US20070105809A1 - Modulators of coagulation factors with enhanced stability - Google Patents
Modulators of coagulation factors with enhanced stability Download PDFInfo
- Publication number
- US20070105809A1 US20070105809A1 US11/605,712 US60571206A US2007105809A1 US 20070105809 A1 US20070105809 A1 US 20070105809A1 US 60571206 A US60571206 A US 60571206A US 2007105809 A1 US2007105809 A1 US 2007105809A1
- Authority
- US
- United States
- Prior art keywords
- composition
- methoxy
- pharmaceutically acceptable
- acceptable salt
- approximately
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 *C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(CO[PH](=O)O)C(C)F)C(C)F)C(C)OC)C(C)F)C(C)OC)C(F)[U])OC.*C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(CO[PH](=O)O)C(C)OC)C(*)OC)C(C)F)C(F)[U])C(*)OC)C(F)[U])OC.*C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(C)(=O)O)C(C)F)C(F)[U])C(C)OC)C(C)OC)OP(=O)(O)OCC(OP(=O)(O)OC(CO)C([H])[3H])C(C)OC.*C(OC)C(COP(C)(=O)O)OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OC)C(C)OC)C(C)O)C([U])OC)C(C)F)C(C)OC)C(F)[U].COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(O)C(C)OC)C([U])OC)C(C)OC.[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH] Chemical compound *C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(CO[PH](=O)O)C(C)F)C(C)F)C(C)OC)C(C)F)C(C)OC)C(F)[U])OC.*C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(CO[PH](=O)O)C(C)OC)C(*)OC)C(C)F)C(F)[U])C(*)OC)C(F)[U])OC.*C(OC)C(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(=O)(O)OC(COP(C)(=O)O)C(C)F)C(F)[U])C(C)OC)C(C)OC)OP(=O)(O)OCC(OP(=O)(O)OC(CO)C([H])[3H])C(C)OC.*C(OC)C(COP(C)(=O)O)OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(OC)C(C)OC)C(C)O)C([U])OC)C(C)F)C(C)OC)C(F)[U].COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)(O)OCC(OP(=O)(O)OCC(OP(=O)(O)OCC(O)C(C)OC)C([U])OC)C(C)OC.[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH].[NaH] 0.000 description 11
- ZQIBRWUPFVYSJZ-UHFFFAOYSA-N CC(C)N(C(C)C)P(C)OCCCCCCNC(=O)C(F)(F)F Chemical compound CC(C)N(C(C)C)P(C)OCCCCCCNC(=O)C(F)(F)F ZQIBRWUPFVYSJZ-UHFFFAOYSA-N 0.000 description 2
- CKQBSPYDLJOTFD-UHFFFAOYSA-N CC(C)N(C(C)C)P(C)OCCCCNC(=O)C(F)(F)F Chemical compound CC(C)N(C(C)C)P(C)OCCCCNC(=O)C(F)(F)F CKQBSPYDLJOTFD-UHFFFAOYSA-N 0.000 description 2
- FYUQULQPTKEEQP-UHFFFAOYSA-N CC(C)N(C(C)C)P(C)OCCCNC(=O)C(F)(F)F Chemical compound CC(C)N(C(C)C)P(C)OCCCNC(=O)C(F)(F)F FYUQULQPTKEEQP-UHFFFAOYSA-N 0.000 description 2
- RKXZWFNRPYUBKI-UHFFFAOYSA-N CNCCOCCOP(C)N(C(C)C)C(C)C Chemical compound CNCCOCCOP(C)N(C(C)C)C(C)C RKXZWFNRPYUBKI-UHFFFAOYSA-N 0.000 description 2
- HTOPJUYWNIBSBB-UHFFFAOYSA-M COCCOCC(COC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC HTOPJUYWNIBSBB-UHFFFAOYSA-M 0.000 description 2
- LWXXPDDMTWMINI-UHFFFAOYSA-N C.C.C.C.C.C.C.C.CC(C)C(=O)ON1C(=O)CCC1=O.CCCC(=O)ON1C(=O)CCC1=O.CCCC(C)C(=O)ON1C(=O)CCC1=O.CNC(=O)OCC(COC(=O)NC)OCCCC(=O)ON1C(=O)CCC1=O.COC(=O)NCCCCC(NC(=O)OC)C(=O)ON1C(=O)CCC1=O.COCCOCC(COC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCCCCC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCNC(=O)CCCC(=O)ON1C(=O)CCC1=O)OCCOC Chemical compound C.C.C.C.C.C.C.C.CC(C)C(=O)ON1C(=O)CCC1=O.CCCC(=O)ON1C(=O)CCC1=O.CCCC(C)C(=O)ON1C(=O)CCC1=O.CNC(=O)OCC(COC(=O)NC)OCCCC(=O)ON1C(=O)CCC1=O.COC(=O)NCCCCC(NC(=O)OC)C(=O)ON1C(=O)CCC1=O.COCCOCC(COC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCCCCC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCNC(=O)CCCC(=O)ON1C(=O)CCC1=O)OCCOC LWXXPDDMTWMINI-UHFFFAOYSA-N 0.000 description 1
- RZOPCLVOTJRMAY-UHFFFAOYSA-N C.C.C.C.COCCOCC(COC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCCCCC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCNC(=O)CCCC(=O)ON1C(=O)CCC1=O)OCCOC Chemical compound C.C.C.C.COCCOCC(COC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCCCCC(=O)ON1C(=O)CCC1=O)OCCOC.COCCOCC(COCCCNC(=O)CCCC(=O)ON1C(=O)CCC1=O)OCCOC RZOPCLVOTJRMAY-UHFFFAOYSA-N 0.000 description 1
- YGZXOHNXCZSBFK-UHFFFAOYSA-M C.C.CCOP(=O)([O-])OC Chemical compound C.C.CCOP(=O)([O-])OC YGZXOHNXCZSBFK-UHFFFAOYSA-M 0.000 description 1
- MGXSZGXPSMLHPK-UHFFFAOYSA-M C.C.CNC(=O)CC(NC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)C(=O)NC Chemical compound C.C.CNC(=O)CC(NC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)C(=O)NC MGXSZGXPSMLHPK-UHFFFAOYSA-M 0.000 description 1
- MZHFQUMQZKPFAX-UHFFFAOYSA-M C.C.COCCOCC(COC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound C.C.COCCOCC(COC(=O)NCCCCOP(=O)([O-])OC)OCCOC MZHFQUMQZKPFAX-UHFFFAOYSA-M 0.000 description 1
- IIPJJDQXOLRZJI-UHFFFAOYSA-M C.C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound C.C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC IIPJJDQXOLRZJI-UHFFFAOYSA-M 0.000 description 1
- HGDUNLJMMJGPKT-UHFFFAOYSA-M C.C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound C.C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOP(=O)([O-])OC)OCCOC HGDUNLJMMJGPKT-UHFFFAOYSA-M 0.000 description 1
- XJLNPRFWRNPQDU-UHFFFAOYSA-M C.C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCOP(=O)([O-])OC Chemical compound C.C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCOP(=O)([O-])OC XJLNPRFWRNPQDU-UHFFFAOYSA-M 0.000 description 1
- TUQKKZXTOIFDBE-UHFFFAOYSA-M C.C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCOP(=O)([O-])OC Chemical compound C.C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCOP(=O)([O-])OC TUQKKZXTOIFDBE-UHFFFAOYSA-M 0.000 description 1
- LAJFXZWSHUMDQV-UHFFFAOYSA-M C.C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCOP(=O)([O-])OC Chemical compound C.C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCOP(=O)([O-])OC LAJFXZWSHUMDQV-UHFFFAOYSA-M 0.000 description 1
- QMTSQJXLZIJGLS-UHFFFAOYSA-M C.C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOP(=O)([O-])OC Chemical compound C.C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOP(=O)([O-])OC QMTSQJXLZIJGLS-UHFFFAOYSA-M 0.000 description 1
- PFKLFVVTDSMSKN-UHFFFAOYSA-N C.CC(=O)ON1C(=O)CCC1=O Chemical compound C.CC(=O)ON1C(=O)CCC1=O PFKLFVVTDSMSKN-UHFFFAOYSA-N 0.000 description 1
- YYLJPLOTZXLWTM-UHFFFAOYSA-N C.CC(C)N1C(=O)C=CC1=O Chemical compound C.CC(C)N1C(=O)C=CC1=O YYLJPLOTZXLWTM-UHFFFAOYSA-N 0.000 description 1
- FEHORNMQAMOJAF-UHFFFAOYSA-N C.CN1C(=O)C=CC1=O Chemical compound C.CN1C(=O)C=CC1=O FEHORNMQAMOJAF-UHFFFAOYSA-N 0.000 description 1
- KMLRWDCCUVGJQA-UHFFFAOYSA-M C.CNC(=O)CC(NC(=O)NCCCCCCOP(=O)([O-])OC)C(=O)NC Chemical compound C.CNC(=O)CC(NC(=O)NCCCCCCOP(=O)([O-])OC)C(=O)NC KMLRWDCCUVGJQA-UHFFFAOYSA-M 0.000 description 1
- MUGOWAORHJGSRY-UHFFFAOYSA-M C.CNC(=O)CC(NC(=O)NCCCOCCOP(=O)([O-])OC)C(=O)NC Chemical compound C.CNC(=O)CC(NC(=O)NCCCOCCOP(=O)([O-])OC)C(=O)NC MUGOWAORHJGSRY-UHFFFAOYSA-M 0.000 description 1
- ZHSHCFYMXYXZSR-UHFFFAOYSA-N C.CNCCCCCCCCCCCCOP(C)N(C(C)C)C(C)C Chemical compound C.CNCCCCCCCCCCCCOP(C)N(C(C)C)C(C)C ZHSHCFYMXYXZSR-UHFFFAOYSA-N 0.000 description 1
- YEVVXOOWMOWOFO-UHFFFAOYSA-M C.COCCOCC(COC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO Chemical compound C.COCCOCC(COC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO YEVVXOOWMOWOFO-UHFFFAOYSA-M 0.000 description 1
- VGCBDTPTULKJBD-UHFFFAOYSA-M C.COCCOCC(COC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound C.COCCOCC(COC(=O)NCCCOP(=O)([O-])OC)OCCOC VGCBDTPTULKJBD-UHFFFAOYSA-M 0.000 description 1
- VEOKVAITZRXHCE-UHFFFAOYSA-M C.COCCOCC(COCCCCCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO Chemical compound C.COCCOCC(COCCCCCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO VEOKVAITZRXHCE-UHFFFAOYSA-M 0.000 description 1
- YDYJTRPDPGUVOR-UHFFFAOYSA-M C.COCCOCC(COCCCCCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound C.COCCOCC(COCCCCCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC YDYJTRPDPGUVOR-UHFFFAOYSA-M 0.000 description 1
- HBAABYYWBYOZBS-UHFFFAOYSA-M C.COCCOCC(COCCCCCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC Chemical compound C.COCCOCC(COCCCCCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC HBAABYYWBYOZBS-UHFFFAOYSA-M 0.000 description 1
- DNSLGCJNDYEDBN-UHFFFAOYSA-M C.COCCOCC(COCCCCCCC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound C.COCCOCC(COCCCCCCC(=O)NCCCOP(=O)([O-])OC)OCCOC DNSLGCJNDYEDBN-UHFFFAOYSA-M 0.000 description 1
- CHAFULGQEUXQIP-UHFFFAOYSA-M C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO Chemical compound C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCCO CHAFULGQEUXQIP-UHFFFAOYSA-M 0.000 description 1
- BTEZNNOEIMVGAF-UHFFFAOYSA-M C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC Chemical compound C.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC BTEZNNOEIMVGAF-UHFFFAOYSA-M 0.000 description 1
- JPLPJPZPUAFEIJ-UHFFFAOYSA-M C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)C1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O Chemical compound C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)C1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O JPLPJPZPUAFEIJ-UHFFFAOYSA-M 0.000 description 1
- SAWBKWRUXYNNAJ-UHFFFAOYSA-M C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC Chemical compound C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC SAWBKWRUXYNNAJ-UHFFFAOYSA-M 0.000 description 1
- LURVQNABOBIGEE-UHFFFAOYSA-M C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCOCCOP(=O)([O-])OC Chemical compound C.COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCOCCOP(=O)([O-])OC LURVQNABOBIGEE-UHFFFAOYSA-M 0.000 description 1
- BZJGIFTUAZIKKD-UHFFFAOYSA-M C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCCCCCCCOP(=O)([O-])OC Chemical compound C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCCCCCCCOP(=O)([O-])OC BZJGIFTUAZIKKD-UHFFFAOYSA-M 0.000 description 1
- CITZBBUBWIMAAG-UHFFFAOYSA-M C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)([O-])OC Chemical compound C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)([O-])OC CITZBBUBWIMAAG-UHFFFAOYSA-M 0.000 description 1
- WJTDILJHXBDCQO-UHFFFAOYSA-M C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOCCOP(=O)([O-])OC Chemical compound C.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOCCOP(=O)([O-])OC WJTDILJHXBDCQO-UHFFFAOYSA-M 0.000 description 1
- OVGUTGCBBUOQQU-UHFFFAOYSA-M C.COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCCCCCOP(=O)([O-])OC Chemical compound C.COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCCCCCOP(=O)([O-])OC OVGUTGCBBUOQQU-UHFFFAOYSA-M 0.000 description 1
- WIYMLKXVVSTCCQ-UHFFFAOYSA-M C.COP(=O)([O-])OCCCCNC(=O)NC(CC(C)=O)C(C)=O Chemical compound C.COP(=O)([O-])OCCCCNC(=O)NC(CC(C)=O)C(C)=O WIYMLKXVVSTCCQ-UHFFFAOYSA-M 0.000 description 1
- JHJVTHXNNCWFAN-UHFFFAOYSA-M C.COP(=O)([O-])OCCCNC(=O)NC(CC(C)=O)C(C)=O Chemical compound C.COP(=O)([O-])OCCCNC(=O)NC(CC(C)=O)C(C)=O JHJVTHXNNCWFAN-UHFFFAOYSA-M 0.000 description 1
- AKHDKUGNELIGFY-UHFFFAOYSA-N C.CSCCCCCCOP(C)N(C(C)C)C(C)C Chemical compound C.CSCCCCCCOP(C)N(C(C)C)C(C)C AKHDKUGNELIGFY-UHFFFAOYSA-N 0.000 description 1
- NAFPXNPPFIAROV-BFMOFHOQSA-N C=C(CN)OP(=O)(OC[C@H]1C[C@@H](C)[C@@H](OC)C1OC)OC1[C@@H](CC)O[C@@H](C)[C@H]1OC.CO[C@H]1C(OP(=O)(O)OC[C@H]2C[C@@H](C)[C@@H](OC)C2O)[C@@H](CO)O[C@H]1C Chemical compound C=C(CN)OP(=O)(OC[C@H]1C[C@@H](C)[C@@H](OC)C1OC)OC1[C@@H](CC)O[C@@H](C)[C@H]1OC.CO[C@H]1C(OP(=O)(O)OC[C@H]2C[C@@H](C)[C@@H](OC)C2O)[C@@H](CO)O[C@H]1C NAFPXNPPFIAROV-BFMOFHOQSA-N 0.000 description 1
- MVARZXYLPQACAY-UHFFFAOYSA-N C=P(C)(OC)OCCCCCCNC(=O)CCCOC(COC(=O)NCCOCCOC)COC(=O)NCOCCOC Chemical compound C=P(C)(OC)OCCCCCCNC(=O)CCCOC(COC(=O)NCCOCCOC)COC(=O)NCOCCOC MVARZXYLPQACAY-UHFFFAOYSA-N 0.000 description 1
- GBBVILIBKXEXPR-UHFFFAOYSA-N CC(=O)OCC(COC(C)=O)OCCCC(=O)ON1C(=O)CCC1=O.COC(=O)NC(CCCCNC(C)=O)C(=O)ON1C(=O)CCC1=O Chemical compound CC(=O)OCC(COC(C)=O)OCCCC(=O)ON1C(=O)CCC1=O.COC(=O)NC(CCCCNC(C)=O)C(=O)ON1C(=O)CCC1=O GBBVILIBKXEXPR-UHFFFAOYSA-N 0.000 description 1
- FHNMAOLQZHUPIJ-UHFFFAOYSA-N CC(C)C(=O)ON1C(=O)CCC1=O Chemical compound CC(C)C(=O)ON1C(=O)CCC1=O FHNMAOLQZHUPIJ-UHFFFAOYSA-N 0.000 description 1
- FSYPIBZNYNCCDY-UHFFFAOYSA-M CC.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)CN1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O Chemical compound CC.COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)CN1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O FSYPIBZNYNCCDY-UHFFFAOYSA-M 0.000 description 1
- AASBXERNXVFUEJ-UHFFFAOYSA-N CCC(=O)ON1C(=O)CCC1=O Chemical compound CCC(=O)ON1C(=O)CCC1=O AASBXERNXVFUEJ-UHFFFAOYSA-N 0.000 description 1
- GNMPQCCQPIPNML-UHFFFAOYSA-N CNC(=N)C(CC(=O)NC)N1C(=O)C=CC1=O Chemical compound CNC(=N)C(CC(=O)NC)N1C(=O)C=CC1=O GNMPQCCQPIPNML-UHFFFAOYSA-N 0.000 description 1
- WSZLQYUFLAJEHL-UHFFFAOYSA-M CNC(=O)CC(NC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)C(=O)NC Chemical compound CNC(=O)CC(NC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)C(=O)NC WSZLQYUFLAJEHL-UHFFFAOYSA-M 0.000 description 1
- HTVKVBFMZHAGFX-UHFFFAOYSA-M CNC(=O)CC(NC(=O)NCCCOCCOP(=O)([O-])OC)C(=O)NC Chemical compound CNC(=O)CC(NC(=O)NCCCOCCOP(=O)([O-])OC)C(=O)NC HTVKVBFMZHAGFX-UHFFFAOYSA-M 0.000 description 1
- IEDDWPJQXVWRFJ-UHFFFAOYSA-N CNC(=O)CC(NC(=O)OC1=CC=C([N+](=O)[O-])C=C1)C(=O)NC Chemical compound CNC(=O)CC(NC(=O)OC1=CC=C([N+](=O)[O-])C=C1)C(=O)NC IEDDWPJQXVWRFJ-UHFFFAOYSA-N 0.000 description 1
- XFSLBSZASWEJDE-UHFFFAOYSA-N CNCCCCCCCCCCCCOP(C)N(C(C)C)C(C)C Chemical compound CNCCCCCCCCCCCCOP(C)N(C(C)C)C(C)C XFSLBSZASWEJDE-UHFFFAOYSA-N 0.000 description 1
- CFRMQMZFOKZPPZ-UHFFFAOYSA-M COC.COCCOCC(COCCCCCCC(=O)NCCCCCCCCCCCCOP(C)(=O)[O-])OCCOC Chemical compound COC.COCCOCC(COCCCCCCC(=O)NCCCCCCCCCCCCOP(C)(=O)[O-])OCCOC CFRMQMZFOKZPPZ-UHFFFAOYSA-M 0.000 description 1
- MVVQCTQPLVPZAC-UHFFFAOYSA-M COC.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCCCCCCCOP(C)(=O)[O-])OCCOC Chemical compound COC.COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCCCCCCCOP(C)(=O)[O-])OCCOC MVVQCTQPLVPZAC-UHFFFAOYSA-M 0.000 description 1
- ALCPWLHNYNUWDQ-UHFFFAOYSA-N COCCOCC(COC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC ALCPWLHNYNUWDQ-UHFFFAOYSA-N 0.000 description 1
- ALCPWLHNYNUWDQ-UHFFFAOYSA-M COCCOCC(COC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC)OCCOC ALCPWLHNYNUWDQ-UHFFFAOYSA-M 0.000 description 1
- MOXIMYXZOAHJHX-UHFFFAOYSA-M COCCOCC(COC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC MOXIMYXZOAHJHX-UHFFFAOYSA-M 0.000 description 1
- LOKXGWNSAXVGQW-UHFFFAOYSA-M COCCOCC(COC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCCOP(=O)([O-])OC)OCCOC LOKXGWNSAXVGQW-UHFFFAOYSA-M 0.000 description 1
- RFFOXWGFMOMTMZ-UHFFFAOYSA-M COCCOCC(COC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COC(=O)NCCCOP(=O)([O-])OC)OCCOC RFFOXWGFMOMTMZ-UHFFFAOYSA-M 0.000 description 1
- GITNTIRPFYJIED-UHFFFAOYSA-M COCCOCC(COCCCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC GITNTIRPFYJIED-UHFFFAOYSA-M 0.000 description 1
- DWPZVZJEMCHAAS-UHFFFAOYSA-N COCCOCC(COCCCCCCC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC Chemical compound COCCOCC(COCCCCCCC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC DWPZVZJEMCHAAS-UHFFFAOYSA-N 0.000 description 1
- OEYFKRPMLLMWOW-UHFFFAOYSA-M COCCOCC(COCCCCCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCCCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC OEYFKRPMLLMWOW-UHFFFAOYSA-M 0.000 description 1
- FSWMZJSZSHLARZ-UHFFFAOYSA-M COCCOCC(COCCCCCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCCCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC FSWMZJSZSHLARZ-UHFFFAOYSA-M 0.000 description 1
- BHNWUJXWDSAASO-UHFFFAOYSA-M COCCOCC(COCCCCCCC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCCCCC(=O)NCCCOP(=O)([O-])OC)OCCOC BHNWUJXWDSAASO-UHFFFAOYSA-M 0.000 description 1
- VIQCWUJIGDMISW-UHFFFAOYSA-N COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC Chemical compound COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCCCCCCCOP(=O)(O)OC)OCCOC VIQCWUJIGDMISW-UHFFFAOYSA-N 0.000 description 1
- BHOAJAOJGBVVPZ-UHFFFAOYSA-M COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCCCOP(=O)([O-])OC)OCCOC BHOAJAOJGBVVPZ-UHFFFAOYSA-M 0.000 description 1
- PFHTZCPOOYLQTE-UHFFFAOYSA-M COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCNC(=O)CCCC(=O)NCCCCOP(=O)([O-])OC)OCCOC PFHTZCPOOYLQTE-UHFFFAOYSA-M 0.000 description 1
- HNIQTXMYFDMOAK-UHFFFAOYSA-M COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOCCOP(=O)([O-])OC)OCCOC HNIQTXMYFDMOAK-UHFFFAOYSA-M 0.000 description 1
- CWWMNYOPQRBDNQ-UHFFFAOYSA-M COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOP(=O)([O-])OC)OCCOC Chemical compound COCCOCC(COCCCNC(=O)CCCC(=O)NCCCOP(=O)([O-])OC)OCCOC CWWMNYOPQRBDNQ-UHFFFAOYSA-M 0.000 description 1
- LOFIFTOQRQFXRO-UHFFFAOYSA-M COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCOP(=O)([O-])OC Chemical compound COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCCOP(=O)([O-])OC LOFIFTOQRQFXRO-UHFFFAOYSA-M 0.000 description 1
- GZLHKMBXELVPNA-UHFFFAOYSA-M COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCOP(=O)([O-])OC Chemical compound COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCCCC(=O)NCCCOP(=O)([O-])OC GZLHKMBXELVPNA-UHFFFAOYSA-M 0.000 description 1
- WCFKKEKWSCFUHC-UHFFFAOYSA-M COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCN1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O Chemical compound COCCOCCNC(=O)OCC(COC(=O)NCOCCOC)OCN1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O WCFKKEKWSCFUHC-UHFFFAOYSA-M 0.000 description 1
- FTYYJFQUOSQGEN-UHFFFAOYSA-M COCCOCCOC(=O)NC(CCCCNC(=O)OCCOCCCO)C(=O)C1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O Chemical compound COCCOCCOC(=O)NC(CCCCNC(=O)OCCOCCCO)C(=O)C1C(=O)CC(SCCCCCCOP(=O)([O-])OC)C1=O FTYYJFQUOSQGEN-UHFFFAOYSA-M 0.000 description 1
- QXRXFINGQZCSPQ-UHFFFAOYSA-M COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCCCCCCCOP(=O)([O-])OC Chemical compound COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCCCCCCCOP(=O)([O-])OC QXRXFINGQZCSPQ-UHFFFAOYSA-M 0.000 description 1
- YIWQDMIJXYYMQF-UHFFFAOYSA-M COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)([O-])OC Chemical compound COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCCCOP(=O)([O-])OC YIWQDMIJXYYMQF-UHFFFAOYSA-M 0.000 description 1
- VESMCXPXHSTSOZ-UHFFFAOYSA-M COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCOP(=O)([O-])OC Chemical compound COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCCOP(=O)([O-])OC VESMCXPXHSTSOZ-UHFFFAOYSA-M 0.000 description 1
- JFSQQHFVNWQWSN-UHFFFAOYSA-M COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOCCOP(=O)([O-])OC Chemical compound COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOCCOP(=O)([O-])OC JFSQQHFVNWQWSN-UHFFFAOYSA-M 0.000 description 1
- YCZQGBGQAUYNCR-UHFFFAOYSA-M COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOP(=O)([O-])OC Chemical compound COCCOCCOC(=O)NCCCCC(NC(=O)OCCOCCOC)C(=O)NCCCOP(=O)([O-])OC YCZQGBGQAUYNCR-UHFFFAOYSA-M 0.000 description 1
- ZNECGHWWWJCWQK-UHFFFAOYSA-M COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC Chemical compound COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCCCCCCCCCCCOP(=O)([O-])OC ZNECGHWWWJCWQK-UHFFFAOYSA-M 0.000 description 1
- AATWZUXKXGLUQT-UHFFFAOYSA-M COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCOCCOP(=O)([O-])OC Chemical compound COCCOCNC(=O)OCC(COC(=O)NCCOCCCO)OCCCC(=O)NCCOCCOP(=O)([O-])OC AATWZUXKXGLUQT-UHFFFAOYSA-M 0.000 description 1
- ZYZSUBOQOKQCLC-UHFFFAOYSA-M COP(=O)([O-])OCCCCCCNC(=O)NC(CC(C)=O)C(C)=O Chemical compound COP(=O)([O-])OCCCCCCNC(=O)NC(CC(C)=O)C(C)=O ZYZSUBOQOKQCLC-UHFFFAOYSA-M 0.000 description 1
- JXRKBXUYUPPDAQ-UHFFFAOYSA-M COP(=O)([O-])OCCCCNC(=O)NC(CC(C)=O)C(C)=O Chemical compound COP(=O)([O-])OCCCCNC(=O)NC(CC(C)=O)C(C)=O JXRKBXUYUPPDAQ-UHFFFAOYSA-M 0.000 description 1
- ZQVCHTLPEJCJJD-UHFFFAOYSA-M COP(=O)([O-])OCCCNC(=O)NC(CC(C)=O)C(C)=O Chemical compound COP(=O)([O-])OCCCNC(=O)NC(CC(C)=O)C(C)=O ZQVCHTLPEJCJJD-UHFFFAOYSA-M 0.000 description 1
- JXBRHSYGTFAQBP-UHFFFAOYSA-N CSCCCCCCOP(C)N(C(C)C)C(C)C Chemical compound CSCCCCCCOP(C)N(C(C)C)C(C)C JXBRHSYGTFAQBP-UHFFFAOYSA-N 0.000 description 1
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/115—Aptamers, i.e. nucleic acids binding a target molecule specifically and with high affinity without hybridising therewith ; Nucleic acids binding to non-nucleic acids, e.g. aptamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/711—Natural deoxyribonucleic acids, i.e. containing only 2'-deoxyriboses attached to adenine, guanine, cytosine or thymine and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/001—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof by chemical synthesis
- C07K14/003—Peptide-nucleic acids (PNAs)
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/02—Applications for biomedical use
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/16—Aptamers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/321—2'-O-R Modification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/322—2'-R Modification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/352—Nature of the modification linked to the nucleic acid via a carbon atom
- C12N2310/3521—Methyl
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/50—Physical structure
- C12N2310/53—Physical structure partially self-complementary or closed
- C12N2310/531—Stem-loop; Hairpin
Definitions
- nucleic acid ligands e.g., aptamers
- vascular endothelial cells lining a blood vessel triggers a hemostatic response through a sequence of events commonly referred to as the coagulation “cascade.”
- the cascade culminates in the conversion of soluble fibrinogen to insoluble fibrin which, together with platelets, forms a localized clot or thrombus which prevents extravascular release of blood components. Wound healing can then occur followed by clot dissolution and restoration of blood vessel integrity and flow.
- Initiation of blood coagulation arises from two distinct pathways: the intrinsic and extrinsic pathways.
- the intrinsic pathway can be triggered in vitro by contact of blood borne factors with artificial negatively charged surfaces such as glass.
- the extrinsic pathway can be initiated in vivo or in vitro when tissue factor (TF), normally sequestered from the circulatory system, comes into contact with blood after injury.
- TF tissue factor
- Blood exposed TF acts as a cofactor for the factor VIIa (“FVIIa”) catalyzed activation of factor IX (“FIX”) and factor X (“FX”). This leads to rapid formation of FXa and thrombin, which subsequently polymerizes to form the fibrin clot.
- FVIIa factor VIIa
- FIX factor IX
- FX factor X
- Both the intrinsic and extrinsic pathways are characterized by the assembly of multiple protein complexes on procoagulant surfaces, which localizes the response to the site of injury (see Mann, K. G.
- Coumarin drugs such as warfarin as well as the glycosaminoglycans, heparin and heparan sulfate, are commonly used as anticoagulants.
- Warfarin a coumarin derivative, acts by competing with vitamin K dependent post-translational modification of prothrombin and other vitamin K-dependent clotting factors. Its action is somewhat slower and longer lasting effect than heparin.
- the coumarin drugs inhibit coagulation by inhibiting the vitamin K-dependent carboxylation reactions necessary to the function of thrombin, and factors VII, IX, and X as well as proteins C and S. These drugs act by inhibiting the reduction of the quinone derivatives of vitamin K to their active hydroquinone forms.
- Heparin binds to, and activates, antithrombin III which then inhibits the serine proteases of the coagulation cascade.
- heparin and LMW heparin suffer drawbacks. Uncontrolled bleeding is a major complication observed in up to 7% of patients receiving continuous infusion up to 14% of patients given intermittent bolus doses.
- the therapeutic range to achieve efficacy without placing the patient at risk for bleeding is narrow, approximately 1 to less than 3 ug heparin/ml plasma. At concentrations greater than 4 ug/ml of heparin, clotting activity is not detectable. Thus, great care must be taken to keep the patient's plasma concentrations within the therapeutic range.
- U.S. Pat. No. 6,001,820 to Hamilton® Hospitals Research Development Inc. provides heparin cofactor II specific catalytic agents which are capable of (1) selectively inactivating thrombin which is bound either to fibrin in a clot or to some other surface, but which has only minimal inhibitory activity against free thrombin; (2) inhibiting the assembly of the intrinsic tenase complex and thereby the activation of Factor X by Factor IXa; and (3) inhibiting the activation of Factor IX by Factor XIa.
- nucleic acids have conventionally been thought of as primarily playing an informational role in biological processes. In the past decade it has become clear that the three dimensional structure of nucleic acids can give them the capacity to interact with and regulate proteins.
- Such nucleic acid ligands or “aptamers” are short DNA or RNA oligomers which can bind to a given ligand with high affinity and specificity.
- the three dimensional structures of aptamers are sufficiently variable to allow aptamers to bind to and act as ligands for virtually any chemical compound, whether monomeric or polymeric. Aptamers have emerged as promising new diagnostic and therapeutic compounds, particularly in cancer therapy and the regulation of blood coagulation.
- Nucleic acid ligands can be identified through methods related to a method termed the Systematic Evolution of Ligands by EXponential enrichment (SELEX).
- SELEX involves selection of protein-binding nucleic acids from a mixture of candidate oligonucleotides and step-wise iterations of binding, partitioning and amplification to achieve the desired criterion of binding affinity and selectivity.
- the SELEX process was first described by Gold and Tuerk in U.S. Pat. No. 5,475,096, and thereafter in U.S. Pat. No. 5,270,163 (see also WO 91/19813; Tuerk et al. (1990) Science 249:505-10).
- Rusconi Kontos and White in WO 02/26932 describe RNA aptamers that bind to coagulation factors, E2F family transcription factors, Ang1, Ang2, and fragments or peptides thereof, transcription factors, autoimmune antibodies and cell surface receptors useful in the modulation of hemostasis and other biologic events. See also Rusconi et al, Thrombosis and Haemostasis 83:841-848 (2000), White et al, J. Clin Invest 106:929-34 (2000), Ishizaki et al, Nat Med 2:1386-1389 (1996), and Lee et al, Nat. Biotechnol. 15:41-45 (1997)).
- PCT Publication No. WO 02/096926 to Duke University describes agents and methods to modulate the biological activity of nucleic acid ligands through the administration of a modulator.
- the publication describes aptamers controlled by modulators that can be nucleic acids.
- the modulatable aptamers are described as being useful in the the treatment of diseases in which it is important to inhibit coagulation, elongation factor 2 activity or angiogenesis.
- the modulatable aptamers to control coagulation include the aptamers to coagulation factors VII or VIIa, VIII or VIIIa, IX or LXa, V or Va, X or Xa, complexes formed with these factors, as well as platelet receptors.
- the modulator can change the binding of the nucleic acid ligand for its target, degrade or otherwise cleave, metabolize or break down the nucleic acid ligand while the ligand is exerting its effect. Modulators can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy.
- aptamers In order for aptamers to be useful therapeutic reagents, they should bind tightly to proteins, inhibit a specified function of that protein if an antagonist is desired and have no harmful side-effects. Unmodified RNA is not realistically used as a therapeutic agent since blood is rich in ribonucleases. Some modification of single-stranded RNA and DNA can produce molecules which are stable in blood and certain known aptamers have 2′F or 2′NH 2 groups within each pyrimidine nucleotide.
- U.S. Pat. Nos. 5,670,633 and 6,005,087 to Cook et al. describe thermally stable 2′-fluoro oligonucleotides that are complementary to an RNA or DNA base sequence.
- U.S. Pat. Nos. 6,222,025 and 5,760,202 to Cook et al. describe the synthesis of 2′-O substituted pyrimidines and oligomers containing the modified pyrimidines.
- EP 0 593 901 B1 discloses oligonucleotide and ribozyme analogues with terminal 3′,3′- and 5′,5′-nucleoside bonds.
- U.S. Pat. No. 6,011,020 to Gold et al. discloses and claims an aptamer modified by polyethylene glycol.
- Improved nucleic acid ligands for anticoagulant therapy are disclosed as well as improved nucleic acid ligands in combination with an antidote that changes the binding of the nucleic acid ligand for its target or that degrades or otherwise cleaves, metabolizes or breaks down the nucleic acid ligand while the ligand is still exerting its effect.
- These improved aptamers provide favorable anticoagulant properties for in vivo applications, including during human or veterinary surgery.
- the anticoagulant function of the improved aptamer is conveniently neutralized on administration of its antidote when desired by the surgeon or orther medical care specialist.
- the aptamers of the invention include additional agents to increase bioavailability and/or decrease degradation of the active agent.
- improved nucleic acid ligands or aptamers to a factor in the blood coagulation cascade include at least one moiety that increases bioavailability of the agent.
- the factors include Factor IX (FIX) or the cleavage product Factor IXa (FIXa).
- the aptamers are ligands to the complex formed by FIXa with Factor VIIIa (FVIIIa), also known as the “intrinsic tenase complex.”
- the aptamers are ligands that inhibit the complex formation between FIXa and FVIIIa.
- the aptamers of the present invention bind to the complex of FIX and FVIIIa and inhibit activation of Factor X (FX).
- the aptamers can interact with FIX, FIXa or a complex formed with FVIIIa in the presence or absence of additional calcium.
- the aptamers can also interact with the factors of the complex at a cell membrane.
- the aptamers bind to the intrinsic tenase complex at the membrane surface.
- the aptamers are linked to a stabilizing moiety on at least one terminus.
- the aptamers are linked to a polymeric agent.
- the aptamers are linked to one or more polyethylene glycol molecules.
- the aptamer and stabilizing agent can be linked at, for example, a 5′ terminus of the nucleic acid sequence.
- the aptamers are linked to multiple polyethylene glycol molecules.
- a single polyethylene glycol molecule can be linked to more than one aptamer to provide improved delivery of the agent.
- the aptamers of the present invention can be comprised of ribonucleotides or deoxyribonucleotides, or a combination thereof.
- the improved aptamers are at least 25 nucleotides long, and typically not longer than 35-40 nucleotides long.
- aptamers are at least 25, 30, 35, or 40 nucleotides in length.
- the sequence of stem 1 includes 5 nucleotides in the 5′-3′ direction.
- stem 1 includes three guanine (G) residues in the 5′-3′ direction.
- the aptamers can include a “suicide position.” In one embodiment, this position becomes single stranded and labile upon binding of the antidote to the improved aptamer and allows for cleavage of the improved aptamer upon binding of the antidote by enzymes in the circulation, such as blood or liver endonucleases, thereby effectively eliminating the active aptamer from circulation.
- the suicide position can be at a guanine in stem 2 that is hydroxylated. In one embodiment, this nucleotide is in a double stranded configuration until bound with an antidote and becomes single stranded and available for cleavage upon binding of the antidote.
- the aptamers include the nucleotide sequence gugg and the complimentary sequence ccac.
- the aptamer to Factor IX comprises the nucleotide sequence: gugga cuauacc gcg uaaugc ugc c uccac t (SeqID 19).
- Another embodiment of the invention includes an antidote oligonucleotide paired with the aptamer of the invention.
- the antidote oligonucleotide can be complementary to at least a portion of the aptamer.
- the antidote can, for example, comprise the following sequences: (5′-3′) sequence: cgcgguauaguccccau (Apt/AD; SEQ ID NO:1); (5′-3′) sequence: cgcgguauaguccc (Apt6/AD; SEQ ID NO:2); (5′-3′) sequence: cgcgguauaguccac (Apt7/AD; SEQ ID NO:3); (5′-3′) sequence: cgcgguauaguccauc (Apt8/AD; SEQ ID NO:4); (5′-3′) sequence: cgcgguauagucag (Apt9/AD; SEQ ID NO:5); (5′-3′)
- the improved aptamer is provided in alternation with an antidote.
- the antidote sequence does not need to be completely complemetary to the improved anticoagulant aptamer as long as the antidote sufficiently binds to or hybridizes to the aptamer to neutralize its activity.
- the aptamer pairs of the present invention include the following sequences: Apta mer Antidote augggga cuauacc gcg uaaugc ugc cgcgguauaguccccau c uccccau t (SEQ ID NO:1) (SEQ ID NO:9) augggga cuauaccgcguaaugcugcc cgcgguauaguccccau uccccau t (SEQ ID NO:1) (SEQ ID NO:10) ggga cuauaccgcguaaugcugcc uccc t cgcgguauaguccc (SEQ ID NO:11) (SEQ ID NO:2) gugga cuauaccgcguaaugcugcc c cgcgguauaguccac uccac t (SEQ ID NO:3) (SEQ ID NO:12) gaugga cuauaccgcguaaugcugcc cgcgguau
- Improved aptamer-antidote pairs that are more stable and bioactive are developed by including secondary modifications on either the aptamer or antidote or both.
- the improved aptamer to Factor IX includes one or more 2′-O-methyl modified nucleotides.
- the improved aptamer contains one or more 2′-O-methyl and one or more 2′-fluoro modifications.
- the aptamer and antidote contain no 2′-fluoro modifications.
- the improved aptamer includes one or more 2′-O-methyl and one or more 2′-fluoro modifications on a stem.
- At least one guanine in stem 2 of an improved aptamer includes a hydroxyl sugar (2′—OH).
- at least one uridine in stem 1 or in stem 2 of the improved aptamer is modified with either a 2′-fluoro or 2′-O-methyl.
- at least one cytidine in stem 2 of the improved aptamer is 2′-fluoro modified.
- the aptamer is linked to one or more polyethylene glycol (PEG) molecule.
- the aptamer is linked to 40 KD PEG using a six carbon amino linker.
- the six carbon amino linker is attached to the PEG through an amino acid attachment.
- the PEG is two twenty KD PEG that are attached to one or more amino acid, such as lysine, which is attached to the six carbon amino linker.
- the aptamer to Factor IXa is the following structure: 5′-O-[[5-[N 2 -(monomethoxy 20K polyethylene glycol carbamoyl)-N 6 -(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycyt
- the aptamer to Factor IXa is of the following structure: where n is approximately 450
- the aptamer to Factor IXa is 10,282.4 Daltons in its protonated form. In another embodiment, the aptamer is in its sodium salt form. In yet another embodiment, the aptamer is PEGylated RB006 and in its sodium salt.
- the improved aptamers and antidotes can also include nucleotides that are modified with water-soluble polymers.
- Such polymers can include a polyethylene glycol, polyamine, polyether, polyanhydride, polyester, or other biodegradable pharmaceutically acceptable polymer.
- the invention includes the use of the improved aptamers to bind to FIX, FIXa, or the intrinsic tenase complex. This binding can be in vitro or in vivo. The result of the binding to FIX, FIXa or the tenase complex can be to inhibit the biological activity of the proteins or complex.
- the improved aptamer inhibits blood coagulation by binding to FIXa, which is derived from the same gene product as FIX.
- the invention includes administering the improved aptamers of the invention to a mammal in need thereof to inhibit blood coagulation.
- Another embodiment of the invention provides methods of using the improved aptamers and antidotes during a therapeutic regime.
- antidotes to the improved aptamers of the invention are provided to a mammal in need thereof to reverse the anticoagulant effects of the improved aptamers.
- Improved aptamers and aptamer-antidote pairs can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy.
- this invention discloses an improved regulatable therapeutic regime in the course of nucleic acid ligand therapy for blood coagulation.
- an antidote is provide that neutralizes the effect of the improved aptamer to turn off anticoagulant activity when desired by the physician or other health care provider.
- the improved aptamers and antidotes to blood coagulation factors are administered in sequential steps, in which the aptamers are administered, the antidotes are used to limit the activity of the improved aptamers, and subsequently the aptamers are re-administered to a patient in need thereof.
- the antidote achieves this neutralization effect by binding to or hybridizing to the improved aptamer.
- the improved aptamers can be administered to patients suffering from or at risk of suffering from a cardiovascular disease or intervention, including surgical intervention, that causes or results in a coagulation-inducing event.
- cardiovascular disease or intervention including surgical intervention, that causes or results in a coagulation-inducing event.
- Examples include acute myocardial infarction (heart attack), cerebrovascular accidents (stroke), ischemia, angioplasty, CABG (coronary artery bypass grafts), cardiopulmonary bypass, thrombosis in the circuit of cardiac bypass apparatus and in patients undergoing renal dialysis, unstable angina, pulmonary embolism, deep vein thrombosis, arterial thrombosis, and disseminated intravascular coagulation.
- the improved aptamers can also be administered to prevent coagulation-induced inflammation. It appears that early inflammation is induced by activation of the coagulation cascade. Therefore, the improved aptamers can be used to treat cardiovascular diseases that include a inflammatory component, for example, atherosclerosis, acute coronary syndrome (ACS), myocardial infarction which may result in reperfusion injury, or to treat adverse events associated with post-angioplasty restenosis.
- cardiovascular diseases that include a inflammatory component, for example, atherosclerosis, acute coronary syndrome (ACS), myocardial infarction which may result in reperfusion injury, or to treat adverse events associated with post-angioplasty restenosis.
- FIG. 1 is a schematic of proposed two dimensional configurations of Apt A, 1-39 described below.
- FIG. 1 a is a schematic of aptamers Apt A and 1-5.
- 1 b is a schematic of aptamers 6-11.
- 1 c is a schematic of aptamers Apt 12-17.
- 1 d is a schematic of aptamers Apt 18-20, 1 e of Apt 21.
- 1 f is a schematic of aptamers Apt 22-29, 1 g of Apt 30-34 and 1 h of Apt 35-39.
- FIG. 2 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A and Apt 1-5 (left panel) and neutralizability by antidote AptA-AD.
- APTT activated partial thromboplastin time
- FIG. 3 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 6-8 (right panel) and neutralizability by antidote (left panel).
- APTT activated partial thromboplastin time
- FIG. 4 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 9-11 (left panel) and neutralizability by antidote (right panel).
- APTT activated partial thromboplastin time
- FIG. 5 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A, 2 and 12-17 (left panel) and neutralizability by antidote (right panel).
- APTT activated partial thromboplastin time
- FIG. 6 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2, 15, 16 and 21 (left panel) and neutralizability by antidote (right panel).
- APTT activated partial thromboplastin time
- FIG. 7 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 16-20 (left panel) and neutralizability by antidote (right panel).
- APTT activated partial thromboplastin time
- FIG. 8 is a graph of results of activated partial thromboplastin time (APTT) test assays of pegylated AptA compared to pegylated Apt 16 and 19 and cholesterol-modified Apt A (chol-A). Left panel is aptamer control of coagulation factor IX and right panel is neutralizability by antidote.
- APTT activated partial thromboplastin time
- FIG. 9 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2, 16 and 22-27 (left panel) and neutralizability by antidote (right panel).
- APTT activated partial thromboplastin time
- FIG. 10 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 30-33 (A, left panel), neutralizability by antidote of Apt 2, 30 and 33 (A, right panel), APTT test assays of aptamers Apt 2, 30, 33 and 34 (B, left panel) and neutralizability by antidote (B, right panel).
- APTT activated partial thromboplastin time
- FIG. 11 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A, 2, 19 and 35-39 (left panel) and neutralizability by antidote of Apt A, 19,35,38 and 39(right panel).
- APTT activated partial thromboplastin time
- FIG. 12 is a graph of results of neutralizability by antidote assays of aptamers Apt 2, 34, 39 and Peg-19.
- FIG. 13 is a graph of the in vitro anticoagulant activity of PEG-Apt39 compared to CH-AptA and PEG-AptA.
- FIG. 14 are graphs of systemic anticoagulant activity ( 14 a ) and neutralizability ( 14 b ) of PEG-Apt39 in swine.
- the change in the value of the respective clotting assays is the difference between the clotting time at the time point and the pre-injection baseline for that animal.
- Whole blood ACT values are shown in the bottom panel, and plasma APTT values in the panel at the top.
- FIG. 15 is graphs of systemic anticoagulant activity ( 14 a ) and neutralizability ( 14 b ) of PEG-Apt39 in swine.
- Data from this experiment is compared to the anticoagulation and neutralization data for PEG-Apt39 presented in FIG. 3 .
- Whole blood ACT values are shown in the left panel, and plasma APTT values in the panel at right.
- FIG. 16 is a graph of systemic anticoagulation of monkeys by Apt39 administration as described in Example 9. The level of anticoagulation in the monkeys was monitored with the APTT. For animals treated with 15 mg/kg, Apt39 data are presented as the mean ⁇ SEM. For animals at the 5 and 30-mg/kg dose levels, data are presented as the mean ⁇ range, as there were only 2 animals at each of these dose levels.
- a “nucleic acid ligand” or “aptamer” is a nucleic acid that can form a three dimensional configuration, which allows it to interact as a ligand with a target molecule.
- the terms refer to oligonucleotides having specific binding regions that are capable of forming complexes with an intended target molecule in an environment wherein other substances in the same environment are not complexed to the oligonucleotide.
- the specificity of the binding is defined in terms of the comparative dissociation constants (K d ) of the aptamer for target as compared to the dissociation constant with respect to the aptamer and other materials in the environment or unrelated molecules in general.
- the K d for the aptamer with respect to the target will be 10-fold , 50-fold, 100-fold, or 200-fold less than the K d with respect to the unrelated material or accompanying material in the environment.
- “Aptamer antidote pair” is meant to include a specified aptamer to a target molecule, and an oligonucleotide that changes the three dimensional configuration of the aptamer so that the aptamer can no longer interact with its target.
- the antidote can be an oligonucleotide complimentary to a portion of the aptamer.
- the antidote can change the conformation of the aptamer to reduce the target binding capacity of the aptamer by 10 to 100%, 20 to 100%, 25%, 40%, 50%, 60%, 70%, 80%, 90% or 100%, or any percentage in the range between 10 and 100% under physiological conditions.
- the antidote can also form a three dimensional structure with binding activity to a target molecule. This target can be the same or different from the target of the aptamer.
- Antidote refers to any pharmaceutically acceptable agent that can bind an aptamer and modify the interaction between that aptamer and modify the interaction between that aptamer and its target molecule (e.g., my modifying the structure of the aptamer) in a desired manner.
- binding activity and “binding affinity” are meant to refer to the tendency of a ligand molecule to bind or not to bind to a target.
- the energy of said interactions are significant in “binding activity” and “binding affinity” because they define the necessary concentrations of interacting partners, the rates at which these partners are capable of associating, and the relative concentrations of bound and free molecules-in a solution.
- the specificity of the binding is defined in terms of the comparative dissociation constants (K d ) of an antidote of a nucleic acid ligand as compared to the dissociation constant with respect to other materials in the environment or unrelated molecules in general.
- Consensus sequence refers to a nucleotide sequence or region (which might or might not be made up of contiguous nucleotides) that is found in one or more regions of at least two nucleic acid sequences.
- a consensus sequence can be as short as three nucleotides long. It also can be made up of one or more noncontiguous sequences, with nucleotide sequences or polymers of up to hundreds of bases long interspersed between the consensus sequences. Consensus sequences can be identified by sequence comparisons between individual nucleic acid species, which comparisons can be aided by computer programs and other, tools for modeling secondary and tertiary structure from sequence information. Generally, the consensus sequence will contain at least about 3 to 20 nucleotides, more commonly from 6 to 10 nucleotides.
- cardiovascular disease and “cardiovascular diseases” are meant to refer to any cardiovascular disease as would be understood by one of ordinary skill in the art.
- cardiovascular diseases include, but are not limited to, atherosclerosis, thrombophilia, embolisms, cardiac infarction (e.g., myocardial infarction), thromboses, angina, stroke, septic shock, hypertension, hyper-cholesterolemia, restenosis and diabetes (and associated diabetic retinopathy).
- Cardiovascular disease can be treated at any stage of progression, such as treatment of early onset cardiovascular disease as well as treatment of advanced cardiovascular disease.
- a therapeutic method directed toward-inhibiting the aggravation of cardiovascular disease by modulating coagulation is also included in the invention.
- the invention provides improved nucleic acid ligands or aptamers that regulate blood coagulation through interaction with specific factors in the blood coagulation cascade.
- the invention also provides improved aptamer-antidote pairs to regulate coagulation.
- the improved aptamers target Factor IX gene products(which include Factor IXa) and thus reduce the non-specific side effects associated with other blood coagulation factor targets.
- Most factors in the coagulation cascade are broad spectrum proteins with a variety of physiological roles (i.e. thrombin).
- tissue factor-bearing cells monocytes, macrophages, endothelial cells.
- FVIIa complexed with tissue factor
- FIX and FX generate a small amount of thrombin from prothrombin (which subsequently activates FV)
- the amplification phase also referred to as the priming phase
- the small amount of thrombin generated activates platelets, causing release of FVa, FXIa and FVIIIa.
- FXa-FVa complex During the final phase of coagulation, propagation, FIXa complexes with FVIIIa, activating FX.
- the FXa-FVa complex in the presence of calcium and phospholipids substrate (prothrombinase complex), leads to a “burst” of thrombin generation.
- thromboin is a broad acting protein with effects throughout the body. Inhibitors of thrombin can therefore have unanticipated side effects in addition to the effects on coagulation. Thrombin not only activates endothelial cells and induces leukocyte infiltration and edema but also activates astrocytes and microglia to propagate the focal inflammation and produce potential neurotoxic effects.
- Factor IXa in particular represents an attractive target because of its participation in both the initiation and propagation phases of coagulation. Iteractive in vitro selection techniques have been used to identify oligonucleotides capable of binding FIXa with high affinity (K d 0.65 ⁇ 0.2 nM). Experimental studies suggest that FIXa may have a critical role in thrombosis, as well as hemostasis. Infusion of purified FIXa into rabbits induces thrombosis (Gitel et al. (1977) PNAS 74:3028-32; Gurewich et al. (1979) Thromb. Rsch. 14:931-940). In contrast, active site-blocked FIXa prevented clot formation and reduced intra-arterial coronary thrombosis (Lowe (2001) Brit. J. Haem. 115:507-513).
- Antibodies to factor IX have also been shown to interfere with the function of the intrinsic tenase complex, the activation of zymogen factor IX by factor XIa and by the tissue factor:factor VIIa complex and potently inhibit activated partial thromboplastin clotting times (APTT) in plasma of guinea pig and rat (Refino, C. J., et al, (1999) Thromb and Haemost, 82:1188-1195; Feuerstein G Z, et al. (1999) Arterioscler Thromb Vasc Biol 19(10):2554-62; Toomey J R, et al. (2000) Thromb Res. 100(1):73-9).
- APTT activated partial thromboplastin clotting times
- the invention provides nucleic acid ligands or aptamers to a factor in the blood coagulation cascade.
- the factors include Factor IX (FIX) or the cleavage product Factor IXa (FIXa).
- the aptamers are ligands to the complex formed by FIXa with Factor VIIIa (FVIIIa), also known as the “intrinsic tenase complex.”
- the aptamers are ligands that inhibit the complex formation between FIXa and FVIIIa.
- the aptamers of the present invention bind to the complex of FIX and FVIIIa and inhibit activation of Factor X (FX).
- the aptamers can interact with FIX, FIXa or a complex formed with FVIIIa in the presence or absence of additional calcium.
- the aptamers can also interact with the factors of the complex at a cell membrane.
- the aptamers bind to the intrinsic tenase complex at the membrane surface.
- the applicants have discovered improved aptamers to gene products of coagulation Factor IX (FIX), and to its cleavage product, Factor IXa (FIXa).
- the nucleic acid ligand includes at least one region that binds to another region in the molecule via Watson-Crick base pairing (stem) and at least one region that does not bind to any other regions of the molecule under physiological conditions (loop).
- the nucleic acid ligand includes two stems (stem 1 and stem 2) and two loops (loop 1 and loop 2).
- stem 1 is one to twenty nucleotides long.
- stem 1 is one to ten nucleotides long.
- stem 1 is seven, six, five, four, three or two nucleotides long.
- stem 2 one to twenty nucleotides long.
- stem 2 is one to ten nucleotides long.
- stem 2 is seven, six, five, four, three or two nucleotides long.
- the aptamers to a Factor IX gene product of the present invention can be comprised of ribonucleotides or deoxyribonucleotides, or a combination thereof.
- the improved aptamers are at least 25 nucleotides long, and typically not longer than 35-40 nucleotides long.
- aptamers are at least 25, 30, 35, or 40 nucleotides in length.
- the sequence of stem 1 includes 5 nucleotides in the 5′-3′ direction.
- stem 1 includes three guanine (G) residues in the 5′-3′ direction.
- the aptamers include the consensus nucleotide sequences gugg and the complimentary sequence ccac.
- sequences can be examined for “consensus sequences.”
- Consensus sequence refers to a nucleotide sequence or region (which might or might not be made up of contiguous nucleotides)that is found in one or more regions of at least two aptamers, the presence of which can be correlated with aptamer-to-target-binding or with aptamer structure.
- a consensus sequence can be as short as three nucleotides long. It also can be made up of one or more noncontiguous sequences. With nucleotide sequences or polymers of hundreds of bases long interspersed between the consensus sequences. Consensus sequences can be identified by sequence comparisons between individual aptamer species, which comparisons can be aided by computer programs and other, tools for modeling secondary and tertiary structure from sequence information. Generally, the consensus sequence will contain at least about 3 to 20 nucleotides, more commonly from 6 to 10 nucleotides. Not all oligonucleotides in a mixture can have the same nucleotide at such position; for example, the consensus sequence can contain a known ratio of particular nucleotides.
- a consensus sequence might consist of a series of four positions wherein the first position in all members of the mixture is A, the second position is 25% A, 35% T and 40% C, the third position is T in all oligonucleotides, and the fourth position is G in 50% of the oligonucleotides and C in 50% of the oligonucleotides.
- the aptamers include the nucleotide sequences of the following Seq ID Nos.: SeqID Code Size Sequence 9 AptA 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 10 Apt1 35 mer (5′-3′) sequence: augggga cuaua ccgcguaaugcugcc uccccau t 9 Apt2 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 9 Apt3 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 9 Apt4 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c
- the aptamer to Factor IX comprises, consists, or consists essentially of, the nucleotide sequence: gugga cuauacc gcg uaaugc ugc c uccac t (SeqID 19).
- the improved aptamers and aptamer-antidote combinations of the present invention are modified by substituting particular sugar residues, by changing the composition of the aptamer and the size of particular regions in the aptamer, and by designing aptamers that can be more effectively inhibited by antidotes.
- the design of aptamers includes an appreciation for the secondary structure of the aptamer (see FIG. 1 ) and the relationship between the secondary structure and the antidote control. Unlike conventional methods of modifying nucleic acids, the design of the improved aptamers to FIX gene products included in the invention must include a consideration of the antidote control.
- Controlled aptamers require that the aptamer be stable in circulation but not so stable that it is not antidote controlled.
- the aptamers can be modified by truncation, but antidotes need to be designed to control each aptamer when truncated. Further, certain modifications, particularly at the interface of the stems and loops cannot be modified from 2′-fluoro or the aptamer can lose activity.
- the design includes decreasing the 2′-hydroxyl content of the aptamer or the antidote, or both. In another embodiment, the design includes decreasing the fluoro content of the aptamer or the antidote, or both. In a further embodiment, the design includes increasing the 0-methyl content of the aptamer or the antidote, or both. In a further embodiment, the design includes decreasing the size of the aptamer. In another embodiment, the size of the antidote is changed in relation to the size of the aptamer. In yet another embodiment, guanine strings are reduced to less than four guanine, or less than three guanine, or less than two guanine or no guanines. However, the joint effect of these changes must meet the challenge of creating an anticoagulant that provides adequate activity but is easily neutralized by the antidote.
- Yet another embodiment includes a method of designing aptamers with a “suicide position” which allows more effective regulation by paired antidotes.
- this position becomes single stranded and labile upon binding of the antidote to the improved aptamer and allows for cleavage of the improved aptamer upon binding of the antidote by enzymes in the circulation, such as blood or liver endonucleases, thereby effectively eliminating the active aptamer from circulation.
- the suicide position can be, in one embodiment, at a guanine in stem 2 that is hydroxylated.
- the aptamer is in a double stranded configuration until bound with an antidote and becomes single stranded and available for cleavage upon binding of the antidote.
- the aptamers to Factor IX include modified nucleotides.
- the aptamer contains one or more 2′-O-methyl groups.
- the aptamer and antidote contain one or more 2′-O-methyl and one or more 2′-fluoro modifications.
- the aptamer and antidote contain no 2′-fluoro modifications.
- the aptamer includes one or more 2′-O-methyl and one or more 2′-fluoro modifications on its stem.
- the aptamers can also include nucleotides that are modified with soluble polymers. Such polymers can include polyethylene glycol, polyamines, polyesters, polyanhydrides, polyethers or other water soluble pharmaceutically acceptable polymer.
- Purines within given aptamer sequence of FIX inhibitor can tolerate substitution of 2′-O-methyl sugars for current 2′hydroxyl sugars (Example 1, FIG. 1 ).
- the aptamers fall into three classes: (1) gain of anticoagulant activity (Apt4); (2) moderate loss of activity (Apt-1, 2, and 3); and (3) severe loss of activity (Apt 5) ( FIG. 2 ).
- Data from Apt-5 indicates that the impact of wholly substituting 2′-O-methyl purines for 2′hydroxyl purines is significantly greater than any individual sector substitution alone ( FIG. 2 ). In the case of this aptamer, it is possible that this suggests potential interaction between sectors, or that impairment caused by substitution within one of the sectors is exacerbated by additional modifications (ie.
- one of the sectors is an Achilles heel).
- the enhanced antidote control exhibited by Apt-1, 2 and 3 suggests that introduction of 2′O-methyl residues within the antidote binding site improves the ability of the antidote oligonucleotide to bind to the aptamer. This is consistent with the increase in thermodynamic stability observed for duplexes containing 2′-O-methyl RNA residues in each strand, and suggests that duplexes of 2′ -O-methyl-2′-O-methyl strands are more thermodynamically stable than duplexes composed of 2′O-methyl-2′fluoro strands.
- An alternative conclusion is that the reduction in activity of Apt-1, 2 and 3 leads to more “free” aptamer in the plasma at any given time, which is thus more readily bound by the antidote oligonucleotide
- aptamers of the present invention can include modified pyrimidine nucleosides. Replacing 2′fluoropyrimidines with 2′-O-methyls within stem 1 improved activity and yielded a compound that tolerates a greater level of substitution. Comparison of the activity of Apt 30 and 33 to Apt 31 and 32 demonstrates that C16 needs to contain a 2′fluoro sugar and G25 a 2′hydroxyl sugar ( FIG. 16 a ). Activity observed between Apt 31 and 32 suggests that remaining positions within stem 2 can contain 2′-O-methyl sugars.
- Apt 31 appears to possess slightly greater potency than Apt 32, indicating that a compound with 2′fluoro at C16, 2′hydroxyl at G25, and the remaining residues 2′-O-methyl may exhibit greater potency than Apt 33.
- Apt 33 is more readily neutralizable than Apt 30, suggesting additional 2′-O-methyl residue within the antidote-binding site of the aptamer improves antidote binding.
- Apt 34 have C16 as a 2′fluoro rather than 2′-O-methyl nucleoside ( FIG. 16 b ).
- At least on guanine in stem 2 of an aptamer includes a hydroxyl sugar (2′—OH).
- at least one uridine in stem 1 or stem 2 is a modified base. This can be either a 2′-fluoro (2′-F) or 2′-O-methyl (2′—OCH 3 ) modification.
- at least one uridine in stem 1 or stem 2 is 2′-O-methyl modified.
- at least one cytidine in stem 2 is modified.
- at least on cytidine in stem 2 is 2′-Fluoro modified.
- aptamers may ensure stability but they do not guarantee adequate pharmacokinetics for aptamers to be therapeutically active.
- aptamers are cleared from plasma within minutes of IV injection, probably through renal excretion. Keeping intact aptamers in the blood from hours to days after injection has been accomplished by conjugating them to larger macromolecules such as polyethyleneglycol (PEG).
- PEG polyethyleneglycol
- aptamer plasma clearance has also been decreased by embedding them in liposomes.
- Nucleic acid aptamers of the present invention can also be modified by varying the stem and loop sizes.
- Two families of aptamers with four, five, or six 2-O-methyl modified base pair stem 1 regions showed varying levels of anticoagulant activity and antidote control (see Example 2, FIGS. 3-5 ).
- Stem 1 mutants FIG. 3
- All stem 1 variants exhibit less activity than the fully 2′-O-methyl purine/2′fluoro pyrimidine compound Apt 5, suggesting that one of the pyrimidines within stem 1 must contain a 2′fluoro sugar for the compound to retain potency.
- stem length may not cause loss of activity.
- 5 base pair stem 1 constructs (Apt 10 and 7) do appear to be more readily antidote controlled than six base pair. Data suggests that a stem 1 of 5 base pairs may be preferable to those composed of 4, 6 or 7 base pairs to enhance antidote neutralization.
- an improved aptamer can also be modified so as to include a single-stranded tail (3′ or 5′) in order to promote association with an oligonucleotide antidote.
- Suitable tails can comprise 1 to 20 nucleotides, preferably, 1-10 nucleotides, more preferably; 1-5 nucleotides and, most preferably, 3-5 nucleotides (e.g., modified nucleotides such as 2′-O-methyl sequences).
- Tailed aptamers can be tested in binding and bioassays (e.g., as described below) to verify that addition of the single-stranded tail does not disrupt the active structure of the aptamer.
- a series of oligonucleotides (for example, 2′-O-methyl oligonucleotides) that can form, for example, 1, 3 or 5 basepairs with the tail sequence can be designed and tested for their ability to associate with the tailed aptamer alone, as well as their ability to increase the rate of dissociation of the aptamer from, or association of the aptamer with, its target molecule. Scrambled sequence controls can be employed to verify that the effects are due to duplex formation and not non-specific effects.
- the aptamers include the nucleotide sequences of any of the following sequences.
- A is 2′OH A; “a” is 2′-O-methyl A; “G” is 2′—OH G; “g” is 2′-O-methyl G; “C” is 2′-Fluoro C; “c” is 2′-O-methyl C; “U” is 2° Fluoro U; “u” is 2′-O-methyl U; and “T” is inverted 2′H T.
- SeqID Code Sequence 20 AptA AUGGGGA CUAUACC GCG UAAUGC UGC C UCC CCAU T 21 Apt1 aUgggga CUAUACCGCGUAAUGCUGCC UCCCCaU T 22 Apt2 AUGGGGA CUaUaCC GCG UAAUGC UGC C UCC CCAU T 23 Apt3 AUGGGGA CUAUACC gCg UAAUGC UgC C UCC CCAU T 24 Apt4
- the aptamer to Factor IXa comprises, consists, or consists essentially of, the nucleotide sequence: gugga CUaUaCC gCg UaaUgC uGc C Uccac T (Apt39; SEQ ID NO: 59).
- the aptamer is linked to one or more polyethylene glycol (PEG) molecules.
- the aptamer is linked to 40 KD PEG using a six carbon amino linker.
- one or more phosphate groups are included between the linker and the nucleic acid sequence.
- the six carbon amino linker is attached to the PEG through an amino acid attachment.
- the PEG is two twenty KD PEG that are attached to one or more amino acid, such as lysine, which is attached to the six carbon amino linker.
- the aptamer to Factor IXa is the following structure: 5′-O-[6-[N 2 -(monomethoxy 20K polyethylene glycol carbamoyl)-N 6 -(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycyti
- the aptamer to Factor IXa is of the following structure: where n is approximately 450
- the molecular weight of the aptamer to Factor IXa is 10,282.4 Daltons in its protonated form.
- the aptamer is in its sodium salt form.
- the aptamer is PEGylated Apt39 and in its sodium salt.
- the oligonucleotide antidotes can be administered directly (e. g., alone or in a liposomal formulation or complexed to a carrier, e.g. PEG)) (see for example, U.S. Pat. No. 6,147,204, U.S. Pat. No. 6,011,020).
- a carrier e.g. PEG
- FIG. 10 shows the activity and neutralizability of a pegylated aptamer with a 5 base pair stem (Apt 19).
- Apt 19 possesses anticoagulant activity very similar to pegylated Apt16 with a 7 base pair stem 1, but ⁇ 90% of its activity can be neutralized with only a 2.5:1 excess of antidote to drug.
- the improved aptamer or antidotes can be attached to a non-immunogenic, high molecular weight compound such as polyethylene glycol (PEG) or other water soluble pharmaceutically acceptable polymer as described herein.
- PEG polyethylene glycol
- the aptamer or antidote is associated with the PEG molecule through covalent bonds. Where covalent attachment is employed, PEG may be covalently bound to a variety of positions on the improved aptamer or antidote.
- an oligonucleotide aptamer or antidote is bonded to the 5′-thiol through a maleimide or vinyl sulfone functionality.
- a plurality of improved aptamers or antidotes can be associated with a single PEG molecule.
- the improved aptamers and antidotes can be the same or different sequences and modifications.
- a plurality of PEG molecules can be attached to each other.
- one or more aptamers or antidotes to the same target or different targets can be associated with each PEG molecule.
- multiple aptamers or antidotes specific for the same target are attached to PEG, there is the possibility of bringing the same targets in close proximity to each other in order to generate specific interactions between the same targets.
- aptamers or antidotes specific for different targets are attached to PEG, there is the possibility of bringing the distinct targets in close proximity to each other in order to generate specific interactions between the targets.
- a drug can also be associated with PEG.
- the complex would provide targeted delivery of the drug, with PEG serving as a Linker.
- Linkers can be selected from, for example, 6-(trifluoroacetamido)hexanol (2-cyanoethyl-N,N-diisopropyl)phosphoramidite of the structure: TFA-amino C4 CED phosphoramidite (available from ChemGenes, cat# CLP-1453) of the structure: 5′-amino modifier C3 TFA (available from Glen Research cat# 10-1923-90) of the structure:
- 5′-amino modifier 5 available from Glen Research cat# 10-1905-90 of the structure:
- 5′thiol-modifier C6 (available from Glen Research cat# 10-1926-90) of the structure:
- the 5′-thiol modified linker is used with PEG-maleimides, PEG-vinylsulfone, PEG-iodoacetamide and PEG-orthopyridyl-disulfide, for example.
- PEGs Polyethylene glycols
- PEGs can be conjugated to biologically active compounds to serve as “inert” carriers to potentially (1) prolong the half-life of the compound in the circulation, (2) alter the pattern of distribution of the compound and/or (3) camouflage the compound, thereby reducing its immunogenic potential and protecting it from enzymatic degradation.
- PEGs can range in size from 5 to 200 KD, with typical PEGs used in pharmaceutical formulations in the 10-60 KD range. Linear chain PEGs of up to about 30 KD can be produced. For PEGs of greater than 30 KD, multiple PEGs can be attached together (multi-arm or ‘branched’ PEGs) to produce PEGs of the desired size.
- 40 KD total molecular weight PEGs that can be used as reagents in producing a PEGylated compound, include, for example, [N 2 -(monometioxy 20K polyethylene glycol carbamoyl)-N 6 -(monomethoxy 20K polyethylene glycol carbamoyl)]-lysine N-hydroxysuccinimide of the structures: (mPEG is a 20 KD monomethoxy PEG).
- Additional PEG reagents that can be used to prepare stabilized compounds of the invention include other branched PEG N-Hydroxysuccinimide (mPEG-NHS) of the general formula: with a 40 KD or 60 KD total molecular weight (where each mPEG is about 20 or about 30 KD).
- mPEG-NHS branched PEG N-Hydroxysuccinimide
- Compounds of this structure are sold, for example, by Nektar Therapeutics as cat# 2Z3Y0L01 and cat# 2Z3Y0V01.
- the branched PEGs can be linked through any appropriate reagent, such as an amino acid, and in certain embodiments are linked via lysine residues or glycerine residues.
- mPEG-SPA non-branched mPEG-Succinimidyl Propionate
- mPEG-SPA non-branched mPEG-Succinimidyl Propionate
- the reactive ester is —O—CH2CH2—C02—NHS.
- Nektar Therapeutics as catalog numbers #2M4M0P01.
- the reagents can also include a branched PEG linked through glycerol, such as the Sunbright TM series from NOF Corporation, Japan. Specific, non-limiting examples of these reagents are:
- the reagents can also include non-branched Succinimidyl alpha-methylbutanoate (mPEG-SMB) of the general formula: in which mPEG is between 10 and 30 KD.
- mPEG-SMB non-branched Succinimidyl alpha-methylbutanoate
- the reactive ester is —O—CH 2 CH 2 CH(CH 3 )—CO 2 —NHS.
- Nektar Therapeutics catalog numbers cat#2M4K0R01.
- PEG reagents can also include nitrophenyl carbonate linked PEGs, such as of the following structure: Compounds of this structure are commercially available, for example from Sunbio, Inc. Compounds including nitrophenyl carbonate can be conjugated to primary amine containing linkers. In this reaction, the 0-nitrophenyl serves as the leaving group, leaving a structure [mPEG] n -NH—CO—NH-linker-ligand.
- PEGs with thiol-reactive groups that can be used with a thiol-modified linker include compounds of the general structure: in which mPEG is about 10, about 20 or about 30 KD. Additionally, the structure can be branched, such as in which each mPEG is about 10, about 20, or about 30 KD and the total mass is about 20, about 40, or about 60 KD.
- Branched PEGs with thiol reactive groups that can be used with a thiol-modified linker, as described above include compounds of the general structure (need structure), in which the branched PEG has a total molecular weight of about 40 or 60 KD (where each mPEG is 20 or 30 KD).
- PEG reagents can also be of the following structure: PEG reagents of this structure are sold, for example, by Sunbio, Inc. PEG-maleimide pegylates thiols of the target compound in which the double bond of the maleimic ring breaks to connect with the thiol.
- the rate of reaction is pH dependent and, in one embodiment, is carried out between pH 6 and 10, or between pH 7 and 9 or about pH 8.
- the aptamers of the invention have the general structure.
- the aptamer has the following structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamer is of the structure:
- Ligand oligonucleotide of SEQ ID NO:59
- n approximately 450
- the aptamers or antidotes of the invention can also include other conjugate groups covalently bound to functional groups such as primary or secondary hydroxyl groups.
- Conjugate groups of the invention include polyamines, polyamides, polyethylene glycols, polyethers, groups that enhance the pharmacodynamic properties of oligomers, and groups that enhance the pharmacokinetic properties of oligomers.
- Groups that enhance the pharmacodynamic properties include groups that improve oligomer bioavailability, enhance oligomer resistance to degradation, and/or strengthen sequence-specific hybridization with RNA.
- the improved aptamers described herein can be manufactured using techniques known in the art.
- U.S. patents have issued describing methods of large scale manufacturing that can be used to manufacture aptamers.
- Caruthers et al. describe in U.S. Pat. Nos. 4,973,679; 4,668,777; and 4,415,732 a class of phosphoramidite compounds that are useful in the manufacture of oligonucleotides.
- Caruthers et al. disclose a method of synthesizing oligonucleotides using an inorganic polymer support. See, e.g., U.S. Pat. Nos. 4,500,707, 4,458,066 and 5,153,319.
- Caruthers et al. discloses a class of nucleoside phosphorodithioates that can be used to manufacture oligonucleotides. See, e.g., U.S. Pat. Nos. 5,278,302, 5,453,496 and 5,602,244.
- manufacture of improved nucleic acid ligands of the invention is a multi-step process involving solid phase chemical synthesis of the oligonucleotide strand; cleavage and deprotection of the crude oligonucleotide; purification by preparative anion exchange chromatography; desalting followed by PEGylation; purification of the PEGylated oligonucleotide by preparative anion exchange chromatography to remove unPEGylated oligonucleotide impurities; ultrafiltration for desalting; concentration and lyophilization of the final product.
- Chemical synthesis via phosphoramidite chemistry involves sequential coupling of activated monomers to an elongating polymer, one terminus of which is covalently attached to a solid support matrix.
- the general steps include protecting a 3′-hydroxy of a nucleoside, coupling a 5′-carbon to a solid support, deprotecting, coupling a phosphopramidate-linked, protected nucleoside to the 3′-hydroxy, deprotecting, sequentially reacting phosphoramidate-linked nucleosides to build the molecule, coupling the terminal 5′-nucleoside with a linker and reacting with an inert carrier, such as a linked PEG.
- the solid phase approach allows for easy purification of the reaction product at each step in the synthesis by simple solvent washing of the solid phase.
- the oligonucleotides are sequentially assembled from the 3′- end towards the 5′- end by deprotecting the 5′- end of the support-bound molecule, allowing the support-bound molecule to react with an incoming tetrazole-activated phosphoramidite monomer, oxidizing the resulting phosphite triester to a phosphate triester, and blocking any unreacted hydroxyl groups by acetylation (capping) to prevent non-sequential coupling with the next incoming monomer to form a “deletion sequence”.
- This sequence of steps is repeated for subsequent coupling reactions until the full-length oligonucleotides are synthesized. Due to the presence of a 3′-3′ linkage at the 3′ end and a C-6 linker for PEGylation at the 5′ end, the synthesis is modified at the first and last step to accommodate these changes.
- the oligonucleotide can then be cleaved from the solid support and protecting groups on the linker are removed.
- the support-attached intermediate is cleaved using basic conditions at an elevated temperature.
- the temperature can be from about 40 to about 55° C., or from about 42 to about 50° C., or from about 45 to about 50° C.
- An exemplary reaction is carried out in ammonium hydroxide and tert-butylamine.
- the protecting groups on the linker are removed by incubation with an appropriate deprotecting reagent at reduced temperature.
- An exemplary deprotecting reagent is triethylamine trishydrofluoride.
- the solid-support and associated oligonucleotide are transferred to a filter funnel, dried under vacuum and transferred to a reaction vessel.
- Ammonium hydroxide (28-30%) and tert-butylamine are added and the mixture is heated to approximately 45° C. for approximately 15 hours to effect cleavage from the solid support, removal of the cyanoethyl phosphate protecting group, deprotection of exocyclic amine protecting groups as well as removal of the trifluoroacetyl group from the linker.
- the sample is cooled and the 2′-TBDMS protecting group is then cleaved by addition of Et 3 N.3HF in DMSO followed by heating the sample to 45° C. for up to 5 hours to yield the crude oligoribonucleotide.
- the mixture is filtered under vacuum to remove the waste solid support.
- the reaction is quenched with glacial acetic acid to provide a pH neutral solution of crude product.
- the cleaved oligonucleotide can be purified using any techniques known in the art.
- the oligonucleotide is purified by ion exchange chromatography.
- the crude oligonucleotide is purified by preparative anion exchange chromatography using column chromatography, for example on a TosoBiosep TSK Gel SuperQ-5PW column.
- the purification can be performed using standard reagents and can be performed at elevated temperature (above room temperature). In a specific embodiment, the purification is performed at approximately 75° C.
- the reagents include increasing concentrations of sodium bromide as an eluting reagent.
- the gradient is made of 10% ACN, 20mM sodium phosphate, pH 8.0 (Buffer A) and 10% ACN, 20 mM sodium phosphate and 1.0M NaBr, pH 8.0 (Buffer B).
- Purification is accomplished by eluting the product from the column through a controlled increase in sodium bromide concentration in the buffer system by increasing the proportion of Buffer B. Fractions are collected and analyzed, for example by UV and IP RP-HPLC. When necessary, the fractions are combined to yield a product pool of the desired purity, desalted by ultrafiltration (e.g. using 1 KD Polyethersulfone (PES) Pall Filtron UF Cassettes) and concentrated at about 40° C. and about 80 mbar pressure using a rotary evaporator. The concentrated product is stored at 2-8 ° C.
- PES Polyethersulfone
- the purified and concentrated intermediate from above is reacted with a PEG reagent as described herein at a pH between about pH 7-10, or between about pH 8-10, or between about pH 8.5-10, or between about pH 8.8-9.8.
- a 40 KD mPEG-linked reagent can be incubated with the purified oligonucleotide at about 25° C. in 0.1M sodium borate buffer (pH about 8.8) for 25-30 min. The reaction is shown in Scheme 2.
- the purified and concentrated intermediate is 5 reacted with an appropriate PEG (i.e. PEG-Mal) at pH from about 6-8 or from about 6.5-7.5.
- PEG i.e. PEG-Mal
- Thiol PEGylation is specific for free thiols on biological reagents using mPEG-MAL.
- Coupling of maleimide to thiol groups is highly specific for thiols at ca. pH 6.5-7.5 in the presence of other functional groups. Reaction with a thiol moiety generates a stable 3-thiosuccinimidyl ether linkage.
- hemophilia B results from deficiencies in factor IX. All patients with hemophilia B have prolonged coagulation time and decreased factor IX clotting activity. Like hemophilia A, there are severe, moderate and mild forms of hemophilia B and reflect the factor IX activity in plasma.
- Antidotes or modulators can include any pharmaceutically acceptable agent that can bind an aptamer and modify the interaction between that aptamer and its target molecule (e.g., by modifying the structure of the aptamer) in a desired manner.
- antidotes include (A) oligonucleotides complementary to at least a portion of the aptamer sequence (including ribozymes or DNAzymes or peptide nucleic acids (PNAs)), (B) nucleic acid binding peptides, polypeptides or proteins (including nucleic acid binding tripeptides (see, generally, Hwang et al.
- the antidote oligonucleotide reverses or neutralizes at least 25%, 50%, 75%, 80% or 90% of the anticoagulant activity of the aptamer.
- the antidote generally has the ability to substantially bind to a nucleic acid ligand in solution at antidote concentrations of less than one 1 ⁇ M, or less than 0.1 ⁇ M and more preferably less than 0.01 ⁇ M. In one embodiment, the antidote reduces the biological activity of the aptamer by 50%.
- the improved antidote of the invention is an oligonucleotide that comprises a sequence complementary to at least a portion of the targeted aptamer sequence. Absolute complementarity is not required.
- the sequence in one embodiment has sufficient complementarity to be able to hybridize with the aptamer. The ability to hybridize will depend on both the degree of complementarity and the length of the antisense nucleic acid.
- the antidote oligonucleotide comprises a sequence complementary to 6-25 consecutive nucleotides of the targeted aptamer, preferably, 8-20 consecutive nucleotides, more preferably, 10-15 consecutive nucleotides.
- the antidote is at least 10-25 nucleotides, at least 15-25, at least 20-25, at least 14, 17 or at least 25 nucleotides long.
- the antidotes of the invention can be DNA or RNA or chimeric mixtures or derivatives or modified versions thereof, single-stranded.
- duplexes by binding of complementary pairs of short oligonucleotides is a fairly rapid reaction with second order association rate constants generally between 1 ⁇ 10 6 and 3 ⁇ 10 5 M 1 s 1 .
- Stability of short duplexes can be highly dependent on the length and base-composition of the duplex.
- the thermodynamic parameters for formation of short nucleic acid duplexes have been rigorously measured, resulting in nearest-neighbor rules for all possible base pairs such that accurate predictions of the free energy, T m and thus half-life of a given oligoribonucleotide duplex can be calculated (e.g., Xia et al. (1998) Biochem. 37:14719; see also Eguchi et al. (1991) Antigensis RNA, Annu. Rev. Biochem. 60:631).
- the present invention provides improved antidotes that specifically and rapidly reverse the anticoagulant and antithrombotic effects of the improved aptamers that target components of the coagulation pathway, in particular the aptamers of FIX and FIXa.
- the antidotes can be administered to reverse the aptamer activity by a physician or other health care provider.
- the improved antidotes according to the present invention are nucleic acids corresponding to the sequences: (5′-3′) sequence: cgcgguauaguccccau (Apt/AD; SEQ ID NO:1); (5′-3′) sequence: cgcgguauaguccc (Apt6/AD SEQ ID NO:2); (5′-3′) sequence: cgcgguauaguccac (Apt7/AD; SEQ ID NO:3); (5′-3′) sequence: cgcgguauaguccauc (Apt8/AD; SEQ ID NO:4); (5′-3′) sequence: cgcgguauagucag (Apt9/AD; SEQ ID NO:5); (5′-3′) sequence: cgcgguauagucagg (Apt10/AD; SEQ ID NO:6); (5′-3′) sequence: cgcgguauagucagag (Apt11/AD; SEQ ID NO:1
- the antidote has the structure 2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxyuridylyl-(3′-5′)-2-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxyuridylyl-(3′,5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxyuridylyl-(3′,5′)-2′-me
- oligonucleotide antidotes of the invention are advantageously targeted at single-stranded regions of the aptamer. This can facilitate nucleation and, therefore, the rate of aptamer activity modulation, and also, generally leads to intermolecular duplexes that contain more base pairs than the targeted aptamer.
- the aptamer to Factor IXa of the present invention may be used to design an antisense oligonucleotide.
- the antisense oligonucleotide hybridizes to the aptamer in vivo and blocks the binding of the aptamer to factor IXa.
- the complimentary oligonucleotides can be “walked” around the aptamer.
- This “walking” procedure includes, after a minimal consensus ligand sequence has been determined for a given improved aptamer, adding random sequence to the minimal consensus ligand sequence and evolving additional contacts with the target, such as in separate but adjacent domains.
- a walking experiment can involve two experiments performed sequentially.
- a new candidate mixture is produced in which each of the members of the candidate mixture has a fixed nucleic acid-region that corresponds to a nucleic acid ligand of interest.
- Each member of the candidate mixture also contains a randomized region of sequences. According to this method it is possible to identify what are referred to as “extended” nucleic acid ligands, which contain regions that can bind to more than one binding domain of a target.
- RNA-like oligonucleotides e.g., 20-fluoro or 20-methoxy
- Alterations in the orientation of the sugar to the base can also affect RNase H activation,.
- backbone modifications influence the ability of oligonucleotides to activate RNase H. Methylphosphonates do not activate it, whereas phosphorothioates are excellent substrates.
- chimeric molecules have been studied as oligonucleotides that bind to RNA and activate RNase H . For example, oligonucleotides comprising wings of 20-methoxy phosphonates and a five-base gap of deoxyoligonucleotides bind to their target RNA and activate RNase H.
- 2′-O-methyl modified antidotes e.g., 2′-O-methyl oligonucleotides
- 2′-O-methyl oligonucleotides about 15 nucleotides in length
- the complementarity of which is staggered by about 5 nucleotides on the aptamer (e.g., oligonucleotides complementary to nucleotides 1-15, 6-20, 11-25, etc.).
- the impact of tertiary structure of the improved aptamer on the efficiency of hybridization is difficult to predict. Assays described in the Examples that follow can be used to assess the ability of the different oligonucleotides to hybridize to a specific aptamer.
- oligonucleotide antidotes can also be determined by conducting kinetic studies using, for example, BIACORE assays. Oligonucleotide antidotes can be selected such that a 5-50 fold molar excess of oligonucleotide, or less, is required to modify the interaction between the aptamer and its target molecule in the desired manner.
- the antidotes of the invention may be conjugated to another molecule, e.g., a peptide, a hybridization-triggered cross-linking agent, a transport agent, a hybridization-triggered cleavage agent, etc.
- another molecule e.g., a peptide, a hybridization-triggered cross-linking agent, a transport agent, a hybridization-triggered cleavage agent, etc.
- the antisense oligonucleotide may optionally comprise at least one modified base moiety, including but not limited to one selected from 5-fluorouracil, 5-fluorocytosine, 5-bromouracil, 5-bromocytosine, 5-chlorouracil, 5-chlorocytosine, 5-iodouracil, 5-iodocytosine, 5-methylcytosine, 5-methyluracil, hypoxanthine, xantine, 4-acetylcytosine, 5-(carboxyhydroxymethyl) uracil, 5-carboxymethylamin-O-methyl thiouridine, 5-carboxymethylamin-O-methyluracil, dihydrouracil, beta-D-galactosylqueosine, inosine, N6-isopentenyladenine, 1-methylguanine, 1-methylinosine, 2,2-dimethylguanine,2-methyladenine, 2-methylguanine, 3-methylcytosine, 6-methylcytosine, N6-
- the antidotes may also include at least one modified sugar moiety selected from the group including, but not limited to, arabinose, 2-fluoroarabinose, xylulose, hexose, 2′-fluororibose, 2′ -O-methylribose, 2′ -O-methoxyethylribose, 2′ -O-propylribose, 2′-O-methylthioethylribose, 2′-O-diethylaminooxyethylribose, 2′-O-(3-aminopropyl)ribose, 2′-O-dimethylaminopropyl)ribose, 2′-O-(methylacetamido)ribose, and 2′-O-(dimethylaminoethyloxyethyl)ribose.
- modified sugar moiety selected from the group including, but not limited to, arabinose, 2-fluo
- the antisense oligonucleotide comprises at least one modified phosphate backbone selected from the group including, but not limited to, a phosphorothioate, a phosphorodithioate, a phosphoramidothioate, a phosphoramidate, a phosphordiainidate, a methylphosphonate, an alkyl phosphotriester, and a formacetal or analog thereof.
- Manufacture of nucleic acid antidotes is a multi-step process involving solid phase chemical synthesis of the oligonucleotide strand; cleavage and deprotection of the crude oligonucleotide; purification by preparative anion exchange chromatography; desalting and lyophilization of the final product.
- Chemical synthesis via phosphoramidite chemistry involves sequential coupling of activated monomers to an elongating polymer, one terminus of which is covalently attached to a solid support matrix.
- the solid phase approach allows for easy purification of the reaction product at each step in the synthesis by simple solvent washing of the support-bound intermediates.
- the oligonucleotide can be sequentially assembled from the 3′-end towards the 5′-end by deprotecting the 5′-end of the support-bound intermediate, allowing the support-bound intermediate to react with an incoming tetrazole-activated phosphoramidite monomer, oxidizing the resulting phosphite triester to a phosphate triester, and blocking any unreacted hydroxyl groups by acetylation to prevent non-sequential coupling with the next incoming monomer to form a “deletion sequence”. This sequence of steps is repeated for subsequent coupling reactions until the full-length oligonucleotides are synthesized.
- the oligonucleotide can then be cleaved from the solid support and protecting groups on the linker are removed.
- the support-attached intermediate is cleaved using basic conditions at an elevated temperature.
- the support-bound crude product is transferred to a filter funnel, dried under vacuum and transferred to a reaction vessel.
- a basic reagent is added to effect cleavage from the solid support, removal of the cyanoethyl phosphate protecting group and deprotection of exocyclic amine protecting groups.
- the reaction temperature can also be increased to enhance cleavage.
- the reagent is 30-40% methylamine and the mixture is heated to approximately 45° C. for up to 90 minutes.
- the mixture is filtered under vacuum to remove the waste solid support.
- the reaction is quenched with glacial acetic acid to provide a pH neutral solution of crude product.
- the cleaved oligonucleotide can be purified using any techniques known in the art.
- the oligonucleotide is purified by ion exchange chromatography.
- the crude product is purified by preparative anion exchange chromatography using, for example, a TosoBiosep TSK Gel SuperQ-5PW column.
- the purification can be performed using standard reagents and can be performed at elevated temperature (above room temperature).
- the reagents include increasing concentrations of sodium bromide as an eluting reagent.
- the gradient is made of 5-35% of Buffer B (10% ACN, 20 mM sodium phosphate and 1.0 M NaBr, pH 8.0) in Buffer A (10% ACN, 20 mM sodium phosphate, pH 8.0).
- Buffer B 10% ACN, 20 mM sodium phosphate and 1.0 M NaBr, pH 8.0
- Buffer A 10% ACN, 20 mM sodium phosphate, pH 8.0.
- the purification can be performed at approximately 70° C. using.
- Purification is accomplished by eluting the product from the column through a controlled increase in sodium bromide concentration in the buffer system. Fractions can be collected and analyzed by UV and AX-HPLC. Selected fractions can be combined to yield a product pool with of the desired purity, desalted by ultrafiltration and stored at 2-8° C. Aliquots can be freeze-dried to a dry powder and stored in glass at ⁇ 15° C. to ⁇ 25° C.
- Improved aptamers or antidotes can also be enzymatic nucleic acids.
- Such a ribozyme or DNAzyme act by first binding to a target RNA or DNA (see Cech U.S. Pat. No. 5,180,818) and then cleaving the target.
- An enzymatic nucleic acid can repeatedly bind and cleave new targets thereby allowing for inactivation of RNA aptamers.
- ribozyme may be advantageous over other technologies because the effective concentration of ribozyme necessary to effect a therapeutic treatment is lower than that of an antisense oligonucleotide.
- a single ribozyme molecule is able to cleave many molecules of target RNA.
- the ribozyme is a highly specific inhibitor, with the specificity of inhibition depending not only on the base pairing mechanism of binding, but also on the mechanism by which the molecule inhibits the expression of the RNA to which it binds.
- the inhibition is caused by cleavage of the RNA target and so specificity is defined as the ratio of the rate of cleavage of the targeted RNA over the rate of cleavage of non-targeted RNA.
- This cleavage mechanism is dependent upon factors additional to those involved in base pairing. Thus, it may be that the specificity of action of a ribozyme is greater than that of antisense oligonucleotide binding the same RNA site.
- DNAzymes Another class of catalytic molecules are called “DNAzymes”. DNAzymes are single-stranded, and cleave both RNA and DNA. A general model for the DNAzyme has been proposed, and is known as the “10-23” model. DNAzymes following the “10-23” model, also referred to simply as “10-23 DNAzymes”, have a catalytic domain of 15 deoxyribonucleotides, flanked by two substrate-recognition domains of seven to nine deoxyribonucleotides each. In vitro analyses show that this type of DNAzyme can effectively cleave its substrate RNA at purine:pyrimidine junctions under physiological conditions. As used herein, “DNAzyme” means a DNA molecule that specifically recognizes and cleaves a distinct target nucleic acid sequence, which may be either DNA or RNA.
- the antidote to the improved aptamers are Peptide Nucleic Acids (PNAs).
- PNAs are compounds that are analogous to oligonucleotides, but differ in composition in that the deoxyribose backbone of oligonucleotide is replaced by a peptide backbone. Each subunit of the peptide backbone is attached to a naturally-occurring or non-naturally-occurring nucleobase.
- PNAs can be advantageous as fast acting antidotes because they bind more tightly to the corresponding improved aptamers than their non-substituted oligonucleotide counterparts.
- PNAs bind to both DNA and RNA and the resulting PNA/DNA or PNA/RNA duplexes are bound tighter than corresponding DNA/DNA or DNA/RNA duplexes as evidenced by their higher melting temperatures (T m ).
- T m melting temperatures
- Another advantage of PNA/DNA(RNA) duplexes is that T m is practically independent of salt concentration. Since PNAs are an analogue of DNA in which the backbone is a pseudopeptide rather than a sugar, they mimic the behaviour of DNA and binds complementary nucleic acid strands.
- PNAs are synthetic polyamides comprised of repeating units of the amino acid, N-(2-aminoethyl)-glycine, to which the nucleobases adenine, cytosine, guanine, thymine and uracil are attached through a methylene carbonyl group.
- Natural and unnatural nucleobases such as pseudo isocytosine, 5-methyl cytosine and 2,6-diaminopurine, inosine, uracil, 5-methylcytosine, thiouracil, 2,6-diaminopurine, bromothymine, azaadenines or azaguanines among many others, also can be incorporated in PNA synthons.
- PNAs are most commonly synthesized from monomers (PNA synthons) protected according to the t-Boclbenzyl protection strategy, wherein the backbone amino group of the growing polymer is protected with the t-butyloxycarbonyl (t-Boc) group and the exocyclic amino groups of the nucleobases, if present, are protected with the benzyloxycarbonyl (benzyl) group.
- PNA synthons protected using the t-Boc/benzyl strategy are now commercially available.
- Morpholino nucleic acids can also be ad disadvantageous in antidote preparation because morpholinos are completely resistant to nucleases and they appear to be free of most or all of the non-antisense effects that plague S-DNAs.
- MNAs are assembled from morpholino subunits, each of which contains one of the four genetic bases (adenine, cytosine, guanine, and thymine) linked to a 6-membered morpholine ring. Subunits of are joined by non-ionic phosphorodiamidate intersubunit linkages to give a MNA.
- MNAs can have substantially better antisense properties than do RNA, DNA, and their analogs having 5-membered ribose or deoxyribose backbone moieties joined by ionic linkages (see wwwgenetools.com/Morpholinosibody_morpholinos.HTML).
- U.S. Pat. No. 6,153,737 to Manoharan et al. is directed to derivatized oligonucleotides wherein the linked nucleosides are functionalized with peptides, proteins, water soluble vitamins or lipid soluble vitamins.
- This disclosure was directed towards antisense therapeutics by modification of oligonucleotides with a peptide or protein sequence that aids in the selective entry of the complex into the nuclear envelope.
- water-soluble and lipid-soluble vitamins can be used to assist in the transfer of the anti-sense therapeutic or diagnostic agent across cellular membranes.
- LNAs Locked nucleic acids
- LNAs are a novel class of DNA analogues that possess certain features that make them prime candidates for improving nucleic acid properties.
- the LNA monomers are bi-cyclic compounds structurally similar to RNA-monomers. LNAs share most of the chemical properties of DNA and RNA, are water-soluble, can be separated by gel electrophoreses, ethanol precipitated, etc. (Tetrahedron, 54, 3607-3630 (1998)).
- introduction of LNA monomers into either DNA or RNA oligos results in high thermal stability of duplexes with complementary DNA or RNA, while, at the same time obeying the Watson-Crick base-pairing rules.
- the stabilized nucleic acid can be a PCO (pseudocyclic oligonucleobase), or a 2′-O,4′-C-ethylene bridged nucleic acid (ENA).
- the invention includes the use of improved aptainers to bind to FIX, FIXa, or the intrinsic tenase complex.
- the binding can be in vitro or in vivo.
- the result of the binding to FIX, FIXa or the tenase complex can be to inhibit the biological activity of the proteins or complex.
- the improved aptamers can be used to treat diseases such as deep venous thrombosis, arterial thrombosis, post surgical thrombosis, coronary artery bypass graft (CABG), percutaneous transdermal coronary angioplastry (PTCA), stroke, tumour metastasis, inflammation, septic chock, hypotension, ARDS, pulmonary embolism, disseminated intravascular coagulation (DIC), vascular restenosis, platelet deposition, myocardial infarction, angiogenesis, or the prophylactic treatment of mammals with atherosclerotic vessels at risk for thrombosis.
- diseases such as deep venous thrombosis, arterial thrombosis, post surgical thrombosis, coronary artery bypass graft (CABG), percutaneous transdermal coronary angioplastry (PTCA), stroke, tumour metastasis, inflammation, septic chock, hypotension, ARDS, pulmonary embolism, disseminated intravascular coagulation (DIC
- the improved aptamer inhibits blood coagulation by binding to FIXa.
- the invention includes administering the aptamer of the invention to a mammal, for example, a human, in need thereof to inhibit blood coagulation.
- Another embodiment of the invention provides methods of using aptamers that are well suited for administration during a therapeutic regime.
- a method of improved regulating coagulation in a mammal in need thereof comprises: (a) administering to a warm-blooded vertebrate or mammal in need thereof, an effective amount of an improved aptamer that selectively binds coagulation pathway FIX, FIXa, or the intrinsic tenase complex, or inhibits a subunit of the intrinsic tenase complex (i.e.
- the warm-blooded vertebrate or mammal is a human.
- mammal is meant to include any human or non-human mammal, including but not limited to porcine, ovine, bovine, rodents, ungulates, pigs, sheep, lambs, goats, cattle, deer, mules, horses, monkeys, dogs, cats, rats, and mice.
- the improved aptamers of the present invention can be used to treat percutaneous coronary interventions where vascular injury occurs at a specific time and place, yielding a sudden but relative brief prothrombotic stimulus. This may also be the case following carotid angioplasty.
- the improved aptamers can be used in extracorporeal circulation utilized in coronary bypass grafting and hemodialysis. The latter condition is somewhat complex because of the inherent thrombogenicity of arteriovenous (AV) shunts.
- AV arteriovenous
- the aptamers of the invention also can be used to treat a venous thromboembolic disease, mechanical heart valve replacement, atrial fibrillation, and conceivably in either primary or secondary prevention of cardiovascular events among patients with prior events, an unfavorable risk profile, documented multibed vascular disease, vascular inflammation (early stages of atherosclerotic vasculopathy).
- a method of treating cardiovascular disease in a warm-blooded vertebrate comprises administering an effective amount of an improved aptamer to a vertebrate subject suffering from cardiovascular disease that selectively binds a coagulation pathway factor IX, IXa, or the intrinsic tenase complex, or inhibits a subunit of the intrinsic tenase complex (i.e. FIX, FIXa, FVIII binding to or activation of FX).
- Administration of the aptamer treats the cardiovascular disease in the vertebrate subject.
- the method can further comprise providing an antidote to reverse the effects of the improved aptamer by administration of an antidote.
- the improved aptamers can be administered to mammals who require blood coagulation therapy.
- the invention provides methods of treating mammals with an aptamer to inhibit blood coagulation.
- the paired antidote can be administered to reverse the effects of the aptamer.
- a benefit of this discovery is that blood coagulation can be controlled in real time and does not rely on the mammal's own metabolism.
- compositions and methods of the present invention are particularly useful for preventing thrombosis in the circuit of cardiac bypass apparatus and in patients undergoing renal dialysis, and for treating patients suffering from or at risk of suffering from thrombus-related cardiovascular conditions, such as unstable angina, acute myocardial infarction (heart attack), cerebrovascular accidents (stroke), pulmonary embolism, deep vein thrombosis, arterial thrombosis, CABG surgery and disseminated intravascular coagulation.
- thrombus-related cardiovascular conditions such as unstable angina, acute myocardial infarction (heart attack), cerebrovascular accidents (stroke), pulmonary embolism, deep vein thrombosis, arterial thrombosis, CABG surgery and disseminated intravascular coagulation.
- the improved aptamers and antidotes of the present invention can inhibit other cardiovascular disease associated with FIX or FIX-regulated cascades.
- Coagulation plays an important part in ischaemic cardiovascular disease. Results of studies have shown that extremes in hypocoagulability protect against ischaemic cardiovascular disease. A mild decrease in coagulability found in hemophiliac patients can have a protective effect against fatal ischaemic heart disease (Sramek A, et al. (2003) Lancet 362(9381):3514 ; Bilora F, et al. (1999) Clin Appl Thromb Hemost. 5(4):232-5.
- the aptamers can be administered to prevent coagulation-induced inflammation. Inflammation is induced by thrombolytic therapy in patients with acute myocardial infarction (AMI), which might contribute to microvascular obstruction and reperfusion injury. Improved aptamers of the present invention can inhibit this early inflammatory response. In one embodiment, methods are provided to reduce the early inflammatory response in mammals that are in need thereof by administering the improved aptamers of the present invention.
- AMD acute myocardial infarction
- the improved aptamers and antidotes of the present invention can be used to inhibit atherosclerosis.
- Some adverse events in atherosclerosis are associated with ruptured plaques, which are a major cause of morbidity and mortality associated with atherosclerosis.
- coagulation factor IX activation peptide and fibrinogen can be positively associated with risk of coronary heart disease (R. Rosenberg et al. (2001) Thromb Haemost 86: 41-50; J A Cooper et al. (2000) Circulation 102: 2816-2822).
- the intrinsic pathway may significantly enhance thrombogenicity of atherosclerotic lesions after removal of the endothelial layer and exposure of SMCs and macrophages to blood flow (Ananyeva N M et al. (2002) Blood 99: 44754485).
- the improved aptamers can also be provided to prevent morbidity in mammals suffering from acute coronary syndrome (ACS) associated with inflammation.
- ACS acute coronary syndrome
- the contact pathway becomes the major pathway for blood clotting.
- CPB cardiopulmonary bypass
- genetic epidemiology and prospective clinical studies have linked the magnitude of the inflammatory response during coronary revascularization procedures with multiple adverse effects of CPB, including renal damage, atrial fibrillation, stroke, gut damage and neuronal damage.
- the inflammatory response induced by activation of the coagulation pathway is mediated by coagulation factors Xa and thrombin, which in addition to their role in blood clot formation, are themselves pro-inflammatory and mitogenic signaling proteins.
- the improved aptamers can also be administered to prevent adverse effects associated with post-angioplasty restenosis.
- the first case is when anticoagulant or antithrombotic treatment leads to hemorrhage, including intracranial or gastrointestinal hemorrhage. While identifying safer target proteins may reduce this risk, the potential for morbidity or mortality from this type of bleeding event is such that the risk can not be overlooked.
- the second case is when emergency surgery is required for patients who have received antithrombotic treatment. This clinical situation arises in a percentage of patients who require emergency coronary artery bypass grafts (CABG) while undergoing percutaneous coronary intervention under the coverage of GPIIb/IIIa inhibitors.
- CABG emergency coronary artery bypass grafts
- the third case is when an anticoagulant nucleic acid ligand is used during a cardiopulmonary bypass procedure. Bypass patients are predisposed to post operative bleeding.
- an antidote e. g., an oligonucleotide antidote of the invention targeted to an anticoagulant or antithrombotic nucleic acid ligand
- an antidote e. g., an oligonucleotide antidote of the invention targeted to an anticoagulant or antithrombotic nucleic acid ligand
- the applicants have discovered improved aptamer-antidote pairs that precisely regulate proteins in the blood coagulation cascade.
- the antidotes of the invention are provided to a mammal in need thereof after the aptamers of the invention to reverse the effects of the aptamers.
- Aptamers and aptamer-antidote pairs can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy.
- this invention discloses an improved regulatable therapeutic regime in the course of nucleic acid ligand therapy for blood coagulation.
- the aptamers are administered to patients undergoing general surgery.
- the aptam rs are administered to patients with cardiovascular disease, which can include coronary heart disease.
- the patients can be undergoing treatment including bypass surgery or percutaneous coronary interventions.
- the mammals who can be treated with the aptamers of the present invention can also include patients who have had physical trauma that requires coagulation therapy.
- Preoperative assessment of the patient can identify drug-induced, acquired, or inherited coagulation defects.
- the main attention in anticoagulant therapy is directed to the perioperative period.
- a further, often overlooked, management strategy in treating major coagulopathies is the consideration of the cost and half-lives of the coagulation factors in individual blood components. Prevention of bleeding has become possible by manipulation of the control of coagulation and inflammatory processes.
- the modulatable improved aptamers and antidote pairs of the present invention are particularly useful in ensuring that, in case of incorrect diagnosis and treatment, treatment can immediately be disabled.
- the symptoms of coronary infarction can closely mimic those of an acute coronary dissection. See Scarabeo et al. (2002) Italian Heart Journal 3: 490494.
- a diagnosis of coronary infarction immediately calls for an anticoagulant, which is counterindicated in acute coronary dissection.
- ICH intracerebral hemorrhage
- Factor IXa is a key intermediary in the intrinsic pathway of coagulation
- targeted inhibition of Factor IXa-dependent coagulation can inhibit microvascular thrombosis in stroke without impairing extrinsic hemostatic mechanisms that limit ICH.
- the improved aptamers and antidotes of the present invention can be used to inhibit stroke associated with cardiovascular disease and surgery.
- the present method for treating cardiovascular disease in a tissue contemplates contacting a tissue in which cardiovascular disease is occurring, or is at risk for occurring, with a composition comprising a therapeutically effective amount of an improved aptamer capable of binding a coagulation factor as well as providing an improved antidote to reverse the effects of the aptamer by administration of an antidote.
- the method comprises administering to a patient a therapeutically effective amount of a physiologically tolerable composition containing the RNA aptamer as well as a method to provide an antidote to reverse the effects of the aptamer by administration of an antidote.
- the dosage ranges for the administration of the antidote depend upon the form of the antidote, and can be assessed by a physician or other health care provider. Generally, the dosage will vary with the age, condition, sex and extent of the disease in the patient and can be determined by one of skill in the art. The individual physician in the event of any complication can also adjust the dosage.
- a therapeutically effective amount is an amount of a antidote sufficient to produce a measurable modulation of the effects of the nucleic acid ligand, including but not limited to a coagulation-modulating amount or an inflammation-modulating amount.
- Preferred modes of administration of the improved aptamers of the present invention are parenteral, intravenous, intradermal, intra-articular, intra-synovial, intrathecal, intra-arterial, intracardiac, intramuscular, subcutaneous, intraorbital, intracapsular, intraspinal, intrasternal, topical, transdermal patch, via rectal, vaginal or urethral suppository, peritoneal, percutaneous, nasal spray, surgical implant, internal surgical paint, infusion pump or via catheter.
- the agent and carrier are administered in a slow release formulation such as an implant, bolus, microparticle, microsphere, nanoparticle or nanosphere.
- the antidotes of the present invention can be preferably administered parenterally by injection or by gradual infusion over time.
- tissue to be treated can typically be accessed in the body by systemic administration and therefore most often treated by intravenous administration of therapeutic compositions, other tissues and delivery techniques are provided where there is a likelihood that the tissue targeted contains the target molecule.
- antidotes of the present invention are typically administered orally, topically to a vascular tissue, intravenously, intraperitoneally, intramuscularly, subcutaneously, intra-cavity, transdermally, and can be delivered by peristaltic techniques.
- compositions can be provided to the individual by a variety of routes such orally, topically to a vascular tissue, intravenously, intraperitoneally, intramuscularly, subcutaneously, intra-cavity, transdermally, and can be delivered by peristaltic techniques.
- routes such orally, topically to a vascular tissue, intravenously, intraperitoneally, intramuscularly, subcutaneously, intra-cavity, transdermally, and can be delivered by peristaltic techniques.
- Representative, non- liming approaches for topical administration to a vascular tissue include (1) coating or impregnating a blood vessel tissue with a gel comprising a nucleic acid ligand, for delivery in vivo, e. g.
- nucleic acid ligand composition by implanting the coated or impregnated vessel in place of a damaged or diseased vessel tissue segment that was removed or by-passed; (2) delivery via a catheter to a vessel in which delivery is desired; (3) pumping a nucleic acid ligand composition into a vessel that is to be implanted into a patient.
- the nucleic acid ligand can be introduced into celis by microinjection, or by liposome encapsulation.
- nucleic acid ligands of the present invention can be administered in a single daily dose, or the total daily dosage can be administered in several divided doses.
- the antidote is provided by any suitable means to alter the effect of the nucleic acid ligand by administration of the antidote.
- the aptamers and antidotes of the invention can be formulated into pharmaceutical compositions that can include, in addition to an improved aptamer, a antidote or modulator, and a pharmaceutically acceptable carrier, diluent or excipient.
- a pharmaceutically acceptable carrier diluent or excipient.
- the precise nature of the composition will depend, at least in part, on the nature of the improved aptamer and antidote and the route of administration. Optimum dosing regimens can be readily established by one skilled in the art and can vary with the improved aptamer, the antidote combination, the patient and the effect sought.
- the therapeutic compositions comprising aptamers and antidotes of the present invention are conventionally administered intravenously, as by injection of a unit dose, for example.
- unit dose when used in reference to a therapeutic composition of the present invention refers to physically discrete units suitable as unitary dosage for the subject, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect in association with the required diluent; i.e. carrier or vehicle.
- compositions are administered in a manner compatible with the dosage formulation, and in a therapeutically effective amount.
- quantity to be administered depends on the subject to be treated, capacity of the subject's system to utilize the active ingredient, and degree of therapeutic effect desired. Precise amounts of active ingredient required to be administered depend on the judgment of the practitioner and are peculiar to each individual.
- suitable dosage ranges for systemic application are disclosed herein and depend on the route of administration. Suitable regimes for administration are also variable, but are typified by an initial administration followed by repeated doses at one or more hour intervals by a subsequent injection or other administration. Alternatively, continuous intravenous infusion sufficient to maintain concentrations in the blood in the ranges specified for in vivo therapies are contemplated.
- compositions comprising an aptamer or antidote of the present invention can be formulated according to known methods such as by the admixture of a pharmaceutically acceptable carrier. Examples of such carriers and methods of formulation can be found in Remington's Pharmaceutical Sciences. To form a pharmaceutically acceptable composition suitable for effective administration, such compositions will contain an effective amount of the aptamer. Such compositions can contain admixtures of more than one aptamers or antidotes.
- the effective amount of an improved aptamer of the invention can vary according to a variety of factors such as the individual's condition, weight, sex and age. Other factors include the mode of administration.
- the compositions will be administered in dosages adjusted for body weight, e. g., dosages ranging from about 0.1 mgikg body weight to about 100 mg/kg body weight.
- the dosages are about 0.5 mg/kg body weight to 50 mg/kg body weight.
- the dosage is between 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, and 1 mg/kg body weight, and any dosage in between.
- Specific dosage units can range from 1 ng to 1 g, but are more conventionally about 0.01 ⁇ g, 0. ⁇ g, 1 ⁇ g, 10 ⁇ g, 100 ⁇ g, 500 ⁇ g, or 1 g or any amount in between.
- the effective amount of antibody being delivered to a patient will vary according to a variety of factors such as the individual's condition, weight, sex, age and amount of nucleic acid ligand administered.
- the antidote ranges from 0.5-50 mg/kg.
- the amount of antidote being delivered ranges from 0.5-10, 0.5-5, 1-10 or 1-5 mg/kg.
- the amount of antidote being delivered is not less than the amount of aptamer being delivered.
- the amount of antidote is from about 1 to about 20 times the amount of aptamer.
- the antidote is about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 times the amount of aptamer delivered to a patient.
- Improved combinations of aptamers and antidotes in pharmaceutical compositions are administered in therapeutically effective amounts, that is, in amounts sufficient to generate a coagulation-modulating response, or in a prophylactically effective amounts, that is in amounts sufficient to prevent a coagulation factor from acting in a coagulation cascade.
- the therapeutically effective amount and prophylactically effective amount can vary according to the modulator.
- the pharmaceutical composition can be administered in single or multiple doses.
- the activity of the improved antidotes is lasting, once the desired level of modulation of the nucleic acid ligand by the antidote is achieved, infusion of the antidote can be terminated, allowing residual antidote to clear the human or animal. This allows for subsequent re-treatment with the nucleic acid ligand as needed.
- subsequent treatment can involve the use of a second, different improved aptamer/antidote pair.
- Antidotes synthesized or identified according to the methods disclosed herein can be used alone at appropriate dosages defined by routine testing in order to obtain optimal modulation of nucleic acid ligand activity in coagulation, while minimizing any potential toxicity.
- co- administration or sequential administration of other agents can be desirable.
- the active agents can be administered concurrently, or they each can be administered at separately staggered times.
- the dosage regimen utilizing the improved aptamers and antidotes of the present invention is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal and hepatic function of the patient; and the particular combination employed.
- a physician of ordinary skill can readily determine and prescribe the effective amount of the aptamer required to prevent, counter or arrest the progress of the condition.
- Optimal precision in achieving concentrations of the combination within the range that yields efficacy without toxicity requires a regimen based on the kinetics of the aptamer and antidote's availability to target sites. This involves a consideration of the distribution, equilibrium, and elimination of the modulator.
- the combinations described in detail can form the active ingredient, and are typically administered in admixture with suitable pharmaceutical diluents, excipients or carriers (collectively referred to herein as “carrier” materials) suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrup, suppositories, gels and the like, and consistent with conventional pharmaceutical practices.
- carrier suitable pharmaceutical diluents, excipients or carriers
- the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture.
- suitable binders include without limitation, starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
- Lubricants used in these dosage forms include, without limitation, sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like.
- the active drug component can be combined in suitably flavored suspending or dispersing agents such as the synthetic and natural gums, for example, tragacanth, acacia, methyl-cellulose and the like.
- suspending or dispersing agents such as the synthetic and natural gums, for example, tragacanth, acacia, methyl-cellulose and the like.
- Other dispersing agents include glycerin and the like.
- sterile suspensions and solutions are desired.
- Isotonic preparations that generally contain suitable preservatives are employed when intravenous administration is desired.
- Topical preparations containing the active drug component can be admixed with a variety of carrier materials well known in the art, such as, e. g., alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, PPG2 mydstyl propionate, and the like, to form, e.g. alcoholic solutions, topical cleansers, cleansing creams, skin gels, skin lotions, and shampoos in cream or gel formulations.
- carrier materials well known in the art, such as, e. g., alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, PPG2 mydstyl propionate, and the like, to form, e.g. alcoholic solutions, topical cleansers, cleansing creams, skin gels, skin lotions, and shampoos in cream or gel formulations.
- the aptamers and antidotes of the present invention can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- the aptamers and antidotes of the present invention can also be coupled with soluble polymers as targetable drug carriers.
- soluble polymers can include polyvinyl-pyrrolidone, pyran copolymer, polyhydroxypropylmethacryl-amidephenol, polyhydroxy-ethylaspartamidephenol, or polyethyl-eneoxidepolylysine substituted with palmitoyl residues.
- the aptamers and antidotes of the present invention can be coupled (preferably via a covalent linkage) to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polyethylene glycol (PEG), polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydro-pyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- PEG polyethylene glycol
- polylactic acid polyepsilon caprolactone
- polyhydroxy butyric acid polyorthoesters
- polyacetals polydihydro-pyrans
- polycyanoacrylates polycyanoacrylates
- cross-linked or amphipathic block copolymers of hydrogels for example, polyethylene glycol (PEG), polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals,
- the complex comprises a liposome with a targeting nucleic acid ligand (s) associated with the surface of the liposome and an encapsulated therapeutic or diagnostic agent.
- a targeting nucleic acid ligand s
- Preformed liposomes can be modified to associate with the nucleic acid ligands.
- a cationic liposome associates through electrostatic interactions with the nucleic acid.
- a nucleic acid attached to a lipophilic compound, such as cholesterol can be added to preformed liposomes whereby the cholesterol becomes associated with the liposomal membrane.
- the nucleic acid can be associated with the liposome during the formulation of the liposome.
- the nucleic acid is associated with the liposome by loading into preformed liposomes.
- ACT Activated Clotting Time Test
- APTT activated partial thromboplastin time
- ACT can be to monitor a patient's coagulation status in connection with clinical procedures, such as those that involve the administration of high doses of heparin (e.g., CPB and PTCA).
- the Activated Partial Thromboplastin Time Test is a common central laboratory test, typically performed using an automated coagulometer, for example Diagnostica Stago's STA coagulometer (MDA/96/23), or another coagulometer produced by this company or otherwise known in the art.
- the test is performed using a plasma sample, in which the intrinsic pathway is activated by the addition of phospholipid, an activator (ellagic acid, kaolin, or micronized silica), and Ca 2+ .
- the bleeding time test can be used for the diagnosis of hemostatic dysfunction, von Willebrand's disease, and vascular disorders. It also can be used to screen for platelet abnormalities prior to surgery. The test is performed by making a small incision on the forearm and wicking away the blood from the wound site. The time it takes for bleeding to stop is recorded and in control subjects is approximately 3.5 minutes. Prolongation of the bleeding time is indicative of qualitative or quantitative platelet defects.
- the Prothrombin Time Test (PT), which was first described by Quick in 1935, measures the tissue factor-induced coagulation time of blood or plasma. It is used as a screening test to evaluate the integrity of the extrinsic coagulation pathway, and is sensitive to coagulation factors I, II, V, VII, and X.
- the test can be performed by adding thromboplastin and Ca 2+ to a patient sample and measuring the time for clot formation. A prolonged clotting time suggests the presence of an inhibitor to, or a deficiency in, one or more of the coagulation factors of the extrinsic pathway. But PT clotting time can also be prolonged for patients on warfarin therapy, or for those with vitamin K deficiency or liver dysfunction.
- the PT test can provide an assessment of the extrinsic coagulation pathway, and is widely used to monitor oral anticoagulation therapy.
- the Thrombin Clotting Time Test measures the rate of a patient's clot formation compared to that of a normal plasma control.
- the test can be performed by adding a standard amount of thrombin to a patient's plasma that has been depleted of platelets, and measuring the time required for a clot to form. This test has been used as an aid in the diagnosis of disseminated intravascular coagulation (DIC) and liver disease.
- DIC disseminated intravascular coagulation
- Complex Tests include specific factor assays based on laboratory tests, such as the APTT, PT, and TCT tests.
- One assay measures the level of the activation Factor IXa or the Factor IXa-antithrombin III complex. These measurements are used to determine the levels of factor IXa or factor VII-tissue mediated complex.
- Assays for activated protein C resistance, antithrombin, protein C deficiency, and protein S deficiency are also part of this group. Asymptomatic individuals who have heterogeneous deficiencies of proteins C and S, and resistance to activated protein C, have significantly elevated levels of the prothrombin fragnent F1.2 compared to controls.
- 2′-Hydroxyl purines were substituted with 2′-O-methyl purines in the 4 secondary structure units of in which purine residues are present: Stem 1 (Apt 1); Loop 1 (Apt 2); Stem 2 (Apt 3); Loop 2 (Apt 4) (see FIG. 1A ).
- Apt 4 showed gain of anticoagulant activity ( FIG. 2 ); Apt 1-3 showed moderate loss of activity; and Apt 5 showed severe loss of activity. Apt 1-3 exhibit enhanced neutralization, suggesting that introduction of 2′ -O-methyl residues within the antidote binding site improves the ability of the antidote oligonucleotide to bind to the aptamer.
- stem 1 variants Two “families” of stem 1 variants were designed (Apt 6-8 and 9-11; FIG. 1B ) consisting of 4, 5, and 6 basepair stems. All constructs were designed in the Apt-2 background. Stem 1 sequences were evaluated for the ability to design complementary antidote oligonucleotides to them such that the antidotes contain minimal secondary structure, and for the ability of the aptamer to assume the proper secondary structure.
- Antidote oligonucleotides were designed specific for Apt 6-11 that bind to their respective target aptamer in the same register as AptA AD (see sequence listings below).
- the anticoagulant activity of Apt 6-11 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the antidote control of Apt 6-11 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down.
- the concentration of Apt 2, and Apt 6-11 was set at 250 nM (as opposed to 125 nM for standard AptA and Apt 2 experiments).
- Apt 6-8 exhibit loss of anticoagulant activity ( FIG. 3 ), however, all exhibit similar activity levels. Thus stem length is not be the main cause for loss of activity.
- the 5 base pair stem 1 constructs (Apt 10 and Apt 7) do appear to be more neutralizable than Apt 2 (FIGS. 3 and 4 ). Data suggests that a stem 1 of 5 base pairs may be preferable to those composed of 4, 6 or 7 base pairs to enhance antidote neutralization.
- the anticoagulant activity of Apt 12-17 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the “neutralizability” of Apt 12-17 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays, and for Apt 13 and 17, the aptamer concentration was fixed at 250 nM.
- Comparison of the anticoagulant activity of Apt 12 with Apt 13 and Apt17 ( FIG. 5 ) demonstrates that the loss of activity observed for Apt6-11 is due to the presence of 2′-O-methyl substitutions at one or more critical residues. Comparison of the anticoagulant activity of Apt14 to Apt12 indicates that the stretch of 4 consecutive guanosines within stem 1 can be altered without a significant impact on anticoagulant activity.
- the anticoagulant activity of Apt 18-20 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the “neutralizability” of Apt 18-20 was evaluated in standard APTT antidote assays over antidote concentrations (Antidote 6, 7 and 8 for 18-20 respectively) ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays.
- Each of the aptamers (Apt 18-20) is a potent anticoagulant, as or more potent than Apt 2 ( FIG. 7 ). Furthermore, all three are readily neutralized by their respective antidote oligonucleotides. Apt 19 was evaluated for anticoagulant activity of a pegylated version.
- PEG Apt 19 and, for comparison, PEG-Apt 16 are used (PEG is a 40 KDa bifunctional polyethylene glycol mPEG2-NHS ester (MW 40 KDa; Nektar/Shearwater 2Z3XOT01, which includes two 20 KD monomethoxy PEGs bound together through a lysine and linked via a 6-carbon linker to a 5′-terminal phosphate on the aptamer), appended to the 5′end via conjugation to a C6 amino linker added to the aptamer during solid phase synthesis).
- FIG. 8 indicates that the length of stem 1 does not affect how 40 KDa PEG addition impacts the activity of AptA and AptA derivatives.
- the anticoagulant activity of PEG Apt 19 and 16 is essentially identical to the anticoagulant activity of both pegylated (PEG AptA) and cholesterol-modified (CH-AptA) versions of the parental AptA sequence.
- PEG Apt 19 is more readily neutralized by its matched antidote (7 AD) than AptA, Apt 16 or any of the PEG or cholesterol-modified versions of these compounds.
- FIG. 8 shows approximately 90% reversal of PEG Apt 19 at 2.5:1 AD:Aptamer. Looked at as an absolute change in APTT rather than on a % reversal basis, the APTT of plasma treated with PEG Apt 19+2.5:1 7AD:Aptamer is only 4-5 seconds above baseline.
- the anticoagulant activity of Apt 22-29 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the “neutralizability” of Apt 22-29 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays, except for Apt 24 in which the aptamer concentration was fixed at 250 nM.
- the anticoagulant activity of Apt 30-33 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the “neutralizability” of Apt 30 and 33 was evaluated in standard APTT antidote assays over antidote concentrations (Apt 14 AD) ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays (see FIG. 10 ).
- Apt 34 had C16 a 2′fluoro rather than 2′-O-methyl ( FIG. 10 b ). Increase in anticoagulant activity (compare Apt 34 to Apt 33). However, substitution did result in a modest loss of “neutralizability”, although 34 still requires a lower excess of antidote to achieve 90% neutralization ( ⁇ 5:1 vs 10:1) than the parental AptA compound. Both results are consistent with an increase in the stability of stem 2 due to 2′-O-methyl substitution.
- the anticoagulant activity of Apt 35-39 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the “neutralizability” of Apt 35, 38 and 39 was evaluated in standard APTT antidote assays over antidote concentrations (Apt 7 AD) ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays ( FIG. 11 ).
- Apt 35-39 Anticoagulant activity of Apt 39 is superior to Apt 19 and all other stem 2-optimization constructs in the Apt 19 background ( FIG. 11 ). Results were consistent with those obtained with Apt 34. In addition, potency of Apt 39 is comparable to parental AptA and greater than Apt 2. The neutralization of Apt 39 by Apt7AD is excellent, and similar to neutralization of Apt 19 ( FIG. 12 ). Again, sugar optimization and truncation of Apt 39 has resulted in a compound neutralized at lower excesses of antidote:drug as compared to parental AptA and Apt 2 ( FIG. 11 ).
- the anticoagulant tested is Apt39 with a 40 KDa polyethylene glycol (PEG) conjugated to the 5′end of the aptamer sequence via a 6-carbon NH2 linker (PEG-Apt39).
- the antidote is Apt7AD.
- the anticoagulant activity of PEG-Apt39 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar.
- the anticoagulant activity of Apt39 was compared to two formulations of the parental AptA, CH-Apt S(5′ cholesterol-modified) and PEG-AptA (5′ 40 KDa PEG-modified).
- the molecular weight of the “aptamer” portion only was used to calculate the concentration of each compound.
- the “neutralizability” of PEG-Apt39 was evaluated in standard APTT antidote assays over antidote concentrations (Apt7AD) ranging from 5 uM and down.
- the aptamer concentration was fixed at 125 nM in these assays.
- the in vitro anticoagulant activity of PEG-Apt39 is essentially equivalent to CH-AptA and PEG-AptA ( FIG. 13 ).
- the dose of PEG-Apt39 was 0.5 mg/kg (dose of aptamer based upon molecular weight of nucleic acid component only; 10,103.2 Da) for each animal.
- the aptamer dose was also 0.5 mg/kg (dose of aptamer based upon molecular weight of nucleic acid component only).
- the dose of the antidote was 3 mg/kg.
- the antidote dose was 5 mg/kg.
- ACT's Activated clotting times
- Aptamer levels in plasma are determined using a sandwich-type hybridization assay with an enzyme-linked immunoassay (ELISA) for detection.
- Quantitation of aptamer employs two oligonucleotide probes, a DNA capture probe, and a 2′Omethyl RNA detection probe.
- the DNA capture probe is 15 nucleotides in length, is complementary to the 3′ terminal 15 nucleotides of the aptamer, and contains a biotin moiety on its 5′terminus, allowing for capture of oligonucleotide complexes containing this probe to an avidin coated surface.
- the 2′Omethyl RNA detection probe is also 15 nucleotides in length, is complementary to the portion of aptamer to which antidote binds, and contains a digoxigenin moiety to enable detection of complexes containing this probe using standard enzyme-linked fluorescence generating enzyme/substrate reagents.
- Quantitation of aptamer is achieved by hybridization of the capture and detection probes to aptamer in plasma and subsequent immobilization of the complex onto the surface of a Neutravidin-coated microtitre plate by way of the 5′-biotin group.
- Measurement of the digoxigenin-labeled 2′-O-methyl RNA probe is performed subsequent to the plate immobilization reaction using an anti-digoxigenin antibody conjugated to alkaline phosphatase, which catalyzes the fluorescence of a substrate. Fluorescence intensity is then measured, the signal of which is directly proportional to the amount of aptamer present in the calibration standards and validation samples.
- aptamer Apt39 SEQ ID NO:88
- Plasma FIX assays were performed to aid in interpretation of the Apt39 APTT dose-response curve in monkey plasma. As shown in Table A, the APTT in monkey plasma is sensitive to the FIX level. However, the magnitude of the response to reduction in the FIX level is modest.
- a 75% reduction in the FIX level results in a 1.4-fold increase in the APTT
- a >95% reduction in the FIX level results in a doubling of the APTT
- a 99.9% reduction in the plasma FIX level yields a 2.5-fold increase in the APTT.
- the four monkeys assigned to Group 1 were subdivided into two groups at Day 13, with two animals receiving a low dose (Group 1a, 5 mg/kg) and two animals receiving a high dose (Group 1b, 30 mg/kg).
- APTT was followed until it returned to baseline in monkeys receiving 5 and 15 mg/kg doses.
- the whole-blood activated clotting time (ACT) data mirrored the APTT data (data not shown).
- the APTT data from animals treated with aptamer followed by antidote 3 hours later are shown in FIG. 17 .
- administration of aptamer at these dose levels resulted in a profound level of anticoagulation, with the mean APTTs at 0.25 and 3 hours post administration consistent with essentially complete FIX inhibition at both dose levels.
- Subsequent administration of Apt7AD rapidly and completely neutralized the anticoagulant effects of Apt39 in the monkey, with the mean APTT returning to baseline within 15 minutes following Apt7AD administration.
- the APTT was followed for 5 days post aptamer administration.
- APTT data collected over this time frame indicate the anticoagulant effects of aptamer were durably neutralized, with no evidence of rebound anticoagulation over 120 hours, or approximately 10 half-lives of aptamer in the monkey ( FIG. 17 ).
- Toxicokinetic data were collected for 24 hours following Apt39 administration in the Group 3 animals (Table D).
- both free aptamer and complexed aptamer plasma concentrations were measured.
- the mean concentration of free aptamer decreased 5,000-10,000 fold, to levels below the Lower Limit of Quantitation (LLOQ) of the assay employed.
- LLOQ Lower Limit of Quantitation
- the mean plasma concentration of complexed aptamer increased from below the LLOQ of the assay to ⁇ 125 to 220 ⁇ g/mL at the 15/30 and 30/60 mg/kg dose levels respectively, indicating the rapid decrease in free Apt39 concentrations was due to binding of Apt7AD.
- the concentration of free aptamer remained below the LLOQ of the assay as long as 3 hours after antidote administration, consistent with the APTT results. At 21 hours after antidote administration, very low levels of Apt39 were detectable in several animals (mean of only 0.17 ⁇ g/mL or lower).
Abstract
The invention provides improved nucleic acid ligands with enhanced stability that inhibit coagulation and improved modulators of the nucleic acids to provide ideal modulators of coagulation. These improved nucleic acids and modulators are particularly useful for inhibiting coagulation in a host undergoing a therapeutic regime such as surgery or coronary artery bypass.
Description
- This application is a continuation-in-part of U.S. application Ser. No. 11/113,378, filed Apr. 22, 2005.
- An improved agent, composition and method to regulate the pharmacological activity of a coagulation factor with nucleic acid ligands (e.g., aptamers) that have improved stability is disclosed.
- Despite substantial efforts to treat and prevent thrombotic events, arterial thrombosis continues to be the major cause of death in adult populations of developed nations. Although numerous medical strategies exist for treating thrombosis, no available agent meets the therapeutic endpoints of both bioavailability and efficacy, while also having a reasonable safety profile (see Feuerstein et al. (1999) Arterioscler. Thromb. Vasc. Biol. 19:2554-2562).
- Under normal circumstances, an injury to vascular endothelial cells lining a blood vessel triggers a hemostatic response through a sequence of events commonly referred to as the coagulation “cascade.” The cascade culminates in the conversion of soluble fibrinogen to insoluble fibrin which, together with platelets, forms a localized clot or thrombus which prevents extravascular release of blood components. Wound healing can then occur followed by clot dissolution and restoration of blood vessel integrity and flow.
- Initiation of blood coagulation arises from two distinct pathways: the intrinsic and extrinsic pathways. The intrinsic pathway can be triggered in vitro by contact of blood borne factors with artificial negatively charged surfaces such as glass. In contrast, the extrinsic pathway can be initiated in vivo or in vitro when tissue factor (TF), normally sequestered from the circulatory system, comes into contact with blood after injury. Blood exposed TF acts as a cofactor for the factor VIIa (“FVIIa”) catalyzed activation of factor IX (“FIX”) and factor X (“FX”). This leads to rapid formation of FXa and thrombin, which subsequently polymerizes to form the fibrin clot. Both the intrinsic and extrinsic pathways are characterized by the assembly of multiple protein complexes on procoagulant surfaces, which localizes the response to the site of injury (see Mann, K. G. et al. (1990) Blood 76: 1).
- Anticoagulant Therapy
- Coumarin drugs, such as warfarin as well as the glycosaminoglycans, heparin and heparan sulfate, are commonly used as anticoagulants. Warfarin, a coumarin derivative, acts by competing with vitamin K dependent post-translational modification of prothrombin and other vitamin K-dependent clotting factors. Its action is somewhat slower and longer lasting effect than heparin. The coumarin drugs inhibit coagulation by inhibiting the vitamin K-dependent carboxylation reactions necessary to the function of thrombin, and factors VII, IX, and X as well as proteins C and S. These drugs act by inhibiting the reduction of the quinone derivatives of vitamin K to their active hydroquinone forms. Because of the mode of action of coumarin drugs, it takes several days for their maximum effect to be realized. Heparin binds to, and activates, antithrombin III which then inhibits the serine proteases of the coagulation cascade. In part due to their potency, heparin and LMW heparin suffer drawbacks. Uncontrolled bleeding is a major complication observed in up to 7% of patients receiving continuous infusion up to 14% of patients given intermittent bolus doses. The therapeutic range to achieve efficacy without placing the patient at risk for bleeding is narrow, approximately 1 to less than 3 ug heparin/ml plasma. At concentrations greater than 4 ug/ml of heparin, clotting activity is not detectable. Thus, great care must be taken to keep the patient's plasma concentrations within the therapeutic range.
- Groups have used antibodies to coagulation factors to regulate the coagulation cascade. For example PCT Publication No.
WO 03/093422 to Schering Aktiengesellschaft discloses antibodies that bind with greater affinity to the factor VIIa/tissue factor (FVIIa/TF) complex than to tissue factor (TF) alone. These antibodies allegedly do not compete for binding to tissue factor with Factor VII and Factor X, and inhibit FX activation. - U.S. Pat. No. 6,001,820 to Hamilton Civic Hospitals Research Development Inc. provides heparin cofactor II specific catalytic agents which are capable of (1) selectively inactivating thrombin which is bound either to fibrin in a clot or to some other surface, but which has only minimal inhibitory activity against free thrombin; (2) inhibiting the assembly of the intrinsic tenase complex and thereby the activation of Factor X by Factor IXa; and (3) inhibiting the activation of Factor IX by Factor XIa.
- Aptamers
- Nucleic acids have conventionally been thought of as primarily playing an informational role in biological processes. In the past decade it has become clear that the three dimensional structure of nucleic acids can give them the capacity to interact with and regulate proteins. Such nucleic acid ligands or “aptamers” are short DNA or RNA oligomers which can bind to a given ligand with high affinity and specificity. As a class, the three dimensional structures of aptamers are sufficiently variable to allow aptamers to bind to and act as ligands for virtually any chemical compound, whether monomeric or polymeric. Aptamers have emerged as promising new diagnostic and therapeutic compounds, particularly in cancer therapy and the regulation of blood coagulation.
- Nucleic acid ligands can be identified through methods related to a method termed the Systematic Evolution of Ligands by EXponential enrichment (SELEX). SELEX involves selection of protein-binding nucleic acids from a mixture of candidate oligonucleotides and step-wise iterations of binding, partitioning and amplification to achieve the desired criterion of binding affinity and selectivity. The SELEX process was first described by Gold and Tuerk in U.S. Pat. No. 5,475,096, and thereafter in U.S. Pat. No. 5,270,163 (see also WO 91/19813; Tuerk et al. (1990) Science 249:505-10).
- A number of third parties have applied for and secured patents covering the identification, manufacture and use of aptamers. As stated above, Gold and Tuerk are generally credited with first developing the SELEX method for isolating aptamers, and their method is described in a number of United States patents including U.S. Pat. Nos. 5,670,637, 5,696,249, 5,843,653, 6,110,900, and 5,270,163. Thomas Bruice et al. reported a process for producing aptamers in U.S. Pat. No. 5,686,242, which differs from the original SELEX process reported by Tuerk and Gold because it employs strictly random oligonucleotides during the screening sequence. The oligonucleotides screened in the '242 patent lack the oligonucleotide primers that are present in oligonucleotides screened in the SELEX process.
- Several patents to Gold et al. contain claims covering aptamers to thrombin. For example, U.S. Pat. No. 5,670,637 contains claims covering aptamers that bind to proteins. U.S. Pat. No. 5,696,249 claims an aptamer produced by the SELEX process. U.S. Pat. Nos. 5,756,291 and 5,582,981 to O'Toole, disclose and claim a method for detecting thrombin using a labeled aptamer that comprises a defined six nucleotide sequence. U.S. Pat. Nos. 5,476,766 and 6,177,557 disclose compounds and methods to identify nuclei acid ligand solutions to thrombin using SELEX.
- Sullenger, Rusconi, Kontos and White in WO 02/26932 describe RNA aptamers that bind to coagulation factors, E2F family transcription factors, Ang1, Ang2, and fragments or peptides thereof, transcription factors, autoimmune antibodies and cell surface receptors useful in the modulation of hemostasis and other biologic events. See also Rusconi et al, Thrombosis and Haemostasis 83:841-848 (2000), White et al, J. Clin Invest 106:929-34 (2000), Ishizaki et al, Nat Med 2:1386-1389 (1996), and Lee et al, Nat. Biotechnol. 15:41-45 (1997)).
- Modulation of Aptamers
- PCT Publication No. WO 02/096926 to Duke University describes agents and methods to modulate the biological activity of nucleic acid ligands through the administration of a modulator. The publication describes aptamers controlled by modulators that can be nucleic acids. The modulatable aptamers are described as being useful in the the treatment of diseases in which it is important to inhibit coagulation,
elongation factor 2 activity or angiogenesis. The modulatable aptamers to control coagulation include the aptamers to coagulation factors VII or VIIa, VIII or VIIIa, IX or LXa, V or Va, X or Xa, complexes formed with these factors, as well as platelet receptors. The modulator can change the binding of the nucleic acid ligand for its target, degrade or otherwise cleave, metabolize or break down the nucleic acid ligand while the ligand is exerting its effect. Modulators can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy. - Maximizing utility of Aptamers
- In order for aptamers to be useful therapeutic reagents, they should bind tightly to proteins, inhibit a specified function of that protein if an antagonist is desired and have no harmful side-effects. Unmodified RNA is not realistically used as a therapeutic agent since blood is rich in ribonucleases. Some modification of single-stranded RNA and DNA can produce molecules which are stable in blood and certain known aptamers have 2′F or 2′NH2 groups within each pyrimidine nucleotide.
- However, there is no way to predict how a particular modification changes aptamers. In particular, when additional limitations are required, as is the case with modulatable aptamers, no techniques exist to predict how one or more modifications can affect the capacity of the aptamer to regulate its ligands and at the same time continue to be regulated by antidote binding.
- Several methods have been developed that modify the base SELEX process to obtain modified aptamers. For example, patents disclose the use of modified nucleotides in the SELEX process to obtain aptamers that exhibit improved properties. U.S. Pat. No. 5,660,985 provides 2′-modified nucleotides that allegedly display enhanced in vivo stability. U.S. Pat. No. 6,083,696 discloses a “blended” SELEX process in which oligonucleotides covalently linked to non-nucleic acid functional units are screened for their capacity to bind a target molecule. Other patents describe post-SELEX modifications to aptamers to decrease their size, increase their stability, or increase target binding affinity (see, e.g., U.S. Pat. Nos. 5,817,785 and 5,648,214).
- In U.S. Pat. No. 5,245,022 Weis et al. disclose an oligonucleotide of about 12-25 bases that is terminally substituted by a polyalkyleneglycol. These modified oligonucleotides are reported to be resistant to exonuclease activity.
- U.S. Pat. Nos. 5,670,633 and 6,005,087 to Cook et al. describe thermally stable 2′-fluoro oligonucleotides that are complementary to an RNA or DNA base sequence. U.S. Pat. Nos. 6,222,025 and 5,760,202 to Cook et al. describe the synthesis of 2′-O substituted pyrimidines and oligomers containing the modified pyrimidines.
EP 0 593 901 B1 discloses oligonucleotide and ribozyme analogues withterminal 3′,3′- and 5′,5′-nucleoside bonds. U.S. Pat. No. 6,011,020 to Gold et al. discloses and claims an aptamer modified by polyethylene glycol. - Currently, a strong need remains to provide methods and compositions to treat patients in need of anticoagulant therapy, and in particular, during surgery or other medical intervention.
- Therefore, it is an object of the present invention to provide methods and compositions to treat patients in need of anticoagulant therapy, and in particular, during surgery or other medical intervention
- It is another object of the present invention to provide more control over the therapeutic effect, pharmacokinetics and duration of activity of anticoagulant therapies.
- Improved nucleic acid ligands for anticoagulant therapy are disclosed as well as improved nucleic acid ligands in combination with an antidote that changes the binding of the nucleic acid ligand for its target or that degrades or otherwise cleaves, metabolizes or breaks down the nucleic acid ligand while the ligand is still exerting its effect. These improved aptamers provide favorable anticoagulant properties for in vivo applications, including during human or veterinary surgery. The anticoagulant function of the improved aptamer is conveniently neutralized on administration of its antidote when desired by the surgeon or orther medical care specialist. In particular, the aptamers of the invention include additional agents to increase bioavailability and/or decrease degradation of the active agent.
- In one aspect of the invention, improved nucleic acid ligands or aptamers to a factor in the blood coagulation cascade are provided that include at least one moiety that increases bioavailability of the agent. In some embodiments, the factors include Factor IX (FIX) or the cleavage product Factor IXa (FIXa). In some embodiments, the aptamers are ligands to the complex formed by FIXa with Factor VIIIa (FVIIIa), also known as the “intrinsic tenase complex.” In some embodiments, the aptamers are ligands that inhibit the complex formation between FIXa and FVIIIa. In a subembodiment, the aptamers of the present invention bind to the complex of FIX and FVIIIa and inhibit activation of Factor X (FX). The aptamers can interact with FIX, FIXa or a complex formed with FVIIIa in the presence or absence of additional calcium. The aptamers can also interact with the factors of the complex at a cell membrane. In one embodiment, the aptamers bind to the intrinsic tenase complex at the membrane surface.
- In particular, the aptamers, such as aptamers to FIXa, are linked to a stabilizing moiety on at least one terminus. In one embodiment, the aptamers are linked to a polymeric agent. In one embodiment, the aptamers are linked to one or more polyethylene glycol molecules. The aptamer and stabilizing agent can be linked at, for example, a 5′ terminus of the nucleic acid sequence. In certain embodiments, the aptamers are linked to multiple polyethylene glycol molecules. In other embodiments, a single polyethylene glycol molecule can be linked to more than one aptamer to provide improved delivery of the agent.
- The aptamers of the present invention can be comprised of ribonucleotides or deoxyribonucleotides, or a combination thereof. In general, the improved aptamers are at least 25 nucleotides long, and typically not longer than 35-40 nucleotides long. In one embodiment, aptamers are at least 25, 30, 35, or 40 nucleotides in length. In specific embodiments, the sequence of
stem 1 includes 5 nucleotides in the 5′-3′ direction. In a sub-embodiment, stem 1 includes three guanine (G) residues in the 5′-3′ direction. - The aptamers can include a “suicide position.” In one embodiment, this position becomes single stranded and labile upon binding of the antidote to the improved aptamer and allows for cleavage of the improved aptamer upon binding of the antidote by enzymes in the circulation, such as blood or liver endonucleases, thereby effectively eliminating the active aptamer from circulation. The suicide position can be at a guanine in
stem 2 that is hydroxylated. In one embodiment, this nucleotide is in a double stranded configuration until bound with an antidote and becomes single stranded and available for cleavage upon binding of the antidote. - In an embodiment, the aptamers include the nucleotide sequence gugg and the complimentary sequence ccac. In one embodiment, the aptamer to Factor IX comprises the nucleotide sequence: gugga cuauacc gcg uaaugc ugc c uccac t (SeqID 19).
- Another embodiment of the invention includes an antidote oligonucleotide paired with the aptamer of the invention. The antidote oligonucleotide can be complementary to at least a portion of the aptamer. The antidote can, for example, comprise the following sequences: (5′-3′) sequence: cgcgguauaguccccau (Apt/AD; SEQ ID NO:1); (5′-3′) sequence: cgcgguauaguccc (Apt6/AD; SEQ ID NO:2); (5′-3′) sequence: cgcgguauaguccac (Apt7/AD; SEQ ID NO:3); (5′-3′) sequence: cgcgguauaguccauc (Apt8/AD; SEQ ID NO:4); (5′-3′) sequence: cgcgguauagucag (Apt9/AD; SEQ ID NO:5); (5′-3′) sequence: cgcgguauagucagg (Apt10/AD; SEQ ID NO:6); (5′-3′) sequence: cgcgguauagucagag (Apt11/AD; SEQ ID NO:7); (5′-3′) sequence: cgcgguauaguccucac (Apt14/AD; SEQ ID NO:8), or any modification or derivative thereof. In certain embodiments, the antidote consists essentially of one of the above sequences, or consists entirely of one of the above sequences.
- In certain embodiments, the improved aptamer is provided in alternation with an antidote. The antidote sequence does not need to be completely complemetary to the improved anticoagulant aptamer as long as the antidote sufficiently binds to or hybridizes to the aptamer to neutralize its activity.
- The aptamer pairs of the present invention include the following sequences:
Apta mer Antidote augggga cuauacc gcg uaaugc ugc cgcgguauaguccccau c uccccau t (SEQ ID NO:1) (SEQ ID NO:9) augggga cuauaccgcguaaugcugcc cgcgguauaguccccau uccccau t (SEQ ID NO:1) (SEQ ID NO:10) ggga cuauaccgcguaaugcugcc uccc t cgcgguauaguccc (SEQ ID NO:11) (SEQ ID NO:2) gugga cuauaccgcguaaugcugcc cgcgguauaguccac uccac t (SEQ ID NO:3) (SEQ ID NO:12) gaugga cuauaccgcguaaugcugcc cgcgguauaguccauc uccauc t (SEQ ID NO:4) (SEQ ID NO:13) cuga cuauaccgcguaaugcugcc ucag t cgcgguauagucag (SEQ ID NO:14) (SEQ ID NO:5) ccuga cuauaccgcguaaugcugcc cgcgguauagucagg ucaggg t (SEQ ID NO:6) (SEQ ID NO:15) cucuga cuauaccgcguaaugcugcc cgcgguauagucagag ucagag t (SEQ ID NO:7) (SEQ ID NO:16) gugagga cuauaccgcguaaugcugcc cgcgguauaguccucac uccucac t (SEQ ID NO:8) (SEQ ID NO:17) gugagga cuauacc gcg uaaugc ugc cgcgguauaguccucac c uccucac t (SEQ ID NO:8) (SEQ ID NO:18) gugga cuauacc gcg uaaugc ugc c cgcgguauaguccac uccac t (SEQ ID NO:3) (SEQ ID NO:19) - Improved aptamer-antidote pairs that are more stable and bioactive are developed by including secondary modifications on either the aptamer or antidote or both. In one embodiment, the improved aptamer to Factor IX includes one or more 2′-O-methyl modified nucleotides. In another embodiment, the improved aptamer contains one or more 2′-O-methyl and one or more 2′-fluoro modifications. In another embodiment, the aptamer and antidote contain no 2′-fluoro modifications. In yet another embodiment, the improved aptamer includes one or more 2′-O-methyl and one or more 2′-fluoro modifications on a stem. In one embodiment, at least one guanine in
stem 2 of an improved aptamer includes a hydroxyl sugar (2′—OH). In another embodiment, at least one uridine instem 1 or instem 2 of the improved aptamer is modified with either a 2′-fluoro or 2′-O-methyl. In another embodiment, at least one cytidine instem 2 of the improved aptamer is 2′-fluoro modified. - In one embodiment, the aptamer is linked to one or more polyethylene glycol (PEG) molecule. In a specific embodiment, the aptamer is linked to 40 KD PEG using a six carbon amino linker. In a more specific embodiment, the six carbon amino linker is attached to the PEG through an amino acid attachment. In a subembodiment, the PEG is two twenty KD PEG that are attached to one or more amino acid, such as lysine, which is attached to the six carbon amino linker.
- In one specific embodiment, the aptamer to Factor IXa is the following structure: 5′-O-[[5-[N2-(monomethoxy 20K polyethylene glycol carbamoyl)-N6-(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)- riboguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-5′)-2′-methoxy-2′-deoxytidylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-3′)-thymidine.
- In another embodiment, the aptamer to Factor IXa is of the following structure:

where n is approximately 450 - In one embodiment, the aptamer to Factor IXa is 10,282.4 Daltons in its protonated form. In another embodiment, the aptamer is in its sodium salt form. In yet another embodiment, the aptamer is PEGylated RB006 and in its sodium salt.
- The improved aptamers and antidotes can also include nucleotides that are modified with water-soluble polymers. Such polymers can include a polyethylene glycol, polyamine, polyether, polyanhydride, polyester, or other biodegradable pharmaceutically acceptable polymer.
- The invention includes the use of the improved aptamers to bind to FIX, FIXa, or the intrinsic tenase complex. This binding can be in vitro or in vivo. The result of the binding to FIX, FIXa or the tenase complex can be to inhibit the biological activity of the proteins or complex.
- In one embodiment, the improved aptamer inhibits blood coagulation by binding to FIXa, which is derived from the same gene product as FIX. The invention includes administering the improved aptamers of the invention to a mammal in need thereof to inhibit blood coagulation. Another embodiment of the invention provides methods of using the improved aptamers and antidotes during a therapeutic regime.
- In one embodiment, antidotes to the improved aptamers of the invention are provided to a mammal in need thereof to reverse the anticoagulant effects of the improved aptamers. Improved aptamers and aptamer-antidote pairs can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy. Thus, this invention discloses an improved regulatable therapeutic regime in the course of nucleic acid ligand therapy for blood coagulation. In one example, an antidote is provide that neutralizes the effect of the improved aptamer to turn off anticoagulant activity when desired by the physician or other health care provider. In another embodiment, the improved aptamers and antidotes to blood coagulation factors are administered in sequential steps, in which the aptamers are administered, the antidotes are used to limit the activity of the improved aptamers, and subsequently the aptamers are re-administered to a patient in need thereof. In one embodiment, the antidote achieves this neutralization effect by binding to or hybridizing to the improved aptamer.
- The improved aptamers can be administered to patients suffering from or at risk of suffering from a cardiovascular disease or intervention, including surgical intervention, that causes or results in a coagulation-inducing event. Examples include acute myocardial infarction (heart attack), cerebrovascular accidents (stroke), ischemia, angioplasty, CABG (coronary artery bypass grafts), cardiopulmonary bypass, thrombosis in the circuit of cardiac bypass apparatus and in patients undergoing renal dialysis, unstable angina, pulmonary embolism, deep vein thrombosis, arterial thrombosis, and disseminated intravascular coagulation.
- The improved aptamers can also be administered to prevent coagulation-induced inflammation. It appears that early inflammation is induced by activation of the coagulation cascade. Therefore, the improved aptamers can be used to treat cardiovascular diseases that include a inflammatory component, for example, atherosclerosis, acute coronary syndrome (ACS), myocardial infarction which may result in reperfusion injury, or to treat adverse events associated with post-angioplasty restenosis.
-
FIG. 1 is a schematic of proposed two dimensional configurations of Apt A, 1-39 described below.FIG. 1 a is a schematic of aptamers Apt A and 1-5. 1 b is a schematic of aptamers 6-11. 1 c is a schematic of aptamers Apt 12-17. 1 d is a schematic of aptamers Apt 18-20, 1 e ofApt 21. 1 f is a schematic of aptamers Apt 22-29, 1 g of Apt 30-34 and 1 h of Apt 35-39. -
FIG. 2 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A and Apt 1-5 (left panel) and neutralizability by antidote AptA-AD. -
FIG. 3 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 6-8 (right panel) and neutralizability by antidote (left panel). -
FIG. 4 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 9-11 (left panel) and neutralizability by antidote (right panel). -
FIG. 5 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A, 2 and 12-17 (left panel) and neutralizability by antidote (right panel). -
FIG. 6 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2, 15, 16 and 21 (left panel) and neutralizability by antidote (right panel). -
FIG. 7 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 16-20 (left panel) and neutralizability by antidote (right panel). -
FIG. 8 is a graph of results of activated partial thromboplastin time (APTT) test assays of pegylated AptA compared to pegylated Apt 16 and 19 and cholesterol-modified Apt A (chol-A). Left panel is aptamer control of coagulation factor IX and right panel is neutralizability by antidote. -
FIG. 9 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2, 16 and 22-27 (left panel) and neutralizability by antidote (right panel). -
FIG. 10 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt 2 and 30-33 (A, left panel), neutralizability by antidote ofApt -
FIG. 11 is a graph of results of activated partial thromboplastin time (APTT) test assays of aptamers Apt A, 2, 19 and 35-39 (left panel) and neutralizability by antidote of Apt A, 19,35,38 and 39(right panel). -
FIG. 12 is a graph of results of neutralizability by antidote assays of aptamers Apt 2, 34, 39 and Peg-19. -
FIG. 13 is a graph of the in vitro anticoagulant activity of PEG-Apt39 compared to CH-AptA and PEG-AptA. -
FIG. 14 are graphs of systemic anticoagulant activity (14 a) and neutralizability (14 b) of PEG-Apt39 in swine. The change in the value of the respective clotting assays is the difference between the clotting time at the time point and the pre-injection baseline for that animal. n=2 for PEG-Apt39 treated animals and n=3 for CH-AptA treated animals. Whole blood ACT values are shown in the bottom panel, and plasma APTT values in the panel at the top. -
FIG. 15 is graphs of systemic anticoagulant activity (14 a) and neutralizability (14 b) of PEG-Apt39 in swine. The change in the value of the respective clotting assays is the difference between the clotting time at the time point and the pre-injection baseline for that animal. n=2 for PEG-Apt39 treated animals. Data from this experiment is compared to the anticoagulation and neutralization data for PEG-Apt39 presented inFIG. 3 . Whole blood ACT values are shown in the left panel, and plasma APTT values in the panel at right. -
FIG. 16 is a graph of systemic anticoagulation of monkeys by Apt39 administration as described in Example 9. The level of anticoagulation in the monkeys was monitored with the APTT. For animals treated with 15 mg/kg, Apt39 data are presented as the mean±SEM. For animals at the 5 and 30-mg/kg dose levels, data are presented as the mean±range, as there were only 2 animals at each of these dose levels. -
FIG. 17 is a graph of the systemic anticoagulation of monkeys with Apt39 and reversal with antidote Apt7AD, as described in Example 9. The level of anticoagulation in the monkeys was monitored with the APTT. Apt7AD was administered at t=3 hours following Apt39 administration. Data are presented as the mean±SEM. - 1. Definitions
- A “nucleic acid ligand” or “aptamer” is a nucleic acid that can form a three dimensional configuration, which allows it to interact as a ligand with a target molecule. The terms refer to oligonucleotides having specific binding regions that are capable of forming complexes with an intended target molecule in an environment wherein other substances in the same environment are not complexed to the oligonucleotide. The specificity of the binding is defined in terms of the comparative dissociation constants (Kd) of the aptamer for target as compared to the dissociation constant with respect to the aptamer and other materials in the environment or unrelated molecules in general. Typically, the Kd for the aptamer with respect to the target will be 10-fold , 50-fold, 100-fold, or 200-fold less than the Kd with respect to the unrelated material or accompanying material in the environment.
- “Aptamer antidote pair” is meant to include a specified aptamer to a target molecule, and an oligonucleotide that changes the three dimensional configuration of the aptamer so that the aptamer can no longer interact with its target. The antidote can be an oligonucleotide complimentary to a portion of the aptamer. The antidote can change the conformation of the aptamer to reduce the target binding capacity of the aptamer by 10 to 100%, 20 to 100%, 25%, 40%, 50%, 60%, 70%, 80%, 90% or 100%, or any percentage in the range between 10 and 100% under physiological conditions. The antidote can also form a three dimensional structure with binding activity to a target molecule. This target can be the same or different from the target of the aptamer.
- “Antidote,” “Regulator” or “Modulator” refers to any pharmaceutically acceptable agent that can bind an aptamer and modify the interaction between that aptamer and modify the interaction between that aptamer and its target molecule (e.g., my modifying the structure of the aptamer) in a desired manner.
- The terms “binding activity” and “binding affinity” are meant to refer to the tendency of a ligand molecule to bind or not to bind to a target. The energy of said interactions are significant in “binding activity” and “binding affinity” because they define the necessary concentrations of interacting partners, the rates at which these partners are capable of associating, and the relative concentrations of bound and free molecules-in a solution. The specificity of the binding is defined in terms of the comparative dissociation constants (Kd) of an antidote of a nucleic acid ligand as compared to the dissociation constant with respect to other materials in the environment or unrelated molecules in general.
- As used herein, “consensus sequence” refers to a nucleotide sequence or region (which might or might not be made up of contiguous nucleotides) that is found in one or more regions of at least two nucleic acid sequences. A consensus sequence can be as short as three nucleotides long. It also can be made up of one or more noncontiguous sequences, with nucleotide sequences or polymers of up to hundreds of bases long interspersed between the consensus sequences. Consensus sequences can be identified by sequence comparisons between individual nucleic acid species, which comparisons can be aided by computer programs and other, tools for modeling secondary and tertiary structure from sequence information. Generally, the consensus sequence will contain at least about 3 to 20 nucleotides, more commonly from 6 to 10 nucleotides.
- The terms “cardiovascular disease” and “cardiovascular diseases” are meant to refer to any cardiovascular disease as would be understood by one of ordinary skill in the art. Nonlimiting examples of particularly contemplated cardiovascular diseases include, but are not limited to, atherosclerosis, thrombophilia, embolisms, cardiac infarction (e.g., myocardial infarction), thromboses, angina, stroke, septic shock, hypertension, hyper-cholesterolemia, restenosis and diabetes (and associated diabetic retinopathy). Cardiovascular disease can be treated at any stage of progression, such as treatment of early onset cardiovascular disease as well as treatment of advanced cardiovascular disease. A therapeutic method directed toward-inhibiting the aggravation of cardiovascular disease by modulating coagulation is also included in the invention.
- 2. Aptamers To Factor IX
- The invention provides improved nucleic acid ligands or aptamers that regulate blood coagulation through interaction with specific factors in the blood coagulation cascade. The invention also provides improved aptamer-antidote pairs to regulate coagulation. The improved aptamers target Factor IX gene products(which include Factor IXa) and thus reduce the non-specific side effects associated with other blood coagulation factor targets. Most factors in the coagulation cascade are broad spectrum proteins with a variety of physiological roles (i.e. thrombin).
- The events which occur between injury and blood clot formation are a carefully regulated and linked series of reactions. In a cell-based model of coagulation, initiation takes place on tissue factor-bearing cells (monocytes, macrophages, endothelial cells). In the presence of FVIIa (complexed with tissue factor), activation of FIX and FX generates a small amount of thrombin from prothrombin (which subsequently activates FV) In the amplification phase (also referred to as the priming phase), the small amount of thrombin generated activates platelets, causing release of FVa, FXIa and FVIIIa. During the final phase of coagulation, propagation, FIXa complexes with FVIIIa, activating FX. The FXa-FVa complex, in the presence of calcium and phospholipids substrate (prothrombinase complex), leads to a “burst” of thrombin generation.
- The cell-based model of anticoagulation has been instrumental in defining coagulation protease targets. Most previous work has focused on blood coagulation through a variety of factors such as trhrombin. Thrombin is a broad acting protein with effects throughout the body. Inhibitors of thrombin can therefore have unanticipated side effects in addition to the effects on coagulation. Thrombin not only activates endothelial cells and induces leukocyte infiltration and edema but also activates astrocytes and microglia to propagate the focal inflammation and produce potential neurotoxic effects.
- The inventor has determined that Factor IXa in particular represents an attractive target because of its participation in both the initiation and propagation phases of coagulation. Iteractive in vitro selection techniques have been used to identify oligonucleotides capable of binding FIXa with high affinity (Kd 0.65±0.2 nM). Experimental studies suggest that FIXa may have a critical role in thrombosis, as well as hemostasis. Infusion of purified FIXa into rabbits induces thrombosis (Gitel et al. (1977) PNAS 74:3028-32; Gurewich et al. (1979) Thromb. Rsch. 14:931-940). In contrast, active site-blocked FIXa prevented clot formation and reduced intra-arterial coronary thrombosis (Lowe (2001) Brit. J. Haem. 115:507-513).
- Antibodies to factor IX have also been shown to interfere with the function of the intrinsic tenase complex, the activation of zymogen factor IX by factor XIa and by the tissue factor:factor VIIa complex and potently inhibit activated partial thromboplastin clotting times (APTT) in plasma of guinea pig and rat (Refino, C. J., et al, (1999) Thromb and Haemost, 82:1188-1195; Feuerstein G Z, et al. (1999) Arterioscler Thromb Vasc Biol19(10):2554-62; Toomey J R, et al. (2000) Thromb Res. 100(1):73-9).
- In one embodiment, the invention provides nucleic acid ligands or aptamers to a factor in the blood coagulation cascade. In some embodiments, the factors include Factor IX (FIX) or the cleavage product Factor IXa (FIXa). In some embodiments, the aptamers are ligands to the complex formed by FIXa with Factor VIIIa (FVIIIa), also known as the “intrinsic tenase complex.” In some embodiments, the aptamers are ligands that inhibit the complex formation between FIXa and FVIIIa. In a subembodiment, the aptamers of the present invention bind to the complex of FIX and FVIIIa and inhibit activation of Factor X (FX). The aptamers can interact with FIX, FIXa or a complex formed with FVIIIa in the presence or absence of additional calcium. The aptamers can also interact with the factors of the complex at a cell membrane. In one embodiment, the aptamers bind to the intrinsic tenase complex at the membrane surface.
- In one embodiment, the applicants have discovered improved aptamers to gene products of coagulation Factor IX (FIX), and to its cleavage product, Factor IXa (FIXa). In one embodiment, the nucleic acid ligand includes at least one region that binds to another region in the molecule via Watson-Crick base pairing (stem) and at least one region that does not bind to any other regions of the molecule under physiological conditions (loop). In a further embodiment, the nucleic acid ligand includes two stems (
stem 1 and stem 2) and two loops (loop 1 and loop 2). In one embodiment,stem 1 is one to twenty nucleotides long. In a further embodiment,stem 1 is one to ten nucleotides long. In a further sub-embodiment, stem 1 is seven, six, five, four, three or two nucleotides long. In another embodiment,stem 2 one to twenty nucleotides long. In a further embodiment,stem 2 is one to ten nucleotides long. In a further sub-embodiment, stem 2 is seven, six, five, four, three or two nucleotides long. - The aptamers to a Factor IX gene product of the present invention can be comprised of ribonucleotides or deoxyribonucleotides, or a combination thereof. In general, the improved aptamers are at least 25 nucleotides long, and typically not longer than 35-40 nucleotides long. In one embodiment, aptamers are at least 25, 30, 35, or 40 nucleotides in length. In specific embodiments, the sequence of
stem 1 includes 5 nucleotides in the 5′-3′ direction. In a sub-embodiment, stem 1 includes three guanine (G) residues in the 5′-3′ direction. - In an embodiment, the aptamers include the consensus nucleotide sequences gugg and the complimentary sequence ccac. When a number of individual, distinct aptamer sequences for a single target molecule have been obtained and sequenced, the sequences can be examined for “consensus sequences.” As used herein, “consensus sequence” refers to a nucleotide sequence or region (which might or might not be made up of contiguous nucleotides)that is found in one or more regions of at least two aptamers, the presence of which can be correlated with aptamer-to-target-binding or with aptamer structure.
- A consensus sequence can be as short as three nucleotides long. It also can be made up of one or more noncontiguous sequences. With nucleotide sequences or polymers of hundreds of bases long interspersed between the consensus sequences. Consensus sequences can be identified by sequence comparisons between individual aptamer species, which comparisons can be aided by computer programs and other, tools for modeling secondary and tertiary structure from sequence information. Generally, the consensus sequence will contain at least about 3 to 20 nucleotides, more commonly from 6 to 10 nucleotides. Not all oligonucleotides in a mixture can have the same nucleotide at such position; for example, the consensus sequence can contain a known ratio of particular nucleotides. For example, a consensus sequence might consist of a series of four positions wherein the first position in all members of the mixture is A, the second position is 25% A, 35% T and 40% C, the third position is T in all oligonucleotides, and the fourth position is G in 50% of the oligonucleotides and C in 50% of the oligonucleotides.
- In specific embodiments, the aptamers include the nucleotide sequences of the following Seq ID Nos.:
SeqID Code Size Sequence 9 AptA 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 10 Apt1 35 mer (5′-3′) sequence: augggga cuaua ccgcguaaugcugcc uccccau t 9 Apt2 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 9 Apt3 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 9 Apt4 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 9 Apt5 35 mer (5′-3′) sequence: augggga cuaua cc gcg uaaugc ugc c uccccau t 11 Apt6 29 mer (5′-3′) sequence: ggga cuauaccg cguaaugcugcc uccc t 12 Apt7 31 mer (5′-3′) sequence: gugga cuauacc gcguaaugcugcc uccac t 13 Apt8 33 mer (5′-3′) sequence: gaugga cuauac cgcguaaugcugcc uccauc t 14 Apt9 29 mer (5′-3′) sequence: cuga cuauaccg cguaaugcugcc ucag t 15 Apt10 31 mer (5′-3′) sequence: ccuga cuauacc gcguaaugcugcc ucagg t 16 Apt11 33 mer (5′-3′) sequence: cucuga cuauac cgcguaaugcugcc ucagag t 10 Apt12 35 mer (5′-3′) sequence: augggga cuaua ccgcguaaugcugcc uccccau t 10 Apt13 35 mer (5′-3′) sequence: augggga cuaua ccgcguaaugcugcc uccccau t 17 Apt14 35 mer (5′-3′) sequence: gugagga cuaua ccgcguaaugcugcc uccucac t 17 Apt15 35 mer (5′-3′) sequence: gugagga cuaua ccgcguaaugcugcc uccucac t 17 Apt16 35 mer (5′-3′) sequence: gugagga cuaua ccgcguaaugcugcc uccucac t 17 Apt17 35 mer (5′-3′) sequence: gugagga cuaua ccgcguaaugcugcc uccucac t 11 Apt18 29 mer (5′-3′) sequence: ggga cuauaccg cguaaugcugcc uccc t 12 Apt19 31 mer (5′-3′) sequence: gugga cuauacc gcguaaugcugcc uccac t 13 Apt20 33 mer (5′-3′) sequence: gaugga cuauac cgcguaaugcugcc uccauc t 18 Apt21 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt22 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt23 35 mer (5′-3′) sequence: gugaggacuauac c gcg uaaugc ugc c uccucac t 18 Apt24 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt25 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 27 Apt26 33 mer (5′-3′) sequence: gugagga cuaua cc gca aucg ugc c uccucac t 28 Apt27 33 mer (5′-3′) sequence: gugagga cuaua cc gca aucg ugc c uccucac t 29 Apt28 33 mer (5′-3′) sequence: gugagga cuaua cc gca aucg ugc c uccucac t 30 Apt29 33 mer (5′-3′) sequence: gugagga cuaua cc gca aucg ugc c uccucac t 18 Apt30 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt31 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt32 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt33 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 18 Apt34 35 mer (5′-3′) sequence: gugagga cuaua cc gcg uaaugc ugc c uccucac t 19 Apt35 31 mer (5′-3′) sequence: gugga cuauacc gcg uaaugc ugc c uccac t 19 Apt36 31 mer (5′-3′) sequence: gugga cuauacc gcg uaaugc ugc c uccac t 19 Apt37 31 mer (5′-3′) sequence: gugga cuauacc gcg uaaugc ugc c uccac t 19 Apt38 31 mer (5′-3′) sequence: gugga cuauacc gcg uaaugc ugc c uccac t 19 Apt39 31 mer (5′-3′) sequence: gugga cuauacc gcg uaaugc ugc c uccac t - In one embodiment, the aptamer to Factor IX comprises, consists, or consists essentially of, the nucleotide sequence: gugga cuauacc gcg uaaugc ugc c uccac t (SeqID 19).
- 3. Modifications
- The improved aptamers and aptamer-antidote combinations of the present invention are modified by substituting particular sugar residues, by changing the composition of the aptamer and the size of particular regions in the aptamer, and by designing aptamers that can be more effectively inhibited by antidotes. The design of aptamers includes an appreciation for the secondary structure of the aptamer (see
FIG. 1 ) and the relationship between the secondary structure and the antidote control. Unlike conventional methods of modifying nucleic acids, the design of the improved aptamers to FIX gene products included in the invention must include a consideration of the antidote control. Controlled aptamers require that the aptamer be stable in circulation but not so stable that it is not antidote controlled. The aptamers can be modified by truncation, but antidotes need to be designed to control each aptamer when truncated. Further, certain modifications, particularly at the interface of the stems and loops cannot be modified from 2′-fluoro or the aptamer can lose activity. - In one embodiment, the design includes decreasing the 2′-hydroxyl content of the aptamer or the antidote, or both. In another embodiment, the design includes decreasing the fluoro content of the aptamer or the antidote, or both. In a further embodiment, the design includes increasing the 0-methyl content of the aptamer or the antidote, or both. In a further embodiment, the design includes decreasing the size of the aptamer. In another embodiment, the size of the antidote is changed in relation to the size of the aptamer. In yet another embodiment, guanine strings are reduced to less than four guanine, or less than three guanine, or less than two guanine or no guanines. However, the joint effect of these changes must meet the challenge of creating an anticoagulant that provides adequate activity but is easily neutralized by the antidote.
- Yet another embodiment includes a method of designing aptamers with a “suicide position” which allows more effective regulation by paired antidotes. In one embodiment, this position becomes single stranded and labile upon binding of the antidote to the improved aptamer and allows for cleavage of the improved aptamer upon binding of the antidote by enzymes in the circulation, such as blood or liver endonucleases, thereby effectively eliminating the active aptamer from circulation. The suicide position can be, in one embodiment, at a guanine in
stem 2 that is hydroxylated. In one embodiment, the aptamer is in a double stranded configuration until bound with an antidote and becomes single stranded and available for cleavage upon binding of the antidote. - The applicants have discovered aptamer-antidote pairs that are stable and bioactive by including secondary modifications on either the aptamer or antidote or both. In specific embodiments, the aptamers to Factor IX include modified nucleotides. In one embodiment, the aptamer contains one or more 2′-O-methyl groups. In another embodiment, the aptamer and antidote contain one or more 2′-O-methyl and one or more 2′-fluoro modifications. In another embodiment, the aptamer and antidote contain no 2′-fluoro modifications. In yet another embodiment, the aptamer includes one or more 2′-O-methyl and one or more 2′-fluoro modifications on its stem. The aptamers can also include nucleotides that are modified with soluble polymers. Such polymers can include polyethylene glycol, polyamines, polyesters, polyanhydrides, polyethers or other water soluble pharmaceutically acceptable polymer.
- Purines within given aptamer sequence of FIX inhibitor can tolerate substitution of 2′-O-methyl sugars for current 2′hydroxyl sugars (Example 1,
FIG. 1 ). Applicants found that the aptamers fall into three classes: (1) gain of anticoagulant activity (Apt4); (2) moderate loss of activity (Apt-1, 2, and 3); and (3) severe loss of activity (Apt 5) (FIG. 2 ). Data from Apt-5 indicates that the impact of wholly substituting 2′-O-methyl purines for 2′hydroxyl purines is significantly greater than any individual sector substitution alone (FIG. 2 ). In the case of this aptamer, it is possible that this suggests potential interaction between sectors, or that impairment caused by substitution within one of the sectors is exacerbated by additional modifications (ie. one of the sectors is an Achilles heel). The enhanced antidote control exhibited by Apt-1, 2 and 3 suggests that introduction of 2′O-methyl residues within the antidote binding site improves the ability of the antidote oligonucleotide to bind to the aptamer. This is consistent with the increase in thermodynamic stability observed for duplexes containing 2′-O-methyl RNA residues in each strand, and suggests that duplexes of 2′ -O-methyl-2′-O-methyl strands are more thermodynamically stable than duplexes composed of 2′O-methyl-2′fluoro strands. An alternative conclusion is that the reduction in activity of Apt-1, 2 and 3 leads to more “free” aptamer in the plasma at any given time, which is thus more readily bound by the antidote oligonucleotide - In another embodiment, aptamers of the present invention can include modified pyrimidine nucleosides. Replacing 2′fluoropyrimidines with 2′-O-methyls within
stem 1 improved activity and yielded a compound that tolerates a greater level of substitution. Comparison of the activity ofApt Apt FIG. 16 a). Activity observed betweenApt stem 2 can contain 2′-O-methyl sugars. In fact,Apt 31 appears to possess slightly greater potency thanApt 32, indicating that a compound with 2′fluoro at C16, 2′hydroxyl at G25, and the remainingresidues 2′-O-methyl may exhibit greater potency thanApt 33. Apt 33 is more readily neutralizable thanApt 30, suggesting additional 2′-O-methyl residue within the antidote-binding site of the aptamer improves antidote binding. Apt 34 have C16 as a 2′fluoro rather than 2′-O-methyl nucleoside (FIG. 16 b). Substitution increased anticoagulant activity (compare Apt 34 to Apt 33) but did result in a modest loss of “neutralizability”, although 34 still requires a lower excess of antidote to achieve 90% neutralization (˜5: 1 vs 10:1) than the parental AptA compound (FIG. 16b ). Both results are consistent with an increase in the stability ofstem 2 due to 2′-O-methyl substitution. It is surprising that others have not obviously pursued 2′-O-methyl substitution of 2′-fluoropyrimidines, as such substitution reduces cost of synthesis and appears to enable increased aptamer modification due to increased stem stability. - In one embodiment, at least on guanine in
stem 2 of an aptamer includes a hydroxyl sugar (2′—OH). In one embodiment, at least one uridine instem 1 orstem 2 is a modified base. This can be either a 2′-fluoro (2′-F) or 2′-O-methyl (2′—OCH3) modification. In one embodiment, at least one uridine instem 1 orstem 2 is 2′-O-methyl modified. In one embodiment, at least one cytidine instem 2 is modified. In one embodiment, at least on cytidine instem 2 is 2′-Fluoro modified. - However, comparison of the anticoagulant activity of
Apt 12 withApt 13 and 17 (FIG. 6 ) demonstrates that the loss of activity observed for Apt 6-11 is due to the presence of 2′-O-methyl substitutions at one or more critical residues (FIG. 7 ). Comparison of the anticoagulant activity ofApt 14 toApt 12 indicates that the stretch of 4 consecutive guanosines withinstem 1 can be altered without a significant impact on anticoagulant activity. Comparison ofApt Apt stem 1 except for the closing A-U pair at the top ofstem 1 enhances activity; and b) demonstrates that the sugar of the U in this base pair must be 2′-fluoro for the aptamer to retain potency; and c) suggests that the sugar of the A in this base pair can be a 2′-O-methyl sugar without a significant impact on anticoagulant activity. In fact,Apt 16 retains essentially full potency. - Data suggests that the antidote can more readily bind the aptamer when
stem 1 is a 2′-O-methyl-2′fluoro stem as opposed to when both strands of the duplex contain largely 2′-O-methyl residues. This is again consistent with the notion that duplexes composed of 2′-O-methyl residues in both strands are more stable than those composed of a largely 2′-O-methyl strand and a largely 2′fluoro strand. The enhanced anticoagulant activity ofApt 16 vs.Apt 15 is also consistent with this. Alternatively, the difference in neutralizability between 14, 15 and 16 could be due to the enhanced potency ofApt 16 compared to these two compounds. Regardless, all are neutralized at least as well as AptA. Based upon the observation that the anticoagulant activity ofApt Apt 21,FIG. 8 ). - Substitution of a 2′-O-methyl sugar at this adenosine residue is well tolerated in the background of a largely 2′-O-methyl stem (
FIG. 9 ). In fact, the potency ofApt 21 is intermediate betweenApt Apt 21 is enhanced as compared to Apt 16 (see especially the 2.5:1 and 5:1 AD:Drug data points inFIG. 9 ). - Sugar modifications may ensure stability but they do not guarantee adequate pharmacokinetics for aptamers to be therapeutically active. In healthy individuals, aptamers are cleared from plasma within minutes of IV injection, probably through renal excretion. Keeping intact aptamers in the blood from hours to days after injection has been accomplished by conjugating them to larger macromolecules such as polyethyleneglycol (PEG). In another embodiment, aptamer plasma clearance has also been decreased by embedding them in liposomes.
- Nucleic acid aptamers of the present invention can also be modified by varying the stem and loop sizes. Two families of aptamers with four, five, or six 2-O-methyl modified base pair stem 1 regions showed varying levels of anticoagulant activity and antidote control (see Example 2,
FIGS. 3-5 ).Stem 1 mutants (FIG. 3 ) exhibit a loss of anticoagulant activity as measured in the APTT assay (FIGS. 4 and 5 ). Allstem 1 variants exhibit less activity than the fully 2′-O-methyl purine/2′fluoropyrimidine compound Apt 5, suggesting that one of the pyrimidines withinstem 1 must contain a 2′fluoro sugar for the compound to retain potency. However, all exhibit similar activity levels suggesting stem length may not cause loss of activity. However, 5 base pair stem 1 constructs (Apt 10 and 7) do appear to be more readily antidote controlled than six base pair. Data suggests that astem 1 of 5 base pairs may be preferable to those composed of 4, 6 or 7 base pairs to enhance antidote neutralization. - For targeting of an antidote, an improved aptamer can also be modified so as to include a single-stranded tail (3′ or 5′) in order to promote association with an oligonucleotide antidote. Suitable tails can comprise 1 to 20 nucleotides, preferably, 1-10 nucleotides, more preferably; 1-5 nucleotides and, most preferably, 3-5 nucleotides (e.g., modified nucleotides such as 2′-O-methyl sequences). Tailed aptamers can be tested in binding and bioassays (e.g., as described below) to verify that addition of the single-stranded tail does not disrupt the active structure of the aptamer. A series of oligonucleotides (for example, 2′-O-methyl oligonucleotides) that can form, for example, 1, 3 or 5 basepairs with the tail sequence can be designed and tested for their ability to associate with the tailed aptamer alone, as well as their ability to increase the rate of dissociation of the aptamer from, or association of the aptamer with, its target molecule. Scrambled sequence controls can be employed to verify that the effects are due to duplex formation and not non-specific effects.
- In specific embodiments, the aptamers include the nucleotide sequences of any of the following sequences. (“A” is 2′OH A; “a” is 2′-O-methyl A; “G” is 2′—OH G; “g” is 2′-O-methyl G; “C” is 2′-Fluoro C; “c” is 2′-O-methyl C; “U” is 2° Fluoro U; “u” is 2′-O-methyl U; and “T” is inverted 2′H T.)
SeqID Code Sequence 20 AptA AUGGGGA CUAUACC GCG UAAUGC UGC C UCC CCAU T 21 Apt1 aUgggga CUAUACCGCGUAAUGCUGCC UCCCCaU T 22 Apt2 AUGGGGA CUaUaCC GCG UAAUGC UGC C UCC CCAU T 23 Apt3 AUGGGGA CUAUACC gCg UAAUGC UgC C UCC CCAU T 24 Apt4 AUGGGGA CUAUACC GCG UaaUgC UGC C UCC CCAU T 25 Apt5 aUgggga CUaUaCC gCg UaaUgC UgC C UCC CCaU T 26 Apt6 ggga CUaUaCCGCGUAAUGCUGCC uccc T 27 Apt7 gugga CUaUaCCGCGUAAUGCUGCC uccac T 28 Apt8 gaugga CUaUaCCGCGUAAUGCUGCC uccauc T 29 Apt9 cuga CUaUaCCGCGUAAUGCUGCC ucag T 30 Apt10 ccuga GUaUaCCGCGUAAUGCUGCC ucagg T 31 Apt11 cucuga CUaUaGCGCGUAAUGGUGCC ucagag T 32 Apt12 aUgggga CUaUaCCGCGUAAUGCUGCC UCGCCaU T 33 Apt13 augggga CUaUaCGGCGUAAUGCUGCC uccccau T 34 Apt14 gUgagga CUaUaCCGCGUAAUGCUGCC UGGUCaC T 35 Apt15 gUgaggA CUaUaGGGCGUAAUGCUGGC UCCUCaC T 36 Apt16 gugaggA CUaUaCCGCGUAAUGCUGCC Uccucac T 37 Apt17 gugagga CUaUaCCGCGUAAUGCUGCC uccucac T 38 Apt18 gggA CUaUaCCGCGUAAUGCUGCC Uccc T 39 Apt19 guggA CUaUaCCGCGUAAUGCUGCC Uccac T 40 Apt20 gauggA CUaUaCCGCGUAAUGCUGCC Uccauc T 41 Apt21 gugagga CUaUaCC GCG UAAUGC UGC C Ucc ucac T 42 Apt22 gugaggA CUaUaCC gCg UAAUGC UgC C Ucc ucac T 43 Apt23 gugaggA CUaUaCC GCG UaaUgC UGC C Ucc ucac T 44 Apt24 gugaggA CUaUaCC gCg UaaUgC UgC C Ucc ucac T 45 Apt25 gugaggA CUaUaCC GCg UaaUgC UgC C Ucc ucac T 46 Apt26 gugaggA CUaUaCC gCa AUCG UgC C Uccuc ac T 47 Apt27 gugaggA CUaUaCC GCA aUCg UGC C Uccuc ac T 48 Apt28 gugaggA CUaUaCC gCa aUCg UgC C Uccuc ac T 49 Apt29 gugaggA CUaUaCC GCa aUCg UgC C Uccuc ac T 50 Apt30 gugagga CUaUaCC gCG UaaUgC UGC C Ucc ucac T 51 Apt31 gugagga CUaUaCC gcg UaaUgC ugc C Ucc ucac T 52 Apt32 gugagga CUaUaCC gcg UaaUgC UgC C Ucc ucac T 53 Apt33 gugagga CUaUaCC gCg UaaUgC UGC C Ucc ucac T 54 Apt34 gugagga CUaUaCC gCg UaaUgC uGc C Ucc ucac T 55 Apt35 gugga CUaUaCC gCG UaaUgC UGC C Uccac T 56 Apt36 gugga CUaUaCC gCG UaaUgC ugc C Uccac T 57 Apt37 gugga CUaUaCC gCG UaaUgC UgC C Uccac T 58 Apt38 gugga CUaUaCC gCg UaaUgC UGC C Uccac T 59 Apt39 gugga CUaUaCC gCg UaaUgC uGc C Uccac T - In one specific embodiment, the aptamer to Factor IXa comprises, consists, or consists essentially of, the nucleotide sequence: gugga CUaUaCC gCg UaaUgC uGc C Uccac T (Apt39; SEQ ID NO: 59).
- In one embodiment, the aptamer is linked to one or more polyethylene glycol (PEG) molecules. In a specific embodiment, the aptamer is linked to 40 KD PEG using a six carbon amino linker. In one embodiment, one or more phosphate groups are included between the linker and the nucleic acid sequence. In a more specific embodiment, the six carbon amino linker is attached to the PEG through an amino acid attachment. In a subembodiment, the PEG is two twenty KD PEG that are attached to one or more amino acid, such as lysine, which is attached to the six carbon amino linker.
- In one specific embodiment, the aptamer to Factor IXa is the following structure: 5′-O-[6-[N2-(monomethoxy 20K polyethylene glycol carbamoyl)-N6-(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-riboguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′deoxcytidylyl-(3′-5′)-2′-methoxy-2′-deoxytidylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-3′)-thymidine.
- In another embodiment, the aptamer to Factor IXa is of the following structure:

where n is approximately 450 - In one embodiment, the molecular weight of the aptamer to Factor IXa is 10,282.4 Daltons in its protonated form. In another embodiment, the aptamer is in its sodium salt form. In yet another embodiment, the aptamer is PEGylated Apt39 and in its sodium salt.
- The oligonucleotide antidotes can be administered directly (e. g., alone or in a liposomal formulation or complexed to a carrier, e.g. PEG)) (see for example, U.S. Pat. No. 6,147,204, U.S. Pat. No. 6,011,020). Surprisingly, the addition of a PEG molecule does not reduce aptamer binding to Factor IXa. A shorted
stem 1 with pegylation do appears to increase neutralizability, providing a potentially more effective therapeutic.FIG. 10 shows the activity and neutralizability of a pegylated aptamer with a 5 base pair stem (Apt 19). Apt 19 possesses anticoagulant activity very similar to pegylated Apt16 with a 7base pair stem 1, but ˜90% of its activity can be neutralized with only a 2.5:1 excess of antidote to drug. - Therefore, in one embodiment, the improved aptamer or antidotes can be attached to a non-immunogenic, high molecular weight compound such as polyethylene glycol (PEG) or other water soluble pharmaceutically acceptable polymer as described herein. In one embodiment, the aptamer or antidote is associated with the PEG molecule through covalent bonds. Where covalent attachment is employed, PEG may be covalently bound to a variety of positions on the improved aptamer or antidote. In another embodiment, an oligonucleotide aptamer or antidote is bonded to the 5′-thiol through a maleimide or vinyl sulfone functionality. In one embodiment, a plurality of improved aptamers or antidotes can be associated with a single PEG molecule. The improved aptamers and antidotes can be the same or different sequences and modifications. In yet a further embodiment, a plurality of PEG molecules can be attached to each other. In this embodiment, one or more aptamers or antidotes to the same target or different targets can be associated with each PEG molecule. In embodiments where multiple aptamers or antidotes specific for the same target are attached to PEG, there is the possibility of bringing the same targets in close proximity to each other in order to generate specific interactions between the same targets. Where multiple aptamers or antidotes specific for different targets are attached to PEG, there is the possibility of bringing the distinct targets in close proximity to each other in order to generate specific interactions between the targets. In addition, in embodiments where there are aptamers or antidotes to the same target or different targets associated with PEG, a drug can also be associated with PEG. Thus the complex would provide targeted delivery of the drug, with PEG serving as a Linker.
- Linkers can be selected from, for example, 6-(trifluoroacetamido)hexanol (2-cyanoethyl-N,N-diisopropyl)phosphoramidite of the structure:

TFA-amino C4 CED phosphoramidite (available from ChemGenes, cat# CLP-1453) of the structure:
5′-amino modifier C3 TFA (available from Glen Research cat# 10-1923-90) of the structure:
- 5′-amino modifier 5 (available from Glen Research cat# 10-1905-90) of the structure:

- MMT: 4-Monometholxytrityl
- 5′-amino modifier C12 (available from Glen Research cat# 10-1912-90) of the structure:

- MMT: 4-Monomethoxytrityl
- 5′thiol-modifier C6 (available from Glen Research cat# 10-1926-90) of the structure:

The 5′-thiol modified linker is used with PEG-maleimides, PEG-vinylsulfone, PEG-iodoacetamide and PEG-orthopyridyl-disulfide, for example. - Polyethylene glycols (PEGs) can be conjugated to biologically active compounds to serve as “inert” carriers to potentially (1) prolong the half-life of the compound in the circulation, (2) alter the pattern of distribution of the compound and/or (3) camouflage the compound, thereby reducing its immunogenic potential and protecting it from enzymatic degradation. PEGs can range in size from 5 to 200 KD, with typical PEGs used in pharmaceutical formulations in the 10-60 KD range. Linear chain PEGs of up to about 30 KD can be produced. For PEGs of greater than 30 KD, multiple PEGs can be attached together (multi-arm or ‘branched’ PEGs) to produce PEGs of the desired size. The general synthesis of compounds with a branched, “mPEG2” attachment (two mPEGs linked via an amino acid) is described in Monfardini, et al. (1995) Bioconjugate Chem. 6:62-69. For ‘branched’ PEGs, ie. compounds that include more than one PEG or mPEG linked to a common reactive group, the PEGs or mPEGS can be linked together through an amino acid such as a lysine or they can be linked via, for example, a glycerine. For branched PEGs in which each mPEG is about 10, about 20, or about 30 KD, the total mass is about 20, about 40 or about 60 KD and the compound is referred to by its total mass (i.e. 40 kD mPEG2 is two linked 20 kD mPEGs). 40 KD total molecular weight PEGs, that can be used as reagents in producing a PEGylated compound, include, for example, [N2-(monometioxy 20K polyethylene glycol carbamoyl)-N6-(monomethoxy 20K polyethylene glycol carbamoyl)]-lysine N-hydroxysuccinimide of the structures:

(mPEG is a 20 KD monomethoxy PEG). The general synthesis of compounds with a branched, “mPEG2” attachment is described in Monfardini, et al. (1995) Bioconjugate Chem. 6:62-69. For ‘branched’ PEGs, ie. compounds that include more than one PEG or mPEG linked to a common reactive group, the PEGs or mPEGS can be linked together through an amino acid such as a lysine. - Additional PEG reagents that can be used to prepare stabilized compounds of the invention include other branched PEG N-Hydroxysuccinimide (mPEG-NHS) of the general formula:

with a 40 KD or 60 KD total molecular weight (where each mPEG is about 20 or about 30 KD). Compounds of this structure are sold, for example, by Nektar Therapeutics as cat# 2Z3Y0L01 and cat# 2Z3Y0V01. As described above, the branched PEGs can be linked through any appropriate reagent, such as an amino acid, and in certain embodiments are linked via lysine residues or glycerine residues. - They can also include non-branched mPEG-Succinimidyl Propionate (mPEG-SPA), of the general formula:

in which mPEG is about 20 KD or about 30 KD. In a specific embodiment, the reactive ester is —O—CH2CH2—C02—NHS. Compounds of this structure are sold by Nektar Therapeutics as catalog numbers #2M4M0P01. - The reagents can also include a branched PEG linked through glycerol, such as the Sunbright TM series from NOF Corporation, Japan. Specific, non-limiting examples of these reagents are:

- The reagents can also include non-branched Succinimidyl alpha-methylbutanoate (mPEG-SMB) of the general formula:

in which mPEG is between 10 and 30 KD. In a subembodiment, the reactive ester is —O—CH2CH2CH(CH3)—CO2—NHS. Compounds of this structure are sold by Nektar Therapeutics as catalog numbers cat#2M4K0R01. - PEG reagents can also include nitrophenyl carbonate linked PEGs, such as of the following structure:

Compounds of this structure are commercially available, for example from Sunbio, Inc. Compounds including nitrophenyl carbonate can be conjugated to primary amine containing linkers. In this reaction, the 0-nitrophenyl serves as the leaving group, leaving a structure [mPEG]n-NH—CO—NH-linker-ligand. - PEGs with thiol-reactive groups that can be used with a thiol-modified linker, as described above, include compounds of the general structure:

in which mPEG is about 10, about 20 or about 30 KD. Additionally, the structure can be branched, such as
in which each mPEG is about 10, about 20, or about 30 KD and the total mass is about 20, about 40, or about 60 KD. Branched PEGs with thiol reactive groups that can be used with a thiol-modified linker, as described above, include compounds of the general structure (need structure), in which the branched PEG has a total molecular weight of about 40 or 60 KD (where each mPEG is 20 or 30 KD). Compounds of this general structure are commercially available, for example from Nektar Therapeufics, sold as cat#2F2M0P01, cat#2D3Y0T01, or cat#2D3Y0V01. PEG reagents can also be of the following structure:
PEG reagents of this structure are sold, for example, by Sunbio, Inc. PEG-maleimide pegylates thiols of the target compound in which the double bond of the maleimic ring breaks to connect with the thiol. The rate of reaction is pH dependent and, in one embodiment, is carried out betweenpH pH pH 8. - In a general embodiment, the aptamers of the invention have the general structure.

- , or a pharmaceutically acceptable salt thereof.
- In one subembodiment, the aptamer has the following structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450,
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450,
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:
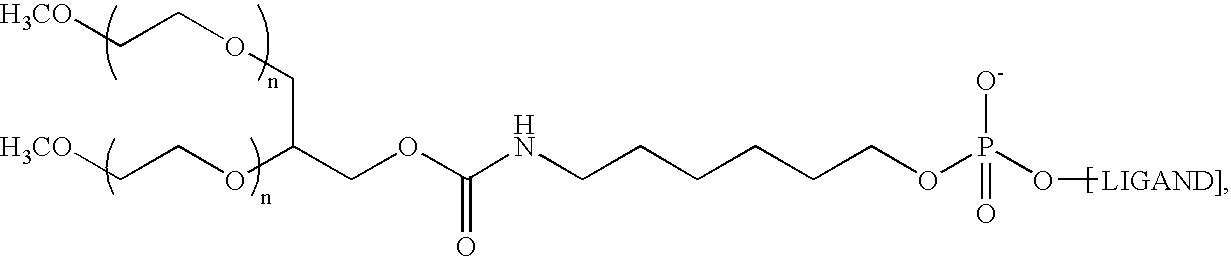
- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In yet another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In yet another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In yet another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In yet another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In yet another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.

- In another subembodiment, the aptamer is of the structure:
- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt thereof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salttherof.
- In another subembodiment, the aptamer is of the structure:

- Ligand=oligonucleotide of SEQ ID NO:59
- n=approximately 450
- , or a pharmaceutically acceptable salt therof.
- The aptamers or antidotes of the invention can also include other conjugate groups covalently bound to functional groups such as primary or secondary hydroxyl groups. Conjugate groups of the invention include polyamines, polyamides, polyethylene glycols, polyethers, groups that enhance the pharmacodynamic properties of oligomers, and groups that enhance the pharmacokinetic properties of oligomers. Groups that enhance the pharmacodynamic properties, in the context of this invention, include groups that improve oligomer bioavailability, enhance oligomer resistance to degradation, and/or strengthen sequence-specific hybridization with RNA.
- 4. Process of Manufacture of Aptamer
- The improved aptamers described herein can be manufactured using techniques known in the art. For example, U.S. patents have issued describing methods of large scale manufacturing that can be used to manufacture aptamers. Caruthers et al., for example, describe in U.S. Pat. Nos. 4,973,679; 4,668,777; and 4,415,732 a class of phosphoramidite compounds that are useful in the manufacture of oligonucleotides. In another series of patents, Caruthers et al. disclose a method of synthesizing oligonucleotides using an inorganic polymer support. See, e.g., U.S. Pat. Nos. 4,500,707, 4,458,066 and 5,153,319. In still another series of patents, Caruthers et al. discloses a class of nucleoside phosphorodithioates that can be used to manufacture oligonucleotides. See, e.g., U.S. Pat. Nos. 5,278,302, 5,453,496 and 5,602,244.
- In a specific embodiment, manufacture of improved nucleic acid ligands of the invention is a multi-step process involving solid phase chemical synthesis of the oligonucleotide strand; cleavage and deprotection of the crude oligonucleotide; purification by preparative anion exchange chromatography; desalting followed by PEGylation; purification of the PEGylated oligonucleotide by preparative anion exchange chromatography to remove unPEGylated oligonucleotide impurities; ultrafiltration for desalting; concentration and lyophilization of the final product.
- Chemical synthesis via phosphoramidite chemistry involves sequential coupling of activated monomers to an elongating polymer, one terminus of which is covalently attached to a solid support matrix. The general steps include protecting a 3′-hydroxy of a nucleoside, coupling a 5′-carbon to a solid support, deprotecting, coupling a phosphopramidate-linked, protected nucleoside to the 3′-hydroxy, deprotecting, sequentially reacting phosphoramidate-linked nucleosides to build the molecule, coupling the
terminal 5′-nucleoside with a linker and reacting with an inert carrier, such as a linked PEG. The solid phase approach allows for easy purification of the reaction product at each step in the synthesis by simple solvent washing of the solid phase. - In one embodiment, the oligonucleotides are sequentially assembled from the 3′- end towards the 5′- end by deprotecting the 5′- end of the support-bound molecule, allowing the support-bound molecule to react with an incoming tetrazole-activated phosphoramidite monomer, oxidizing the resulting phosphite triester to a phosphate triester, and blocking any unreacted hydroxyl groups by acetylation (capping) to prevent non-sequential coupling with the next incoming monomer to form a “deletion sequence”. This sequence of steps is repeated for subsequent coupling reactions until the full-length oligonucleotides are synthesized. Due to the presence of a 3′-3′ linkage at the 3′ end and a C-6 linker for PEGylation at the 5′ end, the synthesis is modified at the first and last step to accommodate these changes.
- The overall cycle is schematically shown in
Scheme 1 for the synthesis of the crude material.


Cleavage and Deprotection - The oligonucleotide can then be cleaved from the solid support and protecting groups on the linker are removed. The support-attached intermediate is cleaved using basic conditions at an elevated temperature. The temperature can be from about 40 to about 55° C., or from about 42 to about 50° C., or from about 45 to about 50° C. An exemplary reaction is carried out in ammonium hydroxide and tert-butylamine. The protecting groups on the linker are removed by incubation with an appropriate deprotecting reagent at reduced temperature. An exemplary deprotecting reagent is triethylamine trishydrofluoride.
- In a specific example, upon completion of synthesis, the solid-support and associated oligonucleotide are transferred to a filter funnel, dried under vacuum and transferred to a reaction vessel. Ammonium hydroxide (28-30%) and tert-butylamine are added and the mixture is heated to approximately 45° C. for approximately 15 hours to effect cleavage from the solid support, removal of the cyanoethyl phosphate protecting group, deprotection of exocyclic amine protecting groups as well as removal of the trifluoroacetyl group from the linker. The sample is cooled and the 2′-TBDMS protecting group is then cleaved by addition of Et3N.3HF in DMSO followed by heating the sample to 45° C. for up to 5 hours to yield the crude oligoribonucleotide. The mixture is filtered under vacuum to remove the waste solid support. The reaction is quenched with glacial acetic acid to provide a pH neutral solution of crude product.

Anion Exchange Purification - The cleaved oligonucleotide can be purified using any techniques known in the art. In one embodiment, the oligonucleotide is purified by ion exchange chromatography.
- In a specific example, the crude oligonucleotide is purified by preparative anion exchange chromatography using column chromatography, for example on a TosoBiosep TSK Gel SuperQ-5PW column. The purification can be performed using standard reagents and can be performed at elevated temperature (above room temperature). In a specific embodiment, the purification is performed at approximately 75° C. In a specific example, the reagents include increasing concentrations of sodium bromide as an eluting reagent. In one embodiment, the gradient is made of 10% ACN, 20mM sodium phosphate, pH 8.0 (Buffer A) and 10% ACN, 20 mM sodium phosphate and 1.0M NaBr, pH 8.0 (Buffer B). Purification is accomplished by eluting the product from the column through a controlled increase in sodium bromide concentration in the buffer system by increasing the proportion of Buffer B. Fractions are collected and analyzed, for example by UV and IP RP-HPLC. When necessary, the fractions are combined to yield a product pool of the desired purity, desalted by ultrafiltration (e.g. using 1 KD Polyethersulfone (PES) Pall Filtron UF Cassettes) and concentrated at about 40° C. and about 80 mbar pressure using a rotary evaporator. The concentrated product is stored at 2-8 ° C.
- PEGylation
- For PEGylation of compounds that react with linkers containing primary amines, the purified and concentrated intermediate from above is reacted with a PEG reagent as described herein at a pH between about pH 7-10, or between about pH 8-10, or between about pH 8.5-10, or between about pH 8.8-9.8. As an example, a 40 KD mPEG-linked reagent can be incubated with the purified oligonucleotide at about 25° C. in 0.1M sodium borate buffer (pH about 8.8) for 25-30 min. The reaction is shown in
Scheme 2.
- For linkers that include free thiol, the purified and concentrated intermediate is 5 reacted with an appropriate PEG (i.e. PEG-Mal) at pH from about 6-8 or from about 6.5-7.5. Thiol PEGylation is specific for free thiols on biological reagents using mPEG-MAL. Coupling of maleimide to thiol groups is highly specific for thiols at ca. pH 6.5-7.5 in the presence of other functional groups. Reaction with a thiol moiety generates a stable 3-thiosuccinimidyl ether linkage.
- 5. Aptamer Antidotes
- It is important to be able to release blood coagulation factors from inhibition. Life threatening diseases can result from over-inhibition of the blood coagulation factors such as Factor IX. For example, hemophilia B results from deficiencies in factor IX. All patients with hemophilia B have prolonged coagulation time and decreased factor IX clotting activity. Like hemophilia A, there are severe, moderate and mild forms of hemophilia B and reflect the factor IX activity in plasma.
- Therefore, another embodiment of the invention includes an antidote paired with the aptamer of the invention. Antidotes or modulators can include any pharmaceutically acceptable agent that can bind an aptamer and modify the interaction between that aptamer and its target molecule (e.g., by modifying the structure of the aptamer) in a desired manner. Examples of such antidotes include (A) oligonucleotides complementary to at least a portion of the aptamer sequence (including ribozymes or DNAzymes or peptide nucleic acids (PNAs)), (B) nucleic acid binding peptides, polypeptides or proteins (including nucleic acid binding tripeptides (see, generally, Hwang et al. (1999) Proc. Natl. Acad. Sci. USA 96:12997), and (C) oligosaccharides (e.g. aminoglycosides (see, generally, Davis et al. (1993)
Chapter 8, p. 185, RNA World, Cold Spring Harbor Laboratory Press, eds. Gestlaad and Atkins; Werstuck et al. (1998) Sciende 282:296; U.S. Pat. Nos. 5,935,776 and 5,534,408). (See also the following which disclose types of antidotes that can be used in accordance with the present invention: Chase et al. (1986)Ann. Rev. Biochem. 56:103, Eichorn et al. (1968) J. Am. Chem. Soc. 90:7323, Dale et al. (1975) Biochemistry 14:2447 and Lippard et al. (1978) Acc. Chem. Res. 11:211). - In one embodiment, the antidote oligonucleotide reverses or neutralizes at least 25%, 50%, 75%, 80% or 90% of the anticoagulant activity of the aptamer. The antidote generally has the ability to substantially bind to a nucleic acid ligand in solution at antidote concentrations of less than one 1 μM, or less than 0.1 μM and more preferably less than 0.01 μM. In one embodiment, the antidote reduces the biological activity of the aptamer by 50%.
- Complementary Oligonucleotides
- In one embodiment, the improved antidote of the invention is an oligonucleotide that comprises a sequence complementary to at least a portion of the targeted aptamer sequence. Absolute complementarity is not required. The sequence in one embodiment has sufficient complementarity to be able to hybridize with the aptamer. The ability to hybridize will depend on both the degree of complementarity and the length of the antisense nucleic acid. Advantageously, the antidote oligonucleotide comprises a sequence complementary to 6-25 consecutive nucleotides of the targeted aptamer, preferably, 8-20 consecutive nucleotides, more preferably, 10-15 consecutive nucleotides. In specific aspects the antidote is at least 10-25 nucleotides, at least 15-25, at least 20-25, at least 14, 17 or at least 25 nucleotides long. The antidotes of the invention can be DNA or RNA or chimeric mixtures or derivatives or modified versions thereof, single-stranded.
- Formation of duplexes by binding of complementary pairs of short oligonucleotides is a fairly rapid reaction with second order association rate constants generally between 1×106 and 3×105 M1s1. Stability of short duplexes can be highly dependent on the length and base-composition of the duplex. The thermodynamic parameters for formation of short nucleic acid duplexes have been rigorously measured, resulting in nearest-neighbor rules for all possible base pairs such that accurate predictions of the free energy, Tm and thus half-life of a given oligoribonucleotide duplex can be calculated (e.g., Xia et al. (1998) Biochem. 37:14719; see also Eguchi et al. (1991) Antigensis RNA, Annu. Rev. Biochem. 60:631).
- In a specific embodiment, the present invention provides improved antidotes that specifically and rapidly reverse the anticoagulant and antithrombotic effects of the improved aptamers that target components of the coagulation pathway, in particular the aptamers of FIX and FIXa. The antidotes can be administered to reverse the aptamer activity by a physician or other health care provider. In specific embodiments, the improved antidotes according to the present invention are nucleic acids corresponding to the sequences: (5′-3′) sequence: cgcgguauaguccccau (Apt/AD; SEQ ID NO:1); (5′-3′) sequence: cgcgguauaguccc (Apt6/AD SEQ ID NO:2); (5′-3′) sequence: cgcgguauaguccac (Apt7/AD; SEQ ID NO:3); (5′-3′) sequence: cgcgguauaguccauc (Apt8/AD; SEQ ID NO:4); (5′-3′) sequence: cgcgguauagucag (Apt9/AD; SEQ ID NO:5); (5′-3′) sequence: cgcgguauagucagg (Apt10/AD; SEQ ID NO:6); (5′-3′) sequence: cgcgguauagucagag (Apt11/AD; SEQ ID NO:7); (5′-3′) sequence: cgcgguauaguccucac (Apt14/AD; SEQ ID NO:8), or any modification or derivative thereof. The antidote sequence can be at least 20%, 50%, 75% or 90% homologous to the sequence of the corresponding aptamer. In one embodiment, the antisense sequence is separately administered.
- In one embodiment, the antidote has the structure 2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxyuridylyl-(3′-5′)-2-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxyuridylyl-(3′,5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′,5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxycytidine.
- Antisense techniques are discussed for example, in Okano, et al. (1991) J. Neurochem. 56:560 and “Oligodeoxynucleotides as Antisense Inhibitors of Gene Expression,” (1988) CRC Press, Boca Raton, Fla. In one embodiment, oligonucleotide antidotes of the invention are advantageously targeted at single-stranded regions of the aptamer. This can facilitate nucleation and, therefore, the rate of aptamer activity modulation, and also, generally leads to intermolecular duplexes that contain more base pairs than the targeted aptamer. The aptamer to Factor IXa of the present invention may be used to design an antisense oligonucleotide. The antisense oligonucleotide hybridizes to the aptamer in vivo and blocks the binding of the aptamer to factor IXa.
- To design an improved antidote to the improved aptamers of the invention, various strategies can be used to determine the optimal binding site. The complimentary oligonucleotides can be “walked” around the aptamer. This “walking” procedure includes, after a minimal consensus ligand sequence has been determined for a given improved aptamer, adding random sequence to the minimal consensus ligand sequence and evolving additional contacts with the target, such as in separate but adjacent domains. A walking experiment can involve two experiments performed sequentially. A new candidate mixture is produced in which each of the members of the candidate mixture has a fixed nucleic acid-region that corresponds to a nucleic acid ligand of interest. Each member of the candidate mixture also contains a randomized region of sequences. According to this method it is possible to identify what are referred to as “extended” nucleic acid ligands, which contain regions that can bind to more than one binding domain of a target.
- Changes in the sugar can affect antidote stability, in part because sugar modifications that result in RNA-like oligonucleotides, e.g., 20-fluoro or 20-methoxy, do not appear to serve as substrates for RNase H. Alterations in the orientation of the sugar to the base can also affect RNase H activation,. Additionally, backbone modifications influence the ability of oligonucleotides to activate RNase H. Methylphosphonates do not activate it, whereas phosphorothioates are excellent substrates. In addition, chimeric molecules have been studied as oligonucleotides that bind to RNA and activate RNase H . For example, oligonucleotides comprising wings of 20-methoxy phosphonates and a five-base gap of deoxyoligonucleotides bind to their target RNA and activate RNase H.
- In one embodiment, 2′-O-methyl modified antidotes (e.g., 2′-O-methyl oligonucleotides) about 15 nucleotides in length can be used, the complementarity of which is staggered by about 5 nucleotides on the aptamer (e.g., oligonucleotides complementary to nucleotides 1-15, 6-20, 11-25, etc.). The impact of tertiary structure of the improved aptamer on the efficiency of hybridization is difficult to predict. Assays described in the Examples that follow can be used to assess the ability of the different oligonucleotides to hybridize to a specific aptamer. The ability of the different oligonucleotide antidotes to increase the rate of dissociation of the aptamer from, or association of the aptamer with, its target molecule can also be determined by conducting kinetic studies using, for example, BIACORE assays. Oligonucleotide antidotes can be selected such that a 5-50 fold molar excess of oligonucleotide, or less, is required to modify the interaction between the aptamer and its target molecule in the desired manner.
- The antidotes of the invention may be conjugated to another molecule, e.g., a peptide, a hybridization-triggered cross-linking agent, a transport agent, a hybridization-triggered cleavage agent, etc.
- The antisense oligonucleotide may optionally comprise at least one modified base moiety, including but not limited to one selected from 5-fluorouracil, 5-fluorocytosine, 5-bromouracil, 5-bromocytosine, 5-chlorouracil, 5-chlorocytosine, 5-iodouracil, 5-iodocytosine, 5-methylcytosine, 5-methyluracil, hypoxanthine, xantine, 4-acetylcytosine, 5-(carboxyhydroxymethyl) uracil, 5-carboxymethylamin-O-methyl thiouridine, 5-carboxymethylamin-O-methyluracil, dihydrouracil, beta-D-galactosylqueosine, inosine, N6-isopentenyladenine, 1-methylguanine, 1-methylinosine, 2,2-dimethylguanine,2-methyladenine, 2-methylguanine, 3-methylcytosine, 6-methylcytosine, N6-adenine, 7-methylguanine, 5-methylamin-O-methyluracil, 5-methoxyamin-O-methyl-2-thiouracil, beta-D-mannosylqueosine, 5′-methoxycarboxymethyluracil, 5-methoxyuracil, 5-methoxycytosine, 2-methylthio-N&isopentenyladenine, uracil oxyacetic acid (v), butoxosine, pseudouracil, queosine, 2-thiocytosine, 5-methyl thiouracil, 2-thiouracil, 4-thiouracil, 5-methyluracil, uracil-5-oxyacetic acid methylester, uracil oxyacetic acid (v), 5-methyl thiouracil, 3-(3-amino-3-N carboxypropyl) urdeil, (acp3)w, and 2,6-diaminopurine.
- The antidotes may also include at least one modified sugar moiety selected from the group including, but not limited to, arabinose, 2-fluoroarabinose, xylulose, hexose, 2′-fluororibose, 2′ -O-methylribose, 2′ -O-methoxyethylribose, 2′ -O-propylribose, 2′-O-methylthioethylribose, 2′-O-diethylaminooxyethylribose, 2′-O-(3-aminopropyl)ribose, 2′-O-dimethylaminopropyl)ribose, 2′-O-(methylacetamido)ribose, and 2′-O-(dimethylaminoethyloxyethyl)ribose. In yet another embodiment, the antisense oligonucleotide comprises at least one modified phosphate backbone selected from the group including, but not limited to, a phosphorothioate, a phosphorodithioate, a phosphoramidothioate, a phosphoramidate, a phosphordiainidate, a methylphosphonate, an alkyl phosphotriester, and a formacetal or analog thereof.
- Manufacturing ofAntidote
- Manufacture of nucleic acid antidotes is a multi-step process involving solid phase chemical synthesis of the oligonucleotide strand; cleavage and deprotection of the crude oligonucleotide; purification by preparative anion exchange chromatography; desalting and lyophilization of the final product.
- Chemical synthesis via phosphoramidite chemistry involves sequential coupling of activated monomers to an elongating polymer, one terminus of which is covalently attached to a solid support matrix. The solid phase approach allows for easy purification of the reaction product at each step in the synthesis by simple solvent washing of the support-bound intermediates. The oligonucleotide can be sequentially assembled from the 3′-end towards the 5′-end by deprotecting the 5′-end of the support-bound intermediate, allowing the support-bound intermediate to react with an incoming tetrazole-activated phosphoramidite monomer, oxidizing the resulting phosphite triester to a phosphate triester, and blocking any unreacted hydroxyl groups by acetylation to prevent non-sequential coupling with the next incoming monomer to form a “deletion sequence”. This sequence of steps is repeated for subsequent coupling reactions until the full-length oligonucleotides are synthesized.
- A specific cycle is shown schematically shown in
Scheme 3 for the synthesis of a crude product.
Cleavage and Deprotection - The oligonucleotide can then be cleaved from the solid support and protecting groups on the linker are removed. The support-attached intermediate is cleaved using basic conditions at an elevated temperature. In a specific example, upon completion of the synthesis, the support-bound crude product is transferred to a filter funnel, dried under vacuum and transferred to a reaction vessel. A basic reagent is added to effect cleavage from the solid support, removal of the cyanoethyl phosphate protecting group and deprotection of exocyclic amine protecting groups. The reaction temperature can also be increased to enhance cleavage. In a specific example, the reagent is 30-40% methylamine and the mixture is heated to approximately 45° C. for up to 90 minutes. The mixture is filtered under vacuum to remove the waste solid support. The reaction is quenched with glacial acetic acid to provide a pH neutral solution of crude product.

Anion Exchange Purification - The cleaved oligonucleotide can be purified using any techniques known in the art. In one embodiment, the oligonucleotide is purified by ion exchange chromatography.
- In a specific example, the crude product is purified by preparative anion exchange chromatography using, for example, a TosoBiosep TSK Gel SuperQ-5PW column. The purification can be performed using standard reagents and can be performed at elevated temperature (above room temperature). In a specific example, the reagents include increasing concentrations of sodium bromide as an eluting reagent. In one embodiment, the gradient is made of 5-35% of Buffer B (10% ACN, 20 mM sodium phosphate and 1.0 M NaBr, pH 8.0) in Buffer A (10% ACN, 20 mM sodium phosphate, pH 8.0). The purification can be performed at approximately 70° C. using. Purification is accomplished by eluting the product from the column through a controlled increase in sodium bromide concentration in the buffer system. Fractions can be collected and analyzed by UV and AX-HPLC. Selected fractions can be combined to yield a product pool with of the desired purity, desalted by ultrafiltration and stored at 2-8° C. Aliquots can be freeze-dried to a dry powder and stored in glass at −15° C. to −25° C.
- Ribozymes and DNAzymes
- Improved aptamers or antidotes can also be enzymatic nucleic acids. Such a ribozyme or DNAzyme act by first binding to a target RNA or DNA (see Cech U.S. Pat. No. 5,180,818) and then cleaving the target. An enzymatic nucleic acid can repeatedly bind and cleave new targets thereby allowing for inactivation of RNA aptamers. There are at least five classes of ribozymes that each display a different type of specificity. In the case of antidotes, this enzymatic activity can complement or substitute for the introduction of a “suicide position” in the improved aptamer.
- The enzymatic nature of a ribozyme may be advantageous over other technologies because the effective concentration of ribozyme necessary to effect a therapeutic treatment is lower than that of an antisense oligonucleotide. A single ribozyme molecule is able to cleave many molecules of target RNA. In addition, the ribozyme is a highly specific inhibitor, with the specificity of inhibition depending not only on the base pairing mechanism of binding, but also on the mechanism by which the molecule inhibits the expression of the RNA to which it binds. That is, the inhibition is caused by cleavage of the RNA target and so specificity is defined as the ratio of the rate of cleavage of the targeted RNA over the rate of cleavage of non-targeted RNA. This cleavage mechanism is dependent upon factors additional to those involved in base pairing. Thus, it may be that the specificity of action of a ribozyme is greater than that of antisense oligonucleotide binding the same RNA site.
- Another class of catalytic molecules are called “DNAzymes”. DNAzymes are single-stranded, and cleave both RNA and DNA. A general model for the DNAzyme has been proposed, and is known as the “10-23” model. DNAzymes following the “10-23” model, also referred to simply as “10-23 DNAzymes”, have a catalytic domain of 15 deoxyribonucleotides, flanked by two substrate-recognition domains of seven to nine deoxyribonucleotides each. In vitro analyses show that this type of DNAzyme can effectively cleave its substrate RNA at purine:pyrimidine junctions under physiological conditions. As used herein, “DNAzyme” means a DNA molecule that specifically recognizes and cleaves a distinct target nucleic acid sequence, which may be either DNA or RNA.
- Adapted Nucleic Acids
- In another aspect of the invention, the antidote to the improved aptamers are Peptide Nucleic Acids (PNAs). PNAs are compounds that are analogous to oligonucleotides, but differ in composition in that the deoxyribose backbone of oligonucleotide is replaced by a peptide backbone. Each subunit of the peptide backbone is attached to a naturally-occurring or non-naturally-occurring nucleobase.
- PNAs can be advantageous as fast acting antidotes because they bind more tightly to the corresponding improved aptamers than their non-substituted oligonucleotide counterparts. PNAs bind to both DNA and RNA and the resulting PNA/DNA or PNA/RNA duplexes are bound tighter than corresponding DNA/DNA or DNA/RNA duplexes as evidenced by their higher melting temperatures (Tm). Another advantage of PNA/DNA(RNA) duplexes is that Tm is practically independent of salt concentration. Since PNAs are an analogue of DNA in which the backbone is a pseudopeptide rather than a sugar, they mimic the behaviour of DNA and binds complementary nucleic acid strands.
- PNAs are synthetic polyamides comprised of repeating units of the amino acid, N-(2-aminoethyl)-glycine, to which the nucleobases adenine, cytosine, guanine, thymine and uracil are attached through a methylene carbonyl group. Natural and unnatural nucleobases, such as pseudo isocytosine, 5-methyl cytosine and 2,6-diaminopurine, inosine, uracil, 5-methylcytosine, thiouracil, 2,6-diaminopurine, bromothymine, azaadenines or azaguanines among many others, also can be incorporated in PNA synthons. PNAs are most commonly synthesized from monomers (PNA synthons) protected according to the t-Boclbenzyl protection strategy, wherein the backbone amino group of the growing polymer is protected with the t-butyloxycarbonyl (t-Boc) group and the exocyclic amino groups of the nucleobases, if present, are protected with the benzyloxycarbonyl (benzyl) group. PNA synthons protected using the t-Boc/benzyl strategy are now commercially available.
- Morpholino nucleic acids (MNAs) can also be adavantageous in antidote preparation because morpholinos are completely resistant to nucleases and they appear to be free of most or all of the non-antisense effects that plague S-DNAs. MNAs are assembled from morpholino subunits, each of which contains one of the four genetic bases (adenine, cytosine, guanine, and thymine) linked to a 6-membered morpholine ring. Subunits of are joined by non-ionic phosphorodiamidate intersubunit linkages to give a MNA. These MNAs can have substantially better antisense properties than do RNA, DNA, and their analogs having 5-membered ribose or deoxyribose backbone moieties joined by ionic linkages (see wwwgenetools.com/Morpholinosibody_morpholinos.HTML).
- U.S. Pat. No. 6,153,737 to Manoharan et al. is directed to derivatized oligonucleotides wherein the linked nucleosides are functionalized with peptides, proteins, water soluble vitamins or lipid soluble vitamins. This disclosure was directed towards antisense therapeutics by modification of oligonucleotides with a peptide or protein sequence that aids in the selective entry of the complex into the nuclear envelope. Similarly, water-soluble and lipid-soluble vitamins can be used to assist in the transfer of the anti-sense therapeutic or diagnostic agent across cellular membranes.
- Locked nucleic acids (LNAs) can also be used to preparet the antidotes of the present invention. LNAs are a novel class of DNA analogues that possess certain features that make them prime candidates for improving nucleic acid properties. The LNA monomers are bi-cyclic compounds structurally similar to RNA-monomers. LNAs share most of the chemical properties of DNA and RNA, are water-soluble, can be separated by gel electrophoreses, ethanol precipitated, etc. (Tetrahedron, 54, 3607-3630 (1998)). However, introduction of LNA monomers into either DNA or RNA oligos results in high thermal stability of duplexes with complementary DNA or RNA, while, at the same time obeying the Watson-Crick base-pairing rules. This high thermal stability of the duplexes formed with LNA oligomers together with the finding that primers containing 3′ located LNA(s) are substrates for enzymatic extensions, e.g. the PCR reaction, makes these compounds suitable for the antidotes of the present invention. For examples of LNAs see U.S. Pat. No. 6,316,198.
- In other embodiments, the stabilized nucleic acid can be a PCO (pseudocyclic oligonucleobase), or a 2′-O,4′-C-ethylene bridged nucleic acid (ENA).
- 6. Methods of Use
- Regulating Coagulation With an Improved Aptamer
- The invention includes the use of improved aptainers to bind to FIX, FIXa, or the intrinsic tenase complex. The binding can be in vitro or in vivo. The result of the binding to FIX, FIXa or the tenase complex can be to inhibit the biological activity of the proteins or complex. The improved aptamers can be used to treat diseases such as deep venous thrombosis, arterial thrombosis, post surgical thrombosis, coronary artery bypass graft (CABG), percutaneous transdermal coronary angioplastry (PTCA), stroke, tumour metastasis, inflammation, septic chock, hypotension, ARDS, pulmonary embolism, disseminated intravascular coagulation (DIC), vascular restenosis, platelet deposition, myocardial infarction, angiogenesis, or the prophylactic treatment of mammals with atherosclerotic vessels at risk for thrombosis.
- In one embodiment, the improved aptamer inhibits blood coagulation by binding to FIXa. The invention includes administering the aptamer of the invention to a mammal, for example, a human, in need thereof to inhibit blood coagulation. Another embodiment of the invention provides methods of using aptamers that are well suited for administration during a therapeutic regime.
- A method of improved regulating coagulation in a mammal in need thereof is provided. In one embodiment, the method comprises: (a) administering to a warm-blooded vertebrate or mammal in need thereof, an effective amount of an improved aptamer that selectively binds coagulation pathway FIX, FIXa, or the intrinsic tenase complex, or inhibits a subunit of the intrinsic tenase complex (i.e. FIX, FIXa, FVIII binding to or activation of FX); (b) modulating the biological activity of the coagulation pathway factor in the warm-blooded vertebrate through the administering of the aptamer in step (a); and (c) providing an improved antidote to reverse the effects of the aptamer. In certain embodiments, the warm-blooded vertebrate or mammal is a human.
- As used herein, the term “mammal” is meant to include any human or non-human mammal, including but not limited to porcine, ovine, bovine, rodents, ungulates, pigs, sheep, lambs, goats, cattle, deer, mules, horses, monkeys, dogs, cats, rats, and mice.
- An important area for consideration is plasma half-life. Modifications can alter the half-life in vivo of the improved aptamers from a few minutes to 12 or more hours. The improved aptamers of the present invention can be used to treat percutaneous coronary interventions where vascular injury occurs at a specific time and place, yielding a sudden but relative brief prothrombotic stimulus. This may also be the case following carotid angioplasty. In another embodiment, the improved aptamers can be used in extracorporeal circulation utilized in coronary bypass grafting and hemodialysis. The latter condition is somewhat complex because of the inherent thrombogenicity of arteriovenous (AV) shunts. The aptamers of the invention also can be used to treat a venous thromboembolic disease, mechanical heart valve replacement, atrial fibrillation, and conceivably in either primary or secondary prevention of cardiovascular events among patients with prior events, an unfavorable risk profile, documented multibed vascular disease, vascular inflammation (early stages of atherosclerotic vasculopathy).
- A method of treating cardiovascular disease in a warm-blooded vertebrate is also provided. The method comprises administering an effective amount of an improved aptamer to a vertebrate subject suffering from cardiovascular disease that selectively binds a coagulation pathway factor IX, IXa, or the intrinsic tenase complex, or inhibits a subunit of the intrinsic tenase complex (i.e. FIX, FIXa, FVIII binding to or activation of FX). Administration of the aptamer treats the cardiovascular disease in the vertebrate subject. The method can further comprise providing an antidote to reverse the effects of the improved aptamer by administration of an antidote.
- The improved aptamers can be administered to mammals who require blood coagulation therapy. The invention provides methods of treating mammals with an aptamer to inhibit blood coagulation. The paired antidote can be administered to reverse the effects of the aptamer. A benefit of this discovery is that blood coagulation can be controlled in real time and does not rely on the mammal's own metabolism.
- The compositions and methods of the present invention are particularly useful for preventing thrombosis in the circuit of cardiac bypass apparatus and in patients undergoing renal dialysis, and for treating patients suffering from or at risk of suffering from thrombus-related cardiovascular conditions, such as unstable angina, acute myocardial infarction (heart attack), cerebrovascular accidents (stroke), pulmonary embolism, deep vein thrombosis, arterial thrombosis, CABG surgery and disseminated intravascular coagulation.
- In addition, the improved aptamers and antidotes of the present invention can inhibit other cardiovascular disease associated with FIX or FIX-regulated cascades. Coagulation plays an important part in ischaemic cardiovascular disease. Results of studies have shown that extremes in hypocoagulability protect against ischaemic cardiovascular disease. A mild decrease in coagulability found in hemophiliac patients can have a protective effect against fatal ischaemic heart disease (Sramek A, et al. (2003) Lancet 362(9381):3514 ; Bilora F, et al. (1999) Clin Appl Thromb Hemost. 5(4):232-5.
- The aptamers can be administered to prevent coagulation-induced inflammation. Inflammation is induced by thrombolytic therapy in patients with acute myocardial infarction (AMI), which might contribute to microvascular obstruction and reperfusion injury. Improved aptamers of the present invention can inhibit this early inflammatory response. In one embodiment, methods are provided to reduce the early inflammatory response in mammals that are in need thereof by administering the improved aptamers of the present invention.
- The improved aptamers and antidotes of the present invention can be used to inhibit atherosclerosis. Some adverse events in atherosclerosis are associated with ruptured plaques, which are a major cause of morbidity and mortality associated with atherosclerosis. In addition to conventional coronary heart disease risk factors, coagulation factor IX activation peptide and fibrinogen can be positively associated with risk of coronary heart disease (R. Rosenberg et al. (2001) Thromb Haemost 86: 41-50; J A Cooper et al. (2000) Circulation 102: 2816-2822). The intrinsic pathway may significantly enhance thrombogenicity of atherosclerotic lesions after removal of the endothelial layer and exposure of SMCs and macrophages to blood flow (Ananyeva N M et al. (2002) Blood 99: 44754485). In addition, the improved aptamers can also be provided to prevent morbidity in mammals suffering from acute coronary syndrome (ACS) associated with inflammation.
- In certain clinical scenarios, the contact pathway becomes the major pathway for blood clotting. These include surgical procedures where the blood products are removed from the body, such as contact of the blood with the cardiopulmonary bypass (CPB) circuit and oxygenator induces an inflammatory state during and post CPB. Genetic epidemiology and prospective clinical studies have linked the magnitude of the inflammatory response during coronary revascularization procedures with multiple adverse effects of CPB, including renal damage, atrial fibrillation, stroke, gut damage and neuronal damage. The inflammatory response induced by activation of the coagulation pathway is mediated by coagulation factors Xa and thrombin, which in addition to their role in blood clot formation, are themselves pro-inflammatory and mitogenic signaling proteins. The improved aptamers can also be administered to prevent adverse effects associated with post-angioplasty restenosis.
- Regulating Coagulation With Improved Aptamer-Antidote Pairs
- Among the many challenges of treating patients with thrombotic disorders or during a coagulation-inducing event is the potential risk of hemorrhage associated with anticoagulant drug therapy. The mechanisms which underlie bleeding risk are complex, but are unquesionably a function of drug variablity (excerss anticoagulant effect for the degree of thrombogenicity or extent of thrombus burden), relatively poor correlation between drug concentration and anticoagulant effect, wide-spread compromise of hemostatic barriers (platelet performance, vascular integrity, multiple phases of coagulation) and limited control of the anticoagulant's behavior.
- At least three clinical scenarios exist in which the ability to rapidly reverse the activity of an antithrombotic or anticoagulant nucleic acid ligand is desirable. The first case is when anticoagulant or antithrombotic treatment leads to hemorrhage, including intracranial or gastrointestinal hemorrhage. While identifying safer target proteins may reduce this risk, the potential for morbidity or mortality from this type of bleeding event is such that the risk can not be overlooked. The second case is when emergency surgery is required for patients who have received antithrombotic treatment. This clinical situation arises in a percentage of patients who require emergency coronary artery bypass grafts (CABG) while undergoing percutaneous coronary intervention under the coverage of GPIIb/IIIa inhibitors. Current practice in this situation is to allow for clearance of the compound (for small molecule antagonists such as eptifibatide), which may take 2-4 hours, or platelet infusion (for Abciximab treatment). The third case is when an anticoagulant nucleic acid ligand is used during a cardiopulmonary bypass procedure. Bypass patients are predisposed to post operative bleeding. In each case, acute reversal of the anticoagulant effects of a compound via an antidote (e. g., an oligonucleotide antidote of the invention targeted to an anticoagulant or antithrombotic nucleic acid ligand) allows for improved, and likely safer, medical control of the anticoagulant or antithrombotic compound.
- The applicants have discovered improved aptamer-antidote pairs that precisely regulate proteins in the blood coagulation cascade. In one embodiment, the antidotes of the invention are provided to a mammal in need thereof after the aptamers of the invention to reverse the effects of the aptamers. Aptamers and aptamer-antidote pairs can be administered in real time as needed based on various factors, including the progress of the patient, as well as the physician's discretion in how to achieve optimal therapy. Thus, this invention discloses an improved regulatable therapeutic regime in the course of nucleic acid ligand therapy for blood coagulation.
- Individuals who are undergoing surgery also require the targeted modulation of coagulation that occurs through the use of the improved aptamers and antidotes of the present invention. In certain embodiments, the aptamers are administered to patients undergoing general surgery. In certain other embodiments, the aptam rs are administered to patients with cardiovascular disease, which can include coronary heart disease. The patients can be undergoing treatment including bypass surgery or percutaneous coronary interventions. The mammals who can be treated with the aptamers of the present invention can also include patients who have had physical trauma that requires coagulation therapy.
- Preoperative assessment of the patient can identify drug-induced, acquired, or inherited coagulation defects. The main attention in anticoagulant therapy is directed to the perioperative period. A further, often overlooked, management strategy in treating major coagulopathies is the consideration of the cost and half-lives of the coagulation factors in individual blood components. Prevention of bleeding has become possible by manipulation of the control of coagulation and inflammatory processes. Additionally, because diagnosis of patients is often difficult, the modulatable improved aptamers and antidote pairs of the present invention are particularly useful in ensuring that, in case of incorrect diagnosis and treatment, treatment can immediately be disabled. For example, the symptoms of coronary infarction can closely mimic those of an acute coronary dissection. See Scarabeo et al. (2002) Italian Heart Journal 3: 490494. A diagnosis of coronary infarction immediately calls for an anticoagulant, which is counterindicated in acute coronary dissection. With the improved aptamer-antidote pairs described herein, a mistake by a health care provider can readily be reversed.
- Agents that restore vascular patency in stroke also increase the risk of intracerebral hemorrhage (ICH). As Factor IXa is a key intermediary in the intrinsic pathway of coagulation, targeted inhibition of Factor IXa-dependent coagulation can inhibit microvascular thrombosis in stroke without impairing extrinsic hemostatic mechanisms that limit ICH. The improved aptamers and antidotes of the present invention can be used to inhibit stroke associated with cardiovascular disease and surgery.
- Administration
- The present method for treating cardiovascular disease in a tissue contemplates contacting a tissue in which cardiovascular disease is occurring, or is at risk for occurring, with a composition comprising a therapeutically effective amount of an improved aptamer capable of binding a coagulation factor as well as providing an improved antidote to reverse the effects of the aptamer by administration of an antidote. Thus, the method comprises administering to a patient a therapeutically effective amount of a physiologically tolerable composition containing the RNA aptamer as well as a method to provide an antidote to reverse the effects of the aptamer by administration of an antidote.
- The dosage ranges for the administration of the antidote depend upon the form of the antidote, and can be assessed by a physician or other health care provider. Generally, the dosage will vary with the age, condition, sex and extent of the disease in the patient and can be determined by one of skill in the art. The individual physician in the event of any complication can also adjust the dosage.
- Generally, a therapeutically effective amount is an amount of a antidote sufficient to produce a measurable modulation of the effects of the nucleic acid ligand, including but not limited to a coagulation-modulating amount or an inflammation-modulating amount.
- Preferred modes of administration of the improved aptamers of the present invention are parenteral, intravenous, intradermal, intra-articular, intra-synovial, intrathecal, intra-arterial, intracardiac, intramuscular, subcutaneous, intraorbital, intracapsular, intraspinal, intrasternal, topical, transdermal patch, via rectal, vaginal or urethral suppository, peritoneal, percutaneous, nasal spray, surgical implant, internal surgical paint, infusion pump or via catheter. In one embodiment, the agent and carrier are administered in a slow release formulation such as an implant, bolus, microparticle, microsphere, nanoparticle or nanosphere.
- The antidotes of the present invention can be preferably administered parenterally by injection or by gradual infusion over time. Although the tissue to be treated can typically be accessed in the body by systemic administration and therefore most often treated by intravenous administration of therapeutic compositions, other tissues and delivery techniques are provided where there is a likelihood that the tissue targeted contains the target molecule. Thus, antidotes of the present invention are typically administered orally, topically to a vascular tissue, intravenously, intraperitoneally, intramuscularly, subcutaneously, intra-cavity, transdermally, and can be delivered by peristaltic techniques. As noted above, the pharmaceutical compositions can be provided to the individual by a variety of routes such orally, topically to a vascular tissue, intravenously, intraperitoneally, intramuscularly, subcutaneously, intra-cavity, transdermally, and can be delivered by peristaltic techniques. Representative, non- liming approaches for topical administration to a vascular tissue include (1) coating or impregnating a blood vessel tissue with a gel comprising a nucleic acid ligand, for delivery in vivo, e. g. by implanting the coated or impregnated vessel in place of a damaged or diseased vessel tissue segment that was removed or by-passed; (2) delivery via a catheter to a vessel in which delivery is desired; (3) pumping a nucleic acid ligand composition into a vessel that is to be implanted into a patient. Alternatively, the nucleic acid ligand can be introduced into celis by microinjection, or by liposome encapsulation. Advantageously, nucleic acid ligands of the present invention can be administered in a single daily dose, or the total daily dosage can be administered in several divided doses. Thereafter, the antidote is provided by any suitable means to alter the effect of the nucleic acid ligand by administration of the antidote.
- Compositions
- The aptamers and antidotes of the invention can be formulated into pharmaceutical compositions that can include, in addition to an improved aptamer, a antidote or modulator, and a pharmaceutically acceptable carrier, diluent or excipient. The precise nature of the composition will depend, at least in part, on the nature of the improved aptamer and antidote and the route of administration. Optimum dosing regimens can be readily established by one skilled in the art and can vary with the improved aptamer, the antidote combination, the patient and the effect sought.
- For standard information on pharmaceutical formulations, see Ansel, et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, Sixth Edition, Williams & Wilkins, 1995. The therapeutic compositions comprising aptamers and antidotes of the present invention are conventionally administered intravenously, as by injection of a unit dose, for example. The term “unit dose” when used in reference to a therapeutic composition of the present invention refers to physically discrete units suitable as unitary dosage for the subject, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect in association with the required diluent; i.e. carrier or vehicle.
- The compositions are administered in a manner compatible with the dosage formulation, and in a therapeutically effective amount. The quantity to be administered depends on the subject to be treated, capacity of the subject's system to utilize the active ingredient, and degree of therapeutic effect desired. Precise amounts of active ingredient required to be administered depend on the judgment of the practitioner and are peculiar to each individual. However, suitable dosage ranges for systemic application are disclosed herein and depend on the route of administration. Suitable regimes for administration are also variable, but are typified by an initial administration followed by repeated doses at one or more hour intervals by a subsequent injection or other administration. Alternatively, continuous intravenous infusion sufficient to maintain concentrations in the blood in the ranges specified for in vivo therapies are contemplated.
- Pharmaceutically useful compositiohs comprising an aptamer or antidote of the present invention can be formulated according to known methods such as by the admixture of a pharmaceutically acceptable carrier. Examples of such carriers and methods of formulation can be found in Remington's Pharmaceutical Sciences. To form a pharmaceutically acceptable composition suitable for effective administration, such compositions will contain an effective amount of the aptamer. Such compositions can contain admixtures of more than one aptamers or antidotes.
- The effective amount of an improved aptamer of the invention can vary according to a variety of factors such as the individual's condition, weight, sex and age. Other factors include the mode of administration. Generally, the compositions will be administered in dosages adjusted for body weight, e. g., dosages ranging from about 0.1 mgikg body weight to about 100 mg/kg body weight. In specific embodiments, the dosages are about 0.5 mg/kg body weight to 50 mg/kg body weight. In specific embodiments, the dosage is between 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, and 1 mg/kg body weight, and any dosage in between. Specific dosage units can range from 1 ng to 1 g, but are more conventionally about 0.01 μg, 0.μg, 1 μg, 10 μg, 100 μg, 500 μg, or 1 g or any amount in between.
- The effective amount of antibody being delivered to a patient will vary according to a variety of factors such as the individual's condition, weight, sex, age and amount of nucleic acid ligand administered. In one embodiment, the antidote ranges from 0.5-50 mg/kg. In another embodiment, the amount of antidote being delivered ranges from 0.5-10, 0.5-5, 1-10 or 1-5 mg/kg. In general, the amount of antidote being delivered is not less than the amount of aptamer being delivered. Typically, the amount of antidote is from about 1 to about 20 times the amount of aptamer. In certain embodiments, the antidote is about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 times the amount of aptamer delivered to a patient.
- Improved combinations of aptamers and antidotes in pharmaceutical compositions are administered in therapeutically effective amounts, that is, in amounts sufficient to generate a coagulation-modulating response, or in a prophylactically effective amounts, that is in amounts sufficient to prevent a coagulation factor from acting in a coagulation cascade. The therapeutically effective amount and prophylactically effective amount can vary according to the modulator. The pharmaceutical composition can be administered in single or multiple doses.
- Because the activity of the improved antidotes is lasting, once the desired level of modulation of the nucleic acid ligand by the antidote is achieved, infusion of the antidote can be terminated, allowing residual antidote to clear the human or animal. This allows for subsequent re-treatment with the nucleic acid ligand as needed. Alternatively, and in view of the specificity of the antidotes of the invention, subsequent treatment can involve the use of a second, different improved aptamer/antidote pair.
- Antidotes synthesized or identified according to the methods disclosed herein can be used alone at appropriate dosages defined by routine testing in order to obtain optimal modulation of nucleic acid ligand activity in coagulation, while minimizing any potential toxicity. In addition, co- administration or sequential administration of other agents can be desirable. For combination treatment with more than one active agent, where the active agents are in separate dosage formulations, the active agents can be administered concurrently, or they each can be administered at separately staggered times.
- The dosage regimen utilizing the improved aptamers and antidotes of the present invention is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal and hepatic function of the patient; and the particular combination employed. A physician of ordinary skill can readily determine and prescribe the effective amount of the aptamer required to prevent, counter or arrest the progress of the condition. Optimal precision in achieving concentrations of the combination within the range that yields efficacy without toxicity requires a regimen based on the kinetics of the aptamer and antidote's availability to target sites. This involves a consideration of the distribution, equilibrium, and elimination of the modulator.
- In the methods of the present invention, the combinations described in detail can form the active ingredient, and are typically administered in admixture with suitable pharmaceutical diluents, excipients or carriers (collectively referred to herein as “carrier” materials) suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrup, suppositories, gels and the like, and consistent with conventional pharmaceutical practices.
- For instance, for oral administration in the form of a tablet or capsule, the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like. Moreover, when desired or necessary, suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include without limitation, starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like. Lubricants used in these dosage forms include, without limitation, sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like. Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like.
- For liquid forms the active drug component can be combined in suitably flavored suspending or dispersing agents such as the synthetic and natural gums, for example, tragacanth, acacia, methyl-cellulose and the like. Other dispersing agents that can be employed include glycerin and the like. For parenteral administration, sterile suspensions and solutions are desired. Isotonic preparations that generally contain suitable preservatives are employed when intravenous administration is desired.
- Topical preparations containing the active drug component can be admixed with a variety of carrier materials well known in the art, such as, e. g., alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, PPG2 mydstyl propionate, and the like, to form, e.g. alcoholic solutions, topical cleansers, cleansing creams, skin gels, skin lotions, and shampoos in cream or gel formulations.
- The aptamers and antidotes of the present invention can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles. Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- The aptamers and antidotes of the present invention can also be coupled with soluble polymers as targetable drug carriers. Such polymers can include polyvinyl-pyrrolidone, pyran copolymer, polyhydroxypropylmethacryl-amidephenol, polyhydroxy-ethylaspartamidephenol, or polyethyl-eneoxidepolylysine substituted with palmitoyl residues. Furthermore, the aptamers and antidotes of the present invention can be coupled (preferably via a covalent linkage) to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polyethylene glycol (PEG), polylactic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydro-pyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels. Cholesterol and similar molecules can be linked to the aptamers to increase and prolong bioavailability.
- In certain embodiments of this invention, the complex comprises a liposome with a targeting nucleic acid ligand (s) associated with the surface of the liposome and an encapsulated therapeutic or diagnostic agent. Preformed liposomes can be modified to associate with the nucleic acid ligands. For example, a cationic liposome associates through electrostatic interactions with the nucleic acid. Alternatively, a nucleic acid attached to a lipophilic compound, such as cholesterol, can be added to preformed liposomes whereby the cholesterol becomes associated with the liposomal membrane. Alternatively, the nucleic acid can be associated with the liposome during the formulation of the liposome. Preferably, the nucleic acid is associated with the liposome by loading into preformed liposomes.
- Certain aspects of the invention can be described in greater detail in the non-limiting Examples that follow.
- Tests of Aptamers for Coagulation
- The following tests are used to assess the capacity of modified aptamers and antidotes to inhibit coagulation factors.
- The Activated Clotting Time Test (ACT) is a screening test that resembles the activated partial thromboplastin time (APTT) test, but is performed using fresh whole blood samples. ACT can be to monitor a patient's coagulation status in connection with clinical procedures, such as those that involve the administration of high doses of heparin (e.g., CPB and PTCA).
- The Activated Partial Thromboplastin Time Test (APTT) is a common central laboratory test, typically performed using an automated coagulometer, for example Diagnostica Stago's STA coagulometer (MDA/96/23), or another coagulometer produced by this company or otherwise known in the art. The test is performed using a plasma sample, in which the intrinsic pathway is activated by the addition of phospholipid, an activator (ellagic acid, kaolin, or micronized silica), and Ca2+.
- The bleeding time test can be used for the diagnosis of hemostatic dysfunction, von Willebrand's disease, and vascular disorders. It also can be used to screen for platelet abnormalities prior to surgery. The test is performed by making a small incision on the forearm and wicking away the blood from the wound site. The time it takes for bleeding to stop is recorded and in control subjects is approximately 3.5 minutes. Prolongation of the bleeding time is indicative of qualitative or quantitative platelet defects.
- The Prothrombin Time Test (PT), which was first described by Quick in 1935, measures the tissue factor-induced coagulation time of blood or plasma. It is used as a screening test to evaluate the integrity of the extrinsic coagulation pathway, and is sensitive to coagulation factors I, II, V, VII, and X. The test can be performed by adding thromboplastin and Ca2+ to a patient sample and measuring the time for clot formation. A prolonged clotting time suggests the presence of an inhibitor to, or a deficiency in, one or more of the coagulation factors of the extrinsic pathway. But PT clotting time can also be prolonged for patients on warfarin therapy, or for those with vitamin K deficiency or liver dysfunction. The PT test can provide an assessment of the extrinsic coagulation pathway, and is widely used to monitor oral anticoagulation therapy.
- The Thrombin Clotting Time Test (TCT) measures the rate of a patient's clot formation compared to that of a normal plasma control. The test can be performed by adding a standard amount of thrombin to a patient's plasma that has been depleted of platelets, and measuring the time required for a clot to form. This test has been used as an aid in the diagnosis of disseminated intravascular coagulation (DIC) and liver disease.
- There are also a number of tests that may be used in the diagnosis of a patient's coagulative status. These fall into two categories: complex tests, some of which are based on the screening tests outlined above, and immunoassays. Complex Tests include specific factor assays based on laboratory tests, such as the APTT, PT, and TCT tests. One assay measures the level of the activation Factor IXa or the Factor IXa-antithrombin III complex. These measurements are used to determine the levels of factor IXa or factor VII-tissue mediated complex. Assays for activated protein C resistance, antithrombin, protein C deficiency, and protein S deficiency are also part of this group. Asymptomatic individuals who have heterogeneous deficiencies of proteins C and S, and resistance to activated protein C, have significantly elevated levels of the prothrombin fragnent F1.2 compared to controls.
- 2′-Hydroxyl purines were substituted with 2′-O-methyl purines in the 4 secondary structure units of in which purine residues are present: Stem 1 (Apt 1); Loop 1 (Apt 2); Stem 2 (Apt 3); Loop 2 (Apt 4) (see
FIG. 1A ). - Procedure: The anticoagulant activity of AptA derivatives Apt 1-5 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar (
FIG. 2 ). The “neutralizability” of Apt1-5 was evaluated in standard APTT antidote assays over AptA antidote concentrations (AptA AD; see sequence listings) ranging from 5 uM and down (FIG. 2 ). For these assays, the concentration of AptA and derivatives was fixed at 125 nM. - Apt 4 showed gain of anticoagulant activity (
FIG. 2 ); Apt 1-3 showed moderate loss of activity; and Apt 5 showed severe loss of activity. Apt 1-3 exhibit enhanced neutralization, suggesting that introduction of 2′ -O-methyl residues within the antidote binding site improves the ability of the antidote oligonucleotide to bind to the aptamer. - Sequence Listings:
1) AptA Length: 35 (5′-3′) sequence: AUGGGGA CUAUACC GCG UAAUGC UGC C UCCCCAU T (SEQ ID NO:20) 1) Apt 1 Length: 35 (5′-3′) sequence: aUgggga CUAUACCGCGUAAUGCUGCC UGCC GaU T (SEQ ID NO:21) 3) Apt 2 Length: 35 (5′-3′) sequence: AUGGGGA CUaUaCC GCG UAAUGG UGC C UCCCCAU T (SEQ ID NO:22) 4) Apt 3 Length: 35 (5′-3′) sequence: AUGGGGA CUAUACC gCg UAAUGC UgC C UCCCCAU T (SEQ ID NO:23) 5) Apt 4 Length: 35 (5′-3′) sequence: AUGGGGA CUAUACC GCG UaaUgC UGC C UCCCCAU T (SEQ ID NO:24) 6) Apt 5 Length: 35 (5′-3′) sequence: aUgggga CUaUaCC gCg UaaUgC UgC C UCCCCaU T (SEQ ID NO:25) 7) AptA AD Length: 17 (5′-3′) sequence: cgcggua uaguccccau (SEQ ID NO:1)
“A”: 2′OH Adenine; “a”: 2′-O-methyl Adenine; “G”: 2′OH Guanine; “g”: 2′-O-methyl Guanine; “C”: 2′Fluro-Cytidine; “c”: 2′-O-methyl Cytidine; “U”: 2′Fluoro-Uridine; “u”: 2′-O-methyl Uridine; “T”: inverted 2′H Thymidine.
- Two “families” of
stem 1 variants were designed (Apt 6-8 and 9-11;FIG. 1B ) consisting of 4, 5, and 6 basepair stems. All constructs were designed in the Apt-2 background.Stem 1 sequences were evaluated for the ability to design complementary antidote oligonucleotides to them such that the antidotes contain minimal secondary structure, and for the ability of the aptamer to assume the proper secondary structure. - Stems were wholly 2′-O-methyl modified. Antidote oligonucleotides were designed specific for Apt 6-11 that bind to their respective target aptamer in the same register as AptA AD (see sequence listings below).
- Experiments: The anticoagulant activity of Apt 6-11 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The antidote control of Apt 6-11 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down. For these assays, the concentration of
Apt 2, and Apt 6-11 was set at 250 nM (as opposed to 125 nM for standard AptA andApt 2 experiments). - Apt 6-8 exhibit loss of anticoagulant activity (
FIG. 3 ), however, all exhibit similar activity levels. Thus stem length is not be the main cause for loss of activity. The 5 base pair stem 1 constructs (Apt 10 and Apt 7) do appear to be more neutralizable than Apt 2 (FIGS. 3 and 4). Data suggests that astem 1 of 5 base pairs may be preferable to those composed of 4, 6 or 7 base pairs to enhance antidote neutralization. - Sequence Listings:
1) Apt 6 Length: 29 (5′-3′): ggga CUaUaCCGCG UAAUGCUGCC uccc T (SEQ ID NO:26) 2) Apt 7 Length: 31 (5′-3′): gugga CUaUaCCGC GUAAUGCUGCC uccac T (SEQ ID NO:27) 3) Apt 8 Length: 33 (5′-3′): gaugga CUaUaCCG CGUAAUGCUGCC uccauc T (SEQ ID NO:28) 4) Apt 9 Length: 29 (5′-3′): cuga CUaUaCCGCG UAAUGCUGCC ucag T (SEQ ID NO:29) 5) Apt 10 Length: 31 (5′-3′): ccuga CUaUaCCGC GUAAUGCUGCC ucagg T (SEQ ID NO:30) 6) Apt 11 Length: 33 (5′-3′): cucuga CUaUaCCG GGUAAUGCUGCC ucagag T (SEQ ID NO:31) 7) Apt 6 AD Length: 14 (5′-3′): cgcgguauaguccc (SEQ ID NO:2) 8) Apt 7 AD Length: 15 (5′-3′): cgcgguauaguccac (SEQ ID NO:3) 9) Apt 8 AD Length: 16 (5′-3′): cgcgguauaguccauc (SEQ ID NO:4) 10) Apt 9 AD Length: 14 (5′-3′): cgcgguauagucag (SEQ ID NO:5) 11) Apt 10 AD Length: 15 (5′-3′): cgcgguauagucagg (SEQ ID NO:6) 12) Apt 11 AD Length: 16 (5′-3′): cgcgguauagucagag (SEQ ID NO:7)
“A”: 2′OH Adenine; “a”: 2′-O-methyl Adenine; “G”: 2′OH Guanine; “g”: 2′-O-methyl Guanine; “C”: 2′Fluro-Cytidine; “c”: 2′-O-methyl Cytidine; “U”: 2′Fluoro-Uridine; “u”: 2′-O-methyl Uridine; “T”: inverted 2′H Thymidine.
- The anticoagulant activity of Apt 12-17 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The “neutralizability” of Apt 12-17 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down. For
Apt Apt - Comparison of the anticoagulant activity of
Apt 12 withApt 13 and Apt17 (FIG. 5 ) demonstrates that the loss of activity observed for Apt6-11 is due to the presence of 2′-O-methyl substitutions at one or more critical residues. Comparison of the anticoagulant activity of Apt14 to Apt12 indicates that the stretch of 4 consecutive guanosines withinstem 1 can be altered without a significant impact on anticoagulant activity. Comparison of Apt15 and 16 withApt stem 1 except for the closing A-U pair at the top ofstem 1 enhances activity; b) demonstrates that the sugar of the U in this base pair must be 2′fluoro for the aptamer to retain potency; and c) suggests that the sugar of the A in this base pair can be a 2′-O-methyl sugar without a significant impact on anticoagulant activity. In fact,Apt 16 retains essentially full potency. - Comparison of the neutralization of Apt14-16 with Apt14/AD suggests that the antidote can more readily bind the aptamer when
stem 1 is a 2′-O-methyl-2′fluoro stem as opposed to when both strands of the duplex contain largely 2′-O-methyl residues. Apt21 was designed in which the sugar of the A at the stop ofstem 1 is 2′-O-methyl substituted (FIG. 1 ). Substitution of a 2′-O-methyl sugar at this adenosine residue is well tolerated in the background of a largely 2′-O-methyl stem (FIG. 6 ). Antidote neutralization ofApt 21 is enhanced as compared to Apt16 (see especially the 2.5:1 and 5:1 AD:Drug data points inFIG. 4 ). - Sequence Listings:
1) Apt2 Length: 35 (5′-3′) sequence: aUgggga CUaUaCCGCGUAAUGCUGCC UCGCCa U T (SEQ ID NO:32) 2) Apt13 Length: 35 (5′-3′) sequence: augggga CUaUaCCGCGUAAUGCUGCC ucccca u T (SEQ ID NO:33) 3) Apt14 Length: 35 (5′-3′) sequence: gUgagga CUaUaCGGGGUAAUGCUGCC UGGUCa C T (SEQ ID NO:34) 4) Apt15 Length: 35 (5′-3′) sequence: gUgaggA CUaUaGCGCGUAAUGCUGGG UCCUGa C T (SEQ ID NO:35) 5) Apt16 Length: 35 (5′-3′) sequence: gugaggA CUaUaCCGCGUAAUGCUGCC Uccuca c T (SEQ ID NO:36) 6) Apt17 Length: 35 (5′-3′) sequence: gugagga CUaUaCCGCGUAAUGCUGCC uccuca c T (SEQ ID NO:37) 7) Apt14AD Length: 17 (5′-3′) sequence: cgcgguaua guccucac (SEQ ID NO:8) 8) Apt21 Length: 35 (5′-3′) sequence: gugagga C UaUaCC GCG UAAUGC UGC C Ucc ucac T (SEQ ID NO:41)
“A”: 2′OH Adenine; “a”: 2′-O-methyl Adenine; “G”: 2′OH Guanine; “g”: 2′-O-methyl Guanine; “C”: 2′Fluro-Cytidine; “c”: 2′-O-methyl Cytidine; “U”: 2′Fluoro-Uridine; “u”: 2′-O-methyl Uridine; “T”: inverted 2′H Thymidine.
- The anticoagulant activity of Apt 18-20 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The “neutralizability” of Apt 18-20 was evaluated in standard APTT antidote assays over antidote concentrations (
Antidote - Each of the aptamers (Apt 18-20) is a potent anticoagulant, as or more potent than Apt 2 (
FIG. 7 ). Furthermore, all three are readily neutralized by their respective antidote oligonucleotides. Apt 19 was evaluated for anticoagulant activity of a pegylated version.PEG Apt 19 and, for comparison, PEG-Apt 16 are used (PEG is a 40 KDa bifunctional polyethylene glycol mPEG2-NHS ester (MW 40 KDa; Nektar/Shearwater 2Z3XOT01, which includes two 20 KD monomethoxy PEGs bound together through a lysine and linked via a 6-carbon linker to a 5′-terminal phosphate on the aptamer), appended to the 5′end via conjugation to a C6 amino linker added to the aptamer during solid phase synthesis).FIG. 8 indicates that the length ofstem 1 does not affect how 40 KDa PEG addition impacts the activity of AptA and AptA derivatives. The anticoagulant activity ofPEG Apt Apt 19,PEG Apt 19 is more readily neutralized by its matched antidote (7 AD) than AptA,Apt 16 or any of the PEG or cholesterol-modified versions of these compounds.FIG. 8 shows approximately 90% reversal ofPEG Apt 19 at 2.5:1 AD:Aptamer. Looked at as an absolute change in APTT rather than on a % reversal basis, the APTT of plasma treated withPEG Apt 19+2.5:1 7AD:Aptamer is only 4-5 seconds above baseline. - Sequence Listings:
1) Apt18 Length: 29 (5′-3′) sequence: gggA CUa UaCCGCGUAAUGCUGCC Uccc T (SEQ ID NO:38) 2) Apt19 Length: 31 (5′-3′) sequence: guggA CU aUaCGGCGUAAUGCUGCC Uccac T (SEQ ID NO:39) 3) Apt20 Length: 33 (5′-3′) sequence: gauggA C UaUaCCGCGUAAUGCUGCC Uccauc T (SEQ ID NO:40) 4) PEG Apt 16Length: 35 (5′-3′) sequence: P-L-guga ggA CUaUaCCGCGUAAUGCUGCC U ccucac T 5) PEG Apt 19Length: 31 (5′-3′) sequence: P-L-gugg ACUaUaCCGCGUAAUGCUGCG Ucca c T
“A”: 2′OH Adenine; “a”: 2′-O-methyl Adenine; “G”: 2′OH Guanine; “g”: 2′-O-methyl Guanine; “C”: 2′Fluro-Cytidine; “c”: 2′-O-methyl Cytidine; “U”: 2′Fluoro-Uridine; “u”: 2′-O-methyl Uridine; “T”: inverted 2′H Thymidine; “P”: mPEG2-NHS ester MW 40 KDa (Nektar/Shearwater 2Z3XOT01); “L”: C6 amino linker
- Two series of variants evaluating the optimal sugar composition for the residues in
stem 2 andloop 2. The first series in the Apt 16 background. The second series in the Apt 16 background, but substituting the tetraloop found in FIXa aptamer 9.20 (see Rusconi et al Nature 419, p. 90-94, 2002 andFIG. 1 ) for the hexanucleotide loop found in AptA. Studies onApt 4 indicated that 2′-O-methyl purine substitution withinloop 2 led to an enhancement in AptA potency, whereas 2′-O-methyl purine substitution withinstem 2 led to a modest loss of potency, and that simultaneous 2′-O-methyl purine substitution withinstem 2 andloop 2 in the context of a 2′-O-methyl purine stem 1 led to a significant loss of AptA potency (Apt 5). Therefore, independently substitute 2′-O-methyl purines in stem 2 (Apt 22, 26) and loop 2 (Apt 23, 27) (FIG. 9 ). Re-evaluated complete 2′-O-methyl substitution of purines withinstem 2 and loop 2 (Apt 24, Apt 28) but leave the G at the base ofstem 2 as a 2′hydroxl (Apt 25, 29) in the event that a 2′hydroxyl is required at this position. - The anticoagulant activity of Apt 22-29 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The “neutralizability” of Apt 22-29 was evaluated in standard APTT antidote assays over antidote concentrations ranging from 5 uM and down. The aptamer concentration was fixed at 125 nM in these assays, except for
Apt 24 in which the aptamer concentration was fixed at 250 nM. - As previously observed with
Apt 3, substitution of 2′-O-methyl purines withinloop 2 leads to enhanced potency (Apt 23, compare Apt 23 to 16) (FIG. 9 ). Likewise, substitution of 2′-O-methyl purines intostem 2 leads to a moderate loss of activity (Apt 22, 24) (FIG. 14 ). Apt 24 is significantly more potent thanApt 5. Maintenance of a 2′hydroxyl on the G residue at the base of stem 2 (Apt 25) does not lead to enhanced activity as compared toApt 24, indicating that a) substitution of a 2-O-methyl sugar at this residue is not the problem withinApt Apt 23 is reduced as compared toApt 16, but still equivalent to AptA. - Sequence Listings:
1) Apt22 Length: 35 (5′-3′) sequence: gugaggA CUaUa CC gCg UAAUGC UgC C Uccucac T (SEQ ID NO:42) 2) Apt23 Length: 35 (5′-3′) sequence: gugaggA CUaUa CC GCG UaaUgC UGC C Uccucac T (SEQ ID NO:43) 3) Apt24 Length: 35 (5′-3′) sequence: gugaggA CUaUa CC gCg UaaUgC UgC C Uccucac T (SEQ ID NO:44) 4) Apt25 Length: 35 (5′-3′) sequence: gugaggA CUaUa CC GCg UaaUgC UgC C Uccucac T (SEQ ID NO:45) 5) Apt26 Length: 33 (5′-3′) sequence: gugaggA CUaUa CC gCa AUCG UgC C Uccucac T (SEQ ID NO:46) 6) Apt27 Length: 33 (5′-3′) sequence: gugaggA CUaUa CC GCA aUCg UGC C Uccucac T (SEQ ID NO:47) 7) Apt28 Length: 33 (5′-3′) sequence: gugaggA CUaUa CC gCa aUCg UgC C Uccucac T (SEQ ID NO:48) 8) Apt29 Length: 33 (5′-3′) sequence: gugaggA CUaUa GG GCa aUCg UgC C Uccucac T (SEQ ID NO:49) - The anticoagulant activity of Apt 30-33 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The “neutralizability” of
Apt FIG. 10 ). - Comparison of the activity of
Apt Apt FIG. 10 a). Activity observed betweenApt stem 2 can contain 2′-O-methyl sugars. In fact,Apt 31 appears to possess slightly greater potency thanApt 32, indicating that a compound with 2′fluoro at C16, 2′hydroxyl at G25, and the remainingresidues 2′-O-methyl may exhibit greater potency thanApt 33. Regardless,Apt 33 exhibits greater activity thanApt 2 and is fairly equivalent to original AptA. Apt 33 is more readily neutralizable thanApt 30, suggesting additional 2′-O-methyl residue within the antidote-binding site of the aptamer improves antidote binding. - Apt 34 had C16 a 2′fluoro rather than 2′-O-methyl (
FIG. 10 b). Increase in anticoagulant activity (compare Apt 34 to Apt 33). However, substitution did result in a modest loss of “neutralizability”, although 34 still requires a lower excess of antidote to achieve 90% neutralization (˜5:1 vs 10:1) than the parental AptA compound. Both results are consistent with an increase in the stability ofstem 2 due to 2′-O-methyl substitution. - Sequence Listings:
1) Apt30 Length: 35 (5′-3′) sequence: gugagga CUaUa GC gCG UaaUgC UGC C Uccucac T (SEQ ID NO:50) 2) Apt31 Length: 35 (5′-3′) sequence: gugagga CUaUa CG gcg UaaUgC ugc C Uccucac T (SEQ ID NO:51) 3) Apt32 Length: 35 (5′-3′) sequence: gugagga CUaUa CC gcg UaaUgC UgC C Uccucac T (SEQ ID NO:52) 4) Apt33 Length: 35 (5′-3′) sequence: gugagga CUaUa CC gCg UaaUgC UGC C Uccucac T (SEQ ID NO:53) 5) Apt34 Length: 35 (5′-3′) sequence: gugagga CUaUa CC gCg UaaUgC uGc C Uccucac T (SEQ ID NO:54) - Apt35-39 compared with original AptA (numbering based upon AptA stem 1):
- 1) Apt 30 to 31: differences are C16, G17, U24, G25, C26.
- 2) Apt 30 to 32: differences are C16, G17, G25.
- 3) Apt 31 to 32: differences are U24, C26.
- The anticoagulant activity of Apt 35-39 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The “neutralizability” of
Apt FIG. 11 ). - Apt 35-39: Anticoagulant activity of
Apt 39 is superior toApt 19 and all other stem 2-optimization constructs in the Apt 19 background (FIG. 11 ). Results were consistent with those obtained withApt 34. In addition, potency ofApt 39 is comparable to parental AptA and greater thanApt 2. The neutralization ofApt 39 by Apt7AD is excellent, and similar to neutralization of Apt 19 (FIG. 12 ). Again, sugar optimization and truncation ofApt 39 has resulted in a compound neutralized at lower excesses of antidote:drug as compared to parental AptA and Apt 2 (FIG. 11 ). - Sequence Listings:
1) Apt35 Length: 31 (5′-3′) sequence: gugga CUaUaCC gCG UaaUgC UGC C Uccac T (SEQ ID NO:55) 2) Apt36 Length: 31 (5′-3′) sequence: gugga CUaUaCC gCG UaaUgC ugc C Uccac T (SEQ ID NO:56) 3) Apt37 Length: 31 (5′-3′) sequence: gugga CUaUaCC gCG UaaUgC UgC C Uccac T (SEQ ID NO:57) 4) Apt38 Length: 31 (5′-3′) sequence: gugga CUaUaCC gCg UaaUgC UGC C Uccac T (SEQ ID NO:58) 5) Apt39 Length: 31 (5′-3′) sequence: gugga CUaUaCC gCg UaaUgC uGc C Uccac T (SEQ ID NO:59) - The anticoagulant tested is Apt39 with a 40 KDa polyethylene glycol (PEG) conjugated to the 5′end of the aptamer sequence via a 6-carbon NH2 linker (PEG-Apt39). The antidote is Apt7AD.
- The anticoagulant activity of PEG-Apt39 was evaluated in standard APTT coagulation assays over compound concentrations ranging from 1 uM to low nanomolar. The anticoagulant activity of Apt39 was compared to two formulations of the parental AptA, CH-Apt S(5′ cholesterol-modified) and PEG-AptA (5′ 40 KDa PEG-modified). For these studies, the molecular weight of the “aptamer” portion only was used to calculate the concentration of each compound. The “neutralizability” of PEG-Apt39 was evaluated in standard APTT antidote assays over antidote concentrations (Apt7AD) ranging from 5 uM and down. The aptamer concentration was fixed at 125 nM in these assays.
- The in vitro anticoagulant activity of PEG-Apt39 is essentially equivalent to CH-AptA and PEG-AptA (
FIG. 13 ). - This study compares the in vivo anticoagulant and antidote neutralization activity of PEG-Ap39 in swine to previous data obtained with CH-AptA with respect to: a) Potency and durability of anticoagulant activity and the b) Neutralization of anticoagulant activity. The three experimental groups (n=2 animals for each) are a) Systemic anticoagulation; b) Systemic anticoagulation and drug neutralization; and c) Systemic anticoagulation, drug neutralization and re-anticoagulation.
- Experiment: Six neonatal piglets (1 week old, 2.5-3.5 kg) were randomly assigned to three groups. Femoral arterial and venous lines were placed in the piglet. The arterial line was used to monitor blood pressure and arterial blood sampling. The venous line was used to administer drugs and test compounds as specified. Temperature of the piglet was monitored with a nasopharyngeal temperature probe.
- The dose of PEG-Apt39 was 0.5 mg/kg (dose of aptamer based upon molecular weight of nucleic acid component only; 10,103.2 Da) for each animal. In the prior experiments with CH-AptA, the aptamer dose was also 0.5 mg/kg (dose of aptamer based upon molecular weight of nucleic acid component only). In experiments in which Apt7 AD was used as an antidote, the dose of the antidote was 3 mg/kg. By comparison, in experiments with CH-AptA in which AptA AD was used, the antidote dose was 5 mg/kg.
- a) Systemic anticoagulation. A pre-injection blood sample was taken prior to injection of PEG-Apt39, the drug was then injected (time of injection is t=0), and blood samples removed at 5, 15, 25, 60, 90, 120 and 150 minutes post injection. Activated clotting times (ACT's) were performed on-site immediately after blood draw on the whole blood in duplicate using the Hemochron 801 junior and glass-activated flip-top tubes per the manufacturers directions. Blood samples were then transferred to citrated vacutainer tubes and stored on ice. Platelet poor plasma was prepared, and APTT and PT assays performed per the standard protocol. (
FIG. 14A ) - The in vivo anticoagulant potency of PEG-Apt39 is superior to CH-AptA. In addition, the loss of anticoagulant activity over time is reduced for PEG-Apt39 vs. CH-AptA. These results are in contrast with the in vitro anticoagulant activity studies, which demonstrate that the anticoagulant activity of PEG- Apt39 and CH-AptA are equivalent in vitro in pooled human plasma.
- b) Systemic anticoagulation and drug neutralization. A pre-injection blood sample was taken prior to injection of PEG- Apt39, the drug was then injected (0.5 mg/kg; time of injection is t=0), and blood samples removed at 5 and 15 minutes post drug injection. At t=15 minutes post drug injection, REGI S7 AD was administered (3 mg/kg), and additional blood samples removed at 25, 60, 90, 120 and 150 minutes post drug injection. Activated clotting times (ACT's) were performed on-site immediately after blood draw on the whole blood in duplicate using the Hemochron 801 junior and glass-activated flip-top tubes per the manufacturers directions. Blood samples were then transferred to citrated vacutainer tubes and stored on ice. Platelet poor plasma was prepared, and APTT and PT assays performed per the standard protocol.
- Essentially complete neutralization of the anticoagulant activity of PEG- Apt39 was achieved within 10 minutes of administration of 3 mg/kg Apt7 AD. The anticoagulant activity remained neutralized throughout the remainder of the experiment (2hr and 5min after initial demonstration of drug neutralization). Thus, the neutralization of PEG- Apt39 appears to be superior to that of CH-AptA, as similar levels of neutralization of PEG- Apt39 can be achieved with a 40% lower dose of antidote (3 mg/kg of Apt7 AD vs. 5 mg/kg REG1 AD). This in vivo data is consistent with in vitro experiments in pooled human plasma in which PEG- Apt39 is more readily neutralized by its matched antidote than any prior formulation of AptA. (
FIG. 14B ) - c) Systemic anticoagulation, drug neutralization and re-anticoagulation. A pre-injection blood sample was taken prior to injection of PEG-Apt39, the drug was then injected (0.5 mg/kg; time of injection is t=0), and blood samples removed at 5 and 15 minutes post drug injection. At t=15 minutes post drug injection, Apt7 AD was administered (3 mg/kg), and additional blood samples removed at 25, and 40 minutes post drug injection. At t45 minutes post drug injection (30 minutes following antidote administration), PEG- Apt39 was re-administered (0.5 mg/kg) and additional blood samples removed at 50, 60, 90, 120, and 150 minutes post drug injection. Activated clotting times (ACT's) were performed on-site immediately after blood draw on the whole blood in duplicate using the Hemochron 801 junior and glass-activated flip-top tubes per the manufacturers directions. Blood samples were then transferred to citrated vacutainer tubes and stored on ice. Platelet poor plasma was prepared, and APTT and PT assays performed per the standard protocol. (
FIG. 15 ) - Re-administration of PEG-Apt39 following neutralization of the initial drug dose is feasible within 30 minutes of administration of the neutralizing antidote. The levels of anticoagulation achieved following administration of the first and second dose appear to be equivalent to each other, suggesting that there is little remaining “free” antidote in the circulation at the time of administration of the second dose of drug.
- Aptamer levels in plasma are determined using a sandwich-type hybridization assay with an enzyme-linked immunoassay (ELISA) for detection. Quantitation of aptamer employs two oligonucleotide probes, a DNA capture probe, and a 2′Omethyl RNA detection probe. The DNA capture probe is 15 nucleotides in length, is complementary to the 3′ terminal 15 nucleotides of the aptamer, and contains a biotin moiety on its 5′terminus, allowing for capture of oligonucleotide complexes containing this probe to an avidin coated surface. The 2′Omethyl RNA detection probe is also 15 nucleotides in length, is complementary to the portion of aptamer to which antidote binds, and contains a digoxigenin moiety to enable detection of complexes containing this probe using standard enzyme-linked fluorescence generating enzyme/substrate reagents.
- Quantitation of aptamer is achieved by hybridization of the capture and detection probes to aptamer in plasma and subsequent immobilization of the complex onto the surface of a Neutravidin-coated microtitre plate by way of the 5′-biotin group. Measurement of the digoxigenin-labeled 2′-O-methyl RNA probe is performed subsequent to the plate immobilization reaction using an anti-digoxigenin antibody conjugated to alkaline phosphatase, which catalyzes the fluorescence of a substrate. Fluorescence intensity is then measured, the signal of which is directly proportional to the amount of aptamer present in the calibration standards and validation samples.
- The in vitro anticoagulant activity of aptamer Apt39 (SEQ ID NO:88) in plasma from cynomolgus monkeys is reflected by concentration-dependent prolongation of time-to-clot in the APTT assay. Plasma FIX assays were performed to aid in interpretation of the Apt39 APTT dose-response curve in monkey plasma. As shown in Table A, the APTT in monkey plasma is sensitive to the FIX level. However, the magnitude of the response to reduction in the FIX level is modest. A 75% reduction in the FIX level results in a 1.4-fold increase in the APTT, a >95% reduction in the FIX level results in a doubling of the APTT, and a 99.9% reduction in the plasma FIX level yields a 2.5-fold increase in the APTT.
TABLE A FIX Activity Assay Standard Curve in Cynomolgus Monkey Plasma % FIX Level APTT Clot Time Fold Increase in Clot Time 100* 35.1 1.0 50 41.9 1.2 25 49.4 1.4 12.5 55.9 1.6 6.25 62.2 1.8 3.13 68.0 1.9 1.56 74.7 2.1 0.78 77.7 2.2 0.39 83.8 2.4 0.098 88.1 2.5
*100% FIX level represents a 1:5 dilution of normal pooled cynomolgus plasma in buffer. Human FIX-deficient plasma (George King Biomedical) was used as the source of FIX-deficient plasma.
- The date in table A indicate that ˜6 μg/mL Apt39 is required to inhibit approximately 90% of plasma FIX activity in monkeys (i.e., this concentration yields a. 1.6-fold increase in the APTT), and that >95% inhibition of plasma FIX activity occurs at Apt39 concentrations of 10-12 μg/mL.
- In vivo Activity ofApt39 and Apt7AD in Cynomolgus Monkeys
- The relationship between the anticoagulant properties of Apt39 and the Apt39/Apt7AD complex and the plasma levels of these compounds was evaluated in monkey. Briefly, 12 monkeys were assigned to three treatment groups.
Group 1 received the anti-FIXa aptamer Apt39,Group 2 received the antidote Apt7AD andGroup 3 was treated with Apt39 and three hours later with Apt7AD. Doses were escalated through two quantities of test articles, with the first dose occurring onDay 4 of the study and the second dose occurring onDay 13. To better understand the dose-response to aptamer, the four monkeys assigned toGroup 1 were subdivided into two groups atDay 13, with two animals receiving a low dose (Group Group 1b, 30 mg/kg). - As shown in
FIG. 16 , administration of Apt39 at doses ranging from 5 to 30 mg/kg resulted in a profound level of anticoagulation in the monkeys. The mean APTT at each dose level exceeded 60 seconds from 0.25 to 24 hours following aptamer administration, which is equivalent to <0.1% normal plasma FIX levels in the monkey. There is a dose-dependent increase in APTT in response to Apt39 administration. However, the dose-response is not immediately evident due to the fact that, up to the 6-hour time point following Apt39 administration, the aptamer plasma level exceeded the concentration at which the in vitro APTT dose-response curve approaches a plateau (˜40-50 μg/mL; see Table B). At times beyond 6 hours after administration, as the aptamer concentration decreases below this level, the dose-response is more apparent. APTT was followed until it returned to baseline in monkeys receiving 5 and 15 mg/kg doses. Mean APTT returned to baseline by 120 hours at the 5-mg/kg dose level and 192 hours at the 15-mg/kg dose level, consistent with both the in vitro APTT dose-response curve (data not shown) and the observed half-life of approximately 12 hours in monkeys (see Table B). The whole-blood activated clotting time (ACT) data mirrored the APTT data (data not shown). - There is an excellent correspondence between the
mean Apt39 concentration 24 hours post administration in theGroup 1a animals and the mean APTT of these animals. The mean aptamer concentration of the animals treated with 5 mg/kg at 24 hours was 15.9 μg/mL and the mean APTT was 61.1 seconds.TABLE B Group 1 Apt39 Plasma Levels (μg/mL) Group 1 Dose Levels (animals/dose level)Time Post 5 mg/ kg 15 mg/ kg 30 mg/kg Injection (hours) (n = 2)* (n = 4) (n = 2)* Pre-dose 0.2 <0.04 0.2 0.25 59.8 179.8 ± 28.9 465.5 3 66.6 145.6 ± 32.5 328.9 6 42.1 101.5 ± 13.4 275.3 24 15.9 51.1 ± 11.2 164.6
*ForDay 13 dosing, animals were split intoGroup 1a (5 mg/kg) and 1b (30 mg/kg). For these dose levels, the average plasma level for the two animals per dose level is reported. The Apt39 present inGroup 1a and 1b animals at the pre-dose time point is residual Apt39 from the 15-mg/kg dose atDay 4. The LLOQ of the assay is <0.04 μg/mL.
- In the
Group 2 animals treated with the antidote only, mean APTT and ACT were not affected by antidote administration at either dose level tested (30 and 60 mg/kg). Toxicokinetic data were collected at several time points over the first 24 hours after administration. As shown in Table C, low, but measurable levels of the antidote were present in plasma from animals receiving antidote at 0.25 hours after injection of 30 mg/kg onDay Day 13. The post-dosing level of the antidote was very low by comparison to the concentration of the aptamer (in Group 1) following IV injection.TABLE C Group 2 Apt7AD Plasma Levels (μg/mL) Time Post Apt39 Group 2 Dose Levels (4 animals/dose) Injection (hours) 30 mg/ kg 60 mg/kg Pre-dose <0.01 <0.01 3.25 0.4 ± 0.1 0.6 ± 0.5 6 0.02 ± 0.01* <0.02*** 24 0.01 ± 0.01** <0.01***
*1 animal at <LLOQ of 0.01 included in calculations
**3 animals at <LLOQ of 0.01 included in calculations
***Average of LLOQs
- The APTT data from animals treated with aptamer followed by
antidote 3 hours later (Group 3) are shown inFIG. 17 . In agreement with the data from animals treated with aptamer only, administration of aptamer at these dose levels resulted in a profound level of anticoagulation, with the mean APTTs at 0.25 and 3 hours post administration consistent with essentially complete FIX inhibition at both dose levels. Subsequent administration of Apt7AD rapidly and completely neutralized the anticoagulant effects of Apt39 in the monkey, with the mean APTT returning to baseline within 15 minutes following Apt7AD administration. In theGroup 3 animals treated with 30/60 mg/kg Apt39/Apt7AD, the APTT was followed for 5 days post aptamer administration. APTT data collected over this time frame indicate the anticoagulant effects of aptamer were durably neutralized, with no evidence of rebound anticoagulation over 120 hours, or approximately 10 half-lives of aptamer in the monkey (FIG. 17 ). - Toxicokinetic data were collected for 24 hours following Apt39 administration in the
Group 3 animals (Table D). ForGroup 3 animals, both free aptamer and complexed aptamer plasma concentrations were measured. Within 15 minutes of antidote administration, the mean concentration of free aptamer decreased 5,000-10,000 fold, to levels below the Lower Limit of Quantitation (LLOQ) of the assay employed. Concomitant with the decrease in free aptamer levels, the mean plasma concentration of complexed aptamer increased from below the LLOQ of the assay to ˜125 to 220 μg/mL at the 15/30 and 30/60 mg/kg dose levels respectively, indicating the rapid decrease in free Apt39 concentrations was due to binding of Apt7AD. The concentration of free aptamer remained below the LLOQ of the assay as long as 3 hours after antidote administration, consistent with the APTT results. At 21 hours after antidote administration, very low levels of Apt39 were detectable in several animals (mean of only 0.17 μg/mL or lower).TABLE D Group 3 Free and Complexed Apt389 Plasma Levels (μg/mL) Group 3 Dose LevelsTime Post Apt39 15/30 mg/kg Apt39 + Apt7AD 30/60 mg/kg Apt39 + Apt7AD Injection (hours) Free Apt39 Complexed Apt39 Free Apt39 Complexed Apt39 Pre-dose <0.04 ND 0.05 ± 0.01 ND 0.25 280.2 ± 64.3 ND 467.6 ± 67 ND 3.0 214.6 ± 31.8 <0.04 488.4 ± 68.6 <0.04 3.25 <0.04 125.1 ± 7.9 <0.04 218.2 ± 27.2 6 <0.04 98.7 ± 20.5 <0.04 184.8 ± 28.9 24 0.14 ± 0.08* 8.3 ± 4.5 <0.04 ± 0.01** 22.3 ± 12
*1 animal at <LLOQ of 0.04 μg/mL included in calculations
**3 animals at <LLOQ of 0.04 μg/mL included in calculations
Apt7AD administered at t = 3 hrs immediately after 3 hr blood draw.
(ND) Not determined.
- The invention has been described with reference to various specific and preferred embodiments and techniques. It should be understood that many variations and modifications may be made while remaining within the spirit and scope of the invention.
Claims (46)
1. A composition comprising a nucleic acid sequence of one of SEQ ID NOs:9-59 linked to a polyethylene glycol molecule at a 5′ terminus via a linker moiety, or a pharmaceutically acceptable salt thereof.
2. The composition of claim 1 of the formula:

wherein ligand is selected from one of SEQ ID NO:9-59 or a pharmaceutically acceptable salt thereof.
3. The composition of claim 1 wherein the composition is produced by conjugating a linker precursor to a nucleic acid molecule to form a nucleic acid linker-composition and further, conjugating a polyethylene glycol precursor moiety to the linker of the nucleic acid-linker composition.
4. The composition of claim 3 in which the linker precursor is selected from the group consisting of:






6-(trifluoroacetamido)hexanol (2-cyanoethyl-N,N-diisopropyl)phosphoramidite of the structure:
TFA-amino C4 CED phosphoramidite of the structure:
5′-amino modifier C3 TFA of the structure:
5′-Amino-Modifier C3-TFA
5′-amino modifier 5 of the structure:
5′-Amino Modifier 5,
5′-amino modifier C12 of the structure:
, and 5′thiol-modifier C6 of the structure:
5. The composition of claim 3 wherein the polyethylene glycol precursor is selected from:


6. The composition of claim 1 wherein the polyethylene glycol has a total mass of about 40 kD.
7. The composition of claim 1 wherein the composition comprises two polyethylene glycol moieties.
8. The composition of claim 7 wherein each moiety has a total mass of about 20 kD.
9. The composition of claim 1 wherein the nucleic acid comprises SEQ ID NO: 19.
10. The composition of claim 1 wherein the nucleic acid comprises SEQ ID NO: 39.
11. The composition of claim 1 comprising the structure:
5′-O-[6-[N2-(monomethoxy 20K polyethylene glycol carbamoyl)-N6-(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanyiyl-(3-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-riboguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-5′)-2′-methoxy-2′-deoxytidylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-3)-thymidine.
12. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
13. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
14. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
15. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
16. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
17. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
18. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
19. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
20. The composition of claim I of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
21. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
22. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
23. The composition of claim 1 of the formula:
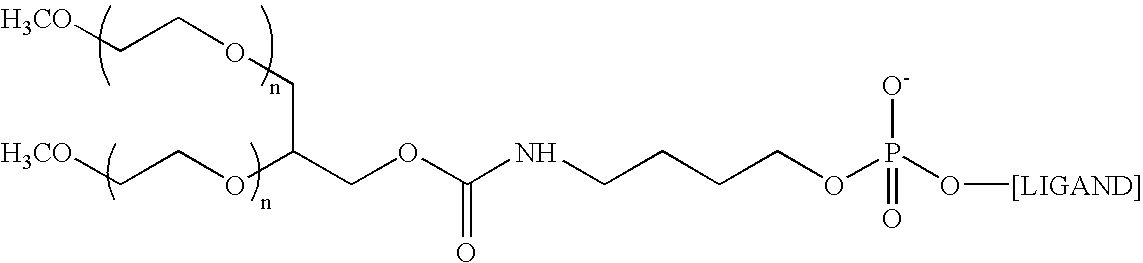
n=approximately 450
, or a pharmaceutically acceptable salt thereof.
24. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
25. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
26. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
27. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
28. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
29. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
30. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
31. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
32. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
33. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
34. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
35. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
36. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt thereof.
37. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
38. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
39. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
40. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
41. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof
42. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
43. The composition of claim 1 of the formula:

n=approximately 450
, or a pharmaceutically acceptable salt therof.
44. A method of regulating blood coagulation comprising contacting a FIXa molecule with a composition comprising a nucleic acid sequence of one of SEQ ID NOs:9-59 linked to a polyethylene glycol molecule at a 5′ terminus via a linker moiety, or a pharmaceutically acceptable salt thereof.
45. The method of claim 44 wherein the aptamer comprises structure:
5′-O-[6-[N2-(monomethoxy 20K polyethylene glycol carbamoyl)-N6-(monomethoxy 20K polyethylene glycol carbamoyl)]-lysylamido]hexyl]-2′-methoxy-2′-deoxguanylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxyguanylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-methoxy-2′-deoxyuridylyl-(3′-5′)-riboguanylyl-(3′-5′)-2′-methoxy-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxycytidylyl-(3′-5′)-2′-fluoro-2′-deoxyuridylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-5′)-2′-methoxy-2′-deoxytidylyl-(3′-5′)-2′-methoxy-2′-deoxyadenylyl-(3′-5′)-2′-methoxy-2′-deoxcytidylyl-(3′-3)-thymidine, or a pharmaceutically acceptable salt thereof.
46. The method of claim 44 further comprising contacting the composition with an antidote oligonucleotide.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/605,712 US20070105809A1 (en) | 2004-04-22 | 2006-11-27 | Modulators of coagulation factors with enhanced stability |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US56487304P | 2004-04-22 | 2004-04-22 | |
| US11/113,378 US7304041B2 (en) | 2004-04-22 | 2005-04-22 | Modulators of coagulation factors |
| US11/605,712 US20070105809A1 (en) | 2004-04-22 | 2006-11-27 | Modulators of coagulation factors with enhanced stability |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/113,378 Continuation-In-Part US7304041B2 (en) | 2004-04-22 | 2005-04-22 | Modulators of coagulation factors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20070105809A1 true US20070105809A1 (en) | 2007-05-10 |
Family
ID=35242272
Family Applications (7)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/113,378 Active US7304041B2 (en) | 2004-04-22 | 2005-04-22 | Modulators of coagulation factors |
| US11/546,190 Expired - Fee Related US7531524B2 (en) | 2004-04-22 | 2006-10-11 | Modulators of coagulation factors with enhanced stability |
| US11/605,712 Abandoned US20070105809A1 (en) | 2004-04-22 | 2006-11-27 | Modulators of coagulation factors with enhanced stability |
| US11/821,183 Active 2025-04-25 US7723315B2 (en) | 2004-04-22 | 2007-06-22 | Modulators of coagulation factors |
| US12/758,565 Expired - Fee Related US8389489B2 (en) | 2004-04-22 | 2010-04-12 | Modulators of coagulation factors |
| US13/751,623 Active US8859518B2 (en) | 2004-04-22 | 2013-01-28 | Modulators of coagulation factors |
| US14/508,609 Abandoned US20150247147A1 (en) | 2004-04-22 | 2014-10-07 | Modulators of coagulation factors |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/113,378 Active US7304041B2 (en) | 2004-04-22 | 2005-04-22 | Modulators of coagulation factors |
| US11/546,190 Expired - Fee Related US7531524B2 (en) | 2004-04-22 | 2006-10-11 | Modulators of coagulation factors with enhanced stability |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/821,183 Active 2025-04-25 US7723315B2 (en) | 2004-04-22 | 2007-06-22 | Modulators of coagulation factors |
| US12/758,565 Expired - Fee Related US8389489B2 (en) | 2004-04-22 | 2010-04-12 | Modulators of coagulation factors |
| US13/751,623 Active US8859518B2 (en) | 2004-04-22 | 2013-01-28 | Modulators of coagulation factors |
| US14/508,609 Abandoned US20150247147A1 (en) | 2004-04-22 | 2014-10-07 | Modulators of coagulation factors |
Country Status (16)
| Country | Link |
|---|---|
| US (7) | US7304041B2 (en) |
| EP (2) | EP2460811A1 (en) |
| JP (1) | JP4718541B2 (en) |
| KR (1) | KR101185050B1 (en) |
| CN (2) | CN1980947B (en) |
| AU (1) | AU2005238490B2 (en) |
| BR (1) | BRPI0508931A (en) |
| CA (1) | CA2563176A1 (en) |
| DK (1) | DK1745062T3 (en) |
| ES (1) | ES2494940T3 (en) |
| IL (1) | IL178564A0 (en) |
| PL (1) | PL1745062T3 (en) |
| PT (1) | PT1745062E (en) |
| SG (1) | SG152262A1 (en) |
| SI (1) | SI1745062T1 (en) |
| WO (1) | WO2005106042A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110190192A1 (en) * | 2009-12-15 | 2011-08-04 | Cebix Inc. | Methods for treating erectile dysfunction in patients with insulin-dependent diabetes |
| US20120178676A1 (en) * | 2010-05-17 | 2012-07-12 | Cebix, Inc. | Pegylated c-peptide |
| WO2012149198A3 (en) * | 2011-04-26 | 2013-03-14 | Regado Biosciences, Inc. | A method for manufacturing pegylated oligonucleotides |
| US20140179621A1 (en) * | 2009-05-01 | 2014-06-26 | Ophthotech Corporation | Methods for Treating or Preventing Ophthalmological Diseases |
| US8962553B2 (en) | 2011-11-17 | 2015-02-24 | Cebix Ab | Method of treating a diabetic subject having a microvascular impairment disorder by a pegylated C-peptide |
| US11273171B2 (en) | 2013-07-12 | 2022-03-15 | Iveric Bio, Inc. | Methods for treating or preventing ophthalmological conditions |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2351855A1 (en) * | 2000-09-26 | 2011-08-03 | Duke University | RNA aptamers and methods for identifying the same |
| PT1401853E (en) * | 2001-05-25 | 2010-12-07 | Univ Duke | Modulators of pharmacological agents |
| SI1745062T1 (en) * | 2004-04-22 | 2014-09-30 | Regado Biosciences, Inc. | Improved modulators of coagulation factors |
| JP4833727B2 (en) * | 2006-05-08 | 2011-12-07 | 藤森工業株式会社 | Thrombus monitoring method and thrombus monitoring apparatus using vascular endothelial cells |
| AU2007267801A1 (en) * | 2006-05-26 | 2007-12-06 | Regado Biosciences, Inc. | Administration of the REG1 anticoagulation system |
| EP2139992A4 (en) * | 2007-03-30 | 2011-08-10 | Univ Duke | A method of modulating the activity of a nucleic acid molecule |
| US20090163437A1 (en) * | 2007-10-16 | 2009-06-25 | Regado Biosciences, Inc. | Steady-state subcutaneous administration of aptamers |
| WO2010045509A2 (en) | 2008-10-15 | 2010-04-22 | Isis Pharmaceuticals, Inc. | Modulation of factor 11 expression |
| EP2368124A4 (en) * | 2008-11-24 | 2012-09-19 | Bayer Healthcare Llc | Method of determining pegylated blood coagulation factor activity in a silica-based activated partial thromboplastin time assay |
| FR2942231B1 (en) * | 2009-02-19 | 2015-03-20 | Lfb Biotechnologies | NUCLEIC ACIDS BINDING SPECIFICALLY TO HUMAN VII / VIIA FACTOR, AND USES THEREOF |
| WO2010121074A1 (en) * | 2009-04-15 | 2010-10-21 | Isis Pharmaceuticals, Inc. | Modulation of inflammatory responses by factor xi |
| US8889645B2 (en) | 2009-06-03 | 2014-11-18 | Regado Biosciences, Inc. | Nucleic acid modulators of glycoprotein VI |
| US8889646B2 (en) | 2009-06-03 | 2014-11-18 | Regado Biosciences, Inc. | Nucleic acid modulators of glycoprotein VI |
| KR20120055529A (en) * | 2009-06-03 | 2012-05-31 | 레가도 바이오사이언스, 인코포레이티드 | Nucleic acid modulators of glycoprotein vi |
| CN102639142A (en) | 2009-09-16 | 2012-08-15 | 杜克大学 | Inhibition of endosomal toll-like receptor activation |
| WO2011059459A1 (en) * | 2009-11-16 | 2011-05-19 | Regado Biosciences, Inc | Physical characterization of oligonucleotides conjugates |
| US20110117660A1 (en) * | 2009-11-16 | 2011-05-19 | Brooks Douglas G | Physical characterization of oligonucleotide conjugates |
| EA201690861A1 (en) * | 2012-10-24 | 2016-08-31 | ОБЩЕСТВО С ОГРАНИЧЕННОЙ ОТВЕТСТВЕННОСТЬЮ "НоваМедика" | NUCLEIN-ACID MODULATORS OF PROTEIN ALPHA2BETA1 |
| FR3011250A1 (en) * | 2013-09-30 | 2015-04-03 | Lab Francais Du Fractionnement | NUCLEIC ACIDS BINDING SPECIFICALLY TO HUMAN IX / IXA FACTOR, AND USES |
| US9545383B2 (en) | 2014-04-01 | 2017-01-17 | Massachusetts Institute Of Technology | Blood clotting control |
| US10066323B2 (en) | 2014-04-16 | 2018-09-04 | Duke University | Electrospun cationic nanofibers and methods of making and using the same |
| WO2016176426A1 (en) | 2015-04-28 | 2016-11-03 | Duke University | Thrombus imaging aptamers and methods of using same |
| CN108463244B (en) | 2015-08-04 | 2022-05-27 | 杜克大学 | Gene-encoded intrinsically disordered stealth polymers for delivery and methods of use thereof |
| US11752213B2 (en) | 2015-12-21 | 2023-09-12 | Duke University | Surfaces having reduced non-specific binding and antigenicity |
| US11467156B2 (en) | 2016-06-01 | 2022-10-11 | Duke University | Nonfouling biosensors |
| CN109982707A (en) | 2016-09-16 | 2019-07-05 | 杜克大学 | VWF ELISA (VWF) targeting agent and its application method |
| JP7288852B2 (en) | 2016-11-23 | 2023-06-08 | アルニラム・ファーマシューティカルズ・インコーポレーテッド | Modified RNA agents with reduced off-target effects |
| WO2018132732A1 (en) | 2017-01-12 | 2018-07-19 | Duke University | Genetically encoded lipid-polypeptide hybrid biomaterials that exhibit temperature triggered hierarchical self-assembly |
| US11554097B2 (en) | 2017-05-15 | 2023-01-17 | Duke University | Recombinant production of hybrid lipid-biopolymer materials that self-assemble and encapsulate agents |
| EP3658168A4 (en) | 2017-06-30 | 2021-07-14 | Duke University | Order and disorder as a design principle for stimuli-responsive biopolymer networks |
| US20210238594A1 (en) * | 2018-05-07 | 2021-08-05 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for improving strand biased |
| JP7438135B2 (en) | 2018-05-09 | 2024-02-26 | アイオーニス ファーマシューティカルズ, インコーポレーテッド | Compounds and methods for reducing expression of FXI |
| EP3829622A4 (en) | 2018-08-02 | 2022-05-11 | Duke University | Dual agonist fusion proteins |
| CN111019100B (en) * | 2018-10-10 | 2022-05-03 | 中国石油化工股份有限公司 | Montmorillonite-modified barrier polyester and preparation method thereof |
| US20220025373A1 (en) * | 2018-10-26 | 2022-01-27 | North Carolina State University | Compositions and methods related to nucleic acid anticoagulants |
| WO2020203626A1 (en) * | 2019-03-29 | 2020-10-08 | 日油株式会社 | Branched degradable polyethylene glycol binder |
| CN110305868B (en) * | 2019-07-03 | 2022-08-02 | 合肥工业大学 | Thrombin circular nucleic acid aptamer and application thereof |
| US11512314B2 (en) | 2019-07-12 | 2022-11-29 | Duke University | Amphiphilic polynucleotides |
| US20230012024A1 (en) * | 2019-12-04 | 2023-01-12 | Duke University | Methods for controlling extracorporeal membrane oxygenation (ecmo) coagulation |
| WO2022178438A1 (en) | 2021-02-22 | 2022-08-25 | Duke University | Non-immunogenic poegma-aptamer conjugates |
Citations (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4415732A (en) * | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4458066A (en) * | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4500707A (en) * | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US4668777A (en) * | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4973649A (en) * | 1988-03-03 | 1990-11-27 | Rhone-Poulenc Chimie | Novel hindered diamine/imido prepolymers |
| US5153319A (en) * | 1986-03-31 | 1992-10-06 | University Patents, Inc. | Process for preparing polynucleotides |
| US5180818A (en) * | 1990-03-21 | 1993-01-19 | The University Of Colorado Foundation, Inc. | Site specific cleavage of single-stranded dna |
| US5245022A (en) * | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5270163A (en) * | 1990-06-11 | 1993-12-14 | University Research Corporation | Methods for identifying nucleic acid ligands |
| US5278302A (en) * | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5475096A (en) * | 1990-06-11 | 1995-12-12 | University Research Corporation | Nucleic acid ligands |
| US5476766A (en) * | 1990-06-11 | 1995-12-19 | Nexstar Pharmaceuticals, Inc. | Ligands of thrombin |
| US5532130A (en) * | 1993-07-20 | 1996-07-02 | Dyad Pharmaceutical Corporation | Methods and compositions for sequence-specific hybridization of RNA by 2'-5' oligonucleotides |
| US5534408A (en) * | 1993-09-24 | 1996-07-09 | University Of Massachusetts Medical Center | 2-deoxystreptamine aminoglycoside inhibition of HIV RRE/Rev binding |
| US5543293A (en) * | 1990-06-11 | 1996-08-06 | Nexstar Pharmaceuticals, Inc. | DNA ligands of thrombin |
| US5552391A (en) * | 1990-01-16 | 1996-09-03 | La Jolla Pharmaceutical Company | Chemically-defined non-polymeric valency platform molecules and conjugates thereof |
| US5582981A (en) * | 1991-08-14 | 1996-12-10 | Gilead Sciences, Inc. | Method for identifying an oligonucleotide aptamer specific for a target |
| US5594121A (en) * | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5595877A (en) * | 1990-06-11 | 1997-01-21 | Nexstar Pharmaceuticals, Inc. | Methods of producing nucleic acid ligands |
| US5602244A (en) * | 1988-05-26 | 1997-02-11 | Competitive Technologies, Inc. | Polynucleotide phosphorodithioate compounds |
| US5645985A (en) * | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5648214A (en) * | 1990-06-11 | 1997-07-15 | University Research Corporation | High-affinity oligonucleotide ligands to the tachykinin substance P |
| US5660985A (en) * | 1990-06-11 | 1997-08-26 | Nexstar Pharmaceuticals, Inc. | High affinity nucleic acid ligands containing modified nucleotides |
| US5670633A (en) * | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5681702A (en) * | 1994-08-30 | 1997-10-28 | Chiron Corporation | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| US5686242A (en) * | 1991-09-05 | 1997-11-11 | Isis Pharmaceuticals, Inc. | Determination of oligonucleotides for therapeutics, diagnostics and research reagents |
| US5723323A (en) * | 1985-03-30 | 1998-03-03 | Kauffman; Stuart Alan | Method of identifying a stochastically-generated peptide, polypeptide, or protein having ligand binding property and compositions thereof |
| US5750729A (en) * | 1995-02-01 | 1998-05-12 | Gilead Sciences,Inc. | Compounds and methods for making and using same |
| US5756291A (en) * | 1992-08-21 | 1998-05-26 | Gilead Sciences, Inc. | Aptamers specific for biomolecules and methods of making |
| US5760202A (en) * | 1995-03-06 | 1998-06-02 | Isis Pharmaceuticals, Inc. | Process for the synthesis of 2'-O-substituted pyrimidines |
| US5780449A (en) * | 1995-11-24 | 1998-07-14 | Crinos Industria Farmacobiologica S.P.A. | Cathepsin G-inhibiting aptamers |
| US5780221A (en) * | 1995-05-03 | 1998-07-14 | Whitehead Institute For Biomedical Research | Identification of enantiomeric ligands |
| US5792613A (en) * | 1996-06-12 | 1998-08-11 | The Curators Of The University Of Missouri | Method for obtaining RNA aptamers based on shape selection |
| US5792608A (en) * | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5807718A (en) * | 1994-12-02 | 1998-09-15 | The Scripps Research Institute | Enzymatic DNA molecules |
| US5817781A (en) * | 1992-06-01 | 1998-10-06 | Gilead Sciences, Inc. | Modified internucleoside linkages (II) |
| US5830653A (en) * | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5839443A (en) * | 1996-05-16 | 1998-11-24 | The Trustees Of Columbia University In The City Of New York | Method for inhibiting thrombosis in a patient whose blood is subjected to extracorporeal circulation |
| US5840867A (en) * | 1991-02-21 | 1998-11-24 | Gilead Sciences, Inc. | Aptamer analogs specific for biomolecules |
| US5859221A (en) * | 1990-01-11 | 1999-01-12 | Isis Pharmaceuticals, Inc. | 2'-modified oligonucleotides |
| US5861254A (en) * | 1997-01-31 | 1999-01-19 | Nexstar Pharmaceuticals, Inc. | Flow cell SELEX |
| US5861501A (en) * | 1995-06-07 | 1999-01-19 | Merck & Co., Inc. | Capped synthetic RNA, analogs, and aptamers |
| US5872232A (en) * | 1990-01-11 | 1999-02-16 | Isis Pharmaceuticals Inc. | 2'-O-modified oligonucleotides |
| US5879917A (en) * | 1994-05-04 | 1999-03-09 | Massachusetts Institute Of Technology | Programmable genotoxic agents and uses therefor |
| US5882870A (en) * | 1998-01-14 | 1999-03-16 | Becton Dickinson And Company | Rapidly reversed thrombin binding oligonucleotide anticoagulants |
| US5891689A (en) * | 1994-04-12 | 1999-04-06 | Innovir Laboratories, Inc. | Heme-bearing microparticles for targeted delivery of drugs |
| US5935776A (en) * | 1992-10-23 | 1999-08-10 | University Of Massachusetts Medical Center | Small molecule inhibition of RNA/ligand binding |
| US5989823A (en) * | 1998-09-18 | 1999-11-23 | Nexstar Pharmaceuticals, Inc. | Homogeneous detection of a target through nucleic acid ligand-ligand beacon interaction |
| US6001820A (en) * | 1995-03-31 | 1999-12-14 | Hamilton Civic Hospitals Research Development Inc. | Compositions and methods for inhibiting thrombogenesis |
| US6004746A (en) * | 1994-07-20 | 1999-12-21 | The General Hospital Corporation | Interaction trap systems for detecting protein interactions |
| US6005087A (en) * | 1995-06-06 | 1999-12-21 | Isis Pharmaceuticals, Inc. | 2'-modified oligonucleotides |
| US6011020A (en) * | 1990-06-11 | 2000-01-04 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US6051388A (en) * | 1998-12-22 | 2000-04-18 | Toxin Alert, Inc. | Method and apparatus for selective biological material detection |
| US6054274A (en) * | 1997-11-12 | 2000-04-25 | Hewlett-Packard Company | Method of amplifying the signal of target nucleic acid sequence analyte |
| US6060056A (en) * | 1991-02-08 | 2000-05-09 | La Jolla Pharmaceutical Company | Composition for inducing humoral anergy to an immunogen comprising a T cell epitope-deficient analog of the immunogen conjugated to a nonimmunogenic valency platform molecule |
| US6083696A (en) * | 1990-06-11 | 2000-07-04 | Nexstar Pharmaceuticals, Inc. | Systematic evolution of ligands exponential enrichment: blended selex |
| US6093555A (en) * | 1993-10-27 | 2000-07-25 | Ribozyme Pharmaceuticals, Inc. | Amido and peptido modified enzymatic nucleic acid molecules |
| US6110462A (en) * | 1999-03-03 | 2000-08-29 | The Scripps Research Institute | Enzymatic DNA molecules that contain modified nucleotides |
| US6110721A (en) * | 1993-11-12 | 2000-08-29 | Gilead Sciences, Inc. | Polypeptides and coagulation therapy |
| US6114038A (en) * | 1998-11-10 | 2000-09-05 | Biocrystal Ltd. | Functionalized nanocrystals and their use in detection systems |
| US6117557A (en) * | 1995-04-19 | 2000-09-12 | Lexmark International, Inc. | Caprolactone ester polyurethane developer roller |
| US6120997A (en) * | 1997-01-13 | 2000-09-19 | The Scripps Research Institute | Nucleic acid binders having an hydroxyamine motif |
| US6127119A (en) * | 1990-06-11 | 2000-10-03 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands of tissue target |
| US6127173A (en) * | 1997-09-22 | 2000-10-03 | Ribozyme Pharmaceuticals, Inc. | Nucleic acid catalysts with endonuclease activity |
| US6136545A (en) * | 1996-09-13 | 2000-10-24 | Roche Diagnostics Gmbh | Homogeneous detection methods for the determination of subpopulations of an analyte |
| US6147204A (en) * | 1990-06-11 | 2000-11-14 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US6150461A (en) * | 1997-05-27 | 2000-11-21 | Hisamitsu Pharmaceutical Co., Inc. | Carriers targettable to organ |
| US6153410A (en) * | 1997-03-25 | 2000-11-28 | California Institute Of Technology | Recombination of polynucleotide sequences using random or defined primers |
| US6153737A (en) * | 1990-01-11 | 2000-11-28 | Isis Pharmaceuticals, Inc. | Derivatized oligonucleotides having improved uptake and other properties |
| US6163714A (en) * | 1998-07-03 | 2000-12-19 | Torsana Diabetes Diagnostics A/S | Optical sensor for in situ measurement of analytes |
| US6171795B1 (en) * | 1999-07-29 | 2001-01-09 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands to CD40ligand |
| US6177557B1 (en) * | 1990-06-11 | 2001-01-23 | Nexstar Pharmaceuticals, Inc. | High affinity ligands of basic fibroblast growth factor and thrombin |
| US6180348B1 (en) * | 1998-04-20 | 2001-01-30 | Weihua Li | Method of isolating target specific oligonucleotide ligands |
| US6183967B1 (en) * | 1995-06-07 | 2001-02-06 | Nexstar Pharmaceuticals | Nucleic acid ligand inhibitors to DNA polymerases |
| US6222025B1 (en) * | 1995-03-06 | 2001-04-24 | Isis Pharmaceuticals, Inc. | Process for the synthesis of 2′-O-substituted pyrimidines and oligomeric compounds therefrom |
| US6258601B1 (en) * | 2000-09-07 | 2001-07-10 | Isis Pharmaceuticals, Inc. | Antisense modulation of ubiquitin protein ligase expression |
| US6316198B1 (en) * | 1999-03-18 | 2001-11-13 | Exiqon A/S | Detection of mutations in genes by specific LNA primers |
| US6316403B1 (en) * | 1996-09-27 | 2001-11-13 | The Trustees Of Columbia University In The City Of New York | Methods for treating an ischemic disorder and improving stroke outcome |
| US6315995B1 (en) * | 1996-09-27 | 2001-11-13 | The Trustees Of Columbia University In The City Of New York | Methods for treating an ischemic disorder and improving stroke outcome |
| US20030083294A1 (en) * | 2001-05-25 | 2003-05-01 | Sullenger Bruce A. | Modulators of pharmacological agents |
| US20030175703A1 (en) * | 2000-09-26 | 2003-09-18 | Sullenger Bruce A. | RNA aptamers and methods for identifying the same |
| US20060004088A1 (en) * | 2002-10-22 | 2006-01-05 | Oct Inc. | Furan derivatives for preventing and curing osteoporosis and pharmaceutical compositions containing the same |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| IE920561A1 (en) | 1991-02-21 | 1992-08-26 | Gilead Sciences | Aptamer specific for thrombin and methods of use |
| AU5162993A (en) | 1992-09-23 | 1994-04-12 | Integrated Dna Technologies, Inc. | Siloxy oligonucleotide analogs |
| DE59306170D1 (en) | 1992-09-24 | 1997-05-22 | Hoechst Ag | Oligoribonucleotide and ribozyme analogs with terminal 3'-3 'and 5'-5' linkages |
| CA2145761C (en) | 1992-09-29 | 2009-12-22 | Larry M. Gold | Nucleic acid ligands and methods for producing the same |
| JP3706186B2 (en) * | 1995-12-25 | 2005-10-12 | 日本トムソン株式会社 | Linear motion rolling guide unit device |
| AU2994997A (en) | 1996-05-07 | 1997-11-26 | T Cell Sciences, Inc. | Aptamers for complement protein c3b |
| US5779917A (en) * | 1996-08-09 | 1998-07-14 | Fluid Technologies, Inc. | Process for separating fluids having different densities |
| JP3143740B2 (en) | 1997-10-29 | 2001-03-07 | 工業技術院長 | RNA with high affinity for HIV Tat protein |
| AU2018999A (en) | 1997-12-31 | 1999-07-19 | Medical College Of Georgia Research Institute, Inc. | Oligomers that bind to ku protein |
| AU3363399A (en) | 1998-03-27 | 1999-10-18 | President And Fellows Of Harvard College | Aptamer based bacterial inhibition systems (abbis) |
| US6163174A (en) * | 1998-05-26 | 2000-12-19 | The University Of Rochester | Digital buffer circuits |
| EP1026243B1 (en) | 1998-08-14 | 2006-02-01 | Japan Science and Technology Agency | NUCLEIC ACID CAPABLE OF BINDING SPECIFICALLY TO Ras TARGET PROTEIN |
| AU2342900A (en) * | 1998-09-23 | 2000-05-01 | Cleveland Clinic Foundation, The | Novel interferon stimulated and repressed genes |
| AU1201600A (en) | 1998-10-08 | 2000-04-26 | University Of Massachusetts | Controlling gene expression in living cells |
| AU6298899A (en) | 1998-10-09 | 2000-05-01 | Ingene, Inc. | Production of ssdna (in vivo) |
| WO2000024912A1 (en) | 1998-10-23 | 2000-05-04 | The Children's Medical Center Corporation | Use of a self-cleaving rna motif to modulate gene expression |
| DE19901008A1 (en) | 1999-01-13 | 2000-07-20 | Deutsches Krebsforsch | Peptides to inhibit HPV E6 proteins |
| DE19901009A1 (en) | 1999-01-13 | 2000-07-20 | Deutsches Krebsforsch | Peptides to inhibit HBV core proteins |
| US6329145B1 (en) * | 1999-02-09 | 2001-12-11 | Gilead Science, Inc. | Determining non-nucleic acid molecule binding to target by competition with nucleic acid ligand |
| CA2483910A1 (en) | 2002-05-01 | 2003-11-13 | Schering Aktiengesellschaft | Novel tissue factor targeted antibodies as anticoagulants |
| JP2005533862A (en) | 2002-07-25 | 2005-11-10 | アーケミックス コーポレイション | Modulated aptamer therapeutics |
| CN1703395A (en) | 2002-08-09 | 2005-11-30 | 特兰斯泰克制药公司 | Aryl and heteroaryl compounds and methods to modulate coagulation |
| JP2006516151A (en) | 2002-11-21 | 2006-06-22 | アーケミックス コーポレイション | Multivalent aptamer therapeutics with improved pharmacodynamic properties and methods for their preparation and use |
| US20040197804A1 (en) | 2002-12-03 | 2004-10-07 | Keefe Anthony D. | Method for in vitro selection of 2'-substituted nucleic acids |
| US20050037394A1 (en) | 2002-12-03 | 2005-02-17 | Keefe Anthony D. | Method for in vitro selection of 2'-substituted nucleic acids |
| EP1660439A2 (en) | 2003-08-08 | 2006-05-31 | Transtech Pharma, Inc. | Aryl and heteroaryl compounds, compositions, and methods of use |
| WO2005052121A2 (en) | 2003-11-21 | 2005-06-09 | Archemix Corp. | Multivalent aptamers |
| WO2005084412A2 (en) | 2004-03-05 | 2005-09-15 | Archemix Corp. | Controlled modulation of the pharmacokinetics and biodistribution of aptamer therapeutics |
| WO2005111238A2 (en) | 2004-04-19 | 2005-11-24 | Archemix Corporation | Aptamer-mediated intracellular delivery of therapeutic oligonucleotides |
| SI1745062T1 (en) * | 2004-04-22 | 2014-09-30 | Regado Biosciences, Inc. | Improved modulators of coagulation factors |
| CA2579374A1 (en) | 2004-09-07 | 2006-03-30 | Archemix Corp. | Aptamers to von willebrand factor and their use as thrombotic disease therapeutics |
| US20070066551A1 (en) | 2004-09-07 | 2007-03-22 | Keefe Anthony D | Aptamer medicinal chemistry |
-
2005
- 2005-04-22 SI SI200531879T patent/SI1745062T1/en unknown
- 2005-04-22 JP JP2007509694A patent/JP4718541B2/en not_active Expired - Fee Related
- 2005-04-22 US US11/113,378 patent/US7304041B2/en active Active
- 2005-04-22 KR KR1020067024518A patent/KR101185050B1/en not_active IP Right Cessation
- 2005-04-22 AU AU2005238490A patent/AU2005238490B2/en not_active Ceased
- 2005-04-22 PL PL05740895T patent/PL1745062T3/en unknown
- 2005-04-22 CN CN2005800199137A patent/CN1980947B/en not_active Expired - Fee Related
- 2005-04-22 PT PT57408957T patent/PT1745062E/en unknown
- 2005-04-22 DK DK05740895.7T patent/DK1745062T3/en active
- 2005-04-22 EP EP11192967A patent/EP2460811A1/en not_active Withdrawn
- 2005-04-22 SG SG200902764-0A patent/SG152262A1/en unknown
- 2005-04-22 WO PCT/US2005/013926 patent/WO2005106042A2/en active Application Filing
- 2005-04-22 CN CN201210545424.7A patent/CN103045601B/en not_active Expired - Fee Related
- 2005-04-22 CA CA002563176A patent/CA2563176A1/en active Pending
- 2005-04-22 EP EP05740895.7A patent/EP1745062B1/en not_active Not-in-force
- 2005-04-22 ES ES05740895.7T patent/ES2494940T3/en active Active
- 2005-04-22 BR BRPI0508931-0A patent/BRPI0508931A/en active Search and Examination
-
2006
- 2006-10-11 US US11/546,190 patent/US7531524B2/en not_active Expired - Fee Related
- 2006-10-15 IL IL178564A patent/IL178564A0/en unknown
- 2006-11-27 US US11/605,712 patent/US20070105809A1/en not_active Abandoned
-
2007
- 2007-06-22 US US11/821,183 patent/US7723315B2/en active Active
-
2010
- 2010-04-12 US US12/758,565 patent/US8389489B2/en not_active Expired - Fee Related
-
2013
- 2013-01-28 US US13/751,623 patent/US8859518B2/en active Active
-
2014
- 2014-10-07 US US14/508,609 patent/US20150247147A1/en not_active Abandoned
Patent Citations (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4458066A (en) * | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4500707A (en) * | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US4668777A (en) * | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4415732A (en) * | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US5723323A (en) * | 1985-03-30 | 1998-03-03 | Kauffman; Stuart Alan | Method of identifying a stochastically-generated peptide, polypeptide, or protein having ligand binding property and compositions thereof |
| US5814476A (en) * | 1985-03-30 | 1998-09-29 | Stuart Kauffman | Process for the production of stochastically-generated transcription or translation products |
| US5153319A (en) * | 1986-03-31 | 1992-10-06 | University Patents, Inc. | Process for preparing polynucleotides |
| US4973649A (en) * | 1988-03-03 | 1990-11-27 | Rhone-Poulenc Chimie | Novel hindered diamine/imido prepolymers |
| US5453496A (en) * | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US5602244A (en) * | 1988-05-26 | 1997-02-11 | Competitive Technologies, Inc. | Polynucleotide phosphorodithioate compounds |
| US5278302A (en) * | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5859221A (en) * | 1990-01-11 | 1999-01-12 | Isis Pharmaceuticals, Inc. | 2'-modified oligonucleotides |
| US6531584B1 (en) * | 1990-01-11 | 2003-03-11 | Isis Pharmaceuticals, Inc. | 2'modified oligonucleotides |
| US5872232A (en) * | 1990-01-11 | 1999-02-16 | Isis Pharmaceuticals Inc. | 2'-O-modified oligonucleotides |
| US6153737A (en) * | 1990-01-11 | 2000-11-28 | Isis Pharmaceuticals, Inc. | Derivatized oligonucleotides having improved uptake and other properties |
| US5670633A (en) * | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5552391A (en) * | 1990-01-16 | 1996-09-03 | La Jolla Pharmaceutical Company | Chemically-defined non-polymeric valency platform molecules and conjugates thereof |
| US5606047A (en) * | 1990-01-16 | 1997-02-25 | La Jolla Pharmaceutical Company | Chemically-defined non-polymeric valency platform molecules and conjugates thereof |
| US5180818A (en) * | 1990-03-21 | 1993-01-19 | The University Of Colorado Foundation, Inc. | Site specific cleavage of single-stranded dna |
| US5817785A (en) * | 1990-06-11 | 1998-10-06 | Nexstar Pharmaceuticals, Inc. | Methods of producing nucleic acid ligands |
| US5958691A (en) * | 1990-06-11 | 1999-09-28 | Nexstar Pharmaceuticals, Inc. | High affinity nucleic acid ligands containing modified nucleotides |
| US6127119A (en) * | 1990-06-11 | 2000-10-03 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands of tissue target |
| US6331398B1 (en) * | 1990-06-11 | 2001-12-18 | Gilead Sciences, Inc. | Nucleic acid ligands |
| US5843653A (en) * | 1990-06-11 | 1998-12-01 | Nexstar Pharmaceuticals, Inc. | Method for detecting a target molecule in a sample using a nucleic acid ligand |
| US5660985A (en) * | 1990-06-11 | 1997-08-26 | Nexstar Pharmaceuticals, Inc. | High affinity nucleic acid ligands containing modified nucleotides |
| US5543293A (en) * | 1990-06-11 | 1996-08-06 | Nexstar Pharmaceuticals, Inc. | DNA ligands of thrombin |
| US5670637A (en) * | 1990-06-11 | 1997-09-23 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands |
| US6147204A (en) * | 1990-06-11 | 2000-11-14 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US5595877A (en) * | 1990-06-11 | 1997-01-21 | Nexstar Pharmaceuticals, Inc. | Methods of producing nucleic acid ligands |
| US5696249A (en) * | 1990-06-11 | 1997-12-09 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands |
| US5270163A (en) * | 1990-06-11 | 1993-12-14 | University Research Corporation | Methods for identifying nucleic acid ligands |
| US6083696A (en) * | 1990-06-11 | 2000-07-04 | Nexstar Pharmaceuticals, Inc. | Systematic evolution of ligands exponential enrichment: blended selex |
| US6110900A (en) * | 1990-06-11 | 2000-08-29 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands |
| US5476766A (en) * | 1990-06-11 | 1995-12-19 | Nexstar Pharmaceuticals, Inc. | Ligands of thrombin |
| US6177557B1 (en) * | 1990-06-11 | 2001-01-23 | Nexstar Pharmaceuticals, Inc. | High affinity ligands of basic fibroblast growth factor and thrombin |
| US5475096A (en) * | 1990-06-11 | 1995-12-12 | University Research Corporation | Nucleic acid ligands |
| US5648214A (en) * | 1990-06-11 | 1997-07-15 | University Research Corporation | High-affinity oligonucleotide ligands to the tachykinin substance P |
| US6011020A (en) * | 1990-06-11 | 2000-01-04 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US5245022A (en) * | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US6060056A (en) * | 1991-02-08 | 2000-05-09 | La Jolla Pharmaceutical Company | Composition for inducing humoral anergy to an immunogen comprising a T cell epitope-deficient analog of the immunogen conjugated to a nonimmunogenic valency platform molecule |
| US5840867A (en) * | 1991-02-21 | 1998-11-24 | Gilead Sciences, Inc. | Aptamer analogs specific for biomolecules |
| US5582981A (en) * | 1991-08-14 | 1996-12-10 | Gilead Sciences, Inc. | Method for identifying an oligonucleotide aptamer specific for a target |
| US5686242A (en) * | 1991-09-05 | 1997-11-11 | Isis Pharmaceuticals, Inc. | Determination of oligonucleotides for therapeutics, diagnostics and research reagents |
| US5594121A (en) * | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5830653A (en) * | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5645985A (en) * | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5792608A (en) * | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5817781A (en) * | 1992-06-01 | 1998-10-06 | Gilead Sciences, Inc. | Modified internucleoside linkages (II) |
| US5756291A (en) * | 1992-08-21 | 1998-05-26 | Gilead Sciences, Inc. | Aptamers specific for biomolecules and methods of making |
| US5935776A (en) * | 1992-10-23 | 1999-08-10 | University Of Massachusetts Medical Center | Small molecule inhibition of RNA/ligand binding |
| US5532130A (en) * | 1993-07-20 | 1996-07-02 | Dyad Pharmaceutical Corporation | Methods and compositions for sequence-specific hybridization of RNA by 2'-5' oligonucleotides |
| US5534408A (en) * | 1993-09-24 | 1996-07-09 | University Of Massachusetts Medical Center | 2-deoxystreptamine aminoglycoside inhibition of HIV RRE/Rev binding |
| US6093555A (en) * | 1993-10-27 | 2000-07-25 | Ribozyme Pharmaceuticals, Inc. | Amido and peptido modified enzymatic nucleic acid molecules |
| US6110721A (en) * | 1993-11-12 | 2000-08-29 | Gilead Sciences, Inc. | Polypeptides and coagulation therapy |
| US5891689A (en) * | 1994-04-12 | 1999-04-06 | Innovir Laboratories, Inc. | Heme-bearing microparticles for targeted delivery of drugs |
| US5879917A (en) * | 1994-05-04 | 1999-03-09 | Massachusetts Institute Of Technology | Programmable genotoxic agents and uses therefor |
| US5882941A (en) * | 1994-05-04 | 1999-03-16 | Massachusette Institute Of Technology | Programmable genotoxic agents and uses therefor |
| US6004746A (en) * | 1994-07-20 | 1999-12-21 | The General Hospital Corporation | Interaction trap systems for detecting protein interactions |
| US5780610A (en) * | 1994-08-30 | 1998-07-14 | Collins; Mark L. | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| US5681702A (en) * | 1994-08-30 | 1997-10-28 | Chiron Corporation | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| US5807718A (en) * | 1994-12-02 | 1998-09-15 | The Scripps Research Institute | Enzymatic DNA molecules |
| US5750729A (en) * | 1995-02-01 | 1998-05-12 | Gilead Sciences,Inc. | Compounds and methods for making and using same |
| US5760202A (en) * | 1995-03-06 | 1998-06-02 | Isis Pharmaceuticals, Inc. | Process for the synthesis of 2'-O-substituted pyrimidines |
| US6222025B1 (en) * | 1995-03-06 | 2001-04-24 | Isis Pharmaceuticals, Inc. | Process for the synthesis of 2′-O-substituted pyrimidines and oligomeric compounds therefrom |
| US6001820A (en) * | 1995-03-31 | 1999-12-14 | Hamilton Civic Hospitals Research Development Inc. | Compositions and methods for inhibiting thrombogenesis |
| US6117557A (en) * | 1995-04-19 | 2000-09-12 | Lexmark International, Inc. | Caprolactone ester polyurethane developer roller |
| US5780221A (en) * | 1995-05-03 | 1998-07-14 | Whitehead Institute For Biomedical Research | Identification of enantiomeric ligands |
| US6005087A (en) * | 1995-06-06 | 1999-12-21 | Isis Pharmaceuticals, Inc. | 2'-modified oligonucleotides |
| US5861501A (en) * | 1995-06-07 | 1999-01-19 | Merck & Co., Inc. | Capped synthetic RNA, analogs, and aptamers |
| US6183967B1 (en) * | 1995-06-07 | 2001-02-06 | Nexstar Pharmaceuticals | Nucleic acid ligand inhibitors to DNA polymerases |
| US6111095A (en) * | 1995-06-07 | 2000-08-29 | Merck & Co., Inc. | Capped synthetic RNA, analogs, and aptamers |
| US5780449A (en) * | 1995-11-24 | 1998-07-14 | Crinos Industria Farmacobiologica S.P.A. | Cathepsin G-inhibiting aptamers |
| US6391300B1 (en) * | 1996-05-16 | 2002-05-21 | The Trustees Of Columbia University In The City Of New York | Method for inhibiting thrombosis in a patient whose blood is subjected to extracorporeal circulation |
| US5839443A (en) * | 1996-05-16 | 1998-11-24 | The Trustees Of Columbia University In The City Of New York | Method for inhibiting thrombosis in a patient whose blood is subjected to extracorporeal circulation |
| US5792613A (en) * | 1996-06-12 | 1998-08-11 | The Curators Of The University Of Missouri | Method for obtaining RNA aptamers based on shape selection |
| US6136545A (en) * | 1996-09-13 | 2000-10-24 | Roche Diagnostics Gmbh | Homogeneous detection methods for the determination of subpopulations of an analyte |
| US6316403B1 (en) * | 1996-09-27 | 2001-11-13 | The Trustees Of Columbia University In The City Of New York | Methods for treating an ischemic disorder and improving stroke outcome |
| US6315995B1 (en) * | 1996-09-27 | 2001-11-13 | The Trustees Of Columbia University In The City Of New York | Methods for treating an ischemic disorder and improving stroke outcome |
| US6120997A (en) * | 1997-01-13 | 2000-09-19 | The Scripps Research Institute | Nucleic acid binders having an hydroxyamine motif |
| US5861254A (en) * | 1997-01-31 | 1999-01-19 | Nexstar Pharmaceuticals, Inc. | Flow cell SELEX |
| US6153410A (en) * | 1997-03-25 | 2000-11-28 | California Institute Of Technology | Recombination of polynucleotide sequences using random or defined primers |
| US6177263B1 (en) * | 1997-03-25 | 2001-01-23 | California Institute Of Technology | Recombination of polynucleotide sequences using random or defined primers |
| US6150461A (en) * | 1997-05-27 | 2000-11-21 | Hisamitsu Pharmaceutical Co., Inc. | Carriers targettable to organ |
| US6127173A (en) * | 1997-09-22 | 2000-10-03 | Ribozyme Pharmaceuticals, Inc. | Nucleic acid catalysts with endonuclease activity |
| US6054274A (en) * | 1997-11-12 | 2000-04-25 | Hewlett-Packard Company | Method of amplifying the signal of target nucleic acid sequence analyte |
| US5882870A (en) * | 1998-01-14 | 1999-03-16 | Becton Dickinson And Company | Rapidly reversed thrombin binding oligonucleotide anticoagulants |
| US6180348B1 (en) * | 1998-04-20 | 2001-01-30 | Weihua Li | Method of isolating target specific oligonucleotide ligands |
| US6163714A (en) * | 1998-07-03 | 2000-12-19 | Torsana Diabetes Diagnostics A/S | Optical sensor for in situ measurement of analytes |
| US6177555B1 (en) * | 1998-09-18 | 2001-01-23 | Nexstar Pharmaceuticals, Inc. | Homogeneous detection of a target through nucleic acid ligand-ligand beacon interaction |
| US5989823A (en) * | 1998-09-18 | 1999-11-23 | Nexstar Pharmaceuticals, Inc. | Homogeneous detection of a target through nucleic acid ligand-ligand beacon interaction |
| US6114038A (en) * | 1998-11-10 | 2000-09-05 | Biocrystal Ltd. | Functionalized nanocrystals and their use in detection systems |
| US6051388A (en) * | 1998-12-22 | 2000-04-18 | Toxin Alert, Inc. | Method and apparatus for selective biological material detection |
| US6110462A (en) * | 1999-03-03 | 2000-08-29 | The Scripps Research Institute | Enzymatic DNA molecules that contain modified nucleotides |
| US6316198B1 (en) * | 1999-03-18 | 2001-11-13 | Exiqon A/S | Detection of mutations in genes by specific LNA primers |
| US6171795B1 (en) * | 1999-07-29 | 2001-01-09 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands to CD40ligand |
| US6258601B1 (en) * | 2000-09-07 | 2001-07-10 | Isis Pharmaceuticals, Inc. | Antisense modulation of ubiquitin protein ligase expression |
| US20030175703A1 (en) * | 2000-09-26 | 2003-09-18 | Sullenger Bruce A. | RNA aptamers and methods for identifying the same |
| US20030083294A1 (en) * | 2001-05-25 | 2003-05-01 | Sullenger Bruce A. | Modulators of pharmacological agents |
| US20060004088A1 (en) * | 2002-10-22 | 2006-01-05 | Oct Inc. | Furan derivatives for preventing and curing osteoporosis and pharmaceutical compositions containing the same |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140179621A1 (en) * | 2009-05-01 | 2014-06-26 | Ophthotech Corporation | Methods for Treating or Preventing Ophthalmological Diseases |
| US20110190192A1 (en) * | 2009-12-15 | 2011-08-04 | Cebix Inc. | Methods for treating erectile dysfunction in patients with insulin-dependent diabetes |
| US20120178676A1 (en) * | 2010-05-17 | 2012-07-12 | Cebix, Inc. | Pegylated c-peptide |
| US20120220542A1 (en) * | 2010-05-17 | 2012-08-30 | Cebix, Inc. | Pegylated c-peptide |
| US8691755B2 (en) * | 2010-05-17 | 2014-04-08 | Cebix Ab | Pegylated C-peptide |
| US8927488B2 (en) * | 2010-05-17 | 2015-01-06 | Cebix, Inc. | Pegylated C-peptide |
| WO2012149198A3 (en) * | 2011-04-26 | 2013-03-14 | Regado Biosciences, Inc. | A method for manufacturing pegylated oligonucleotides |
| US8962553B2 (en) | 2011-11-17 | 2015-02-24 | Cebix Ab | Method of treating a diabetic subject having a microvascular impairment disorder by a pegylated C-peptide |
| US11273171B2 (en) | 2013-07-12 | 2022-03-15 | Iveric Bio, Inc. | Methods for treating or preventing ophthalmological conditions |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7531524B2 (en) | Modulators of coagulation factors with enhanced stability | |
| US8283330B2 (en) | Modulators of pharmacological agents | |
| AU2002312059A1 (en) | Modulators of pharmacological agents | |
| AU2010256511B2 (en) | Nucleic acid modulators of glycoprotein VI | |
| WO2023169548A1 (en) | Lpa inhibitor and use thereof | |
| AU2012244176B8 (en) | Modulators of pharmacological agents | |
| AU2012244176B2 (en) | Modulators of pharmacological agents | |
| ES2351784T3 (en) | MODULATORS OF PHARMACOLOGICAL AGENTS. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: REGADO BIOSCIENCES, INC., NORTH CAROLINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:RUSCONI, CHRISTOPHER P.;REEL/FRAME:018988/0374 Effective date: 20070308 |
|
| AS | Assignment |
Owner name: REGADO BIOSCIENCES, INC., NORTH CAROLINA Free format text: CHANGE OF ADDRESS OF ASSIGNEE;ASSIGNOR:REGADO BIOSCIENCES, INC.;REEL/FRAME:022000/0975 Effective date: 20081218 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |