US20030191020A1 - Autothermal process for the production of olefins - Google Patents
Autothermal process for the production of olefins Download PDFInfo
- Publication number
- US20030191020A1 US20030191020A1 US10/323,175 US32317502A US2003191020A1 US 20030191020 A1 US20030191020 A1 US 20030191020A1 US 32317502 A US32317502 A US 32317502A US 2003191020 A1 US2003191020 A1 US 2003191020A1
- Authority
- US
- United States
- Prior art keywords
- catalyst
- support
- composition
- platinum
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 238000000034 method Methods 0.000 title claims abstract description 140
- 230000008569 process Effects 0.000 title claims abstract description 124
- 150000001336 alkenes Chemical class 0.000 title abstract description 43
- 238000004519 manufacturing process Methods 0.000 title description 5
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical group [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims abstract description 202
- 239000003054 catalyst Substances 0.000 claims abstract description 194
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims abstract description 79
- 229910052751 metal Inorganic materials 0.000 claims abstract description 69
- 239000002184 metal Substances 0.000 claims abstract description 69
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 claims abstract description 51
- 239000003607 modifier Substances 0.000 claims abstract description 26
- -1 rare earth lanthanides Chemical class 0.000 claims abstract description 25
- 239000000395 magnesium oxide Substances 0.000 claims abstract description 24
- 229910052768 actinide Inorganic materials 0.000 claims abstract description 4
- 229910052747 lanthanoid Inorganic materials 0.000 claims abstract description 4
- 150000001255 actinides Chemical class 0.000 claims abstract description 3
- 229910052697 platinum Inorganic materials 0.000 claims description 79
- 239000010949 copper Substances 0.000 claims description 71
- 239000000203 mixture Substances 0.000 claims description 69
- 229910052802 copper Inorganic materials 0.000 claims description 50
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical group [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 48
- 239000011135 tin Substances 0.000 claims description 48
- 229910052718 tin Inorganic materials 0.000 claims description 47
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 claims description 44
- 239000000919 ceramic Substances 0.000 claims description 38
- 239000000835 fiber Substances 0.000 claims description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 33
- 239000006260 foam Substances 0.000 claims description 30
- 229910052787 antimony Inorganic materials 0.000 claims description 25
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 claims description 22
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 claims description 16
- 239000008188 pellet Substances 0.000 claims description 14
- 239000000377 silicon dioxide Substances 0.000 claims description 13
- 238000000151 deposition Methods 0.000 claims description 10
- 238000001354 calcination Methods 0.000 claims description 9
- 229910052746 lanthanum Inorganic materials 0.000 claims description 9
- 229910052709 silver Inorganic materials 0.000 claims description 9
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 8
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 claims description 7
- 239000004332 silver Substances 0.000 claims description 7
- 229910052791 calcium Inorganic materials 0.000 claims description 6
- 239000011575 calcium Substances 0.000 claims description 6
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical group [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 5
- 229910052738 indium Inorganic materials 0.000 claims description 5
- 239000011777 magnesium Substances 0.000 claims description 5
- 229910052726 zirconium Inorganic materials 0.000 claims description 5
- 229910052684 Cerium Inorganic materials 0.000 claims description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 4
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 4
- 229910052788 barium Inorganic materials 0.000 claims description 4
- 229910052749 magnesium Inorganic materials 0.000 claims description 4
- 230000000737 periodic effect Effects 0.000 claims description 4
- 239000011148 porous material Substances 0.000 claims description 4
- 229910052700 potassium Inorganic materials 0.000 claims description 4
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 claims description 4
- 229910010271 silicon carbide Inorganic materials 0.000 claims description 4
- 229910052691 Erbium Inorganic materials 0.000 claims description 3
- 229910052689 Holmium Inorganic materials 0.000 claims description 3
- 229910052765 Lutetium Inorganic materials 0.000 claims description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 claims description 3
- 239000003638 chemical reducing agent Substances 0.000 claims description 3
- UYAHIZSMUZPPFV-UHFFFAOYSA-N erbium Chemical compound [Er] UYAHIZSMUZPPFV-UHFFFAOYSA-N 0.000 claims description 3
- KJZYNXUDTRRSPN-UHFFFAOYSA-N holmium atom Chemical compound [Ho] KJZYNXUDTRRSPN-UHFFFAOYSA-N 0.000 claims description 3
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 claims description 3
- 150000002602 lanthanoids Chemical class 0.000 claims description 3
- OHSVLFRHMCKCQY-UHFFFAOYSA-N lutetium atom Chemical compound [Lu] OHSVLFRHMCKCQY-UHFFFAOYSA-N 0.000 claims description 3
- 239000011591 potassium Substances 0.000 claims description 3
- 229910052581 Si3N4 Inorganic materials 0.000 claims description 2
- 229910000323 aluminium silicate Inorganic materials 0.000 claims description 2
- 239000000391 magnesium silicate Substances 0.000 claims description 2
- 235000012243 magnesium silicates Nutrition 0.000 claims description 2
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 claims description 2
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 claims 2
- 229910052814 silicon oxide Inorganic materials 0.000 claims 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 abstract description 127
- 229910052739 hydrogen Inorganic materials 0.000 abstract description 115
- 239000001257 hydrogen Substances 0.000 abstract description 115
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 abstract description 92
- 238000007254 oxidation reaction Methods 0.000 abstract description 80
- 230000003647 oxidation Effects 0.000 abstract description 79
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 abstract description 77
- 239000005977 Ethylene Substances 0.000 abstract description 77
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 abstract description 72
- 239000001301 oxygen Substances 0.000 abstract description 72
- 229910052760 oxygen Inorganic materials 0.000 abstract description 72
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 abstract description 46
- 229930195733 hydrocarbon Natural products 0.000 abstract description 46
- 150000002430 hydrocarbons Chemical class 0.000 abstract description 46
- 239000004215 Carbon black (E152) Substances 0.000 abstract description 34
- 239000001294 propane Substances 0.000 abstract description 20
- 230000003197 catalytic effect Effects 0.000 abstract description 19
- 230000036961 partial effect Effects 0.000 abstract description 14
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 abstract description 13
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 abstract description 11
- 230000007423 decrease Effects 0.000 abstract description 7
- 239000003498 natural gas condensate Substances 0.000 abstract description 4
- 230000036284 oxygen consumption Effects 0.000 abstract 1
- 229910052761 rare earth metal Inorganic materials 0.000 abstract 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 63
- 238000006243 chemical reaction Methods 0.000 description 58
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 48
- 239000000243 solution Substances 0.000 description 40
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 38
- 229910002092 carbon dioxide Inorganic materials 0.000 description 35
- 230000002829 reductive effect Effects 0.000 description 34
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 33
- 239000007789 gas Substances 0.000 description 31
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 29
- 229910002091 carbon monoxide Inorganic materials 0.000 description 29
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 28
- 239000012188 paraffin wax Substances 0.000 description 27
- 239000001569 carbon dioxide Substances 0.000 description 25
- 238000005470 impregnation Methods 0.000 description 25
- 239000007864 aqueous solution Substances 0.000 description 24
- 229910052757 nitrogen Inorganic materials 0.000 description 24
- 238000011068 loading method Methods 0.000 description 23
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 22
- 150000002431 hydrogen Chemical class 0.000 description 20
- 150000001875 compounds Chemical class 0.000 description 19
- 229910018883 Pt—Cu Inorganic materials 0.000 description 18
- 239000002253 acid Substances 0.000 description 17
- 239000003570 air Substances 0.000 description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 239000011550 stock solution Substances 0.000 description 14
- 229910052593 corundum Inorganic materials 0.000 description 13
- 229910001845 yogo sapphire Inorganic materials 0.000 description 13
- 238000004227 thermal cracking Methods 0.000 description 12
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 11
- 229910052799 carbon Inorganic materials 0.000 description 11
- 238000002485 combustion reaction Methods 0.000 description 11
- 239000003085 diluting agent Substances 0.000 description 11
- 239000002245 particle Substances 0.000 description 11
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 10
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 10
- 238000005336 cracking Methods 0.000 description 10
- 229960003280 cupric chloride Drugs 0.000 description 10
- 239000001119 stannous chloride Substances 0.000 description 10
- 235000011150 stannous chloride Nutrition 0.000 description 10
- 230000008901 benefit Effects 0.000 description 9
- 238000005243 fluidization Methods 0.000 description 9
- 150000001721 carbon Chemical group 0.000 description 8
- 238000010790 dilution Methods 0.000 description 8
- 239000012895 dilution Substances 0.000 description 8
- 239000000446 fuel Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 150000002739 metals Chemical class 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000010453 quartz Substances 0.000 description 8
- 239000000376 reactant Substances 0.000 description 8
- 150000001335 aliphatic alkanes Chemical class 0.000 description 7
- 239000012080 ambient air Substances 0.000 description 7
- 230000008021 deposition Effects 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 230000003068 static effect Effects 0.000 description 7
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 6
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000005839 oxidative dehydrogenation reaction Methods 0.000 description 6
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 229910002090 carbon oxide Inorganic materials 0.000 description 5
- 150000005673 monoalkenes Chemical class 0.000 description 5
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 5
- 229910052759 nickel Inorganic materials 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 229910045601 alloy Inorganic materials 0.000 description 4
- 239000000956 alloy Substances 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000004744 fabric Substances 0.000 description 4
- 239000003863 metallic catalyst Substances 0.000 description 4
- 239000003345 natural gas Substances 0.000 description 4
- 229910052703 rhodium Inorganic materials 0.000 description 4
- 239000010948 rhodium Substances 0.000 description 4
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 229910002621 H2PtCl6 Inorganic materials 0.000 description 3
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 3
- 229910002846 Pt–Sn Inorganic materials 0.000 description 3
- 150000004703 alkoxides Chemical class 0.000 description 3
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 3
- 239000001273 butane Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 150000007942 carboxylates Chemical class 0.000 description 3
- 239000000571 coke Substances 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- JVLRYPRBKSMEBF-UHFFFAOYSA-K diacetyloxystibanyl acetate Chemical compound [Sb+3].CC([O-])=O.CC([O-])=O.CC([O-])=O JVLRYPRBKSMEBF-UHFFFAOYSA-K 0.000 description 3
- KZHJGOXRZJKJNY-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Si]=O.O=[Al]O[Al]=O.O=[Al]O[Al]=O.O=[Al]O[Al]=O KZHJGOXRZJKJNY-UHFFFAOYSA-N 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 125000001475 halogen functional group Chemical group 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- VUZPPFZMUPKLLV-UHFFFAOYSA-N methane;hydrate Chemical compound C.O VUZPPFZMUPKLLV-UHFFFAOYSA-N 0.000 description 3
- 229910052863 mullite Inorganic materials 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 3
- 150000002823 nitrates Chemical class 0.000 description 3
- 230000001590 oxidative effect Effects 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 3
- 238000009834 vaporization Methods 0.000 description 3
- 230000008016 vaporization Effects 0.000 description 3
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 229920000181 Ethylene propylene rubber Polymers 0.000 description 2
- 241000264877 Hippospongia communis Species 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 229910018054 Ni-Cu Inorganic materials 0.000 description 2
- 229910018481 Ni—Cu Inorganic materials 0.000 description 2
- 229910020935 Sn-Sb Inorganic materials 0.000 description 2
- 229910008757 Sn—Sb Inorganic materials 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 229910052785 arsenic Inorganic materials 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229910052797 bismuth Inorganic materials 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- ZMIGMASIKSOYAM-UHFFFAOYSA-N cerium Chemical compound [Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce] ZMIGMASIKSOYAM-UHFFFAOYSA-N 0.000 description 2
- 229910052804 chromium Inorganic materials 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000006356 dehydrogenation reaction Methods 0.000 description 2
- 229910001882 dioxygen Inorganic materials 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 229910052733 gallium Inorganic materials 0.000 description 2
- 229910052732 germanium Inorganic materials 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 229910052745 lead Inorganic materials 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910044991 metal oxide Chemical group 0.000 description 2
- 150000004706 metal oxides Chemical group 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229910052750 molybdenum Inorganic materials 0.000 description 2
- 150000004767 nitrides Chemical class 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 150000002902 organometallic compounds Chemical class 0.000 description 2
- CMOAHYOGLLEOGO-UHFFFAOYSA-N oxozirconium;dihydrochloride Chemical compound Cl.Cl.[Zr]=O CMOAHYOGLLEOGO-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000010944 silver (metal) Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000004230 steam cracking Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- BHSXLOMVDSFFHO-UHFFFAOYSA-N (3-ethylsulfanylphenyl)methanamine Chemical compound CCSC1=CC=CC(CN)=C1 BHSXLOMVDSFFHO-UHFFFAOYSA-N 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical class CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- ODINCKMPIJJUCX-UHFFFAOYSA-N Calcium oxide Chemical compound [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 229910018967 Pt—Rh Inorganic materials 0.000 description 1
- 229910000629 Rh alloy Inorganic materials 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 229910020888 Sn-Cu Inorganic materials 0.000 description 1
- 229910019204 Sn—Cu Inorganic materials 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- 229910006213 ZrOCl2 Inorganic materials 0.000 description 1
- RRJJTVONCGYCIP-UHFFFAOYSA-N [Cu].[Sn].[Pt] Chemical compound [Cu].[Sn].[Pt] RRJJTVONCGYCIP-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical compound C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000001462 antimony Chemical class 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000000292 calcium oxide Substances 0.000 description 1
- 235000012255 calcium oxide Nutrition 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 238000004939 coking Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000011217 control strategy Methods 0.000 description 1
- WBLJAACUUGHPMU-UHFFFAOYSA-N copper platinum Chemical compound [Cu].[Pt] WBLJAACUUGHPMU-UHFFFAOYSA-N 0.000 description 1
- 229910052878 cordierite Inorganic materials 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- JSKIRARMQDRGJZ-UHFFFAOYSA-N dimagnesium dioxido-bis[(1-oxido-3-oxo-2,4,6,8,9-pentaoxa-1,3-disila-5,7-dialuminabicyclo[3.3.1]nonan-7-yl)oxy]silane Chemical compound [Mg++].[Mg++].[O-][Si]([O-])(O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2)O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2 JSKIRARMQDRGJZ-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- JUWSSMXCCAMYGX-UHFFFAOYSA-N gold platinum Chemical compound [Pt].[Au] JUWSSMXCCAMYGX-UHFFFAOYSA-N 0.000 description 1
- 229910052735 hafnium Inorganic materials 0.000 description 1
- 238000007210 heterogeneous catalysis Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000008676 import Effects 0.000 description 1
- 238000009413 insulation Methods 0.000 description 1
- 239000012774 insulation material Substances 0.000 description 1
- 238000005468 ion implantation Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000001282 iso-butane Substances 0.000 description 1
- ICAKDTKJOYSXGC-UHFFFAOYSA-K lanthanum(iii) chloride Chemical compound Cl[La](Cl)Cl ICAKDTKJOYSXGC-UHFFFAOYSA-K 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 150000001247 metal acetylides Chemical class 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- LAIZPRYFQUWUBN-UHFFFAOYSA-L nickel chloride hexahydrate Chemical compound O.O.O.O.O.O.[Cl-].[Cl-].[Ni+2] LAIZPRYFQUWUBN-UHFFFAOYSA-L 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- PXXKQOPKNFECSZ-UHFFFAOYSA-N platinum rhodium Chemical compound [Rh].[Pt] PXXKQOPKNFECSZ-UHFFFAOYSA-N 0.000 description 1
- FHMDYDAXYDRBGZ-UHFFFAOYSA-N platinum tin Chemical compound [Sn].[Pt] FHMDYDAXYDRBGZ-UHFFFAOYSA-N 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- MWWATHDPGQKSAR-UHFFFAOYSA-N propyne Chemical group CC#C MWWATHDPGQKSAR-UHFFFAOYSA-N 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- 238000010517 secondary reaction Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229910052566 spinel group Inorganic materials 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 229910052712 strontium Inorganic materials 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- 229910052716 thallium Inorganic materials 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- 229910000597 tin-copper alloy Inorganic materials 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/42—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with a hydrogen acceptor
- C07C5/48—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with a hydrogen acceptor with oxygen as an acceptor
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/62—Platinum group metals with gallium, indium, thallium, germanium, tin or lead
- B01J23/622—Platinum group metals with gallium, indium, thallium, germanium, tin or lead with germanium, tin or lead
- B01J23/626—Platinum group metals with gallium, indium, thallium, germanium, tin or lead with germanium, tin or lead with tin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/644—Arsenic, antimony or bismuth
- B01J23/6445—Antimony
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/89—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with noble metals
- B01J23/8926—Copper and noble metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/89—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with noble metals
- B01J23/8933—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with noble metals also combined with metals, or metal oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8966—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with noble metals also combined with metals, or metal oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with germanium, tin or lead
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/90—Regeneration or reactivation
- B01J23/96—Regeneration or reactivation of catalysts comprising metals, oxides or hydroxides of the noble metals
-
- B01J35/56—
-
- B01J35/58—
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0238—Impregnation, coating or precipitation via the gaseous phase-sublimation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/02—Boron or aluminium; Oxides or hydroxides thereof
- B01J21/04—Alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/10—Magnesium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
- B01J37/0207—Pretreatment of the support
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
- B01J37/18—Reducing with gases containing free hydrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/02—Boron or aluminium; Oxides or hydroxides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/02—Boron or aluminium; Oxides or hydroxides thereof
- C07C2521/04—Alumina
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- C07C2521/08—Silica
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/10—Magnesium; Oxides or hydroxides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/12—Silica and alumina
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/14—Silica and magnesia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the alkali- or alkaline earth metals or beryllium
- C07C2523/04—Alkali metals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/08—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of gallium, indium or thallium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/14—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of germanium, tin or lead
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/18—Arsenic, antimony or bismuth
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of noble metals
- C07C2523/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of noble metals of the platinum group metals
- C07C2523/42—Platinum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of noble metals
- C07C2523/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of noble metals of the platinum group metals
- C07C2523/44—Palladium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of noble metals
- C07C2523/48—Silver or gold
- C07C2523/50—Silver
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/72—Copper
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2527/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- C07C2527/20—Carbon compounds
- C07C2527/22—Carbides
- C07C2527/224—Silicon carbide
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2400/00—Products obtained by processes covered by groups C10G9/00 - C10G69/14
- C10G2400/20—C2-C4 olefins
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S502/00—Catalyst, solid sorbent, or support therefor: product or process of making
- Y10S502/514—Process applicable either to preparing or to regenerating or to rehabilitating catalyst or sorbent
Definitions
- the present invention relates to the field of catalytic oxidation of hydrocarbons. More particularly, the present invention relates to the catalytic partial oxidation of paraffinic hydrocarbons, such as ethane, propane, and naphtha, to produce olefins, such as ethylene and propylene.
- paraffinic hydrocarbons such as ethane, propane, and naphtha
- Olefins find widespread utility in industrial organic chemistry.
- Ethylene is needed for the preparation of important polymers, such as polyethylene, vinyl plastics, and ethylene-propylene rubbers, and important basic chemicals, such as ethylene oxide, styrene, acetaldehyde, ethyl acetate, and dichloroethane.
- Propylene is needed for the preparation of polypropylene plastics, ethylene-propylene rubbers, and important basic chemicals, such as propylene oxide, cumene, and acrolein.
- Isobutylene is needed for the preparation of methyl tertiary butyl ether.
- Long chain mono-olefins find utility in the manufacture of linear alkylated benzene sulfonates, which are used in the detergent industry.
- Low molecular weight olefins such as ethylene, propylene, and butylene
- thermal cracking pyrolysis/steam cracking
- An ethylene plant typically achieves an ethylene selectivity of about 85 percent calculated on a carbon atom basis at an ethane conversion of about 60 mole percent.
- Undesired coproducts are recycled on the shell side of the cracking furnace to be burned, so as to produce the heat necessary for the process.
- thermal cracking processes for olefin production are highly endothermic. Accordingly, these processes require the construction and maintenance of large, capital intensive, and complex cracking furnaces.
- Catalytic processes are known wherein paraffinic hydrocarbons are oxidatively cracked to form mono-olefins.
- a paraffinic hydrocarbon is contacted with oxygen in the presence of a catalyst consisting of a platinum group metal or mixture thereof deposited on a ceramic monolith support.
- hydrogen may be a component of the feed.
- the process is conducted under autothermal reaction conditions wherein the feed is partially combusted, and the heat produced during combustion drives the endothermic cracking process. Consequently, under these autothermal process conditions there is no external heat source required; however, the catalyst is required to support combustion above the normal fuel-rich limit of flammability.
- Representative references disclosing this type of process include the following U.S. Pat. Nos.
- M. Huff and L. D. Schmidt disclose in the Journal of Physical Chemistry, 97, 1993, 11,815, the production of ethylene from ethane in the presence of air or oxygen under autothermal conditions over alumina foam monoliths coated with platinum, rhodium, or palladium.
- a similar article by M. Huff and L. D. Schmidt in the Journal of Catalysis, 149, 1994, 127-141 discloses the autothermal production of olefins from propane and butane by oxidative dehydrogenation and cracking in air or oxygen over platinum and rhodium coated alumina foam monoliths. The olefin selectivity achieved in these processes is not comparable to that achieved by steam cracking and therefore could be improved.
- U.S. Pat. No. 5,639,929 teaches an autothermal process for the oxidative dehydrogenation of C 2 -C 6 alkanes with an oxygen-containing gas in a fluidized catalyst bed of platinum, rhodium, nickel, or platinum-gold supported on alpha alumina or zirconia.
- Ethane produces ethylene, while higher alkanes produce ethylene, propylene, and iso-butylene.
- the olefin selectivity could be improved.
- This invention is a process for the partial oxidation of paraffinic hydrocarbons to form olefins.
- the process comprises contacting a paraffinic hydrocarbon or mixture thereof with oxygen in the presence of hydrogen and a catalyst.
- the contacting is conducted under autothermal process conditions sufficient to form the olefin.
- the catalyst employed in the process of this invention comprises a Group 8B metal and at least one promoter.
- the process of this invention efficiently produces olefins, particularly mono-olefins, from paraffinic hydrocarbons, oxygen, and hydrogen.
- the process of this invention achieves a higher paraffin conversion and a higher olefin selectivity as compared with prior art catalytic, autothermal processes.
- the process of this invention produces fewer undesirable deep oxidation products, such as carbon monoxide and carbon dioxide, as compared with prior art catalytic, autothermal processes.
- the process of this invention achieves a paraffin conversion and olefin selectivity which are comparable to commercial thermal cracking processes.
- the process produces little, if any, coke, thereby substantially prolonging catalyst lifetime and eliminating the necessity to shut down the reactor to remove coke deposits.
- the process of this invention allows the operator to employ a simple engineering design and control strategy, which eliminates the requirement for a large, expensive, and complex furnace like that used in thermal cracking processes.
- the reactor simply comprises an exterior housing which contains a monolithic support onto which the catalytic components are deposited. Since the residence time of the reactants in the process of this invention is on the order of milliseconds, the reaction zone operates at high volumetric throughput. Accordingly, the reaction zone measures from about one-fiftieth to about one-hundredth the size of a commercially available steam cracker of comparable capacity. The reduced size of the reactor lowers costs and simplifies maintenance procedures. Finally, since the process of this invention is exothermic, the heat produced can be harvested via integrated heat exchangers to generate electricity or steam credits for other processes.
- thermal energy is needed to maintain autothermal process conditions. Without preheating the feedstream, the required thermal energy is totally supplied by the reaction of the feedstream with oxygen, namely, alkane oxidative dehydrogenation to form olefins and water, hydrogen oxidation to form water, and carbon combustion to form carbon monoxide and carbon dioxide. These processes can supply the heat necessary for any endothermic dehydrogenation which takes place to form ethylene and hydrogen.
- the prior art has recognized that a portion of the required thermal energy can be obtained by preheating the feedstream.
- the preheat can be conveniently supplied by condensing high pressure saturated steam, or alternatively, by combusting process off-gas or another fuel source.
- the paraffinic hydrocarbon and oxygen which together comprise the reactant feedstream, are preheated at a temperature greater than about 200° C., but below the onset of reaction of the feedstream components.
- An increased yield of hydrogen in the product stream further improves the economics of the autothermal oxidation process of this invention. Since hydrogen is required for the process, hydrogen in the product should be recycled and any deficit must be replaced by importing hydrogen from an external source. Alternatively, hydrogen can be made from off-gas streams, for example, a water-shift reaction which converts carbon monoxide and water to hydrogen and carbon dioxide. As a consequence of using the high preheat temperature of this invention, the product stream is enriched in hydrogen. Under optimal preheat conditions, the recycled hydrogen substantially eliminates the need to import hydrogen or to derive make-up hydrogen from other sources.
- the autothermal oxidation process of this invention is beneficially conducted in a unique fluidized bed reactor, characterized in that the reactor bed possesses an aspect ratio of less than about 1:1, as measured during operation.
- the aspect ratio is defined as the ratio of the height (or depth) of the reactor bed to its cross-sectional dimension (diameter or width).
- the catalyst comprises a support in the form of pellets or spheres onto which the catalytic components are deposited.
- this invention is a catalyst composition
- a catalyst composition comprising a Group 8B metal and at least one promoter supported on a catalyst support which has been pretreated with at least one support modifier.
- the aforementioned composition is beneficially employed as a catalyst in the autothermal partial oxidation of a paraffinic hydrocarbon to an olefin.
- the catalyst composition beneficially produces an olefin or mixture of olefins at conversions and selectivities which are comparable to those of industrial thermal cracking processes. Accordingly, the catalyst composition of this invention produces low amounts of carbon monoxide and carbon dioxide. Finally, the catalyst composition of this invention advantageously exhibits good catalyst stability.
- the process of this invention involves the partial oxidation of a paraffinic hydrocarbon to form an olefin.
- paraffinic hydrocarbon to form an olefin.
- partial oxidation imply that the paraffin is not substantially oxidized to deep oxidation products, specifically, carbon monoxide and carbon dioxide. Rather, the partial oxidation comprises one or both of oxidative dehydrogenation and cracking to form primarily olefins. It is not known or suggested to what extent or degree either process, oxidative dehydrogenation or cracking, predominates or occurs to the exclusion of the other.
- the partial oxidation process of this invention comprises contacting a paraffinic hydrocarbon with oxygen in the presence of a multi-metallic catalyst and in the presence of a hydrogen co-feed.
- the contacting is conducted under autothermal process conditions sufficient to form the olefin.
- the catalyst which is employed in the process of this invention comprises a Group 8B metal and at least one promoter, optionally supported on a catalyst support.
- the paraffinic hydrocarbon is a paraffin selected from ethane, propane, mixtures of ethane and propane, naphtha, natural gas condensates, and mixtures of the aforementioned hydrocarbons; and the preferred olefins produced are ethylene, propylene, butylene, isobutylene, and butadiene.
- the feedstream comprising the paraffinic hydrocarbon and oxygen is preheated before introducing the feedstream into the autothermal oxidation reactor.
- the preheat temperature is greater than about 200° C., but less than the temperature wherein reaction of the feedstream components begins.
- the upper limit on the preheat temperature is less than about 900° C.
- the reactor comprises an exterior housing which holds the catalyst, the catalyst being provided in the form of a ceramic monolith support onto which the catalytic components, including the Group 8B metal and any promoter(s), have been deposited.
- the reactor comprises a modified fluidized bed characterized by an aspect ratio of less than about 1:1 in operating mode.
- the aspect ratio is the ratio of the height (depth) of the reactor to its cross-sectional dimension (diameter or width).
- the catalyst is provided typically in the form of spheres or granules.
- the catalyst which is employed in the process of this invention comprises a Group 8B metal and at least one promoter supported on a catalytic support which has been pretreated with at least one support modifier.
- the Group 8B metal is a platinum group metal.
- the preferred platinum group metal is platinum.
- the preferred promoter is selected from the elements of Groups 1B, 6B, 3A, 4A, 5A, (equivalent to Groups 11, 6, 13, 14, and 15), and mixtures of the aforementioned elements of the Periodic Table, as referenced by S. R. Radel and M. H. Navidi, in Chemistry, West Publishing Company, New York, 1990.
- the preferred support modifier is selected from Groups 1A, 2A, 3B, 4B, 5B, 6B, 1B, 3A, 4A, 5A (equivalent Groups 1, 2, 3, 4, 5, 6, 11, 13, 14, 15), and the lanthanide rare earths and actinide metals of the Periodic Table, as referenced by S. R. Radel and M. H. Navidi, ibid.
- the platinum group metal is platinum; the promoter is selected from tin, copper, and mixtures thereof; the support is selected from alumina, magnesia, and mixtures thereof; and the modifier is selected from tin, lanthanum, and mixtures thereof.
- paraffinic hydrocarbon any paraffinic hydrocarbon or mixture of paraffinic hydrocarbons can be employed in the process of this invention provided that an olefin, preferably, a mono-olefin, is produced.
- the paraffin contains at least 2 carbon atoms.
- the paraffin contains from 2 to about 25 carbon atoms, more preferably, from 2 to about 15 carbon atoms, and even more preferably, from 2 to about 10 carbon atoms.
- the paraffin can have a linear, branched, or cyclic structure, and can be a liquid or gas at ambient temperature and pressure.
- paraffin can be supplied as an essentially pure paraffinic compound, or mixture of paraffinic compounds, or as a paraffin-containing mixture of hydrocarbons.
- Paraffin feeds which are suitably employed in the process of this invention include, but are not limited to, ethane, propane, butane, pentane, hexane, heptane, octane, and higher homologues thereof, as well as complex higher boiling mixtures of paraffin-containing hydrocarbons, such as naphtha, gas oil, vacuum gas oil, and natural gas condensates. Additional feed components may include methane, nitrogen, carbon monoxide, carbon dioxide, and steam, if so desired. Minor amounts of unsaturated hydrocarbons may also be present.
- the paraffin is selected from ethane, propane, mixtures of ethane and propane, naphtha, natural gas condensates, and mixtures thereof.
- the paraffinic hydrocarbon is contacted with an oxygen-containing gas.
- the gas is molecular oxygen or molecular oxygen diluted with an unreactive gas, such as nitrogen, helium, carbon dioxide, or argon, or diluted with a substantially unreactive gas, such as carbon monoxide or steam.
- an unreactive gas such as nitrogen, helium, carbon dioxide, or argon
- a substantially unreactive gas such as carbon monoxide or steam.
- Any molar ratio of paraffin to oxygen is suitable provided the desired olefin is produced in the process of this invention.
- the process is conducted fuel-rich and above the upper flammability limit.
- the molar ratio of paraffinic hydrocarbon to oxygen varies depending upon the specific paraffin feed and autothermal process conditions employed.
- the molar ratio of paraffinic hydrocarbon to oxygen ranges from about 3 to about 77 times the stoichiometric ratio of hydrocarbon to oxygen for complete combustion to carbon dioxide and water.
- the molar ratio of paraffinic hydrocarbon to oxygen ranges from about 3 to about 13, more preferably, from about 4 to about 11, and most preferably, from about 5 to about 9 times the stoichiometric ratio of hydrocarbon to oxygen for complete combustion to carbon dioxide and water.
- a molar ratio of paraffinic hydrocarbon to oxygen greater than about 0.1:1, preferably, greater than about 0.2:1, and by using a molar ratio of paraffinic hydrocarbon to oxygen usually less than about 3.0:1, preferably, less than about 2.7:1.
- the following ratios are more specific.
- the ethane to oxygen molar ratio is typically greater than about 1.5:1, and preferably, greater than about 1.8:1.
- the ethane to oxygen molar ratio is typically less than about 3.0:1, preferably, less than about 2.7:1.
- propane the propane to oxygen molar ratio is typically greater than about 0.9:1, preferably, greater than about 1.1:1.
- the propane to oxygen molar ratio is typically less than about 2.2:1, preferably, less than about 2.0:1.
- the naphtha to oxygen molar ratio is typically greater than about 0.3:1, preferably, greater than about 0.5:1.
- the naphtha to oxygen molar ratio is typically less than about 1.0:1, preferably, less than about 0.9:1.
- the limits on the molar ratio of paraffinic hydrocarbon to oxygen can be shifted towards higher values.
- the molar ratio of paraffinic hydrocarbon to oxygen is typically greater than about 0.1:1 and less than about 4.0:1.
- the ethane to oxygen molar ratio is typically greater than about 1.5:1, preferably, greater than about 1.8:1, and typically less than about 4.0:1, preferably, less than about 3.2:1.
- the molar ratio of propane to oxygen is typically greater than about 0.9:1, preferably, greater than about 1.1:1, and typically, less than about 3.0:1, and preferably, less than about 2.6:1.
- the molar ratio of naphtha to oxygen is typically greater than about 0.3:1, preferably, greater than about 0.5:1, and typically, less than about 1.4:1, and preferably, less than about 1.3:1.
- hydrogen is co-fed with the paraffin and oxygen to the catalyst.
- the presence of hydrogen in the feedstream beneficially improves the conversion of hydrocarbon and the selectivity to olefins, while reducing the formation of deep oxidation products, such as, carbon monoxide and carbon dioxide.
- the molar ratio of hydrogen to oxygen can vary over any operable range provided that the desired olefin product is produced. Typically, the molar ratio of hydrogen to oxygen is greater than about 0.5:1, preferably, greater than about 0.7:1, and more preferably, greater than about 1.5:1. Typically, the molar ratio of hydrogen to oxygen is less than about 3.2:1, preferably, less than about 3.0:1, and more preferably, less than about 2.7:1.
- the molar ratio of hydrogen to oxygen typically is greater than about 0.1:1, preferably, greater than about 0.7:1, and more preferably, greater than about 1.5:1.
- the molar ratio of hydrogen to oxygen is typically less than about 4.0:1, preferably, less than about 3.2:1, and more preferably, less than about 3.0:1.
- the feed may contain a diluent, which can be any gas or vaporizable liquid which does not interfere with the process of the invention.
- the diluent functions as a carrier of the reactants and products and facilitates the transfer of heat generated by the process.
- the diluent also helps to minimize undesirable secondary reactions and helps to expand the non-flammable regime for mixtures of the paraffin, hydrogen, and oxygen.
- Suitable diluents include nitrogen, argon, helium, carbon dioxide, carbon monoxide, methane, and steam.
- concentration of diluent in the feed can vary over a wide range.
- the concentration of diluent is typically greater than about 0.1 mole percent of the total reactant feed including paraffin, oxygen, hydrogen, and diluent.
- the amount of diluent is greater than about 1 mole percent of the total reactant feed.
- the amount of diluent is less than about 70 mole percent, and preferably, less than about 40 mole percent, of the total reactant feed.
- the catalyst which is employed in the process of this invention beneficially comprises a Group 8B metal and at least one promoter, described hereinbelow, optionally supported on a catalyst support.
- the Group 8B metals include iron, cobalt, nickel, and the platinum group metals, namely, ruthenium, rhodium, palladium, osmium, iridium, and platinum. Mixtures of the aforementioned Group 8B metals may also be used.
- the Group 8B metal is a platinum group metal; preferably, the platinum group metal is platinum.
- the catalyst also comprises at least one promoter, which is suitably defined as any element or elemental ion which is capable of enhancing the performance of the catalyst, as measured, for example, by an increase in the paraffin conversion, an increase in the selectivity to olefin, a decrease in the selectivities to deep oxidation products, such as carbon monoxide and carbon dioxide, and/or an increase in catalyst stability and lifetime.
- promoter does not include the platinum group metals.
- the promoter is selected from the elements of Groups 1B (Cu, Ag, Au), 6B (Cr, Mo, W), 3A (for example, Al, Ga, In, Tl), 4A (for example, Ge, Sn, Pb), and 5A (for example, As, Sb, Bi), and mixtures thereof. More preferably, the promoter is selected from copper, tin, antimony, silver, indium, and mixtures thereof. Most preferably, the promoter is selected from copper, tin, antimony, and mixtures thereof.
- any atomic ratio of Group 8B metal to promoter can be employed in the catalyst, provided the catalyst is operable in the process of this invention.
- the optimal atomic ratio will vary with the specific Group 8B metal and promoter(s) employed.
- the atomic ratio of the Group 8B metal to promoter is greater than 0.10 (1:10), preferably, greater than about 0.13 (1:8), and more preferably, greater than about 0.17 (1:6).
- the atomic ratio of the Group 8B metal to promoter is less than about 2.0 (1:0.5), preferably, less than about 0.33 (1:3), and more preferably, less than about 0.25 (1:4).
- the promoter is used in a gram-atom amount equivalent to or greater than the Group 8B metal, the promoter nonetheless functions to enhance the catalytic effect of the catalyst.
- Compositions prepared with promoter alone, in the absence of Group 8B metal, are typically (but not necessarily always) catalytically inactive in the process.
- the Group 8B metal is catalytically active in the absence of promoter, albeit with lesser activity.
- the catalyst can be suitably employed in the form of a metallic gauze. More specifically, the gauze can comprise an essentially pure Group 8B metal or an alloy of Group 8B metals onto which the promoter is deposited. Suitable gauzes of this type include pure platinum gauze and platinum-rhodium alloy gauze coated with the promoter. The method used to deposit or coat the promoter onto the gauze can be any of the methods described hereinafter. Alternatively, a gauze comprising an alloy of a Group 8B metal and the promoter can be employed. Suitable examples of this type include gauzes prepared from platinum-tin, platinum-copper, and platinum-tin-copper alloys.
- the Group 8B metal and promoter are supported on a catalytic support.
- the loading of the Group 8B metal on the support can be any which provides for an operable catalyst in the process of this invention.
- the loading of the Group 8B metal is greater than about 0.001 weight percent, preferably, greater than about 0.1 weight percent, and more preferably, greater than about 0.2 weight percent, based on the total weight of the Group 8B metal and support.
- the loading of the Group 8B metal is less than about 80 weight percent, preferably, less than about 60 weight percent, and more preferably, less than about 10 weight percent, based on the total weight of the Group 8B metal and the support.
- the catalytic support comprises any material which provides a surface to carry the Group 8B metal, the promoter(s), and any support modifiers.
- the support is thermally and mechanically stable under autothermal process conditions.
- the catalytic support is a ceramic, such as, a refractory oxide, carbide, or nitride.
- Non-limiting examples of suitable ceramics include alumina, silica, silica-aluminas, aluminosilicates, including cordierite, magnesia, magnesium aluminate spinels, magnesium silicates, zirconia, titania, boria, zirconia toughened alumina (ZTA), lithium aluminum silicates, silicon carbide, oxide-bonded silicon carbide, and silicon nitride. Mixtures of the aforementioned refractory oxides, nitrides, and carbides may also be employed, as well as washcoats of the aforementioned materials on a support.
- Preferred ceramics include magnesia, alumina, silica, and amorphous or crystalline combinations of magnesia, alumina and silica, including mullite.
- Alpha ( ⁇ ) and gamma ( ⁇ ) alumina are preferred forms of alumina.
- Preferred combinations of alumina and silica comprise from about 65 to about 100 weight percent alumina and from essentially 0 to about 35 weight percent silica.
- Other refractory oxides, such as boria can be present in smaller amounts in the preferred alumina and silica mixtures.
- Preferred zirconias include zirconia fully stabilized with calcia (FSZ) and zirconia partially stabilized with magnesia (PSZ), available from Vesuvius Hi-Tech Ceramics, Inc.
- FSZ calcia
- PSZ magnesia
- Magnesia is the most preferred support, because it produces fewer cracking products and less carbon monoxide. Moreover, the hydrocarbon conversion and olefin selectivity tend to be higher with magnesia.
- the catalytic support may take a variety of shapes including that of porous or non-porous spheres, granules, pellets, irregularly shaped solid or porous particles, or any other shape which is suitable for catalytic reactors, including fixed bed, transport bed, and fluidized bed reactors.
- the catalyst is a monolith.
- the term “monolith” means any continuous structure, including for example, honeycomb structures, foams, and fibers, including fibers woven into fabrics or made into non-woven mats or thin paper-like sheets. Monoliths do not, in general, contain significant microporosity. Foams have a sponge-like structure. More preferably, the support is a foam or fiber monolith. Fibers tend to possess higher fracture resistance as compared with foams and honeycombs.
- Preferred ceramic foams available from Vesuvius Hi-Tech Ceramics, Inc., comprise magnesia, alpha alumina, zirconia, or mullite with a porosity ranging from about 5 to about 100 pores per linear inch (ppi) (2 to 40 pores per linear cm (ppcm)). Foams having about 45 ppi (18 ppcm) are more preferred.
- porosity refers to channel size or dimension. It is important to note that the foam supports are not substantially microporous structures. Rather, the foams are macroporous, meaning that they are low surface area supports with channels ranging in diameter from about 0.1 mm to about 5 mm.
- the foams are estimated to have a surface area less than about 10 m 2 /g, and preferably, less than about 2 m 2 /g, but greater than about 0.001 m 2 /g.
- Preferred ceramic fibers available from 3M Corporation as NextelTM brand ceramic fibers, typically have a diameter greater than about 1 micron ( ⁇ m), preferably, greater than about 5 ⁇ m. The diameter is suitably less than about 20 ⁇ m, preferably, less than about 15 ⁇ m.
- the length of the fibers is generally greater than about 0.5 inch (1.25 cm), preferably, greater than about 1 inch (2.5 cm), and typically less than about 10 inches (25.0 cm), preferably, less than about 5 inches (12.5 cm).
- the surface area of the fibers is very low, being generally less than about 1 m 2 /g, preferably, less than about 0.3 m 2 /g, but greater than about 0.001 m 2 /g.
- the fibers are not woven like cloth, but instead are randomly intertwined as in a mat or matted rug.
- NextelTM brand 440 fibers which consist of gamma alumina (70 weight percent), silica (28 weight percent), and boria (2 weight percent)
- NextelTM brand 610 fibers which consist of alpha alumina (99 weight percent), silica (0.2-0.3 weight percent) and iron oxide (0.4-0.7 weight percent).
- the deposition of the Group 8B metal and promoter(s) onto the support can be made by any technique known to those skilled in the art, for example, impregnation, ion-exchange, deposition-precipitation, vapor deposition, sputtering, and ion implantation.
- the Group 8B metal is deposited onto the support by impregnation. Impregnation is described by Charles N. Satterfield in Heterogeneous Catalysis in Practice , McGraw-Hill Book Company, New York, 1980, 82-84, incorporated herein by reference.
- the support is wetted with a solution containing a soluble Group 8B compound, preferably, to the point of incipient wetness.
- the contacting temperature typically ranges from about ambient, taken as 23° C., to about 100° C., preferably, from about 23° C. to about 50° C.
- the contacting is conducted usually at ambient pressure.
- suitable Group 8B compounds include the Group 8B nitrates, halides, sulfates, alkoxides, carboxylates, and Group 8B organometallic compounds, such as halo, amino, acetylacetonate, and carbonyl complexes.
- the Group 8B compound is a platinum group halide, more preferably, a chloride, such as chloroplatinic acid.
- the solvent can be any liquid which solubilizes the Group 8B compound.
- Suitable solvents include water, aliphatic alcohols, aliphatic and aromatic hydrocarbons, and halo-substituted aliphatic and aromatic hydrocarbons.
- concentration of the Group 8B compound in the solution generally ranges from about 0.001 molar (M) to about 10 M.
- the support After contacting the support with the solution containing the Group 8B compound, the support may be dried under air at a temperature ranging from about 23° C. to a temperature below the decomposition temperature of the Group 8B compound, typically, a temperature between about 23° C. and about 100° C.
- the deposition of the promoter can be accomplished in a manner analogous to the deposition of the Group 8B metal. Accordingly, if impregnation is used, then the support is wetted with a solution containing a soluble compound of the promoter at a temperature between about 23° C. and about 100° C., preferably, between about 23° C. and about 50° C., at about ambient pressure.
- soluble promoter compounds include promoter halides, nitrates, alkoxides, carboxylates, sulfates, and organometallic compounds, such as amino, halo, and carbonyl complexes.
- Suitable solvents comprise water, aliphatic alcohols, aliphatic and aromatic hydrocarbons, and chloro-substituted aliphatic and aromatic hydrocarbons.
- Certain promoter compounds such as compounds of antimony and tin, may be more readily solubilized in the presence of acid.
- hydrochloric acid 5-25 weight percent
- the concentration of the promoter compound in the solution generally ranges from about 0.01 M to about 10 M.
- the impregnated support may be dried under air at a temperature between about 23° C. and a temperature below the temperature wherein vaporization or decomposition of the promoter compound occurs. Typically, the drying is conducted at a temperature between about 23° C. and about 100° C.
- the Group 8B metal is deposited onto the support first, and thereafter the promoter is deposited onto the support.
- the promoter is deposited first, followed by the deposition of the Group 8B metal.
- the Group 8B metal and the promoter are deposited simultaneously onto the support from the same deposition solution.
- a calcination under oxygen is optional. If performed, the calcination is conducted at a temperature ranging from about 100° C. to below the temperature at which volatilization of the metals becomes significant, typically, less than about 1,100° C. Preferably, the calcination is conducted at a temperature between about 100° C. and about 500° C.
- the fully-loaded support is reduced under a reducing agent, such as hydrogen, carbon monoxide, or ammonia, at a temperature between about 100° C. and about 900° C., preferably between about 125° C. and about 800° C., so as to convert the Group 8B metal substantially to its elemental form.
- a reducing agent such as hydrogen, carbon monoxide, or ammonia
- the promoter may be reduced fully or partially, or not reduced at all, depending upon the specific promoter chosen and the reduction conditions.
- reduction at elevated temperatures may produce alloys of the Group 8B metal and the promoter. Alloys may provide enhanced catalyst stability by retarding vaporization of the promoter during the process of this invention.
- the support is pretreated with a support modifier prior to loading the Group 8B and promoter(s).
- the support modifier can be any metal ion having a charge of +1 or greater.
- the support modifier is selected from Groups 1A (Li, Na, K, Rb, Cs), 2A (for example, Mg, Ca, Sr, Ba), 3B (Sc, Y, La), 4B (Ti, Zr, Hf), 5B (V, Nb, Ta), 6B (Cr, Mo, W), 1B (Cu, Ag, Au), 3A (for example, Al, Ga, In), 4A (for example, Ge, Sn, Pb), 5A (for example, As, Sb, Bi), and the lanthanide rare earths (for example, Ce, Er, Lu, Ho) and actinide elements (specifically Th) of the Periodic Table previously identified.
- the support modifier is selected from calcium, zirconium, tin, lanthanum, potassium, lutetium, erbium, barium, holmium, cerium, antimony, and mixtures thereof. Most preferably, the support modifier is selected from lanthanum, tin, antimony, calcium, and mixtures thereof. Certain elements, such as tin, antimony, and silver, may function as both promoter and support modifier simultaneously.
- the procedure to modify the support comprises contacting the support with a solution containing a soluble compound of the support modifier.
- the contacting can involve ion-exchange or impregnation methods.
- the modification procedure involves submerging the support in the solution such that essentially all of the surface area of the support is contacted with an excess of the solution.
- Compounds suitable for preparing the solution of support modifier include modifier nitrates, halides, particularly the chlorides, alkoxides, carboxylates, and organometallic complexes including amino, halo, alkyl, and carbonyl complexes.
- Suitable solvents include water, aliphatic alcohols, aromatic hydrocarbons, and halo-substituted aliphatic and aromatic hydrocarbons.
- concentration of modifier compound in the solution ranges from about 0.001 M to about 10 M.
- Acidified solutions for example, of hydrochloric acid and diluted solutions thereof, may be beneficially employed.
- the contact time generally ranges from about 1 minute to about 1 day.
- the contacting temperature suitably ranges from about 23° C. to about 100° C., and pressure is generally ambient.
- slurries of mixed oxides containing promoter and/or modifier elements, such as magnesium stannate (Mg 2 SnO 4 ) can be deposited onto the support.
- the modified support is typically calcined, as noted hereinabove, or reduced under a reducing agent, such as hydrogen, at a temperature between about 100° C. and about 900° C., preferably, between about 200° C. and about 800° C.
- a reducing agent such as hydrogen
- the choice of calcination or reduction depends on the element used to pretreat the support. If the element or its oxide is readily vaporizable, the pretreated support is reduced. If the element or its oxide is not readily vaporizable, then the pretreated support is calcined.
- the words “readily vaporizable” may be taken to mean that greater than about 1 weight percent of any metal component in the catalyst is vaporized in a period of about 24 hours under calcination conditions at about 200° C.
- the term “readily vaporizable” may be given a narrower or broader definition, as desired.
- the Group 8B metal and promoter(s) are loaded onto the support. Then, the support is reduced as described hereinbefore. Alternatively, the metal-loaded support may be calcined first and then reduced. Whether the modified support is calcined or not depends again upon the vaporization potential of the modifier metal(s) and promoter(s) employed. Supports modified with metals or metal oxides which tend to vaporize readily are typically not calcined. Supports modified with metals or metal oxides which do not vaporize readily can be calcined.
- the process of this invention is advantageously conducted under autothermal process conditions.
- autothermal process conditions means that the heat generated by reaction of the feed is sufficient to support the catalytic process which converts the paraffin to the olefin. Accordingly, the need for an external heating source to supply the energy for the process can be eliminated.
- the catalysts of the prior art are required to support combustion beyond the normal, fuel-rich limit of flammability. This is not a requirement in the present invention.
- autothermal conditions can also be maintained with a catalyst which does not support combustion beyond the normal, fuel-rich limit of flammability, provided that hydrogen and optionally a preheat are supplied to the process.
- Ignition can be effected by preheating the feed to a temperature sufficient to effect ignition when contacted with the catalyst.
- the feed can be ignited with an ignition source, such as a spark or flame.
- an ignition source such as a spark or flame.
- the reaction-generated heat causes the temperature to take a step change jump to a new steady state level that is herein referred to as the autothermal reaction.
- the paraffin feed does not have to be preheated, so long as the feed contains hydrogen or the catalyst supports combustion beyond the normal, fuel-rich limit of flammability.
- combustion means the reaction of the hydrocarbon with oxygen unaided by hydrogen.
- Preheating the feedstream has certain advantages. The advantages comprise a decrease in oxygen and hydrogen consumed, an increase in the paraffin concentration in the feed, an increase in the operating paraffin to oxygen molar ratio, and a net increase in recycle hydrogen in the product stream.
- catalysts can be used which do not support combustion beyond the normal fuel-rich limit of flammability. These advantages are particularly significant when the preheating is conducted at a temperature greater than about 200° C.
- Suitable preheat temperatures are typically greater than about 40° C., preferably, greater than about 125° C., and even more preferably, greater than about 200° C. In another preferred embodiment, the preheat temperature is greater than about 400° C. Suitable preheat temperatures are typically less than about 900° C., preferably, less than about 800° C., and more preferably less than about 600° C.
- the autothermal process operates at close to the adiabatic temperature (that is, essentially without loss of heat), which is typically greater than about 750° C., and preferably, greater than about 925° C.
- the autothermal process operates at a temperature less than about 1,150° C., and preferably, less than about 1,050° C.
- the temperature at the reactor exit can be measured, for example, by using a Pt/Pt—Rh thin wire thermocouple. With a monolith catalyst, the thermocouple can be sandwiched between the monolith and the downstream radiation shield. Measurement of temperature close to the reactor exit may be complicated by the high temperature involved and the fragility of the thermocouple.
- adiabatic temperature is the temperature of the product stream without any heat loss, that is, when all of the heat generated by the process is used to heat the products.
- the measured temperature is found to be within about 25° C. of the calculated adiabatic temperature.
- the operating pressure is typically equal to or greater than about 1 atmosphere absolute (atm abs) (100 kPa abs). Typically, the pressure is less than about 20 atm abs (2,000 kPa abs), preferably, less than about 10 atm (1,000 kPa abs), and more preferably, less than about 7 atm abs (700 kPa abs).
- gas hourly space velocity calculated as the total flow of the hydrocarbon, oxygen, hydrogen, and optional diluent flows, is greater than about 50,000 ml total feed per ml catalyst per hour (h ⁇ 1 ) measured at standard temperature and pressure (0° C., 1 atm) (STP).
- the GHSV is greater than about 80,000 h ⁇ 1 , and more preferably, greater than 100,000 h ⁇ 1 .
- the gas hourly space velocity is less than about 6,000,000 h ⁇ 1 , preferably, less than about 4,000,000 h ⁇ 1 , more preferably, less than 3,000,000 h ⁇ 1 , measured as the total flow at STP. Gas flows are typically monitored in units of liters per minute at standard temperature and pressure (slpm).
- the residence time of the reactants in the reactor is simply calculated as the inverse of the gas hourly space velocity.
- the residence time is on the order of milliseconds.
- a gas hourly space velocity of 100,000 h ⁇ 1 measured at STP is equivalent to a residence time of 36 milliseconds at STP.
- the process of this invention may be conducted in any reactor designed for use under adiabatic, autothermal process conditions.
- the catalyst is prepared on a monolith support which is sandwiched between two radiation shields inside a reactor housing.
- fixed bed and fluidized bed reactors can be used with catalysts in the form of pellets, spheres, and other particulate shapes. Continuous and intermittent flow of the feedstream are both suitable.
- fluidized bed reactors of the prior art typically possess an aspect ratio in static mode of greater than 1:1, and more preferably, greater than about 5:1. Static mode is defined as the unfluidized or fixed bed configuration.
- Fluidized bed reactors are generally operated in a bubbling, turbulent, or fast-fluidized regime with expanded beds measuring from about 1.5 to 15 times the static depth.
- the aspect ratio in operating mode is greater than about 5:1 to 10:1.
- a catalyst particle size ranging between about 30 and 1,000 microns is satisfactory.
- the oxidation reaction of this invention occurs predominantly at the reactor entry, which in the case of a stationary catalyst is at the front edge of the catalyst.
- the optimal reactor for the process of this invention should possess a large cross-sectional dimension and a short height (or depth).
- a catalyst bed of diameter about 5 to 8 feet (1.5 m to 2.4 m) and a height of about 1 inch (2.5 cm) may be suitably employed.
- catalyst located at the front edge of a stationary bed can deactivate more quickly with time. As a consequence, longer catalyst lifetime and better selectivities can be achieved by circulating particles of the catalyst, rather than using a stationary bed.
- a preferred reactor design for the process of this invention comprises a modified fluidized bed reactor, characterized in that its aspect ratio in operating mode, and preferably also in static mode (unfluidized or fixed bed configuration), is less than 1:1, and more preferably, less than about 0.1:1, but greater than about 0.001:1. Most preferably, the aspect ratio is about 0.01:1.
- This unique fluidized bed is operated above the minimum fluidization flow with an expanded bed on the order of about 2 or 3 times the static depth, and preferably, less than about 1.5 times the static depth.
- “minimum fluidization flow” is defined as the minimum gas velocity at which the catalyst particles are suspended under operating conditions.
- the velocity necessary to achieve minimum fluidization depends upon the density and viscosity of the gas phase and the catalyst particle size and density.
- One skilled in the art would know how to calculate the minimum fluidization flow for any given gas composition and catalyst particle.
- a suitable authority on the subject is found in Fluidization Engineering, by D. Kunii and O. Levenspeil, 2 nd ed., Butterworth-Heineman, 1989, incorporated herein by reference.
- a catalyst particle size of between about 500 and about 850 microns (23-30 US mesh) is suitable for feed velocities of about 0.05 to 5 meters per second (mps) at standard temperature and pressure.
- An advantage of the modified fluidized bed reactor may result from its continuous circulation (fluidization), which results in continuous renewal of catalyst particles at the reactor entry. This configuration produces substantially better product yields than a stationary catalyst.
- an olefin preferably a mono-olefin
- Ethane is converted primarily to ethylene.
- Propane and butane are converted primarily to ethylene and propylene.
- Isobutane is converted primarily to isobutylene and propylene.
- Naphtha and other higher molecular weight paraffins are converted primarily to ethylene and propylene.
- the conversion of paraffinic hydrocarbon in the process of this invention can vary depending upon the specific feed composition, catalyst composition, reactor, and process conditions employed.
- conversion is defined as the mole percentage of paraffinic hydrocarbon in the feed which is converted to products.
- the conversion increases with increasing temperature.
- the conversion does not change significantly over a wide range of high space velocities employed.
- the conversion of paraffinic hydrocarbon is typically greater than about 50 mole percent, preferably, greater than about 60 mole percent, and more preferably, greater than about 70 mole percent.
- selectivity is defined as the percentage of carbon atoms in the converted paraffin feed which react to form a specific product.
- olefin selectivity is calculated as follows: Moles ⁇ ⁇ of ⁇ ⁇ olefin ⁇ ⁇ formed ⁇ Number ⁇ ⁇ of ⁇ ⁇ carbon ⁇ ⁇ atoms ⁇ ⁇ in ⁇ ⁇ olefin Moles ⁇ ⁇ of ⁇ ⁇ paraffin ⁇ ⁇ converted ⁇ Number ⁇ ⁇ of ⁇ ⁇ carbon ⁇ atoms ⁇ ⁇ in ⁇ ⁇ paraffin ⁇ 100
- the olefin selectivity increases with increasing temperature up to a maximum value and declines as the temperature continues to rise. Usually, the olefin selectivity does not change substantially over a wide range of high space velocities employed.
- the olefin selectivity is typically greater than about 50 carbon atom percent, preferably, greater than about 60 carbon atom percent, more preferably, greater than about 70 carbon atom percent, and even more preferably, greater than about 80 carbon atom percent.
- Other products formed in smaller quantities include methane, carbon monoxide, carbon dioxide, propane, butenes, butadiene, propadiene, acetylene, methylacetylene, and C 6+ hydrocarbons.
- Acetylene can be hydrogenated to ethylene downstream to increase the overall selectivity to olefin. At least part of the carbon monoxide, carbon dioxide, and methane formed may be recycled to the reactor.
- Water is also formed in the process of this invention from the reaction of hydrogen or hydrocarbon.
- the presence of hydrogen in the feed minimizes the formation of carbon oxides by reacting with the oxygen to produce water and energy. Accordingly, it is advantageous to recycle the hydrogen in the product stream, obtained from the dehydrogenation of the paraffin, back to the reactor.
- the hydrogen needed to meet the demands of the process essentially equals the hydrogen formed during conversion of the paraffin to olefin. Under these balanced conditions, the hydrogen forms a closed loop wherein there is essentially no demand for additional hydrogen to be added to the feed. Such conditions are more easily met when the feed is preheated and a higher hydrocarbon to oxygen molar ratio is employed.
- a catalyst comprising platinum and tin supported on an alumina monolith was prepared by the following method. Platinum and tin were codeposited on a foam monolith (92 weight percent alpha alumina, 8 weight percent silica; 1.8 cm diameter ⁇ 1 cm thick, 45 ppi (18 ppcm)) by impregnation with an aqueous solution of platinum and tin in a Pt:Sn atomic ratio of 1:5.
- the impregnation solution was prepared from a stock aqueous solution of hexachloroplatinic acid (0.193 M H 2 PtCl 6 ) and a stock aqueous solution of stannous chloride (0.372 M SnCl 2 ) acidified with 5 weight percent hydrochloric acid. Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent.
- the impregnated monolith was dried in ambient air and then reduced under flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cubic foot per hour (cfh) (473 cm 3 /min) using the following temperature profile: 1 h from ambient to 125° C., then 1 h from 125° C. to 300° C.; 1 h from 300° C. to 450° C., held for 30 min at 450° C., and then cooled to room temperature.
- the catalyst was sandwiched between two inert alpha alumina monoliths which acted as radiation shields.
- the monoliths were sealed in a quartz tube using FiberFraxTM brand alumina-silica cloth (FiberFrax is a trademark of and available from Unifrax Corporation), and the reactor was insulated by wrapping the quartz tube with high temperature insulation.
- a feed comprising ethane (2.6 standard liters per minute (slpm)), oxygen (1.3 slpm), hydrogen (2.6 slpm) and nitrogen (1.147 slpm) was fed to the reactor.
- Total flow was 7.647 slpm (GHSV 180,305 h ⁇ 1 ) at 15 volume percent dilution with nitrogen.
- the ethane to oxygen molar ratio was 2:1; the hydrogen to oxygen molar ratio was 2:1.
- the catalyst was operated autothermally and the heat generated by the reaction was sufficient to sustain the process. However, heat was needed initially to ignite the process.
- the procedure for light-off involved establishing the flows of nitrogen, ethane, and hydrogen, then adding the oxygen flow; and then heating the feed to 200° C. until ignition. This procedure ensured a fuel-rich feed for safety considerations.
- the light-off conditions were 7 slpm total gas flow, 2.24 slpm of ethane, 2.24 slpm of hydrogen, 1.12 slpm of oxygen, 1.40 slpm of nitrogen, 20 percent dilution with nitrogen, an ethane to oxygen molar ratio of 2:1, a hydrogen to oxygen molar ratio of 2:1, and a pressure of 1.34 atm abs (136 kPa abs).
- the external heat source was removed, and the gas flow rates and pressure were adjusted to the desired conditions, as shown in Table 1. Pressure was maintained at 1.34 atm abs (136 kPa abs). Shutdown of the reactor was accomplished by turning off oxygen prior to alkane and hydrogen.
- a catalyst comprising platinum and tin supported on a ceramic monolith was active in the partial oxidation of ethane in the presence of hydrogen to produce ethylene.
- the catalyst achieved an ethane conversion of 69.6 percent and an ethylene selectivity of 81.1 percent.
- the ethane conversion and ethylene selectivity achieved were comparable to those obtained from commercial thermal cracking furnaces.
- Very low amounts of carbon monoxide (7.29 percent) and carbon dioxide (0.34 percent) were found, as well as comparable amounts of methane and C3+ products. Carbon monoxide, carbon dioxide, and methane, at least in part, can be recycled to the reactor along with the hydrogen produced in the process.
- the oxidation of ethane was conducted under autothermal process conditions with a catalyst consisting of platinum supported on a ceramic monolith support.
- the catalyst was prepared as in E-1, with the exception that no tin was added to the catalyst.
- the process was conducted first in the absence of hydrogen (CE-1a) as noted hereinafter, and then, in the presence of hydrogen (CE-1b) in a manner similar to E-1.
- Molar ratio of ethane to oxygen was 2:1. This flow adjustment ensured that identical absolute amounts of ethane and oxygen were used with and without hydrogen. The level of nitrogen dilution was adjusted to ensure equivalent ethane conversions with and without hydrogen. Processes were conducted autothermally in the manner described in E-1 with the results shown in Table 1 (CE-1a and CE-1b).
- a catalyst comprising platinum and antimony supported on an alumina monolith was prepared in a manner similar to that described in Example 1.
- the monolith of Example 1 was impregnated with an aqueous solution of platinum and antimony in a Pt:Sb atomic ratio of 1:5.
- the impregnation solution was prepared from a stock aqueous solution of hexachloroplatinic acid (0.193 M H 2 PtCl 6 ) and a stock aqueous solution of antimony triacetate (0.182 M Sb(OAc) 3 ) containing hydrochloric acid sufficient to dissolve the antimony salt.
- Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent.
- the impregnated monolith was dried in ambient air, then reduced under flowing hydrogen in the manner described in E-1 hereinabove.
- the catalyst was tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions as described in E-1 with the results shown in Table 1 hereinabove. It was seen that under autothermal process conditions a catalyst comprising platinum and antimony supported on a ceramic monolith achieved an ethane conversion of 69.5 percent and an ethylene selectivity of 81.5 percent. Selectivities to carbon monoxide and carbon dioxide were low. The results are comparable to those achieved in commercial thermal cracking furnaces.
- a catalyst comprising platinum, tin, and antimony supported on an alumina monolith was prepared in a manner similar to that described in Examples 1 and 2.
- the metals were codeposited by impregnation of the support with an aqueous solution of Pt, Sn, and Sb salts in a Pt:Sn:Sb atomic ratio of 1:5:0.26.
- the impregnation solution was prepared from a stock aqueous solution of platinum hexachloroplatinic acid (0.193 M), a stock aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent), and a stock aqueous solution of antimony triacetate (0.182 M) containing hydrochloric acid (50 weight percent). Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent.
- the impregnated monolith was dried in ambient air and reduced under hydrogen as described in E-1 hereinabove.
- the catalyst was tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions as described in E-1 with the results shown in Table 1 hereinabove. It was seen that a catalyst comprising platinum, tin, and antimony supported on a ceramic monolith achieved an ethane conversion of 68.5 percent and an ethylene selectivity of 80.6 percent. Carbon monoxide and carbon dioxide were produced only at low levels. The results are comparable to those achieved in commercial thermal cracking furnaces.
- the catalyst of E-3 comprising platinum, tin, and antimony on an alumina monolith was evaluated in the oxidation of ethane as described in E-3, with the exception that no hydrogen was used in the feedstream. Process conditions were as described in CE-1a. Results are set forth in Table 1 (CE-3). When E-3 was compared with CE-3 and any of CE 1a and CE-1b, it was seen that the combined use of antimony and tin in the catalyst and hydrogen in the feedstream as shown in E-3 gave the highest conversion of ethane, the highest selectivity to ethylene, and the lowest levels of carbon oxides.
- An aqueous impregnation solution was prepared containing platinum and copper in a Pt:Cu atomic ratio of 1:5.
- the impregnation solution was prepared from stock solutions of chloroplatinic acid (0.193 M H 2 PtCl 6 ) and cupric chloride (1.49 M CuC1 2 ).
- a ceramic fiber mat (NextelTM 440 brand fiber mat, 2 cm square ⁇ 1 cm thick, weighing 0.25 g) was precalcined in air at 900° C., cooled, and then impregnated with the impregnation solution to saturation. Sufficient solution was used to obtain a calculated platinum loading of 16 weight percent in the finished mat.
- the impregnated fiber mat was dried in ambient air, then reduced in flowing hydrogen, as described in E-1.
- the catalyst was sandwiched between two inert foam monoliths (1.8 cm dia by 1 cm thick, 18 ppcm alumina or mullite), wrapped in FiberFraxTM brand alumina-silica cloth, and packed into a quartz tube reactor (Inner Diameter (I.D.) 1.9 cm).
- the feed to the reactor was preheated with a heating tape wrapped around the quartz tube upstream of the catalyst.
- the catalyst zone was not heated, but was insulated with high temperature insulation material to minimize heat losses.
- Ethane, hydrogen, and nitrogen were preheated to 200° C. and fed to the reactor. Oxygen was then introduced to the reactor which resulted in catalyst ignition. Upon ignition the temperature rose within a few seconds to 1000° C., and the reactor operated autothermally.
- Catalyst A comprised platinum and copper in a Pt:Cu atomic ratio of 1:1 supported on an alumina foam monolith.
- the monolith of E-1 was impregnated with an aqueous impregnation solution (1 ml) prepared from stock solutions of hexachloroplatinic acid (0.193 M) and cupric chloride (1.49 M). Sufficient stock solutions were used to achieve a Pt:Cu atomic ratio of 1:1. Calculated platinum loading was 1.2 weight percent.
- the impregnated monolith was dried and reduced under hydrogen in the manner described in E-1.
- Catalyst B comprised platinum and copper in a Pt:Cu atomic ratio 1:1 supported on a NextelTM 440 ceramic fiber mat.
- the catalyst was prepared in the manner described in E-4 hereinabove, with the exception that the amounts of stock solutions used were adjusted to provide the Pt:Cu atomic ratio of 1:1. Calculated platinum loading was 20 weight percent.
- Catalyst C comprised platinum and copper in an atomic ratio of 1:2 supported on a NextelTM 440 ceramic fiber mat.
- the catalyst was prepared in the manner described in E-4 hereinabove, with the exception that the amounts of stock solutions used were adjusted to give the Pt:Cu atomic ratio of 1:2. Calculated platinum loading was 24 weight percent.
- Catalyst D comprised platinum, tin, and copper in an atomic ratio 1:1:1 supported on a NextelTM 440 ceramic fiber mat.
- the catalyst was prepared by calcining the fiber mat at 900° C., cooling, then impregnating the calcined mat to wetness with an impregnation solution prepared from stock solutions of hexachloroplatinic acid (0.193 M), cupric chloride (1.49 M), and stannous chloride (0.372 M) acidified with 5 weight percent hydrochloric acid. Calculated platinum loading was 18 weight percent.
- the impregnated fiber mat was dried in ambient air and reduced under hydrogen, as described in E-1 hereinbefore.
- Catalyst E comprised platinum and tin in an atomic ratio of 1:5 supported on a NextelTM 440 brand ceramic fiber mat.
- the catalyst was prepared by calcining the fiber mat at 900° C., cooling, and impregnating the fiber mat to saturation with an aqueous impregnation solution prepared from stock solutions of hexachloroplatinic acid (0.193 M) and stannous chloride (0.372 M) acidified with hydrochloric acid (5 weight percent). Calculated platinum loading was 8.5 weight percent.
- the impregnated fiber mat was dried and reduced as described in E-1 hereinbefore.
- catalysts comprising platinum and tin, copper, or a mixture thereof, supported on a ceramic foam or fiber mat achieved a high ethane conversion, a high ethylene selectivity, and good catalyst stability in a process of oxidizing ethane to ethylene in the presence of hydrogen.
- Catalysts comprising platinum and copper supported on NextelTM 440 brand ceramic fiber mats were prepared in the manner described in E-4 hereinabove.
- the atomic ratio of platinum to copper was varied from 1:0.1 to 1:5.
- the catalysts were tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions, with the results shown in Table 4.
- TABLE 4 Oxidation of Ethane to Ethylene Variation in Pt/Cu Ratio a Pt:Cu Pt loading TOS b % C 2 H 4 % C 2 H 6 ratio (wt %) h Sel. Conv.
- a catalyst comprising platinum and copper in an atomic ratio of 1:1 supported on a NextelTM 440 brand ceramic fiber mat was prepared in a manner similar to that described in E-4 hereinabove.
- the catalyst was evaluated in the oxidation of ethane to ethylene in the presence of hydrogen under autothermal process conditions.
- the gas hourly space velocity of the total feed was progressively increased at constant pressure with the results shown in Table 5.
- TABLE 5 Oxidation of Ethane to Ethylene Variation in Space Velocity a Flow rate GHSV % C 2 H 4 % C 2 H 6 slpm h ⁇ 1 Sel. Conv. 7 330,100 80.6 54.3 14 660,200 81.0 55.7 21 990,300 81.7 55.5 28 1,320,400 81.6 55.7 35 1,650,500 82.4 53.0 42 1,980,600 81.9 52.4
- catalysts comprising platinum supported on a modified ceramic foam monolith (92 weight percent alpha alumina, 8 weight percent silica; 45 ppi (18 ppcm); 1.8 cm dia by 1 cm thick; average weight 2.8 g).
- the preparation was characterized by first modifying the support with a support modifier, specifically tin or antimony, and then depositing platinum and optionally copper on the modified support.
- a support modifier specifically tin or antimony
- platinum and optionally copper on the modified support.
- the same element (Sn) that modifies the support also functions as a promoter. Details of the preparation were as follows:
- Catalyst A comprised platinum on a tin-modified alumina monolith.
- the monolith of E-1 was impregnated to wetness with an aqueous solution of stannous chloride (0.372 M) containing 5 weight percent hydrochloric acid.
- the impregnated support was air dried and then reduced at 700° C. under flowing hydrogen at a flow rate of 1 cfh (473 cm 3 /min).
- the modified support was impregnated with an aqueous solution (1 ml) of hexachloroplatinic acid (0.193 M), then dried in ambient air and reduced under hydrogen as described in E-1.
- Catalyst B comprised platinum and copper (1:1) on a tin-modified alumina monolith.
- the monolith was impregnated to wetness with an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent).
- the tin-impregnated monolith was air dried and reduced at 700° C. for 2 h in flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cfh (473 cm 3 /min).
- the modified monolith was impregnated with an aqueous solution (1 ml) prepared from stock solutions of hexachloroplatinic acid (0.193 M) and cupric chloride (1.49 M). The impregnated monolith was air dried and reduced under flowing hydrogen as described in E-1.
- Catalyst C comprised platinum and copper (1:5) on a tin-modified alumina monolith.
- the catalyst was prepared as in “B” hereinabove with the exception that sufficient stock solutions were used to give a Pt:Cu atomic ratio of 1:5.
- Catalyst D comprised platinum and copper (1:5) on an antimony-modified alumina monolith.
- the monolith comprised alpha alumina (99.5 weight percent).
- the monolith was impregnated to wetness with a solution of antimony triacetate (0.182 M) dissolved in hydrochloric acid.
- the monolith was air dried and reduced for 2 h at 700° C. under flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cfh (473 cm 3 /min).
- the reduced monolith was impregnated with an aqueous impregnation solution (1 ml) prepared from a stock solution of hexachloroplatinic acid (0.193 M, 1 ml) and a stock solution of cupric chloride (1.49 M, 0.65 ml).
- the monolith was dried in air and reduced as in E-1.
- a catalyst comprising platinum and copper (1:2) on a tin-modified alumina monolith (92 weight percent alumina) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:2.
- This catalyst was evaluated in the oxidation of a natural gas liquid (NGL) feed in the presence of hydrogen under autothermal process conditions.
- NNL natural gas liquid
- the liquid feed composition, an Norwayn condensate comprised on a weight percent basis a mixture of 42.1 percent paraffins, 34.4 percent isoparaffins, 7.3 percent aromatics, 12.3 percent naphthenes, 0.2 percent oxygenates, and the balance (about 3-4 percent) unidentified components.
- the alkanes comprised C 1-19 alkanes having a maximum molar concentration in the C 5-8 range. Feed was preheated at 200° C. Total gas flow was about 8 slpm (GHSV 200,000 h ⁇ 1 ). Process conditions and results are set forth in Table 7. TABLE 7 Oxidation of Natural Gas Liquid Feed (NGL) a,b Liq.
- a catalyst comprising platinum and copper on a ceramic monolith support was capable of oxidizing a natural gas liquid feed in the presence of hydrogen under autothermal conditions to a mixture of low molecular weight olefins, specifically, ethylene, propylene, butylene, and butadiene.
- Catalysts were prepared comprising platinum and copper supported on a modified ceramic foam monolith.
- the preparation was characterized by first modifying the support with tin and optionally a second modifier, and thereafter depositing platinum and copper on the modified support.
- the support comprised a foam monolith, either 92 or 99.5 weight percent alumina [1.8 cm dia ⁇ 1 cm thick; 45 ppi (18 ppcm)]. Preparations were as follows:
- Catalyst A comprising platinum and copper (1:2) on a tin-modified alumina (92 weight percent) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:2.
- Catalyst B comprising platinum and copper (1:5) on a tin-modified alumina (92.0 weight percent) was prepared as in Example 8C.
- Catalyst C comprising platinum and copper (1:4) on a tin-modified alumina (99.5 weight percent) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:4.
- Catalyst D comprising platinum and copper (1:5) on a tin-modified alumina (99.5 weight percent) was prepared as in Example 8C.
- Catalyst E comprised platinum and copper (1:5) on a tin and calcium-modified alumina monolith (99.5 weight percent).
- the monolith was immersed in a saturated aqueous solution of calcium hydroxide for 24 h. Then, the monolith was rinsed several times with distilled water, air dried, and calcined at 900° C. for 1 h.
- the calcined monolith was immersed in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent) for several hours, after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm 3 /min) at 700° C. for 2 h.
- An aqueous impregnation solution having a Pt:Cu atomic ratio of 1:5 was prepared from stock solutions comprising hexachloroplatinic acid (1 ml, 0.193 M) and cupric chloride (0.65 ml, 1.49 M). The monolith was impregnated with the impregnation solution (1 ml). The impregnated monolith was dried in ambient air and reduced as in E-1.
- Catalyst F comprised platinum and copper (1:5) on a tin and zirconium-modified alumina monolith (99.5 weight percent).
- the monolith was immersed for 24 h in an aqueous solution of zirconium oxychloride (ZrOCl 2 , 1M) containing 1 weight percent hydrochloric acid.
- the monolith was rinsed with distilled water, air dried, and calcined at 900° C. for 1 h.
- the calcined monolith was immersed for several hours in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent), after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm 3 /min) at 700° C. for 2 h.
- An impregnation solution containing platinum and copper (1:5) was prepared from stock solutions of hexachloroplatinic acid (1 ml, 0.193 M) and cupric chloride (0.65 ml, 1.49 M). The monolith was impregnated with the impregnation solution (1 ml), dried in air, and reduced as in E-1.
- Catalyst G comprised platinum and copper (1:5) on a tin and lanthanum-modified alumina foam monolith (99.5 weight percent).
- the monolith was immersed for 24 h in an aqueous solution of lanthanum chloride (1 M) containing 1 weight percent hydrochloric acid.
- the monolith was rinsed with distilled water several times, air dried, and then calcined at 900° C. for 1 h.
- the calcined monolith was cooled and then immersed in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent) for several hours, after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm 3 /min) at 700° C. for 2 h.
- the modified monolith was impregnated with an impregnation solution (1 ml) containing platinum and copper (1:5) prepared from the stock solutions noted hereinbefore. The impregnated monolith was reduced under hydrogen as in E-1.
- the catalyst of E-7 was evaluated in the partial oxidation of ethane to ethylene in the presence of hydrogen in the manner described in E-7, with the exception that the pressure in the reactor was varied from about 2 atm abs (200 kPa abs) to about 4 atm abs (400 kPa abs).
- the catalyst comprised platinum and copper in an atomic ratio 1:1 supported on a NextelTM 440 brand fiber mat. Process conditions and results are set forth in Table 9. TABLE 9 Oxidation of Ethane-Variation in Pressure a, b Flow rate GHSV % N 2 Pressure % C 2 H 4 % C 2 H 6 slpm h ⁇ 1 dilution kPa Sel. Conv.
- Catalyst A identical to Catalyst E-1 hereinabove, comprised platinum and tin in an atomic ratio 1:5 supported on an alumina foam monolith (92 weight percent).
- Feed comprised a mixture of ethane (70 volume percent) and propane (30 volume percent) at a nitrogen dilution of 21 percent.
- Table 10 Other process conditions and results are shown in Table 10.
- Catalyst B identical to Catalyst E-3 hereinabove, comprised platinum, tin, and antimony in an atomic ratio of 1:5:0.26 supported on an alumina foam monolith (92 weight percent). Feed comprised propane at a nitrogen dilution of 30 percent. Process conditions and results are shown in Table 10. TABLE 10 Partial Oxidation of Propane to Propylene and Ethylene With Hydrogen and Multi-Metallic Catalyst Mol % Conversion Feed % C 2 H 6 % C 3 H 8 % Carbon Atom Selectivities Conditions a, b Conv Conv C 2 H 4 C 3 H 6 CO CO 2 CH 4 C4+ A a.
- a catalyst comprising platinum and copper on a tin and lanthanum-modified alumina monolith support was prepared as in Example E-10G.
- the catalyst was evaluated in the autothermal partial oxidation of ethane to ethylene under the conditions shown in Table 11.
- the feed comprising ethane, oxygen, hydrogen, and nitrogen was preheated to temperatures ranging from 281° C. to 589° C.
- the molar ratio of ethane to oxygen was raised to 2.7:1 and higher. Results are set forth in Table 11.
- a catalyst was prepared as in Example E-4 hereinabove, with the exception that magnesia pellets (Norton; 3 mm dia ⁇ 5 mm length cylinders) were used in place of the alumina fiber mat support. The magnesia pellets were heated to 1200° C. for 16 h to reduce the surface area to less than 1 m 2 /g. A solution containing platinum and copper in an atomic ratio of 1:5 was prepared using hexachloroplatinic acid and cupric chloride. The pellets were loaded with the solution, dried at 80° C. overnight, and reduced at 450° C. under hydrogen (5 volume percent) in nitrogen. Pt:Cu atomic ratio was 1:5.6. Pt loading, 0.57 weight percent; copper loading, 1.03 weight percent; balance magnesia.
- magnesia pellets Norton; 3 mm dia ⁇ 5 mm length cylinders
- the catalyst particles were sandwiched between two inert alumina monoliths in a quartz tube reactor. Catalyst bed dimensions were 17 mm (dia) by 15 mm (depth). The catalyst was evaluated in the autothermal oxidation of ethane to ethylene in the manner described hereinbefore. Process conditions and results are set forth in Table 12.
- magnesia pellets can be suitably employed as a catalyst support in the autothermal oxidation process of this invention.
- a catalyst comprising platinum and copper on a ceramic monolith was prepared in the manner described in Example E-4, with the exception that a magnesia monolith (Hi-Tech Ceramics, 17 mm dia ⁇ 10 mm width, 45 ppi) was used in place of the alumina fiber mat. Pt:Cu atomic ratio was 1:5, and the total metal loading was 5.67 weight percent.
- the catalyst was evaluated in the autothermal oxidation of ethane to ethylene as described hereinbefore, with the results shown in Table 13.
- magnesia monolith can be suitably employed as a support in the autothermal oxidation process of this invention.
- Alumina beads were used to prepare a catalyst.
- a solution containing platinum, copper, and tin in an atomic ratio of 1:5:5 was prepared by mixing hexachloroplatinic acid (0.659 ml, 0.193 M), cupric chloride (0.427 ml, 1.48 M), and stannous chloride (9.97 ml, 0.064 M, HCl to dissolve).
- Alumina beads (Norton, 590-850 ⁇ m, 28 g) were suspended in the solution with excess deionized water. The mixture was stirred and heated until almost all of the water was evaporated. The resulting solids were dried at 80° C. The total metals loading was 0.5 weight percent.
- the catalyst was loaded into the reactor and reduced under hydrogen (5 volume percent in nitrogen) at 300° C.
- a reactor comprising a quartz tube (19 mm dia) into which the catalyst (6 g) was loaded to a bed height of 1.5 cm (static aspect ratio 0.8).
- a quartz frit was used to support the catalyst and evenly distribute the gas flow.
- the feed was preheated and the reactor insulated in the manner described hereinbefore.
- Ethane, hydrogen, oxygen, and nitrogen were preheated to 275° C. and fed to the reactor at a flow rate which disengaged the particles and circulated them within the bed.
- the flow rate was set for slightly above minimal fluidization at operating conditions (5 slpm).
- the bed expanded to a height of 3.0 cm (operating aspect ratio 1.6). Oxygen was introduced which resulted in catalyst ignition. Upon light-off, the catalyst operated autothermally.
- Example 16 It was found that ethane could be oxidized to ethylene in a fixed bed reactor over a catalyst prepared on alumina pellets.
- Example 17 it was concluded that although both the fixed bed and the modified fluidized bed reactors were suitable, the selectivities were more favorable in the modified fluidized bed reactor. Less methane, carbon monoxide, and carbon dioxide were obtained, and more ethylene was obtained at closely similar conversions.
- a magnesia monolith support (Hi-Tech Ceramics, Inc.; 17 mm dia ⁇ 10 mm thick) was treated with an aqueous solution of tin (IV) chloride (0.24 M), then dried at 90° C. and reduced at about 875° C. under hydrogen (5 percent in nitrogen). Tin loading was 1 weight percent.
- the tin-treated support was impregnated with an aqueous solution of platinum and copper (Pt/Cu atomic ratio 1:5) prepared using solutions of hexachloroplatinic acid (0.19 M) and cupric chloride (1.49 M). Then, the monolith was dried at 80° C. and reduced at 450° C. under the aforementioned hydrogen flow. Pt loading was 3.26 weight percent.
- a solution containing nickel and copper in an atomic ratio of 1:1 was prepared from an aqueous solution of nickel (II) chloride hexahydrate (0.2 M) and an aqueous solution of copper (II) chloride (1.49 M).
- An alumina monolith (99.5 weight percent alumina; 17 mm diameter ⁇ 10 mm length) was loaded with the Ni—Cu solution, dried at 80° C. overnight, and then reduced at 450° C. in hydrogen (5 volume percent) in nitrogen. The total metal loading was 1.48 weight percent.
- the catalyst was evaluated in the autothermal oxidation of ethane to ethylene in the manner previously described. The catalyst required at least 400° C. preheat for ignition.
- a catalyst comprising copper and nickel on an alumina monolith is capable of oxidizing ethane to ethylene under autothermal conditions.
- the catalyst is more active at a higher preheat temperature.
- the catalyst is relatively stable for several hours.
Abstract
A process and catalyst for the partial oxidation of paraffinic hydrocarbons, such as ethane, propane, naphtha, and natural gas condensates, to olefins, such as ethylene and propylene. The process involves contacting a paraffinic hydrocarbon with oxygen in the presence of hydrogen and a catalyst under autothermal process conditions. Preheating the feed decreases oxygen consumption and increases the net hydrogen balance. The catalyst comprises a Group 8B metal, preferably, a platinum group metal, and at least one promoter selected from Groups 1B, 6B, 3A, 4A, and 5A, optionally supported on a catalytic support, such as magnesia or alumina. In preferred embodiments, the support is pretreated with a support modifier selected from Groups 1A, 2A, 3B, 4B, 5B, 6B, 1B, 3A, 4A, 5A, the rare earth lanthanides, and the actinides. A modified fluidized bed reactor is disclosed for the process.
Description
- This application claims the benefit of U.S. application Ser. No. 09/388,219, filed Sep. 1, 1999, U.S. Provisional Application Serial No. 60/099,041, filed Sep. 3, 1998, U.S. Provisional Application Serial No. 60/111,861, filed Dec. 11, 1998, and U.S. Provisional Application Serial No. 60/136,003, filed May 26, 1999.
- The present invention relates to the field of catalytic oxidation of hydrocarbons. More particularly, the present invention relates to the catalytic partial oxidation of paraffinic hydrocarbons, such as ethane, propane, and naphtha, to produce olefins, such as ethylene and propylene.
- Olefins find widespread utility in industrial organic chemistry. Ethylene is needed for the preparation of important polymers, such as polyethylene, vinyl plastics, and ethylene-propylene rubbers, and important basic chemicals, such as ethylene oxide, styrene, acetaldehyde, ethyl acetate, and dichloroethane. Propylene is needed for the preparation of polypropylene plastics, ethylene-propylene rubbers, and important basic chemicals, such as propylene oxide, cumene, and acrolein. Isobutylene is needed for the preparation of methyl tertiary butyl ether. Long chain mono-olefins find utility in the manufacture of linear alkylated benzene sulfonates, which are used in the detergent industry.
- Low molecular weight olefins, such as ethylene, propylene, and butylene, are produced almost exclusively by thermal cracking (pyrolysis/steam cracking) of alkanes at elevated temperatures. An ethylene plant, for example, typically achieves an ethylene selectivity of about 85 percent calculated on a carbon atom basis at an ethane conversion of about 60 mole percent. Undesired coproducts are recycled on the shell side of the cracking furnace to be burned, so as to produce the heat necessary for the process. Disadvantageously, thermal cracking processes for olefin production are highly endothermic. Accordingly, these processes require the construction and maintenance of large, capital intensive, and complex cracking furnaces. The heat required to operate these furnaces at a process temperature of about 900° C. is frequently obtained from the combustion of methane which disadvantageously produces undesirable quantities of carbon dioxide. As a further disadvantage, the crackers must be shut down periodically to remove coke deposits on the inside of the cracking coils.
- Catalytic processes are known wherein paraffinic hydrocarbons are oxidatively cracked to form mono-olefins. In these processes a paraffinic hydrocarbon is contacted with oxygen in the presence of a catalyst consisting of a platinum group metal or mixture thereof deposited on a ceramic monolith support. Optionally, hydrogen may be a component of the feed. The process is conducted under autothermal reaction conditions wherein the feed is partially combusted, and the heat produced during combustion drives the endothermic cracking process. Consequently, under these autothermal process conditions there is no external heat source required; however, the catalyst is required to support combustion above the normal fuel-rich limit of flammability. Representative references disclosing this type of process include the following U.S. Pat. Nos. 4,940,826; 5,105,052; 5,382,741; and 5,625,111. Disadvantageously, substantial amounts of deep oxidation products, such as carbon monoxide and carbon dioxide, are produced, and the selectivity to olefins remains too low when compared with thermal cracking.
- M. Huff and L. D. Schmidt disclose in the Journal of Physical Chemistry, 97, 1993, 11,815, the production of ethylene from ethane in the presence of air or oxygen under autothermal conditions over alumina foam monoliths coated with platinum, rhodium, or palladium. A similar article by M. Huff and L. D. Schmidt in the Journal of Catalysis, 149, 1994, 127-141, discloses the autothermal production of olefins from propane and butane by oxidative dehydrogenation and cracking in air or oxygen over platinum and rhodium coated alumina foam monoliths. The olefin selectivity achieved in these processes is not comparable to that achieved by steam cracking and therefore could be improved.
- U.S. Pat. No. 5,639,929 teaches an autothermal process for the oxidative dehydrogenation of C 2-C6 alkanes with an oxygen-containing gas in a fluidized catalyst bed of platinum, rhodium, nickel, or platinum-gold supported on alpha alumina or zirconia. Ethane produces ethylene, while higher alkanes produce ethylene, propylene, and iso-butylene. Again, the olefin selectivity could be improved.
- C. Yokoyama, S. S. Bharadwaj and L. D. Schmidt disclose in Catalysis Letters, 38, 1996, 181-188, the oxidative dehydrogenation of ethane to ethylene under autothermal reaction conditions in the presence of a bimetallic catalyst comprising platinum and a second metal selected from tin, copper, silver, magnesium, cerium, lanthanum, nickel, cobalt, and gold supported on a ceramic foam monolith. This reference is silent with respect to co-feeding hydrogen in the feedstream. While the use of a catalyst containing platinum and tin and/or copper is better than a catalyst containing a platinum group metal alone, the olefin selectivity should be improved if the process is to be commercialized.
- In view of the above, it would be desirable to discover a catalytic process wherein a paraffinic hydrocarbon is converted to an olefin in a conversion and selectivity comparable to commercial thermal cracking processes. It would be desirable if the catalytic process were to produce only small quantities of deep oxidation products, such as, carbon monoxide and carbon dioxide. It would also be desirable if the process were to achieve low levels of catalyst coking. It would be even more desirable if the process could be easily engineered without the necessity for a large, capital intensive, and complex cracking furnace. Finally, it would be most desirable if the catalyst for the process exhibited good stability.
- This invention is a process for the partial oxidation of paraffinic hydrocarbons to form olefins. The process comprises contacting a paraffinic hydrocarbon or mixture thereof with oxygen in the presence of hydrogen and a catalyst. The contacting is conducted under autothermal process conditions sufficient to form the olefin. The catalyst employed in the process of this invention comprises a Group 8B metal and at least one promoter.
- The process of this invention efficiently produces olefins, particularly mono-olefins, from paraffinic hydrocarbons, oxygen, and hydrogen. Advantageously, the process of this invention achieves a higher paraffin conversion and a higher olefin selectivity as compared with prior art catalytic, autothermal processes. More advantageously, the process of this invention produces fewer undesirable deep oxidation products, such as carbon monoxide and carbon dioxide, as compared with prior art catalytic, autothermal processes. Even more advantageously, in preferred embodiments, the process of this invention achieves a paraffin conversion and olefin selectivity which are comparable to commercial thermal cracking processes. As a further advantage, the process produces little, if any, coke, thereby substantially prolonging catalyst lifetime and eliminating the necessity to shut down the reactor to remove coke deposits.
- Most advantageously, the process of this invention allows the operator to employ a simple engineering design and control strategy, which eliminates the requirement for a large, expensive, and complex furnace like that used in thermal cracking processes. In one preferred embodiment, the reactor simply comprises an exterior housing which contains a monolithic support onto which the catalytic components are deposited. Since the residence time of the reactants in the process of this invention is on the order of milliseconds, the reaction zone operates at high volumetric throughput. Accordingly, the reaction zone measures from about one-fiftieth to about one-hundredth the size of a commercially available steam cracker of comparable capacity. The reduced size of the reactor lowers costs and simplifies maintenance procedures. Finally, since the process of this invention is exothermic, the heat produced can be harvested via integrated heat exchangers to generate electricity or steam credits for other processes.
- As noted hereinbefore, thermal energy is needed to maintain autothermal process conditions. Without preheating the feedstream, the required thermal energy is totally supplied by the reaction of the feedstream with oxygen, namely, alkane oxidative dehydrogenation to form olefins and water, hydrogen oxidation to form water, and carbon combustion to form carbon monoxide and carbon dioxide. These processes can supply the heat necessary for any endothermic dehydrogenation which takes place to form ethylene and hydrogen. The prior art has recognized that a portion of the required thermal energy can be obtained by preheating the feedstream. The preheat can be conveniently supplied by condensing high pressure saturated steam, or alternatively, by combusting process off-gas or another fuel source. Surprisingly, it has now been discovered that a high preheat temperature can be used without loss in olefin selectivity, and further, that a high preheat temperature provides advantages unrecognized heretofore. Accordingly, in another aspect of this invention, the paraffinic hydrocarbon and oxygen, which together comprise the reactant feedstream, are preheated at a temperature greater than about 200° C., but below the onset of reaction of the feedstream components.
- When the high preheat temperatures of this invention are employed, advantageously less oxygen is required in the feedstream. Since the cost of pure oxygen can be a significant cost component of the feedstream, the decrease in oxygen employed translates directly into economic savings. Moreover, since oxygen reacts with hydrogen in the feedstream, the decrease in oxygen employed leads to a decrease in hydrogen consumed and in the waste water produced. As a consequence, more hydrogen is found in the product stream.
- An increased yield of hydrogen in the product stream further improves the economics of the autothermal oxidation process of this invention. Since hydrogen is required for the process, hydrogen in the product should be recycled and any deficit must be replaced by importing hydrogen from an external source. Alternatively, hydrogen can be made from off-gas streams, for example, a water-shift reaction which converts carbon monoxide and water to hydrogen and carbon dioxide. As a consequence of using the high preheat temperature of this invention, the product stream is enriched in hydrogen. Under optimal preheat conditions, the recycled hydrogen substantially eliminates the need to import hydrogen or to derive make-up hydrogen from other sources.
- In a third aspect, the autothermal oxidation process of this invention is beneficially conducted in a unique fluidized bed reactor, characterized in that the reactor bed possesses an aspect ratio of less than about 1:1, as measured during operation. For the purposes of this invention, the aspect ratio is defined as the ratio of the height (or depth) of the reactor bed to its cross-sectional dimension (diameter or width). For use in this fluidized bed, the catalyst comprises a support in the form of pellets or spheres onto which the catalytic components are deposited.
- When operation of the process in the aforementioned unique fluidized bed reactor is compared with operation in a fixed bed reactor, several advantages become apparent. For example, ethylene selectivity improves with use of the fluidized bed, while selectivities to methane and deep oxidation products, such as carbon monoxide and carbon dioxide, decrease. Significantly, the selectivity advantages are achieved at ethane conversions which are comparable to or better than those obtained in a fixed bed reactor.
- In a fourth aspect, this invention is a catalyst composition comprising a Group 8B metal and at least one promoter supported on a catalyst support which has been pretreated with at least one support modifier.
- The aforementioned composition is beneficially employed as a catalyst in the autothermal partial oxidation of a paraffinic hydrocarbon to an olefin. The catalyst composition beneficially produces an olefin or mixture of olefins at conversions and selectivities which are comparable to those of industrial thermal cracking processes. Accordingly, the catalyst composition of this invention produces low amounts of carbon monoxide and carbon dioxide. Finally, the catalyst composition of this invention advantageously exhibits good catalyst stability.
- The process of this invention involves the partial oxidation of a paraffinic hydrocarbon to form an olefin. The words “partial oxidation” imply that the paraffin is not substantially oxidized to deep oxidation products, specifically, carbon monoxide and carbon dioxide. Rather, the partial oxidation comprises one or both of oxidative dehydrogenation and cracking to form primarily olefins. It is not known or suggested to what extent or degree either process, oxidative dehydrogenation or cracking, predominates or occurs to the exclusion of the other.
- The partial oxidation process of this invention comprises contacting a paraffinic hydrocarbon with oxygen in the presence of a multi-metallic catalyst and in the presence of a hydrogen co-feed. The contacting is conducted under autothermal process conditions sufficient to form the olefin. The catalyst which is employed in the process of this invention comprises a Group 8B metal and at least one promoter, optionally supported on a catalyst support. In a preferred embodiment of the process of this invention, the paraffinic hydrocarbon is a paraffin selected from ethane, propane, mixtures of ethane and propane, naphtha, natural gas condensates, and mixtures of the aforementioned hydrocarbons; and the preferred olefins produced are ethylene, propylene, butylene, isobutylene, and butadiene.
- In a preferred aspect of this invention, the feedstream comprising the paraffinic hydrocarbon and oxygen is preheated before introducing the feedstream into the autothermal oxidation reactor. The preheat temperature is greater than about 200° C., but less than the temperature wherein reaction of the feedstream components begins. Preferably, the upper limit on the preheat temperature is less than about 900° C.
- In another preferred embodiment of this invention, the reactor comprises an exterior housing which holds the catalyst, the catalyst being provided in the form of a ceramic monolith support onto which the catalytic components, including the Group 8B metal and any promoter(s), have been deposited.
- In another preferred aspect of this invention, the reactor comprises a modified fluidized bed characterized by an aspect ratio of less than about 1:1 in operating mode. As noted hereinbefore, the aspect ratio is the ratio of the height (depth) of the reactor to its cross-sectional dimension (diameter or width). In this reactor, the catalyst is provided typically in the form of spheres or granules.
- In yet another preferred embodiment, the catalyst which is employed in the process of this invention comprises a Group 8B metal and at least one promoter supported on a catalytic support which has been pretreated with at least one support modifier. Preferably, the Group 8B metal is a platinum group metal. The preferred platinum group metal is platinum. The preferred promoter is selected from the elements of Groups 1B, 6B, 3A, 4A, 5A, (equivalent to Groups 11, 6, 13, 14, and 15), and mixtures of the aforementioned elements of the Periodic Table, as referenced by S. R. Radel and M. H. Navidi, in Chemistry, West Publishing Company, New York, 1990. The preferred support modifier is selected from Groups 1A, 2A, 3B, 4B, 5B, 6B, 1B, 3A, 4A, 5A (equivalent Groups 1, 2, 3, 4, 5, 6, 11, 13, 14, 15), and the lanthanide rare earths and actinide metals of the Periodic Table, as referenced by S. R. Radel and M. H. Navidi, ibid.
- In a most preferred embodiment of the catalyst composition, the platinum group metal is platinum; the promoter is selected from tin, copper, and mixtures thereof; the support is selected from alumina, magnesia, and mixtures thereof; and the modifier is selected from tin, lanthanum, and mixtures thereof.
- Any paraffinic hydrocarbon or mixture of paraffinic hydrocarbons can be employed in the process of this invention provided that an olefin, preferably, a mono-olefin, is produced. The term “paraffinic hydrocarbon,” as used herein, refers to a saturated hydrocarbon. Generally, the paraffin contains at least 2 carbon atoms. Preferably, the paraffin contains from 2 to about 25 carbon atoms, more preferably, from 2 to about 15 carbon atoms, and even more preferably, from 2 to about 10 carbon atoms. The paraffin can have a linear, branched, or cyclic structure, and can be a liquid or gas at ambient temperature and pressure. The paraffin can be supplied as an essentially pure paraffinic compound, or mixture of paraffinic compounds, or as a paraffin-containing mixture of hydrocarbons. Paraffin feeds which are suitably employed in the process of this invention include, but are not limited to, ethane, propane, butane, pentane, hexane, heptane, octane, and higher homologues thereof, as well as complex higher boiling mixtures of paraffin-containing hydrocarbons, such as naphtha, gas oil, vacuum gas oil, and natural gas condensates. Additional feed components may include methane, nitrogen, carbon monoxide, carbon dioxide, and steam, if so desired. Minor amounts of unsaturated hydrocarbons may also be present. Most preferably, the paraffin is selected from ethane, propane, mixtures of ethane and propane, naphtha, natural gas condensates, and mixtures thereof.
- In the process of this invention, the paraffinic hydrocarbon is contacted with an oxygen-containing gas. Preferably, the gas is molecular oxygen or molecular oxygen diluted with an unreactive gas, such as nitrogen, helium, carbon dioxide, or argon, or diluted with a substantially unreactive gas, such as carbon monoxide or steam. Any molar ratio of paraffin to oxygen is suitable provided the desired olefin is produced in the process of this invention. Preferably, the process is conducted fuel-rich and above the upper flammability limit. Generally, the molar ratio of paraffinic hydrocarbon to oxygen varies depending upon the specific paraffin feed and autothermal process conditions employed. Typically, the molar ratio of paraffinic hydrocarbon to oxygen ranges from about 3 to about 77 times the stoichiometric ratio of hydrocarbon to oxygen for complete combustion to carbon dioxide and water. Preferably, the molar ratio of paraffinic hydrocarbon to oxygen ranges from about 3 to about 13, more preferably, from about 4 to about 11, and most preferably, from about 5 to about 9 times the stoichiometric ratio of hydrocarbon to oxygen for complete combustion to carbon dioxide and water. These general limits are usually achieved by employing a molar ratio of paraffinic hydrocarbon to oxygen greater than about 0.1:1, preferably, greater than about 0.2:1, and by using a molar ratio of paraffinic hydrocarbon to oxygen usually less than about 3.0:1, preferably, less than about 2.7:1. For preferred paraffins, the following ratios are more specific. For ethane, the ethane to oxygen molar ratio is typically greater than about 1.5:1, and preferably, greater than about 1.8:1. The ethane to oxygen molar ratio is typically less than about 3.0:1, preferably, less than about 2.7:1. For propane, the propane to oxygen molar ratio is typically greater than about 0.9:1, preferably, greater than about 1.1:1. The propane to oxygen molar ratio is typically less than about 2.2:1, preferably, less than about 2.0:1. For naphtha, the naphtha to oxygen molar ratio is typically greater than about 0.3:1, preferably, greater than about 0.5:1. The naphtha to oxygen molar ratio is typically less than about 1.0:1, preferably, less than about 0.9:1.
- When a high preheat temperature is used, for example, above 200° C., the limits on the molar ratio of paraffinic hydrocarbon to oxygen can be shifted towards higher values. For example, at high preheat the molar ratio of paraffinic hydrocarbon to oxygen is typically greater than about 0.1:1 and less than about 4.0:1. Specifically, at high preheat the ethane to oxygen molar ratio is typically greater than about 1.5:1, preferably, greater than about 1.8:1, and typically less than about 4.0:1, preferably, less than about 3.2:1. At high preheat, the molar ratio of propane to oxygen is typically greater than about 0.9:1, preferably, greater than about 1.1:1, and typically, less than about 3.0:1, and preferably, less than about 2.6:1. At high preheat, the molar ratio of naphtha to oxygen is typically greater than about 0.3:1, preferably, greater than about 0.5:1, and typically, less than about 1.4:1, and preferably, less than about 1.3:1. As an advantageous feature of the process of this invention, hydrogen is co-fed with the paraffin and oxygen to the catalyst. The presence of hydrogen in the feedstream beneficially improves the conversion of hydrocarbon and the selectivity to olefins, while reducing the formation of deep oxidation products, such as, carbon monoxide and carbon dioxide. The molar ratio of hydrogen to oxygen can vary over any operable range provided that the desired olefin product is produced. Typically, the molar ratio of hydrogen to oxygen is greater than about 0.5:1, preferably, greater than about 0.7:1, and more preferably, greater than about 1.5:1. Typically, the molar ratio of hydrogen to oxygen is less than about 3.2:1, preferably, less than about 3.0:1, and more preferably, less than about 2.7:1.
- At high preheat the molar ratio of hydrogen to oxygen typically is greater than about 0.1:1, preferably, greater than about 0.7:1, and more preferably, greater than about 1.5:1. At high preheat the molar ratio of hydrogen to oxygen is typically less than about 4.0:1, preferably, less than about 3.2:1, and more preferably, less than about 3.0:1.
- Optionally, the feed may contain a diluent, which can be any gas or vaporizable liquid which does not interfere with the process of the invention. The diluent functions as a carrier of the reactants and products and facilitates the transfer of heat generated by the process. The diluent also helps to minimize undesirable secondary reactions and helps to expand the non-flammable regime for mixtures of the paraffin, hydrogen, and oxygen. Suitable diluents include nitrogen, argon, helium, carbon dioxide, carbon monoxide, methane, and steam. The concentration of diluent in the feed can vary over a wide range. If a diluent is used, the concentration of diluent is typically greater than about 0.1 mole percent of the total reactant feed including paraffin, oxygen, hydrogen, and diluent. Preferably, the amount of diluent is greater than about 1 mole percent of the total reactant feed. Typically, the amount of diluent is less than about 70 mole percent, and preferably, less than about 40 mole percent, of the total reactant feed.
- The catalyst which is employed in the process of this invention beneficially comprises a Group 8B metal and at least one promoter, described hereinbelow, optionally supported on a catalyst support. The Group 8B metals include iron, cobalt, nickel, and the platinum group metals, namely, ruthenium, rhodium, palladium, osmium, iridium, and platinum. Mixtures of the aforementioned Group 8B metals may also be used. Preferably, the Group 8B metal is a platinum group metal; preferably, the platinum group metal is platinum. The catalyst also comprises at least one promoter, which is suitably defined as any element or elemental ion which is capable of enhancing the performance of the catalyst, as measured, for example, by an increase in the paraffin conversion, an increase in the selectivity to olefin, a decrease in the selectivities to deep oxidation products, such as carbon monoxide and carbon dioxide, and/or an increase in catalyst stability and lifetime. For the purposes of this invention, the term “promoter” does not include the platinum group metals. Preferably, the promoter is selected from the elements of Groups 1B (Cu, Ag, Au), 6B (Cr, Mo, W), 3A (for example, Al, Ga, In, Tl), 4A (for example, Ge, Sn, Pb), and 5A (for example, As, Sb, Bi), and mixtures thereof. More preferably, the promoter is selected from copper, tin, antimony, silver, indium, and mixtures thereof. Most preferably, the promoter is selected from copper, tin, antimony, and mixtures thereof.
- Any atomic ratio of Group 8B metal to promoter can be employed in the catalyst, provided the catalyst is operable in the process of this invention. The optimal atomic ratio will vary with the specific Group 8B metal and promoter(s) employed. Generally, the atomic ratio of the Group 8B metal to promoter is greater than 0.10 (1:10), preferably, greater than about 0.13 (1:8), and more preferably, greater than about 0.17 (1:6). Generally, the atomic ratio of the Group 8B metal to promoter is less than about 2.0 (1:0.5), preferably, less than about 0.33 (1:3), and more preferably, less than about 0.25 (1:4). Although the promoter is used in a gram-atom amount equivalent to or greater than the Group 8B metal, the promoter nonetheless functions to enhance the catalytic effect of the catalyst. Compositions prepared with promoter alone, in the absence of Group 8B metal, are typically (but not necessarily always) catalytically inactive in the process. In contrast, the Group 8B metal is catalytically active in the absence of promoter, albeit with lesser activity.
- The catalyst can be suitably employed in the form of a metallic gauze. More specifically, the gauze can comprise an essentially pure Group 8B metal or an alloy of Group 8B metals onto which the promoter is deposited. Suitable gauzes of this type include pure platinum gauze and platinum-rhodium alloy gauze coated with the promoter. The method used to deposit or coat the promoter onto the gauze can be any of the methods described hereinafter. Alternatively, a gauze comprising an alloy of a Group 8B metal and the promoter can be employed. Suitable examples of this type include gauzes prepared from platinum-tin, platinum-copper, and platinum-tin-copper alloys.
- In another embodiment, the Group 8B metal and promoter are supported on a catalytic support. The loading of the Group 8B metal on the support can be any which provides for an operable catalyst in the process of this invention. In general, the loading of the Group 8B metal is greater than about 0.001 weight percent, preferably, greater than about 0.1 weight percent, and more preferably, greater than about 0.2 weight percent, based on the total weight of the Group 8B metal and support. Preferably, the loading of the Group 8B metal is less than about 80 weight percent, preferably, less than about 60 weight percent, and more preferably, less than about 10 weight percent, based on the total weight of the Group 8B metal and the support. Once the Group 8B metal loading is established, the desired atomic ratio of Group 8B metal to promoter determines the loading of the promoter.
- The catalytic support comprises any material which provides a surface to carry the Group 8B metal, the promoter(s), and any support modifiers. Preferably, the support is thermally and mechanically stable under autothermal process conditions. Preferably, the catalytic support is a ceramic, such as, a refractory oxide, carbide, or nitride. Non-limiting examples of suitable ceramics include alumina, silica, silica-aluminas, aluminosilicates, including cordierite, magnesia, magnesium aluminate spinels, magnesium silicates, zirconia, titania, boria, zirconia toughened alumina (ZTA), lithium aluminum silicates, silicon carbide, oxide-bonded silicon carbide, and silicon nitride. Mixtures of the aforementioned refractory oxides, nitrides, and carbides may also be employed, as well as washcoats of the aforementioned materials on a support. Preferred ceramics include magnesia, alumina, silica, and amorphous or crystalline combinations of magnesia, alumina and silica, including mullite. Alpha (α) and gamma (γ) alumina are preferred forms of alumina. Preferred combinations of alumina and silica comprise from about 65 to about 100 weight percent alumina and from essentially 0 to about 35 weight percent silica. Other refractory oxides, such as boria, can be present in smaller amounts in the preferred alumina and silica mixtures. Preferred zirconias include zirconia fully stabilized with calcia (FSZ) and zirconia partially stabilized with magnesia (PSZ), available from Vesuvius Hi-Tech Ceramics, Inc. Magnesia is the most preferred support, because it produces fewer cracking products and less carbon monoxide. Moreover, the hydrocarbon conversion and olefin selectivity tend to be higher with magnesia. The catalytic support may take a variety of shapes including that of porous or non-porous spheres, granules, pellets, irregularly shaped solid or porous particles, or any other shape which is suitable for catalytic reactors, including fixed bed, transport bed, and fluidized bed reactors. In a preferred form, the catalyst is a monolith. As used herein, the term “monolith” means any continuous structure, including for example, honeycomb structures, foams, and fibers, including fibers woven into fabrics or made into non-woven mats or thin paper-like sheets. Monoliths do not, in general, contain significant microporosity. Foams have a sponge-like structure. More preferably, the support is a foam or fiber monolith. Fibers tend to possess higher fracture resistance as compared with foams and honeycombs. Preferred ceramic foams, available from Vesuvius Hi-Tech Ceramics, Inc., comprise magnesia, alpha alumina, zirconia, or mullite with a porosity ranging from about 5 to about 100 pores per linear inch (ppi) (2 to 40 pores per linear cm (ppcm)). Foams having about 45 ppi (18 ppcm) are more preferred. The term “porosity,” as used herein, refers to channel size or dimension. It is important to note that the foam supports are not substantially microporous structures. Rather, the foams are macroporous, meaning that they are low surface area supports with channels ranging in diameter from about 0.1 mm to about 5 mm. The foams are estimated to have a surface area less than about 10 m 2/g, and preferably, less than about 2 m2/g, but greater than about 0.001 m2/g. Preferred ceramic fibers, available from 3M Corporation as Nextel™ brand ceramic fibers, typically have a diameter greater than about 1 micron (μm), preferably, greater than about 5 μm. The diameter is suitably less than about 20 μm, preferably, less than about 15 μm. The length of the fibers is generally greater than about 0.5 inch (1.25 cm), preferably, greater than about 1 inch (2.5 cm), and typically less than about 10 inches (25.0 cm), preferably, less than about 5 inches (12.5 cm). The surface area of the fibers is very low, being generally less than about 1 m2/g, preferably, less than about 0.3 m2/g, but greater than about 0.001 m2/g. Preferably, the fibers are not woven like cloth, but instead are randomly intertwined as in a mat or matted rug. Most preferred are Nextel™ brand 440 fibers which consist of gamma alumina (70 weight percent), silica (28 weight percent), and boria (2 weight percent) and Nextel™ brand 610 fibers which consist of alpha alumina (99 weight percent), silica (0.2-0.3 weight percent) and iron oxide (0.4-0.7 weight percent).
- The deposition of the Group 8B metal and promoter(s) onto the support can be made by any technique known to those skilled in the art, for example, impregnation, ion-exchange, deposition-precipitation, vapor deposition, sputtering, and ion implantation. In one preferred method, the Group 8B metal is deposited onto the support by impregnation. Impregnation is described by Charles N. Satterfield in Heterogeneous Catalysis in Practice, McGraw-Hill Book Company, New York, 1980, 82-84, incorporated herein by reference. In this procedure, the support is wetted with a solution containing a soluble Group 8B compound, preferably, to the point of incipient wetness. The contacting temperature typically ranges from about ambient, taken as 23° C., to about 100° C., preferably, from about 23° C. to about 50° C. The contacting is conducted usually at ambient pressure. Non-limiting examples of suitable Group 8B compounds include the Group 8B nitrates, halides, sulfates, alkoxides, carboxylates, and Group 8B organometallic compounds, such as halo, amino, acetylacetonate, and carbonyl complexes. Preferably, the Group 8B compound is a platinum group halide, more preferably, a chloride, such as chloroplatinic acid. The solvent can be any liquid which solubilizes the Group 8B compound. Suitable solvents include water, aliphatic alcohols, aliphatic and aromatic hydrocarbons, and halo-substituted aliphatic and aromatic hydrocarbons. The concentration of the Group 8B compound in the solution generally ranges from about 0.001 molar (M) to about 10 M. After contacting the support with the solution containing the Group 8B compound, the support may be dried under air at a temperature ranging from about 23° C. to a temperature below the decomposition temperature of the Group 8B compound, typically, a temperature between about 23° C. and about 100° C.
- The deposition of the promoter can be accomplished in a manner analogous to the deposition of the Group 8B metal. Accordingly, if impregnation is used, then the support is wetted with a solution containing a soluble compound of the promoter at a temperature between about 23° C. and about 100° C., preferably, between about 23° C. and about 50° C., at about ambient pressure. Suitable examples of soluble promoter compounds include promoter halides, nitrates, alkoxides, carboxylates, sulfates, and organometallic compounds, such as amino, halo, and carbonyl complexes. Suitable solvents comprise water, aliphatic alcohols, aliphatic and aromatic hydrocarbons, and chloro-substituted aliphatic and aromatic hydrocarbons. Certain promoter compounds, such as compounds of antimony and tin, may be more readily solubilized in the presence of acid. For example, hydrochloric acid (5-25 weight percent) can be suitably employed. The concentration of the promoter compound in the solution generally ranges from about 0.01 M to about 10 M. Following deposition of the soluble promoter compound or mixture thereof, the impregnated support may be dried under air at a temperature between about 23° C. and a temperature below the temperature wherein vaporization or decomposition of the promoter compound occurs. Typically, the drying is conducted at a temperature between about 23° C. and about 100° C.
- In one method of preparing the catalyst, the Group 8B metal is deposited onto the support first, and thereafter the promoter is deposited onto the support. In an alternative method, the promoter is deposited first, followed by the deposition of the Group 8B metal. In a preferred method of preparing the catalyst, the Group 8B metal and the promoter are deposited simultaneously onto the support from the same deposition solution. In any of these methods, following one or more of the depositions, a calcination under oxygen is optional. If performed, the calcination is conducted at a temperature ranging from about 100° C. to below the temperature at which volatilization of the metals becomes significant, typically, less than about 1,100° C. Preferably, the calcination is conducted at a temperature between about 100° C. and about 500° C.
- As a final step in the preparation of the catalyst, the fully-loaded support is reduced under a reducing agent, such as hydrogen, carbon monoxide, or ammonia, at a temperature between about 100° C. and about 900° C., preferably between about 125° C. and about 800° C., so as to convert the Group 8B metal substantially to its elemental form. The promoter may be reduced fully or partially, or not reduced at all, depending upon the specific promoter chosen and the reduction conditions. In addition, reduction at elevated temperatures may produce alloys of the Group 8B metal and the promoter. Alloys may provide enhanced catalyst stability by retarding vaporization of the promoter during the process of this invention.
- In another preferred embodiment, the support is pretreated with a support modifier prior to loading the Group 8B and promoter(s). The support modifier can be any metal ion having a charge of +1 or greater. Preferably, the support modifier is selected from Groups 1A (Li, Na, K, Rb, Cs), 2A (for example, Mg, Ca, Sr, Ba), 3B (Sc, Y, La), 4B (Ti, Zr, Hf), 5B (V, Nb, Ta), 6B (Cr, Mo, W), 1B (Cu, Ag, Au), 3A (for example, Al, Ga, In), 4A (for example, Ge, Sn, Pb), 5A (for example, As, Sb, Bi), and the lanthanide rare earths (for example, Ce, Er, Lu, Ho) and actinide elements (specifically Th) of the Periodic Table previously identified. More preferably, the support modifier is selected from calcium, zirconium, tin, lanthanum, potassium, lutetium, erbium, barium, holmium, cerium, antimony, and mixtures thereof. Most preferably, the support modifier is selected from lanthanum, tin, antimony, calcium, and mixtures thereof. Certain elements, such as tin, antimony, and silver, may function as both promoter and support modifier simultaneously.
- The procedure to modify the support comprises contacting the support with a solution containing a soluble compound of the support modifier. The contacting can involve ion-exchange or impregnation methods. Preferably, the modification procedure involves submerging the support in the solution such that essentially all of the surface area of the support is contacted with an excess of the solution. Compounds suitable for preparing the solution of support modifier include modifier nitrates, halides, particularly the chlorides, alkoxides, carboxylates, and organometallic complexes including amino, halo, alkyl, and carbonyl complexes. Suitable solvents include water, aliphatic alcohols, aromatic hydrocarbons, and halo-substituted aliphatic and aromatic hydrocarbons. Typically, the concentration of modifier compound in the solution ranges from about 0.001 M to about 10 M. Acidified solutions, for example, of hydrochloric acid and diluted solutions thereof, may be beneficially employed. The contact time generally ranges from about 1 minute to about 1 day. The contacting temperature suitably ranges from about 23° C. to about 100° C., and pressure is generally ambient. Alternatively, slurries of mixed oxides containing promoter and/or modifier elements, such as magnesium stannate (Mg 2SnO4), can be deposited onto the support. The modified support is typically calcined, as noted hereinabove, or reduced under a reducing agent, such as hydrogen, at a temperature between about 100° C. and about 900° C., preferably, between about 200° C. and about 800° C. The choice of calcination or reduction depends on the element used to pretreat the support. If the element or its oxide is readily vaporizable, the pretreated support is reduced. If the element or its oxide is not readily vaporizable, then the pretreated support is calcined. As a guideline, the words “readily vaporizable” may be taken to mean that greater than about 1 weight percent of any metal component in the catalyst is vaporized in a period of about 24 hours under calcination conditions at about 200° C. The term “readily vaporizable” may be given a narrower or broader definition, as desired.
- Following the pretreatment modification, the Group 8B metal and promoter(s) are loaded onto the support. Then, the support is reduced as described hereinbefore. Alternatively, the metal-loaded support may be calcined first and then reduced. Whether the modified support is calcined or not depends again upon the vaporization potential of the modifier metal(s) and promoter(s) employed. Supports modified with metals or metal oxides which tend to vaporize readily are typically not calcined. Supports modified with metals or metal oxides which do not vaporize readily can be calcined.
- The process of this invention is advantageously conducted under autothermal process conditions. The term “autothermal process conditions” means that the heat generated by reaction of the feed is sufficient to support the catalytic process which converts the paraffin to the olefin. Accordingly, the need for an external heating source to supply the energy for the process can be eliminated. In order to maintain autothermal conditions, the catalysts of the prior art are required to support combustion beyond the normal, fuel-rich limit of flammability. This is not a requirement in the present invention. Here, autothermal conditions can also be maintained with a catalyst which does not support combustion beyond the normal, fuel-rich limit of flammability, provided that hydrogen and optionally a preheat are supplied to the process.
- Ignition can be effected by preheating the feed to a temperature sufficient to effect ignition when contacted with the catalyst. Alternatively, the feed can be ignited with an ignition source, such as a spark or flame. Upon ignition, the reaction-generated heat causes the temperature to take a step change jump to a new steady state level that is herein referred to as the autothermal reaction.
- While running autothermally, the paraffin feed does not have to be preheated, so long as the feed contains hydrogen or the catalyst supports combustion beyond the normal, fuel-rich limit of flammability. (The word “combustion,” as used herein, means the reaction of the hydrocarbon with oxygen unaided by hydrogen.) Preheating the feedstream, however, has certain advantages. The advantages comprise a decrease in oxygen and hydrogen consumed, an increase in the paraffin concentration in the feed, an increase in the operating paraffin to oxygen molar ratio, and a net increase in recycle hydrogen in the product stream. In addition, catalysts can be used which do not support combustion beyond the normal fuel-rich limit of flammability. These advantages are particularly significant when the preheating is conducted at a temperature greater than about 200° C. and less than the temperature wherein reaction of the feedstream components begins. Suitable preheat temperatures are typically greater than about 40° C., preferably, greater than about 125° C., and even more preferably, greater than about 200° C. In another preferred embodiment, the preheat temperature is greater than about 400° C. Suitable preheat temperatures are typically less than about 900° C., preferably, less than about 800° C., and more preferably less than about 600° C.
- As a general rule, the autothermal process operates at close to the adiabatic temperature (that is, essentially without loss of heat), which is typically greater than about 750° C., and preferably, greater than about 925° C. Typically, the autothermal process operates at a temperature less than about 1,150° C., and preferably, less than about 1,050° C. Optionally, the temperature at the reactor exit can be measured, for example, by using a Pt/Pt—Rh thin wire thermocouple. With a monolith catalyst, the thermocouple can be sandwiched between the monolith and the downstream radiation shield. Measurement of temperature close to the reactor exit may be complicated by the high temperature involved and the fragility of the thermocouple. Thus, as an alternative, one skilled in the art can calculate the adiabatic temperature at the reactor exit from a knowledge of the preheat temperature and the exit stream composition. The “adiabatic temperature” is the temperature of the product stream without any heat loss, that is, when all of the heat generated by the process is used to heat the products. Typically, the measured temperature is found to be within about 25° C. of the calculated adiabatic temperature.
- The operating pressure is typically equal to or greater than about 1 atmosphere absolute (atm abs) (100 kPa abs). Typically, the pressure is less than about 20 atm abs (2,000 kPa abs), preferably, less than about 10 atm (1,000 kPa abs), and more preferably, less than about 7 atm abs (700 kPa abs).
- Since the products of this process must be removed rapidly from the reaction zone, gas hourly space velocities are very high. The specific gas hourly space velocity employed will depend upon the choice of reactor cross sectional dimension (for example, diameter) and the form and weight of the catalyst particles. Generally, the gas hourly space velocity (GHSV), calculated as the total flow of the hydrocarbon, oxygen, hydrogen, and optional diluent flows, is greater than about 50,000 ml total feed per ml catalyst per hour (h −1) measured at standard temperature and pressure (0° C., 1 atm) (STP). Preferably, the GHSV is greater than about 80,000 h−1, and more preferably, greater than 100,000 h−1. Generally, the gas hourly space velocity is less than about 6,000,000 h−1, preferably, less than about 4,000,000 h−1, more preferably, less than 3,000,000 h−1, measured as the total flow at STP. Gas flows are typically monitored in units of liters per minute at standard temperature and pressure (slpm). The conversion of gas flow from “slpm” units to gas hourly space velocity units (h−1) is made as follows:

- The residence time of the reactants in the reactor is simply calculated as the inverse of the gas hourly space velocity. At the high space velocities employed in the process of this invention, the residence time is on the order of milliseconds. Thus, for example, a gas hourly space velocity of 100,000 h −1 measured at STP is equivalent to a residence time of 36 milliseconds at STP.
- The process of this invention may be conducted in any reactor designed for use under adiabatic, autothermal process conditions. In one preferred design, the catalyst is prepared on a monolith support which is sandwiched between two radiation shields inside a reactor housing. Alternatively, fixed bed and fluidized bed reactors can be used with catalysts in the form of pellets, spheres, and other particulate shapes. Continuous and intermittent flow of the feedstream are both suitable. It is noted that fluidized bed reactors of the prior art typically possess an aspect ratio in static mode of greater than 1:1, and more preferably, greater than about 5:1. Static mode is defined as the unfluidized or fixed bed configuration. Fluidized bed reactors are generally operated in a bubbling, turbulent, or fast-fluidized regime with expanded beds measuring from about 1.5 to 15 times the static depth. Typically, the aspect ratio in operating mode is greater than about 5:1 to 10:1. For full fluidization, a catalyst particle size ranging between about 30 and 1,000 microns is satisfactory.
- It is believed that the oxidation reaction of this invention occurs predominantly at the reactor entry, which in the case of a stationary catalyst is at the front edge of the catalyst. Such a theory should not be binding or limiting of the invention in any manner. In view of this theory, the optimal reactor for the process of this invention should possess a large cross-sectional dimension and a short height (or depth). On a commercial scale, for example, a catalyst bed of diameter about 5 to 8 feet (1.5 m to 2.4 m) and a height of about 1 inch (2.5 cm) may be suitably employed. Additionally, it is believed that catalyst located at the front edge of a stationary bed can deactivate more quickly with time. As a consequence, longer catalyst lifetime and better selectivities can be achieved by circulating particles of the catalyst, rather than using a stationary bed.
- A preferred reactor design for the process of this invention comprises a modified fluidized bed reactor, characterized in that its aspect ratio in operating mode, and preferably also in static mode (unfluidized or fixed bed configuration), is less than 1:1, and more preferably, less than about 0.1:1, but greater than about 0.001:1. Most preferably, the aspect ratio is about 0.01:1. This unique fluidized bed is operated above the minimum fluidization flow with an expanded bed on the order of about 2 or 3 times the static depth, and preferably, less than about 1.5 times the static depth. For the purposes of this invention, “minimum fluidization flow” is defined as the minimum gas velocity at which the catalyst particles are suspended under operating conditions. The velocity necessary to achieve minimum fluidization depends upon the density and viscosity of the gas phase and the catalyst particle size and density. One skilled in the art would know how to calculate the minimum fluidization flow for any given gas composition and catalyst particle. A suitable authority on the subject is found in Fluidization Engineering, by D. Kunii and O. Levenspeil, 2nd ed., Butterworth-Heineman, 1989, incorporated herein by reference. A catalyst particle size of between about 500 and about 850 microns (23-30 US mesh) is suitable for feed velocities of about 0.05 to 5 meters per second (mps) at standard temperature and pressure. An advantage of the modified fluidized bed reactor may result from its continuous circulation (fluidization), which results in continuous renewal of catalyst particles at the reactor entry. This configuration produces substantially better product yields than a stationary catalyst.
- When a paraffinic hydrocarbon is contacted with oxygen under autothermal process conditions in the presence of a co-feed of hydrogen and in the presence of the multi-metallic catalyst described hereinabove, an olefin, preferably a mono-olefin, is produced. Ethane is converted primarily to ethylene. Propane and butane are converted primarily to ethylene and propylene. Isobutane is converted primarily to isobutylene and propylene. Naphtha and other higher molecular weight paraffins are converted primarily to ethylene and propylene.
- The conversion of paraffinic hydrocarbon in the process of this invention can vary depending upon the specific feed composition, catalyst composition, reactor, and process conditions employed. For the purposes of this invention, “conversion” is defined as the mole percentage of paraffinic hydrocarbon in the feed which is converted to products. Generally, at constant pressure and space velocity, the conversion increases with increasing temperature. Typically, at constant temperature and pressure, the conversion does not change significantly over a wide range of high space velocities employed. In this process, the conversion of paraffinic hydrocarbon is typically greater than about 50 mole percent, preferably, greater than about 60 mole percent, and more preferably, greater than about 70 mole percent.
- Likewise, the selectivity to products will vary depending upon the specific feed composition, catalyst composition, reactor, and process conditions employed. For the purposes of this invention, “selectivity” is defined as the percentage of carbon atoms in the converted paraffin feed which react to form a specific product. For example, the olefin selectivity is calculated as follows:
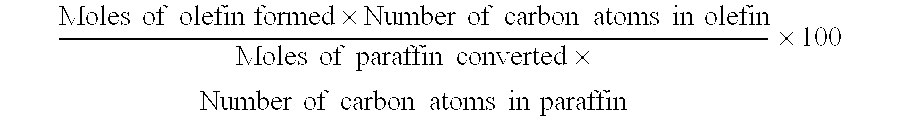
- Generally, the olefin selectivity increases with increasing temperature up to a maximum value and declines as the temperature continues to rise. Usually, the olefin selectivity does not change substantially over a wide range of high space velocities employed. In the process of this invention, the olefin selectivity is typically greater than about 50 carbon atom percent, preferably, greater than about 60 carbon atom percent, more preferably, greater than about 70 carbon atom percent, and even more preferably, greater than about 80 carbon atom percent. Other products formed in smaller quantities include methane, carbon monoxide, carbon dioxide, propane, butenes, butadiene, propadiene, acetylene, methylacetylene, and C 6+hydrocarbons. Acetylene can be hydrogenated to ethylene downstream to increase the overall selectivity to olefin. At least part of the carbon monoxide, carbon dioxide, and methane formed may be recycled to the reactor.
- Water is also formed in the process of this invention from the reaction of hydrogen or hydrocarbon. The presence of hydrogen in the feed minimizes the formation of carbon oxides by reacting with the oxygen to produce water and energy. Accordingly, it is advantageous to recycle the hydrogen in the product stream, obtained from the dehydrogenation of the paraffin, back to the reactor. Optimally, the hydrogen needed to meet the demands of the process essentially equals the hydrogen formed during conversion of the paraffin to olefin. Under these balanced conditions, the hydrogen forms a closed loop wherein there is essentially no demand for additional hydrogen to be added to the feed. Such conditions are more easily met when the feed is preheated and a higher hydrocarbon to oxygen molar ratio is employed.
- The invention will be further clarified by a consideration of the following examples, which are intended to be purely illustrative of the use of the invention. Other embodiments of the invention will be apparent to those skilled in the art from a consideration of this specification or practice of the invention as disclosed herein. Unless otherwise noted, all percentages are given on a mole percent basis. Selectivities are given on a carbon atom percent basis.
- A catalyst comprising platinum and tin supported on an alumina monolith was prepared by the following method. Platinum and tin were codeposited on a foam monolith (92 weight percent alpha alumina, 8 weight percent silica; 1.8 cm diameter×1 cm thick, 45 ppi (18 ppcm)) by impregnation with an aqueous solution of platinum and tin in a Pt:Sn atomic ratio of 1:5. The impregnation solution was prepared from a stock aqueous solution of hexachloroplatinic acid (0.193 M H 2PtCl6) and a stock aqueous solution of stannous chloride (0.372 M SnCl2) acidified with 5 weight percent hydrochloric acid. Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent. The impregnated monolith was dried in ambient air and then reduced under flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cubic foot per hour (cfh) (473 cm3/min) using the following temperature profile: 1 h from ambient to 125° C., then 1 h from 125° C. to 300° C.; 1 h from 300° C. to 450° C., held for 30 min at 450° C., and then cooled to room temperature.
- The catalyst was sandwiched between two inert alpha alumina monoliths which acted as radiation shields. The monoliths were sealed in a quartz tube using FiberFrax™ brand alumina-silica cloth (FiberFrax is a trademark of and available from Unifrax Corporation), and the reactor was insulated by wrapping the quartz tube with high temperature insulation. A feed comprising ethane (2.6 standard liters per minute (slpm)), oxygen (1.3 slpm), hydrogen (2.6 slpm) and nitrogen (1.147 slpm) was fed to the reactor. Total flow was 7.647 slpm (GHSV 180,305 h −1) at 15 volume percent dilution with nitrogen. The ethane to oxygen molar ratio was 2:1; the hydrogen to oxygen molar ratio was 2:1.
- The catalyst was operated autothermally and the heat generated by the reaction was sufficient to sustain the process. However, heat was needed initially to ignite the process. The procedure for light-off involved establishing the flows of nitrogen, ethane, and hydrogen, then adding the oxygen flow; and then heating the feed to 200° C. until ignition. This procedure ensured a fuel-rich feed for safety considerations. The light-off conditions were 7 slpm total gas flow, 2.24 slpm of ethane, 2.24 slpm of hydrogen, 1.12 slpm of oxygen, 1.40 slpm of nitrogen, 20 percent dilution with nitrogen, an ethane to oxygen molar ratio of 2:1, a hydrogen to oxygen molar ratio of 2:1, and a pressure of 1.34 atm abs (136 kPa abs). After light-off, the external heat source was removed, and the gas flow rates and pressure were adjusted to the desired conditions, as shown in Table 1. Pressure was maintained at 1.34 atm abs (136 kPa abs). Shutdown of the reactor was accomplished by turning off oxygen prior to alkane and hydrogen.
- The product gases were analyzed on a Carle gas chromatograph designed for refinery gas analyses of components up to C 6 hydrocarbons. For quantitative determination of concentrations, standards were used for all species except water, which was obtained from an oxygen atom balance. Nitrogen was used as an internal GC calibration standard. Results are set forth in Table 1.
TABLE 1 Oxidation of Ethane to Ethylene with Pt Catalysts Examples vs. Comparative Experimentsa,b % Carbon Atom Selectivities Ex. # % C2H6 Catalyst Conv. C2H4 CO CO2 CH4 C3+ CE-1a 62.3 61.1 22.60 7.75 4.05 4.50 Pt - No H2 CE-1b 62.2 71.1 16.92 1.40 6.10 4.48 Pt - H2 CE-1c 65.9 67.6 17.65 6.40 3.80 4.55 Pt—Sn No H2 E-1 69.6 81.1 7.29 0.34 6.28 4.99 Pt—Sn H2 CE-2 68.0 68.3 17.70 6.40 3.95 3.65 Pt—Sb No H2 E-2 69.5 81.5 7.56 0.27 6.32 4.35 Pt—Sb H2 CE-3 66.5 68.0 17.03 6.57 3.87 4.53 Pt—Sn—Sb No H2 E-3 68.5 80.6 8.27 0.46 6.10 4.57 Pt—Sn—Sb H2 - It was seen that a catalyst comprising platinum and tin supported on a ceramic monolith was active in the partial oxidation of ethane in the presence of hydrogen to produce ethylene. The catalyst achieved an ethane conversion of 69.6 percent and an ethylene selectivity of 81.1 percent. The ethane conversion and ethylene selectivity achieved were comparable to those obtained from commercial thermal cracking furnaces. Very low amounts of carbon monoxide (7.29 percent) and carbon dioxide (0.34 percent) were found, as well as comparable amounts of methane and C3+ products. Carbon monoxide, carbon dioxide, and methane, at least in part, can be recycled to the reactor along with the hydrogen produced in the process.
- The oxidation of ethane was conducted under autothermal process conditions with a catalyst consisting of platinum supported on a ceramic monolith support. The catalyst was prepared as in E-1, with the exception that no tin was added to the catalyst. The process was conducted first in the absence of hydrogen (CE-1a) as noted hereinafter, and then, in the presence of hydrogen (CE-1b) in a manner similar to E-1. For the part of the experiment without hydrogen, the flow rates of the reactant feed were adjusted as follows: ethane (2.6 slpm); oxygen (1.3 slpm); nitrogen (2.1 slpm). Total flow was 6 slpm (GHSV=141,471 h −1) at 35 volume percent nitrogen dilution. Molar ratio of ethane to oxygen was 2:1. This flow adjustment ensured that identical absolute amounts of ethane and oxygen were used with and without hydrogen. The level of nitrogen dilution was adjusted to ensure equivalent ethane conversions with and without hydrogen. Processes were conducted autothermally in the manner described in E-1 with the results shown in Table 1 (CE-1a and CE-1b).
- It was seen that a catalyst consisting of pure platinum on an alumina monolith achieved an ethylene selectivity of 61.1 percent in the absence of hydrogen (CE-1a) and 71.1 percent in the presence of hydrogen (CE-1b) at similar ethane conversions. Thus, the addition of hydrogen improved ethylene selectivity. More significantly, when CE-1a and CE-1b were compared with E-1, it was found that the combined use of hydrogen in the feed and tin in the catalyst resulted in the highest ethane conversion and ethylene selectivity at significantly lower selectivities to carbon oxides.
- The oxidation of ethane was conducted as in E-1 with the exception that no hydrogen was used in the process, and the feed flow rates were adjusted as described in CE 1a. The catalyst used was identical to the catalyst of E1. Results are shown in Table 1 (CE-1c). When the process of E-1, using a catalyst containing platinum and tin, was repeated in the absence of hydrogen, an ethylene selectivity of 67.6 percent was achieved at an ethane conversion of 65.9 percent. When E-1 was compared with CE-1c, it was seen that the combined use of hydrogen in the feed and tin in the catalyst gave the highest ethane conversion, the highest ethylene selectivity, and the lowest selectivities to carbon oxides.
- A catalyst comprising platinum and antimony supported on an alumina monolith was prepared in a manner similar to that described in Example 1. The monolith of Example 1 was impregnated with an aqueous solution of platinum and antimony in a Pt:Sb atomic ratio of 1:5. The impregnation solution was prepared from a stock aqueous solution of hexachloroplatinic acid (0.193 M H 2PtCl6) and a stock aqueous solution of antimony triacetate (0.182 M Sb(OAc)3) containing hydrochloric acid sufficient to dissolve the antimony salt. Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent. The impregnated monolith was dried in ambient air, then reduced under flowing hydrogen in the manner described in E-1 hereinabove.
- The catalyst was tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions as described in E-1 with the results shown in Table 1 hereinabove. It was seen that under autothermal process conditions a catalyst comprising platinum and antimony supported on a ceramic monolith achieved an ethane conversion of 69.5 percent and an ethylene selectivity of 81.5 percent. Selectivities to carbon monoxide and carbon dioxide were low. The results are comparable to those achieved in commercial thermal cracking furnaces.
- The catalyst of E-2 comprising platinum and antimony supported on an alumina monolith was tested in the oxidation of ethane as described in E-2, with the exception that no hydrogen was used in the feedstream. Process conditions were as described in CE-1a. Results are set forth in Table 1 (CE-2).
- When E-2 was compared with CE-2 or any of CE-1a and CE-1b, it was seen that the combined use of antimony in the catalyst and hydrogen in the feedstream as shown in E-2 gave the highest ethane conversion and ethylene selectivity and the lowest levels of carbon oxides.
- A catalyst comprising platinum, tin, and antimony supported on an alumina monolith was prepared in a manner similar to that described in Examples 1 and 2. The metals were codeposited by impregnation of the support with an aqueous solution of Pt, Sn, and Sb salts in a Pt:Sn:Sb atomic ratio of 1:5:0.26. The impregnation solution was prepared from a stock aqueous solution of platinum hexachloroplatinic acid (0.193 M), a stock aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent), and a stock aqueous solution of antimony triacetate (0.182 M) containing hydrochloric acid (50 weight percent). Sufficient impregnation solution was used to obtain a platinum loading of 1.3 weight percent. The impregnated monolith was dried in ambient air and reduced under hydrogen as described in E-1 hereinabove.
- The catalyst was tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions as described in E-1 with the results shown in Table 1 hereinabove. It was seen that a catalyst comprising platinum, tin, and antimony supported on a ceramic monolith achieved an ethane conversion of 68.5 percent and an ethylene selectivity of 80.6 percent. Carbon monoxide and carbon dioxide were produced only at low levels. The results are comparable to those achieved in commercial thermal cracking furnaces.
- The catalyst of E-3 comprising platinum, tin, and antimony on an alumina monolith was evaluated in the oxidation of ethane as described in E-3, with the exception that no hydrogen was used in the feedstream. Process conditions were as described in CE-1a. Results are set forth in Table 1 (CE-3). When E-3 was compared with CE-3 and any of CE 1a and CE-1b, it was seen that the combined use of antimony and tin in the catalyst and hydrogen in the feedstream as shown in E-3 gave the highest conversion of ethane, the highest selectivity to ethylene, and the lowest levels of carbon oxides.
- An aqueous impregnation solution was prepared containing platinum and copper in a Pt:Cu atomic ratio of 1:5. The impregnation solution was prepared from stock solutions of chloroplatinic acid (0.193 M H 2PtCl6) and cupric chloride (1.49 M CuC12). A ceramic fiber mat (Nextel™ 440 brand fiber mat, 2 cm square×1 cm thick, weighing 0.25 g) was precalcined in air at 900° C., cooled, and then impregnated with the impregnation solution to saturation. Sufficient solution was used to obtain a calculated platinum loading of 16 weight percent in the finished mat. The impregnated fiber mat was dried in ambient air, then reduced in flowing hydrogen, as described in E-1.
- The catalyst was sandwiched between two inert foam monoliths (1.8 cm dia by 1 cm thick, 18 ppcm alumina or mullite), wrapped in FiberFrax™ brand alumina-silica cloth, and packed into a quartz tube reactor (Inner Diameter (I.D.) 1.9 cm). The feed to the reactor was preheated with a heating tape wrapped around the quartz tube upstream of the catalyst. The catalyst zone was not heated, but was insulated with high temperature insulation material to minimize heat losses. Ethane, hydrogen, and nitrogen were preheated to 200° C. and fed to the reactor. Oxygen was then introduced to the reactor which resulted in catalyst ignition. Upon ignition the temperature rose within a few seconds to 1000° C., and the reactor operated autothermally. Process conditions and results are shown in Table 2.
TABLE 2 Ethane Oxidation to Ethylene Over Catalyst of Pt/Cu on Fiber Monolith E-4 Run 1a Run 2a Total Feed Flow, slpm 6.75 8.0 GHSV, h−1 318,471 377,448 N2 Dilution 12% 30% Molar Ratio C2H6/O2 2.2 2.0 Molar Ratio H2/O2 2.2 2.0 % C2H6 Conv 70.0 71.2 % CO Sel 7.5 7.8 % CO2 Sel 0.6 0.7 % C2H4 Sel 80.0 80.4 % C2H2 Sel 1.0 1.2 % Total C2H4 Sel 81.0 81.6 - It was found that a catalyst comprising platinum and copper supported on a ceramic fiber monolith achieved an ethane conversion of about 70 percent and an ethylene selectivity of 80 percent. Carbon monoxide and carbon dioxide were produced only at low levels. The results are comparable to those achieved in commercial thermal cracking furnaces.
- Five catalysts were prepared as follows:
- Catalyst A comprised platinum and copper in a Pt:Cu atomic ratio of 1:1 supported on an alumina foam monolith. The monolith of E-1 was impregnated with an aqueous impregnation solution (1 ml) prepared from stock solutions of hexachloroplatinic acid (0.193 M) and cupric chloride (1.49 M). Sufficient stock solutions were used to achieve a Pt:Cu atomic ratio of 1:1. Calculated platinum loading was 1.2 weight percent. The impregnated monolith was dried and reduced under hydrogen in the manner described in E-1.
- Catalyst B comprised platinum and copper in a Pt:Cu atomic ratio 1:1 supported on a Nextel™ 440 ceramic fiber mat. The catalyst was prepared in the manner described in E-4 hereinabove, with the exception that the amounts of stock solutions used were adjusted to provide the Pt:Cu atomic ratio of 1:1. Calculated platinum loading was 20 weight percent.
- Catalyst C comprised platinum and copper in an atomic ratio of 1:2 supported on a Nextel™ 440 ceramic fiber mat. The catalyst was prepared in the manner described in E-4 hereinabove, with the exception that the amounts of stock solutions used were adjusted to give the Pt:Cu atomic ratio of 1:2. Calculated platinum loading was 24 weight percent.
- Catalyst D comprised platinum, tin, and copper in an atomic ratio 1:1:1 supported on a Nextel™ 440 ceramic fiber mat. The catalyst was prepared by calcining the fiber mat at 900° C., cooling, then impregnating the calcined mat to wetness with an impregnation solution prepared from stock solutions of hexachloroplatinic acid (0.193 M), cupric chloride (1.49 M), and stannous chloride (0.372 M) acidified with 5 weight percent hydrochloric acid. Calculated platinum loading was 18 weight percent. The impregnated fiber mat was dried in ambient air and reduced under hydrogen, as described in E-1 hereinbefore.
- Catalyst E comprised platinum and tin in an atomic ratio of 1:5 supported on a Nextel™ 440 brand ceramic fiber mat. The catalyst was prepared by calcining the fiber mat at 900° C., cooling, and impregnating the fiber mat to saturation with an aqueous impregnation solution prepared from stock solutions of hexachloroplatinic acid (0.193 M) and stannous chloride (0.372 M) acidified with hydrochloric acid (5 weight percent). Calculated platinum loading was 8.5 weight percent. The impregnated fiber mat was dried and reduced as described in E-1 hereinbefore.
- The catalysts were tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal reaction conditions. Process conditions and results are set forth in Table 3.
TABLE 3 Oxidation of Ethane to Ethylene - Stability of Catalystsa Flow GHSV % C2H6 TOSb Catalyst slpm h−1 % N2 % C2H4 Sel Conv h A: Pt—Cu 7.33 172,830 20.5 79.1 63.1 2.0 (1:1) 78.9 63.0 13.7 on Al2O3 foam B: Pt—Cu 8.33 392,959 30 82.2 58.5 1 (1:1) 81.1 56.5 8 on fiber mat C: Pt—Cu 8.33 392,959 30 82.5 60.9 0.5 (1:2) 81.6 59.3 17.3 on fiber mat D: Pt—Sn—Cu 8.33 392,959 30 83.7 61.2 0.3 (1:1:1) 81.6 59.3 18 on fiber mat E: Pt—Sn 8.33 392,959 30 83.2 59.4 2 (1:5) 82.0 55.1 18 on fiber mat - It was found that catalysts comprising platinum and tin, copper, or a mixture thereof, supported on a ceramic foam or fiber mat achieved a high ethane conversion, a high ethylene selectivity, and good catalyst stability in a process of oxidizing ethane to ethylene in the presence of hydrogen.
- Catalysts comprising platinum and copper supported on Nextel™ 440 brand ceramic fiber mats were prepared in the manner described in E-4 hereinabove. The atomic ratio of platinum to copper was varied from 1:0.1 to 1:5. The catalysts were tested in the oxidation of ethane to ethylene in the presence of hydrogen and under autothermal process conditions, with the results shown in Table 4.
TABLE 4 Oxidation of Ethane to Ethylene Variation in Pt/Cu Ratioa Pt:Cu Pt loading TOSb % C2H4 % C2H6 ratio (wt %) h Sel. Conv. 1:0.1 21 2.1 79.3 55.1 1:0.5 21 8.5 80.3 56.2 1:1 20 8.0 81.1 56.5 1:2 24 16.3 81.6 59.3 1:3 21 16.5 81.7 59.4 1:5 18 15.5 82.1 59.3 - Samples run out to 8 or less hours would have given slightly lower conversion and selectivity if run out to 16 hours. Thus, it was found that as the atomic ratio of platinum to copper decreased from 1:0.1 to 1:5, the ethane conversion and ethylene selectivity increased. It was also found that at the higher concentrations of copper, the catalyst did not remain lit in the absence of hydrogen.
- A catalyst comprising platinum and copper in an atomic ratio of 1:1 supported on a Nextel™ 440 brand ceramic fiber mat was prepared in a manner similar to that described in E-4 hereinabove. The catalyst was evaluated in the oxidation of ethane to ethylene in the presence of hydrogen under autothermal process conditions. The gas hourly space velocity of the total feed was progressively increased at constant pressure with the results shown in Table 5.
TABLE 5 Oxidation of Ethane to Ethylene Variation in Space Velocitya Flow rate GHSV % C2H4 % C2H6 slpm h−1 Sel. Conv. 7 330,100 80.6 54.3 14 660,200 81.0 55.7 21 990,300 81.7 55.5 28 1,320,400 81.6 55.7 35 1,650,500 82.4 53.0 42 1,980,600 81.9 52.4 - Over a wide range of space velocities tested, it was found that the ethane conversion and ethylene selectivity did not change significantly.
- Four catalysts were prepared comprising platinum supported on a modified ceramic foam monolith (92 weight percent alpha alumina, 8 weight percent silica; 45 ppi (18 ppcm); 1.8 cm dia by 1 cm thick; average weight 2.8 g). The preparation was characterized by first modifying the support with a support modifier, specifically tin or antimony, and then depositing platinum and optionally copper on the modified support. In this example, the same element (Sn) that modifies the support also functions as a promoter. Details of the preparation were as follows:
- Catalyst A comprised platinum on a tin-modified alumina monolith. The monolith of E-1 was impregnated to wetness with an aqueous solution of stannous chloride (0.372 M) containing 5 weight percent hydrochloric acid. The impregnated support was air dried and then reduced at 700° C. under flowing hydrogen at a flow rate of 1 cfh (473 cm 3/min). The modified support was impregnated with an aqueous solution (1 ml) of hexachloroplatinic acid (0.193 M), then dried in ambient air and reduced under hydrogen as described in E-1.
- Catalyst B comprised platinum and copper (1:1) on a tin-modified alumina monolith. The monolith was impregnated to wetness with an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent). The tin-impregnated monolith was air dried and reduced at 700° C. for 2 h in flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cfh (473 cm 3/min). The modified monolith was impregnated with an aqueous solution (1 ml) prepared from stock solutions of hexachloroplatinic acid (0.193 M) and cupric chloride (1.49 M). The impregnated monolith was air dried and reduced under flowing hydrogen as described in E-1.
- Catalyst C comprised platinum and copper (1:5) on a tin-modified alumina monolith. The catalyst was prepared as in “B” hereinabove with the exception that sufficient stock solutions were used to give a Pt:Cu atomic ratio of 1:5.
- Catalyst D comprised platinum and copper (1:5) on an antimony-modified alumina monolith. In this example, the monolith comprised alpha alumina (99.5 weight percent). The monolith was impregnated to wetness with a solution of antimony triacetate (0.182 M) dissolved in hydrochloric acid. The monolith was air dried and reduced for 2 h at 700° C. under flowing hydrogen (5 volume percent in nitrogen) at a flow rate of 1 cfh (473 cm 3/min). The reduced monolith was impregnated with an aqueous impregnation solution (1 ml) prepared from a stock solution of hexachloroplatinic acid (0.193 M, 1 ml) and a stock solution of cupric chloride (1.49 M, 0.65 ml). The monolith was dried in air and reduced as in E-1.
- The catalysts were evaluated in the oxidation of ethane in the presence of hydrogen and under autothermal reaction conditions with the results shown in Table 6.
TABLE 6 Oxidation of Ethane to Ethylene - Modified Supporta % % Flow GHSV C2H6 H2 Pre- C2H4 C2H6 TOSb Catalyst slpm h−1 % N2 O2 O2 heat ° C. Sel. Conv h A: Pt 11 259,364 5 2.1 2.1 150 78.3 68.1 0.5 Sn—Al2O3 150 76.6 67.5 7.5 foam B: Pt—Cu (1:1) 11 259,364 5 2.1 2.1 150 79.8 71.1 0.5 Sn—Al2O3 150 78.8 69.7 15 foam C: Pt—Cu (1:5) 7 159,155 12 2.2 2.2 200 81.4 69.8 5 Sn—Al2O3 200 81.0 69.1 21 foam D: Pt—Cu 7 165,050 10 2.3 2.3 250 82.0 67.8 2.5 (1:5) 250 81.8 67.4 15 Sb—Al2O3 foam - It was found that a catalyst comprising platinum and optionally copper supported on a ceramic foam monolith modified with tin or antimony achieved a high conversion of ethane, a high selectivity to ethylene, and good catalyst stability. Results are comparable to those obtained from a commercial cracking furnace.
- A catalyst comprising platinum and copper (1:2) on a tin-modified alumina monolith (92 weight percent alumina) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:2. This catalyst was evaluated in the oxidation of a natural gas liquid (NGL) feed in the presence of hydrogen under autothermal process conditions. The liquid feed composition, an Algerian condensate, comprised on a weight percent basis a mixture of 42.1 percent paraffins, 34.4 percent isoparaffins, 7.3 percent aromatics, 12.3 percent naphthenes, 0.2 percent oxygenates, and the balance (about 3-4 percent) unidentified components. The alkanes comprised C 1-19 alkanes having a maximum molar concentration in the C5-8 range. Feed was preheated at 200° C. Total gas flow was about 8 slpm (GHSV 200,000 h−1). Process conditions and results are set forth in Table 7.
TABLE 7 Oxidation of Natural Gas Liquid Feed (NGL)a,b Liq. feed O2 H2 N2 CH4 C2H4 C3H6 C4H8 C4H6 C6+ gm/min slpm slpm slpm NGL NGL NGL NGL NGL NGL 3.2 1.2 3.2 2.0 0.17 0.38 0.10 0.026 0.033 0.09 3.2 1.1 3.2 2.0 0.135 0.33 0.14 0.050 0.039 0.11 3.15 1.3 3.2 2.6 0.176 0.39 0.08 0.021 0.031 0.092 3.15 1.2 3.2 2.6 0.149 0.36 0.13 0.043 0.039 0.099 - It was seen that a catalyst comprising platinum and copper on a ceramic monolith support was capable of oxidizing a natural gas liquid feed in the presence of hydrogen under autothermal conditions to a mixture of low molecular weight olefins, specifically, ethylene, propylene, butylene, and butadiene.
- Catalysts were prepared comprising platinum and copper supported on a modified ceramic foam monolith. The preparation was characterized by first modifying the support with tin and optionally a second modifier, and thereafter depositing platinum and copper on the modified support. The support comprised a foam monolith, either 92 or 99.5 weight percent alumina [1.8 cm dia×1 cm thick; 45 ppi (18 ppcm)]. Preparations were as follows:
- Catalyst A comprising platinum and copper (1:2) on a tin-modified alumina (92 weight percent) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:2. Catalyst B comprising platinum and copper (1:5) on a tin-modified alumina (92.0 weight percent) was prepared as in Example 8C. Catalyst C comprising platinum and copper (1:4) on a tin-modified alumina (99.5 weight percent) was prepared in the manner described in Example 8B, with the exception that the atomic ratio of Pt:Cu was adjusted to 1:4. Catalyst D comprising platinum and copper (1:5) on a tin-modified alumina (99.5 weight percent) was prepared as in Example 8C.
- Catalyst E comprised platinum and copper (1:5) on a tin and calcium-modified alumina monolith (99.5 weight percent). The monolith was immersed in a saturated aqueous solution of calcium hydroxide for 24 h. Then, the monolith was rinsed several times with distilled water, air dried, and calcined at 900° C. for 1 h. The calcined monolith was immersed in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent) for several hours, after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm 3/min) at 700° C. for 2 h. An aqueous impregnation solution having a Pt:Cu atomic ratio of 1:5 was prepared from stock solutions comprising hexachloroplatinic acid (1 ml, 0.193 M) and cupric chloride (0.65 ml, 1.49 M). The monolith was impregnated with the impregnation solution (1 ml). The impregnated monolith was dried in ambient air and reduced as in E-1.
- Catalyst F comprised platinum and copper (1:5) on a tin and zirconium-modified alumina monolith (99.5 weight percent). The monolith was immersed for 24 h in an aqueous solution of zirconium oxychloride (ZrOCl 2, 1M) containing 1 weight percent hydrochloric acid. The monolith was rinsed with distilled water, air dried, and calcined at 900° C. for 1 h. The calcined monolith was immersed for several hours in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent), after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm3/min) at 700° C. for 2 h. An impregnation solution containing platinum and copper (1:5) was prepared from stock solutions of hexachloroplatinic acid (1 ml, 0.193 M) and cupric chloride (0.65 ml, 1.49 M). The monolith was impregnated with the impregnation solution (1 ml), dried in air, and reduced as in E-1.
- Catalyst G comprised platinum and copper (1:5) on a tin and lanthanum-modified alumina foam monolith (99.5 weight percent). The monolith was immersed for 24 h in an aqueous solution of lanthanum chloride (1 M) containing 1 weight percent hydrochloric acid. The monolith was rinsed with distilled water several times, air dried, and then calcined at 900° C. for 1 h. The calcined monolith was cooled and then immersed in an aqueous solution of stannous chloride (0.372 M) containing hydrochloric acid (5 weight percent) for several hours, after which the monolith was air dried and reduced under flowing hydrogen (1 cfh; 473 cm 3/min) at 700° C. for 2 h. The modified monolith was impregnated with an impregnation solution (1 ml) containing platinum and copper (1:5) prepared from the stock solutions noted hereinbefore. The impregnated monolith was reduced under hydrogen as in E-1.
- The catalysts were evaluated in the oxidation of ethane in the presence of hydrogen under autothermal reaction conditions with the results shown in Table 8.
TABLE 8 Oxidation of Ethane to Ethylene Catalyst of Pt—Cu on Modified Supporta Pre- % % Flow GHSV % heat C2H4 C2H6 TOSb Catalyst slpm h−1 N2 ° C. Sel. Conv. h A: Pt—Cu (1:2) 7.0 165,050 10 300 79.9 71.3 7.2 Sn—Al2O3 (92%) B: Pt—Cu (1:5) 7.0 165,050 10 300 80.2 71.6 13 Sn—Al2O3 (92.0%) C: Pt—Cu (1:4) 8.3 196,409 5 300 81.0 71.9 4.5 Sn—Al2O3 (99.5%) D: Pt—Cu (1:5) 7.0 165,050 10 300 80.7 72.4 12 Sn—Al2O3 (99.5%) E: Pt—Cu (1:5) 8.3 196,409 5 250 81.5 71.4 0.3 Sn—Ca—Al2O3 (99.5%) F: Pt—Cu (1:5) 8.3 196,409 5 250 81.0 71.7 0.3 Sn—Zr—Al2O3 (99.5%) G: Pt—Cu (1:5) 8.3 196,409 5 250 81.7 71.2 0.5 Sn—La—Al2O3 (99.5%) - It was seen that a catalyst comprising platinum and copper supported on an alumina monolith which had been pretreated with at least one of tin, calcium, zirconium, and lanthanum achieved a high ethane conversion, a high ethylene selectivity, and good stability in the oxidation of ethane to ethylene in the presence of hydrogen. Results of this invention are comparable to those obtained from commercial thermal cracking furnaces.
- The catalyst of E-7 was evaluated in the partial oxidation of ethane to ethylene in the presence of hydrogen in the manner described in E-7, with the exception that the pressure in the reactor was varied from about 2 atm abs (200 kPa abs) to about 4 atm abs (400 kPa abs). As in E-7, the catalyst comprised platinum and copper in an atomic ratio 1:1 supported on a Nextel™ 440 brand fiber mat. Process conditions and results are set forth in Table 9.
TABLE 9 Oxidation of Ethane-Variation in Pressurea, b Flow rate GHSV % N2 Pressure % C2H4 % C2H6 slpm h−1 dilution kPa Sel. Conv. 49 2,310,770 25 214 80.8 55.6 56 2,640,800 25 249 79.6 57.6 61 2,886,017 23 406 76.4 60.2 45 2,122,071 20 406 76.1 62.4 40 1,886,286 10 406 72.9 67.3 - It was seen that as the pressure of the process increased, the ethane conversion increased and the ethylene selectivity decreased.
- Two catalysts were evaluated in the partial oxidation of propane to ethylene and propylene in the presence of hydrogen. Catalyst A, identical to Catalyst E-1 hereinabove, comprised platinum and tin in an atomic ratio 1:5 supported on an alumina foam monolith (92 weight percent). Feed comprised a mixture of ethane (70 volume percent) and propane (30 volume percent) at a nitrogen dilution of 21 percent. Other process conditions and results are shown in Table 10.
- Catalyst B, identical to Catalyst E-3 hereinabove, comprised platinum, tin, and antimony in an atomic ratio of 1:5:0.26 supported on an alumina foam monolith (92 weight percent). Feed comprised propane at a nitrogen dilution of 30 percent. Process conditions and results are shown in Table 10.
TABLE 10 Partial Oxidation of Propane to Propylene and Ethylene With Hydrogen and Multi-Metallic Catalyst Mol % Conversion Feed % C2H6 % C3H8 % Carbon Atom Selectivities Conditionsa, b Conv Conv C2H4 C3H6 CO CO2 CH4 C4+ Aa. 70/30 71.8 94.6 65.2 3.5 12.6 0.6 14.2 3.0 C2H6/C3H8 Bb. 100% C3H8 (1) Comparativeb — 71.9 35.0 22.8 12.3 8.2 14.2 5.1 (2)b — 77.5 41.5 20.7 10.1 1.0 18.0 5.5 (3)b — 67.8 39.5 25.4 8.6 1.1 16.8 5.7 - It was seen that propane is converted primarily to ethylene and propylene in the presence of hydrogen and a multi-metallic catalyst supported on an alumina monolith. When the Comparative Experiment 12-B1 is compared with Examples 12-B2 and 12-B3, it is seen that the total selectivity to ethylene and propylene is higher when hydrogen is co-fed.
- A catalyst comprising platinum and copper on a tin and lanthanum-modified alumina monolith support was prepared as in Example E-10G. The catalyst was evaluated in the autothermal partial oxidation of ethane to ethylene under the conditions shown in Table 11. The feed comprising ethane, oxygen, hydrogen, and nitrogen was preheated to temperatures ranging from 281° C. to 589° C. At preheat temperatures above 400° C., the molar ratio of ethane to oxygen was raised to 2.7:1 and higher. Results are set forth in Table 11.
TABLE 11 Autothermal Oxidation of Ethane to Ethylene Using Preheata T (° C.) % C2H6 % C2H4 % CH4 % CO % CO2 Net C2H6/O2 H2/O2 Preheat Conv Sel Sel Sel Sel H2/C2H4 2.3 2.3 281 70.4 80.00 6.67 8.20 0.55 0.004 2.7 2.7 488 67.7 81.80 6.30 6.90 0.53 0.12 2.7 2.7 538 70.1 81.22 6.66 7.33 0.53 0.18 2.8 2.8 589 69.7 81.46 6.65 7.23 0.53 0.21 - It was found that by preheating the feed to temperatures above 400° C., substantially the same ethane conversion and product selectivities were obtained at higher hydrocarbon to oxygen molar ratios, as were obtained at lower preheat and lower hydrocarbon to oxygen ratios. Compare, for example, the run at 281° C. preheat with the run at 538° C. preheat. The ethane conversion and ethylene selectivity were similar while oxygen usage dropped from 0.88 g O 2 per g ethylene (ethane/oxygen molar ratio of 2.3:1) to 0.76 g O2 per g ethylene (ethane/oxygen molar ratio 2.7:1.) Likewise, the net hydrogen balance per mole ethylene improved from about zero (0.004) at 281° C. to 0.18 at 538° C.
- A catalyst was prepared as in Example E-4 hereinabove, with the exception that magnesia pellets (Norton; 3 mm dia×5 mm length cylinders) were used in place of the alumina fiber mat support. The magnesia pellets were heated to 1200° C. for 16 h to reduce the surface area to less than 1 m 2/g. A solution containing platinum and copper in an atomic ratio of 1:5 was prepared using hexachloroplatinic acid and cupric chloride. The pellets were loaded with the solution, dried at 80° C. overnight, and reduced at 450° C. under hydrogen (5 volume percent) in nitrogen. Pt:Cu atomic ratio was 1:5.6. Pt loading, 0.57 weight percent; copper loading, 1.03 weight percent; balance magnesia.
- The catalyst particles were sandwiched between two inert alumina monoliths in a quartz tube reactor. Catalyst bed dimensions were 17 mm (dia) by 15 mm (depth). The catalyst was evaluated in the autothermal oxidation of ethane to ethylene in the manner described hereinbefore. Process conditions and results are set forth in Table 12.
TABLE 12 Ethane Autothermal Oxidation to Ethylene with Pt/Cu/MgO Pelleted Catalysta Time % C2H6 % C2H4 % CH4 % CO % CO2 h C2H6/O2 H2/O2 Cony Sel Sel Sel Sel Preheat, 250° C.; 8 slpmb 0.9 2.3 2.3 73.6 79.6 7.5 5.4 2.2 4.5 2.3 2.3 73.5 80.0 7.0 5.4 2.0 6.2 2.3 2.3 73.7 79.4 6.9 5.5 2.0 7.2 2.3 2.3 73.7 79.5 7.0 5.6 2.0 Preheat, 275° C.; 6 slpmb; stable after 10 h 10.3 2.3 2.3 73.3 80.0 6.6 5.6 2.2 Preheat, 250° C. at 2.5 h and 275° C. at 3.5 h; 8 slpmb 2.5 2.4 2.4 69.3 82.0 6.4 4.7 1.8 3.5 2.4 2.4 70.8 81.5 6.6 4.9 1.8 - It was found that magnesia pellets can be suitably employed as a catalyst support in the autothermal oxidation process of this invention.
- A catalyst comprising platinum and copper on a ceramic monolith was prepared in the manner described in Example E-4, with the exception that a magnesia monolith (Hi-Tech Ceramics, 17 mm dia×10 mm width, 45 ppi) was used in place of the alumina fiber mat. Pt:Cu atomic ratio was 1:5, and the total metal loading was 5.67 weight percent. The catalyst was evaluated in the autothermal oxidation of ethane to ethylene as described hereinbefore, with the results shown in Table 13.
TABLE 13 Autothermal Oxidation of Ethane to Ethylene Using Pt/Cu on MgO Monolitha Time % C2H6 % C2H4 % CH4 % CO % C02 (h) Conv Sel Sel Sel Sel 0.5 75.1 80.0 6.7 6.0 1.8 1.5 75.1 80.7 6.5 5.9 1.5 2.5 74.6 81.2 6.3 5.9 1.4 4.5 74.5 81.4 6.3 6.1 1.3 5.5 74.4 81.2 6.3 6.1 1.3 6.5 74.5 81.4 6.3 6.2 1.2 - It was found that a magnesia monolith can be suitably employed as a support in the autothermal oxidation process of this invention.
- Alumina beads were used to prepare a catalyst. A solution containing platinum, copper, and tin in an atomic ratio of 1:5:5 was prepared by mixing hexachloroplatinic acid (0.659 ml, 0.193 M), cupric chloride (0.427 ml, 1.48 M), and stannous chloride (9.97 ml, 0.064 M, HCl to dissolve). Alumina beads (Norton, 590-850 μm, 28 g) were suspended in the solution with excess deionized water. The mixture was stirred and heated until almost all of the water was evaporated. The resulting solids were dried at 80° C. The total metals loading was 0.5 weight percent. The catalyst was loaded into the reactor and reduced under hydrogen (5 volume percent in nitrogen) at 300° C.
- A reactor was used comprising a quartz tube (19 mm dia) into which the catalyst (6 g) was loaded to a bed height of 1.5 cm (static aspect ratio 0.8). A quartz frit was used to support the catalyst and evenly distribute the gas flow. The feed was preheated and the reactor insulated in the manner described hereinbefore. Ethane, hydrogen, oxygen, and nitrogen were preheated to 275° C. and fed to the reactor at a flow rate which disengaged the particles and circulated them within the bed. The flow rate was set for slightly above minimal fluidization at operating conditions (5 slpm). The bed expanded to a height of 3.0 cm (operating aspect ratio 1.6). Oxygen was introduced which resulted in catalyst ignition. Upon light-off, the catalyst operated autothermally. Process conditions and results are shown in Table 14.
TABLE 14 Autothermal Oxidation of Ethane to Ethylene Using Modified Fluidized Bed Reactora Time % C2H6 % C2H4 % CH4 % CO % CO2 (h) Conv Sel Sel Sel Sel 4 70.6 82.7 5.8 3.6 0.72 5 70.8 82.8 5.9 3.5 0.71 6 70.5 83.0 5.8 3.4 0.68 7 70.7 83.0 5.9 3.3 0.68 8 70.3 83.1 5.8 3.3 0.67 14.5b 71.1 81.9 6.4 4.0 1.0 36.5b 71.6 81.7 6.4 4.0 0.8 - It was seen that a reactor operating at slightly above minimal fluidization could be used for the autothermal oxidation of ethane to ethylene to achieve high selectivity to ethylene and low selectivities to methane, carbon monoxide, and carbon dioxide. In this laboratory example, the aspect ratio during operation was greater than 1:1, because of the smaller reactor diameter; however, the same results are expected with a commercial scale reactor having a diameter of 1.5 or more meters and the same bed depth of 3 cm during operation, which results in an aspect ratio less than 1:1.
- The catalyst (6 g) from Example 16, prepared with alumina pellets, was evaluated in the oxidation of ethane to ethylene in a fixed bed reactor. The pellets were sandwiched between an inert alumina monolith and a quartz frit to retain the pellets in a fixed bed. Results are set forth in Table 15.
TABLE 15 Autothermal Oxidation of Ethane to Ethylene in Fixed Bed Reactora Time % C2H6 % C2H4 % CH4 % CO % CO2 (h) Conv Sel Sel Sel Sel 2.5 70.8 80.4 6.7 5.8 0.94 3.5 70.6 80.6 6.6 5.9 0.95 4.5 70.4 80.6 6.5 6.0 0.95 5.5 70.2 80.7 6.5 6.0 0.93 - It was found that ethane could be oxidized to ethylene in a fixed bed reactor over a catalyst prepared on alumina pellets. When Example 16 was compared with Example 17, it was concluded that although both the fixed bed and the modified fluidized bed reactors were suitable, the selectivities were more favorable in the modified fluidized bed reactor. Less methane, carbon monoxide, and carbon dioxide were obtained, and more ethylene was obtained at closely similar conversions.
- A magnesia monolith support (Hi-Tech Ceramics, Inc.; 17 mm dia×10 mm thick) was treated with an aqueous solution of tin (IV) chloride (0.24 M), then dried at 90° C. and reduced at about 875° C. under hydrogen (5 percent in nitrogen). Tin loading was 1 weight percent. The tin-treated support was impregnated with an aqueous solution of platinum and copper (Pt/Cu atomic ratio 1:5) prepared using solutions of hexachloroplatinic acid (0.19 M) and cupric chloride (1.49 M). Then, the monolith was dried at 80° C. and reduced at 450° C. under the aforementioned hydrogen flow. Pt loading was 3.26 weight percent.
- The catalyst was evaluated in the oxidation of ethane under autothermal conditions as shown in Table 16.
TABLE 16 Ethane Oxidation Using Pt—Cu on Sn-Treated MgO Supporta TOS % Conv % Sel % Sel % Sel % Sel (h) C2H6 C2H4 CH4 CO CO2 1.33 76.8 79.9 7.1 4.8 1.8 2.66 76.5 80.3 6.9 5.0 1.7 4.00 76.4 80.4 6.7 5.1 1.6 6.66 76.6 80.3 6.7 5.4 1.4 18.66 75.7 80.8 6.5 5.8 1.2 34.25 75.6 80.4 6.5 6.3 1.1 50.33 74.3 81.0 6.3 6.5 1.0 - It was found that the catalyst of Example 18 with a tin-modified magnesia support achieved somewhat higher conversion and higher selectivity than the related catalyst of Example 15 which used an unmodified magnesia support.
- A solution containing nickel and copper in an atomic ratio of 1:1 was prepared from an aqueous solution of nickel (II) chloride hexahydrate (0.2 M) and an aqueous solution of copper (II) chloride (1.49 M). An alumina monolith (99.5 weight percent alumina; 17 mm diameter×10 mm length) was loaded with the Ni—Cu solution, dried at 80° C. overnight, and then reduced at 450° C. in hydrogen (5 volume percent) in nitrogen. The total metal loading was 1.48 weight percent. The catalyst was evaluated in the autothermal oxidation of ethane to ethylene in the manner previously described. The catalyst required at least 400° C. preheat for ignition. Upon ignition, the preheat was reduced, and the catalyst remained ignited under the process conditions employed; however, the catalyst extinguished in the absence of hydrogen in the feedstream.
TABLE 17 Autothemial Oxidation of Ethane Over Ni—Cu/Al2O3 Catalysta,b T Mol % Mol % Mol % Mol % Mol % Run Time preheat C2H6 C2H6 CH4 CO CO2 #19 (h) (° C.) Conv. Sel. Sel Sel Sel (a) 2.5 250 79.7 73.2 7.3 12.3 1.01 (b) 3.5 125 72.9 75.0 6.2 11.7 0.90 (c) 5.5 200 64.4 79.6 5.4 8.7 0.91 (d) 7.4 250 68.4 78.1 5.9 9.2 0.94 (e) 8.2 275 69.5 77.7 6.1 9.3 0.93 (f) 17.5 275 68.6 76.3 6.6 10.9 0.59 (g) 27.0 275 68.1 76.2 6.3 11.4 0.56 - It was observed that a catalyst comprising copper and nickel on an alumina monolith is capable of oxidizing ethane to ethylene under autothermal conditions. As shown in Examples 19(a) versus 19(b) and Examples 19(c)-19(e), the catalyst is more active at a higher preheat temperature. As shown in Examples 19(e)-19(g) the catalyst is relatively stable for several hours.
Claims (16)
1. A catalyst composition comprising a Group 8B metal and at least one promoter supported on a ceramic monolith support which has been pretreated with a support modifier.
2. The composition of claim 1 wherein the Group 8B metal is a platinum group metal.
3. The composition of claim 1 wherein the platinum group metal is platinum.
4. The composition of claim 1 wherein the promoter is selected from Groups 1B, 6B, 3A, 4A, 5A, and mixtures thereof.
5. The composition of claim 1 wherein the promoter is selected from copper, tin, antimony, silver, indium, and mixtures thereof.
6. The composition of claim 1 wherein the support modifier is selected from Groups 1A, 2A, 3B, 4B, 5B, 6B, 1B, 3A, 4A, 5A, the lanthanide rare earths, and the actinide elements of the Periodic Table, and mixtures of the aforementioned elements.
7. The composition of claim 1 wherein the support modifier is selected from calcium, zirconium, tin, lanthanum, potassium, lutetium, erbium, barium, holmium, cerium, silver, and antimony.
8. The composition of claim 1 wherein the ceramic monolith is selected from silica, alumina, silica-aluminas, aluminosilicates, magnesia, magnesium aluminates, magnesium silicates, zirconia, titania, boria, zirconia toughened alumina, lithium aluminum silicates, silicon carbide, silicon nitride, and oxide-bonded silicon carbide.
9. The composition of claim 8 wherein the ceramic monolith comprises from 65 to 100 weight percent alpha alumina or gamma alumina.
10. The composition of claim 1 wherein the support is in the form of a foam, a fiber, or a pellet.
11. The composition of claim 10 wherein the ceramic monolith comprises a foam having from 5 to 100 pores per linear inch (2 to 40 pores per linear cm) and a surface area greater than 0.001 m2/g and less than 10 m2/g.
12. The composition of claim 10 wherein the ceramic monolith is in the form of a fiber having a diameter greater than 1 micron and less than 20 microns, and a surface area greater than 0.001 m2/g and less than 1 m2/g.
13. The composition of claim 10 wherein the ceramic monolith is in the form of pellets having a size between 30 and 1,000 microns.
14. The composition of claim 1 which is prepared by a process comprising (a) contacting a ceramic support with a support modifier to form a pretreated support, (b) optionally, calcining or reducing the pretreated support, (c) depositing onto the pretreated support a Group 8B metal and at least one promoter, (d) optionally, calcining the metal loaded support, and (e) reducing the support with a reducing agent under conditions sufficient to prepare the catalyst composition.
15. The composition of claim 1 wherein the Group 8B metal is platinum; the promoter is selected from copper, tin, antimony, silver, indium, and mixtures thereof; the support is selected from alumina, magnesia, and mixtures thereof; and the modifier is selected from calcium, zirconium, tin, lanthanum, potassium, lutetium, erbium, barium, holmium, cerium, silver, and antimony.
16. The composition of claim 1 wherein the Group 8B metal is platinum; the promoter is selected from tin, copper, and mixtures thereof; the support is selected from alumina, magnesia, and mixtures thereof; and the modifier is selected from tin, lanthanum, and mixtures thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/323,175 US20030191020A1 (en) | 1998-09-03 | 2002-12-17 | Autothermal process for the production of olefins |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9904198P | 1998-09-03 | 1998-09-03 | |
| US11186198P | 1998-12-11 | 1998-12-11 | |
| US13600399P | 1999-05-26 | 1999-05-26 | |
| US09/388,219 US6566573B1 (en) | 1998-09-03 | 1999-09-01 | Autothermal process for the production of olefins |
| US10/323,175 US20030191020A1 (en) | 1998-09-03 | 2002-12-17 | Autothermal process for the production of olefins |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/388,219 Division US6566573B1 (en) | 1998-09-03 | 1999-09-01 | Autothermal process for the production of olefins |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20030191020A1 true US20030191020A1 (en) | 2003-10-09 |
Family
ID=27378717
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/388,219 Expired - Fee Related US6566573B1 (en) | 1998-09-03 | 1999-09-01 | Autothermal process for the production of olefins |
| US09/388,220 Expired - Lifetime US6166283A (en) | 1998-09-03 | 1999-09-01 | On-line synthesis and regenerating of a catalyst used in autothermal oxidation |
| US09/706,464 Expired - Lifetime US6624116B1 (en) | 1998-09-03 | 2000-11-03 | On-line synthesis and regeneration of a catalyst used in autothermal oxidation |
| US10/323,175 Abandoned US20030191020A1 (en) | 1998-09-03 | 2002-12-17 | Autothermal process for the production of olefins |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/388,219 Expired - Fee Related US6566573B1 (en) | 1998-09-03 | 1999-09-01 | Autothermal process for the production of olefins |
| US09/388,220 Expired - Lifetime US6166283A (en) | 1998-09-03 | 1999-09-01 | On-line synthesis and regenerating of a catalyst used in autothermal oxidation |
| US09/706,464 Expired - Lifetime US6624116B1 (en) | 1998-09-03 | 2000-11-03 | On-line synthesis and regeneration of a catalyst used in autothermal oxidation |
Country Status (15)
| Country | Link |
|---|---|
| US (4) | US6566573B1 (en) |
| EP (2) | EP1114013B1 (en) |
| JP (2) | JP2002524429A (en) |
| KR (2) | KR100669000B1 (en) |
| CN (3) | CN1168688C (en) |
| AR (2) | AR020369A1 (en) |
| AT (2) | ATE237569T1 (en) |
| AU (2) | AU754523B2 (en) |
| BR (2) | BR9913611B1 (en) |
| CA (2) | CA2339809C (en) |
| DE (2) | DE69906994T3 (en) |
| ES (2) | ES2192075T5 (en) |
| ID (2) | ID27739A (en) |
| MY (2) | MY124615A (en) |
| WO (2) | WO2000014037A1 (en) |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050090392A1 (en) * | 2003-10-25 | 2005-04-28 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US20050113247A1 (en) * | 2003-11-21 | 2005-05-26 | Conocophillips Company | Copper modified catalysts for oxidative dehydrogenation |
| US20090274597A1 (en) * | 2006-09-08 | 2009-11-05 | David Waller | Method and device for catchment of platinum group metals in a gas stream |
| US20100191031A1 (en) * | 2009-01-26 | 2010-07-29 | Kandasamy Meenakshi Sundaram | Adiabatic reactor to produce olefins |
| US20100197485A1 (en) * | 2008-07-31 | 2010-08-05 | Celanese International Corporation | Catalysts for making ethanol from acetic acid |
| US20110190551A1 (en) * | 2010-02-02 | 2011-08-04 | Celanese International Corporation | Processes for Producing Ethanol from Acetaldehyde |
| US8080694B2 (en) | 2008-07-31 | 2011-12-20 | Celanese International Corporation | Catalyst for gas phase hydrogenation of carboxylic acids having a support modified with a reducible metal oxide |
| US8211821B2 (en) | 2010-02-01 | 2012-07-03 | Celanese International Corporation | Processes for making tin-containing catalysts |
| US8304586B2 (en) | 2010-02-02 | 2012-11-06 | Celanese International Corporation | Process for purifying ethanol |
| US8314272B2 (en) | 2010-02-02 | 2012-11-20 | Celanese International Corporation | Process for recovering ethanol with vapor separation |
| US8338650B2 (en) | 2008-07-31 | 2012-12-25 | Celanese International Corporation | Palladium catalysts for making ethanol from acetic acid |
| US8350098B2 (en) | 2011-04-04 | 2013-01-08 | Celanese International Corporation | Ethanol production from acetic acid utilizing a molybdenum carbide catalyst |
| US20130072738A1 (en) * | 2011-09-20 | 2013-03-21 | Korea Institute Of Science And Technology | SUPPORTED CATALYST FOR DIRECT DEHYDROGENATION OF n-BUTANE AND PREPARING METHOD OF BUTENES FROM n-BUTANE USING THE SAME |
| US8450535B2 (en) | 2009-07-20 | 2013-05-28 | Celanese International Corporation | Ethanol production from acetic acid utilizing a cobalt catalyst |
| US8460405B2 (en) | 2010-02-02 | 2013-06-11 | Celanese International Corporation | Ethanol compositions |
| US8471075B2 (en) | 2008-07-31 | 2013-06-25 | Celanese International Corporation | Processes for making ethanol from acetic acid |
| WO2013030677A3 (en) * | 2011-08-30 | 2013-07-11 | Do Carmo Roberto Werneck | A process for the production of olefins and use thereof |
| US8536382B2 (en) | 2011-10-06 | 2013-09-17 | Celanese International Corporation | Processes for hydrogenating alkanoic acids using catalyst comprising tungsten |
| US8541633B2 (en) | 2010-02-02 | 2013-09-24 | Celanese International Corporation | Processes for producing anhydrous ethanol compositions |
| US8546622B2 (en) | 2008-07-31 | 2013-10-01 | Celanese International Corporation | Process for making ethanol from acetic acid using acidic catalysts |
| US8569549B2 (en) | 2010-02-02 | 2013-10-29 | Celanese International Corporation | Catalyst supports having crystalline support modifiers |
| US8637714B2 (en) | 2008-07-31 | 2014-01-28 | Celanese International Corporation | Process for producing ethanol over catalysts containing platinum and palladium |
| US8658843B2 (en) | 2011-10-06 | 2014-02-25 | Celanese International Corporation | Hydrogenation catalysts prepared from polyoxometalate precursors and process for using same to produce ethanol while minimizing diethyl ether formation |
| US8668750B2 (en) | 2010-02-02 | 2014-03-11 | Celanese International Corporation | Denatured fuel ethanol compositions for blending with gasoline or diesel fuel for use as motor fuels |
| US8680317B2 (en) | 2008-07-31 | 2014-03-25 | Celanese International Corporation | Processes for making ethyl acetate from acetic acid |
| US8680343B2 (en) | 2010-02-02 | 2014-03-25 | Celanese International Corporation | Process for purifying ethanol |
| US8680321B2 (en) | 2009-10-26 | 2014-03-25 | Celanese International Corporation | Processes for making ethanol from acetic acid using bimetallic catalysts |
| US8703868B2 (en) | 2011-11-28 | 2014-04-22 | Celanese International Corporation | Integrated process for producing polyvinyl alcohol or a copolymer thereof and ethanol |
| US8710277B2 (en) | 2009-10-26 | 2014-04-29 | Celanese International Corporation | Process for making diethyl ether from acetic acid |
| US8728179B2 (en) | 2010-02-02 | 2014-05-20 | Celanese International Corporation | Ethanol compositions |
| US8747492B2 (en) | 2010-02-02 | 2014-06-10 | Celanese International Corporation | Ethanol/fuel blends for use as motor fuels |
| US8754267B2 (en) | 2010-05-07 | 2014-06-17 | Celanese International Corporation | Process for separating acetaldehyde from ethanol-containing mixtures |
| US8858659B2 (en) | 2010-02-02 | 2014-10-14 | Celanese International Corporation | Processes for producing denatured ethanol |
| US8907143B2 (en) | 2011-12-15 | 2014-12-09 | Celanese International Corporation | Process for producing ethanol by hydrogenating mixed feeds comprising water |
| US8932372B2 (en) | 2010-02-02 | 2015-01-13 | Celanese International Corporation | Integrated process for producing alcohols from a mixed acid feed |
| US9000234B2 (en) | 2011-12-22 | 2015-04-07 | Celanese International Corporation | Calcination of modified support to prepare hydrogenation catalysts |
| US9233899B2 (en) | 2011-12-22 | 2016-01-12 | Celanese International Corporation | Hydrogenation catalysts having an amorphous support |
| US9415378B2 (en) | 2013-12-02 | 2016-08-16 | Saudi Basic Industries Corporation | Dehydrogenation catalyst, its use and method of preparation |
| CN107790182A (en) * | 2016-08-29 | 2018-03-13 | 中国石油化工股份有限公司 | 1,4 diacetoxy butylene hydrogenation process catalyst |
| CN109821530A (en) * | 2017-11-23 | 2019-05-31 | 中国科学院大连化学物理研究所 | A kind of cobalt-base catalyst and its method for propylene ring oxidation reaction |
| US10968148B2 (en) | 2015-08-28 | 2021-04-06 | University College Cardiff Consultants Limited | Method of producing compound comprising alkenyl group |
| US20220379287A1 (en) * | 2021-05-28 | 2022-12-01 | National University Corporation Hokkaido University | Dehydrogenation catalyst |
| US11779915B2 (en) | 2019-01-15 | 2023-10-10 | IFP Energies Nouvelles | Process for preparing selective hydrogenation catalyst, comprising a step of forming a Ni—Cu alloy in pre-impregnation |
Families Citing this family (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE237569T1 (en) * | 1998-09-03 | 2003-05-15 | Dow Global Technologies Inc | AUTOTHERMAL PROCESS FOR PRODUCING OLEFINS |
| AU3601900A (en) * | 1999-02-22 | 2000-09-04 | Symyx Technologies, Inc. | Compositions comprising nickel and their use as catalyst in oxidative dehydrogenation of alkanes |
| AU777050B2 (en) * | 1999-12-23 | 2004-09-30 | Ineos Europe Limited | Process for the production of olefins |
| JP2003519106A (en) * | 1999-12-23 | 2003-06-17 | ビーピー ケミカルズ リミテッド | Olefin production method |
| GB0006384D0 (en) | 2000-03-16 | 2000-05-03 | Bp Chem Int Ltd | Process for production of olefins |
| GB0010693D0 (en) | 2000-05-03 | 2000-06-28 | Bp Chem Int Ltd | Process for the production of olefins |
| AU2001291563A1 (en) * | 2000-09-18 | 2002-04-02 | Ensyn Group, Inc. | Products produced from rapid thermal processing of heavy hydrocarbon feedstocks |
| US20020085975A1 (en) * | 2001-01-04 | 2002-07-04 | Wambaugh James Allen | Method to equalize heat distribution in reactor tube |
| KR100409083B1 (en) * | 2001-03-12 | 2003-12-11 | 주식회사 엘지화학 | Steam cracking catalyst of hydrocarbons for olefins production |
| US20040102858A1 (en) * | 2001-09-04 | 2004-05-27 | Boris Kesil | Soft-touch gripping mechanism for flat objects |
| US20030065235A1 (en) * | 2001-09-24 | 2003-04-03 | Allison Joe D. | Oxidative dehydrogenation of alkanes to olefins using an oxide surface |
| ZA200209470B (en) * | 2001-12-04 | 2003-06-03 | Rohm & Haas | Improved processes for the preparation of olefins, unsaturated carboxylic acids and unsaturated nitriles from alkanes. |
| GB0203058D0 (en) | 2002-02-09 | 2002-03-27 | Bp Chem Int Ltd | Production of olefins |
| GB0204140D0 (en) | 2002-02-22 | 2002-04-10 | Bp Chem Int Ltd | Production of olefins |
| DE60300995T2 (en) * | 2002-04-04 | 2006-04-20 | Nippon Shokubai Co. Ltd. | Silver catalyst containing a support based on silica-alumina and its use in the preparation of epoxides |
| US7402719B2 (en) * | 2002-06-13 | 2008-07-22 | Velocys | Catalytic oxidative dehydrogenation, and microchannel reactors for catalytic oxidative dehydrogenation |
| AU2003243575A1 (en) * | 2002-06-20 | 2004-01-06 | The Regents Of The University Of California | Supported metal catalyst with improved thermal stability |
| US20040010174A1 (en) * | 2002-07-15 | 2004-01-15 | Conoco Inc. | Oxidative dehydrogenation of hydrocarbons by promoted metal oxides |
| US7014835B2 (en) | 2002-08-15 | 2006-03-21 | Velocys, Inc. | Multi-stream microchannel device |
| US7250151B2 (en) | 2002-08-15 | 2007-07-31 | Velocys | Methods of conducting simultaneous endothermic and exothermic reactions |
| US9192929B2 (en) | 2002-08-15 | 2015-11-24 | Velocys, Inc. | Integrated combustion reactor and methods of conducting simultaneous endothermic and exothermic reactions |
| US20040068148A1 (en) * | 2002-10-08 | 2004-04-08 | Conoco Inc. | Oxidative dehydrogenation of hydrocarbons using catalysts with trace promoter metal loading |
| US20040068153A1 (en) * | 2002-10-08 | 2004-04-08 | Conoco Inc. | Rare earth metals as oxidative dehydrogenation catalysts |
| US6946013B2 (en) | 2002-10-28 | 2005-09-20 | Geo2 Technologies, Inc. | Ceramic exhaust filter |
| US7459224B1 (en) | 2002-11-26 | 2008-12-02 | General Motors Corporation | Methods, apparatus, and systems for producing hydrogen from a fuel |
| US7105148B2 (en) | 2002-11-26 | 2006-09-12 | General Motors Corporation | Methods for producing hydrogen from a fuel |
| US20040158112A1 (en) * | 2003-02-10 | 2004-08-12 | Conocophillips Company | Silicon carbide-supported catalysts for oxidative dehydrogenation of hydrocarbons |
| US20040192546A1 (en) * | 2003-03-27 | 2004-09-30 | Zhongyuan Dang | Catalyst for the low temperature oxidation of methane |
| US7405338B2 (en) * | 2003-04-07 | 2008-07-29 | Velocys | Dehydrogenation reactions in narrow reaction chambers and integrated reactors |
| CA2427722C (en) | 2003-04-29 | 2007-11-13 | Ebrahim Bagherzadeh | Preparation of catalyst and use for high yield conversion of methane to ethylene |
| US7153334B2 (en) * | 2003-05-21 | 2006-12-26 | General Motors Corporation | Fuel reforming system and method of operation |
| US7208647B2 (en) * | 2003-09-23 | 2007-04-24 | Synfuels International, Inc. | Process for the conversion of natural gas to reactive gaseous products comprising ethylene |
| US7183451B2 (en) * | 2003-09-23 | 2007-02-27 | Synfuels International, Inc. | Process for the conversion of natural gas to hydrocarbon liquids |
| JP2005144432A (en) * | 2003-11-18 | 2005-06-09 | Rohm & Haas Co | Catalyst system for converting alkane into alkene and corresponding oxygenated product |
| US20050124840A1 (en) * | 2003-12-05 | 2005-06-09 | Conocophillips Company | Process for the production of olefins from alkanes with carbon monoxide co-feed and/or recycle |
| US7470408B2 (en) | 2003-12-18 | 2008-12-30 | Velocys | In situ mixing in microchannels |
| US8378163B2 (en) | 2004-03-23 | 2013-02-19 | Velocys Corp. | Catalysts having catalytic material applied directly to thermally-grown alumina and catalytic methods using same, improved methods of oxidative dehydrogenation |
| US7468455B2 (en) * | 2004-11-03 | 2008-12-23 | Velocys, Inc. | Process and apparatus for improved methods for making vinyl acetate monomer (VAM) |
| GB0501253D0 (en) * | 2005-01-21 | 2005-03-02 | Bp Chem Int Ltd | Process for the production of olefins |
| GB0501255D0 (en) * | 2005-01-21 | 2005-03-02 | Bp Chem Int Ltd | Process for the production of olefins |
| US8013197B2 (en) * | 2005-02-18 | 2011-09-06 | Synfuels International, Inc. | Absorption and conversion of acetylenic compounds |
| WO2006117509A1 (en) * | 2005-04-29 | 2006-11-09 | Ineos Europe Limited | Process for the production of olefins by autothermal cracking |
| BRPI0502015A (en) * | 2005-06-01 | 2007-01-23 | Petroleo Brasileiro Sa | catalytically selective cracking process of the natural gas liquid fraction to light olefins and other products |
| WO2007003890A2 (en) * | 2005-07-06 | 2007-01-11 | Ineos Europe Limited | Process for the production of linear alpha-olefins |
| WO2007003892A1 (en) * | 2005-07-06 | 2007-01-11 | Ineos Europe Limited | Process for the production of olefins |
| CN100368357C (en) * | 2005-08-15 | 2008-02-13 | 中国石油化工股份有限公司 | Method for producing ethene, propylene by using naphtha |
| US7682578B2 (en) | 2005-11-07 | 2010-03-23 | Geo2 Technologies, Inc. | Device for catalytically reducing exhaust |
| US7682577B2 (en) | 2005-11-07 | 2010-03-23 | Geo2 Technologies, Inc. | Catalytic exhaust device for simplified installation or replacement |
| CN1986505B (en) * | 2005-12-23 | 2010-04-14 | 中国石油化工股份有限公司 | Catalytic conversion process with increased low carbon olefine output |
| US7722828B2 (en) | 2005-12-30 | 2010-05-25 | Geo2 Technologies, Inc. | Catalytic fibrous exhaust system and method for catalyzing an exhaust gas |
| US7829035B2 (en) * | 2006-01-19 | 2010-11-09 | Massachusetts Institute Of Technology | Oxidation catalyst |
| EP1820569A1 (en) * | 2006-01-20 | 2007-08-22 | Ineos Europe Limited | Process for contacting a hydrocarbon and an oxygen-containing gas with a catalyst bed |
| GB2435883A (en) * | 2006-03-10 | 2007-09-12 | Innovene Europ Ltd | Autothermal cracking process for ethylene production |
| US7771669B2 (en) * | 2006-03-20 | 2010-08-10 | Ford Global Technologies, Llc | Soot oxidation catalyst and method of making |
| US7797931B2 (en) * | 2006-03-20 | 2010-09-21 | Ford Global Technologies, Llc | Catalyst composition for diesel particulate filter |
| GB0613676D0 (en) * | 2006-07-10 | 2006-08-16 | Ineos Europe Ltd | Process |
| US7999144B2 (en) | 2006-09-01 | 2011-08-16 | Velocys | Microchannel apparatus and methods of conducting catalyzed oxidative dehydrogenation |
| FR2910347B1 (en) * | 2006-12-22 | 2009-12-04 | Inst Francais Du Petrole | BIMETALLIC OR MULTI-METALLIC CATALYST HAVING OPTIMIZED HYDROGEN ADSORPTION CAPABILITY AND BIMETALLICITY INDEX |
| DE102007006647A1 (en) | 2007-02-06 | 2008-08-07 | Basf Se | Process for the regeneration of a catalyst bed deactivated in the context of a heterogeneously catalyzed partial dehydrogenation of a hydrocarbon |
| US8951929B2 (en) * | 2008-01-16 | 2015-02-10 | Agency For Science, Technology And Research | Catalyst preparation and methods of using such catalysts |
| US7745667B2 (en) * | 2008-04-07 | 2010-06-29 | Velocys | Microchannel apparatus comprising structured walls, chemical processes, methods of making formaldehyde |
| US8237000B2 (en) | 2008-06-19 | 2012-08-07 | Lummus Technology, Inc. | Combined carbon dioxide and oxygen process for ethylbenzene dehydrogenation to styrene |
| KR100963778B1 (en) * | 2008-07-25 | 2010-06-14 | 현대제철 주식회사 | Manufacture method of catalyst for the carbon dioxide reforming of methane, and its reforming reaction |
| IT1395473B1 (en) * | 2008-08-08 | 2012-09-21 | Univ Degli Studi Salerno | SELF-THERMAL CATALYTIC REACTOR WITH FLAT TEMPERATURE PROFILE FOR THE PRODUCTION OF HYDROGEN FROM LIGHT HYDROCARBONS |
| DE102008044946B4 (en) | 2008-08-29 | 2022-06-15 | Evonik Superabsorber Gmbh | Use of foam bodies in oxidation reactors for the production of unsaturated carboxylic acids |
| WO2010040688A1 (en) * | 2008-10-08 | 2010-04-15 | Basf Se | Method for producing an alkylene oxide |
| US20100116714A1 (en) * | 2008-11-12 | 2010-05-13 | Lapinski Mark P | Process and System for the Addition of Promoter Metal In Situ in a Catalytic Reforming Unit |
| US20100150805A1 (en) * | 2008-12-17 | 2010-06-17 | Uop Llc | Highly stable and refractory materials used as catalyst supports |
| CA2655841C (en) | 2009-02-26 | 2016-06-21 | Nova Chemicals Corporation | Supported oxidative dehydrogenation catalyst |
| US8519210B2 (en) * | 2009-04-02 | 2013-08-27 | Lummus Technology Inc. | Process for producing ethylene via oxidative dehydrogenation (ODH) of ethane |
| US8105972B2 (en) * | 2009-04-02 | 2012-01-31 | Lummus Technology Inc. | Catalysts for the conversion of paraffins to olefins and use thereof |
| US8105971B2 (en) * | 2009-04-02 | 2012-01-31 | Lummus Technology Inc. | Process for making catalysts useful for the conversion of paraffins to olefins |
| MY161990A (en) * | 2009-05-20 | 2017-05-31 | Basf Se | Monolith catalyst and use thereof |
| FR2949078B1 (en) * | 2009-08-17 | 2011-07-22 | Inst Francais Du Petrole | PROCESS FOR PREPARING NI / SN-SUPPORTED CATALYST FOR SELECTIVE HYDROGENATION OF POLYUNSATURATED HYDROCARBONS |
| US8314279B2 (en) | 2010-08-30 | 2012-11-20 | The United States of America as reprensted by the Secretary of the Army | Process for selectively making olefins from energy dense alcohols |
| RU2483799C2 (en) * | 2010-12-24 | 2013-06-10 | Российская Федерация, от имени которой выступает Министерство промышленности и торговли Российской Федерации (Минпромторг России) | Method of producing catalyst of methane-bearing hydrocarbon steam conversion |
| CA2752409C (en) | 2011-09-19 | 2018-07-03 | Nova Chemicals Corporation | Membrane-supported catalysts and the process of oxidative dehydrogenation of ethane using the same |
| CN103086821B (en) * | 2011-10-28 | 2015-10-21 | 中国石油化工股份有限公司 | A kind of production method of low-carbon alkene |
| CN103087765B (en) * | 2011-10-28 | 2015-09-16 | 中国石油化工股份有限公司 | A kind of production method of low-carbon alkene |
| US8912110B2 (en) | 2012-03-29 | 2014-12-16 | Uop Llc | Catalyst for conversion of hydrocarbons |
| US9266091B2 (en) | 2012-03-29 | 2016-02-23 | Uop Llc | Reforming catalysts with tuned acidity for maximum aromatics yield |
| US9545610B2 (en) | 2013-03-04 | 2017-01-17 | Nova Chemicals (International) S.A. | Complex comprising oxidative dehydrogenation unit |
| CN103232312B (en) * | 2013-05-07 | 2015-01-07 | 青岛京齐新材料科技有限公司 | Device and process for preparing isobutylene by dehydrogenating isobutane |
| RU2529995C1 (en) * | 2013-07-01 | 2014-10-10 | Федеральное государственное бюджетное учреждение науки Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук | Catalyst and method of producing ethylene and propylene |
| CA2833822C (en) | 2013-11-21 | 2020-08-04 | Nova Chemicals Corporation | Inherently safe odh operation |
| CA2867731C (en) | 2014-10-15 | 2022-08-30 | Nova Chemicals Corporation | High conversion and selectivity odh process |
| CN105582997B (en) * | 2014-10-21 | 2018-05-18 | 中国石油化工股份有限公司 | The method of catalyst of naphtha catalytic cracking production propylene and preparation method thereof and naphtha catalytic cracking production propylene |
| KR101716170B1 (en) * | 2015-11-10 | 2017-03-14 | 희성촉매 주식회사 | A stabilized active metal complex based catalyst for dehydrogenation of light straight-chain hydrocarbons |
| US9815752B2 (en) | 2016-02-12 | 2017-11-14 | King Fahd University Of Petroleum And Minerals | Fluidizable catalyst for oxidative dehydrogenation of alkanes to olefins in an oxygen free environment |
| CA2965062A1 (en) | 2017-04-25 | 2018-10-25 | Nova Chemicals Corporation | Complex comprising odh unit with integrated oxygen separation module |
| US20180333703A1 (en) | 2017-05-17 | 2018-11-22 | Exxonmobil Research And Engineering Company | Metal monolith for use in a reverse flow reactor |
| CN107152356B (en) * | 2017-06-09 | 2023-05-26 | 李秋生 | Front-mounted U-shaped fuel oil catalyst for motor vehicle |
| CN111107929B (en) * | 2017-07-31 | 2023-02-17 | 沙特基础工业全球技术公司 | System and method for dehydrogenating isobutane to isobutylene |
| CN110624554B (en) * | 2018-06-22 | 2022-07-05 | 中国石油天然气股份有限公司 | Catalyst for preparing 1, 3-butadiene and preparation method and application thereof |
| US10974226B2 (en) | 2018-09-24 | 2021-04-13 | Sabic Global Technologies B.V. | Catalytic process for oxidative coupling of methane |
| FR3091660A1 (en) * | 2019-01-15 | 2020-07-17 | IFP Energies Nouvelles | Process for the preparation of a selective hydrogenation catalyst comprising a step of forming a NiCu alloy in post-impregnation |
| FR3091658A1 (en) * | 2019-01-15 | 2020-07-17 | IFP Energies Nouvelles | Process for the preparation of an aromatic hydrogenation catalyst comprising a step of forming an NiCu alloy in post-impregnation |
| FR3091659B1 (en) * | 2019-01-15 | 2023-04-14 | Ifp Energies Now | Process for the preparation of a catalyst for the hydrogenation of aromatics comprising a step of forming a NiCu alloy in pre-impregnation |
| CN111893347B (en) * | 2019-05-06 | 2022-10-21 | 中国石油化工股份有限公司 | A 2 B-type hydrogen storage alloy, preparation method and application thereof, and method for purifying hydrogen containing organic matters |
| US11097257B2 (en) | 2019-09-10 | 2021-08-24 | Saudi Arabian Oil Company | Catalytic hydrocarbon dehydrogenation |
| US11338269B2 (en) | 2019-09-10 | 2022-05-24 | Saudi Arabian Oil Company | Catalytic hydrocarbon dehydrogenation |
| KR20230025654A (en) * | 2020-02-20 | 2023-02-22 | 에코카탈리틱 인크. | Fluid enhancer for oxidative dehydrogenation of hydrocarbons |
| US11161093B1 (en) | 2021-04-16 | 2021-11-02 | King Abdulaziz University | Gold-decorated magnesium silicate catalyst for producing light olefins |
| WO2023244938A1 (en) * | 2022-06-14 | 2023-12-21 | Dow Global Technologies Llc | Methods for making light olefins by dehydrogenation that utilize combustion additives that include transition metals |
| CN117443369A (en) * | 2023-12-26 | 2024-01-26 | 橙雨化学(大连)有限公司 | Modified magnesia-alumina mixed metal oxide carrier and preparation method and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4902664A (en) * | 1987-08-13 | 1990-02-20 | Engelhard Corporation | Thermally stabilized catalysts containing alumina and methods of making the same |
| US5705265A (en) * | 1986-03-24 | 1998-01-06 | Emsci Inc. | Coated substrates useful as catalysts |
Family Cites Families (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE605998A (en) * | 1950-10-30 | |||
| US3143490A (en) * | 1961-07-27 | 1964-08-04 | Standard Oil Co | Hydrocarbon conversion process to produce isoparaffins from olefins |
| US3308181A (en) | 1963-11-04 | 1967-03-07 | Phillips Petroleum Co | Oxidative dehydrogenation with added hydrogen |
| US3270080A (en) * | 1964-06-22 | 1966-08-30 | Petro Tex Chem Corp | Production of unsaturated compounds |
| US3670044A (en) | 1969-07-18 | 1972-06-13 | Phillips Petroleum Co | Catalytic dehydrogenation process |
| US3584060A (en) | 1970-06-08 | 1971-06-08 | Universal Oil Prod Co | Dehydrogenation with a catalytic composite containing platinum,rhenium and tin |
| DK310979A (en) | 1978-08-04 | 1980-02-05 | Energy Equip | PROCEDURE FOR CONTROLING THE OPERATION OF A FLUIDIZED BOTTLE LIFTING PLANT AND USING MEASURES IN THE IMPLEMENTATION OF THE PROCEDURE |
| US4844837A (en) | 1982-09-30 | 1989-07-04 | Engelhard Corporation | Catalytic partial oxidation process |
| US4551574A (en) | 1983-06-02 | 1985-11-05 | Uop Inc. | Indium-containing dehydrogenation catalyst |
| GB8426344D0 (en) | 1984-10-18 | 1984-11-21 | British Petroleum Co Plc | Conversion process |
| US4899003A (en) * | 1985-07-11 | 1990-02-06 | Union Carbide Chemicals And Plastics Company Inc. | Process for oxydehydrogenation of ethane to ethylene |
| US4652687A (en) | 1986-07-07 | 1987-03-24 | Uop Inc. | Process for the dehydrogenation of dehydrogenatable hydrocarbons |
| GB8623482D0 (en) | 1986-09-30 | 1986-11-05 | Johnson Matthey Plc | Catalytic generation of hydrogen |
| US4886932A (en) | 1987-03-30 | 1989-12-12 | Atlantic Richfield Company | Thin bed cofeed reaction system for methane conversion |
| US4886926A (en) | 1988-06-24 | 1989-12-12 | Mobil Oil Corporation | Catalytic dehydrogenation of hydrocarbons over tin-containing crystalline microporous materials |
| US4788371A (en) | 1987-12-30 | 1988-11-29 | Uop Inc. | Catalytic oxidative steam dehydrogenation process |
| GB8805447D0 (en) * | 1988-03-08 | 1988-04-07 | British Petroleum Co Plc | Chemical process |
| US4886928A (en) | 1988-09-26 | 1989-12-12 | Uop | Hydrocarbon dehydrogenation process |
| CA2004219A1 (en) | 1988-11-30 | 1990-05-31 | Michael Dunster | Production of hydrogen from hydrocarbonaceous feedstock |
| US4902849A (en) * | 1989-02-06 | 1990-02-20 | Phillips Petroleum Company | Dehydrogenation process |
| DE3926561A1 (en) * | 1989-08-11 | 1991-02-14 | Basf Ag | PALLADIUM CATALYSTS |
| US5073657A (en) * | 1989-09-19 | 1991-12-17 | Union Carbide Chemicals And Plastics Company Inc. | Vapor phase modifiers for oxidative coupling |
| DE4006346A1 (en) * | 1990-03-01 | 1991-09-05 | Degussa | METHOD FOR THE PRODUCTION OF MONOLITH CATALYSTS WITH IMPROVED THERMOSHOCKE PROPERTIES |
| JPH04371231A (en) | 1991-06-18 | 1992-12-24 | N E Chemcat Corp | Catalyst for purification of exhaust gas |
| GB9117216D0 (en) * | 1991-08-09 | 1991-09-25 | British Petroleum Co Plc | Process for the production of mono-olefins |
| DE4200006A1 (en) * | 1992-01-02 | 1993-07-08 | Tech Hochschule C Schorlemmer | Activating molybdenum-contg. mixed oxide catalyst for propene ammoxidation - by treating with readily-dispersed bismuth-III cpd. at 300-600 deg.C |
| US5436383A (en) | 1992-03-02 | 1995-07-25 | Institut Francais Du Petrole | Process for the dehydrogenation of aliphatic hydrocarbons saturated into olefinic hydrocarbons |
| IT1254988B (en) | 1992-06-23 | 1995-10-11 | Eniricerche Spa | Process for the dehydrogenation of light paraffins in a fluidised bed reactor |
| GB9217685D0 (en) * | 1992-08-20 | 1992-09-30 | British Petroleum Co Plc | Process for the production of mono-olefins |
| US5258567A (en) | 1992-08-26 | 1993-11-02 | Exxon Research And Engineering Company | Dehydrogenation of hydrocarbons |
| GB9226453D0 (en) | 1992-12-18 | 1993-02-10 | Johnson Matthey Plc | Metal oxide catalyst |
| NO179131C (en) | 1993-06-14 | 1996-08-14 | Statoil As | Catalyst, process for its preparation and process for dehydrogenation of light paraffins |
| GB9316955D0 (en) | 1993-08-14 | 1993-09-29 | Johnson Matthey Plc | Improvements in catalysts |
| US5648582A (en) * | 1993-08-20 | 1997-07-15 | Regents Of The University Of Minnesota | Stable, ultra-low residence time partial oxidation |
| US5527979A (en) | 1993-08-27 | 1996-06-18 | Mobil Oil Corporation | Process for the catalytic dehydrogenation of alkanes to alkenes with simultaneous combustion of hydrogen |
| AU3734695A (en) † | 1994-10-27 | 1996-05-23 | Regents Of The University Of Minnesota | Catalytic oxidative dehydrogenation process |
| US5639929A (en) * | 1995-04-17 | 1997-06-17 | Regents Of The University Of Minnesota | Oxidative dehydrogenation process |
| US5677260A (en) | 1995-06-23 | 1997-10-14 | Indian Petrochemicals Corporation Limited | Catalyst composite for dehydrogenation of paraffins to mono-olefins and method for the preparation thereof |
| US5763725A (en) * | 1995-06-27 | 1998-06-09 | Council Of Scientific & Industrial Research | Process for the production of ethylene by non-catalytic oxidative cracking of ethane or ethane rich C2 -C4 paraffins |
| US5658497A (en) | 1995-12-05 | 1997-08-19 | Shell Oil Company | Process for the catalytic partial oxidation of hydrocarbons using a certain catalyst support |
| US5905180A (en) * | 1996-01-22 | 1999-05-18 | Regents Of The University Of Minnesota | Catalytic oxidative dehydrogenation process and catalyst |
| US5654491A (en) | 1996-02-09 | 1997-08-05 | Regents Of The University Of Minnesota | Process for the partial oxidation of alkanes |
| US6254807B1 (en) * | 1998-01-12 | 2001-07-03 | Regents Of The University Of Minnesota | Control of H2 and CO produced in partial oxidation process |
| ATE237569T1 (en) * | 1998-09-03 | 2003-05-15 | Dow Global Technologies Inc | AUTOTHERMAL PROCESS FOR PRODUCING OLEFINS |
| AU757855B2 (en) * | 1998-09-03 | 2003-03-06 | Dow Global Technologies Inc. | Autothermal process for the production of olefins |
-
1999
- 1999-09-01 AT AT99945396T patent/ATE237569T1/en not_active IP Right Cessation
- 1999-09-01 AU AU57999/99A patent/AU754523B2/en not_active Ceased
- 1999-09-01 BR BRPI9913611-2A patent/BR9913611B1/en not_active IP Right Cessation
- 1999-09-01 EP EP99949571A patent/EP1114013B1/en not_active Expired - Lifetime
- 1999-09-01 WO PCT/US1999/019974 patent/WO2000014037A1/en active IP Right Grant
- 1999-09-01 ES ES99945396T patent/ES2192075T5/en not_active Expired - Lifetime
- 1999-09-01 ID IDW20010433D patent/ID27739A/en unknown
- 1999-09-01 BR BR9913487-0A patent/BR9913487A/en not_active Application Discontinuation
- 1999-09-01 MY MYPI99003772A patent/MY124615A/en unknown
- 1999-09-01 DE DE69906994T patent/DE69906994T3/en not_active Expired - Fee Related
- 1999-09-01 CA CA002339809A patent/CA2339809C/en not_active Expired - Fee Related
- 1999-09-01 CA CA002340255A patent/CA2340255C/en not_active Expired - Fee Related
- 1999-09-01 US US09/388,219 patent/US6566573B1/en not_active Expired - Fee Related
- 1999-09-01 KR KR1020017002779A patent/KR100669000B1/en not_active IP Right Cessation
- 1999-09-01 AT AT99949571T patent/ATE228493T1/en not_active IP Right Cessation
- 1999-09-01 JP JP2000568795A patent/JP2002524429A/en active Pending
- 1999-09-01 DE DE69904212T patent/DE69904212T2/en not_active Expired - Lifetime
- 1999-09-01 JP JP2000568797A patent/JP3934878B2/en not_active Expired - Fee Related
- 1999-09-01 CN CNB998106011A patent/CN1168688C/en not_active Expired - Fee Related
- 1999-09-01 ID IDW20010434A patent/ID27645A/en unknown
- 1999-09-01 CN CNB998103330A patent/CN1229313C/en not_active Expired - Fee Related
- 1999-09-01 KR KR1020017002673A patent/KR100591029B1/en not_active IP Right Cessation
- 1999-09-01 AU AU62416/99A patent/AU751440B2/en not_active Ceased
- 1999-09-01 US US09/388,220 patent/US6166283A/en not_active Expired - Lifetime
- 1999-09-01 AR ARP990104389A patent/AR020369A1/en active IP Right Grant
- 1999-09-01 WO PCT/US1999/020061 patent/WO2000014035A2/en active IP Right Grant
- 1999-09-01 CN CNA031585523A patent/CN1519053A/en active Pending
- 1999-09-01 AR ARP990104388A patent/AR020651A1/en unknown
- 1999-09-01 ES ES99949571T patent/ES2183610T3/en not_active Expired - Lifetime
- 1999-09-01 EP EP99945396A patent/EP1109763B2/en not_active Expired - Lifetime
- 1999-09-01 MY MYPI99003777A patent/MY123470A/en unknown
-
2000
- 2000-11-03 US US09/706,464 patent/US6624116B1/en not_active Expired - Lifetime
-
2002
- 2002-12-17 US US10/323,175 patent/US20030191020A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5705265A (en) * | 1986-03-24 | 1998-01-06 | Emsci Inc. | Coated substrates useful as catalysts |
| US4902664A (en) * | 1987-08-13 | 1990-02-20 | Engelhard Corporation | Thermally stabilized catalysts containing alumina and methods of making the same |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070041896A1 (en) * | 2003-10-25 | 2007-02-22 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US7256154B2 (en) * | 2003-10-25 | 2007-08-14 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US20080296535A1 (en) * | 2003-10-25 | 2008-12-04 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US7547659B2 (en) | 2003-10-25 | 2009-06-16 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US7560047B2 (en) | 2003-10-25 | 2009-07-14 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US20050090392A1 (en) * | 2003-10-25 | 2005-04-28 | Korea Institute Of Science And Technology | Structured catalyst for POX reforming of gasoline for fuel-cell powered vehicles applications and a method of preparing the same |
| US20050113247A1 (en) * | 2003-11-21 | 2005-05-26 | Conocophillips Company | Copper modified catalysts for oxidative dehydrogenation |
| US7067455B2 (en) * | 2003-11-21 | 2006-06-27 | Conocophillips Company | Copper modified catalysts for oxidative dehydrogenation |
| US8105975B2 (en) * | 2006-09-08 | 2012-01-31 | Yara International Asa | Method and device for catchment of platinum group metals in a gas stream |
| US20090274597A1 (en) * | 2006-09-08 | 2009-11-05 | David Waller | Method and device for catchment of platinum group metals in a gas stream |
| US9040443B2 (en) | 2008-07-31 | 2015-05-26 | Celanese International Corporation | Catalysts for making ethanol from acetic acid |
| US9024087B2 (en) | 2008-07-31 | 2015-05-05 | Celanese International Corporation | Process for making ethanol from acetic acid using acidic catalysts |
| US8471075B2 (en) | 2008-07-31 | 2013-06-25 | Celanese International Corporation | Processes for making ethanol from acetic acid |
| US8080694B2 (en) | 2008-07-31 | 2011-12-20 | Celanese International Corporation | Catalyst for gas phase hydrogenation of carboxylic acids having a support modified with a reducible metal oxide |
| US8546622B2 (en) | 2008-07-31 | 2013-10-01 | Celanese International Corporation | Process for making ethanol from acetic acid using acidic catalysts |
| US20100197485A1 (en) * | 2008-07-31 | 2010-08-05 | Celanese International Corporation | Catalysts for making ethanol from acetic acid |
| US8637714B2 (en) | 2008-07-31 | 2014-01-28 | Celanese International Corporation | Process for producing ethanol over catalysts containing platinum and palladium |
| US8501652B2 (en) | 2008-07-31 | 2013-08-06 | Celanese International Corporation | Catalysts for making ethanol from acetic acid |
| US8309772B2 (en) | 2008-07-31 | 2012-11-13 | Celanese International Corporation | Tunable catalyst gas phase hydrogenation of carboxylic acids |
| US8993815B2 (en) | 2008-07-31 | 2015-03-31 | Celanese International Corporation | Process for vapor phase hydrogenation |
| US8338650B2 (en) | 2008-07-31 | 2012-12-25 | Celanese International Corporation | Palladium catalysts for making ethanol from acetic acid |
| US8680317B2 (en) | 2008-07-31 | 2014-03-25 | Celanese International Corporation | Processes for making ethyl acetate from acetic acid |
| US8802904B2 (en) | 2008-07-31 | 2014-08-12 | Celanese International Corporation | Processes for making ethanol from acetic acid |
| US8754270B2 (en) | 2008-07-31 | 2014-06-17 | Celanese International Corporation | Process for vapor phase hydrogenation |
| CN102292151A (en) * | 2009-01-26 | 2011-12-21 | 鲁玛斯科技公司 | Adiabatic reactor to produce olefins |
| US8815080B2 (en) | 2009-01-26 | 2014-08-26 | Lummus Technology Inc. | Adiabatic reactor to produce olefins |
| WO2010085550A3 (en) * | 2009-01-26 | 2010-10-21 | Lummus Technology Inc. | Adiabatic reactor to produce olefins |
| US20100191031A1 (en) * | 2009-01-26 | 2010-07-29 | Kandasamy Meenakshi Sundaram | Adiabatic reactor to produce olefins |
| US8450535B2 (en) | 2009-07-20 | 2013-05-28 | Celanese International Corporation | Ethanol production from acetic acid utilizing a cobalt catalyst |
| US8680321B2 (en) | 2009-10-26 | 2014-03-25 | Celanese International Corporation | Processes for making ethanol from acetic acid using bimetallic catalysts |
| US8710277B2 (en) | 2009-10-26 | 2014-04-29 | Celanese International Corporation | Process for making diethyl ether from acetic acid |
| US8211821B2 (en) | 2010-02-01 | 2012-07-03 | Celanese International Corporation | Processes for making tin-containing catalysts |
| US8569203B2 (en) | 2010-02-01 | 2013-10-29 | Celanese International Corporation | Processes for making tin-containing catalysts |
| US8304586B2 (en) | 2010-02-02 | 2012-11-06 | Celanese International Corporation | Process for purifying ethanol |
| US8728179B2 (en) | 2010-02-02 | 2014-05-20 | Celanese International Corporation | Ethanol compositions |
| US20110190551A1 (en) * | 2010-02-02 | 2011-08-04 | Celanese International Corporation | Processes for Producing Ethanol from Acetaldehyde |
| US8314272B2 (en) | 2010-02-02 | 2012-11-20 | Celanese International Corporation | Process for recovering ethanol with vapor separation |
| US8668750B2 (en) | 2010-02-02 | 2014-03-11 | Celanese International Corporation | Denatured fuel ethanol compositions for blending with gasoline or diesel fuel for use as motor fuels |
| US8932372B2 (en) | 2010-02-02 | 2015-01-13 | Celanese International Corporation | Integrated process for producing alcohols from a mixed acid feed |
| US8680343B2 (en) | 2010-02-02 | 2014-03-25 | Celanese International Corporation | Process for purifying ethanol |
| US8344186B2 (en) | 2010-02-02 | 2013-01-01 | Celanese International Corporation | Processes for producing ethanol from acetaldehyde |
| US8704014B2 (en) | 2010-02-02 | 2014-04-22 | Celansese International Corporation | Process for purifying ethanol |
| US8569549B2 (en) | 2010-02-02 | 2013-10-29 | Celanese International Corporation | Catalyst supports having crystalline support modifiers |
| US8426652B2 (en) | 2010-02-02 | 2013-04-23 | Celanese International Corporation | Processes for producing ethanol from acetaldehyde |
| US8541633B2 (en) | 2010-02-02 | 2013-09-24 | Celanese International Corporation | Processes for producing anhydrous ethanol compositions |
| US8747492B2 (en) | 2010-02-02 | 2014-06-10 | Celanese International Corporation | Ethanol/fuel blends for use as motor fuels |
| US8858659B2 (en) | 2010-02-02 | 2014-10-14 | Celanese International Corporation | Processes for producing denatured ethanol |
| US9447005B2 (en) | 2010-02-02 | 2016-09-20 | Celanese International Corporation | Processes for producing anhydrous ethanol compositions |
| US8747493B2 (en) | 2010-02-02 | 2014-06-10 | Celanese International Corporation | Ethanol compositions |
| US8460405B2 (en) | 2010-02-02 | 2013-06-11 | Celanese International Corporation | Ethanol compositions |
| US8754267B2 (en) | 2010-05-07 | 2014-06-17 | Celanese International Corporation | Process for separating acetaldehyde from ethanol-containing mixtures |
| US8350098B2 (en) | 2011-04-04 | 2013-01-08 | Celanese International Corporation | Ethanol production from acetic acid utilizing a molybdenum carbide catalyst |
| WO2013030677A3 (en) * | 2011-08-30 | 2013-07-11 | Do Carmo Roberto Werneck | A process for the production of olefins and use thereof |
| US20130072738A1 (en) * | 2011-09-20 | 2013-03-21 | Korea Institute Of Science And Technology | SUPPORTED CATALYST FOR DIRECT DEHYDROGENATION OF n-BUTANE AND PREPARING METHOD OF BUTENES FROM n-BUTANE USING THE SAME |
| US8658843B2 (en) | 2011-10-06 | 2014-02-25 | Celanese International Corporation | Hydrogenation catalysts prepared from polyoxometalate precursors and process for using same to produce ethanol while minimizing diethyl ether formation |
| US8536382B2 (en) | 2011-10-06 | 2013-09-17 | Celanese International Corporation | Processes for hydrogenating alkanoic acids using catalyst comprising tungsten |
| US8703868B2 (en) | 2011-11-28 | 2014-04-22 | Celanese International Corporation | Integrated process for producing polyvinyl alcohol or a copolymer thereof and ethanol |
| US8907143B2 (en) | 2011-12-15 | 2014-12-09 | Celanese International Corporation | Process for producing ethanol by hydrogenating mixed feeds comprising water |
| US9000234B2 (en) | 2011-12-22 | 2015-04-07 | Celanese International Corporation | Calcination of modified support to prepare hydrogenation catalysts |
| US9233899B2 (en) | 2011-12-22 | 2016-01-12 | Celanese International Corporation | Hydrogenation catalysts having an amorphous support |
| US9415378B2 (en) | 2013-12-02 | 2016-08-16 | Saudi Basic Industries Corporation | Dehydrogenation catalyst, its use and method of preparation |
| US10968148B2 (en) | 2015-08-28 | 2021-04-06 | University College Cardiff Consultants Limited | Method of producing compound comprising alkenyl group |
| CN107790182A (en) * | 2016-08-29 | 2018-03-13 | 中国石油化工股份有限公司 | 1,4 diacetoxy butylene hydrogenation process catalyst |
| CN109821530A (en) * | 2017-11-23 | 2019-05-31 | 中国科学院大连化学物理研究所 | A kind of cobalt-base catalyst and its method for propylene ring oxidation reaction |
| US11779915B2 (en) | 2019-01-15 | 2023-10-10 | IFP Energies Nouvelles | Process for preparing selective hydrogenation catalyst, comprising a step of forming a Ni—Cu alloy in pre-impregnation |
| US20220379287A1 (en) * | 2021-05-28 | 2022-12-01 | National University Corporation Hokkaido University | Dehydrogenation catalyst |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6566573B1 (en) | Autothermal process for the production of olefins | |
| EP1109876B1 (en) | Autothermal process for the production of olefins | |
| JPH07145086A (en) | Catalyst and method for dehydrogenation of alkane | |
| CA2414186A1 (en) | Process for the production of olefins | |
| EP1295637A1 (en) | A catalyst composition comprising a group 8B metal and a prometer on a ceramic support | |
| AU2001267742C1 (en) | Process for the production of olefins | |
| MXPA01002280A (en) | Autothermal process for the production of olefins | |
| MXPA01002286A (en) | On-line synthesis and regeneration of a catalyst used in autothermal oxidation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |