US20100009961A1 - Dipeptidyl peptidase-iv inhibitors - Google Patents
Dipeptidyl peptidase-iv inhibitors Download PDFInfo
- Publication number
- US20100009961A1 US20100009961A1 US12/492,254 US49225409A US2010009961A1 US 20100009961 A1 US20100009961 A1 US 20100009961A1 US 49225409 A US49225409 A US 49225409A US 2010009961 A1 US2010009961 A1 US 2010009961A1
- Authority
- US
- United States
- Prior art keywords
- alkyl
- mixture
- title compound
- afford
- concentrated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- VLJNHYLEOZPXFW-BYPYZUCNSA-N NC(=O)[C@@H]1CCCN1 Chemical compound NC(=O)[C@@H]1CCCN1 VLJNHYLEOZPXFW-BYPYZUCNSA-N 0.000 description 113
- MCEWPPMUTVLMJG-YUPRTTJUSA-N NC(=O)[C@@H]1C[C@@H]2C[C@@H]2N1 Chemical compound NC(=O)[C@@H]1C[C@@H]2C[C@@H]2N1 MCEWPPMUTVLMJG-YUPRTTJUSA-N 0.000 description 112
- 0 [3*]C(C)(C)C(C)C Chemical compound [3*]C(C)(C)C(C)C 0.000 description 91
- JJWLVOIRVHMVIS-UHFFFAOYSA-N CC(C)N Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 17
- KRZCOLNOCZKSDF-UHFFFAOYSA-N NC1=CC=C(F)C=C1 Chemical compound NC1=CC=C(F)C=C1 KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 12
- PLNNJQXIITYYTN-UHFFFAOYSA-N CC(C)C(=O)NN Chemical compound CC(C)C(=O)NN PLNNJQXIITYYTN-UHFFFAOYSA-N 0.000 description 11
- ZXBMIRYQUFQQNX-UHFFFAOYSA-N NNC1=CC=C(F)C=C1 Chemical compound NNC1=CC=C(F)C=C1 ZXBMIRYQUFQQNX-UHFFFAOYSA-N 0.000 description 10
- GUQRKZPMVLRXLT-UHFFFAOYSA-N ONC1CCCCC1 Chemical compound ONC1CCCCC1 GUQRKZPMVLRXLT-UHFFFAOYSA-N 0.000 description 10
- OFLXLNCGODUUOT-UHFFFAOYSA-N CC(=O)NN Chemical compound CC(=O)NN OFLXLNCGODUUOT-UHFFFAOYSA-N 0.000 description 9
- DGMOBVGABMBZSB-UHFFFAOYSA-N CC(C)C(=O)Cl Chemical compound CC(C)C(=O)Cl DGMOBVGABMBZSB-UHFFFAOYSA-N 0.000 description 9
- ODHYIQOBTIWVRZ-UHFFFAOYSA-N CC(C)NO Chemical compound CC(C)NO ODHYIQOBTIWVRZ-UHFFFAOYSA-N 0.000 description 9
- HDZGCSFEDULWCS-UHFFFAOYSA-N CNN Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 9
- CPQCSJYYDADLCZ-UHFFFAOYSA-N CNO Chemical compound CNO CPQCSJYYDADLCZ-UHFFFAOYSA-N 0.000 description 9
- UIVXXFYJRYVRKJ-UHFFFAOYSA-N NNC(=O)C1=CC=C(F)C=C1 Chemical compound NNC(=O)C1=CC=C(F)C=C1 UIVXXFYJRYVRKJ-UHFFFAOYSA-N 0.000 description 9
- CZKLEJHVLCMVQR-UHFFFAOYSA-N O=C(Cl)C1=CC=C(F)C=C1 Chemical compound O=C(Cl)C1=CC=C(F)C=C1 CZKLEJHVLCMVQR-UHFFFAOYSA-N 0.000 description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N [H]N1CCCC1 Chemical compound [H]N1CCCC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- HONIICLYMWZJFZ-UHFFFAOYSA-N C1CNC1 Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1COCCN1 Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- WFDIJRYMOXRFFG-UHFFFAOYSA-N CC(=O)OC(C)=O Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 8
- YBRBMKDOPFTVDT-UHFFFAOYSA-N CC(C)(C)N Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 8
- KJAQRHMKLVGSCG-UHFFFAOYSA-N CC(C)NN Chemical compound CC(C)NN KJAQRHMKLVGSCG-UHFFFAOYSA-N 0.000 description 8
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N CCC(=O)OC(=O)CC Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 8
- DILRJUIACXKSQE-UHFFFAOYSA-N CN(C)CCN Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 8
- AVXURJPOCDRRFD-UHFFFAOYSA-N NO Chemical compound NO AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 8
- XZBIXDPGRMLSTC-UHFFFAOYSA-N [H]C(=O)NN Chemical compound [H]C(=O)NN XZBIXDPGRMLSTC-UHFFFAOYSA-N 0.000 description 8
- NQRYJNQNLNOLGT-UHFFFAOYSA-N [H]N1CCCCC1 Chemical compound [H]N1CCCCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- PVOAHINGSUIXLS-UHFFFAOYSA-N [H]N1CCN(C)CC1 Chemical compound [H]N1CCN(C)CC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 8
- LXQMHOKEXZETKB-UHFFFAOYSA-N CC(C)(O)CN Chemical compound CC(C)(O)CN LXQMHOKEXZETKB-UHFFFAOYSA-N 0.000 description 7
- ROSDSFDQCJNGOL-UHFFFAOYSA-N CNC Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 7
- QUSNBJAOOMFDIB-UHFFFAOYSA-N CCN Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 6
- NYFSRXOWJXAHOO-UHFFFAOYSA-N CC(C)(N)C1=CC=C(F)C=C1 Chemical compound CC(C)(N)C1=CC=C(F)C=C1 NYFSRXOWJXAHOO-UHFFFAOYSA-N 0.000 description 5
- ODZXRBRYQGYVJY-ZCFIWIBFSA-N C[C@@H](N)C1=CC(C(F)(F)F)=CC=C1 Chemical compound C[C@@H](N)C1=CC(C(F)(F)F)=CC=C1 ODZXRBRYQGYVJY-ZCFIWIBFSA-N 0.000 description 4
- QPERNSDCEUTOTE-UHFFFAOYSA-N ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 QPERNSDCEUTOTE-UHFFFAOYSA-N 0.000 description 4
- XJUHMYPLEUPNOF-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 XJUHMYPLEUPNOF-VWLOTQADSA-N 0.000 description 3
- WDHXBNGCBUJEPO-KEJDIYNNSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 WDHXBNGCBUJEPO-KEJDIYNNSA-N 0.000 description 3
- XTIXPIMMHGCRJD-RXMQYKEDSA-N C[C@@H](N)C1=CC(F)=CC(F)=C1 Chemical compound C[C@@H](N)C1=CC(F)=CC(F)=C1 XTIXPIMMHGCRJD-RXMQYKEDSA-N 0.000 description 3
- KWJVCRYUAKXPTJ-MRVPVSSYSA-N C[C@@H](N)C1=CC=CC(C(=O)N(C)C)=C1 Chemical compound C[C@@H](N)C1=CC=CC(C(=O)N(C)C)=C1 KWJVCRYUAKXPTJ-MRVPVSSYSA-N 0.000 description 3
- MYIPCVJUEKPEBT-ZCFIWIBFSA-N C[C@@H](N)C1=CC=CC(C2=NN=NN2)=C1 Chemical compound C[C@@H](N)C1=CC=CC(C2=NN=NN2)=C1 MYIPCVJUEKPEBT-ZCFIWIBFSA-N 0.000 description 3
- KQYBOYXTCLYWST-ZCFIWIBFSA-N C[C@@H](N)C1=CC=CC(S(N)(=O)=O)=C1 Chemical compound C[C@@H](N)C1=CC=CC(S(N)(=O)=O)=C1 KQYBOYXTCLYWST-ZCFIWIBFSA-N 0.000 description 3
- GUMZDWPMXGQNBG-LURJTMIESA-N C[C@H](N)C1=CC=C(C(F)(F)F)C=C1 Chemical compound C[C@H](N)C1=CC=C(C(F)(F)F)C=C1 GUMZDWPMXGQNBG-LURJTMIESA-N 0.000 description 3
- QGCLEUGNYRXBMZ-LURJTMIESA-N C[C@H](N)C1=CC=C(F)C=C1 Chemical compound C[C@H](N)C1=CC=C(F)C=C1 QGCLEUGNYRXBMZ-LURJTMIESA-N 0.000 description 3
- KQYBOYXTCLYWST-LURJTMIESA-N C[C@H](N)C1=CC=CC(S(N)(=O)=O)=C1 Chemical compound C[C@H](N)C1=CC=CC(S(N)(=O)=O)=C1 KQYBOYXTCLYWST-LURJTMIESA-N 0.000 description 3
- WRKGTUUHIURRMW-UHFFFAOYSA-N NC(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 Chemical compound NC(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 WRKGTUUHIURRMW-UHFFFAOYSA-N 0.000 description 3
- SWHLYGWSQYSQBX-UHFFFAOYSA-N NC1(C2=CC=C(F)C=C2)CCCCC1 Chemical compound NC1(C2=CC=C(F)C=C2)CCCCC1 SWHLYGWSQYSQBX-UHFFFAOYSA-N 0.000 description 3
- KTPONJJKCBOJCQ-UHFFFAOYSA-N NC1=CC=C(C2=NN=NN2)C=C1 Chemical compound NC1=CC=C(C2=NN=NN2)C=C1 KTPONJJKCBOJCQ-UHFFFAOYSA-N 0.000 description 3
- KTADHXXSCNRJRT-UHFFFAOYSA-N NC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound NC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 KTADHXXSCNRJRT-UHFFFAOYSA-N 0.000 description 3
- NTNLGQWPYJTABN-LBPRGKRZSA-N N[C@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N[C@H](C1=CC=C(F)C=C1)C1CCCC1 NTNLGQWPYJTABN-LBPRGKRZSA-N 0.000 description 3
- YVWNDABPZGGQFE-UHFFFAOYSA-N O=C1NC2=C(C=CC(Cl)=C2)NC2=C1C=CC=C2 Chemical compound O=C1NC2=C(C=CC(Cl)=C2)NC2=C1C=CC=C2 YVWNDABPZGGQFE-UHFFFAOYSA-N 0.000 description 3
- FSSPGSAQUIYDCN-UHFFFAOYSA-N O=S1(=O)CCCO1 Chemical compound O=S1(=O)CCCO1 FSSPGSAQUIYDCN-UHFFFAOYSA-N 0.000 description 3
- NPRDLWSYONSQQI-UHFFFAOYSA-N [C-]#[N+]C1=CC(CN)=CC=C1 Chemical compound [C-]#[N+]C1=CC(CN)=CC=C1 NPRDLWSYONSQQI-UHFFFAOYSA-N 0.000 description 3
- DONREFRRKVKWDM-IGKIAQTJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DONREFRRKVKWDM-IGKIAQTJSA-N 0.000 description 3
- AJXQPMJGISVOHK-CMOCDZPBSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C)(C)C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C)(C)C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N AJXQPMJGISVOHK-CMOCDZPBSA-N 0.000 description 2
- FEBMJKZBEOTVHM-KGJIALSRSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C)(C)C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C)(C)C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FEBMJKZBEOTVHM-KGJIALSRSA-N 0.000 description 2
- ZUCBPXHMAQFYAI-JTQLQIEISA-N CC(C)(C)[C@@H](N)C1=CC=C(F)C=C1 Chemical compound CC(C)(C)[C@@H](N)C1=CC=C(F)C=C1 ZUCBPXHMAQFYAI-JTQLQIEISA-N 0.000 description 2
- ZUCBPXHMAQFYAI-SNVBAGLBSA-N CC(C)(C)[C@H](N)C1=CC=C(F)C=C1 Chemical compound CC(C)(C)[C@H](N)C1=CC=C(F)C=C1 ZUCBPXHMAQFYAI-SNVBAGLBSA-N 0.000 description 2
- JCGWWAQMNOTXIR-MHZLTWQESA-N CC(C)NC(=N)C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)NC(=N)C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 JCGWWAQMNOTXIR-MHZLTWQESA-N 0.000 description 2
- DKHSXWXHXFRKON-SNVBAGLBSA-N CC(C)[C@@H](N)C1=CC=C(F)C=C1 Chemical compound CC(C)[C@@H](N)C1=CC=C(F)C=C1 DKHSXWXHXFRKON-SNVBAGLBSA-N 0.000 description 2
- DKHSXWXHXFRKON-JTQLQIEISA-N CC(C)[C@H](N)C1=CC=C(F)C=C1 Chemical compound CC(C)[C@H](N)C1=CC=C(F)C=C1 DKHSXWXHXFRKON-JTQLQIEISA-N 0.000 description 2
- UKGBHKUPJJIEEC-UHFFFAOYSA-N CC1(C)C2=CC=CC=C2C(=O)NC2=C1C=CC=C2 Chemical compound CC1(C)C2=CC=CC=C2C(=O)NC2=C1C=CC=C2 UKGBHKUPJJIEEC-UHFFFAOYSA-N 0.000 description 2
- BWZFVQWUKNCKQL-SECBINFHSA-N CC[C@@H](N)C1=CC=C(F)C=C1 Chemical compound CC[C@@H](N)C1=CC=C(F)C=C1 BWZFVQWUKNCKQL-SECBINFHSA-N 0.000 description 2
- BWZFVQWUKNCKQL-VIFPVBQESA-N CC[C@H](N)C1=CC=C(F)C=C1 Chemical compound CC[C@H](N)C1=CC=C(F)C=C1 BWZFVQWUKNCKQL-VIFPVBQESA-N 0.000 description 2
- KZMQLAUEMUGCLY-FQEVSTJZSA-N CC[C@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC[C@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 KZMQLAUEMUGCLY-FQEVSTJZSA-N 0.000 description 2
- XDPOVHYONPMWSX-YFKPBYRVSA-N CC[C@H]1COS(=O)(=O)C1 Chemical compound CC[C@H]1COS(=O)(=O)C1 XDPOVHYONPMWSX-YFKPBYRVSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N CN Chemical compound CN BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- ZWSBTDVURYYJIO-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 ZWSBTDVURYYJIO-UHFFFAOYSA-N 0.000 description 2
- HPUCZJUFYQBSEC-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 HPUCZJUFYQBSEC-UHFFFAOYSA-N 0.000 description 2
- RXMGRNSFGCJVOX-PMERELPUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 RXMGRNSFGCJVOX-PMERELPUSA-N 0.000 description 2
- AWBRTMPJTQRAAE-PMERELPUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 AWBRTMPJTQRAAE-PMERELPUSA-N 0.000 description 2
- WYBKTTANRURPSC-NSHDSACASA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 WYBKTTANRURPSC-NSHDSACASA-N 0.000 description 2
- WYBKTTANRURPSC-LLVKDONJSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N)CC2 WYBKTTANRURPSC-LLVKDONJSA-N 0.000 description 2
- ZEZYBJNAFFTVRS-FZADBTJQSA-N CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 ZEZYBJNAFFTVRS-FZADBTJQSA-N 0.000 description 2
- WUFZOBYKRMYJNS-BAVGOCKISA-N CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 WUFZOBYKRMYJNS-BAVGOCKISA-N 0.000 description 2
- LYZCJWJPKCZQKY-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)S1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)S1)CC2 LYZCJWJPKCZQKY-UHFFFAOYSA-N 0.000 description 2
- ADTDFCKINGZRCO-DFOYWCFBSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 ADTDFCKINGZRCO-DFOYWCFBSA-N 0.000 description 2
- ZBWSFNISUZNHQP-NSHDSACASA-N CN(C)OOSC1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@@H](N)CC2 ZBWSFNISUZNHQP-NSHDSACASA-N 0.000 description 2
- HMQGYUAUZPTJHV-UHFFFAOYSA-N CN1C2=C(C=C(Cl)C=C2)NC(=O)C2=C1C=CC=C2 Chemical compound CN1C2=C(C=C(Cl)C=C2)NC(=O)C2=C1C=CC=C2 HMQGYUAUZPTJHV-UHFFFAOYSA-N 0.000 description 2
- HFNDZALKMIVKEC-UHFFFAOYSA-N CN1N=NC(C2(CCN)C3=CC=C(C(=O)N(C)C)C=C3CCC3=C2C=CC(C(=O)N(C)C)=C3)=N1 Chemical compound CN1N=NC(C2(CCN)C3=CC=C(C(=O)N(C)C)C=C3CCC3=C2C=CC(C(=O)N(C)C)=C3)=N1 HFNDZALKMIVKEC-UHFFFAOYSA-N 0.000 description 2
- JOKRVNJQGJDJDY-SSDOTTSWSA-N CNC(=O)C1=CC([C@@H](C)N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@@H](C)N)=CC=C1 JOKRVNJQGJDJDY-SSDOTTSWSA-N 0.000 description 2
- JOKRVNJQGJDJDY-ZETCQYMHSA-N CNC(=O)C1=CC([C@H](C)N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@H](C)N)=CC=C1 JOKRVNJQGJDJDY-ZETCQYMHSA-N 0.000 description 2
- GRQLXBKHAPJVCW-QFIPXVFZSA-N CNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)C=C1)CC2 GRQLXBKHAPJVCW-QFIPXVFZSA-N 0.000 description 2
- JBAMNGXVZXFWNB-NSHDSACASA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 JBAMNGXVZXFWNB-NSHDSACASA-N 0.000 description 2
- RHIQELMDYRPHPN-JTQLQIEISA-N CNC(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 RHIQELMDYRPHPN-JTQLQIEISA-N 0.000 description 2
- RHIQELMDYRPHPN-SNVBAGLBSA-N CNC(=O)C1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@H](N)CC2 RHIQELMDYRPHPN-SNVBAGLBSA-N 0.000 description 2
- WZVKEQWGVIZCPG-UHFFFAOYSA-N CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(CCN)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(CCN)C2=NNN=N2)S1 WZVKEQWGVIZCPG-UHFFFAOYSA-N 0.000 description 2
- JGYWPAUSQIKCSD-ZETCQYMHSA-N CNC(=O)C1=CC=C([C@H](C)N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@H](C)N)C=C1 JGYWPAUSQIKCSD-ZETCQYMHSA-N 0.000 description 2
- SMLXJMQBLUFBCR-UMFSSWHCSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C SMLXJMQBLUFBCR-UMFSSWHCSA-N 0.000 description 2
- KXJQPBQGNBNIRM-SSDOTTSWSA-N CNOOSC1=CC([C@@H](C)N)=CC=C1 Chemical compound CNOOSC1=CC([C@@H](C)N)=CC=C1 KXJQPBQGNBNIRM-SSDOTTSWSA-N 0.000 description 2
- KXJQPBQGNBNIRM-ZETCQYMHSA-N CNOOSC1=CC([C@H](C)N)=CC=C1 Chemical compound CNOOSC1=CC([C@H](C)N)=CC=C1 KXJQPBQGNBNIRM-ZETCQYMHSA-N 0.000 description 2
- BLQZWAUTCSDXHC-JTQLQIEISA-N CNOOSC1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@@H](N)CC2 BLQZWAUTCSDXHC-JTQLQIEISA-N 0.000 description 2
- BLQZWAUTCSDXHC-SNVBAGLBSA-N CNOOSC1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@H](N)CC2 BLQZWAUTCSDXHC-SNVBAGLBSA-N 0.000 description 2
- XFLHYQRBKUEFKO-KANYKUSLSA-N CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 XFLHYQRBKUEFKO-KANYKUSLSA-N 0.000 description 2
- PQXNJYMDRZNTGA-SSDOTTSWSA-N CNOOSC1=CC=C([C@@H](C)N)C=C1 Chemical compound CNOOSC1=CC=C([C@@H](C)N)C=C1 PQXNJYMDRZNTGA-SSDOTTSWSA-N 0.000 description 2
- PQXNJYMDRZNTGA-ZETCQYMHSA-N CNOOSC1=CC=C([C@H](C)N)C=C1 Chemical compound CNOOSC1=CC=C([C@H](C)N)C=C1 PQXNJYMDRZNTGA-ZETCQYMHSA-N 0.000 description 2
- OOGHWKACDVSHQO-VWQWZFRASA-N COC(O)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound COC(O)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OOGHWKACDVSHQO-VWQWZFRASA-N 0.000 description 2
- BMWVZDDLOXXIRI-NHKCCNDQSA-N C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N BMWVZDDLOXXIRI-NHKCCNDQSA-N 0.000 description 2
- KQYLVIQPGMDYTC-XSXWSVAESA-N C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N KQYLVIQPGMDYTC-XSXWSVAESA-N 0.000 description 2
- AXUOZQBCABYKJM-RXVVDRJESA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N AXUOZQBCABYKJM-RXVVDRJESA-N 0.000 description 2
- DPLIMKBGTYIUCB-ZCFIWIBFSA-N C[C@@H](N)C1=C(C(F)(F)F)C=CC=C1 Chemical compound C[C@@H](N)C1=C(C(F)(F)F)C=CC=C1 DPLIMKBGTYIUCB-ZCFIWIBFSA-N 0.000 description 2
- DIWHJJUFVGEXGS-ZCFIWIBFSA-N C[C@@H](N)C1=C(F)C=CC=C1 Chemical compound C[C@@H](N)C1=C(F)C=CC=C1 DIWHJJUFVGEXGS-ZCFIWIBFSA-N 0.000 description 2
- PFVWEAYXWZFSSK-RXMQYKEDSA-N C[C@@H](N)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 Chemical compound C[C@@H](N)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 PFVWEAYXWZFSSK-RXMQYKEDSA-N 0.000 description 2
- ASNVMKIDRJZXQZ-ZCFIWIBFSA-N C[C@@H](N)C1=CC(F)=CC=C1 Chemical compound C[C@@H](N)C1=CC(F)=CC=C1 ASNVMKIDRJZXQZ-ZCFIWIBFSA-N 0.000 description 2
- TYFVCNMXZGFFAG-MRVPVSSYSA-N C[C@@H](N)C1=CC=C(C(=O)N(C)C)C=C1 Chemical compound C[C@@H](N)C1=CC=C(C(=O)N(C)C)C=C1 TYFVCNMXZGFFAG-MRVPVSSYSA-N 0.000 description 2
- NDMBVGSUFFPAFE-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(C(=O)O)C=C1 Chemical compound C[C@@H](N)C1=CC=C(C(=O)O)C=C1 NDMBVGSUFFPAFE-ZCFIWIBFSA-N 0.000 description 2
- GUMZDWPMXGQNBG-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(C(F)(F)F)C=C1 Chemical compound C[C@@H](N)C1=CC=C(C(F)(F)F)C=C1 GUMZDWPMXGQNBG-ZCFIWIBFSA-N 0.000 description 2
- PXJMJWOLRCACSF-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(C(N)=O)C=C1 Chemical compound C[C@@H](N)C1=CC=C(C(N)=O)C=C1 PXJMJWOLRCACSF-ZCFIWIBFSA-N 0.000 description 2
- QQXOVWASMFSHBM-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(C2=NN=NN2)C=C1 Chemical compound C[C@@H](N)C1=CC=C(C2=NN=NN2)C=C1 QQXOVWASMFSHBM-ZCFIWIBFSA-N 0.000 description 2
- JAHDRLFMQPSWSR-MRVPVSSYSA-N C[C@@H](N)C1=CC=C(SOON(C)C)C=C1 Chemical compound C[C@@H](N)C1=CC=C(SOON(C)C)C=C1 JAHDRLFMQPSWSR-MRVPVSSYSA-N 0.000 description 2
- FKRRXVGMNMPFPE-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(SOON)C=C1 Chemical compound C[C@@H](N)C1=CC=C(SOON)C=C1 FKRRXVGMNMPFPE-ZCFIWIBFSA-N 0.000 description 2
- UTDRNGQVIRUPOC-SSDOTTSWSA-N C[C@@H](N)C1=CC=CC(C#N)=C1 Chemical compound C[C@@H](N)C1=CC=CC(C#N)=C1 UTDRNGQVIRUPOC-SSDOTTSWSA-N 0.000 description 2
- NSRCJWRFTUMEEL-ZCFIWIBFSA-N C[C@@H](N)C1=CC=CC(C(=O)O)=C1 Chemical compound C[C@@H](N)C1=CC=CC(C(=O)O)=C1 NSRCJWRFTUMEEL-ZCFIWIBFSA-N 0.000 description 2
- PJYWNTXHGXEKSG-ZCFIWIBFSA-N C[C@@H](N)C1=CC=CC(C(N)=O)=C1 Chemical compound C[C@@H](N)C1=CC=CC(C(N)=O)=C1 PJYWNTXHGXEKSG-ZCFIWIBFSA-N 0.000 description 2
- PZXVPSLVBOZCKH-MRVPVSSYSA-N C[C@@H](N)C1=CC=CC(S(=O)(=O)N(C)C)=C1 Chemical compound C[C@@H](N)C1=CC=CC(S(=O)(=O)N(C)C)=C1 PZXVPSLVBOZCKH-MRVPVSSYSA-N 0.000 description 2
- CFFJFHJYXBYTIE-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound C[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 CFFJFHJYXBYTIE-OAHLLOKOSA-N 0.000 description 2
- BKMMTJMQCTUHRP-GSVOUGTGSA-N C[C@@H](N)CO Chemical compound C[C@@H](N)CO BKMMTJMQCTUHRP-GSVOUGTGSA-N 0.000 description 2
- KZDZUPGTYYIAAV-SCSAIBSYSA-N C[C@@H]1COS(=O)(=O)C1 Chemical compound C[C@@H]1COS(=O)(=O)C1 KZDZUPGTYYIAAV-SCSAIBSYSA-N 0.000 description 2
- NFAKQWFVEAEEPG-ZCFIWIBFSA-N C[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C Chemical compound C[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C NFAKQWFVEAEEPG-ZCFIWIBFSA-N 0.000 description 2
- JTNUUVSSEKOUGF-NLFFAJNJSA-N C[C@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1CCC[C@H]1C#N JTNUUVSSEKOUGF-NLFFAJNJSA-N 0.000 description 2
- IDMOKZCKSLCOAG-XJFCNFDWSA-N C[C@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N IDMOKZCKSLCOAG-XJFCNFDWSA-N 0.000 description 2
- DPLIMKBGTYIUCB-LURJTMIESA-N C[C@H](N)C1=C(C(F)(F)F)C=CC=C1 Chemical compound C[C@H](N)C1=C(C(F)(F)F)C=CC=C1 DPLIMKBGTYIUCB-LURJTMIESA-N 0.000 description 2
- DIWHJJUFVGEXGS-LURJTMIESA-N C[C@H](N)C1=C(F)C=CC=C1 Chemical compound C[C@H](N)C1=C(F)C=CC=C1 DIWHJJUFVGEXGS-LURJTMIESA-N 0.000 description 2
- PFVWEAYXWZFSSK-YFKPBYRVSA-N C[C@H](N)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 Chemical compound C[C@H](N)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 PFVWEAYXWZFSSK-YFKPBYRVSA-N 0.000 description 2
- ODZXRBRYQGYVJY-LURJTMIESA-N C[C@H](N)C1=CC(C(F)(F)F)=CC=C1 Chemical compound C[C@H](N)C1=CC(C(F)(F)F)=CC=C1 ODZXRBRYQGYVJY-LURJTMIESA-N 0.000 description 2
- XTIXPIMMHGCRJD-YFKPBYRVSA-N C[C@H](N)C1=CC(F)=CC(F)=C1 Chemical compound C[C@H](N)C1=CC(F)=CC(F)=C1 XTIXPIMMHGCRJD-YFKPBYRVSA-N 0.000 description 2
- ASNVMKIDRJZXQZ-LURJTMIESA-N C[C@H](N)C1=CC(F)=CC=C1 Chemical compound C[C@H](N)C1=CC(F)=CC=C1 ASNVMKIDRJZXQZ-LURJTMIESA-N 0.000 description 2
- TYFVCNMXZGFFAG-QMMMGPOBSA-N C[C@H](N)C1=CC=C(C(=O)N(C)C)C=C1 Chemical compound C[C@H](N)C1=CC=C(C(=O)N(C)C)C=C1 TYFVCNMXZGFFAG-QMMMGPOBSA-N 0.000 description 2
- NDMBVGSUFFPAFE-LURJTMIESA-N C[C@H](N)C1=CC=C(C(=O)O)C=C1 Chemical compound C[C@H](N)C1=CC=C(C(=O)O)C=C1 NDMBVGSUFFPAFE-LURJTMIESA-N 0.000 description 2
- PXJMJWOLRCACSF-LURJTMIESA-N C[C@H](N)C1=CC=C(C(N)=O)C=C1 Chemical compound C[C@H](N)C1=CC=C(C(N)=O)C=C1 PXJMJWOLRCACSF-LURJTMIESA-N 0.000 description 2
- QQXOVWASMFSHBM-LURJTMIESA-N C[C@H](N)C1=CC=C(C2=NN=NN2)C=C1 Chemical compound C[C@H](N)C1=CC=C(C2=NN=NN2)C=C1 QQXOVWASMFSHBM-LURJTMIESA-N 0.000 description 2
- JAHDRLFMQPSWSR-QMMMGPOBSA-N C[C@H](N)C1=CC=C(SOON(C)C)C=C1 Chemical compound C[C@H](N)C1=CC=C(SOON(C)C)C=C1 JAHDRLFMQPSWSR-QMMMGPOBSA-N 0.000 description 2
- FKRRXVGMNMPFPE-LURJTMIESA-N C[C@H](N)C1=CC=C(SOON)C=C1 Chemical compound C[C@H](N)C1=CC=C(SOON)C=C1 FKRRXVGMNMPFPE-LURJTMIESA-N 0.000 description 2
- UTDRNGQVIRUPOC-ZETCQYMHSA-N C[C@H](N)C1=CC=CC(C#N)=C1 Chemical compound C[C@H](N)C1=CC=CC(C#N)=C1 UTDRNGQVIRUPOC-ZETCQYMHSA-N 0.000 description 2
- KWJVCRYUAKXPTJ-QMMMGPOBSA-N C[C@H](N)C1=CC=CC(C(=O)N(C)C)=C1 Chemical compound C[C@H](N)C1=CC=CC(C(=O)N(C)C)=C1 KWJVCRYUAKXPTJ-QMMMGPOBSA-N 0.000 description 2
- NSRCJWRFTUMEEL-LURJTMIESA-N C[C@H](N)C1=CC=CC(C(=O)O)=C1 Chemical compound C[C@H](N)C1=CC=CC(C(=O)O)=C1 NSRCJWRFTUMEEL-LURJTMIESA-N 0.000 description 2
- PJYWNTXHGXEKSG-LURJTMIESA-N C[C@H](N)C1=CC=CC(C(N)=O)=C1 Chemical compound C[C@H](N)C1=CC=CC(C(N)=O)=C1 PJYWNTXHGXEKSG-LURJTMIESA-N 0.000 description 2
- MYIPCVJUEKPEBT-LURJTMIESA-N C[C@H](N)C1=CC=CC(C2=NN=NN2)=C1 Chemical compound C[C@H](N)C1=CC=CC(C2=NN=NN2)=C1 MYIPCVJUEKPEBT-LURJTMIESA-N 0.000 description 2
- PZXVPSLVBOZCKH-QMMMGPOBSA-N C[C@H](N)C1=CC=CC(S(=O)(=O)N(C)C)=C1 Chemical compound C[C@H](N)C1=CC=CC(S(=O)(=O)N(C)C)=C1 PZXVPSLVBOZCKH-QMMMGPOBSA-N 0.000 description 2
- BZBIUFNKQBRQJT-JTQLQIEISA-N C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CC2=C1C=CC(C(N)=O)=C2 BZBIUFNKQBRQJT-JTQLQIEISA-N 0.000 description 2
- HLCPWBZNUKCSBN-UHFFFAOYSA-N N#CC1=C(N)C=CC=C1 Chemical compound N#CC1=C(N)C=CC=C1 HLCPWBZNUKCSBN-UHFFFAOYSA-N 0.000 description 2
- ATKRPKHUJKXHOK-DQUNLGLBSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=C(F)C=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=C(F)C=C2 ATKRPKHUJKXHOK-DQUNLGLBSA-N 0.000 description 2
- CUVBFMGTDUQFOA-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 CUVBFMGTDUQFOA-FQEVSTJZSA-N 0.000 description 2
- VRUUQARGUKGQPW-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 VRUUQARGUKGQPW-FQEVSTJZSA-N 0.000 description 2
- RCPQDYIUDYFLSJ-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(Cl)C=C2)CCC2=C1C=CC(Cl)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(Cl)C=C2)CCC2=C1C=CC(Cl)=C2 RCPQDYIUDYFLSJ-FQEVSTJZSA-N 0.000 description 2
- INDDAIKUZWZIFG-JINQPTGOSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2 INDDAIKUZWZIFG-JINQPTGOSA-N 0.000 description 2
- WSVVSYQXMMKPGN-NRFANRHFSA-N N#C[C@@H]1CCCN1C(=O)CNCCCN1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCCN1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2 WSVVSYQXMMKPGN-NRFANRHFSA-N 0.000 description 2
- OQEXZTGABKHXBC-INIZCTEOSA-N N#C[C@@H]1CCCN1C(=O)CNCCN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2 OQEXZTGABKHXBC-INIZCTEOSA-N 0.000 description 2
- LFRYSSZFILOTMY-XUXIUFHCSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CS1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CS1 LFRYSSZFILOTMY-XUXIUFHCSA-N 0.000 description 2
- GYUOJPHVAONXKG-HVTWWXFQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H]1C[C@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)CN1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H]1C[C@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)CN1 GYUOJPHVAONXKG-HVTWWXFQSA-N 0.000 description 2
- PXBFMLJZNCDSMP-UHFFFAOYSA-N NC(=O)C1=C(N)C=CC=C1 Chemical compound NC(=O)C1=C(N)C=CC=C1 PXBFMLJZNCDSMP-UHFFFAOYSA-N 0.000 description 2
- JVSJEGRMACWUTM-VIFPVBQESA-N NC(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound NC(=O)C1=CC2=C(C=C1)[C@@H](N)CC2 JVSJEGRMACWUTM-VIFPVBQESA-N 0.000 description 2
- JVSJEGRMACWUTM-SECBINFHSA-N NC(=O)C1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound NC(=O)C1=CC2=C(C=C1)[C@H](N)CC2 JVSJEGRMACWUTM-SECBINFHSA-N 0.000 description 2
- QIKYZXDTTPVVAC-UHFFFAOYSA-N NC(=O)C1=CC=C(N)C=C1 Chemical compound NC(=O)C1=CC=C(N)C=C1 QIKYZXDTTPVVAC-UHFFFAOYSA-N 0.000 description 2
- GSCPDZHWVNUUFI-UHFFFAOYSA-N NC(=O)C1=CC=CC(N)=C1 Chemical compound NC(=O)C1=CC=CC(N)=C1 GSCPDZHWVNUUFI-UHFFFAOYSA-N 0.000 description 2
- PAPAYUQUJVLXAF-UHFFFAOYSA-N NC1(C2=CC=C(F)C=C2)CCC1 Chemical compound NC1(C2=CC=C(F)C=C2)CCC1 PAPAYUQUJVLXAF-UHFFFAOYSA-N 0.000 description 2
- DBSPGNRTDZEXGR-UHFFFAOYSA-N NC1(C2=CC=C(F)C=C2)CCCC1 Chemical compound NC1(C2=CC=C(F)C=C2)CCCC1 DBSPGNRTDZEXGR-UHFFFAOYSA-N 0.000 description 2
- GUCGQKSPWMDAEX-UHFFFAOYSA-N NC1=C(C2=NN=NN2)C=CC=C1 Chemical compound NC1=C(C2=NN=NN2)C=CC=C1 GUCGQKSPWMDAEX-UHFFFAOYSA-N 0.000 description 2
- CDIDGWDGQGVCIB-UHFFFAOYSA-N NC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 Chemical compound NC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 CDIDGWDGQGVCIB-UHFFFAOYSA-N 0.000 description 2
- FLGISLVJQPPAMV-UHFFFAOYSA-N NC1=CC(C2=NN=NN2)=CC=C1 Chemical compound NC1=CC(C2=NN=NN2)=CC=C1 FLGISLVJQPPAMV-UHFFFAOYSA-N 0.000 description 2
- KQOIBXZRCYFZSO-UHFFFAOYSA-N NC1=CC(F)=CC(F)=C1 Chemical compound NC1=CC(F)=CC(F)=C1 KQOIBXZRCYFZSO-UHFFFAOYSA-N 0.000 description 2
- QZVQQUVWFIZUBQ-UHFFFAOYSA-N NC1=CC=CC(F)=C1 Chemical compound NC1=CC=CC(F)=C1 QZVQQUVWFIZUBQ-UHFFFAOYSA-N 0.000 description 2
- OUGMRQJTULXVDC-UHFFFAOYSA-N NC1C2=CC=CC=C2C2=C1C=CC=C2 Chemical compound NC1C2=CC=CC=C2C2=C1C=CC=C2 OUGMRQJTULXVDC-UHFFFAOYSA-N 0.000 description 2
- BYDPSHUZBXOTOM-UHFFFAOYSA-N NC1C2=CC=CC=C2CC2=C1C=CC=C2 Chemical compound NC1C2=CC=CC=C2CC2=C1C=CC=C2 BYDPSHUZBXOTOM-UHFFFAOYSA-N 0.000 description 2
- VJNGGOMRUHYAMC-UHFFFAOYSA-N NCC1=CC(F)=CC(F)=C1 Chemical compound NCC1=CC(F)=CC(F)=C1 VJNGGOMRUHYAMC-UHFFFAOYSA-N 0.000 description 2
- OMPNJKFUZZAZRS-UHFFFAOYSA-N NCCC(c1n[nH]nn1)(c(c(Cc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O Chemical compound NCCC(c1n[nH]nn1)(c(c(Cc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O OMPNJKFUZZAZRS-UHFFFAOYSA-N 0.000 description 2
- MYMBYWOWQYHPCX-UHFFFAOYSA-N NCCC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 MYMBYWOWQYHPCX-UHFFFAOYSA-N 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N NCCN Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- LMUSKBNHAPBMAD-VIFPVBQESA-N NOOSC1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound NOOSC1=CC2=C(C=C1)[C@@H](N)CC2 LMUSKBNHAPBMAD-VIFPVBQESA-N 0.000 description 2
- LMUSKBNHAPBMAD-SECBINFHSA-N NOOSC1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound NOOSC1=CC2=C(C=C1)[C@H](N)CC2 LMUSKBNHAPBMAD-SECBINFHSA-N 0.000 description 2
- DMSWDNXXNCUYLN-SNVBAGLBSA-N N[C@@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N[C@@H](C1=CC=C(F)C=C1)C1CC1 DMSWDNXXNCUYLN-SNVBAGLBSA-N 0.000 description 2
- URHZIKVJKQVHEV-LLVKDONJSA-N N[C@@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N[C@@H](C1=CC=C(F)C=C1)C1CCC1 URHZIKVJKQVHEV-LLVKDONJSA-N 0.000 description 2
- NTNLGQWPYJTABN-GFCCVEGCSA-N N[C@@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N[C@@H](C1=CC=C(F)C=C1)C1CCCC1 NTNLGQWPYJTABN-GFCCVEGCSA-N 0.000 description 2
- OYCUPNFRMCKIHN-CYBMUJFWSA-N N[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 OYCUPNFRMCKIHN-CYBMUJFWSA-N 0.000 description 2
- ZFBHHXHSPRMSQS-SECBINFHSA-N N[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 ZFBHHXHSPRMSQS-SECBINFHSA-N 0.000 description 2
- HGSPZRYUXCSCBR-SECBINFHSA-N N[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 HGSPZRYUXCSCBR-SECBINFHSA-N 0.000 description 2
- POTIEZMRLKEJOZ-SECBINFHSA-N N[C@@H]1CCC2=C1C=CC(F)=C2 Chemical compound N[C@@H]1CCC2=C1C=CC(F)=C2 POTIEZMRLKEJOZ-SECBINFHSA-N 0.000 description 2
- DMSWDNXXNCUYLN-JTQLQIEISA-N N[C@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N[C@H](C1=CC=C(F)C=C1)C1CC1 DMSWDNXXNCUYLN-JTQLQIEISA-N 0.000 description 2
- URHZIKVJKQVHEV-NSHDSACASA-N N[C@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N[C@H](C1=CC=C(F)C=C1)C1CCC1 URHZIKVJKQVHEV-NSHDSACASA-N 0.000 description 2
- OYCUPNFRMCKIHN-ZDUSSCGKSA-N N[C@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N[C@H](C1=CC=C(F)C=C1)C1CCCCC1 OYCUPNFRMCKIHN-ZDUSSCGKSA-N 0.000 description 2
- SRQPEYLZIUEVIA-MRVPVSSYSA-N N[C@H](CO)C1=CC=C(F)C=C1 Chemical compound N[C@H](CO)C1=CC=C(F)C=C1 SRQPEYLZIUEVIA-MRVPVSSYSA-N 0.000 description 2
- ZFBHHXHSPRMSQS-VIFPVBQESA-N N[C@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N[C@H]1CCC2=C1C=CC(C(=O)O)=C2 ZFBHHXHSPRMSQS-VIFPVBQESA-N 0.000 description 2
- HGSPZRYUXCSCBR-VIFPVBQESA-N N[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 HGSPZRYUXCSCBR-VIFPVBQESA-N 0.000 description 2
- POTIEZMRLKEJOZ-VIFPVBQESA-N N[C@H]1CCC2=C1C=CC(F)=C2 Chemical compound N[C@H]1CCC2=C1C=CC(F)=C2 POTIEZMRLKEJOZ-VIFPVBQESA-N 0.000 description 2
- VYVZFMVKNZQIKD-JTQLQIEISA-N [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N)CC2 VYVZFMVKNZQIKD-JTQLQIEISA-N 0.000 description 2
- VYVZFMVKNZQIKD-SNVBAGLBSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@H](N)CC2 VYVZFMVKNZQIKD-SNVBAGLBSA-N 0.000 description 2
- YRPQEGYDPCXHSA-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(CN)C=C1 Chemical compound [C-]#[N+]C1=CC=C(CN)C=C1 YRPQEGYDPCXHSA-UHFFFAOYSA-N 0.000 description 2
- CBJLBXCGJVTODE-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(N)C=C1 CBJLBXCGJVTODE-UHFFFAOYSA-N 0.000 description 2
- ZAHSUUCITVTJEL-SSDOTTSWSA-N [C-]#[N+]C1=CC=C([C@@H](C)N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@@H](C)N)C=C1 ZAHSUUCITVTJEL-SSDOTTSWSA-N 0.000 description 2
- ZAHSUUCITVTJEL-ZETCQYMHSA-N [C-]#[N+]C1=CC=C([C@H](C)N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@H](C)N)C=C1 ZAHSUUCITVTJEL-ZETCQYMHSA-N 0.000 description 2
- GIHNRKGWULZCLQ-UHFFFAOYSA-N [C-]#[N+]C1=CC=CC(N)=C1 Chemical compound [C-]#[N+]C1=CC=CC(N)=C1 GIHNRKGWULZCLQ-UHFFFAOYSA-N 0.000 description 2
- WBLUTFCFGXJZPO-UHFFFAOYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 WBLUTFCFGXJZPO-UHFFFAOYSA-N 0.000 description 2
- ICTPKWOCFYEJSK-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ICTPKWOCFYEJSK-QFIPXVFZSA-N 0.000 description 2
- OCVBZOIBMOJRTR-NDEPHWFRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 OCVBZOIBMOJRTR-NDEPHWFRSA-N 0.000 description 2
- IWYSQYCBVURELT-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 IWYSQYCBVURELT-VWLOTQADSA-N 0.000 description 2
- SUPXJLUNBSVPDI-DVECYGJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 SUPXJLUNBSVPDI-DVECYGJZSA-N 0.000 description 2
- HIFDUBSUHUAZCZ-RLWLMLJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 HIFDUBSUHUAZCZ-RLWLMLJZSA-N 0.000 description 2
- YMIYEVNBPLHDFI-DVECYGJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 YMIYEVNBPLHDFI-DVECYGJZSA-N 0.000 description 2
- FJBLDGJRJVYMTD-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 FJBLDGJRJVYMTD-QHCPKHFHSA-N 0.000 description 2
- JVDMVCSRSGGEFL-FIBWVYCGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 JVDMVCSRSGGEFL-FIBWVYCGSA-N 0.000 description 2
- FYYJBJZKUATWEH-UHFFFAOYSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 FYYJBJZKUATWEH-UHFFFAOYSA-N 0.000 description 2
- ASISMOUXFRCWHI-UHFFFAOYSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 ASISMOUXFRCWHI-UHFFFAOYSA-N 0.000 description 2
- WVANHZKACHHNMM-UHFFFAOYSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 WVANHZKACHHNMM-UHFFFAOYSA-N 0.000 description 2
- QFXRNENQKPHGDP-WNJJXGMVSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 QFXRNENQKPHGDP-WNJJXGMVSA-N 0.000 description 2
- ITTZWKZTOXMGHB-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ITTZWKZTOXMGHB-NRFANRHFSA-N 0.000 description 2
- GNSNAIOVJVSADZ-SANMLTNESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GNSNAIOVJVSADZ-SANMLTNESA-N 0.000 description 2
- YAJQXPDQECSILL-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 YAJQXPDQECSILL-NRFANRHFSA-N 0.000 description 2
- FGAXBTCINZZHPE-HRFSGMKKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 FGAXBTCINZZHPE-HRFSGMKKSA-N 0.000 description 2
- ZGRZMQGJLZKSSJ-DVECYGJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ZGRZMQGJLZKSSJ-DVECYGJZSA-N 0.000 description 2
- WVOCRFKQPBGWDX-HXOBKFHXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 WVOCRFKQPBGWDX-HXOBKFHXSA-N 0.000 description 2
- RZHCVZRDJKMPFT-OAQYLSRUSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RZHCVZRDJKMPFT-OAQYLSRUSA-N 0.000 description 2
- DIRNIRRBTQDKDZ-IBGZPJMESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 DIRNIRRBTQDKDZ-IBGZPJMESA-N 0.000 description 2
- MOSOFZHWLHTAMD-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 MOSOFZHWLHTAMD-FQEVSTJZSA-N 0.000 description 2
- XZIDRTVFGFWCDJ-WGVOJFRMSA-N B.CC(C)CC1(C2=NOC(C(F)(F)F)=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(=N)NO.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NOC(C(F)(F)F)=N1.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(C[C@H](C)CC(=O)C(F)(F)F)C1=NOC(C(F)(F)F)=N1 Chemical compound B.CC(C)CC1(C2=NOC(C(F)(F)F)=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(=N)NO.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NOC(C(F)(F)F)=N1.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(C[C@H](C)CC(=O)C(F)(F)F)C1=NOC(C(F)(F)F)=N1 XZIDRTVFGFWCDJ-WGVOJFRMSA-N 0.000 description 1
- KPDRGTFRYWBBPA-GVTJAYESSA-N BrC1=C(C2OCCO2)SC=C1.CC1=CC=C(C(=O)O)S1.CCCC1(C#N)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC=C2.COC(=O)C1=C(C=O)SC=C1.COC(=O)C1=CC2=C(CCC3=C(C=CS3)C2C#N)S1.COC(=O)C1=CC2=C(CCC3=C(C=CS3)C2O)S1.COC(=O)C1=CC=C(CBr)S1.COC(=O)C1=CC=C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)S1.O=C(O)C1=CC2=C(CCC3=C(C=CS3)C2=O)S1.O=C(O)C1=CC=C(/C=C\C2=C(C(=O)O)C=CS2)S1.O=C(O)C1=CC=C(/C=C\C2=C(C(=O)O)C=CS2)S1.O=CC1=C(C(=O)O)C=CS1 Chemical compound BrC1=C(C2OCCO2)SC=C1.CC1=CC=C(C(=O)O)S1.CCCC1(C#N)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC=C2.COC(=O)C1=C(C=O)SC=C1.COC(=O)C1=CC2=C(CCC3=C(C=CS3)C2C#N)S1.COC(=O)C1=CC2=C(CCC3=C(C=CS3)C2O)S1.COC(=O)C1=CC=C(CBr)S1.COC(=O)C1=CC=C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)S1.O=C(O)C1=CC2=C(CCC3=C(C=CS3)C2=O)S1.O=C(O)C1=CC=C(/C=C\C2=C(C(=O)O)C=CS2)S1.O=C(O)C1=CC=C(/C=C\C2=C(C(=O)O)C=CS2)S1.O=CC1=C(C(=O)O)C=CS1 KPDRGTFRYWBBPA-GVTJAYESSA-N 0.000 description 1
- FKBLJGMGWNAIBP-UHFFFAOYSA-N BrC1=CC=CC=C1.CC(C)CC(C1=CC=C(Br)C=C1)(C1=CC=C(Br)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)O)C=C1)(C1=CC=C(C(=O)O)C=C1)C1=NNN=N1.COC(=O)C1=CC=C(C(CC(C)C)(C2=CC=C(C(=O)OC)C=C2)C2=NNN=N2)C=C1.N#CC(C1=CC=C(Br)C=C1)C1=CC=C(Br)C=C1.[C-]#[N+]C(CC(C)C)(C1=CC=C(Br)C=C1)C1=CC=C(Br)C=C1.[H]C(=O)C1=CC=C(Br)C=C1 Chemical compound BrC1=CC=CC=C1.CC(C)CC(C1=CC=C(Br)C=C1)(C1=CC=C(Br)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)O)C=C1)(C1=CC=C(C(=O)O)C=C1)C1=NNN=N1.COC(=O)C1=CC=C(C(CC(C)C)(C2=CC=C(C(=O)OC)C=C2)C2=NNN=N2)C=C1.N#CC(C1=CC=C(Br)C=C1)C1=CC=C(Br)C=C1.[C-]#[N+]C(CC(C)C)(C1=CC=C(Br)C=C1)C1=CC=C(Br)C=C1.[H]C(=O)C1=CC=C(Br)C=C1 FKBLJGMGWNAIBP-UHFFFAOYSA-N 0.000 description 1
- ZLGAECKMSRGUJU-UHFFFAOYSA-N BrCC1=CC=CC(CBr)=C1.CC(=O)C1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC(CC(=O)O)CC1=CC=CC(CC(C)CC(=O)O)=C1.CC(CC1=CC=CC(CC(C)C(=O)O)=C1)C(=O)O.CC(N)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC(O)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC1CC(=O)C2=CC3=C(C=C2C1)CC(C)CC3=O.CC1CCC2=C(C(=O)O)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(C(N)=O)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CC#N)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CC(C)N=[N+]=[N-])C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CCN)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=CC3=C(C=C2C1)CC(C)CC3.CCOC(=O)CC(C)(CC1=CC=CC(CC(C)(CC(=O)OCC)C(=O)OCC)=C1)C(=O)OCC.[H]C(=O)C(=O)C1=C2CCC(C)CC2=CC2=C1CCC(C)C2.[H]C(=O)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2 Chemical compound BrCC1=CC=CC(CBr)=C1.CC(=O)C1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC(CC(=O)O)CC1=CC=CC(CC(C)CC(=O)O)=C1.CC(CC1=CC=CC(CC(C)C(=O)O)=C1)C(=O)O.CC(N)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC(O)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2.CC1CC(=O)C2=CC3=C(C=C2C1)CC(C)CC3=O.CC1CCC2=C(C(=O)O)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(C(N)=O)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CC#N)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CC(C)N=[N+]=[N-])C3=C(C=C2C1)CC(C)CC3.CC1CCC2=C(CCN)C3=C(C=C2C1)CC(C)CC3.CC1CCC2=CC3=C(C=C2C1)CC(C)CC3.CCOC(=O)CC(C)(CC1=CC=CC(CC(C)(CC(=O)OCC)C(=O)OCC)=C1)C(=O)OCC.[H]C(=O)C(=O)C1=C2CCC(C)CC2=CC2=C1CCC(C)C2.[H]C(=O)CC1=C2CCC(C)CC2=CC2=C1CCC(C)C2 ZLGAECKMSRGUJU-UHFFFAOYSA-N 0.000 description 1
- DPLXZGYFHJPFSS-HFSRJZRUSA-O C.ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2.N#C[C@@H]1CCCN1C(=O)CNC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.N#C[C@@H]1CCCN1C(=O)C[NH3+].[Cl-] Chemical compound C.ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2.N#C[C@@H]1CCCN1C(=O)CNC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.N#C[C@@H]1CCCN1C(=O)C[NH3+].[Cl-] DPLXZGYFHJPFSS-HFSRJZRUSA-O 0.000 description 1
- YUHCTCLQVZQPEY-UHFFFAOYSA-N C1=C2CCCC2=CC2=C1CCC2.C1=CC2=C(C=C1)CCC2.CC(=O)C1=C2CCCC2=CC2=C1CCC2.CC(CC1=C2CCCC2=CC2=C1CCC2)N=[N+]=[N-].CC(N)CC1=C2CCCC2=CC2=C1CCC2.CC(O)CC1=C2CCCC2=CC2=C1CCC2.N#CCC1=C2CCCC2=CC2=C1CCC2.NCCC1=C2CCCC2=CC2=C1CCC2.O=C(CCCl)C1=CC2=C(C=C1)CCC2.O=C(O)C1=C2CCCC2=CC2=C1CCC2.O=C1CCC2=CC3=C(C=C12)CCC3.OCC1=C2CCCC2=CC2=C1CCC2.[H]C(=O)CC1=C2CCCC2=CC2=C1CCC2 Chemical compound C1=C2CCCC2=CC2=C1CCC2.C1=CC2=C(C=C1)CCC2.CC(=O)C1=C2CCCC2=CC2=C1CCC2.CC(CC1=C2CCCC2=CC2=C1CCC2)N=[N+]=[N-].CC(N)CC1=C2CCCC2=CC2=C1CCC2.CC(O)CC1=C2CCCC2=CC2=C1CCC2.N#CCC1=C2CCCC2=CC2=C1CCC2.NCCC1=C2CCCC2=CC2=C1CCC2.O=C(CCCl)C1=CC2=C(C=C1)CCC2.O=C(O)C1=C2CCCC2=CC2=C1CCC2.O=C1CCC2=CC3=C(C=C12)CCC3.OCC1=C2CCCC2=CC2=C1CCC2.[H]C(=O)CC1=C2CCCC2=CC2=C1CCC2 YUHCTCLQVZQPEY-UHFFFAOYSA-N 0.000 description 1
- HVLKZENSXWWZOR-UHFFFAOYSA-N C1=C2CCCCC2=CC2=C1CCCC2.CC(=O)C1=C2CCCCC2=CC2=C1CCCC2.CC(CC1=C2CCCCC2=CC2=C1CCCC2)N=[N+]=[N-].CC(N)CC1=C2CCCCC2=CC2=C1CCCC2.CC(O)CC1=C2CCCCC2=CC2=C1CCCC2.N#CCC1=C2CCCCC2=CC2=C1CCCC2.NC(=O)C1=C2CCCCC2=CC2=C1CCCC2.NCCC1=C2CCCCC2=CC2=C1CCCC2.O=C(O)C1=C2CCCCC2=CC2=C1CCCC2.[H]C(=O)C(=O)C1=C2CCCCC2=CC2=C1CCCC2.[H]C(=O)CC1=C2CCCCC2=CC2=C1CCCC2 Chemical compound C1=C2CCCCC2=CC2=C1CCCC2.CC(=O)C1=C2CCCCC2=CC2=C1CCCC2.CC(CC1=C2CCCCC2=CC2=C1CCCC2)N=[N+]=[N-].CC(N)CC1=C2CCCCC2=CC2=C1CCCC2.CC(O)CC1=C2CCCCC2=CC2=C1CCCC2.N#CCC1=C2CCCCC2=CC2=C1CCCC2.NC(=O)C1=C2CCCCC2=CC2=C1CCCC2.NCCC1=C2CCCCC2=CC2=C1CCCC2.O=C(O)C1=C2CCCCC2=CC2=C1CCCC2.[H]C(=O)C(=O)C1=C2CCCCC2=CC2=C1CCCC2.[H]C(=O)CC1=C2CCCCC2=CC2=C1CCCC2 HVLKZENSXWWZOR-UHFFFAOYSA-N 0.000 description 1
- NRHPUDDOAHWHAD-UHFFFAOYSA-N C1=CC2=C(C=C1)NC1=C(C=CC=C1)CC2.NCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2.[N-]=[N+]=NCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound C1=CC2=C(C=C1)NC1=C(C=CC=C1)CC2.NCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2.[N-]=[N+]=NCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 NRHPUDDOAHWHAD-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N C1CSCN1 Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- KFCWFXWXRVTLPH-UHFFFAOYSA-N C=CC=C.CC(C)CC1(C#N)C2CCCCC2CC2CCCCC21.CC(C)CC1(C2=NCN=N2)C2CCCCC2CC2CCCCC21.CC(C)CC1(C2=NCN=N2)C2CCCCC2CC2CCCCC21.ClC1C2CCCCC2CC2CCCCC21.N#CC1C2CCCCC2CC2CCCCC21.O=C1C2CC=CCC2C(=O)C2CC=CCC12.O=C1C2CC=CCC2C(O)C2CC=CCC12.O=C1C2CCCCC2CC2CCCCC12.O=C1C=CC(=O)C=C1.OC1C2CCCCC2CC2CCCCC21 Chemical compound C=CC=C.CC(C)CC1(C#N)C2CCCCC2CC2CCCCC21.CC(C)CC1(C2=NCN=N2)C2CCCCC2CC2CCCCC21.CC(C)CC1(C2=NCN=N2)C2CCCCC2CC2CCCCC21.ClC1C2CCCCC2CC2CCCCC21.N#CC1C2CCCCC2CC2CCCCC21.O=C1C2CC=CCC2C(=O)C2CC=CCC12.O=C1C2CC=CCC2C(O)C2CC=CCC12.O=C1C2CCCCC2CC2CCCCC12.O=C1C=CC(=O)C=C1.OC1C2CCCCC2CC2CCCCC21 KFCWFXWXRVTLPH-UHFFFAOYSA-N 0.000 description 1
- LYTKMPLYWOBNFM-UHFFFAOYSA-N C=CCBr.CC1(C)OB(C(Cl)CCCBr)OC1(C)C.CC1(C)OB(C2CCCN2)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)CBr)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)CNCCC2C3=C(C=CC=C3)CCC3=C2C=CC=C3)OC1(C)C.CC1(C)OB(C2CCCN2[Si](C)(C)C)OC1(C)C.CC1(C)OB(CCCBr)OC1(C)C.O=C(CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2)N1CCCC1B(O)O Chemical compound C=CCBr.CC1(C)OB(C(Cl)CCCBr)OC1(C)C.CC1(C)OB(C2CCCN2)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)CBr)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)CNCCC2C3=C(C=CC=C3)CCC3=C2C=CC=C3)OC1(C)C.CC1(C)OB(C2CCCN2[Si](C)(C)C)OC1(C)C.CC1(C)OB(CCCBr)OC1(C)C.O=C(CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2)N1CCCC1B(O)O LYTKMPLYWOBNFM-UHFFFAOYSA-N 0.000 description 1
- CDTZUFSSJJUHCN-IMYKFIQTSA-N C=CCOC(=O)C1C[C@H](NC[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.C=CCOC(=O)[C@@H]1C[C@@H](O)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.CC(C)(C)OC(=O)N[C@@H](CN)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN[C@H]1CC(C(=O)O)N(C(=O)OCC2C3=C(C=CC=C3)C3=C2C=CC=C3)C1)C(=O)N1CCC[C@H]1C#N.O=C(O)[C@@H]1C[C@@H](O)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2 Chemical compound C=CCOC(=O)C1C[C@H](NC[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.C=CCOC(=O)[C@@H]1C[C@@H](O)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.CC(C)(C)OC(=O)N[C@@H](CN)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN[C@H]1CC(C(=O)O)N(C(=O)OCC2C3=C(C=CC=C3)C3=C2C=CC=C3)C1)C(=O)N1CCC[C@H]1C#N.O=C(O)[C@@H]1C[C@@H](O)CN1C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2 CDTZUFSSJJUHCN-IMYKFIQTSA-N 0.000 description 1
- NMLBUQZOVZXPLN-UHFFFAOYSA-N CC(=O)C1=C2CC(C)CC2=CC2=C1CC(C)C2.CC(N)CC1=C2CC(C)CC2=CC2=C1CC(C)C2.CC(O)CC1=C2CC(C)CC2=CC2=C1CC(C)C2.CC1=CC2=C(C=CC=C2)C1.CC1CC2=C(C1)C(C(=O)O)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CC#N)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CC(C)N=[N+]=[N-])=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CCN)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CO)=C1CC(C)CC1=C2.CC1CC2=C(C=C(C(=O)C(C)CCl)C=C2)C1.CC1CC2=C(C=C3C(=O)C(C)CC3=C2)C1.CC1CC2=C(C=CC=C2)C1.CC1CC2=CC3=C(C=C2C1)CC(C)C3.[H]C(=O)CC1=C2CC(C)CC2=CC2=C1CC(C)C2 Chemical compound CC(=O)C1=C2CC(C)CC2=CC2=C1CC(C)C2.CC(N)CC1=C2CC(C)CC2=CC2=C1CC(C)C2.CC(O)CC1=C2CC(C)CC2=CC2=C1CC(C)C2.CC1=CC2=C(C=CC=C2)C1.CC1CC2=C(C1)C(C(=O)O)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CC#N)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CC(C)N=[N+]=[N-])=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CCN)=C1CC(C)CC1=C2.CC1CC2=C(C1)C(CO)=C1CC(C)CC1=C2.CC1CC2=C(C=C(C(=O)C(C)CCl)C=C2)C1.CC1CC2=C(C=C3C(=O)C(C)CC3=C2)C1.CC1CC2=C(C=CC=C2)C1.CC1CC2=CC3=C(C=C2C1)CC(C)C3.[H]C(=O)CC1=C2CC(C)CC2=CC2=C1CC(C)C2 NMLBUQZOVZXPLN-UHFFFAOYSA-N 0.000 description 1
- OCDMAUAZRZKGBE-WKCARCATSA-N CC(C)(C)C(c(cc1)ccc1F)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)NC(OC(C)(C)C)=O)[C@H]2C1)C2=O Chemical compound CC(C)(C)C(c(cc1)ccc1F)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)NC(OC(C)(C)C)=O)[C@H]2C1)C2=O OCDMAUAZRZKGBE-WKCARCATSA-N 0.000 description 1
- UEURFCGKWPELIB-AJNGGQMLSA-N CC(C)(C)CC(=O)N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)CC(=O)N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](N)C(=O)N1CCC[C@H]1C#N UEURFCGKWPELIB-AJNGGQMLSA-N 0.000 description 1
- XDIAMRVROCPPBK-UHFFFAOYSA-N CC(C)(C)CN Chemical compound CC(C)(C)CN XDIAMRVROCPPBK-UHFFFAOYSA-N 0.000 description 1
- YHYWVPNMQLMZGG-BYVDMUCRSA-N CC(C)(C)N=C=O.CC(C)(C)NC(=O)N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N.[H]N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)N=C=O.CC(C)(C)NC(=O)N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N.[H]N1C[C@@H]2C[C@H]1C(=O)N2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N YHYWVPNMQLMZGG-BYVDMUCRSA-N 0.000 description 1
- GGCOHDZWRMNHLT-SANMLTNESA-N CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 Chemical compound CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 GGCOHDZWRMNHLT-SANMLTNESA-N 0.000 description 1
- UNAVXIGYXFZIMX-IHOKFDBFSA-N CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 UNAVXIGYXFZIMX-IHOKFDBFSA-N 0.000 description 1
- ZMXCHAJKATZBMM-ALTZYDRJSA-N CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 Chemical compound CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 ZMXCHAJKATZBMM-ALTZYDRJSA-N 0.000 description 1
- BLIJBTGRDCNMPJ-AOKHHQPISA-N CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CC(C)(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 BLIJBTGRDCNMPJ-AOKHHQPISA-N 0.000 description 1
- BGBHJBKORSSFMW-UHFFFAOYSA-N CC(C)(C)NC(=O)C1=NC=CC=C1CCC1=CC(Cl)=CC=C1.CC1=CC=CN=C1C#N.CC1=CC=CN=C1C(=O)NC(C)(C)C.ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2.N#CC1=NC=CC=C1CCC1=CC(Cl)=CC=C1.O=C1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.OC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2 Chemical compound CC(C)(C)NC(=O)C1=NC=CC=C1CCC1=CC(Cl)=CC=C1.CC1=CC=CN=C1C#N.CC1=CC=CN=C1C(=O)NC(C)(C)C.ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2.N#CC1=NC=CC=C1CCC1=CC(Cl)=CC=C1.O=C1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.OC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2 BGBHJBKORSSFMW-UHFFFAOYSA-N 0.000 description 1
- ZMXCHAJKATZBMM-VJHAGTGTSA-N CC(C)(C)NC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC(C)(C)C)=O)c2c1)=O Chemical compound CC(C)(C)NC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC(C)(C)C)=O)c2c1)=O ZMXCHAJKATZBMM-VJHAGTGTSA-N 0.000 description 1
- MVASBCDDJYRRDP-UHFFFAOYSA-O CC(C)(C)OC(=O)CC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2.OCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[Cl-].[N-]=[N+]=NCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[NH3+]CC=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound CC(C)(C)OC(=O)CC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2.OCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[Cl-].[N-]=[N+]=NCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[NH3+]CC=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 MVASBCDDJYRRDP-UHFFFAOYSA-O 0.000 description 1
- GKULDBKCVYPTBJ-MWXYNOGXSA-O CC(C)(C)OC(=O)N(CC(=O)N1CCC[C@H]1C#N)C(=O)OC(C)(C)C.CCl.N#C[C@@H]1CCCN1C(=O)CBr.N#C[C@@H]1CCCN1C(=O)C[NH3+] Chemical compound CC(C)(C)OC(=O)N(CC(=O)N1CCC[C@H]1C#N)C(=O)OC(C)(C)C.CCl.N#C[C@@H]1CCCN1C(=O)CBr.N#C[C@@H]1CCCN1C(=O)C[NH3+] GKULDBKCVYPTBJ-MWXYNOGXSA-O 0.000 description 1
- UYRZXGQNBKFADZ-NIUDVCLJSA-N CC(C)(C)OC(=O)N1C2C[C@H]2C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C2C[C@H]2C[C@H]1C(N)=O.CCOC(=O)[C@@H]1CC=C(=O)N1C(=O)OC(C)(C)C.CCOC(=O)[C@@H]1CCC(=O)N1.CCOC(=O)[C@@H]1CCC(=O)N1C(=O)OC(C)(C)C.CCOC(=O)[C@@H]1C[C@@H]2CC2N1C(=O)OC(C)(C)C.COC(O)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21.N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)C=O.NC(=O)[C@@H]1C[C@@H]2CC2N1.NC(=O)[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C(N)=O)C[C@@H]2C[C@@H]21.[C-]#[N+][C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N1C2C[C@H]2C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C2C[C@H]2C[C@H]1C(N)=O.CCOC(=O)[C@@H]1CC=C(=O)N1C(=O)OC(C)(C)C.CCOC(=O)[C@@H]1CCC(=O)N1.CCOC(=O)[C@@H]1CCC(=O)N1C(=O)OC(C)(C)C.CCOC(=O)[C@@H]1C[C@@H]2CC2N1C(=O)OC(C)(C)C.COC(O)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21.N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)C=O.NC(=O)[C@@H]1C[C@@H]2CC2N1.NC(=O)[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C(N)=O)C[C@@H]2C[C@@H]21.[C-]#[N+][C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 UYRZXGQNBKFADZ-NIUDVCLJSA-N 0.000 description 1
- YWHKDJBAIMWZCV-VBSGGXNUSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)C(F)(F)F.CC(C)(C)OC(=O)N1CCC[C@H]1C(O)C(F)(F)F.CC(C)(C)OC(=O)N1CCC[C@H]1C(O[Si](C)(C)C)C(F)(F)F.OC(O)([C@@H]1CCCN1)C(F)(F)F.[H]C(=O)[C@@H]1CCCN1C(=O)OC(C)(C)C Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)C(F)(F)F.CC(C)(C)OC(=O)N1CCC[C@H]1C(O)C(F)(F)F.CC(C)(C)OC(=O)N1CCC[C@H]1C(O[Si](C)(C)C)C(F)(F)F.OC(O)([C@@H]1CCCN1)C(F)(F)F.[H]C(=O)[C@@H]1CCCN1C(=O)OC(C)(C)C YWHKDJBAIMWZCV-VBSGGXNUSA-N 0.000 description 1
- VYZXKDQEWRKNIP-KKLBQHSYSA-N CC(C)(C)OC(=O)N1C[C@@H](N)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H](NC(=O)OCC2C3=C(C=CC=C3)C3=C2C=CC=C3)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H](NCC2=CC=CC(F)=C2)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1C(=O)N2CC1=CC=CC(F)=C1.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)O.COC(=O)[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)NC(=O)OC(C)(C)C.Cl.Cl.O=C1[C@@H]2C[C@@H](CN2)N1CC1=CC=CC(F)=C1 Chemical compound CC(C)(C)OC(=O)N1C[C@@H](N)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H](NC(=O)OCC2C3=C(C=CC=C3)C3=C2C=CC=C3)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H](NCC2=CC=CC(F)=C2)C[C@H]1C(=O)O.CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1C(=O)N2CC1=CC=CC(F)=C1.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)O.COC(=O)[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)NC(=O)OC(C)(C)C.Cl.Cl.O=C1[C@@H]2C[C@@H](CN2)N1CC1=CC=CC(F)=C1 VYZXKDQEWRKNIP-KKLBQHSYSA-N 0.000 description 1
- AMVLAZMIFVTXEI-FIICGKRPSA-N CC(C)(C)OC(=O)N1C[C@@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)C[C@H]1C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N1C[C@@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)C[C@H]1C(=O)O.COC(=O)[C@@H]1C[C@@H](O)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1C[C@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)CN1C(=O)OC(C)(C)C.Cl.Cl.O=C1[C@@H]2C[C@@H](CN2)N1CC1=CC=CC(F)=C1 Chemical compound CC(C)(C)OC(=O)N1C[C@@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)C[C@H]1C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N1C[C@@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)C[C@H]1C(=O)O.COC(=O)[C@@H]1C[C@@H](O)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1C[C@H](N2C[C@@H]3C[C@H]2C(=O)N3CC2=CC(F)=CC=C2)CN1C(=O)OC(C)(C)C.Cl.Cl.O=C1[C@@H]2C[C@@H](CN2)N1CC1=CC=CC(F)=C1 AMVLAZMIFVTXEI-FIICGKRPSA-N 0.000 description 1
- UXAWXZDXVOYLII-YUMQZZPRSA-N CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1CN2 Chemical compound CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1CN2 UXAWXZDXVOYLII-YUMQZZPRSA-N 0.000 description 1
- NVJNQTUEIYKZGP-ASJUWNHESA-N CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1CN2.COC(=O)C1CN1C(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2)NC(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)NC(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)OC(C)(C)C)NC(=O)OCC1=CC=CC=C1.N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)NC(=O)OCC1=CC=CC=C1.O=C(N[C@@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)C(=O)O)OCC1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N1C[C@@H]2C[C@H]1CN2.COC(=O)C1CN1C(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2)NC(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)NC(=O)OCC1=CC=CC=C1.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)OC(C)(C)C)NC(=O)OCC1=CC=CC=C1.N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)NC(=O)OCC1=CC=CC=C1.O=C(N[C@@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1)C(=O)O)OCC1=CC=CC=C1 NVJNQTUEIYKZGP-ASJUWNHESA-N 0.000 description 1
- DOJPPYKZRMDZFH-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1.ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1 Chemical compound CC(C)(C)OC(=O)NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1.ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1 DOJPPYKZRMDZFH-UHFFFAOYSA-N 0.000 description 1
- DQQJBEAXSOOCPG-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1CCNC1 Chemical compound CC(C)(C)OC(=O)NC1CCNC1 DQQJBEAXSOOCPG-UHFFFAOYSA-N 0.000 description 1
- NEMXVXVJGXZDRR-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1CNC1 Chemical compound CC(C)(C)OC(=O)NC1CNC1 NEMXVXVJGXZDRR-UHFFFAOYSA-N 0.000 description 1
- LERIMCAEAUDZIG-UHFFFAOYSA-N CC(C)(C)OC(=O)NCCC1(C(=N)NC(=O)OC(C)(C)C)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.COC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCNC(=O)OC(C)(C)C)(C(=N)NC(=O)OC(C)(C)C)C1=C(C=C(C(=O)OC)C=C1)CC2.N=C(N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)(C)OC(=O)NCCC1(C(=N)NC(=O)OC(C)(C)C)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.COC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCNC(=O)OC(C)(C)C)(C(=N)NC(=O)OC(C)(C)C)C1=C(C=C(C(=O)OC)C=C1)CC2.N=C(N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 LERIMCAEAUDZIG-UHFFFAOYSA-N 0.000 description 1
- YRORKLLHSFDQBJ-UHFFFAOYSA-N CC(C)(C)OC(=O)NCCC1(C(=O)NN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CC(C)(C)OC(=O)NCCC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCOC(=O)NC(=O)NNC(=O)C1(CCNC(=O)OC(C)(C)C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)(C)OC(=O)NCCC1(C(=O)NN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CC(C)(C)OC(=O)NCCC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCOC(=O)NC(=O)NNC(=O)C1(CCNC(=O)OC(C)(C)C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 YRORKLLHSFDQBJ-UHFFFAOYSA-N 0.000 description 1
- KLZZPCMFCWTUDW-UHFFFAOYSA-N CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C(=O)O)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)(C)OC(=O)NCCC1(C(=O)O)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C(=O)O)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 KLZZPCMFCWTUDW-UHFFFAOYSA-N 0.000 description 1
- ZOKWJFZTDUNEFZ-UHFFFAOYSA-O CC(C)(C)OC(=O)NCCC1(C(N)=O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=C(F)C=C2.CC(C)(C)OC(=O)NCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=C(F)C=C2.CC1=CC(Br)=CC=C1Br.CC1=CC(C(=O)O)=CC=C1C(=O)O.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC=C(C(=O)OC)C(C)=C1.COC(=O)C1=CC=C(C(=O)OC)C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)=C1.NC(=O)C1=CC2=C(C=C1)C(CC[NH3+])(C(N)=O)C1=C(C=CC(F)=C1)CC2.O=C(O)C1=CC=C(C(=O)O)C(CCC2=CC=C(F)C=C2)=C1.[C-]#[N+]C1(CCN=[N+]=[N-])C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCN=[N+]=[N-])C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=C(F)C=C2.[Cl-] Chemical compound CC(C)(C)OC(=O)NCCC1(C(N)=O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=C(F)C=C2.CC(C)(C)OC(=O)NCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=C(F)C=C2.CC1=CC(Br)=CC=C1Br.CC1=CC(C(=O)O)=CC=C1C(=O)O.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=CC(F)=C1)CC2.COC(=O)C1=CC=C(C(=O)OC)C(C)=C1.COC(=O)C1=CC=C(C(=O)OC)C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)=C1.NC(=O)C1=CC2=C(C=C1)C(CC[NH3+])(C(N)=O)C1=C(C=CC(F)=C1)CC2.O=C(O)C1=CC=C(C(=O)O)C(CCC2=CC=C(F)C=C2)=C1.[C-]#[N+]C1(CCN=[N+]=[N-])C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCN=[N+]=[N-])C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=C(F)C=C2.[Cl-] ZOKWJFZTDUNEFZ-UHFFFAOYSA-O 0.000 description 1
- ONYMDNZWTMCSIK-UHFFFAOYSA-N CC(C)(C)OC(=O)NCCC1(C2=NN=NN2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1/C=C\C(F)=C/2 Chemical compound CC(C)(C)OC(=O)NCCC1(C2=NN=NN2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1/C=C\C(F)=C/2 ONYMDNZWTMCSIK-UHFFFAOYSA-N 0.000 description 1
- ICCGBDJYFIYSPT-UHFFFAOYSA-N CC(C)(C)OC(=O)NCCC1(C2=NN=NN2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCNC(=O)OC(C)(C)C)C1=NN=NN1.Cl.NCCC1(C2=NN=NN2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2 Chemical compound CC(C)(C)OC(=O)NCCC1(C2=NN=NN2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCNC(=O)OC(C)(C)C)C1=NN=NN1.Cl.NCCC1(C2=NN=NN2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2 ICCGBDJYFIYSPT-UHFFFAOYSA-N 0.000 description 1
- VTOXDICKFULCAB-RKIHRLAKSA-N CC(C)(C)OC(=O)NCCC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCN1C(=O)C3=C(C=CC=C3)C1=O)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCNC(=O)OC(C)(C)C)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC=C(C(=O)OC)C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)=C1.O=C(O)C1=CC2=C(C=C1)C(=O)C1=C(C=C(Br)C=C1)CC2.O=C(O)C1=CC=C(C(=O)O)C(/C=C/C2=CC=CC(Br)=C2)=C1.O=C(O)C1=CC=C(C(=O)O)C(CCC2=CC=CC(Br)=C2)=C1.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2 Chemical compound CC(C)(C)OC(=O)NCCC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCN1C(=O)C3=C(C=CC=C3)C1=O)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(CCNC(=O)OC(C)(C)C)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC2=C(C=C1)C(O)C1=C(C=C(C(=O)OC)C=C1)CC2.COC(=O)C1=CC=C(C(=O)OC)C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)=C1.O=C(O)C1=CC2=C(C=C1)C(=O)C1=C(C=C(Br)C=C1)CC2.O=C(O)C1=CC=C(C(=O)O)C(/C=C/C2=CC=CC(Br)=C2)=C1.O=C(O)C1=CC=C(C(=O)O)C(CCC2=CC=CC(Br)=C2)=C1.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2 VTOXDICKFULCAB-RKIHRLAKSA-N 0.000 description 1
- LWAMAJACNUSUQZ-VNJQCMGLSA-N CC(C)(C)OC(=O)N[C@@H](CN)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)O.N#C[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)O.N#C[C@@H]1CCCN1 LWAMAJACNUSUQZ-VNJQCMGLSA-N 0.000 description 1
- VPHKGCYCMIDHHP-ZWMODFHLSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.O=C(O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N.O=C(O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 VPHKGCYCMIDHHP-ZWMODFHLSA-N 0.000 description 1
- LMQZDYQGNPLLFM-NVORGNFDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N LMQZDYQGNPLLFM-NVORGNFDSA-N 0.000 description 1
- QWWIYGWOHVAZIJ-QAETUUGQSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=CC=CS1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=CC=CS1)C(=O)N1CCC[C@H]1C#N QWWIYGWOHVAZIJ-QAETUUGQSA-N 0.000 description 1
- PIARZMUPOWODJF-XSLAGTTESA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=CSC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=CSC=C1)C(=O)N1CCC[C@H]1C#N PIARZMUPOWODJF-XSLAGTTESA-N 0.000 description 1
- BDYPXJCCNBPEJV-LAQRGFTBSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=NC=CN=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=NC=CN=C1)C(=O)N1CCC[C@H]1C#N BDYPXJCCNBPEJV-LAQRGFTBSA-N 0.000 description 1
- IOELHXAHTGKPKK-QWEJMYKISA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1 IOELHXAHTGKPKK-QWEJMYKISA-N 0.000 description 1
- FFOSRCMBOTYATL-QAETUUGQSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CS1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CS1)C(=O)N1CCC[C@H]1C#N FFOSRCMBOTYATL-QAETUUGQSA-N 0.000 description 1
- XPNKVMXAZSUYEU-LAQRGFTBSA-N CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CSC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CSC=C1)C(=O)N1CCC[C@H]1C#N XPNKVMXAZSUYEU-LAQRGFTBSA-N 0.000 description 1
- AGBHEZJVJIKWSJ-ABJCFRMWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1B1OC(C)(C)C(C)(C)O1.CC1(C)OB(C2CCCN2)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)[C@@H](N)CN2C[C@@H]3CC2C(=O)N3CC2=CC(F)=CC=C2)OC1(C)C.N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC=CC(F)=C1)C(=O)N1CCCC1B(O)O Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1B1OC(C)(C)C(C)(C)O1.CC1(C)OB(C2CCCN2)OC1(C)C.CC1(C)OB(C2CCCN2C(=O)[C@@H](N)CN2C[C@@H]3CC2C(=O)N3CC2=CC(F)=CC=C2)OC1(C)C.N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC=CC(F)=C1)C(=O)N1CCCC1B(O)O AGBHEZJVJIKWSJ-ABJCFRMWSA-N 0.000 description 1
- QFQMMENFXDDSKB-HXIUWYKDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1C(=O)C(F)(F)F.CC(O)(O)C1CCCN1.N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1C(=O)C(F)(F)F Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1C(=O)C(F)(F)F.CC(O)(O)C1CCCN1.N[C@@H](CN1C[C@@H]2CC1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCCC1C(=O)C(F)(F)F QFQMMENFXDDSKB-HXIUWYKDSA-N 0.000 description 1
- VKHQEKKXJUPIDA-CQJMVLFOSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N VKHQEKKXJUPIDA-CQJMVLFOSA-N 0.000 description 1
- SNHHALCEMFJFNC-VUXQNLRASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SNHHALCEMFJFNC-VUXQNLRASA-N 0.000 description 1
- GPGVTJPABBITLL-MLCQCVOFSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1)C(=O)N1CCC[C@H]1C#N GPGVTJPABBITLL-MLCQCVOFSA-N 0.000 description 1
- WREGAEJZKYWUQA-UEHMALFGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 WREGAEJZKYWUQA-UEHMALFGSA-N 0.000 description 1
- RAWJOIOWQUDLNP-ZJZGAYNASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1)C(=O)N1CCC[C@H]1C#N RAWJOIOWQUDLNP-ZJZGAYNASA-N 0.000 description 1
- SPEFLJSRZFNHNI-LZFDFFMTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SPEFLJSRZFNHNI-LZFDFFMTSA-N 0.000 description 1
- ZSFSWWUSTBJBMA-QORCZRPOSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1)C(=O)N1CCC[C@H]1C#N ZSFSWWUSTBJBMA-QORCZRPOSA-N 0.000 description 1
- KBWNPXPPQSISAC-ROMXMSMSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KBWNPXPPQSISAC-ROMXMSMSSA-N 0.000 description 1
- CIGAEXVZVIJFEQ-IWFBPKFRSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C#N)C=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C#N)C=CC=C1)C(=O)N1CCC[C@H]1C#N CIGAEXVZVIJFEQ-IWFBPKFRSA-N 0.000 description 1
- BNYOOXMWUABSAS-RMLDPDKQSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C#N)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C#N)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BNYOOXMWUABSAS-RMLDPDKQSA-N 0.000 description 1
- VZRUBYZTHCGFTN-DUPRYPCISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1)C(=O)N1CCC[C@H]1C#N VZRUBYZTHCGFTN-DUPRYPCISA-N 0.000 description 1
- IOOOTSAHWSGFIL-GWKSQZHDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 IOOOTSAHWSGFIL-GWKSQZHDSA-N 0.000 description 1
- XFMKFYATAGTTKN-DUPRYPCISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1)C(=O)N1CCC[C@H]1C#N XFMKFYATAGTTKN-DUPRYPCISA-N 0.000 description 1
- SWOOMZCAOUFQQC-GWKSQZHDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SWOOMZCAOUFQQC-GWKSQZHDSA-N 0.000 description 1
- XTAXAFBBHSDROT-TUFLPTIASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C#N)=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C#N)=CC=C1)C(=O)N1CCC[C@H]1C#N XTAXAFBBHSDROT-TUFLPTIASA-N 0.000 description 1
- QLOLBUIGPYCAIP-NWFSVNAMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C#N)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C#N)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QLOLBUIGPYCAIP-NWFSVNAMSA-N 0.000 description 1
- KJMDHROXZMPCBE-LEAZDLGRSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)C(=O)N1CCC[C@H]1C#N KJMDHROXZMPCBE-LEAZDLGRSA-N 0.000 description 1
- OJJJQYABDRMIMD-JLUSAJDUSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OJJJQYABDRMIMD-JLUSAJDUSA-N 0.000 description 1
- ILCFCIXGYHRFJX-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1)C(=O)N1CCC[C@H]1C#N ILCFCIXGYHRFJX-MUGJNUQGSA-N 0.000 description 1
- JBDHARNGYODTOW-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1)C(=O)N1CCC[C@H]1C#N JBDHARNGYODTOW-MUGJNUQGSA-N 0.000 description 1
- KCRMORAOGBUFLC-WUEZWNDSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KCRMORAOGBUFLC-WUEZWNDSSA-N 0.000 description 1
- PDCYNXRFZVSKQJ-LEAZDLGRSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1)C(=O)N1CCC[C@H]1C#N PDCYNXRFZVSKQJ-LEAZDLGRSA-N 0.000 description 1
- RRUHSUIVNCZMIQ-JLUSAJDUSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RRUHSUIVNCZMIQ-JLUSAJDUSA-N 0.000 description 1
- OUEBENDYUFKEMA-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1)C(=O)N1CCC[C@H]1C#N OUEBENDYUFKEMA-MUGJNUQGSA-N 0.000 description 1
- VELRBKKNCXKSRM-WUEZWNDSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VELRBKKNCXKSRM-WUEZWNDSSA-N 0.000 description 1
- XXMKGNLDPTUXHV-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1)C(=O)N1CCC[C@H]1C#N XXMKGNLDPTUXHV-MUGJNUQGSA-N 0.000 description 1
- XPHHAPWIPSGAPQ-WUEZWNDSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XPHHAPWIPSGAPQ-WUEZWNDSSA-N 0.000 description 1
- XPBWLVVCAGEQNW-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1)C(=O)N1CCC[C@H]1C#N XPBWLVVCAGEQNW-MUGJNUQGSA-N 0.000 description 1
- RRIGKQIBRNHMNA-FALOATQWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RRIGKQIBRNHMNA-FALOATQWSA-N 0.000 description 1
- LWVUTJDATUOOQI-MUGJNUQGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1)C(=O)N1CCC[C@H]1C#N LWVUTJDATUOOQI-MUGJNUQGSA-N 0.000 description 1
- QSWWJPJGLGQFBH-FALOATQWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QSWWJPJGLGQFBH-FALOATQWSA-N 0.000 description 1
- UGKXMTMJVNDFMT-HKDRDPIHSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N UGKXMTMJVNDFMT-HKDRDPIHSA-N 0.000 description 1
- ONMMLKXTKVGSKQ-MLOXMCPPSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ONMMLKXTKVGSKQ-MLOXMCPPSA-N 0.000 description 1
- AOGIKMYVZWNQET-ZCUSHOGSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N AOGIKMYVZWNQET-ZCUSHOGSSA-N 0.000 description 1
- BWAGYPBBXJKPRX-FPDMQFDYSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BWAGYPBBXJKPRX-FPDMQFDYSA-N 0.000 description 1
- MGIBJHBLSLERBI-LSGCGUROSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2)C(=O)N1CCC[C@H]1C#N MGIBJHBLSLERBI-LSGCGUROSA-N 0.000 description 1
- GBTGIFXEXMXYHT-VHSKNJRQSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 GBTGIFXEXMXYHT-VHSKNJRQSA-N 0.000 description 1
- BASBQDVLSVMAST-LRGYGIHKSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=C(Cl)C=CC=C1Cl)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=C(Cl)C=CC=C1Cl)C(=O)N1CCC[C@H]1C#N BASBQDVLSVMAST-LRGYGIHKSA-N 0.000 description 1
- MDLIJGWZOUGNPZ-TUFLPTIASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(C(N)=O)=CC=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(C(N)=O)=CC=C1)C(=O)N1CCC[C@H]1C#N MDLIJGWZOUGNPZ-TUFLPTIASA-N 0.000 description 1
- UYRGEZSEPOCYJS-TUFLPTIASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1)C(=O)N1CCC[C@H]1C#N UYRGEZSEPOCYJS-TUFLPTIASA-N 0.000 description 1
- NZVCXYXRNDVQHD-HHHIRMLJSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 NZVCXYXRNDVQHD-HHHIRMLJSA-N 0.000 description 1
- YFGLQHUWCODYNT-WKKIFSTQSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C(N)=O.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)O Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C#N.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C(N)=O.CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)O YFGLQHUWCODYNT-WKKIFSTQSA-N 0.000 description 1
- WGLLHSDSTBIJLA-RRRPRTLKSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1 WGLLHSDSTBIJLA-RRRPRTLKSA-N 0.000 description 1
- FZSLSCHDYXYXEF-NWFSVNAMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FZSLSCHDYXYXEF-NWFSVNAMSA-N 0.000 description 1
- WDKGOROLFLKVRE-CMOCDZPBSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC=C(NC=O)C=C1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC=C(NC=O)C=C1)C(=O)N1CCC[C@H]1C#N WDKGOROLFLKVRE-CMOCDZPBSA-N 0.000 description 1
- OCDMAUAZRZKGBE-LSBAASHUSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1CCC[C@H]1C#N OCDMAUAZRZKGBE-LSBAASHUSA-N 0.000 description 1
- DNXOKGFAINDMJT-AIVCMVMSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DNXOKGFAINDMJT-AIVCMVMSSA-N 0.000 description 1
- HFEZLYYAKGWUHO-QCCYXRBGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1CCC[C@H]1C#N HFEZLYYAKGWUHO-QCCYXRBGSA-N 0.000 description 1
- YPMCUWZWNKUZLT-BQVBQXJWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YPMCUWZWNKUZLT-BQVBQXJWSA-N 0.000 description 1
- ATYUYXNNFQZTHU-KFEALIJWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1CCC[C@H]1C#N ATYUYXNNFQZTHU-KFEALIJWSA-N 0.000 description 1
- ARDCGILHAZMHLB-OMIIONTOSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ARDCGILHAZMHLB-OMIIONTOSA-N 0.000 description 1
- LZMVXUKGKLBLJA-WBAQKLHDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1CCC[C@H]1C#N LZMVXUKGKLBLJA-WBAQKLHDSA-N 0.000 description 1
- AYOIYTQFXDIOFE-ZYUYTQNWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AYOIYTQFXDIOFE-ZYUYTQNWSA-N 0.000 description 1
- PRPHBZQJAZLHJB-JSLVBRCRSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1CCC[C@H]1C#N PRPHBZQJAZLHJB-JSLVBRCRSA-N 0.000 description 1
- FPWNKXSBUIJSCV-QVSIKBJVSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FPWNKXSBUIJSCV-QVSIKBJVSA-N 0.000 description 1
- RUKVOYNRUCWETD-JIORRVSTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1CCC[C@H]1C#N RUKVOYNRUCWETD-JIORRVSTSA-N 0.000 description 1
- FIJUTPCVWNXADY-USTCYXQMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FIJUTPCVWNXADY-USTCYXQMSA-N 0.000 description 1
- RFIPNTWKGWMHHN-JIORRVSTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1CCC[C@H]1C#N RFIPNTWKGWMHHN-JIORRVSTSA-N 0.000 description 1
- RDTCDBKBUNSTFL-USTCYXQMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RDTCDBKBUNSTFL-USTCYXQMSA-N 0.000 description 1
- BEYBJOFNDACTIK-JIORRVSTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1CCC[C@H]1C#N BEYBJOFNDACTIK-JIORRVSTSA-N 0.000 description 1
- LFYUWGMFISHXFV-USTCYXQMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LFYUWGMFISHXFV-USTCYXQMSA-N 0.000 description 1
- URHNCVXJTICDFA-JIORRVSTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2)C(=O)N1CCC[C@H]1C#N URHNCVXJTICDFA-JIORRVSTSA-N 0.000 description 1
- CJRLOBCLMMIFRS-ZPOKYXLNSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CJRLOBCLMMIFRS-ZPOKYXLNSA-N 0.000 description 1
- RKCNELTTZSJUDI-GZEZKRSXSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1CCC[C@H]1C#N RKCNELTTZSJUDI-GZEZKRSXSA-N 0.000 description 1
- XBHUYGYEQKQDDH-VZRIRPNFSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XBHUYGYEQKQDDH-VZRIRPNFSA-N 0.000 description 1
- OCDMAUAZRZKGBE-QCCYXRBGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1CCC[C@H]1C#N OCDMAUAZRZKGBE-QCCYXRBGSA-N 0.000 description 1
- DNXOKGFAINDMJT-BQVBQXJWSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DNXOKGFAINDMJT-BQVBQXJWSA-N 0.000 description 1
- HFEZLYYAKGWUHO-LSBAASHUSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1CCC[C@H]1C#N HFEZLYYAKGWUHO-LSBAASHUSA-N 0.000 description 1
- YPMCUWZWNKUZLT-AIVCMVMSSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YPMCUWZWNKUZLT-AIVCMVMSSA-N 0.000 description 1
- ATYUYXNNFQZTHU-KEOOTSPTSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1CCC[C@H]1C#N ATYUYXNNFQZTHU-KEOOTSPTSA-N 0.000 description 1
- ARDCGILHAZMHLB-CQXMGPFDSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ARDCGILHAZMHLB-CQXMGPFDSA-N 0.000 description 1
- LZMVXUKGKLBLJA-LROMGURASA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1CCC[C@H]1C#N LZMVXUKGKLBLJA-LROMGURASA-N 0.000 description 1
- AYOIYTQFXDIOFE-MGEHJJQHSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AYOIYTQFXDIOFE-MGEHJJQHSA-N 0.000 description 1
- PRPHBZQJAZLHJB-IRGGMKSGSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1CCC[C@H]1C#N PRPHBZQJAZLHJB-IRGGMKSGSA-N 0.000 description 1
- FPWNKXSBUIJSCV-CVSBKWFLSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FPWNKXSBUIJSCV-CVSBKWFLSA-N 0.000 description 1
- RUKVOYNRUCWETD-KHKYPALMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1CCC[C@H]1C#N RUKVOYNRUCWETD-KHKYPALMSA-N 0.000 description 1
- FIJUTPCVWNXADY-TXTYPXRISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FIJUTPCVWNXADY-TXTYPXRISA-N 0.000 description 1
- RFIPNTWKGWMHHN-KHKYPALMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1CCC[C@H]1C#N RFIPNTWKGWMHHN-KHKYPALMSA-N 0.000 description 1
- RDTCDBKBUNSTFL-TXTYPXRISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RDTCDBKBUNSTFL-TXTYPXRISA-N 0.000 description 1
- BEYBJOFNDACTIK-KHKYPALMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1CCC[C@H]1C#N BEYBJOFNDACTIK-KHKYPALMSA-N 0.000 description 1
- LFYUWGMFISHXFV-TXTYPXRISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LFYUWGMFISHXFV-TXTYPXRISA-N 0.000 description 1
- URHNCVXJTICDFA-KHKYPALMSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2)C(=O)N1CCC[C@H]1C#N URHNCVXJTICDFA-KHKYPALMSA-N 0.000 description 1
- CJRLOBCLMMIFRS-UPNLCGCFSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CJRLOBCLMMIFRS-UPNLCGCFSA-N 0.000 description 1
- RKCNELTTZSJUDI-VRIGVRMUSA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1CCC[C@H]1C#N RKCNELTTZSJUDI-VRIGVRMUSA-N 0.000 description 1
- XBHUYGYEQKQDDH-WKXSYDTISA-N CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C)OC(=O)N[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XBHUYGYEQKQDDH-WKXSYDTISA-N 0.000 description 1
- BKQWEYNMTIDPBM-UMURJCAFSA-N CC(C)(C)OC(NC(CN(C[C@H](C1)N2C(CCc3c4)c3ccc4C(NC)=O)C1C2=O)C(N(CCC1)C1C#N)=O)=O Chemical compound CC(C)(C)OC(NC(CN(C[C@H](C1)N2C(CCc3c4)c3ccc4C(NC)=O)C1C2=O)C(N(CCC1)C1C#N)=O)=O BKQWEYNMTIDPBM-UMURJCAFSA-N 0.000 description 1
- UASWMDLGWOIPTP-VOQBTPAYSA-N CC(C)(C)OC(NC(CN(C[C@H](C1)N2C3c4ccccc4-c4c3cccc4)C1C2O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O Chemical compound CC(C)(C)OC(NC(CN(C[C@H](C1)N2C3c4ccccc4-c4c3cccc4)C1C2O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O UASWMDLGWOIPTP-VOQBTPAYSA-N 0.000 description 1
- FZSLSCHDYXYXEF-PRSMZQBUSA-N CC(C)(C)OC(NC(CN(C[C@H](C1)N2Cc3cc(F)ccc3)[C@@H]1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O Chemical compound CC(C)(C)OC(NC(CN(C[C@H](C1)N2Cc3cc(F)ccc3)[C@@H]1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O FZSLSCHDYXYXEF-PRSMZQBUSA-N 0.000 description 1
- XPHHAPWIPSGAPQ-KRJPCBTMSA-N CC(C)(C)OC(NC(CN(C[C@H](C1)N2c(cc3)ccc3-c3nnn[nH]3)C1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O Chemical compound CC(C)(C)OC(NC(CN(C[C@H](C1)N2c(cc3)ccc3-c3nnn[nH]3)C1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)=O XPHHAPWIPSGAPQ-KRJPCBTMSA-N 0.000 description 1
- RUKVOYNRUCWETD-IMCLBXFUSA-N CC(C)(C)OC(N[C@@H](CN(CC(C1)N2C(CCc3c4)c3ccc4C(O)=O)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CN(CC(C1)N2C(CCc3c4)c3ccc4C(O)=O)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O)=O RUKVOYNRUCWETD-IMCLBXFUSA-N 0.000 description 1
- FUHNENRNZAARLG-UWVGGRQHSA-N CC(C)(C)OC(N[C@@H](CN)C(N(CCC1)[C@@H]1C#N)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CN)C(N(CCC1)[C@@H]1C#N)=O)=O FUHNENRNZAARLG-UWVGGRQHSA-N 0.000 description 1
- YWGPVVGXQNFHIH-OSYRGRAESA-N CC(C)(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC(C)(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N YWGPVVGXQNFHIH-OSYRGRAESA-N 0.000 description 1
- BLBZLKMITWZRFW-UQVNRYHBSA-N CC(C)(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BLBZLKMITWZRFW-UQVNRYHBSA-N 0.000 description 1
- LOABCWLKIRCMLJ-NRWPOFLRSA-N CC(C)(C)[C@@H](CC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N(CCC1)C1C#N)=O Chemical compound CC(C)(C)[C@@H](CC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N(CCC1)C1C#N)=O LOABCWLKIRCMLJ-NRWPOFLRSA-N 0.000 description 1
- JBULSURVMXPBNA-YFKPBYRVSA-N CC(C)(C)[C@@H](N)CO Chemical compound CC(C)(C)[C@@H](N)CO JBULSURVMXPBNA-YFKPBYRVSA-N 0.000 description 1
- IQZLRPPAGVSIRZ-ZCFIWIBFSA-N CC(C)(C)[C@@H]1COS(=O)(=O)C1 Chemical compound CC(C)(C)[C@@H]1COS(=O)(=O)C1 IQZLRPPAGVSIRZ-ZCFIWIBFSA-N 0.000 description 1
- YWGPVVGXQNFHIH-IXKJJVDVSA-N CC(C)(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC(C)(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N YWGPVVGXQNFHIH-IXKJJVDVSA-N 0.000 description 1
- BLBZLKMITWZRFW-SXYSDOLCSA-N CC(C)(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BLBZLKMITWZRFW-SXYSDOLCSA-N 0.000 description 1
- JBULSURVMXPBNA-RXMQYKEDSA-N CC(C)(C)[C@H](N)CO Chemical compound CC(C)(C)[C@H](N)CO JBULSURVMXPBNA-RXMQYKEDSA-N 0.000 description 1
- IQZLRPPAGVSIRZ-LURJTMIESA-N CC(C)(C)[C@H]1COS(=O)(=O)C1 Chemical compound CC(C)(C)[C@H]1COS(=O)(=O)C1 IQZLRPPAGVSIRZ-LURJTMIESA-N 0.000 description 1
- KVIPEWOFNJCSKJ-RNLLHNEJSA-N CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KVIPEWOFNJCSKJ-RNLLHNEJSA-N 0.000 description 1
- KRNYUWVBDHNCHZ-HHQXYKNISA-N CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KRNYUWVBDHNCHZ-HHQXYKNISA-N 0.000 description 1
- KRNYUWVBDHNCHZ-LGEIMJHGSA-N CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N KRNYUWVBDHNCHZ-LGEIMJHGSA-N 0.000 description 1
- KVIPEWOFNJCSKJ-VJANTYMQSA-N CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KVIPEWOFNJCSKJ-VJANTYMQSA-N 0.000 description 1
- JMEQENKBVMPFIN-SATOSQONSA-N CC(C)(CC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(NC)=O)c2ccc3C(NC)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound CC(C)(CC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(NC)=O)c2ccc3C(NC)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O JMEQENKBVMPFIN-SATOSQONSA-N 0.000 description 1
- PQYKTWBSWATECZ-VJHAGTGTSA-N CC(C)(CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(cc2)c(CC3)cc2C(NCC(C)(C)O)=O)c3c1)=O)O Chemical compound CC(C)(CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(cc2)c(CC3)cc2C(NCC(C)(C)O)=O)c3c1)=O)O PQYKTWBSWATECZ-VJHAGTGTSA-N 0.000 description 1
- MUWTUIQUEBHQAR-NRFANRHFSA-N CC(C)(CNC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC(C)(CNC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2)NCC(=O)N1CCC[C@H]1C#N MUWTUIQUEBHQAR-NRFANRHFSA-N 0.000 description 1
- OPCJOXGBLDJWRM-UHFFFAOYSA-N CC(C)(N)CN Chemical compound CC(C)(N)CN OPCJOXGBLDJWRM-UHFFFAOYSA-N 0.000 description 1
- GTDQFRWCZONMEZ-UHFFFAOYSA-N CC(C)(N)CNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound CC(C)(N)CNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 GTDQFRWCZONMEZ-UHFFFAOYSA-N 0.000 description 1
- CBTVGIZVANVGBH-UHFFFAOYSA-N CC(C)(N)CO Chemical compound CC(C)(N)CO CBTVGIZVANVGBH-UHFFFAOYSA-N 0.000 description 1
- DKHSXWXHXFRKON-UHFFFAOYSA-N CC(C)C(c(cc1)ccc1F)N Chemical compound CC(C)C(c(cc1)ccc1F)N DKHSXWXHXFRKON-UHFFFAOYSA-N 0.000 description 1
- FPUDNNNDUNSGRY-JSVRSXMJSA-N CC(C)C(c(cc1)ccc1F)N([C@@H]1CN(CC(C(N(C(C2)[C@@H]2C2)C2C#N)=O)N)C2C1)C2=O Chemical compound CC(C)C(c(cc1)ccc1F)N([C@@H]1CN(CC(C(N(C(C2)[C@@H]2C2)C2C#N)=O)N)C2C1)C2=O FPUDNNNDUNSGRY-JSVRSXMJSA-N 0.000 description 1
- JTBFGHHDXCWYER-UHFFFAOYSA-N CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 JTBFGHHDXCWYER-UHFFFAOYSA-N 0.000 description 1
- UPOHHWKTLWKUBT-UHFFFAOYSA-N CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 UPOHHWKTLWKUBT-UHFFFAOYSA-N 0.000 description 1
- GOPBCSARLOOCBB-UHFFFAOYSA-N CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 GOPBCSARLOOCBB-UHFFFAOYSA-N 0.000 description 1
- PIGGYPZBNDEXRN-UHFFFAOYSA-N CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 PIGGYPZBNDEXRN-UHFFFAOYSA-N 0.000 description 1
- HYRLVTOTIOEABD-MHZLTWQESA-N CC(C)C1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC(C)C1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 HYRLVTOTIOEABD-MHZLTWQESA-N 0.000 description 1
- KQZUYLRQTNWCGI-MHZLTWQESA-N CC(C)C1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC(C)C1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 KQZUYLRQTNWCGI-MHZLTWQESA-N 0.000 description 1
- FMHOIWXOEWXSMZ-WWDWLDGVSA-N CC(C)C1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 FMHOIWXOEWXSMZ-WWDWLDGVSA-N 0.000 description 1
- YVQQGFUSWQQHJW-WWDWLDGVSA-N CC(C)C1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 YVQQGFUSWQQHJW-WWDWLDGVSA-N 0.000 description 1
- XWMCYWXKLCLSTP-GOSISDBHSA-N CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 XWMCYWXKLCLSTP-GOSISDBHSA-N 0.000 description 1
- FRAJYFCDNYPHBB-CQSZACIVSA-N CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 FRAJYFCDNYPHBB-CQSZACIVSA-N 0.000 description 1
- HBWUNGJSERFKFV-CQSZACIVSA-N CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 HBWUNGJSERFKFV-CQSZACIVSA-N 0.000 description 1
- QPPSKIUZFYYOKD-LXFBAYGMSA-N CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 QPPSKIUZFYYOKD-LXFBAYGMSA-N 0.000 description 1
- QRZLJCPDCXMKPF-LXFBAYGMSA-N CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 QRZLJCPDCXMKPF-LXFBAYGMSA-N 0.000 description 1
- HIZFFHFSCYIBCB-HZRDQOKSSA-N CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 HIZFFHFSCYIBCB-HZRDQOKSSA-N 0.000 description 1
- CFORXXSZRLDNAP-HZRDQOKSSA-N CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 CFORXXSZRLDNAP-HZRDQOKSSA-N 0.000 description 1
- GGEYKBNWZSBPFQ-SFHVURJKSA-N CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 GGEYKBNWZSBPFQ-SFHVURJKSA-N 0.000 description 1
- XWMCYWXKLCLSTP-SFHVURJKSA-N CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 XWMCYWXKLCLSTP-SFHVURJKSA-N 0.000 description 1
- FRAJYFCDNYPHBB-AWEZNQCLSA-N CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 FRAJYFCDNYPHBB-AWEZNQCLSA-N 0.000 description 1
- HBWUNGJSERFKFV-AWEZNQCLSA-N CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 HBWUNGJSERFKFV-AWEZNQCLSA-N 0.000 description 1
- QPPSKIUZFYYOKD-FIPFOOKPSA-N CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 QPPSKIUZFYYOKD-FIPFOOKPSA-N 0.000 description 1
- QRZLJCPDCXMKPF-FIPFOOKPSA-N CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 QRZLJCPDCXMKPF-FIPFOOKPSA-N 0.000 description 1
- HIZFFHFSCYIBCB-KHTJFUAPSA-N CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 HIZFFHFSCYIBCB-KHTJFUAPSA-N 0.000 description 1
- CFORXXSZRLDNAP-KHTJFUAPSA-N CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC(C)C1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 CFORXXSZRLDNAP-KHTJFUAPSA-N 0.000 description 1
- WSJXHOPEEPBJTB-UHFFFAOYSA-N CC(C)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)O)C=C1)(C1=CC=C(C(=O)O)C=C1)C1=NNN=N1 Chemical compound CC(C)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1.CC(C)CC(C1=CC=C(C(=O)O)C=C1)(C1=CC=C(C(=O)O)C=C1)C1=NNN=N1 WSJXHOPEEPBJTB-UHFFFAOYSA-N 0.000 description 1
- WQYDZSSLMXOUCO-UHFFFAOYSA-N CC(C)CC1(C(N)=O)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(N)=O.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 Chemical compound CC(C)CC1(C(N)=O)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(N)=O.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 WQYDZSSLMXOUCO-UHFFFAOYSA-N 0.000 description 1
- BRIXKVFCWVFVII-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)C2=C1/C=C\C(C(=O)O)=C/2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)C2=C1/C=C\C(C(=O)O)=C/2 BRIXKVFCWVFVII-UHFFFAOYSA-N 0.000 description 1
- VTSGLSSSAKZNMV-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(OCO)S2)CCC2=C1SC(C(=O)O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(C=C(OCO)S2)CCC2=C1SC(C(=O)O)=C2 VTSGLSSSAKZNMV-UHFFFAOYSA-N 0.000 description 1
- QCGWXEHUADBLIF-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)S2)CCC2=C1SC(C(=O)O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)S2)CCC2=C1SC(C(=O)O)=C2 QCGWXEHUADBLIF-UHFFFAOYSA-N 0.000 description 1
- FXGQBLXMEOOAMD-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC(C(=O)O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC(C(=O)O)=C2 FXGQBLXMEOOAMD-UHFFFAOYSA-N 0.000 description 1
- SOOUNWHGVWPUAW-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC(C(=O)O)=C2.CC1=C(Br)C=C(Cl)S1.CC1=C(C(=O)O)C=C(Cl)S1.CC1=CC=C(Cl)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2(CC(C)C)C2=NNN=N2)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2=O)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2C#N)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2O)S1.ClCC1=CC=C(Cl)S1.O=C(O)C1=C(CCC2=CC=C(Cl)S2)SC(Cl)=C1.O=C1C2=C(CCC3=C1C=C(Cl)S3)SC(Cl)=C2.[C-]#[N+]C1(CC(C)C)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC(C(=O)OC)=C2.[C-]#[N+]C1=CC2=C(CCC3=C(C=C(C#N)S3)C2=O)S1 Chemical compound CC(C)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC(C(=O)O)=C2.CC1=C(Br)C=C(Cl)S1.CC1=C(C(=O)O)C=C(Cl)S1.CC1=CC=C(Cl)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2(CC(C)C)C2=NNN=N2)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2=O)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2C#N)S1.COC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)OC)S3)C2O)S1.ClCC1=CC=C(Cl)S1.O=C(O)C1=C(CCC2=CC=C(Cl)S2)SC(Cl)=C1.O=C1C2=C(CCC3=C1C=C(Cl)S3)SC(Cl)=C2.[C-]#[N+]C1(CC(C)C)C2=C(CCC3=C1C=C(C(=O)OC)S3)SC(C(=O)OC)=C2.[C-]#[N+]C1=CC2=C(CCC3=C(C=C(C#N)S3)C2=O)S1 SOOUNWHGVWPUAW-UHFFFAOYSA-N 0.000 description 1
- YJHIPOKAHVEGGU-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CC2=C1C=CC(C(=O)O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CC2=C1C=CC(C(=O)O)=C2 YJHIPOKAHVEGGU-UHFFFAOYSA-N 0.000 description 1
- AAKGQXWUWGKWJQ-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NNN=N1.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2C#N.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2Cl.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2O.COC(=O)C1=CC=C2C(=O)C3=C(C=C(C(=O)OC)C=C3)CC2=C1.O=C1C2=CC=C(Br)C=C2C(=O)C2=C1C=CC(Br)=C2.O=C1C2=CC=C(Br)C=C2CC2=C1C=CC(Br)=C2.O=C1C2=CC=CC=C2C(=O)C2=C1C=CC=C2.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CC2=C1C=CC(C(=O)OC)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NNN=N1.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2C#N.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2Cl.COC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)OC)=C1)C2O.COC(=O)C1=CC=C2C(=O)C3=C(C=C(C(=O)OC)C=C3)CC2=C1.O=C1C2=CC=C(Br)C=C2C(=O)C2=C1C=CC(Br)=C2.O=C1C2=CC=C(Br)C=C2CC2=C1C=CC(Br)=C2.O=C1C2=CC=CC=C2C(=O)C2=C1C=CC=C2.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CC2=C1C=CC(C(=O)OC)=C2 AAKGQXWUWGKWJQ-UHFFFAOYSA-N 0.000 description 1
- YCCOMNLMZZUSNM-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 YCCOMNLMZZUSNM-UHFFFAOYSA-N 0.000 description 1
- GWUNRDIHDGPFCV-FEJIQUJVSA-N CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2.C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CC(C)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2.C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 GWUNRDIHDGPFCV-FEJIQUJVSA-N 0.000 description 1
- ODBQNCKETCXQDH-UHFFFAOYSA-N CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=CC=C(OC(C)=O)C=C1CC2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NNN=N1.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=CC=C(OC(C)=O)C=C1CC2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NNN=N1.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 ODBQNCKETCXQDH-UHFFFAOYSA-N 0.000 description 1
- SDIKETFRYSONNH-PGLWRLEMSA-N CC(C)CC1(C2=NNN=N2)C2=CC=CC=C2CCC2=C1C=CC=C2.C[C@H](N)CC1(C2=NNN=N2)C2=CC=CC=C2CCC2=C1C=CC=C2.[C-]#[N+]C1(CC(C)C)C2=CC=CC=C2CCC2=C1C=CC=C2.[C-]#[N+]C1C2=CC=CC=C2CCC2=C1C=CC=C2 Chemical compound CC(C)CC1(C2=NNN=N2)C2=CC=CC=C2CCC2=C1C=CC=C2.C[C@H](N)CC1(C2=NNN=N2)C2=CC=CC=C2CCC2=C1C=CC=C2.[C-]#[N+]C1(CC(C)C)C2=CC=CC=C2CCC2=C1C=CC=C2.[C-]#[N+]C1C2=CC=CC=C2CCC2=C1C=CC=C2 SDIKETFRYSONNH-PGLWRLEMSA-N 0.000 description 1
- LUYQIAIKICDCGL-UHFFFAOYSA-N CC(C)CC1(C2=NOC(=O)N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(=N)NO.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NOC(=O)N1.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 Chemical compound CC(C)CC1(C2=NOC(=O)N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C(=N)NO.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2(CC(C)C)C1=NOC(=O)N1.[C-]#[N+]C1(CC(C)C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 LUYQIAIKICDCGL-UHFFFAOYSA-N 0.000 description 1
- MMNOIADFFPUVNF-WFNDYRQKSA-N CC(C)CO.C[C@H](N)CO.C[C@H]1COS(=O)(=O)C1 Chemical compound CC(C)CO.C[C@H](N)CO.C[C@H]1COS(=O)(=O)C1 MMNOIADFFPUVNF-WFNDYRQKSA-N 0.000 description 1
- LHBNAGMYNMBOEZ-UHFKCPIBSA-N CC(C)N(C(C(C[C@H](C)NCC(N(CCC1)C1C#N)=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)=N1)OC1=O Chemical compound CC(C)N(C(C(C[C@H](C)NCC(N(CCC1)C1C#N)=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)=N1)OC1=O LHBNAGMYNMBOEZ-UHFKCPIBSA-N 0.000 description 1
- LAARSLREYBRJOW-SATOSQONSA-N CC(C)N1N=C(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CC2)c3)ccc3C(N)=O)c(cc3)c2cc3C(N)=O)NC1=O Chemical compound CC(C)N1N=C(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CC2)c3)ccc3C(N)=O)c(cc3)c2cc3C(N)=O)NC1=O LAARSLREYBRJOW-SATOSQONSA-N 0.000 description 1
- YEUOFIDBVWOOFK-WYPWLYFUSA-N CC(C)N1N=C(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N(C)C)=O)c2ccc3C(N(C)C)=O)NC1=O Chemical compound CC(C)N1N=C(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N(C)C)=O)c2ccc3C(N(C)C)=O)NC1=O YEUOFIDBVWOOFK-WYPWLYFUSA-N 0.000 description 1
- UPUIVLSSOFZBHH-UMAHROPBSA-N CC(C)N1N=C(C(C[C@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N)=O)c2ccc3C(N)=O)NC1=O Chemical compound CC(C)N1N=C(C(C[C@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N)=O)c2ccc3C(N)=O)NC1=O UPUIVLSSOFZBHH-UMAHROPBSA-N 0.000 description 1
- XKVXJVYDDLJIEQ-UHFFFAOYSA-N CC(C)N1N=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O XKVXJVYDDLJIEQ-UHFFFAOYSA-N 0.000 description 1
- HQVKGNXKPLGUGI-UHFFFAOYSA-N CC(C)N1N=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O HQVKGNXKPLGUGI-UHFFFAOYSA-N 0.000 description 1
- HVJVHBKEOJYHKI-MHZLTWQESA-N CC(C)N1N=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O Chemical compound CC(C)N1N=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O HVJVHBKEOJYHKI-MHZLTWQESA-N 0.000 description 1
- QJKAZMBBGDNDFG-WWDWLDGVSA-N CC(C)N1N=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O QJKAZMBBGDNDFG-WWDWLDGVSA-N 0.000 description 1
- WANZBCDCHIJZQQ-GOSISDBHSA-N CC(C)N1N=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O WANZBCDCHIJZQQ-GOSISDBHSA-N 0.000 description 1
- VVWCBVJKYFLYMS-CQSZACIVSA-N CC(C)N1N=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O VVWCBVJKYFLYMS-CQSZACIVSA-N 0.000 description 1
- KFOKHODTNOZEIZ-LXFBAYGMSA-N CC(C)N1N=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O KFOKHODTNOZEIZ-LXFBAYGMSA-N 0.000 description 1
- YEUOFIDBVWOOFK-HZRDQOKSSA-N CC(C)N1N=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O YEUOFIDBVWOOFK-HZRDQOKSSA-N 0.000 description 1
- WANZBCDCHIJZQQ-SFHVURJKSA-N CC(C)N1N=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O WANZBCDCHIJZQQ-SFHVURJKSA-N 0.000 description 1
- VVWCBVJKYFLYMS-AWEZNQCLSA-N CC(C)N1N=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O VVWCBVJKYFLYMS-AWEZNQCLSA-N 0.000 description 1
- KFOKHODTNOZEIZ-FIPFOOKPSA-N CC(C)N1N=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)NC1=O KFOKHODTNOZEIZ-FIPFOOKPSA-N 0.000 description 1
- YEUOFIDBVWOOFK-KHTJFUAPSA-N CC(C)N1N=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O Chemical compound CC(C)N1N=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)NC1=O YEUOFIDBVWOOFK-KHTJFUAPSA-N 0.000 description 1
- NVTLZKKDRBSFAI-UHFFFAOYSA-N CC(C)N1OC(=O)N=C1C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 NVTLZKKDRBSFAI-UHFFFAOYSA-N 0.000 description 1
- CLHLVMIEKVDWGN-UHFFFAOYSA-N CC(C)N1OC(=O)N=C1C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 CLHLVMIEKVDWGN-UHFFFAOYSA-N 0.000 description 1
- YOAYIHKKTCCNCU-MHZLTWQESA-N CC(C)N1OC(=O)N=C1C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)N1OC(=O)N=C1C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 YOAYIHKKTCCNCU-MHZLTWQESA-N 0.000 description 1
- XNOLVZZWHBOJLG-WWDWLDGVSA-N CC(C)N1OC(=O)N=C1C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 XNOLVZZWHBOJLG-WWDWLDGVSA-N 0.000 description 1
- DQMBZNXMTQEMQQ-GOSISDBHSA-N CC(C)N1OC(=O)N=C1C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 DQMBZNXMTQEMQQ-GOSISDBHSA-N 0.000 description 1
- MIHODVJQYPOWPN-CQSZACIVSA-N CC(C)N1OC(=O)N=C1C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 MIHODVJQYPOWPN-CQSZACIVSA-N 0.000 description 1
- LHBNAGMYNMBOEZ-LXFBAYGMSA-N CC(C)N1OC(=O)N=C1C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 LHBNAGMYNMBOEZ-LXFBAYGMSA-N 0.000 description 1
- TYGIMZXIBQAHAL-HZRDQOKSSA-N CC(C)N1OC(=O)N=C1C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 TYGIMZXIBQAHAL-HZRDQOKSSA-N 0.000 description 1
- DQMBZNXMTQEMQQ-SFHVURJKSA-N CC(C)N1OC(=O)N=C1C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 DQMBZNXMTQEMQQ-SFHVURJKSA-N 0.000 description 1
- MIHODVJQYPOWPN-AWEZNQCLSA-N CC(C)N1OC(=O)N=C1C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 MIHODVJQYPOWPN-AWEZNQCLSA-N 0.000 description 1
- LHBNAGMYNMBOEZ-FIPFOOKPSA-N CC(C)N1OC(=O)N=C1C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 LHBNAGMYNMBOEZ-FIPFOOKPSA-N 0.000 description 1
- TYGIMZXIBQAHAL-KHTJFUAPSA-N CC(C)N1OC(=O)N=C1C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)N1OC(=O)N=C1C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 TYGIMZXIBQAHAL-KHTJFUAPSA-N 0.000 description 1
- XAKBBDDXGAOTIT-UHFFFAOYSA-N CC(C)NC(=N)C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 XAKBBDDXGAOTIT-UHFFFAOYSA-N 0.000 description 1
- VFSKVJRCCPNEDR-UHFFFAOYSA-N CC(C)NC(=N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)NC(=N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 VFSKVJRCCPNEDR-UHFFFAOYSA-N 0.000 description 1
- KPDSXMLNRWDCSI-WWDWLDGVSA-N CC(C)NC(=N)C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 KPDSXMLNRWDCSI-WWDWLDGVSA-N 0.000 description 1
- KAYMECHQKKVTBO-GOSISDBHSA-N CC(C)NC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 KAYMECHQKKVTBO-GOSISDBHSA-N 0.000 description 1
- ANNIFJVIPXDCAJ-CQSZACIVSA-N CC(C)NC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)NC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 ANNIFJVIPXDCAJ-CQSZACIVSA-N 0.000 description 1
- GZBSCADGZLOPQD-LXFBAYGMSA-N CC(C)NC(=N)C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)NC(=N)C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 GZBSCADGZLOPQD-LXFBAYGMSA-N 0.000 description 1
- YJHYUCGYXTYJEC-HZRDQOKSSA-N CC(C)NC(=N)C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 YJHYUCGYXTYJEC-HZRDQOKSSA-N 0.000 description 1
- KAYMECHQKKVTBO-SFHVURJKSA-N CC(C)NC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 KAYMECHQKKVTBO-SFHVURJKSA-N 0.000 description 1
- ANNIFJVIPXDCAJ-AWEZNQCLSA-N CC(C)NC(=N)C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC(C)NC(=N)C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 ANNIFJVIPXDCAJ-AWEZNQCLSA-N 0.000 description 1
- GZBSCADGZLOPQD-FIPFOOKPSA-N CC(C)NC(=N)C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)NC(=N)C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 GZBSCADGZLOPQD-FIPFOOKPSA-N 0.000 description 1
- YJHYUCGYXTYJEC-KHTJFUAPSA-N CC(C)NC(=N)C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CC(C)NC(=N)C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 YJHYUCGYXTYJEC-KHTJFUAPSA-N 0.000 description 1
- GBVGMIUGOLIHOA-SANMLTNESA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 GBVGMIUGOLIHOA-SANMLTNESA-N 0.000 description 1
- YEQQGVVTCSHGHM-QASNXKAYSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 YEQQGVVTCSHGHM-QASNXKAYSA-N 0.000 description 1
- NOCKJVJNWCBRBV-ALTZYDRJSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 NOCKJVJNWCBRBV-ALTZYDRJSA-N 0.000 description 1
- YUUNCJATGKILTE-WEHNTMOPSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 YUUNCJATGKILTE-WEHNTMOPSA-N 0.000 description 1
- VMZNJYDKYXFXJO-DXOOYCRSSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)(C)C)C=C1)CC2 VMZNJYDKYXFXJO-DXOOYCRSSA-N 0.000 description 1
- LAHLVMPUUKVSDI-AMGIVPHBSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 LAHLVMPUUKVSDI-AMGIVPHBSA-N 0.000 description 1
- WDZJKDMIKLQFAY-XCVXUIKGSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 WDZJKDMIKLQFAY-XCVXUIKGSA-N 0.000 description 1
- LAHLVMPUUKVSDI-CUNXSJBXSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 LAHLVMPUUKVSDI-CUNXSJBXSA-N 0.000 description 1
- WDZJKDMIKLQFAY-VIKYFBQLSA-N CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 Chemical compound CC(C)NC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NC(C)C)C=C1)CC2 WDZJKDMIKLQFAY-VIKYFBQLSA-N 0.000 description 1
- KVEYHDSQGVGGRM-KRWDZBQOSA-N CC(C)NC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC(C)C)=C1)C2(C[C@H](C)N)C1=NNN=N1 Chemical compound CC(C)NC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC(C)C)=C1)C2(C[C@H](C)N)C1=NNN=N1 KVEYHDSQGVGGRM-KRWDZBQOSA-N 0.000 description 1
- UKRIGHIIQWWSHT-DVECYGJZSA-N CC(C)NC(C(C[C@@H](C)NCC(N(CCC1)[C@@H]1C#N)=O)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)=N Chemical compound CC(C)NC(C(C[C@@H](C)NCC(N(CCC1)[C@@H]1C#N)=O)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)=N UKRIGHIIQWWSHT-DVECYGJZSA-N 0.000 description 1
- GZBSCADGZLOPQD-UHFKCPIBSA-N CC(C)NC(C(C[C@H](C)NCC(N(CCC1)C1C#N)=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)=N Chemical compound CC(C)NC(C(C[C@H](C)NCC(N(CCC1)C1C#N)=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)=N GZBSCADGZLOPQD-UHFKCPIBSA-N 0.000 description 1
- YBENNYJISNRGSV-DBYWEBACSA-N CC(C)NC(C(C[C@H](C)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)(c1ccc(C(N(C)C)O)cc1CCc1c2)c1ccc2C(N(C)C)=O)=N Chemical compound CC(C)NC(C(C[C@H](C)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O)(c1ccc(C(N(C)C)O)cc1CCc1c2)c1ccc2C(N(C)C)=O)=N YBENNYJISNRGSV-DBYWEBACSA-N 0.000 description 1
- WDZJKDMIKLQFAY-NAGJEVNVSA-N CC(C)NC(c1ccc(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC(C)C)=O)c2c1)=O Chemical compound CC(C)NC(c1ccc(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC(C)C)=O)c2c1)=O WDZJKDMIKLQFAY-NAGJEVNVSA-N 0.000 description 1
- FPUDNNNDUNSGRY-IXKJJVDVSA-N CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N FPUDNNNDUNSGRY-IXKJJVDVSA-N 0.000 description 1
- KUFHLHYVYOYETJ-SXYSDOLCSA-N CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KUFHLHYVYOYETJ-SXYSDOLCSA-N 0.000 description 1
- NUFCEQRIUPBTRH-LSBAASHUSA-N CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N NUFCEQRIUPBTRH-LSBAASHUSA-N 0.000 description 1
- AXLWGXLARAAATP-AIVCMVMSSA-N CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AXLWGXLARAAATP-AIVCMVMSSA-N 0.000 description 1
- DFQGCLACBUDUJV-LITSAYRRSA-N CC(C)[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC(C)[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N DFQGCLACBUDUJV-LITSAYRRSA-N 0.000 description 1
- YYJYBORMRWXDIP-ZIMFAVFZSA-N CC(C)[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YYJYBORMRWXDIP-ZIMFAVFZSA-N 0.000 description 1
- SKRNTNMBWSAMBH-QHCPKHFHSA-N CC(C)[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 SKRNTNMBWSAMBH-QHCPKHFHSA-N 0.000 description 1
- NWYYWIJOWOLJNR-YFKPBYRVSA-N CC(C)[C@@H](N)CO Chemical compound CC(C)[C@@H](N)CO NWYYWIJOWOLJNR-YFKPBYRVSA-N 0.000 description 1
- HDCHLAWQEMJHNX-ZCFIWIBFSA-N CC(C)[C@@H]1COS(=O)(=O)C1 Chemical compound CC(C)[C@@H]1COS(=O)(=O)C1 HDCHLAWQEMJHNX-ZCFIWIBFSA-N 0.000 description 1
- FPUDNNNDUNSGRY-OSYRGRAESA-N CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N FPUDNNNDUNSGRY-OSYRGRAESA-N 0.000 description 1
- KUFHLHYVYOYETJ-UQVNRYHBSA-N CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KUFHLHYVYOYETJ-UQVNRYHBSA-N 0.000 description 1
- NUFCEQRIUPBTRH-QCCYXRBGSA-N CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N NUFCEQRIUPBTRH-QCCYXRBGSA-N 0.000 description 1
- AXLWGXLARAAATP-BQVBQXJWSA-N CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AXLWGXLARAAATP-BQVBQXJWSA-N 0.000 description 1
- YYJYBORMRWXDIP-UUHGVMMKSA-N CC(C)[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C)[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YYJYBORMRWXDIP-UUHGVMMKSA-N 0.000 description 1
- SKRNTNMBWSAMBH-HSZRJFAPSA-N CC(C)[C@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC(C)[C@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 SKRNTNMBWSAMBH-HSZRJFAPSA-N 0.000 description 1
- NWYYWIJOWOLJNR-RXMQYKEDSA-N CC(C)[C@H](N)CO Chemical compound CC(C)[C@H](N)CO NWYYWIJOWOLJNR-RXMQYKEDSA-N 0.000 description 1
- HDCHLAWQEMJHNX-LURJTMIESA-N CC(C)[C@H]1COS(=O)(=O)C1 Chemical compound CC(C)[C@H]1COS(=O)(=O)C1 HDCHLAWQEMJHNX-LURJTMIESA-N 0.000 description 1
- LQZLBXMGPMWOQC-OSMGYRLQSA-N CC(C)c1nnc(C(C[C@@H](C)NCC(N(CCC2)C2C#N)=O)(c(cc2)c(CCc3c4)cc2C(NC)=O)c3ccc4C(NC)=O)[o]1 Chemical compound CC(C)c1nnc(C(C[C@@H](C)NCC(N(CCC2)C2C#N)=O)(c(cc2)c(CCc3c4)cc2C(NC)=O)c3ccc4C(NC)=O)[o]1 LQZLBXMGPMWOQC-OSMGYRLQSA-N 0.000 description 1
- FQBPQBSMWZCRCD-MOJRSFQUSA-N CC(C)c1nnc(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N)=O)c2ccc3C(N)=O)[nH]1 Chemical compound CC(C)c1nnc(C(C[C@@H](C)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c(c(CCc2c3)c4)ccc4C(N)=O)c2ccc3C(N)=O)[nH]1 FQBPQBSMWZCRCD-MOJRSFQUSA-N 0.000 description 1
- KYSSPJIHHZBRMM-PKHIMPSTSA-N CC(C/C1=C2\CCC\C2=C\C2=C1CCC2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC(C/C1=C2\CCC\C2=C\C2=C1CCC2)NCC(=O)N1CCC[C@H]1C#N KYSSPJIHHZBRMM-PKHIMPSTSA-N 0.000 description 1
- WGOGEUGDOBJLPF-GPGVTBSUSA-N CC(C/C1=C2\CCC\C2=C\C2=C1CCC2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(C/C1=C2\CCC\C2=C\C2=C1CCC2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 WGOGEUGDOBJLPF-GPGVTBSUSA-N 0.000 description 1
- BYUOJXXCQQWBOT-UHFFFAOYSA-N CC(CC1)Cc2c1c(CCNCC(N(CCC1)C1C#N)=O)c(CCC(C)C1)c1c2 Chemical compound CC(CC1)Cc2c1c(CCNCC(N(CCC1)C1C#N)=O)c(CCC(C)C1)c1c2 BYUOJXXCQQWBOT-UHFFFAOYSA-N 0.000 description 1
- AKOTVEZNNPCAMO-OCFBWVMOSA-N CC(CC1)Cc2c1cc(CCC(C)C1)c1c2CCNCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound CC(CC1)Cc2c1cc(CCC(C)C1)c1c2CCNCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O AKOTVEZNNPCAMO-OCFBWVMOSA-N 0.000 description 1
- PLUKLSWRTPIMHG-OZBJMMHXSA-N CC(CC1=C2CCCCC2=CC2=C1CCCC2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC(CC1=C2CCCCC2=CC2=C1CCCC2)NCC(=O)N1CCC[C@H]1C#N PLUKLSWRTPIMHG-OZBJMMHXSA-N 0.000 description 1
- CGCSYSPKQCHOLB-MZVKPOCASA-N CC(CC1=C2CCCCC2=CC2=C1CCCC2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC(CC1=C2CCCCC2=CC2=C1CCCC2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CGCSYSPKQCHOLB-MZVKPOCASA-N 0.000 description 1
- SKUDKBZIOKVQJX-UHFFFAOYSA-N CC(O)(O)C1CCCN1.O=C(CBr)N1CCCC1C(=O)C(F)(F)F.O=C(CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2)N1CCCC1C(=O)C(F)(F)F Chemical compound CC(O)(O)C1CCCN1.O=C(CBr)N1CCCC1C(=O)C(F)(F)F.O=C(CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2)N1CCCC1C(=O)C(F)(F)F SKUDKBZIOKVQJX-UHFFFAOYSA-N 0.000 description 1
- MZBCMSOQVAPVPN-ZDNOCTGMSA-N CC(c(cc1)ccc1C#N)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound CC(c(cc1)ccc1C#N)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O MZBCMSOQVAPVPN-ZDNOCTGMSA-N 0.000 description 1
- RAFYFGWRLPCUQZ-QOTQIBIMSA-N CC(c(cc1)ccc1C(N)=O)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound CC(c(cc1)ccc1C(N)=O)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O RAFYFGWRLPCUQZ-QOTQIBIMSA-N 0.000 description 1
- XMQWLBBFUNDFLJ-AVOOJBPPSA-N CC(c1ccc(C(F)(F)F)cc1)N(C1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound CC(c1ccc(C(F)(F)F)cc1)N(C1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O XMQWLBBFUNDFLJ-AVOOJBPPSA-N 0.000 description 1
- CMHUZFDIJVQXQD-CJARGZMBSA-N CC(c1cccc(S(N)(=O)=O)c1)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound CC(c1cccc(S(N)(=O)=O)c1)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O CMHUZFDIJVQXQD-CJARGZMBSA-N 0.000 description 1
- HLJYBSRGNWFLRI-UHFFFAOYSA-N CC.CC(=O)C1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC(N)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC(O)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC1CCC2=C(C1)C(C(=O)O)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(C(N)=O)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CC#N)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CC(C)N=[N+]=[N-])=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CCN)=C1CC(C)CCC1=C2.CC1CCC2=CC3=C(C=C2C1)CC(C)CC3.[H]C(=O)C(=O)C1=C2CC(C)CCC2=CC2=C1CC(C)CC2.[H]C(=O)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2 Chemical compound CC.CC(=O)C1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC(N)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC(O)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2.CC1CCC2=C(C1)C(C(=O)O)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(C(N)=O)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CC#N)=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CC(C)N=[N+]=[N-])=C1CC(C)CCC1=C2.CC1CCC2=C(C1)C(CCN)=C1CC(C)CCC1=C2.CC1CCC2=CC3=C(C=C2C1)CC(C)CC3.[H]C(=O)C(=O)C1=C2CC(C)CCC2=CC2=C1CC(C)CC2.[H]C(=O)CC1=C2CC(C)CCC2=CC2=C1CC(C)CC2 HLJYBSRGNWFLRI-UHFFFAOYSA-N 0.000 description 1
- SSLKOKTYHITTDI-UHFFFAOYSA-N CC.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)C2=C1/C=C\C(C(=O)O)=C/2.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(CC(C)C)(C1=NNN=N1)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(Cl)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(O)C1=C2/C=C(C(=O)OC)\C=C/1.NC1=CC(Cl)=CC=C1C(=O)O.O=C(O)C1=CC=C(Cl)C=C1C1=CC(Cl)=CC=C1C(=O)O.O=C1C2=C(C=C(Cl)C=C2)C2=C1/C=C\C(Cl)=C/2.[C-]#[N+]C1(CC(C)C)C2=C(C=C(C(=O)OC)C=C2)C2=C1/C=C\C(C(=O)OC)=C/2 Chemical compound CC.CC(C)CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)C2=C1/C=C\C(C(=O)O)=C/2.COC(=O)C1=CC2=C(C=C1)C(=O)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(CC(C)C)(C1=NNN=N1)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(Cl)C1=C2/C=C(C(=O)OC)\C=C/1.COC(=O)C1=CC2=C(C=C1)C(O)C1=C2/C=C(C(=O)OC)\C=C/1.NC1=CC(Cl)=CC=C1C(=O)O.O=C(O)C1=CC=C(Cl)C=C1C1=CC(Cl)=CC=C1C(=O)O.O=C1C2=C(C=C(Cl)C=C2)C2=C1/C=C\C(Cl)=C/2.[C-]#[N+]C1(CC(C)C)C2=C(C=C(C(=O)OC)C=C2)C2=C1/C=C\C(C(=O)OC)=C/2 SSLKOKTYHITTDI-UHFFFAOYSA-N 0.000 description 1
- AUDLAUQECLGROK-RUWRTFLUSA-N CC.CC1(C)COC(C2=CC=CS2)=N1.COC(=O)C1=CC(C(C2=CC=CC=C2)(C2=CC=CC=C2)P(Br)C2=CC=CC=C2)=CS1.COC(=O)C1=CC(C)=CS1.COC(=O)C1=CC(CBr)=CS1.COC(=O)C1=CC2=C(S1)C(C#N)C1=C(C=CS1)CC2.COC(=O)C1=CC2=C(S1)C(O)C1=C(C=CS1)CC2.O=C(Cl)C1=CC=CS1.O=C(O)C1=CC(/C=C\C2=C(C(=O)O)SC=C2)=CS1.O=C(O)C1=CC(/C=C\C2=C(C(=O)O)SC=C2)=CS1.O=C(O)C1=CC2=C(S1)C(=O)C1=C(C=CS1)CC2.[H]C(=O)C1=C(C(=O)O)SC=C1.[H]C(=O)C1=C(C(=O)OC)SC=C1.[H]C(=O)C1=C(C2=NC(C)(C)CO2)SC=C1 Chemical compound CC.CC1(C)COC(C2=CC=CS2)=N1.COC(=O)C1=CC(C(C2=CC=CC=C2)(C2=CC=CC=C2)P(Br)C2=CC=CC=C2)=CS1.COC(=O)C1=CC(C)=CS1.COC(=O)C1=CC(CBr)=CS1.COC(=O)C1=CC2=C(S1)C(C#N)C1=C(C=CS1)CC2.COC(=O)C1=CC2=C(S1)C(O)C1=C(C=CS1)CC2.O=C(Cl)C1=CC=CS1.O=C(O)C1=CC(/C=C\C2=C(C(=O)O)SC=C2)=CS1.O=C(O)C1=CC(/C=C\C2=C(C(=O)O)SC=C2)=CS1.O=C(O)C1=CC2=C(S1)C(=O)C1=C(C=CS1)CC2.[H]C(=O)C1=C(C(=O)O)SC=C1.[H]C(=O)C1=C(C(=O)OC)SC=C1.[H]C(=O)C1=C(C2=NC(C)(C)CO2)SC=C1 AUDLAUQECLGROK-RUWRTFLUSA-N 0.000 description 1
- QPLQUGQFIHYRSA-UHFFFAOYSA-N CC1(C)C2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.CC1(C)C2=C(C=CC=C2)C(=O)N(CCN)C2=C1C=CC=C2.CC1(C)C2=C(C=CC=C2)NC(=O)C2=C1C=CC=C2 Chemical compound CC1(C)C2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.CC1(C)C2=C(C=CC=C2)C(=O)N(CCN)C2=C1C=CC=C2.CC1(C)C2=C(C=CC=C2)NC(=O)C2=C1C=CC=C2 QPLQUGQFIHYRSA-UHFFFAOYSA-N 0.000 description 1
- CBGTXCZGYBNLLW-SFHVURJKSA-N CC1(C)C2=C(C=CC=C2)N(CCNCC(=O)N2CCC[C@H]2C#N)C(=O)C2=C1/C=C\C(Cl)=C/2 Chemical compound CC1(C)C2=C(C=CC=C2)N(CCNCC(=O)N2CCC[C@H]2C#N)C(=O)C2=C1/C=C\C(Cl)=C/2 CBGTXCZGYBNLLW-SFHVURJKSA-N 0.000 description 1
- DAWRSBZCKBBSEV-QSAZIKIQSA-N CC1(C)C2=C(C=CC=C2)N(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C(=O)C2=C1/C=C\C(Cl)=C/2 Chemical compound CC1(C)C2=C(C=CC=C2)N(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C(=O)C2=C1/C=C\C(Cl)=C/2 DAWRSBZCKBBSEV-QSAZIKIQSA-N 0.000 description 1
- WRNLRZTXKWPDEP-UHFFFAOYSA-N CC1(C)COS(=O)(=O)C1 Chemical compound CC1(C)COS(=O)(=O)C1 WRNLRZTXKWPDEP-UHFFFAOYSA-N 0.000 description 1
- URYDJJNPNCNYNY-UHFFFAOYSA-N CC1=C(Br)C=CC(Cl)=C1.CC1=C(C(=O)O)C=CC(Cl)=C1.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2C#N.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2Cl.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2O.COC(=O)C1=CC=C2C(=O)C3=C(C=C(C(=O)OC)C=C3)CCC2=C1.O=C(O)C1=C(CCC2=CC(Cl)=CC=C2)C=C(Cl)C=C1.O=C(O)C1=CC=C2C(=O)C3=C(C=C(C(=O)O)C=C3)CCC2=C1.O=C1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.[C-]#[N+]C1=CC=C2C(=O)C3=C(C=C(C#N)C=C3)CCC2=C1 Chemical compound CC1=C(Br)C=CC(Cl)=C1.CC1=C(C(=O)O)C=CC(Cl)=C1.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2C#N.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2Cl.COC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)OC)=C1)C2O.COC(=O)C1=CC=C2C(=O)C3=C(C=C(C(=O)OC)C=C3)CCC2=C1.O=C(O)C1=C(CCC2=CC(Cl)=CC=C2)C=C(Cl)C=C1.O=C(O)C1=CC=C2C(=O)C3=C(C=C(C(=O)O)C=C3)CCC2=C1.O=C1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.[C-]#[N+]C1=CC=C2C(=O)C3=C(C=C(C#N)C=C3)CCC2=C1 URYDJJNPNCNYNY-UHFFFAOYSA-N 0.000 description 1
- VOPDWPZXSAUIKL-UHFFFAOYSA-N CC1=C(Br)SC(Cl)=C1.CC1=C(Br)SC=C1.CC1=C(C(=O)O)SC(Cl)=C1.COC(=O)C1=CC2=C(S1)C(=O)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(C#N)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(CC(C)C)(C1=NNN=N1)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(O)C1=C(C=C(C(=O)OC)S1)CC2.ClCC1=CSC(Br)=C1.O=C(O)C1=C(CCC2=C(Br)SC=C2)C=C(Cl)S1.O=C1C2=C(C=C(Cl)S2)CCC2=C1SC(Br)=C2.O=CC1=CSC(Br)=C1.O=CC1=CSC=C1.OCC1=CSC(Br)=C1.[C-]#[N+]C1(CC(C)C)C2=C(C=C(C(=O)OC)S2)CCC2=C1SC(C(=O)OC)=C2.[C-]#[N+]C1=CC2=C(S1)C(=O)C1=C(C=C(C#N)S1)CC2 Chemical compound CC1=C(Br)SC(Cl)=C1.CC1=C(Br)SC=C1.CC1=C(C(=O)O)SC(Cl)=C1.COC(=O)C1=CC2=C(S1)C(=O)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(C#N)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(CC(C)C)(C1=NNN=N1)C1=C(C=C(C(=O)OC)S1)CC2.COC(=O)C1=CC2=C(S1)C(O)C1=C(C=C(C(=O)OC)S1)CC2.ClCC1=CSC(Br)=C1.O=C(O)C1=C(CCC2=C(Br)SC=C2)C=C(Cl)S1.O=C1C2=C(C=C(Cl)S2)CCC2=C1SC(Br)=C2.O=CC1=CSC(Br)=C1.O=CC1=CSC=C1.OCC1=CSC(Br)=C1.[C-]#[N+]C1(CC(C)C)C2=C(C=C(C(=O)OC)S2)CCC2=C1SC(C(=O)OC)=C2.[C-]#[N+]C1=CC2=C(S1)C(=O)C1=C(C=C(C#N)S1)CC2 VOPDWPZXSAUIKL-UHFFFAOYSA-N 0.000 description 1
- DMVVHZMWPYCQKC-XZATXCMXSA-N CC1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CC1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 DMVVHZMWPYCQKC-XZATXCMXSA-N 0.000 description 1
- UOKDZBQKEBEJNW-HNCPQSOCSA-N CC1=CC2=C(C=C1)[C@H](N)CC2.CN=S(=O)=O Chemical compound CC1=CC2=C(C=C1)[C@H](N)CC2.CN=S(=O)=O UOKDZBQKEBEJNW-HNCPQSOCSA-N 0.000 description 1
- HDCHFXQHMIATBG-UHFFFAOYSA-N CC1=CC=C(C(=O)O)C=C1C(=O)O.CC1=CC=C(Cl)C=C1Cl.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCN1C(=O)C2=C(C=CC=C2)C1=O)C1=NNN=N1.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCNC(=O)OC(C)(C)C)C1=NNN=N1.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2=O.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2C#N.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2O.COC(=O)C1=CC=C(C)C(C(=O)OC)=C1.COC(=O)C1=CC=C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)C(C(=O)OC)=C1.O=C(O)C1=CC=C(CCC2=CC=C(F)C=C2)C(C(=O)O)=C1.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(=O)OC)C=C2 Chemical compound CC1=CC=C(C(=O)O)C=C1C(=O)O.CC1=CC=C(Cl)C=C1Cl.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCN1C(=O)C2=C(C=CC=C2)C1=O)C1=NNN=N1.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2(CCNC(=O)OC(C)(C)C)C1=NNN=N1.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2=O.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2C#N.COC(=O)C1=CC2=C(C=C1)CCC1=C(C=C(F)C=C1)C2O.COC(=O)C1=CC=C(C)C(C(=O)OC)=C1.COC(=O)C1=CC=C(CP(Br)(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)C(C(=O)OC)=C1.O=C(O)C1=CC=C(CCC2=CC=C(F)C=C2)C(C(=O)O)=C1.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(=O)OC)C=C2 HDCHFXQHMIATBG-UHFFFAOYSA-N 0.000 description 1
- ORUZRUNPIBHNFK-UHFFFAOYSA-N CC1=CC=C(F)C=C1Br.CC1=CC=C(F)C=C1C(=O)O.COC(=O)C1=CC(F)=CC=C1C.N#CC1C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.NCCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.O=C1C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.O=C1C2=C(C=CC=C2)C(=O)N1CCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCBr)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2 Chemical compound CC1=CC=C(F)C=C1Br.CC1=CC=C(F)C=C1C(=O)O.COC(=O)C1=CC(F)=CC=C1C.N#CC1C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.NCCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.O=C1C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.O=C1C2=C(C=CC=C2)C(=O)N1CCC1(C2=NNN=N2)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCBr)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=C(F)C=C2)CCC2=C1C=C(F)C=C2 ORUZRUNPIBHNFK-UHFFFAOYSA-N 0.000 description 1
- FJKHLNURLJLPOK-UHFFFAOYSA-N CC1=CC=C(S(=O)(=O)N=O)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)N=O)C=C1 FJKHLNURLJLPOK-UHFFFAOYSA-N 0.000 description 1
- STQWZPIGSROGOE-LAQRGFTBSA-N CC1=CC=C(S(=O)(=O)NC(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)NC(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 STQWZPIGSROGOE-LAQRGFTBSA-N 0.000 description 1
- DTYWKLOYJZKVDL-QIJZPMMNSA-N CC1=CC=C(S(=O)(=O)NC(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)NC(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 DTYWKLOYJZKVDL-QIJZPMMNSA-N 0.000 description 1
- YROPCBAHFYNGGM-UHFFFAOYSA-N CC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 YROPCBAHFYNGGM-UHFFFAOYSA-N 0.000 description 1
- OTSAHJGOGLIHIR-UHFFFAOYSA-N CC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 OTSAHJGOGLIHIR-UHFFFAOYSA-N 0.000 description 1
- UGCOOMSFYFVICU-UHFFFAOYSA-N CC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 UGCOOMSFYFVICU-UHFFFAOYSA-N 0.000 description 1
- TZHLYUGKGAFIFD-UHFFFAOYSA-N CC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=C(C)O2)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NNN=N2)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C(C=C(C(=O)OC)C=C1)CC2 Chemical compound CC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1.CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=C(C)O2)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NNN=N2)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.COC(=O)C1=CC2=C(C=C1)C(C#N)C1=C(C=C(C(=O)OC)C=C1)CC2 TZHLYUGKGAFIFD-UHFFFAOYSA-N 0.000 description 1
- OKEYHVJGZLOFJH-SANMLTNESA-N CC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 OKEYHVJGZLOFJH-SANMLTNESA-N 0.000 description 1
- LVSZKZDWADVUSH-SANMLTNESA-N CC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 LVSZKZDWADVUSH-SANMLTNESA-N 0.000 description 1
- GXWYUQVNRFZGKU-ALTZYDRJSA-N CC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 GXWYUQVNRFZGKU-ALTZYDRJSA-N 0.000 description 1
- HDKQGEBQOHYTFN-ALTZYDRJSA-N CC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 HDKQGEBQOHYTFN-ALTZYDRJSA-N 0.000 description 1
- DCGDQNIDWWZMIJ-MRXNPFEDSA-N CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 DCGDQNIDWWZMIJ-MRXNPFEDSA-N 0.000 description 1
- VDKCQYYYNIUCKB-MRXNPFEDSA-N CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 VDKCQYYYNIUCKB-MRXNPFEDSA-N 0.000 description 1
- VLKVTWRRFLYAHM-GFCCVEGCSA-N CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 VLKVTWRRFLYAHM-GFCCVEGCSA-N 0.000 description 1
- VPXPZNKPBXAWLG-GFCCVEGCSA-N CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 VPXPZNKPBXAWLG-GFCCVEGCSA-N 0.000 description 1
- GFUZLTIMESASRQ-ZBLYBZFDSA-N CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 GFUZLTIMESASRQ-ZBLYBZFDSA-N 0.000 description 1
- IJFSRNUBUFRRRN-ZBLYBZFDSA-N CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 IJFSRNUBUFRRRN-ZBLYBZFDSA-N 0.000 description 1
- YFLITFHBIFAKMG-DRRFWINISA-N CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 YFLITFHBIFAKMG-DRRFWINISA-N 0.000 description 1
- HKXYBZRBVADWKO-DRRFWINISA-N CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 HKXYBZRBVADWKO-DRRFWINISA-N 0.000 description 1
- DCGDQNIDWWZMIJ-INIZCTEOSA-N CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 DCGDQNIDWWZMIJ-INIZCTEOSA-N 0.000 description 1
- VDKCQYYYNIUCKB-INIZCTEOSA-N CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 VDKCQYYYNIUCKB-INIZCTEOSA-N 0.000 description 1
- VLKVTWRRFLYAHM-LBPRGKRZSA-N CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)N1 VLKVTWRRFLYAHM-LBPRGKRZSA-N 0.000 description 1
- VPXPZNKPBXAWLG-LBPRGKRZSA-N CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 VPXPZNKPBXAWLG-LBPRGKRZSA-N 0.000 description 1
- GFUZLTIMESASRQ-IDISGSTGSA-N CC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 Chemical compound CC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)N1 GFUZLTIMESASRQ-IDISGSTGSA-N 0.000 description 1
- IJFSRNUBUFRRRN-IDISGSTGSA-N CC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 IJFSRNUBUFRRRN-IDISGSTGSA-N 0.000 description 1
- YFLITFHBIFAKMG-YMGKFSCVSA-N CC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 Chemical compound CC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)N1 YFLITFHBIFAKMG-YMGKFSCVSA-N 0.000 description 1
- HKXYBZRBVADWKO-YMGKFSCVSA-N CC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 HKXYBZRBVADWKO-YMGKFSCVSA-N 0.000 description 1
- VKDGLNATNCFDSQ-ZHHSLPHTSA-N CC1CC2=C(C1)/C(CC(C)NCC(=O)N1CCC[C@H]1C#N)=C1/CC(C)C/C1=C/2 Chemical compound CC1CC2=C(C1)/C(CC(C)NCC(=O)N1CCC[C@H]1C#N)=C1/CC(C)C/C1=C/2 VKDGLNATNCFDSQ-ZHHSLPHTSA-N 0.000 description 1
- ZSTBMJXGKSILLA-HCUFZIRMSA-N CC1CC2=C(C1)/C(CC(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1/CC(C)C/C1=C/2 Chemical compound CC1CC2=C(C1)/C(CC(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1/CC(C)C/C1=C/2 ZSTBMJXGKSILLA-HCUFZIRMSA-N 0.000 description 1
- RWRFYABILLMMPG-RJYAGPCLSA-N CC1CC2=C(C1)/C(CCNCC(=O)N1CCC[C@H]1C#N)=C1/CC(C)C/C1=C/2 Chemical compound CC1CC2=C(C1)/C(CCNCC(=O)N1CCC[C@H]1C#N)=C1/CC(C)C/C1=C/2 RWRFYABILLMMPG-RJYAGPCLSA-N 0.000 description 1
- SPFPTKYIFSMOBP-YTWAEYCZSA-N CC1CC2=C(C1)/C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1/CC(C)C/C1=C/2 Chemical compound CC1CC2=C(C1)/C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1/CC(C)C/C1=C/2 SPFPTKYIFSMOBP-YTWAEYCZSA-N 0.000 description 1
- CVJMBRQQAIJLHL-MUJSTJFPSA-N CC1CCC2=C(C1)C(CC(C)NCC(=O)N1CCC[C@H]1C#N)=C1CC(C)CCC1=C2 Chemical compound CC1CCC2=C(C1)C(CC(C)NCC(=O)N1CCC[C@H]1C#N)=C1CC(C)CCC1=C2 CVJMBRQQAIJLHL-MUJSTJFPSA-N 0.000 description 1
- WACCVBOCMPFYSL-BZWFJJCDSA-N CC1CCC2=C(C1)C(CC(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1CC(C)CCC1=C2 Chemical compound CC1CCC2=C(C1)C(CC(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1CC(C)CCC1=C2 WACCVBOCMPFYSL-BZWFJJCDSA-N 0.000 description 1
- JZCJOCPDKSBNQG-NGICGMGXSA-N CC1CCC2=C(C1)C(CCNCC(=O)N1CCC[C@H]1C#N)=C1CC(C)CCC1=C2 Chemical compound CC1CCC2=C(C1)C(CCNCC(=O)N1CCC[C@H]1C#N)=C1CC(C)CCC1=C2 JZCJOCPDKSBNQG-NGICGMGXSA-N 0.000 description 1
- AKOTVEZNNPCAMO-GDAYXQJPSA-N CC1CCC2=C(C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1CC(C)CCC1=C2 Chemical compound CC1CCC2=C(C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)=C1CC(C)CCC1=C2 AKOTVEZNNPCAMO-GDAYXQJPSA-N 0.000 description 1
- PWPYALSOVIYYKU-MUJSTJFPSA-N CC1CCC2=C(CC(C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C2C1)CC(C)CC3 Chemical compound CC1CCC2=C(CC(C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C2C1)CC(C)CC3 PWPYALSOVIYYKU-MUJSTJFPSA-N 0.000 description 1
- VZOZWPAHCAOTMA-BZWFJJCDSA-N CC1CCC2=C(CC(C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C2C1)CC(C)CC3 Chemical compound CC1CCC2=C(CC(C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C2C1)CC(C)CC3 VZOZWPAHCAOTMA-BZWFJJCDSA-N 0.000 description 1
- BYUOJXXCQQWBOT-NGICGMGXSA-N CC1CCC2=C(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C2C1)CC(C)CC3 Chemical compound CC1CCC2=C(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C2C1)CC(C)CC3 BYUOJXXCQQWBOT-NGICGMGXSA-N 0.000 description 1
- OLWLAUBTFBJRTR-GDAYXQJPSA-N CC1CCC2=C(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C2C1)CC(C)CC3 Chemical compound CC1CCC2=C(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C2C1)CC(C)CC3 OLWLAUBTFBJRTR-GDAYXQJPSA-N 0.000 description 1
- KLUDQUOLAFVLOL-UHFFFAOYSA-N CCC(=O)OC(C)=O Chemical compound CCC(=O)OC(C)=O KLUDQUOLAFVLOL-UHFFFAOYSA-N 0.000 description 1
- LHXFHRVJUQNCKD-FABOUENISA-N CCC(c(cc1)ccc1F)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound CCC(c(cc1)ccc1F)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O LHXFHRVJUQNCKD-FABOUENISA-N 0.000 description 1
- AXSYVNMBYMPQPQ-UHFFFAOYSA-N CCC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(CCN)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 AXSYVNMBYMPQPQ-UHFFFAOYSA-N 0.000 description 1
- NVRYRYNHQURVSS-UHFFFAOYSA-N CCC1=NN=C(C2(CCN)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 Chemical compound CCC1=NN=C(C2(CCN)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 NVRYRYNHQURVSS-UHFFFAOYSA-N 0.000 description 1
- FXANCBUZCGVRBC-UHFFFAOYSA-N CCC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CCC1=NN=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 FXANCBUZCGVRBC-UHFFFAOYSA-N 0.000 description 1
- GHNGSGBLYNRTJD-SANMLTNESA-N CCC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CCC1=NN=C(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 GHNGSGBLYNRTJD-SANMLTNESA-N 0.000 description 1
- TUOCZMCJRWASII-ALTZYDRJSA-N CCC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 TUOCZMCJRWASII-ALTZYDRJSA-N 0.000 description 1
- UIZJKVDRKNYGND-QGZVFWFLSA-N CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 UIZJKVDRKNYGND-QGZVFWFLSA-N 0.000 description 1
- QXKIRCHGYAIESJ-OAHLLOKOSA-N CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 QXKIRCHGYAIESJ-OAHLLOKOSA-N 0.000 description 1
- UTGYOTFKTSHLHK-CYBMUJFWSA-N CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 UTGYOTFKTSHLHK-CYBMUJFWSA-N 0.000 description 1
- SUJTWVBBVGYVBD-AMGIVPHBSA-N CCC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CCC1=NN=C(C2(C[C@@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 SUJTWVBBVGYVBD-AMGIVPHBSA-N 0.000 description 1
- COBRZPBWSJEUMX-XCVXUIKGSA-N CCC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 COBRZPBWSJEUMX-XCVXUIKGSA-N 0.000 description 1
- UIZJKVDRKNYGND-KRWDZBQOSA-N CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 UIZJKVDRKNYGND-KRWDZBQOSA-N 0.000 description 1
- QXKIRCHGYAIESJ-HNNXBMFYSA-N CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(=O)NC)C=C3)CCC3=C2C=CC(C(=O)NC)=C3)O1 QXKIRCHGYAIESJ-HNNXBMFYSA-N 0.000 description 1
- UTGYOTFKTSHLHK-ZDUSSCGKSA-N CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@H](C)N)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)O1 UTGYOTFKTSHLHK-ZDUSSCGKSA-N 0.000 description 1
- SUJTWVBBVGYVBD-CUNXSJBXSA-N CCC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 Chemical compound CCC1=NN=C(C2(C[C@H](C)NCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)O1 SUJTWVBBVGYVBD-CUNXSJBXSA-N 0.000 description 1
- COBRZPBWSJEUMX-VIKYFBQLSA-N CCC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 Chemical compound CCC1=NN=C(C2(C[C@H](C)NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)O1 COBRZPBWSJEUMX-VIKYFBQLSA-N 0.000 description 1
- REAZJQDJGXUCEX-UHFFFAOYSA-N CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C(N)=O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C(N)=O)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C(N)=S)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=CN2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CCCC1(C#N)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C(N)=O)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C(N)=O)C2=C(C=C(C(=O)OC)C=C2)CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C(N)=S)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=CN2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2.NCCC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 REAZJQDJGXUCEX-UHFFFAOYSA-N 0.000 description 1
- YGZRAZNBRQOXIB-UHFFFAOYSA-N CCCC1(C#N)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1(CCN)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.N=C1NCCC12C1=C(C=CC=C1)CCC1=C2C=CC=C1.N=C1NCCC12C1=C(C=CC=C1)CCC1=C2C=CC=C1.NCCC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound CCCC1(C#N)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1(CCN)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.N=C1NCCC12C1=C(C=CC=C1)CCC1=C2C=CC=C1.N=C1NCCC12C1=C(C=CC=C1)CCC1=C2C=CC=C1.NCCC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 YGZRAZNBRQOXIB-UHFFFAOYSA-N 0.000 description 1
- RMGYARRIUHQUAR-UHFFFAOYSA-N CCCC1(C#N)C2=C(C=CS2)CCC2=C1SC(C(=O)OC)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)O)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)OC)=C2 Chemical compound CCCC1(C#N)C2=C(C=CS2)CCC2=C1SC(C(=O)OC)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)O)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)OC)=C2 RMGYARRIUHQUAR-UHFFFAOYSA-N 0.000 description 1
- HAYBIIYNYQAUBB-UHFFFAOYSA-N CCCC1(C2=NN(C)N=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN(C)N=N2)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=NN2)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=NN2C)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=NN2C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 Chemical compound CCCC1(C2=NN(C)N=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN(C)N=N2)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=NN2)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2.CCCC1(C2=NN=NN2C)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2.CCCC1(C2=NN=NN2C)C2=CC=C(C(=O)OC)C=C2CCC2=C1C=CC(C(=O)OC)=C2 HAYBIIYNYQAUBB-UHFFFAOYSA-N 0.000 description 1
- GKWIELVZIHVBCJ-UHFFFAOYSA-N CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)O)=C2 Chemical compound CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2.CCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)O)=C2 GKWIELVZIHVBCJ-UHFFFAOYSA-N 0.000 description 1
- YSEGVWYJVRZGOT-UHFFFAOYSA-N CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC=C2 Chemical compound CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2.CCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)O)S3)SC=C2 YSEGVWYJVRZGOT-UHFFFAOYSA-N 0.000 description 1
- QINHQKQNNBSUQB-UHFFFAOYSA-N CCCC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 Chemical compound CCCC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 QINHQKQNNBSUQB-UHFFFAOYSA-N 0.000 description 1
- YCKWWDZJDLMBBR-UHFFFAOYSA-N CCCC1(C2=NNN=N2)C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 Chemical compound CCCC1(C2=NNN=N2)C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 YCKWWDZJDLMBBR-UHFFFAOYSA-N 0.000 description 1
- BXHJBUAHKCILBU-UHFFFAOYSA-N CCCN1C(=O)C2=C(C=CC=C2)N(C)C2=CC=C(Cl)C=C21.CN(C1=C(C(=O)O)C=CC=C1)C1=C([N+](=O)[O-])C=C(Cl)C=C1.CN(C1=C(N)C=C(Cl)C=C1)C1=C(C(=O)O)C=CC=C1.CN1C2=CC=C(Cl)C=C2N(CCN)C(=O)C2=C1C=CC=C2.CN1C2=CC=C(Cl)C=C2NC(=O)C2=C1C=CC=C2.CNC1=C(C(=O)O)C=CC=C1.O=[N+]([O-])C1=C(Br)C=CC(Cl)=C1 Chemical compound CCCN1C(=O)C2=C(C=CC=C2)N(C)C2=CC=C(Cl)C=C21.CN(C1=C(C(=O)O)C=CC=C1)C1=C([N+](=O)[O-])C=C(Cl)C=C1.CN(C1=C(N)C=C(Cl)C=C1)C1=C(C(=O)O)C=CC=C1.CN1C2=CC=C(Cl)C=C2N(CCN)C(=O)C2=C1C=CC=C2.CN1C2=CC=C(Cl)C=C2NC(=O)C2=C1C=CC=C2.CNC1=C(C(=O)O)C=CC=C1.O=[N+]([O-])C1=C(Br)C=CC(Cl)=C1 BXHJBUAHKCILBU-UHFFFAOYSA-N 0.000 description 1
- ZSZXMESTNTYWGL-DEOSSOPVSA-N CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 ZSZXMESTNTYWGL-DEOSSOPVSA-N 0.000 description 1
- ZCTLRUTVVRELAI-XYXHBKGXSA-N CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 ZCTLRUTVVRELAI-XYXHBKGXSA-N 0.000 description 1
- JLQHSJMKKJGULW-DXBVXKBHSA-N CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 JLQHSJMKKJGULW-DXBVXKBHSA-N 0.000 description 1
- JREUITUUGXSTFA-QBUXFMCKSA-N CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 JREUITUUGXSTFA-QBUXFMCKSA-N 0.000 description 1
- XUYDYADIOAUCRP-CPJSRVTESA-N CCNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NCC)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NCC)C=C1)CC2 XUYDYADIOAUCRP-CPJSRVTESA-N 0.000 description 1
- JNBWHPJRERCJQK-RKMGBUROSA-N CCNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 Chemical compound CCNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)NCC)C=C1)CC2 JNBWHPJRERCJQK-RKMGBUROSA-N 0.000 description 1
- QLRIUKFTFWDGGC-HNNXBMFYSA-N CCNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NCC)=C1)C2(C[C@H](C)N)C1=NNN=N1 Chemical compound CCNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NCC)=C1)C2(C[C@H](C)N)C1=NNN=N1 QLRIUKFTFWDGGC-HNNXBMFYSA-N 0.000 description 1
- LDCRCYGXLBRBEC-ACDGEEKESA-N CCNC(c1cc(CCc2c(C3(C[C@@H](Cc(cc4)ccc4F)NCC(N([C@@H](C4)[C@@H]4C4)C4C#N)=O)c4nnn[nH]4)ccc(C(NCC)=O)c2)c3cc1)=O Chemical compound CCNC(c1cc(CCc2c(C3(C[C@@H](Cc(cc4)ccc4F)NCC(N([C@@H](C4)[C@@H]4C4)C4C#N)=O)c4nnn[nH]4)ccc(C(NCC)=O)c2)c3cc1)=O LDCRCYGXLBRBEC-ACDGEEKESA-N 0.000 description 1
- FHOUMNCVSPIZLW-GEVJOTHGSA-N CCNC(c1ccc(C(CC(C2CCCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NCC)=O)c2c1)=O Chemical compound CCNC(c1ccc(C(CC(C2CCCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NCC)=O)c2c1)=O FHOUMNCVSPIZLW-GEVJOTHGSA-N 0.000 description 1
- WLJXCUYXPBQMIK-ZARKZKALSA-N CCNC(c1ccc(C(C[C@@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NCC)=O)c2c1)=O Chemical compound CCNC(c1ccc(C(C[C@@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NCC)=O)c2c1)=O WLJXCUYXPBQMIK-ZARKZKALSA-N 0.000 description 1
- OLSQASKSBXCYRA-ZIAMQFMQSA-N CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N OLSQASKSBXCYRA-ZIAMQFMQSA-N 0.000 description 1
- BUJYYPOSWKUEHA-HVTWWXFQSA-N CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BUJYYPOSWKUEHA-HVTWWXFQSA-N 0.000 description 1
- GLHDKNVVEOHUDG-VUBDRERZSA-N CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N GLHDKNVVEOHUDG-VUBDRERZSA-N 0.000 description 1
- LHXFHRVJUQNCKD-OIEMOICNSA-N CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LHXFHRVJUQNCKD-OIEMOICNSA-N 0.000 description 1
- FQVBPJIMGWINEE-BKBUOWHMSA-N CC[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CC2)cc1C(N(C)C)=O)c(cc1)c2cc1C(N(C)C)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound CC[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CC2)cc1C(N(C)C)=O)c(cc1)c2cc1C(N(C)C)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O FQVBPJIMGWINEE-BKBUOWHMSA-N 0.000 description 1
- ZSFNNIDGFOSLDO-UIOOFZCWSA-N CC[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N ZSFNNIDGFOSLDO-UIOOFZCWSA-N 0.000 description 1
- FQVBPJIMGWINEE-NVGWLYTESA-N CC[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FQVBPJIMGWINEE-NVGWLYTESA-N 0.000 description 1
- RWZHFFWESLQSTH-VXKWHMMOSA-N CC[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2)NCC(=O)N1CCC[C@H]1C#N RWZHFFWESLQSTH-VXKWHMMOSA-N 0.000 description 1
- KZMQLAUEMUGCLY-HXUWFJFHSA-N CC[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CC[C@@H](N)CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 KZMQLAUEMUGCLY-HXUWFJFHSA-N 0.000 description 1
- JCBPETKZIGVZRE-SCSAIBSYSA-N CC[C@@H](N)CO Chemical compound CC[C@@H](N)CO JCBPETKZIGVZRE-SCSAIBSYSA-N 0.000 description 1
- XDPOVHYONPMWSX-RXMQYKEDSA-N CC[C@@H]1COS(=O)(=O)C1 Chemical compound CC[C@@H]1COS(=O)(=O)C1 XDPOVHYONPMWSX-RXMQYKEDSA-N 0.000 description 1
- HBCGPOWMEFXCRO-ZDUSSCGKSA-N CC[C@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 Chemical compound CC[C@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 HBCGPOWMEFXCRO-ZDUSSCGKSA-N 0.000 description 1
- OLSQASKSBXCYRA-JOHWZIMRSA-N CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N OLSQASKSBXCYRA-JOHWZIMRSA-N 0.000 description 1
- BUJYYPOSWKUEHA-FSGKZVOOSA-N CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BUJYYPOSWKUEHA-FSGKZVOOSA-N 0.000 description 1
- GLHDKNVVEOHUDG-FADBTOOJSA-N CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N GLHDKNVVEOHUDG-FADBTOOJSA-N 0.000 description 1
- LHXFHRVJUQNCKD-YEWUNFOKSA-N CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LHXFHRVJUQNCKD-YEWUNFOKSA-N 0.000 description 1
- ZSFNNIDGFOSLDO-FTJBHMTQSA-N CC[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound CC[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N ZSFNNIDGFOSLDO-FTJBHMTQSA-N 0.000 description 1
- FQVBPJIMGWINEE-JMDODURXSA-N CC[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound CC[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 FQVBPJIMGWINEE-JMDODURXSA-N 0.000 description 1
- MQYXZIPBYQHDFQ-INIZCTEOSA-N CC[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CC[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1C=CC(C(N)=O)=C2 MQYXZIPBYQHDFQ-INIZCTEOSA-N 0.000 description 1
- JCBPETKZIGVZRE-BYPYZUCNSA-N CC[C@H](N)CO Chemical compound CC[C@H](N)CO JCBPETKZIGVZRE-BYPYZUCNSA-N 0.000 description 1
- JNFQCFDFVUTABP-XNUZUHMRSA-N CCc1nnc(C(C[C@H](C)NCC(N(CCC2)C2C#N)=O)(c(c(CC2)c3)ccc3C(N)=O)c(cc3)c2cc3C(N)=O)[o]1 Chemical compound CCc1nnc(C(C[C@H](C)NCC(N(CCC2)C2C#N)=O)(c(c(CC2)c3)ccc3C(N)=O)c(cc3)c2cc3C(N)=O)[o]1 JNFQCFDFVUTABP-XNUZUHMRSA-N 0.000 description 1
- HMQJDQMUHXGRPB-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 HMQJDQMUHXGRPB-UHFFFAOYSA-N 0.000 description 1
- VJDVNTDIJYIMRB-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 VJDVNTDIJYIMRB-VWLOTQADSA-N 0.000 description 1
- RFMDSKBRGITERE-QQNWGBJXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 RFMDSKBRGITERE-QQNWGBJXSA-N 0.000 description 1
- VVOQPOYVCAROKJ-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 VVOQPOYVCAROKJ-UHFFFAOYSA-N 0.000 description 1
- NRYSOCSZAKHVDZ-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 NRYSOCSZAKHVDZ-UHFFFAOYSA-N 0.000 description 1
- RULAKPSBDPISON-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 RULAKPSBDPISON-UHFFFAOYSA-N 0.000 description 1
- VXQUWHOTWMSILN-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 VXQUWHOTWMSILN-VWLOTQADSA-N 0.000 description 1
- SMFMZMHBIKSSCB-SANMLTNESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 SMFMZMHBIKSSCB-SANMLTNESA-N 0.000 description 1
- NEBKOXHGYDVVHJ-MHZLTWQESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 NEBKOXHGYDVVHJ-MHZLTWQESA-N 0.000 description 1
- UJIOQACBHFQDEM-NDEPHWFRSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 UJIOQACBHFQDEM-NDEPHWFRSA-N 0.000 description 1
- YSANHBYMKVMWJZ-QQNWGBJXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YSANHBYMKVMWJZ-QQNWGBJXSA-N 0.000 description 1
- AQSWAKCXKSKOPH-ALTZYDRJSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 AQSWAKCXKSKOPH-ALTZYDRJSA-N 0.000 description 1
- LESKPZREXBJIIY-WWDWLDGVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 LESKPZREXBJIIY-WWDWLDGVSA-N 0.000 description 1
- LNPBHJRRGTYEHU-XHWIWGEHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 LNPBHJRRGTYEHU-XHWIWGEHSA-N 0.000 description 1
- AAHCMIYCDRDIFV-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 AAHCMIYCDRDIFV-UHFFFAOYSA-N 0.000 description 1
- RVWGVDSPIIZJKQ-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=O)O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(=O)O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 RVWGVDSPIIZJKQ-UHFFFAOYSA-N 0.000 description 1
- PXQUONDVGHVHAT-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(N)=O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C(N)=O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 PXQUONDVGHVHAT-UHFFFAOYSA-N 0.000 description 1
- QRKNUYGUTJFFPE-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QRKNUYGUTJFFPE-UHFFFAOYSA-N 0.000 description 1
- BJUZFTPGFQFMMY-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 BJUZFTPGFQFMMY-UHFFFAOYSA-N 0.000 description 1
- MOHJCLHLESFKND-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 MOHJCLHLESFKND-UHFFFAOYSA-N 0.000 description 1
- JNDUOXYZCGEKTQ-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 JNDUOXYZCGEKTQ-UHFFFAOYSA-N 0.000 description 1
- TXTNUEJFQWRWAT-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 TXTNUEJFQWRWAT-UHFFFAOYSA-N 0.000 description 1
- KMAQBGPLNGMQLF-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 KMAQBGPLNGMQLF-UHFFFAOYSA-N 0.000 description 1
- BXPATZFRWHKKNO-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 BXPATZFRWHKKNO-UHFFFAOYSA-N 0.000 description 1
- UFROFJDICNGNPA-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=CN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=CN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 UFROFJDICNGNPA-UHFFFAOYSA-N 0.000 description 1
- LBYQHTSUJWFUNY-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 LBYQHTSUJWFUNY-UHFFFAOYSA-N 0.000 description 1
- WVFISYZSUFNOIH-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNN=N1)C1=C2/C=C(C(=O)N(C)C)\C=C/1 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNN=N1)C1=C2/C=C(C(=O)N(C)C)\C=C/1 WVFISYZSUFNOIH-UHFFFAOYSA-N 0.000 description 1
- UPOLMSBQRDUWOG-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 UPOLMSBQRDUWOG-DEOSSOPVSA-N 0.000 description 1
- BLNYSCSANWQPSM-PMERELPUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 BLNYSCSANWQPSM-PMERELPUSA-N 0.000 description 1
- RMGWXMQVKZIALJ-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 RMGWXMQVKZIALJ-DEOSSOPVSA-N 0.000 description 1
- BTODJLWZPQRXOK-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 BTODJLWZPQRXOK-DEOSSOPVSA-N 0.000 description 1
- QMZDWMJGXZEGMJ-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 QMZDWMJGXZEGMJ-DEOSSOPVSA-N 0.000 description 1
- GXCORHLPYFTYKW-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 GXCORHLPYFTYKW-VWLOTQADSA-N 0.000 description 1
- GWINPXJNNCWQHV-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 GWINPXJNNCWQHV-VWLOTQADSA-N 0.000 description 1
- GFHFXTKRFYCHTD-PMERELPUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 GFHFXTKRFYCHTD-PMERELPUSA-N 0.000 description 1
- QGLZIGOSIGVDPJ-PMERELPUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 QGLZIGOSIGVDPJ-PMERELPUSA-N 0.000 description 1
- QOUSTQFSJJBBRD-XYXHBKGXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 QOUSTQFSJJBBRD-XYXHBKGXSA-N 0.000 description 1
- YTENIELCPOYTDQ-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YTENIELCPOYTDQ-VWLOTQADSA-N 0.000 description 1
- QCMLVXSRYLZUAL-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 QCMLVXSRYLZUAL-DEOSSOPVSA-N 0.000 description 1
- UVTIZPZWHIONBY-QHCPKHFHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)C2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)C2 UVTIZPZWHIONBY-QHCPKHFHSA-N 0.000 description 1
- PJRUKQDJUJAIOH-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 PJRUKQDJUJAIOH-DEOSSOPVSA-N 0.000 description 1
- ISBPYBURFZMLAO-FQEVSTJZSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N(C)C)C=C1 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N(C)C)C=C1 ISBPYBURFZMLAO-FQEVSTJZSA-N 0.000 description 1
- KDSLFFLWHADKNK-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 KDSLFFLWHADKNK-DXBVXKBHSA-N 0.000 description 1
- QSVHUOVKISWKPD-DPJKJRSDSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QSVHUOVKISWKPD-DPJKJRSDSA-N 0.000 description 1
- RHJZGMKCZDYJQG-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 RHJZGMKCZDYJQG-DXBVXKBHSA-N 0.000 description 1
- NWDCATOCGYYYQL-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 NWDCATOCGYYYQL-DXBVXKBHSA-N 0.000 description 1
- DIVGITLTZWKEGY-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 DIVGITLTZWKEGY-DXBVXKBHSA-N 0.000 description 1
- INGREKBBEMHQHU-QQNWGBJXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 INGREKBBEMHQHU-QQNWGBJXSA-N 0.000 description 1
- BMVLJOLSLWSFOX-CDTKPYNKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 BMVLJOLSLWSFOX-CDTKPYNKSA-N 0.000 description 1
- MZKISYMROMAEES-QQNWGBJXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 MZKISYMROMAEES-QQNWGBJXSA-N 0.000 description 1
- GULKFZZDDZXSNQ-DPJKJRSDSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 GULKFZZDDZXSNQ-DPJKJRSDSA-N 0.000 description 1
- QGBDLTJGHOXNJY-CDTKPYNKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QGBDLTJGHOXNJY-CDTKPYNKSA-N 0.000 description 1
- XOJXTBQFFFHKQB-CDTKPYNKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 XOJXTBQFFFHKQB-CDTKPYNKSA-N 0.000 description 1
- WBEYTWVXQKIKCN-QQNWGBJXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 WBEYTWVXQKIKCN-QQNWGBJXSA-N 0.000 description 1
- BTNKYNVFPNBGPL-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 BTNKYNVFPNBGPL-DXBVXKBHSA-N 0.000 description 1
- WOORIMQOKIWFGE-QBUXFMCKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 WOORIMQOKIWFGE-QBUXFMCKSA-N 0.000 description 1
- MFOIQLFVMDHZML-DXBVXKBHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 MFOIQLFVMDHZML-DXBVXKBHSA-N 0.000 description 1
- QGELQKXPBAZWAP-UMFSSWHCSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)C2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N(C)C)C=C1)C2 QGELQKXPBAZWAP-UMFSSWHCSA-N 0.000 description 1
- RBCVDNPJKIXCSO-RNHFSVANSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N(C)C)C=C1 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N(C)C)C=C1 RBCVDNPJKIXCSO-RNHFSVANSA-N 0.000 description 1
- QJOCCWYLGMOVGE-JSOSNVBQSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QJOCCWYLGMOVGE-JSOSNVBQSA-N 0.000 description 1
- WOKSVNMXQCDPOA-LSJVWSNHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 WOKSVNMXQCDPOA-LSJVWSNHSA-N 0.000 description 1
- QTKPICYXRMREOH-HSZRJFAPSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 QTKPICYXRMREOH-HSZRJFAPSA-N 0.000 description 1
- MQKIRLBFEWSQJM-AREMUKBSSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 MQKIRLBFEWSQJM-AREMUKBSSA-N 0.000 description 1
- OUJJWZYGVSYVBS-HSZRJFAPSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 OUJJWZYGVSYVBS-HSZRJFAPSA-N 0.000 description 1
- SOFGCUGESUOIHX-XMMPIXPASA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 SOFGCUGESUOIHX-XMMPIXPASA-N 0.000 description 1
- BXKRXHMEHCLXEO-RUZDIDTESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 BXKRXHMEHCLXEO-RUZDIDTESA-N 0.000 description 1
- KBNHOQWDLUZGRB-AREMUKBSSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 KBNHOQWDLUZGRB-AREMUKBSSA-N 0.000 description 1
- TYSYDDCLPPSWBY-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 TYSYDDCLPPSWBY-VWLOTQADSA-N 0.000 description 1
- RNFMNICPVOHZGP-LITSAYRRSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 RNFMNICPVOHZGP-LITSAYRRSA-N 0.000 description 1
- IHIGKWCBCNCRQH-BHDXBOSCSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 IHIGKWCBCNCRQH-BHDXBOSCSA-N 0.000 description 1
- YZXIAWAGNZHHLC-LITSAYRRSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YZXIAWAGNZHHLC-LITSAYRRSA-N 0.000 description 1
- PGKQTZIQOFAWTA-BHBYDHKZSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 PGKQTZIQOFAWTA-BHBYDHKZSA-N 0.000 description 1
- WYDLHIQKCSPCAX-QCENPCRXSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 WYDLHIQKCSPCAX-QCENPCRXSA-N 0.000 description 1
- YNCLFBRKCIZGOH-BHDXBOSCSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YNCLFBRKCIZGOH-BHDXBOSCSA-N 0.000 description 1
- JDZISROXZLXHGJ-MOXFVITDSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 JDZISROXZLXHGJ-MOXFVITDSA-N 0.000 description 1
- LIYAWTCJDFRDGK-HVZDWOJPSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 LIYAWTCJDFRDGK-HVZDWOJPSA-N 0.000 description 1
- XIQGGQSYGCNKDH-ZIMFAVFZSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 XIQGGQSYGCNKDH-ZIMFAVFZSA-N 0.000 description 1
- AVRDRSJIJZOYLM-FSRIWSFDSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 AVRDRSJIJZOYLM-FSRIWSFDSA-N 0.000 description 1
- ORXRDVVZZZPZSS-PYFKZVGHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 ORXRDVVZZZPZSS-PYFKZVGHSA-N 0.000 description 1
- QUKWBISXHHEDEX-MEUCRYMQSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QUKWBISXHHEDEX-MEUCRYMQSA-N 0.000 description 1
- QJOCCWYLGMOVGE-CONSDPRKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QJOCCWYLGMOVGE-CONSDPRKSA-N 0.000 description 1
- WOKSVNMXQCDPOA-XDRKNORUSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 WOKSVNMXQCDPOA-XDRKNORUSA-N 0.000 description 1
- QTKPICYXRMREOH-QHCPKHFHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 QTKPICYXRMREOH-QHCPKHFHSA-N 0.000 description 1
- MQKIRLBFEWSQJM-SANMLTNESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 MQKIRLBFEWSQJM-SANMLTNESA-N 0.000 description 1
- OUJJWZYGVSYVBS-QHCPKHFHSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 OUJJWZYGVSYVBS-QHCPKHFHSA-N 0.000 description 1
- SOFGCUGESUOIHX-DEOSSOPVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 SOFGCUGESUOIHX-DEOSSOPVSA-N 0.000 description 1
- BXKRXHMEHCLXEO-VWLOTQADSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 BXKRXHMEHCLXEO-VWLOTQADSA-N 0.000 description 1
- KBNHOQWDLUZGRB-SANMLTNESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 KBNHOQWDLUZGRB-SANMLTNESA-N 0.000 description 1
- TYSYDDCLPPSWBY-RUZDIDTESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N(C)C)\C=C/1)CC2 TYSYDDCLPPSWBY-RUZDIDTESA-N 0.000 description 1
- RNFMNICPVOHZGP-WNJJXGMVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 RNFMNICPVOHZGP-WNJJXGMVSA-N 0.000 description 1
- IHIGKWCBCNCRQH-NYDCQLBNSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 IHIGKWCBCNCRQH-NYDCQLBNSA-N 0.000 description 1
- YZXIAWAGNZHHLC-WNJJXGMVSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YZXIAWAGNZHHLC-WNJJXGMVSA-N 0.000 description 1
- PGKQTZIQOFAWTA-FIBWVYCGSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 PGKQTZIQOFAWTA-FIBWVYCGSA-N 0.000 description 1
- WYDLHIQKCSPCAX-IZEXYCQBSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 WYDLHIQKCSPCAX-IZEXYCQBSA-N 0.000 description 1
- YNCLFBRKCIZGOH-NYDCQLBNSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 YNCLFBRKCIZGOH-NYDCQLBNSA-N 0.000 description 1
- JDZISROXZLXHGJ-UUHGVMMKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 JDZISROXZLXHGJ-UUHGVMMKSA-N 0.000 description 1
- LIYAWTCJDFRDGK-SHDQMJQSSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 LIYAWTCJDFRDGK-SHDQMJQSSA-N 0.000 description 1
- XIQGGQSYGCNKDH-UUHGVMMKSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 XIQGGQSYGCNKDH-UUHGVMMKSA-N 0.000 description 1
- AVRDRSJIJZOYLM-CRNNPVOZSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 AVRDRSJIJZOYLM-CRNNPVOZSA-N 0.000 description 1
- ORXRDVVZZZPZSS-RMVGYSHESA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 ORXRDVVZZZPZSS-RMVGYSHESA-N 0.000 description 1
- QUKWBISXHHEDEX-CLGPCEASSA-N CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N(C)C)C=C1)CC2 QUKWBISXHHEDEX-CLGPCEASSA-N 0.000 description 1
- BXIJXYPHDCCPII-TULKWJGXSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 BXIJXYPHDCCPII-TULKWJGXSA-N 0.000 description 1
- XUBRABNGMGTNEJ-FQFSJJOFSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 XUBRABNGMGTNEJ-FQFSJJOFSA-N 0.000 description 1
- QFQQYRWNJGPEIX-OWUXGNKUSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 QFQQYRWNJGPEIX-OWUXGNKUSA-N 0.000 description 1
- QAYQZZZANQHDGX-TZJWSJTISA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 QAYQZZZANQHDGX-TZJWSJTISA-N 0.000 description 1
- BXIJXYPHDCCPII-FKKDWBCOSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 BXIJXYPHDCCPII-FKKDWBCOSA-N 0.000 description 1
- XUBRABNGMGTNEJ-UMBQZNGKSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 XUBRABNGMGTNEJ-UMBQZNGKSA-N 0.000 description 1
- QFQQYRWNJGPEIX-SSXAXAPESA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 QFQQYRWNJGPEIX-SSXAXAPESA-N 0.000 description 1
- QAYQZZZANQHDGX-YWUQNDAZSA-N CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 QAYQZZZANQHDGX-YWUQNDAZSA-N 0.000 description 1
- ONRXLSQXPLBBBC-INIZCTEOSA-N CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N(C)C)S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N(C)C)S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 ONRXLSQXPLBBBC-INIZCTEOSA-N 0.000 description 1
- DWZDHDLCVKWQSU-GJYPPUQNSA-N CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N(C)C)S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound CN(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N(C)C)S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 DWZDHDLCVKWQSU-GJYPPUQNSA-N 0.000 description 1
- XXMXTPZRQWWCFK-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(CCC3=C(C=CS3)C2(CCN)C2=NNN=N2)S1 Chemical compound CN(C)C(=O)C1=CC2=C(CCC3=C(C=CS3)C2(CCN)C2=NNN=N2)S1 XXMXTPZRQWWCFK-UHFFFAOYSA-N 0.000 description 1
- OUVZHEHRIJVQKO-UHFFFAOYSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=CS1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=CS1)CC2 OUVZHEHRIJVQKO-UHFFFAOYSA-N 0.000 description 1
- FLXIICKXMVJKFG-SFHVURJKSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N(C)C)S1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N(C)C)S1)CC2 FLXIICKXMVJKFG-SFHVURJKSA-N 0.000 description 1
- HYQFIKCOYXZZJW-QYZOEREBSA-N CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N(C)C)S1)CC2 Chemical compound CN(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N(C)C)S1)CC2 HYQFIKCOYXZZJW-QYZOEREBSA-N 0.000 description 1
- ZZURBXVDNWCHHY-UHFFFAOYSA-N CN(C)C(=O)C1=CC=C(C(CCN)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 Chemical compound CN(C)C(=O)C1=CC=C(C(CCN)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 ZZURBXVDNWCHHY-UHFFFAOYSA-N 0.000 description 1
- DKUWZVBTBJWGKL-DEOSSOPVSA-N CN(C)C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 Chemical compound CN(C)C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 DKUWZVBTBJWGKL-DEOSSOPVSA-N 0.000 description 1
- YRFUKTQCWNZWEO-ZODMCCGTSA-N CN(C)C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 Chemical compound CN(C)C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N(C)C)C=C2)C2=NNN=N2)C=C1 YRFUKTQCWNZWEO-ZODMCCGTSA-N 0.000 description 1
- RXZQLLHSZCSUDE-UHFFFAOYSA-N CN(C)C(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NNN=N1 Chemical compound CN(C)C(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NNN=N1 RXZQLLHSZCSUDE-UHFFFAOYSA-N 0.000 description 1
- VDVOACKKBMCCBL-QQNWGBJXSA-N CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)N(C)C)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C Chemical compound CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)N(C)C)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C VDVOACKKBMCCBL-QQNWGBJXSA-N 0.000 description 1
- JSJAIRWSQYSMOH-UHFFFAOYSA-N CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NN=NN1C Chemical compound CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NN=NN1C JSJAIRWSQYSMOH-UHFFFAOYSA-N 0.000 description 1
- PQVQWVDFSFLVLO-UHFFFAOYSA-N CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NNN=N1 Chemical compound CN(C)C(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)N(C)C)=C1)C2(CCN)C1=NNN=N1 PQVQWVDFSFLVLO-UHFFFAOYSA-N 0.000 description 1
- ONRXLSQXPLBBBC-UHFFFAOYSA-N CN(C)C(c1cc(C(CCNCC(N(CCC2)C2C#N)=O)(c2c(CC3)[s]c(C(N(C)C)=O)c2)c2n[nH]nn2)c3[s]1)=O Chemical compound CN(C)C(c1cc(C(CCNCC(N(CCC2)C2C#N)=O)(c2c(CC3)[s]c(C(N(C)C)=O)c2)c2n[nH]nn2)c3[s]1)=O ONRXLSQXPLBBBC-UHFFFAOYSA-N 0.000 description 1
- DWZDHDLCVKWQSU-LQUOLJLFSA-N CN(C)C(c1cc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2c(CC3)[s]c(C(N(C)C)=O)c2)c2n[nH]nn2)c3[s]1)=O Chemical compound CN(C)C(c1cc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2c(CC3)[s]c(C(N(C)C)=O)c2)c2n[nH]nn2)c3[s]1)=O DWZDHDLCVKWQSU-LQUOLJLFSA-N 0.000 description 1
- ADTDFCKINGZRCO-SKMGBCOQSA-N CN(C)C(c1cc(CCc2c(C3(CCNCC(N([C@@H](C4)[C@@H]4C4)C4C#N)=O)c4n[nH]nn4)[s]cc2)c3[s]1)=O Chemical compound CN(C)C(c1cc(CCc2c(C3(CCNCC(N([C@@H](C4)[C@@H]4C4)C4C#N)=O)c4n[nH]nn4)[s]cc2)c3[s]1)=O ADTDFCKINGZRCO-SKMGBCOQSA-N 0.000 description 1
- AVRDRSJIJZOYLM-QDEJEKJNSA-N CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@@H](C3CCC3)NCC(N([C@@H](C3)[C@@H]3C3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O Chemical compound CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@@H](C3CCC3)NCC(N([C@@H](C3)[C@@H]3C3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O AVRDRSJIJZOYLM-QDEJEKJNSA-N 0.000 description 1
- PGKQTZIQOFAWTA-DINWUYGQSA-N CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@H](C3CCC3)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O Chemical compound CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@H](C3CCC3)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O PGKQTZIQOFAWTA-DINWUYGQSA-N 0.000 description 1
- QJOCCWYLGMOVGE-FSRLHOSWSA-N CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@H](Cc(cc3)ccc3F)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O Chemical compound CN(C)C(c1cc(CCc2cc(C(N(C)C)=O)ccc2C2(C[C@H](Cc(cc3)ccc3F)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O QJOCCWYLGMOVGE-FSRLHOSWSA-N 0.000 description 1
- GFHFXTKRFYCHTD-UHFFFAOYSA-N CN(C)C(c1ccc(C(CCNCC(N(CCC2)C2C#N)=O)(c2nnc(-c(cc3)ccc3F)[nH]2)c(cc2)c(CC3)cc2C(N(C)C)=O)c3c1)=O Chemical compound CN(C)C(c1ccc(C(CCNCC(N(CCC2)C2C#N)=O)(c2nnc(-c(cc3)ccc3F)[nH]2)c(cc2)c(CC3)cc2C(N(C)C)=O)c3c1)=O GFHFXTKRFYCHTD-UHFFFAOYSA-N 0.000 description 1
- ORXRDVVZZZPZSS-TXPBDTFASA-N CN(C)C(c1ccc(C(C[C@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N(C)C)=O)c2c1)=O Chemical compound CN(C)C(c1ccc(C(C[C@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N(C)C)=O)c2c1)=O ORXRDVVZZZPZSS-TXPBDTFASA-N 0.000 description 1
- PIZXPPHCJPLZQH-NXQKYAITSA-N CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 PIZXPPHCJPLZQH-NXQKYAITSA-N 0.000 description 1
- JNXJXYVUGOJRMS-HHRCKSQXSA-N CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 JNXJXYVUGOJRMS-HHRCKSQXSA-N 0.000 description 1
- KFVOYTYYQRFCRS-KCNFHIIESA-N CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 KFVOYTYYQRFCRS-KCNFHIIESA-N 0.000 description 1
- ZBWSFNISUZNHQP-LLVKDONJSA-N CN(C)OOSC1=CC2=C(C=C1)[C@H](N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@H](N)CC2 ZBWSFNISUZNHQP-LLVKDONJSA-N 0.000 description 1
- WDIRZKZHPNMLBT-PKKACIRVSA-N CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 WDIRZKZHPNMLBT-PKKACIRVSA-N 0.000 description 1
- PIZXPPHCJPLZQH-BVKOHWRASA-N CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 PIZXPPHCJPLZQH-BVKOHWRASA-N 0.000 description 1
- JNXJXYVUGOJRMS-ODXUMFNNSA-N CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CN(C)OOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 JNXJXYVUGOJRMS-ODXUMFNNSA-N 0.000 description 1
- GCLYNPRAYARXMT-UHFFFAOYSA-N CN1C(=O)C2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.CN1C(=O)C2=C(C=CC=C2)C(CCN)(C2=NNN=N2)C2=C1C=CC=C2.CN1C(=O)C2=C(C=CC=C2)C(CCN2C(=O)C3=C(C=CC=C3)C2=O)(C2=NNN=N2)C2=C1C=CC=C2.Cl.O=C1C2=CC=CC=C2C(=O)C2=C1C=CC=C2.O=C1NC2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC=C2)C(=O)N(C)C2=C1C=CC=C2 Chemical compound CN1C(=O)C2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.CN1C(=O)C2=C(C=CC=C2)C(CCN)(C2=NNN=N2)C2=C1C=CC=C2.CN1C(=O)C2=C(C=CC=C2)C(CCN2C(=O)C3=C(C=CC=C3)C2=O)(C2=NNN=N2)C2=C1C=CC=C2.Cl.O=C1C2=CC=CC=C2C(=O)C2=C1C=CC=C2.O=C1NC2=C(C=CC=C2)C(=O)C2=C1C=CC=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC=C2)C(=O)N(C)C2=C1C=CC=C2 GCLYNPRAYARXMT-UHFFFAOYSA-N 0.000 description 1
- ALCKIXFWXMACHW-LFUZPPSTSA-N CN1C(=O)C2=C(C=CC=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NNN=N2)C2=C1C=CC=C2 Chemical compound CN1C(=O)C2=C(C=CC=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NNN=N2)C2=C1C=CC=C2 ALCKIXFWXMACHW-LFUZPPSTSA-N 0.000 description 1
- AFWBDJICJYTWBU-KRWDZBQOSA-N CN1C2=C(C=CC=C2)N(CCNCC(=O)N2CCC[C@H]2C#N)C(=O)C2=C1/C=C\C(Cl)=C/2 Chemical compound CN1C2=C(C=CC=C2)N(CCNCC(=O)N2CCC[C@H]2C#N)C(=O)C2=C1/C=C\C(Cl)=C/2 AFWBDJICJYTWBU-KRWDZBQOSA-N 0.000 description 1
- FAROCRNITWOJEL-ZWWNWMABSA-N CN1C2=C(C=CC=C2)N(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C(=O)C2=C1/C=C\C(Cl)=C/2 Chemical compound CN1C2=C(C=CC=C2)N(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C(=O)C2=C1/C=C\C(Cl)=C/2 FAROCRNITWOJEL-ZWWNWMABSA-N 0.000 description 1
- BIZWQSSLHYVWCX-PMERELPUSA-N CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NN=NN2)C2=C(C=C(C(=O)N4CCN(C)CC4)C=C2)CC3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NN=NN2)C2=C(C=C(C(=O)N4CCN(C)CC4)C=C2)CC3)CC1 BIZWQSSLHYVWCX-PMERELPUSA-N 0.000 description 1
- CRFZNGXEGMTEAY-ZGAORZAOSA-N CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NN=NN2)C2=C(C=C(F)C=C2)CC3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=NN=NN2)C2=C(C=C(F)C=C2)CC3)CC1 CRFZNGXEGMTEAY-ZGAORZAOSA-N 0.000 description 1
- LUJQAWVPMNFWGG-ROCBYOHGSA-N CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2[C@H](C#N)C[C@@H]4C[C@@H]42)(C2=NN=NN2)C2=C(C=C(C(=O)N4CCN(C)CC4)C=C2)CC3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2[C@H](C#N)C[C@@H]4C[C@@H]42)(C2=NN=NN2)C2=C(C=C(C(=O)N4CCN(C)CC4)C=C2)CC3)CC1 LUJQAWVPMNFWGG-ROCBYOHGSA-N 0.000 description 1
- GHSHOPPZYOTGRG-ILKCWXONSA-N CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2[C@H](C#N)C[C@@H]4C[C@@H]42)(C2=NN=NN2)C2=C(C=C(F)C=C2)CC3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(C=C2)C(CCNCC(=O)N2[C@H](C#N)C[C@@H]4C[C@@H]42)(C2=NN=NN2)C2=C(C=C(F)C=C2)CC3)CC1 GHSHOPPZYOTGRG-ILKCWXONSA-N 0.000 description 1
- YTGSRVXJINTMAA-UHFFFAOYSA-N CN1N=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O Chemical compound CN1N=C(C2(CCN)C3=C(C=C(C(N)=O)C=C3)CCC3=C2C=CC(C(N)=O)=C3)NC1=O YTGSRVXJINTMAA-UHFFFAOYSA-N 0.000 description 1
- VDIAECDEEFPGBL-VWLOTQADSA-N CN1N=NC(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)=N1 Chemical compound CN1N=NC(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=C(C=C(C(=O)N(C)C)C=C3)CCC3=C2C=CC(C(=O)N(C)C)=C3)=N1 VDIAECDEEFPGBL-VWLOTQADSA-N 0.000 description 1
- HZVBNWXVUOXSRL-NRFANRHFSA-N CN1N=NC(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=CC=C(C(N)=O)C=C3CCC3=C2/C=C\C(C(N)=O)=C/3)=N1 Chemical compound CN1N=NC(C2(CCNCC(=O)N3CCC[C@H]3C#N)C3=CC=C(C(N)=O)C=C3CCC3=C2/C=C\C(C(N)=O)=C/3)=N1 HZVBNWXVUOXSRL-NRFANRHFSA-N 0.000 description 1
- RVZWKXHZQHHJIS-QQNWGBJXSA-N CN1N=NC(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=CC=C(C(=O)N(C)C)C=C3CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)=N1 Chemical compound CN1N=NC(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=CC=C(C(=O)N(C)C)C=C3CCC3=C2/C=C\C(C(=O)N(C)C)=C/3)=N1 RVZWKXHZQHHJIS-QQNWGBJXSA-N 0.000 description 1
- WCINZOZOYXZORB-DPLHUUCSSA-N CN1N=NC(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=CC=C(C(N)=O)C=C3CCC3=C2/C=C\C(C(N)=O)=C/3)=N1 Chemical compound CN1N=NC(C2(CCNCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)C3=CC=C(C(N)=O)C=C3CCC3=C2/C=C\C(C(N)=O)=C/3)=N1 WCINZOZOYXZORB-DPLHUUCSSA-N 0.000 description 1
- FOXFBRVETZBSPI-NRFANRHFSA-N CN1N=NN=C1C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 Chemical compound CN1N=NN=C1C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 FOXFBRVETZBSPI-NRFANRHFSA-N 0.000 description 1
- ZTQIYYCIPKXCPF-DPLHUUCSSA-N CN1N=NN=C1C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 Chemical compound CN1N=NN=C1C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 ZTQIYYCIPKXCPF-DPLHUUCSSA-N 0.000 description 1
- NDHYLYNBLIMUEM-UHFFFAOYSA-N CN1OC(=O)N=C1C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CN1OC(=O)N=C1C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 NDHYLYNBLIMUEM-UHFFFAOYSA-N 0.000 description 1
- WUXCDJFFBGGOMT-UHFFFAOYSA-N CNC(=N)C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(CCN)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 WUXCDJFFBGGOMT-UHFFFAOYSA-N 0.000 description 1
- OMBXVQCKZPXIPR-UHFFFAOYSA-N CNC(=N)C1(CCN)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 Chemical compound CNC(=N)C1(CCN)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 OMBXVQCKZPXIPR-UHFFFAOYSA-N 0.000 description 1
- FHVXGIFAEVXQNF-UHFFFAOYSA-N CNC(=N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CNC(=N)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 FHVXGIFAEVXQNF-UHFFFAOYSA-N 0.000 description 1
- XFUIFHYVFODELL-VWLOTQADSA-N CNC(=N)C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CNC(=N)C1(CCNCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 XFUIFHYVFODELL-VWLOTQADSA-N 0.000 description 1
- IQVFDGVMMZYWEC-QQNWGBJXSA-N CNC(=N)C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 IQVFDGVMMZYWEC-QQNWGBJXSA-N 0.000 description 1
- WPKNEQWMRLVUNG-MRXNPFEDSA-N CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 WPKNEQWMRLVUNG-MRXNPFEDSA-N 0.000 description 1
- RJYCDHPRYUAOJR-CQSZACIVSA-N CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 Chemical compound CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 RJYCDHPRYUAOJR-CQSZACIVSA-N 0.000 description 1
- BCZTVSKSONYTNV-GFCCVEGCSA-N CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CNC(=N)C1(C[C@@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 BCZTVSKSONYTNV-GFCCVEGCSA-N 0.000 description 1
- HUXRFMUEJTWBAY-RLWLMLJZSA-N CNC(=N)C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CNC(=N)C1(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 HUXRFMUEJTWBAY-RLWLMLJZSA-N 0.000 description 1
- GUCRMZGIZLOYFR-ZLGCBXFLSA-N CNC(=N)C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 GUCRMZGIZLOYFR-ZLGCBXFLSA-N 0.000 description 1
- WPKNEQWMRLVUNG-INIZCTEOSA-N CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 WPKNEQWMRLVUNG-INIZCTEOSA-N 0.000 description 1
- RJYCDHPRYUAOJR-AWEZNQCLSA-N CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 Chemical compound CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(=O)NC)C=C2)CCC2=C1C=CC(C(=O)NC)=C2 RJYCDHPRYUAOJR-AWEZNQCLSA-N 0.000 description 1
- BCZTVSKSONYTNV-LBPRGKRZSA-N CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound CNC(=N)C1(C[C@H](C)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 BCZTVSKSONYTNV-LBPRGKRZSA-N 0.000 description 1
- HUXRFMUEJTWBAY-LVXARBLLSA-N CNC(=N)C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound CNC(=N)C1(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2 HUXRFMUEJTWBAY-LVXARBLLSA-N 0.000 description 1
- GUCRMZGIZLOYFR-YIPSIMJMSA-N CNC(=N)C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound CNC(=N)C1(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 GUCRMZGIZLOYFR-YIPSIMJMSA-N 0.000 description 1
- IJWWOTMBEUEFSD-MBFMOYRZSA-N CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 IJWWOTMBEUEFSD-MBFMOYRZSA-N 0.000 description 1
- DVSBOCYHDIMZAM-SSRYDLFMSA-N CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 DVSBOCYHDIMZAM-SSRYDLFMSA-N 0.000 description 1
- XZAZZGFOWINYFU-GLBNMMOUSA-N CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 XZAZZGFOWINYFU-GLBNMMOUSA-N 0.000 description 1
- NGBNOPSFXRCBGB-VAVNUEAYSA-N CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound CNC(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 NGBNOPSFXRCBGB-VAVNUEAYSA-N 0.000 description 1
- IJWWOTMBEUEFSD-UAUOSOAOSA-N CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 IJWWOTMBEUEFSD-UAUOSOAOSA-N 0.000 description 1
- DVSBOCYHDIMZAM-HVRBKMHZSA-N CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 DVSBOCYHDIMZAM-HVRBKMHZSA-N 0.000 description 1
- XZAZZGFOWINYFU-RSYUGDCRSA-N CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 XZAZZGFOWINYFU-RSYUGDCRSA-N 0.000 description 1
- NGBNOPSFXRCBGB-XKZOHVQRSA-N CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound CNC(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 NGBNOPSFXRCBGB-XKZOHVQRSA-N 0.000 description 1
- FXAVMOTWFVJPJQ-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 FXAVMOTWFVJPJQ-UHFFFAOYSA-N 0.000 description 1
- VYNZZEANQZOLPM-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 VYNZZEANQZOLPM-UHFFFAOYSA-N 0.000 description 1
- WEVXHRMCFWNXTE-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 WEVXHRMCFWNXTE-UHFFFAOYSA-N 0.000 description 1
- FULUJQMTRCVPQF-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 FULUJQMTRCVPQF-UHFFFAOYSA-N 0.000 description 1
- YOZDZFGMBOYNLR-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 YOZDZFGMBOYNLR-UHFFFAOYSA-N 0.000 description 1
- CIOQRESBCIDIAD-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1)C1=C(C=C(C(=O)NC)C=C1)CC2 CIOQRESBCIDIAD-UHFFFAOYSA-N 0.000 description 1
- STIGBCSVIXXXPT-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 STIGBCSVIXXXPT-UHFFFAOYSA-N 0.000 description 1
- CNPFNRQUTIKKNN-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 CNPFNRQUTIKKNN-UHFFFAOYSA-N 0.000 description 1
- LERDXLIRTQNDSK-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 LERDXLIRTQNDSK-UHFFFAOYSA-N 0.000 description 1
- KTFVHDMXVAZBKT-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 KTFVHDMXVAZBKT-UHFFFAOYSA-N 0.000 description 1
- IXOLFPKAHRIFSO-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 IXOLFPKAHRIFSO-UHFFFAOYSA-N 0.000 description 1
- YYXIWOHKABUJAS-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 YYXIWOHKABUJAS-UHFFFAOYSA-N 0.000 description 1
- IQHFPLJNDQZTCD-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 IQHFPLJNDQZTCD-UHFFFAOYSA-N 0.000 description 1
- PRQMNFOMPXWFQW-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 PRQMNFOMPXWFQW-UHFFFAOYSA-N 0.000 description 1
- CNHQVRZHAZXHNA-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 CNHQVRZHAZXHNA-UHFFFAOYSA-N 0.000 description 1
- FLQHIWVOQMRKIL-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 FLQHIWVOQMRKIL-UHFFFAOYSA-N 0.000 description 1
- PUTOYKVEISMUCK-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 PUTOYKVEISMUCK-UHFFFAOYSA-N 0.000 description 1
- YHDZIFJUQLJNLL-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 YHDZIFJUQLJNLL-UHFFFAOYSA-N 0.000 description 1
- UTYHMRUGWYHOKP-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 UTYHMRUGWYHOKP-UHFFFAOYSA-N 0.000 description 1
- GJCCLICATUWTHM-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 GJCCLICATUWTHM-UHFFFAOYSA-N 0.000 description 1
- UIYSFJBLUIIJTK-UHFFFAOYSA-N CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(CCN)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 UIYSFJBLUIIJTK-UHFFFAOYSA-N 0.000 description 1
- XCDJLERNZMMMAY-CYBMUJFWSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 XCDJLERNZMMMAY-CYBMUJFWSA-N 0.000 description 1
- RWIUGJAUIWLSRZ-MRXNPFEDSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 RWIUGJAUIWLSRZ-MRXNPFEDSA-N 0.000 description 1
- OPGPYGNOALYGKE-QGZVFWFLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 OPGPYGNOALYGKE-QGZVFWFLSA-N 0.000 description 1
- MHAFVDKQNHPPDA-CYBMUJFWSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 MHAFVDKQNHPPDA-CYBMUJFWSA-N 0.000 description 1
- QYJSDGWXVRVOME-CYBMUJFWSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 QYJSDGWXVRVOME-CYBMUJFWSA-N 0.000 description 1
- GNQLQHVUUZOKJO-CYBMUJFWSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1)C1=C(C=C(C(=O)NC)C=C1)CC2 GNQLQHVUUZOKJO-CYBMUJFWSA-N 0.000 description 1
- BDBPEQBIGSNWBM-MRXNPFEDSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 BDBPEQBIGSNWBM-MRXNPFEDSA-N 0.000 description 1
- YLYVIYCHCVMBDZ-CQSZACIVSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 YLYVIYCHCVMBDZ-CQSZACIVSA-N 0.000 description 1
- PKFCZLAVUDKIKV-GOSISDBHSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 PKFCZLAVUDKIKV-GOSISDBHSA-N 0.000 description 1
- XGFUTMVGCYTVDX-MRXNPFEDSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 XGFUTMVGCYTVDX-MRXNPFEDSA-N 0.000 description 1
- HCMTWJOSMLFHQH-CQSZACIVSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HCMTWJOSMLFHQH-CQSZACIVSA-N 0.000 description 1
- QJNXMZNHEBOVOY-QGZVFWFLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 QJNXMZNHEBOVOY-QGZVFWFLSA-N 0.000 description 1
- HWNUKOLQAGKWFT-MRXNPFEDSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HWNUKOLQAGKWFT-MRXNPFEDSA-N 0.000 description 1
- GINUSCQNEQBHQE-MRXNPFEDSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 GINUSCQNEQBHQE-MRXNPFEDSA-N 0.000 description 1
- VKLGSMYHLVJXCB-CQSZACIVSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 VKLGSMYHLVJXCB-CQSZACIVSA-N 0.000 description 1
- LUFMSRAQRFJXCM-CQSZACIVSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 LUFMSRAQRFJXCM-CQSZACIVSA-N 0.000 description 1
- SZPSZUYHIAWHRZ-QGZVFWFLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 SZPSZUYHIAWHRZ-QGZVFWFLSA-N 0.000 description 1
- ZALVHQXMKHVSAM-QGZVFWFLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 ZALVHQXMKHVSAM-QGZVFWFLSA-N 0.000 description 1
- SMSSDIGHGJGALY-CQSZACIVSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 SMSSDIGHGJGALY-CQSZACIVSA-N 0.000 description 1
- HKTQXEPNYXVFEO-CYBMUJFWSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HKTQXEPNYXVFEO-CYBMUJFWSA-N 0.000 description 1
- JBAMNGXVZXFWNB-LLVKDONJSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C2/C=C(C(=O)NC)\C=C/1 JBAMNGXVZXFWNB-LLVKDONJSA-N 0.000 description 1
- XCDJLERNZMMMAY-ZDUSSCGKSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)N)C1=C(C=C(C(=O)NC)C=C1)CC2 XCDJLERNZMMMAY-ZDUSSCGKSA-N 0.000 description 1
- RWIUGJAUIWLSRZ-INIZCTEOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)NC(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 RWIUGJAUIWLSRZ-INIZCTEOSA-N 0.000 description 1
- OPGPYGNOALYGKE-KRWDZBQOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)NC)C=C1)CC2 OPGPYGNOALYGKE-KRWDZBQOSA-N 0.000 description 1
- MHAFVDKQNHPPDA-ZDUSSCGKSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(=O)O)C1=C(C=C(C(=O)NC)C=C1)CC2 MHAFVDKQNHPPDA-ZDUSSCGKSA-N 0.000 description 1
- QYJSDGWXVRVOME-ZDUSSCGKSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C(N)=O)C1=C(C=C(C(=O)NC)C=C1)CC2 QYJSDGWXVRVOME-ZDUSSCGKSA-N 0.000 description 1
- LXSNIKVTSLPFFU-LNHXHEARSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1)C1=C(C=C(NC(C)=O)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1)C1=C(C=C(NC(C)=O)C=C1)CC2 LXSNIKVTSLPFFU-LNHXHEARSA-N 0.000 description 1
- BDBPEQBIGSNWBM-INIZCTEOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)NC)C=C1)CC2 BDBPEQBIGSNWBM-INIZCTEOSA-N 0.000 description 1
- YLYVIYCHCVMBDZ-AWEZNQCLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)NC)C=C1)CC2 YLYVIYCHCVMBDZ-AWEZNQCLSA-N 0.000 description 1
- PKFCZLAVUDKIKV-SFHVURJKSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)NC)C=C1)CC2 PKFCZLAVUDKIKV-SFHVURJKSA-N 0.000 description 1
- XGFUTMVGCYTVDX-INIZCTEOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 XGFUTMVGCYTVDX-INIZCTEOSA-N 0.000 description 1
- HCMTWJOSMLFHQH-AWEZNQCLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HCMTWJOSMLFHQH-AWEZNQCLSA-N 0.000 description 1
- QJNXMZNHEBOVOY-KRWDZBQOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 QJNXMZNHEBOVOY-KRWDZBQOSA-N 0.000 description 1
- HWNUKOLQAGKWFT-INIZCTEOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HWNUKOLQAGKWFT-INIZCTEOSA-N 0.000 description 1
- GINUSCQNEQBHQE-INIZCTEOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 GINUSCQNEQBHQE-INIZCTEOSA-N 0.000 description 1
- VKLGSMYHLVJXCB-AWEZNQCLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 VKLGSMYHLVJXCB-AWEZNQCLSA-N 0.000 description 1
- LUFMSRAQRFJXCM-AWEZNQCLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 LUFMSRAQRFJXCM-AWEZNQCLSA-N 0.000 description 1
- SZPSZUYHIAWHRZ-KRWDZBQOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 SZPSZUYHIAWHRZ-KRWDZBQOSA-N 0.000 description 1
- ZALVHQXMKHVSAM-KRWDZBQOSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)NC)C=C1)CC2 ZALVHQXMKHVSAM-KRWDZBQOSA-N 0.000 description 1
- SMSSDIGHGJGALY-AWEZNQCLSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NN=CN1)C1=C(C=C(C(=O)NC)C=C1)CC2 SMSSDIGHGJGALY-AWEZNQCLSA-N 0.000 description 1
- HKTQXEPNYXVFEO-ZDUSSCGKSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNC(=O)N1)C1=C(C=C(C(=O)NC)C=C1)CC2 HKTQXEPNYXVFEO-ZDUSSCGKSA-N 0.000 description 1
- DGNVZAGZUZDSBP-MBSDFSHPSA-N CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)C=C1)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)C=C1)CC2 DGNVZAGZUZDSBP-MBSDFSHPSA-N 0.000 description 1
- ZHVDLDSMVOVYRV-RUDJEGBNSA-N CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 ZHVDLDSMVOVYRV-RUDJEGBNSA-N 0.000 description 1
- AKWUKBSAVUDICQ-XYYCENQHSA-N CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 AKWUKBSAVUDICQ-XYYCENQHSA-N 0.000 description 1
- BKQWEYNMTIDPBM-HPGPFUFHSA-N CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 BKQWEYNMTIDPBM-HPGPFUFHSA-N 0.000 description 1
- TZSOAQRWIQKGFD-DQKLJIJWSA-N CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 TZSOAQRWIQKGFD-DQKLJIJWSA-N 0.000 description 1
- ZHVDLDSMVOVYRV-VOSLBQETSA-N CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 ZHVDLDSMVOVYRV-VOSLBQETSA-N 0.000 description 1
- AKWUKBSAVUDICQ-UGKANONSSA-N CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 AKWUKBSAVUDICQ-UGKANONSSA-N 0.000 description 1
- BKQWEYNMTIDPBM-WFYLZJEKSA-N CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 BKQWEYNMTIDPBM-WFYLZJEKSA-N 0.000 description 1
- TZSOAQRWIQKGFD-RGOCWXFISA-N CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CNC(=O)C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 TZSOAQRWIQKGFD-RGOCWXFISA-N 0.000 description 1
- OSRNDAHMEHYUDA-SECBINFHSA-N CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(C[C@@H](C)N)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(C[C@@H](C)N)C2=NNN=N2)S1 OSRNDAHMEHYUDA-SECBINFHSA-N 0.000 description 1
- OSRNDAHMEHYUDA-VIFPVBQESA-N CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(C[C@H](C)N)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=C(C(=O)NC)S3)C2(C[C@H](C)N)C2=NNN=N2)S1 OSRNDAHMEHYUDA-VIFPVBQESA-N 0.000 description 1
- IIBAMMXPMTWYIH-UHFFFAOYSA-N CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(CCN)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(CCN)C2=NNN=N2)S1 IIBAMMXPMTWYIH-UHFFFAOYSA-N 0.000 description 1
- AQZZVNASMILUTA-SUSUGVNDSA-N CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(C[C@@H](C)N)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(C[C@@H](C)N)C2=NNN=N2)S1 AQZZVNASMILUTA-SUSUGVNDSA-N 0.000 description 1
- AQZZVNASMILUTA-WFVOFKTRSA-N CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(C[C@H](C)N)C2=NNN=N2)S1 Chemical compound CNC(=O)C1=CC2=C(CCC3=C(C=CS3)C2(C[C@H](C)N)C2=NNN=N2)S1 AQZZVNASMILUTA-WFVOFKTRSA-N 0.000 description 1
- BRKUQHSVNMUBGJ-UHFFFAOYSA-N CNC(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 BRKUQHSVNMUBGJ-UHFFFAOYSA-N 0.000 description 1
- WLGGQMSBQXTTKF-UHFFFAOYSA-N CNC(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=CS1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(CCN)(C1=NNN=N1)C1=C(C=CS1)CC2 WLGGQMSBQXTTKF-UHFFFAOYSA-N 0.000 description 1
- GKBXYQRZKFWTSQ-SECBINFHSA-N CNC(=O)C1=CC2=C(S1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 GKBXYQRZKFWTSQ-SECBINFHSA-N 0.000 description 1
- VZTQURWYADFJPN-SUSUGVNDSA-N CNC(=O)C1=CC2=C(S1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C(C=CS1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(C[C@@H](C)N)(C1=NNN=N1)C1=C(C=CS1)CC2 VZTQURWYADFJPN-SUSUGVNDSA-N 0.000 description 1
- GKBXYQRZKFWTSQ-VIFPVBQESA-N CNC(=O)C1=CC2=C(S1)C(C[C@H](C)N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(C[C@H](C)N)(C1=NNN=N1)C1=C(C=C(C(=O)NC)S1)CC2 GKBXYQRZKFWTSQ-VIFPVBQESA-N 0.000 description 1
- VZTQURWYADFJPN-WFVOFKTRSA-N CNC(=O)C1=CC2=C(S1)C(C[C@H](C)N)(C1=NNN=N1)C1=C(C=CS1)CC2 Chemical compound CNC(=O)C1=CC2=C(S1)C(C[C@H](C)N)(C1=NNN=N1)C1=C(C=CS1)CC2 VZTQURWYADFJPN-WFVOFKTRSA-N 0.000 description 1
- UCDUVJFKWOWTBG-UHFFFAOYSA-N CNC(=O)C1=CC=C(C(CCN)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 Chemical compound CNC(=O)C1=CC=C(C(CCN)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 UCDUVJFKWOWTBG-UHFFFAOYSA-N 0.000 description 1
- FDHCREMHKGLRQU-CYBMUJFWSA-N CNC(=O)C1=CC=C(C(C[C@@H](C)N)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 Chemical compound CNC(=O)C1=CC=C(C(C[C@@H](C)N)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 FDHCREMHKGLRQU-CYBMUJFWSA-N 0.000 description 1
- FDHCREMHKGLRQU-ZDUSSCGKSA-N CNC(=O)C1=CC=C(C(C[C@H](C)N)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 Chemical compound CNC(=O)C1=CC=C(C(C[C@H](C)N)(C2=CC=C(C(=O)NC)C=C2)C2=NNN=N2)C=C1 FDHCREMHKGLRQU-ZDUSSCGKSA-N 0.000 description 1
- JGYWPAUSQIKCSD-SSDOTTSWSA-N CNC(=O)C1=CC=C([C@@H](C)N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@@H](C)N)C=C1 JGYWPAUSQIKCSD-SSDOTTSWSA-N 0.000 description 1
- ICGDRHFXCFSCCN-MBFMOYRZSA-N CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 ICGDRHFXCFSCCN-MBFMOYRZSA-N 0.000 description 1
- IOMBCYWAEMKGQY-SSRYDLFMSA-N CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 IOMBCYWAEMKGQY-SSRYDLFMSA-N 0.000 description 1
- XRDNYZXPGKJWJZ-GLBNMMOUSA-N CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 XRDNYZXPGKJWJZ-GLBNMMOUSA-N 0.000 description 1
- MYBUUCHGLSQJTA-VAVNUEAYSA-N CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound CNC(=O)C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 MYBUUCHGLSQJTA-VAVNUEAYSA-N 0.000 description 1
- ICGDRHFXCFSCCN-UAUOSOAOSA-N CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 ICGDRHFXCFSCCN-UAUOSOAOSA-N 0.000 description 1
- IOMBCYWAEMKGQY-HVRBKMHZSA-N CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 IOMBCYWAEMKGQY-HVRBKMHZSA-N 0.000 description 1
- XRDNYZXPGKJWJZ-RSYUGDCRSA-N CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 XRDNYZXPGKJWJZ-RSYUGDCRSA-N 0.000 description 1
- MYBUUCHGLSQJTA-XKZOHVQRSA-N CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound CNC(=O)C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 MYBUUCHGLSQJTA-XKZOHVQRSA-N 0.000 description 1
- ITFFVMVBYSGWQL-UHFFFAOYSA-N CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(CCN)C1=NNN=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(CCN)C1=NNN=N1 ITFFVMVBYSGWQL-UHFFFAOYSA-N 0.000 description 1
- YMWSXJYSJVLFPK-GFCCVEGCSA-N CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(C[C@@H](C)N)C1=NNN=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(C[C@@H](C)N)C1=NNN=N1 YMWSXJYSJVLFPK-GFCCVEGCSA-N 0.000 description 1
- YMWSXJYSJVLFPK-LBPRGKRZSA-N CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(C[C@H](C)N)C1=NNN=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CC1=C(C=CC(C(=O)NC)=C1)C2(C[C@H](C)N)C1=NNN=N1 YMWSXJYSJVLFPK-LBPRGKRZSA-N 0.000 description 1
- BSFNOXRQWDVUIS-QHCPKHFHSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 BSFNOXRQWDVUIS-QHCPKHFHSA-N 0.000 description 1
- DMFCEZFUMCODIQ-QHCPKHFHSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C DMFCEZFUMCODIQ-QHCPKHFHSA-N 0.000 description 1
- DEQXVEXNXHDNTF-UMFSSWHCSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(CCNCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 DEQXVEXNXHDNTF-UMFSSWHCSA-N 0.000 description 1
- SMHASWVQXYAWEN-DVECYGJZSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 SMHASWVQXYAWEN-DVECYGJZSA-N 0.000 description 1
- AFGWORSFZSBNBQ-DVECYGJZSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C AFGWORSFZSBNBQ-DVECYGJZSA-N 0.000 description 1
- UBAQMPOMTTUVNL-PFCVVTBNSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 UBAQMPOMTTUVNL-PFCVVTBNSA-N 0.000 description 1
- CRROCNXAYLJUAY-PFCVVTBNSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C CRROCNXAYLJUAY-PFCVVTBNSA-N 0.000 description 1
- SMHASWVQXYAWEN-CYFREDJKSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN(C)N=N1 SMHASWVQXYAWEN-CYFREDJKSA-N 0.000 description 1
- AFGWORSFZSBNBQ-CYFREDJKSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)C1=NN=NN1C AFGWORSFZSBNBQ-CYFREDJKSA-N 0.000 description 1
- UBAQMPOMTTUVNL-PUSCERABSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN(C)N=N1 UBAQMPOMTTUVNL-PUSCERABSA-N 0.000 description 1
- CRROCNXAYLJUAY-PUSCERABSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(/C=C\C(C(=O)NC)=C/1)C2(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21)C1=NN=NN1C CRROCNXAYLJUAY-PUSCERABSA-N 0.000 description 1
- NGWOPCVNKIUINJ-UHFFFAOYSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC)=C1)C2(CCN)C1=NNN=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC)=C1)C2(CCN)C1=NNN=N1 NGWOPCVNKIUINJ-UHFFFAOYSA-N 0.000 description 1
- YRXWGVCQEFEUOX-ZDUSSCGKSA-N CNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC)=C1)C2(C[C@H](C)N)C1=NNN=N1 Chemical compound CNC(=O)C1=CC=C2C(=C1)CCC1=C(C=CC(C(=O)NC)=C1)C2(C[C@H](C)N)C1=NNN=N1 YRXWGVCQEFEUOX-ZDUSSCGKSA-N 0.000 description 1
- JVDMVCSRSGGEFL-DINWUYGQSA-N CNC(c1cc(CCc2cc(C(NC)=O)ccc2C2(C[C@H](C3CCCCC3)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O Chemical compound CNC(c1cc(CCc2cc(C(NC)=O)ccc2C2(C[C@H](C3CCCCC3)NCC(N(CCC3)C3C#N)=O)c3nnn[nH]3)c2cc1)=O JVDMVCSRSGGEFL-DINWUYGQSA-N 0.000 description 1
- PDXCWRRCUHYYEC-UHFFFAOYSA-N CNC(c1ccc(C(CCNCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3F)c2c1)=O Chemical compound CNC(c1ccc(C(CCNCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3F)c2c1)=O PDXCWRRCUHYYEC-UHFFFAOYSA-N 0.000 description 1
- AIQYFAXYTBWNMB-IYRGYZHQSA-N CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(C(NO2)=NC2=O)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O Chemical compound CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(C(NO2)=NC2=O)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O AIQYFAXYTBWNMB-IYRGYZHQSA-N 0.000 description 1
- GIUMXENHAZNPIT-QQVDTZSYSA-N CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2n[nH]nn2)c(c(C2)c3)ccc3C(NC)=O)c2c1)=O Chemical compound CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2n[nH]nn2)c(c(C2)c3)ccc3C(NC)=O)c2c1)=O GIUMXENHAZNPIT-QQVDTZSYSA-N 0.000 description 1
- LCKZZJFDBUOSDP-JPIPQCAJSA-N CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnc(-c(cc3)ccc3F)[o]2)c(cc2)c(CC3)cc2C(NC)=O)c3c1)O Chemical compound CNC(c1ccc(C(CCNCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnc(-c(cc3)ccc3F)[o]2)c(cc2)c(CC3)cc2C(NC)=O)c3c1)O LCKZZJFDBUOSDP-JPIPQCAJSA-N 0.000 description 1
- NNGUBCJLUYIRIZ-BNGNXUGRSA-N CNC(c1ccc(C(C[C@H](C2CCCC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O Chemical compound CNC(c1ccc(C(C[C@H](C2CCCC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O NNGUBCJLUYIRIZ-BNGNXUGRSA-N 0.000 description 1
- CEENDCBXLSGVEP-WIOPSUGQSA-N CNC(c1ccc(C(C[C@H](NCC(N(CCC2)[C@@H]2C#N)=O)[N]2(C)O3)(C2=NC3=O)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O Chemical compound CNC(c1ccc(C(C[C@H](NCC(N(CCC2)[C@@H]2C#N)=O)[N]2(C)O3)(C2=NC3=O)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O CEENDCBXLSGVEP-WIOPSUGQSA-N 0.000 description 1
- AFDRHBSSZDFWTF-DINWUYGQSA-N CNC(c1ccc(C(C[C@H](c(cc2)ccc2F)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O Chemical compound CNC(c1ccc(C(C[C@H](c(cc2)ccc2F)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(NC)=O)c2c1)=O AFDRHBSSZDFWTF-DINWUYGQSA-N 0.000 description 1
- AKWUKBSAVUDICQ-KTPDYHTBSA-N CNC(c1ccc([C@H](CC2)N([C@@H]3CN(CC(C(N(CCC4)C4C#N)=O)N)C4C3)C4=O)c2c1)=O Chemical compound CNC(c1ccc([C@H](CC2)N([C@@H]3CN(CC(C(N(CCC4)C4C#N)=O)N)C4C3)C4=O)c2c1)=O AKWUKBSAVUDICQ-KTPDYHTBSA-N 0.000 description 1
- QOYDZKFJXGLRED-DNTWZEFLSA-N CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 QOYDZKFJXGLRED-DNTWZEFLSA-N 0.000 description 1
- SKUOFWZHCYTXLQ-NHUSPQTBSA-N CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 SKUOFWZHCYTXLQ-NHUSPQTBSA-N 0.000 description 1
- UBZJQACIDLFVEU-PPMIAYNCSA-N CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 UBZJQACIDLFVEU-PPMIAYNCSA-N 0.000 description 1
- XFLHYQRBKUEFKO-ZCCHTWKOSA-N CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 XFLHYQRBKUEFKO-ZCCHTWKOSA-N 0.000 description 1
- QOYDZKFJXGLRED-LPTZDLKRSA-N CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 QOYDZKFJXGLRED-LPTZDLKRSA-N 0.000 description 1
- SKUOFWZHCYTXLQ-WYZBVHOTSA-N CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 SKUOFWZHCYTXLQ-WYZBVHOTSA-N 0.000 description 1
- UBZJQACIDLFVEU-KKXZWXNMSA-N CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound CNOOSC1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 UBZJQACIDLFVEU-KKXZWXNMSA-N 0.000 description 1
- ZGGLKFOPCGRIBH-OXXUSTOISA-N CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 ZGGLKFOPCGRIBH-OXXUSTOISA-N 0.000 description 1
- CRAVHIOHDOWZEG-GVJFSJSTSA-N CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 CRAVHIOHDOWZEG-GVJFSJSTSA-N 0.000 description 1
- XJQPVGUUALPTMF-ATPUAQIGSA-N CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 XJQPVGUUALPTMF-ATPUAQIGSA-N 0.000 description 1
- KHURIIWOZWAUJN-LEFRDSTESA-N CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound CNOOSC1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 KHURIIWOZWAUJN-LEFRDSTESA-N 0.000 description 1
- ZGGLKFOPCGRIBH-MHZWBOMTSA-N CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 ZGGLKFOPCGRIBH-MHZWBOMTSA-N 0.000 description 1
- CRAVHIOHDOWZEG-CBOSDZJASA-N CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 CRAVHIOHDOWZEG-CBOSDZJASA-N 0.000 description 1
- XJQPVGUUALPTMF-MKBYFEBXSA-N CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 XJQPVGUUALPTMF-MKBYFEBXSA-N 0.000 description 1
- KHURIIWOZWAUJN-GDGPAFIMSA-N CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound CNOOSC1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 KHURIIWOZWAUJN-GDGPAFIMSA-N 0.000 description 1
- CHTWMYKEBYDYIY-OXXUSTOISA-N CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 CHTWMYKEBYDYIY-OXXUSTOISA-N 0.000 description 1
- RJZYIIOYGCHNLE-GVJFSJSTSA-N CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 RJZYIIOYGCHNLE-GVJFSJSTSA-N 0.000 description 1
- ICGRYAFIYZFPEW-ATPUAQIGSA-N CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 ICGRYAFIYZFPEW-ATPUAQIGSA-N 0.000 description 1
- CFVIHTMMBWPFGJ-LEFRDSTESA-N CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 CFVIHTMMBWPFGJ-LEFRDSTESA-N 0.000 description 1
- CHTWMYKEBYDYIY-MHZWBOMTSA-N CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=CC=C1 CHTWMYKEBYDYIY-MHZWBOMTSA-N 0.000 description 1
- RJZYIIOYGCHNLE-CBOSDZJASA-N CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 RJZYIIOYGCHNLE-CBOSDZJASA-N 0.000 description 1
- ICGRYAFIYZFPEW-MKBYFEBXSA-N CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 ICGRYAFIYZFPEW-MKBYFEBXSA-N 0.000 description 1
- CFVIHTMMBWPFGJ-GDGPAFIMSA-N CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound CNS(=O)(=O)C1=CC([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 CFVIHTMMBWPFGJ-GDGPAFIMSA-N 0.000 description 1
- PJVJBDAUWILEOG-UHFFFAOYSA-N COC(=O)C1=C(S(=O)(=O)Cl)C=CS1 Chemical compound COC(=O)C1=C(S(=O)(=O)Cl)C=CS1 PJVJBDAUWILEOG-UHFFFAOYSA-N 0.000 description 1
- MPLOCHVNHBFPEE-XUXIUFHCSA-N COC(=O)C1=C(S(=O)(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=CS1 Chemical compound COC(=O)C1=C(S(=O)(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=CS1 MPLOCHVNHBFPEE-XUXIUFHCSA-N 0.000 description 1
- PZVLSEBHRSYWKW-QAETUUGQSA-N COC(=O)C1=C(S(=O)(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=CS1 Chemical compound COC(=O)C1=C(S(=O)(=O)N2C[C@@H]3C[C@H]2C(=O)N3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=CS1 PZVLSEBHRSYWKW-QAETUUGQSA-N 0.000 description 1
- YNNTXXLIYWYSJN-AVEISGIFSA-N COC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNN=N1)C1=CC=C(C(=O)CO)C=C1CC2 Chemical compound COC(=O)C1=CC2=C(C=C1)C(C[C@H](C)N)(C1=NNN=N1)C1=CC=C(C(=O)CO)C=C1CC2 YNNTXXLIYWYSJN-AVEISGIFSA-N 0.000 description 1
- QDOKFDPWEQJJCJ-MBSDFSHPSA-N COC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2 Chemical compound COC(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)OC)C=C1)CC2 QDOKFDPWEQJJCJ-MBSDFSHPSA-N 0.000 description 1
- QDIHMVSOSPKTBP-SKPVKVGISA-N COC(=O)C1C[C@@H](O)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1CC(N[C@@H](C)C2=CC=C(F)C=C2)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C)C1=CC=C(F)C=C1)NC(=O)OC(C)(C)C.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)O.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@H]2CC[C@@H]1C2.C[C@H](N)C1=CC=C(F)C=C1.C[C@H](NC1C[C@@H](C(=O)O)N(C(=O)OC(C)(C)C)C1)C1=CC=C(F)C=C1 Chemical compound COC(=O)C1C[C@@H](O)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1CC(N[C@@H](C)C2=CC=C(F)C=C2)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C.COC(=O)[C@H](CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C)C1=CC=C(F)C=C1)NC(=O)OC(C)(C)C.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)O.C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@H]2CC[C@@H]1C2.C[C@H](N)C1=CC=C(F)C=C1.C[C@H](NC1C[C@@H](C(=O)O)N(C(=O)OC(C)(C)C)C1)C1=CC=C(F)C=C1 QDIHMVSOSPKTBP-SKPVKVGISA-N 0.000 description 1
- QKUBKZCXVSLXDF-HYZPQSCBSA-N COC(O)C(=O)N1CCCC1C#N.NC(=O)C1CCCN1.NC(=O)C1CCCN1C(=O)/C=C/C(=O)N1CCCC1C(N)=O.[C-]#[N+]C1CCCN1C(=O)/C=C/C(=O)N1CCCC1C#N.[H]C(=O)C(=O)N1CCCC1C#N Chemical compound COC(O)C(=O)N1CCCC1C#N.NC(=O)C1CCCN1.NC(=O)C1CCCN1C(=O)/C=C/C(=O)N1CCCC1C(N)=O.[C-]#[N+]C1CCCN1C(=O)/C=C/C(=O)N1CCCC1C#N.[H]C(=O)C(=O)N1CCCC1C#N QKUBKZCXVSLXDF-HYZPQSCBSA-N 0.000 description 1
- LYKSQAJGJWPCJS-MSCYWBEUSA-N COC(O)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)C=O.NC(=O)[C@@H]1CCCN1.[C-]#[N+][C@@H]1CCCC1C(=O)/C=C/C(=O)N1CCC[C@H]1C#N Chemical compound COC(O)C(=O)N1CCC[C@H]1C#N.N#C[C@@H]1CCCN1C(=O)C=O.NC(=O)[C@@H]1CCCN1.[C-]#[N+][C@@H]1CCCC1C(=O)/C=C/C(=O)N1CCC[C@H]1C#N LYKSQAJGJWPCJS-MSCYWBEUSA-N 0.000 description 1
- SZOXVBPLJRHDIC-QYOKMDGLSA-N C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SZOXVBPLJRHDIC-QYOKMDGLSA-N 0.000 description 1
- KADQYVHRKZMKSS-XSHPBUEYSA-N C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KADQYVHRKZMKSS-XSHPBUEYSA-N 0.000 description 1
- DUSZVSSSGFKHGQ-LIATYEAPSA-N C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N DUSZVSSSGFKHGQ-LIATYEAPSA-N 0.000 description 1
- NKLLRXPRHHZCRW-JCTQVDHFSA-N C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 NKLLRXPRHHZCRW-JCTQVDHFSA-N 0.000 description 1
- SWWCBGOGLJVOPA-QYOKMDGLSA-N C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SWWCBGOGLJVOPA-QYOKMDGLSA-N 0.000 description 1
- BWZILVKWSVYHOV-XSHPBUEYSA-N C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BWZILVKWSVYHOV-XSHPBUEYSA-N 0.000 description 1
- GSUNCJAYVJXXGV-LIATYEAPSA-N C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N GSUNCJAYVJXXGV-LIATYEAPSA-N 0.000 description 1
- JGAFZZIGIBXOKH-JCTQVDHFSA-N C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JGAFZZIGIBXOKH-JCTQVDHFSA-N 0.000 description 1
- NVPSSFOKKVMYPF-FKWAHNQWSA-N C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N NVPSSFOKKVMYPF-FKWAHNQWSA-N 0.000 description 1
- SASJSKSWFBJPBD-DYLPAERQSA-N C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N SASJSKSWFBJPBD-DYLPAERQSA-N 0.000 description 1
- VHAIBOSHCDVMJE-ASOVEQLKSA-N C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N VHAIBOSHCDVMJE-ASOVEQLKSA-N 0.000 description 1
- GQTZRTMYXRYUTN-NKELOVKVSA-N C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 GQTZRTMYXRYUTN-NKELOVKVSA-N 0.000 description 1
- MMCQRXGYLGRZQD-RFCFSTKCSA-N C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N MMCQRXGYLGRZQD-RFCFSTKCSA-N 0.000 description 1
- PKMQYZMZLIKZER-MEOVIWEUSA-N C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N PKMQYZMZLIKZER-MEOVIWEUSA-N 0.000 description 1
- SXBOTIVPBRPKQE-YYOJEONGSA-N C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N SXBOTIVPBRPKQE-YYOJEONGSA-N 0.000 description 1
- BKDHQVWRWGFHPO-FJPAKYKUSA-N C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BKDHQVWRWGFHPO-FJPAKYKUSA-N 0.000 description 1
- JEUMCGDPFJOMFC-FKWAHNQWSA-N C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N JEUMCGDPFJOMFC-FKWAHNQWSA-N 0.000 description 1
- FZWSIFXBLJIYHL-DYLPAERQSA-N C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N FZWSIFXBLJIYHL-DYLPAERQSA-N 0.000 description 1
- OBYFNJWXGMQIHL-ASOVEQLKSA-N C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N OBYFNJWXGMQIHL-ASOVEQLKSA-N 0.000 description 1
- YVRJIJIYQZBWAB-NKELOVKVSA-N C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YVRJIJIYQZBWAB-NKELOVKVSA-N 0.000 description 1
- DCTOKFRLUNGYOX-RFCFSTKCSA-N C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N DCTOKFRLUNGYOX-RFCFSTKCSA-N 0.000 description 1
- RXAITCMRXGQVLZ-MEOVIWEUSA-N C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RXAITCMRXGQVLZ-MEOVIWEUSA-N 0.000 description 1
- IZRLTKSFGVPRPY-YYOJEONGSA-N C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N IZRLTKSFGVPRPY-YYOJEONGSA-N 0.000 description 1
- VPKRYRUVZMSEHE-FJPAKYKUSA-N C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VPKRYRUVZMSEHE-FJPAKYKUSA-N 0.000 description 1
- HKXOBNCRXNERCJ-LCUYYGDQSA-N C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N HKXOBNCRXNERCJ-LCUYYGDQSA-N 0.000 description 1
- GQPIBNHWVHVUAX-JMMIECQRSA-N C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N GQPIBNHWVHVUAX-JMMIECQRSA-N 0.000 description 1
- DKZOUOPKKOGZSG-GREONPEQSA-N C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DKZOUOPKKOGZSG-GREONPEQSA-N 0.000 description 1
- ZFZVZPXTFSVPAV-KQPGKXOUSA-N C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N ZFZVZPXTFSVPAV-KQPGKXOUSA-N 0.000 description 1
- UUGXZPUWYXRDQZ-CTXVOCDESA-N C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N UUGXZPUWYXRDQZ-CTXVOCDESA-N 0.000 description 1
- HUDDOULVYQZWPR-XSXWSVAESA-N C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HUDDOULVYQZWPR-XSXWSVAESA-N 0.000 description 1
- YCGLTHIWSVNOLG-GLMUNCEMSA-N C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YCGLTHIWSVNOLG-GLMUNCEMSA-N 0.000 description 1
- LGTDSSFZFJXYPH-RFCFSTKCSA-N C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N LGTDSSFZFJXYPH-RFCFSTKCSA-N 0.000 description 1
- HYGJEWPAZCHXHJ-MEOVIWEUSA-N C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N HYGJEWPAZCHXHJ-MEOVIWEUSA-N 0.000 description 1
- PYHFHARSJHSZOM-YYOJEONGSA-N C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N PYHFHARSJHSZOM-YYOJEONGSA-N 0.000 description 1
- XMQWLBBFUNDFLJ-FJPAKYKUSA-N C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XMQWLBBFUNDFLJ-FJPAKYKUSA-N 0.000 description 1
- BPZNTSNKQMOBBW-KQPGKXOUSA-N C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N BPZNTSNKQMOBBW-KQPGKXOUSA-N 0.000 description 1
- RIGHGJMMPNGTDM-CTXVOCDESA-N C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RIGHGJMMPNGTDM-CTXVOCDESA-N 0.000 description 1
- RAFYFGWRLPCUQZ-GLMUNCEMSA-N C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RAFYFGWRLPCUQZ-GLMUNCEMSA-N 0.000 description 1
- BVVJUMXHVXOGHK-KQPGKXOUSA-N C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N BVVJUMXHVXOGHK-KQPGKXOUSA-N 0.000 description 1
- IRYCMEYPZXDLAY-CTXVOCDESA-N C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N IRYCMEYPZXDLAY-CTXVOCDESA-N 0.000 description 1
- HKVZORFGPRWFBK-XSXWSVAESA-N C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HKVZORFGPRWFBK-XSXWSVAESA-N 0.000 description 1
- CGNKCEJOYKBBFM-GLMUNCEMSA-N C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CGNKCEJOYKBBFM-GLMUNCEMSA-N 0.000 description 1
- NNGJVDSDQJAHPZ-MEOVIWEUSA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N NNGJVDSDQJAHPZ-MEOVIWEUSA-N 0.000 description 1
- FNJXIWGTYRXTLK-CSJRWYNFSA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCSC1 Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCSC1 FNJXIWGTYRXTLK-CSJRWYNFSA-N 0.000 description 1
- ARDPKPPVGLWKNL-YIFBHZASSA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ARDPKPPVGLWKNL-YIFBHZASSA-N 0.000 description 1
- FBBIAJNTWTWLPN-XSXWSVAESA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N FBBIAJNTWTWLPN-XSXWSVAESA-N 0.000 description 1
- LCVYFXWJZMCBNB-KNTRFNDTSA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCSC1 Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCSC1 LCVYFXWJZMCBNB-KNTRFNDTSA-N 0.000 description 1
- BBWJUFMDXXKQMW-UMDVZPRXSA-N C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BBWJUFMDXXKQMW-UMDVZPRXSA-N 0.000 description 1
- IEIKZMAFRCHIFW-ZRIKRKEKSA-N C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N IEIKZMAFRCHIFW-ZRIKRKEKSA-N 0.000 description 1
- AHLZYPTYSPLZQZ-OMBLBEMTSA-N C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N AHLZYPTYSPLZQZ-OMBLBEMTSA-N 0.000 description 1
- ZBWIWEWMZZEQJN-CPAPFFTJSA-N C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N ZBWIWEWMZZEQJN-CPAPFFTJSA-N 0.000 description 1
- BECRWEBGAYZHLS-BDOHWBQPSA-N C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BECRWEBGAYZHLS-BDOHWBQPSA-N 0.000 description 1
- RSMDAAJKYGYJNK-ADYLJTCVSA-N C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N RSMDAAJKYGYJNK-ADYLJTCVSA-N 0.000 description 1
- YSASHIUYEJMGCV-KWFCSAJOSA-N C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N YSASHIUYEJMGCV-KWFCSAJOSA-N 0.000 description 1
- LIEVWQLRESWHFM-ITJSPEIASA-N C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N LIEVWQLRESWHFM-ITJSPEIASA-N 0.000 description 1
- JVHTWVICYMAEIA-JAZTXMKBSA-N C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JVHTWVICYMAEIA-JAZTXMKBSA-N 0.000 description 1
- SLAKQEXWMLMQMQ-UAUOSOAOSA-N C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SLAKQEXWMLMQMQ-UAUOSOAOSA-N 0.000 description 1
- AZEPPKCFOGZYMU-HVRBKMHZSA-N C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N AZEPPKCFOGZYMU-HVRBKMHZSA-N 0.000 description 1
- BOHMQZDMCNCNDS-RSYUGDCRSA-N C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N BOHMQZDMCNCNDS-RSYUGDCRSA-N 0.000 description 1
- UXHPBUKCPOTJRI-XKZOHVQRSA-N C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 UXHPBUKCPOTJRI-XKZOHVQRSA-N 0.000 description 1
- KENZREGLGDETOB-LCUYYGDQSA-N C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N KENZREGLGDETOB-LCUYYGDQSA-N 0.000 description 1
- DAQIUQBFBZSHFB-JMMIECQRSA-N C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N DAQIUQBFBZSHFB-JMMIECQRSA-N 0.000 description 1
- PBFXPZJDSDKHCT-NHKCCNDQSA-N C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N PBFXPZJDSDKHCT-NHKCCNDQSA-N 0.000 description 1
- YYMYULHEDIQCMP-GREONPEQSA-N C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YYMYULHEDIQCMP-GREONPEQSA-N 0.000 description 1
- CRBVYFFPLSWYCE-KQPGKXOUSA-N C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N CRBVYFFPLSWYCE-KQPGKXOUSA-N 0.000 description 1
- BTOHCUVCSPXOME-CTXVOCDESA-N C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BTOHCUVCSPXOME-CTXVOCDESA-N 0.000 description 1
- HTMCXSYUWXFYRJ-XSXWSVAESA-N C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HTMCXSYUWXFYRJ-XSXWSVAESA-N 0.000 description 1
- PDMZOYBXYVNNLG-GLMUNCEMSA-N C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PDMZOYBXYVNNLG-GLMUNCEMSA-N 0.000 description 1
- VQFKGROZJRLPBX-KQPGKXOUSA-N C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N VQFKGROZJRLPBX-KQPGKXOUSA-N 0.000 description 1
- RRFZAFARHBSTGF-CTXVOCDESA-N C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RRFZAFARHBSTGF-CTXVOCDESA-N 0.000 description 1
- XKEMHPGMVSVQOC-XSXWSVAESA-N C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N XKEMHPGMVSVQOC-XSXWSVAESA-N 0.000 description 1
- UBCBOPHTTSFLJL-KQPGKXOUSA-N C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N UBCBOPHTTSFLJL-KQPGKXOUSA-N 0.000 description 1
- JEKNPQPBAURCLJ-CTXVOCDESA-N C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N JEKNPQPBAURCLJ-CTXVOCDESA-N 0.000 description 1
- RSOKJWFKWYEUHI-XSXWSVAESA-N C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N RSOKJWFKWYEUHI-XSXWSVAESA-N 0.000 description 1
- QLENTQILYWVQEY-GLMUNCEMSA-N C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QLENTQILYWVQEY-GLMUNCEMSA-N 0.000 description 1
- VYCYUCAHMJJCAE-ZRIKRKEKSA-N C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N VYCYUCAHMJJCAE-ZRIKRKEKSA-N 0.000 description 1
- IJWNWCJODWVLDO-OMBLBEMTSA-N C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N IJWNWCJODWVLDO-OMBLBEMTSA-N 0.000 description 1
- NVPIVAXESTVEOD-CPAPFFTJSA-N C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N NVPIVAXESTVEOD-CPAPFFTJSA-N 0.000 description 1
- YVUIBGQCQNZVLI-BDOHWBQPSA-N C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YVUIBGQCQNZVLI-BDOHWBQPSA-N 0.000 description 1
- RLIBSICPMPDSTN-ADYLJTCVSA-N C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N RLIBSICPMPDSTN-ADYLJTCVSA-N 0.000 description 1
- SUTIINKCOVHNJN-KWFCSAJOSA-N C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N SUTIINKCOVHNJN-KWFCSAJOSA-N 0.000 description 1
- CMHUZFDIJVQXQD-ITJSPEIASA-N C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N CMHUZFDIJVQXQD-ITJSPEIASA-N 0.000 description 1
- PIKJDSQTHXAWIZ-JAZTXMKBSA-N C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PIKJDSQTHXAWIZ-JAZTXMKBSA-N 0.000 description 1
- NYRGQYKVYMFBEM-XNUZUHMRSA-N C[C@@H](CC(C(N)=N)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(C(N)=N)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O NYRGQYKVYMFBEM-XNUZUHMRSA-N 0.000 description 1
- RSUCYIMOZZILNB-GVNKFJBHSA-N C[C@@H](CC(C(N1)=NN(C)C1=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(C(N1)=NN(C)C1=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)NCC(N(CCC1)C1C#N)=O RSUCYIMOZZILNB-GVNKFJBHSA-N 0.000 description 1
- PENVEAZALBWALH-XGLRFROISA-N C[C@@H](CC(C(N1)=NN(C)C1=O)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(C(N1)=NN(C)C1=O)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O PENVEAZALBWALH-XGLRFROISA-N 0.000 description 1
- MFGCTMHBEHLJMT-JINQPTGOSA-N C[C@@H](CC(C(N1)=NNC1=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(C(N1)=NNC1=O)(c(c(CCc1c2)c3)ccc3C(N(C)C)=O)c1ccc2C(N(C)C)=O)NCC(N(CCC1)C1C#N)=O MFGCTMHBEHLJMT-JINQPTGOSA-N 0.000 description 1
- WSXVTLVKDDISJM-GDYWIIFWSA-N C[C@@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 WSXVTLVKDDISJM-GDYWIIFWSA-N 0.000 description 1
- DDNRADHVUJSPRG-UDFARJDRSA-N C[C@@H](CC(c(c(-c1c2)c3)ccc3C(NC)=O)(c1ccc2C(NC)=O)c1n[nH]nn1)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c(c(-c1c2)c3)ccc3C(NC)=O)(c1ccc2C(NC)=O)c1n[nH]nn1)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O DDNRADHVUJSPRG-UDFARJDRSA-N 0.000 description 1
- BLHPSODRCNQNPO-UXGJTCCMSA-N C[C@@H](CC(c1c(CC2)[s]cc1)(c1c2[s]c(C(N)=O)c1)c1n[nH]nn1)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c1c(CC2)[s]cc1)(c1c2[s]c(C(N)=O)c1)c1n[nH]nn1)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O BLHPSODRCNQNPO-UXGJTCCMSA-N 0.000 description 1
- FONMVIWIGDVZGP-SFVWDYPZSA-N C[C@@H](CC(c1c(CC2)cc(C(N)=O)[s]1)(c1c2cc(C(N)=O)[s]1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1c(CC2)cc(C(N)=O)[s]1)(c1c2cc(C(N)=O)[s]1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O FONMVIWIGDVZGP-SFVWDYPZSA-N 0.000 description 1
- LUHCXMOVWBCXAL-MBIQTGHCSA-N C[C@@H](CC(c1c(CC2)cc(C(NC)=O)[s]1)(c1c2cc(C(NC)=O)[s]1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1c(CC2)cc(C(NC)=O)[s]1)(c1c2cc(C(NC)=O)[s]1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O LUHCXMOVWBCXAL-MBIQTGHCSA-N 0.000 description 1
- ZXLZVBKSAZSRHN-CFUYPVJSSA-N C[C@@H](CC(c1n[nH]nn1)(c(c(CC1)c2)ccc2C(C2CCOCC2)=O)c(cc2)c1cc2C(N1CCOCC1)=O)NCC(N(CCC1)[C@@H]1C#N)=O Chemical compound C[C@@H](CC(c1n[nH]nn1)(c(c(CC1)c2)ccc2C(C2CCOCC2)=O)c(cc2)c1cc2C(N1CCOCC1)=O)NCC(N(CCC1)[C@@H]1C#N)=O ZXLZVBKSAZSRHN-CFUYPVJSSA-N 0.000 description 1
- HRTUBSCIKXTIGA-BJQOMGFOSA-N C[C@@H](CC(c1n[nH]nn1)(c(cc1)ccc1C(N)=O)c(cc1)ccc1C(N)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1n[nH]nn1)(c(cc1)ccc1C(N)=O)c(cc1)ccc1C(N)=O)NCC(N(CCC1)C1C#N)=O HRTUBSCIKXTIGA-BJQOMGFOSA-N 0.000 description 1
- CISSOWMWOCJTGW-SVQMELKDSA-N C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)NCC(N(CCC1)C1C#N)=O CISSOWMWOCJTGW-SVQMELKDSA-N 0.000 description 1
- ZELXPDOQFMOWSE-RMRYCNOXSA-N C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O ZELXPDOQFMOWSE-RMRYCNOXSA-N 0.000 description 1
- YQIIDHHZCLCARN-QYFQTTGNSA-N C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O YQIIDHHZCLCARN-QYFQTTGNSA-N 0.000 description 1
- QGIYOAXWGDIGCJ-VJVHLSROSA-N C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c1ccc(C(N(C)C)O)cc1CCc1c2)c1ccc2C(N(C)C)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(-c(cc2)ccc2F)[o]1)(c1ccc(C(N(C)C)O)cc1CCc1c2)c1ccc2C(N(C)C)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O QGIYOAXWGDIGCJ-VJVHLSROSA-N 0.000 description 1
- WVOCRFKQPBGWDX-NVHKAFQKSA-N C[C@@H](CC(c1nnc(C)[o]1)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(C)[o]1)(c(c(CC1)c2)ccc2C(N)=O)c(cc2)c1cc2C(N)=O)NCC(N(CCC1)C1C#N)=O WVOCRFKQPBGWDX-NVHKAFQKSA-N 0.000 description 1
- PBKWOBZOWKHRQQ-UBDBMELISA-N C[C@@H](CC(c1nnc(C)[o]1)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnc(C)[o]1)(c(c(CCc1c2)c3)ccc3C(NC)=O)c1ccc2C(NC)=O)NCC(N(CCC1)C1C#N)=O PBKWOBZOWKHRQQ-UBDBMELISA-N 0.000 description 1
- QDIQNOIUDRAYNG-CTLOQAHHSA-N C[C@@H](CC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N3CCCC3)=O)c1ccc2C(N1CCCC1)=O)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N3CCCC3)=O)c1ccc2C(N1CCCC1)=O)NCC(N(CCC1)C1C#N)=O QDIQNOIUDRAYNG-CTLOQAHHSA-N 0.000 description 1
- KYKTWJFFRXATOA-RBUKOAKNSA-N C[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CC2)cc1C(NC)=O)c(cc1)c2cc1C(NC)=O)NCC(N1[C@H](C)CCC1)=O Chemical compound C[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CC2)cc1C(NC)=O)c(cc1)c2cc1C(NC)=O)NCC(N1[C@H](C)CCC1)=O KYKTWJFFRXATOA-RBUKOAKNSA-N 0.000 description 1
- HGYWOULATVQZIX-BTBXQEIOSA-N C[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(N1CCCC1)=O)c2ccc3C(N1CCCC1)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@@H](CC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(N1CCCC1)=O)c2ccc3C(N1CCCC1)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O HGYWOULATVQZIX-BTBXQEIOSA-N 0.000 description 1
- IKNMUDOGIYBXES-BACLKCEISA-N C[C@@H](CC1(C#N)C2=CC=CC=C2CCC2=C1C=CC=C2)NC(=O)OC(C)(C)C.C[C@H](N)CC1(C(N)=O)C2=CC=CC=C2CCC2=C1C=CC=C2.N#CC1C2=CC=CC=C2CCC2=C1C=CC=C2 Chemical compound C[C@@H](CC1(C#N)C2=CC=CC=C2CCC2=C1C=CC=C2)NC(=O)OC(C)(C)C.C[C@H](N)CC1(C(N)=O)C2=CC=CC=C2CCC2=C1C=CC=C2.N#CC1C2=CC=CC=C2CCC2=C1C=CC=C2 IKNMUDOGIYBXES-BACLKCEISA-N 0.000 description 1
- PPUHTBCZDZMNJI-CPJSRVTESA-N C[C@@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N PPUHTBCZDZMNJI-CPJSRVTESA-N 0.000 description 1
- KFMFKJJQXPAAAE-RKMGBUROSA-N C[C@@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KFMFKJJQXPAAAE-RKMGBUROSA-N 0.000 description 1
- IDMOKZCKSLCOAG-DLLPINGYSA-N C[C@@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N IDMOKZCKSLCOAG-DLLPINGYSA-N 0.000 description 1
- PJMFKJCVBZYYRM-CDOKILDISA-N C[C@@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PJMFKJCVBZYYRM-CDOKILDISA-N 0.000 description 1
- WMNPOYHOYXXSRM-CPJSRVTESA-N C[C@@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N WMNPOYHOYXXSRM-CPJSRVTESA-N 0.000 description 1
- JOZALJGSCOGUHA-RKMGBUROSA-N C[C@@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JOZALJGSCOGUHA-RKMGBUROSA-N 0.000 description 1
- JJISUKAIFUXRCF-CPJSRVTESA-N C[C@@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N JJISUKAIFUXRCF-CPJSRVTESA-N 0.000 description 1
- QIFXVSDUOGCMHC-RXVVDRJESA-N C[C@@H](CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N QIFXVSDUOGCMHC-RXVVDRJESA-N 0.000 description 1
- UWFRQLKIHGXFDB-LVXARBLLSA-N C[C@@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N UWFRQLKIHGXFDB-LVXARBLLSA-N 0.000 description 1
- OVHXMXCZYRAKOT-YIPSIMJMSA-N C[C@@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OVHXMXCZYRAKOT-YIPSIMJMSA-N 0.000 description 1
- QFMZDUXDLUQNEY-FKAPRUDGSA-N C[C@@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N QFMZDUXDLUQNEY-FKAPRUDGSA-N 0.000 description 1
- SEQQCDHOTLNEQG-DKCCFNNGSA-N C[C@@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SEQQCDHOTLNEQG-DKCCFNNGSA-N 0.000 description 1
- RSUCYIMOZZILNB-LVXARBLLSA-N C[C@@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N RSUCYIMOZZILNB-LVXARBLLSA-N 0.000 description 1
- QIPWOWKRBANZMI-YIPSIMJMSA-N C[C@@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QIPWOWKRBANZMI-YIPSIMJMSA-N 0.000 description 1
- CYHQNOPGVDVJRS-LVXARBLLSA-N C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N CYHQNOPGVDVJRS-LVXARBLLSA-N 0.000 description 1
- YFXHYYUYVVJIAR-YIPSIMJMSA-N C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YFXHYYUYVVJIAR-YIPSIMJMSA-N 0.000 description 1
- XBWVRFNNNBZWSH-JTSKRJEESA-N C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N XBWVRFNNNBZWSH-JTSKRJEESA-N 0.000 description 1
- CCTXIUGGKJKWRO-JNFFJFACSA-N C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CCTXIUGGKJKWRO-JNFFJFACSA-N 0.000 description 1
- ZSUIFRSRPFMZJR-DLLPINGYSA-N C[C@@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N ZSUIFRSRPFMZJR-DLLPINGYSA-N 0.000 description 1
- KSWQEWSMTKKMKQ-CDOKILDISA-N C[C@@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KSWQEWSMTKKMKQ-CDOKILDISA-N 0.000 description 1
- QTRPTBIWXKGXTL-DLLPINGYSA-N C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N QTRPTBIWXKGXTL-DLLPINGYSA-N 0.000 description 1
- ZWYOZOFYPZPFIC-MJRXFLOJSA-N C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ZWYOZOFYPZPFIC-MJRXFLOJSA-N 0.000 description 1
- BGCAHXKFQNUNPI-DLLPINGYSA-N C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N BGCAHXKFQNUNPI-DLLPINGYSA-N 0.000 description 1
- NEAOPGFDFSCMCM-MJRXFLOJSA-N C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 NEAOPGFDFSCMCM-MJRXFLOJSA-N 0.000 description 1
- LOJWCTNXGUBVLV-LVXARBLLSA-N C[C@@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N LOJWCTNXGUBVLV-LVXARBLLSA-N 0.000 description 1
- VMFKWKYVZFMFFS-YIPSIMJMSA-N C[C@@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VMFKWKYVZFMFFS-YIPSIMJMSA-N 0.000 description 1
- OZVMKSLUAGGKTA-RKMGBUROSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OZVMKSLUAGGKTA-RKMGBUROSA-N 0.000 description 1
- SCUSHPVWRJTBTG-VIKYFBQLSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SCUSHPVWRJTBTG-VIKYFBQLSA-N 0.000 description 1
- QDIQNOIUDRAYNG-OUTSHDOLSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N QDIQNOIUDRAYNG-OUTSHDOLSA-N 0.000 description 1
- HGYWOULATVQZIX-ICGWNSLASA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 HGYWOULATVQZIX-ICGWNSLASA-N 0.000 description 1
- GRFSBOGZWUIQBV-HVNZXBJASA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N GRFSBOGZWUIQBV-HVNZXBJASA-N 0.000 description 1
- DRZBEDXSNVFJSB-GCQYPMGCSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DRZBEDXSNVFJSB-GCQYPMGCSA-N 0.000 description 1
- UAKNGRPCXBQROA-HVNZXBJASA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1CCC[C@H]1C#N UAKNGRPCXBQROA-HVNZXBJASA-N 0.000 description 1
- JKXHHOJBUFCQIJ-GCQYPMGCSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JKXHHOJBUFCQIJ-GCQYPMGCSA-N 0.000 description 1
- QOKKCJQMDSSTDF-ICGWNSLASA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QOKKCJQMDSSTDF-ICGWNSLASA-N 0.000 description 1
- DIPYSGFSBBEGCL-CUNXSJBXSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N DIPYSGFSBBEGCL-CUNXSJBXSA-N 0.000 description 1
- QYSKKHBFZBSJNY-VIKYFBQLSA-N C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QYSKKHBFZBSJNY-VIKYFBQLSA-N 0.000 description 1
- RADGCIRPMJIFKP-LVXARBLLSA-N C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N RADGCIRPMJIFKP-LVXARBLLSA-N 0.000 description 1
- ATDVHHUQCKDTCV-YIPSIMJMSA-N C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ATDVHHUQCKDTCV-YIPSIMJMSA-N 0.000 description 1
- YSWKZPBRBRQHFI-JTSKRJEESA-N C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N YSWKZPBRBRQHFI-JTSKRJEESA-N 0.000 description 1
- PWKNSSDGOLSQLA-JNFFJFACSA-N C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PWKNSSDGOLSQLA-JNFFJFACSA-N 0.000 description 1
- MFGCTMHBEHLJMT-CPJSRVTESA-N C[C@@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N MFGCTMHBEHLJMT-CPJSRVTESA-N 0.000 description 1
- GGGXQQXXSVOJGZ-RKMGBUROSA-N C[C@@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 GGGXQQXXSVOJGZ-RKMGBUROSA-N 0.000 description 1
- XFCCODCYUKLMMW-LIYOMKHYSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XFCCODCYUKLMMW-LIYOMKHYSA-N 0.000 description 1
- FGUFRQRXAIFWGF-CYFREDJKSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N FGUFRQRXAIFWGF-CYFREDJKSA-N 0.000 description 1
- AFVWSZWFAVVJNK-PUSCERABSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AFVWSZWFAVVJNK-PUSCERABSA-N 0.000 description 1
- NVSWJKWHLUTHLP-CPJSRVTESA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N NVSWJKWHLUTHLP-CPJSRVTESA-N 0.000 description 1
- FDJCPFLAWDPREM-LPHOPBHVSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1CCC[C@H]1C#N FDJCPFLAWDPREM-LPHOPBHVSA-N 0.000 description 1
- VZSCQFDHSBVENO-LDTOTXGLSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VZSCQFDHSBVENO-LDTOTXGLSA-N 0.000 description 1
- DILYYEIPMGFIEN-CUNXSJBXSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1CCC[C@H]1C#N DILYYEIPMGFIEN-CUNXSJBXSA-N 0.000 description 1
- MXDYTQOZYPXIRK-OUTSHDOLSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1CCC[C@H]1C#N MXDYTQOZYPXIRK-OUTSHDOLSA-N 0.000 description 1
- PDXDINZDGVVAPR-KKSFZXQISA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)O)C=C2)CCC2=C1C=CC(C(=O)O)=C2)NCC(=O)N1CCC[C@H]1C#N PDXDINZDGVVAPR-KKSFZXQISA-N 0.000 description 1
- QJDXSVHDYCMQPP-KKSFZXQISA-N C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2)NCC(=O)N1CCC[C@H]1C#N QJDXSVHDYCMQPP-KKSFZXQISA-N 0.000 description 1
- YMAFZYOKVCGWSW-IRXDYDNUSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N YMAFZYOKVCGWSW-IRXDYDNUSA-N 0.000 description 1
- AZSGAOBTAWNIEZ-CLWJZODNSA-N C[C@@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AZSGAOBTAWNIEZ-CLWJZODNSA-N 0.000 description 1
- QRXIJAYYGSJSCL-SEELHOLOSA-N C[C@@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1CCC[C@H]1C#N QRXIJAYYGSJSCL-SEELHOLOSA-N 0.000 description 1
- MRICTLMFYHIRCH-CPJSRVTESA-N C[C@@H](CC1(C2=NOC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NOC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N MRICTLMFYHIRCH-CPJSRVTESA-N 0.000 description 1
- VAMHYVUTZGIXAM-CPJSRVTESA-N C[C@@H](CC1(C2=NOC(C(F)(F)F)=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CC1(C2=NOC(C(F)(F)F)=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N VAMHYVUTZGIXAM-CPJSRVTESA-N 0.000 description 1
- ZBGXQKHGHMIUMH-HKUYNNGSSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N ZBGXQKHGHMIUMH-HKUYNNGSSA-N 0.000 description 1
- CDLNZLNEOFQKBG-MIVVTYSHSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CDLNZLNEOFQKBG-MIVVTYSHSA-N 0.000 description 1
- VKILDGCIGOUAGY-WMZOPIPTSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N VKILDGCIGOUAGY-WMZOPIPTSA-N 0.000 description 1
- LMJGOCVWUKLUHD-FIEAQMJYSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LMJGOCVWUKLUHD-FIEAQMJYSA-N 0.000 description 1
- HMVXNQSKAKPCDI-RDJZCZTQSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N HMVXNQSKAKPCDI-RDJZCZTQSA-N 0.000 description 1
- JFEWGLBNEHJULV-RYUMIWLPSA-N C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JFEWGLBNEHJULV-RYUMIWLPSA-N 0.000 description 1
- GIYCTOSIWIIBGF-KNTRFNDTSA-N C[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@@H](CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1)NCC(=O)N1CCC[C@H]1C#N GIYCTOSIWIIBGF-KNTRFNDTSA-N 0.000 description 1
- HUVQJGPIFWQKBV-HDEQKYRQSA-N C[C@@H](CO)NC(=O)OC(C)(C)C.C[C@H](N)CO.C[C@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C Chemical compound C[C@@H](CO)NC(=O)OC(C)(C)C.C[C@H](N)CO.C[C@H]1COS(=O)(=O)N1C(=O)OC(C)(C)C HUVQJGPIFWQKBV-HDEQKYRQSA-N 0.000 description 1
- NTPOZISPXJYYOJ-FYZOBXCZSA-N C[C@@H](N)C1=CC=C(C(N)=O)C=C1.[He] Chemical compound C[C@@H](N)C1=CC=C(C(N)=O)C=C1.[He] NTPOZISPXJYYOJ-FYZOBXCZSA-N 0.000 description 1
- QGCLEUGNYRXBMZ-ZCFIWIBFSA-N C[C@@H](N)C1=CC=C(F)C=C1 Chemical compound C[C@@H](N)C1=CC=C(F)C=C1 QGCLEUGNYRXBMZ-ZCFIWIBFSA-N 0.000 description 1
- SXCTVOHWPZGYES-OAHLLOKOSA-N C[C@@H](N)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1 Chemical compound C[C@@H](N)CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1 SXCTVOHWPZGYES-OAHLLOKOSA-N 0.000 description 1
- IXKQDVFWFPTATM-LLVKDONJSA-N C[C@@H](N)CC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 Chemical compound C[C@@H](N)CC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 IXKQDVFWFPTATM-LLVKDONJSA-N 0.000 description 1
- DTQPTDBHZMSWPR-OAHLLOKOSA-N C[C@@H](N)CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 DTQPTDBHZMSWPR-OAHLLOKOSA-N 0.000 description 1
- YAQPTDABKFONBY-LLVKDONJSA-N C[C@@H](N)CC1(C(=N)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C(=N)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 YAQPTDABKFONBY-LLVKDONJSA-N 0.000 description 1
- ZUPVWMBVHJLAND-LJQANCHMSA-N C[C@@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 ZUPVWMBVHJLAND-LJQANCHMSA-N 0.000 description 1
- JVQIKVBGUBFSSV-OAHLLOKOSA-N C[C@@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 JVQIKVBGUBFSSV-OAHLLOKOSA-N 0.000 description 1
- MVNBXIVQZDDZOP-OAHLLOKOSA-N C[C@@H](N)CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 MVNBXIVQZDDZOP-OAHLLOKOSA-N 0.000 description 1
- OGCBLVUBNNJWPO-LLVKDONJSA-N C[C@@H](N)CC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 OGCBLVUBNNJWPO-LLVKDONJSA-N 0.000 description 1
- RUHLYDCUNFKPBC-OAHLLOKOSA-N C[C@@H](N)CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 RUHLYDCUNFKPBC-OAHLLOKOSA-N 0.000 description 1
- WBOKDECMAMLRTI-LLVKDONJSA-N C[C@@H](N)CC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WBOKDECMAMLRTI-LLVKDONJSA-N 0.000 description 1
- DGCGRANUVQVFDX-CYBMUJFWSA-N C[C@@H](N)CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound C[C@@H](N)CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 DGCGRANUVQVFDX-CYBMUJFWSA-N 0.000 description 1
- VYKNYTHYQWQQQB-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 VYKNYTHYQWQQQB-OAHLLOKOSA-N 0.000 description 1
- GPXQCKXZGCYQGO-LLVKDONJSA-N C[C@@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 GPXQCKXZGCYQGO-LLVKDONJSA-N 0.000 description 1
- VINYQRWQECUIGS-MRXNPFEDSA-N C[C@@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 VINYQRWQECUIGS-MRXNPFEDSA-N 0.000 description 1
- PATNJQRNBGZPBI-GFCCVEGCSA-N C[C@@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 PATNJQRNBGZPBI-GFCCVEGCSA-N 0.000 description 1
- QLQJXEBZLLAEQF-HXUWFJFHSA-N C[C@@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 QLQJXEBZLLAEQF-HXUWFJFHSA-N 0.000 description 1
- PNZSYFBIBFQGNU-MRXNPFEDSA-N C[C@@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 PNZSYFBIBFQGNU-MRXNPFEDSA-N 0.000 description 1
- VUIODNZCUVLQEW-MRXNPFEDSA-N C[C@@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 VUIODNZCUVLQEW-MRXNPFEDSA-N 0.000 description 1
- VUEKJWSOLGHEKU-GFCCVEGCSA-N C[C@@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 VUEKJWSOLGHEKU-GFCCVEGCSA-N 0.000 description 1
- FMZUDQDSCOFWMB-LJQANCHMSA-N C[C@@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 FMZUDQDSCOFWMB-LJQANCHMSA-N 0.000 description 1
- KHBGPLYBRUABQP-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 KHBGPLYBRUABQP-OAHLLOKOSA-N 0.000 description 1
- WURVYQXFKLKAQY-LJQANCHMSA-N C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 WURVYQXFKLKAQY-LJQANCHMSA-N 0.000 description 1
- VBDDSARZQLFWAA-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 VBDDSARZQLFWAA-OAHLLOKOSA-N 0.000 description 1
- AGXXQVGJXZDXDV-LJQANCHMSA-N C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 AGXXQVGJXZDXDV-LJQANCHMSA-N 0.000 description 1
- WAUYRZGKFJCNJT-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WAUYRZGKFJCNJT-OAHLLOKOSA-N 0.000 description 1
- LMSMDOFIHABGCM-MRXNPFEDSA-N C[C@@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 LMSMDOFIHABGCM-MRXNPFEDSA-N 0.000 description 1
- WGWHBTJFPAABHU-GFCCVEGCSA-N C[C@@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WGWHBTJFPAABHU-GFCCVEGCSA-N 0.000 description 1
- INDTZLLXHMRIIF-OAHLLOKOSA-N C[C@@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 INDTZLLXHMRIIF-OAHLLOKOSA-N 0.000 description 1
- JTUIZEMYDHJACP-LLVKDONJSA-N C[C@@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 JTUIZEMYDHJACP-LLVKDONJSA-N 0.000 description 1
- QRGCHMKVRBIKEM-CYBMUJFWSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1/C=C\C(C(=O)N(C)C)=C/2 QRGCHMKVRBIKEM-CYBMUJFWSA-N 0.000 description 1
- BLKPSCKMCZWUQE-LLVKDONJSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1SC(C(=O)N(C)C)=C2 BLKPSCKMCZWUQE-LLVKDONJSA-N 0.000 description 1
- RZDYVBBTXMXPCM-SECBINFHSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 RZDYVBBTXMXPCM-SECBINFHSA-N 0.000 description 1
- BLDBZFVOBDEPDZ-SSDOTTSWSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 BLDBZFVOBDEPDZ-SSDOTTSWSA-N 0.000 description 1
- LBFUELDENXLZSD-DMPGVRBJSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(=O)N(C)C)=C2 LBFUELDENXLZSD-DMPGVRBJSA-N 0.000 description 1
- NDOJUNVGWCPPKJ-YRFRWJCTSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 NDOJUNVGWCPPKJ-YRFRWJCTSA-N 0.000 description 1
- VJJQXCKIXQEVTD-LLVKDONJSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2 VJJQXCKIXQEVTD-LLVKDONJSA-N 0.000 description 1
- JSIVHMICGBADLD-DMPGVRBJSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(=O)N(C)C)S3)SC=C2 JSIVHMICGBADLD-DMPGVRBJSA-N 0.000 description 1
- GTUDKTYTUIUNBL-SSDOTTSWSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 GTUDKTYTUIUNBL-SSDOTTSWSA-N 0.000 description 1
- BJEKVXKAYWEZGG-YRFRWJCTSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 BJEKVXKAYWEZGG-YRFRWJCTSA-N 0.000 description 1
- HYADHLYJNICLSS-CQSZACIVSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CC2=C1C=CC(C(=O)N(C)C)=C2 HYADHLYJNICLSS-CQSZACIVSA-N 0.000 description 1
- BZBIUFNKQBRQJT-SNVBAGLBSA-N C[C@@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CC2=C1C=CC(C(N)=O)=C2 BZBIUFNKQBRQJT-SNVBAGLBSA-N 0.000 description 1
- VCHPKGQJBZQHAN-XKMJEZSVSA-N C[C@@H](N)CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 Chemical compound C[C@@H](N)CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 VCHPKGQJBZQHAN-XKMJEZSVSA-N 0.000 description 1
- NVVLYQJFPITSMD-CYBMUJFWSA-N C[C@@H](N)CCCC1(C2=NN=NN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1/C=C\C(C(N)=O)=C/2 Chemical compound C[C@@H](N)CCCC1(C2=NN=NN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1/C=C\C(C(N)=O)=C/2 NVVLYQJFPITSMD-CYBMUJFWSA-N 0.000 description 1
- XPQQMSLMTPLNRW-CYBMUJFWSA-N C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)C(C)(C)C2=C1C=CC=C2 Chemical compound C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)C(C)(C)C2=C1C=CC=C2 XPQQMSLMTPLNRW-CYBMUJFWSA-N 0.000 description 1
- FBKUTPLJZGFNJR-LLVKDONJSA-N C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)N(C)C2=C1C=C(Cl)C=C2 Chemical compound C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)N(C)C2=C1C=C(Cl)C=C2 FBKUTPLJZGFNJR-LLVKDONJSA-N 0.000 description 1
- MXXMKNARNNHUMP-SNVBAGLBSA-N C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 Chemical compound C[C@@H](N)CN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 MXXMKNARNNHUMP-SNVBAGLBSA-N 0.000 description 1
- RIGHGJMMPNGTDM-NRQIEAPRSA-N C[C@@H](c(cc1)ccc1C(N)=O)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O Chemical compound C[C@@H](c(cc1)ccc1C(N)=O)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O RIGHGJMMPNGTDM-NRQIEAPRSA-N 0.000 description 1
- OHJQCVOBXSVLTA-LAQZMHDPSA-N C[C@@H](c(cc1)ccc1S(C)(C)[N+]([O-])=O)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)NC(OC(C)(C)C)=O)[C@H]2C1)C2=O Chemical compound C[C@@H](c(cc1)ccc1S(C)(C)[N+]([O-])=O)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)NC(OC(C)(C)C)=O)[C@H]2C1)C2=O OHJQCVOBXSVLTA-LAQZMHDPSA-N 0.000 description 1
- BWZILVKWSVYHOV-LTFFKMIWSA-N C[C@@H](c(cccc1)c1F)N(C1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O Chemical compound C[C@@H](c(cccc1)c1F)N(C1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O BWZILVKWSVYHOV-LTFFKMIWSA-N 0.000 description 1
- KADQYVHRKZMKSS-LTFFKMIWSA-N C[C@@H](c1c(C(F)(F)F)cccc1)N(C1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O Chemical compound C[C@@H](c1c(C(F)(F)F)cccc1)N(C1CN(CC(C(N(CCC2)C2C#N)=O)N)C2C1)C2=O KADQYVHRKZMKSS-LTFFKMIWSA-N 0.000 description 1
- IZRLTKSFGVPRPY-FGMIYAIPSA-N C[C@@H](c1cc(F)ccc1)N(C1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound C[C@@H](c1cc(F)ccc1)N(C1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O IZRLTKSFGVPRPY-FGMIYAIPSA-N 0.000 description 1
- XZAZZGFOWINYFU-NDDQQEFESA-N C[C@@H](c1cccc(C(NC)=O)c1)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound C[C@@H](c1cccc(C(NC)=O)c1)N([C@@H]1CN(CC(C(N(CCC2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O XZAZZGFOWINYFU-NDDQQEFESA-N 0.000 description 1
- NGBNOPSFXRCBGB-NEBITURZSA-N C[C@@H](c1cccc(C(NC)=O)c1)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O Chemical compound C[C@@H](c1cccc(C(NC)=O)c1)N([C@@H]1CN(CC(C(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)NC(OC(C)(C)C)=O)C2C1)C2=O NGBNOPSFXRCBGB-NEBITURZSA-N 0.000 description 1
- LCBBURJSTNEJFB-JNQBIPAQSA-N C[C@@H]1CCCN1C(=O)/C=C/C(=O)N1CCC[C@H]1C(N)=O Chemical compound C[C@@H]1CCCN1C(=O)/C=C/C(=O)N1CCC[C@H]1C(N)=O LCBBURJSTNEJFB-JNQBIPAQSA-N 0.000 description 1
- VODZFHKVIHIHKE-DUNDADLASA-N C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C(N)=O)C[C@@H]2C[C@@H]21 Chemical compound C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H](C(N)=O)C[C@@H]2C[C@@H]21 VODZFHKVIHIHKE-DUNDADLASA-N 0.000 description 1
- SZOXVBPLJRHDIC-WLTNLOOBSA-N C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SZOXVBPLJRHDIC-WLTNLOOBSA-N 0.000 description 1
- KADQYVHRKZMKSS-QASZKCHUSA-N C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N KADQYVHRKZMKSS-QASZKCHUSA-N 0.000 description 1
- DUSZVSSSGFKHGQ-RQDLXRADSA-N C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N DUSZVSSSGFKHGQ-RQDLXRADSA-N 0.000 description 1
- NKLLRXPRHHZCRW-YDHAOPIHSA-N C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=C(C(F)(F)F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 NKLLRXPRHHZCRW-YDHAOPIHSA-N 0.000 description 1
- SWWCBGOGLJVOPA-WLTNLOOBSA-N C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SWWCBGOGLJVOPA-WLTNLOOBSA-N 0.000 description 1
- BWZILVKWSVYHOV-QASZKCHUSA-N C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BWZILVKWSVYHOV-QASZKCHUSA-N 0.000 description 1
- GSUNCJAYVJXXGV-RQDLXRADSA-N C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N GSUNCJAYVJXXGV-RQDLXRADSA-N 0.000 description 1
- JGAFZZIGIBXOKH-YDHAOPIHSA-N C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=C(F)C=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JGAFZZIGIBXOKH-YDHAOPIHSA-N 0.000 description 1
- NVPSSFOKKVMYPF-GHBAERIYSA-N C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N NVPSSFOKKVMYPF-GHBAERIYSA-N 0.000 description 1
- SASJSKSWFBJPBD-ZZVXVNTOSA-N C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N SASJSKSWFBJPBD-ZZVXVNTOSA-N 0.000 description 1
- VHAIBOSHCDVMJE-GQXGXRCNSA-N C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N VHAIBOSHCDVMJE-GQXGXRCNSA-N 0.000 description 1
- GQTZRTMYXRYUTN-JBCRXWMVSA-N C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 GQTZRTMYXRYUTN-JBCRXWMVSA-N 0.000 description 1
- MMCQRXGYLGRZQD-JHOQEBCJSA-N C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N MMCQRXGYLGRZQD-JHOQEBCJSA-N 0.000 description 1
- PKMQYZMZLIKZER-AXMMUHIGSA-N C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N PKMQYZMZLIKZER-AXMMUHIGSA-N 0.000 description 1
- SXBOTIVPBRPKQE-RMQJPPFZSA-N C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N SXBOTIVPBRPKQE-RMQJPPFZSA-N 0.000 description 1
- BKDHQVWRWGFHPO-RAQVNZQDSA-N C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC(C(F)(F)F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BKDHQVWRWGFHPO-RAQVNZQDSA-N 0.000 description 1
- JEUMCGDPFJOMFC-GHBAERIYSA-N C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N JEUMCGDPFJOMFC-GHBAERIYSA-N 0.000 description 1
- FZWSIFXBLJIYHL-ZZVXVNTOSA-N C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N FZWSIFXBLJIYHL-ZZVXVNTOSA-N 0.000 description 1
- OBYFNJWXGMQIHL-GQXGXRCNSA-N C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N OBYFNJWXGMQIHL-GQXGXRCNSA-N 0.000 description 1
- YVRJIJIYQZBWAB-JBCRXWMVSA-N C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC(F)=CC(F)=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YVRJIJIYQZBWAB-JBCRXWMVSA-N 0.000 description 1
- DCTOKFRLUNGYOX-JHOQEBCJSA-N C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N DCTOKFRLUNGYOX-JHOQEBCJSA-N 0.000 description 1
- RXAITCMRXGQVLZ-AXMMUHIGSA-N C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RXAITCMRXGQVLZ-AXMMUHIGSA-N 0.000 description 1
- IZRLTKSFGVPRPY-RMQJPPFZSA-N C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N IZRLTKSFGVPRPY-RMQJPPFZSA-N 0.000 description 1
- VPKRYRUVZMSEHE-RAQVNZQDSA-N C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC(F)=CC=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VPKRYRUVZMSEHE-RAQVNZQDSA-N 0.000 description 1
- HKXOBNCRXNERCJ-WUIWSVLASA-N C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N HKXOBNCRXNERCJ-WUIWSVLASA-N 0.000 description 1
- GQPIBNHWVHVUAX-TWUVWWFLSA-N C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N GQPIBNHWVHVUAX-TWUVWWFLSA-N 0.000 description 1
- BMWVZDDLOXXIRI-HVURCESRSA-N C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N BMWVZDDLOXXIRI-HVURCESRSA-N 0.000 description 1
- DKZOUOPKKOGZSG-GBWMLQNXSA-N C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(C(=O)N(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DKZOUOPKKOGZSG-GBWMLQNXSA-N 0.000 description 1
- ZFZVZPXTFSVPAV-KDSAPBAQSA-N C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N ZFZVZPXTFSVPAV-KDSAPBAQSA-N 0.000 description 1
- UUGXZPUWYXRDQZ-RUZYHRDJSA-N C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N UUGXZPUWYXRDQZ-RUZYHRDJSA-N 0.000 description 1
- HUDDOULVYQZWPR-RESYIPQXSA-N C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HUDDOULVYQZWPR-RESYIPQXSA-N 0.000 description 1
- YCGLTHIWSVNOLG-INACUINTSA-N C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(C(=O)O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YCGLTHIWSVNOLG-INACUINTSA-N 0.000 description 1
- LGTDSSFZFJXYPH-JHOQEBCJSA-N C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N LGTDSSFZFJXYPH-JHOQEBCJSA-N 0.000 description 1
- HYGJEWPAZCHXHJ-AXMMUHIGSA-N C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N HYGJEWPAZCHXHJ-AXMMUHIGSA-N 0.000 description 1
- PYHFHARSJHSZOM-RMQJPPFZSA-N C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N PYHFHARSJHSZOM-RMQJPPFZSA-N 0.000 description 1
- XMQWLBBFUNDFLJ-RAQVNZQDSA-N C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(C(F)(F)F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XMQWLBBFUNDFLJ-RAQVNZQDSA-N 0.000 description 1
- BPZNTSNKQMOBBW-KDSAPBAQSA-N C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N BPZNTSNKQMOBBW-KDSAPBAQSA-N 0.000 description 1
- RIGHGJMMPNGTDM-RUZYHRDJSA-N C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RIGHGJMMPNGTDM-RUZYHRDJSA-N 0.000 description 1
- KQYLVIQPGMDYTC-RESYIPQXSA-N C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N KQYLVIQPGMDYTC-RESYIPQXSA-N 0.000 description 1
- RAFYFGWRLPCUQZ-INACUINTSA-N C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(C(N)=O)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 RAFYFGWRLPCUQZ-INACUINTSA-N 0.000 description 1
- BVVJUMXHVXOGHK-KDSAPBAQSA-N C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N BVVJUMXHVXOGHK-KDSAPBAQSA-N 0.000 description 1
- IRYCMEYPZXDLAY-RUZYHRDJSA-N C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N IRYCMEYPZXDLAY-RUZYHRDJSA-N 0.000 description 1
- HKVZORFGPRWFBK-RESYIPQXSA-N C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HKVZORFGPRWFBK-RESYIPQXSA-N 0.000 description 1
- CGNKCEJOYKBBFM-INACUINTSA-N C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(C2=NN=NN2)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CGNKCEJOYKBBFM-INACUINTSA-N 0.000 description 1
- NNGJVDSDQJAHPZ-AXMMUHIGSA-N C[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2CC1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N NNGJVDSDQJAHPZ-AXMMUHIGSA-N 0.000 description 1
- FBBIAJNTWTWLPN-RESYIPQXSA-N C[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(F)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N FBBIAJNTWTWLPN-RESYIPQXSA-N 0.000 description 1
- IEIKZMAFRCHIFW-YPLPJNLLSA-N C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N IEIKZMAFRCHIFW-YPLPJNLLSA-N 0.000 description 1
- AHLZYPTYSPLZQZ-SVZPGXKISA-N C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N AHLZYPTYSPLZQZ-SVZPGXKISA-N 0.000 description 1
- ZBWIWEWMZZEQJN-XOTSVCCBSA-N C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N ZBWIWEWMZZEQJN-XOTSVCCBSA-N 0.000 description 1
- BECRWEBGAYZHLS-JVIRBJESSA-N C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(SOON(C)C)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 BECRWEBGAYZHLS-JVIRBJESSA-N 0.000 description 1
- RSMDAAJKYGYJNK-QRMGSKBOSA-N C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N RSMDAAJKYGYJNK-QRMGSKBOSA-N 0.000 description 1
- YSASHIUYEJMGCV-DAYRTQLQSA-N C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N YSASHIUYEJMGCV-DAYRTQLQSA-N 0.000 description 1
- LIEVWQLRESWHFM-XTYSOYBWSA-N C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N LIEVWQLRESWHFM-XTYSOYBWSA-N 0.000 description 1
- JVHTWVICYMAEIA-VBDJFXSVSA-N C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=C(SOON)C=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JVHTWVICYMAEIA-VBDJFXSVSA-N 0.000 description 1
- SLAKQEXWMLMQMQ-MBFMOYRZSA-N C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N SLAKQEXWMLMQMQ-MBFMOYRZSA-N 0.000 description 1
- AZEPPKCFOGZYMU-SSRYDLFMSA-N C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N AZEPPKCFOGZYMU-SSRYDLFMSA-N 0.000 description 1
- BOHMQZDMCNCNDS-GLBNMMOUSA-N C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N BOHMQZDMCNCNDS-GLBNMMOUSA-N 0.000 description 1
- UXHPBUKCPOTJRI-VAVNUEAYSA-N C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(C#N)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 UXHPBUKCPOTJRI-VAVNUEAYSA-N 0.000 description 1
- KENZREGLGDETOB-WUIWSVLASA-N C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N KENZREGLGDETOB-WUIWSVLASA-N 0.000 description 1
- DAQIUQBFBZSHFB-TWUVWWFLSA-N C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N DAQIUQBFBZSHFB-TWUVWWFLSA-N 0.000 description 1
- PBFXPZJDSDKHCT-HVURCESRSA-N C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N PBFXPZJDSDKHCT-HVURCESRSA-N 0.000 description 1
- YYMYULHEDIQCMP-GBWMLQNXSA-N C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(C(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YYMYULHEDIQCMP-GBWMLQNXSA-N 0.000 description 1
- CRBVYFFPLSWYCE-KDSAPBAQSA-N C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N CRBVYFFPLSWYCE-KDSAPBAQSA-N 0.000 description 1
- BTOHCUVCSPXOME-RUZYHRDJSA-N C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N BTOHCUVCSPXOME-RUZYHRDJSA-N 0.000 description 1
- HTMCXSYUWXFYRJ-RESYIPQXSA-N C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N HTMCXSYUWXFYRJ-RESYIPQXSA-N 0.000 description 1
- PDMZOYBXYVNNLG-INACUINTSA-N C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(C(=O)O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PDMZOYBXYVNNLG-INACUINTSA-N 0.000 description 1
- VQFKGROZJRLPBX-KDSAPBAQSA-N C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N VQFKGROZJRLPBX-KDSAPBAQSA-N 0.000 description 1
- RRFZAFARHBSTGF-RUZYHRDJSA-N C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N RRFZAFARHBSTGF-RUZYHRDJSA-N 0.000 description 1
- XKEMHPGMVSVQOC-RESYIPQXSA-N C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N XKEMHPGMVSVQOC-RESYIPQXSA-N 0.000 description 1
- KKLDFZHORFBFHI-INACUINTSA-N C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(C(N)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KKLDFZHORFBFHI-INACUINTSA-N 0.000 description 1
- UBCBOPHTTSFLJL-KDSAPBAQSA-N C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N UBCBOPHTTSFLJL-KDSAPBAQSA-N 0.000 description 1
- JEKNPQPBAURCLJ-RUZYHRDJSA-N C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N JEKNPQPBAURCLJ-RUZYHRDJSA-N 0.000 description 1
- RSOKJWFKWYEUHI-RESYIPQXSA-N C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N RSOKJWFKWYEUHI-RESYIPQXSA-N 0.000 description 1
- QLENTQILYWVQEY-INACUINTSA-N C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(C2=NN=NN2)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QLENTQILYWVQEY-INACUINTSA-N 0.000 description 1
- VYCYUCAHMJJCAE-YPLPJNLLSA-N C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N VYCYUCAHMJJCAE-YPLPJNLLSA-N 0.000 description 1
- IJWNWCJODWVLDO-SVZPGXKISA-N C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N IJWNWCJODWVLDO-SVZPGXKISA-N 0.000 description 1
- NVPIVAXESTVEOD-XOTSVCCBSA-N C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N NVPIVAXESTVEOD-XOTSVCCBSA-N 0.000 description 1
- YVUIBGQCQNZVLI-JVIRBJESSA-N C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(S(=O)(=O)N(C)C)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YVUIBGQCQNZVLI-JVIRBJESSA-N 0.000 description 1
- RLIBSICPMPDSTN-QRMGSKBOSA-N C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1C2C[C@H]2C[C@H]1C#N RLIBSICPMPDSTN-QRMGSKBOSA-N 0.000 description 1
- SUTIINKCOVHNJN-DAYRTQLQSA-N C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](N)C(=O)N1CCC[C@H]1C#N SUTIINKCOVHNJN-DAYRTQLQSA-N 0.000 description 1
- CMHUZFDIJVQXQD-XTYSOYBWSA-N C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N CMHUZFDIJVQXQD-XTYSOYBWSA-N 0.000 description 1
- PIKJDSQTHXAWIZ-VBDJFXSVSA-N C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](C1=CC=CC(S(N)(=O)=O)=C1)N1C(=O)[C@@H]2C[C@H]1CN2C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PIKJDSQTHXAWIZ-VBDJFXSVSA-N 0.000 description 1
- XYJJSQVRLRJJNA-FHAONGHWSA-N C[C@H](CC(C(N)=N)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound C[C@H](CC(C(N)=N)(c(c(CCc1c2)c3)ccc3C(N)=O)c1ccc2C(N)=O)NCC(N([C@@H](C1)[C@@H]1C1)C1C#N)=O XYJJSQVRLRJJNA-FHAONGHWSA-N 0.000 description 1
- WSXVTLVKDDISJM-RHYIPFRBSA-N C[C@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC(C1=CC=C(C(=O)N(C)C)C=C1)(C1=CC=C(C(=O)N(C)C)C=C1)C1=NNN=N1)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 WSXVTLVKDDISJM-RHYIPFRBSA-N 0.000 description 1
- ACNZDQOYDUCXAY-GICMACPYSA-N C[C@H](CC(c1c(CC2)[s]c(C(NC)=O)c1)(c1c2[s]c(C(NC)=O)c1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O Chemical compound C[C@H](CC(c1c(CC2)[s]c(C(NC)=O)c1)(c1c2[s]c(C(NC)=O)c1)c1n[nH]nn1)NCC(N(CCC1)C1C#N)=O ACNZDQOYDUCXAY-GICMACPYSA-N 0.000 description 1
- PPUHTBCZDZMNJI-NLFFAJNJSA-N C[C@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N PPUHTBCZDZMNJI-NLFFAJNJSA-N 0.000 description 1
- KFMFKJJQXPAAAE-SDXKGTORSA-N C[C@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C(=N)N)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KFMFKJJQXPAAAE-SDXKGTORSA-N 0.000 description 1
- PJMFKJCVBZYYRM-IUNGDRIPSA-N C[C@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PJMFKJCVBZYYRM-IUNGDRIPSA-N 0.000 description 1
- WMNPOYHOYXXSRM-NLFFAJNJSA-N C[C@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N WMNPOYHOYXXSRM-NLFFAJNJSA-N 0.000 description 1
- JOZALJGSCOGUHA-SDXKGTORSA-N C[C@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JOZALJGSCOGUHA-SDXKGTORSA-N 0.000 description 1
- JJISUKAIFUXRCF-NLFFAJNJSA-N C[C@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N JJISUKAIFUXRCF-NLFFAJNJSA-N 0.000 description 1
- WUTVXLYTERWGQP-SDXKGTORSA-N C[C@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C(N)=O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 WUTVXLYTERWGQP-SDXKGTORSA-N 0.000 description 1
- QIFXVSDUOGCMHC-NQIIRXRSSA-N C[C@H](CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C(N)=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N QIFXVSDUOGCMHC-NQIIRXRSSA-N 0.000 description 1
- MRICTLMFYHIRCH-NLFFAJNJSA-N C[C@H](CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N MRICTLMFYHIRCH-NLFFAJNJSA-N 0.000 description 1
- DAVXMCVRQIWSAQ-SDXKGTORSA-N C[C@H](CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NC(=O)ON2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DAVXMCVRQIWSAQ-SDXKGTORSA-N 0.000 description 1
- UWFRQLKIHGXFDB-RLWLMLJZSA-N C[C@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N UWFRQLKIHGXFDB-RLWLMLJZSA-N 0.000 description 1
- OVHXMXCZYRAKOT-ZLGCBXFLSA-N C[C@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OVHXMXCZYRAKOT-ZLGCBXFLSA-N 0.000 description 1
- QFMZDUXDLUQNEY-NJHZRGNWSA-N C[C@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N QFMZDUXDLUQNEY-NJHZRGNWSA-N 0.000 description 1
- SEQQCDHOTLNEQG-NMKNLEBWSA-N C[C@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SEQQCDHOTLNEQG-NMKNLEBWSA-N 0.000 description 1
- RSUCYIMOZZILNB-RLWLMLJZSA-N C[C@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N RSUCYIMOZZILNB-RLWLMLJZSA-N 0.000 description 1
- QIPWOWKRBANZMI-ZLGCBXFLSA-N C[C@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QIPWOWKRBANZMI-ZLGCBXFLSA-N 0.000 description 1
- CYHQNOPGVDVJRS-RLWLMLJZSA-N C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N CYHQNOPGVDVJRS-RLWLMLJZSA-N 0.000 description 1
- YFXHYYUYVVJIAR-ZLGCBXFLSA-N C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 YFXHYYUYVVJIAR-ZLGCBXFLSA-N 0.000 description 1
- XBWVRFNNNBZWSH-VGSWGCGISA-N C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N XBWVRFNNNBZWSH-VGSWGCGISA-N 0.000 description 1
- CCTXIUGGKJKWRO-MOMMTWFBSA-N C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN(C)N=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CCTXIUGGKJKWRO-MOMMTWFBSA-N 0.000 description 1
- ZSUIFRSRPFMZJR-XJFCNFDWSA-N C[C@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N ZSUIFRSRPFMZJR-XJFCNFDWSA-N 0.000 description 1
- KSWQEWSMTKKMKQ-IUNGDRIPSA-N C[C@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 KSWQEWSMTKKMKQ-IUNGDRIPSA-N 0.000 description 1
- QTRPTBIWXKGXTL-XJFCNFDWSA-N C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N QTRPTBIWXKGXTL-XJFCNFDWSA-N 0.000 description 1
- ZWYOZOFYPZPFIC-DHSBEYIOSA-N C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ZWYOZOFYPZPFIC-DHSBEYIOSA-N 0.000 description 1
- BGCAHXKFQNUNPI-XJFCNFDWSA-N C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N BGCAHXKFQNUNPI-XJFCNFDWSA-N 0.000 description 1
- NEAOPGFDFSCMCM-DHSBEYIOSA-N C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 NEAOPGFDFSCMCM-DHSBEYIOSA-N 0.000 description 1
- LOJWCTNXGUBVLV-RLWLMLJZSA-N C[C@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N LOJWCTNXGUBVLV-RLWLMLJZSA-N 0.000 description 1
- VMFKWKYVZFMFFS-ZLGCBXFLSA-N C[C@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VMFKWKYVZFMFFS-ZLGCBXFLSA-N 0.000 description 1
- NVSWJKWHLUTHLP-NLFFAJNJSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N NVSWJKWHLUTHLP-NLFFAJNJSA-N 0.000 description 1
- OZVMKSLUAGGKTA-SDXKGTORSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 OZVMKSLUAGGKTA-SDXKGTORSA-N 0.000 description 1
- DILYYEIPMGFIEN-AMGIVPHBSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1CCC[C@H]1C#N DILYYEIPMGFIEN-AMGIVPHBSA-N 0.000 description 1
- SCUSHPVWRJTBTG-XCVXUIKGSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 SCUSHPVWRJTBTG-XCVXUIKGSA-N 0.000 description 1
- QDIQNOIUDRAYNG-GIGWZHCTSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N QDIQNOIUDRAYNG-GIGWZHCTSA-N 0.000 description 1
- HGYWOULATVQZIX-VKVQOMGUSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 HGYWOULATVQZIX-VKVQOMGUSA-N 0.000 description 1
- GRFSBOGZWUIQBV-NEEKEDPPSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1CCC[C@H]1C#N GRFSBOGZWUIQBV-NEEKEDPPSA-N 0.000 description 1
- DRZBEDXSNVFJSB-ZFIWFGPPSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 DRZBEDXSNVFJSB-ZFIWFGPPSA-N 0.000 description 1
- UAKNGRPCXBQROA-NEEKEDPPSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1CCC[C@H]1C#N UAKNGRPCXBQROA-NEEKEDPPSA-N 0.000 description 1
- JKXHHOJBUFCQIJ-ZFIWFGPPSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCN(C)CC3)C=C2)CCC2=C1C=CC(C(=O)N1CCN(C)CC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JKXHHOJBUFCQIJ-ZFIWFGPPSA-N 0.000 description 1
- MXDYTQOZYPXIRK-GIGWZHCTSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1CCC[C@H]1C#N MXDYTQOZYPXIRK-GIGWZHCTSA-N 0.000 description 1
- QOKKCJQMDSSTDF-VKVQOMGUSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QOKKCJQMDSSTDF-VKVQOMGUSA-N 0.000 description 1
- QYSKKHBFZBSJNY-XCVXUIKGSA-N C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2)C2=C(C=C(C(=O)NC(C)(C)C)C=C2)CCC2=C1C=CC(C(=O)NC(C)(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QYSKKHBFZBSJNY-XCVXUIKGSA-N 0.000 description 1
- RADGCIRPMJIFKP-RLWLMLJZSA-N C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N RADGCIRPMJIFKP-RLWLMLJZSA-N 0.000 description 1
- ATDVHHUQCKDTCV-ZLGCBXFLSA-N C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 ATDVHHUQCKDTCV-ZLGCBXFLSA-N 0.000 description 1
- YSWKZPBRBRQHFI-VGSWGCGISA-N C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1CCC[C@H]1C#N YSWKZPBRBRQHFI-VGSWGCGISA-N 0.000 description 1
- PWKNSSDGOLSQLA-MOMMTWFBSA-N C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NN=NN2C)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PWKNSSDGOLSQLA-MOMMTWFBSA-N 0.000 description 1
- MFGCTMHBEHLJMT-NLFFAJNJSA-N C[C@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1/C=C\C(C(=O)N(C)C)=C/2)NCC(=O)N1CCC[C@H]1C#N MFGCTMHBEHLJMT-NLFFAJNJSA-N 0.000 description 1
- GGGXQQXXSVOJGZ-SDXKGTORSA-N C[C@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 GGGXQQXXSVOJGZ-SDXKGTORSA-N 0.000 description 1
- AXUOZQBCABYKJM-NQIIRXRSSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N AXUOZQBCABYKJM-NQIIRXRSSA-N 0.000 description 1
- XFCCODCYUKLMMW-CXLKWVGHSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)C2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XFCCODCYUKLMMW-CXLKWVGHSA-N 0.000 description 1
- FGUFRQRXAIFWGF-DVECYGJZSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N FGUFRQRXAIFWGF-DVECYGJZSA-N 0.000 description 1
- AFVWSZWFAVVJNK-PFCVVTBNSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)C=C2)CC2=C1C=CC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AFVWSZWFAVVJNK-PFCVVTBNSA-N 0.000 description 1
- FDJCPFLAWDPREM-APWZRJJASA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1CCC[C@H]1C#N FDJCPFLAWDPREM-APWZRJJASA-N 0.000 description 1
- VZSCQFDHSBVENO-XCLNPWKQSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C(C(=O)N(C)C)=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 VZSCQFDHSBVENO-XCLNPWKQSA-N 0.000 description 1
- WQKZRORKQBDSTL-DHZNBDHHSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C=C\2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C=C\2)NCC(=O)N1CCC[C@H]1C#N WQKZRORKQBDSTL-DHZNBDHHSA-N 0.000 description 1
- XWCWHEUIZZYVNH-GLOOUZNYSA-N C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(C=C(C(=O)N(C)C)S2)CCC2=C1S/C=C\2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 XWCWHEUIZZYVNH-GLOOUZNYSA-N 0.000 description 1
- YMAFZYOKVCGWSW-SJORKVTESA-N C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N YMAFZYOKVCGWSW-SJORKVTESA-N 0.000 description 1
- AZSGAOBTAWNIEZ-WWNBULGVSA-N C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C(/C(=O)N(C)C)S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 AZSGAOBTAWNIEZ-WWNBULGVSA-N 0.000 description 1
- CCCHFZWATNAGNH-WQXGNCJSSA-N C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C\S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C\S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1CCC[C@H]1C#N CCCHFZWATNAGNH-WQXGNCJSSA-N 0.000 description 1
- PKYFXODDGWMOCR-GGRSRCBSSA-N C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C\S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2=C(CCC3=C1/C=C\S3)SC(C(=O)N(C)C)=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 PKYFXODDGWMOCR-GGRSRCBSSA-N 0.000 description 1
- QRXIJAYYGSJSCL-RLRQHFORSA-N C[C@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1CCC[C@H]1C#N QRXIJAYYGSJSCL-RLRQHFORSA-N 0.000 description 1
- UMRNTYCSMGIJNQ-XMJVQLNRSA-N C[C@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 UMRNTYCSMGIJNQ-XMJVQLNRSA-N 0.000 description 1
- ZBGXQKHGHMIUMH-MJGOQNOKSA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N ZBGXQKHGHMIUMH-MJGOQNOKSA-N 0.000 description 1
- CDLNZLNEOFQKBG-CVJORQDNSA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)C(C)(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 CDLNZLNEOFQKBG-CVJORQDNSA-N 0.000 description 1
- VKILDGCIGOUAGY-AEFFLSMTSA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N VKILDGCIGOUAGY-AEFFLSMTSA-N 0.000 description 1
- LMJGOCVWUKLUHD-VBWKBDAESA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)N(C)C2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 LMJGOCVWUKLUHD-VBWKBDAESA-N 0.000 description 1
- HMVXNQSKAKPCDI-WBVHZDCISA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1CCC[C@H]1C#N HMVXNQSKAKPCDI-WBVHZDCISA-N 0.000 description 1
- JFEWGLBNEHJULV-WQLIQUDQSA-N C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 Chemical compound C[C@H](CN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2)NCC(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 JFEWGLBNEHJULV-WQLIQUDQSA-N 0.000 description 1
- IXKQDVFWFPTATM-NSHDSACASA-N C[C@H](N)CC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 Chemical compound C[C@H](N)CC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 IXKQDVFWFPTATM-NSHDSACASA-N 0.000 description 1
- YAQPTDABKFONBY-NSHDSACASA-N C[C@H](N)CC1(C(=N)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C(=N)N)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 YAQPTDABKFONBY-NSHDSACASA-N 0.000 description 1
- ZUPVWMBVHJLAND-IBGZPJMESA-N C[C@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 ZUPVWMBVHJLAND-IBGZPJMESA-N 0.000 description 1
- JVQIKVBGUBFSSV-HNNXBMFYSA-N C[C@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C(=N)NC2=CC=C(F)C=C2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 JVQIKVBGUBFSSV-HNNXBMFYSA-N 0.000 description 1
- MVNBXIVQZDDZOP-HNNXBMFYSA-N C[C@H](N)CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C(=O)O)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 MVNBXIVQZDDZOP-HNNXBMFYSA-N 0.000 description 1
- OGCBLVUBNNJWPO-NSHDSACASA-N C[C@H](N)CC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C(=O)O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 OGCBLVUBNNJWPO-NSHDSACASA-N 0.000 description 1
- WBOKDECMAMLRTI-NSHDSACASA-N C[C@H](N)CC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WBOKDECMAMLRTI-NSHDSACASA-N 0.000 description 1
- RUHLYDCUNFKPBC-HNNXBMFYSA-N C[C@H](N)CC1(C(N)=O)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C(N)=O)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 RUHLYDCUNFKPBC-HNNXBMFYSA-N 0.000 description 1
- GPXQCKXZGCYQGO-NSHDSACASA-N C[C@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 GPXQCKXZGCYQGO-NSHDSACASA-N 0.000 description 1
- VINYQRWQECUIGS-INIZCTEOSA-N C[C@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 VINYQRWQECUIGS-INIZCTEOSA-N 0.000 description 1
- PATNJQRNBGZPBI-LBPRGKRZSA-N C[C@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NC(=O)ON2C)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 PATNJQRNBGZPBI-LBPRGKRZSA-N 0.000 description 1
- QLQJXEBZLLAEQF-FQEVSTJZSA-N C[C@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 QLQJXEBZLLAEQF-FQEVSTJZSA-N 0.000 description 1
- PNZSYFBIBFQGNU-INIZCTEOSA-N C[C@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 PNZSYFBIBFQGNU-INIZCTEOSA-N 0.000 description 1
- VUIODNZCUVLQEW-INIZCTEOSA-N C[C@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 VUIODNZCUVLQEW-INIZCTEOSA-N 0.000 description 1
- VUEKJWSOLGHEKU-LBPRGKRZSA-N C[C@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NN(C)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 VUEKJWSOLGHEKU-LBPRGKRZSA-N 0.000 description 1
- FMZUDQDSCOFWMB-IBGZPJMESA-N C[C@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 FMZUDQDSCOFWMB-IBGZPJMESA-N 0.000 description 1
- KHBGPLYBRUABQP-HNNXBMFYSA-N C[C@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 KHBGPLYBRUABQP-HNNXBMFYSA-N 0.000 description 1
- WURVYQXFKLKAQY-IBGZPJMESA-N C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 WURVYQXFKLKAQY-IBGZPJMESA-N 0.000 description 1
- AGXXQVGJXZDXDV-IBGZPJMESA-N C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 AGXXQVGJXZDXDV-IBGZPJMESA-N 0.000 description 1
- WAUYRZGKFJCNJT-HNNXBMFYSA-N C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WAUYRZGKFJCNJT-HNNXBMFYSA-N 0.000 description 1
- LMSMDOFIHABGCM-INIZCTEOSA-N C[C@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 LMSMDOFIHABGCM-INIZCTEOSA-N 0.000 description 1
- WGWHBTJFPAABHU-LBPRGKRZSA-N C[C@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NN=CN2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 WGWHBTJFPAABHU-LBPRGKRZSA-N 0.000 description 1
- INDTZLLXHMRIIF-HNNXBMFYSA-N C[C@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(=O)N(C)C)C=C2)CCC2=C1C=CC(C(=O)N(C)C)=C2 INDTZLLXHMRIIF-HNNXBMFYSA-N 0.000 description 1
- JTUIZEMYDHJACP-NSHDSACASA-N C[C@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NNC(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 JTUIZEMYDHJACP-NSHDSACASA-N 0.000 description 1
- RZDYVBBTXMXPCM-VIFPVBQESA-N C[C@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 RZDYVBBTXMXPCM-VIFPVBQESA-N 0.000 description 1
- BLDBZFVOBDEPDZ-ZETCQYMHSA-N C[C@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 BLDBZFVOBDEPDZ-ZETCQYMHSA-N 0.000 description 1
- NDOJUNVGWCPPKJ-UOCSPZAXSA-N C[C@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 NDOJUNVGWCPPKJ-UOCSPZAXSA-N 0.000 description 1
- GTUDKTYTUIUNBL-ZETCQYMHSA-N C[C@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 GTUDKTYTUIUNBL-ZETCQYMHSA-N 0.000 description 1
- BJEKVXKAYWEZGG-UOCSPZAXSA-N C[C@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 BJEKVXKAYWEZGG-UOCSPZAXSA-N 0.000 description 1
- CFFJFHJYXBYTIE-HNNXBMFYSA-N C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 CFFJFHJYXBYTIE-HNNXBMFYSA-N 0.000 description 1
- JFXLEHGDNPWGEZ-KRWDZBQOSA-N C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCC3)C=C2CCC2=C1C=CC(C(=O)N1CCC1)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCC3)C=C2CCC2=C1C=CC(C(=O)N1CCC1)=C2 JFXLEHGDNPWGEZ-KRWDZBQOSA-N 0.000 description 1
- ONZPKZYJYHWNKP-IBGZPJMESA-N C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCOCC3)C=C2CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCOCC3)C=C2CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 ONZPKZYJYHWNKP-IBGZPJMESA-N 0.000 description 1
- FOABBWZXYPCHEO-NSHDSACASA-N C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 Chemical compound C[C@H](N)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1C=CC(C(=O)O)=C2 FOABBWZXYPCHEO-NSHDSACASA-N 0.000 description 1
- VYKNYTHYQWQQQB-HNNXBMFYSA-N C[C@H](N)CC1(C2=NOC(=O)N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NOC(=O)N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 VYKNYTHYQWQQQB-HNNXBMFYSA-N 0.000 description 1
- ZWWFIHUURARWOM-HNNXBMFYSA-N C[C@H](N)CC1(C2=NOC(C(F)(F)F)=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 Chemical compound C[C@H](N)CC1(C2=NOC(C(F)(F)F)=N2)C2=CC=C(C(=O)N(C)C)C=C2CCC2=C1C=CC(C(=O)N(C)C)=C2 ZWWFIHUURARWOM-HNNXBMFYSA-N 0.000 description 1
- XPQQMSLMTPLNRW-ZDUSSCGKSA-N C[C@H](N)CN1C(=O)C2=C(C=CC=C2)C(C)(C)C2=C1C=CC=C2 Chemical compound C[C@H](N)CN1C(=O)C2=C(C=CC=C2)C(C)(C)C2=C1C=CC=C2 XPQQMSLMTPLNRW-ZDUSSCGKSA-N 0.000 description 1
- FBKUTPLJZGFNJR-NSHDSACASA-N C[C@H](N)CN1C(=O)C2=C(C=CC=C2)N(C)C2=C1C=C(Cl)C=C2 Chemical compound C[C@H](N)CN1C(=O)C2=C(C=CC=C2)N(C)C2=C1C=C(Cl)C=C2 FBKUTPLJZGFNJR-NSHDSACASA-N 0.000 description 1
- MXXMKNARNNHUMP-JTQLQIEISA-N C[C@H](N)CN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 Chemical compound C[C@H](N)CN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 MXXMKNARNNHUMP-JTQLQIEISA-N 0.000 description 1
- SASJSKSWFBJPBD-LMXOXSAVSA-N C[C@H](c1cc(C(F)(F)F)cc(C(F)(F)F)c1)N([C@@H]1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)N)C2C1)C2=O Chemical compound C[C@H](c1cc(C(F)(F)F)cc(C(F)(F)F)c1)N([C@@H]1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)N)C2C1)C2=O SASJSKSWFBJPBD-LMXOXSAVSA-N 0.000 description 1
- DVSBOCYHDIMZAM-INTGFUSFSA-N C[C@H](c1cccc(C(NC)=O)c1)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)N)[C@H]2C1)C2=O Chemical compound C[C@H](c1cccc(C(NC)=O)c1)N(C1CN(C[C@@H](C(N(CCC2)[C@@H]2C#N)=O)N)[C@H]2C1)C2=O DVSBOCYHDIMZAM-INTGFUSFSA-N 0.000 description 1
- CFMRGTABHMHURB-UHFFFAOYSA-N C[N]1(C(OC=C[CH]2)=C2[IH][IH]c2c1cccc2)N=C Chemical compound C[N]1(C(OC=C[CH]2)=C2[IH][IH]c2c1cccc2)N=C CFMRGTABHMHURB-UHFFFAOYSA-N 0.000 description 1
- YEGTVLBXSLDPEM-UHFFFAOYSA-N ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2 Chemical compound ClC1=CC2=C(C=C1)C(Cl)C1=C(C=CC=N1)CC2 YEGTVLBXSLDPEM-UHFFFAOYSA-N 0.000 description 1
- MPIOODLEGYTMSX-UHFFFAOYSA-N ClC1=CC=C2C(=C1)CCC1=C(C=CC(Cl)=C1)C2Cl.N#CC1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.O=C1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.OC1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 Chemical compound ClC1=CC=C2C(=C1)CCC1=C(C=CC(Cl)=C1)C2Cl.N#CC1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.O=C1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2.OC1C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 MPIOODLEGYTMSX-UHFFFAOYSA-N 0.000 description 1
- ADVNSTOCRFWYGM-UHFFFAOYSA-N ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 ADVNSTOCRFWYGM-UHFFFAOYSA-N 0.000 description 1
- RTSFFDGPQSVNKL-UHFFFAOYSA-N ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound ClC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 RTSFFDGPQSVNKL-UHFFFAOYSA-N 0.000 description 1
- JZBZEMFEHUPKMH-UHFFFAOYSA-N ClC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 Chemical compound ClC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 JZBZEMFEHUPKMH-UHFFFAOYSA-N 0.000 description 1
- DHBAKPVOZGAEDO-UHFFFAOYSA-N ClC1C2=C(C=CC=C2)CCC2=C1N=CC=C2.OC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.OC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 Chemical compound ClC1C2=C(C=CC=C2)CCC2=C1N=CC=C2.OC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2.OC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 DHBAKPVOZGAEDO-UHFFFAOYSA-N 0.000 description 1
- NLGZBEQXRSLWNZ-ONGXEEELSA-N FC1=CC(N2C[C@@H]3C[C@H]2CN3)=CC=C1 Chemical compound FC1=CC(N2C[C@@H]3C[C@H]2CN3)=CC=C1 NLGZBEQXRSLWNZ-ONGXEEELSA-N 0.000 description 1
- LRQUOXCURHVSKK-UHFFFAOYSA-N N#CC(CCC1)N1C(CNCCC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(N1CCCC1)=O)c2ccc3C(N1CCCC1)=O)=O Chemical compound N#CC(CCC1)N1C(CNCCC(c1nnn[nH]1)(c(cc1)c(CCc2c3)cc1C(N1CCCC1)=O)c2ccc3C(N1CCCC1)=O)=O LRQUOXCURHVSKK-UHFFFAOYSA-N 0.000 description 1
- WEXPITJBJBQYOC-ZOZOJSHZSA-N N#CC(C[C@H]1[C@@H]2C1)N2C(CNCCC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N3CCCCC3)=O)c1ccc2C(N1CCCCC1)=O)=O Chemical compound N#CC(C[C@H]1[C@@H]2C1)N2C(CNCCC(c1nnn[nH]1)(c(c(CCc1c2)c3)ccc3C(N3CCCCC3)=O)c1ccc2C(N1CCCCC1)=O)=O WEXPITJBJBQYOC-ZOZOJSHZSA-N 0.000 description 1
- XNAXNGCPAKXUOT-FJSNOKNSSA-N N#CC(C[C@H]1[C@@H]2C1)N2C(CNCCc1c(CCCC2)c2cc2c1CCCC2)=O Chemical compound N#CC(C[C@H]1[C@@H]2C1)N2C(CNCCc1c(CCCC2)c2cc2c1CCCC2)=O XNAXNGCPAKXUOT-FJSNOKNSSA-N 0.000 description 1
- MNEARZNVFZKCGX-BOXHHOBZSA-N N#CC1(CCN)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#C[C@@H]1CCCN1C(=O)CNCCC1(C#N)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#CC1(CCN)C2=C(C=CC=C2)CCC2=C1C=CC=C2.N#C[C@@H]1CCCN1C(=O)CNCCC1(C#N)C2=C(C=CC=C2)CCC2=C1C=CC=C2 MNEARZNVFZKCGX-BOXHHOBZSA-N 0.000 description 1
- OUVWVQHVDBTSJW-JKBNYCFLSA-N N#CC1=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=CC=C1 Chemical compound N#CC1=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=CC=C1 OUVWVQHVDBTSJW-JKBNYCFLSA-N 0.000 description 1
- PYEFFADJXVUFRV-OVWQWFNUSA-N N#CC1=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=CC=C1 Chemical compound N#CC1=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=CC=C1 PYEFFADJXVUFRV-OVWQWFNUSA-N 0.000 description 1
- GYRYFXAYNGXQBW-RNLLHNEJSA-N N#CC1=CC(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound N#CC1=CC(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=CC=C1 GYRYFXAYNGXQBW-RNLLHNEJSA-N 0.000 description 1
- UYBFBVHIJCRVMJ-RQLZSBSESA-N N#CC1=CC(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound N#CC1=CC(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 UYBFBVHIJCRVMJ-RQLZSBSESA-N 0.000 description 1
- IGVSYWJRUSINTG-OEXJRPJJSA-N N#CC1=CC=CC(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=C1 Chemical compound N#CC1=CC=CC(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)=C1 IGVSYWJRUSINTG-OEXJRPJJSA-N 0.000 description 1
- TVFVBRWVACZGNB-XSLAGTTESA-N N#CC1=CC=CC(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=C1 Chemical compound N#CC1=CC=CC(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)=C1 TVFVBRWVACZGNB-XSLAGTTESA-N 0.000 description 1
- FTGVWSKYBMZSOE-UHFFFAOYSA-N N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 FTGVWSKYBMZSOE-UHFFFAOYSA-N 0.000 description 1
- LSESYRQTTONDGG-UHFFFAOYSA-N N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C1C2=C(C=CC=C2)C(=O)N1CCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[C-]#[N+]C1(CCBr)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#CC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C1C2=C(C=CC=C2)C(=O)N1CCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[C-]#[N+]C1(CCBr)C2=C(C=CC=C2)CCC2=C1C=CC=C2.[C-]#[N+]C1(CCN2C(=O)C3=C(C=CC=C3)C2=O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 LSESYRQTTONDGG-UHFFFAOYSA-N 0.000 description 1
- PCEXXSAEUXFOCB-UHFFFAOYSA-N N#CCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C(CBr)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#CCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2.NCCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C(CBr)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 PCEXXSAEUXFOCB-UHFFFAOYSA-N 0.000 description 1
- QTPVJKRRQFSVMM-FPAZSGHZSA-N N#C[C@@H]1CCCN1C(=O)CBr.NC(=O)[C@@H]1CCCN1 Chemical compound N#C[C@@H]1CCCN1C(=O)CBr.NC(=O)[C@@H]1CCCN1 QTPVJKRRQFSVMM-FPAZSGHZSA-N 0.000 description 1
- YQIAAZYDNMZVJG-SFHVURJKSA-N N#C[C@@H]1CCCN1C(=O)CNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 YQIAAZYDNMZVJG-SFHVURJKSA-N 0.000 description 1
- CYCDNMUOTDBJAH-KEKNWZKVSA-N N#C[C@@H]1CCCN1C(=O)CNC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 Chemical compound N#C[C@@H]1CCCN1C(=O)CNC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 CYCDNMUOTDBJAH-KEKNWZKVSA-N 0.000 description 1
- DSPYWKUOYCNLFI-QHCPKHFHSA-N N#C[C@@H]1CCCN1C(=O)CNC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1 Chemical compound N#C[C@@H]1CCCN1C(=O)CNC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)CC1 DSPYWKUOYCNLFI-QHCPKHFHSA-N 0.000 description 1
- MAXTYMXTBANHNC-NRFANRHFSA-N N#C[C@@H]1CCCN1C(=O)CNC1CN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 Chemical compound N#C[C@@H]1CCCN1C(=O)CNC1CN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 MAXTYMXTBANHNC-NRFANRHFSA-N 0.000 description 1
- UQPNYPSDPFHCHL-IBGZPJMESA-N N#C[C@@H]1CCCN1C(=O)CNCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCC(=O)N1C2=C(C=CC=C2)CCC2=C1C=CC=C2 UQPNYPSDPFHCHL-IBGZPJMESA-N 0.000 description 1
- COVMMFFQOCVEKN-KRWDZBQOSA-N N#C[C@@H]1CCCN1C(=O)CNCC/C1=C2\CCC\C2=C\C2=C1CCC2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCC/C1=C2\CCC\C2=C\C2=C1CCC2 COVMMFFQOCVEKN-KRWDZBQOSA-N 0.000 description 1
- WBNKZKGIXIDXIA-IBGZPJMESA-N N#C[C@@H]1CCCN1C(=O)CNCC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2 WBNKZKGIXIDXIA-IBGZPJMESA-N 0.000 description 1
- KDRWKKLMQDSMSV-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCC=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCC=C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 KDRWKKLMQDSMSV-FQEVSTJZSA-N 0.000 description 1
- CPAVROWHRUJYLE-SANMLTNESA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 CPAVROWHRUJYLE-SANMLTNESA-N 0.000 description 1
- LRQUOXCURHVSKK-NDEPHWFRSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 LRQUOXCURHVSKK-NDEPHWFRSA-N 0.000 description 1
- RGPGYHVGRZIVNV-PMERELPUSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 RGPGYHVGRZIVNV-PMERELPUSA-N 0.000 description 1
- XWOPMVAMVZAKSW-NDEPHWFRSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 XWOPMVAMVZAKSW-NDEPHWFRSA-N 0.000 description 1
- SOEFKUPHSNEEBW-QASNXKAYSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 SOEFKUPHSNEEBW-QASNXKAYSA-N 0.000 description 1
- NKEISVOQERRRDL-YJJLJQPASA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 NKEISVOQERRRDL-YJJLJQPASA-N 0.000 description 1
- FWSMKVOLVNJWBT-ZGAORZAOSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 FWSMKVOLVNJWBT-ZGAORZAOSA-N 0.000 description 1
- SDCKHSQHLMXABA-YJJLJQPASA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 SDCKHSQHLMXABA-YJJLJQPASA-N 0.000 description 1
- KDZMBGABOWUMPY-DQUNLGLBSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(N)=O)=C2 KDZMBGABOWUMPY-DQUNLGLBSA-N 0.000 description 1
- XNHTVQOKSNFOBI-DQUNLGLBSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=CC(F)=C2)CCC2=C1C=C(C(N)=O)C=C2 XNHTVQOKSNFOBI-DQUNLGLBSA-N 0.000 description 1
- ZKVHUFYHCCCCMV-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=CC=C2)CCC2=C1C=CC=C2 ZKVHUFYHCCCCMV-FQEVSTJZSA-N 0.000 description 1
- JEQQPXJMMQVEAB-LTEHYTPHSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 JEQQPXJMMQVEAB-LTEHYTPHSA-N 0.000 description 1
- FNFGRDLSTLWLPJ-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1(O)C2=C(C=CC=C2)CCC2=C1C=CC=C2 FNFGRDLSTLWLPJ-FQEVSTJZSA-N 0.000 description 1
- TYNUQGXNNBGACU-IBGZPJMESA-N N#C[C@@H]1CCCN1C(=O)CNCCC1=C2CCCCC2=CC2=C1CCCC2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1=C2CCCCC2=CC2=C1CCCC2 TYNUQGXNNBGACU-IBGZPJMESA-N 0.000 description 1
- RTSZHGBMZPPLGY-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 RTSZHGBMZPPLGY-FQEVSTJZSA-N 0.000 description 1
- GSKGWCLUKFCYPM-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 GSKGWCLUKFCYPM-FQEVSTJZSA-N 0.000 description 1
- OUGWJDGQYAKZDK-FQEVSTJZSA-N N#C[C@@H]1CCCN1C(=O)CNCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCN1C2=C(C=CC=C2)CCC2=C1C=CC=C2 OUGWJDGQYAKZDK-FQEVSTJZSA-N 0.000 description 1
- NKVJCFJULFRNOO-HSTJUUNISA-N N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=C(Cl)C=C2)CCC2=C1/N=C\C=C/2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=C(Cl)C=C2)CCC2=C1/N=C\C=C/2 NKVJCFJULFRNOO-HSTJUUNISA-N 0.000 description 1
- RVZJHNWBAOBVEO-BDQAORGHSA-N N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2.NCCNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=CC=C2)CCC2=C1/C=C\C=C/2.NCCNC1C2=C(C=CC=C2)CCC2=C1C=CC=C2 RVZJHNWBAOBVEO-BDQAORGHSA-N 0.000 description 1
- JWWPGABHWVYERR-HSTJUUNISA-N N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=CC=C2)CCC2=C1/N=C\C=C/2 Chemical compound N#C[C@@H]1CCCN1C(=O)CNCCNC1C2=C(C=CC=C2)CCC2=C1/N=C\C=C/2 JWWPGABHWVYERR-HSTJUUNISA-N 0.000 description 1
- CLMIKSLMQOMPKN-ABHRYQDASA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=NC=CN=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)C1=NC=CN=C1 CLMIKSLMQOMPKN-ABHRYQDASA-N 0.000 description 1
- RMQXNQKPTZCPHG-SDADXPQNSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1=CC=CS1 RMQXNQKPTZCPHG-SDADXPQNSA-N 0.000 description 1
- VXBYNLLVAZNPNY-XQVMISGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 VXBYNLLVAZNPNY-XQVMISGQSA-N 0.000 description 1
- CUVXXOWURRTZED-WBYKHBECSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2C(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2 CUVXXOWURRTZED-WBYKHBECSA-N 0.000 description 1
- GLPLTOMMEVDJJR-JURCDPSOSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CSC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C(=O)[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CSC=C1 GLPLTOMMEVDJJR-JURCDPSOSA-N 0.000 description 1
- MHYQTKFCEYBSAY-CLTRGCNDSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1 MHYQTKFCEYBSAY-CLTRGCNDSA-N 0.000 description 1
- WCJVONFXYVVQDI-CLTRGCNDSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC=C(C(N)=O)C=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC=C(C(N)=O)C=C1 WCJVONFXYVVQDI-CLTRGCNDSA-N 0.000 description 1
- CCCXZMJCKAIPJC-CLTRGCNDSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC=CC(C(N)=O)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC=CC(C(N)=O)=C1 CCCXZMJCKAIPJC-CLTRGCNDSA-N 0.000 description 1
- VPHGVKOBWZHHOP-MLCQCVOFSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 VPHGVKOBWZHHOP-MLCQCVOFSA-N 0.000 description 1
- XOUZLNNNHROJGO-MUGJNUQGSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1 XOUZLNNNHROJGO-MUGJNUQGSA-N 0.000 description 1
- HZLDPZTXSZFGAT-TUFLPTIASA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1 HZLDPZTXSZFGAT-TUFLPTIASA-N 0.000 description 1
- VKDNMNLLPGXGQX-CMOCDZPBSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1 VKDNMNLLPGXGQX-CMOCDZPBSA-N 0.000 description 1
- OQJMDOHHQNZWQQ-HPZZRFSDSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1 OQJMDOHHQNZWQQ-HPZZRFSDSA-N 0.000 description 1
- MOGKXOHKGYNKHE-HPZZRFSDSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1 MOGKXOHKGYNKHE-HPZZRFSDSA-N 0.000 description 1
- KBMIIKJFDBFSFP-HJWJTTGWSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 KBMIIKJFDBFSFP-HJWJTTGWSA-N 0.000 description 1
- VHBFGSXDRXOMMP-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1 VHBFGSXDRXOMMP-QAETUUGQSA-N 0.000 description 1
- SACJGEGYKKGEAY-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1 SACJGEGYKKGEAY-QAETUUGQSA-N 0.000 description 1
- MGIYKCOOANCKJO-HJWJTTGWSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1 MGIYKCOOANCKJO-HJWJTTGWSA-N 0.000 description 1
- YOKNSXPIYFOTGF-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1 YOKNSXPIYFOTGF-QAETUUGQSA-N 0.000 description 1
- VYVBIVGFFLUUKU-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1 VYVBIVGFFLUUKU-QAETUUGQSA-N 0.000 description 1
- PYPNYWJMILRCGK-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1 PYPNYWJMILRCGK-QAETUUGQSA-N 0.000 description 1
- CTQYJOUJGALPDA-QAETUUGQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1 CTQYJOUJGALPDA-QAETUUGQSA-N 0.000 description 1
- WCYDWMCNINSTMC-DODAUVCRSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2 WCYDWMCNINSTMC-DODAUVCRSA-N 0.000 description 1
- DQUTUXKVLKUKGW-TZYAJKAJSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2 DQUTUXKVLKUKGW-TZYAJKAJSA-N 0.000 description 1
- FRFIBFKYJAOCAR-NBMBROAQSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 FRFIBFKYJAOCAR-NBMBROAQSA-N 0.000 description 1
- WVXGQUBYRMJKFU-LUVWLHFXSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=C(Cl)C=CC=C1Cl Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=C(Cl)C=CC=C1Cl WVXGQUBYRMJKFU-LUVWLHFXSA-N 0.000 description 1
- SHFAPODWDIDEIY-UQVNRYHBSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1 SHFAPODWDIDEIY-UQVNRYHBSA-N 0.000 description 1
- DPGAUDUNWBAHQC-KNOXWWKRSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1 DPGAUDUNWBAHQC-KNOXWWKRSA-N 0.000 description 1
- VSLTZGFUCHUVHV-XZCBWCRCSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1 VSLTZGFUCHUVHV-XZCBWCRCSA-N 0.000 description 1
- HYRGMNMOIZMHNS-QCCYXRBGSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 HYRGMNMOIZMHNS-QCCYXRBGSA-N 0.000 description 1
- VLFRZFZNUBTJSX-HNULKUCHSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 VLFRZFZNUBTJSX-HNULKUCHSA-N 0.000 description 1
- GKSQENNCDGJUDL-HNULKUCHSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2 GKSQENNCDGJUDL-HNULKUCHSA-N 0.000 description 1
- ZBJGFHZHIMGTPV-HNULKUCHSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 ZBJGFHZHIMGTPV-HNULKUCHSA-N 0.000 description 1
- LQWRSWUBDBYBAQ-HNULKUCHSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2 LQWRSWUBDBYBAQ-HNULKUCHSA-N 0.000 description 1
- HDWSAOKQXWEKPD-GYWCDFCBSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2 HDWSAOKQXWEKPD-GYWCDFCBSA-N 0.000 description 1
- SHFAPODWDIDEIY-SXYSDOLCSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1 SHFAPODWDIDEIY-SXYSDOLCSA-N 0.000 description 1
- DPGAUDUNWBAHQC-YFNVTMOMSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1 DPGAUDUNWBAHQC-YFNVTMOMSA-N 0.000 description 1
- VSLTZGFUCHUVHV-VUBDRERZSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1 VSLTZGFUCHUVHV-VUBDRERZSA-N 0.000 description 1
- HYRGMNMOIZMHNS-LSBAASHUSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1 HYRGMNMOIZMHNS-LSBAASHUSA-N 0.000 description 1
- VLFRZFZNUBTJSX-LPBINRMYSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2 VLFRZFZNUBTJSX-LPBINRMYSA-N 0.000 description 1
- GKSQENNCDGJUDL-LPBINRMYSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2 GKSQENNCDGJUDL-LPBINRMYSA-N 0.000 description 1
- ZBJGFHZHIMGTPV-LPBINRMYSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 ZBJGFHZHIMGTPV-LPBINRMYSA-N 0.000 description 1
- LQWRSWUBDBYBAQ-LPBINRMYSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2 LQWRSWUBDBYBAQ-LPBINRMYSA-N 0.000 description 1
- HDWSAOKQXWEKPD-ZESOOZHZSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2 HDWSAOKQXWEKPD-ZESOOZHZSA-N 0.000 description 1
- WNJCGAZSPSKHKR-DTIVQEBISA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=[SH]C=CC1.N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=[SH]C=CC1)NC(=O)OCC1=CC=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=[SH]C=CC1.N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C(=O)CC1=[SH]C=CC1)NC(=O)OCC1=CC=CC=C1 WNJCGAZSPSKHKR-DTIVQEBISA-N 0.000 description 1
- BJSKSYMJWDQHMT-XSLAGTTESA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2C1=CC=CC(F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2C1=CC=CC(F)=C1 BJSKSYMJWDQHMT-XSLAGTTESA-N 0.000 description 1
- YURHNOBNQGHBBY-OVWQWFNUSA-N N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1 YURHNOBNQGHBBY-OVWQWFNUSA-N 0.000 description 1
- RVHRSNBFLJCZJH-QORCZRPOSA-N N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C1=CC=CC(F)=C1)NC(=O)OCC1=CC=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2C1=CC=CC(F)=C1)NC(=O)OCC1=CC=CC=C1 RVHRSNBFLJCZJH-QORCZRPOSA-N 0.000 description 1
- KSNNSMOXAJHCBK-LCXINAFSSA-N N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)NC(=O)OCC1=CC=CC=C1 Chemical compound N#C[C@@H]1CCCN1C(=O)[C@H](CN1C[C@@H]2C[C@H]1CN2S(=O)(=O)C1=CC=CC(F)=C1)NC(=O)OCC1=CC=CC=C1 KSNNSMOXAJHCBK-LCXINAFSSA-N 0.000 description 1
- FCIGPWDQPPHXES-WXKOBWSBSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C(C1=CC=C(F)C=C1)C1=CC=C(F)C=C1 FCIGPWDQPPHXES-WXKOBWSBSA-N 0.000 description 1
- DSVRMKHIYYYIKY-RVLREDAYSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCC1 DSVRMKHIYYYIKY-RVLREDAYSA-N 0.000 description 1
- HVMPLFZIUOXHRK-UJXMJKKGSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCC1 HVMPLFZIUOXHRK-UJXMJKKGSA-N 0.000 description 1
- IGLVLKQTTBXGGV-CGWVLFOHSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1(C2=CC=C(F)C=C2)CCCCC1 IGLVLKQTTBXGGV-CGWVLFOHSA-N 0.000 description 1
- LWDZVINZQGISHW-FTGQOQSCSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C(N)=O)C=CC=C1 LWDZVINZQGISHW-FTGQOQSCSA-N 0.000 description 1
- NGABDKYPGISSKQ-FTGQOQSCSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=C(C2=NN=NN2)C=CC=C1 NGABDKYPGISSKQ-FTGQOQSCSA-N 0.000 description 1
- ZJOLSIOUVNQKJW-ABUQZBSZSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 ZJOLSIOUVNQKJW-ABUQZBSZSA-N 0.000 description 1
- ZUGREBQOZRAHBD-LAGQRLGTSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C(N)=O)=CC=C1 ZUGREBQOZRAHBD-LAGQRLGTSA-N 0.000 description 1
- WKKLLOCWVHQGRU-LAGQRLGTSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(C2=NN=NN2)=CC=C1 WKKLLOCWVHQGRU-LAGQRLGTSA-N 0.000 description 1
- IFKAMRLPPFCVFZ-ABUQZBSZSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC(F)=CC(F)=C1 IFKAMRLPPFCVFZ-ABUQZBSZSA-N 0.000 description 1
- POIGLNUGCMZWNK-LAGQRLGTSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C(N)=O)C=C1 POIGLNUGCMZWNK-LAGQRLGTSA-N 0.000 description 1
- GOLWIEWSULHYMN-LAGQRLGTSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(C2=NN=NN2)C=C1 GOLWIEWSULHYMN-LAGQRLGTSA-N 0.000 description 1
- WBIFXMFFIVAUPY-TWBZXQIOSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=C(F)C=C1 WBIFXMFFIVAUPY-TWBZXQIOSA-N 0.000 description 1
- QCARWGPIMORYHF-TWBZXQIOSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1=CC=CC(F)=C1 QCARWGPIMORYHF-TWBZXQIOSA-N 0.000 description 1
- BZJXAZGCFXWILO-YYPMGOIXSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)C2=C1C=CC=C2 BZJXAZGCFXWILO-YYPMGOIXSA-N 0.000 description 1
- BWRNOUKFHSDXQO-RXIGVHFMSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CC2=C1C=CC=C2 BWRNOUKFHSDXQO-RXIGVHFMSA-N 0.000 description 1
- WWMVEQOGSQLJOL-IXTQLCFBSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2C1C2=C(C=CC=C2)CCC2=C1C=CC=C2 WWMVEQOGSQLJOL-IXTQLCFBSA-N 0.000 description 1
- VQNSIGYZWRJDRA-OSYRGRAESA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CC1 VQNSIGYZWRJDRA-OSYRGRAESA-N 0.000 description 1
- JGLUKSNMPSMHJG-IKBUCZQCSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCC1 JGLUKSNMPSMHJG-IKBUCZQCSA-N 0.000 description 1
- LUQGOTSIYJXRBR-UXWSAQBWSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCC1 LUQGOTSIYJXRBR-UXWSAQBWSA-N 0.000 description 1
- PFNSBWHFPYGQDJ-KXRNXPQJSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H](C1=CC=C(F)C=C1)C1CCCCC1 PFNSBWHFPYGQDJ-KXRNXPQJSA-N 0.000 description 1
- YEPWGEFXXBOUQW-ABSDHOTOSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(=O)O)=C2 YEPWGEFXXBOUQW-ABSDHOTOSA-N 0.000 description 1
- IGWXFYPZYLWRRR-ABSDHOTOSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C(N)=O)=C2 IGWXFYPZYLWRRR-ABSDHOTOSA-N 0.000 description 1
- AIVJLDOGDGLJKV-ABSDHOTOSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 AIVJLDOGDGLJKV-ABSDHOTOSA-N 0.000 description 1
- MJWJOASUEQHBJQ-DPYNCLDUSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(F)=C2 MJWJOASUEQHBJQ-DPYNCLDUSA-N 0.000 description 1
- BQNVBJYCKGGWAO-MOGVHSFPSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@@H]1CCC2=C1C=CC(SOON)=C2 BQNVBJYCKGGWAO-MOGVHSFPSA-N 0.000 description 1
- VQNSIGYZWRJDRA-IXKJJVDVSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CC1 VQNSIGYZWRJDRA-IXKJJVDVSA-N 0.000 description 1
- JGLUKSNMPSMHJG-GUEKSBCBSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCC1 JGLUKSNMPSMHJG-GUEKSBCBSA-N 0.000 description 1
- LUQGOTSIYJXRBR-FBFCCFGDSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCC1 LUQGOTSIYJXRBR-FBFCCFGDSA-N 0.000 description 1
- PFNSBWHFPYGQDJ-KROWKFIASA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H](C1=CC=C(F)C=C1)C1CCCCC1 PFNSBWHFPYGQDJ-KROWKFIASA-N 0.000 description 1
- YEPWGEFXXBOUQW-NHFYAVKLSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(=O)O)=C2 YEPWGEFXXBOUQW-NHFYAVKLSA-N 0.000 description 1
- IGWXFYPZYLWRRR-NHFYAVKLSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C(N)=O)=C2 IGWXFYPZYLWRRR-NHFYAVKLSA-N 0.000 description 1
- AIVJLDOGDGLJKV-NHFYAVKLSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(C1=NN=NN1)=C2 AIVJLDOGDGLJKV-NHFYAVKLSA-N 0.000 description 1
- MJWJOASUEQHBJQ-CGSMEUATSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(F)=C2 MJWJOASUEQHBJQ-CGSMEUATSA-N 0.000 description 1
- BQNVBJYCKGGWAO-JSQIEDONSA-N N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2 Chemical compound N#C[C@@H]1C[C@@H]2CC2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2[C@H]1CCC2=C1C=CC(SOON)=C2 BQNVBJYCKGGWAO-JSQIEDONSA-N 0.000 description 1
- HJHKSFVFRJFFCS-WWMYMODYSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCC/C1=C2\CCC\C2=C\C2=C1CCC2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCC/C1=C2\CCC\C2=C\C2=C1CCC2 HJHKSFVFRJFFCS-WWMYMODYSA-N 0.000 description 1
- GISOSJQEGCEQAB-ALTZYDRJSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 GISOSJQEGCEQAB-ALTZYDRJSA-N 0.000 description 1
- SOKGYQDCJDHEOS-XHWIWGEHSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 SOKGYQDCJDHEOS-XHWIWGEHSA-N 0.000 description 1
- WEXPITJBJBQYOC-ROCBYOHGSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCCCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 WEXPITJBJBQYOC-ROCBYOHGSA-N 0.000 description 1
- LCHYPTSYXUIQRH-XHWIWGEHSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(C(=O)N3CCOCC3)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 LCHYPTSYXUIQRH-XHWIWGEHSA-N 0.000 description 1
- YGHWTEYQLKFIGU-WEHNTMOPSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCC1)=C2 YGHWTEYQLKFIGU-WEHNTMOPSA-N 0.000 description 1
- PDXFBHXXHLGMAC-RHJKKGJFSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCC1)=C2 PDXFBHXXHLGMAC-RHJKKGJFSA-N 0.000 description 1
- GUOSVDLQUKEVEJ-ILKCWXONSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCCCC1)=C2 GUOSVDLQUKEVEJ-ILKCWXONSA-N 0.000 description 1
- XWWFLXUXEWCWDG-RHJKKGJFSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(=O)N1CCOCC1)=C2 XWWFLXUXEWCWDG-RHJKKGJFSA-N 0.000 description 1
- PEUFUMMUZPHSGQ-KGZLQQJFSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NN=NN2)C2=C(C=C(F)C=C2)CCC2=C1C=CC(C(N)=O)=C2 PEUFUMMUZPHSGQ-KGZLQQJFSA-N 0.000 description 1
- MHMCUDHARAVFQK-QTEQDKRBSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 MHMCUDHARAVFQK-QTEQDKRBSA-N 0.000 description 1
- FMHPOPQBKRWSQL-KEYKHBFASA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 FMHPOPQBKRWSQL-KEYKHBFASA-N 0.000 description 1
- XNAXNGCPAKXUOT-MSYCTHLASA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1=C2CCCCC2=CC2=C1CCCC2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCC1=C2CCCCC2=CC2=C1CCCC2 XNAXNGCPAKXUOT-MSYCTHLASA-N 0.000 description 1
- ZZGFGALLTMAHOD-ZJTDPSCFSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)CNCCN1C(=O)C2=C(/C=C\C(Cl)=C/2)NC2=C1C=CC=C2 ZZGFGALLTMAHOD-ZJTDPSCFSA-N 0.000 description 1
- NAUMSTWVSSKVTM-SFMIWVIHSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)[C@@H](N)CN1CC2C[C@H]1C(=O)N2CC1=CC(F)=CC(F)=C1 NAUMSTWVSSKVTM-SFMIWVIHSA-N 0.000 description 1
- CGAOLKDQEGBMLJ-PFFQMSPKSA-N N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1 Chemical compound N#C[C@@H]1C[C@@H]2C[C@@H]2N1C(=O)[C@@H](N)CN1C[C@@H]2C[C@H]1C(=O)N2CC1=CC(F)=CC=C1 CGAOLKDQEGBMLJ-PFFQMSPKSA-N 0.000 description 1
- ALSCEGDXFJIYES-YFKPBYRVSA-N N#C[C@H]1NCCC1 Chemical compound N#C[C@H]1NCCC1 ALSCEGDXFJIYES-YFKPBYRVSA-N 0.000 description 1
- XVLPHPRGHSSWTF-UHFFFAOYSA-N N=C(NC1=CC=C(F)C=C1)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound N=C(NC1=CC=C(F)C=C1)C1(CCN)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 XVLPHPRGHSSWTF-UHFFFAOYSA-N 0.000 description 1
- VLJPJSMVKAZASC-UHFFFAOYSA-O NC(=O)C1=CC2=C(C=C1)C(CC[NH3+])(C1=NN=NN1)C1=C(/C=C(F)\C=C/1)CC2.[Cl-] Chemical compound NC(=O)C1=CC2=C(C=C1)C(CC[NH3+])(C1=NN=NN1)C1=C(/C=C(F)\C=C/1)CC2.[Cl-] VLJPJSMVKAZASC-UHFFFAOYSA-O 0.000 description 1
- WUMOGCPQUNVQMA-UHFFFAOYSA-N NC(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.NCCC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.NCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C(O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.OC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 Chemical compound NC(=O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.NCCC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.NCCC1C2=C(C=CC=C2)CCC2=C1C=CC=C2.O=C(O)CC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2.OC1C2=C(C=CC=C2)C=CC2=C1C=CC=C2 WUMOGCPQUNVQMA-UHFFFAOYSA-N 0.000 description 1
- URHZIKVJKQVHEV-UHFFFAOYSA-N NC(C1CCC1)c(cc1)ccc1F Chemical compound NC(C1CCC1)c(cc1)ccc1F URHZIKVJKQVHEV-UHFFFAOYSA-N 0.000 description 1
- BZJXAZGCFXWILO-AKBLBVQISA-N NC(CN(C[C@H](C1)N2C3c4ccccc4-c4c3cccc4)C1C2=O)C(N(C(C1)C1C1)C1C#N)=O Chemical compound NC(CN(C[C@H](C1)N2C3c4ccccc4-c4c3cccc4)C1C2=O)C(N(C(C1)C1C1)C1C#N)=O BZJXAZGCFXWILO-AKBLBVQISA-N 0.000 description 1
- CGAOLKDQEGBMLJ-FPVUZAAOSA-N NC(CN(C[C@H](C1)N2Cc3cc(F)ccc3)[C@@H]1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O Chemical compound NC(CN(C[C@H](C1)N2Cc3cc(F)ccc3)[C@@H]1C2=O)C(N([C@@H](C1)[C@@H]1C1)C1C#N)=O CGAOLKDQEGBMLJ-FPVUZAAOSA-N 0.000 description 1
- LKKVUBORUGIXGL-NRWPOFLRSA-N NC(c1ccc(C(C[C@H](C2CC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O Chemical compound NC(c1ccc(C(C[C@H](C2CC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O LKKVUBORUGIXGL-NRWPOFLRSA-N 0.000 description 1
- BQDSHYXTIGOBQV-ANWICMFUSA-N NC(c1ccc(C(C[C@H](C2CCC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O Chemical compound NC(c1ccc(C(C[C@H](C2CCC2)NCC(N(CCC2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O BQDSHYXTIGOBQV-ANWICMFUSA-N 0.000 description 1
- KTGZBTINFAMMDA-UWKPJLDSSA-N NC(c1ccc(C(C[C@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O Chemical compound NC(c1ccc(C(C[C@H](C2CCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O KTGZBTINFAMMDA-UWKPJLDSSA-N 0.000 description 1
- RXQIBOWOHSKWIT-HGGCJPGQSA-N NC(c1ccc(C(C[C@H](C2CCCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O Chemical compound NC(c1ccc(C(C[C@H](C2CCCCC2)NCC(N([C@@H](C2)[C@@H]2C2)C2C#N)=O)(c2nnn[nH]2)c(c(CC2)c3)ccc3C(N)=O)c2c1)=O RXQIBOWOHSKWIT-HGGCJPGQSA-N 0.000 description 1
- OBMKZINZPBARIK-UHFFFAOYSA-N NC1(CO)CC1 Chemical compound NC1(CO)CC1 OBMKZINZPBARIK-UHFFFAOYSA-N 0.000 description 1
- VRDXDROAZSDMSA-UHFFFAOYSA-N NC1(CO)CCC1 Chemical compound NC1(CO)CCC1 VRDXDROAZSDMSA-UHFFFAOYSA-N 0.000 description 1
- PDNZJLMPXLQDPL-UHFFFAOYSA-N NC1(CO)CCCC1 Chemical compound NC1(CO)CCCC1 PDNZJLMPXLQDPL-UHFFFAOYSA-N 0.000 description 1
- VDPLLINNMXFNQX-UHFFFAOYSA-N NC1(CO)CCCCC1 Chemical compound NC1(CO)CCCCC1 VDPLLINNMXFNQX-UHFFFAOYSA-N 0.000 description 1
- HNUZOURJSROOBL-UHFFFAOYSA-N NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 Chemical compound NC1CCN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 HNUZOURJSROOBL-UHFFFAOYSA-N 0.000 description 1
- VSTTXWATQKCPGB-UHFFFAOYSA-N NC1CN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 Chemical compound NC1CN(C2C3=C(C=CC=C3)CCC3=C2C=CC=C3)C1 VSTTXWATQKCPGB-UHFFFAOYSA-N 0.000 description 1
- VLVLNNQMURDGPM-UHFFFAOYSA-N NCC1=C(Cl)C=CC=C1Cl Chemical compound NCC1=C(Cl)C=CC=C1Cl VLVLNNQMURDGPM-UHFFFAOYSA-N 0.000 description 1
- QVSVMNXRLWSNGS-UHFFFAOYSA-N NCC1=CC(F)=CC=C1 Chemical compound NCC1=CC(F)=CC=C1 QVSVMNXRLWSNGS-UHFFFAOYSA-N 0.000 description 1
- HYMGBEFLJSAWKE-UHFFFAOYSA-N NCCC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 Chemical compound NCCC(C1=CC=C(C(N)=O)C=C1)(C1=CC=C(C(N)=O)C=C1)C1=NNN=N1 HYMGBEFLJSAWKE-UHFFFAOYSA-N 0.000 description 1
- CIKLLQAOKNDGLZ-UHFFFAOYSA-N NCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C(N)=O)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 CIKLLQAOKNDGLZ-UHFFFAOYSA-N 0.000 description 1
- LSNJQTKZKCMZJN-UHFFFAOYSA-N NCCC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C2=NC(=O)ON2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 LSNJQTKZKCMZJN-UHFFFAOYSA-N 0.000 description 1
- MBAQPSIMYDIWLW-UHFFFAOYSA-N NCCC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C2=NC(=O)ON2C2CCCCC2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 MBAQPSIMYDIWLW-UHFFFAOYSA-N 0.000 description 1
- QTBDEHPFZZVUMV-UHFFFAOYSA-N NCCC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C2=NN(C3=CC=C(F)C=C3)C(=O)N2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 QTBDEHPFZZVUMV-UHFFFAOYSA-N 0.000 description 1
- ZNNKAVMOTQOMQV-UHFFFAOYSA-N NCCC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 Chemical compound NCCC1(C2=NN=C(C3=CC=C(F)C=C3)O2)C2=C(C=C(C(N)=O)C=C2)CCC2=C1C=CC(C(N)=O)=C2 ZNNKAVMOTQOMQV-UHFFFAOYSA-N 0.000 description 1
- SJVAMQKVMZIPBH-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 Chemical compound NCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)C=C2)C2=C1/C=C\C(C(N)=O)=C/2 SJVAMQKVMZIPBH-UHFFFAOYSA-N 0.000 description 1
- YHOAEUDHCROQSP-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 Chemical compound NCCC1(C2=NNN=N2)C2=C(C=C(C(N)=O)S2)CCC2=C1SC(C(N)=O)=C2 YHOAEUDHCROQSP-UHFFFAOYSA-N 0.000 description 1
- PIVZEDRLSBDCMQ-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 Chemical compound NCCC1(C2=NNN=N2)C2=C(C=CS2)CCC2=C1SC(C(N)=O)=C2 PIVZEDRLSBDCMQ-UHFFFAOYSA-N 0.000 description 1
- GRKOMKPEHMWVBS-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 Chemical compound NCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC(C(N)=O)=C2 GRKOMKPEHMWVBS-UHFFFAOYSA-N 0.000 description 1
- GFLKYYDUBNSQRW-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 Chemical compound NCCC1(C2=NNN=N2)C2=C(CCC3=C1C=C(C(N)=O)S3)SC=C2 GFLKYYDUBNSQRW-UHFFFAOYSA-N 0.000 description 1
- AFXMCUGEZASXIB-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 Chemical compound NCCC1(C2=NNN=N2)C2=CC=C(Cl)C=C2CCC2=C1C=CC(Cl)=C2 AFXMCUGEZASXIB-UHFFFAOYSA-N 0.000 description 1
- YRNFBKRKNSNYMH-UHFFFAOYSA-N NCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 Chemical compound NCCC1(C2=NNN=N2)C2CCCCC2CC2CCCCC21 YRNFBKRKNSNYMH-UHFFFAOYSA-N 0.000 description 1
- SHUFOVPKMHBKAM-UHFFFAOYSA-N NCCN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 Chemical compound NCCN1C(=O)C2=C(C=CC=C2)NC2=C1C=C(Cl)C=C2 SHUFOVPKMHBKAM-UHFFFAOYSA-N 0.000 description 1
- BRAAHGXIMQXBLA-UHFFFAOYSA-N NCCNC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2 Chemical compound NCCNC1C2=C(C=C(Cl)C=C2)CCC2=C1N=CC=C2 BRAAHGXIMQXBLA-UHFFFAOYSA-N 0.000 description 1
- LPPXROCVYGCGCQ-UHFFFAOYSA-N NCCNC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 Chemical compound NCCNC1C2=C(C=CC=C2)CCC2=C1N=CC=C2 LPPXROCVYGCGCQ-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N NCCO Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- DPGAUDUNWBAHQC-OVQOIFFZSA-N N[C@@H](CN(CC(C1)N2C(C3CCC3)c(cc3)ccc3F)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O Chemical compound N[C@@H](CN(CC(C1)N2C(C3CCC3)c(cc3)ccc3F)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O DPGAUDUNWBAHQC-OVQOIFFZSA-N 0.000 description 1
- VSLTZGFUCHUVHV-DEJLTHIQSA-N N[C@@H](CN(CC(C1)N2C(C3CCCC3)c(cc3)ccc3F)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O Chemical compound N[C@@H](CN(CC(C1)N2C(C3CCCC3)c(cc3)ccc3F)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O VSLTZGFUCHUVHV-DEJLTHIQSA-N 0.000 description 1
- WCYDWMCNINSTMC-UKGTVQCMSA-N N[C@@H](CN(CC(C1)N2C3c4ccccc4-c4c3cccc4)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O Chemical compound N[C@@H](CN(CC(C1)N2C3c4ccccc4-c4c3cccc4)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O WCYDWMCNINSTMC-UKGTVQCMSA-N 0.000 description 1
- KBMIIKJFDBFSFP-ZIDRKRCISA-N N[C@@H](CN(CC(C1)N2c3cc(C(F)(F)F)cc(C(F)(F)F)c3)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O Chemical compound N[C@@H](CN(CC(C1)N2c3cc(C(F)(F)F)cc(C(F)(F)F)c3)[C@@H]1C2=O)C(N(CCC1)[C@@H]1C#N)=O KBMIIKJFDBFSFP-ZIDRKRCISA-N 0.000 description 1
- KUOUXSFZQGHGKP-JKQORVJESA-N N[C@@H](CN(C[C@@H]1C2)[C@@H]2CN1C(CC1=SC=CC1)=O)C(N(CCC1)[C@@H]1C#N)=O Chemical compound N[C@@H](CN(C[C@@H]1C2)[C@@H]2CN1C(CC1=SC=CC1)=O)C(N(CCC1)[C@@H]1C#N)=O KUOUXSFZQGHGKP-JKQORVJESA-N 0.000 description 1
- SRQPEYLZIUEVIA-QMMMGPOBSA-N N[C@@H](CO)C1=CC=C(F)C=C1 Chemical compound N[C@@H](CO)C1=CC=C(F)C=C1 SRQPEYLZIUEVIA-QMMMGPOBSA-N 0.000 description 1
- LLCCGHDKJXAQLS-YFKPBYRVSA-N N[C@@H](CO)C1CC1 Chemical compound N[C@@H](CO)C1CC1 LLCCGHDKJXAQLS-YFKPBYRVSA-N 0.000 description 1
- PUBFSGQLWVPDER-LURJTMIESA-N N[C@@H](CO)C1CCC1 Chemical compound N[C@@H](CO)C1CCC1 PUBFSGQLWVPDER-LURJTMIESA-N 0.000 description 1
- REQSKJYQZWAJFK-ZETCQYMHSA-N N[C@@H](CO)C1CCCC1 Chemical compound N[C@@H](CO)C1CCCC1 REQSKJYQZWAJFK-ZETCQYMHSA-N 0.000 description 1
- VJJORIDDPKCJTI-QMMMGPOBSA-N N[C@@H](CO)C1CCCCC1 Chemical compound N[C@@H](CO)C1CCCCC1 VJJORIDDPKCJTI-QMMMGPOBSA-N 0.000 description 1
- NHOCAYQCFORHNF-SECBINFHSA-N N[C@@H](CO)CC1=CC=C(F)C=C1 Chemical compound N[C@@H](CO)CC1=CC=C(F)C=C1 NHOCAYQCFORHNF-SECBINFHSA-N 0.000 description 1
- YGLYXCIDXJENDL-SECBINFHSA-N N[C@H](CC1)c(cc2)c1cc2S(N)(=O)=O Chemical compound N[C@H](CC1)c(cc2)c1cc2S(N)(=O)=O YGLYXCIDXJENDL-SECBINFHSA-N 0.000 description 1
- LLCCGHDKJXAQLS-RXMQYKEDSA-N N[C@H](CO)C1CC1 Chemical compound N[C@H](CO)C1CC1 LLCCGHDKJXAQLS-RXMQYKEDSA-N 0.000 description 1
- PUBFSGQLWVPDER-ZCFIWIBFSA-N N[C@H](CO)C1CCC1 Chemical compound N[C@H](CO)C1CCC1 PUBFSGQLWVPDER-ZCFIWIBFSA-N 0.000 description 1
- REQSKJYQZWAJFK-SSDOTTSWSA-N N[C@H](CO)C1CCCC1 Chemical compound N[C@H](CO)C1CCCC1 REQSKJYQZWAJFK-SSDOTTSWSA-N 0.000 description 1
- VJJORIDDPKCJTI-MRVPVSSYSA-N N[C@H](CO)C1CCCCC1 Chemical compound N[C@H](CO)C1CCCCC1 VJJORIDDPKCJTI-MRVPVSSYSA-N 0.000 description 1
- NHOCAYQCFORHNF-VIFPVBQESA-N N[C@H](CO)CC1=CC=C(F)C=C1 Chemical compound N[C@H](CO)CC1=CC=C(F)C=C1 NHOCAYQCFORHNF-VIFPVBQESA-N 0.000 description 1
- QIQITDHWZYEEPA-UHFFFAOYSA-N O=C(Cl)C1=CC=CS1 Chemical compound O=C(Cl)C1=CC=CS1 QIQITDHWZYEEPA-UHFFFAOYSA-N 0.000 description 1
- TXJKATOSKLUITR-UHFFFAOYSA-N O=C(Cl)C1=NC=CN=C1 Chemical compound O=C(Cl)C1=NC=CN=C1 TXJKATOSKLUITR-UHFFFAOYSA-N 0.000 description 1
- AJYXPNIENRLELY-UHFFFAOYSA-N O=C(Cl)CC1=CC=CS1 Chemical compound O=C(Cl)CC1=CC=CS1 AJYXPNIENRLELY-UHFFFAOYSA-N 0.000 description 1
- OKYSUJVCDXZGKE-UHFFFAOYSA-N O=S(=O)(Cl)C1=CC=CC(F)=C1 Chemical compound O=S(=O)(Cl)C1=CC=CC(F)=C1 OKYSUJVCDXZGKE-UHFFFAOYSA-N 0.000 description 1
- VNNLHYZDXIBHKZ-UHFFFAOYSA-N O=S(=O)(Cl)C1=CC=CS1 Chemical compound O=S(=O)(Cl)C1=CC=CS1 VNNLHYZDXIBHKZ-UHFFFAOYSA-N 0.000 description 1
- YSPWSQNKRBSICH-UHFFFAOYSA-N O=S(=O)(Cl)C1=CSC=C1 Chemical compound O=S(=O)(Cl)C1=CSC=C1 YSPWSQNKRBSICH-UHFFFAOYSA-N 0.000 description 1
- QOWBLFJEAHHORF-UHFFFAOYSA-N O=S1(=O)CC2(CC2)CO1 Chemical compound O=S1(=O)CC2(CC2)CO1 QOWBLFJEAHHORF-UHFFFAOYSA-N 0.000 description 1
- TUHYDUGULYFEBZ-UHFFFAOYSA-N O=S1(=O)CC2(CCC2)CO1 Chemical compound O=S1(=O)CC2(CCC2)CO1 TUHYDUGULYFEBZ-UHFFFAOYSA-N 0.000 description 1
- YGFCLRSGMHQUPV-UHFFFAOYSA-N O=S1(=O)CC2(CCCC2)CO1 Chemical compound O=S1(=O)CC2(CCCC2)CO1 YGFCLRSGMHQUPV-UHFFFAOYSA-N 0.000 description 1
- YVTLCIQVULZLFI-UHFFFAOYSA-N O=S1(=O)CC2(CCCCC2)CO1 Chemical compound O=S1(=O)CC2(CCCCC2)CO1 YVTLCIQVULZLFI-UHFFFAOYSA-N 0.000 description 1
- LWKZBQLKBUPUKF-QMMMGPOBSA-N O=S1(=O)C[C@@H](C2=CC=C(F)C=C2)CO1 Chemical compound O=S1(=O)C[C@@H](C2=CC=C(F)C=C2)CO1 LWKZBQLKBUPUKF-QMMMGPOBSA-N 0.000 description 1
- JZIGCIVOEMHEPD-LURJTMIESA-N O=S1(=O)C[C@@H](C2CC2)CO1 Chemical compound O=S1(=O)C[C@@H](C2CC2)CO1 JZIGCIVOEMHEPD-LURJTMIESA-N 0.000 description 1
- KLGZWCBZVGJTOZ-ZETCQYMHSA-N O=S1(=O)C[C@@H](C2CCC2)CO1 Chemical compound O=S1(=O)C[C@@H](C2CCC2)CO1 KLGZWCBZVGJTOZ-ZETCQYMHSA-N 0.000 description 1
- GOXXLAUMUTZUIL-QMMMGPOBSA-N O=S1(=O)C[C@@H](C2CCCC2)CO1 Chemical compound O=S1(=O)C[C@@H](C2CCCC2)CO1 GOXXLAUMUTZUIL-QMMMGPOBSA-N 0.000 description 1
- XIFBWZFUAXWVFC-VIFPVBQESA-N O=S1(=O)C[C@@H](C2CCCCC2)CO1 Chemical compound O=S1(=O)C[C@@H](C2CCCCC2)CO1 XIFBWZFUAXWVFC-VIFPVBQESA-N 0.000 description 1
- NMQQSRZLBMEPCN-VIFPVBQESA-N O=S1(=O)C[C@@H](CC2=CC=C(F)C=C2)CO1 Chemical compound O=S1(=O)C[C@@H](CC2=CC=C(F)C=C2)CO1 NMQQSRZLBMEPCN-VIFPVBQESA-N 0.000 description 1
- LWKZBQLKBUPUKF-MRVPVSSYSA-N O=S1(=O)C[C@H](C2=CC=C(F)C=C2)CO1 Chemical compound O=S1(=O)C[C@H](C2=CC=C(F)C=C2)CO1 LWKZBQLKBUPUKF-MRVPVSSYSA-N 0.000 description 1
- JZIGCIVOEMHEPD-ZCFIWIBFSA-N O=S1(=O)C[C@H](C2CC2)CO1 Chemical compound O=S1(=O)C[C@H](C2CC2)CO1 JZIGCIVOEMHEPD-ZCFIWIBFSA-N 0.000 description 1
- KLGZWCBZVGJTOZ-SSDOTTSWSA-N O=S1(=O)C[C@H](C2CCC2)CO1 Chemical compound O=S1(=O)C[C@H](C2CCC2)CO1 KLGZWCBZVGJTOZ-SSDOTTSWSA-N 0.000 description 1
- GOXXLAUMUTZUIL-MRVPVSSYSA-N O=S1(=O)C[C@H](C2CCCC2)CO1 Chemical compound O=S1(=O)C[C@H](C2CCCC2)CO1 GOXXLAUMUTZUIL-MRVPVSSYSA-N 0.000 description 1
- XIFBWZFUAXWVFC-SECBINFHSA-N O=S1(=O)C[C@H](C2CCCCC2)CO1 Chemical compound O=S1(=O)C[C@H](C2CCCCC2)CO1 XIFBWZFUAXWVFC-SECBINFHSA-N 0.000 description 1
- NMQQSRZLBMEPCN-SECBINFHSA-N O=S1(=O)C[C@H](CC2=CC=C(F)C=C2)CO1 Chemical compound O=S1(=O)C[C@H](CC2=CC=C(F)C=C2)CO1 NMQQSRZLBMEPCN-SECBINFHSA-N 0.000 description 1
- KOOCXEPDDCFBDZ-CMOCDZPBSA-N [C-]#[N+]C1=CC(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 Chemical compound [C-]#[N+]C1=CC(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)=CC=C1 KOOCXEPDDCFBDZ-CMOCDZPBSA-N 0.000 description 1
- GULGSVKUZUYHLP-SICDYIMMSA-N [C-]#[N+]C1=CC(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 Chemical compound [C-]#[N+]C1=CC(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)=CC=C1 GULGSVKUZUYHLP-SICDYIMMSA-N 0.000 description 1
- SLIXYTZHFZUAQM-ZWKXJTFZSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 SLIXYTZHFZUAQM-ZWKXJTFZSA-N 0.000 description 1
- PLNILPBLANSILQ-XYYCENQHSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 PLNILPBLANSILQ-XYYCENQHSA-N 0.000 description 1
- SECXUCFOKLSHBV-HPGPFUFHSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 SECXUCFOKLSHBV-HPGPFUFHSA-N 0.000 description 1
- DHOMGIQABMWIGV-OAYPFKJJSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 DHOMGIQABMWIGV-OAYPFKJJSA-N 0.000 description 1
- SLIXYTZHFZUAQM-FZRUSQKFSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1C3C[C@H]3C[C@H]1C#N)CC2 SLIXYTZHFZUAQM-FZRUSQKFSA-N 0.000 description 1
- PLNILPBLANSILQ-UGKANONSSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](N)C(=O)N1CCC[C@H]1C#N)CC2 PLNILPBLANSILQ-UGKANONSSA-N 0.000 description 1
- SECXUCFOKLSHBV-WFYLZJEKSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C#N)CC2 SECXUCFOKLSHBV-WFYLZJEKSA-N 0.000 description 1
- DHOMGIQABMWIGV-HSSJXSNNSA-N [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 Chemical compound [C-]#[N+]C1=CC2=C(C=C1)[C@H](N1C(=O)[C@@H]3C[C@H]1CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)CC2 DHOMGIQABMWIGV-HSSJXSNNSA-N 0.000 description 1
- STEYRXANPAQSHQ-RNLLHNEJSA-N [C-]#[N+]C1=CC=C(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(CN2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 STEYRXANPAQSHQ-RNLLHNEJSA-N 0.000 description 1
- LOXIAUXPLSRGCJ-CMOCDZPBSA-N [C-]#[N+]C1=CC=C(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(CN2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 LOXIAUXPLSRGCJ-CMOCDZPBSA-N 0.000 description 1
- CPLGIOPATSSOCQ-RDTQZZCZSA-N [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 CPLGIOPATSSOCQ-RDTQZZCZSA-N 0.000 description 1
- AWRYFXJVQKEFPT-XSLAGTTESA-N [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 AWRYFXJVQKEFPT-XSLAGTTESA-N 0.000 description 1
- YTTDFNRRGUWPRO-TUFLPTIASA-N [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 YTTDFNRRGUWPRO-TUFLPTIASA-N 0.000 description 1
- RKLGIPYNFSFZPB-HHHIRMLJSA-N [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound [C-]#[N+]C1=CC=C(N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 RKLGIPYNFSFZPB-HHHIRMLJSA-N 0.000 description 1
- WLBAXBKXVMOCIQ-DGFOKANRSA-N [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 WLBAXBKXVMOCIQ-DGFOKANRSA-N 0.000 description 1
- KZHKXIWKYLARTD-SSRYDLFMSA-N [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 KZHKXIWKYLARTD-SSRYDLFMSA-N 0.000 description 1
- PQLXFORTEZJPOV-GLBNMMOUSA-N [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 PQLXFORTEZJPOV-GLBNMMOUSA-N 0.000 description 1
- GDIARGJUDPOANG-HSQVRJBSSA-N [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 GDIARGJUDPOANG-HSQVRJBSSA-N 0.000 description 1
- KZHKXIWKYLARTD-MJYLKRGBSA-N [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3CC2CN3C[C@H](N)C(=O)N2CCC[C@H]2C#N)C=C1 KZHKXIWKYLARTD-MJYLKRGBSA-N 0.000 description 1
- WLBAXBKXVMOCIQ-VGDVNGIFSA-N [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](N)C(=O)N2C3C[C@H]3C[C@H]2C#N)C=C1 WLBAXBKXVMOCIQ-VGDVNGIFSA-N 0.000 description 1
- PQLXFORTEZJPOV-RSYUGDCRSA-N [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2CCC[C@H]2C#N)C=C1 PQLXFORTEZJPOV-RSYUGDCRSA-N 0.000 description 1
- GDIARGJUDPOANG-ZPMVUVLNSA-N [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 Chemical compound [C-]#[N+]C1=CC=C([C@H](C)N2C(=O)[C@@H]3C[C@H]2CN3C[C@H](NC(=O)OC(C)(C)C)C(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C=C1 GDIARGJUDPOANG-ZPMVUVLNSA-N 0.000 description 1
- XIZCKQLQGNIBKU-LASXONHFSA-N [C-]#[N+][C@@H]1CCCN1C(=O)/C=C/C(=O)N1CCC[C@H]1C Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)/C=C/C(=O)N1CCC[C@H]1C XIZCKQLQGNIBKU-LASXONHFSA-N 0.000 description 1
- QOZQDUMKYMHAPU-MBSDFSHPSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C(N)=O)C2=CC=CC=C2CCC2=C1/C=C\C=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C(N)=O)C2=CC=CC=C2CCC2=C1/C=C\C=C/2 QOZQDUMKYMHAPU-MBSDFSHPSA-N 0.000 description 1
- YWXQLCLARWYDPJ-LKJQRHSJSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)C3CCOCC3)C=C2CCC2=C1/C=C\C(C(=O)N1CCOCC1)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)C3CCOCC3)C=C2CCC2=C1/C=C\C(C(=O)N1CCOCC1)=C/2 YWXQLCLARWYDPJ-LKJQRHSJSA-N 0.000 description 1
- MJMGWMQJHUUWDE-ZTOMLWHTSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCC3)C=C2CCC2=C1/C=C\C(C(=O)N1CCC1)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)N3CCC3)C=C2CCC2=C1/C=C\C(C(=O)N1CCC1)=C/2 MJMGWMQJHUUWDE-ZTOMLWHTSA-N 0.000 description 1
- ORGDGFIIXDMKKT-DCFHFQCYSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)NCC)C=C2CCC2=C1/C=C\C(C(=O)NCC)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)NCC)C=C2CCC2=C1/C=C\C(C(=O)NCC)=C/2 ORGDGFIIXDMKKT-DCFHFQCYSA-N 0.000 description 1
- XXALIBMACONIDN-HJPURHCSSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1/C=C\C(C(=O)O)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)O)C=C2CCC2=C1/C=C\C(C(=O)O)=C/2 XXALIBMACONIDN-HJPURHCSSA-N 0.000 description 1
- GIXOTAVABHUCBH-BVZFJXPGSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)OC)C=C2CCC2=C1/C=C\C(C(=O)OC)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(=O)OC)C=C2CCC2=C1/C=C\C(C(=O)OC)=C/2 GIXOTAVABHUCBH-BVZFJXPGSA-N 0.000 description 1
- DVDAMHOSSACNSY-HJPURHCSSA-N [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 Chemical compound [C-]#[N+][C@@H]1CCCN1C(=O)CN[C@@H](C)CC1(C2=NNN=N2)C2=CC=C(C(N)=O)C=C2CCC2=C1/C=C\C(C(N)=O)=C/2 DVDAMHOSSACNSY-HJPURHCSSA-N 0.000 description 1
- QYTXWXRLCSQBMK-HTZGQBTDSA-N [C-]#[N+][C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H]2C[C@H]2C[C@H]1C Chemical compound [C-]#[N+][C@@H]1C[C@@H]2C[C@@H]2N1C(=O)/C=C/C(=O)N1[C@H]2C[C@H]2C[C@H]1C QYTXWXRLCSQBMK-HTZGQBTDSA-N 0.000 description 1
- UERMJOKSHMUMSK-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 UERMJOKSHMUMSK-QHCPKHFHSA-N 0.000 description 1
- JMEQENKBVMPFIN-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 JMEQENKBVMPFIN-UMFSSWHCSA-N 0.000 description 1
- QQWVCBOBUSORFV-UHFFFAOYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 QQWVCBOBUSORFV-UHFFFAOYSA-N 0.000 description 1
- TZLGASOVCMYKBC-UHFFFAOYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 TZLGASOVCMYKBC-UHFFFAOYSA-N 0.000 description 1
- YVONOFZIGSXZBJ-UHFFFAOYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 YVONOFZIGSXZBJ-UHFFFAOYSA-N 0.000 description 1
- STDZAXODGFAJIR-UHFFFAOYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 STDZAXODGFAJIR-UHFFFAOYSA-N 0.000 description 1
- IKRJDKHSSASFCU-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IKRJDKHSSASFCU-QHCPKHFHSA-N 0.000 description 1
- XYECRHLLKCSVEU-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 XYECRHLLKCSVEU-DEOSSOPVSA-N 0.000 description 1
- TYMLBAHQIGXADC-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 TYMLBAHQIGXADC-VWLOTQADSA-N 0.000 description 1
- IRPOTHQRZNWZKS-SANMLTNESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IRPOTHQRZNWZKS-SANMLTNESA-N 0.000 description 1
- INASAZTVWNTVQT-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 INASAZTVWNTVQT-UMFSSWHCSA-N 0.000 description 1
- QPJXBGLJYDIRQE-DXBVXKBHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 QPJXBGLJYDIRQE-DXBVXKBHSA-N 0.000 description 1
- KNSLWCIPVPFTOS-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 KNSLWCIPVPFTOS-QQNWGBJXSA-N 0.000 description 1
- SYYAZQMFRLQTKV-ALTZYDRJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SYYAZQMFRLQTKV-ALTZYDRJSA-N 0.000 description 1
- DCAIVIFFSYLDDH-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DCAIVIFFSYLDDH-VWLOTQADSA-N 0.000 description 1
- RVNXZFHXAJVFOE-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 RVNXZFHXAJVFOE-QHCPKHFHSA-N 0.000 description 1
- RCNBNRCQFCJCBA-NDEPHWFRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 RCNBNRCQFCJCBA-NDEPHWFRSA-N 0.000 description 1
- ZSKQDJSZFGGTKW-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ZSKQDJSZFGGTKW-QFIPXVFZSA-N 0.000 description 1
- NSAPICUFAVZFJG-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NSAPICUFAVZFJG-QFIPXVFZSA-N 0.000 description 1
- AMAIYOCDPOGQHE-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 AMAIYOCDPOGQHE-QFIPXVFZSA-N 0.000 description 1
- JAZZPYWAHLLUCP-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 JAZZPYWAHLLUCP-VWLOTQADSA-N 0.000 description 1
- NLJQGOJXDXORDR-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NLJQGOJXDXORDR-QHCPKHFHSA-N 0.000 description 1
- BBPGNQZKHHCHOH-NDEPHWFRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BBPGNQZKHHCHOH-NDEPHWFRSA-N 0.000 description 1
- HUKJJNPWPABHQQ-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 HUKJJNPWPABHQQ-VWLOTQADSA-N 0.000 description 1
- BXWVWTRYGGSARS-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BXWVWTRYGGSARS-QHCPKHFHSA-N 0.000 description 1
- NSSVTYLOJAYKPO-VWLOTQADSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NSSVTYLOJAYKPO-VWLOTQADSA-N 0.000 description 1
- HRBBBZMBGHITLN-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 HRBBBZMBGHITLN-DEOSSOPVSA-N 0.000 description 1
- MCEBHTYHKYDZRD-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 MCEBHTYHKYDZRD-DEOSSOPVSA-N 0.000 description 1
- DXMQNQHNIXTTQP-NDEPHWFRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DXMQNQHNIXTTQP-NDEPHWFRSA-N 0.000 description 1
- CVHVCLKEYANPCP-NDEPHWFRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 CVHVCLKEYANPCP-NDEPHWFRSA-N 0.000 description 1
- XEDABNLEGDKYKC-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 XEDABNLEGDKYKC-DEOSSOPVSA-N 0.000 description 1
- FHDPHQJXXUUKAQ-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 FHDPHQJXXUUKAQ-QHCPKHFHSA-N 0.000 description 1
- PDXCWRRCUHYYEC-LWAJAQLZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 PDXCWRRCUHYYEC-LWAJAQLZSA-N 0.000 description 1
- UQBCOYJHXPDJFT-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 UQBCOYJHXPDJFT-QFIPXVFZSA-N 0.000 description 1
- YKDWRPSQHMNJRS-NRFANRHFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 YKDWRPSQHMNJRS-NRFANRHFSA-N 0.000 description 1
- ZTSUBELRYCXUJZ-SFHVURJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 ZTSUBELRYCXUJZ-SFHVURJKSA-N 0.000 description 1
- NLWWXQJZLSGSQP-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NLWWXQJZLSGSQP-SLSDLSHTSA-N 0.000 description 1
- YFFPTQDGCSAPOJ-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YFFPTQDGCSAPOJ-QQNWGBJXSA-N 0.000 description 1
- GOMVATFEHTXBNA-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 GOMVATFEHTXBNA-UMFSSWHCSA-N 0.000 description 1
- PXFPRNKAXJTALX-VUAYVDPTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 PXFPRNKAXJTALX-VUAYVDPTSA-N 0.000 description 1
- PDNVEAKOPUHPNA-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 PDNVEAKOPUHPNA-SLSDLSHTSA-N 0.000 description 1
- HXJFCPREAYHEJI-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 HXJFCPREAYHEJI-SLSDLSHTSA-N 0.000 description 1
- AIQYFAXYTBWNMB-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AIQYFAXYTBWNMB-SLSDLSHTSA-N 0.000 description 1
- LMBIEJDNMZAFFO-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 LMBIEJDNMZAFFO-QQNWGBJXSA-N 0.000 description 1
- AYTSJDINKXIJAV-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AYTSJDINKXIJAV-UMFSSWHCSA-N 0.000 description 1
- AWQWJUIRPBGGAI-RDLJJFNOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AWQWJUIRPBGGAI-RDLJJFNOSA-N 0.000 description 1
- HUSRPSSOBRCOMP-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 HUSRPSSOBRCOMP-QQNWGBJXSA-N 0.000 description 1
- KSZPHJNNRJWQIS-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 KSZPHJNNRJWQIS-UMFSSWHCSA-N 0.000 description 1
- AAEXDPAGYLDYFQ-VUAYVDPTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AAEXDPAGYLDYFQ-VUAYVDPTSA-N 0.000 description 1
- ZXVITHZYBJAGLF-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 ZXVITHZYBJAGLF-QQNWGBJXSA-N 0.000 description 1
- OUKSURQBMQSQSP-QQNWGBJXSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 OUKSURQBMQSQSP-QQNWGBJXSA-N 0.000 description 1
- DWXMEVCYFHBNPL-DXBVXKBHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DWXMEVCYFHBNPL-DXBVXKBHSA-N 0.000 description 1
- QIVQQSDWLQNNSF-DXBVXKBHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 QIVQQSDWLQNNSF-DXBVXKBHSA-N 0.000 description 1
- CCWWQHCULYZFKT-RDLJJFNOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CCWWQHCULYZFKT-RDLJJFNOSA-N 0.000 description 1
- ADZYHSNSFDRJHQ-RDLJJFNOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 ADZYHSNSFDRJHQ-RDLJJFNOSA-N 0.000 description 1
- CWIBUZUUWVNEGV-DXBVXKBHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CWIBUZUUWVNEGV-DXBVXKBHSA-N 0.000 description 1
- JTJFCVFOLUXWCJ-UMFSSWHCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 JTJFCVFOLUXWCJ-UMFSSWHCSA-N 0.000 description 1
- MQYRSKHLBYUSPX-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 MQYRSKHLBYUSPX-SLSDLSHTSA-N 0.000 description 1
- CVRPPDPVQVWSNN-ADBPKCOUSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 CVRPPDPVQVWSNN-ADBPKCOUSA-N 0.000 description 1
- IVXJPQXFYOYYIC-SLSDLSHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IVXJPQXFYOYYIC-SLSDLSHTSA-N 0.000 description 1
- GIUMXENHAZNPIT-DPLHUUCSSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 GIUMXENHAZNPIT-DPLHUUCSSA-N 0.000 description 1
- MEUVCTSXCKAQKO-STSQHVNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 MEUVCTSXCKAQKO-STSQHVNTSA-N 0.000 description 1
- YRXWGVCQEFEUOX-CYBMUJFWSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 YRXWGVCQEFEUOX-CYBMUJFWSA-N 0.000 description 1
- NYRGQYKVYMFBEM-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NYRGQYKVYMFBEM-JPYJTQIMSA-N 0.000 description 1
- ZRHHXFGDSAILBY-RLWLMLJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ZRHHXFGDSAILBY-RLWLMLJZSA-N 0.000 description 1
- DWKBDGDYVUKWIW-MNNSJKJDSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DWKBDGDYVUKWIW-MNNSJKJDSA-N 0.000 description 1
- BWOJBLUIUNZILT-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BWOJBLUIUNZILT-JPYJTQIMSA-N 0.000 description 1
- SMIHNOUDCSGVCA-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 SMIHNOUDCSGVCA-JPYJTQIMSA-N 0.000 description 1
- UVPQVLUCZWHQMV-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 UVPQVLUCZWHQMV-JPYJTQIMSA-N 0.000 description 1
- BDUNSRJTNCLONN-RLWLMLJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BDUNSRJTNCLONN-RLWLMLJZSA-N 0.000 description 1
- WPIKXGHAKIVCNR-DVECYGJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 WPIKXGHAKIVCNR-DVECYGJZSA-N 0.000 description 1
- QTWOHPLHKHGMKP-BTYSJIOQSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 QTWOHPLHKHGMKP-BTYSJIOQSA-N 0.000 description 1
- LXNZOBHSEPTMGU-RLWLMLJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 LXNZOBHSEPTMGU-RLWLMLJZSA-N 0.000 description 1
- PENVEAZALBWALH-DVECYGJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PENVEAZALBWALH-DVECYGJZSA-N 0.000 description 1
- MOEFEJFFXVWKEJ-MNNSJKJDSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 MOEFEJFFXVWKEJ-MNNSJKJDSA-N 0.000 description 1
- LQZLBXMGPMWOQC-RLWLMLJZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 LQZLBXMGPMWOQC-RLWLMLJZSA-N 0.000 description 1
- RZDZAFAEMPERID-CLOONOSVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 RZDZAFAEMPERID-CLOONOSVSA-N 0.000 description 1
- PBKWOBZOWKHRQQ-CLOONOSVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PBKWOBZOWKHRQQ-CLOONOSVSA-N 0.000 description 1
- OUQCALSCDFDRQR-MNNSJKJDSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 OUQCALSCDFDRQR-MNNSJKJDSA-N 0.000 description 1
- NTAJHAWNASPKGG-MNNSJKJDSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NTAJHAWNASPKGG-MNNSJKJDSA-N 0.000 description 1
- PLVSTLGJVDUQAP-NLFFAJNJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PLVSTLGJVDUQAP-NLFFAJNJSA-N 0.000 description 1
- DGNVZAGZUZDSBP-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DGNVZAGZUZDSBP-JPYJTQIMSA-N 0.000 description 1
- KPMJDYFVOZYRFN-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 KPMJDYFVOZYRFN-JPYJTQIMSA-N 0.000 description 1
- RBVOSIMVZCCEEA-VGSWGCGISA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 RBVOSIMVZCCEEA-VGSWGCGISA-N 0.000 description 1
- LGQWSPLUELRBGR-APWZRJJASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 LGQWSPLUELRBGR-APWZRJJASA-N 0.000 description 1
- QOESXUJJGGPCMR-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 QOESXUJJGGPCMR-HLCOLNNTSA-N 0.000 description 1
- BHFMPMYZKBDFRW-ZLGCBXFLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BHFMPMYZKBDFRW-ZLGCBXFLSA-N 0.000 description 1
- IJYZJNVWXKUMGG-PFCVVTBNSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IJYZJNVWXKUMGG-PFCVVTBNSA-N 0.000 description 1
- NPBZHJDQODGSDI-IMVAWRIHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NPBZHJDQODGSDI-IMVAWRIHSA-N 0.000 description 1
- HNCJDTLLBBJWCS-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 HNCJDTLLBBJWCS-HLCOLNNTSA-N 0.000 description 1
- KECCBBWFLRLGDD-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 KECCBBWFLRLGDD-HLCOLNNTSA-N 0.000 description 1
- VMNBCJOHVCNPQC-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 VMNBCJOHVCNPQC-HLCOLNNTSA-N 0.000 description 1
- FEDJPMWHUPKBTH-ZLGCBXFLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 FEDJPMWHUPKBTH-ZLGCBXFLSA-N 0.000 description 1
- BCARHSOASONIRZ-PFCVVTBNSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BCARHSOASONIRZ-PFCVVTBNSA-N 0.000 description 1
- XLBFAVDHYWGXKH-WKECTITQSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 XLBFAVDHYWGXKH-WKECTITQSA-N 0.000 description 1
- NPUVHHLESGNYRE-ZLGCBXFLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NPUVHHLESGNYRE-ZLGCBXFLSA-N 0.000 description 1
- YUFDHIVRXICRCA-PFCVVTBNSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YUFDHIVRXICRCA-PFCVVTBNSA-N 0.000 description 1
- COJRUNZDXWFFAC-IMVAWRIHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 COJRUNZDXWFFAC-IMVAWRIHSA-N 0.000 description 1
- TXXREBHZNHSEMM-ZLGCBXFLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 TXXREBHZNHSEMM-ZLGCBXFLSA-N 0.000 description 1
- IDIZRPVPASFHQL-ZLGCBXFLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IDIZRPVPASFHQL-ZLGCBXFLSA-N 0.000 description 1
- LNVPOGSLDAFVOJ-RFRFBGJFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 LNVPOGSLDAFVOJ-RFRFBGJFSA-N 0.000 description 1
- YPHFYHARCLZTSN-RFRFBGJFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YPHFYHARCLZTSN-RFRFBGJFSA-N 0.000 description 1
- LUEPMPMQVQKQKZ-UWPUCTATSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 LUEPMPMQVQKQKZ-UWPUCTATSA-N 0.000 description 1
- YQIIDHHZCLCARN-UWPUCTATSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YQIIDHHZCLCARN-UWPUCTATSA-N 0.000 description 1
- MXIXRGLCYGXCMC-SDXKGTORSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 MXIXRGLCYGXCMC-SDXKGTORSA-N 0.000 description 1
- RCZUJWVTWREGQL-PFCVVTBNSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 RCZUJWVTWREGQL-PFCVVTBNSA-N 0.000 description 1
- SUJKENGVFFHXJR-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SUJKENGVFFHXJR-HLCOLNNTSA-N 0.000 description 1
- SKJWFZTTZYHCCI-HLCOLNNTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SKJWFZTTZYHCCI-HLCOLNNTSA-N 0.000 description 1
- GEGMZCJRFIMKMZ-MOMMTWFBSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 GEGMZCJRFIMKMZ-MOMMTWFBSA-N 0.000 description 1
- DDNRADHVUJSPRG-RSOWOLOUSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 DDNRADHVUJSPRG-RSOWOLOUSA-N 0.000 description 1
- SBZBKJREBIHEHP-RPWUZVMVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SBZBKJREBIHEHP-RPWUZVMVSA-N 0.000 description 1
- DXUMSLOZZNCKES-INADMSBESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DXUMSLOZZNCKES-INADMSBESA-N 0.000 description 1
- GYIDYVRQVKZESE-WDYNHAJCSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 GYIDYVRQVKZESE-WDYNHAJCSA-N 0.000 description 1
- BGWKAHIJQXUTAX-JIPLTUAOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BGWKAHIJQXUTAX-JIPLTUAOSA-N 0.000 description 1
- DYRCBUBOQOZBAF-OAQYLSRUSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DYRCBUBOQOZBAF-OAQYLSRUSA-N 0.000 description 1
- AIDYLQGZZNUVNU-OAQYLSRUSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 AIDYLQGZZNUVNU-OAQYLSRUSA-N 0.000 description 1
- CAWIKPCTAKMYRP-XMMPIXPASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 CAWIKPCTAKMYRP-XMMPIXPASA-N 0.000 description 1
- FZXGCQKRXDEIMC-OAQYLSRUSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 FZXGCQKRXDEIMC-OAQYLSRUSA-N 0.000 description 1
- MHWLIGXAWQSDRB-JOCHJYFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 MHWLIGXAWQSDRB-JOCHJYFZSA-N 0.000 description 1
- FJBLDGJRJVYMTD-HSZRJFAPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 FJBLDGJRJVYMTD-HSZRJFAPSA-N 0.000 description 1
- WPWNUBPRYCLHNI-XMMPIXPASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 WPWNUBPRYCLHNI-XMMPIXPASA-N 0.000 description 1
- ANWOLDXBHGNBHM-SFHVURJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ANWOLDXBHGNBHM-SFHVURJKSA-N 0.000 description 1
- VKPVMHDTQZFWIP-QHCPKHFHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 VKPVMHDTQZFWIP-QHCPKHFHSA-N 0.000 description 1
- TZHXNYYVOLADTE-RPLLCQBOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 TZHXNYYVOLADTE-RPLLCQBOSA-N 0.000 description 1
- DONREFRRKVKWDM-RPLLCQBOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DONREFRRKVKWDM-RPLLCQBOSA-N 0.000 description 1
- AFDRHBSSZDFWTF-BHBYDHKZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AFDRHBSSZDFWTF-BHBYDHKZSA-N 0.000 description 1
- CBEMHROLMGKXBG-RPLLCQBOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CBEMHROLMGKXBG-RPLLCQBOSA-N 0.000 description 1
- BDQSYTNTTGRSQG-LBNVMWSVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BDQSYTNTTGRSQG-LBNVMWSVSA-N 0.000 description 1
- NNGUBCJLUYIRIZ-LITSAYRRSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NNGUBCJLUYIRIZ-LITSAYRRSA-N 0.000 description 1
- JVDMVCSRSGGEFL-BHBYDHKZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 JVDMVCSRSGGEFL-BHBYDHKZSA-N 0.000 description 1
- BJADJIOPJDVDOB-YFQNEARWSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BJADJIOPJDVDOB-YFQNEARWSA-N 0.000 description 1
- WREKYHOFMYQWRS-CUFBHMEESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 WREKYHOFMYQWRS-CUFBHMEESA-N 0.000 description 1
- CMPPNWGOQTYOGV-XSVIJKGOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CMPPNWGOQTYOGV-XSVIJKGOSA-N 0.000 description 1
- YRNYZQBFRCSRDZ-CUFBHMEESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YRNYZQBFRCSRDZ-CUFBHMEESA-N 0.000 description 1
- AVSVNBGYRHQURE-BVIJCPSYSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AVSVNBGYRHQURE-BVIJCPSYSA-N 0.000 description 1
- CWVCHHGDLZTVHI-ZIMFAVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CWVCHHGDLZTVHI-ZIMFAVFZSA-N 0.000 description 1
- WODQWRGJZMYWBF-FSRIWSFDSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 WODQWRGJZMYWBF-FSRIWSFDSA-N 0.000 description 1
- NYRGQYKVYMFBEM-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NYRGQYKVYMFBEM-MBSDFSHPSA-N 0.000 description 1
- ZRHHXFGDSAILBY-LVXARBLLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ZRHHXFGDSAILBY-LVXARBLLSA-N 0.000 description 1
- SUPXJLUNBSVPDI-CYFREDJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 SUPXJLUNBSVPDI-CYFREDJKSA-N 0.000 description 1
- DWKBDGDYVUKWIW-ZTOMLWHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DWKBDGDYVUKWIW-ZTOMLWHTSA-N 0.000 description 1
- BWOJBLUIUNZILT-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BWOJBLUIUNZILT-MBSDFSHPSA-N 0.000 description 1
- SMIHNOUDCSGVCA-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 SMIHNOUDCSGVCA-MBSDFSHPSA-N 0.000 description 1
- UVPQVLUCZWHQMV-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 UVPQVLUCZWHQMV-MBSDFSHPSA-N 0.000 description 1
- BDUNSRJTNCLONN-LVXARBLLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 BDUNSRJTNCLONN-LVXARBLLSA-N 0.000 description 1
- WPIKXGHAKIVCNR-CYFREDJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 WPIKXGHAKIVCNR-CYFREDJKSA-N 0.000 description 1
- QTWOHPLHKHGMKP-IADCTJSHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 QTWOHPLHKHGMKP-IADCTJSHSA-N 0.000 description 1
- LXNZOBHSEPTMGU-LVXARBLLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 LXNZOBHSEPTMGU-LVXARBLLSA-N 0.000 description 1
- PENVEAZALBWALH-CYFREDJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PENVEAZALBWALH-CYFREDJKSA-N 0.000 description 1
- MOEFEJFFXVWKEJ-ZTOMLWHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 MOEFEJFFXVWKEJ-ZTOMLWHTSA-N 0.000 description 1
- HIFDUBSUHUAZCZ-LVXARBLLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 HIFDUBSUHUAZCZ-LVXARBLLSA-N 0.000 description 1
- LQZLBXMGPMWOQC-LVXARBLLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 LQZLBXMGPMWOQC-LVXARBLLSA-N 0.000 description 1
- RZDZAFAEMPERID-DFBJGRDBSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 RZDZAFAEMPERID-DFBJGRDBSA-N 0.000 description 1
- PBKWOBZOWKHRQQ-DFBJGRDBSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PBKWOBZOWKHRQQ-DFBJGRDBSA-N 0.000 description 1
- OUQCALSCDFDRQR-ZTOMLWHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 OUQCALSCDFDRQR-ZTOMLWHTSA-N 0.000 description 1
- NTAJHAWNASPKGG-ZTOMLWHTSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 NTAJHAWNASPKGG-ZTOMLWHTSA-N 0.000 description 1
- PLVSTLGJVDUQAP-CPJSRVTESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 PLVSTLGJVDUQAP-CPJSRVTESA-N 0.000 description 1
- YMIYEVNBPLHDFI-CYFREDJKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 YMIYEVNBPLHDFI-CYFREDJKSA-N 0.000 description 1
- KPMJDYFVOZYRFN-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 KPMJDYFVOZYRFN-MBSDFSHPSA-N 0.000 description 1
- RBVOSIMVZCCEEA-JTSKRJEESA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 RBVOSIMVZCCEEA-JTSKRJEESA-N 0.000 description 1
- LGQWSPLUELRBGR-LPHOPBHVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 LGQWSPLUELRBGR-LPHOPBHVSA-N 0.000 description 1
- AITLENNQIIMRDM-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C=N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C=N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 AITLENNQIIMRDM-MBSDFSHPSA-N 0.000 description 1
- QOESXUJJGGPCMR-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 QOESXUJJGGPCMR-IAIYQMRASA-N 0.000 description 1
- BHFMPMYZKBDFRW-YIPSIMJMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BHFMPMYZKBDFRW-YIPSIMJMSA-N 0.000 description 1
- IJYZJNVWXKUMGG-PUSCERABSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IJYZJNVWXKUMGG-PUSCERABSA-N 0.000 description 1
- NPBZHJDQODGSDI-WCBIRZFKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NPBZHJDQODGSDI-WCBIRZFKSA-N 0.000 description 1
- HNCJDTLLBBJWCS-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 HNCJDTLLBBJWCS-IAIYQMRASA-N 0.000 description 1
- KECCBBWFLRLGDD-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 KECCBBWFLRLGDD-IAIYQMRASA-N 0.000 description 1
- VMNBCJOHVCNPQC-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 VMNBCJOHVCNPQC-IAIYQMRASA-N 0.000 description 1
- FEDJPMWHUPKBTH-YIPSIMJMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 FEDJPMWHUPKBTH-YIPSIMJMSA-N 0.000 description 1
- BCARHSOASONIRZ-PUSCERABSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BCARHSOASONIRZ-PUSCERABSA-N 0.000 description 1
- XLBFAVDHYWGXKH-CBMHMQAMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 XLBFAVDHYWGXKH-CBMHMQAMSA-N 0.000 description 1
- NPUVHHLESGNYRE-YIPSIMJMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NPUVHHLESGNYRE-YIPSIMJMSA-N 0.000 description 1
- YUFDHIVRXICRCA-PUSCERABSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YUFDHIVRXICRCA-PUSCERABSA-N 0.000 description 1
- COJRUNZDXWFFAC-WCBIRZFKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 COJRUNZDXWFFAC-WCBIRZFKSA-N 0.000 description 1
- TXXREBHZNHSEMM-YIPSIMJMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 TXXREBHZNHSEMM-YIPSIMJMSA-N 0.000 description 1
- IDIZRPVPASFHQL-YIPSIMJMSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 IDIZRPVPASFHQL-YIPSIMJMSA-N 0.000 description 1
- LNVPOGSLDAFVOJ-JCPPJRSHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 LNVPOGSLDAFVOJ-JCPPJRSHSA-N 0.000 description 1
- YPHFYHARCLZTSN-JCPPJRSHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YPHFYHARCLZTSN-JCPPJRSHSA-N 0.000 description 1
- LUEPMPMQVQKQKZ-YRQCHJOISA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 LUEPMPMQVQKQKZ-YRQCHJOISA-N 0.000 description 1
- YQIIDHHZCLCARN-YRQCHJOISA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YQIIDHHZCLCARN-YRQCHJOISA-N 0.000 description 1
- MXIXRGLCYGXCMC-RKMGBUROSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 MXIXRGLCYGXCMC-RKMGBUROSA-N 0.000 description 1
- RCZUJWVTWREGQL-PUSCERABSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 RCZUJWVTWREGQL-PUSCERABSA-N 0.000 description 1
- SUJKENGVFFHXJR-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SUJKENGVFFHXJR-IAIYQMRASA-N 0.000 description 1
- SKJWFZTTZYHCCI-IAIYQMRASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SKJWFZTTZYHCCI-IAIYQMRASA-N 0.000 description 1
- GEGMZCJRFIMKMZ-JNFFJFACSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])C)C=C1)C2 GEGMZCJRFIMKMZ-JNFFJFACSA-N 0.000 description 1
- DDNRADHVUJSPRG-WDEBQMKVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])C)C=C1 DDNRADHVUJSPRG-WDEBQMKVSA-N 0.000 description 1
- SBZBKJREBIHEHP-ZEQRLZLVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 SBZBKJREBIHEHP-ZEQRLZLVSA-N 0.000 description 1
- QSNKLAWOLNYNCA-OKHJTSFASA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 QSNKLAWOLNYNCA-OKHJTSFASA-N 0.000 description 1
- DXUMSLOZZNCKES-HKQJZWTQSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 DXUMSLOZZNCKES-HKQJZWTQSA-N 0.000 description 1
- GYIDYVRQVKZESE-VMPREFPWSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 GYIDYVRQVKZESE-VMPREFPWSA-N 0.000 description 1
- BGWKAHIJQXUTAX-LZMGPXDLSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BGWKAHIJQXUTAX-LZMGPXDLSA-N 0.000 description 1
- DYRCBUBOQOZBAF-NRFANRHFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 DYRCBUBOQOZBAF-NRFANRHFSA-N 0.000 description 1
- AIDYLQGZZNUVNU-NRFANRHFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 AIDYLQGZZNUVNU-NRFANRHFSA-N 0.000 description 1
- CAWIKPCTAKMYRP-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 CAWIKPCTAKMYRP-DEOSSOPVSA-N 0.000 description 1
- FZXGCQKRXDEIMC-NRFANRHFSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 FZXGCQKRXDEIMC-NRFANRHFSA-N 0.000 description 1
- MHWLIGXAWQSDRB-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 MHWLIGXAWQSDRB-QFIPXVFZSA-N 0.000 description 1
- WPWNUBPRYCLHNI-DEOSSOPVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 WPWNUBPRYCLHNI-DEOSSOPVSA-N 0.000 description 1
- ANWOLDXBHGNBHM-GOSISDBHSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 ANWOLDXBHGNBHM-GOSISDBHSA-N 0.000 description 1
- VKPVMHDTQZFWIP-HSZRJFAPSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])C)\C=C/1)CC2 VKPVMHDTQZFWIP-HSZRJFAPSA-N 0.000 description 1
- TZHXNYYVOLADTE-IGKIAQTJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 TZHXNYYVOLADTE-IGKIAQTJSA-N 0.000 description 1
- AFDRHBSSZDFWTF-FIBWVYCGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AFDRHBSSZDFWTF-FIBWVYCGSA-N 0.000 description 1
- CBEMHROLMGKXBG-IGKIAQTJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CBEMHROLMGKXBG-IGKIAQTJSA-N 0.000 description 1
- BDQSYTNTTGRSQG-LSYYVWMOSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BDQSYTNTTGRSQG-LSYYVWMOSA-N 0.000 description 1
- NNGUBCJLUYIRIZ-WNJJXGMVSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 NNGUBCJLUYIRIZ-WNJJXGMVSA-N 0.000 description 1
- BJADJIOPJDVDOB-JGPKDBRGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 BJADJIOPJDVDOB-JGPKDBRGSA-N 0.000 description 1
- WREKYHOFMYQWRS-JGPKDBRGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 WREKYHOFMYQWRS-JGPKDBRGSA-N 0.000 description 1
- CMPPNWGOQTYOGV-URQSHKLJSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CMPPNWGOQTYOGV-URQSHKLJSA-N 0.000 description 1
- YRNYZQBFRCSRDZ-JGPKDBRGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 YRNYZQBFRCSRDZ-JGPKDBRGSA-N 0.000 description 1
- AVSVNBGYRHQURE-AKPJXSIGSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 AVSVNBGYRHQURE-AKPJXSIGSA-N 0.000 description 1
- CWVCHHGDLZTVHI-UUHGVMMKSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 CWVCHHGDLZTVHI-UUHGVMMKSA-N 0.000 description 1
- WODQWRGJZMYWBF-CRNNPVOZSA-N [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])C)C=C1)CC2 WODQWRGJZMYWBF-CRNNPVOZSA-N 0.000 description 1
- UBXIYYPCAULXIP-AWEZNQCLSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 UBXIYYPCAULXIP-AWEZNQCLSA-N 0.000 description 1
- CBZYBPGGEXBEJL-KEYYUXOJSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 CBZYBPGGEXBEJL-KEYYUXOJSA-N 0.000 description 1
- ACNZDQOYDUCXAY-CABCVRRESA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 ACNZDQOYDUCXAY-CABCVRRESA-N 0.000 description 1
- JLKJJRHNFGROIR-BSXFFOKHSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 JLKJJRHNFGROIR-BSXFFOKHSA-N 0.000 description 1
- ACNZDQOYDUCXAY-GJZGRUSLSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 ACNZDQOYDUCXAY-GJZGRUSLSA-N 0.000 description 1
- JLKJJRHNFGROIR-KRXQYRFLSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])C)S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 JLKJJRHNFGROIR-KRXQYRFLSA-N 0.000 description 1
- PODWGVHNGYEFDD-JRQMZCAUSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 PODWGVHNGYEFDD-JRQMZCAUSA-N 0.000 description 1
- VDRLZYYVBBZILE-OKSUZUROSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 VDRLZYYVBBZILE-OKSUZUROSA-N 0.000 description 1
- KJKZEXXOLVRQAA-JTTBUGBHSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 KJKZEXXOLVRQAA-JTTBUGBHSA-N 0.000 description 1
- KATIHPBVJVRHGP-UZDJSJLNSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 KATIHPBVJVRHGP-UZDJSJLNSA-N 0.000 description 1
- KJKZEXXOLVRQAA-QZOKSUHYSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 KJKZEXXOLVRQAA-QZOKSUHYSA-N 0.000 description 1
- KATIHPBVJVRHGP-WBJXPBQYSA-N [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N(C)C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 KATIHPBVJVRHGP-WBJXPBQYSA-N 0.000 description 1
- WFOOKFORXSGHAH-INIZCTEOSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 WFOOKFORXSGHAH-INIZCTEOSA-N 0.000 description 1
- FLFONDBJALQJFU-GZWBLTSWSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 FLFONDBJALQJFU-GZWBLTSWSA-N 0.000 description 1
- JUDJXGCOWSMJEC-IKGGRYGDSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 JUDJXGCOWSMJEC-IKGGRYGDSA-N 0.000 description 1
- SYHNLJLZODBRRD-BWDOMIEYSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 SYHNLJLZODBRRD-BWDOMIEYSA-N 0.000 description 1
- LUHCXMOVWBCXAL-PBHICJAKSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 LUHCXMOVWBCXAL-PBHICJAKSA-N 0.000 description 1
- DLWDOQWKDMLZHY-DGVNKONRSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 DLWDOQWKDMLZHY-DGVNKONRSA-N 0.000 description 1
- MVBSNWDTZXRYED-ZSGPHXLJSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 MVBSNWDTZXRYED-ZSGPHXLJSA-N 0.000 description 1
- QSHBFZORTYFWHR-CUMGEXOGSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 QSHBFZORTYFWHR-CUMGEXOGSA-N 0.000 description 1
- LUHCXMOVWBCXAL-YOEHRIQHSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 LUHCXMOVWBCXAL-YOEHRIQHSA-N 0.000 description 1
- DLWDOQWKDMLZHY-OKJDAMARSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 DLWDOQWKDMLZHY-OKJDAMARSA-N 0.000 description 1
- MVBSNWDTZXRYED-NBMRYCAZSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])C)S1)CC2 MVBSNWDTZXRYED-NBMRYCAZSA-N 0.000 description 1
- QSHBFZORTYFWHR-MSNALIEZSA-N [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N(C)C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 QSHBFZORTYFWHR-MSNALIEZSA-N 0.000 description 1
- RHDBFYCOJHDRGR-QFIPXVFZSA-N [H]N(C)C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(CCNCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 RHDBFYCOJHDRGR-QFIPXVFZSA-N 0.000 description 1
- VPVXORLBBBXIAC-OIBXWCBGSA-N [H]N(C)C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 VPVXORLBBBXIAC-OIBXWCBGSA-N 0.000 description 1
- LKBOTBLYVAFFFP-JPYJTQIMSA-N [H]N(C)C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 LKBOTBLYVAFFFP-JPYJTQIMSA-N 0.000 description 1
- PGQSSRFYVDMKJZ-LFUSFCIXSA-N [H]N(C)C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 PGQSSRFYVDMKJZ-LFUSFCIXSA-N 0.000 description 1
- LKBOTBLYVAFFFP-MBSDFSHPSA-N [H]N(C)C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 LKBOTBLYVAFFFP-MBSDFSHPSA-N 0.000 description 1
- PGQSSRFYVDMKJZ-UHVVYFEBSA-N [H]N(C)C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N(C)C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])C)C=C2)C2=NNN=N2)C=C1 PGQSSRFYVDMKJZ-UHVVYFEBSA-N 0.000 description 1
- JJEALADNNZKJJA-SANMLTNESA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 JJEALADNNZKJJA-SANMLTNESA-N 0.000 description 1
- YLBHNHKDUGNMET-IHOKFDBFSA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 YLBHNHKDUGNMET-IHOKFDBFSA-N 0.000 description 1
- SYWSKNMUZHIXSZ-AOKHHQPISA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 SYWSKNMUZHIXSZ-AOKHHQPISA-N 0.000 description 1
- FVILOHAGLDBETF-AMGIVPHBSA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 FVILOHAGLDBETF-AMGIVPHBSA-N 0.000 description 1
- MEKLMKFSBZWWSM-XCVXUIKGSA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 MEKLMKFSBZWWSM-XCVXUIKGSA-N 0.000 description 1
- FVILOHAGLDBETF-CUNXSJBXSA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 FVILOHAGLDBETF-CUNXSJBXSA-N 0.000 description 1
- MEKLMKFSBZWWSM-VIKYFBQLSA-N [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 Chemical compound [H]N(CC(C)(C)O)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC(C)(C)O)C=C1)CC2 MEKLMKFSBZWWSM-VIKYFBQLSA-N 0.000 description 1
- PTIWJVXQFROLQG-VWLOTQADSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 PTIWJVXQFROLQG-VWLOTQADSA-N 0.000 description 1
- GFNAFPQFZOAFHQ-QQNWGBJXSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 GFNAFPQFZOAFHQ-QQNWGBJXSA-N 0.000 description 1
- RIXYOTYRZBZNMC-UHFFFAOYSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 RIXYOTYRZBZNMC-UHFFFAOYSA-N 0.000 description 1
- ZGEQNLQOJXJAIV-UHFFFAOYSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 ZGEQNLQOJXJAIV-UHFFFAOYSA-N 0.000 description 1
- GIMWDUHSKRZGNU-VWLOTQADSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 GIMWDUHSKRZGNU-VWLOTQADSA-N 0.000 description 1
- PLCKMDGJTGNTLH-SANMLTNESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 PLCKMDGJTGNTLH-SANMLTNESA-N 0.000 description 1
- PRKVNRFLRXZTHN-MHZLTWQESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 PRKVNRFLRXZTHN-MHZLTWQESA-N 0.000 description 1
- RTUHDWOJHXPQNO-NDEPHWFRSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 RTUHDWOJHXPQNO-NDEPHWFRSA-N 0.000 description 1
- RGJOJEUYZXCHJF-QQNWGBJXSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 RGJOJEUYZXCHJF-QQNWGBJXSA-N 0.000 description 1
- FBBAMJYLBVTIQW-ALTZYDRJSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 FBBAMJYLBVTIQW-ALTZYDRJSA-N 0.000 description 1
- AXVFHULIZBRDMS-WWDWLDGVSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 AXVFHULIZBRDMS-WWDWLDGVSA-N 0.000 description 1
- YXYDLFCBNZJYTI-XHWIWGEHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 YXYDLFCBNZJYTI-XHWIWGEHSA-N 0.000 description 1
- QLRIUKFTFWDGGC-OAHLLOKOSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 QLRIUKFTFWDGGC-OAHLLOKOSA-N 0.000 description 1
- XUYDYADIOAUCRP-NLFFAJNJSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 XUYDYADIOAUCRP-NLFFAJNJSA-N 0.000 description 1
- JNBWHPJRERCJQK-SDXKGTORSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 JNBWHPJRERCJQK-SDXKGTORSA-N 0.000 description 1
- VUWYBEZPIYPJAD-FTJBHMTQSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 VUWYBEZPIYPJAD-FTJBHMTQSA-N 0.000 description 1
- DCGYHIUWRGQJST-JMDODURXSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 DCGYHIUWRGQJST-JMDODURXSA-N 0.000 description 1
- ZXXQEQGEVYTQGQ-JSOSNVBQSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 ZXXQEQGEVYTQGQ-JSOSNVBQSA-N 0.000 description 1
- LDCRCYGXLBRBEC-LSJVWSNHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 LDCRCYGXLBRBEC-LSJVWSNHSA-N 0.000 description 1
- IPESEQVXMNLERH-HSZRJFAPSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 IPESEQVXMNLERH-HSZRJFAPSA-N 0.000 description 1
- SCTAPHSHIDXXSO-HSZRJFAPSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 SCTAPHSHIDXXSO-HSZRJFAPSA-N 0.000 description 1
- IOEIMHFULQSUKD-AREMUKBSSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 IOEIMHFULQSUKD-AREMUKBSSA-N 0.000 description 1
- SWRMTXWJKQYKKR-HSZRJFAPSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 SWRMTXWJKQYKKR-HSZRJFAPSA-N 0.000 description 1
- GOJSWNNZBLHZNZ-XMMPIXPASA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 GOJSWNNZBLHZNZ-XMMPIXPASA-N 0.000 description 1
- QISSPDHYBVNVIM-RUZDIDTESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 QISSPDHYBVNVIM-RUZDIDTESA-N 0.000 description 1
- WJBYICCILTUIPU-AREMUKBSSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 WJBYICCILTUIPU-AREMUKBSSA-N 0.000 description 1
- QYHULKHVTJKTQE-FQEVSTJZSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 QYHULKHVTJKTQE-FQEVSTJZSA-N 0.000 description 1
- UZXSHUGDEBOMBH-VWLOTQADSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 UZXSHUGDEBOMBH-VWLOTQADSA-N 0.000 description 1
- QFXRNENQKPHGDP-LITSAYRRSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 QFXRNENQKPHGDP-LITSAYRRSA-N 0.000 description 1
- TZKXXFVOZKGJOE-LITSAYRRSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 TZKXXFVOZKGJOE-LITSAYRRSA-N 0.000 description 1
- IEUDOGXBYSYSJK-BHDXBOSCSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 IEUDOGXBYSYSJK-BHDXBOSCSA-N 0.000 description 1
- USPGTUFRPUHHSM-LITSAYRRSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 USPGTUFRPUHHSM-LITSAYRRSA-N 0.000 description 1
- GMKIMRCKECQNGR-BHBYDHKZSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 GMKIMRCKECQNGR-BHBYDHKZSA-N 0.000 description 1
- BHLNIHZFOONAKG-QCENPCRXSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 BHLNIHZFOONAKG-QCENPCRXSA-N 0.000 description 1
- SAAKMPDFWWWVNW-BHDXBOSCSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 SAAKMPDFWWWVNW-BHDXBOSCSA-N 0.000 description 1
- YYHZZSVGPKUGTB-MOXFVITDSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 YYHZZSVGPKUGTB-MOXFVITDSA-N 0.000 description 1
- TTXMNEQUWCQQCE-ZIMFAVFZSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 TTXMNEQUWCQQCE-ZIMFAVFZSA-N 0.000 description 1
- VZZKBBAGDYFXBL-HVZDWOJPSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 VZZKBBAGDYFXBL-HVZDWOJPSA-N 0.000 description 1
- WKHCBDLZCZZARO-ZIMFAVFZSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 WKHCBDLZCZZARO-ZIMFAVFZSA-N 0.000 description 1
- KWNJWRFGRXDSRT-FSRIWSFDSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 KWNJWRFGRXDSRT-FSRIWSFDSA-N 0.000 description 1
- WLJXCUYXPBQMIK-PYFKZVGHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 WLJXCUYXPBQMIK-PYFKZVGHSA-N 0.000 description 1
- FHOUMNCVSPIZLW-MEUCRYMQSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 FHOUMNCVSPIZLW-MEUCRYMQSA-N 0.000 description 1
- DCGYHIUWRGQJST-NVGWLYTESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 DCGYHIUWRGQJST-NVGWLYTESA-N 0.000 description 1
- ZXXQEQGEVYTQGQ-CONSDPRKSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 ZXXQEQGEVYTQGQ-CONSDPRKSA-N 0.000 description 1
- LDCRCYGXLBRBEC-XDRKNORUSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 LDCRCYGXLBRBEC-XDRKNORUSA-N 0.000 description 1
- IPESEQVXMNLERH-QHCPKHFHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 IPESEQVXMNLERH-QHCPKHFHSA-N 0.000 description 1
- SCTAPHSHIDXXSO-QHCPKHFHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 SCTAPHSHIDXXSO-QHCPKHFHSA-N 0.000 description 1
- IOEIMHFULQSUKD-SANMLTNESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 IOEIMHFULQSUKD-SANMLTNESA-N 0.000 description 1
- SWRMTXWJKQYKKR-QHCPKHFHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 SWRMTXWJKQYKKR-QHCPKHFHSA-N 0.000 description 1
- GOJSWNNZBLHZNZ-DEOSSOPVSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 GOJSWNNZBLHZNZ-DEOSSOPVSA-N 0.000 description 1
- QISSPDHYBVNVIM-VWLOTQADSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 QISSPDHYBVNVIM-VWLOTQADSA-N 0.000 description 1
- WJBYICCILTUIPU-SANMLTNESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 WJBYICCILTUIPU-SANMLTNESA-N 0.000 description 1
- QYHULKHVTJKTQE-HXUWFJFHSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 QYHULKHVTJKTQE-HXUWFJFHSA-N 0.000 description 1
- UZXSHUGDEBOMBH-RUZDIDTESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])CC)\C=C/1)CC2 UZXSHUGDEBOMBH-RUZDIDTESA-N 0.000 description 1
- TZKXXFVOZKGJOE-WNJJXGMVSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 TZKXXFVOZKGJOE-WNJJXGMVSA-N 0.000 description 1
- IEUDOGXBYSYSJK-NYDCQLBNSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 IEUDOGXBYSYSJK-NYDCQLBNSA-N 0.000 description 1
- USPGTUFRPUHHSM-WNJJXGMVSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 USPGTUFRPUHHSM-WNJJXGMVSA-N 0.000 description 1
- GMKIMRCKECQNGR-FIBWVYCGSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 GMKIMRCKECQNGR-FIBWVYCGSA-N 0.000 description 1
- BHLNIHZFOONAKG-IZEXYCQBSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 BHLNIHZFOONAKG-IZEXYCQBSA-N 0.000 description 1
- SAAKMPDFWWWVNW-NYDCQLBNSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 SAAKMPDFWWWVNW-NYDCQLBNSA-N 0.000 description 1
- YYHZZSVGPKUGTB-UUHGVMMKSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 YYHZZSVGPKUGTB-UUHGVMMKSA-N 0.000 description 1
- TTXMNEQUWCQQCE-UUHGVMMKSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 TTXMNEQUWCQQCE-UUHGVMMKSA-N 0.000 description 1
- VZZKBBAGDYFXBL-SHDQMJQSSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 VZZKBBAGDYFXBL-SHDQMJQSSA-N 0.000 description 1
- WKHCBDLZCZZARO-UUHGVMMKSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 WKHCBDLZCZZARO-UUHGVMMKSA-N 0.000 description 1
- KWNJWRFGRXDSRT-CRNNPVOZSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 KWNJWRFGRXDSRT-CRNNPVOZSA-N 0.000 description 1
- WLJXCUYXPBQMIK-RMVGYSHESA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 WLJXCUYXPBQMIK-RMVGYSHESA-N 0.000 description 1
- FHOUMNCVSPIZLW-CLGPCEASSA-N [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 Chemical compound [H]N(CC)C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CC)C=C1)CC2 FHOUMNCVSPIZLW-CLGPCEASSA-N 0.000 description 1
- OXSUBHZDNCNFAJ-NDEPHWFRSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 OXSUBHZDNCNFAJ-NDEPHWFRSA-N 0.000 description 1
- AZXOEMCNMKAWOW-YJJLJQPASA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 AZXOEMCNMKAWOW-YJJLJQPASA-N 0.000 description 1
- XBYIPZKYPKWDRN-XHWIWGEHSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 XBYIPZKYPKWDRN-XHWIWGEHSA-N 0.000 description 1
- QDJDTMDHUUBXHE-RHJKKGJFSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(F)C=C1)CC2 QDJDTMDHUUBXHE-RHJKKGJFSA-N 0.000 description 1
- NBOQTWNVZGEDDG-GIGWZHCTSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 NBOQTWNVZGEDDG-GIGWZHCTSA-N 0.000 description 1
- PWINTBTZPVTIEE-VKVQOMGUSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 PWINTBTZPVTIEE-VKVQOMGUSA-N 0.000 description 1
- NBOQTWNVZGEDDG-OUTSHDOLSA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 NBOQTWNVZGEDDG-OUTSHDOLSA-N 0.000 description 1
- PWINTBTZPVTIEE-ICGWNSLASA-N [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 Chemical compound [H]N(CCN(C)C)C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])CCN(C)C)C=C1)CC2 PWINTBTZPVTIEE-ICGWNSLASA-N 0.000 description 1
- WKEKIHMWIVCJMU-UHFFFAOYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)N)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 WKEKIHMWIVCJMU-UHFFFAOYSA-N 0.000 description 1
- NLQUABBIZYHWBU-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 NLQUABBIZYHWBU-NRFANRHFSA-N 0.000 description 1
- PPAXAYZKTSLHKK-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC(C)(C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PPAXAYZKTSLHKK-DPLHUUCSSA-N 0.000 description 1
- CGMQWZQZPUEHBB-UHFFFAOYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 CGMQWZQZPUEHBB-UHFFFAOYSA-N 0.000 description 1
- HNLNKZWMOPVFCU-UHFFFAOYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 HNLNKZWMOPVFCU-UHFFFAOYSA-N 0.000 description 1
- KHLTYKWPAWSYCW-UHFFFAOYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 KHLTYKWPAWSYCW-UHFFFAOYSA-N 0.000 description 1
- PYLJPYPJMKFCNK-UHFFFAOYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(N)CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 PYLJPYPJMKFCNK-UHFFFAOYSA-N 0.000 description 1
- CWJPGUMERKRGSB-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 CWJPGUMERKRGSB-NRFANRHFSA-N 0.000 description 1
- HEDQKYSRTIOIPV-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 HEDQKYSRTIOIPV-QFIPXVFZSA-N 0.000 description 1
- OUFYCVWHRRTVPW-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 OUFYCVWHRRTVPW-QHCPKHFHSA-N 0.000 description 1
- VLHSDBKTOYIRGP-DEOSSOPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3CCC[C@H]3C#N)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 VLHSDBKTOYIRGP-DEOSSOPVSA-N 0.000 description 1
- RRJDIVBYGWXBFG-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RRJDIVBYGWXBFG-DPLHUUCSSA-N 0.000 description 1
- KSYGEXLIUVRVGT-SLSDLSHTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 KSYGEXLIUVRVGT-SLSDLSHTSA-N 0.000 description 1
- BPFZLPFESLEAET-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BPFZLPFESLEAET-UMFSSWHCSA-N 0.000 description 1
- NTTWQENJVCJUCL-DXBVXKBHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CC1(NCC(=O)N3[C@H](C#N)C[C@@H]4C[C@@H]43)CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 NTTWQENJVCJUCL-DXBVXKBHSA-N 0.000 description 1
- GFMDZVVRDADAPB-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GFMDZVVRDADAPB-FQEVSTJZSA-N 0.000 description 1
- GFHGBTBVXAVWJS-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GFHGBTBVXAVWJS-QHCPKHFHSA-N 0.000 description 1
- QTVXINSKMVQIHW-SANMLTNESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 QTVXINSKMVQIHW-SANMLTNESA-N 0.000 description 1
- CFGKNJUUCXNKBC-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 CFGKNJUUCXNKBC-FQEVSTJZSA-N 0.000 description 1
- ZKGXXTDOQHIIJV-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ZKGXXTDOQHIIJV-FQEVSTJZSA-N 0.000 description 1
- NLWFXRFYRPLIHH-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 NLWFXRFYRPLIHH-FQEVSTJZSA-N 0.000 description 1
- FTQVXQUHGAGNET-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 FTQVXQUHGAGNET-QHCPKHFHSA-N 0.000 description 1
- GNISIPXSUWKRLS-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GNISIPXSUWKRLS-NRFANRHFSA-N 0.000 description 1
- SBZSXVYPNBFBGZ-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 SBZSXVYPNBFBGZ-QHCPKHFHSA-N 0.000 description 1
- YRFRRIFECHBNBW-SANMLTNESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 YRFRRIFECHBNBW-SANMLTNESA-N 0.000 description 1
- VZQQRVWZDXLINU-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 VZQQRVWZDXLINU-QHCPKHFHSA-N 0.000 description 1
- MXZJQMLSULINKG-QHCPKHFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 MXZJQMLSULINKG-QHCPKHFHSA-N 0.000 description 1
- UBHDHPAKLNYUMS-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 UBHDHPAKLNYUMS-QFIPXVFZSA-N 0.000 description 1
- UJRRTLHEYODONC-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 UJRRTLHEYODONC-QFIPXVFZSA-N 0.000 description 1
- XOZSSIMZIOSXLD-SANMLTNESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 XOZSSIMZIOSXLD-SANMLTNESA-N 0.000 description 1
- NSMVIYFEGXJHKJ-SANMLTNESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 NSMVIYFEGXJHKJ-SANMLTNESA-N 0.000 description 1
- ITWTYVILNCXQNK-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ITWTYVILNCXQNK-QFIPXVFZSA-N 0.000 description 1
- QOPGOPFRRCWBOH-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 QOPGOPFRRCWBOH-NRFANRHFSA-N 0.000 description 1
- QAGDOKCEJSTMRR-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 QAGDOKCEJSTMRR-FQEVSTJZSA-N 0.000 description 1
- SOWXNGWJVKBNSP-IBGZPJMESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 SOWXNGWJVKBNSP-IBGZPJMESA-N 0.000 description 1
- WKKLSBWDBBKRRS-INIZCTEOSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 WKKLSBWDBBKRRS-INIZCTEOSA-N 0.000 description 1
- DRZCFRVFQHXOLW-QTEQDKRBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 DRZCFRVFQHXOLW-QTEQDKRBSA-N 0.000 description 1
- JDTGMYROECLNIW-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 JDTGMYROECLNIW-UMFSSWHCSA-N 0.000 description 1
- QCSQHKBNFRTFEH-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QCSQHKBNFRTFEH-DPLHUUCSSA-N 0.000 description 1
- HTAWQOZXCNJEJZ-IXLXJOPFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 HTAWQOZXCNJEJZ-IXLXJOPFSA-N 0.000 description 1
- NPEGHSWHHZJCPA-QTEQDKRBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 NPEGHSWHHZJCPA-QTEQDKRBSA-N 0.000 description 1
- PJNWDVVPRBCXMR-QTEQDKRBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PJNWDVVPRBCXMR-QTEQDKRBSA-N 0.000 description 1
- FCWFTGBJMGORRV-QTEQDKRBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 FCWFTGBJMGORRV-QTEQDKRBSA-N 0.000 description 1
- XBXYCGBPOPDBFW-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XBXYCGBPOPDBFW-UMFSSWHCSA-N 0.000 description 1
- QBZQFOPOVLPZDJ-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QBZQFOPOVLPZDJ-DPLHUUCSSA-N 0.000 description 1
- UHLIIEVJEAMENZ-AZIXZEHSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 UHLIIEVJEAMENZ-AZIXZEHSSA-N 0.000 description 1
- LAARSLREYBRJOW-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 LAARSLREYBRJOW-UMFSSWHCSA-N 0.000 description 1
- WCFGOUYNPILLMD-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 WCFGOUYNPILLMD-DPLHUUCSSA-N 0.000 description 1
- BVEUSILJTAKSOW-IXLXJOPFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BVEUSILJTAKSOW-IXLXJOPFSA-N 0.000 description 1
- YHZKCMJMNXYUQU-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 YHZKCMJMNXYUQU-UMFSSWHCSA-N 0.000 description 1
- IGTSKVKKGRVVEF-UMFSSWHCSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 IGTSKVKKGRVVEF-UMFSSWHCSA-N 0.000 description 1
- UQCNLLWQBBENCJ-SLSDLSHTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 UQCNLLWQBBENCJ-SLSDLSHTSA-N 0.000 description 1
- IRVWBDOHSAMPAT-SLSDLSHTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 IRVWBDOHSAMPAT-SLSDLSHTSA-N 0.000 description 1
- BNDFDVKVXJMSIK-AZIXZEHSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BNDFDVKVXJMSIK-AZIXZEHSSA-N 0.000 description 1
- YXHXICMIOIRDFP-AZIXZEHSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 YXHXICMIOIRDFP-AZIXZEHSSA-N 0.000 description 1
- OVVPGHSAMWPWJV-SLSDLSHTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 OVVPGHSAMWPWJV-SLSDLSHTSA-N 0.000 description 1
- UERHDDDZXYQQJO-DPLHUUCSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 UERHDDDZXYQQJO-DPLHUUCSSA-N 0.000 description 1
- GSQMLMHOTXHWOX-QTEQDKRBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 GSQMLMHOTXHWOX-QTEQDKRBSA-N 0.000 description 1
- RCYDXPZFKDNOQO-DXIQSLLYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 RCYDXPZFKDNOQO-DXIQSLLYSA-N 0.000 description 1
- SMJNDXIZKMTWII-XFQAVAEZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 SMJNDXIZKMTWII-XFQAVAEZSA-N 0.000 description 1
- KQLNPFLDCWCOMY-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 KQLNPFLDCWCOMY-IERDGZPVSA-N 0.000 description 1
- UVLJGAUIJRJDMV-VGSWGCGISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 UVLJGAUIJRJDMV-VGSWGCGISA-N 0.000 description 1
- VEJRVGODLDHFHL-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 VEJRVGODLDHFHL-IERDGZPVSA-N 0.000 description 1
- PETYYJQDSUCHPI-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 PETYYJQDSUCHPI-IERDGZPVSA-N 0.000 description 1
- RCOSSJSTQCSFRV-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RCOSSJSTQCSFRV-IERDGZPVSA-N 0.000 description 1
- OJAHMOUEXBSDRD-DVECYGJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 OJAHMOUEXBSDRD-DVECYGJZSA-N 0.000 description 1
- RXORAHJLSACFMB-VGSWGCGISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RXORAHJLSACFMB-VGSWGCGISA-N 0.000 description 1
- JUYUJLFKVJJXKX-ZBLYBZFDSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JUYUJLFKVJJXKX-ZBLYBZFDSA-N 0.000 description 1
- OFBSLOLVZAHJNB-DVECYGJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 OFBSLOLVZAHJNB-DVECYGJZSA-N 0.000 description 1
- WVAZIWDSBYRZIY-VGSWGCGISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 WVAZIWDSBYRZIY-VGSWGCGISA-N 0.000 description 1
- LNOFSBSYGZMASM-HRFSGMKKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 LNOFSBSYGZMASM-HRFSGMKKSA-N 0.000 description 1
- USXSZMKGCKXKBS-DVECYGJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 USXSZMKGCKXKBS-DVECYGJZSA-N 0.000 description 1
- TZVQQEMCCYIXJY-HXOBKFHXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 TZVQQEMCCYIXJY-HXOBKFHXSA-N 0.000 description 1
- DEYHVDKZPDHHFQ-HRFSGMKKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 DEYHVDKZPDHHFQ-HRFSGMKKSA-N 0.000 description 1
- CISSOWMWOCJTGW-HRFSGMKKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 CISSOWMWOCJTGW-HRFSGMKKSA-N 0.000 description 1
- JNFQCFDFVUTABP-JPYJTQIMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JNFQCFDFVUTABP-JPYJTQIMSA-N 0.000 description 1
- IXWBWTFSZZOISX-VGSWGCGISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 IXWBWTFSZZOISX-VGSWGCGISA-N 0.000 description 1
- QJDXSVHDYCMQPP-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QJDXSVHDYCMQPP-IERDGZPVSA-N 0.000 description 1
- HSHIJPCJWGHMIR-IERDGZPVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 HSHIJPCJWGHMIR-IERDGZPVSA-N 0.000 description 1
- JFSZHFHTNZJZKA-QRWLVFNGSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 JFSZHFHTNZJZKA-QRWLVFNGSA-N 0.000 description 1
- MNTOXLNHEJKAHF-PBHICJAKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 MNTOXLNHEJKAHF-PBHICJAKSA-N 0.000 description 1
- XYJJSQVRLRJJNA-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XYJJSQVRLRJJNA-CEFQEXJZSA-N 0.000 description 1
- JDDVIENDNAPLDA-PFCVVTBNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 JDDVIENDNAPLDA-PFCVVTBNSA-N 0.000 description 1
- KNAXJBYVBVSDQQ-MOMMTWFBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 KNAXJBYVBVSDQQ-MOMMTWFBSA-N 0.000 description 1
- QVCQNBXDGULZJW-CTXJSGFVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QVCQNBXDGULZJW-CTXJSGFVSA-N 0.000 description 1
- MWTVGKIIELWKKC-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MWTVGKIIELWKKC-CEFQEXJZSA-N 0.000 description 1
- WWRCSNDNAFFODD-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 WWRCSNDNAFFODD-CEFQEXJZSA-N 0.000 description 1
- SXOIKXUHJDODKY-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 SXOIKXUHJDODKY-CEFQEXJZSA-N 0.000 description 1
- YEPRZNMKVZNBTR-PFCVVTBNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 YEPRZNMKVZNBTR-PFCVVTBNSA-N 0.000 description 1
- BWYRQALDHWKCQB-MOMMTWFBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BWYRQALDHWKCQB-MOMMTWFBSA-N 0.000 description 1
- VYKZUVDKDREQTK-QSWJDQLRSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 VYKZUVDKDREQTK-QSWJDQLRSA-N 0.000 description 1
- UPUIVLSSOFZBHH-PFCVVTBNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 UPUIVLSSOFZBHH-PFCVVTBNSA-N 0.000 description 1
- ZCLMDEYWRKSRCH-MOMMTWFBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZCLMDEYWRKSRCH-MOMMTWFBSA-N 0.000 description 1
- XJXXMQLUVNQFPA-CTXJSGFVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XJXXMQLUVNQFPA-CTXJSGFVSA-N 0.000 description 1
- FQBPQBSMWZCRCD-PFCVVTBNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 FQBPQBSMWZCRCD-PFCVVTBNSA-N 0.000 description 1
- GSSSNADZJILFHI-PFCVVTBNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 GSSSNADZJILFHI-PFCVVTBNSA-N 0.000 description 1
- SNAIYVOINBWGLA-BCASAEOVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 SNAIYVOINBWGLA-BCASAEOVSA-N 0.000 description 1
- PYWPFZPSXFBUPB-BCASAEOVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PYWPFZPSXFBUPB-BCASAEOVSA-N 0.000 description 1
- QLIXBVJNHSLHNZ-VFICZSPWSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QLIXBVJNHSLHNZ-VFICZSPWSA-N 0.000 description 1
- ZELXPDOQFMOWSE-VFICZSPWSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZELXPDOQFMOWSE-VFICZSPWSA-N 0.000 description 1
- OPJXNOGZFMIVLN-HLCOLNNTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 OPJXNOGZFMIVLN-HLCOLNNTSA-N 0.000 description 1
- MKJLKVLHGZISCO-MOMMTWFBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MKJLKVLHGZISCO-MOMMTWFBSA-N 0.000 description 1
- GMHHOXPWDCUAKX-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 GMHHOXPWDCUAKX-CEFQEXJZSA-N 0.000 description 1
- MABBLHJNSSSHDN-CEFQEXJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MABBLHJNSSSHDN-CEFQEXJZSA-N 0.000 description 1
- GQHHYWUDIGINIO-CJDBZMTKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 GQHHYWUDIGINIO-CJDBZMTKSA-N 0.000 description 1
- ULWLNHJSNRRKSS-LTSIINAVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 ULWLNHJSNRRKSS-LTSIINAVSA-N 0.000 description 1
- RWZHFFWESLQSTH-YADHBBJMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RWZHFFWESLQSTH-YADHBBJMSA-N 0.000 description 1
- QSFFHKODILGSTH-APDHKMKFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QSFFHKODILGSTH-APDHKMKFSA-N 0.000 description 1
- FXWSLUBCVSOISB-SXOMAYOGSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 FXWSLUBCVSOISB-SXOMAYOGSA-N 0.000 description 1
- USEFOFMZHKMQLD-RUFVNJLVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 USEFOFMZHKMQLD-RUFVNJLVSA-N 0.000 description 1
- JQJCZVGJGXWNGE-LJQANCHMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JQJCZVGJGXWNGE-LJQANCHMSA-N 0.000 description 1
- COEZOGRADNXDTK-LJQANCHMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 COEZOGRADNXDTK-LJQANCHMSA-N 0.000 description 1
- RWHVDOIQRIWIBA-JOCHJYFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RWHVDOIQRIWIBA-JOCHJYFZSA-N 0.000 description 1
- DIRNIRRBTQDKDZ-LJQANCHMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 DIRNIRRBTQDKDZ-LJQANCHMSA-N 0.000 description 1
- QLBGAYZHCQXEBH-HXUWFJFHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 QLBGAYZHCQXEBH-HXUWFJFHSA-N 0.000 description 1
- GLEKWJPNUJZBCE-JOCHJYFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GLEKWJPNUJZBCE-JOCHJYFZSA-N 0.000 description 1
- ZSJQUNHWAKNRIB-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ZSJQUNHWAKNRIB-NRFANRHFSA-N 0.000 description 1
- LOABCWLKIRCMLJ-WIOPSUGQSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 LOABCWLKIRCMLJ-WIOPSUGQSA-N 0.000 description 1
- VXXADGJFNRJXQW-WIOPSUGQSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 VXXADGJFNRJXQW-WIOPSUGQSA-N 0.000 description 1
- ZQQSQMCEHOMISR-LBNVMWSVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZQQSQMCEHOMISR-LBNVMWSVSA-N 0.000 description 1
- LKKVUBORUGIXGL-WIOPSUGQSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 LKKVUBORUGIXGL-WIOPSUGQSA-N 0.000 description 1
- BQDSHYXTIGOBQV-JYFHCDHNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BQDSHYXTIGOBQV-JYFHCDHNSA-N 0.000 description 1
- RNDVETCUGMNFKH-RPLLCQBOSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RNDVETCUGMNFKH-RPLLCQBOSA-N 0.000 description 1
- WXKIWTQCOUBNEN-LBNVMWSVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 WXKIWTQCOUBNEN-LBNVMWSVSA-N 0.000 description 1
- CBIHZTDPWAKXLZ-FIKUDORNSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 CBIHZTDPWAKXLZ-FIKUDORNSA-N 0.000 description 1
- PRIPMTRHPKAUFP-GETZNYBYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PRIPMTRHPKAUFP-GETZNYBYSA-N 0.000 description 1
- MRNLBRSEXWSHRG-IIFRUBFJSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MRNLBRSEXWSHRG-IIFRUBFJSA-N 0.000 description 1
- AEZZDBBYGKWHRD-GETZNYBYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 AEZZDBBYGKWHRD-GETZNYBYSA-N 0.000 description 1
- XKUBDRWIAVVJBG-GJHDXROTSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XKUBDRWIAVVJBG-GJHDXROTSA-N 0.000 description 1
- KTGZBTINFAMMDA-CUFBHMEESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 KTGZBTINFAMMDA-CUFBHMEESA-N 0.000 description 1
- RXQIBOWOHSKWIT-BVIJCPSYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RXQIBOWOHSKWIT-BVIJCPSYSA-N 0.000 description 1
- KQLNPFLDCWCOMY-KKSFZXQISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)N)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 KQLNPFLDCWCOMY-KKSFZXQISA-N 0.000 description 1
- UKRIGHIIQWWSHT-CYFREDJKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 UKRIGHIIQWWSHT-CYFREDJKSA-N 0.000 description 1
- UVLJGAUIJRJDMV-JTSKRJEESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 UVLJGAUIJRJDMV-JTSKRJEESA-N 0.000 description 1
- FGAXBTCINZZHPE-DCFHFQCYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=N)NC1=CC=C(F)C=C1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 FGAXBTCINZZHPE-DCFHFQCYSA-N 0.000 description 1
- VEJRVGODLDHFHL-KKSFZXQISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(=O)O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 VEJRVGODLDHFHL-KKSFZXQISA-N 0.000 description 1
- PETYYJQDSUCHPI-KKSFZXQISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C(N)=O)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 PETYYJQDSUCHPI-KKSFZXQISA-N 0.000 description 1
- RCOSSJSTQCSFRV-KKSFZXQISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RCOSSJSTQCSFRV-KKSFZXQISA-N 0.000 description 1
- OJAHMOUEXBSDRD-CYFREDJKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C(C)C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 OJAHMOUEXBSDRD-CYFREDJKSA-N 0.000 description 1
- RXORAHJLSACFMB-JTSKRJEESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RXORAHJLSACFMB-JTSKRJEESA-N 0.000 description 1
- JUYUJLFKVJJXKX-IDISGSTGSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NC(=O)ON1C1CCCCC1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JUYUJLFKVJJXKX-IDISGSTGSA-N 0.000 description 1
- OFBSLOLVZAHJNB-CYFREDJKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C(C)C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 OFBSLOLVZAHJNB-CYFREDJKSA-N 0.000 description 1
- WVAZIWDSBYRZIY-JTSKRJEESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 WVAZIWDSBYRZIY-JTSKRJEESA-N 0.000 description 1
- LNOFSBSYGZMASM-DCFHFQCYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 LNOFSBSYGZMASM-DCFHFQCYSA-N 0.000 description 1
- ZGRZMQGJLZKSSJ-CYFREDJKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ZGRZMQGJLZKSSJ-CYFREDJKSA-N 0.000 description 1
- USXSZMKGCKXKBS-CYFREDJKSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C(C)C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 USXSZMKGCKXKBS-CYFREDJKSA-N 0.000 description 1
- TZVQQEMCCYIXJY-SBUREZEXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 TZVQQEMCCYIXJY-SBUREZEXSA-N 0.000 description 1
- WVOCRFKQPBGWDX-SBUREZEXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 WVOCRFKQPBGWDX-SBUREZEXSA-N 0.000 description 1
- DEYHVDKZPDHHFQ-DCFHFQCYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 DEYHVDKZPDHHFQ-DCFHFQCYSA-N 0.000 description 1
- CISSOWMWOCJTGW-DCFHFQCYSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 CISSOWMWOCJTGW-DCFHFQCYSA-N 0.000 description 1
- JNFQCFDFVUTABP-MBSDFSHPSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=C(CC)O1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JNFQCFDFVUTABP-MBSDFSHPSA-N 0.000 description 1
- IXWBWTFSZZOISX-JTSKRJEESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=CN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 IXWBWTFSZZOISX-JTSKRJEESA-N 0.000 description 1
- HSHIJPCJWGHMIR-KKSFZXQISA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNC(=O)N1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 HSHIJPCJWGHMIR-KKSFZXQISA-N 0.000 description 1
- JFSZHFHTNZJZKA-YWZLYKJASA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 JFSZHFHTNZJZKA-YWZLYKJASA-N 0.000 description 1
- MNTOXLNHEJKAHF-YOEHRIQHSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 MNTOXLNHEJKAHF-YOEHRIQHSA-N 0.000 description 1
- XYJJSQVRLRJJNA-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)N)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XYJJSQVRLRJJNA-YYBAZUPLSA-N 0.000 description 1
- JDDVIENDNAPLDA-PUSCERABSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 JDDVIENDNAPLDA-PUSCERABSA-N 0.000 description 1
- KNAXJBYVBVSDQQ-JNFFJFACSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 KNAXJBYVBVSDQQ-JNFFJFACSA-N 0.000 description 1
- QVCQNBXDGULZJW-YPBXTBBASA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=N)NC1=CC=C(F)C=C1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QVCQNBXDGULZJW-YPBXTBBASA-N 0.000 description 1
- MWTVGKIIELWKKC-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(=O)O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MWTVGKIIELWKKC-YYBAZUPLSA-N 0.000 description 1
- WWRCSNDNAFFODD-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C(N)=O)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 WWRCSNDNAFFODD-YYBAZUPLSA-N 0.000 description 1
- SXOIKXUHJDODKY-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 SXOIKXUHJDODKY-YYBAZUPLSA-N 0.000 description 1
- YEPRZNMKVZNBTR-PUSCERABSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C(C)C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 YEPRZNMKVZNBTR-PUSCERABSA-N 0.000 description 1
- BWYRQALDHWKCQB-JNFFJFACSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BWYRQALDHWKCQB-JNFFJFACSA-N 0.000 description 1
- VYKZUVDKDREQTK-YEHBYBCVSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NC(=O)ON1C1CCCCC1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 VYKZUVDKDREQTK-YEHBYBCVSA-N 0.000 description 1
- UPUIVLSSOFZBHH-PUSCERABSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C(C)C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 UPUIVLSSOFZBHH-PUSCERABSA-N 0.000 description 1
- ZCLMDEYWRKSRCH-JNFFJFACSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZCLMDEYWRKSRCH-JNFFJFACSA-N 0.000 description 1
- XJXXMQLUVNQFPA-YPBXTBBASA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN(C3=CC=C(F)C=C3)C(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XJXXMQLUVNQFPA-YPBXTBBASA-N 0.000 description 1
- FQBPQBSMWZCRCD-PUSCERABSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 FQBPQBSMWZCRCD-PUSCERABSA-N 0.000 description 1
- GSSSNADZJILFHI-PUSCERABSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C(C)C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 GSSSNADZJILFHI-PUSCERABSA-N 0.000 description 1
- SNAIYVOINBWGLA-XVDAFTJMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 SNAIYVOINBWGLA-XVDAFTJMSA-N 0.000 description 1
- PYWPFZPSXFBUPB-XVDAFTJMSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PYWPFZPSXFBUPB-XVDAFTJMSA-N 0.000 description 1
- QLIXBVJNHSLHNZ-WPQAOXEXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 QLIXBVJNHSLHNZ-WPQAOXEXSA-N 0.000 description 1
- ZELXPDOQFMOWSE-WPQAOXEXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(C3=CC=C(F)C=C3)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZELXPDOQFMOWSE-WPQAOXEXSA-N 0.000 description 1
- OPJXNOGZFMIVLN-IAIYQMRASA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=C(CC)O1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 OPJXNOGZFMIVLN-IAIYQMRASA-N 0.000 description 1
- MKJLKVLHGZISCO-JNFFJFACSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=CN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MKJLKVLHGZISCO-JNFFJFACSA-N 0.000 description 1
- GMHHOXPWDCUAKX-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 GMHHOXPWDCUAKX-YYBAZUPLSA-N 0.000 description 1
- MABBLHJNSSSHDN-YYBAZUPLSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNC(=O)N1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MABBLHJNSSSHDN-YYBAZUPLSA-N 0.000 description 1
- GQHHYWUDIGINIO-YBFPIEQSSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(C=C(C(=O)N([H])[H])C=C1)C2 GQHHYWUDIGINIO-YBFPIEQSSA-N 0.000 description 1
- ULWLNHJSNRRKSS-DJPHASSASA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C2C=C(C(=O)N([H])[H])C=C1 ULWLNHJSNRRKSS-DJPHASSASA-N 0.000 description 1
- FXWSLUBCVSOISB-SVBPBHIXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1CCC[C@H]1C#N)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 FXWSLUBCVSOISB-SVBPBHIXSA-N 0.000 description 1
- USEFOFMZHKMQLD-AMWHEABUSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](CC1=CC=C(F)C=C1)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 USEFOFMZHKMQLD-AMWHEABUSA-N 0.000 description 1
- JQJCZVGJGXWNGE-IBGZPJMESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 JQJCZVGJGXWNGE-IBGZPJMESA-N 0.000 description 1
- COEZOGRADNXDTK-IBGZPJMESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C(C)C)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 COEZOGRADNXDTK-IBGZPJMESA-N 0.000 description 1
- RWHVDOIQRIWIBA-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RWHVDOIQRIWIBA-QFIPXVFZSA-N 0.000 description 1
- QLBGAYZHCQXEBH-FQEVSTJZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 QLBGAYZHCQXEBH-FQEVSTJZSA-N 0.000 description 1
- RZHCVZRDJKMPFT-NRFANRHFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 RZHCVZRDJKMPFT-NRFANRHFSA-N 0.000 description 1
- GLEKWJPNUJZBCE-QFIPXVFZSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)C1CCCCC1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 GLEKWJPNUJZBCE-QFIPXVFZSA-N 0.000 description 1
- MQYXZIPBYQHDFQ-MRXNPFEDSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 MQYXZIPBYQHDFQ-MRXNPFEDSA-N 0.000 description 1
- ZSJQUNHWAKNRIB-OAQYLSRUSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](N)CC1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(/C=C(C(=O)N([H])[H])\C=C/1)CC2 ZSJQUNHWAKNRIB-OAQYLSRUSA-N 0.000 description 1
- LOABCWLKIRCMLJ-DHLKQENFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 LOABCWLKIRCMLJ-DHLKQENFSA-N 0.000 description 1
- VXXADGJFNRJXQW-DHLKQENFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 VXXADGJFNRJXQW-DHLKQENFSA-N 0.000 description 1
- ZQQSQMCEHOMISR-LSYYVWMOSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 ZQQSQMCEHOMISR-LSYYVWMOSA-N 0.000 description 1
- LKKVUBORUGIXGL-DHLKQENFSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 LKKVUBORUGIXGL-DHLKQENFSA-N 0.000 description 1
- BQDSHYXTIGOBQV-OZXSUGGESA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 BQDSHYXTIGOBQV-OZXSUGGESA-N 0.000 description 1
- RNDVETCUGMNFKH-IGKIAQTJSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RNDVETCUGMNFKH-IGKIAQTJSA-N 0.000 description 1
- WXKIWTQCOUBNEN-LSYYVWMOSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1CCC[C@H]1C#N)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 WXKIWTQCOUBNEN-LSYYVWMOSA-N 0.000 description 1
- CBIHZTDPWAKXLZ-HRHCJDDBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 CBIHZTDPWAKXLZ-HRHCJDDBSA-N 0.000 description 1
- PRIPMTRHPKAUFP-HRHCJDDBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C(C)C)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 PRIPMTRHPKAUFP-HRHCJDDBSA-N 0.000 description 1
- MRNLBRSEXWSHRG-STJBOXKPSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1=CC=C(F)C=C1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 MRNLBRSEXWSHRG-STJBOXKPSA-N 0.000 description 1
- AEZZDBBYGKWHRD-HRHCJDDBSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 AEZZDBBYGKWHRD-HRHCJDDBSA-N 0.000 description 1
- XKUBDRWIAVVJBG-DTBHQADXSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 XKUBDRWIAVVJBG-DTBHQADXSA-N 0.000 description 1
- KTGZBTINFAMMDA-JGPKDBRGSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 KTGZBTINFAMMDA-JGPKDBRGSA-N 0.000 description 1
- RXQIBOWOHSKWIT-AKPJXSIGSA-N [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(C=C1)C(C[C@H](NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)C1CCCCC1)(C1=NN=NN1)C1=C(C=C(C(=O)N([H])[H])C=C1)CC2 RXQIBOWOHSKWIT-AKPJXSIGSA-N 0.000 description 1
- YPIATAWRQOFUDY-LBPRGKRZSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 YPIATAWRQOFUDY-LBPRGKRZSA-N 0.000 description 1
- VJMAYSYWJGGYEV-XUJVJEKNSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 VJMAYSYWJGGYEV-XUJVJEKNSA-N 0.000 description 1
- YUAOBLVUXNTQNC-OLZOCXBDSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 YUAOBLVUXNTQNC-OLZOCXBDSA-N 0.000 description 1
- OEGDMODYONFULD-FOCJUVANSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 OEGDMODYONFULD-FOCJUVANSA-N 0.000 description 1
- YUAOBLVUXNTQNC-STQMWFEESA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 YUAOBLVUXNTQNC-STQMWFEESA-N 0.000 description 1
- OEGDMODYONFULD-ZWUHOBOKSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C(/C(=O)N([H])[H])S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 OEGDMODYONFULD-ZWUHOBOKSA-N 0.000 description 1
- YDOYAFSKAKACHR-JHAYDQECSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 YDOYAFSKAKACHR-JHAYDQECSA-N 0.000 description 1
- OQDSZJUQVGBYJL-GQDBIGQUSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 OQDSZJUQVGBYJL-GQDBIGQUSA-N 0.000 description 1
- RNFQFDQODXWDQT-SAXCZAFJSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 RNFQFDQODXWDQT-SAXCZAFJSA-N 0.000 description 1
- BLHPSODRCNQNPO-DRNMOZGOSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 BLHPSODRCNQNPO-DRNMOZGOSA-N 0.000 description 1
- RNFQFDQODXWDQT-GWHQMRIRSA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)C2=NNN=N2)S1 RNFQFDQODXWDQT-GWHQMRIRSA-N 0.000 description 1
- BLHPSODRCNQNPO-HSEBKYPISA-N [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 Chemical compound [H]N([H])C(=O)C1=CC2=C(CCC3=C(/C=C\S3)C2(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)C2=NNN=N2)S1 BLHPSODRCNQNPO-HSEBKYPISA-N 0.000 description 1
- MYGMSEZSZHQDGM-AWEZNQCLSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 MYGMSEZSZHQDGM-AWEZNQCLSA-N 0.000 description 1
- GSMSQVRFPFFDGY-UEDXYCIISA-N [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 GSMSQVRFPFFDGY-UEDXYCIISA-N 0.000 description 1
- INBLWYSKZOZWHV-ILXRZTDVSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 INBLWYSKZOZWHV-ILXRZTDVSA-N 0.000 description 1
- FBWKTWVMMYFUSN-CCBOZREWSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(CCNCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 FBWKTWVMMYFUSN-CCBOZREWSA-N 0.000 description 1
- FONMVIWIGDVZGP-DOMZBBRYSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 FONMVIWIGDVZGP-DOMZBBRYSA-N 0.000 description 1
- KXLBNXLOIQRAHM-PQDXNVRXSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 KXLBNXLOIQRAHM-PQDXNVRXSA-N 0.000 description 1
- XBIBHGJRGCMOON-FWYOQMDTSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 XBIBHGJRGCMOON-FWYOQMDTSA-N 0.000 description 1
- VEKHLWLBSZKBOV-VBTFFKHLSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 VEKHLWLBSZKBOV-VBTFFKHLSA-N 0.000 description 1
- FONMVIWIGDVZGP-WFASDCNBSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 FONMVIWIGDVZGP-WFASDCNBSA-N 0.000 description 1
- KXLBNXLOIQRAHM-OZIOYMQNSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1CCC[C@H]1C#N)(C1=NNN=N1)C1=C(/C=C\S1)CC2 KXLBNXLOIQRAHM-OZIOYMQNSA-N 0.000 description 1
- XBIBHGJRGCMOON-MWNCTCPHSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C(/C(=O)N([H])[H])S1)CC2 XBIBHGJRGCMOON-MWNCTCPHSA-N 0.000 description 1
- VEKHLWLBSZKBOV-URFSCYHZSA-N [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 Chemical compound [H]N([H])C(=O)C1=CC2=C(S1)C(C[C@H](C)NCC(=O)N1[C@H](C#N)C[C@@H]3C[C@@H]31)(C1=NNN=N1)C1=C(/C=C\S1)CC2 VEKHLWLBSZKBOV-URFSCYHZSA-N 0.000 description 1
- OWMULFQMWMYULX-QMMLZNLJSA-N [H]N([H])C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(CCNCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 OWMULFQMWMYULX-QMMLZNLJSA-N 0.000 description 1
- HRTUBSCIKXTIGA-IERDGZPVSA-N [H]N([H])C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 HRTUBSCIKXTIGA-IERDGZPVSA-N 0.000 description 1
- ACJNCVPBFUFVMJ-ALALXSPKSA-N [H]N([H])C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(C[C@@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 ACJNCVPBFUFVMJ-ALALXSPKSA-N 0.000 description 1
- HRTUBSCIKXTIGA-KKSFZXQISA-N [H]N([H])C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2CCC[C@H]2C#N)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 HRTUBSCIKXTIGA-KKSFZXQISA-N 0.000 description 1
- ACJNCVPBFUFVMJ-XSLZUNGDSA-N [H]N([H])C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 Chemical compound [H]N([H])C(=O)C1=CC=C(C(C[C@H](C)NCC(=O)N2[C@H](C#N)C[C@@H]3C[C@@H]32)(C2=CC=C(C(=O)N([H])[H])C=C2)C2=NNN=N2)C=C1 ACJNCVPBFUFVMJ-XSLZUNGDSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to pyrrolidine and thiazolidine-based inhibitors of dipeptidyl peptidase-IV (DPP-IV) and to methods for treating diabetes, particularly Type-2 diabetes as well as impaired glucose tolerance, impaired glucose homeostasis and complications associated with diabetes by inhibiting DPP-IV with such cyclic amido and cyclic ureido pyrrolidine and thiazolidine inhibitors.
- DPP-IV dipeptidyl peptidase-IV
- Diabetes results from the occurrence of one or more of several causative factors, and is characterized by an abnormal elevation in levels of plasma glucose (hyperglycemia). Persistent or uncontrolled hyperglycemia results in an increased probability of premature morbidity and mortality. Abnormal glucose homeostasis is usually associated with changes in the lipid, lipoprotein and apolipoprotein metabolism, or due to other metabolic and hemodynamic diseases.
- Type-2 diabetes mellitus or noninsulin dependent diabetes mellitus are especially at increased risk of suffering from macrovascular and microvascular complications, including coronary heart disease, stroke, peripheral vascular disease, hypertension, nepHropathy, neuropathy and retinopathy.
- Therapeutic control of glucose homeostasis, lipid metabolism and hypertension are critical in the clinical management and treatment of Type-2 diabetes mellitus.
- glitazone-type compounds i.e. 5-benzylthiazolidine-2,4-diones
- glitazones are agonists of the peroxisome proliferator activated receptor (PPAR), which is attributed to be responsible for their improved insulin sensitization.
- PPAR peroxisome proliferator activated receptor
- serious side effects e.g. liver toxicity
- some glitazones such as, for example, troglitazone.
- DPP-IV dipeptidyl peptidase-IV
- DPP-4 dipeptidyl peptidase-IV
- DP-IV dipeptidyl peptidase-IV
- DPP-IV is a membrane bound non-classical serine aminodipeptidase which is located in a variety of tissues (intestine, liver, lung, kidney) as well as on circulating T-lymphocytes (where the enzyme is known as CD-26). It is responsible for the metabolic cleavage of certain endogenous peptides (GLP-1(7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (e.g. GHRH, NPY, GLP-2, VIP) in vitro.
- GLP-1(7-36) certain endogenous peptides
- glucagon glucagon
- proteolytic activity against a variety of other peptides e.g. GHRH, NPY, GLP-2, VIP
- GLP-1 (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine.
- GLP-1(7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP-1(7-36) are expected to be beneficial in the prevention and treatment of Type-2 diabetes, and potentially obesity.
- GLP-1(7-36) continuous infusion
- DPP-IV has been shown to be the primary degrading enzyme of GLP-1(7-36) in vivo.
- GLP-1(7-36) is degraded by DPP-IV efficiently to GLP-1(9-36), which has been speculated to act as a physiological antagonist to GLP-1(7-36). Inhibition of DPP-IV in vivo should, therefore, potentiate endogenous levels of GLP-1(7-36) and attenuate formation of its antagonist GLP-1(9-36) and serve to ameliorate the diabetic condition.
- GLP-1 and GIP are incretins that are produced upon ingestion of food, and which stimulate production of insulin.
- Inhibition of DPP-IV causes decreased inactivation of the incretins, which in turn, results in an increase in their effectiveness in stimulating pancreatic production of insulin.
- DPP-IV inhibition therefore, results in an increase in the level of serum insulin. Since the incretins are produced upon consumption of food only, DPP-IV inhibition is not expected to increase insulin levels when not required, thereby precluding excessive lowering of blood sugar (hypoglycemia). Inhibition of DPP-IV, is therefore, is expected to increase insulin levels without increasing the risk of hypoglycemia, thereby lowering deleterious side effects associated with currently used insulin secretagogues.
- DPP-IV inhibitors have not been studied extensively as therapeutics for diseases other than diabetes, they are expected to have other potential therapeutic utilities.
- the present invention relates to a class of pyrrolidine-based inhibitors of dipeptidyl peptidase-IV (DPP-IV).
- DPP-IV dipeptidyl peptidase-IV
- the present invention provides a new class of pyrrolidine and thiazolidine DPP-IV inhibiting compounds (“DPP-IV inhibitors”).
- One aspect of the present invention includes a compound of formula (I):
- E and G are independently 6-membered aryl, or 5-membered heteroaryl or 6-membered heteroaryl;
- E may be substituted with one or more R 1 groups
- G may be substituted with one or more R 2 groups
- X and Y are divalent and are each independently: a bond, CR 4 R 5 , O, NR 4 , S, S ⁇ O, S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), S( ⁇ O) 2 N(R 4 ), C ⁇ N—OR 4 , —C(R 4 R 5 )C(R 4 R 5 )—, —C(R 4 R 5 ) C(R 4 R 5 )C(R 4 R 5 )—, —C(R 4 R 5 )C(R 4 R 5 )C(R 4 R 5 )C(R 4 R 5 )—, —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 )NR 4 —, —C(R 4 R 5 )O—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4
- R 1 and R 2 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O
- R 3 is absent or is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R a is hydrogen, CN, NO 2 , alkyl, haloalkyl, S(O) t NR 4 R 5 , S(O) t R 4 , C(O)OR 4 , C(O)R 4 , or C(O)NR 4 R 5 ; each occurrence of R 4 , R 5 , R 20 and R 21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl
- R 50 is, in each occurrence, R 20 , CN, NO 2 , S(O) t NR 20 R 21 , S(O) t R 20 , C(O)OR 20 , C(O)R 20 C( ⁇ NR a )NR 20 R 21 , C( ⁇ NR 20 )NR 21 R a , C( ⁇ NOR 20 )R 21 or C(O)NR 20 R 21 ;
- each occurrence of R 7 and R 8 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl
- R 9 is H or C 1-6 alkyl
- R 10 is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4 R 5
- R 11 and R 12 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O
- R 13a and R 13b are each independently R 5 or together are ⁇ O;
- R 14a and R 14b are each independently R 5 or together are ⁇ O;
- R 13c and R 14c are each independently R 5 ;
- Q a is CH or N
- U is —C(O)—, —C( ⁇ NR 4 )—, —(CR 4 R 5 —) p , NR 50 , S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), N(R 4 )(C ⁇ O), S( ⁇ O) 2 N(R 4 ), N(R 4 )S( ⁇ O) 2 , C ⁇ N—OR 4 , —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 ) p NR 50 —, N(R 50 )C(R 4 R 5 ) p , —O—C(R 4 R 5 )—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4 )—, —(C ⁇ NR a )—, N(C ⁇ O)NR NR 5 , N(C ⁇ O)
- W is —CH 2 —, —S—, —CHF— or —CF 2 —;
- Z is C or N
- n 1, or 2;
- n 0, 1, or 2;
- p 0 to 6;
- t 0, 1, or 2.
- Another aspect of the present invention includes a method of preparing a compound of the following formula:
- step (b) dehydrating the carboxamides of the compound from step (a) to cyano to provide a compound of formula:
- a further aspect of the present invention provides a method of preparing a compound of the following formula:
- step (b) dehydrating the carboxamide in the compound from step (a) to provide a compound of formula:
- Another aspect of the present invention provides a compound of formula A compound of formula (I):
- A is:
- E and G are independently selected from 6-membered aryl, 5-membered heteroaryl, 6-membered heteroaryl, and 5-6-membered saturated or partially saturated carbocyclic or heterocyclic rings;
- E may be substituted with one or more R 1 groups
- G may be substituted with one or more R 2 groups
- R 1 and R 2 are independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)
- R 3 is absent or is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R a is hydrogen, CN, NO 2 , alkyl, haloalkyl, S(O) t NR 4 R 5 , S(O) t R 4 , C(O)OR 4 , C(O)R 4 , or C(O)NR 4 R 5 ;
- each occurrence of R 4 , R 5 , R 20 and R 21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R 4 and R 5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may be optionally containing a heteroatom selected from O, S, or NR 50 and the 3- to 8-
- R 50 is, in each occurrence, R 20 , CN, NO 2 , S(O) t NR 20 R 21 , S(O) t R 20 , C(O)OR 20 , C(O)R 20 C( ⁇ NR a )NR 20 R 21 , C( ⁇ NR 20 )NR 21 R a , C( ⁇ NOR 20 )R 21 or C(O)NR 20 R 21 ;
- each occurrence of R 7 and R 8 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl
- R 9 is H or C 1-6 alkyl
- R 10 is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4 R 5
- R 11 and R 12 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (CO—C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R 13a and R 13b are each independently R 5 or together are ⁇ O;
- R 14a and R 14b are each independently R 5 or together are ⁇ O;
- R 13c and R 14c are each independently R 5 ;
- Q a is CH or N
- U is —C(O)—, —C( ⁇ NR 4 )—, —(CR 4 R 5 —) p , NR 50 , S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), N(R 4 )(C ⁇ O), S( ⁇ O) 2 N(R 4 ), N(R 4 )S( ⁇ O) 2 , C ⁇ N—OR 4 , —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 ) p NR 50 —, N(R 50 )C(R 4 R 5 ) p —, —O—C(R 4 R 5 )—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4 )—, —(C ⁇ NR a )—, N(C ⁇ O)NR 4 NR 5 , N(C ⁇
- W is —CH 2 —, —S—, —CHF— or —CF 2 —;
- Z is C or N
- n 1, or 2;
- n 0, 1, or 2;
- p 0 to 6;
- t 0, 1, or 2
- R 1 or R 2 is present and is:
- Another aspect of the present invention provides a compound of formula A compound of formula (I):
- A is:
- E, G, and M include a three ring system wherein M shares two carbon atoms with each of E and G;
- E, G and M are each independently selected from a 5-7-membered saturated or partially saturated carbocyclic ring, a 5-7 membered saturated or partially saturated heterocyclic ring, a 5-6-membered aromatic ring, and a 5-6-membered heteroaromatic ring;
- E may be substituted with one or more R 1 groups
- G may be substituted with one or more R 2 groups
- R 1 and R 2 are independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)
- R 3 is absent or is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R a is hydrogen, CN, NO 2 , alkyl, haloalkyl, S(O) t NR 4 R 5 , S(O) t R 4 , C(O)OR 4 , C(O)R 4 , or C(O)NR 4 R 5 ;
- each occurrence of R 4 , R 5 , R 20 and R 21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R 4 and R 5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may be optionally containing a heteroatom selected from O, S, or NR 50 and the 3- to 8-
- R 50 is, in each occurrence, R 20 , CN, NO 2 , S(O) t NR 20 R 21 , S(O) t R 20 , C(O)OR 20 , C(O)R 20 C( ⁇ NR a )NR 20 R 21 , C( ⁇ NR 20 )NR 21 R a , C( ⁇ NOR 20 )R 21 or C(O)NR 20 R 21 ;
- each occurrence of R 7 and R 8 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl
- R 9 is H or C 1-6 alkyl
- R 10 is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4 R 5
- R 11 and R 12 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (CO—C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R 13a and R 13b are each independently R 5 or together are ⁇ O;
- R 14a and R 14b are each independently R 5 or together are ⁇ O;
- R 13c and R 14c are each independently R 5 ;
- Q a is CH or N
- U is —C(O)—, —C( ⁇ NR 4 )—, —(CR 4 R 5 —) p , NR 50 , S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), N(R 4 )(C ⁇ O), S( ⁇ O) 2 N(R 4 ), N(R 4 )S( ⁇ O) 2 , C ⁇ N—OR 4 , —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 ) p NR 50 —, N(R 50 )C(R 4 R 5 ) p —, —O—C(R 4 R 5 )—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4 )—, —(C ⁇ NR a )—, N(C ⁇ O)NR 4 NR 5 , N(C ⁇
- W is —CH 2 —, —S—, —CHF— or —CF 2 —;
- Z is C or N
- n 1, or 2;
- n 0, 1, or 2;
- p 0 to 6;
- t 0, 1, or 2
- R 1 or R 2 is present and is:
- Compounds of the present invention having one or more optically active carbons can exist as racemates and racemic mixtures, diasteromeric mixtures and individual diastereomers, enantiomeric mixtures and single enantiomers, tautomers, atropisomers, and rotamers, with all isomeric forms being included in the present invention.
- Compounds described in this invention containing olefinic double bonds include both E and Z geometric isomers.
- Also included in this invention are all salt forms, polymorphs, hydrates and solvates. All of the above mentioned compounds are included within the scope of the invention.
- the present invention also provides methods of inhibiting the DPP-IV enzyme.
- the present invention further provides methods of treatment or prevention of diseases in which the dipeptidyl peptidase-IV enzyme is involved, such as diabetes and particularly Type-2 diabetes.
- the present invention also provides methods for obtaining the DPP-IV inhibiting compounds and pharmaceutical compositions comprising them either singly or in combination with one or more additional therapeutic agents for the prevention or treatment of DPP-IV enzyme medicated diseases, particularly Type-2 diabetes.
- alkyl or “alk”, as used herein alone or as part of another group, denote optionally substituted, straight and branched chain saturated hydrocarbon groups, preferably having 1 to 10 carbons in the normal chain, most preferably lower alkyl groups.
- exemplary unsubstituted such groups include methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4-trimethylpentyl, nonyl, decyl, undecyl, dodecyl and the like.
- substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkenyl, alkynyl, aryl (e.g., to form a benzyl group), cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH 2 —CO—), substituted carbamoyl ((R 4 )(R 5 )N—CO— wherein R 4 or R 5 are as defined below, except that at least one of R 4 or R 5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- groups halo, alkoxy, alkylthio, alkenyl, alkynyl, aryl (e.g., to form a benzyl group), cycloal
- lower alk or “lower alkyl” as used herein, denote such optionally substituted groups as described above for alkyl having 1 to 4 carbon atoms in the normal chain.
- alkoxy denotes an alkyl group as described above bonded through an oxygen linkage (—O—).
- alkenyl denotes optionally substituted, straight and branched chain hydrocarbon groups containing at least one carbon to carbon double bond in the chain, and preferably having 2 to 10 carbons in the normal chain.
- exemplary unsubstituted such groups include ethenyl, propenyl, isobutenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl, and the like.
- substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH 2 —CO—), substituted carbamoyl ((R 4 )(R 5 )N—CO— wherein R 4 or R 5 are as defined below, except that at least one of R 4 or R 5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- alkynyl denotes optionally substituted, straight and branched chain hydrocarbon groups containing at least one carbon to carbon triple bond in the chain, and preferably having 2 to 10 carbons in the normal chain.
- exemplary unsubstituted such groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl, and the like.
- substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH 2 —CO—), substituted carbamoyl ((R 4 )(R 5 )N—CO— wherein R 4 or R 5 are as defined below, except that at least one of R 4 or R 5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- cycloalkyl denotes optionally substituted, saturated cyclic hydrocarbon ring systems, including bridged ring systems, desirably containing 1 to 3 rings and 3 to 9 carbons per ring.
- exemplary unsubstituted such groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclododecyl, and adamantyl.
- substituents include, but are not limited to, one or more alkyl groups as described above, or one or more groups described above as alkyl substituents.
- aromatic or “aryl”, as used herein alone or as part of another group, denote optionally substituted, homocyclic aromatic groups, preferably containing 1 or 2 rings and 6 to 12 ring carbons.
- exemplary unsubstituted such groups include, but are not limited to, phenyl, biphenyl, and naphthyl.
- substituents include, but are not limited to, one or more nitro groups, alkyl groups as described above or groups described above as alkyl substituents.
- heterocycle or “heterocyclic system” denotes a heterocyclyl, heterocyclenyl, or heteroaryl group as described herein, which contains carbon atoms and from 1 to 4 heteroatoms independently selected from the group consisting of N, O and S and including any bicyclic or tricyclic group in which any of the above-defined heterocyclic rings is fused to one or more heterocycle, aryl or cycloalkyl groups.
- the nitrogen and sulfur heteroatoms may optionally be oxidized.
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom.
- heterocycles include, but are not limited to, 1H-indazole, 2-pyrrolidonyl, 2H,6H-1,5,2-dithiazinyl, 2H-pyrrolyl, 3H-indolyl, 4-piperidonyl, 4aH-carbazole, 4H-quinolizinyl, 6H-1,2,5-thiadiazinyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolinyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazalonyl, carbazolyl, 4aH-carbazolyl, b-carbolinyl, chromanyl, chromenyl, cinnolinyl,
- Heterocyclenyl denotes a non-aromatic monocyclic or multicyclic hydrocarbon ring system of about 3 to about 10 atoms, desirably about 4 to about 8 atoms, in which one or more of the carbon atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur atoms, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond.
- Ring sizes of rings of the ring system may include 5 to 6 ring atoms.
- the designation of the aza, oxa or thia as a prefix before heterocyclenyl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom.
- heterocyclenyl may be optionally substituted by one or more substituents as defined herein.
- the nitrogen or sulpHur atom of the heterocyclenyl may also be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide.
- Heterocyclenyl as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A.
- Exemplary monocyclic azaheterocyclenyl groups include, but are not limited to, 1,2,3,4-tetrahydrohydropyridine, 1,2-dihydropyridyl, 1,4-dihydropyridyl, 1,2,3,6-tetrahydropyridine, 1,4,5,6-tetrahydropyrimidine, 2-pyrrolinyl, 3-pyrrolinyl, 2-imidazolinyl, 2-pyrazolinyl, and the like.
- Exemplary oxaheterocyclenyl groups include, but are not limited to, 3,4-dihydro-2H-pyran, dihydrofuranyl, and fluorodihydrofuranyl.
- An exemplary multicyclic oxaheterocyclenyl group is 7-oxabicyclo[2.2.1]heptenyl.
- Heterocyclyl or “heterocycloalkyl,” denotes a non-aromatic saturated monocyclic or multicyclic ring system of about 3 to about 10 carbon atoms, desirably 4 to 8 carbon atoms, in which one or more of the carbon atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur. Ring sizes of rings of the ring system may include 5 to 6 ring atoms.
- the designation of the aza, oxa or thia as a prefix before heterocyclyl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom.
- the heterocyclyl may be optionally substituted by one or more substituents which may be the same or different, and are as defined herein.
- the nitrogen or sulpHur atom of the heterocyclyl may also be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide.
- Heterocyclyl as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and “J. Am. Chem. Soc.”, 82:5566 (1960).
- Exemplary monocyclic heterocyclyl rings include, but are not limited to, piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, 1,3-dioxolanyl, 1,4-dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, and the like.
- Heteroaryl denotes an aromatic monocyclic or multicyclic ring system of about 5 to about 10 atoms, in which one or more of the atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur. Ring sizes of rings of the ring system include 5 to 6 ring atoms.
- the “heteroaryl” may also be substituted by one or more substituents which may be the same or different, and are as defined herein.
- the designation of the aza, oxa or thia as a prefix before heteroaryl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom.
- a nitrogen atom of a heteroaryl may be optionally oxidized to the corresponding N-oxide.
- Heteroaryl as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and “J. Am. Chem. Soc.”, 82:5566 (1960).
- heteroaryl and substituted heteroaryl groups include, but are not limited to, pyrazinyl, thienyl, isothiazolyl, oxazolyl, pyrazolyl, furazanyl, pyrrolyl, 1,2,4-thiadiazolyl, pyridazinyl, quinoxalinyl, phthalazinyl, imidazo[1,2-a]pyridine, imidazo[2,1-b]thiazolyl, benzofurazanyl, azaindolyl, benzimidazolyl, benzothienyl, thienopyridyl, thienopyrimidyl, pyrrolopyridyl, imidazopyridyl, benzoazaindole, 1,2,3-triazinyl, 1,2,4-triazinyl, 1,3,5-triazinyl, benzthiazolyl, dioxolyl, furanyl, imidazolyl,
- amino denotes the radical —NH 2 wherein one or both of the hydrogen atoms may be replaced by an optionally substituted hydrocarbon group.
- exemplary amino groups include, but are not limited to, n-butylamino, tert-butylamino, methylpropylamino and ethyldimethylamino.
- cycloalkylalkyl denotes a cycloalkyl-alkyl group wherein a cycloalkyl as described above is bonded through an alkyl, as defined above. Cycloalkylalkyl groups may contain a lower alkyl moiety. Exemplary cycloalkylalkyl groups include, but are not limited to, cyclopropylmethyl, cyclopentylmethyl, cyclohexylmethyl, cyclopropylethyl, cyclopentylethyl, cyclohexylpropyl, cyclopropylpropyl, cyclopentylpropyl, and cyclohexylpropyl.
- arylalkyl denotes an aryl group as described above bonded through an alkyl, as defined above.
- heteroarylalkyl denotes a heteroaryl group as described above bonded through an alkyl, as defined above.
- halogen as used herein alone or as part of another group, denote chlorine, bromine, fluorine, and iodine.
- haloalkyl denotes a halo group as described above bonded though an alkyl, as defined above. Fluoroalkyl is an exemplary group.
- aminoalkyl denotes an amino group as defined above bonded through an alkyl, as defined above.
- the pHrase “bicyclic fused ring system wherein at least one ring is partially saturated” denotes an 8- to 13-membered fused bicyclic ring group in which at least one of the rings is non-aromatic.
- the ring group has carbon atoms and optionally 1-4 heteroatoms independently selected from N, O and S.
- Illustrative examples include, but are not limited to, indanyl, tetrahydronaphthyl, tetrahydroquinolyl and benzocycloheptyl.
- tricyclic fused ring system wherein at least one ring is partially saturated denotes a 9- to 18-membered fused tricyclic ring group in which at least one of the rings is non-aromatic.
- the ring group has carbon atoms and optionally 1-7 heteroatoms independently selected from N, O and S.
- Illustrative examples include, but are not limited to, fluorene, 10,11-dihydro-5H-dibenzo[a,d]cycloheptene and 2,2a,7,7a-tetrahydro-1H-cyclobuta[a]indene.
- pharmaceutically acceptable salts refers to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as, but not limited to, hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as, but not limited to, acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- inorganic acids such as, but not limited to, hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like
- organic acids such as, but not limited to
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two.
- Organic solvents include, but are not limited to, nonaqueous media like ethers, ethyl acetate, ethanol, isopropanol, or acetonitrile. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, Pa., 1990, p. 1445, the disclosure of which is hereby incorporated by reference.
- pHrase “pharmaceutically acceptable” denotes those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication commensurate with a reasonable benefit/risk ratio.
- Substituted is intended to indicate that one or more hydrogens on the atom indicated in the expression using “substituted” is replaced with a selection from the indicated group(s), provided that the indicated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- a substituent is keto (i.e., ⁇ O) group, then 2 hydrogens on the atom are replaced.
- moieties of a compound of the present invention are defined as being unsubstituted, the moieties of the compound may be substituted.
- the moieties of the compounds of the present invention may be optionally substituted with one or more groups independently selected from, but not limited to:
- cleave or “cleaving” means splitting a complex molecule into at least two separate molecules. “Cleavage products” are the separate molecules which result from cleaving.
- metabolic refers to a composition which results from a metabolic process. Examples of the results of metabolism on the compounds of the present invention include addition of —OH, hydrolysis, and cleavage.
- polymorphs refers to the various crystalline structures of the compounds of the present invention. This may include, but is not limited to, crystal morphologies (and amorphous materials), all crystal lattice forms, and all salts. Salts of the present invention can be crystalline and may exist as more than one polymorpH. Each polymorpH forms another aspect of the invention. Hydrates as well as anhydrous forms of the salt are also encompassed by the invention.
- Troc is 2-(trimethylsilyl)ethoxycarbonyl
- Et is ethyl (—CH 2 CH 3 ) or ethylene (—CH 2 CH 2 —).
- Me is methyl (—CH 3 ) or methylene (—CH 2 —).
- Boc is tert-butyloxycarbonyl.
- pharmaceutically-acceptable tricyclic moiety is meant to include, but is not limited to, benzocycloheptapyridyl, benzodiazepinyl, and benzozapinyl
- the DPP-IV inhibiting compounds are used in the manufacture of a medicament for the treatment of a disease mediated by an DPP-IV enzyme.
- DPP-IV inhibiting compounds of the present invention are used in combination with another disease modifying drug.
- diseases modifying drugs include, but are not limited to: (a) other dipeptidyl peptidase IV (DPP-IV) inhibitors such as Vildagliptin (Novartis), Sitagliptin (Merck&Co.), Saxagliptin (BMS); (b) insulin sensitizers including (i) PPAR ⁇ agonists such as the glitazones (e.g.
- the DPP-IV inhibiting compounds of the present invention are used in the treatment diseases or symptoms mediated by an DPP-IV enzyme.
- diseases or symptoms mediated by a DPP-IV enzyme include, but are not limited to, Type II (Type-2) Diabetes and Related Disorders, such as hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its 30 sequelae, vascular restenosis, irritable bowel syndrome, inflammatory bowel disease, including Crohn's disease and ulcerative colitis, other inflammatory conditions, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nepHropathy, neuropathy, cataracts, glaucoma, glomerulosclerosis, foot ulcerations and unlcerative colitis, altered gastrointestinal motility, Syndrome X, ovarian hyperandrogenism, polycystic ovarian
- Syndrome X also known as Metabolic Syndrome
- obesity is thought to promote insulin resistance, diabetes, dyslipidemia, hypertension, and increased cardiovascular risk, growth hormone deficiency, neutropenia, neuronal disorders, tumor invasion and metastasis, benign prostatic hypertrophy, gingivitis, osteoporosis, frailty of aging, intestinal injury, benign prostatic hypertrophy (BPH), and sperm motility/male contraception.
- BPH benign prostatic hypertrophy
- the DPP-IV inhibiting compounds of the present invention are useful for the prevention, delay of progression or the treatment of an early cardiac or early cardiovascular diseases or damages, renal diseases or damages, heart Failure, or heart Failure associated diseases like (i) cardiovascular diseases or damages e.g.
- cardiac hypertrophy cardiac remodelling after myocardial infarction, pulmonary congestion and cardiac fibrosis in dilated or in hypertrophic cardiomyopathy
- cardiomyopathy such as dilated cardiomyopathy or hypertrophic cardiomyopathy, mesanglial hypertrophy, or diabetic cardiomyopathy, left or right ventricular hypertrophy, arrhythmia, cardiac dysrhythmia, syncopy, angina pectoris, cardiac bypass reocclusion, intermittent claudication, diastolic and/or systolic dysfunction, diabetic myopathy, stroke prevention in congestive heart failure, hypertrophic medial thickening in arteries and/or large vessels, mesenteric vasculature hypertrophy or artherosclerosis, preferably artherosclerosis in mammalian patients with hypertension of diabetes; (ii) renal diseases or damages like renal hyperfiltration such as after portal renal ablation, proteinuria in chronic renal disease, renal arteriopathy as a consequence of hypertension, nepHrosclerosis, hypertens
- the DPP-IV inhibiting compounds of the present invention are used for the prevention, the delay of the onset, the delay of progression or the treatment of neurodegenerative disorders, cognitive disorders and for improving memory (both short term and long term) and learning ability
- the (i) neurodegenerative disorder is dementia, senile dementia, schizopHrenia, mild cognitive impairment, Alzheimer related dementia, Huntington's chores, tardive dyskinesia, hyperkinesias, mania, Morbus Parkinson, Steel-Richard syndrome, Down's syndrome, myasthenia gravis, nerve and brain trauma, vascular amyloidosis, cerebral haemorrhage I with amyloidosis, brain inflammation, Friedrich ataxia, acute confusion disorders, acute confusion disorders with apoptotic necrocytosis, amyotrophic lateral sclerosis, glaucoma, and Alzheimer's disease; (ii) cognitive disorders like cognitive deficits associated with schizopHrenia, age-induced memory impairment, cognitive deficits associated
- the DPP-IV inhibiting compounds of the present invention are used for stimulating an immune response in a subject having or at risk of having cancer
- the cancer is selected from the group consisting of basal cell carcinomas including cancers of the binary tract, bladder, urinary system, bone, brain, breast, cervical, endometrial, ovarian, uterine, choriocarcinoma, central nervous system, colon and rectal cancers, connective tissue cancer, cancer of the digestive system, esophageal, gastric, stomach, larynx, liver, pancreatic, colorectal, renal cancers; cancers of the urinary system; cancers of eye, head and neck, oral cavity, skin, prostate; cancers of biliary tract, testicular, thyroid; intra-epithelial neoplasm, leukemia, acute myeloid leukemia, acute lymphoid leukemia, chronic myeloid leukemia, chronic lymphoid leukemia; and other cancers of the respiratory system, lung, small cell lung, non-
- the DPP-IV inhibiting compounds of the present invention are useful for the treatment or prophylaxis of chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
- chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
- the DPP-IV inhibiting compounds of the present invention may be useful in the treatment of pain, neuropathic pain, rheumatoid pain, osteoarthritis pain, anesthesia adjunct in mammalian patients undergoing surgery, chronic pain in advanced cancer, treatment of refractory diarrhea, biliary pain caused by gallstones.
- the DPP-IV inhibiting compounds of the present invention are useful for the treatment of mammalian patients undergoing islet/pancreas transplantation, for the prevention or the delay of transplant rejection, or allograft rejection in transplantation, for improving pancreatic function by increasing the number and size of pancreatic beta-cells in the treatment of Type 1 diabetes patients, and for improving pancreatic function by increasing the number and size of pancreatic beta-cells in general.
- the DPP-IV inhibiting compounds of the present invention are useful for the treatment of mammalian patients with acne, skin disorders (e.g. pigmentation disorders or psoriasis), scleroderma, mycoses; anxiety, anxiety neurosis, major depression disorder, drug abuse, alcohol addiction, insomnia, chronic fatigue, sleep apnea; anorexia nervosa; epilepsy; migrane; encephalomyelitis; osteoarthritis, osteoporosis, calcitonin-induced osteoporosis; male and female sexual dysfunction, infertility; Type 1 diabetes; immunosuppression, HIV infection; hematopoiesis, anemia; and for weight reduction.
- skin disorders e.g. pigmentation disorders or psoriasis
- scleroderma mycoses
- anxiety anxiety neurosis
- major depression disorder drug abuse
- alcohol addiction insomnia
- chronic fatigue sleep apnea
- anorexia nervosa epilepsy
- the DPP-IV inhibiting compounds of the present invention are useful for the prevention, delay of progression or treatment of (i) bacterial infections from Escherichia coli, Staphylococcus, Streptoococcus, Pseudomonas, Clostridium difficile infection, Legionella, Pneumococcus, HaemopHilus, Klebsiella, Enterobacter, Citrobacter, Neisseria, Shigella, Salmonella, Listeria, Pasteurella, Streptobacillus, Spirillum, Treponema, Actinomyces, Borrelia, Corynebacterium, Nocardia, Gardnerella, Campylobacter, Spirochaeta, Proteus, Bacteriodes, Helicobacter pylori , and anthrax infection; (ii) mycobacterial infection from tuberculosis and leprosy; (iii) viral infection from HIV, Herpes simplex virus 1, Herpes simplex virus 2,
- the compounds from this invention are suitable for oral, sublingual, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (aerosol inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient.
- parenteral including subcutaneous, intramuscular, and intravenous
- ocular ophthalmic
- pulmonary aserosol inhalation
- nasal administration although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient.
- the compounds from this invention are conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- the DPP-IV inhibiting compounds of the present invention are synthesized by the general method shown in Schemes 1-14.
- bromotoluene derivatives were treated with n-butyllithium and heated, followed by treatment with dry-ice in an appropriate solvent to afford the desired compound.
- the acid can be prepared by Grignard reaction followed by treatment with dry-ice in an appropriate solvent. Esterification of the compound followed by NBS bromination and subsequent conversion to the phosphonium salt in a suitable solvent and heating affords the desired compound.
- Wittig reaction of the phosphonium salt with a suitable aldehyde in an appropriate solvent and heating, followed by saponification of the ester moiety and subsequent catalytic hydrogenation affords the desired compound. Cyclisation of the compound with polyphosphoric acid in sulfolane and heating affords the desired compound after purification.
- R 1 ⁇ COOMe the tricyclic product from the polyphosphoric acid step was treated with thionylchloride in an alcohol. Reduction of the ketone with a metal hydride in an appropriate solvent yields the compound after purification. Treatment of the alcohol with thionylchloride in a suitable solvent affords the final desired compound.
- bromotoluene derivatives are treated with Magnesium in a Grignard reaction followed by treatment with dry-ice in an appropriate solvent to yield the desired acid. This acid is then treated with sec-butyllithium in an appropriate solvent at lower temperature. The anion is added at lower temperature to a solution of a commercially available benzylchloride in an appropriate solvent to afford the desired compound. Cyclisation of the compound with polyphosphoric acid in sulfolane and heating affords the desired compound.
- the tricyclic product from the polyphosphoric acid step with R 1 ⁇ R 2 ⁇ Cl was treated with KCN, a Pd-catalyst, a suitable ligand and a suitable base in an appropriate solvent to afford the dicyano compound, which was converted to the diacid by treatment with base in a suitable solvent.
- Ester formation using thionylchloride in an alcohol and reduction of the ketone with a metal hydride in an appropriate solvent yields the compound after purification.
- Treatment of the alcohol with thionylchloride in a suitable solvent affords the final desired compound.
- prolinamide is treated with fumarylchloride in an appropriate solvent to afford the desired compound. This compound is then treated with oxalylchloride in dimethylformamide to afford the desired compound after purification.
- the coupling product of prolinamide with fumarylchloride can be treated with trifluoroacetic acid anhydride in a suitable solvent to afford the desired compound. Ozonolysis of this compound at ⁇ 78° C. in a suitable solvent, followed by reductive workup affords the desired final compound as a mixture of the aldehyde and its methyl hemiacetal.
- Substituted or unsubstituted tricyclic chlorides are treated in an appropriate solvent with an excess of suitable amines to afford the desired product after purification.
- the reaction product contains additional amino protecting groups like Boc, they are cleaved by acid treatment to afford the desired compound.
- Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative yields the final desired product after purification.
- the amines are treated with a suitable aldehyde (D-CHO) via reductive amination to afford the final compound after purification.
- Substituted or unsubstituted tricycles containing a nitrogen at the doubly benzylic position are treated with bromoacetylbromide and heated to afford the desired compounds. Treating these compounds with sodium azide or sodium cyanide in a suitable solvent and heating affords the desired azido or cyano compounds after purification. Catalytic hydrogenation or reduction with Lithium aluminium hydride in a suitable solvent affords the desired amine compounds. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative yields the final desired product after purification.
- Substituted or unsubstituted tricyclic ketones with Y ⁇ C(R 4 ) ⁇ C(R 5 ) are treated with malonic acid at elevated temperatures to afford the desired product after purification. These compounds are converted to the corresponding amides by treatment with isobutylchloroformate and ammonia. The amides are then converted to the desired amine products with Y ⁇ C(R 4 ) ⁇ C(R 5 ) by reduction with lithium aluminium hydride or to the desired amine products with Y ⁇ C(R 4 R 5 )C(R 4 R 5 ) by reduction with lithium aluminium hydride followed by catalytic hydrogenation with a suitable catalyst. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired product after purification.
- Treating tricyclic ketones in a Reformatskij reaction affords the desired product after purification.
- Reduction with LiAlH 4 in a suitable solvent affords the alcohol products with R 3 ⁇ OH after purification.
- Activation of one of the hydroxyl groups with sulfonylchlorides in a suitable solvent followed by treatment with NaN 3 affords the desired compounds after purification.
- Reduction of the azide reaction products with a catalyst in a suitable solvent affords the desired amine compounds after purification.
- Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired products after purification.
- Treating the amines with R 3 ⁇ OH with acid in a suitable solvent yields the desired unsaturated amine products.
- Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired products after purification.
- Substituted or unsubstituted suberylchlorides are treated in a suitable solvent with a slight excess of AgCN and heated to afford the desired product after purification.
- the nitrile containing compound is then treated with sodium hydride in a suitable solvent and heated.
- the mixture is then treated at rt with a suitable dibromoalkene and heated to give an intermediate which after treatment with sodium azide or potassium phthalimide in an appropriate solvent and heating affords the desired compound after purification.
- Treating the mixture after the addition of sodium hydride at rt with a suitable sulfamidate in an appropriate solvent affords the desired Teoc-protected compound after heating for several hours and subsequent purification.
- the Teoc protecting group is removed by treatment with acid to afford the desired amine compounds.
- the ester moieties are removed by treatment with base in an appropriate solvent to afford the desired dicarboxylic acid derivatives after purification.
- a commercially available hydroxyl-proline derivative is treated with base and alkylated with allylbromide in an appropriate solvent to afford the allyl-protected amino acid after purification.
- This compound is then treated at ⁇ 30° C. with an appropriate base, triflic anhydride and then an appropriately protected diamino acid in an appropriate solvent to afford the desired compound after purification.
- the compound is treated with EDCI and base in an appropriate solvent to afford the desired compound after purification. Cleavage of Fmoc protecting group by treatment with an suitable base affords the desired product.
- the free amine is then treated in the presence of an suitable polymer supported base with sulfonyl chlorides, acid chlorides or isocyanates to afford the desired compounds after purification. Removal of the Boc-protecting group with acid in a suitable solvent affords the final desired compounds after purification.
- the primary amine is treated in an appropriate solvent with aldehydes or ketones in a reductive amination reaction to afford the desired products.
- the commercially available N-Boc-protected hydroxy amino acid ester can be treated with trifluoroacetic acid anhydride.
- the nucleopHilic displacement reaction of the triflate with commercially available amines affords the desired products, after saponification of the ester moiety with base and purification.
- These compounds are then treated with EDCl and a base in an suitable solvent to afford the cyclic amides after purification. These compounds are converted to the desired products by removing the Boc-protection group.
- the commercially available bridged piperazine derivate is treated with a commercially available aziridine ester in an appropriate solvent to afford the desired compound after purification.
- the desired product reacts in presence of a base with an acid chloride or sulfonic acid chloride to yield the desired products after purification.
- the free acids are treated with EDCI in the presence of an appropriate base and a suitable amine derivative to afford the desired compounds after purification.
- the Cbz-protecting group is then removed by treatment with TMSI and subsequent purification to afford the desired final compounds.
- each of the structures of “B” bonds to the “A” structures on its left side and to the “D” structures on its right side as each is depicted below.
- the compound A-B-D chooses an “A” which includes the following:
- A is desirably
- the “B” structures are chosen from:
- B is one of structure (a), (b), (c), and (d). More desirably, B is structure (b)
- the “D” structures are chosen from:
- E, G, and M represent a three ring system wherein M shares two carbon atoms with each of E and G;
- E and G are each independently selected from 6-membered aryl, 5-membered heteroaryl; 6-membered heteroaryl; a 5-7-membered saturated or partially saturated carbocyclic ring; and a 5-7 membered saturated or partially saturated heterocyclic ring; desirably E and G are substituted phenyl; M is a 5-7-membered saturated or partially saturated carboxylic or heterocyclic ring, or a 5-6-membered aromatic or heteroaromatic ring.
- E may be substituted with one or more R 1 groups
- G may be substituted with one or more R 2 groups
- X and Y are divalent and are each independently: a bond, CR 4 R 5 , O, NR 4 , S, S ⁇ O, S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), S( ⁇ O) 2 N(R 4 ), C ⁇ N—OR 4 , —C(R 4 R 5 )C(R 4 R 5 )—, —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 )NR 4 —, —C(R 4 R 5 )O—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4 )—, —(C ⁇ NR a )—, N(C ⁇ O)NR 4 NR 5 , N(C ⁇ O)R 4 , N(C ⁇ O)OR 4 , NS( ⁇ O) 2 NR 4 NR 5 ,
- R 1 and R 2 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O
- R 3 is absent or is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R 3 is absent or is —H, —OH, —CO 2 H, —CN, —CONR 4 R 5 , R 5 , aryl, NH(C ⁇ O)R 4 , NH(SO 2 )R 4 , heteroaryl —SO 3 H, —PO 3 H 2 , —CONR 4 R 5 , R 5 , aryl, NH(C ⁇ O)R 4 , or NH(SO 2 )R 4 , and more desirably, R 3 is —CONR 4 R 5 or tetrazolyl.
- R a is hydrogen, CN, NO 2 , alkyl, haloalkyl, S(O) t NR 4 R 5 , S(O) t R 4 , C(O)OR 4 , C(O)R 4 , or C(O)NR 4 R 5 ;
- each occurrence of R 4 , R 5 , R 20 and R 21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R 4 and R 5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may optionally contain a heteroatom selected from O, S, or NR 50 and the 3- to 8-membere
- R 50 is, in each occurrence, R 20 , CN, NO 2 , S(O) t NR 20 R 21 , S(O) t R 20 , C(O)OR 20 , C(O)R 20 C( ⁇ NR a )NR 20 R 21 , C( ⁇ NR 20 )NR 21 R a , C( ⁇ NOR 20 )R 21 or C(O)NR 20 R 21 ;
- each occurrence of R 7 and R 8 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl
- R 9 is H or C 1-6 alkyl, desirably H.
- R 10 is halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (CO—C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (C 0 -C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (CO—C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C
- R 11 and R 12 are each independently: halogen, CF 3 , COR 4 , OR 4 , NR 4 R 5 , NO 2 , CN, SO 2 OR 4 , CO 2 R 4 , CONR 4 R 5 , CO 2 H, SO 2 NR 4 R 5 , S(O) t R 4 , SO 3 H, OC(O)R 4 , OC(O)NR 4 R 5 , NR 4 C(O)R 5 , NR 4 CO 2 R 5 , (C 0 -C 6 )-alkyl-C( ⁇ NR a )NHR 4 , (C 0 -C 6 )-alkyl-C( ⁇ NR 4 )NHR a , (CO—C 6 )-alkyl-NR 4 C( ⁇ NR 4 )NR 4 R 5 , (C 0 -C 6 )-alkyl-C(O)OR 4 , (C 0 -C 6 )-alkyl-C(O)NR 4
- R 13a and R 13b are each independently R 5 or together are ⁇ O;
- R 14a and R 14b are each independently R 5 or together are ⁇ O;
- R 13c and R 14c are each independently R 5 ;
- Q a is CH or N
- Q b is CH or N
- U is —C(O)—, —C( ⁇ NR 4 )—, —(CR 4 R 5 —) p , NR 50 , S( ⁇ O) 2 , C( ⁇ O), (C ⁇ O)N(R 4 ), N(R 4 )(C ⁇ O), S( ⁇ O) 2 N(R 4 ), N(R 4 )S( ⁇ O) 2 , C ⁇ N—OR 4 , —C(R 4 ) ⁇ C(R 5 )—, —C(R 4 R 5 ) p NR 50 —, N(R 50 )C(R 4 R 5 ) p —, —O—C(R 4 R 5 )—, —C(R 4 R 5 )S( ⁇ O) t —, —(C ⁇ O)O—, —(C ⁇ NR a )N(R 4 )—, —(C ⁇ NR a )—, N(C ⁇ O)NR 4 NR 5 , N(C ⁇
- W is —CH 2 —, —S—, —CHF— or —CF 2 —;
- Z is C or N
- n 1, or 2;
- n 0, 1, or 2;
- p 0 to 6;
- t 0, 1, or 2.
- Compounds of the present invention having one or more optically active carbons can exist as racemates and racemic mixtures, diasteromeric mixtures and individual diastereomers, enantiomeric mixtures and single enantiomers, tautomers, atropisomers, and rotamers, with all isomeric forms being included in the present invention.
- Compounds described in this invention containing olefinic double bonds include both E and Z geometric isomers.
- Also included in this invention are all salt forms, polymorphs, hydrates and solvates. All of the above mentioned compounds are included within the scope of the invention.
- the DPP-IV inhibition activity of the DPP-IV inhibitor compounds of the present invention may be measured using any suitable assay known in the art. A standard in vitro assay for measuring DPP-IV inhibitor activity is described.
- prolinamide (5 g) was first treated with bromacetylbromide (4.2 ml) in CH 2 Cl 2 and then with trifluoracetic acid anhydride in CH 2 Cl 2 as described in WO 98/19998 to afford the title compound (7.85 g; 83%).
- 1 HNMR ⁇ (CDCl 3 ) 2.05-2.40 (m, 4H), 3.51-3.70 (m, 2H), 3.80-3.85 (m, 2H), 4.70-4.86 (m, 1H).
- L-prolinamide 25 g was dissolved in CH 2 Cl 2 (1200 ml) and triethylamine (30 ml) and 4-dimethylaminopyridine (1.9 g) added. The mixture was cooled to 0° C. and treated with fumaryl chloride (11.7 ml). The dark mixture was stirred at rt for 16 h and cooled to 0° C. TFAA (77 ml) was added dropwise under stirring and the solution allowed to warm to rt over 6 hours. The reaction mixture was stirred at rt for 1 to 2 days. Ice (500 g) was added followed by cautious addition of sat. NaHCO 3 (600 ml).
- step E The title compound of step E (370 mg) above was dissolved in toluene (5 ml) and cooled to ⁇ 15° C. Thionyl chloride (286 mg) was slowly added and the reaction was allowed to come to RT and run overnight. The solution was neutralized with triethylamine and directly used in the next step.
- the dark filtrate was poured into a separatory funnel and the phases were separated with the aid of additional Et 2 O (100 ml).
- the aqueous phase was extracted with Et 2 O/CHCl 3 1:1 (2 ⁇ 100 ml).
- the combined organic phases were washed with 10% NH 4 OH solution (4 ⁇ 110 ml, until the basic phase was no longer blue), with H 2 O (100 ml), and brine (100 ml).
- the organic phase was separated, dried over MgSO 4 and concentrated.
- the residue was mixed with NaOH (24.8 g) and diethylene glycol (275 ml) was added together with a few drops of H 2 O. The mixture was heated at 215-220° C. overnight.
- the bromoethyl compound (1.42 g) was dissolved in anhydrous DMF (18 ml) and treated with potassium phthalimide (1.96 g). The suspension was stirred at 80° C. overnight. The solvent was removed and the residue partitioned between EtOAc (50 ml), H 2 O (50 ml) and brine (50 ml).
- Step D If one were to follow a similar procedure as described in Preparative Example 54, but using 3-fluorobenzaldehyde in Step A and omitting Step D, one would obtain the desired compound.
- hydroxylamine hydrochloride (401 mg) was suspended in anhydrous MeOH (14 ml) and a 5.5 M solution of sodium methoxide in MeOH (0.946 ml) added. This mixture was stirred at rt for 45 min and the title compound from Preparative Example 61 Step A (1400 mg) was added. The resulting mixture was heated in a closed vessel at 100° C. overnight and subsequently allowed to cool down to rt. Due to incomplete conversion, hydroxylamine hydrochloride (401 mg) and a 5.5 M solution of sodium methoxide in MeOH (0.946 ml) were added and the mixture was heated again at 100° C. for 20 h.
- N-hydroxyamidine product from Preparative Example 66 Step A (300 mg) was dissolved in anhydrous dichloromethane (5 ml), the solution cooled down to 0° C. and triethylamine (147 ⁇ l) and trifluoroacetic anhydride (103 ⁇ l) added. The reaction mixture was stirred at rt overnight. Due to incomplete conversion, triethylamine (221 ⁇ l) and trifluoroacetic anhydride (155 ⁇ l) were added at 0° C. and stirring was continued at rt for 3 d. Dichloromethane (9 ml) and water (10 ml) were added to the stirred mixture.
- Tosylmethyl isocyanide was dissolved in DMSO (10 ml) under N 2 at 10° C. and KOtBu (1.36 g) was added. The mixture was stirred for 5 min and MeOH (0.173 ml) was added. The title compound from Step B above (0.8 g) was immediately added to the mixture. After 10 min dibromoethane (1.51 ml) was added and stirring was continued for 1 h at rt. The mixture was diluted with EtOAc (10 ml) and sat. NH 4 Cl (30 ml) was added. The organic phase was separated and the aqueous phase was extracted with EtOAc (2 ⁇ 50 ml).
- N-cyclohexylcarbodiimde-N′-methyl polystyrene resin (1.9 g) was suspended in 5 ml dichloromethane and agitated for 5 Min.
- the mixture was agitated for 16 h, filtered and the resin washed with 2 ⁇ 5 ml dichloromethane and 5 ml methanol.
- the combined filtrates were concentrated and the residue purified by flash chromatography (silica, CH 2 Cl 2 /MeOH, 9:1) to afford the title compound (500 mg; 91%).
- Example numbers 336-399 were intentionally excluded.
- Example numbers 435-499 were intentionally excluded.
- Example numbers 536-599 were intentionally excluded.
- Step A Step B Product 601 300 CH 3 NH 2 NH 3 602 300 NH 3 603 300 NH 3 604 61 NH 3 NH 3 605 61 CH 3 NH 2 NH 3 606 61 NH 3 607 61 NH 3 608 65 NH 3 NH 3 609 65 CH 3 NH 2 NH 3 610 65 NH 3 611 65 NH 3 612 300 NH 3 CH 3 NH 2 613 300 CH 3 NH 2 CH 3 NH 2 614 300 CH 3 NH 2 615 300 CH 3 NH 2 616 61 NH 3 CH 3 NH 2 617 61 CH 3 NH 2 CH 3 NH 2 618 61 CH 3 NH 2 619 61 CH 3 NH 2 620 65 NH 3 CH 3 NH 2 621 65 CH 3 NH 2 CH 3 NH 2 622 65 CH 3 NH 2 623 65 CH 3 NH 2 624 300 NH 3 (CH 3 ) 2 NH 625 300 CH 3 NH 2 (CH 3 ) 2 NH 625 300 CH
- Example numbers 636-679 were intentionally excluded.
- Example numbers 688-699 were intentionally excluded.
- Example numbers 736-779 were intentionally excluded.
- Example numbers 789-799 were intentionally excluded.
Abstract
The present invention relates generally to pyrrolidine and thiazolidine DPP-IV inhibitor compounds. The present invention also provides synthetic methods for preparation of such compounds, methods of inhibiting DPP-IV using such compounds and pharmaceutical formulations containing them for treatment of DPP-IV mediated diseases, in particular, Type-2 diabetes.
Description
- This application is a continuation of U.S. application Ser. No. 11/409,481, filed Apr. 21, 2006, now U.S. Pat. No. 7,553,861, the entire contents of which are incorporated herein by reference.
- The present invention relates to pyrrolidine and thiazolidine-based inhibitors of dipeptidyl peptidase-IV (DPP-IV) and to methods for treating diabetes, particularly Type-2 diabetes as well as impaired glucose tolerance, impaired glucose homeostasis and complications associated with diabetes by inhibiting DPP-IV with such cyclic amido and cyclic ureido pyrrolidine and thiazolidine inhibitors.
- Diabetes results from the occurrence of one or more of several causative factors, and is characterized by an abnormal elevation in levels of plasma glucose (hyperglycemia). Persistent or uncontrolled hyperglycemia results in an increased probability of premature morbidity and mortality. Abnormal glucose homeostasis is usually associated with changes in the lipid, lipoprotein and apolipoprotein metabolism, or due to other metabolic and hemodynamic diseases.
- Patients afflicted with Type-2 diabetes mellitus or noninsulin dependent diabetes mellitus (NIDDM), are especially at increased risk of suffering from macrovascular and microvascular complications, including coronary heart disease, stroke, peripheral vascular disease, hypertension, nepHropathy, neuropathy and retinopathy. Therapeutic control of glucose homeostasis, lipid metabolism and hypertension are critical in the clinical management and treatment of Type-2 diabetes mellitus.
- The currently available therapeutics for treating available Type-2 diabetes, although effective, have recognized limitations. Compounds based on sulfonylureas (e.g. tolbutamide, glipizide, etc.), which stimulate the pancreatic beta-cells to secrete more insulin, are limited by the development of inhibitor resistant tissues, causing them to become inefficient or ineffective, even at high doses. Biguanide compounds, on the other hand, increase insulin sensitivity so as to cause correction of hyperglycemia to some extent. However, clinically used biguanides such as phenformin and metformin can induce side-effects such as lactic acidosis, nausea and diarrhea.
- The more recent glitazone-type compounds (i.e. 5-benzylthiazolidine-2,4-diones) substantially increase insulin sensitivity in muscle, liver and adipose tissue resulting in either partial or complete correction of the elevated plasma levels of glucose without occurrence of hypoglycemia. Currently used glitazones are agonists of the peroxisome proliferator activated receptor (PPAR), which is attributed to be responsible for their improved insulin sensitization. However, serious side effects (e.g. liver toxicity) have been known to occur with some glitazones such as, for example, troglitazone. Compounds that are inhibitors of the dipeptidyl peptidase-IV (“DPP-IV”, “DPP-4” or “DP-IV”) enzyme are also under investigation as drugs that may be useful in the treatment of diabetes, and particularly Type-2 diabetes. See for example, WO 97/40832, WO 98/19998, and U.S. Pat. No. 5,939,560.
- DPP-IV is a membrane bound non-classical serine aminodipeptidase which is located in a variety of tissues (intestine, liver, lung, kidney) as well as on circulating T-lymphocytes (where the enzyme is known as CD-26). It is responsible for the metabolic cleavage of certain endogenous peptides (GLP-1(7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (e.g. GHRH, NPY, GLP-2, VIP) in vitro.
- The usefulness of DPP-IV inhibitors in the treatment of Type-2 diabetes is based on the fact that DPP-IV in vivo readily inactivates glucagon like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP). GLP-1 (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine. GLP-1(7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP-1(7-36) are expected to be beneficial in the prevention and treatment of Type-2 diabetes, and potentially obesity. To support this claim, exogenous administration of GLP-1(7-36) (continuous infusion) in diabetic patients has demonstrated efficacy in this patient population. GLP-1(7-36) is degraded rapidly in vivo and has been shown to have a short half-life in vivo (t1/2 of about 1.5 min). Based on a study of genetically bred DPP-IV KO mice and on in vivo/in vitro studies with selective DPP-IV inhibitors, DPP-IV has been shown to be the primary degrading enzyme of GLP-1(7-36) in vivo. GLP-1(7-36) is degraded by DPP-IV efficiently to GLP-1(9-36), which has been speculated to act as a physiological antagonist to GLP-1(7-36). Inhibition of DPP-IV in vivo should, therefore, potentiate endogenous levels of GLP-1(7-36) and attenuate formation of its antagonist GLP-1(9-36) and serve to ameliorate the diabetic condition.
- GLP-1 and GIP are incretins that are produced upon ingestion of food, and which stimulate production of insulin. Inhibition of DPP-IV causes decreased inactivation of the incretins, which in turn, results in an increase in their effectiveness in stimulating pancreatic production of insulin. DPP-IV inhibition therefore, results in an increase in the level of serum insulin. Since the incretins are produced upon consumption of food only, DPP-IV inhibition is not expected to increase insulin levels when not required, thereby precluding excessive lowering of blood sugar (hypoglycemia). Inhibition of DPP-IV, is therefore, is expected to increase insulin levels without increasing the risk of hypoglycemia, thereby lowering deleterious side effects associated with currently used insulin secretagogues. Although DPP-IV inhibitors have not been studied extensively as therapeutics for diseases other than diabetes, they are expected to have other potential therapeutic utilities.
- The present invention relates to a class of pyrrolidine-based inhibitors of dipeptidyl peptidase-IV (DPP-IV). In particular, the present invention provides a new class of pyrrolidine and thiazolidine DPP-IV inhibiting compounds (“DPP-IV inhibitors”).
- One aspect of the present invention includes a compound of formula (I):
-
A-B-D (I) - and all stereoisomers, diastereomers, racemic mixtures and pharmaceutically acceptable salts thereof and all polymorphs; wherein A is:
-


- wherein
- E and G are independently 6-membered aryl, or 5-membered heteroaryl or 6-membered heteroaryl;
- E may be substituted with one or more R1 groups;
- G may be substituted with one or more R2 groups;
- X and Y are divalent and are each independently: a bond, CR4R5, O, NR4, S, S═O, S(═O)2, C(═O), (C═O)N(R4), S(═O)2N(R4), C═N—OR4, —C(R4R5)C(R4R5)—, —C(R4R5) C(R4R5)C(R4R5)—, —C(R4R5)C(R4R5)C(R4R5)C(R4R5)—, —C(R4)═C(R5)—, —C(R4R5)NR4—, —C(R4R5)O—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, NS(═O)2R4; or aryl, heteroaryl, cycloalkyl or heterocyclic ring, all of which may be optionally substituted;
- R1 and R2 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all of which may be optionally substituted;
- R3 is absent or is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocyclyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all of which may be optionally substituted;
- Ra is hydrogen, CN, NO2, alkyl, haloalkyl, S(O)tNR4R5, S(O)tR4, C(O)OR4, C(O)R4, or C(O)NR4R5; each occurrence of R4, R5, R20 and R21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R4 and R5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may optionally contain a heteroatom selected from O, S, or NR50 and the 3- to 8-membered ring may be optionally substituted;
- R50 is, in each occurrence, R20, CN, NO2, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(═NRa)NR20R21, C(═NR20)NR21Ra, C(═NOR20)R21 or C(O)NR20R21;
- each occurrence of R7 and R8 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C1-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- R9 is H or C1-6 alkyl;
- R10 is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C1-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, B(OH)2, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- R11 and R12 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- R13a and R13b are each independently R5 or together are ═O;
- R14a and R14b are each independently R5 or together are ═O;
- R13c and R14c are each independently R5;
- Qa is CH or N;
- U is —C(O)—, —C(═NR4)—, —(CR4R5—)p, NR50, S(═O)2, C(═O), (C═O)N(R4), N(R4)(C═O), S(═O)2N(R4), N(R4)S(═O)2, C═N—OR4, —C(R4)═C(R5)—, —C(R4R5)pNR50—, N(R50)C(R4R5)p, —O—C(R4R5)—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR NR5, N(C═O)R4, N(C═O)ORa, NS(═O)2NR4NR4, NS(═O)2R4, or an optionally substituted aryl, heteroaryl, cycloalkyl or heterocyclic ring, all of which may be optionally substituted;
- W is —CH2—, —S—, —CHF— or —CF2—;
- Z is C or N;
- m is 1, or 2;
- n is 0, 1, or 2;
- p is 0 to 6;
- q is 0 to 6; and
- t is 0, 1, or 2.
- Another aspect of the present invention includes a method of preparing a compound of the following formula:
-

- comprising (a) coupling prolinamide with fumarylchloride to provide a compound of the following formula:
-

- (b) dehydrating the carboxamides of the compound from step (a) to cyano to provide a compound of formula:
-

- and (c) cleaving the C═C bond with an oxidizing agent either: (1) in the presence of methanol, and then adding a reducing agent to the reaction mixture, or (2) and reacting the cleavage products with a reducing agent and subsequently adding methanol to the cleavage product mixture.
- A further aspect of the present invention provides a method of preparing a compound of the following formula:
-

- comprising: (a) coupling a compound of formula:
-

- with fumaryl chloride to provide a compound of formula
-

- (b) dehydrating the carboxamide in the compound from step (a) to provide a compound of formula:
-

- and (c) cleaving the C═C bond with an oxidizing agent either: (1) in the presence of methanol, and then adding a reducing agent to the reaction mixture, or (2) and reacting the cleavage products with a reducing agent and subsequently adding methanol to the cleavage product mixture.
- Another aspect of the present invention provides a compound of formula A compound of formula (I):
-
A-B-D (I) - wherein A is:
-

- B is:
-

- and
- D is:
-

- wherein
- E and G are independently selected from 6-membered aryl, 5-membered heteroaryl, 6-membered heteroaryl, and 5-6-membered saturated or partially saturated carbocyclic or heterocyclic rings;
- E may be substituted with one or more R1 groups;
- G may be substituted with one or more R2 groups;
- R1 and R2 are independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- R3 is absent or is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- Ra is hydrogen, CN, NO2, alkyl, haloalkyl, S(O)tNR4R5, S(O)tR4, C(O)OR4, C(O)R4, or C(O)NR4R5;
- each occurrence of R4, R5, R20 and R21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R4 and R5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may be optionally containing a heteroatom selected from O, S, or NR50 and the 3- to 8-membered ring may be optionally substituted;
- R50 is, in each occurrence, R20, CN, NO2, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(═NRa)NR20R21, C(═NR20)NR21Ra, C(═NOR20)R21 or C(O)NR20R21;
- each occurrence of R7 and R8 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C1-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R9 is H or C1-6alkyl;
- R10 is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, B(OH)2, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R11 and R12 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (CO—C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R13a and R13b are each independently R5 or together are ═O;
- R14a and R14b are each independently R5 or together are ═O;
- R13c and R14c are each independently R5;
- Qa is CH or N;
- U is —C(O)—, —C(═NR4)—, —(CR4R5—)p, NR50, S(═O)2, C(═O), (C═O)N(R4), N(R4)(C═O), S(═O)2N(R4), N(R4)S(═O)2, C═N—OR4, —C(R4)═C(R5)—, —C(R4R5)pNR50—, N(R50)C(R4R5)p—, —O—C(R4R5)—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, NS(═O)2R4, or an optionally substituted aryl, heteroaryl, cycloalkyl or heterocyclic ring, all of which may be optionally substituted;
- W is —CH2—, —S—, —CHF— or —CF2—;
- Z is C or N;
- m is 1, or 2;
- n is 0, 1, or 2;
- p is 0 to 6;
- q is 0 to 6; and
- t is 0, 1, or 2
- wherein: when E and G are both phenyl either:
- (1) at least one of R1 or R2 is present and is:
- CF3, COR4, OR4, NR4R5, NO2, CN, SO20R4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, (C5-20)alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted; and wherein OR4 is alkoxy, OR4 is (C5-20) alkoxy; or (2) and when B is (b) R7 and R8 are not selected from hydrogen, hydroxy, hydroxymethyl, and phenyl; or (3) and when B is (b) or (f), R9 is: C1-6 alkyl.
- Another aspect of the present invention provides a compound of formula A compound of formula (I):
-
A-B-D (I) - wherein A is:
-

-
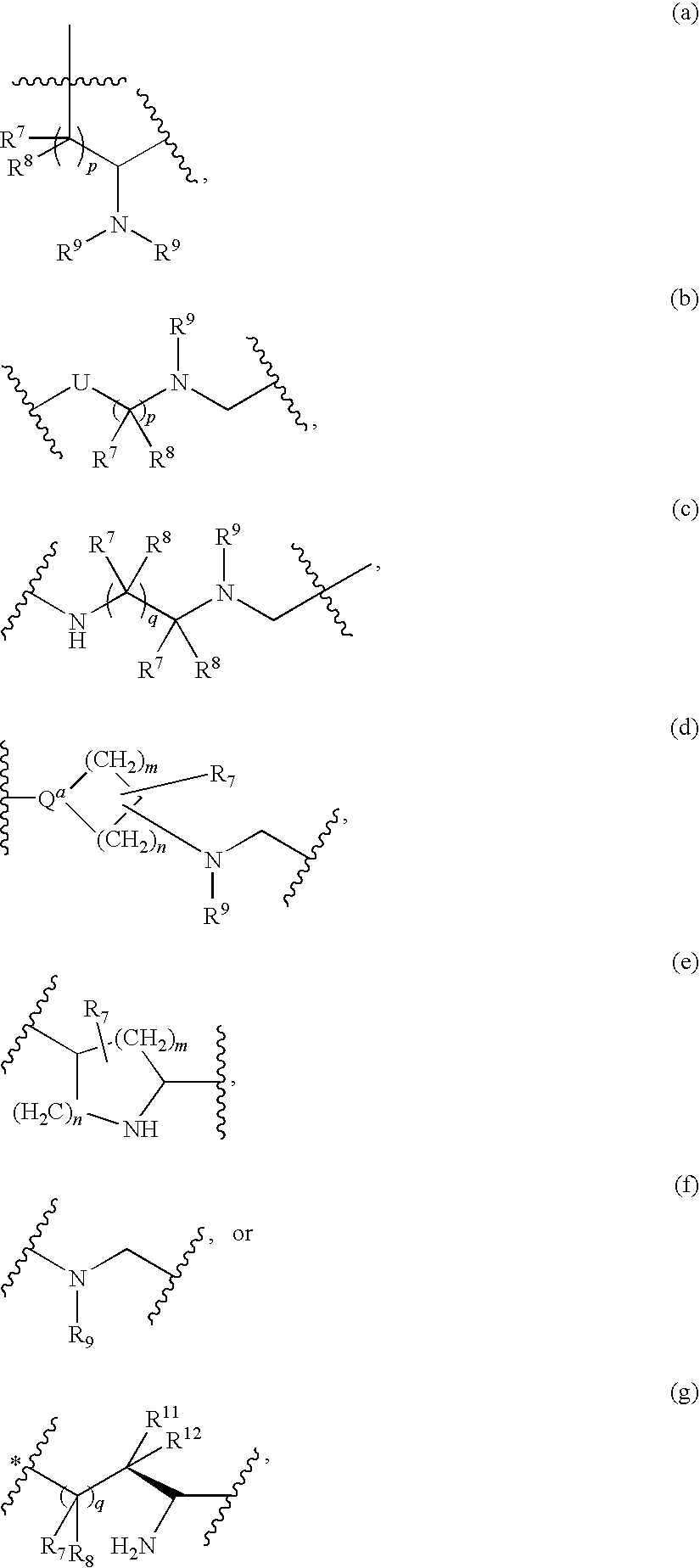
- and
- D is:
-

- wherein
- E, G, and M include a three ring system wherein M shares two carbon atoms with each of E and G;
- E, G and M are each independently selected from a 5-7-membered saturated or partially saturated carbocyclic ring, a 5-7 membered saturated or partially saturated heterocyclic ring, a 5-6-membered aromatic ring, and a 5-6-membered heteroaromatic ring;
- E may be substituted with one or more R1 groups;
- G may be substituted with one or more R2 groups;
- R1 and R2 are independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- R3 is absent or is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O) t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted;
- Ra is hydrogen, CN, NO2, alkyl, haloalkyl, S(O)tNR4R5, S(O)tR4, C(O)OR4, C(O)R4, or C(O)NR4R5;
- each occurrence of R4, R5, R20 and R21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R4 and R5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may be optionally containing a heteroatom selected from O, S, or NR50 and the 3- to 8-membered ring may be optionally substituted;
- R50 is, in each occurrence, R20, CN, NO2, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(═NRa)NR20R21, C(═NR20)NR21Ra, C(═NOR20)R21 or C(O)NR20R21;
- each occurrence of R7 and R8 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R9 is H or C1-6 alkyl;
- R10 is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(CO—C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, B(OH)2, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R11 and R12 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (CO—C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R13a and R13b are each independently R5 or together are ═O;
- R14a and R14b are each independently R5 or together are ═O;
- R13c and R14c are each independently R5;
- Qa is CH or N;
- U is —C(O)—, —C(═NR4)—, —(CR4R5—)p, NR50, S(═O)2, C(═O), (C═O)N(R4), N(R4)(C═O), S(═O)2N(R4), N(R4)S(═O)2, C═N—OR4, —C(R4)═C(R5)—, —C(R4R5)pNR50—, N(R50)C(R4R5)p—, —O—C(R4R5)—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, NS(═O)2R4, or an optionally substituted aryl, heteroaryl, cycloalkyl or heterocyclic ring, all of which may be optionally substituted;
- W is —CH2—, —S—, —CHF— or —CF2—;
- Z is C or N;
- m is 1, or 2;
- n is 0, 1, or 2;
- p is 0 to 6;
- q is 0 to 6; and
- t is 0, 1, or 2
- wherein: when E and G are both phenyl either:
- (1) at least one of R1 or R2 is present and is:
- CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (CO—C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, (C5-20)alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted; and wherein OR4 is alkoxy, OR4 is (C5-20) alkoxy; or (2) and when B is (b) R7 and R8 are not selected from hydrogen, hydroxy, hydroxymethyl, and phenyl; or (3) and when B is (b) or (f), R9 is: C1-6 alkyl.
- Compounds of the present invention having one or more optically active carbons can exist as racemates and racemic mixtures, diasteromeric mixtures and individual diastereomers, enantiomeric mixtures and single enantiomers, tautomers, atropisomers, and rotamers, with all isomeric forms being included in the present invention. Compounds described in this invention containing olefinic double bonds include both E and Z geometric isomers. Also included in this invention are all salt forms, polymorphs, hydrates and solvates. All of the above mentioned compounds are included within the scope of the invention.
- The present invention also provides methods of inhibiting the DPP-IV enzyme.
- The present invention further provides methods of treatment or prevention of diseases in which the dipeptidyl peptidase-IV enzyme is involved, such as diabetes and particularly Type-2 diabetes.
- The present invention also provides methods for obtaining the DPP-IV inhibiting compounds and pharmaceutical compositions comprising them either singly or in combination with one or more additional therapeutic agents for the prevention or treatment of DPP-IV enzyme medicated diseases, particularly Type-2 diabetes.
- The terms “alkyl” or “alk”, as used herein alone or as part of another group, denote optionally substituted, straight and branched chain saturated hydrocarbon groups, preferably having 1 to 10 carbons in the normal chain, most preferably lower alkyl groups. Exemplary unsubstituted such groups include methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4-trimethylpentyl, nonyl, decyl, undecyl, dodecyl and the like. Exemplary substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkenyl, alkynyl, aryl (e.g., to form a benzyl group), cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH2—CO—), substituted carbamoyl ((R4)(R5)N—CO— wherein R4 or R5 are as defined below, except that at least one of R4 or R5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- The terms “lower alk” or “lower alkyl” as used herein, denote such optionally substituted groups as described above for alkyl having 1 to 4 carbon atoms in the normal chain.
- The term “alkoxy” denotes an alkyl group as described above bonded through an oxygen linkage (—O—).
- The term “alkenyl”, as used herein alone or as part of another group, denotes optionally substituted, straight and branched chain hydrocarbon groups containing at least one carbon to carbon double bond in the chain, and preferably having 2 to 10 carbons in the normal chain. Exemplary unsubstituted such groups include ethenyl, propenyl, isobutenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl, and the like. Exemplary substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH2—CO—), substituted carbamoyl ((R4)(R5)N—CO— wherein R4 or R5 are as defined below, except that at least one of R4 or R5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- The term “alkynyl”, as used herein alone or as part of another group, denotes optionally substituted, straight and branched chain hydrocarbon groups containing at least one carbon to carbon triple bond in the chain, and preferably having 2 to 10 carbons in the normal chain. Exemplary unsubstituted such groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl, and the like. Exemplary substituents may include, but are not limited to, one or more of the following groups: halo, alkoxy, alkylthio, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, hydroxy or protected hydroxy, carboxyl (—COOH), alkyloxycarbonyl, alkylcarbonyloxy, alkylcarbonyl, carbamoyl (NH2—CO—), substituted carbamoyl ((R4)(R5)N—CO— wherein R4 or R5 are as defined below, except that at least one of R4 or R5 is not hydrogen), amino, heterocyclo, mono- or dialkylamino, or thiol (—SH).
- The term “cycloalkyl”, as used herein alone or as part of another group, denotes optionally substituted, saturated cyclic hydrocarbon ring systems, including bridged ring systems, desirably containing 1 to 3 rings and 3 to 9 carbons per ring. Exemplary unsubstituted such groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclododecyl, and adamantyl. Exemplary substituents include, but are not limited to, one or more alkyl groups as described above, or one or more groups described above as alkyl substituents.
- The terms “ar” or “aryl”, as used herein alone or as part of another group, denote optionally substituted, homocyclic aromatic groups, preferably containing 1 or 2 rings and 6 to 12 ring carbons. Exemplary unsubstituted such groups include, but are not limited to, phenyl, biphenyl, and naphthyl. Exemplary substituents include, but are not limited to, one or more nitro groups, alkyl groups as described above or groups described above as alkyl substituents.
- The term “heterocycle” or “heterocyclic system” denotes a heterocyclyl, heterocyclenyl, or heteroaryl group as described herein, which contains carbon atoms and from 1 to 4 heteroatoms independently selected from the group consisting of N, O and S and including any bicyclic or tricyclic group in which any of the above-defined heterocyclic rings is fused to one or more heterocycle, aryl or cycloalkyl groups. The nitrogen and sulfur heteroatoms may optionally be oxidized. The heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure. The heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom.
- Examples of heterocycles include, but are not limited to, 1H-indazole, 2-pyrrolidonyl, 2H,6H-1,5,2-dithiazinyl, 2H-pyrrolyl, 3H-indolyl, 4-piperidonyl, 4aH-carbazole, 4H-quinolizinyl, 6H-1,2,5-thiadiazinyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolinyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazalonyl, carbazolyl, 4aH-carbazolyl, b-carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-1,5,2-dithiazinyl, dihydrofuro[2,3-b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, isatinoyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinylperimidinyl, oxindolyl, phenanthridinyl, phenanthrolinyl, phenarsazinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, pteridinyl, piperidonyl, 4-piperidonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, carbolinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, xanthenyl.
- “Heterocyclenyl” denotes a non-aromatic monocyclic or multicyclic hydrocarbon ring system of about 3 to about 10 atoms, desirably about 4 to about 8 atoms, in which one or more of the carbon atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur atoms, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond. Ring sizes of rings of the ring system may include 5 to 6 ring atoms. The designation of the aza, oxa or thia as a prefix before heterocyclenyl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom. The heterocyclenyl may be optionally substituted by one or more substituents as defined herein. The nitrogen or sulpHur atom of the heterocyclenyl may also be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide. “Heterocyclenyl” as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and “J. Am. Chem. Soc.”, 82:5566 (1960), the contents all of which are incorporated by reference herein. Exemplary monocyclic azaheterocyclenyl groups include, but are not limited to, 1,2,3,4-tetrahydrohydropyridine, 1,2-dihydropyridyl, 1,4-dihydropyridyl, 1,2,3,6-tetrahydropyridine, 1,4,5,6-tetrahydropyrimidine, 2-pyrrolinyl, 3-pyrrolinyl, 2-imidazolinyl, 2-pyrazolinyl, and the like. Exemplary oxaheterocyclenyl groups include, but are not limited to, 3,4-dihydro-2H-pyran, dihydrofuranyl, and fluorodihydrofuranyl. An exemplary multicyclic oxaheterocyclenyl group is 7-oxabicyclo[2.2.1]heptenyl.
- “Heterocyclyl,” or “heterocycloalkyl,” denotes a non-aromatic saturated monocyclic or multicyclic ring system of about 3 to about 10 carbon atoms, desirably 4 to 8 carbon atoms, in which one or more of the carbon atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur. Ring sizes of rings of the ring system may include 5 to 6 ring atoms. The designation of the aza, oxa or thia as a prefix before heterocyclyl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom. The heterocyclyl may be optionally substituted by one or more substituents which may be the same or different, and are as defined herein. The nitrogen or sulpHur atom of the heterocyclyl may also be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide.
- “Heterocyclyl” as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and “J. Am. Chem. Soc.”, 82:5566 (1960). Exemplary monocyclic heterocyclyl rings include, but are not limited to, piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, 1,3-dioxolanyl, 1,4-dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, and the like.
- “Heteroaryl” denotes an aromatic monocyclic or multicyclic ring system of about 5 to about 10 atoms, in which one or more of the atoms in the ring system is/are hetero element(s) other than carbon, for example nitrogen, oxygen or sulfur. Ring sizes of rings of the ring system include 5 to 6 ring atoms. The “heteroaryl” may also be substituted by one or more substituents which may be the same or different, and are as defined herein. The designation of the aza, oxa or thia as a prefix before heteroaryl define that at least a nitrogen, oxygen or sulfur atom is present respectively as a ring atom. A nitrogen atom of a heteroaryl may be optionally oxidized to the corresponding N-oxide. Heteroaryl as used herein includes by way of example and not limitation those described in Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and “J. Am. Chem. Soc.”, 82:5566 (1960). Exemplary heteroaryl and substituted heteroaryl groups include, but are not limited to, pyrazinyl, thienyl, isothiazolyl, oxazolyl, pyrazolyl, furazanyl, pyrrolyl, 1,2,4-thiadiazolyl, pyridazinyl, quinoxalinyl, phthalazinyl, imidazo[1,2-a]pyridine, imidazo[2,1-b]thiazolyl, benzofurazanyl, azaindolyl, benzimidazolyl, benzothienyl, thienopyridyl, thienopyrimidyl, pyrrolopyridyl, imidazopyridyl, benzoazaindole, 1,2,3-triazinyl, 1,2,4-triazinyl, 1,3,5-triazinyl, benzthiazolyl, dioxolyl, furanyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, morpholino, oxadiazolyl, oxazinyl, oxiranyl, piperazinyl, piperidinyl, pyranyl, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, pyrrolidinyl, quinazolinyl, quinolinyl, tetrazinyl, tetrazolyl, 1,3,4-thiadiazolyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, thiatriazolyl, thiazinyl, thiazolyl, thienyl, 5-thioxo-1,2,4-diazolyl, thiomorpholino, thiophenyl, thiopyranyl, triazolyl and triazolonyl.
- The term “amino” denotes the radical —NH2 wherein one or both of the hydrogen atoms may be replaced by an optionally substituted hydrocarbon group. Exemplary amino groups include, but are not limited to, n-butylamino, tert-butylamino, methylpropylamino and ethyldimethylamino.
- The term “cycloalkylalkyl” denotes a cycloalkyl-alkyl group wherein a cycloalkyl as described above is bonded through an alkyl, as defined above. Cycloalkylalkyl groups may contain a lower alkyl moiety. Exemplary cycloalkylalkyl groups include, but are not limited to, cyclopropylmethyl, cyclopentylmethyl, cyclohexylmethyl, cyclopropylethyl, cyclopentylethyl, cyclohexylpropyl, cyclopropylpropyl, cyclopentylpropyl, and cyclohexylpropyl.
- The term “arylalkyl” denotes an aryl group as described above bonded through an alkyl, as defined above.
- The term “heteroarylalkyl” denotes a heteroaryl group as described above bonded through an alkyl, as defined above.
- The term “heterocyclylalkyl,” or “heterocycloalkylalkyl,” denotes a heterocyclyl group as described above bonded through an alkyl, as defined above.
- The terms “halogen”, “halo”, or “hal”, as used herein alone or as part of another group, denote chlorine, bromine, fluorine, and iodine.
- The term “haloalkyl” denotes a halo group as described above bonded though an alkyl, as defined above. Fluoroalkyl is an exemplary group.
- The term “aminoalkyl” denotes an amino group as defined above bonded through an alkyl, as defined above.
- The pHrase “bicyclic fused ring system wherein at least one ring is partially saturated” denotes an 8- to 13-membered fused bicyclic ring group in which at least one of the rings is non-aromatic. The ring group has carbon atoms and optionally 1-4 heteroatoms independently selected from N, O and S. Illustrative examples include, but are not limited to, indanyl, tetrahydronaphthyl, tetrahydroquinolyl and benzocycloheptyl.
- The pHrase “tricyclic fused ring system wherein at least one ring is partially saturated” denotes a 9- to 18-membered fused tricyclic ring group in which at least one of the rings is non-aromatic. The ring group has carbon atoms and optionally 1-7 heteroatoms independently selected from N, O and S. Illustrative examples include, but are not limited to, fluorene, 10,11-dihydro-5H-dibenzo[a,d]cycloheptene and 2,2a,7,7a-tetrahydro-1H-cyclobuta[a]indene.
- The term “pharmaceutically acceptable salts” refers to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as, but not limited to, hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as, but not limited to, acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two. Organic solvents include, but are not limited to, nonaqueous media like ethers, ethyl acetate, ethanol, isopropanol, or acetonitrile. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, Pa., 1990, p. 1445, the disclosure of which is hereby incorporated by reference.
- The pHrase “pharmaceutically acceptable” denotes those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication commensurate with a reasonable benefit/risk ratio.
- “Substituted” is intended to indicate that one or more hydrogens on the atom indicated in the expression using “substituted” is replaced with a selection from the indicated group(s), provided that the indicated atom's normal valency is not exceeded, and that the substitution results in a stable compound. When a substituent is keto (i.e., ═O) group, then 2 hydrogens on the atom are replaced.
- Unless moieties of a compound of the present invention are defined as being unsubstituted, the moieties of the compound may be substituted. In addition to any substituents provided above, the moieties of the compounds of the present invention may be optionally substituted with one or more groups independently selected from, but not limited to:
- C1-C4 alkyl;
- C2-C4 alkenyl;
- C2-C4 alkynyl;
- CF3;
- halo;
- OH;
- O—(C1-C4 alkyl);
- OCH2F;
- OCHF2;
- OCF3;
- COCF3;
- OC(O)—(C1-C4 alkyl);
- OC(O)NH—(C1-C4 alkyl);
- OC(O)N(C1-C4 alkyl)2;
- OC(S)NH—(C1-C4 alkyl);
- OC(S)N(C1-C4 alkyl)2;
- ONO2;
- SH;
- S—(C1-C4 alkyl);
- S(O)—(C1-C4 alkyl);
- S(O)2—(C1-C4 alkyl);
- SC(O)—(C1-C4 alkyl);
- SC(O)O—(C1-C4 alkyl);
- NH2;
- N(H)—(C1-C4 alkyl);
- N(C1-C4 alkyl)2;
- N(H)C(O)—(C1-C4 alkyl);
- N(CH3)C(O)—(C1-C4 alkyl);
- N(H)C(O)—CF3;
- N(CH3)C(O)—CF3;
- N(H)C(S)—(C1-C4 alkyl);
- N(CH3)C(S)—(C1-C4 alkyl);
- N(H)S(O)2—(C1-C4 alkyl);
- N(H)C(O)NH2;
- N(H)C(O)NH—(C1-C4 alkyl);
- N(CH3)C(O)NH—(C1-C4 alkyl);
- N(H)C(O)N(C1-C4 alkyl)2;
- N(CH3)C(O)N(C1-C4 alkyl)2;
- N(H)S(O)2NH2);
- N(H)S(O)2NH—(C1-C4 alkyl);
- N(CH3)S(O)2NH—(C1-C4 alkyl);
- N(H)S(O)2N(C1-C4 alkyl)2;
- N(CH3)S(O)2N(C1-C4 alkyl)2;
- N(H)C(O)O—(C1-C4 alkyl);
- N(CH3)C(O)O—(C1-C4 alkyl);
- N(H)S(O)2O—(C1-C4 alkyl);
- N(CH3)S(O)2O—(C1-C4 alkyl);
- N(CH3)C(S)NH—(C1-C4 alkyl);
- N(CH3)C(S)N(C1-C4 alkyl)2;
- N(CH3)C(S)O—(C1-C4 alkyl);
- N(H)C(S)NH2;
- NO2;
- CO2H;
- CO2—(C1-C4 alkyl);
- C(O)N(H)OH;
- C(O)N(CH3)OH:
- C(O)N(CH3)OH;
- C(O)N(CH3)O—(C1-C4 alkyl);
- C(O)N(H)—(C1-C4 alkyl);
- C(O)N(C1-C4 alkyl)2;
- C(S)N(H)—(C1-C4 alkyl);
- C(S)N(C1-C4 alkyl)2;
- C(NH)N(H)—(C1-C4 alkyl);
- C(NH)N(C1-C4 alkyl)2;
- C(NCH3)N(H)—(C1-C4 alkyl);
- C(NCH3)N(C1-C4 alkyl)2;
- C(O)—(C1-C4 alkyl);
- C(NH)—(C1-C4 alkyl);
- C(NCH3)—(C1-C4 alkyl);
- C(NOH)—(C1-C4 alkyl);
- C(NOCH3)—(C1-C4 alkyl);
- CN;
- CHO;
- CH2OH;
- CH2O—(C1-C4 alkyl);
- CH2NH2;
- CH2N(H)—(C1-C4 alkyl);
- CH2N(C1-C4 alkyl)2;
- aryl;
- heteroaryl;
- cycloalkyl; and
- heterocyclyl.
- The term “cleave” or “cleaving” means splitting a complex molecule into at least two separate molecules. “Cleavage products” are the separate molecules which result from cleaving.
- The term “metabolite” refers to a composition which results from a metabolic process. Examples of the results of metabolism on the compounds of the present invention include addition of —OH, hydrolysis, and cleavage.
- The term “polymorphs” refers to the various crystalline structures of the compounds of the present invention. This may include, but is not limited to, crystal morphologies (and amorphous materials), all crystal lattice forms, and all salts. Salts of the present invention can be crystalline and may exist as more than one polymorpH. Each polymorpH forms another aspect of the invention. Hydrates as well as anhydrous forms of the salt are also encompassed by the invention.
- “Teoc” is 2-(trimethylsilyl)ethoxycarbonyl
- “Et” is ethyl (—CH2CH3) or ethylene (—CH2CH2—).
- “Me” is methyl (—CH3) or methylene (—CH2—).
- “Boc” is tert-butyloxycarbonyl.
- “PHCH2” is benzyl.
- The term “pharmaceutically-acceptable tricyclic moiety” is meant to include, but is not limited to, benzocycloheptapyridyl, benzodiazepinyl, and benzozapinyl
- In another embodiment of the present invention, the DPP-IV inhibiting compounds are used in the manufacture of a medicament for the treatment of a disease mediated by an DPP-IV enzyme.
- In another aspect, the DPP-IV inhibiting compounds of the present invention are used in combination with another disease modifying drug. Examples of other disease modifying drugs include, but are not limited to: (a) other dipeptidyl peptidase IV (DPP-IV) inhibitors such as Vildagliptin (Novartis), Sitagliptin (Merck&Co.), Saxagliptin (BMS); (b) insulin sensitizers including (i) PPARγ agonists such as the glitazones (e.g. troglitazone, pioglitazone, edaglitazone, rosiglitazone, and the like) and other PPAR ligands, including PPARα/γ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), and PPARα agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (ii) biguanides such as metformin and phenformin, and (iii) protein tyrosine phosphatase-1B (PTP-1B) inhibitors; (c) insulin or insulin mimetics; (d) incretin and incretin mimetics such as (i) Exenatide available from Amylin Pharmaceuticals, (i) amylin and amylin mimetics such as pramlintide acetate, available as Symlin®, (iii) GLP-1, GLP-1 mimetics, and GLP-1 receptor agonists, (iv) GIP, GIP mimetics and GIP receptor agonists; (e) sulfonylureas and other insulin secretagogues, such as tolbutamide, glyburide, glipizide, glimepiride, meglitinides, and repaglinide; (f) α-glucosidase inhibitors (such as acarbose and miglitol); (g) glucagon receptor antagonists; (h) PACAP, PACAP mimetics, and PACAP receptor agonists; (i) cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, cerivastatin, fluvastatin, atorvastatin, itavastatin, and rosuvastatin, and other statins), (ii) sequestrants such as cholestyramine, colestipol and dialkylaminoalkyl derivatives of a cross-linked dextran, (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPARα agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) PPARα/γ dual agonists such as muraglitazar (BMS) and tesaglitazar (AstraZeneca), (vi) inhibitors of cholesterol absorption, such as beta-sitosterol and ezetimibe, (vii) acyl CoA:cholesterol acyltransferase inhibitors such as avasimibe, and (viii) anti-oxidants such as probucol; (J) PPARδ agonists such as GW-501516 from GSK; (k) anti-obesity compounds such as fenfluramine, dexfenfluramine, phentemine, sibutramine, orlistat, neuropeptide Y1 or Y5 antagonists, MTP inhibitors, squalene synthase inhibitor, lipoxygenase inhibitor, ACAT inhibitor, Neuropeptide Cannabinoid CB-1 receptor antagonists, CB-1 receptor inverse agonists and antagonists, fatty acid oxidation inhibitors, appetite suppressants (1) adrenergic receptor agonists, melanocortin receptor agonists, in particular—melanocortin-4 receptor agonists, ghrelin antagonists, and melanin-concentrating hormone (MCH) receptor antagonists; (m) ileal bile acid transporter inhibitors; (n) agents intended for use in inflammatory conditions such as aspirin, non steroidal anti-inflammatory drugs, glucocorticoids, azalfidine, and selective cyclooxygenase-2 inhibitors; (o) antihypertensive agents such as ACE inhibitors (enalapril, lisinopril, captopril, quinapril, fosinoprol, ramipril, spirapril, tandolapril), angiotensin-II (AT-1) receptor blockers (losartan, candesartan, irbesartan, valsartan, telmisartan, eprosartan), beta blockers and calcium channel blockers; and (p) glucokinase activators (GKAs); (q) agents which can be used for the prevention, delay of progression or treatment of neurodegenerative disorders, cognitive disorders or a drug for improving memory such as anti-inflammatory drugs, antioxidants, neuroprotective agents, glutamate receptor antagonists, acetylcholine esterase inhibitors, butyrylcholinesterase inhibitors, MAO inhibitors, dopamine agonists or antagonists, inhibitors of gamma and beta secretases, inhibitors of amyloid aggregation, amyloid beta peptide, antibodies to amyloid beta peptide, inhibitors of acetylcholinesterase, glucokinase activators, agents directed at modulating GABA, NMDA, cannabinoid, AMPA, kainate, phosphodiesterase (PDE), PKA, PKC, CREB or nootropic systems; (r) leukocyte growth promotors intended for the treatment and prevention of reduced bone marrow production, infectious diseases, hormone dependent disorders, inflammatory diseases, HIV, allergies, leukocytopenia, and rheumatism; (s) SGLT2 inhibitor; (t) glycogen phosphorylase inhibitor; (u) aP2 inhibitors; (v) aminopeptidase N inhibitor (w) vasopeptidase inhibitors like neprilysin inhibitors and/or ACE inhibitors or dual NEP/ACE inhibitor; (x) growth hormone secretagogue for enhancing growth hormone levels and for treating growth retardation/dwarfism or metabolic disorders or where the disorder is an injury, or a wound in need of healing, or a mammalian patient recovering from surgery; (y) 5-HT 3 or 5-HT 4 receptor modulators (tegaserod, cisapride, nor-cisapride, renzapride, zacopride, mosapride, prucalopride, buspirone, norcisapride, cilansetron, ramosetron, azasetron, ondansetron, etc.); (Za) aldose reductase inhibitors; (Zb) sorbitol dehydrogenase inhibitors; (Zc) AGE inhibitors; (Zd) erythropoietin agonist such as EPO, EPO mimetics, and EPO receptor agonists.
- In a further aspect, the DPP-IV inhibiting compounds of the present invention are used in the treatment diseases or symptoms mediated by an DPP-IV enzyme. Examples of diseases or symptoms mediated by a DPP-IV enzyme include, but are not limited to, Type II (Type-2) Diabetes and Related Disorders, such as hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its 30 sequelae, vascular restenosis, irritable bowel syndrome, inflammatory bowel disease, including Crohn's disease and ulcerative colitis, other inflammatory conditions, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nepHropathy, neuropathy, cataracts, glaucoma, glomerulosclerosis, foot ulcerations and unlcerative colitis, altered gastrointestinal motility, Syndrome X, ovarian hyperandrogenism, polycystic ovarian syndrome, premenstrual syndrome, other disorders where insulin resistance is a component. In Syndrome X, also known as Metabolic Syndrome, obesity is thought to promote insulin resistance, diabetes, dyslipidemia, hypertension, and increased cardiovascular risk, growth hormone deficiency, neutropenia, neuronal disorders, tumor invasion and metastasis, benign prostatic hypertrophy, gingivitis, osteoporosis, frailty of aging, intestinal injury, benign prostatic hypertrophy (BPH), and sperm motility/male contraception.
- In a further aspect, the DPP-IV inhibiting compounds of the present invention are useful for the prevention, delay of progression or the treatment of an early cardiac or early cardiovascular diseases or damages, renal diseases or damages, heart Failure, or heart Failure associated diseases like (i) cardiovascular diseases or damages e.g. cardiac hypertrophy, cardiac remodelling after myocardial infarction, pulmonary congestion and cardiac fibrosis in dilated or in hypertrophic cardiomyopathy, cardiomyopathy such as dilated cardiomyopathy or hypertrophic cardiomyopathy, mesanglial hypertrophy, or diabetic cardiomyopathy, left or right ventricular hypertrophy, arrhythmia, cardiac dysrhythmia, syncopy, angina pectoris, cardiac bypass reocclusion, intermittent claudication, diastolic and/or systolic dysfunction, diabetic myopathy, stroke prevention in congestive heart failure, hypertrophic medial thickening in arteries and/or large vessels, mesenteric vasculature hypertrophy or artherosclerosis, preferably artherosclerosis in mammalian patients with hypertension of diabetes; (ii) renal diseases or damages like renal hyperfiltration such as after portal renal ablation, proteinuria in chronic renal disease, renal arteriopathy as a consequence of hypertension, nepHrosclerosis, hypertensive nepHrosclerosis or mesanglial hypertrophy; (iii) Heart Failure to be treated is secondary to idiopathic dilated cardiomyopathy and/or coronary ischemic disease;
- In another aspect, the DPP-IV inhibiting compounds of the present invention are used for the prevention, the delay of the onset, the delay of progression or the treatment of neurodegenerative disorders, cognitive disorders and for improving memory (both short term and long term) and learning ability wherein the (i) neurodegenerative disorder is dementia, senile dementia, schizopHrenia, mild cognitive impairment, Alzheimer related dementia, Huntington's chores, tardive dyskinesia, hyperkinesias, mania, Morbus Parkinson, Steel-Richard syndrome, Down's syndrome, myasthenia gravis, nerve and brain trauma, vascular amyloidosis, cerebral haemorrhage I with amyloidosis, brain inflammation, Friedrich ataxia, acute confusion disorders, acute confusion disorders with apoptotic necrocytosis, amyotrophic lateral sclerosis, glaucoma, and Alzheimer's disease; (ii) cognitive disorders like cognitive deficits associated with schizopHrenia, age-induced memory impairment, cognitive deficits associated with psychosis, cognitive impairment associated with diabetes, cognitive deficits associated with post-stroke, memory defects associated hypoxia, cognitive and attention deficits associated with senile dementia, attention deficits disorders, memory problems associated with mild cognitive impairment, impaired cognitice function associated with vascular dementia, cognitive problems associated with brain tumors, Pick's disease, cognitive deficits due to autism, cognitive deficits post electroconvulsive therapy, cognitive deficits associated with traumatic brain injury, amnesic disorders, deliriums, vitamin deficiency, dementias, impaired cognitive function associated with Parkinson's disease, attention-deficit disorders; (iii) prevention of memory impairment as a result of Alzheimer disease, Creutzfeld-Jakob disease, Pick disease, Huntington disease, AIDS, brain injury, brain aneurysm, epilepsy, stroke, toxicant exposure, mental retardation in children, Huntington's disease; (iv) to improve learning speed and potential in educational and rehabilitation contexts.
- In another aspect, the DPP-IV inhibiting compounds of the present invention are used for stimulating an immune response in a subject having or at risk of having cancer wherein the cancer is selected from the group consisting of basal cell carcinomas including cancers of the binary tract, bladder, urinary system, bone, brain, breast, cervical, endometrial, ovarian, uterine, choriocarcinoma, central nervous system, colon and rectal cancers, connective tissue cancer, cancer of the digestive system, esophageal, gastric, stomach, larynx, liver, pancreatic, colorectal, renal cancers; cancers of the urinary system; cancers of eye, head and neck, oral cavity, skin, prostate; cancers of biliary tract, testicular, thyroid; intra-epithelial neoplasm, leukemia, acute myeloid leukemia, acute lymphoid leukemia, chronic myeloid leukemia, chronic lymphoid leukemia; and other cancers of the respiratory system, lung, small cell lung, non-small cell lung; lymphoma, Hodgkin's lymphoma, Non-Hodgkin's lymphoma; melanoma, myeloma, neuroblastoma, retinoblastoma, fibrosarcoma (bone or connective tissue sarcoma), rhabdomyosarcoma; and other cancers including neoplastic conditions, adipose cell tumors, adipose cell carcinomas, such as liposarcoma;
- In a further aspect, the DPP-IV inhibiting compounds of the present invention are useful for the treatment or prophylaxis of chronic inflammatory diseases such as autoimmune disorders like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, allergies or asthma.
- In another aspect, the DPP-IV inhibiting compounds of the present invention may be useful in the treatment of pain, neuropathic pain, rheumatoid pain, osteoarthritis pain, anesthesia adjunct in mammalian patients undergoing surgery, chronic pain in advanced cancer, treatment of refractory diarrhea, biliary pain caused by gallstones.
- In a further aspect, the DPP-IV inhibiting compounds of the present invention are useful for the treatment of mammalian patients undergoing islet/pancreas transplantation, for the prevention or the delay of transplant rejection, or allograft rejection in transplantation, for improving pancreatic function by increasing the number and size of pancreatic beta-cells in the treatment of Type 1 diabetes patients, and for improving pancreatic function by increasing the number and size of pancreatic beta-cells in general.
- Furthermore, the DPP-IV inhibiting compounds of the present invention are useful for the treatment of mammalian patients with acne, skin disorders (e.g. pigmentation disorders or psoriasis), scleroderma, mycoses; anxiety, anxiety neurosis, major depression disorder, drug abuse, alcohol addiction, insomnia, chronic fatigue, sleep apnea; anorexia nervosa; epilepsy; migrane; encephalomyelitis; osteoarthritis, osteoporosis, calcitonin-induced osteoporosis; male and female sexual dysfunction, infertility; Type 1 diabetes; immunosuppression, HIV infection; hematopoiesis, anemia; and for weight reduction.
- In a further aspect, the DPP-IV inhibiting compounds of the present invention are useful for the prevention, delay of progression or treatment of (i) bacterial infections from Escherichia coli, Staphylococcus, Streptoococcus, Pseudomonas, Clostridium difficile infection, Legionella, Pneumococcus, HaemopHilus, Klebsiella, Enterobacter, Citrobacter, Neisseria, Shigella, Salmonella, Listeria, Pasteurella, Streptobacillus, Spirillum, Treponema, Actinomyces, Borrelia, Corynebacterium, Nocardia, Gardnerella, Campylobacter, Spirochaeta, Proteus, Bacteriodes, Helicobacter pylori, and anthrax infection; (ii) mycobacterial infection from tuberculosis and leprosy; (iii) viral infection from HIV, Herpes simplex virus 1, Herpes simplex virus 2, Cytomegalovirus, hepatitis A virus, hepatitis B virus, hepatitis C virus, human papilloma virus, Epstein Barr virus, rotavirus, adenovirus, influenza A virus, respiratory syncytial virus, varicella-zoster virus, small pox, monkey pox and SARS; (iv) fungal infection from candidiasis, ringworm, histoplasmosis, blastomycosis, paracoccidioidomycosis, cryptococcosis, aspergillosis, chromomycosis, mycetoma infections, pseudallescheriasis, Tinea versicolor infection; (v) parasite infection from amebiasis, Trypanosoma cruzi, Fascioliasis, Leishmaniasis, Plasmodium, Onchocerciasis, Paragonimiasis, Trypanosoma brucei, Pneumocystis, Trichomonas vaginalis, Taenia, Hymenolepsis, Echinococcus, Schistosomiasis, neurocysticerosis, Necator americanus, and Trichuris trichuria.
- The compounds from this invention are suitable for oral, sublingual, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (aerosol inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient. The compounds from this invention are conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- The DPP-IV inhibiting compounds of the present invention are synthesized by the general method shown in Schemes 1-14.
- Generic Schemes
- General synthetic schemes for the preparation of tricyclic building blocks of this invention:
-

- Commercially available bromotoluene derivatives were treated with n-butyllithium and heated, followed by treatment with dry-ice in an appropriate solvent to afford the desired compound. Alternatively, the acid can be prepared by Grignard reaction followed by treatment with dry-ice in an appropriate solvent. Esterification of the compound followed by NBS bromination and subsequent conversion to the phosphonium salt in a suitable solvent and heating affords the desired compound. Wittig reaction of the phosphonium salt with a suitable aldehyde in an appropriate solvent and heating, followed by saponification of the ester moiety and subsequent catalytic hydrogenation affords the desired compound. Cyclisation of the compound with polyphosphoric acid in sulfolane and heating affords the desired compound after purification. For R1═COOMe the tricyclic product from the polyphosphoric acid step was treated with thionylchloride in an alcohol. Reduction of the ketone with a metal hydride in an appropriate solvent yields the compound after purification. Treatment of the alcohol with thionylchloride in a suitable solvent affords the final desired compound. In order to obtain the compounds with R1═R2═COOMe, the tricyclic product from the polyphosphoric acid step with R1═COOH and R2=Br was treated with CuCN in a suitable solvent, followed by saponification of the nitrile to the acid. Ester formation using thionylchloride in an alcohol and reduction of the ketone with a metal hydride in an appropriate solvent yields the compound after purification. Treatment of the alcohol with thionylchloride in a suitable solvent affords the final desired compound.
- Alternative synthetic scheme for the preparation of tricyclic building blocks of this invention:
-

- Commercially available bromotoluene derivatives are treated with Magnesium in a Grignard reaction followed by treatment with dry-ice in an appropriate solvent to yield the desired acid. This acid is then treated with sec-butyllithium in an appropriate solvent at lower temperature. The anion is added at lower temperature to a solution of a commercially available benzylchloride in an appropriate solvent to afford the desired compound. Cyclisation of the compound with polyphosphoric acid in sulfolane and heating affords the desired compound. To obtain the compounds with R1═R2═COOMe, the tricyclic product from the polyphosphoric acid step with R1═R2═Cl was treated with KCN, a Pd-catalyst, a suitable ligand and a suitable base in an appropriate solvent to afford the dicyano compound, which was converted to the diacid by treatment with base in a suitable solvent. Ester formation using thionylchloride in an alcohol and reduction of the ketone with a metal hydride in an appropriate solvent yields the compound after purification. Treatment of the alcohol with thionylchloride in a suitable solvent affords the final desired compound.
- General synthetic scheme for the preparation of aldehyde building blocks of this invention:
-

- Commercially available prolinamide is treated with fumarylchloride in an appropriate solvent to afford the desired compound. This compound is then treated with oxalylchloride in dimethylformamide to afford the desired compound after purification. Alternatively, the coupling product of prolinamide with fumarylchloride can be treated with trifluoroacetic acid anhydride in a suitable solvent to afford the desired compound. Ozonolysis of this compound at −78° C. in a suitable solvent, followed by reductive workup affords the desired final compound as a mixture of the aldehyde and its methyl hemiacetal.
- Treatment of 2-Aza-bicyclo[3.1.0]hexane-3-carboxylic acid amide, prepared according to WO 01/68603, in the same manner as described above yields the desired final compound containing a cyclopropyl moiety at the 4,5-position of the pyrrolidine moiety.
- General synthetic scheme for the preparation of tricyclic compounds of this invention with R3═H:
-

- The reaction of substituted or unsubstituted tricyclic chlorides with an amino derivative in a suitable solvent as described above affords the desired final product after purification. Substituted or unsubstituted tricyclic chlorides are treated in an appropriate solvent with an excess of suitable amines to afford the desired product after purification. In case the reaction product contains additional amino protecting groups like Boc, they are cleaved by acid treatment to afford the desired compound. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative yields the final desired product after purification. Alternatively, the amines are treated with a suitable aldehyde (D-CHO) via reductive amination to afford the final compound after purification.
- General synthetic scheme for the preparation of tricyclic compounds of this invention with Z=N:
-

- Substituted or unsubstituted tricycles containing a nitrogen at the doubly benzylic position are treated with bromoacetylbromide and heated to afford the desired compounds. Treating these compounds with sodium azide or sodium cyanide in a suitable solvent and heating affords the desired azido or cyano compounds after purification. Catalytic hydrogenation or reduction with Lithium aluminium hydride in a suitable solvent affords the desired amine compounds. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative yields the final desired product after purification.
- General synthetic scheme for the preparation of tricyclic compounds of this invention having H, OH or no substituent at R3
-

- Substituted or unsubstituted tricyclic ketones with Y═C(R4)═C(R5) are treated with malonic acid at elevated temperatures to afford the desired product after purification. These compounds are converted to the corresponding amides by treatment with isobutylchloroformate and ammonia. The amides are then converted to the desired amine products with Y═C(R4)═C(R5) by reduction with lithium aluminium hydride or to the desired amine products with Y═C(R4R5)C(R4R5) by reduction with lithium aluminium hydride followed by catalytic hydrogenation with a suitable catalyst. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired product after purification.
- Treating tricyclic ketones in a Reformatskij reaction affords the desired product after purification. Reduction with LiAlH4 in a suitable solvent affords the alcohol products with R3═OH after purification. Activation of one of the hydroxyl groups with sulfonylchlorides in a suitable solvent followed by treatment with NaN3 affords the desired compounds after purification. Reduction of the azide reaction products with a catalyst in a suitable solvent affords the desired amine compounds after purification. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired products after purification.
- Treating the amines with R3═OH with acid in a suitable solvent yields the desired unsaturated amine products. Using these amines for a nucleopHilic displacement reaction in a suitable solvent with a suitable bromo derivative described above yields the final desired products after purification.
- General synthetic schemes (7-9) for the preparation of tricyclic compounds of this invention with R3=nitrile, amide, tetrazolyl or N-alkyl-tetrazolyl
-

- Substituted or unsubstituted suberylchlorides are treated in a suitable solvent with a slight excess of AgCN and heated to afford the desired product after purification. The nitrile containing compound is then treated with sodium hydride in a suitable solvent and heated. The mixture is then treated at rt with a suitable dibromoalkene and heated to give an intermediate which after treatment with sodium azide or potassium phthalimide in an appropriate solvent and heating affords the desired compound after purification. Treating the mixture after the addition of sodium hydride at rt with a suitable sulfamidate in an appropriate solvent affords the desired Teoc-protected compound after heating for several hours and subsequent purification.
- Catalytic hydrogenation of compounds with R′═N3 in a suitable solvent and in the presence of a slight excess of acid affords the free amine compounds. Coupling of these amines with a suitable aldehyde (CHO-D) via reductive amination and subsequent purification affords the final desired compounds with R3═CN.
-

- Catalytic hydrogenation of compounds with R3═CN and R′═N3 in a suitable solvent and in the presence of a slight excess of acid affords the free amine compounds. Treatment of the hydrogenation products with sulphuric acid affords the desired compounds after purification. In case R1═R2≠COOH, the amines are reacted with a suitable aldehyde (D-CHO) in an appropriate solvent to yield the desired final compounds with R3═CONH2 and R1═R2≠COOH, CONR4R5, COOMe. In case R1═COOH, the amines are treated with Boc2O in a suitable solvent to afford the Boc-protected amines. These compounds are then treated with ethylchloroformate, followed by treatment with an amine to yield the desired compounds after purification. The compounds are then treated with acid, followed by reaction with a suitable aldehyde (D-CHO) in an appropriate solvent to yield the desired final compounds with R3═CONH2 and R1═CONR4R5 after purification.
- The compounds with R3═CN and R′═N-phthaloyl are treated with an excess of trimethylsilyl azide and Bu2SnO in an appropriate solvent and heating to afford the desired compounds with R3=tetrazolyl and R′═N-phthaloyl. In case R1═R2≠COOH, the compounds are treated with hydrazine hydrate at elevated temperature in an appropriate solvent to yield the desired amines with R3=tetrazoyl. The reaction of these amines with a suitable aldehyde (D-CHO) in an appropriate solvent affords the desired final compound with R3=tetrazoyl and R1═R2≠COOH, CONR4R5, COOMe after purification. In case R1═COOMe, the compounds are treated with an appropriate amine in a suitable solvent to afford the free amine compounds. Protection of the amines with Boc2O affords the Boc-protected products after purification. Saponification of the ester moieties affords the desired fNH-Boc-protected carboxylic acid derivatives. The acid derivates are then treated with ethylchloroformate, followed by an amine to afford the desired products after acid treatment. The reaction of these amines with a suitable aldehyde (D-CHO) in an appropriate solvent affords the desired final compound with R3=tetrazoyl and R1═CONR4R5 after purification.
-

- The NH Teoc-protected compounds with R3═CN and R1═R2═COOMe or R1═R2=Hal were treated with hydroxylamine hydrochloride and an excess of base at elevated temperatures in an appropriate solvent to afford the desired compounds with R3═CONH2 after purification. The same NH Teoc protected compounds are also reacted with sodium azide and ammonium chloride in a suitable solvent to yield the desired compounds with R3=tetrazoyl after purification. Further reaction of the compound with R3=tetrazoyl with methyl iodide and base in a suitable solvent leads to the formation of the desired compound with R3═N-Me-tetrazoyl after purification. For the compounds with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═COOMe, Hal, the Teoc protecting group is removed by treatment with acid to afford the desired amine compounds. The reaction of these amines with a suitable aldehyde (D-CHO) in an appropriate solvent affords the desired final compound with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═COOMe, Hal after purification. For the compounds with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═COOMe, the ester moieties are removed by treatment with base in an appropriate solvent to afford the desired dicarboxylic acid derivatives after purification. Treatment of these compounds with ethylchloroformate, followed by an amine yields the desired amine compounds with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═CONR4R5 after purification. Cleavage of the Teoc protecting group with acid affords the corresponding amine compounds. The reaction of these amines with a suitable aldehyde (D-CHO) in an appropriate solvent affords the desired final compounds with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═CONR4R5 after purification. To obtain the desired final compounds with R3=tetrazoyl, N-Me-tetrazoyl and R1═R2═COOH after purification, the amide formation steps 2 and 3 are omitted.
- General synthetic scheme for the preparation of tricyclic compounds of this invention with R3=heteroaryl (e.g., oxadiazolone or trifluororoxadiazole)
-

- The NH Teoc-protected compounds with R3═CN and R1═R2═COOMe were treated with hydroxylamine hydrochloride and a base at elevated temperatures, followed by diethylcarbonate in an appropriate solvent to afford the desired compounds with R3=oxadiazolone after purification. In case trifluoroacetic acid anhydride and base are used in a suitable solvent for step 2 of the above scheme, the desired compounds with R3═CF3-oxadiazole are obtained after purification. The compounds with R3=oxadiazolone and R3═CF3-oxadiazole are then treated with base to afford the dicarboxylic acid derivatives. These acids are treated with ethylchloroformate, followed by an amine to afford the desired NH-Teoc protected compounds with R3=oxadiazolone, CF3-oxadiazole and R1═R2═CONR4R5 after purification. Cleavage of the Teoc protecting group with acid affords the corresponding amine compounds. The reaction of these amines with a suitable aldehyde (D-CHO) in an appropriate solvent affords the desired final compounds with R3=oxadiazolone, CF3-oxadiazole and R1═R2═CONR4R5 after purification.
- General synthetic scheme for the preparation of tricyclic compounds of this invention with R3=tetrazole and Y═CONR4
-

- Anthraquinone derivatives are treated with sodium azide and sulphuric acid in a suitable solvent to yield the desired compounds. These compounds are then treated with alkyl halides and base in a suitable solvent to obtain the desired compounds after purification. Reaction of theses compounds with tosylmethyl isocyanide and base in a suitable solvent, followed by treatment with dibromoethane and potassium phthalimide affords the desired compounds with R3=CN and R′═N-phthaloyl after purification. The reaction of these compounds with trimethylsilyl-azide and dibutyltin oxide in a suitable solvent affords the compounds with R3=tetrazoyl and R′═N-phthaloyl. Cleavage of the protecting group with hydrazine hydrate affords the desired amines, which are reacted with a suitable aldehyde (D-CHO) in an appropriate solvent to afford the desired final compound with R3=tetrazoyl. The desired final compound with R3=tetrazoyl and R4═H can be obtained by omitting the alkylation step with alkyl halides in the above scheme.
- General synthetic scheme for the preparation of compounds with bridged piperazinones of this invention with R14a,b=(═O)
-

- A commercially available hydroxyl-proline derivative is treated with base and alkylated with allylbromide in an appropriate solvent to afford the allyl-protected amino acid after purification. This compound is then treated at −30° C. with an appropriate base, triflic anhydride and then an appropriately protected diamino acid in an appropriate solvent to afford the desired compound after purification. After cleavage of the ester moiety with palladium(0) in an appropriate solvent, the compound is treated with EDCI and base in an appropriate solvent to afford the desired compound after purification. Cleavage of Fmoc protecting group by treatment with an suitable base affords the desired product. The free amine is then treated in the presence of an suitable polymer supported base with sulfonyl chlorides, acid chlorides or isocyanates to afford the desired compounds after purification. Removal of the Boc-protecting group with acid in a suitable solvent affords the final desired compounds after purification.
- Starting with the enantiomers of the amino acid derivatives above, and proceeding through the general procedures as described above, the enantiomeric piperazinone derivatives can be made.
- General synthetic scheme for the preparation of compounds with bridged piperazinones of this invention with R13a,b=(═O)
-
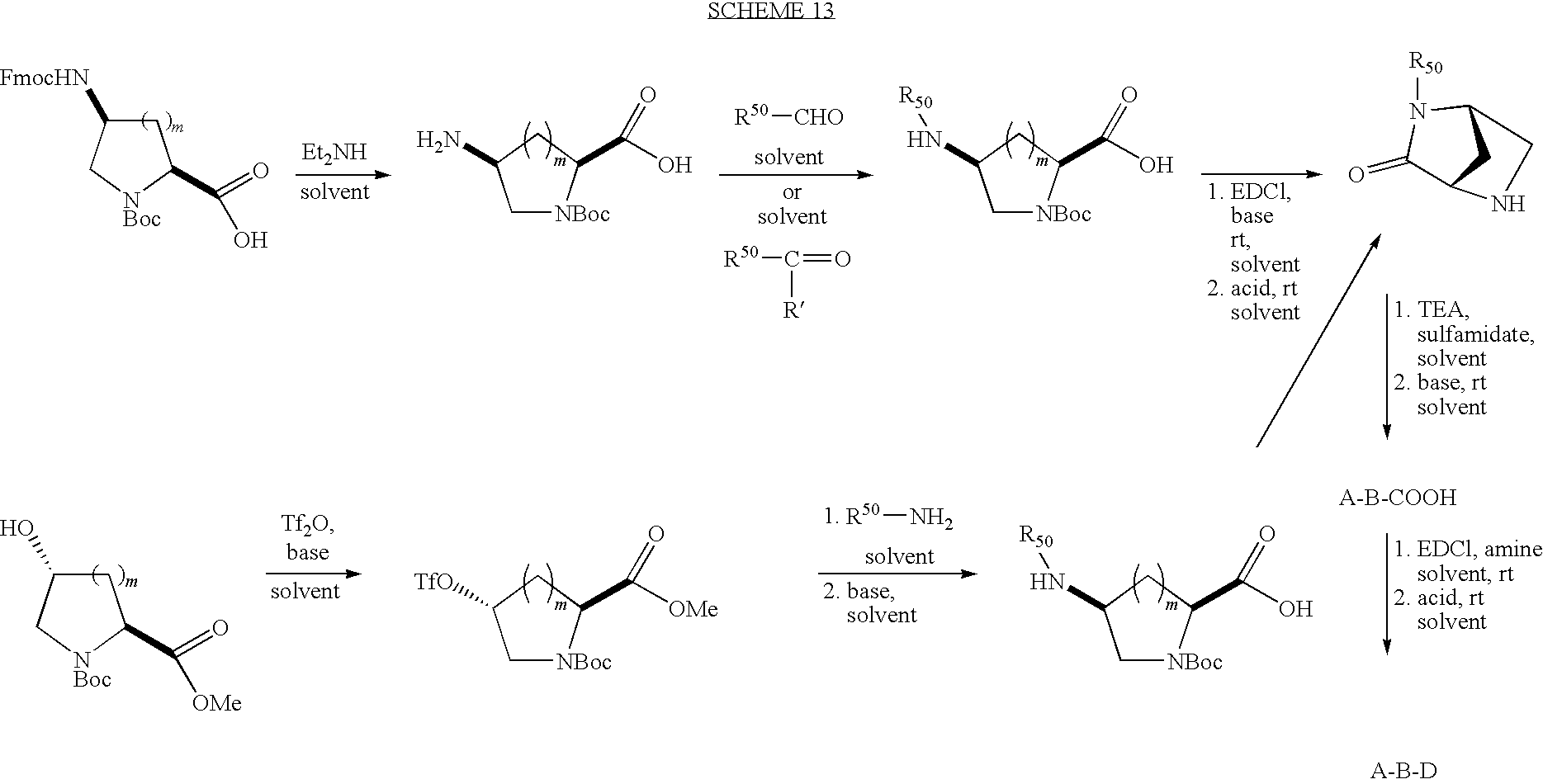
- After removing the Fmoc group of the commercially available amino acid with Et2NH, the primary amine is treated in an appropriate solvent with aldehydes or ketones in a reductive amination reaction to afford the desired products. Alternatively, the commercially available N-Boc-protected hydroxy amino acid ester can be treated with trifluoroacetic acid anhydride. The nucleopHilic displacement reaction of the triflate with commercially available amines affords the desired products, after saponification of the ester moiety with base and purification. These compounds are then treated with EDCl and a base in an suitable solvent to afford the cyclic amides after purification. These compounds are converted to the desired products by removing the Boc-protection group. These compounds are then reacted in a suitable solvent with a cyclic sulfamidate, derived from a serine derivative, in the presence of base. Saponification of the ester of the reaction product with a suitable base yields the desired acid compounds after purification. Further treatment of the free acids with EDCI in the presence of an appropriate base and a suitable amine derivative, followed by acidic removal of the Boc-protecting group yields the desired compounds after purification.
- Starting with the enantiomers of the amino acid and amine derivatives above, and proceeding through the general procedures as described above, the enantiomeric piperazinone derivatives can be made.
- General synthetic scheme for the preparation of compounds with bridged piperazines of this invention with R13a,b and R14a,b═H
-

- The commercially available bridged piperazine derivate is treated with a commercially available aziridine ester in an appropriate solvent to afford the desired compound after purification. After acidic removal of the Boc-protection group, the desired product reacts in presence of a base with an acid chloride or sulfonic acid chloride to yield the desired products after purification. After basic saponification, the free acids are treated with EDCI in the presence of an appropriate base and a suitable amine derivative to afford the desired compounds after purification. The Cbz-protecting group is then removed by treatment with TMSI and subsequent purification to afford the desired final compounds.
- Starting with the enantiomers of the amine and aziridine derivatives above, and proceeding through the general procedures as described above, the enantiomeric piperazine derivatives can be made.
- As can be seen by the generic schemes, each of the structures of “B” bonds to the “A” structures on its left side and to the “D” structures on its right side as each is depicted below. The compound A-B-D chooses an “A” which includes the following:
-

- A is desirably
-

- The “B” structures are chosen from:
-

- Desirably, B is one of structure (a), (b), (c), and (d). More desirably, B is structure (b)
- The “D” structures are chosen from:
-

- The substituents are selected as follows:
- E, G, and M represent a three ring system wherein M shares two carbon atoms with each of E and G;
- E and G are each independently selected from 6-membered aryl, 5-membered heteroaryl; 6-membered heteroaryl; a 5-7-membered saturated or partially saturated carbocyclic ring; and a 5-7 membered saturated or partially saturated heterocyclic ring; desirably E and G are substituted phenyl; M is a 5-7-membered saturated or partially saturated carboxylic or heterocyclic ring, or a 5-6-membered aromatic or heteroaromatic ring.
- E may be substituted with one or more R1 groups;
- G may be substituted with one or more R2 groups;
- X and Y are divalent and are each independently: a bond, CR4R5, O, NR4, S, S═O, S(═O)2, C(═O), (C═O)N(R4), S(═O)2N(R4), C═N—OR4, —C(R4R5)C(R4R5)—, —C(R4)═C(R5)—, —C(R4R5)NR4—, —C(R4R5)O—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, NS(═O)2R4; or aryl, heteroaryl, cycloalkyl or heterocyclic ring, all may be optionally substituted;
- R1 and R2 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all of which may be optionally substituted. Desirably, R1 and R2 may be defined independently as —H, —F, —Cl, —CONR4R5, —CO2H, —CN or —SO2NR4R5R2.
- R3 is absent or is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all of which may be optionally substituted. Desirably, R3 is absent or is —H, —OH, —CO2H, —CN, —CONR4R5, R5, aryl, NH(C═O)R4, NH(SO2)R4, heteroaryl —SO3H, —PO3H2, —CONR4R5, R5, aryl, NH(C═O)R4, or NH(SO2)R4, and more desirably, R3 is —CONR4R5 or tetrazolyl.
- Ra is hydrogen, CN, NO2, alkyl, haloalkyl, S(O)tNR4R5, S(O)tR4, C(O)OR4, C(O)R4, or C(O)NR4R5;
- each occurrence of R4, R5, R20 and R21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl and aminoalkyl are all optionally substituted, or R4 and R5 when taken together with the nitrogen to which they are attached complete a 3- to 8-membered ring containing carbon atoms and may optionally contain a heteroatom selected from O, S, or NR50 and the 3- to 8-membered ring may be optionally substituted. Desirably, R4 and R5 are each independently —H or alkyl.
- R50 is, in each occurrence, R20, CN, NO2, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(═NRa)NR20R21, C(═NR20)NR21Ra, C(═NOR20)R21 or C(O)NR20R21;
- each occurrence of R7 and R8 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C1-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted. Desirably, R7 and R8 are independently H or alkyl.
- R9 is H or C1-6 alkyl, desirably H.
- R10 is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (CO—C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (CO—C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, B(OH)2, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl are all optionally substituted. Desirably R10 is CN.
- R11 and R12 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (CO—C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl and aminoalkyl all may be optionally substituted;
- R13a and R13b are each independently R5 or together are ═O;
- R14a and R14b are each independently R5 or together are ═O;
- R13c and R14c are each independently R5;
- Qa is CH or N;
- Qb is CH or N;
- U is —C(O)—, —C(═NR4)—, —(CR4R5—)p, NR50, S(═O)2, C(═O), (C═O)N(R4), N(R4)(C═O), S(═O)2N(R4), N(R4)S(═O)2, C═N—OR4, —C(R4)═C(R5)—, —C(R4R5)pNR50—, N(R50)C(R4R5)p—, —O—C(R4R5)—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, NS(═O)2R4, or an optionally substituted aryl, heteroaryl, cycloalkyl or heterocyclic ring, all of which may be optionally substituted. Desirably, U is CH2.
- W is —CH2—, —S—, —CHF— or —CF2—;
- Z is C or N;
- m is 1, or 2;
- n is 0, 1, or 2;
- p is 0 to 6;
- q is 0 to 6; and
- t is 0, 1, or 2.
- Compounds of the present invention having one or more optically active carbons can exist as racemates and racemic mixtures, diasteromeric mixtures and individual diastereomers, enantiomeric mixtures and single enantiomers, tautomers, atropisomers, and rotamers, with all isomeric forms being included in the present invention. Compounds described in this invention containing olefinic double bonds include both E and Z geometric isomers. Also included in this invention are all salt forms, polymorphs, hydrates and solvates. All of the above mentioned compounds are included within the scope of the invention.
- The DPP-IV inhibition activity of the DPP-IV inhibitor compounds of the present invention may be measured using any suitable assay known in the art. A standard in vitro assay for measuring DPP-IV inhibitor activity is described.
- The synthesis of DPP-IV inhibiting compounds of the invention and their biological activity assay are described in the following examples which are not intended to be limiting in any way.
- All reagents and solvents were obtained from commercial sources and used without further purification. Proton (1H) spectra were recorded on a 250 MHz NMR spectrometer in deuterated solvents. Chromatography was performed using Roth silica gel (Si 60, 0.06-0.2 mm) and suitable organic solvents as indicated in specific examples. For flash chromatography Roth silica gel (Si 60, 0.04-0.063 mm) was used. Thin layer chromatography (TLC) was carried out on silica gel plates with UV detection. Preparative thin layer chromatography (Prep-TLC) was conducted with 0.5 mm or 1 mm silica gel plates (Merck Si 60, F254) and the solvents indicated in the specific examples.
-

- Commercially available prolinamide (5 g) was first treated with bromacetylbromide (4.2 ml) in CH2Cl2 and then with trifluoracetic acid anhydride in CH2Cl2 as described in WO 98/19998 to afford the title compound (7.85 g; 83%). 1HNMR δ (CDCl3) 2.05-2.40 (m, 4H), 3.51-3.70 (m, 2H), 3.80-3.85 (m, 2H), 4.70-4.86 (m, 1H).
-

- Commercially available L-prolinamide (25 g) was dissolved in CH2Cl2 (1200 ml) and triethylamine (30 ml) and 4-dimethylaminopyridine (1.9 g) added. The mixture was cooled to 0° C. and treated with fumaryl chloride (11.7 ml). The dark mixture was stirred at rt for 16 h and cooled to 0° C. TFAA (77 ml) was added dropwise under stirring and the solution allowed to warm to rt over 6 hours. The reaction mixture was stirred at rt for 1 to 2 days. Ice (500 g) was added followed by cautious addition of sat. NaHCO3 (600 ml). After the evolution of gas had ceased, the organic phase was separated and washed with sat. NaHCO3 (350 ml), H2O (350 ml), and brine (200 ml). The organic phase was dried over MgSO4 and concentrated to afford the title compound (28.6 g; 98%).
- 1HNMR δ (CDCl3) 2.12-2.30 (m, 8H), 3.58-3.69 (m, 2H), 3.73-3.89 (m, 2H), 4.72-4.83 (m, 2H), 7.26 (s, 2H).
- The title compound from Step A above (9.6 g) was dissolved in CHCl3 (90 ml) and MeOH (90 ml) and cooled to −78° C. At −78° C. a slow flow of ozone (originating from an O2 cylinder) was passed through the mixture for 3 h. The mixture was purged with N2 and dimethylsulfide (6 ml) added. The mixture was stirred for 1 h, allowed to reach rt and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 100:0->92:8) to afford the title compound as a mixture of the aldehyde and methoxy hemiacetal in a ratio of ˜1:9 (8.9 g; 69%).
- 1HNMR δ (D2O) 2.10-2.38 (m, 4H), 3.32 (s, 3H), 3.60-3.84 (m, 2H), 4.72-4.81 (m, 1H), 5.5 (s, 9/10H), 7.9 (s, 1/10H).
-

- Commercially available 2-cyano-3-methylpyridine (25 g) was dissolved in t-butanol (50 ml) and stirred at 80° C. Concentrated sulphuric acid (25 ml) was slowly added over a period of 45 minutes. After complete addition of the acid stirring was continued at 80° C. for 1 h. The reaction was diluted with water (50 ml) and toluene (125 ml). The pH was adjusted to 10 with 25% aqueous ammonia (110 ml). The separated organic phase was concentrated in vacuum affording the desired product (27 g, 90%).
- 1HNMR δ (CDCl3) 1.4 (s, 9H), 2.7 (s, 3H), 7.2-7.3 (m, 1H), 7.6 (m, 1H), 8.1 (s br, 1H), 8.4 (m, 1H)
- The title compound of Step A (12 g) above was dissolved in THF (150 ml) and cooled to −64° C. n-Butyllithium (1.6 M in hexane, 77 ml) was added over a period of 30 min. After addition of sodium bromide (0.6 g) stirring was continued for 30 min at −64° C. m-Chlorobenzylchloride (11 g) was added while the temperature was kept below −55° C. The mixture was stirred for 2 hours at −60° C. and for further 2 h at −110° C. Subsequently, the reaction was quenched with water (100 ml) and concentrated. The aqueous phase was extracted with chloroform (3×100 ml). The combined organic phase was dried over MgSO4 and concentrated in vacuum affording the title compound (22 g; 82%).
- 1HNMR δ (CDCl3) 1.4 (s, 9H), 2.9-3.0 (m, 2H), 3.4-3.5 (m, 2H), 7.0-7.4 (m, 6H), 8.0 (s br, 1H), 8.4 (m, 1H)
- The title compound of Step B (21.5 g) above was dissolved in phosphorus oxychloride (80 ml) and refluxed for 5 h. The reaction was concentrated and neutralized with 50% aqueous NaOH. The solid was separated and washed with hot isopropanol to afford the title compound (10.4 g; 63%)
- 1HNMR δ (CDCl3) 2.9-3.0 (m, 2H), 3.0-3.2 (m, 2H), 7.0-7.3 (m, 4H), 7.3-7.4 (m, 1H), 7.4-7.5 (m, 1H), 8.5-8.6 (m, 1H)
- The title compound of Step C (10 g) above was dissolved in trifluorosulfonic acid (80 ml) and stirred at 60° C. for 1 h. At rt 6 N aqueous HCl (80 ml) was dropwise added. The reaction was refluxed for 1 h and subsequently, poured on ice. After neutralization with 50% aqueous NaOH the precipitate was separated, washed with water and recrystallized from isopropanol/water (3.1) affording the title compound. The mother liquor was concentrated and the residue washed with water and chloroform to afford additional title compound (9.4 g; 94%).
- 1HNMR δ (MeOD-d4) 3.3-3.4 (m, 2H), 3.4-3.5 (m, 2H), 7.5 (m, 2H), 8.1-8.2 (m, 2H), 8.7 (d, 1H), 8.9 (d, 1H)
- The title compound of Step D (700 mg) above was dissolved in MeOH (10 ml) and cooled to 0° C. NaBH4 (95 mg) was added in one portion. The mixture was allowed to warm to RT and stirred for 1 h. The reaction was acidified with 1 N HCl and subsequently, brought to pH 12 with 1 N NaOH. The mixture was poured in water (100 ml) and extracted with CHCl3 (100 ml). The organic phase was dried over MgSO4 and concentrated affording the title compound (705 mg; 100%).
- 1HNMR δ (MeOD-d4) 3.0-3.4 (m, 4H), 6.1 (s, 1H), 7.1.7.3 (m, 3H), 7.5-7.6 (m, 2H), 8.3.8.4 (m, 1H)
- The title compound of step E (370 mg) above was dissolved in toluene (5 ml) and cooled to −15° C. Thionyl chloride (286 mg) was slowly added and the reaction was allowed to come to RT and run overnight. The solution was neutralized with triethylamine and directly used in the next step.
-

- The title compound from Preparative Example 3 Step E (285 mg) was dissolved in ethanol (10 ml) and 10% Pd/C (100 mg) and ammonium formiate (916 mg) were added. The mixture was refluxed for 2 h. Subsequently, the reaction was treated with water (20 ml) and extracted twice with chloroform (50 ml). The combined organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane 1:4) to afford the title compound (200 mg; 82%).
- 1HNMR δ (MeOD-d4) 2.9-3.1 (m, 2H), 3.3-3.6 (m, 2H), 6.3 (s, 1H), 7.0-7.3 (m, 4H), 7.4 (m, 1H), 7.8 (m, 1H), 8.3 (m, 1H)
- The title compound of Step A (200 mg) above was dissolved in toluene (5 ml) and cooled to −15° C. Thionyl chloride (235 mg) was slowly added and the reaction was allowed to come to RT and run overnight. The solution was neutralized with triethylamine directly used.
-

- To a cooled solution (12° C.) of commercially available ethylenediamine (30 ml) was added within 5 min commercially available dibenzosuberylchloride (3.3 g). The mixture was stirred at rt for 1 h and then K2CO3 (5.8 g) was added. After an additional 30 min at rt, the mixture as filtered, the salts washed with 5 ml ethylenediamine and the filtrates concentrated. The residue was dissolved in 80 ml EtOAc, 20 ml H2O and 5 ml NH4OH-solution (25%). The organic phase was separated, dried over MgSO4 and concentrated to afford the title compound (3.4 g; 93%; MH+=253).
- The title compounds from Preparative Example 6 to 9 were prepared according to the procedure described in Preparative Example 5 using the chlorides and amines as indicated in the Table below. In case the chlorides did not dissolve in the amines after 10 Min, CH3CN or THF was added until a clear solution was obtained.
-
Preparative 1. Yield Example Chloride Amine Product 2. MH+ 6 
NH4OH 
1. 61% 2. 1H-NMR δ (CDCl3) 2.0 (s, 2H), 3.10-3.24 (m, 2H), 3.31-3.45 (m, 2H), 5.43 (s, 1H), 7.10-7.19 (m, 6H), 7.36-7.41 (m, 2H) 7 


1. 97% 2. 281 8 


1. 60% 2. 288 9 


1. 78% 2. 1H-NMR δ (CD3OD) 2.6-2.8 (m, 4H), 3.0-3.2 (m, 2H), 3.3-3.6 (m, 2H), 5.2 (s, 1H), 7.1-7.2 (m, 4H), 7.3-7.4 (m, 1H), 7.5 (m, 1H), 8.2-8.3 (m, 1H) -

- Commercially available dibenzosuberylchloride (300 mg) and 4-N-Boc-amino-piperidine (290 mg) were suspended in CH3CN (10 ml). After 10 min K2CO3 (545 mg) was added and the mixture was stirred at rt for 3 h. The mixture was diluted with EtOAc (30 ml) and H2O (15 ml), the organic phase separated, dried over MgSO4 and concentrated to afford the title compound (460 mg; 89%; MH+=393).
- The title compound from Step A above (460 mg) was dissolved in a solution of 4 M HCl in dioxane (20 ml). The mixture was stirred at rt for 2 h and concentrated to afford the title compound (335 mg; 97%; MH+=293).
- The title compounds from Preparative Example 11 and 12 were prepared according to the procedure described in Preparative Example 10 using the chlorides and amines as indicated in the Table below.
-
Preparative 1. Yield Example Chloride Amine Product 2. MH+ 11 


1. 64% 2. 279 12 


1. 56% 2. 265 -

- To a suspension of AgCN (4.7 g) in CH3CN (60 ml) under nitrogen was added at rt a solution of commercially available dibenzosuberylchloride (6 g) in CH3CN (60 ml) and benzene (10 ml). The mixture was heated at reflux for 2 h, cooled to rt and filtered. The salts were washed with 20 ml CH3CN and the filtrates concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane, 1:9) to afford the title compound (5 g; 87%; MNa+=242).
- A suspension of LiAlH4 (360 mg) in Et2O (20 ml) was slowly treated with a solution of AlCl3 (950 mg) in Et2O (20 ml). The mixture was stirred at rt for 10 min and then the title compound from Step A above (1.03 g) was added within 5 min. The mixture was stirred at rt for 10 min and then refluxed for 8 h. After the addition of H2O (20 ml) and 25% NH4OH (6 ml), the mixture was filtered and the salts washed with H2O (20 ml) and Et2O (10 ml). The organic phase was separated, dried over MgSO4 and concentrated to afford the title compound (157 mg; 15%; MH+=224).
-

- To a solution of commercially available iminodibenzyl (5 g) in toluene (25 ml) was added commercially available bromoacetylbromide (4.35 ml). The mixture was heated under reflux for 2 h 30 Min, cooled and concentrated. A portion of the crude product (800 mg) was dissolved in DMA (6 ml) and treated with NaN3 (815 mg). The mixture was heated at 60-70° C. overnight and diluted with EtOAc (30 ml) and H2O (10 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was treated with EtOAc/cyclohexane (1:9) (2 ml), sonicated for 2 min and the solvents removed by syringe. The residue was dried to afford the title compound (483 mg; 69%; MH+=279).
- The title compound from Step A above (483 mg) was dissolved in MeOH (25 ml) and 10% Pd/C (100 mg) added. The mixture was hydrogenated for 1 h, filtered and the catalyst washed with MeOH (10 ml). The filtrates were concentrated and the residue purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (415 mg; 95%; MH+=253).
- To a suspension of LiAlH4 (242 mg) in THF (6 ml) was added a solution of the title compound from Step B above (322 mg) in THF (6 ml). The mixture was heated under reflux for 2 h 30 min. The mixture was cooled to 0° C., quenched with H2O (0.3 ml) and diluted with 15% NH4OH-solution (0.3 ml) and H2O (0.8 ml). The mixture was stirred at rt for 45 Min, filtered and the salts washed with THF (8 ml). The filtrates were concentrated and the residue purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (79 mg; 26%; MH+=239).
-

- A mixture of commercially available dibenzosuberenol (1.5 g) and malonic acid (830 mg) was heated at 160-170° C. for 2 h. A mixture of H2O (5 ml) and 0.1 M HCl (5 ml) was added and the mixture cooled to rt. The mixture was diluted with EtOAc (100 ml) and H2O (10 ml), the organic phase separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/acetone, 98:2->CH2Cl2/acetone, 9:1) to afford the title compound (775 mg; 43%; MNa+=273).
- A mixture of title compound from Step A above (775 mg) and triethylamine (0.59 ml) in THF (20 ml) was cooled to −40° C. and treated with isobutylchloroformate. After stirring at −40° C. for 1 h, the mixture was filtered and the salts washed with THF (5 ml). The filtrates were then treated at 0° C. with 25% NH4OH (15 ml) for 1 h 30 min. The mixture was diluted with EtOAc (60 ml), the organic phase separated, dried over MgSO4 and concentrated. The residue was treated with CHCl3 (1.5 ml), the solvent removed by syringe and the residue dried to afford the title compound (677 mg; 88%; MH+=250).
- To a suspension of LiAlH4 (513 mg) in THF (15 ml) was added a solution of the title compound from Step B above (677 mg) in THF (25 ml). The mixture was heated under reflux for 2 h. The mixture was cooled to 0° C., quenched with H2O (0.65 ml) and diluted with 4 M NaOH-solution (2.5 ml). The mixture was stirred at rt for 45 Min, filtered and the salts washed with THF (15 ml). The filtrates were concentrated and the residue purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (560 mg; 88%; MH+=236).
- The title compound from Step C above (350 mg) was dissolved in MeOH (15 ml) and 10% Pd/C (300 mg) and 1 M HCl (1.5 ml) were added. The mixture was hydrogenated overnight, filtered and the catalyst washed with MeOH (10 ml). The filtrates were concentrated and the residue dissolved in EtOAc (30 ml) and sat. NaHCO3 (10 ml). The organic phase was separated and the aqueous phase extracted with EtOAc (20 ml). The combined organic phase was dried over MgSO4 and concentrated to afford the title compound (232 mg; 66%; MH+=238).
-

- The intermediate from Preparative Example 14 Step A (1 g) was dissolved in DMA (6 ml) and treated with NaCN (368 mg). The mixture was heated at 60-70° C. overnight and diluted with EtOAc (50 ml) and H2O (15 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/acetone, 98:2) to afford the title compound (282 mg; 34%; MH+=263).
- To a suspension of LiAlH4 (123 mg) in THF (6 ml) was added a solution of the title compound from Step A above (282 mg) in THF (6 ml). The mixture was heated at 50° C. for 2 h, cooled to 0° C. and treated with H2O (0.2 ml) and 4 M NaOH (0.8 ml). The mixture was stirred at rt for 45 Min, treated with MgSO4 and filtered. The filtrate was concentrated and the residue purified by chromatography on silica (CH2Cl2/MeOH, 95:5->CH2Cl2/MeOH, 9:1) to afford the title compound (32 mg; 12%; MH+=253).
-

- To a suspension of magnesium (701 mg) in Et2O (7 ml) was slowly added ethylbromide (2.15 ml). After the formation of the Grignard reagent, the mixture was cooled to 5° C. and a solution of diethylamine (3 ml) in Et2O (5 ml) was slowly added. The mixture was refluxed for 30 Min, cooled to 5° C. and treated with a mixture of commercially available dibenzosuberone (3 g) and tert-butylacetate (1.95 ml) in Et2O (15 ml). The mixture was heated under reflux for 2 h, cooled to rt and poured onto ice-water containing an excess of NH4Cl. The mixture was extracted with CH2Cl2 (3×100 ml), the organic phase dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane, 1:9) to afford the title compound (3.5 g; 75%; MNa+=347).
- To a suspension of LiAlH4 (346 mg) in THF (12 ml) was added a solution of the title compound from Step A above (2 g) in THF (12 ml). The mixture was heated under reflux for 2 h, cooled to 0° C. and treated 4 M NaOH (4.5 ml). The mixture was stirred at rt for 45 min and filtered. The filtrate was concentrated and the residue dissolved in EtOAc (100 ml), H2O (10 ml) and sat. NH4Cl (10 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane, 3:7) to afford the title compound (937 mg; 60%; MNa+=277).
- The title compound from Step B above (937 mg) was dissolved in benzene (1.5 ml) and pyridine (1.5 ml). The mixture was cooled to 5° C. and treated with a solution of p-tosylchloride in benzene (1.5 ml). The mixture was stirred at rt for 7 h, diluted with EtOAc (40 ml) and washed with 0.1 M HCl (10 ml), sat. NaHCO3 (10 ml) and brine (10 ml). The organic phase was separated, dried over MgSO4 and concentrated. The crude intermediate was dissolved in DMA (9 ml) and treated with NaN3 (1.2 g). The mixture was heated at 70° C. overnight and the DMA removed. The residue was dissolved in EtOAc (50 ml), sat. NaHCO3 (10 ml) and brine (10 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane, 1:4) to afford the title compound (704 mg; 68%; MNa+=302).
- The title compound from Step C above (200 mg) was dissolved in MeOH (8 ml) and 10% Pd/C (40 mg) added. The mixture was hydrogenated for 1 h 30 Min, filtered and the catalyst washed with MeOH (10 ml). The filtrates were concentrated to afford the title compound (175 mg; 96%; MH+=254).
- The title compound from Step D above (75 mg) was dissolved in EtOH (1 ml) and a 4 M solution of HCl in dioxane (1 ml) added. The mixture was stirred at rt for 12 h and concentrated. The residue was dissolved in EtOAc (20 ml) and sat. NaHCO3 (5 ml). The organic phase was separated, dried over MgSO4 and concentrated to afford the title compound (67 mg; 96%; M+—NH3=219).
-

- The title compound from Preparative Example 13 Step A (1.1 g) was dissolved in THF (5 ml) and added to a suspension of NaH (132 mg) in THF (5 ml). The mixture was heated under reflux for 1 h, cooled to rt and treated with 1,2-dibromoethane (0.9 ml) in THF (1 ml). The mixture was heated under reflux for 4 h, cooled to rt and filtered. The salts were washed with THF (5 ml) and the filtrates concentrated. The residue was dissolved in DMA (12 ml) and treated with NaN3 (1.6 g). The mixture was heated at 60-70° C. overnight and the DMA removed. The residue was dissolved in EtOAc (40 ml) and H2O (10 ml), the organic phase separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane, 1:9) to afford the title compound (1.14 g; 78%; MH+=289).
- The title compound from Step A above (510 mg) was dissolved in MeOH (20 ml) and 10% Pd/C (150 mg) and 2 M HCl (0.9 ml) added. The mixture was hydrogenated for 1 h 30 Min, filtered and the catalyst washed with MeOH (10 ml). The filtrates were concentrated and the residue purified by chromatography on silica (CH2Cl2/MeOH, 95:5 to CH2Cl2/MeOH, 4:1) to afford a mixture of the title compound and the cyclic amidine (450 mg; 96%; MH+=263).
- The title compounds from Step B above (350 mg) were treated with 2 ml 57% H2SO4. The mixture was heated at 100° C. for 3 h, cooled to rt and diluted with H2O (10 ml). The mixture was made alkaline (pH˜11) by adding 10% NaOH and extracted with EtOAc (3×30 ml). The organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 9:1 to CH2Cl2/MeOH (7 M NH3), 9:1) to afford a mixture of the title compound and the cyclic amidine (223 mg; 60%; MH+=281).
-

- Commercially available (S)-2-aminopropan-1-ol (2.0 g) was dissolved in CH2Cl2 (20 ml) and Boc2O (6.4 g) was added. After stirring for 4 h at room temperature the solvent was removed to afford the title compound (4.7 g, 99%).
- 1H-NMR δ (CDCl3): 1.10 (s, 3H), 1.50 (s, 9H), 2.40 (s, 1H), 3.45-3.70 (m, 2H), 3.75-3.80 (m, 1H), 4.80 (s, 1H).
- Imidazole (4.1 g) was dissolved in CH2Cl2 (50 ml) and cooled to 0° C. Thionyl chloride (1.3 ml) dissolved in CH2Cl2 (10 ml) was added dropwise and the resulting suspension was allowed to warm to rt. Stirring was continued for 1 h at rt and then the mixture was cooled to −78° C. A solution of the title compound from Step A above (1.8 g) in CH2Cl2 (50 ml) was added over a period of 1 h and the resulting mixture was allowed to warm to rt and stirred overnight. The mixture was filtered through celite and the filter aid was washed well with CH2Cl2. The organic phase was diluted with CH2Cl2, washed with water and brine, dried over MgSO4, filtered and concentrated to a volume of approx. 100 ml.
- A solution of NaIO4 (4.3 g) in water (100 ml) was added and the mixture was cooled to 0° C. Ru(IV)O2 hydrate (150 mg) was added and the black suspension was stirred for 2 h at 0° C. It was then warmed to rt and stirred overnight. The mixture was filtered through celite and the filtrate was extracted with CH2Cl2. The combined organic phase was washed with brine, dried and filtered. Treatment of the filtrate with activated charcoal (2 g) for 30 min removed traces of ruthenium. The mixture was filtered again and evaporated to yield the title compound (1.5 g, 63%).
- 1H-NMR δ (CDCl3): 1.45 (s, 3H), 1.49 (s, 9H), 4.14 (dd, 1H), 4.29-4.42 (m, 1H), 4.61 (dd, 1H).
- The title compound from Preparative Example 20 was prepared according to the procedure described in Preparative Example 19 using the aminoalcohol as indicated in the Table below.
-
Prepar- ative Exam- 1. Yield ple Aminoalcohol Product 2. 1H-NMR 20 

1. 69% 2. 1H-NMR δ (CDCl3): 1.45 (s, 3 H), 1.49 (s, 9 H), 4.14 (dd, 1 H), 4.29-4.42 (m, 1 H), 4.61 (dd, 1 H). -

- To a stirred solution of the commercially available 2-(S)-amino propanol (17.4 g) in water (200 ml) was added a solution of triethylamine (32 ml) in dioxane (200 ml). To the solution was added commercially available 1-[2-(Trimethylsilyl)ethoxy-carbonyloxy]pyrrolidin-2,5-dione (60 g). The mixture was stirred at rt overnight, then diluted with water (200 ml), acidified with 1 N HCl, and extracted with Et2O (2×500 ml). The combined organic phase was washed with brine, dried over MgSO4 and evaporated to afford the title compound (44.2 g; 87%).
- 1H-NMR δ (CDCl3): 0.02 (s, 9H), 0.90-1.05 (m, 2H), 1.20 (d, 3H), 2.80 (br s, 1H), 3.40-3.80 (m, 3H), 4.10-4.20 (m, 2H), 4.85 (s, 1H).
- Imidazole (96 g) was dissolved in CH2Cl2 (1200 ml) and cooled to 0° C. Thionyl chloride (30.8 ml) was diluted with CH2Cl2 (600 ml) and added dropwise. The resulting suspension was allowed to warm to rt. Stirring was continued for 1 h at rt and then the mixture was cooled to −78° C. A solution of the title compound from Step A above (44.2 g) in CH2Cl2 (1200 ml) was added over a period of 1 h and the resulting mixture was allowed to warm to rt and stirred overnight. The mixture was filtered through celite, the filter aid was washed well with CH2Cl2. The organic phase was washed with water (2×700 ml), dried over MgSO4, filtered and concentrated to a volume of approx. 1000 ml.
- A solution of NaIO4 (100 g) in water (1000 ml) was added and the mixture was cooled to 0° C. RuO2×H2O (1 g) was added and the black suspension was stirred for 2 h at 0° C. It was then warmed to rt and stirred overnight. The phases were separated and the organic phase was treated with granulated charcoal (˜20 g). The mixture was stirred for approx. 1 h, filtered through celite and the filtrate was dried with MgSO4, filtered and evaporated to yield the title compound (50.7 g, 89%).
- 1H-NMR δ (CDCl3): 0.02 (s, 9H), 1.00-1.15 (m, 2H), 1.50 (d, 3H), 4.15 (dd, 1H), 4.35-4.45 (m, 3H), 4.65 (dd, 1H).
- Following a similar procedure as that described in Preparative Example 21 but using the aminoalcohols as indicated in the Table below, the title compounds were obtained.
-
Prepar- ative Exam- 1. Yield ple Aminoalcohol Product 2. 1H-NMR 22 

1. 58% 2. 1H-NMR δ (CDCl3): 0.02 (s, 9 H), 1.00-115 (m, 2 H), 4.00-4.10 (m, 2 H), 4.25-4.40 (m, 2 H), 4.55-4.65 (m, 2 H). 23 

1. 32% (M + Na)+ = 318 - If one were to follow a similar procedure as that described in Preparative Example 21 but using the aminoalcohols as indicated in the Table below, one would obtain the desired products.
-
Prepar- ative Example Aminoalcohol Product 24 

25 

26 

27 

28 

29 

30 

31 

32 

33 

34 

35 

36 

37 

38 

39 

40 

41 

42 

43 

44 

45 

46 

-

- A suspension of NaH (132 mg) in THF (10 ml) was added to a solution of Preparative Example 13 Step A (1.1 g) in THF (20 ml) and heated at 60° C. for 1 h. Then the mixture was cooled to 0° C. and a solution of Preparative Example 19 (1.2 g) in THF (10 ml) was added. The suspension was heated at 60° C. for 4 h and then diluted with ethyl acetate. The organic phase was washed with water, brine and dried over MgSO4. Removal of the solvents and column chromatography (EtOAc/hexane, 1:4) afford the title compound (1.7 g, 90%, MH+=377).
- The title compound from Step A above (1.5 g) was dissolved in 57% H2SO4 and the solution was heated at 100° C. for 2 h. The mixture was diluted with water and extracted with ethyl acetate. The organic phase was discarded and 50%-aqueous KOH solution added to the aqueous phase until pH>8. The aqueous phase was extracted with ethyl acetate (2×75 ml). The organic phase was washed with water, brine, dried over MgSO4 and evaporated to afford the title compound. (600 mg, 53%).
- 1H-NMR δ (CDCl3): 0.95 (d, 3H), 1.82 (s, 2H), 2.37-2.58 (m, 2H), 2.82-2.92 (m, 1H), 3.18 (s, 4H), 5.60 (s, 2H), 7.08-7.24 (m, 6H), 7.40-7.48 (m, 2H).
- The title compound was prepared according to the procedure described in Preparative Example 47 using the sulfamidate from Preparative Example 20 as indicated in the Table below.
-
Preparative 1. Yield Example Nitrile Sulfamidate Product 2. 1H-NMR 48 


1. 80% 2. 1H-NMR δ (CDCl3): 0.95 (d, 3 H), 1.82 (s, 2 H), 2.37-2.58 (m, 2 H), 2.82-2.92 (m, 1 H), 3.18 (s, 4 H), 5.60 (s, 2 H), 7.08-7.24 (m, 6 H), 7.40-7.48 (m, 2 H). -

- Commercially available 2,5-dibromotoluene (8.28 ml) was dissolved in hexane (90 ml) and treated with a 1.6 M solution of butyllithium in hexane (160 ml). The mixture was heated at 60° C. for 20 h, cooled to rt and poured onto a mixture of dry ice in Et2O (750 ml). The mixture was allowed to warm to rt, filtered and the precipitate washed with 90 ml Et2O. The precipitate was titrated with 140 ml glacial acetic acid to afford the title compound (10 g; 92%).
- 1H-NMR δ (DMSO-d6) 2.58 (s, 3H), 7.80-7.90 (m, 3H)
- The title compound from Step A above (13 g) was suspended in MeOH (300 ml) and slowly treated with thionyl chloride (15.7 ml). The mixture was heated under reflux for 2 h to become a clear solution. The solvents were concentrated to afford the title compound (13.3 g; 88%; MH+=209).
- The title compound from Step B above (13.3 g) was dissolved in CCl4 (500 ml) and commercially available N-bromosuccinimide (10.7 g) added. The mixture was heated to 80° C. and commercially available AIBN (327 mg) added. The mixture was then irradiated with a 100 W light bulb and heated at 100-105° C. for 2 h 30 min. The cooled mixture was filtered and the precipitate washed with 50 ml CCl4. The filtrates were concentrated and the residue dissolved in CH3CN (180 ml). The mixture was treated with triphenylphosphine (16 g) and heated under reflux for 3 h. The mixture was concentrated to ˜100 ml and Et2O (500 ml) added. The mixture was allowed to stand at rt for 30 Min, filtered and the precipitate washed with Et2O (30 ml) to afford the title compound (20 g; 57%).
- The title compound from Step C above (20 g) was suspended in CH3CN (160 ml) and commercially available 4-Fluorobenzaldehyde (5.4 ml) added. The mixture was then treated with commercially available DBN (10 ml) and heated at 100° C. for 1 h. The mixture was concentrated to half its volume and poured into H2O (150 ml). The mixture was extracted with EtOAc (2×150 ml), the organic phase washed with 5% HCl (2×75 ml), dried over MgSO4 and concentrated. The residue was suspended in H2O (240 ml) and MeOH (20 ml) and KOH (20 g) added. The mixture was heated at 100° C. for 16 h, cooled to rt and washed with CH2Cl2 (3×75 ml). The aqueous phase was acidified (pH˜1) by adding conc. HCl, filtered, the precipitate washed with H2O (20 ml) and air-dried. The residue was dissolved in MeOH (900 ml) and 10% Pd/C (1.5 g) added. The mixture was hydrogenated for 1 h, filtered, the catalyst washed with MeOH (50 ml) and concentrated to afford the title compound (8.6 g; 82%; MH+=289).
- The title compound from Step D above (1.44 g) was suspended in sulfolane (9 ml) and treated with polyphosphoric acid (30 g). The mixture was heated under N2 at 170-175° C. for 3 h and poured onto ice-water (150 ml). The mixture was stirred at rt for 1 h, extracted with EtOAc (2×150 ml), dried over MgSO4 and concentrated. The residue was dissolved in MeOH (20 ml) and treated with thionyl chloride (1 ml). The mixture was heated under reflux for 1 h and concentrated. The residue was dissolved in Et2O (100 ml) and washed with sat. NaHCO3 (30 ml) and brine (30 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2) to afford the title compound (960 mg; 67%; MH+=285).
- The title compound from Step E (1420 mg) was dissolved in CHCl3 (20 ml) and MeOH (20 ml) and treated with NaBH4 (230 mg). The mixture was stirred at rt for 1 h and poured onto ice-water (150 ml). The mixture was extracted with EtOAc (2×150 ml), the organic phase dried over MgSO4 and concentrated to afford the title compound (1420 mg; 99%, M++Na=309).
- The title compound from Step F above (1420 mg) was dissolved in THF (20 ml) and treated with thionyl chloride (0.91 ml). The mixture was stirred at rt for 16 h and concentrated without heating. The residue was dissolved in CH3CN (17 ml) and treated with AgCN (785 mg). The mixture was heated at 90° C. for 2 h 30 Min, filtered and the salts washed with CH3CN (40 ml). The filtrates were concentrated and the residue purified by chromatography on silica (CH2Cl2) to afford the title compound (1160 mg; 79%; MH+=296).
- The title compound from Step G above (1327 mg) was dissolved in degassed THF (15 ml) and added to a suspension of NaH (119 mg) in degassed THF (5 ml). The mixture was heated at 90° C. for 1 h 15 min and cooled to rt. The mixture was then treated with 1,2-dibromoethane (0.81 ml) in THF (1 ml) and the mixture was heated at 90° C. for 4 h 30 min. The mixture was cooled to rt, diluted with 100 ml EtOAc, 10 ml brine and 10 ml sat. NH4Cl. The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in DMA (10 ml) and treated with NaN3 (720 mg). The mixture was heated at 60° C. for 16 h and diluted with EtOAc (100 ml) and brine (15 ml). The organic phase was separated, washed with 0.1 m HCl (15 ml) and brine (15 ml). The organic phase was dried over MgSO4, concentrated and the residue purified by chromatography on silica (EtOAc/cyclohexane, 1:4) to afford the title compound (931 mg; 57%; MH+=365).
- The title compound from Step H above (1050 mg) was dissolved in MeOH (40 ml). The mixture was treated with concentrated HCl (0.25 ml) and 10% Pd/C (250 mg). The mixture was hydrogenated for 1 h, filtered and the catalyst washed with MeOH (20 ml). The filtrates were concentrated to afford a mixture of the title compound and the cyclic amidine in a 9:1 ratio (950 mg; 97%; MH+=339).
- The title compounds from Step I above (950 mg) were treated with 57% H2SO4 (5 ml) and heated under N2 at 90° C. for 3 h. The mixture was cooled, diluted with H2O (80 ml) and made alkaline (pH˜10) by adding 50% NaOH. The mixture was washed with EtOAc (20 ml) and the aqueous phase diluted with dioxane (40 ml). The mixture was treated with an excess of Boc2O and stirred at rt for 16 h while the pH was kept at pH˜10.0. The mixture was acidified to pH˜4.0 by adding 1 M HCl and extracted with EtOAc (2×150 ml). The organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to elute the cyclic amidine side product, followed by CH2Cl2/MeOH (4:1) to afford the title compound (282 mg, 23%; MNa+=465).
- The title compound from Step J above (135 mg) was dissolved in THF (6 ml) and triethylamine (0.056 ml). The mixture was cooled to −40° C. and treated with ethyl chloroformate (0.031 ml). The mixture was stirred at −40° C. for 1 h, diluted with 4 ml THF and treated at 0° C. with 33% aqueous ammonia solution (10 ml). The mixture was stirred at 0° C. for 1 h and then 1 h at rt. The mixture was diluted with EtOAc (80 ml) and washed with brine (25 ml), sat. NH4Cl (25 ml and brine (25 ml). The organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (97 mg, 72%, MNa+=464).
- The title compound from Step K above (94 mg) was treated with 4 M solution of HCl in dioxane (2.5 ml) and the flask was agitated for 30 min. The mixture was concentrated and the residue dissolved in 5 ml H2O. The mixture was filtered through a Millex VV (0.1 μM) filter unit and the filtrate concentrated to afford the title compound (65.8 mg, 82%, MH+=342).
-

- The title compound from Preparative Example 13 Step A (3.3 g) was dissolved in THF (5 ml) and slowly added to a suspension of NaH (540 mg) in THF (10 ml). The mixture was heated at reflux for 30 min, cooled to rt and treated with 1,2-dibromoethane (4 ml). The reaction was stirred at 60° C. overnight, cooled to rt and filtered. The solvent was removed affording the title compound (4.8 g; 98%)
- 1HNMR δ CDCl3 2.9-3.2 (m, 6H), 3.2-3.4 (m, 2H), 7.1-7.3 (m, 6H), 7.9-8.0 (m, 2H)
- The title compound from Step A above (1.5 g) and potassium phthalimide (13.8 g) were suspended in DMF (20 ml) and stirred at 100° C. overnight. The precipitate was removed and the reaction was concentrated in vacuum. Chromatography of the residue on silica (EtOAc/cyclohexane) afforded the title compound (1.4 g; 78%).
- 1HNMR δ CDCl3 2.8-2.9 (m, 2H), 3.0-3.2 (m, 2H), 3.4-3.6 (m, 2H), 3.6-3.8 (m, 2H), 7.1-7.3 (m, 6H), 7.6-7.7 (m, 2H), 7.7-7.8 (m, 2H), 7.9-8.0 (m, 2H)
- The title compound from Step B above (1.40 g) was dissolved in toluene (30 ml) and treated with dibutyltin oxide (446 mg) and trimethylsilylazide (2.3 ml). The mixture was heated under a N2 atmosphere at 90° C. overnight. Additional dibutyltin oxide (200 mg) and trimethylsilylazide (2.3 ml) were added and the reaction was continued for 24 h at 90° C. The solvent was removed and the residue was treated with EtOAc (30 ml) and 1 N HCl (30 ml) at 50° C. for 1 h. The phases were separated and the organic phase was concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (600 mg, 39%, MH+=436).
- The title compound from Step C above (200 mg) was dissolved in ethanol (5 ml) and treated with hydrazine hydrate (100 mg) at rt. The solution was heated at 80° C. for 2 h and then stirred for 1 h at rt. The reaction was filtered and the filtrate was concentrated. The residue was treated with CHCl3 and filtered again. The filtrate was concentrated to afford the title compound (60 mg, 43%, MH+=306).
-

- Commercially available 2-bromo-4-fluorotoluene (5 g) was diluted with diethyl ether (10 ml). About ⅓ of the resulting solution was added to magnesium turnings (761 mg) which were overlayed with Et2O (25 ml). The remaining 2-bromo-4-fluorotoluene solution was added dropwise after the reaction started. The reaction was kept at reflux for 2 h. The Grignard reagent was poured onto a mixture of crushed dry ice in Et2O (750 ml). The resulting mixture was allowed to warm to rt. The solvent was removed, the resulting residue was treated with EtOAc (100 ml) and extracted with aqueous 1 N HCl (100 ml). The organic phase was dried over MgSO4, filtered and concentrated to afford the title compound (2.3 g; 56%).
- 1H-NMR δ CDCl3 2.5 (s, 3H), 7.0-7.2 (m, 2H), 7.7 (m, 1H)
- The title compound from Step A above (2.3 g) was dissolved in THF (50 ml). Methyl iodide (0.95 ml) and N,N-diisopropylethylamine (3.2 ml) were added. The reaction was stirred at rt for 2 h. The reaction mixture was filtered and concentrated to afford the title compound (2.3 g; 90%).
- 1H-NMR δ CDCl3 2.6 (s, 3H), 3.9 (s, 3H), 7.0-7.2 (m, 2H), 7.6-7.7 (m, 1H)
- The title compound from Step B above (8.9 g) and commercially available N-bromosuccinimide (14 g) were suspended in CCl4 (500 ml). The mixture was heated to 80° C. and AIBN (270 mg) added. The mixture was irradiated with a 100 W light bulb and heated at 100-105° C. for 3.5 h. The cooled mixture was filtered. The filtrate was concentrated and the residue dissolved in CH3CN (150 ml). The mixture was treated with triphenylphosphine (14 g), heated under reflux for 3 h and then concentrated. The residue was suspended in CH3CN (160 ml) and treated with commercially available 3-fluorobenzaldehyde (6.5 g) and DBN (13 ml). The mixture was heated under reflux for 3 h. The reaction was concentrated to half its volume and poured into H2O (150 ml). The mixture was extracted with EtOAc (3×150 ml), the organic phase separated and concentrated. The residue was suspended in 1:1 H2O/MeOH-mixture (100 ml) and treated with KOH (30 g). The mixture was stirred at 60° C. overnight, cooled to rt and washed with CHCl3 (3×100 ml). The aqueous phase was acidified (pH˜1) by adding conc. HCl and extracted with EtOAc. The organic phase was separated and concentrated. The crude residue was suspended in sulfolane (20 ml) and treated with polyphosphoric acid (25 g). The mixture was heated under N2 at 200° C. for 2 h, poured onto ice-water (150 ml) and stirred at rt overnight. The mixture was extracted with EtOAc and concentrated. The residue was dissolved in Et2O and extracted with H2O. The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/Cyclohexane) to afford the title compound (4.0 g; 31%; MH+=245).
- The title compound from Step C above (5.4 g) was dissolved in CHCl3 (5 ml) and MeOH (30 ml) and treated with NaBH4 (1.4 g). The mixture was stirred at rt for 1 h and concentrated. The residue was suspended in CHCl3 (50 ml) and extracted with aqueous HCl (50 ml; pH=1). The organic phase was separated, concentrated, then resuspended in toluene and concentrated again. The residue was dissolved in toluene (50 ml). SOCl2 (3.94 ml) was added at 0° C. The reaction was stirred overnight at RT. The solvent was removed and the remaining material was suspended in toluene and concentrated. The residue was dissolved in CH3CN (50 ml) and treated with AgCN (2.96 g). The mixture was heated at reflux for 2 h and then stirred at 60° C. overnight. The mixture was filtered and the filtrate concentrated. The residue was purified by chromatography on silica (EtOAc/Cyclohexane) to afford the title compound (4.4 g; 78%).
- 1H-NMR δ CDCl3 3.1-3.2 (m, 4H), 5.3 (s, 1H), 6.7-6.9 (m, 3H), 7.0-7.2 (m, 2H), 7.4 (m, 1H)
- The title compound from Step D above (1.5 g) was dissolved in THF (5 ml) and slowly added at rt to a suspension of NaH (212 mg) in THF (10 ml). The mixture was heated at 60° C. for 30 min, then cooled to 0° C. and treated with 1,2-dibromoethane (2.3 ml). The reaction was stirred at 60° C. for 3 h, cooled to rt and filtered. The filtrate was concentrated to afford the title compound (2.1 g; 99%).
- 1H-NMR δ CDCl3 2.8-3.0 (m, 4H), 3.0-3.2 (m, 2H), 3.2-3.4 (m, 2H), 6.8-7.2 (m, 4H), 7.6 (m, 1H), 7.8-7.9 (m, 1H)
- The title compound from Step E above (2.1 g) and potassium phthalimide (5.4 g) were suspended in DMF (30 ml) and stirred at 60° C. overnight. The solvent was removed and the residue dissolved in CHCl3, filtrated and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (1.91 g; 76%)
- 1HNMR δ CDCl3 2.8-3.2 (m, 4H), 3.4-3.6 (m, 2H), 3.7-3.9 (m, 2H), 6.8-7.0 (m, 3H), 7.1-7.2 (m, 1H), 7.7-8.0 (m, 6H)
- The title compound from Step F (1.90 g) was dissolved in toluene (20 ml) and treated with dibutyltin oxide (553 mg) and trimethylsilylazide (3.7 ml). The mixture was heated under a N2 atmosphere at 90° C. for 4 d. The reaction was quenched with aqueous 1 N HCl (20 ml) and stirred for 1 h at 50° C. The phases were separated, the aqueous phase was extracted with toluene and the combined organic phase concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (600 mg, 33%, MH+=472).
- The title compound from Step G above (300 mg) was dissolved in ethanol (5 ml) and treated with hydrazine hydrate (127 mg). The solution was stirred at 80° C. for 2 h and subsequently stirred for 1 h at rt. The solvent was removed and the residue treated with 1 N HCl (20 ml) and CHCl3 (10 ml). The aqueous phase was separated, filtered and concentrated affording the title compound (240 mg, 100% MH+=342).
-

- Commercially available 2,4-dichlorotoluene (24.6 g) and dry copper(I) cyanide (50 g) in N-methylpyrrolidone (130 ml) were heated under reflux (200-216° C.) for 4 d. While hot (110° C.), the mixture was poured into a flask containing 33% aq. NH4OH solution (390 ml) and toluene (100 ml) and stirred to break up the lumps. After the mixture was cooled to rt, Et2O (100 ml) was added and filtered through cloth. The precipitate was washed (2×100 ml Et2O/CHCl3 1:1). The dark filtrate was poured into a separatory funnel and the phases were separated with the aid of additional Et2O (100 ml). The aqueous phase was extracted with Et2O/CHCl3 1:1 (2×100 ml). The combined organic phases were washed with 10% NH4OH solution (4×110 ml, until the basic phase was no longer blue), with H2O (100 ml), and brine (100 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was mixed with NaOH (24.8 g) and diethylene glycol (275 ml) was added together with a few drops of H2O. The mixture was heated at 215-220° C. overnight. The cooled mixture was diluted with H2O (220 ml) and acidified to pH 1 with 10% aq. HCl. The suspension was filtered and the precipitate washed with 0.1 N HCl (50 ml). The solid was crystallised from glacial acetic acid to afford the title compound (18.4 g, 78%; MH+=181).
- Following a similar procedure as that described in Preparative Example 49 Step B, the title compound from Step A above (22.1 g) was reacted to afford the title compound (30.0 g, 100%).
- 1H-NMR (CDCl3) δ: 2.65 (s, 3H), 3.91 (s, 3H), 3.92 (s, 3H), 7.32 (d, 1H), 8.04 (dd, 1H), 8.56 (d, 1H).
- Following a similar procedure as that described in Preparative Example 49 Step C, the title compound from Step B above (30.0 g) was reacted. Differing from the cited example, the final mixture was allowed to stand over the weekend to form the precipitate. After filtration, the crude title compound was obtained (38.0 g, 100%; [M-Br]+=469).
- Following a similar procedure as that described in Preparative Example 49 Step D, the title compound from Step C above (38.0 g) was reacted. Differing from the cited example, the hydrogenation was run for 2 days. (29.2 g, 77%; MH+=289).
- Following a similar procedure as that described in Preparative Example 49 Step E, the title compound from Step D above (4.32 g) was reacted and the title compound obtained (1.77 g, 41%; MH+=285).
- Following a similar procedure as that described in Preparative Example 49 Step F, the title compound from Step E above (2.39 g) was reacted and the title compound obtained (2.45 g, 100%; MNa+=309).
- Following a similar procedure as that described in Preparative Example 49 Step G, the title compound from Step F above (3.07 g) was reacted and the title compound was obtained (2.17 g, 69%; MH+=296).
- The title compound from Step G above (2.17 g) was dissolved in THF (30 ml) and added to a suspension of NaH (250 mg) in THF (9 ml). The mixture was heated at 90° C. for 1 h 15 min and cooled to rt. The mixture was then treated with 1,2-dibromoethane (1.6 ml) in THF (3.7 ml) and the mixture was heated at 90° C. for 4 h 30 min. The mixture was cooled to rt, diluted with 200 ml EtOAc, 20 ml brine and 20 ml sat. NH4Cl. The organic phase was separated, dried over MgSO4 and the residue purified by chromatography on silica (CH2Cl2) to afford the bromoethyl intermediate (1.42 g, 50%; [MNH4]+=419) and starting material (636 mg, 24%). The bromoethyl compound (1.42 g) was dissolved in anhydrous DMF (18 ml) and treated with potassium phthalimide (1.96 g). The suspension was stirred at 80° C. overnight. The solvent was removed and the residue partitioned between EtOAc (50 ml), H2O (50 ml) and brine (50 ml). The aqueous phase was extracted with EtOAc (2×50 ml) and the combined organic phase dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH) to afford the title compound (1525 mg; 92%; MH+=469).
- The title compound from Step H above (1475 mg) was dissolved in anhydrous toluene (25 ml) and treated with dibutyltin oxide (784 mg) and trimethylsilylazide (8.3 ml). The mixture was heated under a N2 atmosphere at 90° C. for 3 days. The solvent was removed, the residue dissolved in MeOH (10 ml) and concentrated. The residue was partitioned between EtOAc (100 ml) and 10%. NaHCO3 (100 ml). The aqueous phase was extracted with EtOAc (2×70 ml) and the combined organic phase dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH) to afford the title compound (1216 mg, 75%, MH+=512).
- The title compound from Step I above (1216 mg) was dissolved in anhydrous MeOH (14 ml) and Et3N (0.66 ml). The mixture was cooled to 5° C. and N,N′-dimethylamino-propylamine (0.71 ml) added. The mixture was stirred at rt for 25 h and subsequently evaporated, toluene (10 ml) added, evaporated again and dried in HV. The residue was dissolved in dioxane (8 ml) and H2O (8 ml). To the slightly turbid solution was added Boc2O (2.6 g) and Et3N (1.2 ml) and the mixture was stirred at rt overnight. After evaporation of the solvent, H2O (20 ml) was added and the solution acidified to pH˜4.0 by adding 1 M HCl and the aqueous solution extracted with EtOAc (3×50 ml). The combined organic phase was washed with brine (15 ml), separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH) to afford the title compound (567 mg, 50%, MNa+=504).
-

- The title compound from Preparative Example 52 (215 mg) was dissolved in THF (4 ml) and 33% NH4OH solution (40 ml) was added. The solution was stirred in a closed vessel at 80° C. overnight. The reaction mixture was allowed to cool to rt and subsequently evaporated to dryness. The crude product, which consisted of a mixture of the amide (MNa+=489) and the free acid (MNa+=490), was dissolved in anhydrous THF (8.5 ml) and triethylamine (0.28 ml) added. The ensuing precipitate was dissolved by adding anhydrous CH3CN (6 ml). The mixture was cooled to −40° C. and ethylchloroformate (0.17 ml) was slowly added. The mixture was stirred at −25° C. for 1 h and allowed to warm to 0° C. At 0° C. 7 M NH3/MeOH-solution (10 ml) was added and the mixture was stirred at 0° C. for 30 min and for 1 h at rt. The mixture was concentrated and the residue dissolved in H2O (14 ml) and THF (3 ml). The pH was adjusted to pH˜4.0 by adding 0.1 N HCl and the aqueous phase—after addition of brine (10 ml)—extracted with EtOAc containing 10% THF (4×33 ml) and CH2Cl2 containing 10% THF (1×25 ml)). The combined organic phase was washed with brine (15 ml), dried over MgSO4 and concentrated to afford the title compound (241 mg; 100%, MNa+=489).
- The title compound from Step A above (240 mg) was suspended/dissolved in CH2Cl2/MeOH 4:1 (5 ml) and a 4 M solution of HCl in dioxane (7 ml) added after which a clear solution was obtained. The mixture was stirred at rt for 3 h and concentrated. The residue was partitioned between EtOAc containing 10% THF (25 ml) and 0.01 N HCl (25 ml). The organic phase was extracted with H2O (25 ml) and 0.01 N HCl (25 ml). The combined aqueous phase was concentrated to afford the title compound (162 mg, 90%, MH+=367).
-

- The title compound from Preparative Example 49 Step C (47.6 g) was suspended in CH3CN (350 ml) and commercially available 3-bromobenzaldehyde (13.9 ml) added. After the addition of DBN (24 ml), the mixture was heated at 100° C. for 1 h. The mixture was cooled and the precipitate collected by filtration to afford the trans-olefin (7.5 g). The mother liquor was concentrated to half its volume and poured into H2O (300 ml). The mixture was extracted with EtOAc (2×300 ml), the organic phase washed with 5% HCl (2×80 ml), dried over MgSO4 and concentrated. To this residue was added the trans olefin from above and the mixture was suspended in H2O (500 ml), MeOH (60 ml) and dioxane (60 ml). After the addition of KOH (47 g), the mixture was heated at 60° C. for 16 h, cooled to rt and washed with CH2Cl2 (3×100 ml). The aqueous phase was made acidic (pH˜1) by adding conc. HCl, filtered, the precipitate washed with H2O (150 ml) and air-dried to afford the title compound as a mixture of cis/trans-olefins (26.5 g; 88%; MH+=347).
- The title compound from Step A above (6 g) was dissolved in MeOH (450 ml) and EtOAc (150 ml). After the addition of a suspension of 5% Pt/C (2.5 g) in 10% HCl (5 ml) and MeOH (10 ml), the mixture was hydrogenated for 6 h. The mixture was filtered, the catalyst washed with MeOH (60 ml) and the filtrates evaporated to afford the title compound (5.5 g, 91%).
- 1HNMR δ (DMSO-d6) δ 2.81-2.90 (m, 2H), 3.13-3.27 (m, 2H), 7.23-7.32 (m, 2H), 7.39-7.45 (m, 1H), 7.51 (s, 1H), 7.85-7.95 (m, 3H)
- The title compound from Step B above (4 g) was suspended in sulfolane (9 ml) and treated with polyphosphoric acid (30 g). The mixture was heated under N2 at 175-180° C. for 2 h 30 min and poured into ice-water (250 ml). The mixture was stirred at rt overnight and the precipitate collected by filtration to afford the crude title compound (3.56 g; 94%; MH+=331).
- The title compound from Step C above (3.5 g) was dissolved in N-methyl pyrrolidone (25 ml) and CuCN (900 mg) added. The mixture was heated at 200° C. for 8 h, cooled to rt and diluted with H2O (200 ml) and 1 M HCl (50 ml). The mixture was extracted with EtOAc (3×100 ml) and the combined organic phase washed with H2O (100 ml) and brine (100 ml). The organic phase was dried over MgSO4 and evaporated. The residue was dissolved in dioxane (50 ml) and conc. HCl (50 ml) added. The mixture was heated at 90° C. for 18 h and the solvents evaporated. The residue was suspended in MeOH (75 ml), treated with SOCl2 (1.5 ml) and heated under reflux for 1 h 30 min. The mixture was concentrated to half its volume, diluted with Et2O (300 ml) and washed with sat. NaHCO3 (80 ml) and brine (80 ml). The organic phase was separated, dried over MgSO4 and evaporated. The residue was purified by chromatography on silica (EtOAc/hexane, 1:4) to afford the title compound (1040 mg; 27%; MH+=325).
- The title compound from Step D above (1040 mg) was dissolved in CHCl3 (15 ml) and MeOH (15 ml) and the NaBH4 (150 mg) added. The mixture was stirred at rt for 1 h, diluted with ice water (80 ml) and extracted with EtOAc (2×100 ml). The organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/acetone, 98:2->CH2Cl2/acetone, 95:5) to afford the title compound (817 mg, 78%, MNa+=349).
- The title compound from Step E above (817 mg) was dissolved in THF (10 ml) and treated with SOCl2 (0.46 ml). The mixture was stirred at rt overnight and the solvents evaporated. The residue was dissolved in CH3CN (10 ml) and benzene (5 ml) and added to a suspension of AgCN (406 mg) in CH3CN (10 ml). The mixture was heated at 90° C. for 5 h, filtered and the salts washed with CH3CN (10 ml). The filtrates were evaporated and the residue purified by chromatography on silica (CH2Cl2/acetone, 98:2) to afford the title compound (572 mg, 68%, MH+=336).
- The title compound from Step F above (676 mg) was suspended in THF (20 ml) and DMF (5 ml) and treated under a N2 atmosphere with NaH (106 mg). The mixture was heated at ˜95° C. for 75 Min, cooled to rt and treated with a solution of 1,2-dibromoethane (0.7 ml) in THF (3 ml). The mixture was then heated at 95° C. for 10 h, cooled to rt and treated with sat. NH4Cl (15 ml) and EtOAc (100 ml). The organic phase was separated, washed with brine (15 ml), dried over MgSO4 and concentrated. The residue was dissolved in DMA (8 ml) and treated with potassium phthalimide (554 mg). The mixture was heated at 60° C. overnight, the solvent removed and the residue dissolved in EtOAc (50 ml) and H2O (15 ml). The organic phase was separated, washed with brine (15 ml) and concentrated. The residue was purified by chromatography on silica (CH2Cl2/acetone, 98:2) to afford the title compound (740 mg, 72%, MNH4 +=526).
- The title compound from Step G above (600 mg) was suspended in toluene (5 ml) and treated with dibutyltin oxide (138 mg) and trimethylsilylazide (1.45 ml). The mixture was heated under a N2 atmosphere at 90-95° C. for 3 d and the solvent evaporated. The residue was suspended in MeOH (10 ml) and the solvent evaporated. The residue was dissolved in EtOAc (30 ml) water (10 ml). The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 95:5) to afford the title compound (415 mg, 68%, MH+=552).
- The title compound from Step H above (415 mg) was dissolved in MeOH (6 ml) and triethylamine (0.23 ml). The mixture was cooled to 0° C. and 3-dimethylaminopropylamine (0.23 ml) added. The mixture was stirred at 0° C. for 10 min and at rt overnight. The mixture was concentrated, dissolved in MeOH (10 ml), again concentrated and dried in HV. The residue was dissolved in dioxane (5 ml) and H2O (5 ml) and the pH adjusted to pH=8-9 by adding 1 M KOH. The mixture was then treated with Boc2O (870 mg) and stirred overnight. The mixture was adjusted to pH=4 by adding 1 M HCl and diluted with EtOAc (150 ml). The organic phase was separated and the aqueous phase extracted with EtOAc (2×75 ml). The combined organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica gel (CH2Cl2/MeOH, 95:5->4:1) to afford the title compound (227 mg, 58%, MH+=522).
- The title compound from Step I above (227 mg) was dissolved in dioxane (10 ml) and 1 M KOH (3.75 ml) added. The mixture was stirred at rt overnight and the pH adjusted to pH=4 by adding 1 M HCl. The mixture was extracted with EtOAc, containing 10% THF (2×150 ml). The organic phase was separated, dried over MgSO4 and concentrated to afford the title compound (177 mg, 82%; MH+=494).
-

- If one were to follow a similar procedure as described in Preparative Example 54, but using 3-fluorobenzaldehyde in Step A and omitting Step D, one would obtain the desired compound.
-

- The title compound from Preparative Example 54 (177 mg) was dissolved in THF (6 ml) and triethylamine (0.2 ml) added. The precipitate was dissolved/suspended by adding CH3CN (3 ml). The mixture was cooled to −40° C. and ethylchloroformate (0.1 ml) was slowly added. The mixture was stirred at −25° C. for 1 h and allowed to warm to 0° C. At 0° C. 7 M NH3/MeOH-solution (7 ml) was added and the mixture was stirred at 0° C. for 30 min and 1 h at rt. The mixture was concentrated and the residue dissolved in H2O (10 ml) and THF (2 ml). The pH was adjusted to pH˜4.0 by adding 100 mM HCl and the aqueous phase extracted with EtOAc (4×30 ml) containing 10% THF. The organic phase was dried over MgSO4 and concentrated to afford the title compound (110 mg; 62%, MNa+=514).
- The title compound from Step A above (103 mg) was dissolved in THF (2 ml) and a 4 M solution of HCl in dioxane (5 ml) added. The mixture was stirred at rt for 2 h and concentrated. The residue was dissolved in H2O (20 ml) and washed with EtOAc (2×8 ml). The aqueous phase was concentrated, the residue dissolved in 50 mM HCl (6 ml) and filtered through a Millex VV (0.1 μM) filter unit. The filtrate was concentrated to afford the title compound (90 mg, 94%, MH+=392).
-

- If one were to follow a similar procedure as described in Preparative Example 56, but using the title compound from Preparative Example 55, one would obtain the desired compound.
-

- A suspension of NaH (66 mg) in THF (10 ml) was added to a solution of the title compound from Preparative Example 13 Step A (0.57 g) in THF (20 ml) and heated at 65° C. for 1 h. Then the mixture was cooled to 0° C. and a solution of Preparative Example 21 (0.74 g) in THF (10 ml) was added. The suspension was heated at 65° C. for 5 h and then diluted with ethyl acetate. The organic phase was washed with water, brine and dried over MgSO4. Removal of the solvents and column chromatography (EtOAc/hexane, 1:4) afford the title compound (630 mg, 58%, MH+=421).
- The title compound from Step A above (632 mg) was dissolved in DMF (10 ml) and treated with NaN3 (1.2 g) and NH4Cl (963 mg). The mixture was heated under a N2 atmosphere at 110° C. for 3 d and the solvent evaporated. Column chromatography (CH2Cl2/MeOH, 9:1) afford the title compound (350 mg, 51%, MH+=464).
- The title compound from Step B above (350 mg) was dissolved in THF (10 ml) and treated with TBAF.3H2O. The mixture was stirred at rt for 4 h and the solvent evaporated. Preparative TLC using CH2Cl2/MeOH (4:1) afford the title compound (121 mg, 50%, MH+=320).
-

- Commercially available 2-Brom-5-chlor-toluene (123 g) was diluted with Et2O (70 ml) and 10% of this solution was added to a mixture of Mg (15.2 g) and iodine (3 crystals) in Et2O (250 ml). After the Grignard reaction had started, the remaining starting material was added at such a rate to maintain gentle reflux. After the complete addition of the starting material, the mixture was heated at 60° C. oil-bath temperature for 45 Min. The mixture was then cooled to rt and poured onto a mixture of dry-ice in Et2O (1800 ml). The mixture was allowed to warm to rt over a period of 2 h and the solvent removed. The residue was dissolved with EtOAc (1200 ml) and washed with 3 N HCl (3×1000 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated to afford the title compound (94.3 g, 92%)
- 1HNMR δ (DMSO-d6) 2.51 (s, 3H), 7.33 (dd, 1H), 7.39 (d, 1H), 7.81 (d, 1H), 12.9 (br-s, 1H)
- The title compound from Step A above (47 g) was dissolved in THF (500 ml) and the mixture cooled to −60° C. At −60° C. a 1.3 M solution of sec-BuLi (455 ml) in hexane was slowly added as to keep the internal temperature below −30° C. The precipitate began to dissolve after the addition of more than half of the sec-BuLi solution. After the complete addition of sec-BuLi, the deep red solution was stirred at −50° C. for 1 h. The anion solution was then transferred via canula to a cooled (−40° C.) solution of commercially available 3-chlor-benzylbromide (62.3 g) in THF (150 ml). The addition of the anion was at such a rate as to maintain −40° C. during the addition. After the addition of the anion was completed, the mixture was stirred at −40° C. for 1 h and was then allowed to warm to rt over a period of 3 h. The reaction was quenched by adding 2 M NaOH (1000 ml) and the THF removed in vacuo. The remaining solution was extracted with cyclohexane (2×500 ml) and the aqueuous phase acidified to PH=1 by adding conc. HCl. The mixture was extracted with EtOAc (3×400 ml), the organic phase dried over MgSO4, filtered and concentrated to afford the title compound (71 g, 87%).
- 1HNMR δ (acetone-d6) 2.83-2.91 (m, 2H), 3.22-3.31 (m, 2H), 7.13-7.40 (m, 6H), 7.98 (d, 1H).
- The title compound from Step B above (71 g) was suspended in sulfolane (250 ml) and PPA (700 g) added. The mixture was stirred with a mechanical stirrer and heated at 170° C. oil-bath temperature for 9 h. The hot mixture (˜120° C.) was then poured onto crushed-ice (4000 g) and stirred overnight. The precipitate was allowed to settle for 30 Min and the aqueous phase decanted. The residue was dissolved in Et2O (1500 ml) and washed with 1 M NaOH (2×500 ml). The organic phase was dried over MgSO4, filtered and concentrated to afford the title compound (50 g, 75%).
- 1HNMR δ (CDCl3) 3.16 (s, 4H), 7.23 (d, 2H), 7.32 (dd, 2H), 8.0 (d, 2H)
- The title compound from Step C above (25 g) was dissolved in toluene (160 ml) and added to a mixture of KCN (11.7 g), dipiperidinomethane (7.26 ml), sulfolane (2 ml) and 1,4-Bis-(diphenylphosphino)-butane (6 g). The mixture was degassed by sonication under a stream of nitrogen and then palladium(II)-acetate (1.6 g) was added. The mixture was then heated in a sealed glass reaction vessel at 160° C. oil-bath temperature for 18 h. The mixture was cooled to rt, diluted with CH2Cl2 (800 ml) and washed with H2O (300 ml) and brine (300 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated. The residue was diluted with EtOAc (90 ml) and sonicated. The suspension was then treated with cyclohexane (400 ml) and allowed to stand for 30 Min. The precipitate was collected by filtration and air-dried to afford the title compound (18 g, 77%, MH+=259).
- The title compound from Step D above (18 g) was suspended in EtOH (75 ml) and H2O (20 ml) and the KOH (19.3 g) added. The mixture was heated at 100° C. oil-bath temperature for 12 h, concentrated and the residue dissolved in H2O (500 ml). The aqueous phase was acidified to pH=1 by adding conc. HCl and the precipitate collected by filtration and air-dried to afford the title compound (19.5 g, 95%, MH+=297).
- The title compound from Step E above (19.5 g) was suspended in MeOH (600 ml) and treated with thionyl chloride (29 ml). The mixture was then heated at 90° C. oil-bath temperature for 3 h, the hot mixture filtered and concentrated. The residue was dissolved in CH2Cl2 (800 l) and washed with sat. NaHCO3 (200 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated to afford the title compound (18.8 g, 88%, MH+=325).
- The title compound from Step F above (18.8 g) was dissolved in CHCl3 (250 ml) and MeOH (250 ml). The mixture was then treated with NaBH4 (2.47 g) in small portions. After the complete addition of the reducing agent, the mixture was stirred at rt for 1 h. The mixture was poured into ice-water (800 ml), the organic phase separated and the aqueous phase extracted with EtOAc (300 ml). The combined organic phase was dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (CH2Cl2 to CH2Cl2/acetone, 98:2 to CH2Cl2/acetone, 95:5) to afford the title compound (11.9 g, 63%, MNa+=349).
- The title compound from Step G above (11.9 g) was dissolved in THF (150 ml) and the mixture cooled to 0° C. At 0° C. thionyl chloride (6.5 ml) was added and the mixture was allowed to warm to rt overnight. The solvent was then removed in vacuo to afford the crude title compound.
- 1HNMR δ (CDCl3) 2.93-3.05 (m, 2H), 3.70-3.80 (m, 2H), 3.90 (s, 6H), 6.10 (s, 1H), 7.40 (d, 2H), 7.78-7.86 (m, 4H).
- The title compound from Step H above was dissolved in CH3CN (300 ml) and benzene (95 ml). After the addition of AgCN (5.9 g) the mixture was heated at 95° C. oil-bath temperature for 2 h 45 Min. The mixture was filtered while hot and the salts washed with CH2Cl2 (100 ml). The filtrate was concentrated and the residue purified by chromatography on silica (CH2Cl2/acetone, 98:2) to afford the title compound (11.3 g, 92%, MH+=336).
-

- The title compound from Preparative Example 59 Step C (9.5 g) was dissolved in CHCl3 (100 ml) and MeOH (60 ml) at 0° C. The mixture was then treated with NaBH4 (1.64 g) in small portions. After the complete addition of the reducing agent, the mixture was stirred at rt for 3 h. Water (50 ml) was added and the mixture was concentrated to half of its volume and extracted with EtOAc (2×150 ml). The combined organic layers were washed with water (50 ml), brine (50 ml), dried over MgSO4 and concentrated. The crude product was used without further purification (9 g, 90%, MNa+=301).
- The crude title compound from Step A above (9 g) was dissolved in THF (100 ml) and the mixture was cooled to 0° C. At 0° C. thionyl chloride (7.1 ml) was added and the mixture was allowed to warm to rt overnight. The solvent was then removed in vacuo to afford the title compound (9.2 g).
- The title compound from Step B above (9.2 g) was dissolved in CH3CN (180 ml) and benzene (60 ml). After the addition of solid AgCN (5.2 g) the mixture was heated at 90° C. oil-bath temperature for 2.5 h. The mixture was filtered while hot through celite and the salts washed with CH2Cl2 (200 ml). The filtrate was concentrated to give the crude title compound (8.66 g, 93%, MH+=288).
-

- The title compound from Preparative Example 59 (3.8 g) was suspended in THF (50 ml) and DMF (35 ml). The mixture was treated under a N2 atmosphere with NaH (408 mg) and the mixture was heated at 95° C. oil-bath temperature for 90 Min, cooled to rt and treated with the title compound from Preparative Example 21 (4.78 g). The mixture was then heated at 90-95° C. for 4 h, cooled to rt and quenched with sat. NH4Cl (75 ml) and brine (90 ml). The organic phase was separated and the aqueous layer extracted with EtOAc (2×50 ml). The combined organic phase was dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 95:5) to afford the title compound (5 g, 82%, MH+=537).
- The title compound from Step A above (5 g) was dissolved in DMA (90 ml) and treated with NaN3 (5.9 g) and NH4Cl (4.8 g). The mixture was heated under a N2 atmosphere at 100-105° C. for 50 h. The cooled mixture concentrated and the residue dissolved in EtOAc (600 ml) and H2O (200 ml). The aqueous layer was acidified to pH=4 by adding 1 M HCl and the organic phase separated. The aqueous phase was extracted with EtOAc (2×80 ml) and the combined organic extracts washed with 100 mM HCl (200 ml) and brine (200 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH 9:1->4:1) to afford the title compound (4 g, 74%, MH+=580).
- The title compound from Step B above (4 g) was dissolved in dioxane (153 ml). After the addition of 1 M KOH (42.5 ml), the mixture was stirred at rt overnight. The mixture was concentrated and then 43 ml 1 M HCl added. The precipitate was dissolved in EtOAc (100 ml) and H2O (100 ml) and the organic phase separated. The aqueous phase was extracted with EtOAc (100 ml) and the organic phase combined. The solvent was then removed to afford the title compound (3.9 g, quanta, MH+=552).
- Following a similar procedure as that described in Preparative Example 61 but using the sulfamidates and compounds from the Preparative Examples as indicated in the Table below, the title compounds were obtained.
-
Preparative Preparative Example Example Sulfamidate Title compound MH+ 62 59 

538 63 59 

566 64 60 

475 - If one were to treat the title compound from Preparative Example 59 according to the procedures described in Preparative Example 61, but using the sulfamidate as indicated in the table below, one would obtain the title compound.
-
Preparative Preparative Example Example Sulfamidate Title compound 65 59 

-

- The title compound from Preparative Example 61 Step A (1000 mg) was suspended in MeOH (10 ml) and hydroxylamine hydrochloride (517 mg) and a 5.5 M solution of sodium methoxide in MeOH (1.4 ml) added. The mixture was heated in a pressure bottle at 110° C. for 12 h and then the solvent removed. The residue was purified by chromatography on silica (cyclohexane/EtOAc 1:3->1:1) to afford the title compound (210 mg, 20%, MH+=570).
- The title compound from Step A above (180 mg) was dissolved in MeOH (10 ml) and sodium methoxide (233 mg) and diethyl carbonate (1130 mg) added. The mixture was heated at 110° C. in a pressure bottle overnight. The solvent was removed and the residue purified by chromatography on silica (CHCl3) to afford the title compound (110 mg, 58%, M+−27=568).
- The title compound from Step B above (110 mg) was dissolved in THF (25 ml) and treated with 1M KOH (6 ml). After stirring at rt overnight, 1M HCl (2.8 ml) was added and the solvents removed to afford the crude title compound (105 mg, quant., M+−27=540).
-

- Hydroxylamine hydrochloride (401 mg) was suspended in anhydrous MeOH (14 ml) and a 5.5 M solution of sodium methoxide in MeOH (0.946 ml) added. This mixture was stirred at rt for 45 min and the title compound from Preparative Example 61 Step A (1400 mg) was added. The resulting mixture was heated in a closed vessel at 100° C. overnight and subsequently allowed to cool down to rt. Due to incomplete conversion, hydroxylamine hydrochloride (401 mg) and a 5.5 M solution of sodium methoxide in MeOH (0.946 ml) were added and the mixture was heated again at 100° C. for 20 h. After cooling down to rt, the salts were filtered off and washed with EtOAc (15 ml) and CHCl3 (15 ml). The united organic phases were evaporated and the residue purified by chromatography on silica (cyclohexane/EtOAc 8:2->6:4) to afford the title compound from Preparative Example 66 Step A (300 mg, 20%, MH+=570) and the title compound (1130 g, 74%, MNa+=577).
- The title compound from Step A above (1380 g) was dissolved in THF (30 ml) and treated with 1M KOH (9 ml). After stirring at rt overnight, 1M KOH (9 ml) was added and stirring continued for 22 h. The reaction mixture was acidified with 4 M HCl to pH 2-3, extracted with EtOAc/THF 10/1 (4×40 ml) and the combined organic extracts washed with brine (20 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated to afford the title compound (1220 mg, quant., M+−27=499, MNa+=549).
-

- The N-hydroxyamidine product from Preparative Example 66 Step A (300 mg) was dissolved in anhydrous dichloromethane (5 ml), the solution cooled down to 0° C. and triethylamine (147 μl) and trifluoroacetic anhydride (103 μl) added. The reaction mixture was stirred at rt overnight. Due to incomplete conversion, triethylamine (221 μl) and trifluoroacetic anhydride (155 μl) were added at 0° C. and stirring was continued at rt for 3 d. Dichloromethane (9 ml) and water (10 ml) were added to the stirred mixture. After 5 min, the separated organic phase was washed with brine (5 ml), dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc 8:2->7:3) to afford the title compounds A (267 mg, 68%, MNa+=766) and B (36 mg, 10%, MNa+=670).
- The title compounds A (267 mg; MNa+=766) and B (36 mg, MNa+=670) from Step A above were dissolved in dioxane (11 ml) and water added (11 ml). The resulting suspension was treated with 1M NaOH (3.6 ml). After stirring at rt overnight, the reaction mixture was acidified with 1M HCl to pH 2-3, extracted with EtOAc (4×40 ml) and the combined organic phases dried over MgSO4, filtered and concentrated to afford the title compound (282 mg, quant., MNa+=642).
-

- To the title compound of Preparative Example 61 Step A (500 mg) in anhydrous DMF (10 ml) was added K2CO3 (123 mg). After cooling down to 0° C., methyl iodide (75 μl) was added dropwise to the stirred mixture. After 10 min, the mixture was allowed to rt and stirred overnight. The reaction mixture was cooled down to 0° C., diluted with acidified saturated aq. NaCl solution (pH 2-3) and added to stirred EtOAc (150 ml). The separated organic phase was washed with brine (2×25 ml), dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc 8:2->7:3) to afford the title compounds: the 1-Me-tetrazole (170 mg, 33%, MH+=580) and the 2-Me-tetrazole (163 mg, 32%, MH+=580).
- The title compounds from Step A above (170 mg of the 1-Me-tetrazole and 163 mg of the 2-Me-tetrazole) were separately dissolved in dioxane (5.5 ml) and treated with 1M KOH (1.5 ml) each. After stirring at rt for 3 h, the reaction mixtures were concentrated to ⅓ of their volumes and the pH adjusted to 3 with 1M HCl. The resulting aq. suspension was extracted with EtOAc (3×25 ml) and the combined organic phases dried over MgSO4, filtered and concentrated to afford the title compounds: the 1-Me-tetrazole (171 mg, quant., M+−27=524) and the 2-Me-tetrazole (172 mg, quant., M+−27=524).
-

- The title compound from Preparative Example 61 (2 g) was dissolved in THF (75 ml) and CH3CN (75 ml) and triethylamine (4 ml) added. The mixture was cooled to −40° C. and ethylchloroformate (2.3 ml) was slowly added. The mixture was stirred at −25° C. for 1 h, filtered and the salts washed with 35 ml THF. The filtrate was placed in a cooling bath (−20° C.) and a 33%-solution of NH4OH (30 ml) was added. The mixture was stirred at −20° C. for 30 min and 15 min at rt. Since LC-MS indicated that the conversion was not complete, the mixture was concentrated. The reaction was repeated using the same reaction conditions. After the second run LC-MS indicated that the reaction was completed. The mixture was concentrated to afford the crude title compound together with salts from the reaction (MNa+=572).
- The crude title compound from Step A above was suspended in CHCl3 (25 ml) and the mixture cooled to 0° C. At 0° C. TFA (25 ml) was added and stirring at 0° C. was continued for 2 h. The mixture was concentrated and the residue dissolved in H2O (15 ml). The pH was adjusted to pH=7.0 by adding 10% NaOH and the neutral solution loaded onto a RP-column (Merck; silica gel 60 RP-18, 40-63 μM). The column was washed with H2O to remove the salts, followed by CH3CN/H2O (1:1) to elute the title compound (1.3 g, 88%, MH+=406).
- Treating the compounds from the Preparative Examples with the amines as indicated in the Table below, according to a modified procedure as described in Preparative Example 70, the title compounds were obtained as HCl-salts.
- Modifications:
- Step A The crude mixture from Step A was dissolved in H2O and the pH adjusted to pH=4.0 by adding 1 M HCl. The mixture was then extracted with EtOAc, the organic phase separated, dried over MgSO4, filtered and the solvents removed.
- Step B The residue after removal of the Teoc protecting group was diluted with 1M HCl and the aqueous phase washed with EtOAc. Concentration of the aqueous phase afforded the title compound as HCl-salt.
-
Preparative Preparative Example Example Amines Title compound MH+ 71 61 

462 72 61 

434 73 61 

462 74 61 

490 75 61 

486 76 61 

546 77 62 

420 78 62 

447 79 63 NH3 
420 80 66 

478 81 67 

437 82 68 

530 83 69 1-Me-tetrazole 

406 84 69 2-Me-tetrazole 

406 85 61 Step B none 
436 86 61 none 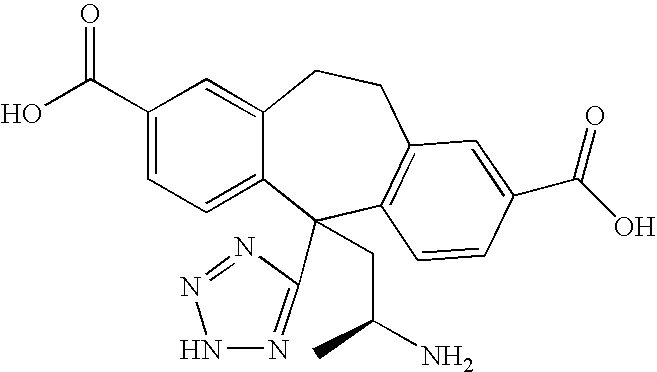
408 87 64 none 
374 -

- Commercially available anthraquinone (8.0 g) was suspended in CHCl3 (100 ml) and conc. H2SO4 (20 ml) was added. The resulting biphasic system was rapidly stirred and NaN3 (3.1 g) was added in portions at rt. The mixture was stirred for 1 h at rt and at 30-40° C. (water bath) for another 3 h. After the addition of ice water (80 ml), the precipitate was collected by filtration and dried to afford the title compound (8.40 g; 97%; MH+=224).
- The title compound from Step A above (8.0 g) was dissolved in DMSO (140 ml) under N2 at 10° C. After the addition of KOtBu (5.7 g), the mixture was stirred for 15 min at that temperature. After the addition of CH3I (4.2 ml), the mixture was allowed to warm to rt and stirred for 2 h. After the addition of 1 M HCl (130 ml) and EtOAc (100 ml), the organic phase was separated and the aqueous phase extracted with EtOAc (2×50 ml). The combined organic phase was washed with H2O (50 ml), brine (50 ml), dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (4.88 g; 61%; MH+=238).
- Tosylmethyl isocyanide was dissolved in DMSO (10 ml) under N2 at 10° C. and KOtBu (1.36 g) was added. The mixture was stirred for 5 min and MeOH (0.173 ml) was added. The title compound from Step B above (0.8 g) was immediately added to the mixture. After 10 min dibromoethane (1.51 ml) was added and stirring was continued for 1 h at rt. The mixture was diluted with EtOAc (10 ml) and sat. NH4Cl (30 ml) was added. The organic phase was separated and the aqueous phase was extracted with EtOAc (2×50 ml). The combined organic phase was washed with H2O (50 ml), brine (50 ml), dried over MgSO4 and concentrated. The residue was dissolved in DMF (40 ml) and potassium phthalimide (3.13 g) added. The resulting mixture was heated to 60° C. for 3 h and concentrated. The residue was suspended in CHCl3 and filtered. The filtrate was concentrated and the residue purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (612 mg; 43%; MH+=422).
- The title compound from Step C above (0.6 g) was dissolved in toluene (30 ml) under N2 and dibutyltin oxide (1.68 g) and trimethylsilylazide (8.9 ml) were added. The mixture was then heated at 75° C. for 24 h. The mixture was concentrated, the residue suspended in EtOAc (40 ml) and 1 M HCl (40 ml) and stirred for 2 h at rt. MeOH (10 ml) was added and the organic phase was separated. The aqueous phase was extracted with EtOAc (3×20 ml) and the combined organic phase was washed with brine (20 ml), dried over MgSO4 and evaporated. The residue was purified by chromatography on silica (MeOH/CH2Cl2) to afford the title compound (565 mg; 84%; MH+=465).
- The title compound from Step D above (0.22 g) was dissolved in EtOH (7 ml) and CHCl3 (3 ml) and the mixture was heated to 80° C. Hydrazine monohydrate (0.108 g) was added and the mixture was stirred at 80° C. for 1 h. The mixture was allowed to cool to rt within 1 h. The precipitate was removed by filtration and washed with EtOH. The filtrate was concentrated and dissolved in CHCl3 (20 ml) and 1 M HCl (10 ml). The aqueous phase was separated, filtered and evaporated to afford the title compound (85 mg; 48%; MH+=335).
-

- To a solution of the commercially available L-pyroglutamic acid ethylester (15.7 g) in methylene chloride (90 ml) was sequentially added at rt di-tert-butyldicarbonate (24 g) and a catalytic amount of DMAP (120 mg). After stirring for 6 h at rt the reaction mixture was quenched with saturated brine and extracted with methylene chloride. (3×30 ml). The organic phase was dried over MgSO4, concentrated and the residue purified by flash chromatography on silica (CH2Cl2) to afford the title compound (16.3 g, 63%, MNa+=280).
- A solution of the title compound from Step A above (16.3 g) in toluene (100 ml) was cooled to −78° C. and triethylborohydride (67 ml of a 1.0 M solution in THF) was added dropwise over 90 minutes. After 3 h, 2,6 lutidine (43 ml) was added dropwise followed by DMAP(20 mg). To this mixture was added TFAA (11 ml) and the reaction was allowed to come to ambient temperature over 2 h. The mixture was diluted with ethyl acetate and water and the organics were washed with 3 N HCl, water, aqueous bicarbonate and brine. The organic phase was dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc 5:1) to afford the title compound (10.9 g, 72%, MNa+=264).
- A solution of the title compound from Step B above (3.5 g) in 1,2 dichloroethane (75 ml) was cooled to −15° C. and Et2Zn (25 mL of a 1.0 M solution in THF) was added dropwise. To this mixture was added drop wise ClCH2I (4.5 ml) over 30 minutes. After stirring for 18 h at −15° C. the mixture was quenched with saturated aqueous bicarbonate and the solvent was evaporated and the reaction was taken up in ethyl acetate and washed with brine. The organic phase was dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc 4:1) to afford the diastereomerically pure title compound (1.5 g, 41%, MNa+=278).
- A solution of the title compound from Step C above (1.4 g) in MeOH (40 ml) and THF (20 ml) was treated with 1 N LiOH (10 ml) and stirred overnight at rt. The reaction mixture was acidified to pH 4.5 with 2 N HCl and stirred for 15 min at rt. The mixture was then extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and evaporated to afford the title compound (1.2 g, 96%, MNa+=250).
- To a solution of the title compound from Step D above (1.2 g) in THF (20 ml) was added at −15° C. 4-methylmorpholine (710 μl) and then isobutyl chloroformate (780 μl) over 5 minutes and stirred then for 30 minutes. The reaction mixture was cooled to −30° C. and treated with a solution of NH3 in dioxane (25 ml, 0.5 M in dioxane). The reaction mixture was stirred for 30 minutes, warmed to rt and stirred overnight. The reaction mixture was acidified to pH 4.5 with 10% aqueous citric acid and extracted with ether (3×50 ml). The organic phase was dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc 1:10) to afford the title compound (1.0 g, 84%, MNa+=248).
- To a stirred solution o of the title compound from Step E above (0.9 g) in methylene chloride (5 ml) was sequentially added at 0° C. TFA (5 ml). After stirring for 12 h at 0° C. the reaction mixture was concentrated under reduced pressure to afford the title compound (0.9 g, 100%, MH+=127).
- The title compound from Step F above (450 mg) was dissolved in CH2Cl2 (12 ml) and triethylamine (0.4 ml). The mixture was cooled to 0° C. and DMAP (25 mg) was added followed by fumarylchloride (0.099 ml). The mixture was stirred at 0° C. and allowed to warm to rt overnight. The mixture was concentrated to afford the crude title compound (MH+=333).
- To a cooled (0° C.) solution of DMF (4 ml) was carefully added oxalylchloride (0.32 ml). After the addition was completed, the mixture was stirred at 0° C. for 5 min. Then pyridine (0.6 ml) was added followed by a solution of the crude title compound from Step G above in DMF (2 ml) and CH2Cl2 (4 ml). The mixture was then stirred at 0° C. for 2 h. The mixture was concentrated and the residue partitioned between EtOAc (50 ml) and brine (25 ml). The organic phase was separated and the aqueous phase extracted with EtOAc (2×25 ml). The combined organic phase was dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 95:5) to afford the title compound (250 mg, 92%, MH+=297).
- The title compound from Step H above (328 mg) was dissolved in CHCl3 (3 ml) and MeOH (3 ml). The mixture was then treated with ozone according to Preparative Example 2 Step C to afford the title compound (350 mg, 80%, MH+=165 (aldehyde); MH+=219 (hemiacetal)).
-

- To a stirred solution of potassium hydroxide (1.2 g) in ethanol (10 mL) was sequentially added at rt the commercial available bis(tert.-butyldicarbonyl)amine (4.5 g). After stirring for 1 h at rt the reaction mixture was quenched with ether and the precipitate was filtered and washed with ether (3×10 mL) to afford the title compound (3.4 g)
- The title compound from Step A above (95 mg) was dissolved in CHCl3 (2.25 ml) and 1,3-dimethoxybenzene (0.18 ml) added. To the mixture was then added TFA (0.75 ml) and the mixture was stirred at rt for 1 h 30 min. The mixture was concentrated, dissolved in CH3CN (3 ml) and concentrated again. The residue was dissolved in 100 mM HCl (3 ml) and EtOAc (3 ml). The aqueous phase was separated, washed with EtOAc (2 ml) and concentrated. The residue was suspended in CH3CN (1.5 ml), sonicated for 1 min and the CH3CN removed by syringe. The residue was then dried in HV to afford the title compound (42 mg, 84%, MH+=154).
-

- To a solution of the commercial available Boc-Fmoc-protected amino acid (1.05 g) in methanol (25 ml) was added diethyl amine (1.5 ml). After stirring for 2.5 h at room temperature the reaction mixture was concentrated, and the residue was dissolved in water (50 ml) and Et2O (50 ml). The organic phase was extracted with water (3×50 ml) and the combined aqueous extracts were concentrated. The residue was used for the next step without any further purification.
- To a solution of the title compound from Step A above (530 mg) and 3-fluorobenzaldehyde (245 μl) in 15 ml of methanol was added NaBH3CN (150 mg), and the mixture was stirred at 25° C. overnight. The mixture was concentrated, and the residue was dissolved in EtOAc (50 ml). The organic layer was extracted with water (3×50 ml) and the combined aqueous extracts were concentrated. The residue was used for the next step without any further purification.
- To a stirring solution of the title compound from Step B above (760 mg) in DMF (20 ml) was added HOBt (470 mg) followed by EDCI (670 mg) and DMAP (30 mg). N-methyl morpholine (440 μl) was added and stirring was continued at rt overnight. The solvent was removed in vacuo, the residue diluted with EtOAc and then washed with saturated aqueous NaHCO3. The organic phase was dried over MgSO4, concentrated and the residue purified by flash chromatography on silica (CH2Cl2/acetone, 9:1) to afford the title compound (430 mg, 60% over 3 steps, MH+=321).
- The title compound from Step C above (760 mg) was dissolved in EtOAc (6 ml) and a solution of 4 M HCl in dioxane (6 ml) was added. After 2 h the mixture was triturated with aqueous NaHCO3 to pH 7.5 and stirred for 15 min at rt. After evaporation of the solvent, the crude product was purified by flash chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (420 mg, 80%, MH+=221).
- To a solution of the title compound from Step D above (85 mg) in THF (5 ml) was added triethylamine (80 μl) and the mixture was stirred for 1 h at 50° C. Then the sulfamidate (240 mg.), prepared according to WO 03/037327, was added in one portion at −15° C. and the mixture was stirred at ambient temperature over 2 d. After the addition of 1 M NH4HCO3 solution (5 ml), the mixture was stirred for 30 min. Then an excess saturated NaHCO3 solution was added and stirring was continued for another 15 min. The mixture was then partitioned between EtOAc and water and the aqueous phase extracted with EtOAc. The combined organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica (CH2Cl2/acetone, 9:1) to afford the title compound (135 mg, 79%, MH+=422).
- A solution of the title compound from Step E above (135 mg) in MeOH (2.5 ml) and THF (5 ml) was treated with 1 N LiOH (1.5 ml) and stirred overnight at rt. The reaction mixture was acidified to pH 4.5 with 2 N HCl and stirred for 15 min at rt. The mixture was then extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and evaporated to afford the title compound (125 mg, 96%, MH+=408).
-

- A solution of commercially available N-Boc-trans-4-hydroxyl-L-proline ester (2.93 g) in CH2Cl2 (20 ml) was cooled to −30° C. and treated with DIEA (4.8 ml). After the addition of triflic anhydride (2.2 ml), the mixture was stirred at −30° C. for 60 min and then treated with a solution of the commercially available amine in CH2Cl2 (20 ml). The mixture was allowed to warm to rt overnight. The mixture was diluted with CH2Cl2 (20 ml), washed with 0.5 M Na2CO3 (2×50 ml) and brine (50 ml). The organic phase was dried over MgSO4 and concentrated to leave a residue, which was purified by chromatography on silica (CH2Cl2/acetone, 4:1) to afford the title compound (2.22 g, 75%, MH+=367).
- A solution of the title compound from Step A above (700 mg) in MeOH (24 ml) and THF (12 ml) was treated with 1 N LiOH (6 ml) and stirred overnight at rt. The reaction mixture was acidified to pH 4.5 with 1 N HCl and stirred for 15 min at rt. The mixture was then extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and evaporated to afford the title compound (665 mg, 95%, MH+=353).
- To a stirring solution of the title compound from Step B above (665 mg) in DMF (15 ml) was added HOBt (390 mg) followed by EDCI (560 mg) and DMAP (30 mg). N-methyl morpholine (420 μl) was added and stirring was continued at rt overnight. The solvent was removed in vacuo, the residue diluted with EtOAc and then washed with saturated aqueous NaHCO3. The organic phase was dried over MgSO4, concentrated and the residue purified by flash chromatography on silica (CH2Cl2/acetone, 9:1) to afford the title compound (556 mg, 87%, MH+=335).
- The title compound from Step C above (760 mg) was dissolved in EtOAc (4 ml) and a solution of 4 M HCl in dioxane (4 ml) was added. After 2 h the mixture was triturated with aqueous NaHCO3 to pH 7.5 and stirred for 15 min at rt. After evaporation of the solvent, the crude residue was purified by flash chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (300 mg, 77%, MH+=235).
- To a solution of the title compound from Step D above (290 mg) in THF (5 ml) was added triethyl amine (280 μl) and the mixture was stirred for 1 h at 50° C. Then the sulfamidate (590 mg.), prepared according to WO 03/037327, was added in one portion at −15° C. and the mixture was stirred at ambient temperature over 2 d. After the addition of 1 M NH4HCO3 solution (5 ml), the mixture was stirred for 30 min. Then an excess saturated NaHCO3 solution was added and stirring was continued for another 15 min. The mixture was then partitioned between EtOAc and water and the aqueous phase extracted with EtOAc. The combined organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica (CH2Cl2/acetone, 4:1) to afford the title compound (163 mg, 30%, MH+=436).
- A solution of the title compound from Step E above (163 mg) in MeOH (2.5 ml) and THF (5 ml) was treated with 1 N LiOH (1.5 ml) and stirred overnight at rt. The reaction mixture was acidified to pH 4.5 with 2 N HCl and stirred for 15 min at rt. The mixture was then extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and evaporated to afford the title compound (140 mg, 96%, MH+=422).
-

- To a stirring solution of the title compound from Preparative Example 91 (25 mg) in DMF (3 ml) was added HOBt (15 mg), followed by EDCI (20 mg) and DMAP (3 mg). Commercially available (S)-Pyrrolidine-2-carbonitrile hydrochloride (15 mg) was added after 1 h, followed by N-methyl morpholine (20 μl). The mixture was stirred at rt overnight, the solvent removed in vacuo, and the residue was diluted with EtOAc. The mixture was washed with saturated aqueous NaHCO3, separated, dried over MgSO4 and concentrated. The residue was purified by flash chromatography on silica (CH2Cl2/acetone, 9:1) to afford the title compound (17 mg, 59%, MH+=486).
- To a stirring solution of the title compound Preparative Example 91 (125 mg) in DMF (5 ml) was HOBt (46 mg), followed by EDCI (65 mg) and DMAP (5 mg). After 1 h commercially available L-proline amide (68 mg) and N-methyl morpholine (100 μl) were added and stirring was continued at rt overnight. The solvent was removed in vacuo, the residue diluted with EtOAc and washed with saturated aqueous NaHCO3. The organic phase was separated, dried over MgSO4 and concentrated. The residue was purified by flash chromatography on silica (CH2Cl2/acetone, 4:1) to afford the title compound (137 mg; 88%; MH+=504).
- To a solution of the title compound from Step B above (137 mg) in pyridine (7 ml) was added imidazole (41 mg). At −30° C. POCl3 (102 μl) was slowly added to the mixture and the mixture was allowed to reach rt over a period of 1 h. Then the solvent was removed and the residue diluted with 1 N HCl and Et2O. The organic phase was separated, dried over MgSO4 and evaporated. The residue was purified by column chromatography on silica (CH2Cl2/acetone, 4:1) to afford the title compound (72 mg, 55%, MH+=486).
- Following a similar procedure as that described in Preparative Examples 92 and 93, except using the amines and amides as indicated in the Table below, the following compound were prepared. For Preparative Examples 105 and 106 the conversion of the nitrile to the carboxamide with subsequent saponification of the ester moiety was done according to Preparative Example 91 Step F with 3M Na2CO3 and H2O2.
-
Preparative 1. Yield Example Amide Amine Product 2. MH+ 94 


1. 55% 2. 498 95 


1. 90% 2. 537 96 


1. 71% 2. 493 97 


1. 70% 2. 504 98 


1. 73% 2. 516 99 


1. 65% 2. 493 100 


1. 54% 2. 505 101 


1. 78% 2. 493 102 


1. 56% 2. 500 103 


1. 65% 2. 512 104 


1. 71% 2. 514 105 


1. 68% 2. 511 106 


1. 56% 2. 511 107 


1. 62% 2. 526 108 


1. 2. -

- A solution of commercially available N-Boc-trans-4-hydroxyl-L-proline methyl ester (370 mg) in CH2Cl2 (2 ml) was cooled to −30° C. and treated with DIEA (600 μl). After the addition of triflic anhydride (280 μl), the mixture was stirred at −30° C. for 60 min and then treated with a solution of the title compound from Preparative Example 91 Step D in CH2Cl2 (2 ml). The mixture was allowed to warm to rt overnight. The mixture was diluted with CH2Cl2 (10 ml), washed with 0.5 M Na2CO3 (2×10 ml) and brine (10 ml). The organic phase was dried over MgSO4 and concentrated to leave a residue, which was purified by chromatography on silica ((CH2Cl2/acetone, 4:1), 4:1) to afford the title compound (225 mg, 33%, MH+=448).
- A solution of the title compound from Step A above (225 mg) in MeOH (4 ml) and THF (8 ml) was treated with 1 N LiOH (2 ml) and stirred overnight at rt. The reaction mixture was acidified to pH 4.5 with 1 N HCl and stirred for 15 min at rt. The mixture was then extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and evaporated to afford the title compound (91 mg, 40%, MH+=434).
- To a stirring solution of the title compound from Step B above (91 mg) in DMF (3 ml) was added HOBt (40 mg), followed by EDCI (60 mg) and DMAP (10 mg). Commercially available (S)-Pyrrolidine-2-carbonitrile hydrochloride (35 mg) was added after 1 h, followed by N-methyl morpholine (66 μl). The mixture was stirred at rt overnight, the solvent removed in vacuo, and the residue was diluted with EtOAc. The mixture was washed with saturated aqueous NaHCO3, separated, dried over MgSO4 and concentrated. The residue was purified by flash chromatography on silica (CH2Cl2/acetone, 1:1) to afford the title compound (50 mg, 47%, MH+=512).
-

- The title compound from Preparative Example 91 Step D (305 mg) was dissolved in THF (2 ml) was added triethyl amine (63 μl) and the mixture was stirred for 1 h at 50° C. Then the title compound from Preparative Example 19 (100 mg) was added in one portion at −15° C. and the mixture was stirred at ambient temperature overnight. After the addition of 1 M NH4HCO3 solution (5 ml), the mixture was stirred for 30 min. Then an excess saturated NaHCO3 solution was added and stirring was continued for another 15 min. The mixture was then partitioned between EtOAc and water and the aqueous phase extracted with EtOAc. The combined organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica (CH2Cl2/acetone, 4:1) to afford the title compound (58 mg, 57%, MH+=378).
- The title compound from Step A above (58 mg) was dissolved in EtOAc (2 ml) and a solution of 4 M HCl in dioxane (2 ml) was added. After 2 h the mixture was evaporated to afford the title compound (48 mg, quant., MH+=278).
-

- Commercially available N-cyclohexylcarbodiimde-N′-methyl polystyrene resin (1.9 g) was suspended in 5 ml dichloromethane and agitated for 5 Min. The commercially available amino acid (468 mg) and amine (86 mg), prepared from the commercially available hydrochloride by adding 1 eq. pyridine, were dissolved in 1.5 ml dimethylformamide and added to the above resin. The mixture was agitated for 16 h, filtered and the resin washed with 2×5 ml dichloromethane and 5 ml methanol. The combined filtrates were concentrated and the residue purified by flash chromatography (silica, CH2Cl2/MeOH, 9:1) to afford the title compound (500 mg; 91%).
- 1H-NMR (CDCl3): δ 1.45 (9H, s), 2.05-2.30 (4H, m), 3.25-3.40 (1H, m), 3.50-3.70 (2H, m), 3.80-3.90 (1H, m), 4.15-4.25 (1H, m), 4.30-4.40 (2H, m), 4.55-4.65 (1H, m), 4.70-4.80 (1H, m), 5.50-5.60 (2H, m), 7.25-7.40 (4H, m), 7.55-7.65 (2H, m), 7.70-7.80 (2H, m).
- The title compound from Step A above (500 mg) was dissolved in dichloromethane (10 ml) and treated with diethylamine (10 ml). After 2 h the mixture was concentrated and the residue was purified by flash chromatography (silica, CH2Cl2/MeOH, 4:1) to afford the title compound (224 mg; 80%).
- 1H-NMR (CDCl3): δ 1.45 (9H, s), 1.70 (2H, s), 2.05-2.30 (4H, m), 2.95-3.05 (2H, m), 3.70-3.85 (2H, m), 4.35-4.50 (1H, m), 4.75-4.85 (1H, m), 5.50-5.60 (1H, m).
-

- A solution of commercially available N-Fmoc-trans-4-hydroxyl-L-proline (4.5 g) in aqueous ethanol (80%, 45 ml) was titrated with a solution of Cs2CO3 (2.3 g) in water (18 ml) to pH 7. The solvents were evaporated and the residue dried in vacuo. The caesium salt was suspended in dry DMF (45 ml), cooled to 0° C. and treated with allyl bromide (11.5 ml) by dropwise addition over 10 min. After 30 min the solution was allowed to reach rt and stirring was continued for another 3 h. The reaction mixture was filtered and concentrated. The residue was purified by chromatography on silica (EtOAc/cyclohexane) to afford the title compound (4.5 g, 90%, MH+=394).
- The title compound from Step A above (2.5 g) in CH2Cl2 (60 ml) was cooled to −30° C. and treated with DIEA (2.5 ml). After the addition of triflic anhydride (1.2 ml), the mixture was stirred at −30° C. for 60 min and then treated with a solution of Preparative Example 84 (1.17 g) in CH2Cl2 (15 ml). The mixture was allowed to warm to 0° C., stirred at 0° C. for 12 h and refluxed for additional 4 h. The mixture was diluted with CH2Cl2 (50 ml), washed with 0.5 M Na2CO3 (2×25 ml) and brine (25 ml). The organic phase was dried over MgSO4 and concentrated to leave a residue, which was purified by chromatography on silica (EtOAc/cyclohexane, 7:3) to afford the title compound (1.41 g, 50%, MH+=658).
- To the title compound from Step B above (1.8 g) in THF (120 ml) was added dimedone (1.27 g) and Pd(PPh3)4 (422 mg). The reaction mixture was stirred at room temperature for 19 h. Following removal of the solvent under reduced pressure, chromatography on silica (CH2Cl2/MeOH 9:1) afforded the title compound (1.42 g, 84%, MH+=618).
- To a solution of the title compound from Step C above (1.42 g) in CH2Cl2 (70 ml) was added HOBT (405 mg) followed by EDCI (575 mg) and N-methyl-morpholine (0.33 ml). After being stirred at ambient temperature for 24 h, the solvent was evaporated to give a viscous residue, which was partitioned between EtOAc and ammonium acetate buffer (pH 6). The aqueous phase was extracted with ethyl acetate (3×100 ml) and the combined organic phase dried over MgSO4 and concentrated to afford the title compound (1.35 g, MNH4 +=617).
- To a solution of the title compound from Step D above (1.35 g) in acetonitrile (100 ml) was added diethyl amine (10 ml). After stirring for 2.5 h at rt, the reaction mixture was concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 9:1) to afford the title compound (712 mg; 85%, MH+=378).
-

- To a solution of the title compound from Preparative Example 112 (13 mg) in CH2Cl2 (0.8 ml) was added piperidino methyl polystyrene resin (65 mg) and 3-fluorobenzene-1-sulfonyl chloride (5.5 μl). After shaking at rt for 3 h, tris-(2-aminoethyl)amine polystyrene resin (30 mg) was added and agitated for additional 1 h at rt. The mixture was filtered, the resin washed with CH2Cl2 (5 ml) and methanol (1 ml) and the combined filtrates evaporated. Purification by chromatography on silica (CH2Cl2/MeOH 9:1) afforded the title compound (13 mg, 71%, MNH4 +=553).
- Following a similar procedure as that described in Preparative Example 113, except using the sulfonic acid chlorides as indicated in the Table below, the following compounds were prepared.
-
Preparative Sulfonic 1. Yield Example acid chloride Product 2. MH+ 114 

1. 69 2. 541 (MNH4 +) 115 

1. 92 2. 546 (MNa+) 116 

1. 89 2. 604 (MNa+) - Following a similar procedure as that described in Preparative Example 113, except using the acid chlorides as indicated in the Table below, the following compounds were prepared.
-
Preparative Acid 1. Yield Example chloride Product 2. MH+ 117 

1. 100 2. 488 (MH+) 118 

1. 49 2. 519 (MNH4 +) 119 

1. 70 2. 506 (MNa+) -

- To a solution of the title compound from Preparative Example 112 (20 mg) in CH2Cl2 (0.8 ml) was added tert.-butyl isocyanate (5.8 mg). After stirring at room temperature for 3 h the solvent was evaporated. Purification by chromatography (CH2Cl2/acetone 1:1) afford the title compound (16 mg, 63%, MH+=477).
- Following a similar procedure as that described in Preparative Example 120, except using the isocyanate as indicated in the Table below, the following compound was prepared.
-
Preparative 1. Yield Example Isocyanate Product 2. MH+ 121 

1. 69 2. 592 (MNH4 +) -

- The title compound from Preparative Example 15 Step A (13 mg) was dissolved in CH2Cl2 (0.7 ml) and added to N-cyclohexylcarbodiimide, N′-methyl polystyrene resin (120 mg). The mixture was agitated for 15 min and then treated with a solution of the title compound from Preparative Example 112 (0.54 ml, 7.5 mM CH2Cl2). After shaking at rt for 12 h, the mixture was filtered and the resin washed with CH2Cl2 (5 ml). The filtrates were concentrated in vacuo to afford the title compound (30 mg, 95%, MNa+=632).
-

- Commercially available 2,5-diaza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (400 mg) and aziridine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester (431 mg) were dissolved in toluene (5 ml). The mixture was stirred at rt overnight and then for 5 h at 80° C. The solvent was removed and the residue purified by chromatography on silica (CH2Cl2/acetone 9:1) to afford the title compound (468 mg, 58%, MH+=434).
- The title compound from Step A above (245 mg) was dissolved in dioxane (5 ml) and a solution of 4 M HCl in dioxane (5 ml) was added. The mixture was stirred for 2 h at rt and the solvents removed to afford the title compound (208 mg, 100%, MH+=334).
- To the title compound from Step B above (130 mg) were added CH2Cl2 (10 ml) and pyridine (1 ml). After the addition of commercially available thiophen-2-yl-acetyl chloride (61 mg) the reaction mixture was stirred at rt overnight. The solvent was removed and the residue purified by chromatography on silica (CH2Cl2/acetone 9:1) to afford the title compound (90 mg, 57%, MH+=458).
- The title compound from Step C above (130 mg) was dissolved in THF (4 ml) and methanol (2 ml). After the addition of 1 M aqueous LiOH-solution (1 ml), the mixture was stirred for 4 h at rt. The solvents were removed and the residue dissolved in water and acidified with 1 M HCl to pH˜4. The mixture was extracted with EtOAc, the organic phase washed with brine, dried over MgSO4 and concentrated to yield the title compound (75 mg, 86%, MH+=444).
- The title compound from Step D above (75 mg) was dissolved in DMF (5 ml). After the addition of EDCI (38 mg), HOBt (27 mg), N-methylmorpholine (0.15 ml) and DMAP (10 mol %), the mixture was stirred for 1 h at rt. Then commercially available 2-(S)-cyanopyrrolidine hydrochloride was added and the mixture was stirred overnight at rt. The solvent was removed and the residue dissolved in EtOAc, washed with brine, dried over MgSO4. and concentrated. The residue was purified by chromatography on silica (cyclohexane/EtOAc, 7:3) to afford the title compound (27 mg, 30%, MH+=522).
- Following a similar procedure as that described in Preparative Example 123, except using the piperazine derivatives and sulfonic acid chlorides as indicated in the Table below, the following compounds were prepared.
-
Piperazine Sulfonic 1. Yield Example derivative Acid chloride Product 2. MH+ 124 


1. 73% 2. 556 125 
none 
1. 27% 2. 492 - Preparative Examples 126-129 have been intentionally excluded.
-

- Commercially available 2-formyl-pyrrolidine-1-carboxylic acid tert-butyl ester (330 mg) in anhydrous THF (5 ml) was cooled to 0° C. and trimethyl-trifluoromethylsilane (300 μl) added, followed by addition of tetrabutylammoniumfluoride (60 μl; 1 M in THF). The reaction mixture was allowed to warm to rt and then stirred for 1 h. After dilution with diethyl ether, the organic phase was washed with brine and the aqueous phase extracted with diethyl ether. The combined organic phases were dried (MgSO4) and evaporated to afford the title compounds as a 1:1 mixture of alcohol and TMS ether (490 mg, 97%, [MH-Boc]+=242 (TMS ether); [MH-Boc]+=170 (alcohol)).
- The title compounds from Step A above (721 mg) in dichloromethane (5 ml) were added to Dess Martin periodinane (2.32 g) in dichloromethane (15 ml) with stirring. Trifluoroacetic acid (410 μl) was added dropwise and the turbid reaction mixture stirred for 17 h at rt, after which it was directly coated on silica and purified by column chromatography (silica, cyclohexane/EtOAc 90:10->80:20) to afford the title compound (301 mg, 45%, [MH-Boc]+=168).
- To the title compound from Step B above (106 mg) in dioxane (500 μl) was added 4 M HCl in dioxane (500 μl) and the resulting mixture stirred for 16 h at rt. Diethyl ether was added (2 ml) and the suspension filtered. The precipitate was dried and the title compound obtained as its HCl salt (81 mg, 91%, MH+=186).
- Preparative Examples 131-199 have been intentionally excluded.
- If one were to follow a similar procedure as that described in Preparative Example 61 and in Preparative Example 44, except using the sulfamidates as indicated in the Table below in Step A of Preparative Example 61, one would obtain the title compounds, listed in the following Table in the “product” column.
-
Preparative Preparative Example Example Sulfamidate Product 200 24 
201 25 
202 26 
203 27 
204 28 
205 29 
206 30 
207 31 
208 32 
209 33 
210 34 
211 35 
212 36 
213 37 
214 38 
215 39 
216 40 
217 41 
218 42 
219 43 
220 44 
221 45 
222 46 
223 24 
224 23 
225 25 
226 26 
227 27 
228 28 
229 29 
230 30 
231 31 
232 32 
233 33 
234 34 
235 35 
236 36 
237 37 
238 38 
239 39 
240 40 
241 41 
242 42 
243 43 
244 44 
245 45 
246 46 
247 24 
248 23 
249 25 
250 26 
251 27 
252 28 
253 29 
254 30 
255 31 
256 32 
257 33 
258 34 
259 35 
260 36 
261 37 
262 38 
263 39 
264 40 
265 41 
266 42 
267 43 
268 44 
269 45 
270 46 
271 24 
272 23 
273 25 
274 26 
275 27 
276 28 
277 29 
278 30 
279 31 
280 32 
281 33 
282 34 
283 35 
284 36 
285 37 
286 38 
287 39 
288 40 
289 41 
290 42 
291 43 
292 44 
293 45 
294 46 
- Examples 295-299 have been intentionally excluded.
-

- If one were to treat the compound from Preparative Example 59 with the sulfimidate from Preparative Example 22 according to the procedure described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step A above with NaN3 as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step B above with acetic acid anhydride in pyridine at 100° C. for 2 h one would obtain, after the removal of the pyridine under reduced pressure and after column chromatography, the title compound.
- If one were to treat the title compound from Step A above according to the procedures described in Preparative Example 70 one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 300, except using the appropriate intermediate from the Preparative Examples and anhydrides or acid chlorides and amines as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Preparative Acid Chloride/ Example Example Anhydride Amine Product 301 300 
NH3 
302 300 
NH3 
303 300 
NH3 
304 61 
NH3 
305 61 
NH3 
306 61 
NH3 
307 61 
NH3 
308 65 
NH3 
309 65 
NH3 
310 65 
NH3 
311 65 
NH3 
312 300 
CH3NH2 
313 300 
CH3NH2 
314 300 
CH3NH2 
315 300 
CH3NH2 
316 61 
CH3NH2 
317 61 
CH3NH2 
318 61 
CH3NH2 
319 61 
CH3NH2 
320 65 
CH3NH2 
321 65 
CH3NH2 
322 65 
CH3NH2 
323 65 
CH3NH2 
324 300 
(CH3)2NH 
325 300 
(CH3)2NH 
326 300 
(CH3)2NH 
327 300 
(CH3)2NH 
328 61 
(CH3)2NH 
329 61 
(CH3)2NH 
330 61 
(CH3)2NH 
331 61 
(CH3)2NH 
332 65 
(CH3)2NH 
333 65 
(CH3)2NH 
334 65 
(CH3)2NH 
335 65 
(CH3)2NH 
- Example numbers 336-399 were intentionally excluded.
- If one were to follow a similar procedure as that described in Preparative Example 66, except using the appropriate intermediate from the Preparative Examples and hydroxylamine hydrochlorides and amines as indicated in the Table below and treat the products according to Preparative Example 70, one would obtain the desired amine product.
-
Preparative Preparative Hydroxylamine Example Example hydrochloride Amine Product 400 300 
NH3 
401 300 
NH3 
402 300 
NH3 
403 300 
NH3 
404 61 
NH3 
405 61 
NH3 
406 61 
NH3 
407 61 
NH3 
408 65 
NH3 
409 65 
NH3 
410 65 
NH3 
411 65 
NH3 
412 300 
CH3NH2 
413 300 
CH3NH2 
414 300 
CH3NH2 
415 300 
CH3NH2 
416 61 
CH3NH2 
417 61 
CH3NH2 
418 61 
CH3NH2 
419 61 
CH3NH2 
420 65 
CH3NH2 
421 65 
CH3NH2 
422 65 
CH3NH2 
423 65 
CH3NH2 
424 300 
(CH3)2NH 
425 300 
(CH3)2NH 
426 300 
(CH3)2NH 
427 300 
(CH3)2NH 
428 61 
(CH3)2NH 
429 61 
(CH3)2NH 
430 61 
(CH3)2NH 
431 65 
(CH3)2NH 
432 65 
(CH3)2NH 
433 65 
(CH3)2NH 
434 65 
(CH3)2NH 
- Example numbers 435-499 were intentionally excluded.
-

- If one were to treat the compound from Preparative Example 300 Step A with conc. HCl in acetic acid according to the procedure described in Preparative Example 49 Step J, one would obtain the title compound.
- If one were to treat the title compound from Step A above according to the procedure described in Preparative Example 70 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step B above according to the procedure described in Preparative Example 70 Step A but using hydrazine instead of an amine, one would obtain the title compound.
- If one were to stir the title compound from Step C above with 1 eq. ethyl isocyanate in DMA one would obtain after removing of DMA and the title compound.
- If one were to treat the title compound from Step D above with a 2% aqueous NaOH at 100° C. for several hours one would obtain after neutralisation, precipitation and recrystallisation from ethanol the title compound.
- If one were to treat the title compound from Step E above according to the procedure described in Preparative Example 70 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 500, except using the appropriate intermediate from the Preparative Examples and hydrazines and amines as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Preparative Example Example Hydrazine Amine Product 501 300 
NH3 
502 300 
NH3 
503 300 
NH3 
504 61 N2H4 NH3 
505 61 
NH3 
506 61 
NH3 
507 61 
NH3 
508 65 N2H4 NH3 
509 65 
NH3 
510 65 
NH3 
511 65 NH3 
512 300 N2H4 CH3NH2 
513 300 
CH3NH2 
514 300 
CH3NH2 
515 300 
CH3NH2 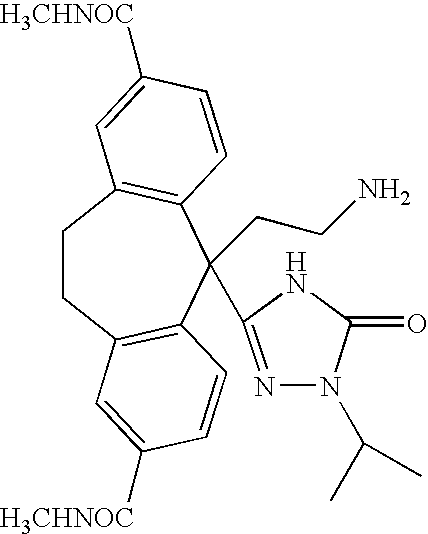
516 61 N2H4 CH3NH2 
517 61 
CH3NH2 
518 61 
CH3NH2 
519 61 
CH3NH2 
520 65 N2H4 CH3NH2 
521 65 
CH3NH2 
522 65 
CH3NH2 
523 65 
CH3NH2 
524 300 N2H4 (CH3)2NH 
525 300 
(CH3)2NH 
526 300 
(CH3)2NH 
527 300 
(CH3)2NH 
528 61 N2H4 (CH3)2NH 
529 61 
(CH3)2NH 
530 61 
(CH3)2NH 
531 61 
(CH3)2NH 
532 65 N2H4 (CH3)2NH 
533 65 
(CH3)2NH 
534 65 
(CH3)2NH 
535 65 
(CH3)2NH 
- Example numbers 536-599 were intentionally excluded.
-

- If one were to treat the intermediate from Preparative Example 300 Step A with dry HCl gas in EtOH/CHCl3 at 0° C. and set aside for 10 days, one would obtain after removal of the solvents the imidate hydrochloride. If one were to treat the imidate hydrochloride with NH3 in dry EtOH and heat it to reflux for 7 h, one would obtain, after filtration and evaporation of the filtrate followed by recrystallization, the title compound.
- If one were to treat the title compound from Step A above with Boc2O according to the procedure described in Preparative Example 49 Step J but without the acid treatment, one would obtain the title compound.
- If one were to treat the title compound from Step B above according to Preparative Example 61 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step C above according to the procedures described in Preparative Example 70, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 600 except using the amines and appropriate intermediate from the Preparative Examples as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Preparative Amine Amine Example Example Step A Step B Product 601 300 CH3NH2 NH3 
602 300 
NH3 
603 300 
NH3 
604 61 NH3 NH3 
605 61 CH3NH2 NH3 
606 61 
NH3 
607 61 
NH3 
608 65 NH3 NH3 
609 65 CH3NH2 NH3 
610 65 
NH3 
611 65 
NH3 
612 300 NH3 CH3NH2 
613 300 CH3NH2 CH3NH2 
614 300 
CH3NH2 
615 300 
CH3NH2 
616 61 NH3 CH3NH2 
617 61 CH3NH2 CH3NH2 
618 61 
CH3NH2 
619 61 
CH3NH2 
620 65 NH3 CH3NH2 
621 65 CH3NH2 CH3NH2 
622 65 
CH3NH2 
623 65 
CH3NH2 
624 300 NH3 (CH3)2NH 
625 300 CH3NH2 (CH3)2NH 
626 300 
(CH3)2NH 
627 300 
(CH3)2NH 
628 61 NH3 (CH3)2NH 
629 61 CH3NH2 (CH3)2NH 
630 61 
(CH3)2NH 
631 61 
(CH3)2NH 
632 65 NH3 (CH3)2NH 
633 65 CH3NH2 (CH3)2NH 
634 65 
(CH3)2NH 
635 65 
(CH3)2NH 
- Example numbers 636-679 were intentionally excluded.
- If one were to follow a similar procedure as that described in Preparative Example 67 and 70, except using the appropriate intermediate from the Preparative Examples and amines as indicated in the Table below, one would obtain the desired amine product.
-
Prepa- rative Preparative Example Example Amine Product 680 300 NH3 
681 61 NH3 
682 65 NH3 
683 300 CH3NH2 
684 61 CH3NH2 
685 65 CH3NH2 
686 300 (CH3)2NH 
687 65 (CH3)2NH 
- Example numbers 688-699 were intentionally excluded.
-

- If one were to treat the compound from Preparative Example 300 Step A with hydroxylamine hydrochloride and base according to Preparative Example 67 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step A above according to Preparative Example 67 Step B, one would obtain the title compound.
- If one were to treat the title compound from step B above with Lawesson's Reagent in toluene and heat the mixture to reflux for 4 h, one would obtain after column chromatography the title compound.
- If one were to treat the title compound from Step C above with formic acid hydrazide (Pellizzari-Synthesis), one would obtain the title compound.
- If one were to treat the title compound from Step D above according to the procedures described in Preparative Example 70, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 700, except using the appropriate intermediate from the Preparative Examples, acid hydrazides and amines as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Preparative Acid Example Example hydrazide Amine Product 701 300 
NH3 
702 300 
NH3 
703 300 
NH3 
704 61 
NH3 
705 61 
NH3 
706 61 
NH3 
707 61 
NH3 
708 65 
NH3 
709 65 
NH3 
710 65 
NH3 
711 65 
NH3 
712 300 
CH3NH2 
713 300 
CH3NH2 
714 300 
CH3NH2 
715 300 
CH3NH2 
716 61 
CH3NH2 
717 61 
CH3NH2 
718 61 
CH3NH2 
719 61 
CH3NH2 
720 65 
CH3NH2 
721 65 
CH3NH2 
722 65 
CH3NH2 
723 65 
CH3NH2 
724 300 
(CH3)2NH 
725 300 
(CH3)2NH 
726 300 
(CH3)2NH 
727 300 
(CH3)2NH 
728 61 
(CH3)2NH 
729 61 
(CH3)2NH 
730 61 
(CH3)2NH 
731 61 
(CH3)2NH 
732 65 
(CH3)2NH 
733 65 
(CH3)2NH 
734 65 
(CH3)2NH 
735 65 
(CH3)2NH 
- Example numbers 736-779 were intentionally excluded.
-

- If one were to treat the starting material, which was obtained by treating the title compound from Preparative Example 300 Step A according to the procedures described in Preparative Example 500 Step A-C, according to the procedure described in Preparative Example 70 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 780, except using the appropriate intermediate from the Preparative Examples and amines as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Preparative Example Example Amine Product 781 61 NH3 
782 65 NH3 
783 300 CH3NH2 
784 61 CH3NH2 
785 65 CH3NH2 
786 300 (CH3)2NH 
787 61 (CH3)2NH 
788 65 (CH3)2NH 
- Example numbers 789-799 were intentionally excluded.
-

- If one were to treat commercial available N methyl anthranilic acid with 2 eq. of 2-bromo-5-chloronitrobenzene, 10 eq. of potassium carbonate and a catalytic amount of copper powder in 3-methylbutan-1-ol under reflux for several hours one would obtain, after removing of the volatile compound by steam distillation, acidification of the residue with 2 M HCl, precipitation and recrystallisation of the precipitate from ethanol, the title compound.
- If one were to treat the title compound from Step A above with 7 eq. of sodium dithionite in 2 M aqueous ammonia at 80° C. one would obtain, after filtration, acidification of the filtrate with glacial acetic acid to pH 4, precipitation and recrystallisation from methanol, the title compound.
- If one were to reflux the title compound from Step B above in xylene under Dean Stark conditions one would obtain, after evaporation of the solvent, washing of the residue with 2 M aqueous ammonia and recrystallisation from acetone, the title compound.
- If one were to treat the title compound from Step C above with the sulfamidate from Preparative Example 22 according to Preparative Example 61 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above with TFA as described in Preparative Example 70 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 800, except using the diazepines and sulfamidates as indicated in the Table below, one would obtain the desired amine product.
-
Preparative Example Diazepine Sulfamidate Product 801 
22 
802 
21 
803 
24 
804 
21 
805 
24 
- Examples 806-809 have been intentionally excluded.
-

- If one were to treat commercially available 10,10-dimethyl-10H-anthracen-9-one and concentrated sulphuric acid in chloroform in a flask equipped with reflux condenser with sodium azide at room temperature, followed by heating this mixture at 50° C. and subsequently pouring it on crushed ice followed by neutralization with conc. aqueous ammonia, separation and evaporation of the organic phase, one would obtain the title compound.
- If one were to treat the title compound from Step A above with the sulfamidate from Preparative Example 22 as described in Preparative Example 800, one would obtain the title compound.
- If one were to follow a similar procedure as described in Preparative Example 810, except using the azepines and sulfamidates as indicated in the able below, one would obtain the desired amine product.
-
Preparative Example Azepine Sulfamidate Product 811 
24 
812 
21 
- Examples 813-829 have been intentionally excluded.
-


- If one were to add a solution of commercially available 2-amino-2-methyl-1-propanol in methylene chloride to a solution of commercially available 2-thiophenecarbonyl chloride in methylene chloride dropwise while maintaining the temperature below 20° C., subsequently stir the mixture at room temperature for 2 h and wash with water, dry the organic layer (MgSO4) and evaporate, suspend the residue in toluene and add thionyl chloride dropwise with stirring while maintaining the temperature below 30° C., subsequently continue the stirring overnight, evaporate the toluene, dissolve the residue in water, basify with 1 N aqueous NaOH and extract with ether, then, after drying (MgSO4) and evaporation of the solvent, followed by distillation, one would obtain the title compound.
- If one were to add commercial -nBuLi in hexane to the title compound from Step AA above in ether at −78° C., stir the mixture under argon for 0.25 h, add DMF, allow the mixture to slowly warm to room temperature and leave the mixture at this temperature for 18 h, subsequently add water and ether, separate the organic solution, wash with water, brine and dry the solution (MgSO4), then, after evaporation of the solvent, followed by chromatographic purification, one would obtain the title compound.
- If one were to boil the title compound from Step BB above under reflux with 4M aqueous hydrochloric acid under argon atmosphere for 14 h, saturate the cooled solution with NaCl, extract repeatedly with ethyl acetate, dry the combined organic extracts (MgSO4), then, after evaporation of the solvent, followed by recrystallization from ethyl acetate/hexane, one would obtain the title compound.
- If one were to treat the title compound from Step CC above in methanol dropwise with an ethereal solution of diazomethane at −15° C., followed by careful removal of all volatiles, then one would obtain the title compound.
- If one were to add commercially available methyl 4-methylthiophene-2-carboxylate to N-bromosuccinimide, benzoyl peroxide and tetrachloromethane and would heat the mixture under reflux for 4 h followed by filtration and evaporation of the solvent, one would obtain the title compound.
- If one were to treat the title compound from Step A above with triphenylphosphine according to Preparative Example 51 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step B above with the thiophene aldehyde from Step DD as described in Preparative Example 54 Step A, one would obtain the title compound.
- If one were to treat a suspension of the title compound from Step C above, hydroiodic acid and red phosphorus at 140° C. for 18 h, followed by cooling and pouring the reaction mixture into an ice/water mixture, subsequent filtration, washing of the precipitate with water, dissolving the precipitate in refluxing conc. ammonia and subsequent filtration, acidification of the filtrate with conc. aqueous hydrochloric acid and extraction of the aqueous phase with dichloromethane, washing of the organic phase with water and drying (MgSO4) followed by evaporation of the solvent, one would obtain the title compound.
- If one were to treat a suspension of the title compound from Step D above with polyphosphoric acid at 170° C., followed by cooling to 30° C., pouring into water, extraction with diethyl ether, washing with 1N aqueous sodium hydroxide solution and drying (MgSO4) followed by evaporation of the solvent, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 59 Step H and Step I, one would obtain the title compound.
- If one were to treat the title compound from Step G above with the compound from Preparative Example 22 as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step I above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 830 as described in Preparative Example 71 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 830, except using the sulfamidates in Step H, and treat the product obtained according to Preparative Example 831 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salts.
-
Preparative Example Sulfamidate Amine Title compound 831 21 NH3 
832 24 NH3 
833 22 NH3 
834 21 CH3NH2 
835 24 CH3NH2 
836 22 CH3NH2 
837 24 (CH3)2NH 
838 22 (CH3)2NH 
- Examples 839 to 849 have been intentionally excluded.
-


- If one were to treat commercially available thiophene-3-carbaldehyde with bromine and aluminium trichloride in dichloromethane and heat the reaction mixture for 2 h, subsequently pouring it into water, followed by extraction with ether, washing of the organic phase successively with aqueous 1N NaOH solution and water until neutral, then, after drying (MgSO4) and evaporation of the solvent, followed by distillation, one would obtain the title compound.
- If one were to treat a solution of the title compound from Step AA above in tetrahydrofuran with NaBH4 for 1 h and quench the reaction by the addition of saturated aqueous ammonium chloride solution followed by dilution with ethyl acetate, separation of the organic layer, washing with H2O and brine, then, after drying (MgSO4) and evaporation of the solvent, one would obtain the title compound.
- If one were to treat a solution of the title compound from Step BB above in chloroform with thionyl chloride at room temperature for 4 h, subsequently pouring it into water, followed by extraction with chloroform, washing of the organic phase with water, then, after drying (MgSO4) and evaporation of the solvent, one would obtain the title compound.
- If one were to treat commercially available 2-bromo-3-methylthiophene in acetic acid with N-chlorosuccinimide and stir the reaction mixture for about 2 h, then refluxing it for 1 h, subsequently pouring it into water, followed by extraction with ether, washing of the organic phase successively with aqueous 1N NaOH solution and water until neutral, then, after drying (MgSO4) and evaporation of the solvent, followed by distillation, one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 59 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step B above with the title compound from Step CC above, as described in Preparative Example 59 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 59 Step D, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step E and Step F, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 59 Step H and Step I, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step I above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step J above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 851 as described in Preparative Example 71 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 850, except using the sulfamidates in Step I, and treat the product obtained according to Preparative Example 851 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 852 21 NH3 
853 24 NH3 
854 22 NH3 
855 21 CH3NH2 
856 24 CH3NH2 
857 22 CH3NH2 
858 24 (CH3)2NH 
859 22 (CH3)2NH 
- Examples 860-899 have been intentionally excluded.
-

- If one were to add a solution of commercially available 2-(3bromo-2-thienyl)-1,3-dioxolane in dry diethylether with stirring to 1.05 N butyl lithium in diethylether at −70° C., followed by addition of the mixture to solid CO2 covered with diethylether. Hydrolysis, followed by extraction with diluted aqueous sodium hydroxide, acidification, then extraction with diethylether afford the title compound.
- If one were to add H2SO4 and methanol to a solution of the title compound from step AA above in dichloroethane, one would obtain the title compound.
- If one were to treat a solution of commercially available 5-methylthiophene-2-carboxylic acid in benzene and methanol at 0° C. dropwise with 2.0 M trimethylsilyldiazo-methane in hexanes, one would obtain the methyl ester. If one were to treat a solution of that ester intermediate in CCl4 with NBS and 2,2′-azobisisobutyronitrile (AIBN) and heat the solution to reflux for 2 h, followed by cooling down to room temperature, filtration and concentration in vacuo one would obtain the title compound.
- If one were to treat the title compound from Step A above with triphenylphosphine according to Preparative Example 49 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step B above with the title compound from Step BB above as described in Preparative Example 54 Step A, one would obtain the title compound.
- If one were to heat a mixture of the title compound from Step C, red phosphorous and hydroiodic acid in acetic acid at 110° C. for 1 h, one would obtain a solution after filtration of the hot mixture. After cooling to room temperature and pouring in ice water one would obtain the title compound by suction.
- If one were to heat a mixture of the title compound from Step D above and polyphosphoric acid at 115° C. for 1.5 h one would obtain a mixture, which was poured on ice. After extraction with Ether washing the organic phases with water, drying (MgSO4) and removing of the solvent one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 59 Step H and Step I, one would obtain the title compound.
- If one were to treat the title compound from Step G above with the compound from Preparative Example 22 as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step I above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 900 as described in Preparative Example 71 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 900, except using the sulfamidates in Step H, and treat the product obtained according to Preparative Example 901 with the amines as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 902 21 NH3 
903 24 NH3 
904 22 NH3 
905 21 CH3NH2 
906 24 CH3NH2 
907 22 CH3NH2 
908 24 (CH3)2NH 
909 22 (CH3)2NH 
- Examples 910-919 have been intentionally excluded.
-

- If one were to add a solution of bromine in CHCl3 slowly to an ice-cooled solution of commercially available 2-chloro-5-methylthiophene in CHCl3 one would obtain a reaction mixture which was stirred for 2 h at room temperature, and subsequently poured into H2O. If one were to extract than the mixture with dichloromethane combine the organic extracts dry filter and evaporate the solvent, one would obtain a yellow/brown oil.
- If one were to treat the title compound from Step A above as described in Preparative Example 59 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step B above with commercially available 2-chloro-5-chloromethyl-thiophene as described in Preparative Example 59 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 59 Step D, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step E and Step F, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 59 Step H and Step I, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step I above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step J above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 920 as described in Preparative Example 71 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 920, except using the sulfamidates in Step I, and treat the product obtained according to Preparative Example 921 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 922 21 NH3 
923 24 NH3 
924 22 NH3 
925 21 CH3NH2 
926 24 CH3NH2 
927 22 CH3NH2 
928 24 (CH3)2NH 
929 22 (CH3)2NH 
- Examples 930-999 have been intentionally excluded.
- If one were to follow similar procedure as described in Preparative Examples 92 and 93, except using the amides and amines as indicated in the Table below, the following title compound would be obtained.
-
Prep Example Amide Amines Title compound 1000 


1001 


1002 


1003 


1004 


1005 


1006 


1007 


1008 


1009 


1010 


1011 


1012 


1013 


1014 


1015 


1016 


1017 


1018 


1019 


1020 


1021 


1022 


1023 


1024 


1025 


1026 


1027 


1028 


1029 


1030 


1031 


1032 


1033 


1034 


1035 


1036 


1037 


1038 


1039 


1040 


1041 


1042 


1043 


1044 


1045 


1046 


1047 


1048 


1049 


1050 


1051 


1052 


1053 


1054 


1055 


1056 


1057 


1058 


1059 


1060 


1061 


1062 


1063 


1064 


1065 


1066 


1067 


1068 


1069 


1070 


1071 


1072 


1073 


1074 


1075 


1076 


1077 


1078 


1079 


1080 


1081 


1082 


1083 


1084 


1085 


1086 


1087 


1088 


1089 


1090 


1091 


1092 


1093 


1094 


1095 


1096 


1097 


1098 


1099 


1100 


1101 


1102 


1103 


1104 


1105 


1106 


1107 


1108 


1109 


1110 


1111 


1112 


1113 


1114 


1115 


1116 


1117 


1118 


1119 


1120 


1121 


1122 


1123 


1124 


1125 


1126 


1127 


1128 


1129 


1130 


1131 


1132 


1133 


1134 


1135 


1136 


1137 


1138 


1139 


1140 


1141 


1142 


1143 


1144 


1145 


1146 


1147 


1148 


1149 


1150 


1151 


1152 


1153 


1154 


1155 


1156 


1157 


1158 


1159 


1160 


1161 


1162 


1163 


1164 


1165 


1166 


1167 


1168 


1169 


1170 


1171 


1172 


1173 


1174 


1175 


1176 


1177 


1178 


1179 


1180 


1181 


1182 


1183 


1184 


1185 


1186 


1187 


1188 


1189 


1190 


1191 


1192 


1193 


1194 


1195 


1196 


1197 


1198 


1199 


1200 


1201 


1202 


1203 


1204 


1205 


1206 


1207 


1208 


1209 


- Examples 1210-1299 have been intentionally excluded.
-

- If one were to treat commercially available anthraquinone with 1.5-2 equivalents of bromine and some iodine at 160° C., and then treat the mixture with aqueous sodium hydroxide at reflux, one would obtain the title compound, after crystallisation from glacial acetic acid.
- If one were to treat the title compound from Step A above with hot concentrated H2SO4, treat the obtained solution with Al powder at rt and stir the mixture at rt for 3 h, one would obtain the title compound, after aqueous workup and chromatography on silica gel.
- If one were to treat the title compound from Step B above as described in Preparative Example 59 Step D, Step E and Step F, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 59 Step H, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step I, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 1300 as described in Preparative Example 71 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 1300, except using the sulfamidates in Step G, and treat the product obtained according to Preparative Example 1301 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 1302 21 NH3 
1303 24 NH3 
1304 22 NH3 
1305 21 CH3NH2 
1306 24 CH3NH2 
1307 22 CH3NH2 
1308 24 (CH3)2NH 
1309 22 (CH3)2NH 
- Examples 1310-1349 have been intentionally excluded.
-

- If one were to treat a solution of commercially available 4-chloroanthranilic acid in water and concentrated hydrochloric acid at 0° C. with a solution of sodium nitrate in water over 45 min and stir the resulting mixture at 0° C. for 1 h, one would obtain the diazonium salt solution after filtration. If one were to treat a solution of commercially available hydroxylamine hydrochloride in water at 10° C. with an aqueous solution of sodium hydroxide and carefully pour the mixture into an aqueous solution of hydrated copper(II) sulfate and concentrated ammonia solution, one would obtain a blue solution after filtration. If one were to carefully add the diazonium salt solution from above to the blue solution over a period of 1 h and then heat the mixture at reflux, followed by the addition of concentrated hydrochloric acid, one would obtain a precipitate after 3 h. If one were to collect the precipitate by filtration, wash it with water and dissolved it in a solution of sodium bicarbonate, one would obtain a clear solution after treatment with charcoal and filtration. If one were to add an excess of 6 M aqueous hydrochloric acid and collect the precipitate, one would obtain the title compound after crystallisation from EtOH.
- If one were to treat the title compound of Step A above at 400° C. for twenty-five minutes and then sublime the mixture at 250° C. under a pressure of 2 mm, one would obtain the title compound after crystallization from benzene.
- If one were to treat the title compound from Step B above as described in Preparative Example 59 Step D, Step E and Step F, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 59 Step H, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 59 Step I, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 1350 as described in Preparative Example 71 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 1350, except using the sulfamidates in Step G, and treat the product obtained according to Preparative Example 1351 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 1352 21 NH3 
1353 24 NH3 
1354 22 NH3 
1355 21 CH3NH2 
1356 24 CH3NH2 
1357 22 CH3NH2 
1358 24 (CH3)2NH 
1359 22 (CH3)2NH 
- Examples 1360-1399 have been intentionally excluded.
-

- If one were to treat commercially available 4-bromo benzaldehyde dissolved in ether at 0° C. over a period of two hours portion-wise with KCN and concentrated HCl and maintain the temperature of the reaction below 10° C., followed by stirring for 1 h after complete addition, while permitting the temperature to rise to 15° C., subsequently the resultant two-phase system is filtered off and washed with ether, separating the combined organic solutions one would obtain the intermediate after washing with saturated aqueous sodium bisulfide, drying over MgSO4, and concentrating in vacuo. If one were to dilute the residue with benzene and slowly add this mixture over a period of one hour to concentrated H2SO4, which would maintained under stirring in an ice bath at a temperature below 15° C. until completion of the addition, followed by stirring for an additional hour, allowing the mixture to warm to room temperature one would obtain after pouring the reaction mixture onto ice and the mixture is being extracted with benzene, the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step B above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step D, Step E and Step F, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 61 Step C, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 1400 as described in Preparative Example 71 Step A one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Preparative Example 71 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 1400, except using the sulfamidates in Step B, and treat the product obtained according to Preparative Example 1401 with the amine as indicated in the table below, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Amine Title compound 1402 21 NH3 
1403 24 NH3 
1404 22 NH3 
1405 21 CH3NH2 
1406 24 CH3NH2 
1407 22 CH3NH2 
1408 24 (CH3)2NH 
1409 22 (CH3)2NH 
- Examples 1410-1449 have been intentionally excluded.
-

- If one were to add commercially available diethylmethylmalonate to a solution of sodium ethoxide in EtOH, and then add a solution of α,α′-dibromo-m-xylene in benzene to the above solution and boil the mixture at reflux for 1 h, one would obtain the title compound after distillation and crystallisation.
- If one were to treat the title compound from Step A above with aqueous-ethanolic potassium hydroxide, one would obtain the crude tetracarboxylic acid. If one were to decarboxylate the crude tetracarboxylic acid at 210° C., one would obtain the title compound.
- If one were to convert the title compound from Step B above to its bis-acid chloride with thionyl chloride in benzene and treat the bis-acid chloride with a solution of diazomethane in ether, one would obtain the diazoketone intermediate after 12 h and evaporation of the solvents. If one were to treat the diazoketone with benzyl alcohol-γ-collidine (1:1) in an oil-bath maintained at 180° C. for 10 Min, one would obtain the crude title compound. If one were to treat the crude title compound with MeOH and HCl, one would obtain the dimethylester. If one were to treat the diemthylester with KOH in EtOH, one would obtain the title compound.
- If one were to treat the title compound from Step C above with phosphorus pentachloride in benzene for 1 h and warm the mixture on a steam-bath for 5 min, one would obtain the crude bis-acid chloride. If one were to dissolve the bis-acid chloride in nitrobenzene, add a solution of aluminium chloride in nitrobenzene at 0° C. and then allow the mixture to stand at rt for 6 h, one would obtain the title compound, after removal of the nitrobenzene by steam distillation and crystallisation of the residue with EtOH.
- If one were to treat the title compound from Step D above with hydrazine hydrate and potassium hydroxide in diethylene glycol for 4 h at 180° C., followed by purification by chromatography on alumina one would obtain the title compound.
- If one were to treat the title compound from Step E with 10 eq. of aluminium chloride by adding the compound to the reagent in tetrachloroethane at low temperature, add dropwise 2.0 eq. of acetic anhydride to the mixture, pour onto ice and hydrochloric acid and extract with an appropriate solvent, wash with water, evaporate, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step F above with selenium dioxide in water and dioxane and refluxed for 4 h, followed by removal of precipitated selenium one would obtain after recrystallizaiton the title compound.
- If one were to treat the title compound from Step G above with hydrogen peroxide and drop wise with 10% NaOH in ethanol at 80° C., followed by dilution with water, treatment with norite, filtration and acidifying with HCl, one would obtain after recrystallization the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 70 Step A, one would obtain the title compound
- If one were to treat the title compound from Step I above as described in Preparative Example 93 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step J above as described in Preparative Example 13 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step K above with diisobutylaluminum hydride in CH2Cl2 at −78° C., add 10% aq AcOH, extract with ether:hexane, wash with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step L above with 1.2 eq. commercially available methylmagnesium bromide in Et2O at room temperature, heat the mixture to reflux, add ice and half concentrated hydrochlorid acid, extract with Et2O, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step M above with methylsulfonyl chloride and triethylamine in CH2Cl2 at 0° C., evaporate, add water and ethyl acetate to the residue, extract with ethyl acetate, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate and then the obtained intermediate with NaN3 in DMA as described in Preparative Example 17 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step N above as described in Preparative Example 17 Step D, one would obtain the title compound.
-

- If one were to treat the title compound from Preparative Example 1450 Step E with 10 eq. of aluminium chloride by adding the compound to the reagent in tetrachloroethane at low temperature, add dropwise 2.0 eq. of acetic anhydride to the mixture, pour onto ice and hydrochloric acid and extract with an appropriate solvent, wash with water, evaporate, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step F above with selenium dioxide in water and dioxane and refluxed for 4 h, followed by removal of precipitated selenium one would obtain after recrystallizaiton the title compound.
- If one were to treat the title compound from Step G above with hydrogen peroxide and drop wise with 10% NaOH in ethanol at 80° C., followed by dilution with water, treatment with norite, filtration and acidifying with HCl, one would obtain after recrystallization the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 70 Step A, one would obtain the title compound
- If one were to treat the title compound from Step Iabove as described in Preparative Example 93 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step J above as described in Preparative Example 13 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step K above with diisobutylaluminium hydride in CH2Cl2 at −78° C., add 10% aq AcOH, extract with ether:hexane, wash with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step L above with 1.2 eq. commercially available methylmagnesium bromide in Et2O at room temperature, heat the mixture to reflux, add ice and half concentrated hydrochlorid acid, extract with Et2O, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step M above with methylsulfonyl chloride and triethylamine in CH2Cl2 at 0° C., evaporate, add water and ethyl acetate to the residue, extract with ethyl acetate, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate and then the obtained intermediate with NaN3 in DMA as described in Preparative Example 17 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step N above as described in Preparative Example 17 Step D, one would obtain the title compound.
-

- If one were to treat commercially available 1,2,3,4,5,6,7,8-octahydro-anthracene with 10 eq. of aluminium chloride by adding the compound to the reagent in tetrachloroethane at low temperature, add dropwise 2.0 eq. of acetic anhydride to the mixture, pour onto ice and hydrochloric acid and extract with an appropriate solvent, wash with water, evaporate, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step A above with selenium dioxide in water and dioxane and refluxed for 4 h, followed by removal of precipitated selenium one would obtain after recrystallization the title compound.
- If one were to treat the title compound from Step B above with hydrogen peroxide and drop wise with 10% NaOH in ethanol at 80° C., followed by dilution with water, treatment with norite, filtration and acidifying with HCl, one would obtain after recrystallization the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 70 Step A, one would obtain the title compound
- If one were to treat the title compound from Step D above as described in Preparative Example 93 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 13 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step F above with diisobutylaluminium hydride in CH2Cl2 at −78° C., add 10% aq AcOH, extract with ether:hexane, wash with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step G above with 1.2 eq. commercially available methylmagnesium bromide in Et2O at room temperature, heat the mixture to reflux, add ice and half concentrated hydrochlorid acid, extract with Et2O, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step H above with methylsulfonyl chloride and triethylamine in CH2Cl2 at 0° C., evaporate, add water and ethyl acetate to the residue, extract with ethyl acetate, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate and then the obtained intermediate with NaN3 in DMA as described in Preparative Example 17 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step I above as described in Preparative Example 17 Step D, one would obtain the title compound.
-

- If one were to treat commercially available 2-methyl-1H-indene and with 0.01 eq of platinum oxide in tetrahydrofuran and hydrogenate at 20-30 psi for 10-15 h at room temperature, filter the mixture through a pad of Celite, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step A above with 1.0 eq. of 3-chloro-2-methyl-propionyl chloride and 3.0 eq. of aluminum chloride in nitromethane at room temperature, decompose the mixture with ice and hydrochloric acid, dilute with water, filter, dissolve the solid in benzene and wash with dilute hydrochloric acid, evaporate, purify with a Soxhlet extractor, one would obtain the title compound.
- If one were to treat the title compound from Step B above with concentrated sulphuric acid by adding the compound in small portions to the acid at low temperature, heat on the steam-bath, pour onto ice and extract with benzene and water, evaporate, distillate at reduced pressure, recrystallize from petroleum ether, sublimate, one would obtain the title compound.
- If one were to treat the title compound from Step C above with amalgamated zinc, water, acetic acid, toluene, hydrochloric acid, separate the organic layer, evaporate, distillate at reduced pressure, recrystallize, one would obtain the title compound.
- If one were to treat the title compound from Step D with 10 eq. of aluminium chloride by adding the compound to the reagent in tetrachloroethane at low temperature, add dropwise 2.0 eq. of acetic anhydride to the mixture, pour onto ice and hydrochloric acid and extract with an appropriate solvent, wash with water, evaporate, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step E with an aqueous solution of potassium hypochlorite prepared from bleaching powder in methanol, separate the precipitate formed by filtration, acidify the filtrate, separate the precipitate formed by filtration, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 70 Step A, one would obtain the title compound
- If one were to treat the title compound from Step G above as described in Preparative Example 93 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step H above with diisobutylaluminium hydride in CH2Cl2 at −78° C., add 10% aq AcOH, extract with ether:hexane, wash with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 13 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step I above with 1.2 eq. commercially available methylmagnesium bromide in Et2O at room temperature, heat the mixture to reflux, add ice and half concentrated hydrochlorid acid, extract with Et2O, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step K above with methylsulfonyl chloride and triethylamine in CH2Cl2 at 0° C., evaporate, add water and ethyl acetate to the residue, extract with ethyl acetate, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate and then the obtained intermediate with NaN3 in DMA as described in Preparative Example 17 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step L above as described in Preparative Example 17 Step D, one would obtain the title compound.
-

- If one were to treat commercially available indane with 1.0 eq. of 3-chloro-propionyl chloride and 3.0 eq. of aluminum chloride in nitromethane at room temperature, decompose the mixture with ice and hydrochloric acid, dilute with water, filter, dissolve the solid in benzene and wash with dilute hydrochloric acid, evaporate, purify with a Soxhlet extractor, one would obtain the title compound.
- If one were to treat the title compound from Step A above with concentrated sulphuric acid by adding the compound in small portions to the acid at low temperature, heat on the steam-bath, pour onto ice and extract with benzene and water, evaporate, distillate at reduced pressure, recrystallize from petroleum ether, sublimate, one would obtain the title compound.
- If one were to treat the title compound from Step B above with amalgamated zinc, water, acetic acid, toluene, hydrochloric acid, separate the organic layer, evaporate, distillate at reduced pressure, recrystallize, one would obtain the title compound.
- If one were to treat the title compound from Step D with 10 eq. of aluminium chloride by adding the compound to the reagent in tetrachloroethane at low temperature, add dropwise 2.0 eq. of acetic anhydride to the mixture, pour onto ice and hydrochloric acid and extract with an appropriate solvent, wash with water, evaporate, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step D with an aqueous solution of potassium hypochlorite prepared from bleaching powder in methanol, separate the precipitate formed by filtration, acidify the filtrate, separate the precipitate formed by filtration, recrystallize from methanol, one would obtain the title compound.
- If one were to treat the title compound from Step E above as described in Preparative Example 70 Step A, one would obtain the title compound
- If one were to treat the title compound from Step F above as described in Preparative Example 93 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step G above with diisobutylaluminium hydride in CH2Cl2 at −78° C., add 10% aq AcOH, extract with ether:hexane, wash with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 13 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step H above with 1.2 eq. commercially available methylmagnesium bromide in Et2O at room temperature, heat the mixture to reflux, add ice and half concentrated hydrochloride acid, extract with Et2O, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate, purify the crude product through chromatography on silica gel, one would obtain the title compound.
- If one were to treat the title compound from Step J above with methylsulfonyl chloride and triethylamine in CH2Cl2 at 0° C., evaporate, add water and ethyl acetate to the residue, extract with ethyl acetate, wash the organic layer with H2O, sat. aq NaHCO3, and brine, dry over Na2SO4, evaporate and then the obtained intermediate with NaN3 in DMA as described in Preparative Example 17 Step C, one would obtain the title compound.
- If one were to treat the title compound from Step K above as described in Preparative Example 17 Step D, one would obtain the title compound.
- Examples 1455-1499 have been intentionally excluded.
-

- If one were to treat commercially available 1,4-benzoquinone with buta-1,3-diene in benzene at 100° C. in an autoclave, separate the precipitate, wash it with methanol, one would obtain the title compound.
- If one were to treat the title compound from Step A above with LiAlH4 in THF at rt for 15 min and then heat to reflux for 50 min, one would obtain after removal of the solvent, followed by aqueous workup and column chromatography the title compound.
- If one were to treat the title compound from Step B above with methanesulfonyl chloride in pyridine at 0° C. for 24 h, one would obtain after pouring into an ice/water mixture followed by extraction with benzene and subsequently washing the organic phase with water, cold 5% sulphuric acid, water, 2% sodium bicarbonate solution, brine and finally evaporation to dryness, the methansulfonate intermediate. If one were to treat the methansulfonate intermediate with LiAlH4 in THF and heat to reflux for 24 h, one would obtain after removal of the solvent, followed by aqueous workup the alcohol intermediate.
- If one were to treat the alcohol intermediate with CrO3 in pyridine at 40° C. for 9 h, one would obtain after pouring into water, followed by extraction with CCl4 and subsequently drying the organic phase and evaporating to dryness, followed by column chromatography and crystallization the alkene intermediate. If one were to treat the alkene intermediate with Pd/C in ethanol at 10 bar H2 and room temperature, separate the crude product from the reaction mixture and then the obtained intermediate with CrO3 in aqueous acetic acid and water, neutralize the mixture, extract with Et2O, recrystallize from THF/CH2Cl2, one would obtain the title compound.
- If one were to treat the title compound from Step C above as described in Preparative Example 59 Step G, one would obtain the title compound.
- If one were to treat the title compound from Step D above as described in Preparative Example 59 Step H, one would obtain the title compound.
- If one were to treat the title compound from Step E with NaCN in 90% ethanol under reflux, add water, extract with CHCl3, wash the organic layer with 5% sulphuric acid, sat. aq NaHCO3, water, brine, dry over Na2SO4, distillate, one would obtain the title compound.
- If one were to treat the title compound from Step F above as described in Preparative Example 61 Step A, one would obtain the title compound.
- If one were to treat the title compound from Step G above as described in Preparative Example 61 Step B, one would obtain the title compound.
- If one were to treat the title compound from Step H above as described in Preparative Example 70 Step B, one would obtain the title compound.
- If one were to follow a similar procedure as that described in Preparative Example 1500, except using the sulfamidates in Step G, one would obtain the desired title compound as HCl salt.
-
Preparative Example Sulfamidate Title compound 1501 22 
1502 24 
-

- The title compound from Preparative Example 5 (378 mg) and 419 mg K2CO3 were suspended in 3 ml THF and cooled to 0° C. A solution of Preparative Example 1 (109 mg) in 1 ml THF was slowly added and the reaction mixture stirred at 0° C. for 2 h and then at rt overnight. The mixture was diluted with 30 ml EtOAc and 10 ml H2O, the organic phase separated, dried over MgSO4 and concentrated. The residue was purified by chromatography on silica (CH2Cl2/MeOH, 4:1) to afford the title compound (66 mg; 39%; MH+=389).
- Following a similar procedure as that described in Example 1, except using the compounds from the Preparative Examples indicated in the Table below, the following compounds were prepared.
-
Compound Compound Preparative Preparative 1. Yield Example Example Example Product 2. MH+ 2 1 6 
1. 17% 2. 346 3 1 7 
1. 8% 2. 417 4 1 13 
1. 19% 2. 360 5 1 14 Step B 
1. 18% 2. 389 6 1 14 
1. 15% 2. 375 7 1 15 Step C 
1. 8% 2. 372 8 1 15 
1. 8% 2. 374 9 1 16 
1. 16% 2. 389 10 1 17 Step D 
1. 7% 2. 390 11 1 17 
1. 8% 2. 372 12 1 10 
1. 16% 2. 429 13 1 11 
1. 19% 2. 415 14 1 12 
1. 19% 2. 401 -

- An aliquot of the title compound of Preparative Example 3 was taken and the solvent removed. The residue (67 mg) was dissolved in DMF (2 ml) and triethylamine (0.1 ml). The title compound from Preparative Example 90 (71 mg) was added and the mixture was stirred at 60° C. for 2 h. The solvent was removed and the residue was purified by preparative TLC (CHCl3/MeOH (+0.1% Triethylamine), 4:1) to afford the title compound (12 mg; 13%; MH+=381).
-

- The title compound from Preparative Example 18 Step B (100 mg) and Preparative Example 2 (68 mg) were dissolved in 2 ml EtOH and 1 ml H2O. The pH of the solution was adjusted to pH˜6 by adding 0.1 M HCl-solution and the mixture was stirred at rt for 10 min. After the addition of NaCNBH3 (24 mg) the pH was maintained at pH˜6 by the addition of 0.1 M HCl and the mixture was stirred at rt overnight. The mixture was diluted with 30 ml EtOAc and 15 ml sat. NaHCO3/brine (1:1), the organic phase separated, dried over MgSO4 and concentrated. The residue was purified by Prep TLC(CH2Cl2/MeOH, 95:5) to afford the title compound (25.9 mg; 17%; MH+=399).
- Following a similar procedure as described in Example 16 by dissolving the amine in a EtOH/H2O— or MeOH/H2O-mixture and adjusting the pH to pH˜6-8 by either 0.1 M HCl, 3 M NaOAc or 1 M NaOH, except using the compounds from the Preparative Examples indicated in the Table below, the following compounds were prepared. In case the reaction was not completed after 24 h as judged by HPLC, additional aldehyde from Preparative Example 2 or 89 and NaCNBH3 were added, and the reaction was continued for another 1-3 days.
- For the products obtained, the following purification methods were employed:
- Method A: chromatography on silica using CH2Cl2/MeOH-mixtures; or
- Method B: product was precipitated from the reaction mixture by adding 1 M HCl to pH 1-3 and the precipitate washed with MeOH; or
- Method C: reaction mixture was concentrated to half its volume and the crude product purified by reverse phase HPLC (21.5×250 mm, Phenomenex, Luna C-18 (2), 5 μM; flow=15 ml/min or 10×250 mm, Phenomenex, Luna C-18 (2), 5 μM; flow=3 ml/min) using acetonitrile (solvent B; 0.1% formic acid) and H2O (solvent A; 0.1% formic acid) as eluents and a suitable gradient, ramping solvent B from 0% to 100% over a period of 18 min.
-
Compound Compound Preparative Preparative Purification 1. Yield Example Example Example Method Product 2. MH+ 17 2 18 A 
1. 17% 2. 417 18 2 47 A 
1. 41% 2. 431 19 2 48 A 
1. 18% 2. 431 20 2 8 A 
1. 25% 2. 424 21 2 9 A 
1. 18% 2. 390 22 2 49 A 
1. 21% 2. 478 23 2 50 B 
1. 30% 2. 442 24 2 51 B 
1. 5% 2. 478 25 2 87 B 
1. 46% 2. 510 26 2 110 A 
1. 15% 2. 414 27 2 70 C 
1. 36% 2. 542 28 2 72 C 
1. 14% 2. 570 29 2 71 C 
1. 38% 2. 598 30 2 73 C 
1. 21% 2. 598 31 2 74 C 
1. 8% 2. 626 32 2 75 C 
1. 58% 2. 622 33 2 76 C 
1. 9% 2. 682 34 2 56 C 
1. 11% 2. 528 35 2 77 C 
1. 7% 2. 556 36 2 78 C 
1. 10% 2. 584 37 2 79 C 
1. 12% 2. 556 38 2 80 C 
1. 43% 2. 614 39 2 81 C 
1. 2% 2. 573 40 2 82 C 
1. 26% 2. 666 41 2 83 C 
1. 12% 2. 542 42 2 84 C 
1. 10% 2. 542 43 2 85 C 
1. 60% 2. 572 44 2 86 C 
1. 28% 2. 544 45 2 52 C 
1. 14% 2. 503 46 2 88 C 
1. 2% 2. 471 47 89 56 C 
1. 9% 2. 540 -

- The title compound from Preparative Example 93 (16 mg) was dissolved in a mixture of H2O (3 ml) and a solution of 4 M HCl in dioxane (3 ml). After 20 h the reaction mixture was diluted with toluene. The organic layer was evaporated to afford the title compound (14 mg; 99%; MH+=386).
- Following a similar procedure as that described in Example 48, except using the compounds from the Preparative Examples indicated in the Table below, the following compound was prepared.
-
Compound Preparative 1. Yield Example Example Product 2. MH+ 49 95 
1. 77% 2. 436 50 96 
1. 92% 2. 393 51 97 
1. 89% 2. 404 52 98 
1. 96% 2. 416 53 99 
1. 57% 2. 393 54 100 
1. 95% 2. 404 55 101 
1. 93% 2. 393 56 102 
1. 98% 2. 400 57 108 
1. 96% 2. 400 58 103 
1. 95% 2. 412 59 104 
1. 95% 2. 414 60 105 
1. 92% 2. 411 61 106 
1. 95% 2. 411 62 107 
1. 81% 2. 426 63 109 
1. 85% 2. 412 64 94 
1. 95% 2. 398 -

- The title compound from Preparative Example 113 (13 mg) was treated with 4 M HCl in dioxane as described in Example 47 to afford the title compound (11.2 mg, 98%, MH+=436).
- Following a similar procedure as that described in Example 65, except using the compounds from the Preparative Examples indicated in the Table below, the following compounds were prepared.
-
Compound Preparative 1. Yield Example Example Product 2. MH+ 66 114 
1. 100 2. 424 67 115 
1. 33 2. 424 68 116 
1. 40 2. 482 69 117 
1. 85 2. 388 70 118 
1. 96 2. 402 71 119 
1. 84 2. 384 72 122 
1. 30 2. 510 73 112 Step D 
1. 50 2. 500 74 121 
1. 97 2. 475 75 120 
1. 100 2. 377 -

- The title compound from Preparative Example 123 (27 mg) was dissolved in dichloromethane (2 ml) and trimethylsilyl iodine (21 mg) was added. The mixture was stirred for 1 h at room temperature. After removal of the solvent the residue was purified by preparative TLC to afford the desired compound (CHCl3/MeOH, 4 mg, 20%, MH+=388).
- Following a similar procedure as that described in Example 76, except using the compounds from the Preparative Examples as indicated in the Table below, the following compounds were prepared.
-
Preparative 1. Yield Example Example Product 2. MH+ 77 124 
1. 10% 2. 422 78 125 
1. 11% 2. 358 - Examples 79-99 have been intentionally excluded.
- If one were to follow the procedures outlined in Preparative Example 71 and Examples 28 or 29 but using the amines, carboxylic acids and aldehydes from the Preparative Examples as indicated in the Table below, one would obtain the indicated Product.
-
Example Carboxylic # Amine Acid Aldehyde Product 100 
Prep Ex 62 Prep Ex 2 
101 
Prep Ex 62 Prep Ex 2 
102 
Prep Ex 62 Prep Ex 2 
103 
Prep Ex 62 Prep Ex 2 
104 
Prep Ex 62 Prep Ex 2 
105 
Prep Ex 62 Prep Ex 2 
106 
Prep Ex 62 Prep Ex 2 
107 
Prep Ex 62 Prep Ex 2 
108 
Prep Ex 62 Prep Ex 2 
109 
Prep Ex 62 Prep Ex 2 
110 NH3 Prep Ex 55 Prep Ex 2 
111 MeNH2 Prep Ex 55 Prep Ex 2 
112 (Me)2NH Prep Ex 55 Prep Ex 2 
113 
Prep Ex 55 Prep Ex 2 
114 
Prep Ex 55 Prep Ex 2 
115 
Prep Ex 55 Prep Ex 2 
116 
Prep Ex 55 Prep Ex 2 
117 
Prep Ex 55 Prep Ex 2 
118 
Prep Ex 55 Prep Ex 2 
119 
Prep Ex 55 Prep Ex 2 
120 
Prep Ex 55 Prep Ex 2 
121 
Prep Ex 55 Prep Ex 2 
122 
Prep Ex 55 Prep Ex 2 
123 
Prep Ex 65 Prep Ex 2 
124 
Prep Ex 65 Prep Ex 2 
125 
Prep Ex 65 Prep Ex 2 
126 
Prep Ex 65 Prep Ex 2 
127 
Prep Ex 65 Prep Ex 2 
128 
Prep Ex 65 Prep Ex 2 
129 
Prep Ex 65 Prep Ex 2 
130 
Prep Ex 65 Prep Ex 2 
131 
Prep Ex 65 Prep Ex 2 
132 
Prep Ex 61 Prep Ex 2 
133 
Prep Ex 61 Prep Ex 2 
134 
Prep Ex 61 Prep Ex 2 
135 
Prep Ex 61 Prep Ex 2 
136 
Prep Ex 61 Prep Ex 2 
137 
Prep Ex 61 Prep Ex 2 
138 MeNH2 Prep Ex 62 Prep Ex 89 
139 (Me)2NH Prep Ex 62 Prep Ex 89 
140 
Prep Ex 62 Prep Ex 89 
141 
Prep Ex 62 Prep Ex 89 
142 
Prep Ex 62 Prep Ex 89 
143 
Prep Ex 62 Prep Ex 89 
144 
Prep Ex 62 Prep Ex 89 
145 
Prep Ex 62 Prep Ex 89 
146 
Prep Ex 62 Prep Ex 89 
147 
Prep Ex 62 Prep Ex 89 
148 
Prep Ex 62 Prep Ex 89 
149 
Prep Ex 62 Prep Ex 89 
150 NH3 Prep Ex 55 Prep Ex 89 
151 MeNH2 Prep Ex 55 Prep Ex 89 
152 (Me)2NH Prep Ex 55 Prep Ex 89 
153 
Prep Ex 55 Prep Ex 89 
154 
Prep Ex 55 Prep Ex 89 
155 
Prep Ex 55 Prep Ex 89 
156 
Prep Ex 55 Prep Ex 89 
157 
Prep Ex 55 Prep Ex 89 
158 
Prep Ex 55 Prep Ex 89 
159 
Prep Ex 55 Prep Ex 89 
160 
Prep Ex 55 Prep Ex 89 
161 
Prep Ex 55 Prep Ex 89 
162 
Prep Ex 55 Prep Ex 89 
163 
Prep Ex 65 Prep Ex 89 
164 
Prep Ex 65 Prep Ex 89 
165 
Prep Ex 65 Prep Ex 89 
166 
Prep Ex 65 Prep Ex 89 
167 
Prep Ex 65 Prep Ex 89 
168 
Prep Ex 65 Prep Ex 89 
169 
Prep Ex 65 Prep Ex 89 
170 
Prep Ex 65 Prep Ex 89 
171 
Prep Ex 65 Prep Ex 89 
172 NH3 Prep Ex 61 Prep Ex 89 
173 MeNH2 Prep Ex 61 Prep Ex 89 
174 (Me)2NH Prep Ex 61 Prep Ex 89 
175 
Prep Ex 61 Prep Ex 89 
176 
Prep Ex 61 Prep Ex 89 
177 
Prep Ex 61 Prep Ex 89 
178 
Prep Ex 61 Prep Ex 89 
179 
Prep Ex 61 Prep Ex 89 
180 
Prep Ex 61 Prep Ex 89 
181 
Prep Ex 61 Prep Ex 89 
182 
Prep Ex 61 Prep Ex 89 
183 
Prep Ex 61 Prep Ex 89 
184 
Prep Ex 61 Prep Ex 89 
- Examples 185-199 have been intentionally excluded.
- If one were to follow the procedures outlined in Examples 28 or 29 except using the compounds from the Preparative Examples as indicated in the Table below, one would obtain the indicated Product.
-
Preparative Preparative Example Example Example Product 200 200 2 
201 201 2 
202 202 2 
203 203 2 
204 204 2 
205 205 2 
206 206 2 
207 207 2 
208 208 2 
209 209 2 
210 210 2 
211 211 2 
212 212 2 
213 213 2 
214 214 2 
215 215 2 
216 216 2 
217 217 2 
218 218 2 
219 219 2 
220 220 2 
221 221 2 
222 222 2 
223 223 2 
224 224 2 
225 225 2 
226 226 2 
227 227 2 
228 228 2 
229 229 2 
230 230 2 
231 231 2 
232 232 2 
233 233 2 
234 234 2 
235 235 2 
236 236 2 
237 237 2 
238 238 2 
239 239 2 
240 240 2 
241 241 2 
242 242 2 
243 243 2 
244 244 2 
245 245 2 
246 246 2 
247 247 2 
248 248 2 
249 249 2 
250 250 2 
251 251 2 
252 252 2 
253 253 2 
254 254 2 
255 255 2 
256 256 2 
257 257 2 
258 258 2 
259 259 2 
260 260 2 
261 261 2 
262 262 2 
263 263 2 
264 264 2 
265 265 2 
266 266 2 
267 267 2 
268 268 2 
269 269 2 
270 270 2 
271 271 2 
272 272 2 
273 273 2 
274 274 2 
275 275 2 
276 276 2 
277 277 2 
278 278 2 
279 279 2 
280 280 2 
281 281 2 
282 282 2 
283 283 2 
284 284 2 
285 285 2 
286 286 2 
287 287 2 
288 288 2 
289 289 2 
290 290 2 
291 291 2 
292 292 2 
293 293 2 
294 294 2 
295 200 89 
296 201 89 
297 202 89 
298 203 89 
299 204 89 
300 205 89 
301 206 89 
302 207 89 
303 208 89 
304 209 89 
305 210 89 
306 211 89 
307 212 89 
308 213 89 
309 214 89 
310 215 89 
311 216 89 
312 217 89 
313 218 89 
314 219 89 
315 220 89 
316 221 89 
317 222 89 
318 223 89 
319 224 89 
320 225 89 
321 226 89 
322 227 89 
323 228 89 
324 229 89 
325 230 89 
326 231 89 
327 232 89 
328 233 89 
329 234 89 
330 235 89 
331 236 89 
332 237 89 
333 238 89 
334 239 89 
335 240 89 
336 241 89 
337 242 89 
338 243 89 
339 244 89 
340 245 89 
341 246 89 
342 247 89 
343 248 89 
344 249 89 
345 250 89 
346 251 89 
347 252 89 
348 253 89 
349 254 89 
350 255 89 
351 256 89 
352 257 89 
353 258 89 
354 259 89 
355 260 89 
356 261 89 
357 262 89 
358 263 89 
359 264 89 
360 265 89 
361 266 89 
362 267 89 
363 268 89 
364 269 89 
365 270 89 
366 271 89 
367 272 89 
368 273 89 
369 274 89 
370 275 89 
371 276 89 
372 277 89 
373 278 89 
374 279 89 
375 280 89 
376 281 89 
377 282 89 
378 283 89 
379 284 89 
380 285 89 
381 286 89 
382 287 89 
383 288 89 
384 289 89 
385 290 89 
386 291 89 
387 292 89 
388 293 89 
389 294 89 
- Examples 390-399 have been intentionally excluded.
- If one were to follow the procedures outlined in Examples 28 or 29 except using the compounds from the Preparative Examples as indicated in the Table below, one would obtain the indicated Product.
-
Preparative Preparative Example Example Example Product 400 300 2 
401 301 2 
402 302 2 
403 303 2 
404 304 2 
405 305 2 
406 306 2 
407 307 2 
408 308 2 
409 309 2 
410 310 2 
411 311 2 
412 312 2 
413 313 2 
414 314 2 
415 315 2 
416 316 2 
417 317 2 
418 318 2 
419 319 2 
420 320 2 
421 321 2 
422 322 2 
423 323 2 
424 324 2 
425 325 2 
426 326 2 
427 327 2 
428 328 2 
429 329 2 
430 330 2 
431 331 2 
432 332 2 
433 333 2 
434 334 2 
435 335 2 
436 400 2 
437 401 2 
438 402 2 
439 403 2 
440 404 2 
441 405 2 
442 406 2 
443 407 2 
444 408 2 
445 409 2 
446 410 2 
447 411 2 
448 412 2 
449 413 2 
450 414 2 
451 415 2 
452 416 2 
453 417 2 
454 418 2 
455 419 2 
456 420 2 
457 421 2 
458 422 2 
459 423 2 
460 424 2 
461 425 2 
462 426 2 
463 427 2 
464 428 2 
465 429 2 
466 430 2 
467 431 2 
468 432 2 
469 433 2 
470 434 2 
471 500 2 
472 501 2 
473 502 2 
474 503 2 
475 504 2 
476 505 2 
477 506 2 
478 507 2 
479 508 2 
480 509 2 
481 510 2 
482 511 2 
483 512 2 
484 513 2 
485 514 2 
486 515 2 
487 516 2 
488 517 2 
489 518 2 
490 519 2 
491 520 2 
492 521 2 
493 522 2 
494 523 2 
495 524 2 
496 525 2 
497 526 2 
498 527 2 
499 528 2 
500 529 2 
501 530 2 
502 531 2 
503 532 2 
504 533 2 
505 534 2 
506 535 2 
507 600 2 
508 601 2 
509 602 2 
510 603 2 
511 604 2 
512 605 2 
513 606 2 
514 607 2 
515 608 2 
516 609 2 
517 610 2 
518 611 2 
519 612 2 
520 613 2 
521 614 2 
522 615 2 
523 616 2 
524 617 2 
525 618 2 
526 619 2 
527 620 2 
528 621 2 
529 622 2 
530 623 2 
531 624 2 
532 625 2 
533 626 2 
534 627 2 
535 628 2 
536 629 2 
537 630 2 
538 631 2 
539 632 2 
540 633 2 
541 634 2 
542 635 2 
543 680 2 
544 681 2 
545 682 2 
546 683 2 
547 684 2 
548 685 2 
549 686 2 
550 687 2 
551 700 2 
552 701 2 
553 702 2 
554 703 2 
555 704 2 
556 705 2 
557 706 2 
558 707 2 
559 708 2 
560 709 2 
561 710 2 
562 711 2 
563 712 2 
564 713 2 
565 714 2 
566 715 2 
567 716 2 
568 717 2 
569 718 2 
570 719 2 
571 720 2 
572 721 2 
573 722 2 
574 723 2 
575 724 2 
576 725 2 
577 726 2 
578 727 2 
579 728 2 
580 729 2 
581 730 2 
582 731 2 
583 732 2 
584 733 2 
585 734 2 
586 735 2 
587 780 2 
588 781 2 
589 782 2 
590 783 2 
591 784 2 
592 785 2 
593 786 2 
594 787 2 
595 788 2 
- Examples 596-599 have been intentionally excluded.
- If one were to follow the procedures outlined in Examples 28 or 29 except using the compounds from the Preparative Examples as indicated in the Table below, one would obtain the indicated Product.
-
Preparative Preparative Example Example Example Product 600 336 89 
601 337 89 
602 338 89 
603 339 89 
604 340 89 
605 341 89 
606 342 89 
607 343 89 
608 344 89 
609 345 89 
610 346 89 
611 347 89 
612 348 89 
613 349 89 
614 350 89 
615 351 89 
616 352 89 
617 353 89 
618 354 89 
619 355 89 
620 356 89 
621 357 89 
622 358 89 
623 359 89 
624 360 89 
625 361 89 
626 362 89 
627 363 89 
628 364 89 
629 365 89 
630 366 89 
631 367 89 
632 368 89 
633 369 89 
634 370 89 
635 371 89 
636 435 89 
637 436 89 
638 437 89 
639 438 89 
640 439 89 
641 440 89 
642 441 89 
643 442 89 
644 443 89 
645 444 89 
646 445 89 
647 446 89 
648 447 89 
649 448 89 
650 449 89 
651 450 89 
652 451 89 
653 452 89 
654 453 89 
655 454 89 
656 455 89 
657 456 89 
658 457 89 
659 458 89 
660 459 89 
661 460 89 
662 461 89 
663 462 89 
664 463 89 
665 464 89 
666 465 89 
667 466 89 
668 467 89 
669 468 89 
670 469 89 
671 536 89 
672 537 89 
673 538 89 
674 539 89 
675 540 89 
676 541 89 
677 542 89 
678 543 89 
679 544 89 
680 545 89 
681 546 89 
682 547 89 
683 548 89 
684 549 89 
685 550 89 
686 551 89 
687 552 89 
688 553 89 
689 554 89 
690 555 89 
691 556 89 
692 557 89 
693 558 89 
694 559 89 
695 560 89 
696 561 89 
697 562 89 
698 563 89 
699 564 89 
700 565 89 
701 566 89 
702 567 89 
703 568 89 
704 569 89 
705 570 89 
706 571 89 
707 636 89 
708 637 89 
709 638 89 
710 639 89 
711 640 89 
712 641 89 
713 642 89 
714 643 89 
715 644 89 
716 645 89 
717 646 89 
718 647 89 
719 648 89 
720 649 89 
721 650 89 
722 651 89 
723 652 89 
724 653 89 
725 654 89 
726 655 89 
727 656 89 
728 657 89 
729 658 89 
730 659 89 
731 660 89 
732 661 89 
733 662 89 
734 663 89 
735 664 89 
736 665 89 
737 666 89 
738 667 89 
739 668 89 
740 669 89 
741 670 89 
742 671 89 
743 688 89 
744 689 89 
745 690 89 
746 691 89 
747 692 89 
748 693 89 
749 694 89 
750 695 89 
751 736 89 
752 737 89 
753 738 89 
754 739 89 
755 740 89 
756 741 89 
757 742 89 
758 743 89 
759 744 89 
760 745 89 
761 746 89 
762 747 89 
763 748 89 
764 749 89 
765 750 89 
766 751 89 
767 752 89 
768 753 89 
769 754 89 
770 755 89 
771 756 89 
772 757 89 
773 758 89 
774 759 89 
775 760 89 
776 761 89 
777 762 89 
778 763 89 
779 764 89 
780 765 89 
781 766 89 
782 767 89 
783 768 89 
784 769 89 
785 770 89 
786 771 89 
787 789 89 
788 790 89 
789 791 89 
790 792 89 
791 793 89 
792 794 89 
793 795 89 
794 796 89 
795 797 89 
- Examples 796-799 have been intentionally excluded.
- If one were to follow a similar procedure as that described in Examples 27 or 28, and treat the title compounds from the Preparative Examples in the table below as described in Preparative Example 69 and 71, except using the amines as indicated in the Table below, one would obtain the desired product.
-
Preparative Preparative Example Example Example Amine Product 800 61 Step B 2 NH3 
801 62 2 NH3 
802 65 2 NH3 
803 61 Step B 2 NH3 
804 62 2 NH3 
805 65 2 NH3 
806 61 Step B 2 CH3NH2 
807 62 2 CH3NH2 
808 65 2 CH3NH2 
809 61 Step B 2 CH3NH2 
810 62 2 CH3NH2 
811 65 2 CH3NH2 
812 61 Step B 2 (CH3)2NH 
813 65 2 (CH3)2NH 
814 61 Step B 2 (CH3)2NH 
815 65 2 (CH3)2NH 
816 61 Step B 89 NH3 
817 62 89 NH3 
818 65 89 NH3 
819 61 Step B 89 NH3 
820 62 89 NH3 
821 65 89 NH3 
822 61 Step B 89 CH3NH2 
823 62 89 CH3NH2 
824 65 89 CH3NH2 
825 61 Step B 89 CH3NH2 
826 62 89 CH3NH2 
827 65 89 CH3NH2 
828 61 Step B 89 (CH3)2NH 
829 62 89 (CH3)2NH 
830 65 89 (CH3)2NH 
831 61 Step B 89 (CH3)2NH 
832 62 89 (CH3)2NH 
833 65 89 (CH3)2NH 
- Examples 834-999 have been intentionally excluded.
- If one were to follow the procedures outlined in Examples 28 or 29 except using the compounds from the Preparative Examples as indicated in the Table below, one would obtain the indicated Product.
-
Preparative Preparative Example Example Example Product 1000 801 2 
1001 804 2 
1002 805 2 
1003 800 2 
1004 802 2 
1005 803 2 
1006 801 89 
1007 804 89 
1008 805 89 
1009 800 89 
1010 802 89 
1011 803 89 
1012 810 2 
1013 812 2 
1014 811 2 
1015 810 89 
1016 812 89 
1017 811 89 
1018 831 2 
1019 832 2 
1020 833 2 
1021 834 2 
1022 835 2 
1023 836 2 
1024 837 2 
1025 838 2 
1026 839 2 
1027 851 2 
1028 852 2 
1029 853 2 
1030 854 2 
1031 855 2 
1032 856 2 
1033 857 2 
1034 858 2 
1035 859 2 
1036 901 2 
1037 902 2 
1038 903 2 
1039 904 2 
1040 905 2 
1041 906 2 
1042 907 2 
1043 908 2 
1044 909 2 
1045 921 2 
1046 922 2 
1047 923 2 
1048 924 2 
1049 925 2 
1050 926 2 
1051 927 2 
1052 928 2 
1053 929 2 
1054 831 89 
1055 832 89 
1056 833 89 
1057 834 89 
1058 835 89 
1059 836 89 
1060 837 89 
1061 838 89 
1062 839 89 
1063 851 89 
1064 852 89 
1065 853 89 
1066 854 89 
1067 855 89 
1068 856 89 
1069 857 89 
1070 858 89 
1071 859 89 
1072 901 89 
1073 902 89 
1074 903 89 
1075 904 89 
1076 905 89 
1077 906 89 
1078 907 89 
1079 908 89 
1080 909 89 
1081 921 89 
1082 922 89 
1083 923 89 
1084 924 89 
1085 925 89 
1086 926 89 
1087 927 89 
1088 928 89 
1089 929 89 
1090 1301 2 
1091 1302 2 
1092 1303 2 
1093 1304 2 
1094 1305 2 
1095 1306 2 
1096 1307 2 
1097 1308 2 
1098 1309 2 
1099 1351 2 
1100 1352 2 
1101 1353 2 
1102 1354 2 
1103 1355 2 
1104 1356 2 
1105 1357 2 
1106 1358 2 
1107 1359 2 
1108 1401 2 
1109 1402 2 
1110 1403 2 
1111 1404 2 
1112 1405 2 
1113 1406 2 
1114 1407 2 
1115 1408 2 
1116 1409 2 
1117 1301 89 
1118 1302 89 
1119 1303 89 
1120 1304 89 
1121 1305 89 
1122 1306 89 
1123 1307 89 
1124 1308 89 
1125 1309 89 
1126 1351 89 
1127 1352 89 
1128 1353 89 
1129 1354 89 
1130 1355 89 
1131 1356 89 
1132 1357 89 
1133 1358 89 
1134 1359 89 
1135 1401 89 
1136 1402 89 
1137 1403 89 
1138 1404 89 
1139 1405 89 
1140 1406 89 
1141 1407 89 
1142 1408 89 
1143 1409 89 
1144 1450 Step K 2 
1145 1450 Step O 2 
1146 1451 Step F 2 
1147 1451 Step J 2 
1148 1452 Step F 2 
1149 1452 Step J 2 
1150 1453 Step J 2 
1151 1453 Step M 2 
1152 1454 Step I 2 
1153 1454 Step L 2 
1154 1500 2 
1155 1501 2 
1156 1502 2 
1157 1450 Step K 89 
1158 1450 Step O 89 
1159 1451 Step F 89 
1160 1451 Step J 89 
1161 1452 Step F 89 
1162 1452 Step J 89 
1163 1453 Step J 89 
1164 1453 Step M 89 
1165 1454 Step I 89 
1166 1454 Step L 89 
1167 1500 89 
1168 1501 89 
- Examples 1169-1499 have been intentionally excluded.
- If one were to follow a similar procedure as that described in Preparative Example 48, except using the compounds from the Preparative Examples as indicated in the Table below, one would obtain the desired amine product.
-
Compound Preparative Example Example Product 1500 1000 
1501 1001 
1502 1002 
1503 1003 
1504 1004 
1505 1005 
1506 1006 
1507 1007 
1508 1008 
1509 1009 
1510 1010 
1511 1011 
1512 1012 
1513 1013 
1514 1014 
1515 1015 
1516 1016 
1517 1017 
1518 1018 
1519 1019 
1520 1020 
1521 1021 
1522 1022 
1523 1023 
1524 1024 
1525 1025 
1526 1026 
1527 1027 
1528 1028 
1529 1029 
1530 1030 
1531 1031 
1532 1032 
1533 1033 
1534 1034 
1535 1035 
1536 1036 
1537 1037 
1538 1038 
1539 1039 
1540 1040 
1541 1041 
1542 1042 
1543 1043 
1544 1044 
1545 1045 
1546 1046 
1547 1047 
1548 1048 
1549 1049 
1550 1050 
1551 1051 
1552 1052 
1553 1053 
1554 1054 
1555 1055 
1556 1056 
1557 1057 
1558 1058 
1559 1059 
1560 1060 
1561 1061 
1562 1062 
1563 1063 
1564 1064 
1565 1065 
1566 1066 
1567 1067 
1568 1068 
1569 1069 
1570 1070 
1571 1071 
1572 1072 
1573 1073 
1574 1074 
1575 1075 
1576 1076 
1577 1077 
1578 1078 
1579 1079 
1580 1080 
1581 1081 
1582 1082 
1583 1083 
1584 1084 
1585 1085 
1586 1086 
1587 1087 
1588 1088 
1589 1089 
1590 1090 
1591 1091 
1592 1092 
1593 1093 
1594 1094 
1595 1095 
1596 1096 
1597 1097 
1598 1098 
1599 1099 
1600 1100 
1601 1101 
1602 1102 
1603 1103 
1604 1104 
1605 1105 
1606 1106 
1607 1107 
1608 1108 
1609 1109 
1610 1110 
1611 1111 
1612 1112 
1613 1113 
1614 1114 
1615 1115 
1616 1116 
1617 1117 
1618 1118 
1619 1119 
1620 1120 
1621 1121 
1622 1122 
1623 1123 
1624 1124 
1625 1125 
1626 1126 
1627 1127 
1628 1128 
1629 1129 
1630 1130 
1631 1131 
1632 1132 
1633 1133 
1634 1134 
1635 1135 
1636 1136 
1637 1137 
1638 1138 
1639 1139 
1640 1140 
1641 1141 
1642 1142 
1643 1143 
1644 1144 
1645 1145 
1646 1146 
1647 1147 
1648 1148 
1649 1149 
1650 1150 
1651 1151 
1652 1152 
1653 1153 
1654 1154 
1655 1155 
1656 1156 
1657 1157 
1658 1158 
1659 1159 
1660 1160 
1661 1161 
1662 1162 
1663 1163 
1664 1164 
1665 1165 
1666 1166 
1667 1167 
1668 1168 
1669 1169 
1670 1170 
1671 1171 
1672 1172 
1673 1173 
1674 1174 
1675 1175 
1676 1176 
1677 1177 
1678 1178 
1679 1179 
1680 1180 
1681 1181 
1682 1182 
1683 1183 
1684 1184 
1685 1185 
1686 1186 
1687 1187 
1688 1188 
1689 1189 
1690 1190 
1691 1191 
1692 1192 
1693 1193 
1694 1194 
1695 1195 
1696 1196 
1697 1197 
1698 1198 
1699 1199 
1700 1200 
1701 1201 
1702 1202 
1703 1203 
1704 1204 
1705 1205 
1706 1206 
1707 1207 
1708 1208 
1709 1209 
- Examples 1710-1799 have been intentionally excluded.
-

- If one were to treat allyl bromide with 1.0 eq. catechol borane, heat the mixture at 100° C., distillate at reduced pressure, treat the intermediate with 2.0 eq. pinacol in THF at 0° C. and room temperature, evaporate, dissolve in hexane and remove pinacol by filtration, distillate at reduced pressure, one would obtain the title compound.
- If one were to dissolve methylene chloride (1.0 eq.) in THF and then slowly add 1.54 N nBuLi in hexane (1.1 eq.) at −100° C., and would then add the title compound from Step A above (1.0 equ.), dissolved in THF, cooled to the freezing point of the solution, to the reaction mixture, followed by adding a suspension of zinc chloride (0.55 eq.) in THF, cooled to 0° C., in several portions to the reaction mixture, subsequently allowing the mixture to slowly warm to room temperature and to stir overnight, then, after evaporation of the solvent and redissolving the residue in hexane and washing with water, discarding insoluble material, drying (MgSO4) and evaporation of the solvent, followed by distillation, one would obtain the title compound.
- If one were to treat a fresh prepared LiHMDS solution in THF with 1 eq. of the title compound from Step B at −78° C., one would obtain after stirring overnight at rt, filtering of the precipitant and distillation of the filtrate the title compound as an oil.
- If one were to treat the title compound from Step C above with 3 eq. of a 4 M HCl solution in dioxane at −78° C., one would obtain after stirring for 1 hour at rt and evaporation of the solvent the title compound as a HCl salt.
- If one were to treat the title compound from Step D above with bromo acetyl bromide as described in Example 1, one would obtain the title compound.
- If one were to treat the title compound from Step E above with the title compound from Preparative Example 15 as described in Example 1, one would obtain the title compound.
- If one were to treat the title compound from Step F above with 6.0 eq. diethanolamine in THF at room temperature, add Et2O to the mixture, separate the precipitate by filtration, dissolve the solid in an appropriate solvent and add Dowex AG 50-X8, filtrate and evaporate the filtrate, one would obtain the title compound.
- Examples 1801-1849 have been intentionally excluded.
-

- If one were to treat the title compound from Preparative Example 92 with the title compound from Example 1800, Step D, as described in Preparative Example 93, one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Example 48, one would obtain the title compound. If one were to use a reverse phase HPLC separation (5-pm Nucleosil C18 HPLC column, acetonitrile:H2O: 0.1% TFA), one could obtain the individual diastereomers.
- If one were to treat the title compound from Step B above with 6.0 eq. diethanolamine in THF at room temperature, add Et2O to the mixture, separate the precipitate by filtration, dissolve the solid in an appropriate solvent and add Dowex AG 50-X8, filtrate and evaporate the filtrate, one would obtain the title compound.
- Examples 1851-1899 have been intentionally excluded.
-

- If one were to treat the title compound from Preparative Example 130 with bromoacetyl bromide as described in Preparative Example 1, one would obtain the title compound.
- If one were to treat the title compound from Step A above with the title compound from Preparative Example 15 as described in Example 1, one would obtain the title compound. Examples 1901-1949 have been intentionally excluded.
-

- If one were to treat title compound from Preparative Example 130 with the title compound from Preparative Example 92 as described in Preparative Example 93, one would obtain the title compound.
- If one were to treat the title compound from Step A above as described in Example 48, one would obtain the title compound.
- The inhibitory activity of compounds against DPP-IV can be determined by in vitro assay systems, which are themselves well established in the art. The assay results given in Table 5 were obtained according to the following method, employing a modified version of the assay described by Leiting et al., in an article entitled “Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII” in Biochem. J. Vol. 371, pages 525-532 (2003):
- DPP-IV activity was determined fluorometrically with Gly-Pro-AMC (where AMC stands for 7-amido-4-methylcoumarin, Bachem AG, Switzerland) as substrate. The reaction mixture contained 10 μl of 1 ng/μl DPP-IV (R&D Systems GmbH, Germany) and 80 μl of 25 mM Tris/HCl buffer, pH 8.0. Compounds were supplied as DMSO stock solutions and diluted in assay buffer to a maximal DMSO concentration of 1% in the assay. Prior to start of the reaction, the mixture was incubated for 30 min at room temperature. The reaction was started by addition of 10 μl of 100 μM substrate solution.
- The fluorescence intensity was measured at excitation and emission wavelengths of 355 and 460 nm, respectively, in a FluoStar Galaxy Multiwell Plate (BMG Labtech, Germany). Fluorescence was determined 3 and 4 minutes after start of reaction and increase in fluorescence was used for determination of enzymatic activity. IC(50) values of tested compounds were determined via plotting enzymatic activity versus concentration of test compound and determining the concentration of test compound which yields a 50% inhibition of enzymatic activity.
- K(i) values were calculated using the Michaelis-Menten equation for competitive inhibition:
-
IC(50)=K(i)(1+[S]/Km) - As set forth in Table A, K(i) for each compound corresponds to A is K(i)<6 nM, B is K(i) 6-50 nM, C is K(i) from 51-500 nM and D is K(i) from 0.5-30 μM.
-
TABLE A Activity Data for Inhibition of DPP-IV Activity Example (K(i)) 1 C 2 D 3 D 4 D 5 D 6 C 7 C 8 C 9 C 10 C 11 C 12 C 13 C 14 D 15 D 16 C 17 B 18 A 19 B 20 C 21 C 22 A 23 B 24 A 25 B 26 C 27 A 28 A 29 A 30 A 31 B 32 A 33 A 34 A 35 A 36 B 37 B 38 B 39 B 40 D 41 B 42 C 43 A 44 A 45 B 46 D 47 A 48 A 49 A 50 B 51 A 52 A 53 A 54 A 55 A 56 A 57 A 58 A 59 A 60 A 61 A 62 A 63 A 64 A 65 B 66 B 67 A 68 B 69 B 70 B 71 B 72 A 73 B 74 C 75 C 76 B 77 A 78 B - All patents, patent applications, and published references cited herein are hereby incorporated by reference in their entirety. While this invention has been particularly shown and described with references to preferred embodiments thereof, it will be understood by those skilled in the art that various changes in form and details may be made therein without departing from the scope of the invention encompassed by the appended claims.
Claims (24)
1. A compound of formula (I):
A-B-D (I)
A-B-D (I)
or a pharmaceutically acceptable salt thereof,
wherein A is:

B is:

D is:

Y is divalent and is: a bond, CR4R5, O, NR4, S, S═O, S(═O)2, C(═O), (C═O)N(R4), S(═O)2N(R4), C═N—OR4, —C(R4R5)C(R4R5)—, —C(R4R5) C(R4R5)C(R4R5)—, —C(R4R5)C(R4R5)C(R4R5)C(R4R5)—, —C(R4)═C(R5)—, —C(R4R5)NR4—, —C(R4R5)O—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, or NS(═O)2R4;
R1 and R2 are independently: hydrogen, —F, —Cl, —CONR4R5, or —CO2R4;
R3 is CONR4R5, tetrazolyl or oxadiazolonyl;
Ra is hydrogen, CN, NO2, alkyl, haloalkyl, S(O)tNR4R5, S(O)tR4, C(O)OR4, C(O)R4, or C(O)NR4R5;
each occurrence of R4 and R5 are each independently: hydrogen or alkyl, or R4 and R5 when taken together with a nitrogen to which they are attached form a 3- to 8-membered ring containing carbon atoms and may optionally contain a heteroatom selected from O, S, or NR50;
R50 is, in each occurrence, R20, CN, NO2, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(═NRa)NR20R21, C(═NR20)NR21Ra, C(═NOR20)R21 or C(O)NR20R21;
each occurrence of R20 and R21 are each independently: hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl or aminoalkyl;
each occurrence of R7 and R8 are each independently: halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C1-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl, wherein alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl;
R9 is H or C1-6 alkyl;
R10 is halogen, CF3, COR4, OR4, NR4R5, NO2, CN, SO2OR4, CO2R4, CONR4R5, CO2H, SO2NR4R5, S(O)tR4, SO3H, OC(O)R4, OC(O)NR4R5, NR4C(O)R5, NR4CO2R5, (C0-C6)-alkyl-C(═NRa)NHR4, (C0-C6)-alkyl-C(═NR4)NHRa, (C0-C6)-alkyl-NR4C(═NR4)NR4R5, (C0-C6)-alkyl-C(O)OR4, (C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)—NH—CN, O—(C0-C6)-alkyl-C(O)NR4R5, S(O)t—(C0-C6)-alkyl-C(O)OR4, S(O)t—(C0-C6)-alkyl-C(O)NR4R5, (C0-C6)-alkyl-C(O)NR4—(C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4R5, (C0-C6)-alkyl-NR4—C(O)R5, (C0-C6)-alkyl-NR4—C(O)OR4, (C0-C6)-alkyl-NR4—C(O)—NR4R5, (C0-C6)-alkyl-NR4—SO2NR4R5, (C0-C6)-alkyl-NR4—SO2R4, hydrogen, B(OH)2, alkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, fluoroalkyl, heterocycloalkylalkyl, haloalkyl, alkenyl, alkynyl, aryl, heteroaryl, arylalkyl, heteroarylalkyl, alkoxyalkyl or aminoalkyl;
Qb is CH or N;
U is —C(O)—, —C(═NR4)—, —(CR4R5—)p, NR50, S(═O)2, C(═O), (C═O)N(R4), N(R4)(C═O), S(═O)2N(R4), N(R4)S(═O)2, C═N—OR4, —C(R4)═C(R5)—, —C(R4R5)pNR50—, N(R50)C(R4R5)p—, —O—C(R4R5)—, —C(R4R5)S(═O)t—, —(C═O)O—, —(C═NRa)N(R4)—, —(C═NRa)—, N(C═O)NR4NR5, N(C═O)R4, N(C═O)OR4, NS(═O)2NR4NR5, or NS(═O)2R4;
W is —CH2—, —S—, —CHF— or —CF2—;
Z is C;
p is 0 to 6; and
t is 0, 1, or 2.
2. A compound of claim 1 , or a pharmaceutically acceptable salt thereof, wherein:
Qb is CH;
U is (—CH2—)p;
p is 1;
R7 and R8 are each independently H or alkyl; and
R9 is H.
3. A compound of claim 1 , or a pharmaceutically acceptable salt thereof, wherein:
D is:

4. A compound of claim 1 , or a pharmaceutically acceptable salt, thereof, wherein:
Y is —CH2—CH2—.
5. A compound of claim 1 , or a pharmaceutically acceptable salt thereof, wherein:
Y is —CH2—CH2—;
D is

Qb is CH;
U is (—CH2—)p;
p is 1; and
R9 is H.
6. A compound of claim 1 , or a pharmaceutically acceptable salt, thereof, wherein:
A is:

7. A compound of claim 1 , or a pharmaceutically acceptable salt, thereof, wherein:
A is:

Y is —CH2—CH2—;
D is

Qb is CH.
8. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
9. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
10. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
11. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
12. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
13. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
14. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
15. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
16. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
17. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
18. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
19. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
20. A compound according to the following formula:

or a pharmaceutically acceptable salt thereof.
21. A pharmaceutical composition comprising a compound in accordance with claim 1 , or a pharmaceutically acceptable salt thereof.
22. A method of treating type-2 diabetes comprising administering to a patient in need thereof an effective amount of a compound in accordance with claim 1 , or a pharmaceutically acceptable salt thereof.
23. A pharmaceutical composition comprising a compound in accordance with claim 7 , or a pharmaceutically acceptable salt thereof.
24. A method of treating type-2 diabetes comprising administering to a patient in need thereof an effective amount of a compound in accordance with claim 7 , or a pharmaceutically acceptable salt thereof.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/492,254 US20100009961A1 (en) | 2005-04-22 | 2009-06-26 | Dipeptidyl peptidase-iv inhibitors |
| US13/006,363 US8076330B2 (en) | 2005-04-22 | 2011-01-13 | Dipeptidyl peptidase-IV inhibitors |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US67415105P | 2005-04-22 | 2005-04-22 | |
| US11/409,481 US7553861B2 (en) | 2005-04-22 | 2006-04-21 | Dipeptidyl peptidase-IV inhibitors |
| US12/492,254 US20100009961A1 (en) | 2005-04-22 | 2009-06-26 | Dipeptidyl peptidase-iv inhibitors |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/409,481 Continuation US7553861B2 (en) | 2005-04-22 | 2006-04-21 | Dipeptidyl peptidase-IV inhibitors |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/006,363 Continuation US8076330B2 (en) | 2005-04-22 | 2011-01-13 | Dipeptidyl peptidase-IV inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20100009961A1 true US20100009961A1 (en) | 2010-01-14 |
Family
ID=36694223
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/409,481 Expired - Fee Related US7553861B2 (en) | 2005-04-22 | 2006-04-21 | Dipeptidyl peptidase-IV inhibitors |
| US12/492,254 Abandoned US20100009961A1 (en) | 2005-04-22 | 2009-06-26 | Dipeptidyl peptidase-iv inhibitors |
| US13/006,363 Expired - Fee Related US8076330B2 (en) | 2005-04-22 | 2011-01-13 | Dipeptidyl peptidase-IV inhibitors |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/409,481 Expired - Fee Related US7553861B2 (en) | 2005-04-22 | 2006-04-21 | Dipeptidyl peptidase-IV inhibitors |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/006,363 Expired - Fee Related US8076330B2 (en) | 2005-04-22 | 2011-01-13 | Dipeptidyl peptidase-IV inhibitors |
Country Status (19)
| Country | Link |
|---|---|
| US (3) | US7553861B2 (en) |
| EP (1) | EP1888562B1 (en) |
| JP (2) | JP4568361B2 (en) |
| KR (1) | KR20080000665A (en) |
| CN (1) | CN101277949A (en) |
| AP (1) | AP2007004234A0 (en) |
| AU (2) | AU2006239929B2 (en) |
| BR (1) | BRPI0608469A2 (en) |
| CA (1) | CA2599419A1 (en) |
| CR (1) | CR9462A (en) |
| EA (1) | EA200702208A1 (en) |
| ES (1) | ES2477868T3 (en) |
| IL (1) | IL186607A0 (en) |
| MA (1) | MA29387B1 (en) |
| MX (1) | MX2007011453A (en) |
| NO (1) | NO20075973L (en) |
| TN (1) | TNSN07244A1 (en) |
| WO (1) | WO2006116157A2 (en) |
| ZA (1) | ZA200708179B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122025A2 (en) | 2011-03-04 | 2012-09-13 | Intrexon Corporation | Vectors conditionally expressing protein |
| WO2018049014A1 (en) * | 2016-09-07 | 2018-03-15 | Trustees Of Tufts College | Dash inhibitors, and uses related thereto |
| US11559537B2 (en) | 2017-04-07 | 2023-01-24 | Trustees Of Tufts College | Combination therapies using caspase-1 dependent anticancer agents and PGE2 antagonists |
Families Citing this family (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7407955B2 (en) | 2002-08-21 | 2008-08-05 | Boehringer Ingelheim Pharma Gmbh & Co., Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| DE102004054054A1 (en) | 2004-11-05 | 2006-05-11 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Process for preparing chiral 8- (3-amino-piperidin-1-yl) -xanthines |
| BRPI0608469A2 (en) * | 2005-04-22 | 2010-01-05 | Alantos Pharmaceuticals Holding Inc | dipeptidyl peptidase-iv inhibitors |
| AU2006306420A1 (en) * | 2005-10-25 | 2007-05-03 | Merck Sharp & Dohme Corp. | Combination of a dipeptidyl peptidase-4 inhibitor and an anti-hypertensive agent for the treatment of diabetes and hypertension |
| EP1962827A4 (en) * | 2005-12-16 | 2011-02-16 | Merck Sharp & Dohme | Pharmaceutical compositions of combinations of dipeptidyl peptidase-4 inhibitors with metformin |
| PE20110235A1 (en) | 2006-05-04 | 2011-04-14 | Boehringer Ingelheim Int | PHARMACEUTICAL COMBINATIONS INCLUDING LINAGLIPTIN AND METMORPHINE |
| EP1852108A1 (en) | 2006-05-04 | 2007-11-07 | Boehringer Ingelheim Pharma GmbH & Co.KG | DPP IV inhibitor formulations |
| NZ600394A (en) | 2006-05-04 | 2014-04-30 | Boehringer Ingelheim Int | Polymorphs of a dpp-iv enzyme inhibitor |
| US20090269313A1 (en) * | 2006-07-19 | 2009-10-29 | Diakine Therapeutics, Inc. | Encapsulation system |
| AU2008210988B2 (en) * | 2007-02-01 | 2012-09-06 | Allocure, Inc. | Potentiation of stem cell homing and treatment of organ dysfunction or organ failure |
| KR20120030570A (en) | 2007-04-03 | 2012-03-28 | 미쓰비시 타나베 파마 코퍼레이션 | Combined use of dipeptidyl peptidase iv inhibitor compound and sweetener |
| US20110112069A1 (en) * | 2007-08-17 | 2011-05-12 | Boehringer Ingelheim International Gmbh | Purin derivatives for use in the treatment of fab-related diseases |
| WO2009037719A1 (en) | 2007-09-21 | 2009-03-26 | Lupin Limited | Novel compounds as dipeptidyl peptidase iv (dpp iv) inhibitors |
| CL2008003653A1 (en) | 2008-01-17 | 2010-03-05 | Mitsubishi Tanabe Pharma Corp | Use of a glucopyranosyl-derived sglt inhibitor and a selected dppiv inhibitor to treat diabetes; and pharmaceutical composition. |
| WO2009099172A1 (en) * | 2008-02-07 | 2009-08-13 | Takeda Pharmaceutical Company Limited | Pharmaceutical product |
| PE20091730A1 (en) | 2008-04-03 | 2009-12-10 | Boehringer Ingelheim Int | FORMULATIONS INVOLVING A DPP4 INHIBITOR |
| US20090264476A1 (en) * | 2008-04-18 | 2009-10-22 | Mckelvey Craig | CB-1 receptor modulator formulations |
| UY32030A (en) | 2008-08-06 | 2010-03-26 | Boehringer Ingelheim Int | "TREATMENT FOR DIABETES IN INAPPROPRIATE PATIENTS FOR THERAPY WITH METFORMIN" |
| BRPI0916997A2 (en) | 2008-08-06 | 2020-12-15 | Boehringer Ingelheim International Gmbh | DPP-4 INHIBITOR AND ITS USE |
| NZ604091A (en) | 2008-08-15 | 2014-08-29 | Boehringer Ingelheim Int | Purin derivatives for use in the treatment of fab-related diseases |
| US20200155558A1 (en) | 2018-11-20 | 2020-05-21 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients with insufficient glycemic control despite therapy with an oral antidiabetic drug |
| EP2382216A1 (en) | 2008-12-23 | 2011-11-02 | Boehringer Ingelheim International GmbH | Salt forms of organic compound |
| TW201036975A (en) | 2009-01-07 | 2010-10-16 | Boehringer Ingelheim Int | Treatment for diabetes in patients with inadequate glycemic control despite metformin therapy |
| AR075204A1 (en) | 2009-01-29 | 2011-03-16 | Boehringer Ingelheim Int | DPP-4 INHIBITORS AND PHARMACEUTICAL COMPOSITIONS THAT INCLUDE THEM, USEFUL TO TREAT METABOLIC DISEASES IN PEDIATRIC PATIENTS, PARTICULARLY MELLITUS DIABETES TYPE 2 |
| NZ594044A (en) | 2009-02-13 | 2014-08-29 | Boehringer Ingelheim Int | Antidiabetic medications comprising a dpp-4 inhibitor (linagliptin) optionally in combination with other antidiabetics |
| WO2010146597A1 (en) | 2009-06-18 | 2010-12-23 | Lupin Limited | 2-amino-2- [8-(dimethyl carbamoyl)- 8-aza- bicyclo [3.2.1] oct-3-yl]-exo- ethanoyl derivatives as potent dpp-iv inhibitors |
| AR077642A1 (en) | 2009-07-09 | 2011-09-14 | Arena Pharm Inc | METABOLISM MODULATORS AND THE TREATMENT OF DISORDERS RELATED TO THE SAME |
| KR20230021159A (en) | 2009-11-27 | 2023-02-13 | 베링거 인겔하임 인터내셔날 게엠베하 | Treatment of genotyped diabetic patients with dpp-iv inhibitors such as linagliptin |
| US20130109703A1 (en) | 2010-03-18 | 2013-05-02 | Boehringer Ingelheim International Gmbh | Combination of a GPR119 Agonist and the DPP-IV Inhibitor Linagliptin for Use in the Treatment of Diabetes and Related Conditions |
| WO2011127051A1 (en) | 2010-04-06 | 2011-10-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| CN102946875A (en) | 2010-05-05 | 2013-02-27 | 贝林格尔.英格海姆国际有限公司 | Combination therapy |
| WO2011161161A1 (en) | 2010-06-24 | 2011-12-29 | Boehringer Ingelheim International Gmbh | Diabetes therapy |
| EA201390421A1 (en) | 2010-09-22 | 2013-09-30 | Арена Фармасьютикалз, Инк. | GPR119 RECEPTOR MODULATORS AND TREATMENT OF RELATED DISORDERS |
| US9034883B2 (en) | 2010-11-15 | 2015-05-19 | Boehringer Ingelheim International Gmbh | Vasoprotective and cardioprotective antidiabetic therapy |
| US20140018371A1 (en) | 2011-04-01 | 2014-01-16 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| US20140066369A1 (en) | 2011-04-19 | 2014-03-06 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| US20140051714A1 (en) | 2011-04-22 | 2014-02-20 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| US20140038889A1 (en) | 2011-04-22 | 2014-02-06 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| NZ618698A (en) | 2011-07-15 | 2015-08-28 | Boehringer Ingelheim Int | Substituted quinazolines, the preparation thereof and the use thereof in pharmaceutical compositions |
| WO2013025882A2 (en) | 2011-08-16 | 2013-02-21 | Mt. Sinai School Of Medicine | Tricyclic compounds as anticancer agents |
| WO2013055910A1 (en) | 2011-10-12 | 2013-04-18 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US9555001B2 (en) | 2012-03-07 | 2017-01-31 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition and uses thereof |
| US20130303462A1 (en) | 2012-05-14 | 2013-11-14 | Boehringer Ingelheim International Gmbh | Use of a dpp-4 inhibitor in podocytes related disorders and/or nephrotic syndrome |
| WO2013174767A1 (en) | 2012-05-24 | 2013-11-28 | Boehringer Ingelheim International Gmbh | A xanthine derivative as dpp -4 inhibitor for use in modifying food intake and regulating food preference |
| WO2014074668A1 (en) | 2012-11-08 | 2014-05-15 | Arena Pharmaceuticals, Inc. | Modulators of gpr119 and the treatment of disorders related thereto |
| US9796717B2 (en) | 2013-02-19 | 2017-10-24 | Icahn School Of Medicine At Mount Sinai | Tricyclic heterocycles as anticancer agents |
| AU2014230096C1 (en) * | 2013-03-15 | 2019-08-29 | Boehringer Ingelheim International Gmbh | Use of linagliptin in cardio- and renoprotective antidiabetic therapy |
| JP6615109B2 (en) | 2014-02-28 | 2019-12-04 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Medical use of DPP-4 inhibitors |
| EP3116857B1 (en) | 2014-03-11 | 2019-02-06 | Icahn School of Medicine at Mount Sinai | Constrained tricyclic sulfonamides |
| US9937186B2 (en) | 2014-03-11 | 2018-04-10 | Icahn School Of Medicine At Mount Sinai | Sulfonamides derived from tricyclyl-2-aminocycloalkanols as anticancer agents |
| CN105330579B (en) * | 2014-08-08 | 2019-08-30 | 杭州雷索药业有限公司 | Ezetimibe and its derivative are treating or preventing the application in epilepsy |
| CN104356048B (en) * | 2014-11-02 | 2016-08-24 | 浙江医药高等专科学校 | Cyclohexane-carboxylic acid amide derivatives, Preparation Method And The Use |
| CN104356046B (en) * | 2014-11-02 | 2017-04-05 | 浙江医药高等专科学校 | Cyclohexane-carboxylic acid amide derivatives that cycloalkyl replaces and application thereof |
| KR20220070057A (en) | 2015-03-09 | 2022-05-27 | 인테크린 테라퓨틱스, 아이엔씨. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| WO2017044569A1 (en) | 2015-09-09 | 2017-03-16 | Icahn School Of Medicine At Mount Sinai | Heterocyclic constrained tricyclic sulfonamides as anti-cancer agents |
| CA2997769A1 (en) | 2015-09-09 | 2017-03-16 | Icahn School Of Medicine At Mount Sinai | Heterocyclic constrained tricyclic sulfonamides as anti-cancer agents |
| KR20180067561A (en) * | 2015-09-28 | 2018-06-20 | 제넨테크, 인크. | Therapeutic compounds and methods for their use |
| CN105343093A (en) * | 2015-12-14 | 2016-02-24 | 上海壹志医药科技有限公司 | Pharmacological purpose of ciprofloxacin |
| JP2019517542A (en) | 2016-06-10 | 2019-06-24 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Combination of linagliptin and metformin |
| US10918647B2 (en) | 2016-07-26 | 2021-02-16 | University Of Southern California | Selective bromodomain inhibition of fungal Bdf1 |
| MX2019005572A (en) * | 2016-11-15 | 2019-08-14 | Lg Chemical Ltd | Medicinal complex for treating type 2 diabetes and diabetic dyslipidemia. |
| MX2019011867A (en) | 2017-04-03 | 2020-01-09 | Coherus Biosciences Inc | Pparî³ agonist for treatment of progressive supranuclear palsy. |
| KR101983298B1 (en) * | 2018-06-11 | 2019-05-29 | 연세대학교 산학협력단 | Pharmaceutical coomposition for preventing or treating inflammasome mediated inflammatory disease |
| EP4295864A3 (en) * | 2018-09-28 | 2024-03-13 | Research Institute at Nationwide Children's Hospital | Phenylpropionic acid derivatives for modulating pathogen activity |
| WO2022003405A1 (en) * | 2020-07-03 | 2022-01-06 | Savoi Guilherme | One-pot process to obtain a pyrrolidine-2-carbonitrile intermediate compound and industrial scale telescopic process to prepare (2s)-1-[n-(3-hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile (vildagliptin) using same |
| US11814367B2 (en) | 2021-03-15 | 2023-11-14 | Maze Therapeutics, Inc. | Inhibitors of glycogen synthase 1 (GYS1) and methods of use thereof |
| CN113040090A (en) * | 2021-04-13 | 2021-06-29 | 南京医科大学 | Method for constructing animal model of autism and corresponding application |
| JP7055517B1 (en) | 2021-12-22 | 2022-04-18 | 株式会社ブンリ | Separator |
Citations (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3297709A (en) * | 1965-11-19 | 1967-01-10 | American Home Prod | Certain substituted tetrazole derivatives |
| US5391556A (en) * | 1992-02-13 | 1995-02-21 | Karl Thomae Gmbh | Benzimidazolyl derivatives, pharmaceutical compositions containing these compounds and processes for preparing them |
| US5407950A (en) * | 1990-06-07 | 1995-04-18 | Zeria Pharmaceutical Co., Ltd. | Arylalkanoylamine derivative and drug containing the same |
| US5482920A (en) * | 1992-06-17 | 1996-01-09 | Ciba-Geigy Corporation | Triazinyl compounds with herbicidal activity |
| US5492916A (en) * | 1993-12-23 | 1996-02-20 | Merck & Co., Inc. | Di- and tri-substituted piperidines, pyrrolidines and hexahydro-1H-azepines promote release of growth hormone |
| US5492920A (en) * | 1993-12-10 | 1996-02-20 | Merck & Co., Inc. | Piperidine, pyrrolidine and hexahydro-1H-azepines promote release of growth hormone |
| US5494919A (en) * | 1993-11-09 | 1996-02-27 | Merck & Co., Inc. | 2-substituted piperidines, pyrrolidines and hexahydro-1H-azepines promote release of growth hormone |
| US5514505A (en) * | 1995-05-15 | 1996-05-07 | Xerox Corporation | Method for obtaining improved image contrast in migration imaging members |
| US5618808A (en) * | 1992-07-01 | 1997-04-08 | Pfizer, Inc. | Benzothiazepine and benzoxazepine derivatives as cholecystokinin receptor antagonists |
| US5627285A (en) * | 1992-02-27 | 1997-05-06 | Bayer Aktiengesellschaft | Sulphonylbenzyl-substituted imidazolylpropenoic acid derivatives |
| US5714471A (en) * | 1995-01-06 | 1998-02-03 | Sibia Neurosciences, Inc. | Peptide and peptide analog protease inhibitors |
| US6011155A (en) * | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6017929A (en) * | 1994-02-17 | 2000-01-25 | Zeria Pharmaceutical Co., Ltd. | Cholinesterase activator |
| US6051575A (en) * | 1996-10-30 | 2000-04-18 | Schering Corporation | Piperazino derivatives as neurokinin antagonists |
| US6057314A (en) * | 1995-12-21 | 2000-05-02 | Biochem Pharma Inc. | Low molecular weight bicyclic thrombin inhibitors |
| US6180698B1 (en) * | 1997-02-28 | 2001-01-30 | Candescent Technologies Corporation | Polycarbonate-containing liquid chemical formulation and method for making polycarbonate film |
| US6191161B1 (en) * | 1995-08-17 | 2001-02-20 | Chinoin Gyogyszer Es Vegyeszeti | Prolylendopeptidase inhibitors |
| US6201132B1 (en) * | 1993-12-03 | 2001-03-13 | Ferring B.V. | Inhibitors of DP-mediated processes, compositions, and therapeutic methods thereof |
| US6207668B1 (en) * | 1996-04-03 | 2001-03-27 | Hoechst Schering | Substituted pyridines/pyrimidines, their preparation, and their use as pesticides |
| US6228885B1 (en) * | 1996-12-19 | 2001-05-08 | Isagro S.P.A. | Fungicidal compositions based on (N-phenylacetyl-N-2,6-XYLYL)methyl alaninate |
| US6337340B1 (en) * | 1998-08-14 | 2002-01-08 | Gpi Nil Holdings, Inc. | Carboxylic acids and isosteres of heterocyclic ring compounds having multiple heteroatoms for vision and memory disorders |
| US20020006899A1 (en) * | 1998-10-06 | 2002-01-17 | Pospisilik Andrew J. | Use of dipeptidyl peptidase IV effectors for lowering blood pressure in mammals |
| US20020016318A1 (en) * | 2000-07-06 | 2002-02-07 | American Home Products Corporation | Methods of treating breast disorders |
| US20020019377A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combination therapy for inhibiting sphincter incontinence |
| US20020019391A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combinations of statins and estrogenic agents |
| US20020019392A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Methods of treating neuropeptide Y-related conditions |
| US20020019433A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Methods of treating excessive intraocular pressure |
| US20020019373A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combinations of bisphosphonates and estrogenic agents |
| US20020022617A1 (en) * | 2000-07-06 | 2002-02-21 | American Home Products Corporation | Methods for increasing nitric oxide synthase activity |
| US20020022618A1 (en) * | 2000-07-06 | 2002-02-21 | American Home Products Corporation | Methods of inhibiting sphincter incontinence |
| US20020022619A1 (en) * | 2000-05-01 | 2002-02-21 | Rajagopal Bakthavatachalam | Tricyclic fused pyridine and pyrimidine derivatives as CRF antagonists |
| US20020025952A1 (en) * | 2000-07-06 | 2002-02-28 | American Home Products Corporation | Combinations of statins, estrogens and estrogenic agents |
| US20020028792A1 (en) * | 2000-07-06 | 2002-03-07 | American Home Products Corporation | Combinations of bisphosphonates, estrogens and estrogenic agents |
| US20020028800A1 (en) * | 2000-07-06 | 2002-03-07 | American Home Products Corporation | Combination therapy for prosthesis-related bone degeneration |
| US6358887B1 (en) * | 1995-02-13 | 2002-03-19 | Bayer Aktiengesellschaft | 2-Phenyl-substituted heterocyclic 1,3-ketonols as herbicides and pesticides |
| US6369051B1 (en) * | 2000-07-06 | 2002-04-09 | American Home Products Corporation | Combinations of SSRI and estrogenic agents |
| US20020045641A1 (en) * | 1998-06-03 | 2002-04-18 | Hamilton Gregory S. | Multiple heteroatom containing heterocyclic ring compounds substituted with carboxylic acids and isosteres thereof |
| US6380398B2 (en) * | 2000-01-04 | 2002-04-30 | Novo Nordisk A/S | Therapeutically active and selective heterocyclic compounds that are inhibitors of the enzyme DPP-IV |
| US6380246B1 (en) * | 1995-05-09 | 2002-04-30 | Bayer Aktiengesellschaft | Alkyl-dihalogenated phenyl-substituted ketoenols useful as pesticides and herbicides |
| US20020052514A1 (en) * | 1998-06-03 | 2002-05-02 | Hamilton Gregory S. | N-linked sulfonamides of N-heterocyclic carboxylic acids or carboxylic acid isosteres |
| US6387893B1 (en) * | 1999-09-30 | 2002-05-14 | Merck & Co., Inc. | Spirotricyclic substituted azacycloalkane derivatives and uses thereof |
| US20030008905A1 (en) * | 2000-03-31 | 2003-01-09 | Hans-Ulrich Demuth | Method for the improvement of islet signaling in diabetes mellitus and for its prevention |
| US6518299B1 (en) * | 2000-10-20 | 2003-02-11 | Biocryst Pharmaceuticals, Inc. | Substituted pyrrolidine compounds useful as neuraminidase inhibitors |
| US20030032625A1 (en) * | 2001-03-29 | 2003-02-13 | Topo Target Aps | Succinimide and maleimide derivatives and their use as topoisomerase II catalytic inhibitors |
| US6521644B1 (en) * | 1999-03-23 | 2003-02-18 | Ferring Bv | Compositions for promoting growth |
| US6537987B1 (en) * | 1996-06-20 | 2003-03-25 | Pfizer Inc. | 4,1-benzoxazepines or 4,1-benzothiazepines and their use as squalene synthetase inhibitors |
| US20030064935A1 (en) * | 2001-04-11 | 2003-04-03 | Gougoutas Jack Z. | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US20030069169A1 (en) * | 2001-03-02 | 2003-04-10 | Macor John E. | Co-administration of melanocortin receptor agonist and phosphodiesterase inhibitor for treatment of cyclic-AMP associated disorders |
| US20030073701A1 (en) * | 2001-03-31 | 2003-04-17 | Thompson Lorin A. | Succinoylamino heterocycles as inhibitors of a beta protein production |
| US20030078247A1 (en) * | 2001-05-15 | 2003-04-24 | De Nanteuil Guillaume | Alpha-amino-acid compounds |
| US6559163B2 (en) * | 2000-08-16 | 2003-05-06 | Neurogen Corporation | 2,4-substituted quinoline derivatives |
| US20030087950A1 (en) * | 2001-03-28 | 2003-05-08 | Denanteuil Guillaume | New alpha-amino acid sulphonyl compounds |
| US20030092732A1 (en) * | 2001-03-02 | 2003-05-15 | Guixue Yu | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US20030096796A1 (en) * | 1999-03-15 | 2003-05-22 | Axys Pharmaceuticals, Inc. | Novel compounds and compositions as protease inhibitors |
| US20030096857A1 (en) * | 1999-11-30 | 2003-05-22 | Evans David Michael | Novel antidiabetic agents |
| US20040002519A1 (en) * | 2002-03-22 | 2004-01-01 | Damon Robert Edson | Combination of organic compounds |
| US20040002495A1 (en) * | 2002-05-20 | 2004-01-01 | Philip Sher | Lactam glycogen phosphorylase inhibitors and method of use |
| US6673815B2 (en) * | 2001-11-06 | 2004-01-06 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| US20040009877A1 (en) * | 2000-06-13 | 2004-01-15 | Reiner Fischer | Hetaryl-substituted heterocycles |
| US20040058876A1 (en) * | 2002-09-18 | 2004-03-25 | Torsten Hoffmann | Secondary binding site of dipeptidyl peptidase IV (DP IV) |
| US20040063935A1 (en) * | 2000-10-06 | 2004-04-01 | Kosuke Yasuda | Aliphatic nitrogenous five-membered ring compounds |
| US20040072892A1 (en) * | 2000-11-10 | 2004-04-15 | Hiroshi Fukushima | Cyanopyrrolidine derivatives |
| US20040072819A1 (en) * | 1995-09-13 | 2004-04-15 | Takeda Chemical Industries, Ltd. | Benzoxazepine compounds, their production and use |
| US20040077761A1 (en) * | 1998-12-30 | 2004-04-22 | Roberto Pavez Aranguiz | Filler-containing polymer dispersion, method for its production and use |
| US20040077601A1 (en) * | 2002-07-09 | 2004-04-22 | Point Therapeutics, Inc. | Methods and compositions relating to isoleucine boroproline compounds |
| US20040077645A1 (en) * | 2001-02-24 | 2004-04-22 | Frank Himmelsbach | Xanthine derivatives,production and use thereof as medicament |
| US20040082497A1 (en) * | 2000-04-26 | 2004-04-29 | Evans David Michael | Inhibitors of dipeptidyl peptidase IV |
| US20040082607A1 (en) * | 2001-02-02 | 2004-04-29 | Satoru Oi | Fused heterocyclic compounds |
| US20040092604A1 (en) * | 2001-12-03 | 2004-05-13 | Madera Ann Marie | Amino-tetralin derivatives as muscarinic receptor antagonists |
| US20050004205A1 (en) * | 2001-10-23 | 2005-01-06 | Evans David M | Novel dipeptidyl peptidase iv (dp-iv) inhibitors as anti-diabetic agents |
| US20050014699A1 (en) * | 2000-05-23 | 2005-01-20 | Siegfried Ansorge | Combinations of enzyme inhibitor-containing preparations and the use thereof |
| US20050014747A1 (en) * | 2003-04-18 | 2005-01-20 | Emily Reinhard | Dihydrothiazine prodrugs of thiazolium agents |
| US20050020677A1 (en) * | 2001-07-04 | 2005-01-27 | Jukka Gynther | Compounds having prolyl oligopeptidase inhibitory activity, methods for their preparation and their use |
| US20050038020A1 (en) * | 2003-08-01 | 2005-02-17 | Hamann Lawrence G. | Adamantylglycine-based inhibitors of dipeptidyl peptidase IV and methods |
| US20050043299A1 (en) * | 2001-10-23 | 2005-02-24 | Ferring B. V. | Inhibitors of dipeptidyl peptidase iv |
| US20050042614A1 (en) * | 2001-10-31 | 2005-02-24 | Hughes Thomas Edward | Methods to treat diabetes and related conditions based on polymorphisms in the tcf-1 gene |
| US6861440B2 (en) * | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| US20050058635A1 (en) * | 2003-05-05 | 2005-03-17 | Hans-Ulrich Demuth | Use of effectors of glutaminyl and glutamate cyclases |
| US20050070482A1 (en) * | 2001-11-26 | 2005-03-31 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| US20050070719A1 (en) * | 2001-12-26 | 2005-03-31 | Sergei Belyakov | Inhibitors of dipeptidyl peptidase iv |
| US20050085497A1 (en) * | 2003-09-25 | 2005-04-21 | Saleem Ahmad | HMG-CoA reductase inhibitors and method |
Family Cites Families (113)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3607194A1 (en) * | 1986-03-05 | 1987-09-10 | Merck Patent Gmbh | Carbamoylphenothiazines |
| DE3633485A1 (en) | 1986-10-02 | 1988-04-07 | Bayer Ag | Substituted 2-pyri(mi)dyl-2'-pyridyl (thio)ethers |
| FR2643371B1 (en) | 1989-02-17 | 1993-11-05 | Roussel Uclaf | NOVEL 2-AMINO PENTANEDIOIC ACID DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| EP0419683A4 (en) | 1989-04-13 | 1992-03-11 | Japan Tobacco Inc. | New amino acid derivatives having prolylendopeptidase inhibitor activity |
| IT1232350B (en) | 1989-08-04 | 1992-01-28 | Poli Ind Chimica Spa | NEW ERGOLINICS DERIVED FROM PROLINA AND RELATED STRUCTURES WITH DOPAMINERGIC ACTIVITY. |
| JP2518965B2 (en) | 1990-09-21 | 1996-07-31 | 三井東圧化学株式会社 | Novel pyridine derivative and antitumor effect enhancer containing the same as an active ingredient |
| IL100428A0 (en) | 1990-12-21 | 1992-09-06 | Ciba Geigy | Pyrimidinyl-and triazinyl-salicylamides,their preparation and their use as herbicides and plant growth regulants |
| DE4141399A1 (en) | 1991-12-16 | 1993-06-17 | Bayer Ag | 4- (SUBSTITUTED) AMINO-3-ARYLPYRROLINONE DERIVATIVES |
| DE4206045A1 (en) | 1992-02-27 | 1993-09-02 | Bayer Ag | SULFONYLBENZYL SUBSTITUTED PYRIDONE |
| US5569662A (en) | 1992-03-23 | 1996-10-29 | Pfizer Inc. | Quinuclidine derivatives as substance P antagonists |
| US5254453A (en) * | 1992-04-16 | 1993-10-19 | Eastman Kodak Company | Process for preparing narrow size distribution small tabular grains |
| DE69313649D1 (en) | 1992-06-19 | 1997-10-09 | Black James Foundation | Bicyclooctan und bicycloheptan derivate |
| DE4301105A1 (en) | 1993-01-18 | 1994-07-21 | Agfa Gevaert Ag | Color photographic recording material |
| DE4301106A1 (en) | 1993-01-18 | 1994-07-21 | Agfa Gevaert Ag | Color photographic recording material |
| AU691829B2 (en) | 1993-07-16 | 1998-05-28 | Merck & Co., Inc. | Benzoxazinone and benzopyrimidinone piperidinyl tocolytic oxytocin receptor antagonists |
| FR2711992A1 (en) | 1993-11-03 | 1995-05-12 | Lipha | New heterocyclic derivatives, process of preparation and pharmaceutical composition containing them |
| JP3599403B2 (en) | 1995-02-24 | 2004-12-08 | 山之内製薬株式会社 | A new method for producing benzazepine derivatives |
| US5795894A (en) | 1995-05-02 | 1998-08-18 | Schering Corporation | Piperazino derivatives as neurokinn antagonists |
| US5563014A (en) | 1995-05-15 | 1996-10-08 | Xerox Corporation | Migration imaging members |
| US5767097A (en) * | 1996-01-23 | 1998-06-16 | Icn Pharmaceuticals, Inc. | Specific modulation of Th1/Th2 cytokine expression by ribavirin in activated T-lymphocytes |
| TW397821B (en) | 1996-04-19 | 2000-07-11 | American Home Produits Corp | 3-[4-(2-phenyl-indole-1-ylmethyl)-phenyl]-acrylamides and 2-phenyl-1-[4-(amino-1-yl-alk-1-ynyl)-benzyl]-1H-indol-5-ol as well as pharmaceutical compositions of estrogenic agents thereof |
| DE122010000020I1 (en) | 1996-04-25 | 2010-07-08 | Prosidion Ltd | Method for lowering the blood glucose level in mammals |
| US5914150A (en) * | 1997-02-28 | 1999-06-22 | Candescent Technologies Corporation | Formation of polycarbonate film with apertures determined by etching charged-particle tracks |
| US6500885B1 (en) | 1997-02-28 | 2002-12-31 | Candescent Technologies Corporation | Polycarbonate-containing liquid chemical formulation and methods for making and using polycarbonate film |
| US6319902B1 (en) | 1997-08-22 | 2001-11-20 | Shionogi & Co., Ltd. | Peptide derivatives having thiazolyl-alanine residue |
| JP2001516767A (en) | 1997-09-24 | 2001-10-02 | アムジエン・インコーポレーテツド | Prevention and treatment of hearing loss using sensory neurotrophic compounds |
| EP1052994A2 (en) | 1998-02-02 | 2000-11-22 | Trustees Of Tufts College | Use of dipeptidylpetidase inhibitors to regulate glucose metabolism |
| US6583170B1 (en) | 1998-05-15 | 2003-06-24 | Wyeth | 2-Phenyl-1-[4-(amino-1-yl-alk-1-ynyl)-benzyl]-1H-indol-5-ol and estrogen formulations |
| US6331537B1 (en) | 1998-06-03 | 2001-12-18 | Gpi Nil Holdings, Inc. | Carboxylic acids and carboxylic acid isosteres of N-heterocyclic compounds |
| DE19826972A1 (en) | 1998-06-18 | 1999-12-23 | Univ Magdeburg Tech | Inhibiting keratinocyte activation and proliferation, for treatment of dermatological disorders such as psoriasis or actinic precancerous states |
| US20030176357A1 (en) | 1998-10-06 | 2003-09-18 | Pospisilik Andrew J. | Dipeptidyl peptidase IV inhibitors and their uses for lowering blood pressure levels |
| CO5150173A1 (en) | 1998-12-10 | 2002-04-29 | Novartis Ag | COMPOUNDS N- (REPLACED GLYCLE) -2-DIPEPTIDYL-IV PEPTIDASE INHIBITING CYANOPIRROLIDINS (DPP-IV) WHICH ARE EFFECTIVE IN THE TREATMENT OF CONDITIONS MEDIATED BY DPP-IV INHIBITION |
| WO2000061556A1 (en) | 1999-04-09 | 2000-10-19 | Meiji Seika Kaisha, Ltd. | Nitrogen-containing heterocyclic compounds and benamide compounds and drugs containing the same |
| DE19940130A1 (en) | 1999-08-24 | 2001-03-01 | Probiodrug Ges Fuer Arzneim | New effectors of Dipeptidyl Peptidase IV for topical use |
| IL139197A0 (en) | 1999-10-29 | 2001-11-25 | Pfizer Prod Inc | Use of corticotropin releasing factor antagonists and related compositions |
| US20040152745A1 (en) | 1999-11-12 | 2004-08-05 | Guilford Pharmaceuticals, Inc. | Dipeptidyl peptidase IV inhibitors and methods of making and using dipeptidyl peptidase IV inhibitors |
| ATE385421T1 (en) | 2000-01-21 | 2008-02-15 | Novartis Pharma Gmbh | COMPOSITIONS CONSISTING OF DIPEPTIDYLPEPTIDASE-IV INHIBITORS AND ANTIDIABETICS |
| US7064145B2 (en) | 2000-02-25 | 2006-06-20 | Novo Nordisk A/S | Inhibition of beta cell degeneration |
| GB0010183D0 (en) | 2000-04-26 | 2000-06-14 | Ferring Bv | Inhibitors of dipeptidyl peptidase IV |
| US6432969B1 (en) | 2000-06-13 | 2002-08-13 | Novartis Ag | N-(substituted glycyl)-2 cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US7078397B2 (en) | 2000-06-19 | 2006-07-18 | Smithkline Beecham Corporation | Combinations of dipeptidyl peptidase IV inhibitors and other antidiabetic agents for the treatment of diabetes mellitus |
| EP1300155A1 (en) | 2000-07-11 | 2003-04-09 | Shionogi & Co., Ltd. | Enteric preparations containing physiologically active peptides |
| JP4101053B2 (en) | 2000-08-10 | 2008-06-11 | 田辺三菱製薬株式会社 | Proline derivative and its pharmaceutical use |
| JPWO2002051836A1 (en) | 2000-12-27 | 2004-04-22 | 協和醗酵工業株式会社 | Dipeptidyl peptidase-IV inhibitor |
| DE10100053A1 (en) | 2001-01-02 | 2002-08-22 | Keyneurotek Ag I G | Use of enzyme inhibitors of dipeptidyl peptidase IV and aminopeptidase N and pharmaceutical preparations therefrom for the prevention and / or therapy of ischemia-related acute and chronic neurodegenerative processes and diseases |
| JP2004520330A (en) | 2001-01-02 | 2004-07-08 | インスティテュート ヒューア メディツィンテクノロギー マグデブルク ゲーエムベーハー イーエムテーエム | Treatment and prevention of arteriosclerosis, treatment and prevention of allergic reactions of the type Gel-Coombs type I, and treatment of skin diseases with follicular and epidermal hyperkeratosis and accelerated proliferation of keratinocytes Of enzyme inhibitor and its drug compound for drug and prevention |
| JP2002265439A (en) | 2001-03-08 | 2002-09-18 | Mitsubishi Pharma Corp | Cyanopyrrolidine derivative and its use for medicine |
| GB0109146D0 (en) | 2001-04-11 | 2001-05-30 | Ferring Bv | Treatment of type 2 diabetes |
| JP2005500270A (en) | 2001-05-29 | 2005-01-06 | ギルフォード ファーマシュウティカルズ インコーポレイテッド | Method for treating nerve injury that has occurred as a result of surgery |
| JP2004535433A (en) | 2001-06-20 | 2004-11-25 | メルク エンド カムパニー インコーポレーテッド | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| GB0115517D0 (en) | 2001-06-25 | 2001-08-15 | Ferring Bv | Novel antidiabetic agents |
| US20030130199A1 (en) | 2001-06-27 | 2003-07-10 | Von Hoersten Stephan | Dipeptidyl peptidase IV inhibitors and their uses as anti-cancer agents |
| US7368421B2 (en) | 2001-06-27 | 2008-05-06 | Probiodrug Ag | Use of dipeptidyl peptidase IV inhibitors in the treatment of multiple sclerosis |
| ES2296962T3 (en) | 2001-06-27 | 2008-05-01 | Smithkline Beecham Corporation | PIRROLIDINS AS INHIBITORS OF DIPEPTIDIL PEPTIDASA. |
| US7183290B2 (en) | 2001-06-27 | 2007-02-27 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| US20030135023A1 (en) | 2001-06-27 | 2003-07-17 | Hans-Ulrich Demuth | Peptide structures useful for competitive modulation of dipeptidyl peptidase IV catalysis |
| DE10150203A1 (en) | 2001-10-12 | 2003-04-17 | Probiodrug Ag | Use of dipeptidyl peptidase IV inhibitor in treatment of cancer |
| EP1406873B1 (en) | 2001-06-27 | 2007-09-26 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| WO2003015768A2 (en) | 2001-08-16 | 2003-02-27 | Probiodrug Ag | Use of inhibitors of proline endopeptidase to modulate inositol (1,4,5) triphosphate concentration dependent on intracellular signal cascades |
| JP4337138B2 (en) | 2001-08-31 | 2009-09-30 | 味の素株式会社 | Novel diarylalkene derivatives and novel diarylalkane derivatives |
| CA2460512A1 (en) | 2001-09-14 | 2003-03-27 | Mitsubishi Pharma Corporation | Thiazolidine derivatives and medicinal use thereof |
| US20030125304A1 (en) | 2001-11-09 | 2003-07-03 | Hans-Ulrich Demuth | Substituted amino ketone compounds |
| US20030114389A1 (en) | 2001-11-13 | 2003-06-19 | Webb Randy Lee | Combination of organic compounds |
| US20050101638A1 (en) * | 2002-11-08 | 2005-05-12 | Webb Randy L. | Combination of organic compounds |
| ATE349435T1 (en) | 2002-02-28 | 2007-01-15 | Prosidion Ltd | GLUTAMINE-BASED DPIV INHIBITORS |
| HUP0200849A2 (en) | 2002-03-06 | 2004-08-30 | Sanofi-Synthelabo | N-aminoacetyl-pyrrolidine-2-carbonitrile derivatives, pharmaceutical compositions containing them and process for producing them |
| JP4329381B2 (en) * | 2002-04-04 | 2009-09-09 | 田辺三菱製薬株式会社 | Pharmaceutical composition |
| JP4329382B2 (en) | 2002-04-04 | 2009-09-09 | 田辺三菱製薬株式会社 | Pharmaceutical composition |
| US20040106802A1 (en) * | 2002-04-08 | 2004-06-03 | Torrent Pharmaceuticals Ltd. | Novel compounds and therapeutic uses thereof |
| EP1492777A1 (en) * | 2002-04-08 | 2005-01-05 | Torrent Pharmaceuticals Ltd | Thiazolidine-4-carbonitriles and analogues and their use as dipeptidyl-peptidase inhibitors |
| JP2004026820A (en) | 2002-05-09 | 2004-01-29 | Taisho Pharmaceut Co Ltd | Dipeptidyl peptidase iv inhibitor |
| NZ536832A (en) | 2002-06-03 | 2006-11-30 | Novartis Ag | The use of substituted cyanopyrrolidines and combination preparations containing them for treating hyperlipidemia and associated diseases |
| HUP0202001A2 (en) | 2002-06-14 | 2005-08-29 | Sanofi-Aventis | Azabicyclo-octane and nonane derivatives with ddp-iv inhibiting activity |
| TW200401635A (en) | 2002-07-23 | 2004-02-01 | Yamanouchi Pharma Co Ltd | 2-Cyano-4-fluoropyrrolidine derivative or salt thereof |
| EP1543023B1 (en) | 2002-09-18 | 2010-03-17 | Prosidion Limited | Secondary binding site of dipeptidyl peptidase iv (dp iv) |
| US7238724B2 (en) | 2002-09-19 | 2007-07-03 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US20040121964A1 (en) | 2002-09-19 | 2004-06-24 | Madar David J. | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7262207B2 (en) | 2002-09-19 | 2007-08-28 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| DE60332856D1 (en) | 2002-10-23 | 2010-07-15 | Bristol Myers Squibb Co | GLYCINNITRIL BASED DIPEPTIDYLPEPTIDASE IV INHIBITORS |
| EP1562925B1 (en) | 2002-11-07 | 2007-01-03 | Merck & Co., Inc. | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7482337B2 (en) | 2002-11-08 | 2009-01-27 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Xanthine derivatives, the preparation thereof and their use as pharmaceutical compositions |
| DE10254304A1 (en) | 2002-11-21 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New xanthine derivatives, their production and their use as medicines |
| EP1422293A1 (en) | 2002-11-25 | 2004-05-26 | F. Hoffmann-La Roche Ag | Crystal structure of dipeptidyl peptidase IV |
| US20040121994A1 (en) | 2002-12-20 | 2004-06-24 | Anderson Steven N. | Novel amides that activate soluble guanylate cyclase |
| US20040192680A1 (en) | 2002-12-20 | 2004-09-30 | Anderson Steven N. | Novel amides that activate soluble guanylate cyclase |
| JP2004244412A (en) | 2003-01-20 | 2004-09-02 | Kotobuki Seiyaku Kk | 2-cyanopyrrolidine derivative having substituent at 4-position, method for producing the same, and medicament containing the same |
| WO2004076433A1 (en) * | 2003-02-28 | 2004-09-10 | Aic | Dipeptidyl peptidase inhibitors |
| DE10309005A1 (en) | 2003-03-01 | 2004-09-09 | Morphochem Aktiengesellschaft für kombinatorische Chemie | New conjugates comprising a dipeptidyl peptidase inhibitor linked to a neural endopeptidase inhibitor, e.g. useful for treating diabetes, obesity, growth hormone deficiency, immunosuppression, HIV infection |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| WO2004099157A1 (en) | 2003-05-07 | 2004-11-18 | Pfizer Products Inc. | Cannabinoid receptor ligands and uses thereof |
| WO2004103993A1 (en) | 2003-05-14 | 2004-12-02 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| JP2006527194A (en) * | 2003-06-06 | 2006-11-30 | メルク エンド カムパニー インコーポレーテッド | Condensed indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7235662B2 (en) | 2003-06-11 | 2007-06-26 | Bristol-Myers Squibb Company | Modulators of the glucocorticoid receptor and method |
| NZ544221A (en) | 2003-06-20 | 2009-05-31 | Hoffmann La Roche | Pyrido[2,1-a]-isoquinoline derivatives as DPP-IV inhibitors |
| JP2005023038A (en) | 2003-07-04 | 2005-01-27 | Taisho Pharmaceut Co Ltd | Therapeutic agent for chronic renal disease |
| JP2005047853A (en) | 2003-07-29 | 2005-02-24 | Fuji Photo Film Co Ltd | Method for producing nitrogen-containing heterocyclic compound |
| DE10337074A1 (en) * | 2003-08-12 | 2005-03-17 | Keyneurotek Ag | Use of the inhibitors of enzymes with activities of aminopeptidase N and / or dipeptidyl peptidase IV and pharmaceutical preparations thereof for the therapy and prevention of chronic neurodegenerative diseases |
| HU227684B1 (en) * | 2003-08-29 | 2011-11-28 | Sanofi Aventis | Adamantane and azabicyclo-octane and nonane derivatives and their use as dpp-iv inhibitors |
| US7205409B2 (en) | 2003-09-04 | 2007-04-17 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| WO2005033099A2 (en) * | 2003-10-03 | 2005-04-14 | Glenmark Pharmaceuticals Ltd. | Novel dipeptidyl peptidase iv inhibitors; processes for their preparation and compositions thereof |
| US20050153885A1 (en) | 2003-10-08 | 2005-07-14 | Yun Anthony J. | Treatment of conditions through modulation of the autonomic nervous system |
| BRPI0415500A (en) * | 2003-10-17 | 2007-04-10 | Incyte Corp | cyclic hydroxamates substituted as matrix metalloproteinase inhibitors |
| AU2004285532A1 (en) | 2003-10-31 | 2005-05-12 | Alza Corporation | Administration of levodopa and carbidopa |
| TW200528440A (en) | 2003-10-31 | 2005-09-01 | Fujisawa Pharmaceutical Co | 2-cyanopyrrolidinecarboxamide compound |
| US20050171112A1 (en) | 2003-11-03 | 2005-08-04 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| JP2007509898A (en) | 2003-11-03 | 2007-04-19 | プロビオドルグ エージー | Useful combinations for the treatment of neurological disorders |
| JP2005139107A (en) | 2003-11-05 | 2005-06-02 | Taisho Pharmaceut Co Ltd | Dipeptidyl peptidase iv inhibitor |
| DE602004011394T2 (en) | 2003-12-08 | 2009-01-08 | F. Hoffmann-La Roche Ag | thiazole |
| US7326728B2 (en) | 2004-01-16 | 2008-02-05 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, AP-1, and/or NF-κβ activity and use thereof |
| US7230002B2 (en) | 2004-02-03 | 2007-06-12 | Glenmark Pharmaceuticals Ltd. | Dipeptidyl peptidase IV inhibitors; processes for their preparation and compositions thereof |
| US7304086B2 (en) | 2004-02-05 | 2007-12-04 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| EP1729774A4 (en) * | 2004-03-09 | 2009-05-06 | Nat Health Research Institutes | Pyrrolidine compounds |
| BRPI0608469A2 (en) * | 2005-04-22 | 2010-01-05 | Alantos Pharmaceuticals Holding Inc | dipeptidyl peptidase-iv inhibitors |
-
2006
- 2006-04-21 BR BRPI0608469-9A patent/BRPI0608469A2/en not_active IP Right Cessation
- 2006-04-21 WO PCT/US2006/015200 patent/WO2006116157A2/en active Application Filing
- 2006-04-21 AU AU2006239929A patent/AU2006239929B2/en not_active Ceased
- 2006-04-21 US US11/409,481 patent/US7553861B2/en not_active Expired - Fee Related
- 2006-04-21 EA EA200702208A patent/EA200702208A1/en unknown
- 2006-04-21 MX MX2007011453A patent/MX2007011453A/en active IP Right Grant
- 2006-04-21 ES ES06758484.7T patent/ES2477868T3/en active Active
- 2006-04-21 CA CA002599419A patent/CA2599419A1/en not_active Abandoned
- 2006-04-21 AP AP2007004234A patent/AP2007004234A0/en unknown
- 2006-04-21 KR KR1020077026980A patent/KR20080000665A/en not_active Application Discontinuation
- 2006-04-21 JP JP2008507950A patent/JP4568361B2/en not_active Expired - Fee Related
- 2006-04-21 EP EP06758484.7A patent/EP1888562B1/en active Active
- 2006-04-21 ZA ZA200708179A patent/ZA200708179B/en unknown
- 2006-04-21 CN CNA2006800134175A patent/CN101277949A/en active Pending
-
2007
- 2007-06-26 TN TNP2007000244A patent/TNSN07244A1/en unknown
- 2007-10-11 MA MA30289A patent/MA29387B1/en unknown
- 2007-10-11 IL IL186607A patent/IL186607A0/en unknown
- 2007-10-22 CR CR9462A patent/CR9462A/en not_active Application Discontinuation
- 2007-11-22 NO NO20075973A patent/NO20075973L/en not_active Application Discontinuation
-
2009
- 2009-06-26 US US12/492,254 patent/US20100009961A1/en not_active Abandoned
-
2010
- 2010-02-23 JP JP2010036861A patent/JP5198494B2/en not_active Expired - Fee Related
- 2010-04-15 AU AU2010201511A patent/AU2010201511B2/en not_active Ceased
-
2011
- 2011-01-13 US US13/006,363 patent/US8076330B2/en not_active Expired - Fee Related
Patent Citations (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3297709A (en) * | 1965-11-19 | 1967-01-10 | American Home Prod | Certain substituted tetrazole derivatives |
| US5407950A (en) * | 1990-06-07 | 1995-04-18 | Zeria Pharmaceutical Co., Ltd. | Arylalkanoylamine derivative and drug containing the same |
| US5391556A (en) * | 1992-02-13 | 1995-02-21 | Karl Thomae Gmbh | Benzimidazolyl derivatives, pharmaceutical compositions containing these compounds and processes for preparing them |
| US5627285A (en) * | 1992-02-27 | 1997-05-06 | Bayer Aktiengesellschaft | Sulphonylbenzyl-substituted imidazolylpropenoic acid derivatives |
| US5482920A (en) * | 1992-06-17 | 1996-01-09 | Ciba-Geigy Corporation | Triazinyl compounds with herbicidal activity |
| US5618808A (en) * | 1992-07-01 | 1997-04-08 | Pfizer, Inc. | Benzothiazepine and benzoxazepine derivatives as cholecystokinin receptor antagonists |
| US5494919A (en) * | 1993-11-09 | 1996-02-27 | Merck & Co., Inc. | 2-substituted piperidines, pyrrolidines and hexahydro-1H-azepines promote release of growth hormone |
| US6201132B1 (en) * | 1993-12-03 | 2001-03-13 | Ferring B.V. | Inhibitors of DP-mediated processes, compositions, and therapeutic methods thereof |
| US5492920A (en) * | 1993-12-10 | 1996-02-20 | Merck & Co., Inc. | Piperidine, pyrrolidine and hexahydro-1H-azepines promote release of growth hormone |
| US5492916A (en) * | 1993-12-23 | 1996-02-20 | Merck & Co., Inc. | Di- and tri-substituted piperidines, pyrrolidines and hexahydro-1H-azepines promote release of growth hormone |
| US5622973A (en) * | 1993-12-23 | 1997-04-22 | Merck & Co., Inc. | Treatment of osteoporosis with substituted piperidines, pyrrolidines and hexahydro-1H-azepines in combination with bisphosphonates |
| US6017929A (en) * | 1994-02-17 | 2000-01-25 | Zeria Pharmaceutical Co., Ltd. | Cholinesterase activator |
| US5714471A (en) * | 1995-01-06 | 1998-02-03 | Sibia Neurosciences, Inc. | Peptide and peptide analog protease inhibitors |
| US6015879A (en) * | 1995-01-06 | 2000-01-18 | Sibia Neurosciences, Inc. | Peptide and peptide analog protease inhibitors |
| US6051684A (en) * | 1995-01-06 | 2000-04-18 | Sibia Neurosciences Inc. | Methods of treating neurodegenerative disorders using protease inhibitors |
| US20030045432A1 (en) * | 1995-02-13 | 2003-03-06 | Reiner Fischer | Phenyl-substituted cyclic ketoenols |
| US6358887B1 (en) * | 1995-02-13 | 2002-03-19 | Bayer Aktiengesellschaft | 2-Phenyl-substituted heterocyclic 1,3-ketonols as herbicides and pesticides |
| US6380246B1 (en) * | 1995-05-09 | 2002-04-30 | Bayer Aktiengesellschaft | Alkyl-dihalogenated phenyl-substituted ketoenols useful as pesticides and herbicides |
| US5514505A (en) * | 1995-05-15 | 1996-05-07 | Xerox Corporation | Method for obtaining improved image contrast in migration imaging members |
| US6191161B1 (en) * | 1995-08-17 | 2001-02-20 | Chinoin Gyogyszer Es Vegyeszeti | Prolylendopeptidase inhibitors |
| US20040072819A1 (en) * | 1995-09-13 | 2004-04-15 | Takeda Chemical Industries, Ltd. | Benzoxazepine compounds, their production and use |
| US6057314A (en) * | 1995-12-21 | 2000-05-02 | Biochem Pharma Inc. | Low molecular weight bicyclic thrombin inhibitors |
| US6207668B1 (en) * | 1996-04-03 | 2001-03-27 | Hoechst Schering | Substituted pyridines/pyrimidines, their preparation, and their use as pesticides |
| US6537987B1 (en) * | 1996-06-20 | 2003-03-25 | Pfizer Inc. | 4,1-benzoxazepines or 4,1-benzothiazepines and their use as squalene synthetase inhibitors |
| US6051575A (en) * | 1996-10-30 | 2000-04-18 | Schering Corporation | Piperazino derivatives as neurokinin antagonists |
| US6011155A (en) * | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6228885B1 (en) * | 1996-12-19 | 2001-05-08 | Isagro S.P.A. | Fungicidal compositions based on (N-phenylacetyl-N-2,6-XYLYL)methyl alaninate |
| US6180698B1 (en) * | 1997-02-28 | 2001-01-30 | Candescent Technologies Corporation | Polycarbonate-containing liquid chemical formulation and method for making polycarbonate film |
| US20020045641A1 (en) * | 1998-06-03 | 2002-04-18 | Hamilton Gregory S. | Multiple heteroatom containing heterocyclic ring compounds substituted with carboxylic acids and isosteres thereof |
| US20040082622A1 (en) * | 1998-06-03 | 2004-04-29 | Guilford Pharmaceuticals Inc. | N-linked sulfonamides of N-heterocyclic carboxylic acids or carboxylic acid isosteres |
| US20020052514A1 (en) * | 1998-06-03 | 2002-05-02 | Hamilton Gregory S. | N-linked sulfonamides of N-heterocyclic carboxylic acids or carboxylic acid isosteres |
| US20020052510A1 (en) * | 1998-06-03 | 2002-05-02 | Gregory S. Hamilton | N-linked sulfonamides of n-heterocyclic carboxylic acids or carboxylic acid isosteres |
| US6337340B1 (en) * | 1998-08-14 | 2002-01-08 | Gpi Nil Holdings, Inc. | Carboxylic acids and isosteres of heterocyclic ring compounds having multiple heteroatoms for vision and memory disorders |
| US20020006899A1 (en) * | 1998-10-06 | 2002-01-17 | Pospisilik Andrew J. | Use of dipeptidyl peptidase IV effectors for lowering blood pressure in mammals |
| US20040077761A1 (en) * | 1998-12-30 | 2004-04-22 | Roberto Pavez Aranguiz | Filler-containing polymer dispersion, method for its production and use |
| US20030096796A1 (en) * | 1999-03-15 | 2003-05-22 | Axys Pharmaceuticals, Inc. | Novel compounds and compositions as protease inhibitors |
| US6521644B1 (en) * | 1999-03-23 | 2003-02-18 | Ferring Bv | Compositions for promoting growth |
| US6387893B1 (en) * | 1999-09-30 | 2002-05-14 | Merck & Co., Inc. | Spirotricyclic substituted azacycloalkane derivatives and uses thereof |
| US20030096857A1 (en) * | 1999-11-30 | 2003-05-22 | Evans David Michael | Novel antidiabetic agents |
| US6380398B2 (en) * | 2000-01-04 | 2002-04-30 | Novo Nordisk A/S | Therapeutically active and selective heterocyclic compounds that are inhibitors of the enzyme DPP-IV |
| US20030008905A1 (en) * | 2000-03-31 | 2003-01-09 | Hans-Ulrich Demuth | Method for the improvement of islet signaling in diabetes mellitus and for its prevention |
| US20050014703A1 (en) * | 2000-03-31 | 2005-01-20 | Hans-Ulrich Demuth | Method for the improvement of islet signaling in diabetes mellitus and for its prevention |
| US20040082497A1 (en) * | 2000-04-26 | 2004-04-29 | Evans David Michael | Inhibitors of dipeptidyl peptidase IV |
| US20020022619A1 (en) * | 2000-05-01 | 2002-02-21 | Rajagopal Bakthavatachalam | Tricyclic fused pyridine and pyrimidine derivatives as CRF antagonists |
| US20050014699A1 (en) * | 2000-05-23 | 2005-01-20 | Siegfried Ansorge | Combinations of enzyme inhibitor-containing preparations and the use thereof |
| US20040009877A1 (en) * | 2000-06-13 | 2004-01-15 | Reiner Fischer | Hetaryl-substituted heterocycles |
| US20020019433A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Methods of treating excessive intraocular pressure |
| US20020028792A1 (en) * | 2000-07-06 | 2002-03-07 | American Home Products Corporation | Combinations of bisphosphonates, estrogens and estrogenic agents |
| US6369051B1 (en) * | 2000-07-06 | 2002-04-09 | American Home Products Corporation | Combinations of SSRI and estrogenic agents |
| US6509332B2 (en) * | 2000-07-06 | 2003-01-21 | Wyeth | Methods of treating excessive intraocular pressure |
| US20020042432A1 (en) * | 2000-07-06 | 2002-04-11 | American Home Products Corporation | Combinations of SSRI and estrogenic agents |
| US20020019391A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combinations of statins and estrogenic agents |
| US20020022618A1 (en) * | 2000-07-06 | 2002-02-21 | American Home Products Corporation | Methods of inhibiting sphincter incontinence |
| US20020022617A1 (en) * | 2000-07-06 | 2002-02-21 | American Home Products Corporation | Methods for increasing nitric oxide synthase activity |
| US20020019373A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combinations of bisphosphonates and estrogenic agents |
| US20020016318A1 (en) * | 2000-07-06 | 2002-02-07 | American Home Products Corporation | Methods of treating breast disorders |
| US6358991B2 (en) * | 2000-07-06 | 2002-03-19 | American Home Products Corporation | Methods of treating neuropeptide Y-related conditions |
| US20020025952A1 (en) * | 2000-07-06 | 2002-02-28 | American Home Products Corporation | Combinations of statins, estrogens and estrogenic agents |
| US20020019392A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Methods of treating neuropeptide Y-related conditions |
| US20020028800A1 (en) * | 2000-07-06 | 2002-03-07 | American Home Products Corporation | Combination therapy for prosthesis-related bone degeneration |
| US20020019377A1 (en) * | 2000-07-06 | 2002-02-14 | American Home Products Corporation | Combination therapy for inhibiting sphincter incontinence |
| US6376486B1 (en) * | 2000-07-06 | 2002-04-23 | American Home Products Corporation | Methods of inhibiting sphincter incontinence |
| US6559163B2 (en) * | 2000-08-16 | 2003-05-06 | Neurogen Corporation | 2,4-substituted quinoline derivatives |
| US20040048867A1 (en) * | 2000-08-16 | 2004-03-11 | Neurogen Corporation | 2,4-Substituted pyridine derivatives |
| US20050054678A1 (en) * | 2000-10-06 | 2005-03-10 | Kosuke Yasuda | Nitrogenous five-membered ring compounds |
| US20040063935A1 (en) * | 2000-10-06 | 2004-04-01 | Kosuke Yasuda | Aliphatic nitrogenous five-membered ring compounds |
| US6518299B1 (en) * | 2000-10-20 | 2003-02-11 | Biocryst Pharmaceuticals, Inc. | Substituted pyrrolidine compounds useful as neuraminidase inhibitors |
| US20040072892A1 (en) * | 2000-11-10 | 2004-04-15 | Hiroshi Fukushima | Cyanopyrrolidine derivatives |
| US20040082607A1 (en) * | 2001-02-02 | 2004-04-29 | Satoru Oi | Fused heterocyclic compounds |
| US20040087587A1 (en) * | 2001-02-24 | 2004-05-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Xanthine derivatives, the preparation thereof and their use as pharmaceutical compositions |
| US20040077645A1 (en) * | 2001-02-24 | 2004-04-22 | Frank Himmelsbach | Xanthine derivatives,production and use thereof as medicament |
| US6713487B2 (en) * | 2001-03-02 | 2004-03-30 | Bristol-Myers Squibb Co. | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US20030092732A1 (en) * | 2001-03-02 | 2003-05-15 | Guixue Yu | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US20030096827A1 (en) * | 2001-03-02 | 2003-05-22 | Guixue Yu | Compounds useful as modulators of melanocortin receptors and pharmaceutical compositions comprising same |
| US20030069169A1 (en) * | 2001-03-02 | 2003-04-10 | Macor John E. | Co-administration of melanocortin receptor agonist and phosphodiesterase inhibitor for treatment of cyclic-AMP associated disorders |
| US20030087950A1 (en) * | 2001-03-28 | 2003-05-08 | Denanteuil Guillaume | New alpha-amino acid sulphonyl compounds |
| US6716843B2 (en) * | 2001-03-28 | 2004-04-06 | Les Laboratoires Servier | Alpha-amino acid sulphonyl compounds |
| US20030032625A1 (en) * | 2001-03-29 | 2003-02-13 | Topo Target Aps | Succinimide and maleimide derivatives and their use as topoisomerase II catalytic inhibitors |
| US20030073701A1 (en) * | 2001-03-31 | 2003-04-17 | Thompson Lorin A. | Succinoylamino heterocycles as inhibitors of a beta protein production |
| US20030064935A1 (en) * | 2001-04-11 | 2003-04-03 | Gougoutas Jack Z. | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US6706742B2 (en) * | 2001-05-15 | 2004-03-16 | Les Laboratories Servier | Alpha-amino-acid compounds |
| US20030078247A1 (en) * | 2001-05-15 | 2003-04-24 | De Nanteuil Guillaume | Alpha-amino-acid compounds |
| US20050020677A1 (en) * | 2001-07-04 | 2005-01-27 | Jukka Gynther | Compounds having prolyl oligopeptidase inhibitory activity, methods for their preparation and their use |
| US20050043299A1 (en) * | 2001-10-23 | 2005-02-24 | Ferring B. V. | Inhibitors of dipeptidyl peptidase iv |
| US20050004205A1 (en) * | 2001-10-23 | 2005-01-06 | Evans David M | Novel dipeptidyl peptidase iv (dp-iv) inhibitors as anti-diabetic agents |
| US6861440B2 (en) * | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| US20050042614A1 (en) * | 2001-10-31 | 2005-02-24 | Hughes Thomas Edward | Methods to treat diabetes and related conditions based on polymorphisms in the tcf-1 gene |
| US6673815B2 (en) * | 2001-11-06 | 2004-01-06 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| US20050070482A1 (en) * | 2001-11-26 | 2005-03-31 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| US20040092604A1 (en) * | 2001-12-03 | 2004-05-13 | Madera Ann Marie | Amino-tetralin derivatives as muscarinic receptor antagonists |
| US20050070719A1 (en) * | 2001-12-26 | 2005-03-31 | Sergei Belyakov | Inhibitors of dipeptidyl peptidase iv |
| US20040002519A1 (en) * | 2002-03-22 | 2004-01-01 | Damon Robert Edson | Combination of organic compounds |
| US20040002495A1 (en) * | 2002-05-20 | 2004-01-01 | Philip Sher | Lactam glycogen phosphorylase inhibitors and method of use |
| US20040077601A1 (en) * | 2002-07-09 | 2004-04-22 | Point Therapeutics, Inc. | Methods and compositions relating to isoleucine boroproline compounds |
| US20040058876A1 (en) * | 2002-09-18 | 2004-03-25 | Torsten Hoffmann | Secondary binding site of dipeptidyl peptidase IV (DP IV) |
| US20050014747A1 (en) * | 2003-04-18 | 2005-01-20 | Emily Reinhard | Dihydrothiazine prodrugs of thiazolium agents |
| US20050058635A1 (en) * | 2003-05-05 | 2005-03-17 | Hans-Ulrich Demuth | Use of effectors of glutaminyl and glutamate cyclases |
| US20050038020A1 (en) * | 2003-08-01 | 2005-02-17 | Hamann Lawrence G. | Adamantylglycine-based inhibitors of dipeptidyl peptidase IV and methods |
| US20050085497A1 (en) * | 2003-09-25 | 2005-04-21 | Saleem Ahmad | HMG-CoA reductase inhibitors and method |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122025A2 (en) | 2011-03-04 | 2012-09-13 | Intrexon Corporation | Vectors conditionally expressing protein |
| EP3450568A2 (en) | 2011-03-04 | 2019-03-06 | Intrexon Corporation | Vectors conditionally expressing protein |
| WO2018049014A1 (en) * | 2016-09-07 | 2018-03-15 | Trustees Of Tufts College | Dash inhibitors, and uses related thereto |
| US11096924B2 (en) | 2016-09-07 | 2021-08-24 | Trustees Of Tufts College | Combination therapies using immuno-dash inhibitors and PGE2 antagonists |
| US11583516B2 (en) | 2016-09-07 | 2023-02-21 | Trustees Of Tufts College | Dash inhibitors, and uses related thereto |
| US11559537B2 (en) | 2017-04-07 | 2023-01-24 | Trustees Of Tufts College | Combination therapies using caspase-1 dependent anticancer agents and PGE2 antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| AP2007004234A0 (en) | 2007-12-31 |
| MA29387B1 (en) | 2008-04-01 |
| JP2008538574A (en) | 2008-10-30 |
| BRPI0608469A2 (en) | 2010-01-05 |
| KR20080000665A (en) | 2008-01-02 |
| US20110112051A1 (en) | 2011-05-12 |
| AU2010201511B2 (en) | 2012-02-16 |
| JP5198494B2 (en) | 2013-05-15 |
| NO20075973L (en) | 2008-01-21 |
| EP1888562B1 (en) | 2014-06-18 |
| WO2006116157A3 (en) | 2007-04-19 |
| US8076330B2 (en) | 2011-12-13 |
| MX2007011453A (en) | 2008-02-12 |
| CN101277949A (en) | 2008-10-01 |
| US20060270701A1 (en) | 2006-11-30 |
| TNSN07244A1 (en) | 2008-11-21 |
| IL186607A0 (en) | 2008-01-20 |
| WO2006116157A2 (en) | 2006-11-02 |
| WO2006116157A9 (en) | 2007-03-01 |
| EA200702208A1 (en) | 2008-04-28 |
| EP1888562A2 (en) | 2008-02-20 |
| JP2010150273A (en) | 2010-07-08 |
| ZA200708179B (en) | 2009-12-30 |
| AU2010201511A1 (en) | 2010-05-06 |
| AU2006239929A1 (en) | 2006-11-02 |
| CA2599419A1 (en) | 2006-11-02 |
| CR9462A (en) | 2008-04-16 |
| JP4568361B2 (en) | 2010-10-27 |
| US7553861B2 (en) | 2009-06-30 |
| ES2477868T3 (en) | 2014-07-18 |
| AU2006239929B2 (en) | 2011-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8076330B2 (en) | Dipeptidyl peptidase-IV inhibitors | |
| US11639353B2 (en) | Cyclobutanes- and azetidine-containing mono and spirocyclic compounds as αV integrin inhibitors | |
| CN107949559B (en) | Bicyclic fused heteroaryl or aryl compounds as IRAK4 modulators | |
| JP2020509004A (en) | [1,2,4] -Triazolo [1,5-A] -pyrimidinyl derivatives substituted with piperidine, morpholine or piperazine as OGA inhibitors | |
| TWI481609B (en) | Substituted piperidine spiro pyrrolidinone and piperidinone, preparation and therapeutic use thereof | |
| JP2019501899A (en) | Modified apelin receptor (APJ) agonists and uses thereof | |
| CA2673375A1 (en) | 1-aminomethyl-l-phenyl-cyclohexane derivatives as ddp-iv inhibitors | |
| WO2007055418A1 (en) | Aza-substituted spiro derivative | |
| TWI395582B (en) | Dicyclic azaalkane derivatives,preparing method and use thereof | |
| KR20150082617A (en) | Dihydropyrazole gpr40 modulators | |
| AU2012292036A1 (en) | Phenyl-3-aza-bicyclo[3.1.0]hex-3-yl-methanones and the use thereof as medicament | |
| US8304411B2 (en) | Dicycloazaalkane derivates, preparation processes and medical uses thereof | |
| JP6483105B2 (en) | Piperazine derivatives and their use as medicaments | |
| JP6488320B2 (en) | Pyrrolidine GPR40 modulator for the treatment of diseases such as diabetes | |
| TW201016699A (en) | Novel derivatives | |
| WO2009033360A1 (en) | Benzamide derivatives, their preparation and uses in medicine thereof | |
| NZ718251B2 (en) | Piperazine derivatives and the use thereof as medicament | |
| TW200914010A (en) | Novel compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO PAY ISSUE FEE |