the buprenorphine may be administered transdermally, parenterally, sublingually, orally, buccally, rectally, etc. Oral bioavailability of buprenorphine is very low (estimated as 15%). In order to better control plasma concentrations of buprenorphine within the concentrations desired in the herein-described inventive methods, it is preferred that the buprenorphine is administered via a transdermal delivery system or via continuous infusion.
- the method comprises applying a transdermal delivery system containing buprenorphine as the active ingredient onto the skin of patients which provide a release rate of buprenorphine over about a 72 hour dosing interval such that a maximum plasma concentration from about 20 pg/ml to about 850 pg/ml is attained (depending upon the dosage levels needed to maintain analgesia in the particular patients), and then maintaining the transdermal delivery systems on the skin of the patients for at least an additional 24 hour interval during which the plasma concentrations of buprenorphine in the patients are maintained above minimum effective concentrations of the drug and the patients continue to experience effective pain management during this additional dosing interval.
- the invention is further directed to a method of effectively treating pain in humans, comprising administering buprenorphine transdermally to human patients such that mean relative release rates are achieved as follows: a mean relative release rate of from about 3 ug/hr to about 86 ug/hr from initiation of the dosing interval until about 72 hours thereafter; and a mean relative release rate of about 0.3 ug/hr to about 9 ug/hr from about 72 hours after the initiation of the dosing interval until at least about 120 hour hours after the initiation of the dosing interval.
- the mean relative release rate of about 0.3 ug/hr to about 9 ug/hr is maintained from about 72 hours after the initiation of the dosing interval until at least about 168 hours after the initiation of the dosing interval.
- the present invention is further related to a method of effectively treating pain in humans, comprising administering buprenorphine transdermally to human patients such that a mean relative release rate from about 3 ug/hr to about 86 ug/hr of buprenorphine is achieved until about 72 hours after the application of a transdermal delivery system, and thereafter providing (either with the same transdermal delivery system or upon removal of the system and replacement with a different transdermal delivery system) a mean relative release rate of about 0.3 ug/hr to about 9 ug/hr from about 72 hours after the intimation of the dosing interval until at least about 120 hours after the initiation of the dosing interval, and preferably until at least about 168 hours after the initiation of the dosing interval.
- the method comprises the application of a transdermal delivery system which is designed to be provide analgesia for about 72 hours, and which provides a release rate of the drug when applied to the skin which generally follows first order pharmacokinetics over that 72 hour period, and further comprises taking advantage of the fact that such transdermal delivery systems typically provide a dramatic drop-off in the release rate of buprenorphine after the first 72 hours, but nevertheless provide a relatively small but sufficient release of buprenorphine to maintain analgesia and desirable plasma concentrations in the patients over a further period of time of at least, e.g., preferably at least 48 hours, by leaving the transdermal delivery system in contact with the skin of the patient for such additional desired dosing interval, which may be as long as, e.g., an additional 96 hours or more.
- a transdermal delivery system which is designed to be provide analgesia for about 72 hours, and which provides a release rate of the drug when applied to the skin which generally follows first order pharmacokinetics over that
- transdermal dosage systems exhibit substantially zero order release after about the initial 72 hour dosage interval, and therefore are capable of maintaining effective plasma concentrations of buprenorphine for a much longer period than previously reported in the prior art.
- inventive method also contemplates the possibility of utilizing a first transdermal delivery system which provides the desired substantially first order kinetics, and thereafter the removal of the first transdermal delivery system and its replacement with a second system which provides the desired substantially zero order pharmacokinetics for a prolonged period of time (e.g., at least about 24 hours, preferably at least about 48 hours, and most preferably about 96 hours).
- This second system may be a second transdermal delivery system which provides the afore-mentioned mean relative release rate of about 0.3 ug/hr to about 9 ug/hr.
- the second system may even utilize a different mode of administration, for example, continuous infusion.
- the present invention is also related, in part, to a method of effectively treating pain in patients, comprising applying onto the skin of the patients a transdermal delivery system containing buprenorphine which transdermal delivery system delivers the buprenorphine substantially according to first order kinetics to provide a mean plasma concentration from about 24 to about 850 pg/ml about 3 days after application, and then maintaining the transdermal buprenorphine formulation in contact with the skin of the human patient for about 2 to about 6 additional days without removing the transdermal formulation, such that the patient continues to receive effective analgesia from the transdermal buprenorphine formulation.
- the invention also provides, in certain preferred embodiments, an improvement in a method of treating pain in human patients by applying a 3 day transdermal delivery system containing buprenorphine onto the skin of the patient and maintaining the transdermal delivery system in contact with the skin for a 3 day dosing interval, the transdermal delivery system containing an amount of buprenorphine sufficient to provide effective analgesia in the patient for about 3 days, the improvement comprising maintaining the transdermal dosage form in contact with the patient's skin for at least 2 to about 6 additional days beyond the 3 day dosing interval.
- the present invention also relates to a method of treating opioid addiction by administering buprenorphine transdermally to human patients which provides a release rate of the drug when applied to the skin which generally follows first order pharmacokinetics over a 72 hour period, such that the addict attains a buprenorphine plasma concentration from about 1000 to about 10,000 ⁇ g/ml, and preferably from about 5000 to about 8000 ⁇ g/ml, about 72 hours after application of a buprenorphine transdermal delivery system, and thereafter maintaining the transdermal delivery system in contact with the skin of the addict such that a mean relative release rate of buprenorphine approximating zero order kinetics over an additional dosing interval of at least about 48 hours, to provide the desired therapeutic effect (detoxification).
- the transdermal delivery system is maintained in contact with the addict's skin for about 7 days.
- analgesia is defined for purposes of the present invention as a satisfactory reduction in or elimination of pain, along with the process of a tolerable level of side effects, as determined by the human patient.
- effective pain management means for purposes of the present invention as the objective evaluation of a human patient's response (pain experienced versus side effects) to analgesic treatment by a physician as well as subjective evaluation of therapeutic treatment by the patient undergoing such treatment.
- effective analgesia will vary according to many factors, including individual patient variations.
- breakthrough pain means pain which the patient experiences despite the fact that the patient is being administered generally effective amounts of, e.g., an opioid analgetic such as buprenorphine.
- rescue refers to a dose of an analgesic which is administered to a patient experiencing breakthrough pain.
- first order pharmacokinetics is defined as plasma concentrations which increase over a specified time period.
- zero order pharmacokinetics contemplates an amount of drug released from a buprenorphine formulation which substantially maintains plasma concentrations at a relatively constant level.
- a relatively constant plasma concentration is defined as a concentration which does not decrease more than about 30% over a 48 hour time period.
- mean relative release rate is determined from the amount of drug released per unit time from the transdermal delivery system through the skin and into the bloodstream of a human patient.
- Mean relative release rate may be expressed, e.g, as ⁇ g drug/cm 2 /hr.
- a transdermal delivery system that releases 1.2 mg of buprenorphine over a time period of 72 hours is considered to have a relative release rate of 16.67 ⁇ g/hr.
- relative release rates may change between any particular time points within a particular dosing interval, and the term therefore only reflects the overall release rate during the particular dosing interval.
- relative release rate should be considered synonomous with the term “flux rate”.
- sustained release is defined for purposes of the present invention as the release of the drug (opioid analgesic) from the transdermal formulation at such a rate that blood (e.g., plasma) concentrations (levels) are maintained within the therapeutic range (above the minimum effective analgesic concentration or “MEAC”) but below toxic levels over a period of time of about 3 days or longer.
- blood e.g., plasma
- concentrations levels
- MEAC minimum effective analgesic concentration
- steady state means that the blood plasma concentration curve for a given drug has been substantially repeated from dose to dose.
- minimum effective analgesic concentration is defined for purposes of this invention as the minimum effective therapeutic blood plasma level of the drug at which at least some pain relief is achieved in a given patient. It will be well understood by those skilled in the medical art that pain measurement is highly subjective and great individual variations may occur among patients.
- buprenorphine shall include buprenorphine base, pharmaceutically acceptable salts thereof, stereoisomers thereof, ethers and esters thereof, and mixtures thereof.
- overage means for the purposes of the present invention the amount of buprenorphine contained in a transdermal delivery system which is not delivered to the patient.
- the overage is necessary for creating a concentration gradient by means of which the active agent (e.g., buprenorphine) migrates through the layers of the transdermal dosage form to the desired site on a patient's skin.
- FIG. 1 is a graphical representation of the mean plasma concentration (pg/ml) versus time (days) for Example 1;
- FIG. 2 is a graphical representation of pharmacodynamic variables versus time (days) for Example 1;
- FIG. 3 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example A;
- FIG. 4 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example B (intravenous concentrations divided by 100);
- FIG. 5 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example C;
- FIG. 6 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example A;
- FIG. 7 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example B;
- FIG. 8 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example C;
- FIG. 9 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example D;
- FIG. 10 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example E;
- FIG. 11 is a graphical representation of the plasma concentration (pg/ml) over time (hours) for Comparative Example F;
- FIG. 12 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example D;
- FIG. 13 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example E.
- FIG. 14 is a graphical representation pharmacodynamic variables versus time (hours) for Comparative Example F.
- strong analgesics encompasses, inter alia, several classes of opioid analgesics, including the partial agonists.
- Parenteral buprenorphine (a Schedule V drug under the Controlled Substances Act) is the only example of a partial agonist opioid analgesic currently marketed in the United States.
- Partial agonists provide several therapeutic advantages in many patients when compared to morphine-like agonists and mixed agonists-antagonists.
- buprenorphine unlike the mixed agonists-antagonists (e.g., pentazocine, butorphanol, nalbuphine), buprenorphine is devoid of psychotomimetic adverse reactions; in comparison with agonists (e.g., morphine and fentanyl), the dose-responsive relationship for respiratory depression with buprenorphine is relatively low and the abuse liability of buprenorphine is less.
- mixed agonists-antagonists e.g., pentazocine, butorphanol, nalbuphine
- agonists e.g., morphine and fentanyl
- buprenorphine The chemical of name of buprenorphine is 21-cyclopropyl-7-[(S)-1-hydroxy-1,2,2-trimethylpropyl]-6,14-endo-ethano-6,7,8,14-tetrahydrooripavine.
- the molecular weight of buprenorphine base is 467.7; the empirical formula is C 29 H 4 1NO 4 .
- buprenorphine The structural formula of buprenorphine is shown below:
- Buprenorphine is an opioid partial agonist and shares many of the actions, such as analgesia, of opioid agonists.
- a “ceiling effect” to analgesia i.e., no additional analgesia with increasing dose
- Buprenorphine is considered in the art to be a partial agonist at ⁇ opioid receptors in the central nervous system (“CNS”) and peripheral tissues. It is further thought that buprenorphine binds with high affinity to ⁇ and ⁇ 1 receptors, and, with lower affinity, to ⁇ receptors.
- the intrinsic agonist activity at the ⁇ receptor seems to be limited and most evidence suggests that buprenorphine has antagonist activity at ⁇ receptors.
- the lack of ⁇ agonism accounts for buprenorphine's freedom from the dysphoric and psychotomimetic effects often seen with agonist/antagonist drugs.
- Other studies suggest that the opioid antagonist effects of buprenorphine may be mediated via an interaction with ⁇ opioid receptors.
- buprenorphine binds slowly with, and dissociates slowly from, the ⁇ receptor.
- the high affinity of buprenorphine for the ⁇ receptor and its slow binding to, and dissociation from, the receptor is thought to possibly account for the prolonged duration of analgesia, and in part, for the limited physical dependence potential observed with the drug.
- the high affinity binding may also account for the fact that buprenorphine can block the ⁇ agonist effects of other administered opioids.
- buprenorphine produces dose-related analgesia.
- analgesia appears to result from a high affinity of buprenorphine for ⁇ and possibly ⁇ opioid receptors in the CNS.
- the drug may also alter the pain threshold (threshold of afferent nerve endings to noxious stimuli).
- the analgesic potency of parenteral buprenorphine appears to be about 25 to about 50 times that of parenteral morphine, about 200 times that of pentazocine, and about 600 times that of meperidine.
- Buprenorphine may produce sex-related differences in analgesia, with females requiring substantially less drug than males to produce adequate analgesia.
- Buprenorphine has a low abuse liability compared to full agonist opioids. Although infrequent, however, buprenorphine may also produce limited physical dependence, and signs and symptoms of mild withdrawal may appear following discontinuance of prolonged therapy with the drug alone. Due to buprenorphine's slow binding with and slow dissociation from the ⁇ receptor, elimination of the drug from the CNS is prolonged following abrupt discontinuance; consequently, signs and symptoms of acute withdrawal are less intense than those produced by morphine and are delayed in appearance.
- buprenorphine produces many of the subjective and objective effects of opioids; however, the drug may not be a satisfactory substitute for opioid agonists in all patients physically dependent on opioids. Tolerance to the opioid agonist activity of the drug reportedly develops rarely, if at all.
- Buprenorphine may produce psychological dependence.
- Buprenorphine is a partial opioid agonist with behavioral and psychic effects similar to morphine. Unlike pentazocine, however, buprenorphine rarely causes psychotomimetic effects. Like other opioid agonists, buprenorphine may produce increases in cerebrospinal fluid pressure.
- the pharmacokinetics of buprenorphine administered parenterally and sublingually are known.
- Intravenous administration of a single dose of about 0.3 mg of buprenorphine has been shown to provide mean peak plasma drug concentrations of about 18 ng/ml which occur within about 2 minutes; plasma concentrations declined to about 9 and about 0.4 ng/ml after about 5 minutes and about 3 hours, respectively.
- mean peak plasma buprenorphine concentrations of about 3.6 ng/ml occur within about 2 to about 5 minutes and decline to about 0.4 ng/ml after about 3 hours.
- plasma concentrations of buprenorphine are similar following intravenous or intramuscular injection.
- a parenteral solution of buprenorphine hydrochloride (0.3 mg buprenorphine/ml) is commercially available as Buprenex® (Reckitt & Colman) for intramuscular and intravenous administration.
- the usual adult dose (over age 13) is 0.3 mg IM or IV every 6 to 8 hours as needed for moderate to severe pain.
- the pediatric dose in patients age 2 to 12 is 2-6 mcg/kg of body weight every 4-6 hours.
- the increased frequency of administration in the pediatric population is believed to be caused by increased clearance of buprenorphine compared to the adult population.
- the mean duration of analgesia generally is six hours following single intramuscular or intravenous doses of 0.2 to 0.3 mg or 2 to 4 ⁇ g/kg; however, in some studies, the mean duration of analgesia reportedly ranged from 4 to 10 hours following single intramuscular doses of 0.2 to 0.6 mg and 2 to 24 hours following single intravenous doses of 0.3 mg or 2 to 15 ⁇ g/kg.
- the mean peak plasma buprenorphine concentration, time to peak concentration, and systemic availability for a 0.4 mg and 0.8 mg single-dose sublingual dose of buprenorphine has been reported by Cowan, Alan and Lewis John, W., Buprenorphine: combating Drug Abuse With a Unique Opioids, Wiley-Liss, Inc., New York, pp. 137-147 (1995), hereby incorporated by reference in its entirety.
- the Cmax was reported as 0.50 ⁇ 0.06 ng/ml; the Tmax was reported 210 ⁇ 40 minutes; and a systemic availability of 57.7% ⁇ 6.
- the Cmax was reported at 1.04 ⁇ 0.27 ng/ml; the Tmax was reported 192 ⁇ 49 minutes; and a systemic availability of 54.1% ⁇ 12.7.
- transdermal delievery system e.g., a transdermal patch
- the total buprenorphine administered over a 24 hour period would be about 0.6 mg
- the sublingual equivalent dose over the same period would be about 1.2 mg
- the total buprenorphine administered over a 24 hour period would be about 1.2 mg
- the sublingual equivalent dose over the same period would be about 2.4 mg.
- buprenorphine Distribution of buprenorphine into human body tissues and fluids has not been well characterized. Following oral or intramuscular administration in rats, buprenorphine distributes into the liver, brain, placenta, and GI tract; highest concentrations were attained in the liver within 10 or 40 minutes following oral or intramuscular administration, respectively. The hepatic extraction ratio of buprenorphine is approximately 1. The drug and its metabolites are distributed into bile. Following intravenous administration in humans, the drug rapidly distributes into cerebro spinal fluid (“CSF”) (within several minutes). CSF buprenorphine concentrations appear to be approximately 15% to 25% of concurrent plasma concentrations. Buprenorphine is approximately 96% bound to plasma proteins, mainly to and ⁇ globulins; the drug does not appear to bind substantially to albumin.
- CSF cerebro spinal fluid
- Buprenorphine is almost completely metabolized in the liver, principally by N-dealkylation, to form norbuprenorphine (N-dialkylbuprenorphine); buprenorphine and norbuprenorphine also undergo conjugation with glucuronic acid.
- norbuprenorphine may have weak analgesic activity; however, studies to determine the analgesic activity of the metabolites of buprenorphine have not been performed.
- Buprenorphine and its metabolites are excreted principally in feces via biliary elimination and also in urine.
- Buprenorphine is excreted in feces mainly as unchanged drug; small amounts of norbuprenorphine are also excreted in feces. The drug and its metabolites are believed to undergo enterohepatic circulation. Norbuprenorphine appears to be excreted principally in urine at a slower rate than the parent drug. Total plasma clearance of buprenorphine reportedly is approximately 1.28 l/minute in conscious postoperative patients. Limited data indicate that there is considerable interindividual variability in buprenorphine pharmacokinetics in children; however, clearance of the drug appears to be increased in children (e.g., those 5 to 7 years of age) compared with that in adults. Optimal dosing interval of buprenorphine may have to be decreased in pediatric patients.
- analgesic plasma opioid concentrations in patients is very complicated and involves a host of considerations, including the inherent chemical and physical properties of the opioid itself. Further considerations include in-vivo metabolism, individual patient response and tolerance. Generally, however, there is a “minimally effective analgesic concentration” (MEAC) in plasma for a particular opioid below which no analgesia is provided. There is relationship between plasma opioid levels and analgesia. Higher plasma levels are generally associated with greater pain relief, and (possibly) greater incidence and severity of side effects.
- MEAC minimumally effective analgesic concentration
- the buprenorphine is administered in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 0.3 to about 113 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 3 to about 296 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 7 to about 644 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 13 to about 753 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 16 to about 984 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 20 to about 984 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from
- this method further comprises maintaining the dosing of buprenorphine during the at least next 48 hours in accordance with zero under kinetics.
- the mean plasma concentrations are maintained after the 72 hour dosing interval as follows: a mean plasma concentration from about 23 to about 1052 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 23 to about 1052 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 22 to about 970 pg/ml at about 144 hours after initiation of the dosing interval; and a mean plasma concentration from about 19 to about 841 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate from about 3 ug/hr to about 86 ug/hr is preferably maintained from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate is preferably maintained from about 0.3 ug/hr to about 9 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval.
- the administration of buprenorphine is accomplished via a mode selected from the group consisting of transdermally, continuous infusion, and a mixture of transdermally and continuous infusion.
- the administration is accomplished by applying a transdermal delivery system to the skin of a patient, and maintaining said transdermal delivery system in contact with the patient's skin for at least 5 days.
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 1 to about 28 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 14 to about 74 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 30 to about 161 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 51 to about 188 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 62 to about 246 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 79 to about 246 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from about 85 to about 263 pg/m
- buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 77 to about 263 pg/ml over at least the next 48 hours.
- the plasma concentrations are maintained after the 72 hour dosing interval as follows: a mean plasma concentration from about 92 to about 263 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 94 to about 263 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 86 to about 243 pg/ml at about 144 hours after initiation of the dosing interval; and a mean plasma concentration from about 77 to about 210 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate of from about 13 ug/hr to about 21 ug/hr is maintained from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and that a mean relative release rate of about 1 ug/hr to about 2 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval is maintained (e.g., about 168 hours after initiation for a seven-day dosing interval).
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 0.3 to about 7 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 4 to about 19 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 7 to about 40 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 13 to about 47 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 16 to about 62 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 21 to about 62 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from about 20 to about 66 pg/ml at about 72
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 19 to about 66 pg/ml over at least the next 48 hours.
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained as follows: a mean plasma concentration from about 23 to about 66 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 23 to about 66 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 22 to about 61 pg/ml at about 144 hours after initiation of the dosing interval; and a mean plasma concentration from about 19 to about 53 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate is maintained from about 3 ug/hr to about 5 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 0.3 ug/hr to about 0.6 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven-day dosing interval).
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 0.7 to about 14 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 7 to about 37 pg/m at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 15 to about 80 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 25 to about 94 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 31 to about 123 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 40 to about 123 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from about 42 to about 132 pg/ml at about 72
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 38 to about 132 pg/ml over at least the next 48 hours.
- the buprenorphine is further administered in a manner such that the mean plasma concentrations are maintained as follows: a mean plasma concentration from about 46 to about 132 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 47 to about 132 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 43 to about 121 pg/ml at about 144 hours after initiation of the dosing interval; and a mean plasma concentration from about 38 to about 105 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate from about 6 ug/hr to about 11 ug/hr is maintained from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 0.7 ug/hr to about 1 ug/hr is maintained from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven day dosing interval).
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 3 to about 57 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 28 to about 148 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 59 to about 322 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 102 to about 377 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 124 to about 492 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 159 to about 492 ml at about 60 hours; after initiation of the dosing interval; a mean plasma concentration from about 169 to about 526 pg
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 153 to about 526 pg/ml over at least the next 48 hours.
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained as follows: a mean plasma concentration from about 184 to about 526 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 187 to about 526 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 173 to about 485 pg/ml at about 144 hours after initiation of the dosing interval; a mean plasma concentration from about 153 to about 420 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate from about 26 ug/hr to about 43 ug/hr is maintained from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 2 ug/hr to about 4 ug/hr is maintained from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven-day dosing interval).
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 4 to about 85 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 42 to about 222 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 89 to about 483 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 152 to about 565 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 186 to about 738 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 238 to about 738 pg/ml at 60 hours after initiation of the dosing interval; a mean plasma concentration from about 254 to about 789 pg
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 230 to about 789 pg/ml over at least the next 48 hours.
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained as follows: a mean plasma concentration from about 276 to about 789 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 281 to about 789 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 259 to about 727 pg/ml at about 144 hours after initiation of the dosing interval; a mean plasma concentration from about 230 to about 630 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate of from about 38 ug/hr to about 64 ug/hr is maintained from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 4 ug/hr to about 7 ug/hr is maintained from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after the initiation of a seven-day dosing interval).
- buprenorphine is administered to human patients in a manner such that the following mean plasma concentrations are achieved over a 72 hour dosing interval: a mean plasma concentration from about 5 to about 113 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 55 to about 296 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 118 to about 644 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 203 to about 753 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 247 to about 984 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 317 to about 984 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from about 339 to about 1052
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained from about 306 to about 1052 pg/ml over at least the next 48 hours.
- the buprenorphine is administered in a manner such that the mean plasma concentrations are maintained as follows: a mean plasma concentration from about 369 to about 1052 pg/ml at about 96 hours after initiation of the dosing interval; a mean plasma concentration from about 374 to about 1052 pg/ml at about 120 hours after initiation of the dosing interval; a mean plasma concentration from about 346 to about 970 pg/ml at about 144 hours after initiation of the dosing interval; a mean plasma concentration from about 306 to about 841 pg/ml at about 168 hours after initiation of the dosing interval (for a seven day dosing interval).
- a mean relative release rate of from about 51 ug/hr to about 86 ug/hr is maintained from the initiation of the dosing interval until about 72 hours after the initiation of the, e.g., dosing interval; and a mean relative release rate of about 5 ug/hr to about 9 ug/hr is maintained from about 72 hours after the initiation of the dosing interval until the end of the dosing interval, e.g., about 168 hours after the initiation of a seven-day dosing interval (e.g., about 168 hours after the initiation of a seven-day dosing interval).
- the method comprises the administration of buprenorphine transdermally to human patients according to very different relative release rates for the first 3 day portion of the dosing interval (indicative of substantially first order release), and the additional at least 2 day long portion of the dosing interval (substantially zero order release) such that mean relative release rates are achieved over the dosing interval as follows: a mean relative release rate of from about 3 ug/hr to about 86 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 0.3 ug/hr to about 9 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after the initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 3 ug/hr to about 5 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 0.3 ug/hr to about 0.6 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 6 ug/hr to about 11 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of dosing interval; and a mean relative release rate of about 0.7 ug/hr to about 1 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 13 ug/hr to about 21 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 1 ug/hr to about 2 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 26 ug/hr to about 43 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 3 ug/hr to about 4 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after the initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 39 ug/hr to about 64 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 4 ug/hr to about 7 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval (e.g., about 168 hours after the initiation of a seven-day dosing interval).
- the mean relative release rates achieved over the dosing interval are as follows: a mean relative release rate of from about 51 ug/hr to about 86 ug/hr from the initiation of the dosing interval until about 72 hours after the initiation of the dosing interval; and a mean relative release rate of about 5 ug/hr to about 9 ug/hr from about 72 hours after the initiation of the dosing interval until the end of the dosing interval, e.g., about 168 hours after the initiation of the dosing interval.
- the method of the present invention may be accomplished by any mode of administration useful for buprenorphine known to those skilled in the art. However, certain modes of administration are more practical than others. Preferably, the mode of administration is via continuous infusion, through the oral mucosa, or most preferably, transdermally.
- the pattern of plasma concentrations seen through time in this invention can be achieved by using the injectable, parenteral form of, e.g., buprenorphine hydrochloride suitably diluted in an intravenous infusion solution.
- the infusion rate would be controlled by a programmable infusion pump, to provide the desired plasma profile.
- the mode of administration of the buprenorphine is transdermal.
- Transdermal delivery of active agents is measured in terms of “relative release rate” or “flux”, i.e., the rate of penetration of the active agent through the skin of an individual.
- M represents the cumulative amount of drug entering the blood stream.
- the variables dM and dt represent the change in cumulative amount of drug entering the blood stream and change in time, respectively.
- transdermal delivery systems in order to maintain a desired flux rate for a desired dosing period, it is necessary to include an overage of active agent in the transdermal delivery system in an amount that is substantially greater than the amount to be delivered to the patient over the desired time period. For example, to maintain the desired flux rate for a three day time period, it is considered necessary to include much greater than 100% of a three day dose of an active agent in a transdermal delivery system. This overage is necessary for creating a concentration gradient by means of which the active agent migrates through the layers of the transdermal delivery system to the desired site on a patient's skin. The remainder of the active agent remains in the transdermal delivery system.
- transdermal delivery system It is only the portion of active agent that exits the transdermal delivery system that becomes available for absorption into the skin. The total amount of active agent absorbed into the patient's blood stream is less than the total amount available. The amount of overage to be included in a transdermal delivery system is dependent on these and other factors known to the skilled artisan.
- a transdermal delivery system containing a sufficient amount of opioid, e.g., buprenorphine, to provide a desired relative release rate for up to 3 days, and after single administration (application) of the transdermal dosage form, leaving the dosage form on the skin for approximately a 5 to 8 day time period, thereby resulting in the flux being maintained over the prolonged period and effective blood plasma levels and pain management being maintained over the prolonged period.
- the desired flux is maintained at least about 5, preferably at least 8 days after application of the transdermal delivery system. If the transdermal delivery system is removed 3 days after its administration, no analgesia is present a short time after removal.
- analgesia is maintained over the prolonged period of contact, but the patient continues to experience analgesia.
- inclusion of the aforementioned overage of buprenorphine provides analgesia for at least about twice the expected 3 day dosing interval.
- transdermal delivery system Any type of transdermal delivery system may be used in accordance with the methods of the present invention so long as the desired pharmacokinetic and pharmacodynamic response(s) are attained over at least 3 days, e.g., from about 5 to about 8 days.
- Preferable transdermal delivery systems include e.g., transdermal patches, transdermal plasters, transdermal discs, iontophoretic transdermal devices and the like.
- Transdermal dosage forms used in accordance with the invention preferably include a backing layer made of pharmaceutically acceptable material which is impermeable to the buprenorphine.
- the backing layer preferably serves as a protective cover for the active agent, e.g. buprenorphine and may also provide a support function.
- materials suitable for making the backing layer are films of high and low density polyethylene, polypropylene, polyvinylchloride, polyurethane, polyesters such as poly(ethylene phthalate), metal foils, metal foil laminates of such suitable polymer films, textile fabrics, if the components of the reservoir cannot penetrate the fabric due to their physical properties and the like.
- the materials used for the backing layer are laminates of such polymer films with a metal foil such as aluminum foil.
- the backing layer can be any appropriate thickness which will provide the desired protective and support functions. A suitable thickness will be from about 10 to about 200 microns. Desirable materials and thickness will be apparent to the skilled artisan.
- the transdermal dosage forms used in accordance with the invention contain a polymer matrix layer.
- the polymers used to form the biologically acceptable polymer matrix are those capable of forming thin walls or coatings through which pharmaceuticals can pass at a controlled rate.
- a non-limiting list of exemplary materials for inclusion in the polymer matrix includes polyethylene, polypropylene, ethylene/propylene copolymers, ethylene/ethylacrylate copolymers, ethylenevinyl acetate copolymers, silicones, rubber, rubber-like synthetic homo-, co- or block polymers, polyacrylic esters and the copolymers thereof, polyurethanes, polyisobutylene, chlorinated polyethylene, polyvinylchloride, vinyl chloride-vinyl acetate copolymer, polymethacrylate polymer (hydrogel), polyvinylidene chloride, poly(ethylene terephthalate), ethylene-vinyl alcohol copolymer, ethylene-vinyloxyethanol copolymer, silicones including silicone copolymers such as polysiloxane-polymethacrylate copolymers, cellulose polymers (e.g., ethyl cellulose, and cellulose esters), polycarbonates, polytetrafluor
- Preferred materials for inclusion in the polymer matrix layer are silicone elastomers of the general polydimethylsiloxane structures, (e.g., silicone polymers). Preferred silicone polymers cross-link and are pharmaceutically acceptable. Other preferred materials for inclusion in the polymer matrix layer include: silicone polymers that are cross-linkable copolymers having dimethyl and/or dimethylvinyl siloxane units which can be crosslinked using a suitable peroxide catalyst.
- polymers consisting of block copolymers based on styrene and 1,3-dienes (particularly linear styrene-isoprene-block copolymers of styrene-butadiene-block copolymers), polyisobutylenes, polymers based on acrylate and/or methacrylate.
- the polymer matrix layer may optionally include a pharmaceutically acceptable crosslinking agent.
- Suitable crosslinking agents include, e.g., tetrapropoxy silane.
- Preferred transdermal delivery systems used in accordance with the methods of the present invention include an adhesive layer to affix the dosage form to the skin of the patient for a desired period of administration, e.g., about 5 to about 8 days. If the adhesive layer of the dosage form fails to provide adhesion for the desired period of time, it is possible to maintain contact between the dosage form with the skin by, for instance, affixing the dosage form to the skin of the patient with an adhesive tape, e.g., surgical tape.
- an adhesive tape e.g., surgical tape.
- adhesion of the dosage form to the skin of the patient is achieved solely by the adhesive layer of the dosage form or in connection with a peripheral adhesive source, such as surgical tape, provided that the dosage form is adhered to the patient's skin for the requisite administration period.
- the adhesive layer preferably includes using any adhesive known in the art that is pharmaceutically compatible with the dosage form and preferably hypoallergenic, such as polyacrylic adhesive polymers, acrylate copolymers (e.g., polyacrylate) and polyisobutylene adhesive polymers.
- the adhesive is a pressure-sensitive contact adhesive, which is preferably hypoallergenic.
- the transdermal dosage forms which can be used in accordance with the present invention may optionally include permeation enhancing agent.
- Permeation enhancing agents are compounds which promote penetration and/or absorption of the buprenorphine into the blood stream of the patient.
- a non-limiting list of permeation enhancing agents includes polyethylene glycols, surfactants, and the like.
- permeation of buprenorphine may be enhanced by occlusion of the dosage form after application to the desired site on the patient with, e.g. an occlusive bandage. Permeation may also be enhanced by removing hair from the application site by, e.g. clipping, shaving or use of a depilatory agent.
- Another permeation enhancer is heat. It is thought that heat enhancement can be induced by, among other things, using a radiating heat form, such as an infrared lamp, onto the application site after application of the transdermal dosage form.
- Other means of enhancing permeation of buprenorphine such as the use of iontophoretic means are also contemplated to be within the scope of the present invention.
- a preferred transdermal dosage form which may be used in accordance with the present invention includes a non-permeable backing layer made, for example, of polyester; an adhesive layer made, for example of a polyacrylate; and a matrix containing the buprenorphine and other desirable pharmaceutical aids such as softeners, permeability enhancers, viscosity agents and the like.
- the active agent may be included in the device in a drug reservoir, drug matrix or drug/adhesive layer.
- the active agent is buprenorphine or a pharmaceutically acceptable salt thereof.
- Suitable transdermal delivery systems also include a softening agent.
- Suitable softening agents include higher alcohols such as dodecanol, undecanol, octanol, esters of carboxylic acids, wherein the alcohol component may also be a polyethoxylated alcohol, diesters of dicarboxylic acids, such as di-n-butyladiapate, and triglycerides particularly medium-chain triglycerides of the caprylic/capric acids or coconut oil, have proved to be particularly suitable.
- suitable softeners are multivalent alcohols, for example, levulinic acid, cocprylic acids glycerol and 1,2-propanediol which can also be etherified by polyethylene glycols.
- a buprenorphine solvent may also be included in the transdermal delivery systems of the present invention.
- the solvents dissolve the buprenorphine to a sufficient extent thereby avoiding complete salt formation.
- suitable solvents include those with at least one acidic group. Particularly suitable are monoesters of dicarboxylic acids such as monomethylglutarate and monomethyladipate.
- compositions which may be included in the reservoir or matrix include: solvents, for example alcohols such as isopropanol; permeation enhancing agents such as those described above; and viscosity agents, such as cellulose derivatives, natural or synthetic gums, such as guar gum, and the like.
- solvents for example alcohols such as isopropanol
- permeation enhancing agents such as those described above
- viscosity agents such as cellulose derivatives, natural or synthetic gums, such as guar gum, and the like.
- the transdermal dosage form includes a removable protective layer.
- the removable protective layer is removed prior to application, and consists of the materials used for the production of the backing layer described above provided that they are rendered removable, for example, by a silicone treatment.
- Other removable protective layers for example, are polyletra-fluoroethylene, treated paper, allophane, polyvinyl chloride, and the like.
- the removable protective layer is in contact with the adhesive layer and provides a convenient means of maintaining the integrity of the adhesive layer until the desired time of application.
- composition of the transdermal dosage forms used in accordance with the invention and the type of device used are not considered critical to the method of the invention, provided that the device delivers the active agent, e.g. buprenorphine, for the desired time period and at the desired flux rate and/or the desired delivery rate of the transdermal dosage form.
- the active agent e.g. buprenorphine
- Such buprenorphine transdermal delivery systems may be a laminated composite having an impermeable backing layer containing buprenorphine, and optionally, a permeation enhancer combined with a pressure-sensitive adhesive.
- a preferred transdermal dosage form in accordance with the '711 patent includes: (i) a polyester backing layer which is impermeable to buprenorphine; (ii) a polyacrylate adhesive layer; (iii) a separating polyester layer; and (iv) a matrix containing buprenorphine, a solvent for the buprenorphine, a softener and a polyacrylate adhesive.
- the buprenorphine solvent may or may not be present in the final formulation.
- the transdermal delivery device described therein includes a backing layer which is impermeable to the active substance, a pressure-sensitive adhesive reservoir layer and optionally, a removable protective layer.
- the reservoir layer includes about 10 to about 98%-wt polymeric material, about 0.1 to about 40%-wt softener, about 0.1 to about 30%-wt buprenorphine.
- a solvent for the buprenorphine base or pharmaceutically acceptable salt thereof may be included as about 0.1 to about 30%-wt.
- the transdermal delivery system is prepared in accordance with Example 1 appended hereto.
- the transdermal delivery system was prepared in accordance with the disclosure of International Patent Application No. WO 96/19975 (Hille, et. al.; assigned to LTS Lohmann Therapie-Systeme GMBH), hereby incorporated by reference.
- the buprenorphine transdermal delivery device contains resorption-promoting auxiliary substances. The resorption-promoting auxiliary substance forms an undercooled mass.
- the delivery system contains 10% buprenorphine base, 10-15% acid (such as levulinic acid), about 10% softener (such as oleyoleate); 55-70% polyacrylate; and 0-10% polyvinylpyrollidone (PVP).
- 10% buprenorphine base 10-15% acid (such as levulinic acid), about 10% softener (such as oleyoleate); 55-70% polyacrylate; and 0-10% polyvinylpyrollidone (PVP).
- the nominal delivery rate of buprenorphine from such patches will be, e.g., from about 12.5 to about 100 ug/hr.
- the total of buprenorphine included in the transdermal patch is about 5 mg
- the active surface area is about 6.25 cm 2
- the patch size may be, e.g., about 19.4 cm 2 .
- the total of buprenorphine included in the transdermal patch is about 10 mg
- the active surface area is about 12.5 cm 2 and the patch size may be, e.g., about 30.6 cm 2 .
- the total of buprenorphine included in the transdermal patch is about 20 mg
- the active surface area is about 25 cm 2 and the patch size may be, e.g., about 51.8 cm 2 .
- the total of buprenorphine included in the transdermal patch is about 30 mg
- the active surface area is about 37.5 cm 2 and the patch size may be, e.g., about 69.8 cm 2 .
- the total of buprenorphine included in the transdermal patch is about 40 mg
- the active surface area is about 50 cm 2 and the patch size may be, e.g., about 87.8 cm 2 .
- the transdermal delivery system has been designed to be adhered to the patient for only three days and is expected to release analgetically effective doses of buprenorphine for only about 3 days.
- the transdermal delivery device is maintained in contact with the skin of the patient for a much longer time period, e.g., from about 5 to about 8 days, without any change in the formulation of the transdermal device itself. It has surprisingly been found that analgesia is maintained for this extended period of time (the time beyond the useful life designed for the transdermal formulation).
- the buprenorphine transdermal delivery system may be a plaster such as that described in U.S. Pat. No. 5,225,199 to Hidaka et al., hereby incorporated by reference.
- Such plasters include a film layer including a polyester film of about 0.5 to about 4.9 ⁇ m thickness, about 8 to about 85 g/mm strength, respectively in the two directions intersecting substantially at right angles, about 30 to about 150% elongation, in the two directions intersecting substantially at right angles and an elongation ratio of A to B of about 1.0 to about 5.0, wherein A and B represent data in two directions intersecting at right angles, and A is greater than B and wherein said polyester film includes about 0.01 to about 1.0% by weight, based on the total weight of the polyester film, of solid fine particles in which the average particle size is about 0.001 to about 3.0 ⁇ m and an adhesive layer which is composed of an adhesive containing transdermally absorbable drugs; wherein the adhesive layer is laminated on said film layer over the surface in about 2
- the transdermal delivery system used in the present invention may also be prepared in accordance with U.S. Pat. No. 5,069,909 (Sharma et al.), hereby incorporated by reference.
- This patent describes a laminated composite for administering buprenorphine transdermally to treat pain.
- the composite includes an impermeable backing layer providing a protective covering for the composite which may be made from an elastomeric polymer such as polyurethane, polyether amide, or copolyester and may be about 15-250 microns in thickness.
- the composite further includes a reservoir lamina composed of buprenorphine (base or HCl) in n amount of 1-12% by weight and a pressure-sensitive adhesive, e.g., polyisobutylene, or a silicone adhesive such as silastic, or an acrylate adhesive, and 2-35% permeation enhancer (comprising propylene glycol monolaurate in combination with capric acid or oleic acid).
- a pressure-sensitive adhesive e.g., polyisobutylene, or a silicone adhesive such as silastic, or an acrylate adhesive
- permeation enhancer comprising propylene glycol monolaurate in combination with capric acid or oleic acid.
- the transdermal delivery system used in the present invention may also be prepared in accordance with U.S. Pat. No. 4,806,341 (Chien et al.), hereby incorporated by reference.
- This patent describes a transdermal morphinan narcotic analgesic or antagonist (including buprenorphine) pharmaceutical polymer matrix dosage unit having a backing layer which is substantially impervious to the buprenorphine, and a polymer matrix disc layer which is adhered to the backing layer and which has microdispersed therein effective dosage amounts of the buprenorphine.
- the polymer matrix may be a silicon polymer or copolymer, such as methyl silicone polymer or copolymer, or methylvinyl silicone polymer or copolymer.
- the polymer matrix layer preferably has dispersed therein a skin permeation enhancing agent such as isopropyl myristate, azone, or a combination of ethyl caprylate and capryl alcohol.
- compositions for the transdermal delivery of buprenorphine comprise buprenorphine in a carrier of a polar solvent material selected from the group consisting of C 3 -C 4 diols, C 3 -C 6 triols, and mixtures thereof, and a polar lipid material selected from the group consisting of fatty alcohol esters, fatty acid esters, and mixtures thereof; wherein the polar solvent material and the lipid material are present in a weight ratio of solvent material:lipid material of from 60:40 to about 99:1.
- the transdermal delivery system used in the present invention may also be that described in U.S. Pat. No. 4,588,580 (Gale, et. al.), hereby incorporated by reference.
- That system comprises a reservoir for the drug having a skin proximal, material releasing surface area in the range of about 5-100 cm 2 and containing between 0.1 and 50% by weight of a skin permeable form of the buprenorphine.
- the reservoir contains an aqueous gel comprising up to about 47-95% ethanol, 1-10% gelling agent, 0.1-10% buprenorphine, and release rate controlling means disposed in the flow path of the drug to the skin which limits the flux of the buprenorphine from the system through the skin.
- the release rate controlling means is more permeable to the buprenorphine than to the ethanol, and may be for example low density polyethylene (LDPE), ehtylene-vinyl acetate (EVA) copolymers, heat sealable polyesters, and elastomeric polyester block copolymers, such as HYTREL® from DuPont.
- LDPE low density polyethylene
- EVA ehtylene-vinyl acetate copolymers
- heat sealable polyesters elastomeric polyester block copolymers
- HYTREL® elastomeric polyester block copolymers
- the present invention may also be accomplished via the use of a sustained oral mucosal delivery system.
- a sustained oral mucosal delivery system Such a system is described by McQuinn, R. L. et al., “Sustained Oral Mucosal Delivery in Human Volunteers J. Controlled Release; (34) 1995 (243-250).
- oral mucosal patches were prepared by homogeneously mixing buprenorphine free base (8%), Carbopol 934 (52%), polyisobutylene (35%) and polyisoprene (5%, w/w) via a two-roll mill and then compressing the mixture to the appropriate thickness.
- a membrane backing ethylcellulose
- circular disks 0.5 cm 2
- the backing was included in order to retard drug release from one side of the disk and to prohibit adhesion to opposing side tissues.
- Each soft, flexible disk was approximately 0.6 mm thick and contained 2.9 mg buprenorphine. These patches were worn by the subjects for 12 hours. Gum and lip application was tested, although adhesion at the gum site was considered superior. After the initial appearance of serum buprenorphine ( ⁇ 25 pg/ml), levels generally increased relatively rapidly and persisted until the patch was removed. After the patch was removed, buprenorphine levels fell promptly and were at a relatively low (but measureable) level by 24 hours post-dose. It was estimated that 0.42 ⁇ 0.18 mg were delivered via the gum treatment. From this discussion, it is apparent that an oral mucosal patch can be prepared which will provide plasma concentrations considered desirable according to the present invention.
- a significantly higher incidence in side effects such as nausea, vomiting or drowsiness would normally be expected when high blood levels of opioid analgesics are administered.
- the present invention by maintaining a lower blood level of drug over the 7 day dosing period while maintaining effective pain management, has a lower incidence of side effects.
- a much higher plasma concentration is seen in patients over the same period of time when a new transdermal delivery device of the same strength is put on every three days, and therefore increased side effects are expected with each new 3 day transdermal application.
- hysteresis In general, upon administration of an opioid analgesic, there is a lag time or “hysteresis”, between the pharmacodynamic effects and the time course of opioid plasma concentration levels. Generally, peak plasma level concentrations are often attained prior to exhibition of the maximum pharmacotherapeutic or side effect response. It has been surprisingly discovered that the method according to the present invention provides a “reverse hysteresis”, i.e. the rise in plasma concentrations follow the appearance and rise of certain of the pharmacodynamic events and side effects.
- a seven day pharmacokinetic/pharmacodynamic study was conducted on 24 healthy human patients.
- the subjects were comprised of approximately an equal number of male and female subjects.
- the buprenorphine was administered via a transdermal patch which is described in WO 96/19975.
- transdermal patch is prepared in accordance with the disclosure of WO 96/19975 for Example 1 therein as follows:
- Example 1 The formulation utilized for Example 1 is substantially the same as that described in Example 3 of WO 96/19975, which is prepared in accordance with Example 1 and is stated therein to include 10% buprenorphine, 10% levulinic acid, 10% polyvinylpyrollidone, 10% oleyloeate, and 60% polyacrylate.
- the total of buprenorphine included in the transdermal patch is about 10 mg
- the active surface area is about 12.5 cm 2
- the patch size may be, e.g., about 30.6 cm 2 .
- the dosing regimen was one (1) patch containing 10 mg buprenorphine base/patch reservoir applied to the subject's skin and maintained in contact with the skin for a time period of seven (7) days.
- the adhesive patch with the medication being tested was placed on the right midaxillary line at the level of the 5th intercostal space at approximately 0800 hr on day 1.
- the skin was washed with lukewarm soapy water, then rinsed with clear water and left to air dry. The skin was not rubbed while it was being washed. The application site was relatively hairless. Hair was not clipped or shaven.
- the patches were removed at approximately 0800 hr on day 8. Following patch removal, the patch site was not washed or rubbed until the last blood collection for that treatment period was over. Each patch was placed unfolded onto its release liner and the patch/release liner unit was placed back in the correct pouch, which was then sent to a bioanalytical laboratory for residual buprenorphine assay.
- Blood sampling (10 ml at each time point) started on day 1, and continued thereafter at the following times: 1 hr (pre-dose) and at regular intervals thereafter during the 7 day dosing interval.
- Patch site skin observations of the patch sites were performed by the investigator/staff rating the quality of the skin at the site of the actual medication reservoir of the patch at 0 hr (prior to patch placement) and 30 minutes after patch removal.
- the rating scale was as follows:
- AUC (O-last) (pg.hr/ml)—the area under the curve from time zero to the time of last non-zero plasma buprenorphine concentration, calculated by the linear trapezoidal method;
- the mean plasma concentrations are further depicted in FIG. 1 (concentration pg/ml vs. time (days)). It is apparent from the pharmacokinetic results obtained with respect to Example 1 that the mean blood plasma concentrations rose steadily and peaked at about the 3-day time point during the dosing interval (e.g., about 72 hours after application of the patch), and thereafter surprisingly remained relatively steady throughout the remaining portion of the dosing interval (e.g., to about the 7-day time point, 168 hours after initiation of the dosing interval). Further, it is apparent from the buprenorphine plasma concentrations that first order kinetics were present during the first 72 hours of the dosing interval, and substantially zero order kinetics were present thereafter.
- VAS visual analog scale
- Table 4 provides a summary of the amount of drug which was measured as remaining in the patches which were removed from the subjects after 7 days.
- the buprenorphine transdermal delivery system (patch) used in the Comparative Examples A and C contained 20 mg buprenorphine base, and is prepared in accordance with Example 1. It was contemplated that the buprenorphine patch of Comparative Examples A and C would provide approximately double the dose and approximately double the relative release rate as compared to the buprenorphine patch of Example 1. For Comparative Examples A and C, it was contemplated that approximately 1.2 mg buprenorphine would be released from the patch per day, which is equivalent to an intravenous dose of 0.3 mg every 6 hours.
- the reference buprenorphine intravenous injection (Comparative Example B) was 0.3 mg (Temgesic®) Injectable 0.3 mg/ml, [1 ml/vial]).
- the buprenorphine transdermal delivery system (single dose) was adhered to a relatively hairless area of a subject's right thorax at the level of the fifth intercostal space in the midaxillary line at approximately 8 am on day 1 and removed at approximately 8 am on day 4.
- buprenorphine intravenous (IV) injection 0.3 mg was infused over 2 minutes at approximately 8 am on day 1 through an in-dwelling cannula in the right anticubital vein.
- the buprenorphine intravenous 0.3 mg blood sampling was conducted as follows: Day 1: 0, 1, 2, 3, 5, 10, 15, 20, 25, 30, 45 minutes and 1, 1.5, 2, 3, 4, 5, 6, 10, 12, 24 hr; arterial blood sampling (left radial artery) for the first 4 hours; venous blood sampling from 2 hours post-dose to 24 hours post-dose. Therefore arterial and venous blood sampling occurred simultaneously 2, 3 and 4 hours post-dose.
- the buprenorphine transdermal delivery system (3 sequential applications), was adhered to a relatively hairless area of a subject's right thorax at the level of the fifth intercostal space in the midaxillary line at approximately 8 am on day 1 and removed at approximately 8 am on day 4.
- the second buprenorphine transdermal delivery system 50 ⁇ g/hr was placed just adjacent to the first patch after the first was removed on day 4 at approximately 8 am and removed on day 7 at approximately 8 am.
- the third buprenorphine transdermal delivery system 50 ⁇ g/hr was placed just adjacent to the second patch but not in the same place as the first patch after the second patch is removed on day 7 at approximately 8 am and removed on day 10 at approximately 8 am.
- Plasma concentration data was analyzed using standard noncompartmental and compartmental techniques to derive pharmacokinetic parameters.
- various exploratory methods including fitting the intravenous data to pharmacokinetic models to determine which model best describes the data, and deconvolution analysis to determine the absorption rate was employed.
- Other parameters such as clearance, volumes of distribution, absorption rate, amount absorbed and bioavailability were determined by either standard noncompartmental or compartmental analysis or exploratory methods.
- the intravenous data was also analyzed utilizing compartmental modeling techniques.
- VAS visual analog scale
- FIGS. 3-5 The pharmacokinetic results (concentration in pg/ml vs. hours) for Comparative Examples A-C are depicted in FIGS. 3-5 , respectively.
- FIG. 4 depicts the plasma concentration obtained divided by 100.
- the pharmacodynamic results (PD variables (VAS)) for Comparative Examples A-C C, B, A are depicted in FIGS. 6 - 8 6 , 7 , 8 , respectively.
- the bioequivalence between a buprenorphine transdermal delivery system in accordance with Example 1 is compared to identically prepared patches having different sizes and therefore different amounts of buprenorphine contained therein.
- Comparative Example D utilized a patch identical in size and containing the same amount of buprenorphine as Example 1.
- the total of buprenorphine included in the transdermal patch is 10 mg, the active surface area is 12.5 cm 2 and the patch size is 30.6 cm 2 .
- Comparative Example E two patches are utilized, each patch including total of buprenorphine of about 5 mg, and having an active surface area of 6.25 cm 2 and a patch size of 19.4 cm 2 .
- Comparative Example F allows for the determination of the dose proportionality of a buprenorphine transdermal delivery system (patch) having twice the dose as compared to Example 1.
- the total of buprenorphine included in the transdermal patch is 20 mg, the active surface area is 25 cm 2 and the patch size is 51.8 cm 2 .
- the study was conducted via a 3-way cross-over design. The patches were left in place for 72 hours and then removed.
- Table 11 provides a summary of mean plasma buprenorphine concentrations (pg/ml) at each sampling time for Comparative Example D:
- Table 13 provides a summary of mean plasma buprenorphine concentrations for Comparative Example E:
- Table 15 provides a summary of mean plasma buprenorphine concentrations for Comparative Example F:
- Table 16 provides a summary of the dose-corrected pharmacokinetic parameters for Comparative Example F. The values are calculated based on a Cmax value which is one-half the actual reported value:
- Table 17 provides a summary of the buprenorphine patch residuals for each of Comparative Examples D-F:
- Table 18 provides a direct comparison of the plasma concentrations obtained from Example 1 (a 10 mg buprenorphine patch maintained in contact with the subjects' skin for 7 days) to Comparative Example A (20 mg buprenorphine patch left on the subjects' skin for only 3 days, then removed) to Comparative Example C (three sequential applications of a 20 mg buprenorphine patch left on the subjects' skin for only 3 days, then removed).
- Example 1 a 10 mg buprenorphine patch maintained in contact with the subjects' skin for 7 days
- Comparative Example A (20 mg buprenorphine patch left on the subjects' skin for only 3 days, then removed
- Comparative Example C three sequential applications of a 20 mg buprenorphine patch left on the subjects' skin for only 3 days, then removed.
- the plasma concentrations of Comparative Examples A and C are also presented at 50% concentrations for each time interval:
- Example 18 shows that, surprisingly, plasma levels effective to provide analgesia were present in Example 1 (patch remained on skin for 7 days) even 7 days after application of the patch; whereas in Comparative Example A (patch removed after 3 days), blood levels fell dramatically once the patch was removed, such that plasma levels which would be indicative of ineffective treatment for the dosage of buprenorphine occurred not long after patch removal.
- Comparative Example C it is apparent that the plasma levels obtained from 3-day sequential administration of the buprenorphine patch resulted in significant increases in Cmax levels during each day dosing interval. This fact is confirmed by the graph of plasma concentration over time for Comparative Example C provided in FIG. 3 .
- Example 1 the plasma level for Example 1 remained substantially level over the time-frame of 72 hours-168 hours after patch application.
- the results indicate that the method of the present invention has the surprising benefit of reducing total plasma concentrations of buprenorphine required to allow patients to experience effective analgesia.
- Comparative Example C it is apparent that side effects were significantly reduced according to the method of Example 1, during the 7-day dosage interval. Further benefits are obtained from the invention with respect to modes of administration other than transdermally where the large plasma concentration peaks obtained in the prior art, e.g., through intravenous dosing, can be avoided.
- Comparative Example B a Cmax in excess of about 30,000 pg/ml was obtained.
- Table 19 provides a direct comparison of the plasma concentrations of Example 1 (a 10 mg buprenorphine patch maintained in contact with the subjects' skin for 7 days) to Comparative Example D (same 10 mg buprenorphine patch left on the subjects' skin for only 3 days, then removed) to Comparative Example E (two 5 mg buprenorphine patches left on the subject' skin for only 3 days, then removed):
- the total amount of buprenorphine released for Example 1 (1.40 mg) may be expressed as 0.2 mg buprenorphine administered per day, when averaged over the seven day dosing interval.
- Comparative Example E D (same patch over 3 days) released a total of 1.23 mg, which may be expressed as 0.41 mg buprenorphine administered per day. Comparing the cumulative amount released for Example 1 as compared to Comparative Example D, it can be seen that the present invention results in one-half the dose (mg/patch/day) which would be administered based on prior art methodology.
- the results indicate that over the first 72 hours the buprenorphine is released substantially according to first order kinetics, whereas during the 72-168 hour time period after administration, the buprenorphine is released substantially according to zero order kinetics. This is confirmed from the plasma concentration curve provided for Example 1 in FIG. 1 .
- Example 2 the method of the present invention is accomplished via a different mode of administration, i.e., intravenous infusion.
- the pattern of plasma concentrations seen through time in this invention can be achieved by using an intravenous infusion using the injectable, parenteral form of, e.g., buprenorphine hydrochloride suitably diluted in an intravenous infusion solution.
- the infusion rate would be controlled by a programable infusion pump, to provide the desired plasma concentration profile.
- the rate of infusion through time can be determined and adjusted based upon pharmacodynamic parameters such as pupil size (pupilometry) or pain relief (analgesia) or by the results of a suitable bioassay to determine the plasma buprenorphine concentrations at any particular point in time.
- mean plasma concentrations are obtained as follows: a mean plasma concentration from about 1 to about 28 pg/ml at about 6 hours after initiation of the dosing interval; a mean plasma concentration from about 14 to about 74 pg/ml at about 12 hours after initiation of the dosing interval; a mean plasma concentration from about 30 to about 161 pg/ml at about 24 hours after initiation of the dosing interval; a mean plasma concentration from about 51 to about 188 pg/ml at about 36 hours after initiation of the dosing interval; a mean plasma concentration from about 62 to about 246 pg/ml at about 48 hours after initiation of the dosing interval; a mean plasma concentration from about 79 to about 246 pg/ml at about 60 hours after initiation of the dosing interval; a mean plasma concentration from about 85 to about 263 pg/ml at about 72 hours after initiation of the dosing interval; a mean plasma concentration from about 92 to about 263
Abstract
Description
-
- Deff=apparent diffusion coefficient M/√t=2·CO·√{square root over (Deff/π)}
- CO=initial drug concentration in the transdermal delivery system
- CS=saturation concentration
- t=time
- Assumptions: perfect sink; diffusion of dissolved drug is rate controlling; therefore Q=const.√t
-
- Assumptions: perfect sink; diffusion of dissolved drug is rate controlling; Mt≦0.4M0 therefore Q=cost.√t
Amount released per area unit Q=const (zero order kinetics)
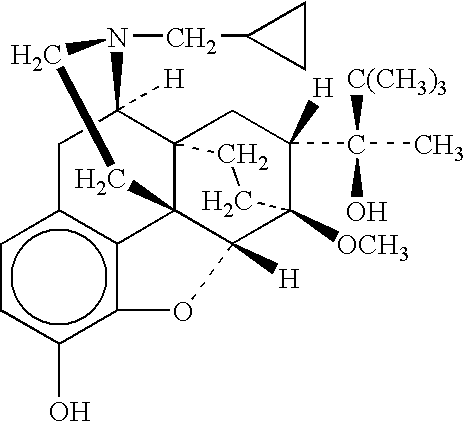
dM/dt=J=P*C
where J is the skin flux, P is the permeability coefficient and C is the concentration gradient across the membrane, assumed to be the same as the donor concentration. M represents the cumulative amount of drug entering the blood stream. The variables dM and dt represent the change in cumulative amount of drug entering the blood stream and change in time, respectively.
-
- 1.139 g of a 47.83 w/% polyacrylate solution with a selfnetting acrylate copolymers containing 2-ethylhexylacrylates, vinyl acetates, acrylic acid (dissolving agent:ethylacetate:heptan:isopropanol:toluol:acetylacetonate in the ratio of 37:26:26:4:1), 100 g laevulinic acid, 150 g oleyloleate, 100 g polyvinylpyrollidone, 150 g ethanol, 200 g ethyl acetate and 100 g buprenorphine base are homogenized. The mixture is stirred for about 2 hours and then examined visually to determine if all solid substances have been dissolved. One has to control the evaporation loss by method of weighing back and makes up for the solvent with the help of ethylacetate, if necessary. Thereafter, the mixture is put onto a 420 mm wide, transparent polyester foil, since the surface weight of the dried layer of paste is 80 g per m2. The polyester foil which can be dissolved again with treatment of silicone serves as a protective layer. The solvent is removed by drying with heated air which is led over a moist lane. With this treatment of warmth not only do solvents evaporate but the the laevulinic acid melts as well. Thereafter, the sealing film is covered with a
polyester foil 15 μab. A surface of about 16 cm2 is cut with the help of the appropriate cutting tool, and the rims that have been left between the individual systems are removed.
- 1.139 g of a 47.83 w/% polyacrylate solution with a selfnetting acrylate copolymers containing 2-ethylhexylacrylates, vinyl acetates, acrylic acid (dissolving agent:ethylacetate:heptan:isopropanol:toluol:acetylacetonate in the ratio of 37:26:26:4:1), 100 g laevulinic acid, 150 g oleyloleate, 100 g polyvinylpyrollidone, 150 g ethanol, 200 g ethyl acetate and 100 g buprenorphine base are homogenized. The mixture is stirred for about 2 hours and then examined visually to determine if all solid substances have been dissolved. One has to control the evaporation loss by method of weighing back and makes up for the solvent with the help of ethylacetate, if necessary. Thereafter, the mixture is put onto a 420 mm wide, transparent polyester foil, since the surface weight of the dried layer of paste is 80 g per m2. The polyester foil which can be dissolved again with treatment of silicone serves as a protective layer. The solvent is removed by drying with heated air which is led over a moist lane. With this treatment of warmth not only do solvents evaporate but the the laevulinic acid melts as well. Thereafter, the sealing film is covered with a
-
- Erythema: 0=No visible redness; 1=Very slight redness (just perceptible); 2=Slight but well-defined redness; 3=Moderately intense redness; 4=Severe erythema (dark red discoloration of the skin).
- Edema: 0=No visible reactions; 1=Very mild edema (just perceptible); 2=Mild edema (corners of the area are well defined due to noticeable swelling); 3=Moderate edema (up to 1 mm swelling in diameter); 4=Severe edema (more than 1 mm swelling in diameter, protruding over the edges of the patch).
| TABLE 1 | |||||
| HOURS1 | MEAN2 | STD. DEV.3 | |
||
| 6 | 1.76 | 6.20 | 352.77 | ||
| 12 | 18.47 | 26.00 | 140.78 | ||
| 18 | 39.45 | 36.16 | 91.67 | ||
| 24 | 58.94 | 44.66 | 75.76 | ||
| 30 | 67.69 | 48.78 | 72.06 | ||
| 36 | 82.44 | 53.02 | 64.32 | ||
| 42 | 107.61 | 65.43 | 60.81 | ||
| 48 | 104.69 | 60.69 | 57.97 | ||
| 54 | 105.81 | 66.68 | 63.02 | ||
| 60 | 112.93 | 63.02 | 55.81 | ||
| 66 | 129.25 | 64.37 | 49.80 | ||
| 72 | 130.55 | 64.16 | 49.14 | ||
| 78 | 122.83 | 54.97 | 44.75 | ||
| 84 | 129.03 | 51.50 | 39.92 | ||
| 90 | 139.50 | 68.26 | 48.93 | ||
| 96 | 146.70 | 62.76 | 42.78 | ||
| 102 | 130.19 | 57.68 | 44.31 | ||
| 108 | 135.49 | 67.72 | 49.98 | ||
| 114 | 150.24 | 71.69 | 47.72 | ||
| 120 | 136.22 | 63.62 | 46.70 | ||
| 126 | 130.25 | 57.77 | 44.35 | ||
| 132 | 124.78 | 52.82 | 42.34 | ||
| 138 | 138.55 | 58.34 | 42.11 | ||
| 144 | 115.23 | 48.30 | 41.92 | ||
| 150 | 116.30 | 49.04 | 42.16 | ||
| 156 | 120.07 | 50.88 | 42.38 | ||
| 162 | 117.66 | 52.71 | 44.80 | ||
| 168 | 102.00 | 49.92 | 48.94 | ||
| 1hours after administration of dose (e.g., application of patch) | |||||
| 2mean blood plasma concentration for the 24 test subjects (pg/ml) | |||||
| 3standard deviation of mean blood plasma concentrations | |||||
| 4coefficient of variation (%) | |||||
| TABLE 2 | |||||
| GEOMETRIC | |||||
| MEAN | STD. DEV. | MEAN | CV % | ||
| AUC (0-168 hrs) | 17740.68 | 7503.50 | 16263.88 | 42.30 |
| Cmax (pg/ml) | 184.80 | 68.84 | 171.78 | 37.25 |
| Tmax (hrs) | 110.50 | 26.48 | 23.96 | |
| TABLE 3 |
| SUMMARY OF SEVERITY FOR THE MOST COMMONLY |
| REPORTED (>=10% OF SUBJECTS) ADVERSE EVENTS |
| (RELATED TO TREATMENT) |
| (N = 24) |
| MILD | MODERATE | SEVERE | TOTAL |
| N | (%) | N | (%) | N | (%) | N | (%) | ||
| |
3 | 12.5 | 0 | 0.0 | 0 | 0.0 | 3 | 12.5 |
| DIZZINESS | 8 | 33.3 | 0 | 0.0 | 0 | 0.0 | 8 | 33.3 |
| HEADACHE | 7 | 29.2 | 0 | 0.0 | 0 | 0.0 | 7 | 29.2 |
| |
6 | 25.0 | 0 | 0.0 | 0 | 0.0 | 6 | 25.0 |
| |
20 | 83.3 | 0 | 0.0 | 0 | 0.0 | 20 | 83.3 |
| SOMNOLENCE | 11 | 45.8 | 0 | 0.0 | 0 | 0.0 | 11 | 45.8 |
| |
2 | 8.3 | 1 | 4.2 | 0 | 0.0 | 3 | 12.5 |
| TABLE 4 | ||
| (mg) | ||
| AMOUNT LEFT IN PATCH | |||
| MEAN | 8.59 | ||
| SE | 0.11 | ||
| % RELEASED (ASSAY) | |||
| MEAN | 14.02 | ||
| SE | 1.08 | ||
-
- AUC(O-last): pg-hr/ml—The area under the curve, as calculated by the linear trapezoidal method, up to the last observed value;
- AUCinf: pg-hr/ml—The area under the curve, calculated using the linear trapezoidal method;
- Cmax: pg/ml—Maximum measured plasma buprenorphine over the time span specified;
- Tmax: hrs—Time of the maximum measured plasma buprenorphine; when the maximum value occurs in more than one time point, Tmax is defined as the first time point with this value;
- T(½)elm: The plasma half life of buprenorphine elimination, defined as ln2/Kelm, where Kelm is the apparent first order elimination constant. The elimination rate constant was obtained from the slope of the terminal portion of the plasma-concentration time curve determined by regression analysis techniques;
- T(½)abs: The absorption half life of transdermal buprenorphine elimination, defined as ln2/Kabs, where Kabs is the apparent first order absorption constant. Absorption rate was calculated only for the transdermal buprenorphine;
- Cl: ml/min or 1/hr—Total clearance characterizes the clearing of the hypothetical plasma volume of drug per unit time;
- Vd: 1 or 1/kg—Hypothetical volumes in which the drug is distributed in the body; and
- Absorption Rate: μg/hr—The rate at which buprenorphine enters the systemic circulation.
| TABLE 5 |
| Comparative Example A |
| MEAN PLASMA | |||||
| HOUR | CONC. (pg/ml) | STD. | CV % | ||
| 2 | 2.04 | 5.87 | 287.10 | ||
| 3 | 7.96 | 16.28 | 204.47 | ||
| 4 | 14.84 | 18.63 | 125.51 | ||
| 6 | 23.49 | 25.81 | 109.85 | ||
| 8 | 42.34 | 37.91 | 89.52 | ||
| 10 | 72.03 | 71.36 | 99.07 | ||
| 12 | 85.96 | 68.69 | 79.90 | ||
| 16 | 133.89 | 103.43 | 77.25 | ||
| 24 | 175.58 | 120.17 | 68.44 | ||
| 30 | 169.15 | 108.65 | 64.23 | ||
| 36 | 200.16 | 134.45 | 67.17 | ||
| 48 | 251.10 | 156.66 | 62.39 | ||
| 60 | 250.11 | 125.01 | 49.98 | ||
| 72 | 286.50 | 131.58 | 45.92 | ||
| 78 | 168.73 | 61.26 | 36.30 | ||
| 84 | 114.68 | 52.72 | 45.97 | ||
| 96 | 90.75 | 39.12 | 43.11 | ||
| 108 | 56.82 | 25.66 | 45.17 | ||
| 120 | 44.85 | 23.80 | 53.06 | ||
| 132 | 30.40 | 21.87 | 71.95 | ||
| 144 | 29.14 | 20.27 | 69.58 | ||
| TABLE 6 |
| Comparative Example C |
| MEAN PLASMA | |||||
| HOUR | CONC. (pg/ml) | STD. | CV % | ||
| 2 | 0.54 | 2.63 | 489.90 | ||
| 3 | 5.70 | 13.18 | 231.23 | ||
| 4 | 10.33 | 14.64 | 141.71 | ||
| 6 | 28.84 | 31.19 | 108.13 | ||
| 8 | 54.62 | 65.83 | 120.52 | ||
| 10 | 78.92 | 81.23 | 102.93 | ||
| 12 | 95.14 | 75.70 | 79.57 | ||
| 16 | 162.26 | 114.80 | 70.75 | ||
| 24 | 218.57 | 153.58 | 70.27 | ||
| 30 | 206.10 | 141.70 | 68.75 | ||
| 36 | 205.08 | 110.76 | 54.01 | ||
| 48 | 265.04 | 123.66 | 46.66 | ||
| 60 | 256.18 | 133.48 | 52.11 | ||
| 72 | 306.02 | 152.77 | 49.92 | ||
| 74 | 278.22 | 135.14 | 48.57 | ||
| 75 | 245.91 | 112.66 | 45.82 | ||
| 76 | 237.01 | 83.41 | 35.19 | ||
| 78 | 213.54 | 94.42 | 44.22 | ||
| 80 | 215.45 | 103.75 | 48.15 | ||
| 82 | 216.00 | 107.68 | 49.85 | ||
| 84 | 210.52 | 107.67 | 51.14 | ||
| 88 | 219.77 | 110.46 | 50.26 | ||
| 96 | 269.91 | 134.61 | 49.87 | ||
| 102 | 205.54 | 102.03 | 49.64 | ||
| 108 | 225.11 | 87.97 | 39.08 | ||
| 120 | 310.27 | 153.57 | 49.50 | ||
| 132 | 300.34 | 157.05 | 52.29 | ||
| 144 | 305.99 | 159.75 | 52.21 | ||
| 146 | 301.39 | 141.37 | 46.91 | ||
| 147 | 289.96 | 132.91 | 45.84 | ||
| 148 | 287.68 | 151.93 | 52.81 | ||
| 150 | 260.04 | 130.19 | 50.07 | ||
| 152 | 236.61 | 119.77 | 50.62 | ||
| 154 | 284.15 | 158.84 | 55.90 | ||
| 156 | 271.83 | 145.11 | 53.38 | ||
| 160 | 303.46 | 182.37 | 60.10 | ||
| 168 | 340.71 | 209.87 | 61.60 | ||
| 174 | 302.22 | 179.74 | 59.47 | ||
| 180 | 322.67 | 183.63 | 56.91 | ||
| 192 | 395.95 | 220.27 | 55.63 | ||
| 204 | 344.83 | 201.90 | 58.55 | ||
| 216 | 415.33 | 229.92 | 55.36 | ||
| 216.25 | 388.64 | 186.67 | 48.03 | ||
| 216.50 | 390.97 | 208.34 | 53.29 | ||
| 216.75 | 392.63 | 188.89 | 48.11 | ||
| 217 | 399.51 | 197.86 | 49.53 | ||
| 218 | 312.65 | 173.12 | 55.37 | ||
| 219 | 295.17 | 148.13 | 50.18 | ||
| 222 | 201.37 | 85.54 | 42.48 | ||
| 228 | 173.89 | 75.96 | 43.68 | ||
| 240 | 119.13 | 48.99 | 41.13 | ||
| 252 | 84.21 | 49.61 | 58.91 | ||
| 264 | 72.33 | 37.86 | 52.42 | ||
| 276 | 50.18 | 25.83 | 51.47 | ||
| 288 | 43.06 | 26.61 | 61.79 | ||
| TABLE 7 |
| Comparative Example B |
| MEAN PLASMA | |||||
| HOUR | CONC. (pg/ml) | STD. DEV | CV % | ||
| 0.02 | 14812.04 | 11319.10 | 76.42 | ||
| 0.03 | 31052.04 | 16156.81 | 52.03 | ||
| 0.05 | 24547.00 | 16461.86 | 67.06 | ||
| 0.08 | 6418.80 | 1976.26 | 30.79 | ||
| 0.17 | 3360.76 | 2457.58 | 73.13 | ||
| 0.25 | 1747.96 | 465.81 | 26.65 | ||
| 0.33 | 1210.08 | 219.28 | 18.12 | ||
| 0.42 | 1050.00 | 242.10 | 23.06 | ||
| 0.50 | 931.52 | 207.25 | 22.25 | ||
| 0.75 | 692.92 | 175.29 | 25.30 | ||
| 1.00 | 584.40 | 148.93 | 25.48 | ||
| 1.50 | 457.44 | 131.44 | 28.73 | ||
| 2.00 | 335.12 | 79.36 | 23.68 | ||
| 3.00 | 238.80 | 63.03 | 26.39 | ||
| 4.00 | 170.87 | 49.84 | 29.17 | ||
| TABLE 8 | ||
| Cmax Values for Comparative Examples A-C | ||
| Comparative | Comparative | Cmax (pg/ml) - Comparative |
| Example A | Example C | Example B |
| Mean | 318.20 | 477.33 | 38635.56 |
| Std. Dev. | 151.24 | 216.92 | 14499.55 |
| Geometric | 291.13 | 435.50 | 35251.92 |
| Mean | |||
| CV % | 47.53 | 45.44 | 37.53 |
| TABLE 9 | |||
| Tmax Prior to Patch Removal (hrs) | Tmax (hrs) | ||
| Comparative | Comparative | Comparative | |
| Example A | Example C | Example B | |
| Mean | 61.92 | 168.39 | 0.04 |
| (out of 72 hrs total) | (out of 260 hrs total) | ||
| Std. Dev. | 13.27 | 42.68 | 0.01 |
| CV % | 21.43 | 25.35 | 26.26 |
| TABLE 10 | ||||
| Comparative | Comparative | Comparative | ||
| Example A | Example C | Example B | ||
| Mean | 18,829.13 | 65,217.25 | 3,699.91 | ||
| Std. Dev. | 9,136.12 | 31,124.37 | 526.64 | ||
| Geometric | 16,760.39 | 57,794.90 | 3,666.65 | ||
| Mean | |||||
| CV % | 48.52 | 47.72 | 14.23 | ||
| TABLE 11 | |||||
| MEAN PLASMA | |||||
| HOUR | CONC. (pg/ml) | STD. | CV % | ||
| 3 | 1.92 | 8.82 | 458.26 | ||
| 6 | 22.69 | 30.98 | 136.54 | ||
| 9 | 38.54 | 48.79 | 126.62 | ||
| 12 | 59.22 | 62.92 | 106.24 | ||
| 16 | 89.85 | 78.93 | 87.84 | ||
| 24 | 128.70 | 72.79 | 56.55 | ||
| 30 | 125.99 | 84.68 | 67.21 | ||
| 36 | 143.07 | 78.40 | 54.80 | ||
| 48 | 196.72 | 101.50 | 51.59 | ||
| 60 | 182.72 | 82.61 | 45.21 | ||
| 72 | 169.95 | 65.04 | 38.27 | ||
| 84 | 122.19 | 41.69 | 34.12 | ||
| 96 | 83.30 | 35.56 | 42.69 | ||
| 108 | 55.09 | 30.82 | 55.94 | ||
| 120 | 41.63 | 20.74 | 49.82 | ||
| 132 | 27.14 | 25.47 | 93.84 | ||
| 144 | 17.54 | 20.09 | 114.51 | ||
| TABLE 12 | ||
| ARITHMETIC | GEOMETRIC | |
| PARAMETER | MEAN (SE) | MEAN (SE) |
| AUC (0-Infinity) | 16278.05 | (1246.6) | 15255.84 | (1272.5) |
| AUC (0-Last) | 14446.10 | (1292.0) | 13162.96 | (1340.6) |
| Cmax (pg/ml) | 229.87 | (19.29) | 214.47 | (17.92) |
| T ½ Elim. (hrs) | 30.53 | (2.80) | ||
| Tmax (hrs) | 67.02 | (3.14) | ||
| TABLE 13 | |||||
| MEAN PLASMA | |||||
| HOUR | CONC. (pg/ml) | STD. | CV % | ||
| 3 | 1.63 | 7.29 | 447.21 | ||
| 6 | 19.61 | 33.28 | 169.70 | ||
| 9 | 29.09 | 44.04 | 151.40 | ||
| 12 | 44.43 | 56.91 | 128.09 | ||
| 16 | 59.77 | 66.25 | 110.86 | ||
| 24 | 110.49 | 98.86 | 89.48 | ||
| 30 | 107.58 | 86.83 | 80.71 | ||
| 36 | 116.36 | 83.01 | 71.34 | ||
| 48 | 154.35 | 83.40 | 54.03 | ||
| 60 | 151.22 | 90.70 | 59.98 | ||
| 72 | 145.20 | 62.84 | 43.28 | ||
| 84 | 106.91 | 38.86 | 36.35 | ||
| 96 | 82.61 | 34.87 | 42.21 | ||
| 108 | 44.83 | 26.74 | 59.65 | ||
| 120 | 29.68 | 24.26 | 81.73 | ||
| 132 | 22.52 | 24.42 | 108.44 | ||
| 144 | 9.24 | 17.28 | 186.93 | ||
| TABLE 14 | ||
| ARITHMETIC | GEOMETRIC | |
| PARAMETER | MEAN (SE) | MEAN (SE) |
| AUC (0-Infinity) | 13450.96 | (1326.8) | 12315.56 | (1142.0) |
| AUC (0-Last) | 12026.65 | (1318.7) | 10796.23 | (1110.3) |
| Cmax (pg/ml) | 199.10 | (17.50) | 186.49 | (14.69) |
| T ½ Elim. (hrs) | 25.82 | (1.51) | ||
| Tmax (hrs) | 68.26 | (3.18) | ||
| TABLE 15 | |||||
| MEAN PLASMA | |||||
| HOURS | CONC. (pg/ml) | STD. DEV. | |
||
| 3 | 5.23 | 13.21 | 252.44 | ||
| 6 | 34.49 | 55.11 | 159.80 | ||
| 9 | 58.67 | 91.17 | 155.40 | ||
| 12 | 94.52 | 111.07 | 117.51 | ||
| 16 | 137.07 | 118.65 | 86.56 | ||
| 24 | 195.58 | 148.53 | 75.94 | ||
| 30 | 201.51 | 142.24 | 70.59 | ||
| 36 | 229.52 | 154.25 | 67.20 | ||
| 48 | 283.35 | 124.06 | 43.78 | ||
| 60 | 314.17 | 173.81 | 55.32 | ||
| 72 | 306.60 | 124.57 | 40.63 | ||
| 84 | 209.66 | 62.84 | 29.97 | ||
| 96 | 143.30 | 43.88 | 30.62 | ||
| 108 | 113.53 | 70.33 | 61.95 | ||
| 120 | 78.71 | 37.46 | 47.59 | ||
| 132 | 75.29 | 47.92 | 63.64 | ||
| 144 | 44.45 | 32.26 | 72.57 | ||
| TABLE 16 | ||
| ARITHMETIC | GEOMETRIC | |
| PARAMETER | MEAN (SE) | MEAN (SE) |
| AUC (0-Infinity) | 14761.59 | (1469.7) | 13540.78 | (1228.3) |
| AUC (0-Last) | 12558.04 | (1313.9) | 11456.76 | (1067.0) |
| Cmax (pg/ml) | 191.84 | (16.93) | 179.60 | (14.23) |
| T ½ Elim. (hrs) | 26.59 | (1.52) | ||
| Tmax (hrs) | 72.37 | (1.89) | ||
| TABLE 17 | ||
| SUMMARY OF BUPRENORPHINE PATCH RESIDUALS | ||
| Ex. D | Ex. F | Ex. E |
| AMOUNT LEFT IN PATCH (mg) | |||||
| N | 27 | 27 | 52 | ||
| MEAN | 8.76 | 18.31 | 4.75 | ||
| SE | 0.07 | 0.15 | 0.03 | ||
| % RELEASED (ASSAY) | |||||
| N | 27 | 27 | 52 | ||
| MEAN | 12.31 | 10.84 | 8.43 | ||
| SE | 0.67 | 0.73 | 0.53 | ||
| TABLE 18 |
| COMPARISON OF PLASMA CONCENTRATIONS |
| COMPARATIVE | COMPARATIVE | |
| EXAMPLE C | EXAMPLE A |
| HOUR/ | Ex. 1 | MEAN | MEAN | ||
| (DAY) | MEAN | MEAN | (½ DOSE) | MEAN | (½ DOSE) |
| 24 (1) | 58.94 | 218.57 | 109.29 | 175.58 | 87.79 |
| 48 (2) | 104.69 | 265.04 | 132.52 | 251.10 | 125.55 |
| 72 (3) | 130.55 | 306.02 | 153.01 | 286.50 | 143.25 |
| 96 (4) | 146.70 | 269.91 | 134.96 | 90.75 | 45.38 |
| 120 (5) | 136.22 | 310.27 | 155.14 | 44.85 | 22.43 |
| 144 (6) | 115.23 | 305.99 | 153.00 | 29.14 | 14.57 |
| 168 (7) | 102.00 | 340.71 | 170.36 | ||
| 192 (8) | 395.95 | 197.98 | |||
| TABLE 19 |
| COMPARISON OF PLASMA CONCENTRATIONS (PG/ML) |
| Hours | Ex. 1 | Ex. D | Ex. E |
| (Post-Application) | MEAN CONC. | MEAN CONC. | MEAN CONC. |
| 3 | 1.92 | 1.63 | |
| 6 | 1.76 | 22.69 | 19.61 |
| 9 | 38.54 | 29.09 | |
| 12 | 18.47 | 59.22 | 44.43 |
| 16 | 89.85 | 59.77 | |
| 24 | 58.94 | 128.70 | 110.49 |
| 30 | 67.69 | 125.99 | 107.58 |
| 36 | 82.44 | 143.07 | 116.36 |
| 48 | 104.69 | 196.72 | 154.35 |
| 60 | 112.93 | 182.72 | 151.22 |
| 72 | 130.55 | 169.95 | 145.20 |
| 84 | 129.03 | 122.19 | 106.91 |
| 96 | 146.70 | 83.30 | 82.61 |
| 108 | 135.49 | 55.09 | 44.83 |
| 120 | 136.22 | 41.63 | 29.68 |
| 132 | 124.78 | 27.14 | 22.52 |
| 144 | 115.23 | 17.54 | 9.24 |
| TABLE 20 |
| BUPRENORPHINE PATCH RELEASE RATES |
| cum. amt. | RR [mg/ | RR [mg/ | RRnorm | ||
| Patch | released | patch/day] | patch/day] | [mg/cm2/ | |
| strength | Example | [mg] | 3 days appl. | 7 days appl. | day] |
| 5 MG | E | 0.44 mg | 0.146 | — | 0.0234 |
| 10 MG | D | 1.23 mg | 0.410 | — | 0.0328 |
| 20 MG | F | 2.52 mg | 0.742 | — | 0.0297 |
| 20 MG | A, C | 3.21 mg | 1.090 | — | 0.0437 |
| 10 MG | 1 | 1.40 mg | — | 0.200 | 0.160 |
Claims (60)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/799,610 USRE41571E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorphine |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3891997P | 1997-02-24 | 1997-02-24 | |
| US08/939,068 US5968547A (en) | 1997-02-24 | 1997-09-29 | Method of providing sustained analgesia with buprenorphine |
| US3310605A | 2005-01-11 | 2005-01-11 | |
| US11/799,610 USRE41571E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorphine |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/939,068 Reissue US5968547A (en) | 1997-02-24 | 1997-09-29 | Method of providing sustained analgesia with buprenorphine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE41571E1 true USRE41571E1 (en) | 2010-08-24 |
Family
ID=26715643
Family Applications (15)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/939,068 Ceased US5968547A (en) | 1997-02-24 | 1997-09-29 | Method of providing sustained analgesia with buprenorphine |
| US09/311,997 Ceased US6231886B1 (en) | 1997-02-24 | 1999-05-14 | Methods of providing sustained treatment with opioids |
| US09/756,419 Ceased US6344212B2 (en) | 1997-02-24 | 2001-01-08 | Method of providing sustained analgesia with buprenorphine |
| US10/033,056 Abandoned US20030091631A1 (en) | 1997-02-24 | 2001-12-27 | Method of providing sustained analgesia with buprenorphine |
| US10/402,288 Abandoned US20030198675A1 (en) | 1997-02-24 | 2003-03-28 | Method of providing sustained analgesia with buprenorphine |
| US11/442,512 Abandoned US20060216340A1 (en) | 1997-02-24 | 2006-05-26 | Method of providing sustained analgesia with buprenorphine |
| US11/799,610 Expired - Lifetime USRE41571E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorphine |
| US11/799,611 Expired - Lifetime USRE41489E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorphine |
| US11/799,608 Expired - Lifetime USRE41408E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorpine |
| US12/888,298 Abandoned US20110288112A1 (en) | 1997-02-24 | 2010-09-22 | Method of Providing Sustained Analgesia With Buprenorphine |
| US13/663,033 Abandoned US20130197020A1 (en) | 1997-02-24 | 2012-10-29 | Method of Providing Sustained Analgesia With Buprenorphine |
| US14/080,168 Abandoned US20140073663A1 (en) | 1997-02-24 | 2013-11-14 | Method of providing sustained analgesia with buprenorphine |
| US14/331,966 Abandoned US20150157625A1 (en) | 1997-02-24 | 2014-07-15 | Method of providing sustained analgesia with buprenorphine |
| US14/847,211 Abandoned US20160220559A1 (en) | 1997-02-24 | 2015-09-08 | Method of providing sustained analgesia with buprenorphine |
| US15/351,879 Expired - Fee Related US9642850B2 (en) | 1997-02-24 | 2016-11-15 | Method of providing sustained analgesia with buprenorphine |
Family Applications Before (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/939,068 Ceased US5968547A (en) | 1997-02-24 | 1997-09-29 | Method of providing sustained analgesia with buprenorphine |
| US09/311,997 Ceased US6231886B1 (en) | 1997-02-24 | 1999-05-14 | Methods of providing sustained treatment with opioids |
| US09/756,419 Ceased US6344212B2 (en) | 1997-02-24 | 2001-01-08 | Method of providing sustained analgesia with buprenorphine |
| US10/033,056 Abandoned US20030091631A1 (en) | 1997-02-24 | 2001-12-27 | Method of providing sustained analgesia with buprenorphine |
| US10/402,288 Abandoned US20030198675A1 (en) | 1997-02-24 | 2003-03-28 | Method of providing sustained analgesia with buprenorphine |
| US11/442,512 Abandoned US20060216340A1 (en) | 1997-02-24 | 2006-05-26 | Method of providing sustained analgesia with buprenorphine |
Family Applications After (8)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/799,611 Expired - Lifetime USRE41489E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorphine |
| US11/799,608 Expired - Lifetime USRE41408E1 (en) | 1997-02-24 | 2007-05-01 | Method of providing sustained analgesia with buprenorpine |
| US12/888,298 Abandoned US20110288112A1 (en) | 1997-02-24 | 2010-09-22 | Method of Providing Sustained Analgesia With Buprenorphine |
| US13/663,033 Abandoned US20130197020A1 (en) | 1997-02-24 | 2012-10-29 | Method of Providing Sustained Analgesia With Buprenorphine |
| US14/080,168 Abandoned US20140073663A1 (en) | 1997-02-24 | 2013-11-14 | Method of providing sustained analgesia with buprenorphine |
| US14/331,966 Abandoned US20150157625A1 (en) | 1997-02-24 | 2014-07-15 | Method of providing sustained analgesia with buprenorphine |
| US14/847,211 Abandoned US20160220559A1 (en) | 1997-02-24 | 2015-09-08 | Method of providing sustained analgesia with buprenorphine |
| US15/351,879 Expired - Fee Related US9642850B2 (en) | 1997-02-24 | 2016-11-15 | Method of providing sustained analgesia with buprenorphine |
Country Status (22)
| Country | Link |
|---|---|
| US (15) | US5968547A (en) |
| EP (9) | EP2301493A1 (en) |
| JP (1) | JP2000511936A (en) |
| KR (1) | KR100703101B1 (en) |
| CN (2) | CN1252705A (en) |
| AT (3) | ATE336212T1 (en) |
| AU (2) | AU2004218685B2 (en) |
| BR (1) | BR9815439A (en) |
| CA (1) | CA2276170C (en) |
| CZ (2) | CZ299414B6 (en) |
| DE (2) | DE69835584T2 (en) |
| DK (4) | DK2305194T3 (en) |
| ES (4) | ES2380023T3 (en) |
| HK (3) | HK1096847A1 (en) |
| HU (3) | HUP1500370D0 (en) |
| IL (4) | IL131561A0 (en) |
| NO (11) | NO322515B1 (en) |
| NZ (1) | NZ337386A (en) |
| PL (1) | PL196086B1 (en) |
| PT (4) | PT2305194E (en) |
| SI (3) | SI2305194T1 (en) |
| WO (1) | WO1998036728A2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150157625A1 (en) * | 1997-02-24 | 2015-06-11 | Purdue Pharma L.P. | Method of providing sustained analgesia with buprenorphine |
| WO2016142877A1 (en) | 2015-03-10 | 2016-09-15 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| US9662326B2 (en) | 2012-04-17 | 2017-05-30 | Purdue Pharma L.P. | Systems for treating an opioid-induced adverse pharmacodynamic response |
| US9849124B2 (en) | 2014-10-17 | 2017-12-26 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
Families Citing this family (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020114827A1 (en) * | 1995-07-28 | 2002-08-22 | Jie Zhang | Methods and apparatus for improved administration of analgesics |
| US20040265353A1 (en) * | 1995-07-28 | 2004-12-30 | Zars, Inc. | Systems and methods for treating panic attacks |
| US20030211157A1 (en) * | 1996-05-06 | 2003-11-13 | Simon David Lew | Semi-sol delivery blend for water soluble molecules |
| US20040024006A1 (en) * | 1996-05-06 | 2004-02-05 | Simon David Lew | Opioid pharmaceutical compositions |
| DE19746191C2 (en) * | 1997-10-18 | 2000-05-18 | Lohmann Therapie Syst Lts | Method of using an active ingredient-containing patch to combat or alleviate addiction |
| US6375957B1 (en) | 1997-12-22 | 2002-04-23 | Euro-Celtique, S.A. | Opioid agonist/opioid antagonist/acetaminophen combinations |
| KR100417489B1 (en) | 1997-12-22 | 2004-02-05 | 유로-셀티크 소시에떼 아노뉨 | Opioid agonist/antagonist combinations |
| SE9803240D0 (en) * | 1998-09-24 | 1998-09-24 | Diabact Ab | A pharmaceutical composition having a rapid action |
| IL151058A0 (en) * | 2000-02-08 | 2003-04-10 | Euro Celtique Sa | Controlled-release compositions containing opioid agonist and antagonist |
| US6716449B2 (en) | 2000-02-08 | 2004-04-06 | Euro-Celtique S.A. | Controlled-release compositions containing opioid agonist and antagonist |
| DK1299104T3 (en) | 2000-02-08 | 2009-08-03 | Euro Celtique Sa | Oral opioid agonist formulations secured against forgery |
| WO2001064149A1 (en) * | 2000-02-29 | 2001-09-07 | Jie Zhang | Improved transdermal drug patch |
| KR100452972B1 (en) * | 2000-05-16 | 2004-10-14 | 주식회사 삼양사 | Hydrogel composition for transdermal drug |
| AU2001262683A1 (en) * | 2000-06-02 | 2001-12-17 | Teijin Limited | Buprenorphine-containing patch |
| GB0021317D0 (en) * | 2000-08-30 | 2000-10-18 | Queen Mary & Westfield College | Transdermal pharmaceutical delivery composition |
| US7018649B2 (en) * | 2000-10-23 | 2006-03-28 | Euro-Celtique, S.A. | Felodipine transdermal device and methods |
| DE60141596D1 (en) | 2000-10-23 | 2010-04-29 | Euro Celtique Sa | TRANSDERMALES SYSTEM FOR THE ADMINISTRATION OF LORATADINE AND ITS USE |
| DK1328273T3 (en) * | 2000-10-23 | 2008-10-20 | Euro Celtique Sa | Device and methods for transdermal administration of felodipine |
| US6682757B1 (en) | 2000-11-16 | 2004-01-27 | Euro-Celtique, S.A. | Titratable dosage transdermal delivery system |
| US20020106407A1 (en) * | 2000-12-11 | 2002-08-08 | Dennis Coleman | Method and apparatus for treating breakthrough pain |
| EP1368023B1 (en) * | 2001-02-16 | 2005-12-28 | Grünenthal GmbH | Utilization of buprenorphine in urinary incontinence therapy |
| WO2002083135A2 (en) * | 2001-02-16 | 2002-10-24 | Grünenthal GmbH | Use of buprenorphine for the manufacture of a transdermal delivery device for the treatment of urinary incontinence, especially urge incontinence |
| CA2439859A1 (en) * | 2001-03-02 | 2002-09-12 | Donna-Donigi Gale | N-but-3-enyl norbuprenorphine and its use as analgesic |
| US20030065002A1 (en) | 2001-05-11 | 2003-04-03 | Endo Pharmaceuticals, Inc. | Abuse-resistant controlled-release opioid dosage form |
| US7968119B2 (en) * | 2001-06-26 | 2011-06-28 | Farrell John J | Tamper-proof narcotic delivery system |
| JP4256259B2 (en) | 2001-07-18 | 2009-04-22 | ユーロ−セルティーク エス.エイ. | Pharmaceutical formulation of oxycodone and naloxone |
| US20030044458A1 (en) | 2001-08-06 | 2003-03-06 | Curtis Wright | Oral dosage form comprising a therapeutic agent and an adverse-effect agent |
| US20030157168A1 (en) | 2001-08-06 | 2003-08-21 | Christopher Breder | Sequestered antagonist formulations |
| AU2002323032B2 (en) | 2001-08-06 | 2005-02-24 | Euro-Celtique S.A. | Opioid agonist formulations with releasable and sequestered antagonist |
| US20030068375A1 (en) | 2001-08-06 | 2003-04-10 | Curtis Wright | Pharmaceutical formulation containing gelling agent |
| US7645813B2 (en) * | 2001-08-10 | 2010-01-12 | Cosmed Pharmaceutical Co., Ltd. | Pressure-sensitive adhesive for the skin and tapes or sheets for the skin made by using the same |
| AU2002361709A1 (en) * | 2001-12-17 | 2003-06-30 | Aryx Therapeutics | Analgesic delivery systems and methods of use |
| US20040033253A1 (en) * | 2002-02-19 | 2004-02-19 | Ihor Shevchuk | Acyl opioid antagonists |
| AU2003224742A1 (en) * | 2002-03-20 | 2003-10-08 | Euro-Celtique S.A. | Method of administering buprenorphine to treat depression |
| CA2708900C (en) | 2002-04-05 | 2019-06-04 | Purdue Pharma | Pharmaceutical preparation containing oxycodone and naloxone |
| US20050106249A1 (en) * | 2002-04-29 | 2005-05-19 | Stephen Hwang | Once-a-day, oral, controlled-release, oxycodone dosage forms |
| ES2665999T3 (en) * | 2002-05-31 | 2018-04-30 | Titan Pharmaceuticals, Inc. | Implantable polymeric device for sustained release of buprenorphine |
| PT1513532E (en) | 2002-06-10 | 2007-05-31 | Euro Celtique Sa | Disposal systems of transdermal delivery devices to prevent misuse of the active agents contained therein |
| EP1526848B1 (en) * | 2002-08-09 | 2007-07-18 | Grünenthal GmbH | Opioid-receptor antagonists in transdermal systems having buprenorphine |
| WO2004017941A2 (en) * | 2002-08-20 | 2004-03-04 | Euro-Celtique, S.A. | Transdermal dosage form comprising an active agent and a salt and free-baseform of an antagonist |
| AU2003270778B2 (en) | 2002-09-20 | 2009-10-08 | Alpharma Pharmaceuticals, Llc | Sequestering subunit and related compositions and methods |
| AU2003272601B2 (en) * | 2002-09-20 | 2009-05-07 | Alpharma Pharmaceuticals, Llc | Sustained-release opioid formulations and methods of use |
| DE60329515D1 (en) * | 2002-10-25 | 2009-11-12 | Euro Celtique Sa | ANALOGUE AND PRODRUGS OF BUPRENORPHINE |
| US7413748B2 (en) * | 2002-12-13 | 2008-08-19 | Purdue Pharma L.P. | Transdermal buprenorphine to treat pain in sickle cell crisis |
| EP1913938A1 (en) | 2002-12-13 | 2008-04-23 | Euro-Celtique S.A. | Transdermal buprenorphine dosage regimen for analgesia |
| DE60322102D1 (en) * | 2002-12-13 | 2008-08-21 | Euro Celtique Sa | TRANSDERMALES BUPRENORPHINE DOSING SCHEME FOR PAIN RELIEF |
| DK1610791T3 (en) * | 2003-03-31 | 2011-05-09 | Titan Pharmaceuticals Inc | Implantable polymer dopamine agonist sustained release device |
| US20040202717A1 (en) | 2003-04-08 | 2004-10-14 | Mehta Atul M. | Abuse-resistant oral dosage forms and method of use thereof |
| MY135852A (en) | 2003-04-21 | 2008-07-31 | Euro Celtique Sa | Pharmaceutical products |
| US7879357B2 (en) * | 2003-04-28 | 2011-02-01 | Bayer Schering Pharma Ag | Pharmaceutical composition in the form of a hydrogel for transdermal administration of active ingredients |
| EP2316440A1 (en) | 2003-04-30 | 2011-05-04 | Purdue Pharma L.P. | Transdermal dosage form comprising an active agent component and an adverse agent component at the distal site of the active agent layer and one fluid communication between the surface of the active agent and the adverse agent |
| US8790689B2 (en) | 2003-04-30 | 2014-07-29 | Purdue Pharma L.P. | Tamper resistant transdermal dosage form |
| WO2004103317A2 (en) * | 2003-05-15 | 2004-12-02 | Euro-Celtique S.A. | Transdermal buprenorphine dosage regimen for treatment of diarrhea |
| PL1638492T3 (en) * | 2003-07-25 | 2008-06-30 | Euro Celtique Sa | Preoperative treatment of post operative pain |
| PL1646328T3 (en) | 2003-07-25 | 2008-03-31 | Euro Celtique Sa | Treatment of dependence withdrawal |
| US20080020028A1 (en) * | 2003-08-20 | 2008-01-24 | Euro-Celtique S.A. | Transdermal dosage form comprising an active agent and a salt and a free-base form of an adverse agent |
| PL2351555T3 (en) | 2004-02-23 | 2017-06-30 | Euro-Celtique S.A. | Abuse resistance opioid transdermal delivery device |
| US7220842B2 (en) * | 2004-04-05 | 2007-05-22 | Dade Behring Inc. | Immunoassays for buprenorphine and norbuprenorphine |
| EP1604666A1 (en) | 2004-06-08 | 2005-12-14 | Euro-Celtique S.A. | Opioids for the treatment of the Chronic Obstructive Pulmonary Disease (COPD) |
| PT1765292T (en) | 2004-06-12 | 2017-12-29 | Collegium Pharmaceutical Inc | Abuse-deterrent drug formulations |
| EP1702558A1 (en) | 2005-02-28 | 2006-09-20 | Euro-Celtique S.A. | Method and device for the assessment of bowel function |
| US20060281775A1 (en) | 2005-06-14 | 2006-12-14 | Applied Pharmacy Services, Inc. | Two-component pharmaceutical composition for the treatment of pain |
| EP1743638A1 (en) | 2005-07-15 | 2007-01-17 | Laboratorios Del Dr. Esteve, S.A. | Pharmaceutical formulations of substituted pyrazoline compounds |
| US8535714B2 (en) | 2006-01-06 | 2013-09-17 | Acelrx Pharmaceuticals, Inc. | Small volume oral transmucosal dosage forms containing sufentanil for treatment of pain |
| US8202535B2 (en) * | 2006-01-06 | 2012-06-19 | Acelrx Pharmaceuticals, Inc. | Small-volume oral transmucosal dosage forms |
| US8357114B2 (en) | 2006-01-06 | 2013-01-22 | Acelrx Pharmaceuticals, Inc. | Drug dispensing device with flexible push rod |
| US8252329B2 (en) * | 2007-01-05 | 2012-08-28 | Acelrx Pharmaceuticals, Inc. | Bioadhesive drug formulations for oral transmucosal delivery |
| US8252328B2 (en) * | 2006-01-06 | 2012-08-28 | Acelrx Pharmaceuticals, Inc. | Bioadhesive drug formulations for oral transmucosal delivery |
| US8753308B2 (en) | 2006-01-06 | 2014-06-17 | Acelrx Pharmaceuticals, Inc. | Methods for administering small volume oral transmucosal dosage forms using a dispensing device |
| US8865743B2 (en) | 2006-01-06 | 2014-10-21 | Acelrx Pharmaceuticals, Inc. | Small volume oral transmucosal dosage forms containing sufentanil for treatment of pain |
| US9066847B2 (en) | 2007-01-05 | 2015-06-30 | Aceirx Pharmaceuticals, Inc. | Storage and dispensing devices for administration of oral transmucosal dosage forms |
| US9289583B2 (en) * | 2006-01-06 | 2016-03-22 | Acelrx Pharmaceuticals, Inc. | Methods for administering small volume oral transmucosal dosage forms using a dispensing device |
| AU2007238685B2 (en) * | 2006-04-13 | 2012-09-13 | Teva Pharmaceuticals International Gmbh | Transdermal methods and systems for the delivery of anti-migraine compounds |
| MX2008016372A (en) | 2006-06-19 | 2009-05-28 | Alpharma Inc | Pharmaceutical compositions. |
| EP1897543A1 (en) | 2006-08-30 | 2008-03-12 | Euro-Celtique S.A. | Buprenorphine- wafer for drug substitution therapy |
| DE102006054732B4 (en) * | 2006-11-21 | 2010-12-30 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system with ion-pair microreservoirs |
| DE102006054731B4 (en) * | 2006-11-21 | 2013-02-28 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system for administration of the active ingredient buprenorphine and use thereof in pain therapy |
| WO2008120562A1 (en) * | 2007-04-02 | 2008-10-09 | Toyo Boseki Kabushiki Kaisha | Therapeutic tablet for postherpetic neuralgia and method of treating postherpetic neuralgia |
| CN101820838B (en) * | 2007-08-07 | 2013-06-05 | 阿塞尔Rx制药有限公司 | Compositions and methods for procedural sedation and analgesia using oral transmucosal dosage forms |
| US20090082383A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched buprenorphine |
| US8623418B2 (en) | 2007-12-17 | 2014-01-07 | Alpharma Pharmaceuticals Llc | Pharmaceutical composition |
| US20110097395A1 (en) * | 2008-03-08 | 2011-04-28 | Najib Babul | Oral Pharmaceutical Compositions of Buprenorphine and Method of Use |
| AU2009271271A1 (en) * | 2008-06-23 | 2010-01-21 | Biodelivery Sciences International, Inc. | Multidirectional mucosal delivery devices and methods of use |
| US8945592B2 (en) | 2008-11-21 | 2015-02-03 | Acelrx Pharmaceuticals, Inc. | Sufentanil solid dosage forms comprising oxygen scavengers and methods of using the same |
| PT2405915T (en) | 2009-03-10 | 2019-01-29 | Euro Celtique Sa | Immediate release pharmaceutical compositions comprising oxycodone and naloxone |
| US8548623B2 (en) | 2009-03-18 | 2013-10-01 | Acelrx Pharmaceuticals, Inc. | Storage and dispensing devices for administration of oral transmucosal dosage forms |
| US8475832B2 (en) | 2009-08-07 | 2013-07-02 | Rb Pharmaceuticals Limited | Sublingual and buccal film compositions |
| US20110037455A1 (en) * | 2009-08-17 | 2011-02-17 | Oren Navot | System for measuring electrical power |
| US10668060B2 (en) | 2009-12-10 | 2020-06-02 | Collegium Pharmaceutical, Inc. | Tamper-resistant pharmaceutical compositions of opioids and other drugs |
| DE102010024105A1 (en) * | 2010-06-17 | 2011-12-22 | Grünenthal GmbH | Transdermal administration of memantine |
| US9168251B2 (en) * | 2010-09-03 | 2015-10-27 | Zoetis Belgium S.A | High dose buprenorphine compositions and use as analgesic |
| EP2606895A4 (en) * | 2010-10-12 | 2014-04-02 | Univ Wuhan | Transdermal absorption patch of antiviral drug and its preparation method |
| AU2011331511C1 (en) * | 2010-11-17 | 2018-01-18 | Hexal Ag | Transdermal therapeutic system comprising buprenorphine |
| DE102011076653A1 (en) | 2011-05-27 | 2012-11-29 | Acino Ag | Transdermal therapeutic system containing buprenorphine and an alpha hydroxy acid |
| US10813925B2 (en) | 2011-09-19 | 2020-10-27 | Carmel—Haifa University Economic Corporation Ltd. | Buprenorphine for the treatment of acute suicidality |
| CN104114161A (en) * | 2011-12-12 | 2014-10-22 | Lts勒曼治疗系统股份公司 | Transdermal delivery system comprising buprenorphine |
| AU2012358308A1 (en) * | 2011-12-21 | 2014-07-24 | Biodelivery Sciences International, Inc. | Transmucosal drug delivery devices for use in chronic pain relief |
| ES2663412T3 (en) * | 2012-01-31 | 2018-04-12 | Grünenthal GmbH | Pharmaceutical patch for transdermal administration of (1R, 4R) -6'-fluor-N, N-dimethyl-4-phenyl-4 ', 9'-dihydro-3'H-spiro [cyclohexane-1,1'-pyran [3,4-b] indole] -4-amine |
| CN107582544A (en) * | 2012-08-24 | 2018-01-16 | 英特格拉斯疗法有限公司 | For strengthening the Chemical composition that and method of therapeutic agent transdermal delivery |
| AU2013205080B2 (en) * | 2012-12-12 | 2016-07-07 | Lts Lohmann Therapie-Systeme Ag | Transdermal Delivery System |
| CN104955516B (en) * | 2012-12-28 | 2019-01-22 | 帝国制药美国公司 | Extend buprenorphine percutaneous delivering compositions and its application method |
| AP2015008920A0 (en) | 2013-06-04 | 2015-12-31 | Lohmann Therapie Syst Lts | Transdermal delivery system |
| MY183489A (en) | 2013-07-23 | 2021-02-22 | Euro Celtique Sa | A combination of oxycodone and naloxone for use in treating pain in patients suffering from pain and a disease resulting in intestinal dysbiosis and/or increasing the risk for intestinal bacterial translocation |
| DE102014013448A1 (en) | 2014-09-16 | 2016-03-17 | Alfred E. Tiefenbacher (Gmbh & Co. Kg) | Transdermal therapeutic system comprising buprenorphine |
| MY187877A (en) | 2014-12-23 | 2021-10-26 | Acelrx Pharmaceuticals Inc | Systems, devices and methods for dispensing oral transmucosal dosage forms |
| US9656441B2 (en) | 2015-01-08 | 2017-05-23 | Alfred E. Tiefenbacher ( Gmbh & Co. Kg) | Transdermal patch |
| US9737530B1 (en) | 2016-06-23 | 2017-08-22 | Collegium Pharmaceutical, Inc. | Process of making stable abuse-deterrent oral formulations |
| CA3092757A1 (en) * | 2018-03-13 | 2019-09-19 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system comprising a silicone acrylic hybrid polymer |
| WO2021163529A2 (en) * | 2020-02-13 | 2021-08-19 | Biodelivery Sciences International, Inc. | Methods of treatment with buprenorphine |
| EP4238669A1 (en) | 2022-03-02 | 2023-09-06 | Recutech S.r.o. | Method of manufacturing a heat-humidity exchange plate of an enthalpy air-to-air exchanger, a heat-humidity exchange plate and an enthalpy exchanger |
Citations (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3996934A (en) | 1971-08-09 | 1976-12-14 | Alza Corporation | Medical bandage |
| US4058599A (en) | 1973-02-24 | 1977-11-15 | Bayer Aktiengesellschaft | Ethyleneimine inactivated organisms |
| US4060084A (en) | 1976-09-07 | 1977-11-29 | Alza Corporation | Method and therapeutic system for providing chemotherapy transdermally |
| US4119713A (en) | 1977-02-09 | 1978-10-10 | Sam Carosio | Analgesic and anti-inflammatory composition |
| US4262003A (en) | 1975-12-08 | 1981-04-14 | Alza Corporation | Method and therapeutic system for administering scopolamine transdermally |
| US4379454A (en) | 1981-02-17 | 1983-04-12 | Alza Corporation | Dosage for coadministering drug and percutaneous absorption enhancer |
| DE3526339A1 (en) | 1984-07-23 | 1986-01-30 | Alza Corp., Palo Alto, Calif. | TRANSDERMAL DELIVERY SYSTEM FOR THE ADMINISTRATION OF FENTANYL |
| US4582835A (en) | 1983-12-06 | 1986-04-15 | Reckitt & Colman Products Limited | Analgesic compositions |
| US4661492A (en) * | 1984-11-30 | 1987-04-28 | Reckitt & Colman Products Limited | Analgesic compositions |
| US4673679A (en) | 1986-05-14 | 1987-06-16 | E. I. Du Pont De Nemours And Company | Use of prodrugs of 3-hydroxymorphinans to prevent bitter taste upon buccal, nasal or sublingual administration |
| US4784855A (en) | 1986-06-16 | 1988-11-15 | Otsuka Pharmaceutical Co., Ltd. | Pharmaceutical compositions and suppository |
| US4806341A (en) | 1985-02-25 | 1989-02-21 | Rutgers, The State University Of New Jersey | Transdermal absorption dosage unit for narcotic analgesics and antagonists and process for administration |
| US4879297A (en) | 1987-06-01 | 1989-11-07 | Warner-Lambert Company | Fatty acids and their small chain esters as penetration enhancers in aqueous systems |
| US4906463A (en) | 1986-12-22 | 1990-03-06 | Cygnus Research Corporation | Transdermal drug-delivery composition |
| US4908027A (en) | 1986-09-12 | 1990-03-13 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| EP0368409A2 (en) | 1988-11-10 | 1990-05-16 | Norwich Eaton Pharmaceuticals, Inc. | Compositions for the transdermal delivery of buprenorphine salts |
| US4938759A (en) | 1986-09-02 | 1990-07-03 | Alza Corporation | Transdermal delivery device having a rate controlling adhesive |
| US4945103A (en) | 1989-01-17 | 1990-07-31 | Michael Cohen | Method of treating pre-menstrual syndrome |
| US4956171A (en) | 1989-07-21 | 1990-09-11 | Paco Pharmaceutical Services, Inc. | Transdermal drug delivery using a dual permeation enhancer and method of performing the same |
| US4983395A (en) | 1987-11-12 | 1991-01-08 | Theratech Inc. | Device for administering an active agent to the skin or mucosa |
| US4994278A (en) | 1988-03-04 | 1991-02-19 | Noven Pharmaceuticals, Inc. | Breathable backing |
| EP0430019A2 (en) | 1989-11-29 | 1991-06-05 | LTS Lohmann Therapie-Systeme GmbH & Co. KG | Transdermal therapeutic system containing buprenorphine as an active component |
| EP0432945A1 (en) | 1989-12-12 | 1991-06-19 | Warner-Lambert Company | A transdermal delivery system for treatment of cocaine and heroin addiction |
| US5026556A (en) | 1988-11-10 | 1991-06-25 | Norwich Eaton Pharmaceuticals, Inc. | Compositions for the transdermal delivery of pharmaceutical actives |
| US5028435A (en) | 1989-05-22 | 1991-07-02 | Advanced Polymer Systems, Inc. | System and method for transdermal drug delivery |
| US5069909A (en) | 1990-06-20 | 1991-12-03 | Cygnus Therapeutic Systems | Transdermal administration of buprenorphine |
| US5091186A (en) | 1989-08-15 | 1992-02-25 | Cygnus Therapeutic Systems | Biphasic transdermal drug delivery device |
| US5090405A (en) | 1987-08-26 | 1992-02-25 | Bayer Aktiengesellschaft | Water-hardening polymer preparations |
| US5132115A (en) | 1986-04-17 | 1992-07-21 | Karin Wolter | Planar therapeutic system, process for its production and utilization |
| US5149538A (en) * | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| US5176916A (en) | 1990-04-18 | 1993-01-05 | Nitto Electric Industrial Co., Ltd. | Medical adhesives |
| US5225199A (en) | 1990-04-24 | 1993-07-06 | Teijin Limited | Pharmaceutical plasters |
| US5238933A (en) | 1991-10-28 | 1993-08-24 | Sri International | Skin permeation enhancer compositions |
| US5240711A (en) | 1989-11-29 | 1993-08-31 | Lts Lohmann Therapie-Systeme Gmbh & Co. Kg | Transdermal therapeutic system comprising as active component buprenorphine |
| US5306503A (en) | 1990-06-25 | 1994-04-26 | Lts Lohmann Therapie-Systeme Gmbh & Co. Kg | Patch with a high content of softening ingredients |
| US5336210A (en) | 1989-02-28 | 1994-08-09 | Teijin Limited | Plaster agent |
| US5342623A (en) | 1986-09-12 | 1994-08-30 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| US5352457A (en) | 1990-10-05 | 1994-10-04 | Ethical Pharmaceuticals Limited | Transdermal device |
| WO1995001167A2 (en) | 1993-06-25 | 1995-01-12 | Alza Corporation | Incorporating poly-n-vinyl amide in a transdermal system |
| PL166095B1 (en) | 1991-04-03 | 1995-03-31 | Warszawskie Zaklady Farma | Method of obtaining buprenophine |
| DE3546830C2 (en) | 1984-07-23 | 1995-07-20 | Alza Corp | Device for delivering fentanyl across the skin |
| WO1995020393A1 (en) | 1994-01-27 | 1995-08-03 | Schering Corporation | Use of mometasone furoate for treating airway passage and lung diseases |
| US5486362A (en) | 1991-05-07 | 1996-01-23 | Dynagen, Inc. | Controlled, sustained release delivery system for treating drug dependency |
| WO1996019975A1 (en) | 1994-12-24 | 1996-07-04 | Lts Lohmann Therapie-Systeme Gmbh | Transdermal resorption of active substances from supercooled masses |
| WO1997004835A1 (en) | 1995-07-28 | 1997-02-13 | Novartis Ag | Transdermal system |
| US5613958A (en) | 1993-05-12 | 1997-03-25 | Pp Holdings Inc. | Transdermal delivery systems for the modulated administration of drugs |
| US5635203A (en) | 1994-09-29 | 1997-06-03 | Alza Corporation | Transdermal device having decreased delamination |
| US5683711A (en) | 1993-04-20 | 1997-11-04 | Hexal Pharma Gmbh | Active ingredient patch |
| US5700480A (en) | 1993-01-23 | 1997-12-23 | Lts Lohman Therapie-Systeme Gmbh & Co. Kg | Transdermal therapeutic system comprising galanthamine as active component |
| WO1997048380A1 (en) | 1996-06-20 | 1997-12-24 | Astra Aktiebolag (Publ) | ADMINISTRATION REGIMEN OF H+, K+-ATPase INHIBITORS |
| EP0819438A2 (en) | 1996-07-19 | 1998-01-21 | Nitto Denko Corporation | Buprenorphine percutaneous absorption preparation |
| EP0821957A2 (en) | 1996-08-01 | 1998-02-04 | Eli Lilly And Company | Use of 3-(4-hexyloxy-1,2,5-thiadiazol-3-yl)-1,2,5,6-tetrahydro-1-methylpyridine (xanomeline) for treating substance abuse |
| US5732717A (en) | 1995-08-15 | 1998-03-31 | Eli Lilly And Company | Method for treating substance abuse withdrawal |
| WO1998026780A2 (en) | 1996-12-16 | 1998-06-25 | Lts Lohmann Therapie-Systeme Gmbh | Flat medicament preparation for the application and release of buprenorphine or a pharmacologically comparable substance in the buccal cavity, and method of producing the same |
| US5785991A (en) | 1995-06-07 | 1998-07-28 | Alza Corporation | Skin permeation enhancer compositions comprising glycerol monolaurate and lauryl acetate |
| WO1998036728A2 (en) | 1997-02-24 | 1998-08-27 | Euro-Celtique, S.A. | Sustained analgesia achieved with buprenorphine |
| US5814032A (en) | 1994-01-21 | 1998-09-29 | Nitto Denko Corporation | Percutaneous administration tape preparation |
| US5817665A (en) | 1993-03-02 | 1998-10-06 | John S. Nagle | Composition and method of treating depression using naloxone or naltrexone in combination with a serotonin reuptake inhibitor |
| US5834010A (en) | 1995-04-26 | 1998-11-10 | Theratech, Inc. | Triacetin as a penetration enhancer for transdermal delivery of a basic drug |
| US5837289A (en) | 1996-07-23 | 1998-11-17 | Grasela; John C. | Transdermal delivery of medications using a combination of penetration enhancers |
| WO1999012529A2 (en) | 1997-09-05 | 1999-03-18 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system comprising a reservoir-type pressure-sensitive adhesive layer and a back layer with uni-directional resilience |
| US5900420A (en) | 1997-06-19 | 1999-05-04 | Cole; William L. | Method for treating chronic fatigue syndrome and fibromyalgia with buprenorphine |
| WO1999032120A1 (en) | 1997-12-22 | 1999-07-01 | Euro-Celtique, S.A. | A method of preventing abuse of opioid dosage forms |
| US5919473A (en) | 1997-05-12 | 1999-07-06 | Elkhoury; George F. | Methods and devices for delivering opioid analgesics to wounds via a subdermal implant |
| US5942530A (en) | 1997-08-28 | 1999-08-24 | Eli Lilly And Company | Method for treating pain |
| US5989585A (en) | 1995-10-23 | 1999-11-23 | Hexal Ag | Transdermal therapeutic system (TTS) containing vitamin E for the treatment of drug dependency |
| US6004969A (en) | 1996-04-15 | 1999-12-21 | National Science Council | Transdermal delivery of buprenorphine preparations |
| US6024976A (en) | 1988-03-04 | 2000-02-15 | Noven Pharmaceuticals, Inc. | Solubility parameter based drug delivery system and method for altering drug saturation concentration |
| US6024974A (en) | 1995-01-06 | 2000-02-15 | Noven Pharmaceuticals, Inc. | Composition and methods for transdermal delivery of acid labile drugs |
| DE19840758A1 (en) | 1998-09-07 | 2000-03-09 | Liedtke Pharmed Gmbh | Treatment of pain or depression comprises systemic administration of oxytocin |
| US6165499A (en) | 1996-03-25 | 2000-12-26 | Lts Lohmann Therapie-Systeme Gmbh | Transdermal therapeutic system with small application-area thickness and great flexibility, and production process |
| US6210699B1 (en) | 1999-04-01 | 2001-04-03 | Watson Pharmaceuticals, Inc. | Oral transmucosal delivery of drugs or any other ingredients via the inner buccal cavity |
| US6242456B1 (en) | 1998-03-09 | 2001-06-05 | Trustees Of Tufts College | Treatment of stereotypic, self-injurious and compulsive behaviors in man and animals using antagonists of NMDA receptors |
| US6280766B1 (en) | 1997-10-18 | 2001-08-28 | Lts Lehman Therapie-Systeme Gmbh | Method for transdermal application of active substances at high constant dosage |
| US6391294B1 (en) | 1997-08-21 | 2002-05-21 | Reckitt Benckiser Healthcare (Uk) Limited | In situ formation of polymeric material |
| US6436977B1 (en) | 1999-09-29 | 2002-08-20 | Pfizer Inc. | Dosing regimens for lasofoxifene |
| US20020137761A1 (en) | 2001-03-23 | 2002-09-26 | Crain Stanley M. | Methods for increasing analgesic potency and attenuating adverse excitatory effects of bimodally -acting opioid agonists by inhibiting GM1-ganglioside |
| US6554851B1 (en) | 1999-05-07 | 2003-04-29 | Scimed Life Systems, Inc. | Methods of sealing an injection site |
| US20030104976A1 (en) | 2001-07-23 | 2003-06-05 | Gudarz Davar | Analgesic methods using endothelin receptor ligands |
| US20030114475A1 (en) | 2001-10-31 | 2003-06-19 | Addiction Therapies, Inc. | Methods for the treatment of addiction |
| US6595956B1 (en) | 1998-03-23 | 2003-07-22 | Joseph Gross | Drug delivery device |
| EP1422230A1 (en) | 2002-11-25 | 2004-05-26 | Chi Mei Foundation Medical Center | Novel ester derivatives of buprenorphine and their preparation processes, and long acting analgestic pharmaceutical compositions |
| US20040146647A1 (en) | 2001-06-04 | 2004-07-29 | Fixter Gregory Peter Wade | Patterning method |
| US6787149B1 (en) | 1996-12-12 | 2004-09-07 | El Khoury And Stein Ltd. | Topical application of opioid analgesic drugs such as morphine |
| US20040241218A1 (en) | 2001-05-01 | 2004-12-02 | Lino Tavares | Abuse resistant opioid containing transdermal systems |
| US6865444B2 (en) | 2001-05-22 | 2005-03-08 | Euro-Celtique S.A. | Container and method for dispensing transdermal dosage forms |
| US7011843B2 (en) | 1997-10-01 | 2006-03-14 | Lts Lohmann-Therapie Systeme Ag | Method for protecting a human being against health impairment by ingestion of a transdermal therapeutic system |
| US7056527B2 (en) | 2000-02-25 | 2006-06-06 | Teijin Limited | Patches containing buprenorphine hydrochloride |
| US7270830B2 (en) | 2002-12-13 | 2007-09-18 | Purdue Pharma L.P. | Transdermal buprenorphine dosage regimen for analgesia |
Family Cites Families (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3330750A1 (en) * | 1983-08-26 | 1985-03-14 | Chemische Werke Hüls AG, 4370 Marl | METHOD FOR GENERATING ACETYLENE AND SYNTHESIS OR REDUCING GAS FROM COAL IN AN ARC PROCESS |
| US4636539A (en) | 1984-01-30 | 1987-01-13 | Loctite (Ireland) Limited | Instant adhesive composition utilizing calixarene accelerators |
| US4601893A (en) | 1984-02-08 | 1986-07-22 | Pfizer Inc. | Laminate device for controlled and prolonged release of substances to an ambient environment and method of use |
| US4626539A (en) | 1984-08-10 | 1986-12-02 | E. I. Dupont De Nemours And Company | Trandermal delivery of opioids |
| US4834978A (en) | 1984-10-01 | 1989-05-30 | Biotek, Inc. | Method of transdermal drug delivery |
| US4698062A (en) | 1985-10-30 | 1987-10-06 | Alza Corporation | Medical device for pulsatile transdermal delivery of biologically active agents |
| USRE33093E (en) | 1986-06-16 | 1989-10-17 | Johnson & Johnson Consumer Products, Inc. | Bioadhesive extruded film for intra-oral drug delivery and process |
| US5091405A (en) * | 1987-01-05 | 1992-02-25 | E. I. Du Pont De Nemours And Company | Insecticidal pyrazolines |
| US5171576A (en) | 1987-03-09 | 1992-12-15 | Alza Corporation | Prevention of contact allergy by coadministration of a corticosteroid with a sensitizing drug |
| US4915954A (en) | 1987-09-03 | 1990-04-10 | Alza Corporation | Dosage form for delivering a drug at two different rates |
| US5236714A (en) | 1988-11-01 | 1993-08-17 | Alza Corporation | Abusable substance dosage form having reduced abuse potential |
| US5075341A (en) | 1989-12-01 | 1991-12-24 | The Mclean Hospital Corporation | Treatment for cocaine abuse |
| US5688547A (en) | 1990-08-03 | 1997-11-18 | American Cyanamid Company | Meal replacement composition and method of weight control |
| CN1036343C (en) | 1990-11-10 | 1997-11-05 | 天津市计划生育研究所 | Synthesis, products and its application for luleinizing hormone releasing hormone and antagonistic znalogue |
| WO1992019226A1 (en) | 1991-05-07 | 1992-11-12 | Dynagen, Inc. | A controlled, sustained release delivery system for treating drug dependency |
| US5225440A (en) * | 1991-09-13 | 1993-07-06 | The United States Of America As Represented By The Department Of Health And Human Services | Attenuation of the opioid withdrawal syndrome by inhibitors of nitric oxide synthase |
| US5229130A (en) | 1991-12-20 | 1993-07-20 | Cygnus Therapeutics Systems | Vegetable oil-based skin permeation enhancer compositions, and associated methods and systems |
| US5958459A (en) | 1991-12-24 | 1999-09-28 | Purdue Pharma L.P. | Opioid formulations having extended controlled released |
| US5478577A (en) | 1993-11-23 | 1995-12-26 | Euroceltique, S.A. | Method of treating pain by administering 24 hour oral opioid formulations exhibiting rapid rate of initial rise of plasma drug level |
| US5272149A (en) | 1992-05-05 | 1993-12-21 | Stalling Reginald W | Symptom controlled receptor substitution for addiction withdrawl |
| WO1993023019A1 (en) | 1992-05-11 | 1993-11-25 | Sri International | Transdermal drug delivery systems and related compositions and methods of use |
| ATE185694T1 (en) | 1992-05-13 | 1999-11-15 | Alza Corp | OXYBUTYNIN FOR TRANSDERMAL ADMINISTRATION |
| WO1993025168A1 (en) | 1992-06-11 | 1993-12-23 | Theratech, Inc. | The use of glycerin in moderating transdermal drug delivery |
| US6096756A (en) | 1992-09-21 | 2000-08-01 | Albert Einstein College Of Medicine Of Yeshiva University | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by morphine and other bimodally-acting opioid agonists |
| US5580876A (en) * | 1992-09-21 | 1996-12-03 | Albert Einstein College Of Medicine Of Yeshiva University, A Division Of Yeshiva University | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by morphine and other bimodally-acting opioid agonists |
| DE4237453C1 (en) | 1992-11-06 | 1993-08-19 | Lts Lohmann Therapie-Systeme Gmbh & Co Kg, 5450 Neuwied, De | |
| WO1995005138A1 (en) | 1993-08-16 | 1995-02-23 | Cygnus Therapeutic Systems | Contact adhesive extends wear time on skin |
| US5662933A (en) | 1993-09-09 | 1997-09-02 | Edward Mendell Co., Inc. | Controlled release formulation (albuterol) |
| CN1138864A (en) | 1994-01-20 | 1996-12-25 | 阿克佐诺贝尔公司 | New compsn. of glycoprotein isoforms having follicle stimulating |
| JP2819236B2 (en) * | 1994-05-06 | 1998-10-30 | 日東電工株式会社 | Transdermal formulation |
| US5965161A (en) | 1994-11-04 | 1999-10-12 | Euro-Celtique, S.A. | Extruded multi-particulates |
| US20020006438A1 (en) | 1998-09-25 | 2002-01-17 | Benjamin Oshlack | Sustained release hydromorphone formulations exhibiting bimodal characteristics |
| US5468457A (en) | 1994-12-22 | 1995-11-21 | Osram Sylvania Inc. | Method of making tungsten-copper composite oxides |
| FR2739731B1 (en) | 1995-10-09 | 1998-01-02 | Axon Cable Sa | METHOD FOR CONNECTING COAXIAL MICRO-CABLES TO THE TRACKS OF A PRINTED CIRCUIT |
| US5792740A (en) | 1996-03-08 | 1998-08-11 | Firmenich Sa | Fragrant macrocyclic lactones |
| US6271240B1 (en) | 1996-05-06 | 2001-08-07 | David Lew Simon | Methods for improved regulation of endogenous dopamine in prolonged treatment of opioid addicted individuals |
| US20010006677A1 (en) | 1996-10-29 | 2001-07-05 | Mcginity James W. | Effervescence polymeric film drug delivery system |
| AT405158B (en) * | 1996-12-04 | 1999-06-25 | Steyr Daimler Puch Ag | METHOD FOR AUTOMATICALLY CONTROLLING THE LOCKING CLUTCHES OF AN ALL-WHEEL DRIVE VEHICLE AND VEHICLE WITH ARTICULATED STEERING AND LOCKABLE DIFFERENTIALS |
| KR20000070757A (en) | 1997-02-07 | 2000-11-25 | 에버트 챨스 디 | Composition and method for supplementing testosterone in women with symptoms of testosterone deficiency |
| WO1998048811A1 (en) | 1997-04-30 | 1998-11-05 | Human Pheromone Sciences, Inc. | Method of fixing fragrances in fragrance composition and other compositions |
| RS49982B (en) * | 1997-09-17 | 2008-09-29 | Euro-Celtique S.A., | Synergistic analgesic combination of opioid analgesic and cyclooxygenase-2 inhibitor |
| US6210705B1 (en) | 1997-12-15 | 2001-04-03 | Noven Pharmaceuticals, Nc. | Compositions and methods for treatment of attention deficit disorder and attention deficit/hyperactivity disorder with methylphenidate |
| ES2237104T3 (en) | 1998-04-29 | 2005-07-16 | Virotex Corporation | CARRIER DEVICE FOR PHARMACEUTICAL PRODUCTS SUITABLE FOR THE CONTRIBUTION OF PHARMACEUTICAL COMPOUNDS TO MUCOUS SURFACES. |
| KR100383252B1 (en) | 1998-12-17 | 2003-07-16 | 주식회사 삼양사 | Transdermal Dosage Compositions Containing Buprenoline and Patches Comprising the Same |
| US6552024B1 (en) | 1999-01-21 | 2003-04-22 | Lavipharm Laboratories Inc. | Compositions and methods for mucosal delivery |
| IT1305303B1 (en) | 1999-03-10 | 2001-05-04 | Farmigea Spa | USE OF NIAOULI ESSENTIAL OIL AS A PROMOTER FOR TRANSDERMAL PERMEAZION. |
| US6716449B2 (en) * | 2000-02-08 | 2004-04-06 | Euro-Celtique S.A. | Controlled-release compositions containing opioid agonist and antagonist |
| US7067116B1 (en) | 2000-03-23 | 2006-06-27 | Warner-Lambert Company Llc | Fast dissolving orally consumable solid film containing a taste masking agent and pharmaceutically active agent at weight ratio of 1:3 to 3:1 |
| US6503894B1 (en) | 2000-08-30 | 2003-01-07 | Unimed Pharmaceuticals, Inc. | Pharmaceutical composition and method for treating hypogonadism |
| EP1315502B1 (en) | 2000-08-30 | 2010-03-17 | Unimed Pharmaceuticals, LLC | Method for treating erectile dysfunction and increasing libido in men |
| US6682757B1 (en) * | 2000-11-16 | 2004-01-27 | Euro-Celtique, S.A. | Titratable dosage transdermal delivery system |
| CA2439859A1 (en) * | 2001-03-02 | 2002-09-12 | Donna-Donigi Gale | N-but-3-enyl norbuprenorphine and its use as analgesic |
| EP2266718B1 (en) | 2001-04-23 | 2016-06-22 | Euro-Celtique S.A. | Disposal system for transdermal dosage form |
| AU2003224742A1 (en) * | 2002-03-20 | 2003-10-08 | Euro-Celtique S.A. | Method of administering buprenorphine to treat depression |
| US7413748B2 (en) * | 2002-12-13 | 2008-08-19 | Purdue Pharma L.P. | Transdermal buprenorphine to treat pain in sickle cell crisis |
| WO2004103317A2 (en) * | 2003-05-15 | 2004-12-02 | Euro-Celtique S.A. | Transdermal buprenorphine dosage regimen for treatment of diarrhea |
| PL1646328T3 (en) * | 2003-07-25 | 2008-03-31 | Euro Celtique Sa | Treatment of dependence withdrawal |
| PL1638492T3 (en) | 2003-07-25 | 2008-06-30 | Euro Celtique Sa | Preoperative treatment of post operative pain |
| AU2004266731A1 (en) | 2003-08-21 | 2005-03-03 | Transoral Pharmaceuticals, Inc. | Compositions for delivering 5-HT agonists across the oral mucosa and methods of use thereof |
| EP1877094A1 (en) | 2005-05-03 | 2008-01-16 | Innozen, Inc. | Edible film for transmucosal delivery of nutritional supplements |
| JP5586151B2 (en) | 2005-12-13 | 2014-09-10 | バイオデリバリー サイエンシーズ インターナショナル インコーポレイティッド | Abuse-resistant transmucosal drug delivery device |
| US8252329B2 (en) | 2007-01-05 | 2012-08-28 | Acelrx Pharmaceuticals, Inc. | Bioadhesive drug formulations for oral transmucosal delivery |
| US20080096900A1 (en) | 2006-06-26 | 2008-04-24 | Amgen Inc. | Methods for treating atherosclerosis |
| PL2054031T3 (en) | 2006-07-21 | 2016-09-30 | Transmucosal delivery devices with enhanced uptake | |
| EP1897543A1 (en) | 2006-08-30 | 2008-03-12 | Euro-Celtique S.A. | Buprenorphine- wafer for drug substitution therapy |
| MX2009003372A (en) | 2006-10-02 | 2009-09-24 | Labtec Gmbh | Non-mucoadhesive film dosage forms. |
| WO2008042508A1 (en) | 2006-10-03 | 2008-04-10 | University Of Southern California | Grp78 as a predictor of responsiveness to therapeutic agents |
| GB2447016A (en) | 2007-03-01 | 2008-09-03 | Reckitt Benckiser Healthcare | Buprenorphine/naloxone compositions |
| WO2008115811A2 (en) | 2007-03-16 | 2008-09-25 | Endo Pharmaceuticals, Inc. | Transdermal delivery form disposal systems and methods |
| AU2009271271A1 (en) | 2008-06-23 | 2010-01-21 | Biodelivery Sciences International, Inc. | Multidirectional mucosal delivery devices and methods of use |
| AU2011331511C1 (en) | 2010-11-17 | 2018-01-18 | Hexal Ag | Transdermal therapeutic system comprising buprenorphine |
-
1997
- 1997-09-29 US US08/939,068 patent/US5968547A/en not_active Ceased
-
1998
- 1998-02-24 CZ CZ0300999A patent/CZ299414B6/en not_active IP Right Cessation
- 1998-02-24 HU HUP1500370 patent/HUP1500370D0/en unknown
- 1998-02-24 PT PT10185240T patent/PT2305194E/en unknown
- 1998-02-24 IL IL13156198A patent/IL131561A0/en unknown
- 1998-02-24 EP EP10185235A patent/EP2301493A1/en not_active Withdrawn
- 1998-02-24 ES ES05011213T patent/ES2380023T3/en not_active Expired - Lifetime
- 1998-02-24 ES ES06118876T patent/ES2423925T3/en not_active Expired - Lifetime
- 1998-02-24 BR BR9815439-7A patent/BR9815439A/en not_active Application Discontinuation
- 1998-02-24 SI SI9830937T patent/SI2305194T1/en unknown
- 1998-02-24 NZ NZ337386A patent/NZ337386A/en not_active IP Right Cessation
- 1998-02-24 CZ CZ2015-477A patent/CZ2015477A3/en unknown
- 1998-02-24 DE DE69835584T patent/DE69835584T2/en not_active Expired - Lifetime
- 1998-02-24 PT PT98906678T patent/PT964677E/en unknown
- 1998-02-24 PT PT05011213T patent/PT1570823E/en unknown
- 1998-02-24 EP EP98906678A patent/EP0964677B1/en not_active Revoked
- 1998-02-24 CN CN98804261A patent/CN1252705A/en active Pending
- 1998-02-24 JP JP10536980A patent/JP2000511936A/en active Pending
- 1998-02-24 CN CN2005101286952A patent/CN1827107B/en not_active Expired - Lifetime
- 1998-02-24 EP EP10185240A patent/EP2305194B1/en not_active Revoked
- 1998-02-24 ES ES98906678T patent/ES2271988T3/en not_active Expired - Lifetime
- 1998-02-24 PT PT61188769T patent/PT1731152E/en unknown
- 1998-02-24 EP EP10185230A patent/EP2305191A1/en not_active Withdrawn
- 1998-02-24 AT AT98906678T patent/ATE336212T1/en active
- 1998-02-24 DK DK10185240.8T patent/DK2305194T3/en active
- 1998-02-24 AT AT05011213T patent/ATE538765T1/en active
- 1998-02-24 EP EP10185233A patent/EP2305192A1/en not_active Withdrawn
- 1998-02-24 AT AT10185240T patent/ATE556682T1/en active
- 1998-02-24 PL PL335306A patent/PL196086B1/en unknown
- 1998-02-24 DE DE05011213T patent/DE05011213T1/en active Pending
- 1998-02-24 DK DK06118876.9T patent/DK1731152T3/en active
- 1998-02-24 SI SI9830933T patent/SI1570823T1/en unknown
- 1998-02-24 CA CA002276170A patent/CA2276170C/en not_active Expired - Lifetime
- 1998-02-24 ES ES10185240T patent/ES2387752T3/en not_active Expired - Lifetime
- 1998-02-24 SI SI9830859T patent/SI0964677T1/en unknown
- 1998-02-24 EP EP10185237A patent/EP2305193A1/en not_active Withdrawn
- 1998-02-24 DK DK05011213.5T patent/DK1570823T3/en active
- 1998-02-24 HU HU1200599A patent/HU230608B1/en unknown
- 1998-02-24 EP EP05011213A patent/EP1570823B1/en not_active Revoked
- 1998-02-24 DK DK98906678T patent/DK0964677T3/en active
- 1998-02-24 WO PCT/US1998/003584 patent/WO1998036728A2/en active Application Filing
- 1998-02-24 EP EP06118876.9A patent/EP1731152B1/en not_active Revoked
- 1998-02-24 HU HU0002342A patent/HU230374B1/en unknown
- 1998-02-24 EP EP10185241A patent/EP2301494A1/en not_active Withdrawn
- 1998-02-24 KR KR1019997007713A patent/KR100703101B1/en active IP Right Grant
-
1999
- 1999-05-14 US US09/311,997 patent/US6231886B1/en not_active Ceased
- 1999-08-24 IL IL131561A patent/IL131561A/en not_active IP Right Cessation
- 1999-08-24 NO NO994087A patent/NO322515B1/en not_active IP Right Cessation
-
2001
- 2001-01-08 US US09/756,419 patent/US6344212B2/en not_active Ceased
- 2001-12-27 US US10/033,056 patent/US20030091631A1/en not_active Abandoned
-
2003
- 2003-03-28 US US10/402,288 patent/US20030198675A1/en not_active Abandoned
-
2004
- 2004-10-08 AU AU2004218685A patent/AU2004218685B2/en not_active Expired
-
2006
- 2006-05-26 US US11/442,512 patent/US20060216340A1/en not_active Abandoned
- 2006-09-28 NO NO20064395A patent/NO329734B1/en not_active IP Right Cessation
-
2007
- 2007-02-12 HK HK07101623.6A patent/HK1096847A1/en not_active IP Right Cessation
- 2007-05-01 US US11/799,610 patent/USRE41571E1/en not_active Expired - Lifetime
- 2007-05-01 US US11/799,611 patent/USRE41489E1/en not_active Expired - Lifetime
- 2007-05-01 US US11/799,608 patent/USRE41408E1/en not_active Expired - Lifetime
- 2007-06-12 HK HK07106323.8A patent/HK1103621A1/en not_active IP Right Cessation
- 2007-08-22 NO NO20074292A patent/NO329838B1/en not_active IP Right Cessation
-
2008
- 2008-12-19 AU AU2008261134A patent/AU2008261134B2/en not_active Expired
-
2009
- 2009-10-11 IL IL201437A patent/IL201437A/en not_active IP Right Cessation
-
2010
- 2010-07-18 IL IL207067A patent/IL207067A/en not_active IP Right Cessation
- 2010-08-23 NO NO20101190A patent/NO332248B1/en unknown
- 2010-09-22 US US12/888,298 patent/US20110288112A1/en not_active Abandoned
-
2011
- 2011-07-19 HK HK11107538.1A patent/HK1153377A1/en not_active IP Right Cessation
- 2011-10-24 NO NO20111441A patent/NO333374B1/en not_active IP Right Cessation
-
2012
- 2012-05-14 NO NO20120563A patent/NO333139B1/en unknown
- 2012-10-29 US US13/663,033 patent/US20130197020A1/en not_active Abandoned
-
2013
- 2013-02-26 NO NO20130298A patent/NO334290B1/en unknown
- 2013-11-14 US US14/080,168 patent/US20140073663A1/en not_active Abandoned
- 2013-11-18 NO NO20131533A patent/NO337094B1/en active Protection Beyond IP Right Term
-
2014
- 2014-07-15 US US14/331,966 patent/US20150157625A1/en not_active Abandoned
-
2015
- 2015-09-08 US US14/847,211 patent/US20160220559A1/en not_active Abandoned
- 2015-10-26 NO NO20151450A patent/NO340067B1/en active Protection Beyond IP Right Term
-
2016
- 2016-07-18 NO NO2016013C patent/NO2016013I1/en unknown
- 2016-11-15 US US15/351,879 patent/US9642850B2/en not_active Expired - Fee Related
-
2017
- 2017-04-04 NO NO2017011C patent/NO2017011I1/en unknown
Patent Citations (102)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3996934A (en) | 1971-08-09 | 1976-12-14 | Alza Corporation | Medical bandage |
| US4058599A (en) | 1973-02-24 | 1977-11-15 | Bayer Aktiengesellschaft | Ethyleneimine inactivated organisms |
| US4262003A (en) | 1975-12-08 | 1981-04-14 | Alza Corporation | Method and therapeutic system for administering scopolamine transdermally |
| US4060084A (en) | 1976-09-07 | 1977-11-29 | Alza Corporation | Method and therapeutic system for providing chemotherapy transdermally |
| US4119713A (en) | 1977-02-09 | 1978-10-10 | Sam Carosio | Analgesic and anti-inflammatory composition |
| US4379454A (en) | 1981-02-17 | 1983-04-12 | Alza Corporation | Dosage for coadministering drug and percutaneous absorption enhancer |
| US4582835A (en) | 1983-12-06 | 1986-04-15 | Reckitt & Colman Products Limited | Analgesic compositions |
| US4588580B2 (en) | 1984-07-23 | 1999-02-16 | Alaz Corp | Transdermal administration of fentanyl and device therefor |
| US4588580A (en) | 1984-07-23 | 1986-05-13 | Alza Corporation | Transdermal administration of fentanyl and device therefor |
| US4588580B1 (en) | 1984-07-23 | 1989-01-03 | ||
| DE3526339A1 (en) | 1984-07-23 | 1986-01-30 | Alza Corp., Palo Alto, Calif. | TRANSDERMAL DELIVERY SYSTEM FOR THE ADMINISTRATION OF FENTANYL |
| GB2165148A (en) | 1984-07-23 | 1986-04-09 | Alza Corp | Transdermal administration of fentanyl |
| DE3546830C2 (en) | 1984-07-23 | 1995-07-20 | Alza Corp | Device for delivering fentanyl across the skin |
| US4661492A (en) * | 1984-11-30 | 1987-04-28 | Reckitt & Colman Products Limited | Analgesic compositions |
| US4806341A (en) | 1985-02-25 | 1989-02-21 | Rutgers, The State University Of New Jersey | Transdermal absorption dosage unit for narcotic analgesics and antagonists and process for administration |
| US5132115A (en) | 1986-04-17 | 1992-07-21 | Karin Wolter | Planar therapeutic system, process for its production and utilization |
| US4673679A (en) | 1986-05-14 | 1987-06-16 | E. I. Du Pont De Nemours And Company | Use of prodrugs of 3-hydroxymorphinans to prevent bitter taste upon buccal, nasal or sublingual administration |
| US4784855A (en) | 1986-06-16 | 1988-11-15 | Otsuka Pharmaceutical Co., Ltd. | Pharmaceutical compositions and suppository |
| US4938759A (en) | 1986-09-02 | 1990-07-03 | Alza Corporation | Transdermal delivery device having a rate controlling adhesive |
| US4908027A (en) | 1986-09-12 | 1990-03-13 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| US5344656A (en) | 1986-09-12 | 1994-09-06 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| US5342623A (en) | 1986-09-12 | 1994-08-30 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| US5462745A (en) | 1986-09-12 | 1995-10-31 | Alza Corporation | Subsaturated transdermal therapeutic system having improved release characteristics |
| US4906463A (en) | 1986-12-22 | 1990-03-06 | Cygnus Research Corporation | Transdermal drug-delivery composition |
| US4879297A (en) | 1987-06-01 | 1989-11-07 | Warner-Lambert Company | Fatty acids and their small chain esters as penetration enhancers in aqueous systems |
| US5090405A (en) | 1987-08-26 | 1992-02-25 | Bayer Aktiengesellschaft | Water-hardening polymer preparations |
| US4983395A (en) | 1987-11-12 | 1991-01-08 | Theratech Inc. | Device for administering an active agent to the skin or mucosa |
| US6024976A (en) | 1988-03-04 | 2000-02-15 | Noven Pharmaceuticals, Inc. | Solubility parameter based drug delivery system and method for altering drug saturation concentration |
| US4994278A (en) | 1988-03-04 | 1991-02-19 | Noven Pharmaceuticals, Inc. | Breathable backing |
| US5026556A (en) | 1988-11-10 | 1991-06-25 | Norwich Eaton Pharmaceuticals, Inc. | Compositions for the transdermal delivery of pharmaceutical actives |
| EP0368409A2 (en) | 1988-11-10 | 1990-05-16 | Norwich Eaton Pharmaceuticals, Inc. | Compositions for the transdermal delivery of buprenorphine salts |
| US4945103A (en) | 1989-01-17 | 1990-07-31 | Michael Cohen | Method of treating pre-menstrual syndrome |
| US5336210A (en) | 1989-02-28 | 1994-08-09 | Teijin Limited | Plaster agent |
| US5028435A (en) | 1989-05-22 | 1991-07-02 | Advanced Polymer Systems, Inc. | System and method for transdermal drug delivery |
| US4956171A (en) | 1989-07-21 | 1990-09-11 | Paco Pharmaceutical Services, Inc. | Transdermal drug delivery using a dual permeation enhancer and method of performing the same |
| US5091186A (en) | 1989-08-15 | 1992-02-25 | Cygnus Therapeutic Systems | Biphasic transdermal drug delivery device |
| EP0430019A2 (en) | 1989-11-29 | 1991-06-05 | LTS Lohmann Therapie-Systeme GmbH & Co. KG | Transdermal therapeutic system containing buprenorphine as an active component |
| US5240711A (en) | 1989-11-29 | 1993-08-31 | Lts Lohmann Therapie-Systeme Gmbh & Co. Kg | Transdermal therapeutic system comprising as active component buprenorphine |
| EP0432945A1 (en) | 1989-12-12 | 1991-06-19 | Warner-Lambert Company | A transdermal delivery system for treatment of cocaine and heroin addiction |
| US5176916A (en) | 1990-04-18 | 1993-01-05 | Nitto Electric Industrial Co., Ltd. | Medical adhesives |
| US5225199A (en) | 1990-04-24 | 1993-07-06 | Teijin Limited | Pharmaceutical plasters |
| US5069909A (en) | 1990-06-20 | 1991-12-03 | Cygnus Therapeutic Systems | Transdermal administration of buprenorphine |
| US5306503A (en) | 1990-06-25 | 1994-04-26 | Lts Lohmann Therapie-Systeme Gmbh & Co. Kg | Patch with a high content of softening ingredients |
| US5352457A (en) | 1990-10-05 | 1994-10-04 | Ethical Pharmaceuticals Limited | Transdermal device |
| PL166095B1 (en) | 1991-04-03 | 1995-03-31 | Warszawskie Zaklady Farma | Method of obtaining buprenophine |
| US5486362A (en) | 1991-05-07 | 1996-01-23 | Dynagen, Inc. | Controlled, sustained release delivery system for treating drug dependency |
| US5149538A (en) * | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| US5238933A (en) | 1991-10-28 | 1993-08-24 | Sri International | Skin permeation enhancer compositions |
| US5700480A (en) | 1993-01-23 | 1997-12-23 | Lts Lohman Therapie-Systeme Gmbh & Co. Kg | Transdermal therapeutic system comprising galanthamine as active component |
| US5817665A (en) | 1993-03-02 | 1998-10-06 | John S. Nagle | Composition and method of treating depression using naloxone or naltrexone in combination with a serotonin reuptake inhibitor |
| US5683711A (en) | 1993-04-20 | 1997-11-04 | Hexal Pharma Gmbh | Active ingredient patch |
| US5830505A (en) | 1993-04-20 | 1998-11-03 | Hexal Pharma Gmbh | Active ingredient patch |
| US5613958A (en) | 1993-05-12 | 1997-03-25 | Pp Holdings Inc. | Transdermal delivery systems for the modulated administration of drugs |
| WO1995001167A2 (en) | 1993-06-25 | 1995-01-12 | Alza Corporation | Incorporating poly-n-vinyl amide in a transdermal system |
| US5814032A (en) | 1994-01-21 | 1998-09-29 | Nitto Denko Corporation | Percutaneous administration tape preparation |
| WO1995020393A1 (en) | 1994-01-27 | 1995-08-03 | Schering Corporation | Use of mometasone furoate for treating airway passage and lung diseases |
| US5635203A (en) | 1994-09-29 | 1997-06-03 | Alza Corporation | Transdermal device having decreased delamination |
| WO1996019975A1 (en) | 1994-12-24 | 1996-07-04 | Lts Lohmann Therapie-Systeme Gmbh | Transdermal resorption of active substances from supercooled masses |
| US6264980B1 (en) | 1994-12-24 | 2001-07-24 | Lts Lohmann Therapie-Systeme Gmbh | Transdermal resorption of active substances from supercooled masses of levulic acid |
| US6024974A (en) | 1995-01-06 | 2000-02-15 | Noven Pharmaceuticals, Inc. | Composition and methods for transdermal delivery of acid labile drugs |
| US5834010A (en) | 1995-04-26 | 1998-11-10 | Theratech, Inc. | Triacetin as a penetration enhancer for transdermal delivery of a basic drug |
| US5785991A (en) | 1995-06-07 | 1998-07-28 | Alza Corporation | Skin permeation enhancer compositions comprising glycerol monolaurate and lauryl acetate |
| US5843468A (en) | 1995-06-07 | 1998-12-01 | Alza Corporation | Skin permeation enhancer compositions comprising glycerol monolaurate and lauryl acetate |
| WO1997004835A1 (en) | 1995-07-28 | 1997-02-13 | Novartis Ag | Transdermal system |
| US5732717A (en) | 1995-08-15 | 1998-03-31 | Eli Lilly And Company | Method for treating substance abuse withdrawal |
| US5989585A (en) | 1995-10-23 | 1999-11-23 | Hexal Ag | Transdermal therapeutic system (TTS) containing vitamin E for the treatment of drug dependency |
| US6165499A (en) | 1996-03-25 | 2000-12-26 | Lts Lohmann Therapie-Systeme Gmbh | Transdermal therapeutic system with small application-area thickness and great flexibility, and production process |
| US6004969A (en) | 1996-04-15 | 1999-12-21 | National Science Council | Transdermal delivery of buprenorphine preparations |
| WO1997048380A1 (en) | 1996-06-20 | 1997-12-24 | Astra Aktiebolag (Publ) | ADMINISTRATION REGIMEN OF H+, K+-ATPase INHIBITORS |
| EP0819438A2 (en) | 1996-07-19 | 1998-01-21 | Nitto Denko Corporation | Buprenorphine percutaneous absorption preparation |
| US5837289A (en) | 1996-07-23 | 1998-11-17 | Grasela; John C. | Transdermal delivery of medications using a combination of penetration enhancers |
| EP0821957A2 (en) | 1996-08-01 | 1998-02-04 | Eli Lilly And Company | Use of 3-(4-hexyloxy-1,2,5-thiadiazol-3-yl)-1,2,5,6-tetrahydro-1-methylpyridine (xanomeline) for treating substance abuse |
| US6787149B1 (en) | 1996-12-12 | 2004-09-07 | El Khoury And Stein Ltd. | Topical application of opioid analgesic drugs such as morphine |
| WO1998026780A2 (en) | 1996-12-16 | 1998-06-25 | Lts Lohmann Therapie-Systeme Gmbh | Flat medicament preparation for the application and release of buprenorphine or a pharmacologically comparable substance in the buccal cavity, and method of producing the same |
| US6344212B2 (en) | 1997-02-24 | 2002-02-05 | Euro-Celtique S.A. | Method of providing sustained analgesia with buprenorphine |
| US6231886B1 (en) | 1997-02-24 | 2001-05-15 | Robert F. Reder | Methods of providing sustained treatment with opioids |
| US5968547A (en) | 1997-02-24 | 1999-10-19 | Euro-Celtique, S.A. | Method of providing sustained analgesia with buprenorphine |
| WO1998036728A2 (en) | 1997-02-24 | 1998-08-27 | Euro-Celtique, S.A. | Sustained analgesia achieved with buprenorphine |
| US5919473A (en) | 1997-05-12 | 1999-07-06 | Elkhoury; George F. | Methods and devices for delivering opioid analgesics to wounds via a subdermal implant |
| US5900420A (en) | 1997-06-19 | 1999-05-04 | Cole; William L. | Method for treating chronic fatigue syndrome and fibromyalgia with buprenorphine |
| US6391294B1 (en) | 1997-08-21 | 2002-05-21 | Reckitt Benckiser Healthcare (Uk) Limited | In situ formation of polymeric material |
| US5942530A (en) | 1997-08-28 | 1999-08-24 | Eli Lilly And Company | Method for treating pain |
| WO1999012529A2 (en) | 1997-09-05 | 1999-03-18 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system comprising a reservoir-type pressure-sensitive adhesive layer and a back layer with uni-directional resilience |
| US7011843B2 (en) | 1997-10-01 | 2006-03-14 | Lts Lohmann-Therapie Systeme Ag | Method for protecting a human being against health impairment by ingestion of a transdermal therapeutic system |
| US6280766B1 (en) | 1997-10-18 | 2001-08-28 | Lts Lehman Therapie-Systeme Gmbh | Method for transdermal application of active substances at high constant dosage |
| WO1999032120A1 (en) | 1997-12-22 | 1999-07-01 | Euro-Celtique, S.A. | A method of preventing abuse of opioid dosage forms |
| US6242456B1 (en) | 1998-03-09 | 2001-06-05 | Trustees Of Tufts College | Treatment of stereotypic, self-injurious and compulsive behaviors in man and animals using antagonists of NMDA receptors |
| US6595956B1 (en) | 1998-03-23 | 2003-07-22 | Joseph Gross | Drug delivery device |
| DE19840758A1 (en) | 1998-09-07 | 2000-03-09 | Liedtke Pharmed Gmbh | Treatment of pain or depression comprises systemic administration of oxytocin |
| US6210699B1 (en) | 1999-04-01 | 2001-04-03 | Watson Pharmaceuticals, Inc. | Oral transmucosal delivery of drugs or any other ingredients via the inner buccal cavity |
| US20010051186A1 (en) | 1999-04-01 | 2001-12-13 | Acharya Ramesh N. | Oral transmucosal delivery of drugs or any other ingredients via the inner buccal cavity |
| US6554851B1 (en) | 1999-05-07 | 2003-04-29 | Scimed Life Systems, Inc. | Methods of sealing an injection site |
| US6436977B1 (en) | 1999-09-29 | 2002-08-20 | Pfizer Inc. | Dosing regimens for lasofoxifene |
| US7056527B2 (en) | 2000-02-25 | 2006-06-06 | Teijin Limited | Patches containing buprenorphine hydrochloride |
| US20020137761A1 (en) | 2001-03-23 | 2002-09-26 | Crain Stanley M. | Methods for increasing analgesic potency and attenuating adverse excitatory effects of bimodally -acting opioid agonists by inhibiting GM1-ganglioside |
| US20040241218A1 (en) | 2001-05-01 | 2004-12-02 | Lino Tavares | Abuse resistant opioid containing transdermal systems |
| US6865444B2 (en) | 2001-05-22 | 2005-03-08 | Euro-Celtique S.A. | Container and method for dispensing transdermal dosage forms |
| US20040146647A1 (en) | 2001-06-04 | 2004-07-29 | Fixter Gregory Peter Wade | Patterning method |
| US20030104976A1 (en) | 2001-07-23 | 2003-06-05 | Gudarz Davar | Analgesic methods using endothelin receptor ligands |
| US20030114475A1 (en) | 2001-10-31 | 2003-06-19 | Addiction Therapies, Inc. | Methods for the treatment of addiction |
| EP1422230A1 (en) | 2002-11-25 | 2004-05-26 | Chi Mei Foundation Medical Center | Novel ester derivatives of buprenorphine and their preparation processes, and long acting analgestic pharmaceutical compositions |
| US7270830B2 (en) | 2002-12-13 | 2007-09-18 | Purdue Pharma L.P. | Transdermal buprenorphine dosage regimen for analgesia |
Non-Patent Citations (712)
| Title |
|---|
| 7.2. Buprenorphine, MNH/PAD/87.11, pp. 29-63 (undated). |
| A review of data for the scientific community in preparation for the WHO 2002 critical review of buprenorphine: Scientific, medical and policy support for the continuing of Buprenorphine in Schedule III of the 1971 Convention on Psychotropic Substance. Dec. 13, 2001 (30 pp.). |
| Abse et al., "The poppy: therapeutic potential in cases of demential with depression" Ann. NY Acad. Sci.398: 79-83 (1982). |
| Adams, Expert Report on Buprenorphine (undated). |
| Addendum to a review of data for the scientific community in preparation for the WHO 2002 critical review of buprenorphine: Data on reactions possibly related to abuse of Buprenorphine reported to the WHO collaborating centre for international drug monitoring, Uppsala Sweden and Data on seizures from the NFLIS. Feb. 26, 2002 (11 pp.). |
| Adriaensen et al. A long-term open, clinical and pharmacokinetic assessment of sublingual buprenorphine in patients suffering from chronic pain. Acta Anaesthesiol Belg. Mar. 1985; 36(1)33-40. |
| Agar et al., Buprenorphine: "field trials" of a new drug. Qual Health Res. Jan. 2001;11(1):69-84. |
| Agin M, Kazierad DJ, Abel R, et al., Assessing QT variability in healthy volunteers. J. Clin. Pharmacol. 2003; 43:1028. |
| Ahmadi, J. et al., Treatment of heroin dependence. German Journal of Psychiatry 2004;7(2): 1-5. |
| Ahmedzai, S, "New approached to pain control in patients with cancer" Eur. J. Cancer 33(Suppl. 6): S8-S14 (1997). |
| Akatsuka et al., "The relief of postoperative pain by suppositories of buprenorphine or NSAID" Masui 45(3):298-303 (1996). (Abstract). |
| Al-Gommer O. Sexual dysfunctions in male opiate users: A comparative study of heroin, methadone, and buprenorphine. Addictive Disorders and their Treatment 2007;6(3): 137-43. |
| Amass et al. A preliminary investigation of outcome following gradual or rapid buprenorphine detoxification. J Addict Dis. 1994; 13:33-45. |
| Amass et al. Alternate-day dosing during buprenorphine treatment of opioid dependence. Life Sci. 1994;54:1215-28. |
| Amass et al. Detectability of buprenorphine dose alterations in opioid-dependent humans. NIDA Res Monogr. 1993; 132:335. |
| Ang-Lee K. Single dose of 24 milligrams of buprenorphine for heroin detoxification: An open-label study of five inpatients. Journal of psychoactive drugs 2007; 38(4):505-12. |
| Annual report on the state of the drugs problem in the European Union by EMCDDA. 1999 (33 pp.). |
| Arditti et al. [Buprenorphine abuse in a series of 50 drug addicts hospitalized at a drug dependence evaluation hospital center of Marseille] Therapie. 1992; 47:561-2. [in French w/ English transl]. |
| Ashcroft et al., Buprenorphine TDS: Comparison with sublingual buprenorphine in osteoarthritic pain. 10th World Congress on Pain, Aug. 19, 2002 Abstract 510-P144. |
| Auriacombe et al., Buprenorphine prescribed by general practitioners—A safe means of increasing patient access to treatment?, in Maintenance Treatment in Heroin Addiction—Evidence at the Crossroads (Wall and Haga, eds.), 2003 (5 pp.). |
| Bailey et al., Package inserts and other dosage guidelines are especially useful with new analgesics and new analgesic delivery systems. Anesth Analg. Dec. 1992;75(6):873-5. |
| Baker JR. Effect of buprenorphine and antiretroviral agents on the QT interval in opioid-dependent patients. Annals of Pharmacotherapy 2006: 40:392-6. |
| Banerjee et al. Haematological changes in buprenorphine-treated mice. Folia Biol (Krakow). 1997;45(3-4):157-62. |
| Banks. Overdosage of buprenorphine: case report. N Z Med J. 1979; 89:255-6. |
| Banys et al. An open trial of low dose buprenorphine in treating methadone withdrawal. J Subst Abuse Treat. 1994;11:9-15. |
| Barrett et al. The pharmacokinetics and physiological effects of buprenophine infusion in premature neonates. Br J Clin Pharmacol. Sep. 1993;36(3):215-9. |
| Barron et al. Prenatal buprenorphine exposure and sexually dimorphic nonreproductive behaviors in rats. Pharmacol Biochem Behav, Oct. 1997;58(2):337-43. |
| Barry, "Reflections on Transdermal Drug Delivery", Pharmaceutical Science & Technology Today 2(2):41-43 (1999). |
| Basu et al. Buprenorphine dependence: a new addiction in India. Disabil Impair. 1990; 3:142-6. |
| Bates' Guide to Physical Examination and History Taking, 6th ed., Bickley t al., eds., Lippincott Williams & Wilkins Publishers, 1995, pp. 276-280. |
| Bauer, K.H. et al. Pharmazuetische Technologie, (1986) pp. 362-365. |
| Bauer, KH (ed.) Pharmaceutische Technologie pp. 362-365 (1993). |
| Baumevieille et al. Abuse of prescription medicines in southwestern France. Ann Pharmacother 1997; 31:847-50. |
| Becker et al., Transdermal buprenorphine: Abuse potential assessment in non-opioid-dependent volunteers. CPDD Jun. 16-21, 2001 Abstract 40 (w/ PowerPoint presentation). |
| Bedi et al. Abuse Liability of Buprenorphine-A study Among Experienced Drug Users. Indian J Physiol. Pharmacol. 1998;42(1), 95-100. |
| Bedi et al. Abuse Liability of Buprenorphine—A study Among Experienced Drug Users. Indian J Physiol. Pharmacol. 1998;42(1), 95-100. |
| Bégaud et al., of the Joint Ministerial Mission for Combating Drugs and Drug Addiction Information Office, Evaluation of Subutex® availability in the treatment of drug users. Summary review of the literature and available data and proposals for a research program. Jun. 1998 (83 pp.). |
| Bell et al., Evaluation of transdermal fentanyl for multi-day analgesia in postoperative patients. Anesth Analg. 1989; Abstract S22. |
| Bellamy N, Buchanan WW, Goldsmith CH, Campbell J. Stitt LW. Validation study of WOMAC: a health status instrument for measuring clinically important subject relevant outcomes to antirheumatic drug therapy in subjects with osteoarthritis of the hip or knee. J. Rheumatol 1988; 15:1833-1840. |
| Bellamy N, Campbell J, Hill J, Band P. A comparative study of telephone versus onsite completion of the WOMAC 3.0 Osteoarthritis Index. J Rheumatol 2002; 29:783-786. |
| Bellamy N. WOMAC Osteoarthritis Index, a user's guide. London, Ontario, Canada: Victoria Hospital Corporation, 1995. |
| Benos. [A case of secondary buprenorphine (Temgesic®) dependence] Der Nervenarzt. 1983;54:259-61. [in German w/ English trans]. |
| Bentley et al., "Age and fentanyl pharmacokinetics" Anesth. Analg. 61: 968-971 (1982). |
| Berrocoso et al., "Differential role of 5-HT1A and 5-HT1B receptors on the antiociceptive and antidepressant effect of tramdol in mice" Psychopharmacology 188(1): 111-118 (2006). |
| Biagini et al. Evaluation of cutaneous responses and lung function from exposure to opiate compounds among ethical narcotics-manufacturing workers. J Allergy Clin Immunol. Jan. 1992; 89(1 Pt 1):108-18. |
| Bickel et al. A clinical trial of buprenorphine: comparison with methadone in the detoxification of heroin addicts. Clin Pharmacol Ther. 1988;43:72-8. |
| Bickel et al. A clinical trial of buprenorphine: I. Comparison with methadone in the detoxification of heroin addicts. II. Examination of its opioid blocking properties. NIDA Res Monogr. 1987;76:182-8. |
| Bickel et al. Buprenorphine treatment of opioid dependence: a review. Exp Clin Psychopharmacology 1995; 3:477-89. |
| Bickel et al. Buprenorphine: dose-related blockade of opioid challenge effects in opioid dependent humans. J Pharmacol Exp Ther. Oct. 1988;247(1):47-53. |
| Bickel et al. Effects of adding behavioral treatment to opioid detoxification with buprenorphine. J Consult Clin Psychol. Oct. 1997;65(5):803-10. |
| Bigelow (w/ Introduction by Blaine). Assessment of buprenorphine in a drug discrimination procedure in humans. Buprenorphine: An alternative Treatment for Opioid Dependence. NIDA Res Monogr. 1992;121:28-37. |
| Bigelow et al. Abuse liability assessment of buprenorphine-naloxone combinations. NIDA Res Monogr. 1987;76:145-9. |
| Bigelow GE. Forward. In: Cowan A, Lewis JW, eds. Buprenorphine: Combating Drug Abuse With a Unique Opioid. New York, NY: Wiley-Liss: 1995:xi-xiii. |
| Bigelow, Buprenorphine: Combatting drug abuse with a unique opioid. 1995;(Foreword):xi-Xiii. |
| Bigelow, Human drug abuse liability assessment: opioids and analgesics. Br J Addict. 1991; 86:1615-23. |
| Blaine, Introduction, NIDA Res. Monogr. 1992;1-4. |
| Bliesener N, et al., Plasma Testosterone and Sexual Function in Men Receiving Buprenorphine Maintenance for Opioid Dependence. The Journal of Clinical Endocrinology & Metabolism 90(1):203-6, 2005. |
| Boas et al., Clinical actions of fentanyl and buprenorphine the significance of receptor binding. Br. J. Anaesth. 1985, 57:192-6. |
| Bodkin et al., "Buprenorphine treatment of refractory depression" J. Clin. Psychoparmacology 15(1): 49-57 (1995). |
| Boger RH. Renal impairment: a challenge for opioid treatment? The role of buprenorphine. Palliative medicine 2006; 20:S17-S23. |
| Bonica, JJ, "Past and current status of pain research and therapy" Semin. Anesth. 5:82-99 (1986). |
| Brema et al., "Oral tramadol and buprenorphine in tumor pain. An Italian multicentre trial" Int. J. Clin. Pharm. Rex. 16(4/5) : 109-116 (1996). |
| Brenn et al., "Epidural analgesia in children with cerebral palsy" 45(12): 1156-1161 (1998). |
| Briefing document, cardiovascular and renal drug products advisory committee. May 29, 2003. Division of reproductive and urologic drug products. Apr. 29, 2003. Available at: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3956B1_01_FDA-alfuzosin.pdf. |
| Brown, Jr., of American Society of American Society of Addiction Medicine, letter to DEA Administrator dated May 17, 2002 in response to notice published in the Federal Register of Mar. 21, 2002. |
| Bruce RD. Pharmacokinetic interactions between buprenorphine and antiretroviral medications. Clinical Infectious Diseases 2006; 43 (Suppl 4):S216-S223. |
| BTDS List of Studies (undated). |
| BTDS Outstanding Issues—Awaiting FDA response from Oct. 2, 1999-Mar. 20, 2000. |
| BTDS Planned FDA Submissions/Interactions for 2000. |
| Buchwald et al., "Quantitative structure-metabolism relationships: Steric and non-steric effects in the enzymatic hydrolysis of noncongener carboxylic esters" J. Med. Chem. 42:5160-5168 (1999). |
| Budd, K., "Experience with partial agonists in the treatment of cancer pain" in: Doyle, D (ed.), Opioids in the Treatment of Cancer Pain, Royal Society of Medicine Services International Congress and Symposium Series No. 146 (1990). |
| Budd. High dose buprenorphine for postoperative analgesia. Anaesthia. Sep. 1981;36(9):900-3. |
| Bullingham et al. Buprenorphine kinetics. Clin Pharmacol Ther. Nov. 1980;28(5)667-72. |
| Bullingham et al. Clinical pharmacokinetics of narcotics agonist-antagonist drugs. Clin Pharmacokinet. Jul.-Aug. 1983;8(4):332-43. |
| Bullingham et al. Sublingual buprenorphine used postoperatively: ten hour plasma drug concentration analysis. Br J Clin Pharmacol. May 1982;13(5):665-73. |
| Bundgaard, H (ed.) Design of Prodrugs Elsevier: Amsterdam, New York 1985. |
| Bupreniorphine TDS Pre-NDA Meeting. Flux rate of Buprenorphine transdermal delivery systems (BTDS) (undated). |
| Buprenorphine (Annex 3) (18 pp.) (undated). |
| Buprenorphine DEA Review DocumentScheduling under the CSA. Feb. 2002 (26 pp.). |
| Buprenorphine is now a controlled drug. Drug Ther Bull. Oct. 16, 1989;27(21):84. |
| Buprenorphine prescription withdrawn in Norway, available at http://www.drugscope.org.uk. |
| Buprenorphine Product Photo, Norspan(TM) 5, 10 & 20. |
| Buprenorphine Product Photo, Norspan™ 5, 10 & 20. |
| Buprenorphine RTECS Info. In: RTCES Registry No. 52485-79-7. Jan 13, 2000. |
| Buprenorphine RTECS Record. In: RTCES Registry No. 52485-79-7. Apr. 25, 2000. |
| Buprenorphine TDS Pre-NDA Meeting. Flux rate analysis of Buprenorphine transdermal delivery systems (BTDS) Jun. 9, 1999. |
| Buprenorphine transdermal system (IND 50,272) meeting minutes of Nov. 18, 1998. |
| Buprenorphine transdermal system (IND 50,273) meeting minutes dated Jan. 23, 1997. |
| Buprenorphine transdermal system (IND 50,273) meeting minutes of Jul. 14, 1999 ( 8 pp.). |
| Buprenorphine transdermal system (IND 50,273) meeting minutes of May 16, 1996. |
| Buprenorphine transdermal system (IND 50,273) nonclinical video conference of Feb. 24, 1997 ( 4 pp.). |
| Buprenorphine: Differential Interaction with Opiate Receptor Subtypes in Vivo, Wolfgang Sadée, et al., The Journal of Pharmacology and Experimental Therapeutics, Copyright® 1982, vol. 223, No. 1, pp.. 157-162. |
| Burke et al., "Increased rates of drug abuse and dependence after onset of mood or anxiety disorders in adolescence" Hospital& Community Psychiatry 45(5): 455 (1994) (Abstract only). |
| Bushnell et al., Choosing the right analgesic. A guide to selection. Drugs, Sep. 1993;46(3):394-408. |
| Callaway, E., "Buprenorphine for depression: The un-adoptable orphan" Biol. Psychiatry 39: 989-990 (1996). |
| Callesen et al., "Prospective study of chronic pain after groin hernia repair" Br. J. Surg. 86: 1528-1531 (1999). |
| Caplan et al., Transdermal fentanyl: An overview of clinical progress in Opioids in Anesthesia II (Estafanous, ed.) , 1991(21):267-73. |
| Capogna et al., "Intrathecal buprenorphine for postoperative analgesia in the elderly patient" Anaesthesia 43: 128-130 (1988). |
| Capurso et al., Matrix transdermal technology: focus on a buprenorphine transdermal system. ASHP Midyear Clinical Meeting, Dec. 2001, p. 231D Abstract (w/ PowerPoint presentation). |
| Carl et al. Pain relief after major abdominal surgery: a double-blind controlled comparison of sublingual buprenorphine, intramuscular buprenorphine, and intramuscular meperidine. Anesth Analg. Feb. 1987; 66(2):142-6. |
| Catapres TTS® Product Information, Physicians' Desk Reference (1998) pp. 610-612. |
| Catapres TTS® Product Information, Physicians' Desk Reference 1998, pp. 610-612. |
| Cathelin et al, "Comparison between the side-effects of buprenorphine and morphine in conscious man" Anesth. Analg. (Paris) 37(5-6): 283-293 (1980). English abstract. (Original in French). |
| Cervera et al. [Addiction to buprenorphine] Rev Clin Esp 1989; 184:159. Letter. [in Spanish w/ English transl]. |
| Chapters 17 and 18 discussion in Opioids in Anesthesiia II (Estafanous, ed.) 1991:223-38. |
| Charuvastra et al., Buprenorphine versus placebo taste test. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Cherny NJ, Chang V, Frager G, Ingham JM, Tiseo PJ, Popp B, Portenoy RK, Foley KM. Opioid Pharmacotherapy in the Management of Cancer Pain: A Survey of Strategies Used by Pain Physicians for the Selection of Analgesic Drugs and Routes of Administration. Cancer 1995; 76:1288-1293. |
| Cherny, J., "New strategies in opioid therapy for cancer pain" J. Oncol. Management 9: 8-15 (2000). |
| Cheskin et al. A controlled comparison of buprenorphine and clonidine for acute detoxification from opiods. Drug Alchol Depend 1994;36:115-21. |
| Chien YW "Developmental Concepts and Practice in Transdermat Therapeutic Systems" in : Chien, YW (ed.), Transdermal Controlled SystemicMedications New York: Marcel Dekker, Inc. pp. 25-44. (1987). |
| Chien, pp. 31-44, col. 8 and 9. |
| Cirado et al., "Reduction of isoflurane MAC with buprenorphine and morphine in rats" Laboratory Animals 34(3): 252-9 (2000). |
| Ciraulo DA. Pharmacokinetics and pharmacodynamics of multiple sublingual buprenorphine tablets in dose-escalation trials. Journal of Clinical Pharmacology 2006;46(2):179-92. |
| Clarification of pharmtox requirements for NDA (IND 50,273) meeting minutes with sponsor of Apr. 13, 1999. |
| Clausen et al. Legal opioid consumption in Denmark 1981-1983, Eur J Clin Pharmacol. 1995;48(5):321-5. |
| Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med. 1994;23(2):129-138. |
| Climara® Product Information, Physicians' Desk Reference 1998, pp. 672-676. |
| Clinical chronology for BTDS NDA from May 10, 1996-Feb. 4, 2000. |
| Clinical documentation (Part IV), vols. 6-37 Table of Contents from Grünethal GmbH as of Dec. 13, 1999. |
| CMC chronology for BTDS NDA from Jun. 26, 1997-Mar. 2, 2000. |
| Coli et al., "Evaluation of the effectiveness of NSAIDs in the prevention of postoperative pain. Comparison between pre-and postoperative administration of sodium naproxen in orthopedic surgery" Minerva Anestesiol. 59(10): 531-535 (1993). (Abstract). |
| Colloidal Dispersions, p. 267. |
| Colucci, History of Buprenorphine. PowerPoint presentation presented in Dec. 2002. |
| Comparison of the analgesic efficacy and safety of buprenorphine in the form of a sublingual tablet and a transdermal therapeutic system (TTS 50) in chronic pain, Grunenthal; GmbH—Medical Department, Germany, Report No. WIS-BUP 03, May 20, 1999. |
| Compton et al., What dose of buprenorphine reduces opiate use? A double-blind dose-ranging study. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Controlled Substance Staff—Background material for peripheral and central nervous system advisory committee. Risk management plans for recently approved drugs. Mar. 15, 2001. |
| Coop et al. Ring constrained analogs of Buprenorphine. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Cowan et al. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol. Aug. 1977;60(4):537-45. |
| Cowan et al. The animal pharmacology of buprenorphine, an oripavine analgesic agent. Br J Pharmacol. Aug. 1977;60(4):547-54. |
| Cowan. Update on the general pharmacology of buprenorphine. Buprenorphine: Combatting Drug Abuse with a Unique Opioid, 1995;31-47. |
| Cranmer K, Landau CJ, Friedman My Turner NG, Ripa SR. The safety and tolerability 0 of 7-day buprenorphine TDS in the analgesic management of pain in the elderly—a 6-month evaluation. www.ASCP/com.education/postersandpapersam05.com. Poster 34. (Study BUP3002S). |
| Cranmer, K.W. et al., Transdermal buprenorphine (BTDS) on associated health outcomes in the elderly. Presented at the 11th World Congress on Pain, Sydney, Australia, Aug. 21-26, 2005: Abstr 691-P297. (Study sponsored by Purdue Pharma L.P.). |
| Crook et al., "The prevalance of pain complaints in a general population" Pain 18: 299-314 (1984). |
| Curran et al., "Recognition and management of depression in a substance use disorder treatment population" American Journal of Drug& Alchol Abuse 33(4): 563-569 (Abstract only). |
| Currie et al., "Comorbidity of major depression with substance use disorders" Canadian Journal of Psychiatry 50(10): 660-666 (2005). |
| Cymbalta® (duloxetine hydrochloride) Delayed-release Capsules) Package Insert. |
| Dahan A. Opioid effects on respiratory function and analgesia: New data on buprenorphine and fentanyl in a new human model [abstract]. 3rd Research Forum of the European Association for Palliative Care, Stesa, Italy, 2004. |
| D'Ambrosio P, McCarberg By Landau CJ, Hsu Y, Colucci R, Ripa S. Conversion from Vicodin® to buprenorphine transdermal system in subjects with osteoarthritis pain. J Pain 2006; 7(4) (Suppl 2):S5 1. Abstract 801. (Study BUP3018). |
| D'Arcy, [Drug reactions and interactions after drug reactions and interactions] J Pharm Belg. Sep-Oct. 1988;43(5):401-4. [in French w/ partial English transl]. |
| Daut RL, Cleeland,CS, Flanery RC. Development of the Wisconsin Brief Pain Questionaire to assess pain in cancer and other diseases. Pain 1983; 17:197-210. |
| Davids et al., "Buprenorphine in the treatment of opioid dependence" European Neuropsycharmacology 14: 209-216 (2004). |
| Davies et al. Pharmacokinetics of opioids in renal dysfunction. Clin Pharmacokinet. Dec. 1996;31(6):410-22. |
| Davis et al., "Major depression and comorbid substance use disorders" Current Opinions in Psychiatry 21(1):14-18 (2008). |
| Dayer et al., "Pharmacology of tramadol" Drugs 53(Suppl. 2): 18-24 (1997). English abstract. (Original in French). |
| Dean et al., "Depressive symptoms during buprenorphine vs. methadone maintenance: Findings from a randomized, controlled trial in opioid dependence" European Psychiatry 19:510-513 (2004). |
| Decision revoking the European Patent EP-B-964677, dated Jul. 24, 2009. |
| Declaration of Prof. Jonathan Hadgraft dated Apr. 22, 2009. |
| Declaration of Prof. Jonathan Hadgraft dated Jul. 9, 2004. |
| Declaration of Prof. Jonathan Hadgraft dated Oct. 11, 2007. |
| Declaration of Prof. Jonathan Hadgraft dated Oct. 8, 2004. |
| Definition of "Aldehyde," printed on Sep. 1, 2009 from Answers.com. |
| Definition of "Photoinitiator," printed Sep. 1, 2009 from Answers.com. |
| Definition of "Polymerize," printed on Sep. 1, 2009 from Answers.com. |
| Definition of "Tannin," printed on Sep. 1, 2009 from Wikipedia.org., the free encyclopedia. |
| Dertwinkel et al. Clinical status of opioid tolerance in long-term therapy of chronic noncancer pain. In: Progress in Pain Research and Management. Opioid Sensitivity of Chronic Noncancer Pain, Kalso et al., eds. 1999; 14:129-41. |
| Desjardins et al., "The injectable cyclooxegenase-2 specific inhibitor parecoxib sodium has analgesic efficacy when administered preoperatively" Anesth. Analg. 93: 721-727 (2001). |
| Determination of the absolute bioavailability of buprenorphine from a transdermal therapeutic system with 2 different loadings (20 and 40 mg) in comparison to an intravenous administration in an open 3-way crossovere trial with 24 healthy male volunteers. Gruenenthal GmbH—Research Centre, Germany. Report No. FO-PK 391, May 6, 1996. |
| Determination of the analgesic efficacy of three buprenorphine dosages versus placebo in a transdermal therapeutic system (TTS) in patients with tumour pain and patients with chronic pain of non-tumour-related origin. Grunenthal GmbH—Medical Department, Germany. Report No. WIS-BUP 01, Jun. 17, 1999. |
| Determination of the analgesic of three buprenorphine dosages versus placebo in a transdermal therapeutic system (TTS) in patients with tumour pain and patients with chronic non-tumour related pain. Grunenthal GmbH—Medical Department, Germany. Report No. WIS-BUP 02, Jun. 1, 1999. |
| Determination of the pharmacokinetic paramenters of buprenorphine from a transdermal therapeutic system with 3 different loadings (20, 30, and 40 mg) in an open, balanced 3-parallel group study in 54 healthy volunteers. Chrysalis Clin. Pharmcol Services GmbH, Germany. Final Report PK 402 Integrated PK, Jun. 30, 1997. |
| Determination of the plasma concentration of buprenorphine from a transdermal therapeutic system with three different loadings in a patient population. Gruenenthal GmbH—Research Centre, Germany. Report No. WIS-BUP 02 PK, Jan. 20, 1999. |
| Diamant et al. Outpatient opiate detoxification treatment with buprenorphine. Preliminary investigation. Eur Addict Res. 1998;4:198-202. |
| Dini et al. [Controlled study of the analgesic effect and tolerability of buprenorphine in cancer patients] Minerva Med. Jan. 28, 1986;77(3-4):93-104, [in Italian w/ English abstract]. |
| Dionne et al., "Evaluation of preoperative ibuprofen for postoperative pain after removal of third molars" Oral Surgery 45: 851-856 (1978). |
| Dionne et al., "Suppression of postoperative pain by preoperative administration of ibuprofen in comparison to placebo, acetaminophen, and acetaminophen plus codeine" J. Clin. Pharmacol. 23: 37-43 (1983). |
| Driscoll, CE Primary Care 14(2): 337-352 (1987). |
| Dum et al. In vivo receptor binding of the opiate partial agonist, buprenorphine, correlated with its agonistic and antagonistic actions, Br J Pharmacol. Nov. 1981;74(3):627-33. |
| Dum et al., "Opioids and Motivation" Interdisciplinary Science Reviews 12(2): 180-190 (1987). |
| Duragesic® [package insert]. Titusville, NJL Janssen Pharmaceutica; 2001. |
| Eder at al., "Buprenorphin in der Schwangerschaft" Psychiat. Prax. 28: 267-269 (2001). (XP009068360). (Abstract). |
| Eissenberg et al. Buprenorphine's physical dependence potential: antagonist-precitated withdrawal in humans. J Pharmacol Exp Ther. 1996; 276:449-59. |
| Eissenberg et al. Controlled opioid withdrawal evaluation during 72 h dose omission in buprenorphine-maintained patients. Drug Alchol Depend. Apr. 14, 1997;45(1-2):81-91. |
| Eissenberg T et al., Buprenorphine's physical dependence potential: Antagonist-precipitated withdrawal in humans. J. Pharmacol. Exp. Ther. 276: 449-459, 1996. |
| Eke et al., "An open comparative study of dispersible piroxicam versus soluble acetylsalicylic acid for the treatment of osteoarticular painful attack during sickle cell crisis" Tropical Medicine and International Health 5(2): 81-84 (2000). |
| El-Tahtawy et al., 7-Day bioavailability of buprenorphine from a novel transdermal system in demographic subgroups. 13th Annual ACCP Meeting Abstracts, Sep. 23-25, 2001, p. 1027, Abstract 56 (w/ PowerPoint presentation). |
| Emrich et al. "Antidepressant effects of buprenorphine" Lancet 2: 709 (1982). |
| Emrich et al., "Current perspectives in the pharmacopsychiatry of depression and mania" Neuropharmacology 22(3 Special No.): 385-388 (1983). |
| Emrich et al., "Possible antidepressive effects of opioids: action of buprenorphine" Ann. NY Acad. Sci. 398: 108-112 (1982). |
| Emrich, HM, in: "Typical and Atypical Antidepressants: Clinical Practice" Costa et al., (Eds.), Raven Press: New York, 1982 pp. 77 et seq. |
| Escher, M. et al., Pharmacokinetics and analgesic effects of intravenous buprenorphine (abstract). Clinical Pharmacology and Therapeutics 2005; 77(2 Suppl): 51. |
| Etchepare F, Coutaux, A, Edel Y, Bourgeois P. Enterobacter cloacae spondylodiscitis through misuse of high-dose intravenous buprenorphine, La Presse Medicale 2005; 34(10):729-731. |
| Everhart et al., "Subnanogram-concentration measurement of buprenorphine in human plasma by electron-capture capillary gas chromatography: application to pharmacokinetics of sublingual buprenorphine" Clin. Chem. 43(12): 2292-2302 (1997). |
| Excerpt of Review for Approval issued Aug. 7, 1990 by the FDA for Duragesic®. |
| Extein et al., "Deficient prolactin response to morphine in depressed patients" Am. J. Psychiatry 137:845-846 (1980). |
| Extein et al., "Methadone and morphine in depression" Psychopharmacol. Bull. 17(1): 29-33 (1981). |
| Faponle, AF, "Management of pain after surgery—A short review" Nigerian Journal of Medicine 10(3): 112-115 (Jul./Sep. 2001). |
| Faroqui et al. Buprenophine, benzodiazepines and respiratory depression. Anaesthesia. Oct. 1983;38(10):1002. |
| Feinberg et al., "The effect of morphine on symptoms of endogenous depression" in: Harris, (ed.)Problems of Drug Dependence, 1992 National Institute on Drug Abuse Research Monograph 43 pp. 245-250 (1982). |
| Fentanyl Published Information (undated). |
| Ferrell et al., "Principles of pain management in older people" Compr. Ther. 17: 53-58 (1991). |
| Ferrell, BA, "Pain evaluation and management in the nursing home" Ann. Int. Med. 123(9): 681-687 91995). |
| Fincham. Cardiopulmonary arrest and subsequent death administration of buprenorphine in an elderly female: a case report. J. Geriatric Drug Ther. 1989;3(3):103-5. |
| Fischer et al. Buprenorphine maintenance in pregnant opiate addicts. Eur Addict Res. 1998;4(Suppl 1):32-6. |
| Fischer et al., "Treatment of opioid-dependent pregnant women with buprenorphine" Addiction 95(2): 239-244 (2000). |
| Fiset et al., Biopharmaceutics of a new transdermal fentanyl device. Anesthesiology. Sep. 1995;83(3):459-69. |
| Fleiss JL. Statistical Methods for Rates and Proportions. 2nd ed. New York, NY: John Wiley & Sons; 1981:33-45, 272-273 (Table A.3). |
| Fletcher D, "Prevention of postoperative pain" Ann. Fr. Anesth. Reanim. 17(6): 622-632 (1998). (Abstract). |
| Flitz J. Effects of intermittent hemodialysis on buprenorphine and norbuprenorphine plasma concentrations in chronic pain patients treated with transdermal buprenorphine. European Journal of Pain 2006;10(8):743-8. |
| Follow-up treatment with buprenorphine TTS 50 after completion of the double-blind phase of studies. Grunenthal GmbH—Medical Department, Germany. Report No. WIS-BUP FU, May 25, 1999. |
| Forth. [another analgesic unmasked as an addictive substance: a vicious circle] MMW Munch Med Wochenschr. Sep 30, 1983; 125(39):834. [in German w/ English transl]. |
| Francaviglia et al. Subarachnoid buprenorphine administered by implantable micropumps. Acta Neurochir (Wien), 1990;102(1-2):62-8. |
| Franklin et al. Risk assessment in dermatoxicology. In: Marzulli et al. (Eds.) Dermatotoxicology (4th ed.) 1991. 1991;30:713-47. |
| Fraser. Clinical toxicology of drugs used in the treatment of opiate dependency. Clinics in Laboratory Medicne, Jun. 1990; 10(2):375-86. |
| Friend et al., Simple alkyl esters as skin permeation enhancers. J. Controlled Release 1988 9:33-41. |
| Friend et al., Simple alkyl esters as skin permeation enhancers. J. Controlled Release 1998 9:33-41. |
| Friend et al., Transdermal delivery of levonorgestrel I: Alkanols as permeation enhancers in vitro, J. Controlled Release 1988 7:243-50. |
| Fudala et al. A multi-site efficacy evaluation of a buprenorphine/naloxone product for opiate dependence treatment. NIDA Res Monogr. Problems of Drug Dependent: Proceedings of the 60th annual Scientific Meeting; 1998:105. |
| Fudala et al. Clinical efficacy studies of buprenorphine for the treatment of opiate dependence. Buprenorphine: Combatting Drug Abuse with a Unique Opioid. 1995:213-39. |
| Fudala et al. Outpatient comparison of buprenorphine and methadone maintenance. II. Effects on cocaine usage, retention time in study and missed clinic visits. NIDA Res Monogr. 1991;105:587-8. |
| Fudala et al. Use of buprenorphine in the treatment of opioid addiction. II. Physiologic and behavioral effects of daily and alternate-day administration and abrupt withdrawl. Clin Pharmacol Ther. 1990; 47:525-34. |
| Fujimura et al. Influences of bathing and hot weather on the pharmacokinetics of a new transdermal clonidine, M-5041T. J Clin Pharmacol. Oct. 1996;36(10):892-6. |
| Fukaze et al., Precipitation of morphine withdrawal by buprenorphine and butorphanol in male cynomolgus monkeys. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Fullerton et al. Prolonged nausea and vomiting associated with buprenorphine. Pharmacotherapy 1991;11:90-93. |
| Gasfriend et al., Long-term effects of buprenorphine for treatment of combined opiate and cocaine dependence. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Gaulier J-M, et al., Ingestion of High-Dose Buprenorphine by a 4 Year-Old Child. Journal ofToxicology-Clinical Toxicology 42(7):993-5, 2004. |
| Gautschi OP, Zellweger R. Images in emergency medicine. Extensive groin abscess and myositis after intravenous cubital buprenorphine injection. Annals of Emergency Medicine 2006; 48(6):656-859. |
| Gebhart et al, Opiod modulation of visceral pain. In: Progress in Pain Research and Management. Opioid Sensitivity of Chronic Noncancer Pain. Kalso et al., eds. 1999;225-35. |
| Gerra et al., "Buprenorphine treatment outcome in dually diagnosed heroin dependent patients: A retrospective study" Progress in Neuro-Psychopharmacology & Biological Psychiatry 30:265-272 (2006). |
| Gerra et al., "Buprenorphine versus methadone for opioid dependence: predictor variables for treatment outcome" Drug and Alcohol Dependence 75: 37-45 (2004). |
| Gerra G. Naltrexone and buprenorphine combination in the treatment of opioid dependence. Journal of psychopharmacology 2006;20(6):806-14. |
| Gibaldi, Prolonged-Release medication in Biopharmaceutics and Clinical Pharmacokinetics. 1984;3rd ed.: 113-30. |
| Glasper et al., "Induction of patients with moderately severe methadone dependence onto buprenorphine" Addiction Biology 10: 149-155 (Jun. 2005). |
| Gold et al., "Antimanic, antidepressant, and antipanic effects of opiates: clinical, neuroanatomical, and biochemical evidence" Ann. NY Acad. Sci. 398: 140-150 (1982). |
| Gold et al., "Clinical evidence of antidepressant and antipanic effects of opiates" Am. J. Psychiatry 136: 982-983 (1979). |
| Goldstein, "Methadone for depression" Biol. Psychiatry 19 1272-1273 (1984). |
| Golianu, B., "Pediatric acute pain management" Pediatr. Clin. North Am. 47(3): 559-587 (2000). |
| Goodman & Gillman's The Pharmacological Basis of Therapeutics Hardman, JG (ed.), McGraw-Hill Professional Publishing, 2001, pp. 530-532. |
| Goodman & Gilman's The Pharmacological Basis of therapeuticHardman, JG (ed.) McGraw-Hill Professional Publishing 2001, pp. 31-32. |
| Goodman & Gilman's The Pharmacological Basis of Therapeutics Hardman, JG(ed.) McGraw-Hill Professional Publishing 2001, p. 586. |
| Gould. Buprenorphine causes pulmonary edema just like all othr mu-opioid narcotics. Upper airway obstruction, negative alveolar pressure. Chest. May 1995;107(5):1478. |
| Gourlay. Different opioids—same actions? In: Progress in Pain Research and Management. Opioid Sensitivity of Chronic Noncancer Pain. Kalso et al., eds. 1999; 14:97-115. |
| Griffin et al., "High-dose intravenous methylprednisolone therapy for pain in children and adolescents with sickle cell disease" N. Engl. J. Med. 330(11): 733-737 (1994). |
| Grond et al. Clinical pharmacokinetics of transdermal opioids: focus on transdermal fentanyl. Clin Pharmacokinet. Jan. 2000;38(1):59-89. |
| Grond, S et al., Pain 69: 191-198 (1997). |
| Guidline for Industry—Clinical Safety Data management: Definitions and Standards for Expedited Reporting ICH-E2A, Mar. 1995, pp. 5-7. |
| Guilbaud et al. Antinociceptive effect of opioid susbstances in different models of inflammatory pain. In: Progress in Pain Research and Management. Opioid Sensitivity of Chronic Noncancer Pain. Kalso et al., eds. 1999; 14:201-23. |
| Guo et al., Bioadhesive buccal polymer patches for buprenorphine controlled delivery: Solubility consideration. Proceed Intern Symp Control Rel Bioact Mater. 1995. |
| Hadgraft, In vitro testing of dermal and transdermal products. Mar. 28, 2001 (6 pages). |
| Hale et al., Analgesic efficacy of buprenorphine transdermal system vs. Oxy/APAP in patients with chronic low back pain. The Gerontologist, vol. 41, Special Issue 1, Oct. 2001, Program Abstracts from the Gerontological Society of America 54th Annual Scientific Meeting, Nov. 15-18, 2001, p. 25 (w /PowerPoint presentation first presented on May 7, 2002 and revised on Oct. 30, 2002). |
| Hale et al., Dose proportionality and the dose response of buprenorphine transdermal system in patients with chronic pain. 13th Annual ACCP Meeting Abstracts, Sep. 23-25, 2001, p. 1027, Abstract 58(w/ PowerPoint presentation). |
| Hale et al., Long-term use of buprenorphine transdermal system (BTDS) in patients with chronic pain. PowerPoint presentation presented May 2002. |
| Hale et al., Treatment of patients with chronic low back pain with buprenorphine transdermal system (BTDS) compared with hydrocodone/acetaminophen. National Clinical Symposium of the American College of Nurse Practitioners, Oct. 20, 2001 Abstract (w/ PowerPoint presentation). |
| Hale ME, Ahdieh H, Ma T, Rauck R, for the Oxymorphone ER Study Group. Efficacy and Safety of OPANA ER (Oxymorphone Extended Release) for Relief of Moderate to Servere Chronic Low Back Pain in Opioid-Experienced Patients: A 12-Week, Randomized, Doubleblind, Placebo-controlled Study. J Pain 2007; 8(2):175-184. |
| Halpern SD, Karlawish JHT, Berlin JA. The Continuing Unethical Conduct of Underpowered Clinical Trials. JAMA 2002; 288:358-362. |
| Hambrook JM, Rance MJ. The interaction of buprenorphine with opiate receptor: lipophilicity as a determining factor in drug-receptor kinetics. In: Kosterlitz HW, editor. Opiates and endogenous opioid peptide. Amsterdam: Elsevier/North Holland, Biomedical Press; 1976. p. 295-301. |
| Hand et al. Buprenorphine disposition in patients with renal impairment: single and continuous dosing, with special reference to metabolites. Br J Anaesth. Mar. 1990;64(3):276-82. |
| Hand et al. Radioimmunoassay of buprenorphine in urine: studies in patients and in a drug clinic. J Anal Toxicol. Mar-Apr. 1989;13(2):100-4. |
| Hannibal et al., "Preoperative wound infiltration with bulpivacaine reduces early and late opioid requirement after hysterectomy" Anesth. Analg. 83: 376-381 (1996). |
| Harris S, Hoelscher D, Kristensen A, O'Keefe S, Schemera A. Effects of bupreorphine transdermal system 10 mg and 2 ×20 mg on QT intervals in healthy subjects. Clin Pharmacol ner 2006; 79(2):P35. (Study BUP1011). |
| Harvey-Clark et al., Transdermal fentanyl compared with parenteral buprenorphine in post-surgical pain in swine: a case study. Lab Anim. Oct. 2000;34(4):386-98. |
| Hawks et al., Buprenorphine-naloxone combination drug for the treatment of drug addiction. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Hayes, of FDA, Comments dated Mar. 31, 1997 regarding buprentrphine patch (IND 50,273), Mar. 31, 1997 (2 pp.). |
| Heel et al. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979;17:81-110. |
| Heel et al., Curr.Ther. 5: 29-33 (1979). |
| Heel RC, Brogden RN, Speight TM, A very GS. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979; 17:81-110. |
| Heilman, K. Therapeutische Systeme (1984) pp. 26-27 and pp. 48-53. |
| Heilman, N Therapeutische System , 4th ed., Stuttgart: Enke pp. 67-77 and 83-86 (1984). |
| Heilman, N Therapeutische Systems , 4th ed., Stuttgart: Enke pp. 104-106 (1984). |
| Henrion. [Assessment of the effectiveness of measures taken in France to reduce the risk of intravanenous heroin addiction] Bull Acad Natl Med. Jun.-Jul. 1997;181(6):1177-85; discussion 1186-9, [in French w/ English transl]. |
| Hernandez-Mora et al. [Paroxysmal atrial fibrillation after ingestion of buprenorphine.] Rev Clin Esp. Jun. 1988;183(2):99-100. [in Spanish w/ English transl]. |
| Herve S, et al., Acute hepatitis due to buprenorphine administration. European Journal of Gastroenterology & Hepatology 16(10):1033-7, 2004. |
| Hexal AG Opposition against EP-B-964677, dated May 15, 2007. (in German, w/ Engl. translation). |
| Hihuchi, William I., Ph.D. et al., "Particle Phenomena and Coarse Dispersions" Chapter 21, p. 294. |
| Hirschauer et al. [Is buprenorphine hepatotoxic?] Gastroenterol Clin Biol. Jun. 1989;13(6-7):636. [in French w/ English transl]. |
| Hoffman D, Freidman M, Colucci S, Richards, P, Zhang P. Validation of the Brief Pain Inventory for subjects with osteoarthritis. J Pain 2002; 3(2):(suppl)3. |
| Hogan & Hartson's comments dated May 22, 2002 on 67 FR 13114 of Mar. 21, 2002. |
| Hogan & Hartson's response dated May 9, 2002 to 67 FR 17074 of Apr. 9, 2002. |
| Hogan & Hartson's supplemental filing dated Apr. 10, 2002 in support of citizen petition filed on Dec. 11, 2001. |
| Holdsworth et al. Transdermal fentanyl disposition in elderly subjects. Gerontology. 1994;40(1):32-7. |
| Holloway, M. (Erickson, D.), "Rx for addiction" Sci. Am. 264(3): 94-103 (1991). |
| Holmes. Buprenorphine side effects. N Z Med J. Mar. 14, 1984;97(751):166. |
| Hoskin et al. Opioid agonist-antagonist drug in acute and chronic pain states. Drugs 1991;41:326-44. |
| Hoskin PJ, Hanks GW. Opioid agonist-antogonist drugs in acute and chronic pain states. Drugs. 1991; 41(3):326-344. |
| Huang et al., "Atmospheric pressure ionization mass spectrometry" Anal. Chem. 62(13): 713A-725A (1990). |
| Huguet-Levet. [Buprenorphine: its ambiguity] Ann Pharm Fr. 1995;53(3):124-30. [in French w/ English transl]. |
| Human immunodeficiency virus/acquired immunodeficiency syndrome in the context of drug abuse. Report of the Executive Director, United Nations Economic and Social Council. Jan. 30, 2003 (12 pp.). |
| Human Pharmacolinetics of Intravenous, Sublingual and Buccal Buprenorphine*, James J. Kuhlman, Jr., et al., Journal of Analytical Toxicology, vol. 20, Oct. 1996. |
| Hyman, Phelps & McNamara P.C.'s comments and objections dated Jul. 5, 2002 to the citizen petition. |
| Hyman, Phelps & McNamara P.C.'s comments dated May 22, 2002 on behalf of Purdue Pharma L.P. to 67 Fed. Reg. 13,114 of Mar. 21, 2002. |
| Hyman, Phelps & McNamara P.C.'s response dated May 9, 2002 on behalf of Purdue Pharma L.P. to 67 Fed. Reg. 17074 of Apr. 9, 2002. |
| ICH harmonized tripartite guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs, E14, Final Draft May 12, 2005. Available at [http://www.ich.org/cache/compo/475-272-1.httml #E14]. |
| Imoto et al., Transdermal prodrug concepts: permeation of buprenorphine and its alkyl esters through hairless mouse skin and influence of vehicles. Biol Pharm Bull. Feb. 1996;19(2):263-7. |
| Inagaki et al., "Mode and site of analgesic action of epidural buprenorphine in humans" Anesth. Analg. 830: 530-536 (1996). |
| International Search Report, PCT/US2003/039793, dated Mar. 31, 2004, 2 ppgs. |
| Introduction to the Pharmacology of Opioids. (Apr. 23, 2009). |
| Jacobsen et al., "Age-related changes in sebaceous wax ester secretion rates in men and women" J. Invest. Dermatol. 85: 483-485 (1985). |
| Jaffe et al., Opioid Analgesics and Antagonists. The Pharmacological Basis of Therapeutic (8th ed. Pergamon Press) 1990, Chap. 21, pp. 485-521. |
| Jagadheesan K and Muihead D., Possible manic potential of buprenorphine [letter]. Australian and New Zealand Journal of Psychiatry 37*8):560-1, 2004. |
| Jakobovits SL, Mcdonough M, Chen RY. Buprenorphine-associated gastroparesis during in-patient heroin detoxification. Addiction 2007; 101: 490-491. |
| Jamison. Comprehensive pretreatment and outcome assessment for chronic opioid therapy in nonmalignant pain. J Pain Symptom Manage. Apr. 1996;11(4):231-41. |
| Japanese Supreme Court, Third Petty Branch decision dated Jan. 16, 2007. (English translation). |
| Jasinski et al. Abuse liability assessment in human subjects. Trends Pharmacol Sci. 1984;5:196-200. |
| Jasinski et al. Human pharmacology and abuse potential of the analgesic buprenorphine: a potential agent for treating narcotic addiction. Arch Gen Psychiatry. 1978;35:501-16. |
| Jasinski et al. Laboratory studies of buprenorphine in opioid abusers. In: Cowan A, Lewis JW, eds.. Buprenorphine: combatting drug abuse with a unique opioid. New York: Wiley-Liss. 1995:189-211. |
| Jasinski et al. Progress report from the NIDA Addiction Research Center, Baltimore, Maryland, NIDA Res Monogr. Mar. 1984;49:69-76. |
| Jasinski et al. Progress report of the NIDA Addiction Research Center, Baltimore, Maryland, 1982. NIDA Res Monogr. Apr. 1983;43:92-8. |
| Jasinski et al. Progress report of the NIDA Addiction Research Center, NIDA Res Monogr. 1982;41:45-52. |
| Jasinski et al. Sublingual versus subcutaneous buprenorphine in opiate abusers. Clinical Pharmacol Ther. 1989;45:513-9. |
| Jeal et al., Transdermal fentanyl. A review of its pharmacological properties and therapeutic efficacy in pain control. Drugs Jan. 1997, 53(1):109-38. Review. |
| Jernite et al., "Burprenorphine and pregnancy. Analysis of 24 cases" Arch. Pediatr. 6:1179-1185 (1999). |
| Jin-Jie, G. "Synthesis of biodegradable polyurethane foams condensed tannin and bark of Acacia mearnsii" Bull. Kyushu Univ. For. 79: 21-85 (1998). |
| Johnson et al. A controlled trial of buprenorphine treatment for opiod dependence. JAMA. 1992;267:2750-5. |
| Johnson et al., Daily versus alternate-day dosing of buprenorphine in the outpatient treatment of opioid dependence. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Johnson RE, Fudala PJ, Payne R. Buprenorphine: considerations for pain management. JPain Symptom Manage 2005;29:297-326. |
| Jones et al., Buprenorphine and naloxone interactions in heroin-dependent volunteers. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Journal of Pain and Symptom Management. Apr. 1992 7(S3) (whole journal). |
| Juhlin-Dannefelt et al., "Premedication with sublingual buprenorphine for out-patient arthroscopy: Reduced need for postoperative pethidine but higher incidence of nausea" Acta Anaesthesio. Scand. 39: 633-636 (1995). |
| Kahila H. A prospective study on buprenorphine use during pregnancy: Effects on maternal and neonatal outcome. Acta Obstetricia et Gynecologica Scandinavica 2007; 86(2): 185-90. |
| Kahila H. Brain magnetic resonance imaging of infants exposed prenatally to buprenorphine. Acta Radiologica 2007; 48(2):228-31. |
| Kaiko et al., Transdermal buprenorphine. Memorial Sloan Kettering Manuscript, Chapter 15 (w/ PowerPoint Final presentation presented May 2, 2003). |
| Kakinohana et al., "Pre-emptive analgesia with intravenous ketamine reduces postoperative pain in young patients after appendectomy: A randomized control study" Masui 49(10): 1092-1096 (2000). ( Abstract). |
| Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. New York: John Wiley and Sons (2nd edition). 2002, pp. 22-23. |
| Kalso. Route of opioid administration—does it make a difference? In: Progress in Pain Research and Management, Opioid Sensitivity of Chronic Noncancer Pain. Kalso et al., eds. 1999; 14:117-28. |
| Kareti et al. Effects of buprenorphine, a new narcotic agonist-antagonist analgesic on the EEG, power spectrum and behavior of the rat. Neuropharmacology. Feb. 1980;19(2):195-201. |
| Katchman AN, McGroary KA, Kilborn MJ, et al., Influence of opioid agonists on cardiac human ether-a-go-go-related gene K(+) currents, 2002. J Pharmacol Exp Ther 303:688-94. |
| Kenny et al., Clin. Geriatr. 8: 1-4 (2000). |
| Keup et al. [Potential for buprenorphine abuse] MMW Munch Med Wochenschr. Sep. 30, 1983;125(39):835-7. [in German w/ English transl]. |
| Keup. [Buprenorphine (Temgesic®) abuse and depenence] Suchtgefahren. 1983;29:193-4. [in German w/ English transl]. |
| Kinney et al., AACN's Clinical Reference Clinical Reference for Critical Care Nursing, Mosby, 4th ed., 1998, pp. 285-287. |
| Kintz, Deaths involving buprenorphine: A compendium of French cases. Forensic Sci Int. Sep. 15, 2001; 121(1-2):65-9. |
| Kokki et al., "Comparison of pre-and postoperative administration of ketoprofen for analgesia after tonsillectomy in children" Pediatric Anesthesia 12(2): 162-167 (2002). |
| Kolloch RE, Mehlburger L, Schumacher H, Göbel Bo. Efficacy and safety of two different galenic formulations of a transdermal clonidine system in the treatment of hypertension. Clin Auton Res. 1993;3:373-378. |
| Korte, Titration with TTS fentanyl systems for previously uncontrolled cancer pain, and Lipman's response thereto. Anesth Analg. Sep. 1994;79(3):612-4. |
| Kosten et al., "Depressive symptoms during buprenorphine treatment of opioid abusers" Journal of Substance Abuse Treatment 7: 51-54 (1990). |
| Kosten T, et al., Depression Predicts Higher Rates of Heroin Use on Desipramine with Buprenorphine than with Methadone. The American Journal of Addictions 13:191-201, 2004. |
| Krantz, M.J. et al., Effects of buprenorphine on cardiac repolarization in a patient with methadone-related torsade de pointes. Pharmacotherapy 2005; 25(4): 611-614. |
| Kuhlman, J.J., et al., "Human pharmacokinetics of Intravenous, sublingual and buccal buprenorphine" J. Anal. Toxicol. v 20 (1996). |
| Kuhlman, Jr., et al., Human pharmacokinetics of intravenous, sublingual, and buccal buprenorphine. J Anal Toxicol. Oct. 1996: 20(6);369-78. |
| Kumar, Chemists selling illegal drugs to be booked in The times of India, Aug. 16, 2000. |
| Landau, C.J. et al., The safety and tolerability of buprenorphine 7-day transdermal system in patients with nonmalignant pain syndromes responsive to opioids. Presented at the 11th World Congress on Pain, Sydney Australia, Aug. 21-26, 2005: Abstr. 690-P296 (Study sponsored by Purdue Pharma L.P.). |
| Landau, CJ et al., "Buprenorphine transdermal delivery system in adults with persistent noncancer-related pain syndromes who require opioid therapy: a multicenter, 5 week run-in and randomized, double-blind maintenance-of-analgesia study" Clinical Therapeutics 29(10): 2179-2193 (2007). |
| Lange et al. Safety and side-effects of buprenorphine in the clinical management of heroin addiction. Drug Alchol Depend. Aug. 1990;26(1):19-28. |
| Lasseter et al., Systemic pharmacokinetic (PK) study of buprenorphine (B) in mild to moderate chronic hepatic impairment (CHI). Amer Soc for Clin Pharmacol and Therapeut. PI-4. |
| Lasseter et al., Systemic pharmacokinetic (PK) Study of buprenorphine (B) in mild to moderate chronic hepatic impairment (CHI). Amer. Society for Clinical Pharmacology and Therapeutics 69(2) 2(PI-4) Feb. 2001 Abstract (w/ PowerPoint presentation presented Mar. 2001). |
| Law et al. The feasibility of abrupt methadone-buprenorphine transfer in British opiate addicts in an outpatient setting. Addiction Bio. 1997;2:191-200. |
| Leander. Buprenorphine has potent kappa opioid receptor antagonist activity. Neuropharmacol. 1987; 26:1445-7. |
| Leander. Buprenorphine is a potent kappa-opioid receptor antagonist in pigeons and mice. Eur J Pharmacol. Jul. 14, 1988; 151(3):457-61. |
| Lehmann et al., "Treatment of depression with dexedrine and demerol" Curr. Therapeutic Res. Clin. Exp. 13(1): 42-49 (1971). |
| Lemens HJM, Wada DR, Munera C, El-Tahtawy A, Stanski DR. Enriched analgesic efficacy studies: an assessment by clinical trial simulation. Contemp Clin Trials, 2006;27; 165-1 73. |
| Letter from counsel for Acino AG (previously Novosis AG), dated Apr. 17, 2009, to EPO re EP-B-964677 opposition proceedings. (in German, w/Engl. translation). |
| Letter from counsel for Purdue Pharma LP, dated Apr. 23, 2009, to EPO re EP-B-964677 opposition proceedings (English). |
| Letter from counsel for Purdue Pharma LP, dated Dec. 20, 2007, to EPO re EP-B-964677 opposition proceedings (English). |
| Letter from counsel for Purdue Pharma LP, dated Jun. 4, 2009, to EPO re EP-B-964677 opposition proceedings (English). |
| Lewis et al. Buprenorphine—background to its development as a treatment for opiate dependence. NIDA Res Monogr. 1992;121:5-11. |
| Lewis et al. The pharmacology and abuse potential of buprenorphine: a new antagonist analgesic. Advances in substance abuse. 1983;3:103-54. |
| Lewis. Buprenorphine, Drug Alcohol Depend. 1985; 14:363-72. |
| Lewis. Clinical pharmacology of buprenorphine in relation to its use as an analgesic, Buprenorphine: Combatting Drug Abuse with a Unique Opioid. 1995;151-63. |
| Lichtenstein et al., "Disaggregating pain and its effect on physical functional limitations" J. Gerontol. 53(5): M361-M371 (1998). |
| Liguori et al., Modification of respiratory effects of levorphanol by nalbuphine, butorphanol, and buprenorphine in rhesus monkeys. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Ling et al. A controlled trial comparing buprenorphine and methadone maintenance in opiod dependence. Arch Gen Psychiatry. 1996 May;53(5):401-7. |
| Ling et al. Buprenorphine maintenance treatment of opiate dependence: a multicenter, randomized clinical trial. Addiction. 1998;93:475-86. |
| Ling et al. Methadyl acetate and methadone as maintenance treatments for heroin addicts. A veterans administration cooperative study. Arch Gen Psychiatry, Jun. 1976;33(6):709-20. |
| Ling et al. Substitution pharmacotherapies for opioid addiction: from methadone to LAAM and buprenorphine, J Psychoactive Drugs, Apr.-Jun. 1994;26(2):119-28. |
| Lintzeris N. Interactions on mixing diazepam with methadone or buprenorphine in maintenance patients. Journal of clinical psychopharmacology 2006; 26(3):274-283. |
| Litchfield, Buprenorphine in oral surgery. A comparison with fentanyl. SAAD Dig. Oct. 1986;6(8):182-6. |
| Liu et al. Rapid detoxification of heroin dependence by buprenorphine. Acta Pharmacol. Sin. 1997;18:112-4. |
| Lizasoain et al. Buprenorphine: bell-shaped dose-response curve for its antagonist effects. Gen Pharmacol. 1991;22(2):297-300. |
| Lloyd-Jones et al. Plasma concentration and disposition of buprenorphine after intraveneous and intramuscular doses to baboons. Eur J Drug Metab Pharmacokinet. 1980;5(4):233-9. |
| Lovell et al., "Type I and III collagen content and fibre distribution in normal human skin during ageing" Br. J. Dermatol. 117: 419-328 (1987). |
| Luukinen et al., "Prognosis of diastolic and systolic orthostatic hypotension in older persons" Arch. Int. Med. 159: 273-280 (1999). |
| Macdonald et al. Psychomotor effects of ketorolac in comparison with buprenorphine and diclofenac. Br J Clin Pharmacol. 1989; 27;453-9. |
| Mackenzie et al. Influence of pretreatment with a monomine oxidese inhibitor (Phenelzine) on the effects of buprenorphine and pethidine in the conscious rabbit. Br J Anaesth. 1988;60:216-21. |
| Mann et al., Buprenorphine alone or in combination with naltrexone for inpatient medically supervised opiate withdrawal. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Marek, GJ "Behavioral evidence for μ-opioid and 5-HT2A receptor interactions" European Journal of Pharmacology 474: 77-83 (2003). |
| Markou et al., "Neurobiological similarities in depression and drug dependence: a self-medication hypothesis" Neuropsychopharmagology 18(3): 135-174 (1998). |
| Markowitz et al., "Venlafaxine-tramadol similarities" Med. Hypotheses 51(2): 167-168 (1998). |
| Marquardt et al., Fentanyl remaining in a transdermal system following three days of continuous use. Ann Pharmacother Oct. 1995, 29(10):969-71. |
| Marquet et al. [In utero exposure to Subutex induces no or mild withdrawal syndromes in the newborn] Therapie. 1998;53-178. [in German w/ English abstract]. |
| Marquet et al. Buprenorphine withdrawal syndrome in a newborn. Clin Pharmacol Ther. 1997;62:569-71. |
| Martin et al. The effects of morphine-and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517-32. |
| Martin. History and development of mixed opioid agonists, partial agonists and antagonist . Br J Clin Pharmacol. 1979;7:273S-9S. |
| Matussek et al., "Investigations with the specific μ-opiate receptor antagonist fentanyl in depressive patients: Growth hormone, prolactin, cortisol, noradrenaline and euphoric responses" Neuropsychobiology 21: 1-8 (1989). |
| Mauryama S, Nomura Y, Fukushige T, Eguchi T, Nishi J, Yoshinaga M, Kawano Y. Suspected Takotsubo cardiomyopathy caused by withdrawal of buprenorphine in a child. Circulation Journal 2006; 70:509-511. |
| Max et al. Single-dose analgesic comparisons. Advances in pain research and therapy. 1991;18:55-95. |
| McCance-Katz EF, Interactions between buprenorphine and antiretrovirals. I. The nonnucleoside reverse-transcriptase inhibitors efavirenz and delaviridine. Clinical infectious Diseases 2006; 43(Suppl 4):S224-S234. |
| McCance-Katz EF. Interaction between buprenorphine and atazanavir or atazanavir/ritonavir. Drug and Alcohol Dependence 2007; 91(2-3):269-78. |
| McCance-Katz EF. Interactions between buprenorphine and antiretrovirals. II. The protease inhibitors nelfinavir, lopinavir/ritonavir, and ritonavir. Clinical infectious Diseases 2006; 43 (Supp 4):S235-S246. |
| McCance-Katz, of American Academy of Addiction Psychiatry, letter to DEA Administration dated May 15, 2002 in response to proposed rule: Rescheduling of Buprenorphine from Schedule V to Schedule III, published in the Federal Register of Mar. 21, 2002. |
| McMahon FG, Jain AK, Vargas R, Fillingim J. A double-blind comparison of transdermal clonidine and oral captopril is essential hypertension. Clin.Ther. 1990; 12:88-100. |
| McNeal, of FDA, Comments dated Aug. 30, 1996 on the protocols submitted on Aug. 5, 1996 in Ser. No. #006 of IND 50,273. |
| McNeal, of FDA, Medical Review on IND 40,273, dated May 10, 1996. |
| McNicholas et al., Buprenorphine clinical practice guidelines. Field review draft. Nov. 17, 2000. |
| McQuay et al. Buprenorphine kinetics in humans. In: Buprenorphine: Combatting Drug Abuse with a Unique Opioid. 1995;137-47. |
| McQuay et al. Buprenorphine kinetics. Advances in Pain Res and Ther. 1986. 8:271-83. |
| McQuay et al. Clinical effects of buprenorphine during and after operation. Br J Anaesth. Oct. 1980;52(10):1013-9. |
| McQuay et al. Delayed respiratory depression. A case report and a new hypothesis. Acta Anaesthesiol Belg. 1979;30 Suppl:245-7. |
| McQuinn, R.I., et al., "Sustained oral mucosal delivery in human volunteers of buprenorphine from a thin non-eroding mucoadhesive polymeric disk" J. Contr. Rel. (1995). |
| McQuinn, R.L. et al., Sustained oral mucosal delivery in human volunteers of buprenophine from a thin non-croding mucoadhesive polymeric disk. Journal of Controlled Release 1995, 34:243-50. |
| Mello et al. Behavioral pharmacology of buprenorphine. Drug Alchol Depend. Feb. 1985;14(3-4):283-303. |
| Mello et al. Buprenorphine effects on human heroin self-administration: an operant analysis. J Pharmacol Exp Ther. 1982;223:30-9. |
| Mello et al. Buprenorphine self-administration by rhesus monkey. Pharmacol Biochem Behav. 1981;15:215-25. |
| Mello et al. Buprenorphine suppresses cocaine self-administration by rhesus monkeys. Science. 1989;245:859-62. |
| Mello et al. Buprenorphine suppresses heroin use by heroin addicts. Science. 1980;207:657-9. |
| Mello et al. Buprenorphine's effects on cocaine self-administration: preclinical studies. In: Buprenorphine Combatting Drug Abuse With A Unique Opioid. 1995;249-50. |
| Mello et al., "Buprenorphine treatment of opiate and cocaine abuse: Clinical and preclinical studies" Harvard Rev. Psychiatry 1: 168-183 (1993). |
| Melon et al., "Buprenorphine. Haemodynamic study" Anesth. Analg. (Paris) 37(3-4): 121-125 (1980). English abstract. (Original in French). |
| Mendelson et al. Bioavailability of sublingual buprenorphine. J Clin Pharmacol. 1997;37:31-7. |
| Mendelson et al. Buprenorphine and naloxone interactions in methadone maintenance patients. Biol Psychiatry. 1997;41:1095-101. |
| Mendelson et al. Buprenorphine and naloxone interactions in opiate-dependent volunteers. Clin Pharmacol Ther. Jul. 1996;60(1):105-14. |
| Mendelson et al. Buprenorphine attenuates the effects of cocaine on adrenocorticotropin (ACTH) secretion and mood states in man. Neuropsychopharmacology. 1992;7:157-62. |
| Mendelson et al. Human laboratory studies of buprenorphine. NIDA Res Monogr. 1992;121:38-60. |
| Mendelson et al., "Dose proportionality of 4, 8, 16 and 32 mg sublingual buprenorphine solutions" American Society for Clinical Pharmacology and Therapeutics 65(2): 154 Abstract PII-28 (1999). |
| Mendelson et al., Buprenorphine treatment improves brain perfusiion abnormalities in men with concurrent cocaine and heroin dependence: A spect brain imaging analysis. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Mercadante et al., "Alternatives to oral opioids for cancer pain" Oncology 13(2):215-220 (1999). |
| Mercadante, "Opioid rotation in cancer pain: rationale and clinical aspects" Cancer 86:1 1856-66 (1999). |
| Michaels et al., Drug permeation through human skin: Theory and in vitro experimental measurement. AIChE J. 1975;221(5):985-96. |
| Miller, Memorandum to Brogden, Napp Pharmaceuticals Ltd., re Evaluation of clinical data received from Grunenthal buprenorphine TTS, Nov. 11, 1999 ( 5 pp.). |
| Minutes of the oral proceedings before the Opposition Division held Jun. 23, 2009 re: EP-B-964677. |
| Mitaka et al. Comparison of hemodynamic effects of morphine, butorphanol, buprenorphine and pentzocine on ICU patients. Bull Tokyo Med Dent Univ. Jun. 1985;32(2):31-9. |
| Moa et al. Sublingual buprenorphine as postoperative analgesic: a double-blind comparison with pethidine. Acta Anaesthesiol Scand. 1990;34:68-71. |
| Mobily et al., "An epidemiologic analysis of pain in the elderly" J. Aging Health 6: 139-154 (1994). |
| Mok et al. Multidose/observational, comparative clinical analgetic evaluation of buprenorphine, J Clin Pharmacol. Jul. 1981;21(7):323-9. |
| Mongan et al., "Buprenorphine responders" Biol. Psychiatry 28: 1078-1080 (1990). |
| Mongan, Mary Louise Hack The Effects of Low Dose Buprenorphine on Selected Psychiatric Patients San Francisco State University, San Francisco, CA. 1992. 105 ppg. |
| Moore et al. Reversal of postoperative hyperglycaemia by buprenorphine. Lancet. Sep 13, 1980;2(8194):597-8. |
| Moragas et al., "Image analysis of dermal collagen changes during skin aging" Analyt. Quant. Cytol. Histol. 20: 493-499 (1998). |
| Morrison, Psychoactive substance use an related behaviours of 135 regular illicit drug users in Scotland. Drug Alchol Depend. 1989;23:95-101. |
| Mukhtar et al. Cutaneous cytochrome P-450. Drug. Metabolism Revs. 1989;20(204):657-73. |
| Mukhtar et al. Cytochrome P-450 dependent metabolism of testosterone in rat skin. Biochem Biophys Res Commun. Jun. 15, 1987;145(2):749-53. |
| Müller et al., Intra-and postoperative interactions between the 2 opioids fentanyl and buprenorphine. Anaesthesist. Apr. 1986, 35(4):219-25. (in German, w/ English abstract). |
| Müller et al., Intra—and postoperative interactions between the 2 opioids fentanyl and buprenorphine. Anaesthesist. Apr. 1986, 35(4):219-25. (in German, w/ English abstract). |
| Muriel, C. et al., Effectiveness and tolerability of the buprenorphine transdermal system in patients with moderate to severe chronic pain: a multicenter, open-label, uncontrolled, prospective, observational clinical study. Clinical Therapeutics 2005; 27(4): 451-462. (Study sponsored by Grünenthal GmbH). |
| Nagle, CJ et al., Current Anaesthesia and Care 1: 247-252 (1990). |
| Nanovskaya et al., "Transplacental transfer and methabolism of buprenorphine" J. Pharmacol. Exp. Ther. 300(1): 26-33 (2002). |
| Nasar et al. An open study of sub-lingual buprenorphine in the treatment of chronic pain in the elderly. Curr Med Res Opin. 1986;10(4):251-5. |
| Neri, S. et al., Randomized clinical trial to compare the effects of methadone and buprenorphine on the immune system in drug abusers. Psychopharmacology 2005; 179(3): 700-704. |
| New Approaches to Pain Management. Progress in Palliative Care. Meeting Report. Oct. 12-13, 2000; 100-101. |
| Nielsen, S.; Taylor, D.A. The effect of buprenorphine and benzodiazepines on respiration in the rat. Drug and Alchol Dependence 2005;79:95-101. |
| Nigam et al. Buprenorphine in opiate withdrawal: a comparison with clonidine. J Subst Abuse Treat. Jul.-Aug. 1993;10(4):391-4. |
| Nizamie et al. Buprenorphine abuse: a case report. Indian J Psychiatry. 1990;32:198-200. |
| Noble et al., "Protection of endogenous enkephalin catabolism as natural approach to novel analgesic and antidepressant drugs" Expert Opinion on Therapeutic Targets 11(2): 145-159 (Nov. 2007). |
| Nolan et al., "Anaesthesia and pain management in cerebral palsy" Anaesthesia 55(1): 32-41 (2000). |
| Non-Clinical Chronology for BTDS NDA from May 2, 1996-Apr. 2000. |
| Noveck et al., Pharmacokinetics of buprenorphine transdermal system (BTDS 10) employing the LPS pyrogen model. Clinical Pharmacology & Therapeutics 69(2):3 200 2001 Abstract PI-7 (w/ PowerPoint presentation). |
| Noveck, R. et al., Lack of effect of inhibitor ketoconazole on transdermally administered buprenorphine. Presented at the 106th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, Orlando, FL., Mar. 2-5, 2005. (Study sponsored by Purdue Pharma L.P.). |
| Novosis AG Opposition against EP-B-964677, dated May 15, 2007. (in German, w/ Engl. translation). |
| Nyhuis et al., "Opiate receptors in ECT-resistant depression" European Neuropsychopharmacology 15(3): S420 (2005). |
| Obel et al. Buprenorphine-supplemented anaesthia. Influence of dose on duration of analgesia after cholecystectomy. Br J Anaesth. Mar. 1985;57(3):271-4. |
| O'Brien. Drug adiction and abuse. In: Goodman & Gilman's The Pharmacological Basis of Therapeutics. 9th ed. 1996;Chp. 24:557-577. |
| O'Connor et al. Buprenorphine abuse among opiate addicts. Brit J Addict. 1998; 83:1085-7. |
| O'Connor et al. Rapid and ultrarapid detoxification techniques. JAMA 1998;279:229-34. |
| O'Connor et al. Three methods of opioid detoxification in a primary care setting: a randomized trial. Ann Intern Med. 1997;127:526-30. |
| O'Connor et al., A pilot study of primary care-based buprenorphine maintenance. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Oda et al., "Fentanyl inhibits metabolism of midazolam: competitive inhibition of CYP3A4 in vivtro " Br. J. Anaesthesia 82(6): 900-903 (1999). |
| Office Actins issued in the prosecution of U.S. Appl. No. 11/033,108 to Reder et al., filed Jan. 11, 2005. |
| Office Actions dated Dec. 15, 2008, and Feb. 18, 2009 and Apr. 6, 2009, issued in the prosecution of U.S. Appl. No. 10/476,601 to Tavares et al. filed Nov. 20, 2003. |
| Office Actions dated Sep. 19, 2008 and Feb. 9, 2009, issued in the prosecution of U.S. Appl. No. 10/394,425 to Kaiko et al. filed Mar. 20, 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 09/08/939,068 to Reder et al., filed Sep. 29, 1997. |
| Office Actions issued in the prosecution of U.S. Appl. No. 09/311,997 to Reder et al., filed Jun. 14, 1999. |
| Office Actions issued in the prosecution of U.S. Appl. No. 09/756,419 to Reder et al., filed Jan. 18, 2001. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/033,056 to Reder et al., filed Dec. 27, 2001. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/394,425 to Kaiko et al., filed Mar. 27, 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/402,288 to Reder et al., filed Mar. 28, 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/476,601 to Tavares et al., filed Nov. 20, 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/736,043 to Reidenberg et al., filed Dec. 15, 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 10/736,049 toReder et al., filed Dec. 15. 2003. |
| Office Actions issued in the prosecution of U.S. Appl. No. 11/033,106 to Reder et al., filed Jan. 11, 2005. |
| Office Actions issued in the prosecution of U.S. Appl. No. 11/033,107 to Reder et al., filed Jan. 11, 2005. |
| Ohtani et al. Kinetics of respiratory depression in rats induced by buprenorphine and its metabolite, norbuprenorphine, J Pharmacol Exp Ther. Apr. 1997;281(1):428-33. |
| O'Keeffe, of Reckitt Benckiser Pharmaceuticals Inc. letter to DEA Administrator dated Apr. 18, 2002 in response to proposed rule: Rescheduling of buprenorphine from Schedule V to Schedule III, published in the Federal Register on Mar. 21, 2002. |
| Oliveto et al., "Desipramine, amantadine, or fluoxetine in buprenorphine-maintained cocaine users" Journal of Substance Abuse Treatment 12(6): 423-428 (1995). |
| Olley et al. Plasma levels of opioid material in man following sublingual and intravenous administration of buprenorphine: exogenous/endogenous opioid interaction? J Pharm Pharmacol. 1988;40:667-7. |
| O'Neill. The cognitive and psychomotor effects of opioid drugs in cancer management. Cancer Surv. 1994;21:67-84. |
| Opana® ER( (Oxymorphone Hydrochloride) Extended Release Tablets)) Package Insert. |
| Opioid agonist-antagonist analgesics. In: WHO Expert Committee on Drug Dependence. 25th Report. Geneva: World Health Organization Technical Report Series; 1989;775 16-24. |
| Orwin et al. A double blind comparison of buprenorphine and morphine in conscious subjects following administration by the intramuscular route. Acta Anaesthesiol Belg. 1976;27:171-81. |
| Orwin. Pharmacological aspects in man. Pain:vNew Perspect. Meas. Manage. (Symp) 1977;141-59. |
| Ouellete. Bupreniorphine and morphine efficacy in postoperative pain: a double-blind multiple-dose study. J Clin Pharmacol. Apr. 1982;22(4):165-72. |
| Ouellete. Comparison of analgesic activity of buprenorphine hydrochloride and morphine in patients with moderate to severe pain postoperatively. Surg Gynecol Obstet. Sep. 1984;159(3):201-6. |
| Ouellette et al. Comparison of buprenorphine and morphine: a multicenter, multidose study in patients with severe postoperative pain. Contemp. Surg. 1986; 28:55,57-59,62-64. |
| Overweg-Van Kintz et al. [Failing pain suppression during sublingual use of buprenorphine] Ned Tijdschr Geneeskd. 1987;131(44)1973-4. [in Dutch w/ English transl]. |
| Paetzold et al., "Buprenorphine: therapeutical use in opioid-dependence, depression and schizophrenia" Nervenheilkunde 19(3): 143-150 (2000). (w/ English abstract. |
| Pani et al., "Buprenorphine: A controlled clinical trial in the treatment of opioid dependence" Drug and Alcohol Dependence 60:39-50 (2000). |
| Parmelee, P.A., "Assessment of pain in the elderly" Annual Review of Gerontology and Geriatrics 14: 281-301 (1994). |
| Parran et al. A bupreniorphine stabilization and rapid-taper protocol for the detoxification of opioid dependent patients. Am J Addict. 1994;3:306-13. |
| Pathre et al. Generalised seizure following sublingual buprenorphine. J Assoc Physicans India. Apr. 1994;42(4):327-8. |
| Patterson S, Agin M, Anziano R, et al., Investigating drug-induced QT and QTc prolongation in the clinic: a review of statistical design and analysis considerations. Report from the pharmaceutical research and manufacturers of America QT statistics expert team. Drug Information Journal 2003:39:243-265. |
| Pausawasdi et al. comparison of buprenorphine and morphine for immediate postoperative pain relief in Thai patients. J Med Assoc Thai. May 1984;67(5):284-9. |
| Payne et al., Guidelines for the clinical use of transdermal fentanyl. Anticancer Drugs. Apr. 1995, 6 Suppl 3:50-3. |
| Payne et al., The role of transdermal fentanyl in the management of cancer pain in Opioids in Anesthesia II (Estafanous. ed.), 1991(18):215-22. |
| Pechnick et al. The effects of the acute administration of buprenorphine hydrochloride on the release of anterior pituitary hormones in the rat: evidence for the involvement of multiple opiate receptors. Life Sci. Nov. 18, 1985;37(20): 1861-8. |
| Pedersen et al. Peroperative buprenorphine: do high dosages shorten analgesia postoperatively? Acta Anaesthesiol Scand. Nov. 1986;30(8):660-3. |
| Peng et al., A review of the use of fentanyl analgesia in the management of acute pain in adults. Anaesthesiology 1999, 90:576-99. |
| Pereira et al., "Analgesic effects of diclofenac suppository and injection after postoperative administration" Int. J. Clin. Pharm. Res. 19(2): 47-51 (1999). |
| Perttunen et al., "Chronic pain after thoracic surgery: a follow-up study" Acta Anaesthesiol. Scand. 43: 563-567 (1999). |
| Petti, Postoperative pain relief with pentazocine and acetaminophen: comparison with other analgesic combinations and placebo. Clin Ther. 1985;8(1):126-33. |
| Pharmaceuticals: Restrictions in Use and Availability, Mar. 2001 (up to p. 5 re Buprenorphine). |
| Pharmacolinetic evaluation of transdermal burenorphine in man, I.R. Wilding, et al., International Journal of Pharmaceutics 132 (1996) 81-87. |
| Philipz, J. et al., Pharmacokinetics of transdermal buprenorphine (Transtec®) in patients with renal insufficiency. Presented at the 9th Congress of the European Association for Palliative Care, Aachen, Germany, Apr. 8-10, 2005. (Study sponsored by Grünenthal GmbH). |
| Physician's Desk Reference 49th ed., 1995, entry on Duragesic®. |
| Physician's Desk Reference 50th ed., 1996, entry on Duragesic®. |
| Pickar et al., "Behavioral and biological effects of acute beta-endorphin injection in schizophrenic and depressed patients" Am. J. Psychiatry 138: 160-166 (1981). |
| Pilot study on the dermal tolerability and adhesion of a buprenorphine path and the absorption of the active substance over an application period of 72 hours in 6 healthy volunteers, Final Report, 1992. |
| Pirnay S, et al., A critical review of the causes of death among post-mortem toxicological investigations: analysis of 34 buprenorphine-associated and 35 methadone-associated deaths. Addiction 99:978-88, 2004. |
| Ponsoda et al. The effects of buprenorphine on the metabolism of human hepatocytes. Toxic. In vitro. 1991;5(3):219-24. |
| Pontani et al. Disposition in the rat of buprenorphine administered parenterally and as a subcultaneous implant. Xenobiotica. Apr. 1985; 15(4):287-97. |
| Portenoy et al. Acute and chronic pain in Substance Abuse. A Comprehensive Textbook (Lowinson et al., eds.), 2nd ed. 2005. 52:691-721. |
| Portenoy et al., Transdermal fentanyl for cancer pain. Repeated dose pharmacokinetics. Anesthesiology. Jan. 1993;78(1):36-43. |
| Potter, Fundamentals of Nursing, Mosby, 4th ed., 1997 p. 633. |
| Preston et al. Abuse liability studies of opioid agonist-antagonists in humans. Drug Alcohol Depend. 1991;28:49-82. |
| Preston et al. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. Jan. 1991;27(1):7-17. |
| Preston et al. Diazepam and methadone interactions in methadone maintenance. Clin Pharmacol Ther. Oct. 1984;36(4):534-41. |
| Preston et al. Discrimination of agonist-antagonist opioids in humans trained on two-choice saline-hydromorphone discrimination. J Pharmacol Exp Ther. 1992;261:62-71. |
| Preston et al. Drug discrimination assessment of agonist-antagonist opioids in humans: a three-choice saline-hydromorphone-butorphanol procedure. J Pharmacol Exp Ther. 1994;271:48-60. |
| Preston et al. Drug discrimination in human postaddicts: agonist-antagonist opioids. J Pharmacol Exp Ther. 1989;250:184-96. |
| Preston et al. Effects of sublingually given naloxone in opioid-dependent human volunteers. Drug Alchol Depend. 1990;25:27-34. |
| Preston et al., Abuse liability evaluation of buprenorphine in buprenorphine-treated patients. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Price et al. A psychophysical analysis of experimental factors that selectively influence the affective dimension of pain. Pain. Apr. 1980;8(2):137-49. |
| Product Monograph for Duragesic® (2008). |
| Quang-Cantagrel N.D., Wallace M.S., Magnuson S.K. Opioid Substitution to Improve the Effectiveness of Chronic Noncancer Pain Control: A Chart Review: Anest Anag 2000; 90:933-937. |
| Quigley et al. A case of buprenorphine abuse. Med J Aust. 1984; 140:425-6. |
| Rainey. Abuse of buprenorphine. N Z Med J. 1986;99-72. |
| Raja SN, Haythornthwaite, JA, Pappagallo M, Clark MR, Travison TG, Sabeen S, Royall RM, and Max MB. Opioids versus antidepressants in postherpetic neuralgia: A randomized, placebo-controlled trial. Neurology 59: 1015-1021, 2002. |
| Razzetti AJ, Carr W, Landau CJ, Munera C, Ripa SR, Sessler N. Effectiveness of 7-day buprenorphine transdermal system in the management of chronic nonmalignant pain syndromes. J Pain 2005; 6(3) Suppl I:542. (Study BUP3201). |
| Reddy L, Kranjnik M, Zylicz Z. Transdermal buprenorphine may be effective in the treatment of pruritus in primary biliary cirrhosis. Journal of Pain and Symptom Management 2007; 34(5):455-456. |
| Regier et al., "Comorbidity of mental disorders with alcohol and other drug abuse. Results from Epidemiological Catchment Area (ECA) Study" JAMA 264(19):2549-2550 (990) (Abstract only). |
| Regini et al. [Buprenorphine withdrawal syndrome in a neonate. Which treatment?] Ped Med Chir. 1998; 20:67-9. [in Italian with English transl]. |
| Reidenberg et al., Absolute bioavailability of a novel buprenorphine transdermal system (BTDS) applied for 7 days. J. Clin Pharmacol. 2001 41(9):1014-1033, Abstract 55, p. 1026 (w/ PowerPoint presentation presented in the ACCP Meeting, Sep. 2001). |
| Reidenberg et al., Daily pharmacokinetic performance of a buprenorphine transdermal system (BTDS) for up to 7 days. J. Clin Pharmacol. 2001 41(9):1014-1033, Abstract 57, p. 1027 (w/ PowerPoint presentation). |
| Reidenberg et al., Pharmacokinetics and safety of buprenorphine transdermal system (BTDS) for 7-Day application comparing healthy elderly and young adult subjects. Am Pain Soc, 19th Ann. Sci. Meet., Nov. 2-5, 2000 Abstract 776 (w/ PowerPoint presentation). |
| Reidenberg et al., Physiologic effects of buprenorphine transdermal system (BTDS) dose escalation in the young, healthy elderly and elderly hypertensive subjects, FASEB J. 154), Mar. 2001 Abstract 457.3 (w/ PowerPoint presentation presented Apr. 2001). |
| Reisine et al. Opioid analgesics and antagonists. In: Molinoff PB, Ruddon RW, Gilman AG, editors. Goodman and Gilman's The pharmacological basis of therapeutics. 9th ed. 1996; Chp. 23:521-55. |
| Reisinger, [Value of comparing buprenorphine with methadone] Ann Med Interne (Paris), Nov. 1994; 145 Suppl 3:23-5. [in French w/ English transl]. |
| Reisinger. Buprenorphine as a new treatment for heroin dependence. Drug Alcohol Depend. Dec. 1985;16(3):257-62. |
| Reisinger. Results from experience with buprenorphine replacement in outpatients in Belgium. Ann. Med. Interne. 1994;145(Supp.3):46-47. |
| Reisinger. Treatment of four pregnant heroin addicts with buprenorphine: history and outcome. NIDA Res Monogr 1995;162-261. |
| Report of the commission on the evaluation of pain. In: Soc. Security Bull. 1986. |
| Report of the International Narcotics Control Board for 2001, United Nations Publication. |
| Response to the Critical Review (undated). |
| Reynaud et al. Six deaths linked to concomitant use of buprenorphine and benzodiazepines. Addiction 1998;93:1385-92. |
| Richard et al. [Vertiginous syndrome: side effect of buprenorphine?] Cah Anesthesiol. 1988; 36:641-2. [in French with English transl]. |
| Richert et al. [Drug dependence on buprenorphine] MMW Munch Wochenschr 1983; 125:1195-8 [. in German w/ English transl]. |
| Rigas et al., "Transdermal fentanyl: Practical use in the hospital and the home" ASHP Midyear Clinical Meeting v.27 (1992). |
| Rigas et al., Transdermal fentanyl: Practical use in the hospital and the home (undated). |
| Risbo et al. Sublingual buprenorphine for premedication and postoperative pain relief in orthopaedic surgery. Acta Anaesthesiol Scand. Feb. 1985;29(2):180-2. |
| Robbie. A trial of sublingual bupreniorphine in cancer pain. Br J Clin Pharmacol. 1979;7 Suppl 3:315S-317S. |
| Robertson et al. Buprenorphine: dangerous drug or overlooked therapy? Br Med J. 1986;292:1465. |
| Robinson et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33:81-6. |
| Rolandi et al. Changes in pituitary secretion induced by an agonist-antagonist opioid drug, buprenorphine. Acta Endocrinol (Copenh). Nov. 1983;104(3):257-60. |
| Romero et al., "Opioid peptide receptor studies. 12. Buprenorphine is a potent and selective μ/κ antagonist in the [35S]-GTP-γ-S functional binding assay" Synapse 34: 83-94 (1999). |
| Rooke et al., "Maximal skin blood flow is decreased in elderly men" J. Appl. Physiol. 77: 11-14 (1994). |
| Rosen et al. Buprenorphine: duration of blockade of effects of intramuscular hydromorphone. Drug Alcohol Depend. 1994;35:141-9. |
| Rosen et al. Effects of acute bupreniorphine on responses to intranasal cocaine: a pilot study. Am J Drug Alcohol Abuse. 1993;19:451-64. |
| Rosenfeldt et al. Haemodynamic effects of buprenorphine after heart surgery. Br Med J. Dec. 9, 1978;2(6152):1602-3. |
| Rothman et al. Buprenorphine: a review of the binding literature. In: Buprenorphine: Combatting Drug Abuse with a Unique Opioid. 1995;19-29. |
| Rothman et al., "An open-label study of a functional opioid κ antagonist in the treatment of opioid dependence" Journal of Substance Abuse Treatment 18: 277-281 (2000). |
| Roy et al., "Controlled transdermal delivery of fentanyl: characterizations of pressure-sensitive adhesives for matrix patch design, " J Pharm Sci. May 1996, 85(5):491-5. |
| Roy et al., "Controlled transdermal delivery of fentanyl: characterizations of pressure-sensitive adhesives for matrix patch design," J Pharm Sci. May 1996, 85(5):491-5. |
| Roy et al., Transdermal delivery of buprenorphine through cadaver skin. J Pharma Sci 1994, 83(2):126-30. |
| Roy, S.D. et al., Transdermal delivery of buprenorphine through cadaver skin J. Pharm. Sci. 83(2): 126-30 (1994). |
| Roy, SD et al., J. Pharmaceutical Sciences 83: 1723-1728 (1994). |
| Rozenbaum H, Birkhauser M, DeNooyer C, et al., Comparison of two estradiol transdermal systems (Oesclim® 50 and Estraderm TTS® 50). II. Local skin tolerability. Maturitas. 1996; 25:175-185. |
| Russo and Brose, Ann. Rev. Med. 49: 123-133 (1998). |
| Saarialho-Kere et al. Psychomotor, respiratory, and neuroendocrinological effects of buprenorphine and amitriptyline in healthy volunteers. Eur J Clin Pharmacol. 1987;33:139-46. |
| Sadee et al., Buprenorphine: Differential interaction with opiate receptor subtypes in vivo. The Journal of Pharmacology and Experimental Therapeutics, Copyright © 1982, 223(1):157-62. |
| Sadée, W. et al., "Buprenorphine: Differential interaction with opiate receptor subtypes in vivo" J. Pharm. Exp. Ther. 223(1) 1982. |
| Sakol et al. Buprenorphine and temazepam abuse by drug takers in Glasgow—an increase. Brit J Addict. 1989;84:439-41. |
| San et al. [Prevalence of buprenorphine consumption in heroin addicts undergoing treatment] Med Clin (Barc). 1989; 93:645-8. [In Spanish w/ English transl]. |
| San et al. Assessment and management of opioid withdrawal symptoms in buprenorphine-dependent subjects. Br J Addict. Jan. 1992;871):55-62. |
| San et al. Consumption of buprenorphine and other drugs among heroin addicts under ambulatory treatment: results from cross-sectional studies in 1988 and 1990. Addiction. 1993; 88:1341-9. |
| San Molina et al. [Addiction to buprenorphine] Rev Clin Esp. 1987; 181:288-9. [in Spanish w/ English transl]. |
| Sandler et al., A double-blind, placebo-controlled trial of transdermal fentanyl after abdominal hysterectomy. Analgesic, respiratory, and pharmacokinetic effects. Anesthesiology Nov. 1994, 81(5):1169-80; discussion 26A. |
| Schmid-Grendelmeier P. A comparison of the skin irritation potential of transdermal fentanyl versus transdermal buprenorphnie in middle-aged to elderly healthy volunteers. Current Medical Research and Opinion 2006;22(3)501-9. |
| Schmidt et al. Postoperative pain relief with naloxone. Severe respiratory depression and pain after high dose buprenorphine. Anaesthesia. 1985;40:583-6. |
| Schnoll SH, Smith MY, Colucci RD, Munoz A. Development of a denominator for calculating rates of opioid abuse. CPDD Annual Meeting Abstracts. 2004. (Study BUP3018 and non-BTDS studies). |
| Schott, H., Colloidal Dispersions, Remington's Pharmaceutical Sciences, 16th ed., ppg. 266-293 (1980). |
| Schottenfeld et al. Buprenorphine vs. methadone maintenance treatment for concurrent opioid dependence and cocaine abuse. Arch Gen Psychiatry. 1997;54:713-20. |
| Schriek P. Treatment of cancer-related pain with transdermal buprenorphine: a report of three cases. European Support Care Cancer 2004; 12:882-884. |
| Schuh et al. Buprenorphine, morphine, and naloxone effects during ascending morphine maintenance in humans. J Pharmacol Exp Ther. 1996; 278:836-46. |
| Schuh, A comparison of buprenorphine's and naltrexone'opioid blockade abilities. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Sear et al. Buprenorphine for postoperative analgesia. Br J Anaesth. Jan. 1979;51(1):71. |
| Seet RCS, Lim ECH. Intravenous use of buprenorphine tablets associated with rhabdomyolysis and compressive sciatic neuropathy. Annals of Emergency Medicine 2006; 47(4):396-397. |
| Seet RCS, Rathakrishnan R, Chan BP, Lim ECH. Diffuse-cystic leucoencephalopathy after buprenorphine injection. Journal of Neurology, Neurosurgery and Psychiatry 2005; 76(6):890-891. |
| Segal et al. Buprenorphine: what interests the national institute on drug abuse? Buprenorphine: In: Combatting Drug Abuse with a Unique Opioid. 1995;309-20. |
| Segal et al., A double blind, multicenter clinical trial comparing four doses of buprenorphine. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Segui et al. [Buprenorphine consumption, an indicator of poor prognosis in the course of drug dependencies] Actas Luso Esp Neurol Psiquiatr Ciene Afines. Jan.-Feb. 1992;20(1):17-22. [in Spanish w/ English transl]. |
| Segui et al. [Data regarding buprenorphine consumption by drug-addicted individuals] Rev Clin Esp. 1989; 185-271-2. [in Spanish with English transl]. |
| Segui et al. [Prevalence of buprenorphine consumption in a sample of outpatient drug abusers] Rev Clin Esp. Jun. 1991;189(1):14-7. [in Spanish w/ English transl]. |
| Segui et al. [Subgroups of addicted buprenorphine-consuming patients.] An Med Interna. Jan. 1991;8(1):18-22. [in Spanish w/ English transl]. |
| Seidenari et al., "Echographic evaluation with image analysis of normal skin: variation according to age and sex" Skin Pharmacol. 7:201-209 (1994). |
| Seifert et al., "Detoxification of opiate addicts with multiple drug abuse: a comparison of buprenorphine vs. methadone" Pharmacopsychiatry 35: 159-164 (2000). |
| Seifert et al., "Mood and affect during detoxification of opiate addicts: A comparison of buprenorphine versus methadone" Addiction Biology, 10: 157-164 (Jun. 2005). |
| Sekar et al. Buprenorphine benzodiazepines and prolonged respiratory depression. Anaesthia. 1987;42:567-8. |
| Seow et al. Buprenorphine: a new maintenance opiate? Med J Aust. Apr. 14. 1986;144(8):407-11. |
| Sganzerla et al. Analgesic and hemodynamic effects of buprenorphine in acute infarction of the heart. Jpn Heart J. Jan. 1987;28(1):63-71. |
| Shah, Sr. Research Scientist, Center for Drug Evaluation and Research, Food and Drug Administration, Rockville, MD, In vitro release of special/novel dosage forms: What is its value? (undated). |
| Shannon et al. Morphine-like discriminative stimulus effects of buprenorphine and demethoxybuprenorphine in rats: quantitive antagonism by naloxone. J Pharmacol Exp Ther. 1984; 229:768-74. |
| Shannon MJ, Kivitz A, Landau CJ, Sessler NE, Xia Y, Ripa SR. Buprenorphine transdermal system in chronic pain due to osteoarthritis. Arch Phys Med Rehabil2005; 86(9):e32. (Study BUP3012). |
| Shapira et al., "Treatment of refractory major depression with tramadol monotherapy" Am. J. Psychiatry 62(3):205-206 (2001). |
| Sheikh et al., "Geriatric Depression Scale: Recent evidence and development of a shorter version" Clin. Gerontol. 5: 165-173 (1986). |
| Shuster. Fluoroquinolones and tendon rupture or tendinitis. Buprenorphine-induced hypertension and tachycardia: rare but serious. Hospital Pharmacy. 1996;31(1):41-2. |
| Singh et al. Cases of buprenorphine abuse in India. Acta Psychiatr Scand. 1992;86:46-8. |
| Singh et al., Cases of buprenorphine abuse in India. Acta Psychiatr Scand. Jul. 1992;86(1):46-8. |
| Singh J, Grover S, Basu D. Very high-dose intravenous buprenorphine dependence: A case report. German J Psychiatry 2004; 7:58-59. |
| Sittl, R. et al., Equipotent doses of transdermal fentanyl and transdermal buprenorphine in patients with cancer and noncancer pain: Results of a retrospective cohort study. Clinical Therapeutics 2005; 27(2):225-237. (Study sponsored by Grünenthal GmbH). |
| Sjovall.Use of midazolam and buprenorphine in combination anaesthia. Ann Clin Res. Aug. 1983;15(4):151-5. |
| Slides on Buprenorphine (21 pp.) (undated). |
| Smith, Grunenthal buprenorphine transdermal system. Review of pharmacokinetic studies, 11/10/199 ( 7 pp.). |
| Sorrell, DC Nursing 24:30 (1994). |
| Southam, MA Anti-Cancer Drugs 6(suppl. 3): 29-34 (1995). |
| Soyka M, Penning R, Wittchen U. Fatal poisoning in methadone and buprenorphine treated patients—are there differences? Pharmacopsychiatry 2006; 39:85-87. |
| Soyka, M. et al., Less impairment on one portion of a driving-relevant psychomotor battery in buprenorphine-maintained that in methadone-maintained patients: results of a randomized clinical trial. Journal of Clinical Psychopharmacology 2005; 25(5):490-493. |
| Spitzer et al. Scientific approach to the assessment and management of activity-related sphinal disorders. Spine. 1987;12(7):S9-S59. |
| Sporer KA, Buprenorphine: A Primer for Emergency Physicians. Annals of Emergency Medicine 43(5):580-4, 2004. |
| Spyker DA, Hale ME, Lederman My Creanga DL, Coles C, Reder RF, Long-term use of buprenorphine transdermal system (BTDS) in patients with chronic pain. JAm Geriatr Soc 2002; 50(4 Suppl):S66. Abstract P162. (Study BP96-0103). |
| Spyker DA, Hale ME, Munera CL, Wright C. Treatment of patients with chronic low back pain with buprenorphine transdermal system (BTDS) compared with hydrocodone/acetaminophen. Pain Mgmt 2001: PF2001-85. (Study BP98-1201). |
| Spyker et al., Analgesic efficacy and safety of buprenorphine transdermal system (BTDS) in patients with osteoarthritis. The Journal of Pain 3(2, Supp. 1):12 Abstract 645 (w/ PowerPoint presentation). |
| Spyker et al., Clin. Pharmacol. Ther. 67(2): 145, Abstract PII-12. |
| Spyker et al., Effect Size (ES) Meta-analysis Approach to Noinferiority Clinical Trials. Clinical Pharmacology & Therapeutics 69(2):33 Abstract PII-3 (w/ PowerPoint presentation presented Mar. 5, 2001). |
| Spyker et al., Effectiveness and safety of buprenorphine transdermal system (BTDS) compared with hydrocodone/acetaminophen in the treatment of patients with chronic low back pain. J. Pain 2002 3(Suppl.1):14 Abstract 653 (w/ PowerPoint presentation). |
| Spyker et al., Effectiveness of buprenorphine transdermal system (BTDS) compared with oxycodone/acetaminophen and placebo in the treatment of patients with chronic back pain. Morial Convention Center, Oct. 15, 2001 Abstract (w/PowerPoint presentation). |
| Spyker et al., Transdermal buprenorphine system (BTDS) in patient-controlled Analgesia (PCA). PowerPoint presentation presented Mar. 2000. |
| Stanway, GW, "A preliminary investigation comparing pre-operative morphine and buprenorphine for postoperative analgesia and sedation in cats" Veterinary Anaesthesia and Analgesia 29:29-35 (2002). |
| Staritz. Pharmacology of the spincter of Oddi. Endoscopy. Aug. 1988;20 Suppl 1:171-4. |
| Steinberg et al., Acute toxic delirium in a patient using transdermal fentanyl. Anesth Analg. Dec. 1992;75(6):1014-6. |
| Steiner, D et al., "The efficacy and safety of buprenorphine transdermal system (BTDS) in subjects with moderate to severe low back pain" Presentation #305, PowerPoint Presentation at American Pain Society Annual Meeting, May 6-9, 2009, San Diego, CA. |
| Stellato et al. Human basophil/mast cell releasability. IX. Heterogeneity of the effects of opioids on mediator release. Anesthesiology. Nov. 1992;77(5):932-40. |
| Stewart et al., eds., "Medical Outcomes Study Pain Evaluation" in: Measuring Functioning and Well-Being—The Medical Outcomes Study Approach, Durham and London: Duke University Press, 1992. |
| Stewart. Effect of scheduling of buprenorphine (Temgesic) on drug abuse patterns in Glasgow. BMJ. Apr. 20, 1991;302(6782):969. |
| Stinchcomb et al. Permeation of buprenorphine and its 3-alkyl-ester prodrugs through human skin. Pharm Res. Oct. 1996;13(10):1519-23. |
| Stinchcomb et al., "A solubility and related physicochemical property of buprenorphine and its 3-alkyl esters" Pharmaceutical Research 12(10): 1526-1529 (1995). |
| Stinchcomb et al., Permeation of buprenorphine and its 3-alkyl-ester prodrugs through human skin. Pharm Res. Oct. 1996;13(10):1519-23. |
| Strain et al. Acute effects of buprenorphine, hydromorphone, and naloxone in methadone-maintained volunteers. J Pharmacol Exp Ther. 1992;261:985-93. |
| Strain et al. Buprenorphine effects in methadone-maintained volunteers: effects at two hours after methadone. J Pharmacol Exp Ther. 1995;272:628-38. |
| Strain et al. Comparison of buprenorphine and methadone in the treatment of opioid dependence. Am J Psychiatry. 1994;151:1025-30. |
| Strain et al. The effects of buprenorphine in buprenorphine-maintained volunteers. Psychopharmacol. 1997;129:329-38. |
| Strang. Abuse of buprenorphine (Temgesic) by snorting. BMJ. Apr. 20, 1991;302(6782):969. |
| Strang. Abuse of buprenorphine. Lancet. Sep. 28, 1985;2(8457):725. |
| Streisand. Transdermal-mucosal sedative and analgesic delivery. West. J Med. 1990;153:310. |
| Su. Further demonstration of κ opioid binding sites in the brain: evidence of heterogeneity. J Pharmacol Exp Ther. 1985;232:144-8. |
| Subramaniam et al., "Baseline depressive symptoms predict poor substance use outcome following adolescent residential treatment" Journal of the American Academy of Child& Adolescent Psychiatry 46(8): 1062-1069 (2007) (Abstract only). |
| Summerfield et al. Buprenorphine in end stage renal failure. Anaesthesia. Sep. 1985;40(9):914. |
| Summons to attend oral proceedings pursuant to Rule 115(1) EPC re EP-B-964677 opposition proceedings dated Dec. 29, 2008. |
| Supplementary partial European Search Report for EP Application No. 03721427, dated Mar. 31, 2006. |
| Swain et al. Primary addiction study. UM952. In: Minutes of the Committee on Problems of Drug dependence, Washington (DC), National Academy of Sciences, National Research Council. 1975;791. |
| Swendsen et al., "The comorbidity of depression and substance use disorders" Clin. Psychol. Rev. 20(2): 173-189 (2000) (Abstract only). |
| Tallarida et al. Theory and statistics of detecting synergism between two active drugs: cocaine and buprenorphine. Psychoparmacology (Berl). Oct. 1997;133(4):378-82. |
| Tanaka et al., "Preoperative fluribiprofen provides pain relief after laparascopic cholecystectomy" Masui 46(5): 679-683 (1997). Abstract). |
| Tantucci et al. Acute respiratory effects of sublingual buprenorphine: comparison with intramuscular morphine. Int J Clin Pharmacol Ther Toxicol. Jun. 1992;30(6):202-7. |
| Tauzin-Fin et al., "Effect of balanced analgesia with buprenorphine on pain response and general anesthesia requirement during lithotripsy procedures" European Journal of Anaesthesiology 15: 147-152 (1998). |
| Tebbett. Analysis of bupreniorphine by high-performance liquid chromatography. J Chromatogr. Nov. 22, 1985;347(3):411-3. |
| Teoh et al. Acute interactions of buprenorphine with intravenous cocaine and morphine: an investigational new drug phase I safety evaluation. Clin Psychopharmacol. 1993;13:87-99. |
| Teoh et al. Buprenorphine effects on morphine—and cocaine-induced subjective responses by drug-dependent men. J Clin Psychopharmacol. Feb. 1994;14(1):15-27. |
| Thammakumpee et al. Noncardiogenic pulmonary edema induced by sublingual buprenorphine. Chest. Jul. 1994;106(1):306-8. |
| Tharp et al. Functional heterogeneity of human mast cells from different anatomic sites: in vitro responses to morphine sulfate. J Allergy Clin Immunol. Apr. 1987;79(4):646-53. |
| The Safety of High Doses of Buprenorphine. Table of Contents and Introduction (undated). |
| The Safety of High Doses of Burprenorphine. Tables of Contents and Introduction. Feb. 22, 2002. |
| Thompson et al., "Perioperative pharmacokinetics of transdermal fentanyl in elderly and young adult patients" Br. J. Anaesth. 81: 152-154 (1998). |
| Thorn et al. Prolonged respiratory depression caused by sublingual buprenorphine. Lancet. Jan. 23, 1988;1(8578):179-80. |
| Tigerstedt et al. Double-blind, multiple-dose comparison of buprenorphine and morphine in postoperative pain. Acta Anaesthesiol Scand. Dec. 1980;24(6):462-8. |
| Torrens et al., "Buprenorphine versus heroin dependence: Comparison of toxicologic and psychopathologic characteristics" Am. J. Psychiatry 150(5): 822-824 (1993). |
| Touzeau et al. Benzodiazepines and methadone: a dangerous combination? Ann Med Interne (Paris). Nov. 1994;145 Suppl 3:19-22. [in French w/ English transl]. |
| Tracqui et al. [Prison, drugs and death: two deaths due to overdoses in a prison environment] J Med Leg Droit Med. 1998a;41:185-92. [in French w/ English transl]. |
| Tracqui et al. Buprenorphine-related deaths among drug addicts in France: a report on 20 fatalities. J Anal Toxicol. 1998c;22:430-4. |
| Transdermal Delivery of Buprenorphine through Cadaver Skin, Samir D. Roy, et al., Journal of Pharmaceutical Sciences, vol. 83, No. 2, Feb. 1994, pp. 126-130. |
| Transtec Summary of Product Characteristics, Oct. 2003. |
| Turk DC, Melzack R. The measurement of pain and the assessment of people experiencing pain. In: Turk DC, Melzack R, eds. Handbook of Pain Assessment. New York, NY: The Guilford Press; 1992:3-12. |
| Uehlinger et al. Comparison of buprenorphine and methadone in the treatment of opioid dependence. Swiss multicentre study. Eur Addict Res. 1998;4 Suppl 1:13-8. |
| Umbricht et al. Safety of buprenorphine: ceilling for cardio-respiratory effects at high IV doses. NIDA Res Monogr. 1998;179:225. |
| Using buprenorphine for office-based treatment of opiate addiction.Reccommendations to the CSAT of the SAMHSA, from CSAT's National Advisory Council, approved by the CSAT's National Advisory Council on Sep. 15, 1999 (28 pp.). |
| Van Buskirk et al., Scale-up of adhesive transdermal drug delivery systems. Pharmaceutical Research 1997, 14(7). |
| Van Buskirk, G.A. et al., "Scale-up of adhesive transdermal drug delivery systems" Pharmaceutical Research 14(7) (1997). |
| Van Loveren et al. Assessment of immunotoxicity of buprenorphine. Lab Anim. Oct. 1994;28(4):355-63. |
| Vanakoski et al. Exposure to high ambient temperature increases absorption and plasma concentrations of transdermal nicotine. Clin Pharmacol Ther. Sep. 1996;60(3):308-15. |
| Varey. The safety of buprenorphine (Temgesic). N Z Med J. Jan. 24, 1990;103(882):24. |
| Varga et al., "The effect of codeine on involutional and senile depression" Ann. NY Acad. Sci. 398: 103-105 (1982). |
| Vargas et al. Buprenorphine: a case of abuse [letter] [in Spanish]. An Med Interna. 1987;4:366. |
| Ventafridda et al. Chronic analgesic study on buprenorphine action in cancer pain. Comnparison with pentazocine. Arzneimittelforschung. 1983;33(4):587-90. |
| Verhaeverbeke et al., "Drug-induced orthostatic hypotension in the elderly: avoiding its onser" Drug Safety 17: 108-118 (1997). |
| Vibbert et al., eds., "Modified Brief Pain Inventory," in: The 1995 Medical Outcomes and Guidelines Sourcebook, New York: Faulkner & Gray, Inc., 1994, pp. 269-270. |
| Vignau. Preliminary assessment of a 10-day rapid detoxification programme using high dosage buprenorphine. Eur Addict Res 1998;4 Suppl. 1:29-31. |
| Villiger, Binding of buprenorphine to opiate receptors. Regulation by guanyl nucleotides and metal ions. Neuropharmacology. Mar. 1984;23(3):373-5. |
| Vocci. Basis for the reccommendation for rescheduling of buprenorphine into Schedule IV of the Controlled Substances Act. FDA Document 2726A; Jul. 31, 1980; 1-8. |
| Waal. Buprenorphine (Temgesic)—new agent of abuse. Tidsskr Nor Laegeforen 1989; 109:1326-7. [in Norwegian w/ English transl]. |
| Wall, PD, et al., Textbook of Pain 2nd ed., New York: Churchill Livingstone, pp. 686-701 (1989). |
| Wallenstein, "Crossover Trials in Clinical Analgesic Assays: Studies of Buprenorphine and Morphine" Pharmacotherapy 6(5):228-235 (1986). |
| Walsh et al. Clinical pharmacology of buprenorphine: ceilling effects at high doses. Clin Pharmacol Ther. 1994;55:569-80. |
| Walsh et al. The acute effects of high dose buprenorphine in non-dependent humans. NIDA Res. Monogr. 1992;119:245. |
| Walsh et al., "Acute administration of buprenorphine in humans: Partial agonist and blockade effects" The Journal of Pharmacology and Experimental Therapeutics 274(1): 361-372 (1995). |
| Walter et al. Absorption, distribution, metabolism, and excretion of buprenorphine in animals and humans. In: Burprenorphine: Combatting Drug Abuse with a Unique Opiod. 1995;113-35. |
| Walter et al. Preclinical evaluation of buprenorphine. Research and Clinical Forums. 1997;19(2):17-23. |
| Wang et al. The study of analgesics following single and repeated doses. J Clin Pharmacol. Feb.-Mar. 1981;21(2):121-5. |
| Wang et al., Negative opiates in urine of patients on buprenorphine study. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Watanabe et al. Rectal absorption and mucosal irritation of rectal gels containing buprenorphine hydrochloride prepared with water-soluble dietary fibers, xanthan gum and locust bean gum. J Controlled Release. 1996;38:29-37, |
| Weber et al., "Current and historic concepts of opiate treatment in psychiatric disorders" Int. Clin. Psychopharmacology 3: 255-266 (1988). |
| Weinberg et al. Sublingual absorption of selected opioid analgesics Clin Pharmacol. Ther. 1988;44:335-42. |
| Weiss et al., "Analysis of the diminished skin perfusion in elderly people by laser doppler flowmetry" Age Ageing 21:237-241 (1992). |
| WHO Critical review of psychoactive substances, 33rd Expert Committee on Drug Dependence, Sep. 17-20, 2002. |
| WHO Expert Committee on Drug Dependence 25th Report. WHO technical Report Series. 1989. |
| WHO Expert Committee on Drug Dependence 32nd Report. WHO Technical Report Series. 2001. |
| WHO Expert Committee on Drug Dependence 33rd Report. WHO Technical Report Series. 2003. |
| Wiesenfeld-Hallin et al. Opioid sensitivity in antinociception: Role of anti-opioid systems with emphasis on cholecystokinin and NMDA receptors. In: Progress in Pain Research and Management. Opioid Sensitivity of Chronic Noncancer Pain. Kalso et al., eds. 1999;14:237-52. |
| Wilding et al., Pharmacokinetic evaluation of transdermal buprenorphine in man. International Journal of Pharmaceutics 1996, 132:81-7. |
| Wilding, I.R. et al., "Pharmacokinetic evaluation of transdermal buprenorphine in man" Intl. J. Pharmaceutics 132:81-7 (1996). |
| Williams et al., "Case-finding for depression in primary care: a randomized trial" Am. J. Med. 106(1):36-43 (1999). |
| Wodak, Additional commentary on a proposed review of the classification of buprenorphine in Schedule III of the 1971 Convention on Psychotropic substances (undated). |
| Woodroffe, MA et al., Canadian Family Physician 43: 268-272 (1997). |
| Woods et al. Behavioral pharmacology of buprenorphine: issues relevant to its treating drug abuse. NIDA Res Monogr. 1992;121:12-27. |
| Woods et al., "Efficacy of nalbuphine as a parenteral analgesic for the treatment of painful episodes in children with sickle cell disease" J. Assoc. Acad. Minor Phys. 1(3): 90-92 (1990). |
| World Medical Association Declaration of Helsinki, Ethical Principals for Medical Research Involving Human Subjects. 2004. |
| Wright C, Zalman M-A, Haddox JD, Kramer ED, Colucci RD, D'Ambrosio P., Systematic assessment of abuse or diversion in a clinical trial of analgesics. CPDD Annual Meeting Abstracts. 2006. (Study BUP3018). |
| Wright et al. Acute physical dependence in humans: repeated naloxone-precipitated withdrawal after a single dose of methadone. Drug Alcohol Depend. 1991;27:139-48. |
| Yanagita et al. Dependence potential of buprenorphine studied in rhesus monkeys. NIDA Res Monogr. 1982;41:208-14. |
| Yashiki et al., "Dual mass spectrometry of trifluoracetyl derivatives of opioid bases" GC-MS News 13(4): 101-106 (1985). |
| Yassen A. Mechanism-based pharmacokinetic-pharmacodynamic modeling of the reversal of buprenorphine-induced respiratory depression by naloxone: a study in healthy volunteers. Clinical Pharmacokinetics 2007; 46(11):965-80. |
| Yassen A. Mechanisms-based PK/PD modeling of the respiratory depressant effect of buprenorphine and fentanyl in healthy volunteers. Clinical Pharmacology & Therapeutics 2007;8(1):50-8. |
| Yaster et al., "Epidural analgesia in the management of severe vaso-occlusive sickle cell crisis" Pediatrics 93(2): 310-315 (1994). |
| Yaster et al., "The management of pain in sickle cell disease" Pediatr. Clin. North Am. 47(3):699-710 (2000). |
| Zacny et al. Comparing the subjective, psychomotor and physiological effects of intravenous buprenorphine and morphine in healthy volunteers. J Pharmacol Exp Ther. 1997;282:1187-97. |
| Zacny. A review of the effects of opioids on psychomotor and cognitive functioning in humans. Experimental and Clin. Psychopharmacol. 1995;3(4):432-66. |
| Ziedonis et al., Depression in cocaine abusing opioid addicts treated with buprenorphine versus methadone. CPDD 1994 Annual Scientific Meeting Abstracts. |
| Zola et al. Comparative effects and analgesic efficacy of the agonist-antagonist opioids. Drug Intell Clin Pharm. Jun. 1983;17(6):411-7. |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9642850B2 (en) * | 1997-02-24 | 2017-05-09 | Purdue Pharma L.P. | Method of providing sustained analgesia with buprenorphine |
| US20150157625A1 (en) * | 1997-02-24 | 2015-06-11 | Purdue Pharma L.P. | Method of providing sustained analgesia with buprenorphine |
| US20170056393A1 (en) * | 1997-02-24 | 2017-03-02 | Purdue Pharma L.P. | Method of providing sustained analgesia with buprenorphine |
| US9872856B2 (en) | 2012-04-17 | 2018-01-23 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9855262B2 (en) | 2012-04-17 | 2018-01-02 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9867817B2 (en) | 2012-04-17 | 2018-01-16 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US10398690B2 (en) | 2012-04-17 | 2019-09-03 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9884059B2 (en) | 2012-04-17 | 2018-02-06 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9662326B2 (en) | 2012-04-17 | 2017-05-30 | Purdue Pharma L.P. | Systems for treating an opioid-induced adverse pharmacodynamic response |
| US9931337B2 (en) | 2012-04-17 | 2018-04-03 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US10226457B2 (en) | 2014-10-17 | 2019-03-12 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9849124B2 (en) | 2014-10-17 | 2017-12-26 | Purdue Pharma L.P. | Systems and methods for treating an opioid-induced adverse pharmacodynamic response |
| US9926329B2 (en) | 2015-03-10 | 2018-03-27 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| US10278967B2 (en) | 2015-03-10 | 2019-05-07 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| WO2016142877A1 (en) | 2015-03-10 | 2016-09-15 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| US10406152B2 (en) | 2015-03-10 | 2019-09-10 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| US10835528B2 (en) | 2015-03-10 | 2020-11-17 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
| US10874662B2 (en) | 2015-03-10 | 2020-12-29 | Rhodes Technologies | Acetate salt of buprenorphine and methods for preparing buprenorphine |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9642850B2 (en) | Method of providing sustained analgesia with buprenorphine | |
| WO1998036728A9 (en) | Sustained analgesia achieved with buprenorphine | |
| AU2012244363B2 (en) | Method of providing sustained analgesia with buprenorphine | |
| AU2013204227B2 (en) | Method of providing sustained analgesia with buprenorphine | |
| US20040228906A1 (en) | Use of buprenorphine for the manufacture of a transdermal delivery device for the treatment of urinary incontinence, especially urge incontinence | |
| EP1323421A1 (en) | Use of buprenorphine for the manufacture of a transdermal delivery device for the treatment of urinary incontinence, especially urge incontinence |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: PURDUE PHARMA L.P., CONNECTICUT Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:EURO-CELTIQUE S.A.;REEL/FRAME:021269/0778 Effective date: 20060418 |
|
| PTEF | Application for a patent term extension |
Free format text: PRODUCT NAME: BUTRANS (BUPRENORPHINE BASE); REQUESTED FOR 5 YEARS Filing date: 20100826 Expiry date: 20170929 |
|
| FPAY | Fee payment |
Year of fee payment: 12 |
|
| CC | Certificate of correction | ||
| SULP | Surcharge for late payment | ||
| AS | Assignment |
Owner name: PURDUE PHARMA L.P., CONNECTICUT Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:THE P.F. LABORATORIES, INC.;REEL/FRAME:050412/0030 Effective date: 20190910 |