US20030190360A1 - Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment - Google Patents
Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment Download PDFInfo
- Publication number
- US20030190360A1 US20030190360A1 US10/099,462 US9946202A US2003190360A1 US 20030190360 A1 US20030190360 A1 US 20030190360A1 US 9946202 A US9946202 A US 9946202A US 2003190360 A1 US2003190360 A1 US 2003190360A1
- Authority
- US
- United States
- Prior art keywords
- dosage form
- delayed release
- core
- oral solid
- solid dosage
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- HXZNLQDRFLNONT-UHFFFAOYSA-N CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36CCCCCCCCC%28C%34C%34C%28C%28C%34C%34C%28C%28C%34C%34C%28C%36C%34%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 Chemical compound CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36CCCCCCCCC%28C%34C%34C%28C%28C%34C%34C%28C%28C%34C%34C%28C%36C%34%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 HXZNLQDRFLNONT-UHFFFAOYSA-N 0.000 description 14
- YDTXLBSBZPSEOU-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C(C%49C%42C(C%49%48)C%42C%48CC%49C%48C%48C%49C%49C%48C%48C%49C%49C%48C%48C%49C%49C%50C%51C%52C%53C%54C%55C%56CCC%56C%55C%54C%53C%52C%51C%50C%48%49C%42%44)C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C(C%49C%42C(C%49%48)C%42C%48CC%49C%48C%48C%49C%49C%48C%48C%49C%49C%48C%48C%49C%49C%50C%51C%52C%53C%54C%55C%56CCC%56C%55C%54C%53C%52C%51C%50C%48%49C%42%44)C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 YDTXLBSBZPSEOU-UHFFFAOYSA-N 0.000 description 3
- SULFMGWYGFWQGS-UHFFFAOYSA-N CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C%49C(C%50CCCCCCC%44C%44%51C%52C%53C%54C%55C%56C%57C%58CCC%58C%57C%56C%55C%54C%53C%52C%44%52C%44C%53C%54C%55C%56C%57C(C%58CCCCCCC%52C%52%59C%60C%61C%62C%63C%64C%65C%66CCC%66C%65C%64C%63C%62C%61C%60C%52%60CCCCCCCCC%52C%58C%58C%52C%52C%58C%58C%52C%52C%58C%58C%52C%60C%58%59)C%50C%57C%56C%55C%54C%53C%44%51)C%42C%49C%48C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 Chemical compound CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C%49C(C%50CCCCCCC%44C%44%51C%52C%53C%54C%55C%56C%57C%58CCC%58C%57C%56C%55C%54C%53C%52C%44%52C%44C%53C%54C%55C%56C%57C(C%58CCCCCCC%52C%52%59C%60C%61C%62C%63C%64C%65C%66CCC%66C%65C%64C%63C%62C%61C%60C%52%60CCCCCCCCC%52C%58C%58C%52C%52C%58C%58C%52C%52C%58C%58C%52C%60C%58%59)C%50C%57C%56C%55C%54C%53C%44%51)C%42C%49C%48C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 SULFMGWYGFWQGS-UHFFFAOYSA-N 0.000 description 3
- XBZHHHDQLHFBEC-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%42CC%49C%48C%47C%46C%45C%44C%36C%36C%42C%44C%45C%46C%47C%48CCC%48C%47C%46C%45C%44C%42C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%42CC%49C%48C%47C%46C%45C%44C%36C%36C%42C%44C%45C%46C%47C%48CCC%48C%47C%46C%45C%44C%42C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 XBZHHHDQLHFBEC-UHFFFAOYSA-N 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N C Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- KACFMTPWZQGVSR-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%18CC%25C%24C%23C%22C%21C%20C%12C%12C%18C%20C%21C%22C%23C%24CCC%24C%23C%22C%21C%20C%18C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%18CC%25C%24C%23C%22C%21C%20C%12C%12C%18C%20C%21C%22C%23C%24CCC%24C%23C%22C%21C%20C%18C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 KACFMTPWZQGVSR-UHFFFAOYSA-N 0.000 description 1
- NJUAOAJLNFUTTN-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CC%27C%28CC%29C%28C%28C%29C%29C%28C%28C%29C%29C%28C%28C%29C%29C%28C%28%30C%31C%32C%33C%34C%35C%36C(C%37CCCCCCC%28C%12(C%20C%21C%22C%23C%24C%25C%27%26)C%12C%20C%21C%22C%23C%24C%37C%18C%24C%23C%22C%21C%20C%12%19)C%12C%18CCCCCCC(C%19%20C%21C%22C%23C%24C%25C%26C%27CCC%27C%26C%25C%24C%23C%22C%21C%19%21C%19C%22C%23C%24C%25C%26C(C%27CCCCCCC%21C%21%28C%37C%38C%39C%40C%41C%42C%43CCC%43C%42C%41C%40C%39C%38C%37C%21%37C%21C%38C%39C%40C%41C(C%42C%27C(C%42%41)C%27C%41CC%42C%41C%41C%42C%42C%41C%41C%42C%42C%41C%41C%42C%42C%43C%44C%45C%46C%47C%48C%49CCC%49C%48C%47C%46C%45C%44C%43C%41%42C%27%37)C%40C%39C%38C%21%28)C%18C%26C%25C%24C%23C%22C%19%20)C%29%30C%31C%32C%33C%34C%35C%12%36)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CC%27C%28CC%29C%28C%28C%29C%29C%28C%28C%29C%29C%28C%28C%29C%29C%28C%28%30C%31C%32C%33C%34C%35C%36C(C%37CCCCCCC%28C%12(C%20C%21C%22C%23C%24C%25C%27%26)C%12C%20C%21C%22C%23C%24C%37C%18C%24C%23C%22C%21C%20C%12%19)C%12C%18CCCCCCC(C%19%20C%21C%22C%23C%24C%25C%26C%27CCC%27C%26C%25C%24C%23C%22C%21C%19%21C%19C%22C%23C%24C%25C%26C(C%27CCCCCCC%21C%21%28C%37C%38C%39C%40C%41C%42C%43CCC%43C%42C%41C%40C%39C%38C%37C%21%37C%21C%38C%39C%40C%41C(C%42C%27C(C%42%41)C%27C%41CC%42C%41C%41C%42C%42C%41C%41C%42C%42C%41C%41C%42C%42C%43C%44C%45C%46C%47C%48C%49CCC%49C%48C%47C%46C%45C%44C%43C%41%42C%27%37)C%40C%39C%38C%21%28)C%18C%26C%25C%24C%23C%22C%19%20)C%29%30C%31C%32C%33C%34C%35C%12%36)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 NJUAOAJLNFUTTN-UHFFFAOYSA-N 0.000 description 1
- QHWQQGGCMFRORM-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C%49C(C%50CCCCCCC%44C%44%51C%52C%53C%54C%55C%56C%57C%58CCC%58C%57C%56C%55C%54C%53C%52C%44%52C%44C%53C%54C%55C%56C%57C(C%58CCCCCCC%52C%52%59C%60C%61C%62C%63C%64C%65C%66CCC%66C%65C%64C%63C%62C%61C%60C%52%60C%52C%61C%62C%63C%64C%65C(C%66CCCCCCC%60C%60%67C%68C%69C%70C%71C%72C%73C%66CC%73C%72C%71C%70C%69C%68C%60C%60C%66C%68C%69C%70C%71C%72CCC%72C%71C%70C%69C%68C%66C%60%67)C%58C%65C%64C%63C%62C%61C%52%59)C%50C%57C%56C%55C%54C%53C%44%51)C%42C%49C%48C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C%33C(C%34CCCCCCC%28C%28%35C%36C%37C%38C%39C%40C%41C%42CCC%42C%41C%40C%39C%38C%37C%36C%28%36C%28C%37C%38C%39C%40C%41C(C%42CCCCCCC%36C%36%43C%44C%45C%46C%47C%48C%49C%50CCC%50C%49C%48C%47C%46C%45C%44C%36%44C%36C%45C%46C%47C%48C%49C(C%50CCCCCCC%44C%44%51C%52C%53C%54C%55C%56C%57C%58CCC%58C%57C%56C%55C%54C%53C%52C%44%52C%44C%53C%54C%55C%56C%57C(C%58CCCCCCC%52C%52%59C%60C%61C%62C%63C%64C%65C%66CCC%66C%65C%64C%63C%62C%61C%60C%52%60C%52C%61C%62C%63C%64C%65C(C%66CCCCCCC%60C%60%67C%68C%69C%70C%71C%72C%73C%66CC%73C%72C%71C%70C%69C%68C%60C%60C%66C%68C%69C%70C%71C%72CCC%72C%71C%70C%69C%68C%66C%60%67)C%58C%65C%64C%63C%62C%61C%52%59)C%50C%57C%56C%55C%54C%53C%44%51)C%42C%49C%48C%47C%46C%45C%36%43)C%34C%41C%40C%39C%38C%37C%28%35)C%26C%33C%32C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 QHWQQGGCMFRORM-UHFFFAOYSA-N 0.000 description 1
- WADSTEAYQWLTDF-UHFFFAOYSA-N C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C(C%33C%26C(C%33%32)C%26C%32CC%33C%32C%32C%33C%33C%32C%32C%33C%33C%32C%32C%33C%33C%34C%35C%36C%37C%38C%39C%40CCC%40C%39C%38C%37C%36C%35C%34C%32%33C%26%28)C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 Chemical compound C1CCCCC23C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C24C2C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28C%20C%29C%30C%31C%32C(C%33C%26C(C%33%32)C%26C%32CC%33C%32C%32C%33C%33C%32C%32C%33C%33C%32C%32C%33C%33C%34C%35C%36C%37C%38C%39C%40CCC%40C%39C%38C%37C%36C%35C%34C%32%33C%26%28)C%31C%30C%29C%20%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C(CCC1)C9C8C7C6C5C23 WADSTEAYQWLTDF-UHFFFAOYSA-N 0.000 description 1
- NWANJWDRVBUHMZ-UHFFFAOYSA-N CC12CCCCCCC(C3C4C5CCCCCCC(C67C8C9C%10C%11C%12C%13C%14CCC%14C%13C%12C%11C%10C9C8C68C6C9C%10C%11C%12C%13C(C%14CCCCCCC8C8%15C%16C%17C%18C%19C%20C%21C%22CCC%22C%21C%20C%19C%18C%17C%16C8%16C8C%17C%18C%19C%20C(C%21C%14C(C%21%20)C%14C%20CC%21C%20C%20C%21C%21C%20C%20C%21C%21C%20C%20C%21C%21C%22C%23C%24C%25C%26C%27C%28CCC%28C%27C%26C%25C%24C%23C%22C%20%21C%14%16)C%19C%18C%17C8%15)C5C%13C%12C%11C%10C9C67)C56C7C8C9C%10C%11C%12C%13CCC%13C%12C%11C%10C9C8C7C51C1C5C7C8C9C3C4C9C8C7C5C16)C1C3C4CCCCCCC56C(C7CC8C7C7C8C8C7C7C8C8C7C7C8C8C7C57C5C9C%10C%11C%12C%13C4C4C%14CCCCCCC(C%15%16C%17C%18C%19C%20C%21C%22C%23CCC%23C%22C%21C%20C%19C%18C%17C%15%17CCCCCCCCC%15C%14C%14C%15C%15C%14C%14C%15C%15C%14C%14C%15C%17C%14%16)C87C5C9C%10C%11C%12C4%13)C4CC5C4C4C5C5C4C4C5C5C4C4C5C5C4C64C6C7C8C9C%10C3C1C%10C9C8C7C6C524 Chemical compound CC12CCCCCCC(C3C4C5CCCCCCC(C67C8C9C%10C%11C%12C%13C%14CCC%14C%13C%12C%11C%10C9C8C68C6C9C%10C%11C%12C%13C(C%14CCCCCCC8C8%15C%16C%17C%18C%19C%20C%21C%22CCC%22C%21C%20C%19C%18C%17C%16C8%16C8C%17C%18C%19C%20C(C%21C%14C(C%21%20)C%14C%20CC%21C%20C%20C%21C%21C%20C%20C%21C%21C%20C%20C%21C%21C%22C%23C%24C%25C%26C%27C%28CCC%28C%27C%26C%25C%24C%23C%22C%20%21C%14%16)C%19C%18C%17C8%15)C5C%13C%12C%11C%10C9C67)C56C7C8C9C%10C%11C%12C%13CCC%13C%12C%11C%10C9C8C7C51C1C5C7C8C9C3C4C9C8C7C5C16)C1C3C4CCCCCCC56C(C7CC8C7C7C8C8C7C7C8C8C7C7C8C8C7C57C5C9C%10C%11C%12C%13C4C4C%14CCCCCCC(C%15%16C%17C%18C%19C%20C%21C%22C%23CCC%23C%22C%21C%20C%19C%18C%17C%15%17CCCCCCCCC%15C%14C%14C%15C%15C%14C%14C%15C%15C%14C%14C%15C%17C%14%16)C87C5C9C%10C%11C%12C4%13)C4CC5C4C4C5C5C4C4C5C5C4C4C5C5C4C64C6C7C8C9C%10C3C1C%10C9C8C7C6C524 NWANJWDRVBUHMZ-UHFFFAOYSA-N 0.000 description 1
- MQXIUCMWWWKWFU-UHFFFAOYSA-N CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28CCCCCCCCC%20C%26C%26C%20C%20C%26C%26C%20C%20C%26C%26C%20C%28C%26%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 Chemical compound CC1C2C3CCCCCCC(C45C6C7C8C9C%10C%11C%12CCC%12C%11C%10C9C8C7C6C4(C)C4C6C7C8C9C1C2C9C8C7C6C45)C12C4C5C6C7C8C9C%10CCC%10C9C8C7C6C5C4C14C1C5C6C7C8C9C(C%10CCCCCCC4C4%11C%12C%13C%14C%15C%16C%17C%18CCC%18C%17C%16C%15C%14C%13C%12C4%12C4C%13C%14C%15C%16C%17C(C%18CCCCCCC%12C%12%19C%20C%21C%22C%23C%24C%25C%26CCC%26C%25C%24C%23C%22C%21C%20C%12%20C%12C%21C%22C%23C%24C%25C(C%26CCCCCCC%20C%20%27C%28C%29C%30C%31C%32C%33C%34CCC%34C%33C%32C%31C%30C%29C%28C%20%28CCCCCCCCC%20C%26C%26C%20C%20C%26C%26C%20C%20C%26C%26C%20C%28C%26%27)C%18C%25C%24C%23C%22C%21C%12%19)C%10C%17C%16C%15C%14C%13C4%11)C3C9C8C7C6C5C12 MQXIUCMWWWKWFU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
- A61K9/2826—Sugars or sugar alcohols, e.g. sucrose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2813—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
Definitions
- the present invention relates to a chronotherapeutic dosage form containing a therapeutically effective amount of a drug.
- the present invention is further related to methods of preparing such formulations, and to methods of treatment utilizing such formulations.
- Chronotherapy takes into consideration a person's biological rhythms in determining the timing—and sometimes the amount—of medication to optimize desired effects of a drug(s) and minimize the undesired effects.
- the synchronization of medication levels to the biological rhythms of disease activity is playing an increasing role in the management of common cardiovascular conditions such as hypertension, elevated cholesterol, angina, stroke and ischemic heart disease, according to experts in this new and ever-expanding field. For example, in humans, at 1 am post-surgical death is most likely; at 2 am peptic ulcers flare up; at 3 am blood pressure bottoms out; at 4 am asthma is at its worst.
- Oral controlled release delivery systems may also be capable of passing over the entire tract of the small intestine, including the duodenum, jejunum, and ileum, so that the active ingredients can be released directly in the colon, if such site specific delivery is desired.
- One means of accomplishing this is by providing a coating surrounding the active pharmaceutical formulation core so as to preserve the integrity of the formulation while it is passing through the gastric tract.
- the high acidity of the gastric tract and presence of proteolytic and other enzymes therein generates a highly digestive environment that readily disintegrates pharmaceutical formulations that do not possess some type of gastro-resistance protection. This disintegration would typically have a detrimental effect upon the sustained release of the active agent.
- Such coated pharmaceutical formulations in addition to slowing the release rate of the active agent contained within the core of the tablet, can also effectuate a delay in the release of the active ingredient for a desired period of time such that the dissolution of the active drug core can be delayed.
- coated pharmaceutical delivery systems for delayed release can be found in U.S. Pat. Nos. 4,863,742 (Panoz et al.) and 5,891,474 (Busetti et al.), as well as in European Patent Applications Nos. 366 621, 572 942 and 629 398.
- the therapeutically active drug core is coated with at least one and potentially several layers of coating, wherein the layers of coating have a direct effect upon the timed release of the active drug within the tablet core into the system of the patient.
- the present invention is directed in part to an oral dosage form which comprises a core comprising a therapeutically effective amount of a steroidal drug, and a compression coating material applied to the core, the compression coating having a delayed release material comprising one or more natural or synthetic gums which are compression coated onto its surface such that the release of the drug from the dosage form is delayed for a desired time period after oral administration of the dosage form to a mammal (e.g., human patient).
- a mammal e.g., human patient
- the compression coating comprises a mixture (e.g., matrix) of xanthan gum, locust bean gum, and a pharmaceutically acceptable saccharide, e.g., a monosaccharide, a disaccharide, a polyhydric alcohol, or a combination of any of the foregoing.
- the core is an immediate release core comprising the drug together with one or more pharmaceutically acceptable excipients.
- the invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto said core, the delayed release material comprising one or more natural or synthetic gums, the compression coating delaying the release of said drug from said dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- the invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and an agglomerated delayed release material compression coated onto the core, the agglomerated delayed release material comprising a gum selected from, e.g., a homopolysaccharide, a heteropolysaccharide, and a mixture of a homopolysaccharide and a heteropolysaccharide, together with a pharmaceutically acceptable excipient, the compression coating delaying the release of said drug from the dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution.
- a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and an agglomerated delayed release material compression coated onto the core, the agglomerated delayed release material comprising a gum selected from, e.g., a homopolys
- the invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid and a disintegrant, and a delayed release material compression coated onto the core, said delayed release material comprising one or more natural or synthetic gums, said compression coating delaying the release of the drug from the dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution, the disintegrant being included in the core in an amount effective to cause the release of at least about 50 percent of the drug into said aqueous solution within one hour after said predetermined period of time.
- the invention is further directed in part to a delayed release oral solid tablet, comprising a tablet core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto the core, the delayed release material comprising one or more natural or synthetic gums, the gums comprising from about 6.5 percent to about 83 percent of the tablet by weight, said compression coating delaying the release of said drug from the dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- the invention is further directed to a chronotherapeutic, delayed release oral solid dosage form comprising a core comprising from about 0.01 mg to about 40 mg glucocorticosteroid, and a delayed release material compression coated onto the core, the delayed release material comprising one or more natural or synthetic gums, the compression coating comprising from about 75 to about 94 percent by weight of the oral solid dosage form, and the ratio of the core to gum in the compression coating being from about 1:0.37 to about 1:5, by weight, the compression coating delaying the release of said glucocorticosteroid from the dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- the invention is further directed in part to a method of preparing a chronotherapeutic oral solid dosage form of a glucocorticosteroid, comprising preparing a core comprising a therapeutically effective amount of a glucocorticosteroid and from about 5 to about 20% disintegrant, by weight of the core, preparing a granulate of a delayed release material comprising one or more natural or synthetic gums, compression coating the granulate onto said core, the compression coating delaying the release of said glucocorticosteroid from the dosage form until after a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- the method further comprises preparing the granulate of delayed release material by wet granulating one or more natural or synthetic gums together with at least one pharmaceutically acceptable excipient, and drying the resultant granulate to obtain agglomerated particles of the delayed release material.
- the method further comprises granulating the glucocorticosteroid, the disintegrant, and a pharmaceutically acceptable inert diluent prior to the compression coating step.
- the disintegrant is a superdisintegrant incorporated in the core in an amount effective to cause the release of at least about 50 percent of the glucocorticosteroid into the aqueous solution within one hour upon completion of the time period for delayed release.
- the oral dosage form provides a lag time (delayed release of drug) from about 2 to about 18 hours, after oral administration to, e.g., a human subject or patient.
- the oral dosage form releases at least about 50 percent of the drug(s) contained in the core within about one hour, and preferably at least about 80 percent of the drug(s) contained in the core within about one or two hours, after the end of the lag time provided by the compression coating.
- the oral dosage form provides a lag time of from about 5 to about 8 hours with a full release by about 8 to about 12 hours, after oral administration, e.g., to a human patient.
- the oral dosage form provides a lag time of about 6 to about 7 hours with full release by about 8 to about 9 hours, after oral administration of the dosage form.
- the oral dosage form provides a lag time of about 6 to about 7 hours, followed by full release of the drug by about 7 to about 8 hours after oral administration.
- the formulation provides a lag time from about 9 to about 12 hours, with full release by about 11 to about 13 hours after oral administration, preferably a lag time of about 10 to about 11 hours followed by full release at about 11 to about 12 hours after oral administration of the dosage form.
- the formulation provides a lag time of, e.g., about 3-12 hours, with full release of the drug from the dosage form after 24 hours.
- delayed release it is meant for purposes of the present invention that the release of the drug is delayed and the drug is not substantially released from the formulation until after a certain period of time, e.g., such that the drug is not released into the bloodstream of the patient immediately upon ingestion by the patient of the tablet but rather only after a specific period of time, e.g., a 4 hour to a 9 hour delay.
- delayed release is synonomous with “timed delay” or a release of drug after a lag time, or a programmed release.
- sustained release it is meant for purposes of the present invention that, once the drug is released from the formulation, it is released at a controlled rate such that therapeutically beneficial blood levels (but below toxic levels) of the medicament are maintained over an extended period of time from the start of drug release, e.g., providing a release over a time period, e.g., from about 4 to about 24 hours from the point of drug release after the lag time, onward.
- environment fluid is meant for purposes of the present invention to encompass, e.g., an aqueous solution (e.g., an in-vitro dissolution bath) or gastrointestinal fluid.
- USP apparatus type II used herein is described e.g., in the United States Pharmacopeia XXV (2002).
- the present invention may be employed to achieve the time-delayed release of a steroid, e.g., a glucocorticosteroid, and in certain embodiments to provide a controlled-release pharmaceutical formulation for one or more glucocorticosteroids that are desirously delivered over a predetermined period of time.
- the formulations of the present invention provide the time-delayed release of the drug and may be useful for the treatment of conditions that are desirously treated with that class of drug, e.g., arthritis.
- the formulations of the present invention are useful for the treatment of morning pathologiessuch as arthritis, the symptoms of which are generally more acute in the morning as the patient awakens from sleep.
- These conditions may be treated by administering the time-delayed release formulation according to the present invention to the patient prior to sleeping, such that the delivery of the drug is achieved at about the time the patient awakens, or preferably the drug has been delivered from the dosage form (and absorbed from the gastrointestinal tract) to an extent that it has achieved a therapeutic effect, thereby alleviating the symptoms of the morning pathology.
- the formulations of the present invention comprise a core comprising the drug(s) and a compression coating over the core that comprises one or more natural or synthetic pharmaceutically acceptable gums.
- the compression coating comprises a combination of a heteropolysaccharide gum (e.g., xanthan gum) and a homopolysaccharide gum (e.g., locust bean gum), together with a pharmaceutically acceptable saccharide (e.g., lactose, dextrose, mannitol, etc.).
- the gum(s) are wet granulated together with the optional saccharide(s) to form agglomerated particles comprising a mixture of, e.g., xanthan gum, locust bean gum and dextrose.
- the goal of the compression coating of the present invention is to delay the release of the active agent, for a predetermined period of time, referred to in the art as a “lag time.”
- the release of the active agent is delayed for, or has a lag time of, about 2 to about 12 hours after administration of the dosage form.
- the core comprising the active agent can be formulated for either immediate release or sustained release of the active agent.
- Formulations for both immediate release and sustained release of active agents are well known to those skilled in the art.
- the core when the core comprising the drug is formulated for immediate release, the core can be prepared by any suitable tableting technique known to those skilled in the art.
- the drug may be admixed with excipient(s) and formed into a tablet core using a conventional tableting press or using conventional wet granulation techniques.
- ingredients for the core are dry blended in a V-blender and compressed on a rotary tablet press into tablet cores.
- the ingredients for the core can be wet granulated, dried and thereafter compressed into tablet cores.
- the core should be compressed to a degree of hardness such that they do not chip or come apart during further processing, such as during the coating process.
- the cores can be compressed to 50 mg weight and 2 to 8, preferably 4 to 8, most preferably 4-5 kP hardness.
- tablet core size should range from 1 ⁇ 8 inch to 5 ⁇ 8 inch, preferably from 1 ⁇ 8 inch to 1 ⁇ 2 inch, more preferably from ⁇ fraction (3/16) ⁇ inch to 1 ⁇ 4 inch.
- all or part of the excipient in the core may comprise a pre-manufactured direct compression diluent.
- pre-manufactured direct compression diluents include Emcocel® (microcrystalline cellulose, N.F.), Emdex® (dextrates, N.F.), and Tab-Fine® (a number of direct-compression sugars including sucrose, fructose and dextrose), all of which are commercially available from Penwest Pharmaceuticals Co., Patterson, N.Y.).
- lactose N.F. anhydrous direct tableting
- Elcems® G-250 powdered cellulose), N.F.
- Fast-Flo Lactose® Lactose, N.F., spray dried
- the directly compressible inert diluent which is used in the core of the present invention is an augmented microcrystalline cellulose as disclosed in U.S. Pat. No 5,585,115, issued Dec. 17, 1996, and entitled “PHARMACEUTICAL EXCIPIENT HAVING IMPROVED COMPRESSIBILITY”, hereby incorporated by reference in its entirety.
- the augmented microcrystalline cellulose described therein is commercially available under the tradename Prosolv® from Penwest Pharmaceuticals Co.
- PROSOLV SMCC 50 is a silicified microcrystalline. This particular grade has a median particle size (by sieve analysis) in the region of 50 ⁇ m.
- PROSOLV SMCC 90 is a silicified microcrystalline cellulose. This grade has a median particle size (by sieve analysis) in the region of 90 ⁇ m.
- the core comprising the drug can be formulated as a sustained release core for the sustained release of the drug.
- the core can be prepared in a number of ways known in the art.
- the drug can be incorporated in a sustained release matrix and thereafter compressed into a core, or a sustained release material can be coated onto the immediate release core to provide for the sustained release of the drug, or a combination of the compressed sustained release matrix and sustained release coating on the core can be used.
- spheroids comprising the active agent, or multiparticulates with sustained release coatings and comprising the drug may be compressed with optional binders and other excipients into a sustained release core.
- the matrix formulations are generally prepared using standard techniques well known in the art. Typically, they are prepared by dry blending a sustained release material, diluent, active agent, and optional other excipients followed by granulating the mixture until proper granulation is obtained. The granulation is done by methods known in the art. Typically with a wet granulation, the wet granules are dried in a fluid bed dryer, sifted and ground to appropriate size. Lubricating agents are mixed with the dried granulation to obtain the final core formulation.
- a controlled release excipient that is comprised of a gelling agent such as synergistic heterodisperse polysaccharides (e.g., a heteropolysaccharide such as xanthan gum) preferably in combination with a polysaccharide gum capable of cross-linking with the heteropolysarcharide (e.g., locust bean gum) is capable of processing into oral solid dosage forms using either direct compression, following addition of drug and lubricant powder, conventional wet granulation, or a combination of the two
- a gelling agent such as synergistic heterodisperse polysaccharides (e.g., a heteropolysaccharide such as xanthan gum) preferably in combination with a polysaccharide gum capable of cross-linking with the heteropolysarcharide (e.g., locust bean gum) is capable of processing into oral solid dosage forms using either direct compression, following addition of drug and lubricant powder, conventional wet granulation, or a
- the core comprises a sustained release matrix such as those disclosed in our foregoing patents.
- the core in addition to the active agent, the core comprises a sustained release excipient comprising a gelling agent comprising a heteropolysaccharide gum and a homopolysaccharide gum capable of cross-linking said heteropolysaccharide gum when exposed to an environmental fluid, and an inert pharmaceutical diluent.
- the ratio of the heteropolysaccharide gum to the homopolysaccharide gum is from about 1:3 to about 3:1, and the ratio of active agent to gelling agent is preferably from about 1:3 to about 1:8.
- the resulting core preferably provides a therapeutically effective blood level of the active agent for at least about 4 hours, and in certain preferred embodiments, for about 24 hours.
- the sustained release excipient further comprises an effective amount of a pharmaceutically acceptable ionizable gel strength enhancing agent, such as those described hereinafter, to provide a sustained release of the active when the core is exposed to an environmental fluid.
- the sustained release excipient may be further modified by incorporation of a hydrophobic material which slows the hydration of the gums without disrupting the hydrophilic matrix.
- the sustained release excipient can be modified to provide for bi- or multi-phasic release profiles of the active agent by the inclusion of a pharmaceutically acceptable surfactant or wetting agent in the core.
- the sustained release excipient comprises only one of the aforementioned gums.
- the sustained release excipient comprises a different pharmaceutically acceptable gum.
- sustained release materials may be used for the sustained release matrix cores of the inventive formulations.
- suitable sustained-release materials include hydrophilic and/or hydrophobic materials, such as sustained release polymers gums, acrylic resins, protein derived materials, waxes, shellac, and oils such as hydrogenated castor oil, hydrogenated vegetable oil.
- Preferred sustained-release polymers include alkylcelluloses such as ethylcellulose, acrylic and methacrylic acid polymers and copolymers; and cellulose ethers, especially hydroxyalkylcelluloses (especially hydroxypropylmethylcellulose) and carboxyalkylcelluloses.
- Preferred waxes include for example natural and synthetic waxes, fatty acids, fatty alcohols, and mixtures of the same (e.g., beeswax, carnauba wax, stearic acid and stearyl alcohol). Certain embodiments utilize mixtures of any of the foregoing sustained release materials in the matrix of the core. However, any pharmaceutically acceptable hydrophobic or hydrophilic sustained-release material which is capable of imparting sustained-release of the active agent may be used in accordance with the present invention.
- the core may be formulated to provide for the sustained release of the active agent through the use of an immediate release core (as previously described) with a sufficient amount of a hydrophobic coating to provide for the sustained release of the active agent from the immediate release core.
- the hydrophobic coating may be applied to the core using methods and techniques known to those skilled in the art. Examples of suitable coating devices include fluid bed coaters, pan coaters, etc.
- hydrophobic materials which may be used in such hydrophobic coatings include for-example, alkylcelluloses (e.g., ethylcellulose), copolymers of acrylic and methacrylic acid esters, waxes, shellac, zein, hydrogenated vegetable oil, mixtures thereof, and the like.
- alkylcelluloses e.g., ethylcellulose
- copolymers of acrylic and methacrylic acid esters e.g., ethylcellulose
- waxes e.g., ethylcellulose
- zein zein
- hydrogenated vegetable oil mixtures thereof, and the like.
- the cores may be formulated for sustained release of the active agent by using a combination of the sustained release matrix and sustained release coating.
- the sustained release cores e.g, sustained release matrix, sustained release coated, or combination thereof
- the immediate release cores may also contain suitable quantities of additional excipients, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are conventional in the pharmaceutical art.
- the oral dosage form includes one or more disintegrants preferably incorporated in the core.
- the rate of release of drug is an immediate pulse effect.
- a controlled profile may be produced.
- Suitable disintegrants are known to those skilled in the art, and include for example sodium starch glycolate (commercially available as Explotab® from Penwest Pharmaceuticals Co.).
- the mechanism of disintegration is based on swelling, wicking, and deformation of the disintegrants.
- a compressed tablet When a compressed tablet is placed in aqueous solution, water can be quickly absorbed, and the swelling of the disintegrant breaks apart tablets quickly.
- the rate of release of the active agent is an immediate pulse effect.
- a controlled profile may be produced.
- disintegrants for use in the present invention include, for example, starch, veegum, crospovidone, cellulose, kaolin, microcrystalline cellulose (e.g., Avicel PH101 & PH102), crosslinked polyvinyl pyrrolidone (e.g., Kollidon CL), and mixtures thereof.
- the disintegrant is a superdisintegrant, such as, for example, croscarmellose sodium, crospovidone, crosslinked carboxy methyl cellulose, sodium starch glycolate, and mixtures thereof.
- superdisintegrants can be incorporated at lower levels than regular disintegrants to increase the water content.
- Some brand named superdisintegrants for use in the present invention include, Ac-Di-Sol®, Primojel®, Explotab®, and Crospovidone®.
- the core of the present invention includes a wicking agent in addition to or as an alternative to a disintegrant.
- Wicking agents such as those materials already mentioned as disintegrants (e.g. microcrystalline cellulose) may be included if necessary to enhance the speed of water uptake.
- Other materials suitable for acting as wicking agents include, but are not limited to, colloidal silicon dioxide, kaolin, titanium dioxide, fumed silicon dioxide, alumina, niacinamide, sodium lauryl sulfate, low molecular weight polyvinyl pyrrolidone, m-pyrol, bentonite, magnesium aluminum silicate, polyester, polyethylene, mixtures thereof, and the like.
- the one or more disintegrant(s) in the core is included in an amount from about 5 to about 20 percent, preferably from about 6 to about 10 percent, most preferably about 8 percent by weight of the core.
- the one or more disintegrant(s) in the core are included in an amount from about 0.1 to about 5 percent, preferably from about 0.3 to about 2 percent, by weight of the tablet (entire formulation).
- the core containing active drug is completely surrounded or substantially surrounded by a compression coating.
- the compression coating preferably delays the release of the pharmaceutically active agent for a predetermined period of time, which time is dependent upon the formulation of the coating and the thickness of the coating layer.
- the appropriate time period for the release of the active ingredient can be determined prior to the preparation of the formulation, and the formulation can be designed by applying the appropriate thickness and composition of the coating to achieve the desired time delay prior to release of the active ingredient and the desired release rate of the active ingredient following the time delay.
- the compression coating comprises a natural or synthetic gum which can function as a gelling agent, causing the core to be surrounded by the gel when the compression coated tablet is exposed to an environmental fluid (e.g., water or gastrointestinal fluid) and thereby causing the drug to be released after diffusion of the environmental fluid through the compression coating, the dissolution of the drug into the environmental fluid, and the egress of the dissolved drug into the fluid surrounding the compression coated tablet.
- an environmental fluid e.g., water or gastrointestinal fluid
- gums for use in the compression coating include, for example and without limitation, heteropolysaccharides such as xanthan gum(s), homopolysaccharides such as locust bean gum, galactans, mannans, vegetable gums such as alginates, gum karaya, pectin, agar, tragacanth, accacia, carrageenan, tragacanth, chitosan, agar, alginic acid, other polysaccharide gums (e.g. hydrocolloids), and mixtures of any of the foregoing.
- heteropolysaccharides such as xanthan gum(s), homopolysaccharides such as locust bean gum, galactans, mannans, vegetable gums such as alginates, gum karaya, pectin, agar, tragacanth, accacia, carrageenan, tragacanth, chitosan, agar, alginic acid, other polys
- the compression coating comprises a heteropolysaccharide such as xanthan gum, a homopolysaccharide such as locust bean gum, or a mixture of one or more hetero- and one or more homopolysaccharide(s).
- Heterodisperse excipients previously disclosed as a sustained release tablet matrix in our U.S. Pat. Nos. 4,994,276, 5,128,143, and 5,135,757, maybe utilized in the compression coatings of the present invention.
- a gelling agent of both hetero- and homo- polysaccharides which exhibit synergism e.g., the combination of two or more polysaccharide gums producing a higher viscosity and faster hydration than that which would be expected by either of the gums alone, the resultant gel being faster-forming and more rigid, may be used in the compression coatings of the present invention.
- heteropolysaccharide as used in the present invention is defined as a water-soluble polysaccharide containing two or more kinds of sugar units, the heteropoly-saccjaride having a branched or helical configuration, and having excellent water-wicking properties and immense thickening properties.
- An especially preferred heteropolysaccharide is xanthan gum, which is a high molecular weight (>10 6 ) heteropolysaccharide.
- Other preferred heteropolysaccharides include derivatives of xanthan gum, such as deacylated xanthan gum, the carboxymethyl ether, and the propylene glycol ester.
- the homopolysaccharide materials used in the present invention that are capable of cross-linking with the heteropolysaccharide include the galactomannans, i.e., polysaccharides that are composed solely of mannose and galactose.
- a possible mechanism for the interaction between the galactomannan and the heteropolysaccharide involves the interaction between the helical regions of the heteropolysaccharide and the unsubstituted mannose regions of the galactomannan.
- Galactomannans that have higher proportions of unsubstituted mannose regions have been found to achieve more interaction with the heteropolysaccharide.
- locust bean gum which has a higher ratio of mannose to galactose, is especially preferred as compared to other galactomannans, such as guar and hydroxypropyl guar.
- the heteropolysaccharide comprises from about 1 to about 50 percent and the homopolysaccharide material comprises from about 50 to about 1 percent by weight of the compression coating.
- the ratio of heteropolysaccharide to homopolysaccharide material is from about 1:3 to 3:1, preferably from about 2:3 to 3:2, or 1:1.
- the compression coating comprises from about 5 to about 70 percent or more by weight of a hydrophilic material (e.g., gums).
- a hydrophilic material e.g., gums.
- the higher the percentage of gums in the compression coating the longer the delay of the release or “lag time” of the active agent.
- the percent of gums in the compression coating corresponds to a delayed release of the active agent which is independent of pH.
- the compression coating is less than about 25% gums, preferably comprising about 5 to about 15% gums
- the delayed release is more independent of pH than a compression coating comprising greater than about 25% gums (e.g., 30, 40, or 50% gums).
- the compression coating also includes pharmaceutically acceptable excipients, for example, a saccharide such as a monosaccharide, a disaccharide or a polyhydric alcohol, and/or mixtures of any of the foregoing, or microcrystalline cellulose or a starch.
- suitable such excipients include sucrose, dextrose, lactose, fructose, xylitol, sorbitol, mannitol, starches, mixtures thereof and the like.
- a soluble pharmaceutical excipient such as lactose, dextrose, sucrose, mannitol, or mixtures thereof is included in the materials to be used in the compression coating.
- the gum(s) is wet granulated with the pharmaceutically acceptable excipient prior to its use as a compression coating on the surface of the inner cores of the invention.
- the compression coating may comprise, e.g., up to about 95% pharmaceutically acceptable excipient(s), by weight.
- the amount of gum(s) contained in the compression coating is from about 1 percent to about 90 percent by weight, preferably from about 6.5 percent to about 83 percent of the total tablet, by weight.
- the ingredients of the compression (delayed release) coating without utilizing a wet granulation step. If the mixture is to be manufactured without a wet granulation step, and the final mixture is to be compression coated onto a pre-formed tablet core, it is preferred that all or part of the pharmaceutically acceptable excipient(s) should impart sufficient compressibility to provide a pharmaceutically acceptable product.
- properties and characteristics of a specific excipient system prepared according to the present invention may be dependent in part on the individual characteristics, e.g., of the homo- and heteropolysaccharide constituents, in terms of polymer solubility, glass transition temperatures etc., as well as on the synergism both between different homo- and heteropolysaccharides and between the homo- and heteropolysaccharides and the inert saccharide constituent(s) in modifying dissolution fluid-excipient interactions.
- a release-modifying agent as described in our previous patents directed to the use of these materials in sustained release matrices can also be utilized in the compression coating.
- release-modifying agents and pre-manufactured excipients disclosed in our U.S. Pat. Nos. 5,455,046; 5,512,297; 5,554,387; 5,667,801; 5,846,563; 5,773,025; 6,048,548; 5,662,933; 5,958,456; 5,472,711; 5,670,168; and 6,039,980 may be utilized in the compression coatings of the present invention.
- the release-modifying agent may comprise an ionizable gel-strength enhancing agent.
- the ionizable gel strength-enhancing agent that is optionally used in conjunction with the present invention may be monovalent or multivalent metal cations.
- the preferred salts are the inorganic salts, including various alkali metal and/or alkaline earth metal sulfates, chlorides, borates, bromides, citrates, acetates, lactates, etc.
- suitable ionizable gel strength enhancing agent include calcium sulfate, sodium chloride, potassium sulfate, sodium carbonate, lithium chloride, tripotassium phosphate, sodium borate, potassium bromide, potassium fluoride, sodium bicarbonate, calcium chloride, magnesium chloride, sodium citrate, sodium acetate, calcium lactate, magnesium sulfate and sodium fluoride. Multivalent metal cations may also be utilized.
- the preferred ionizable gel strength-enhancing agents are bivalent. Particularly preferred salts are calcium sulfate and sodium chloride.
- the ionizable gel strength enhancing agents of the present invention are added in an amount effective to obtain a desirable increased gel strength due to the cross-linking of the gelling agent (e.g., the heteropolysaccharide and homopolysaccharide gums).
- the ionizable gel strength-enhancing agent is included in the delayed release excipient of the present invention in an amount from about 1 to about 20% by weight of the delayed release excipient, and in an amount 0.5% to about 16% by weight of the final dosage form.
- the inclusion of an ionizable gel strength-enhancing agent not only delays the release of the active, but also provides for a sustained release of the active agent.
- the (delayed release) compression coating coated onto the core comprises from about 1 to about 90 percent by weight of a gelling agent comprising a heteropolysaccharide gum and a homopolysaccharide gum, from about 0 to about 20 percent by weight of an ionizable gel strength enhancing agent, and from about 10 to about 95 percent by weight of an pharmaceutically acceptable excipient.
- the compression coating material comprises from about 5 to about 75 percent gelling agent (gum), from about 0 to about 15 percent ionizable gel strength enhancing agent, and from about 30 to about 95 percent pharmaceutically acceptable excipient (e.g., an inert diluent).
- the compression coating material comprises from about 7.5 to about 50 percent gelling agent, from about 0 to about 10 percent ionizable gel strength enhancing agent, and from about 30 to about 95 percent pharmaceutically acceptable excipient.
- Surfactants that may be used in the present invention generally include pharmaceutically acceptable anionic surfactants, cationic surfactants, amphoteric (amphipathic/amphophilic) surfactants, and non-ionic surfactants.
- Suitable pharmaceutically acceptable anionic surfactants include, for example, monovalent alkyl carboxylates, acyl lactylates, alkyl ether carboxylates, N-acyl sarcosinates, polyvalent alkyl carbonates, N-acyl glutamates, fatty acid-polypeptide condensates, sulfuric acid esters, alkyl sulfates (including sodium lauryl sulfate (SLS)), ethoxylated alkyl sulfates, ester linked sulfonates (including docusate sodium or dioctyl sodium succinate (DSS)), alpha olefin sulfonates, and phosphated ethoxylated alcohols.
- SLS sodium la
- Suitable pharmaceutically acceptable cationic surfactants include, for example, monoalkyl quaternary ammonium salts, dialkyl quaternary ammonium compounds, amidoamines, and aminimides.
- Suitable pharmaceutically acceptable amphoteric (amphipathic/amphophilic) surfactants include, for example, N-substituted alkyl amides, N-alkyl betaines, sulfobetaines, and N-alkyl ⁇ -aminoproprionates.
- surfactants for use in conjunction with the present invention include polyethyleneglycols as esters or ethers.
- examples include polyethoxylated castor oil, polyethoxylated hydrogenated castor oil, polyethoxylated fatty acid from castor oil or polyethoxylated fatty acid from hydrogenated castor oil.
- Commercially available surfactants that can be used are known under trade names Cremophor, Myi, Polyoxyl 40 stearate, Emerest 2675, Lipal 395 and PEG 3350.
- release-modifying pharmaceutically acceptable agents that may be added in appropriate quantities for their particular ability to modify dissolution rates include, for example: stearic acid, metallic stearates, stearyl alcohol, hydrogenated cotton seed oil, sodium chloride and certain disintegrants that are described below.
- the quantity of such release-modifying agent employed depends on the release characteristics required and the nature of the agent.
- the level of release-modifying agents used may be from about 0.1 to about 25%, preferably from about 0.5 to about 10% by weight of the total composition.
- the compression coating includes a pH-modifying agent.
- the pH-modifying agent may be present in the compression coating from about 1% to about 10% by weight of the final dosage form.
- the pH-modifying agent is an organic acid such as citric acid, succinic acid, fumaric acid, malic acid, maleic acid, glutaric acid or lactic acid.
- the release of drug occurs when aqueous environmental fluid (e.g., water or gastrointestinal fluid, etc. surrounding the dosage form) diffuses through the compression coating of the dosage form, resulting in hydration of the core and dissolving the drug, which then can pass into the fluid surrounding the core.
- aqueous environmental fluid e.g., water or gastrointestinal fluid, etc. surrounding the dosage form
- the delayed release of the drug is varied by increasing the thickness of the compression coating (increased lag time) or by decreasing the thickness of the compressing coating (decreased lag time).
- the delayed release may also be varied, e.g., by changing the gum(s) included in the delayed release compression coating, selecting a particular combination of gums, by including or not including an pharmaceutically acceptable excipient, such as a saccharide (including polysaccharides) or a combination of saccharide(s) (or polysaccharides) in the compression coating, by changing or by adding additional agents to the compression coating which cause the compression coating to further delay the diffusion of water (or gastrointestinal fluid) through the compression coating (e.g., matrix) into the inner core (thereby allowing hydration of the inner core).
- an pharmaceutically acceptable excipient such as a saccharide (including polysaccharides) or a combination of saccharide(s) (or polysaccharides) in the compression coating
- the compression force used to apply the compression coating may be used to alter the release rate of the active ingredient.
- release can be modified via the use of an extragranular excipient addition to the compression coating.
- ingredients may comprise, for example, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, and the like.
- the delayed release of the drug may further be varied by utilizing a further coating (i) between the core and the compression coating; (ii) over the compression coating; or (iii) both between the core and the compression coating and over the compression coating.
- Such coatings may comprise, for example a hydrophilic polymer (such as hydroxypropylmethylcellulose) and/or a hydrophobic polymer (such as an acrylic polymer, a copolymer of acrylic and methacrylic acid esters, an alkylcellulose such as ethylcellulose, etc.).
- a hydrophilic polymer such as hydroxypropylmethylcellulose
- a hydrophobic polymer such as an acrylic polymer, a copolymer of acrylic and methacrylic acid esters, an alkylcellulose such as ethylcellulose, etc.
- the dissolution rates of the present invention may be further modified by incorporation of a hydrophobic material in the compression coating, which slows the hydration of the gums without disrupting the hydrophilic matrix. This is accomplished in alternate embodiments of the present invention by granulating the delayed release excipient with a solution or dispersion of a hydrophobic material prior to the compression coating of the core.
- the hydrophobic polymer may be selected from an alkylcellulose such as ethylcellulose, other hydrophobic cellulosic materials, polymers or copolymers derived from acrylic or methacrylic acid esters, copolymers of acrylic and methacrylic acid esters, zein, waxes, shellac, hydrogenated vegetable oils, and any other pharmaceutically acceptable hydrophobic material known to those skilled in the art.
- the solvent for the hydrophobic material may be an aqueous or organic solvent, or mixtures thereof.
- the amount of hydrophobic material incorporated into the delayed release excipient is that which is effective to slow the hydration of the gums without disrupting the hydrophilic matrix formed upon exposure to an environmental fluid.
- the hydrophobic material is included in the compression coating in an amount from about 1 to about 20 percent by weight.
- the compression coating may also contain suitable quantities of, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are described hereinafter and are which are conventional in the pharmaceutical art.
- the combination of the gum gelling agent e.g., a mixture of xanthan gum and locust bean gum
- the pharmaceutical excipient(s) with or without a release modifying agent, provides a ready-to-use compression coating product in which a formulator need only apply the material onto the core by compression coating to provide the desired chronotherapeutic dosage forms.
- the compression coating may comprise a physical admix of the gums along with a soluble excipient such as compressible sucrose, lactose, dextrose, etc., although it is preferred to granulate or agglomerate the gums with a plain pharmaceutically acceptable excipient (i.e., crystalline) sucrose, lactose, dextrose, mannitol, etc., to form a delayed release excipient for use in the compression coating.
- a soluble excipient such as compressible sucrose, lactose, dextrose, etc.
- a plain pharmaceutically acceptable excipient i.e., crystalline sucrose, lactose, dextrose, mannitol, etc.
- the gums and optional pharmaceutical excipients used in the compression coating are preferably prepared according to any agglomeration technique to yield an acceptable excipient product.
- a moistening agent such as water, propylene glycol, glycerol, alcohol or the like is added to prepare a moistened mass.
- the moistened mass is dried. The dried mass is then milled with conventional equipment into granules. Thereafter, the excipient product is ready to use.
- the pre-manufactured delayed release excipient is preferably free-flowing and directly compressible. Accordingly, the excipient may be directly compressed onto a pre-formed inner core of a therapeutically active medicament to form coated tablets.
- the delayed release coating mixture in an amount sufficient to make a uniform coating onto a preformed tablet core, is subjected to tableting in a conventional production scale tableting machine at normal compression pressure, i.e., about 2000-1600 lbs/sq in. However, the mixture should not be compressed to such a degree that there is subsequent difficulty in its hydration when exposed to gastric fluid.
- the average particle size of the granulated delayed release excipient of the present invention ranges from about 50 microns to about 400 microns and preferably from about 185 microns to about 265 microns.
- the particle size of the granulation is not narrowly critical, the important parameter being that the average particle size of the granules must permit the formation of a directly compressible excipient which forms a coating over pharmaceutically active tablet cores.
- the desired tap and bulk densities of the granulation of the present invention are normally between from about 0.3 to about 0.8 g/ml, with an average density of from about 0.5 to about 0.7 g/ml.
- the compression coatings of the present invention preferably have uniform packing characteristics over a range of different particle size distributions and are capable of processing onto the preformed tablet core using direct compression, following the addition of a lubricant.
- one or more pharmaceutically acceptable lubricants be added to the compression coating materials (preferably pre-agglomerated) prior to the mixture being compression coated onto the surface of the core.
- suitable lubricants for use in the core and compression coating of the invention include, for example and without limitation, talc, stearic acid, vegetable oil, calcium stearate, zinc stearate, magnesium stearate, etc.
- an effective amount of any generally accepted pharmaceutical lubricant, including calcium or magnesium soaps is preferably added to the mixture of ingredients prior to compression of the mixture onto the solid preformed tablet core.
- An especially preferred lubricant is sodium stearyl fumarate, NF, commercially available under the trade name Pruv® from Penwest Pharmaceuticals Co.
- the present invention is further directed towards a method of manufacturing the delayed release solid oral dosage forms (e.g., tablets) of the present invention.
- the steps for preparation of a delayed release oral solid dosage form of the present invention may include the following:
- Preparation of delayed release (compression) coating may be accomplished, e.g., as follows:
- film coat the final dosage form (if desired).
- steps 4 & 10 are combined in a single unit operation when using e.g., a Dry-Cota Press as described hereinafter.
- a functional coating of the tablet cores may be possible using the Dry-Cota Press if a modification is made to the press to add a core tablet feeder system.
- a Manesty Dry-Cota press press consists of two side by side interconnected tablet presses where the core is made on one press then mechanically transferred to the next press for compression coating.
- Each “press” has an independent powder feed mechanism so that core blend is loaded on one machine and coating blend on the other. Mechanical transfer arms rotate between the machines to remove cores from one press and transfer them to the coating press.
- Other and more modem types of presses which may be used e.g. Elizabeth Hata HT-AP44-MSU-C, Killian RUD, Fette PT 4090
- the compression coating surrounds the entire core
- the compression coating substantially surrounds, but does not entirely surround the tablet core. In such instances, the release of drug from the tablet core will occur first from that portion of the inner core to which the compression is not applied.
- compression coating is not applied to the same thickness around the entire inner core, thereby creating areas of the compressed dosage form that release drug earlier (and later) than other areas. This may be accomplished, e.g, by having the core to which the compression coating is applied not being centered in the press.
- the tablets formed from the compression coating of the core are from about 4 to about 25 kP, preferably about 5 to about 15 kP, most preferably about 8 to about 9 kP hardness.
- the diameter may be up to 5 ⁇ 8 inch or greater, and for caplet shaped compression coated tablets the diameter may be up to 3 ⁇ 4 inch or greater.
- the average flow of the (non-compression) coatings prepared in accordance with the present invention is from about 25 to about 40 g/sec.
- the compression coated tablet may then be further overcoated with an enteric coating material or a hydrophobic material.
- enteric polymers include cellulose acetate phthalate, hydroxypropyl-methylcellulose phthalate, polyvinylacetate phthalate, methacrylic acid copolymer, shellac, hydroxypropyhnethylcellulose succinate, cellulose acetate trimellitate, and mixtures of any of the foregoing.
- An example of a suitable commercially available enteric material is available under the trade name Eudragit® L30D55.
- the dosage form may be coating with a hydrophilic coating in addition to or instead of the above-mentioned enteric coating or hydrophobic coating.
- a hydrophilic coating is hydroxypropylmethylcellulose (e.g., Opadry®, commercially available from Colorcon, West Point, Pa.).
- the optional enteric and/or hydrophobic and/or hydrophilic coatings may be alternatively or additionally applied as an intermediate layer(s) between the core and the compression coating.
- the optional enteric and/or hydrophobic and/or hydrophilic coatings may be applied in any pharmaceutically acceptable manner known to those skilled in the art.
- the coating is applied via a fluidized bed or in a coating pan.
- the coated tablets may be dried, e.g., at about 60-70° C. for about 3-4 hours in a coating pan.
- the solvent for the hydrophobic polymer or enteric coating may be organic, aqueous, or a mixture of an organic and an aqueous solvent.
- the organic solvents may be, e.g., isopropyl alcohol, ethanol, and the like, with or without water.
- a support platform is applied to the tablets manufactured in accordance with the present invention.
- Suitable support platforms are well known to those skilled in the art.
- An example of suitable support platforms is set forth, e.g., in U.S. Pat. No. 4,839,177, hereby incorporated by reference.
- the support platform partially coats the tablet, and consists of a polymeric material insoluble in aqueous liquids.
- the support platform may, for example, be designed to maintain its impermeability characteristics during the transfer of the therapeutically active medicament.
- the support platform may be applied to the tablets, e.g., via compression coating onto part of the tablet surface, by spray coating the polymeric materials comprising the support platform onto all or part of the tablet surface, or by immersing the tablets in a solution of the polymeric materials.
- the support platform may have a thickness of, e.g., about 2 mm if applied by compression, and about 10 ⁇ if applied via spray-coating or immersion-coating.
- a hydrophobic polymer or enteric coating is applied to the tablets over the delayed release coating, the tablets are coated to a weight gain from about 1 to about 20%, and in certain embodiments preferably from about 5% to about 10%.
- Materials useful in the hydrophobic coatings and support platforms of the present invention include derivatives of acrylic acid (such as esters of acrylic acid, methacrylic acid, and copolymers thereof) celluloses and derivatives thereof (such as ethylcellulose), polyvinylalcohols, and the like.
- the cores and/or compression coatings may also contain suitable quantities of, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are conventional in the pharmaceutical art.
- Suitable binders for use in the present invention include for example and without limitation, povidone, polyvinylpyrrolidone, xanthan gum, cellulose gums such as carboxymethylcellulose, methyl cellulose, hydroxypropyhnethylcellulose, hydroxycellulose, gelatin, starch, and pregelatinized starch.
- glidants for use in the present invention include talc, silicon dioxide, and cornstarch.
- the tablet core includes an additional dose of the drug (or a therapeutically effective dose of a different drug) included in either the (optional) hydrophobic or enteric coating, or in an additional (optional) overcoating coated on the outer surface of the tablet core (without the hydrophobic or enteric coating) or as an additional coating layer coated on the surface of the base coating(s) comprising the compression coating and, if applicable, hydrophobic and/or enteric coating material.
- an additional dose of the drug included in either the (optional) hydrophobic or enteric coating, or in an additional (optional) overcoating coated on the outer surface of the tablet core (without the hydrophobic or enteric coating) or as an additional coating layer coated on the surface of the base coating(s) comprising the compression coating and, if applicable, hydrophobic and/or enteric coating material.
- This may be desired when, for example, a loading dose of the drug is needed to provide therapeutically effective blood levels of the active agent when the formulation is first exposed to gastric fluid.
- the active agent is a glucocorticoid.
- Glucocorticoids have a very favorable effect on the symptoms of rheumatoid arthritis, e.g. morning stiffness, joint pain and joint swelling.
- Glucocorticoids for use in the present invention include for example, prednisolone, prednisone, cortisone, hydrocortisone, methylprednisolone, betamethasone, dexamethasone, triamcinolone, mixtures thereof, and pharmaceutically acceptable salts thereof.
- Prednisolone is particularly preferred.
- Prednisolone is a potent pharmaceutical agent which has been commercially available for many years.
- Prednisolone is characterized by pronounced anti-inflammatory activity, when administered locally or systemically.
- Prednisolone is known as an anti-inflammatory and anti-rheumatic drug.
- the prednisolone is in an amount of from about 0.1 to about 20 mg, preferably from about 1 to about 6 mg, and in most preferred embodiments about 1, 2 or 5 mg.
- Equivalent doses of other glucocorticoids can be calculated based on the following chart: Approximate Equivalent Glucocorticoid Dose (mg) Cortisone 25 Hydrocortisone 20 Prednisone 5 Prednisolone 5 Triamcinolone 4 Methylprednisolone 4 Dexamethasone 0.75 Betamethasone 0.6-0.75
- the chronotherapeutic formulations are preferably orally administered to the patient at bedtime (e.g., at about 9 or 10 p.m.) and have a lag time of about 5 or 6 hours, so that, e.g., a substantial portion of the drug in the compression coated delayed release oral dosage form is released, e.g., between 2-3 a.m., or between 3-4 a.m., and the drug is absorbed from the gastrointestinal tract and provides therapeutic efficacy at a time which correlates with the peak of the manifestations of the disease state.
- the benefits of chronotherapeutics include safety and more efficient treatment than conventional therapies. This is achieved by delivering more medication when risk of disease is greater, and delivering less medication when potential for disease symptoms are less likely.
- Other benefits to the patient include an increased quality of life and a once-a-day drug delivery system to increase patient compliance.
- the chronotherapeutic formulations are preferably orally administered to the patient at bedtime (e.g., at about 9 or 10 p.m.) and have a lag time of about 5 or 6 hours, so that, e.g., a substantial portion of the drug in the compression coated delayed release oral dosage form is released, e.g., between 2-3 a.m., or between 3-4 a.m., and the drug is absorbed from the gastrointestinal tract and provides therapeutic efficacy at a time which correlates with the peak of the manifestations of the disease state.
- the total tablet weight is from about 220 mg to about 900 mg; and the core weight is preferably from about 50 mg to about 170 mg.
- the core is from about 5 to about 23 percent, most preferably about 18 to about 20 percent by weight of the total tablet weight.
- the compression coating is preferably from about 150 mg to about 850 mg.
- the compression coating is from about 75 to about 94 percent by weight, most preferably from about 78 to 80 percent, by weight of the total tablet.
- the ratio of the core to gum (in the compression coating) is from about 1:0.37 to about 1:5, preferably from about 1:0.37 to about 1:1.12, most preferably from about 1:0.75.
- the ratio of the core to compression coating material (all ingredients) is preferably from about 1:2 to about 1:9, and in certain embodiments more preferably about 1:4.
- the cores comprising the active agent are typically compression coated with the coating formulation by hand on a rotary tablet press.
- a rotary tablet press In such a process, roughly half the outer core material is first added to the die.
- An inner core tablet is typically centered on the powder bed and is covered with the other half of the outer coating powder.
- compression coating may be accomplished via automated tablet presses for commercialization. Prior to compression coating with any tablet press, preferably 0.75% Pruv® (sodium stearyl fumarate, NF) or another suitable lubricant is added to the compression coating material(s).
- the coatings are indicate by the gums, for example, 50% xanthan gum (XG), the coating comprises 50% xanthan gum diluted with dextrose; and for example 50% locust bean gum (LBG), the coating comprises 50% locust bean gum diluted with dextrose, etc.
- XG xanthan gum
- LBG locust bean gum
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 1: TABLE 1 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Dextrose 70 4. Water* q.s.
- the granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- LOD loss on drying
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 2: TABLE 2 Component Percentage 1. Xanthan Gum 25 2. Locust Bean Gum 25 3. Dextrose 35 4. Calcium Sulfate Dihydrate 10 5. Ethylcellulose 5 5. Alcohol, SD3A, anhydrous* 20 6. Water* q.s.
- a slurry of hydrophobic polymer (ethylcellulose) is prepared by dissolving ethyl cellulose in ethyl alcohol.
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 3: TABLE 3 Component Percentage 1. Xanthan Gum 15 2. Locust Bean Gum 15 3. Dextrose 60 4. Calcium Sulfate Dihydrate 10 5. Water* q.s.
- the granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than abou-t 10% by weight (e.g., 4-7% LOD).
- LOD loss on drying
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 4: TABLE 4 Component Percentage 1. Xanthan Gum 16 2. Locust Bean Gum 24 3. Dextrose 60 4. Water* q.s.
- Example 1 The same process for Example 1 is used to prepare the delayed release coating of Example 4.
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 5: TABLE 5 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Dextrose 45 4. Calcium Sulfate Dihydrate 5 5. Water* q.s.
- Example 3 The same process for Example 3 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 5.
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 6: TABLE 6 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Dextrose 65 4. Calcium Sulfate Dihydrate 5 5. Water* q.s.
- Example 3 The same process for Example 3 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 6.
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 7: TABLE 7 Component Percentage 1. Xanthan Gum 10 2. Locust Bean Gum 15 3. Dextrose 75 4. Water* q.s.
- Example 7 The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 7.
- a delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 8: TABLE 8 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose 80 4. Water* q.s.
- Example 8 The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 8.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 9: TABLE 9 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Lactose 50 4. Water* q.s.
- Example 1 The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 5, substituting lactose for dextrose.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 10: TABLE 10 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Mannitol 45 4. Hydroxypropylmethylcellulose 5 5. Water* q.s.
- the granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- LOD loss on drying
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 11: TABLE 11 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Mannitol 70 4. Water* q.s.
- Example 1 The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 11, substituting marmitol for dextrose.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 12: TABLE 12 Component Percentage 1. Xanthan Gum 69 2. Locust Bean Gum 13.5 3. Mannitol 77.5 4. Water* q.s.
- Example 10 The same process for Example 10 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 12.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 13: TABLE 13 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Mannitol 80 4. Water* q.s.
- Example 12 The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 13.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 14: TABLE 14 Component Percentage 1. Xanthan Gum 6 2. Locust Bean Gum 9 3. Mannitol 85 4. Water* q.s.
- Example 12 The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 14.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 15: TABLE 15 Component Percentage 1. Xanthan Gum 4 2. Locust Bean Gum 6 3. Mannitol 90 4. Alcohol, SD3A, anhydrous* — 5. Water* q.s.
- Example 12 The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 15.
- a delayed release coating is prepared having the following formulation listed in Table 16: TABLE 16 Component Percentage 1. Xanthan Gum 3 2. Locust Bean Gum 4.5 3. Mannitol 92.5 4. Water* q.s.
- Example 12 The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 16.
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 17: TABLE 17 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose 40 4. Microcrystalline Cellulose 40 5. Water* q.s.
- the granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- LOD loss on drying
- a delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 18: TABLE 18 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose — 4. Microcrystalline Cellulose 80 5. Water* q.s.
- Example 1 The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 18, substituting microcrystalline cellulose for dextrose.
- a prednisolone core composition was prepared having the ingredients set forth in Table 19: TABLE 19 Component Percent amt. (mg) 1. Prednisolone 2.0 1.0 2. Prosolv SMCC TM 50 32.75 16.4 3. Prosolv SMCC TM 90 50 25.0 4. Explotab ® 10 5.0 5. Sodium carboxymethylcellulose 5 2.5 6. Pruv 0.25 0.1 Total 100 50 Core size and shape 3/16′′ Round SC*
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 20: TABLE 20 Component Percent amt. (mg) 1. Prednisolone 10 5 2. Prosolv SMCC TM 50 NA NA 3. Prosolv SMCC TM 90 81.75 40.875 4. Explotal ® 6 3 5. Sodium carboxymethylcellulose NA NA 6. Pruv 0.25 0.125 7. PVP 2 1 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round FF*
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 21: TABLE 21 Component Percent amt. (mg) 1. Prednisolone 10 5 2. Prosolv SMCC TM 50 NA NA 3. Prosolv SMCC TM 90 75.75 37.375 4. Explotab ® 10 5 5. Sodium carboxymethylcellulose 5 2.5 6. Pruv 0.25 0.1 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round FF
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 22: TABLE 22 Component Percent amt. (mg) 1. Prednisolone 2 1.0 2. Prosolv SMCC TM 50 40 20.0 3. Prosolv SMCC TM 90 47.75 23.9 4. Explotab ® 6 3.0 5. PVP 2 1.0 6. Talc 2 1.0 7. Pruv 0.25 0.1 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 23: TABLE 23 Component Percent amt. (mg) 1. Prednisolone 2 3.4 2. Prosolv SMCC TM 50 40 68 3. Prosolv SMCC TM 90 47.75 81.75 4. Explotab ® 6 10.2 5. Sodium carboxymethylcellulose 2 3.4 6. Talc 2 3.4 7. Pruv 0.25 0.425 Total 100 170 Core size and shape 1 ⁇ 4′′ Round FF
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 24: TABLE 24 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 47.75 23.875 4. Explotab ® 6 3 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 6. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 24.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 25: TABLE 25 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 55.75 27.85 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 25, without the inclusion of Explotab® and sodium carboxymethylcellulose.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 26: TABLE 26 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 52.75 26.375 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 26, without the inclusion of sodium carboxymethylcellulose.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 27: TABLE 27 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 51.75 25.875 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose 1 0.5 6. Talc 2 1 6. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 27.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 28: TABLE 28 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 53.75 26.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 28, without the inclusion of Explotab®.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 29: TABLE 29 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 47.75 23.875 4. Explotab ® 2 1 5. Sodium carboxymethylcellulose 6 3 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 29.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 30: TABLE 30 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 49.75 24.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 6 3 6. Talc 2 1 7 Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 30, without the inclusion of Explotab®.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 31: TABLE 31 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 26 13 3. Prosolv SMCC TM 90 49.75 24.875 4. Explotab ® 20 10 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 31, without the inclusion of sodium carboxymethylcellulose.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 32: TABLE 32 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 26 13 3. Prosolv SMCC TM 90 49.75 24.875 4. Explotab ® 20 10 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 32, without the inclusion of sodium carboxymethylcellulose.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 33: TABLE 33 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 52.75 26.375 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 33, without the inclusion sodium carboxymethylcellulose.
- a prednisolone core composition was prepared having the formulation set forth in Table 34: TABLE 34 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 51.75 25.875 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose 1 0.5 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 34.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 35: TABLE 35 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 53.75 26.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 35, without the inclusion of Explotab®.
- a prednisolone core composition was prepared having the formulation ingredients set forth in Table 36: TABLE 36 Component Percent amt. (mg) 1. Prednisolone 4 2 2. Prosolv SMCC TM 50 40 20 3. Prosolv SMCC TM 90 45.75 22.875 4. Explotab ® 6 3 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape ⁇ fraction (3/16) ⁇ ′′ Round SC
- Example 23 The same process for Example 23 is used to prepare the core of Example 36.
- Example 37-39 prednisolone tablets were prepared having a core formulation as described in Example 21 and coating formulation as described in Example 3.
- the tablet formulations of Examples 37-39 are listed in Table 37 below: TABLE 37 Ex. 37 Ex. 38 Ex. 39 Component amt. (mg) amt. (mg) amt. (mg) 1.
- Example 37 The tablets of Examples 37-39 resulted the following lag times (time when prednisolone is release from the tablet) and full release times (time when all of prednisolone is released from the tablet) listed in Table 39: TABLE 39 Example 37 Example 38 Example 39 Lag time (hours) 6-7 8-9 10-12 Full release (hours) 8-9 10-12 14-16
- Examples 40-42 prednisolone tablets were prepared having a core formulation as described in Example 21 and coating formulation as described Example 2.
- the tablet formulations of Examples 40-42 are listed in Table 40 below: TABLE 40 Ex. 40 Ex. 41 Ex. 42 Component Amt. (mg) amt. (mg) amt. (mg) 1.
- Example 40-42 were tested using USP apparatus type III with 250 mL DI water at 15 dips per minute (dpm) giving the following results listed in Table 41: TABLE 41 Time (hours) 0 2 4 6 8 10 12 16 Example 40 0 — 22.7 40.6 100 — — — % dissolved Example 41 0 0 0 0 0 0 100 100 % dissolved Example 42 0 0 0 0 0 0 19.5 19.5 % dissolved
- Example 40 The tablets of Examples 40-42 resulted in the following lag times (time when prednisolone is release from the tablet) and full release times (time when all of prednisolone is released from the tablet) listed in Table 42: TABLE 42
- Example 40 Example 41
- Example 42 Lag time 6-7 10-11 10-11 (hours)
- Full release 7-8 11-12 11-12 (hours)
- Example 43 various delayed release coating formulations were prepared in order to determine the effect of the gum percentage in the coating formulation on the time of release and the rate of release of the active agent within the tablet core.
- Example 44 various examples of delayed release compression coating formulations were prepared in order to determine the effect of ratio of the drug within the tablet to the gum within the coating formulation on the time of release and the rate of release of the active agent within the tablet core.
- Example 45 various lots of delayed release coating formulations were prepared in order to determine the effect of the thickness of the sustained release coating on the time of release and the rate of release of the active agent within the tablet core.
- Example 8 coating were faster releasing than tablets with 250 mg Example 8 coating. It was thus observed that, the thickness of coating is increased in the tablet, a corresponding increase in lag time is observed. The conclusion reached is that tablets with a thicker coating showed a longer lag time before release of the drug.
- Example 46 the effect of the addition of an extragranular excipient(s) to the delayed release coating of Example 2 was measured.
- the types of excipient added were Microcrystalline cellulose, Polyvinylpyrrolidone and Polyethylene glycol, and these excipients were added in levels of 0, 5% and 10%.
- Example 24 The ingredients of the inner core formulation of this Example is shown in Example 24 above, and the ingredients of the various delayed release coating granulation of this Example are shown in Example 2 above.
- the amounts or percentages of extragranular excipients added are set forth in Tables 46a and 46b below: TABLE 46a addition of addition of Control 5% PVP 10% PVP Formulation: % mg/tab % mg/tab % mg/tab Core (Ex. 24) 22.72% 50 22.73% 50 22.73% 50 Delayed release coating (Ex.
- Example 47 a scaled up production of the sustained release coating was done in order to determine whether tablets produced at production scale exhibit release profiles similar to those of tablets produced in laboratory scale.
- the core-coated tablets were produced on a production press at Elizabeth Hata.
- a HT-AP44MSU-C 44 station core coating press was used to press the tablets.
- the inner cores of the tablets were made with a 1 ⁇ 4′′ round flat face with a beveled edge, tableted to 170 mg and 8-10 kP.
- the final tablets were made 9 mm round concave, and were tableted to 525 and 560 mg, 8-10 kP.
- the blends for the production scale were composed as follows: Inner Core blend: Outer Coating blend: 2% Prednisolone Example 2 w/0.75% sodium stearyl 40% Prosolv SMCC 50 fumarate 47.75% Prosolv SMCC 90 6% Croscarmellose Sodium 2% Sodium Starch Glycolate 2% Talc 0.25% Sodium Stearyl Fumarate
- the press speed varied from 9 to 12 rpm.
- Examples 48-57 prednisolone tablets were prepared having core formulations with different amounts of disintegrants and coating formulations as described Example 10. Each tablet had the same core weight, same coating weight, and the same total tablet weight.
- the tablet formulations of Examples 48-57 are listed in Tables 48 and 49 below: TABLE 48 Component Ex. 48 Ex. 49 Ex. 50 Ex. 51 Ex. 52 Core formulation Ex. 25 core Ex. 28 core Ex. 26 core Ex. 27 core Ex. 31 core used Disintegrant none 2% Ac-Di-Sol 3% Explotab 3% Explotab and 6% Ac-Di-Sol 2% Ac-Di-Sol amt. (mg) amt. (mg) amt. (mg) amt. (mg) 1. Core 50 50 50 50 50 2.
- Example 10 450 450 450 450 450 450 450 coating Total tablet 500 500 500 500 weight
- Examples 58-60 prednisolone tablets were prepared having core formulations of Example 24 and coating formulations as described Examples 2, 4, and a combination Examples 2 and 4 (25% of Example 2 and 75% of Example 4). Each tablet had the same core weight, same coating weight, and the same total tablet weight.
- the tablet formulations of Examples 58-60 are listed in Table 51 below: TABLE 51 Ex. 58 Ex. 59 Ex. 60 Component amt. (mg) Amt. (mg) amt. (mg) 1. Core (Ex. 24) 50 50 50 2.
- Coating — — 250 consisting of 25% of Example 2 and 75% of Example 4 coatings Total tablet 300 300 300 weight
- Example 61 prednisolone tablets having the formulations described in Examples 59 were subjected to dissolution variations in pH, ionic strength, and dip rates. The pH evaluated was 1.5, 7.5 and pH change.
- the pH change method uses increasing pH from one dissolution vessel to the next to simulate the transport of the dosage form through the gastrointestinal tract. Initially the pH is 1.5 for 1 hour. The pH of the second station is 3.5 for two additional hours and when the third station is 5.5 for an additional 2 hours. Finally the last three stations are at pH 7.5. The time length for the last three stations can vary depending on the expected release for the dosage form.
- Example 62 a 2 mg prednisolone core composition was prepared similarly to Example 24, increasing the amount of prednisolone in the core and decreasing the amount of Prosolve SMCC 90, and having the following formulation listed in Table 57: TABLE 57 Component Percent amt. (mg) 1. Prednisolone 4 2.0 2. Prosolv SMCC TM 50 40 20.0 3. Prosolv SMCC TM 90 45.75 22.875 4. Explotab ® 6 3.0 5. Sodium carboxymethylcellulose 2 1.0 6. Talc 2 1.0 7. Pruv 0.25 0.125 Total 100 50 Core size and shape 3/16 Round SC
- Example 63-68 the core formulation of Example 62 was coated with coatings prepared in accordance with Examples 11, 12, 13, 14, 15, and 16.
- the formulations of Examples are listed in Table 58 below: TABLE 58 Ex. 63 Ex. 64 Ex. 65 Ex. 66 Ex. 67 Ex. 68 amt. amt. amt. amt. amt. amt. amt. amt. amt. amt. Component (mg) (mg) (mg) (mg) (mg) 1.
- Ex. 16 — — — — 250 coating Total tablet 300 300 300 300 300 300 300 300 300 300 300 300 300 300 300 300 300
- Dissolution testing was done on each formulation using USP apparatus 3 with 250 ml of media and 15 dpm. Two dissolution methods using different media (1) DI water and (2) pH change were performed. Table 59 provides the DI water dissolution results, and Table 60 provides the pH change (0.1M) dissolution results. TABLE 59 Time Ex. 63 Ex. 64 Ex. 65 Ex. 66 Ex. 67 Ex.
- Example 69 other formulations were prepared and tested using USP apparatus Type, 3, with 250 ml of the dissolution media and dips per minute as indicated in the Table 62.
- the particular dissolution media are defined as follows: DI water: USP purified water; pH change NI (“no ion”) pH change method as described in Example 61, or pH change: without the use of ions to change adjust the pH; pH change (0.1 M): pH change method as described in Example 61 with the use of salts to give an ionic strength of 0.1 molar; pH 7.5: dissolution media having a pH of 7.5; pH 7.5 (0.1 M): dissolution media having a pH of 7.5 and ionic strength of 0.1 M; SGI: simulated gastric fluid; Peanut oil pH 7.5: peanut oil with a pH of 7.5;
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Heart & Thoracic Surgery (AREA)
- Inorganic Chemistry (AREA)
- Cardiology (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pulmonology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
A chronotherapeutic pharmaceutical formulation comprising a core containing an active agent (e.g., a drug) and a delayed release compression coating comprising a natural or synthetic gum applied onto the surface of the core.
Description
- This application claims priority from U.S. Provisional Application No. 60/275,382, filed Mar., 31, 2001, the disclosure of which is hereby incorporated by reference in its entirety.
- The present invention relates to a chronotherapeutic dosage form containing a therapeutically effective amount of a drug. The present invention is further related to methods of preparing such formulations, and to methods of treatment utilizing such formulations.
- Coordinating biological rhythms (chronobiology) with medical treatment is called chronotherapy. Chronotherapy takes into consideration a person's biological rhythms in determining the timing—and sometimes the amount—of medication to optimize desired effects of a drug(s) and minimize the undesired effects. The synchronization of medication levels to the biological rhythms of disease activity is playing an increasing role in the management of common cardiovascular conditions such as hypertension, elevated cholesterol, angina, stroke and ischemic heart disease, according to experts in this new and ever-expanding field. For example, in humans, at 1 am post-surgical death is most likely; at 2 am peptic ulcers flare up; at 3 am blood pressure bottoms out; at 4 am asthma is at its worst. When one wakes up, hay fever is at its most tormenting, and in the morning hours, as ones blood pressure rises to meet the day, one is most likely to suffer a heart attack or stroke. Rheumatoid arthritis improves through the day, but osteoarthritis grows worse. Alcohol is least toxic to the body at around 5 pm: cocktail hour.
- The first application of chronotherapy, in the 1960s, was a synthetic corticosteroid tablet (Medrol, Upjohn). Clinicians found that when used in the morning, the drug was more effective and caused fewer adverse reactions. Another example of a commercial product employing chronotherapy is the bronchodilator, Uniphyl®, a long-acting theophylline preparation manufactured by Purdue Frederick (approved by the FDA in 1989). Taken once a day at dinner to control night-time asthma symptoms. Uniphyl causes theophylline blood levels to reach their peak and improve lung function during the difficult morning hours
- Oral controlled release delivery systems may also be capable of passing over the entire tract of the small intestine, including the duodenum, jejunum, and ileum, so that the active ingredients can be released directly in the colon, if such site specific delivery is desired. One means of accomplishing this is by providing a coating surrounding the active pharmaceutical formulation core so as to preserve the integrity of the formulation while it is passing through the gastric tract. The high acidity of the gastric tract and presence of proteolytic and other enzymes therein generates a highly digestive environment that readily disintegrates pharmaceutical formulations that do not possess some type of gastro-resistance protection. This disintegration would typically have a detrimental effect upon the sustained release of the active agent. Such coated pharmaceutical formulations, in addition to slowing the release rate of the active agent contained within the core of the tablet, can also effectuate a delay in the release of the active ingredient for a desired period of time such that the dissolution of the active drug core can be delayed. Examples of coated pharmaceutical delivery systems for delayed release can be found in U.S. Pat. Nos. 4,863,742 (Panoz et al.) and 5,891,474 (Busetti et al.), as well as in European Patent Applications Nos. 366 621, 572 942 and 629 398. In the delayed release tablets described in each of these references, the therapeutically active drug core is coated with at least one and potentially several layers of coating, wherein the layers of coating have a direct effect upon the timed release of the active drug within the tablet core into the system of the patient.
- It is considered desirable by those skilled in the art to provide an oral controlled release delivery system that is adaptable to deliver a drug(s) such that release rates and drug plasma profiles can be matched to physiological and chronotherapeutic requirements.
- It is an object of the present invention to provide an oral pharmaceutical dosage form that releases a steroidal drug(s) into the body of a patient at a predetermined time after oral ingestion of the dosage form by the patient.
- It is a further object of the present invention to provide an oral pharmaceutical dosage form that provides a delayed release of a steroidal drug(s) into the gastrointestinal tract of a patient at a predetermined time after oral ingestion of the dosage form.
- It is a further object of certain embodiments of the present invention to provide an oral pharmaceutical dosage form having a core containing steroidal drug, the core being compression coated with a coating that provides a delayed release of the drug from the dosage form after the dosage form is orally administered to a patient.
- It is a further object of certain embodiments of the present invention to provide an oral pharmaceutical dosage form having a drug-containing core that is compression coated with a coating which provides a delayed release of the steroidal drug when the dosage form is orally administered to a patient.
- It is a further object of certain embodiments of the present invention to provide a dosage form which allows time-specific dosing for arthritis, which is typically more symptomatic in the early morning corresponding to circadian rhythms.
- It is a further object of certain embodiments of the present invention to provide a dosage form which provides a delayed release of steroidal drug from the dosage form, followed by a sustained release of the drug thereafter as the dosage form travels through the gastrointestinal tract.
- It is a further object of certain embodiments of the present invention to provide a compression coated dosage form having an immediate release layer of a glucosteroid drug(s) overcoating a compression coated core which provides a delayed release of a glucocorticosteroid drug(s) from the dosage form; the core optionally providing a sustained release of the glucocorticosteroid drug thereafter as the dosage form travels through the gastrointestinal tract.
- It is a further object of certain embodiments of the present invention to provide an oral dosage form which provides site-specific delivery of a steroidal drug (e.g., to the colon).
- It is a further object of certain embodiments of the present invention to develop an oral dosage form which provides programmed release of a steroidal drug.
- It is a further object of certain embodiments of the present invention to develop an oral dosage form which provides pulsatile release of a steroidal drug.
- In accordance with the above-mentioned objects of the invention, the present invention is directed in part to an oral dosage form which comprises a core comprising a therapeutically effective amount of a steroidal drug, and a compression coating material applied to the core, the compression coating having a delayed release material comprising one or more natural or synthetic gums which are compression coated onto its surface such that the release of the drug from the dosage form is delayed for a desired time period after oral administration of the dosage form to a mammal (e.g., human patient).
- In certain preferred embodiments, the compression coating comprises a mixture (e.g., matrix) of xanthan gum, locust bean gum, and a pharmaceutically acceptable saccharide, e.g., a monosaccharide, a disaccharide, a polyhydric alcohol, or a combination of any of the foregoing. In certain preferred embodiments, the core is an immediate release core comprising the drug together with one or more pharmaceutically acceptable excipients.
- The invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto said core, the delayed release material comprising one or more natural or synthetic gums, the compression coating delaying the release of said drug from said dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- The invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and an agglomerated delayed release material compression coated onto the core, the agglomerated delayed release material comprising a gum selected from, e.g., a homopolysaccharide, a heteropolysaccharide, and a mixture of a homopolysaccharide and a heteropolysaccharide, together with a pharmaceutically acceptable excipient, the compression coating delaying the release of said drug from the dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution.
- The invention is further directed in part to a delayed release oral solid dosage form comprising a core comprising a therapeutically effective amount of a glucocorticosteroid and a disintegrant, and a delayed release material compression coated onto the core, said delayed release material comprising one or more natural or synthetic gums, said compression coating delaying the release of the drug from the dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution, the disintegrant being included in the core in an amount effective to cause the release of at least about 50 percent of the drug into said aqueous solution within one hour after said predetermined period of time.
- The invention is further directed in part to a delayed release oral solid tablet, comprising a tablet core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto the core, the delayed release material comprising one or more natural or synthetic gums, the gums comprising from about 6.5 percent to about 83 percent of the tablet by weight, said compression coating delaying the release of said drug from the dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- The invention is further directed to a chronotherapeutic, delayed release oral solid dosage form comprising a core comprising from about 0.01 mg to about 40 mg glucocorticosteroid, and a delayed release material compression coated onto the core, the delayed release material comprising one or more natural or synthetic gums, the compression coating comprising from about 75 to about 94 percent by weight of the oral solid dosage form, and the ratio of the core to gum in the compression coating being from about 1:0.37 to about 1:5, by weight, the compression coating delaying the release of said glucocorticosteroid from the dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
- The invention is further directed in part to a method of preparing a chronotherapeutic oral solid dosage form of a glucocorticosteroid, comprising preparing a core comprising a therapeutically effective amount of a glucocorticosteroid and from about 5 to about 20% disintegrant, by weight of the core, preparing a granulate of a delayed release material comprising one or more natural or synthetic gums, compression coating the granulate onto said core, the compression coating delaying the release of said glucocorticosteroid from the dosage form until after a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution. In certain preferred embodiments, the method further comprises preparing the granulate of delayed release material by wet granulating one or more natural or synthetic gums together with at least one pharmaceutically acceptable excipient, and drying the resultant granulate to obtain agglomerated particles of the delayed release material. In certain embodiments the method further comprises granulating the glucocorticosteroid, the disintegrant, and a pharmaceutically acceptable inert diluent prior to the compression coating step.
- In certain preferred embodiments, the disintegrant is a superdisintegrant incorporated in the core in an amount effective to cause the release of at least about 50 percent of the glucocorticosteroid into the aqueous solution within one hour upon completion of the time period for delayed release.
- In certain embodiments, the oral dosage form provides a lag time (delayed release of drug) from about 2 to about 18 hours, after oral administration to, e.g., a human subject or patient.
- In certain preferred embodiments, the oral dosage form releases at least about 50 percent of the drug(s) contained in the core within about one hour, and preferably at least about 80 percent of the drug(s) contained in the core within about one or two hours, after the end of the lag time provided by the compression coating.
- In certain embodiments, the oral dosage form provides a lag time of from about 5 to about 8 hours with a full release by about 8 to about 12 hours, after oral administration, e.g., to a human patient.
- In certain preferred embodiments, the oral dosage form provides a lag time of about 6 to about 7 hours with full release by about 8 to about 9 hours, after oral administration of the dosage form.
- In certain other preferred embodiments, the oral dosage form provides a lag time of about 6 to about 7 hours, followed by full release of the drug by about 7 to about 8 hours after oral administration.
- In yet other embodiments, the formulation provides a lag time from about 9 to about 12 hours, with full release by about 11 to about 13 hours after oral administration, preferably a lag time of about 10 to about 11 hours followed by full release at about 11 to about 12 hours after oral administration of the dosage form.
- In yet other embodiments, the formulation provides a lag time of, e.g., about 3-12 hours, with full release of the drug from the dosage form after 24 hours.
- By “delayed release” it is meant for purposes of the present invention that the release of the drug is delayed and the drug is not substantially released from the formulation until after a certain period of time, e.g., such that the drug is not released into the bloodstream of the patient immediately upon ingestion by the patient of the tablet but rather only after a specific period of time, e.g., a 4 hour to a 9 hour delay. For purposes of the present invention, delayed release is synonomous with “timed delay” or a release of drug after a lag time, or a programmed release.
- By “sustained release” it is meant for purposes of the present invention that, once the drug is released from the formulation, it is released at a controlled rate such that therapeutically beneficial blood levels (but below toxic levels) of the medicament are maintained over an extended period of time from the start of drug release, e.g., providing a release over a time period, e.g., from about 4 to about 24 hours from the point of drug release after the lag time, onward.
- The term “environmental fluid” is meant for purposes of the present invention to encompass, e.g., an aqueous solution (e.g., an in-vitro dissolution bath) or gastrointestinal fluid.
- The term USP apparatus type II used herein is described e.g., in the United States Pharmacopeia XXV (2002).
- The present invention may be employed to achieve the time-delayed release of a steroid, e.g., a glucocorticosteroid, and in certain embodiments to provide a controlled-release pharmaceutical formulation for one or more glucocorticosteroids that are desirously delivered over a predetermined period of time. The formulations of the present invention provide the time-delayed release of the drug and may be useful for the treatment of conditions that are desirously treated with that class of drug, e.g., arthritis. For example, the formulations of the present invention are useful for the treatment of morning pathologiessuch as arthritis, the symptoms of which are generally more acute in the morning as the patient awakens from sleep. These conditions may be treated by administering the time-delayed release formulation according to the present invention to the patient prior to sleeping, such that the delivery of the drug is achieved at about the time the patient awakens, or preferably the drug has been delivered from the dosage form (and absorbed from the gastrointestinal tract) to an extent that it has achieved a therapeutic effect, thereby alleviating the symptoms of the morning pathology.
- The formulations of the present invention comprise a core comprising the drug(s) and a compression coating over the core that comprises one or more natural or synthetic pharmaceutically acceptable gums. In certain especially preferred embodiments, the compression coating comprises a combination of a heteropolysaccharide gum (e.g., xanthan gum) and a homopolysaccharide gum (e.g., locust bean gum), together with a pharmaceutically acceptable saccharide (e.g., lactose, dextrose, mannitol, etc.). In certain preferred embodiments, the gum(s) are wet granulated together with the optional saccharide(s) to form agglomerated particles comprising a mixture of, e.g., xanthan gum, locust bean gum and dextrose.
- The goal of the compression coating of the present invention is to delay the release of the active agent, for a predetermined period of time, referred to in the art as a “lag time.” In certain embodiments, the release of the active agent is delayed for, or has a lag time of, about 2 to about 12 hours after administration of the dosage form.
- The core comprising the active agent can be formulated for either immediate release or sustained release of the active agent. Formulations for both immediate release and sustained release of active agents are well known to those skilled in the art.
- In the present invention, when the core comprising the drug is formulated for immediate release, the core can be prepared by any suitable tableting technique known to those skilled in the art. For example, the drug may be admixed with excipient(s) and formed into a tablet core using a conventional tableting press or using conventional wet granulation techniques. According certain preferred embodiments of the present invention, ingredients for the core are dry blended in a V-blender and compressed on a rotary tablet press into tablet cores. Alternatively, in certain embodiments, the ingredients for the core can be wet granulated, dried and thereafter compressed into tablet cores. Preferably, the core should be compressed to a degree of hardness such that they do not chip or come apart during further processing, such as during the coating process. In certain embodiments, the cores can be compressed to 50 mg weight and 2 to 8, preferably 4 to 8, most preferably 4-5 kP hardness. In addition, tablet core size should range from ⅛ inch to ⅝ inch, preferably from ⅛ inch to ½ inch, more preferably from {fraction (3/16)} inch to ¼ inch.
- In certain embodiments, wherein the core is manufactured without a wet granulation step, and the final mixture is to be compressed into a tablet core, all or part of the excipient in the core may comprise a pre-manufactured direct compression diluent. Examples of such pre-manufactured direct compression diluents include Emcocel® (microcrystalline cellulose, N.F.), Emdex® (dextrates, N.F.), and Tab-Fine® (a number of direct-compression sugars including sucrose, fructose and dextrose), all of which are commercially available from Penwest Pharmaceuticals Co., Patterson, N.Y.). Other direct compression diluents include anhydrous lactose (Lactose N.F., anhydrous direct tableting) from Sheffield Chemical, Union, N.J. 07083; Elcems® G-250 (powdered cellulose), N.F.) from Degussa, D-600 Frankfurt (Main) Germany; Fast-Flo Lactose® (Lactose, N.F., spray dried) from Foremost Whey Products, Banaboo, Wis. 53913; Maltrin® (Agglomerated maltodextrin) from Grain Processing Corp., Muscatine, Iowa 52761; Neosorb 60® (Sorbitol, N.F., direct-compression from Roquet Corp., 645 5th Ave., New York, N.Y. 10022; Nu-Tab® (Compressible sugar, N.F.) from Ingredient Technology, Inc., Pennsauken, N.J. 08110; Polyplasdone XL® (Crospovidone, N.F., cross-linked polyvinylpyrrolidone) from GAF Corp., New York, N.Y. 10020; Primojel® (Sodium starch glycolate, N.F., carboxymethyl starch) from Generichem Corp., Little Falls, N.J. 07424; Solka Floc® (Cellulose floc) from Penwest Pharmaceuticals Co., Patterson, N.Y. 10512; Spray-dried lactose® (Lactose N.F., spray dried) from Foremost Whey Products, Baraboo, Wis. 53913 and DMV Corp., Vehgel, Holland; and Sta-Rx 1500® (Starch 1500) (Pregelatinized starch, N.F., compressible) from Colorcon, Inc., West Point, Pa. 19486. In certain embodiments of the present invention, the directly compressible inert diluent which is used in the core of the present invention is an augmented microcrystalline cellulose as disclosed in U.S. Pat. No 5,585,115, issued Dec. 17, 1996, and entitled “PHARMACEUTICAL EXCIPIENT HAVING IMPROVED COMPRESSIBILITY”, hereby incorporated by reference in its entirety. The augmented microcrystalline cellulose described therein is commercially available under the tradename Prosolv® from Penwest Pharmaceuticals Co. PROSOLV SMCC 50 is a silicified microcrystalline. This particular grade has a median particle size (by sieve analysis) in the region of 50 μm. PROSOLV SMCC 90 is a silicified microcrystalline cellulose. This grade has a median particle size (by sieve analysis) in the region of 90 μm.
- Alternatively, in certain embodiments, the core comprising the drug can be formulated as a sustained release core for the sustained release of the drug. When the core comprising the drug is formulated for sustained release, the core can be prepared in a number of ways known in the art. For example, the drug can be incorporated in a sustained release matrix and thereafter compressed into a core, or a sustained release material can be coated onto the immediate release core to provide for the sustained release of the drug, or a combination of the compressed sustained release matrix and sustained release coating on the core can be used. Additionally, spheroids comprising the active agent, or multiparticulates with sustained release coatings and comprising the drug, may be compressed with optional binders and other excipients into a sustained release core.
- When the core of the present invention comprises a sustained release matrix, the matrix formulations are generally prepared using standard techniques well known in the art. Typically, they are prepared by dry blending a sustained release material, diluent, active agent, and optional other excipients followed by granulating the mixture until proper granulation is obtained. The granulation is done by methods known in the art. Typically with a wet granulation, the wet granules are dried in a fluid bed dryer, sifted and ground to appropriate size. Lubricating agents are mixed with the dried granulation to obtain the final core formulation.
- In our U.S. Pat. Nos. 4,994,276; 5,128,143; 5,135,757; 5,455,046; 5,512,297; 5,554,387; 5,667,801; 5,846,563; 5,773,025; 6,048,548; 5,662,933; 5,958,456; 5,472,711; 5,670,168; and 6,039,980, all of which are hereby incorporated by reference, we reported that a controlled release excipient that is comprised of a gelling agent such as synergistic heterodisperse polysaccharides (e.g., a heteropolysaccharide such as xanthan gum) preferably in combination with a polysaccharide gum capable of cross-linking with the heteropolysarcharide (e.g., locust bean gum) is capable of processing into oral solid dosage forms using either direct compression, following addition of drug and lubricant powder, conventional wet granulation, or a combination of the two. These systems (controlled release excipients) are commercially available under the trade name TIMERx® from Penwest Pharmaceuticals Co., Patterson, N.Y., which is the assignee of the present invention.
- In certain embodiments of the present invention, wherein the core provides for the sustained release of the active agent, the core comprises a sustained release matrix such as those disclosed in our foregoing patents. For example, in certain embodiments of the present invention, in addition to the active agent, the core comprises a sustained release excipient comprising a gelling agent comprising a heteropolysaccharide gum and a homopolysaccharide gum capable of cross-linking said heteropolysaccharide gum when exposed to an environmental fluid, and an inert pharmaceutical diluent. Preferably, the ratio of the heteropolysaccharide gum to the homopolysaccharide gum is from about 1:3 to about 3:1, and the ratio of active agent to gelling agent is preferably from about 1:3 to about 1:8. The resulting core preferably provides a therapeutically effective blood level of the active agent for at least about 4 hours, and in certain preferred embodiments, for about 24 hours. In certain preferred embodiments, the sustained release excipient further comprises an effective amount of a pharmaceutically acceptable ionizable gel strength enhancing agent, such as those described hereinafter, to provide a sustained release of the active when the core is exposed to an environmental fluid. The sustained release excipient (with or without the optional ionizable gel strength enhancing agent) may be further modified by incorporation of a hydrophobic material which slows the hydration of the gums without disrupting the hydrophilic matrix. In addition, in certain embodiments, the sustained release excipient can be modified to provide for bi- or multi-phasic release profiles of the active agent by the inclusion of a pharmaceutically acceptable surfactant or wetting agent in the core. Alternatively, the sustained release excipient comprises only one of the aforementioned gums. In yet other embodiments, the sustained release excipient comprises a different pharmaceutically acceptable gum.
- In addition to the above, other sustained release materials may be used for the sustained release matrix cores of the inventive formulations. A non-limiting list of suitable sustained-release materials which may be included in a sustained-release matrix according to the present invention include hydrophilic and/or hydrophobic materials, such as sustained release polymers gums, acrylic resins, protein derived materials, waxes, shellac, and oils such as hydrogenated castor oil, hydrogenated vegetable oil. Preferred sustained-release polymers include alkylcelluloses such as ethylcellulose, acrylic and methacrylic acid polymers and copolymers; and cellulose ethers, especially hydroxyalkylcelluloses (especially hydroxypropylmethylcellulose) and carboxyalkylcelluloses. Preferred waxes include for example natural and synthetic waxes, fatty acids, fatty alcohols, and mixtures of the same (e.g., beeswax, carnauba wax, stearic acid and stearyl alcohol). Certain embodiments utilize mixtures of any of the foregoing sustained release materials in the matrix of the core. However, any pharmaceutically acceptable hydrophobic or hydrophilic sustained-release material which is capable of imparting sustained-release of the active agent may be used in accordance with the present invention.
- Alternatively, in certain embodiments of the present invention, the core may be formulated to provide for the sustained release of the active agent through the use of an immediate release core (as previously described) with a sufficient amount of a hydrophobic coating to provide for the sustained release of the active agent from the immediate release core. The hydrophobic coating may be applied to the core using methods and techniques known to those skilled in the art. Examples of suitable coating devices include fluid bed coaters, pan coaters, etc. Examples of hydrophobic materials which may be used in such hydrophobic coatings include for-example, alkylcelluloses (e.g., ethylcellulose), copolymers of acrylic and methacrylic acid esters, waxes, shellac, zein, hydrogenated vegetable oil, mixtures thereof, and the like.
- Additionally, the cores may be formulated for sustained release of the active agent by using a combination of the sustained release matrix and sustained release coating. The sustained release cores (e.g, sustained release matrix, sustained release coated, or combination thereof), and the immediate release cores, may also contain suitable quantities of additional excipients, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are conventional in the pharmaceutical art.
- Specific examples of pharmaceutically acceptable diluents and excipients that may be used in formulating the cores are described in the Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), incorporated by reference herein.
- In certain preferred embodiments, the oral dosage form includes one or more disintegrants preferably incorporated in the core. When such an agent is included in the core, the rate of release of drug (after the initial delay caused by the compression coating) is an immediate pulse effect. In certain embodiments, when no disintegrant is present, a controlled profile may be produced. Suitable disintegrants are known to those skilled in the art, and include for example sodium starch glycolate (commercially available as Explotab® from Penwest Pharmaceuticals Co.).
- The mechanism of disintegration is based on swelling, wicking, and deformation of the disintegrants. When a compressed tablet is placed in aqueous solution, water can be quickly absorbed, and the swelling of the disintegrant breaks apart tablets quickly. In one embodiment in which the therapeutic active drug is formulated for immediate release, when a disintegrant is present in the core of the tablet, the rate of release of the active agent is an immediate pulse effect. In certain embodiments in which the therapeutic active drug is formulated for immediate release, when no disintegrant is present, a controlled profile may be produced.
- Examples of such disintegrants for use in the present invention include, for example, starch, veegum, crospovidone, cellulose, kaolin, microcrystalline cellulose (e.g., Avicel PH101 & PH102), crosslinked polyvinyl pyrrolidone (e.g., Kollidon CL), and mixtures thereof. In certain preferred embodiments, the disintegrant is a superdisintegrant, such as, for example, croscarmellose sodium, crospovidone, crosslinked carboxy methyl cellulose, sodium starch glycolate, and mixtures thereof. Superdisintegrants can be incorporated at lower levels than regular disintegrants to increase the water content. Some brand named superdisintegrants for use in the present invention include, Ac-Di-Sol®, Primojel®, Explotab®, and Crospovidone®.
- In certain embodiments, the core of the present invention includes a wicking agent in addition to or as an alternative to a disintegrant. Wicking agents such as those materials already mentioned as disintegrants (e.g. microcrystalline cellulose) may be included if necessary to enhance the speed of water uptake. Other materials suitable for acting as wicking agents include, but are not limited to, colloidal silicon dioxide, kaolin, titanium dioxide, fumed silicon dioxide, alumina, niacinamide, sodium lauryl sulfate, low molecular weight polyvinyl pyrrolidone, m-pyrol, bentonite, magnesium aluminum silicate, polyester, polyethylene, mixtures thereof, and the like.
- In certain embodiments, the one or more disintegrant(s) in the core is included in an amount from about 5 to about 20 percent, preferably from about 6 to about 10 percent, most preferably about 8 percent by weight of the core. In terms of whole tablet weight (e.g., core plus compression coating), the one or more disintegrant(s) in the core are included in an amount from about 0.1 to about 5 percent, preferably from about 0.3 to about 2 percent, by weight of the tablet (entire formulation).
- According to the present invention, the core containing active drug is completely surrounded or substantially surrounded by a compression coating. The compression coating preferably delays the release of the pharmaceutically active agent for a predetermined period of time, which time is dependent upon the formulation of the coating and the thickness of the coating layer. The appropriate time period for the release of the active ingredient can be determined prior to the preparation of the formulation, and the formulation can be designed by applying the appropriate thickness and composition of the coating to achieve the desired time delay prior to release of the active ingredient and the desired release rate of the active ingredient following the time delay.
- Preferably, the compression coating comprises a natural or synthetic gum which can function as a gelling agent, causing the core to be surrounded by the gel when the compression coated tablet is exposed to an environmental fluid (e.g., water or gastrointestinal fluid) and thereby causing the drug to be released after diffusion of the environmental fluid through the compression coating, the dissolution of the drug into the environmental fluid, and the egress of the dissolved drug into the fluid surrounding the compression coated tablet.
- In certain embodiments, gums for use in the compression coating include, for example and without limitation, heteropolysaccharides such as xanthan gum(s), homopolysaccharides such as locust bean gum, galactans, mannans, vegetable gums such as alginates, gum karaya, pectin, agar, tragacanth, accacia, carrageenan, tragacanth, chitosan, agar, alginic acid, other polysaccharide gums (e.g. hydrocolloids), and mixtures of any of the foregoing. Further examples of specific gums which may be useful in the compression coatings of the invention include, but are not limited to, acacia catechu, salai guggal, indian bodellum, copaiba gum, asafetida, cambi gum, Enterolobium cyclocarpum, mastic gum, benzoin gum, sandarac, gambier gum, butea frondosa (Flame of Forest Gum), myrrh, konjak mannan, guar gum, welan gum, gellan gum, tara gum, locust bean gum, carageenan gum, glucomannan, galactan gum, sodium alginate, tragacanth, chitosan, xanthan gum, deacetylated xanthan gum, pectin, sodium polypectate, gluten, karaya gum, tamarind gum, ghatti gum, Accaroid/Yacca/Red gum, dammar gum, juniper gum, ester gum, ipil-ipil seed gum, gum talha (acacia seyal), and cultured plant cell gums including those of the plants of the genera: acacia, actinidia, aptenia, carbobrotus, chickorium, cucumis, glycine, hibiscus, hordeum, letuca, lycopersicon, malus, medicago, mesembryanthemum, oryza, panicum, phalaris, phleum, poliathus, polycarbophil, sida, solanum, trifolium, trigonella, Afzelia africana seed gum, Treculia africana gum, detarium gum, cassia gum, carob gum, Prosopis africana gum, Colocassia esulenta gum, Hakea gibbosa gum, khaya gum, scleroglucan, zea, mixtures of any of the foregoing, and the like.
- In certain especially preferred embodiments, the compression coating comprises a heteropolysaccharide such as xanthan gum, a homopolysaccharide such as locust bean gum, or a mixture of one or more hetero- and one or more homopolysaccharide(s). Heterodisperse excipients, previously disclosed as a sustained release tablet matrix in our U.S. Pat. Nos. 4,994,276, 5,128,143, and 5,135,757, maybe utilized in the compression coatings of the present invention. For example, in certain embodiments of the present invention, a gelling agent of both hetero- and homo- polysaccharides which exhibit synergism, e.g., the combination of two or more polysaccharide gums producing a higher viscosity and faster hydration than that which would be expected by either of the gums alone, the resultant gel being faster-forming and more rigid, may be used in the compression coatings of the present invention.
- The term “heteropolysaccharide” as used in the present invention is defined as a water-soluble polysaccharide containing two or more kinds of sugar units, the heteropoly-saccjaride having a branched or helical configuration, and having excellent water-wicking properties and immense thickening properties.
- An especially preferred heteropolysaccharide is xanthan gum, which is a high molecular weight (>10 6) heteropolysaccharide. Other preferred heteropolysaccharides include derivatives of xanthan gum, such as deacylated xanthan gum, the carboxymethyl ether, and the propylene glycol ester.
- The homopolysaccharide materials used in the present invention that are capable of cross-linking with the heteropolysaccharide include the galactomannans, i.e., polysaccharides that are composed solely of mannose and galactose. A possible mechanism for the interaction between the galactomannan and the heteropolysaccharide involves the interaction between the helical regions of the heteropolysaccharide and the unsubstituted mannose regions of the galactomannan. Galactomannans that have higher proportions of unsubstituted mannose regions have been found to achieve more interaction with the heteropolysaccharide. Hence, locust bean gum, which has a higher ratio of mannose to galactose, is especially preferred as compared to other galactomannans, such as guar and hydroxypropyl guar.
- In certain preferred embodiments, the heteropolysaccharide comprises from about 1 to about 50 percent and the homopolysaccharide material comprises from about 50 to about 1 percent by weight of the compression coating. In certain preferred embodiments, the ratio of heteropolysaccharide to homopolysaccharide material is from about 1:3 to 3:1, preferably from about 2:3 to 3:2, or 1:1.
- In a certain preferred embodiment, the compression coating comprises from about 5 to about 70 percent or more by weight of a hydrophilic material (e.g., gums). In certain preferred embodiments of the present invention, the higher the percentage of gums in the compression coating, the longer the delay of the release or “lag time” of the active agent.
- In certain embodiments, the percent of gums in the compression coating corresponds to a delayed release of the active agent which is independent of pH. For example, in certain preferred embodiments, when the compression coating is less than about 25% gums, preferably comprising about 5 to about 15% gums, the delayed release is more independent of pH than a compression coating comprising greater than about 25% gums (e.g., 30, 40, or 50% gums).
- In certain preferred embodiments, the compression coating also includes pharmaceutically acceptable excipients, for example, a saccharide such as a monosaccharide, a disaccharide or a polyhydric alcohol, and/or mixtures of any of the foregoing, or microcrystalline cellulose or a starch. Examples of suitable such excipients include sucrose, dextrose, lactose, fructose, xylitol, sorbitol, mannitol, starches, mixtures thereof and the like. In certain embodiments, it is preferred that a soluble pharmaceutical excipient such as lactose, dextrose, sucrose, mannitol, or mixtures thereof is included in the materials to be used in the compression coating. In certain preferred embodiments, the gum(s) is wet granulated with the pharmaceutically acceptable excipient prior to its use as a compression coating on the surface of the inner cores of the invention. The compression coating may comprise, e.g., up to about 95% pharmaceutically acceptable excipient(s), by weight.
- In certain embodiments, the amount of gum(s) contained in the compression coating is from about 1 percent to about 90 percent by weight, preferably from about 6.5 percent to about 83 percent of the total tablet, by weight.
- In certain embodiments, it is possible to dry mix the ingredients of the compression (delayed release) coating without utilizing a wet granulation step. If the mixture is to be manufactured without a wet granulation step, and the final mixture is to be compression coated onto a pre-formed tablet core, it is preferred that all or part of the pharmaceutically acceptable excipient(s) should impart sufficient compressibility to provide a pharmaceutically acceptable product. The properties and characteristics of a specific excipient system prepared according to the present invention may be dependent in part on the individual characteristics, e.g., of the homo- and heteropolysaccharide constituents, in terms of polymer solubility, glass transition temperatures etc., as well as on the synergism both between different homo- and heteropolysaccharides and between the homo- and heteropolysaccharides and the inert saccharide constituent(s) in modifying dissolution fluid-excipient interactions.
- In certain embodiments of the invention where the compression coating comprises a heteropolysaccharide, a homopolysaccharide, or both, a release-modifying agent as described in our previous patents directed to the use of these materials in sustained release matrices can also be utilized in the compression coating. Such release-modifying agents and pre-manufactured excipients disclosed in our U.S. Pat. Nos. 5,455,046; 5,512,297; 5,554,387; 5,667,801; 5,846,563; 5,773,025; 6,048,548; 5,662,933; 5,958,456; 5,472,711; 5,670,168; and 6,039,980 may be utilized in the compression coatings of the present invention.
- Thus, for example, the release-modifying agent may comprise an ionizable gel-strength enhancing agent. The ionizable gel strength-enhancing agent that is optionally used in conjunction with the present invention may be monovalent or multivalent metal cations. The preferred salts are the inorganic salts, including various alkali metal and/or alkaline earth metal sulfates, chlorides, borates, bromides, citrates, acetates, lactates, etc. Specific examples of suitable ionizable gel strength enhancing agent include calcium sulfate, sodium chloride, potassium sulfate, sodium carbonate, lithium chloride, tripotassium phosphate, sodium borate, potassium bromide, potassium fluoride, sodium bicarbonate, calcium chloride, magnesium chloride, sodium citrate, sodium acetate, calcium lactate, magnesium sulfate and sodium fluoride. Multivalent metal cations may also be utilized. However, the preferred ionizable gel strength-enhancing agents are bivalent. Particularly preferred salts are calcium sulfate and sodium chloride. The ionizable gel strength enhancing agents of the present invention are added in an amount effective to obtain a desirable increased gel strength due to the cross-linking of the gelling agent (e.g., the heteropolysaccharide and homopolysaccharide gums). In alternate embodiments, the ionizable gel strength-enhancing agent is included in the delayed release excipient of the present invention in an amount from about 1 to about 20% by weight of the delayed release excipient, and in an amount 0.5% to about 16% by weight of the final dosage form. In certain embodiments, the inclusion of an ionizable gel strength-enhancing agent not only delays the release of the active, but also provides for a sustained release of the active agent.
- In certain embodiments of the present invention, the (delayed release) compression coating coated onto the core comprises from about 1 to about 90 percent by weight of a gelling agent comprising a heteropolysaccharide gum and a homopolysaccharide gum, from about 0 to about 20 percent by weight of an ionizable gel strength enhancing agent, and from about 10 to about 95 percent by weight of an pharmaceutically acceptable excipient. In other embodiments, the compression coating material comprises from about 5 to about 75 percent gelling agent (gum), from about 0 to about 15 percent ionizable gel strength enhancing agent, and from about 30 to about 95 percent pharmaceutically acceptable excipient (e.g., an inert diluent). In yet other embodiments, the compression coating material comprises from about 7.5 to about 50 percent gelling agent, from about 0 to about 10 percent ionizable gel strength enhancing agent, and from about 30 to about 95 percent pharmaceutically acceptable excipient.
- Surfactants that may be used in the present invention generally include pharmaceutically acceptable anionic surfactants, cationic surfactants, amphoteric (amphipathic/amphophilic) surfactants, and non-ionic surfactants. Suitable pharmaceutically acceptable anionic surfactants include, for example, monovalent alkyl carboxylates, acyl lactylates, alkyl ether carboxylates, N-acyl sarcosinates, polyvalent alkyl carbonates, N-acyl glutamates, fatty acid-polypeptide condensates, sulfuric acid esters, alkyl sulfates (including sodium lauryl sulfate (SLS)), ethoxylated alkyl sulfates, ester linked sulfonates (including docusate sodium or dioctyl sodium succinate (DSS)), alpha olefin sulfonates, and phosphated ethoxylated alcohols.
- Suitable pharmaceutically acceptable cationic surfactants include, for example, monoalkyl quaternary ammonium salts, dialkyl quaternary ammonium compounds, amidoamines, and aminimides.
- Suitable pharmaceutically acceptable amphoteric (amphipathic/amphophilic) surfactants, include, for example, N-substituted alkyl amides, N-alkyl betaines, sulfobetaines, and N-alkyl β-aminoproprionates.
- Other suitable surfactants for use in conjunction with the present invention include polyethyleneglycols as esters or ethers. Examples include polyethoxylated castor oil, polyethoxylated hydrogenated castor oil, polyethoxylated fatty acid from castor oil or polyethoxylated fatty acid from hydrogenated castor oil. Commercially available surfactants that can be used are known under trade names Cremophor, Myi, Polyoxyl 40 stearate, Emerest 2675, Lipal 395 and PEG 3350.
- Other release-modifying pharmaceutically acceptable agents that may be added in appropriate quantities for their particular ability to modify dissolution rates include, for example: stearic acid, metallic stearates, stearyl alcohol, hydrogenated cotton seed oil, sodium chloride and certain disintegrants that are described below.
- The quantity of such release-modifying agent employed depends on the release characteristics required and the nature of the agent. For a delayed release formulation according to the invention, the level of release-modifying agents used may be from about 0.1 to about 25%, preferably from about 0.5 to about 10% by weight of the total composition.
- In certain other embodiments of the invention, the compression coating includes a pH-modifying agent. The pH-modifying agent may be present in the compression coating from about 1% to about 10% by weight of the final dosage form. In preferred embodiments, the pH-modifying agent is an organic acid such as citric acid, succinic acid, fumaric acid, malic acid, maleic acid, glutaric acid or lactic acid.
- In certain preferred embodiments, the release of drug occurs when aqueous environmental fluid (e.g., water or gastrointestinal fluid, etc. surrounding the dosage form) diffuses through the compression coating of the dosage form, resulting in hydration of the core and dissolving the drug, which then can pass into the fluid surrounding the core.
- In certain preferred embodiments, the delayed release of the drug (lag time) is varied by increasing the thickness of the compression coating (increased lag time) or by decreasing the thickness of the compressing coating (decreased lag time). The delayed release may also be varied, e.g., by changing the gum(s) included in the delayed release compression coating, selecting a particular combination of gums, by including or not including an pharmaceutically acceptable excipient, such as a saccharide (including polysaccharides) or a combination of saccharide(s) (or polysaccharides) in the compression coating, by changing or by adding additional agents to the compression coating which cause the compression coating to further delay the diffusion of water (or gastrointestinal fluid) through the compression coating (e.g., matrix) into the inner core (thereby allowing hydration of the inner core). In addition, the compression force used to apply the compression coating may be used to alter the release rate of the active ingredient. Also, release can be modified via the use of an extragranular excipient addition to the compression coating. Such ingredients may comprise, for example, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, and the like.
- The delayed release of the drug may further be varied by utilizing a further coating (i) between the core and the compression coating; (ii) over the compression coating; or (iii) both between the core and the compression coating and over the compression coating. Such coatings may comprise, for example a hydrophilic polymer (such as hydroxypropylmethylcellulose) and/or a hydrophobic polymer (such as an acrylic polymer, a copolymer of acrylic and methacrylic acid esters, an alkylcellulose such as ethylcellulose, etc.). In such circumstances, the release of drug from the dosage form may not only be occurring as fluid diffuses through the compression coating; erosion of the further coatings described in this paragraph may also delay the release of drug.
- The dissolution rates of the present invention (with or without the optional release modifying agents mentioned above) may be further modified by incorporation of a hydrophobic material in the compression coating, which slows the hydration of the gums without disrupting the hydrophilic matrix. This is accomplished in alternate embodiments of the present invention by granulating the delayed release excipient with a solution or dispersion of a hydrophobic material prior to the compression coating of the core. The hydrophobic polymer may be selected from an alkylcellulose such as ethylcellulose, other hydrophobic cellulosic materials, polymers or copolymers derived from acrylic or methacrylic acid esters, copolymers of acrylic and methacrylic acid esters, zein, waxes, shellac, hydrogenated vegetable oils, and any other pharmaceutically acceptable hydrophobic material known to those skilled in the art. The solvent for the hydrophobic material may be an aqueous or organic solvent, or mixtures thereof. The amount of hydrophobic material incorporated into the delayed release excipient is that which is effective to slow the hydration of the gums without disrupting the hydrophilic matrix formed upon exposure to an environmental fluid. In certain preferred embodiments of the present invention, the hydrophobic material is included in the compression coating in an amount from about 1 to about 20 percent by weight.
- The compression coating may also contain suitable quantities of, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are described hereinafter and are which are conventional in the pharmaceutical art.
- In preferred embodiments where the materials to be included in the compression coating are pre-manufactured, the combination of the gum gelling agent (e.g., a mixture of xanthan gum and locust bean gum) with the pharmaceutical excipient(s), with or without a release modifying agent, provides a ready-to-use compression coating product in which a formulator need only apply the material onto the core by compression coating to provide the desired chronotherapeutic dosage forms. The compression coating may comprise a physical admix of the gums along with a soluble excipient such as compressible sucrose, lactose, dextrose, etc., although it is preferred to granulate or agglomerate the gums with a plain pharmaceutically acceptable excipient (i.e., crystalline) sucrose, lactose, dextrose, mannitol, etc., to form a delayed release excipient for use in the compression coating. The granulate form has certain advantages including the fact that it can be optimized for flow and compressibility.
- The gums and optional pharmaceutical excipients used in the compression coating are preferably prepared according to any agglomeration technique to yield an acceptable excipient product. In wet granulation techniques, the desired amounts of the hydrophilic material (e.g., heteropolysaccharide gum and/or the homopolysaccharide gum) and the inert diluent are mixed together and thereafter a moistening agent such as water, propylene glycol, glycerol, alcohol or the like is added to prepare a moistened mass. Next, the moistened mass is dried. The dried mass is then milled with conventional equipment into granules. Thereafter, the excipient product is ready to use.
- The pre-manufactured delayed release excipient is preferably free-flowing and directly compressible. Accordingly, the excipient may be directly compressed onto a pre-formed inner core of a therapeutically active medicament to form coated tablets. The delayed release coating mixture, in an amount sufficient to make a uniform coating onto a preformed tablet core, is subjected to tableting in a conventional production scale tableting machine at normal compression pressure, i.e., about 2000-1600 lbs/sq in. However, the mixture should not be compressed to such a degree that there is subsequent difficulty in its hydration when exposed to gastric fluid.
- The average particle size of the granulated delayed release excipient of the present invention ranges from about 50 microns to about 400 microns and preferably from about 185 microns to about 265 microns. The particle size of the granulation is not narrowly critical, the important parameter being that the average particle size of the granules must permit the formation of a directly compressible excipient which forms a coating over pharmaceutically active tablet cores. The desired tap and bulk densities of the granulation of the present invention are normally between from about 0.3 to about 0.8 g/ml, with an average density of from about 0.5 to about 0.7 g/ml.
- The compression coatings of the present invention preferably have uniform packing characteristics over a range of different particle size distributions and are capable of processing onto the preformed tablet core using direct compression, following the addition of a lubricant.
- In addition to being (optionally) used in the tablet core, in certain embodiments it is preferred that one or more pharmaceutically acceptable lubricants be added to the compression coating materials (preferably pre-agglomerated) prior to the mixture being compression coated onto the surface of the core. Examples of suitable lubricants for use in the core and compression coating of the invention include, for example and without limitation, talc, stearic acid, vegetable oil, calcium stearate, zinc stearate, magnesium stearate, etc. Preferably, an effective amount of any generally accepted pharmaceutical lubricant, including calcium or magnesium soaps is preferably added to the mixture of ingredients prior to compression of the mixture onto the solid preformed tablet core. An especially preferred lubricant is sodium stearyl fumarate, NF, commercially available under the trade name Pruv® from Penwest Pharmaceuticals Co.
- In certain embodiments, the present invention is further directed towards a method of manufacturing the delayed release solid oral dosage forms (e.g., tablets) of the present invention. In certain preferred embodiments, the steps for preparation of a delayed release oral solid dosage form of the present invention may include the following:
- Preparation of inner core formulation:
- 1. (A) Wet granulate active ingredient (e.g., drug) together with optional excipients, followed by drying and milling as necessary to obtain a granulate; or (B) Dry blend the active together with optional excipients using geometric dilution as necessary to obtain a granulate;
- 2. Optionally, extragranularly add excipients to the material prepared in Step 1 with appropriate blending;
- 3. Preferably, lubricate powder blend prepared in Step 1 or 2:
- 4. Compress core using powder blend prepared in Step 3 with an appropriate press.
- 5. Optionally, applying a functional film coating onto the tablet cores prepared in Step 4;
- Preparation of delayed release (compression) coating may be accomplished, e.g., as follows:
- 6. (A) Wet granulate a gum(s) (e.g., a heteropolysaccharide gum and a homopoly-saccharide gum) together with optional excipients to form a delayed release material (agglomerated particles), and then dry the delayed release material; or (B) Dry blend a gum(s) together with optional excipients to form a delayed release material (granulate);
- 7. Preferably, mill the delayed release material prepared in Step 6;
- 8. Preferably, lubricate the delayed release material prepared in Step 6 or 7; Coating of inner core:
- 9. Compression coat the delayed release material prepared in Steps 6-8 over the tablet cores prepared in Step 1-5;
- 10. Optionally, film coat the final dosage form (if desired).
- In certain embodiments, steps 4 & 10 are combined in a single unit operation when using e.g., a Dry-Cota Press as described hereinafter. A functional coating of the tablet cores may be possible using the Dry-Cota Press if a modification is made to the press to add a core tablet feeder system.
- A Manesty Dry-Cota press press consists of two side by side interconnected tablet presses where the core is made on one press then mechanically transferred to the next press for compression coating. Each “press” has an independent powder feed mechanism so that core blend is loaded on one machine and coating blend on the other. Mechanical transfer arms rotate between the machines to remove cores from one press and transfer them to the coating press. Other and more modem types of presses which may be used (e.g. Elizabeth Hata HT-AP44-MSU-C, Killian RUD, Fette PT 4090) have a dual feed system for coating blend and pre-made cores. This configuration is more flexible, in that cores can be pan coated with a functional or cosmetic coating before compression coating. In addition, this allows multiple compression coating layers to be achieved by recycling tablets that have already been compression coated. Both types of presses have mechanisms to center the tablet within the coating both vertically and radially. One of ordinary skill would understand that other tablet presses may be used to provide for the final dosage forms of the present invention.
- Although typically the compression coating surrounds the entire core, in certain embodiments of the present invention, the compression coating substantially surrounds, but does not entirely surround the tablet core. In such instances, the release of drug from the tablet core will occur first from that portion of the inner core to which the compression is not applied. In other embodiments of the invention, compression coating is not applied to the same thickness around the entire inner core, thereby creating areas of the compressed dosage form that release drug earlier (and later) than other areas. This may be accomplished, e.g, by having the core to which the compression coating is applied not being centered in the press.
- For best results, the tablets formed from the compression coating of the core are from about 4 to about 25 kP, preferably about 5 to about 15 kP, most preferably about 8 to about 9 kP hardness. In certain preferred embodiments, for round compression coated tablets the diameter may be up to ⅝ inch or greater, and for caplet shaped compression coated tablets the diameter may be up to ¾ inch or greater. The average flow of the (non-compression) coatings prepared in accordance with the present invention is from about 25 to about 40 g/sec.
- In certain embodiments of the present invention, the compression coated tablet may then be further overcoated with an enteric coating material or a hydrophobic material. Examples of suitable enteric polymers include cellulose acetate phthalate, hydroxypropyl-methylcellulose phthalate, polyvinylacetate phthalate, methacrylic acid copolymer, shellac, hydroxypropyhnethylcellulose succinate, cellulose acetate trimellitate, and mixtures of any of the foregoing. An example of a suitable commercially available enteric material is available under the trade name Eudragit® L30D55.
- In further embodiments, the dosage form may be coating with a hydrophilic coating in addition to or instead of the above-mentioned enteric coating or hydrophobic coating. An example of a suitable material that may be used for such a hydrophilic coating is hydroxypropylmethylcellulose (e.g., Opadry®, commercially available from Colorcon, West Point, Pa.).
- In still further embodiments, the optional enteric and/or hydrophobic and/or hydrophilic coatings may be alternatively or additionally applied as an intermediate layer(s) between the core and the compression coating.
- The optional enteric and/or hydrophobic and/or hydrophilic coatings may be applied in any pharmaceutically acceptable manner known to those skilled in the art. For example, in one embodiment, the coating is applied via a fluidized bed or in a coating pan. For example, the coated tablets may be dried, e.g., at about 60-70° C. for about 3-4 hours in a coating pan. The solvent for the hydrophobic polymer or enteric coating may be organic, aqueous, or a mixture of an organic and an aqueous solvent. The organic solvents may be, e.g., isopropyl alcohol, ethanol, and the like, with or without water.
- In additional embodiments of the present invention, a support platform is applied to the tablets manufactured in accordance with the present invention. Suitable support platforms are well known to those skilled in the art. An example of suitable support platforms is set forth, e.g., in U.S. Pat. No. 4,839,177, hereby incorporated by reference. In that patent, the support platform partially coats the tablet, and consists of a polymeric material insoluble in aqueous liquids. The support platform may, for example, be designed to maintain its impermeability characteristics during the transfer of the therapeutically active medicament. The support platform may be applied to the tablets, e.g., via compression coating onto part of the tablet surface, by spray coating the polymeric materials comprising the support platform onto all or part of the tablet surface, or by immersing the tablets in a solution of the polymeric materials.
- The support platform may have a thickness of, e.g., about 2 mm if applied by compression, and about 10 μ if applied via spray-coating or immersion-coating. Generally, in embodiments of the invention wherein a hydrophobic polymer or enteric coating is applied to the tablets over the delayed release coating, the tablets are coated to a weight gain from about 1 to about 20%, and in certain embodiments preferably from about 5% to about 10%.
- Materials useful in the hydrophobic coatings and support platforms of the present invention include derivatives of acrylic acid (such as esters of acrylic acid, methacrylic acid, and copolymers thereof) celluloses and derivatives thereof (such as ethylcellulose), polyvinylalcohols, and the like.
- As mentioned above, the cores and/or compression coatings may also contain suitable quantities of, e.g., lubricants, binders, granulating aids, diluents, colorants, flavorants and glidants which are conventional in the pharmaceutical art.
- Examples of suitable binders for use in the present invention include for example and without limitation, povidone, polyvinylpyrrolidone, xanthan gum, cellulose gums such as carboxymethylcellulose, methyl cellulose, hydroxypropyhnethylcellulose, hydroxycellulose, gelatin, starch, and pregelatinized starch.
- Examples of suitable glidants for use in the present invention include talc, silicon dioxide, and cornstarch.
- In certain embodiments of the present invention, the tablet core includes an additional dose of the drug (or a therapeutically effective dose of a different drug) included in either the (optional) hydrophobic or enteric coating, or in an additional (optional) overcoating coated on the outer surface of the tablet core (without the hydrophobic or enteric coating) or as an additional coating layer coated on the surface of the base coating(s) comprising the compression coating and, if applicable, hydrophobic and/or enteric coating material. This may be desired when, for example, a loading dose of the drug is needed to provide therapeutically effective blood levels of the active agent when the formulation is first exposed to gastric fluid. The loading dose of drug included in the coating layer may be, e.g., from about 10% to about 40% of the total amount of drug included in the formulation.
- Chronobiological patterns have been observed with arthritis pain. People with osteoarthritis (the most common form of arthritis) tend to have less pain in the morning and more at night. But for people with rheumatoid arthritis, the pain usually peaks in the morning and decreases as the day wears on. Recent animal studies showing that joint inflammation in rats fluctuates over a 24-hour period support these observations by both patients and physicians. Potential drug candidates in this therapeutic area include (for all forms of arthritis) standard treatment, glucocorticosteroids, etc. Preferably, the dosages should be timed to ensure that the highest blood levels of the drug coincide with peak pain. For osteoarthritis—the optimal time for the glucocorticosteroid would be around noon or mid-afternoon. For rheumatoid arthritis—the optimal time for the glucocorticosteroid to be taken is after the evening meal.
- In certain preferred embodiments, the active agent is a glucocorticoid. Glucocorticoids have a very favorable effect on the symptoms of rheumatoid arthritis, e.g. morning stiffness, joint pain and joint swelling. Glucocorticoids for use in the present invention include for example, prednisolone, prednisone, cortisone, hydrocortisone, methylprednisolone, betamethasone, dexamethasone, triamcinolone, mixtures thereof, and pharmaceutically acceptable salts thereof. Prednisolone is particularly preferred. Prednisolone is a potent pharmaceutical agent which has been commercially available for many years. Prednisolone is characterized by pronounced anti-inflammatory activity, when administered locally or systemically. Prednisolone is known as an anti-inflammatory and anti-rheumatic drug. Preferably, when the active agent is prednisolone, the prednisolone is in an amount of from about 0.1 to about 20 mg, preferably from about 1 to about 6 mg, and in most preferred embodiments about 1, 2 or 5 mg.
- Equivalent doses of other glucocorticoids can be calculated based on the following chart:
Approximate Equivalent Glucocorticoid Dose (mg) Cortisone 25 Hydrocortisone 20 Prednisone 5 Prednisolone 5 Triamcinolone 4 Methylprednisolone 4 Dexamethasone 0.75 Betamethasone 0.6-0.75 - In certain preferred embodiments of the invention where the manifestations of the disease state to be treated (e.g., pain from arthritis) are greatest upon awakening, the chronotherapeutic formulations are preferably orally administered to the patient at bedtime (e.g., at about 9 or 10 p.m.) and have a lag time of about 5 or 6 hours, so that, e.g., a substantial portion of the drug in the compression coated delayed release oral dosage form is released, e.g., between 2-3 a.m., or between 3-4 a.m., and the drug is absorbed from the gastrointestinal tract and provides therapeutic efficacy at a time which correlates with the peak of the manifestations of the disease state.
- The benefits of chronotherapeutics include safety and more efficient treatment than conventional therapies. This is achieved by delivering more medication when risk of disease is greater, and delivering less medication when potential for disease symptoms are less likely. Other benefits to the patient include an increased quality of life and a once-a-day drug delivery system to increase patient compliance.
- In certain preferred embodiments of the invention where the manifestations of the disease state to be treated (e.g., pain from arthritis) are greatest upon awakening, the chronotherapeutic formulations are preferably orally administered to the patient at bedtime (e.g., at about 9 or 10 p.m.) and have a lag time of about 5 or 6 hours, so that, e.g., a substantial portion of the drug in the compression coated delayed release oral dosage form is released, e.g., between 2-3 a.m., or between 3-4 a.m., and the drug is absorbed from the gastrointestinal tract and provides therapeutic efficacy at a time which correlates with the peak of the manifestations of the disease state.
- In preferred embodiments of the present invention, the total tablet weight is from about 220 mg to about 900 mg; and the core weight is preferably from about 50 mg to about 170 mg. Preferably, the core is from about 5 to about 23 percent, most preferably about 18 to about 20 percent by weight of the total tablet weight. The compression coating is preferably from about 150 mg to about 850 mg. Preferably, the compression coating is from about 75 to about 94 percent by weight, most preferably from about 78 to 80 percent, by weight of the total tablet. Preferably, the ratio of the core to gum (in the compression coating) is from about 1:0.37 to about 1:5, preferably from about 1:0.37 to about 1:1.12, most preferably from about 1:0.75. The ratio of the core to compression coating material (all ingredients) is preferably from about 1:2 to about 1:9, and in certain embodiments more preferably about 1:4.
- In the appended examples, the cores comprising the active agent are typically compression coated with the coating formulation by hand on a rotary tablet press. In such a process, roughly half the outer core material is first added to the die. An inner core tablet is typically centered on the powder bed and is covered with the other half of the outer coating powder. However, one skilled in the art will appreciate that compression coating may be accomplished via automated tablet presses for commercialization. Prior to compression coating with any tablet press, preferably 0.75% Pruv® (sodium stearyl fumarate, NF) or another suitable lubricant is added to the compression coating material(s). In certain examples wherein the coatings are indicate by the gums, for example, 50% xanthan gum (XG), the coating comprises 50% xanthan gum diluted with dextrose; and for example 50% locust bean gum (LBG), the coating comprises 50% locust bean gum diluted with dextrose, etc.
- The following examples illustrate various aspects of the present invention. They are not to be construed to limit the claims in any manner whatsoever.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 1:
TABLE 1 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Dextrose 70 4. Water* q.s. - Process:
- 1. The requisite amounts of xanthan gum, locust bean gum, and dextrose are dry blended in a high speed mixer/granulator for 3 minutes.
- 2. Water (125-150 ml) is added to the dry blended mixture, and granulated for another 3 minutes.
- 3. The granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 2:
TABLE 2 Component Percentage 1. Xanthan Gum 25 2. Locust Bean Gum 25 3. Dextrose 35 4. Calcium Sulfate Dihydrate 10 5. Ethylcellulose 5 5. Alcohol, SD3A, anhydrous* 20 6. Water* q.s. - Process:
- 1. The requisite amounts of xanthan gum, locust bean gum, calcium sulfate, and dextrose are dry blended in a high speed mixer/granulator for 3 minutes.
- 2. A slurry of hydrophobic polymer (ethylcellulose) is prepared by dissolving ethyl cellulose in ethyl alcohol.
- 3. The slurry is added to the dry blended mixture, and granulated for another 3 minutes.
- 4. The granulation was then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- 5.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 3:
TABLE 3 Component Percentage 1. Xanthan Gum 15 2. Locust Bean Gum 15 3. Dextrose 60 4. Calcium Sulfate Dihydrate 10 5. Water* q.s. - Process:
- 1. The requisite amounts of xanthan gum, locust bean gum, calcium sulfate, and dextrose are dry blended in a high speed mixer/granulator for 3 minutes.
- 2. Water (125-150 ml) is added to the dry blended mixture, and granulated for another 3 minutes.
- 3. The granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than abou-t 10% by weight (e.g., 4-7% LOD).
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 4:
TABLE 4 Component Percentage 1. Xanthan Gum 16 2. Locust Bean Gum 24 3. Dextrose 60 4. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release coating of Example 4.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 5:
TABLE 5 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Dextrose 45 4. Calcium Sulfate Dihydrate 5 5. Water* q.s. - Process:
- The same process for Example 3 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 5.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 6:
TABLE 6 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Dextrose 65 4. Calcium Sulfate Dihydrate 5 5. Water* q.s. - Process:
- The same process for Example 3 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 6.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 7:
TABLE 7 Component Percentage 1. Xanthan Gum 10 2. Locust Bean Gum 15 3. Dextrose 75 4. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 7.
- A delayed release material to be used in the compression coatings of the invention is prepared having the following formulation listed in Table 8:
TABLE 8 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose 80 4. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 8.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 9:
TABLE 9 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Lactose 50 4. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 5, substituting lactose for dextrose.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 10:
TABLE 10 Component Percentage 1. Xanthan Gum 20 2. Locust Bean Gum 30 3. Mannitol 45 4. Hydroxypropylmethylcellulose 5 5. Water* q.s. - Process:
- 1. The requisite amounts of xanthan gum, locust bean gum, mannitol, and hydroxypropylmethylcellulose are dry blended in a high speed mixer/granulator for 3 minutes.
- 2. Water (125-150 ml) is added to the dry blended mixture, and granulated for another 3 minutes.
- 3. The granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 11:
TABLE 11 Component Percentage 1. Xanthan Gum 12 2. Locust Bean Gum 18 3. Mannitol 70 4. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 11, substituting marmitol for dextrose.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 12:
TABLE 12 Component Percentage 1. Xanthan Gum 69 2. Locust Bean Gum 13.5 3. Mannitol 77.5 4. Water* q.s. - Process:
- The same process for Example 10 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 12.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 13:
TABLE 13 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Mannitol 80 4. Water* q.s. - Process:
- The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 13.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 14:
TABLE 14 Component Percentage 1. Xanthan Gum 6 2. Locust Bean Gum 9 3. Mannitol 85 4. Water* q.s. - Process:
- The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 14.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 15:
TABLE 15 Component Percentage 1. Xanthan Gum 4 2. Locust Bean Gum 6 3. Mannitol 90 4. Alcohol, SD3A, anhydrous* — 5. Water* q.s. - Process:
- The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 15.
- A delayed release coating is prepared having the following formulation listed in Table 16:
TABLE 16 Component Percentage 1. Xanthan Gum 3 2. Locust Bean Gum 4.5 3. Mannitol 92.5 4. Water* q.s. - Process:
- The same process for Example 12 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 16.
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 17:
TABLE 17 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose 40 4. Microcrystalline Cellulose 40 5. Water* q.s. - Process:
- 1. The requisite amounts of xanthan gum, locust bean gum, dextrose, and microcrystalline cellulose are dry blended in a high speed mixer/granulator for 3 minutes.
- 2. Water (125-150 ml) is added to the dry blended mixture, and granulated for another 3 minutes.
- 3. The granulation is then dried in a fluid bed dryer to a LOD (loss on drying) of less than about 10% by weight (e.g., 4-7% LOD).
- A delayed release material to be used in the compression coatings of the invention is prepared having the formulation listed in Table 18:
TABLE 18 Component Percentage 1. Xanthan Gum 8 2. Locust Bean Gum 12 3. Dextrose — 4. Microcrystalline Cellulose 80 5. Water* q.s. - Process:
- The same process for Example 1 is used to prepare the delayed release material to be used in the compression coatings of the invention in Example 18, substituting microcrystalline cellulose for dextrose.
- A prednisolone core composition was prepared having the ingredients set forth in Table 19:
TABLE 19 Component Percent amt. (mg) 1. Prednisolone 2.0 1.0 2. Prosolv SMCC ™ 50 32.75 16.4 3. Prosolv SMCC ™ 90 50 25.0 4. Explotab ® 10 5.0 5. Sodium carboxymethylcellulose 5 2.5 6. Pruv 0.25 0.1 Total 100 50 Core size and shape 3/16″ Round SC* - Process:
- 1. Blend the requisite amounts of prednisolone and Prosolv™ SMCC 50 in a V-blender for 5 to 10 minutes.
- 2. Add the requisite amounts of Prosolv™ SMCC 90, Explotab® and sodium carboxymethylcellulose to the blend and continue blending for another 5 minutes.
- 3. Add the requisite amount of Pruv to the mixture and blend for an additional 5 minutes. 4. Compress the tablet cores using tablet press.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 20:
TABLE 20 Component Percent amt. (mg) 1. Prednisolone 10 5 2. Prosolv SMCC ™ 50 NA NA 3. Prosolv SMCC ™ 90 81.75 40.875 4. Explotal ® 6 3 5. Sodium carboxymethylcellulose NA NA 6. Pruv 0.25 0.125 7. PVP 2 1 Total 100 50 Core size and shape {fraction (3/16)}″ Round FF* - Process:
- 1. Blend the requisite amounts of prednisolone, Prosolv™ SMCC 90, Explotab® for 5 to 10 minutes.
- 2. Add the requisite amount of Pruv and PVP to the mixture and blend for an additional 5 minutes.
- 3. Compress the tablet cores using tablet press.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 21:
TABLE 21 Component Percent amt. (mg) 1. Prednisolone 10 5 2. Prosolv SMCC ™ 50 NA NA 3. Prosolv SMCC ™ 90 75.75 37.375 4. Explotab ® 10 5 5. Sodium carboxymethylcellulose 5 2.5 6. Pruv 0.25 0.1 Total 100 50 Core size and shape {fraction (3/16)}″ Round FF - Process:
- 1. Blend the requisite amounts of prednisolone, Prosolv™ SMCC 90, Explotab®, and sodium carboxymethylcellulose for 5 to 10 minutes.
- 2. Add the requisite amount of Pruv and PVP to the mixture and blend for an additional 5 minutes.
- 3. Compress the tablet cores using tablet press.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 22:
TABLE 22 Component Percent amt. (mg) 1. Prednisolone 2 1.0 2. Prosolv SMCC ™ 50 40 20.0 3. Prosolv SMCC ™ 90 47.75 23.9 4. Explotab ® 6 3.0 5. PVP 2 1.0 6. Talc 2 1.0 7. Pruv 0.25 0.1 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- 1. Blend the requisite amounts of prednisolone and Prosolv™ SMCC 50 in a V-blender for 5 to 10 minutes.
- 2. Add the requisite amounts of Prosolv™ SMCC 90, Explotab®, PVP, and talc to the blend and continue blending for another 5 minutes.
- 3. Add the requisite amount of Pruv to the mixture and blend for an additional 5 minutes.
- 4. Compress the tablet cores using tablet press.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 23:
TABLE 23 Component Percent amt. (mg) 1. Prednisolone 2 3.4 2. Prosolv SMCC ™ 50 40 68 3. Prosolv SMCC ™ 90 47.75 81.75 4. Explotab ® 6 10.2 5. Sodium carboxymethylcellulose 2 3.4 6. Talc 2 3.4 7. Pruv 0.25 0.425 Total 100 170 Core size and shape ¼″ Round FF - Process:
- 1. Blend the requisite amounts of prednisolone and Prosolv™ SMCC 50 in a V-blender for 5 to 10 minutes.
- 2. Add the requisite amounts of Prosolv™ SMCC 90, Explotab®, sodium carboxymethylcellulose, and talc to the blend and continue blending for another 5 minutes.
- 3. Add the requisite amount of Pruv to the mixture and blend for an additional 5 minutes.
- 4. Compress the tablet cores using tablet press.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 24:
TABLE 24 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 47.75 23.875 4. Explotab ® 6 3 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 6. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 24.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 25:
TABLE 25 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 55.75 27.85 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 25, without the inclusion of Explotab® and sodium carboxymethylcellulose.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 26:
TABLE 26 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 52.75 26.375 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 26, without the inclusion of sodium carboxymethylcellulose.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 27:
TABLE 27 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 51.75 25.875 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose 1 0.5 6. Talc 2 1 6. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 27.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 28:
TABLE 28 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 53.75 26.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 28, without the inclusion of Explotab®.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 29:
TABLE 29 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 47.75 23.875 4. Explotab ® 2 1 5. Sodium carboxymethylcellulose 6 3 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 29.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 30:
TABLE 30 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 49.75 24.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 6 3 6. Talc 2 1 7 Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 30, without the inclusion of Explotab®.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 31:
TABLE 31 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 26 13 3. Prosolv SMCC ™ 90 49.75 24.875 4. Explotab ® 20 10 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 31, without the inclusion of sodium carboxymethylcellulose.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 32:
TABLE 32 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 26 13 3. Prosolv SMCC ™ 90 49.75 24.875 4. Explotab ® 20 10 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 32, without the inclusion of sodium carboxymethylcellulose.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 33:
TABLE 33 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 52.75 26.375 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose NA NA 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 33, without the inclusion sodium carboxymethylcellulose.
- A prednisolone core composition was prepared having the formulation set forth in Table 34:
TABLE 34 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 51.75 25.875 4. Explotab ® 3 1.5 5. Sodium carboxymethylcellulose 1 0.5 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 34.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 35:
TABLE 35 Component Percent amt. (mg) 1. Prednisolone 2 1 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 53.75 26.875 4. Explotab ® NA NA 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 35, without the inclusion of Explotab®.
- A prednisolone core composition was prepared having the formulation ingredients set forth in Table 36:
TABLE 36 Component Percent amt. (mg) 1. Prednisolone 4 2 2. Prosolv SMCC ™ 50 40 20 3. Prosolv SMCC ™ 90 45.75 22.875 4. Explotab ® 6 3 5. Sodium carboxymethylcellulose 2 1 6. Talc 2 1 7. Pruv 0.25 0.125 Total 100 50 Core size and shape {fraction (3/16)}″ Round SC - Process:
- The same process for Example 23 is used to prepare the core of Example 36.
- In Examples 37-39, prednisolone tablets were prepared having a core formulation as described in Example 21 and coating formulation as described in Example 3. The tablet formulations of Examples 37-39 are listed in Table 37 below:
TABLE 37 Ex. 37 Ex. 38 Ex. 39 Component amt. (mg) amt. (mg) amt. (mg) 1. Example 21 core 50 50 50 2. Example 3 coating 150 250 350 Total tablet weight 200 300 400 Size and shape of tablet {fraction (3/32)}″ round ⅜″ round ⅜″ round standard standard standard concave concave concave Compression force 8-9 kP 8-9 kP 8-9 kP - Process:
- 1. Weigh out the requisite amount of immediate release cores and set aside.
- 2. Blend 0.75% by weight of Pruv® sodium stearyl fumarate, NF, (commercially available from the Edward Mendell Co., Inc.) with the requisite amount of coating for 5 minutes
- 3. Weigh out approximately half of the compression coating.
- 4. Pour the lower layer of the compression coating into the lower punch of the die.
- 5. Place the immediate release core in the center of the compression coating.
- 6. Pour the top layer of into the die.
- 7. Rotate the punch station for compression.
- 8. Weigh out finished tablets to ensure proper weight.
- The tablets of Examples 37-39 were tested using USP apparatus type III with 250 mL DI water at 15 dips per minute (dpm) giving the following results listed in Table 38:
TABLE 38 Time (hours) 0 2 4 6 8 10 12 16 Example 37 0 0 0 0 18.6 — — — % dissolved Example 38 0 0 0 0 0 35.6 100 100 % dissolved Example 39 0 0 0 0 0 0 6.8 100 % dissolved - The tablets of Examples 37-39 resulted the following lag times (time when prednisolone is release from the tablet) and full release times (time when all of prednisolone is released from the tablet) listed in Table 39:
TABLE 39 Example 37 Example 38 Example 39 Lag time (hours) 6-7 8-9 10-12 Full release (hours) 8-9 10-12 14-16 - In Examples 40-42, prednisolone tablets were prepared having a core formulation as described in Example 21 and coating formulation as described Example 2. The tablet formulations of Examples 40-42 are listed in Table 40 below:
TABLE 40 Ex. 40 Ex. 41 Ex. 42 Component Amt. (mg) amt. (mg) amt. (mg) 1. Example 21 core 50 50 50 2. Example 2 coating 150 250 350 Total tablet weight 200 300 400 Size and shape of tablet {fraction (9/32)}″ round ⅜″ round ⅜″ round standard standard standard concave concave concave Compression force 8-9 kP 8-9 kP 8-9 kP - Process:
- The tablets of Examples 40-42 are prepared using the same process as examples 37-39.
- The tablets of Examples 40-42 were tested using USP apparatus type III with 250 mL DI water at 15 dips per minute (dpm) giving the following results listed in Table 41:
TABLE 41 Time (hours) 0 2 4 6 8 10 12 16 Example 40 0 — 22.7 40.6 100 — — — % dissolved Example 41 0 0 0 0 0 0 100 100 % dissolved Example 42 0 0 0 0 0 0 19.5 19.5 % dissolved - The tablets of Examples 40-42 resulted in the following lag times (time when prednisolone is release from the tablet) and full release times (time when all of prednisolone is released from the tablet) listed in Table 42:
TABLE 42 Example 40 Example 41 Example 42 Lag time 6-7 10-11 10-11 (hours) Full release 7-8 11-12 11-12 (hours) - As can be seen, as total tablet weight increases (due to increase in coating weight), lag time and the corresponding release time also tend to increase.
- In Example 43, various delayed release coating formulations were prepared in order to determine the effect of the gum percentage in the coating formulation on the time of release and the rate of release of the active agent within the tablet core.
- The ingredients of the various delayed release coating granulations of this example having varying gum percentages are as follows:
TABLE 43a Ex. 2 Coating Ex. 3 coating Ex. 8 coating Xanthan Formulation: % mg/tab % Mg/tab % mg/tab % mg/tab 1. a. Core (Ex. 24 core) — — — — 22.73% 50 22.73% 50 b. Core (Ex. 22 core) 22.73% 50 22.73% 50 — — — — 2. Ex. 2 coating 76.69% 168.725 — — — — — — 3. Ex. 3 coating — — 76.69% 168.725 — — — — 4. Ex. 8 coating — — — — 76.69% 168.725 — — 5. Xanthan Gum — — — — — — 76.69% 168.7 6. Sodium Stearyl Fumarate 0.58% 1.275 0.58% 1.275 0.58% 1.275 0.58% 1.275 Tablet weight (mg) 220 220 220 220 Tablet hardness (kP) 8-9 8-9 8-9 8-9 - The effects of the different percentages of gums within the delayed release compression coating are set forth in the table below.
TABLE 43b Xanthan Gum coating Ex. 8 coating Ex. 2 coating Ex. 3 coating 220 mg tablet 220 mg tablet 220 mg tablet 220 mg tablet Time Normalized Time Normalized Time Normalized Time Normalized (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean 0 0.0 0 0.0 0 0.0 0 0.0 1 0.0 1 28.4 4 0.0 2 0.0 2 0.0 2 100.0 6 0.0 4 0.0 3 0.0 3 100.0 8 0.0 6 33.3 4 66.7 4 100.0 10 0.0 8 50.0 5 100.0 5 100.0 12 33.3 10 100.0 6 100.0 6 100.0 16 100.0 12 100.0 - As shown in the Table 43b, the formulation with 20% gums released the active drug faster than did the formulations with 30% or 50% gums. The results followed the rand order for % gums with granulated delayed release examples. Xanthan gum (ungranulated) did not track (e.g., provide the same delayed release) as with the other delayed release coatings in this example (which had granulated gums).
- As can be seen from the results set forth above, as the amount of gum with respect to drug in the formulation is increased, a corresponding increase in lag time before release of the drug is observed.
- In Example 44, various examples of delayed release compression coating formulations were prepared in order to determine the effect of ratio of the drug within the tablet to the gum within the coating formulation on the time of release and the rate of release of the active agent within the tablet core.
- The ingredients of the various delayed release coating granulations are shown in the examples above. The effects of the different drug to gum ratios are set forth in the table below.
TABLE 44 Xanthan Gum Xanthan Gum Ex. 8 coating coating coating Ex. 2 coating Ex. 3 coating 220 mg tablet 220 mg tablet 300 mg tablet 220 mg tablet 220 mg tablet Time Normalized Time Normalized Time Normalized Time Normalized Time Normalized (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 1 28.4 1 0.0 3 0.0 4 0.0 2 0.0 2 100.0 2 0.0 4 0.0 6 0.0 4 0.0 3 100.0 3 0.0 5 38.8 8 0.0 6 33.3 4 100.0 4 66.7 6 100.0 10 0.0 8 50.0 5 100.0 5 100.0 7 100.0 12 33.3 10 100.0 6 100.0 6 100.0 8 100.0 16 100.0 12 100.0 - In this Example, the gums of the Ex. 8 coating (Drug:Gum ratio of 1:33.75) showed faster release time than both Ex. 3 coating (Drug:Gum ratio of 1:50.6) and Ex. 2 coating (Drug:Gum ratio of 1:84.4). Xanthan gum followed rank order with itself (e.g., the larger tablet having more total gum had a longer delay) but not with the granulated materials (Ex. 8, 2, and 3 coatings). It was observed that, as the amount of gum relative to drug is increased, a corresponding increase in lag time is observed. The conclusion reached is that increasing gum to drug ratio increased (longer) release lag time before release of the drug.
- In Example 45, various lots of delayed release coating formulations were prepared in order to determine the effect of the thickness of the sustained release coating on the time of release and the rate of release of the active agent within the tablet core.
- The ingredients of the various inner core formulations are shown in the examples above, and the ingredients of the various delayed release coating granulations are shown in Table 45. The effects of the delayed release coating thickness are set forth in the table below.
TABLE 45 Xanthan Gum Xanthan Gum Ex. 3 coating Ex. 3 coating Ex. 2 coating coating coating Ex. 8 coating Ex. 8 coating 300 mg tablet 200 mg tablet 300 mg tablet 220 mg tablet 300 mg tablet 220 mg tablet 300 mg tablet Time Normal- Time Normal- Time Normal- Time Normal- Time Normal- Time Normal- Time Normal- (hours) ized Mean (hours) ized Mean (hours) ized Mean (hours) ized Mean (hours) ized Mean (hours) ized Mean (hours) ized Mean 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 4 100.0 4 49.0 4 0.0 1 0.0 3 0.0 1 28.4 1 0.0 6 100.0 6 100.0 6 0.0 2 0.0 4 0.0 2 100.0 2 50.0 8 100.0 8 100.0 8 17.9 3 0.0 5 38.8 3 100.0 3 100.0 10 100.0 10 100.0 10 51.7 4 66.7 6 100.0 4 100.0 4 100.0 12 100.0 12 100.0 12 100.0 5 100.0 7 100.0 5 100.0 5 100.0 16 100.0 16 100.0 16 100.0 6 100.0 8 100.0 6 100.0 6 100.0 - In this example, it was observed that tablets with 170 mg of Example 8 coating were faster releasing than tablets with 250 mg Example 8 coating. It was thus observed that, the thickness of coating is increased in the tablet, a corresponding increase in lag time is observed. The conclusion reached is that tablets with a thicker coating showed a longer lag time before release of the drug.
- In Example 46, the effect of the addition of an extragranular excipient(s) to the delayed release coating of Example 2 was measured. In this example, the types of excipient added were Microcrystalline cellulose, Polyvinylpyrrolidone and Polyethylene glycol, and these excipients were added in levels of 0, 5% and 10%.
- The ingredients of the inner core formulation of this Example is shown in Example 24 above, and the ingredients of the various delayed release coating granulation of this Example are shown in Example 2 above. The amounts or percentages of extragranular excipients added are set forth in Tables 46a and 46b below:
TABLE 46a addition of addition of Control 5% PVP 10% PVP Formulation: % mg/tab % mg/tab % mg/tab Core (Ex. 24) 22.72% 50 22.73% 50 22.73% 50 Delayed release coating (Ex. 2) 76.69% 168.725 73% 160.225 68.97% 151.725 Polyvinylpyrolidone K-30 Na na 3.86% 8.5 7.73% 17 Polyethylene Glycol, 6000 Na na na na Na na Microcrystalline Cellulose Na na na na Na na Sodium Stearyl Fumarate 0.58% 1.275 0.58% 1.275 0.58% 1.275 Tablet weight (mg) 220 220 220 Tablet hardness (kP) 8-9 8-9 8-9 -
TABLE 46b addition of addition of addition of addition of 5% PEG 10% PEG 5% MCC 10% MCC Formulation: % mg/tab % mg/tab % mg/tab % mg/tab Core (Ex. 24) 22.73% 50 22.73% 50 22.73% 50 22.73% 50 Delayed release coating 73% 160.225 68.97% 151.725 73% 160.225 68.97% 151.725 (Ex. 2) Polyvinylpyrolidone K-30 Na na Na na na na na na Polyethylene Glycol, 6000 3.86% 8.5 0.077272 17 na na na na Microcrystalline Cellulose Na na Na na 3.86% 8.5 7.73% 17 Sodium Stearyl Fumarate 0.58% 1.275 0.58% 1.275 0.58% 1.275 0.58% 1.275 Tablet weight (mg) 220 220 220 220 Tablet hardness (kP) 8-9 8-9 8-9 8-9 - The effects of the addition of extragranular excipient to the sustained release coating are set forth in the table below.
TABLE 46c Addition of 5% Addition of 10% Addition of 5% Addition of 10% Addition of 5% Addition of 10% Control PVP PVP MCC MCC PEG PEG 220 mg tablet 220 mg tablet 220 mg tablet 220 mg tablet 220 mg tablet 220 mg tablet 220 mg tablet Time Normalized Time Normalized Time Normalized Time Normalized Time Normalized Time Normalized Time Normalized (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean (hrs) Mean 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 0 0.0 4 0.0 2 0.0 2 0.0 8 0.0 2 0.0 8 0.0 8 0.0 6 0.0 4 0.0 4 0.0 9 0.0 4 0.0 9 1.8 9 0.0 8 0.0 6 0.0 6 0.0 10 0.0 6 0.0 10 1.8 10 22.1 10 0.0 8 0.0 8 0.0 11 0.0 8 0.0 11 41.8 11 83.3 12 33.3 10 0.0 10 16.7 12 0.0 10 0.0 12 61.8 12 100.0 16 100.0 12 16.7 14 100.0 14 42.4 14 100.0 - In this example, it was observed that the addition of 5% and 10% Polyethylene Glycol 6000 served to slightly speed the release of active agent. Tablets with 5% PVP K-30 also show slightly shorter lag time compared to the control. The release speed and lag time of tablets with 10% PVP K-30 were unchanged. Tablets made with 10% MCC showed no change in lag time, although they were slower with 5% MCC. The conclusion reached is that lag time can be varied by the addition of extragranular additives into the sustained release coating.
- In Example 47, a scaled up production of the sustained release coating was done in order to determine whether tablets produced at production scale exhibit release profiles similar to those of tablets produced in laboratory scale.
- In this example, the core-coated tablets were produced on a production press at Elizabeth Hata. A HT-AP44MSU-C 44 station core coating press was used to press the tablets. The inner cores of the tablets were made with a ¼″ round flat face with a beveled edge, tableted to 170 mg and 8-10 kP. The final tablets were made 9 mm round concave, and were tableted to 525 and 560 mg, 8-10 kP.
- The blends for the production scale were composed as follows:
Inner Core blend: Outer Coating blend: 2% Prednisolone Example 2 w/0.75% sodium stearyl 40% Prosolv SMCC 50 fumarate 47.75% Prosolv SMCC 90 6% Croscarmellose Sodium 2% Sodium Starch Glycolate 2% Talc 0.25% Sodium Stearyl Fumarate - In production of the tablets, the press speed varied from 9 to 12 rpm.
- The effects of the scaled up production of the delayed release coating are shown in the data points for which are set forth in Table 47 below.
TABLE 47 Production scale Production Scale 525 mg tablet weight 560 mg tablet weight Time Normalized Time Normalized (hours) Mean (hours) Mean 0.0 0 0.0 13.5 23.7 12 10.1 14 38.2 13 10.7 14.5 65.0 14 35.4 15 72.9 15 65.0 15.5 85.7 16 79.0 16 100.0 17 100.0 - In this example, it was observed that tablets produced at production scale exhibit similar release profiles to tablets produced at laboratory scale. Accordingly, the conclusion reached is that formulation and production technology can be successfully scaled up.
- In Examples 48-57, prednisolone tablets were prepared having core formulations with different amounts of disintegrants and coating formulations as described Example 10. Each tablet had the same core weight, same coating weight, and the same total tablet weight. The tablet formulations of Examples 48-57 are listed in Tables 48 and 49 below:
TABLE 48 Component Ex. 48 Ex. 49 Ex. 50 Ex. 51 Ex. 52 Core formulation Ex. 25 core Ex. 28 core Ex. 26 core Ex. 27 core Ex. 31 core used Disintegrant none 2% Ac-Di-Sol 3% Explotab 3% Explotab and 6% Ac-Di-Sol 2% Ac-Di-Sol amt. (mg) amt. (mg) amt. (mg) amt. (mg) amt. (mg) 1. Core 50 50 50 50 50 2. Example 10 450 450 450 450 450 coating Total tablet 500 500 500 500 500 weight -
TABLE 49 Component Ex. 53 Ex. 54 Ex. 55 Ex. 56 Ex. 57 Core Ex. 29 core Ex. 29 core Ex. 31 core Ex. 30 core Ex. 32 core formulation used Disintegrant 2% Explotab V17 2% Explotab 6% Ac-Di-Sol 20% Explotab 20% Explotab and 6% V17 and 6% Ac-Di-Sol Ac-Di-Sol amt. (mg) amt. (mg) Amt. (mg) amt. (mg) amt. (mg) 1. Core 50 50 50 50 50 2. Example 450 450 450 450 450 10 coating Total tablet 500 500 500 500 500 weight - The tablets of Examples 48-57 were tested using USP dissolution apparatus type III with 250 mL DI water at 15 dips per minute (dpm) giving the following results listed in Table 50:
TABLE 50 Time (hours) 0 3 4 4.5 5 5.5 6 6.5 7 8 Example 48 0 0 0 — 0 — 8.3 — 12 12 % dissolved Example 49 0 0 0 — 73.7 — 100 — 100 100 % dissolved Example 50 0 0 0 — 78.3 — 100 — 100 100 % dissolved Example 51 0 0 0 — 14.9 — 100 — 100 100 % dissolved Example 52 0 0 0 — 0 — 65.8 — 100 100 % dissolved Example 53 0 0 0 — 2.7 — 96.8 — 100 100 % dissolved Example 54 0 — 9.2 56.9 100 100 100 100 — — % dissolved Example 55 0 — 17.3 50.6 64.6 66.7 100 100 — — % dissolved Example 56 0 0 0 0 14.9 40.8 60.6 — — — % dissolved Example 57 0 0 0 — 0 — 66.7 — 100 100 % dissolved - The results indicated that the inclusion of a disintegrant in the core can lead to a more rapid release of the active agent from the formulation.
- In Examples 58-60, prednisolone tablets were prepared having core formulations of Example 24 and coating formulations as described Examples 2, 4, and a combination Examples 2 and 4 (25% of Example 2 and 75% of Example 4). Each tablet had the same core weight, same coating weight, and the same total tablet weight. The tablet formulations of Examples 58-60 are listed in Table 51 below:
TABLE 51 Ex. 58 Ex. 59 Ex. 60 Component amt. (mg) Amt. (mg) amt. (mg) 1. Core (Ex. 24) 50 50 50 2. Example 2 250 — — coating 3. Example 4 — 250 — coating 4. Coating — — 250 consisting of 25% of Example 2 and 75% of Example 4 coatings Total tablet 300 300 300 weight - Tablets having the formulations described in Examples 58-60 were subjected to dissolution testing using the USP apparatus type 3 with 250 ml DI water at 15 dips per minute. The results are set forth in Table 52 below.
TABLE 52 Ex. 58 Ex. 59 Ex. 60 Time % Time % Time % (hr) Dissolved (hr) Dissolved (hr) Dissolved 0 0 0 0 0 0 8 3 2 0 4 0.3 10 6.9 3 0 6 4 12 23.8 4 59.4 8 39.6 14 52.3 5 99.6 10 99.4 16 99 6 100 12 100 20 100 7 100 14 100 - In Example 61, prednisolone tablets having the formulations described in Examples 59 were subjected to dissolution variations in pH, ionic strength, and dip rates. The pH evaluated was 1.5, 7.5 and pH change.
- The pH change method uses increasing pH from one dissolution vessel to the next to simulate the transport of the dosage form through the gastrointestinal tract. Initially the pH is 1.5 for 1 hour. The pH of the second station is 3.5 for two additional hours and when the third station is 5.5 for an additional 2 hours. Finally the last three stations are at pH 7.5. The time length for the last three stations can vary depending on the expected release for the dosage form.
- The results are set forth in Tables 53 and 54 below. The results indicate that the dissolution profiles for a formulation prepared in accordance with Example 59 where the dissolution media pH and ionic strength were varied.
TABLE 53 pH 1.5 pH 7.5 pH 7.5 pH 7.5 pH Change Time (0.25 M) (0.01 M) (0.1 M) (0.25 M) (0.1 M) 0 0 0 0 0 0 1 0 0 0 0 0 3 9.3 98.1 5.9 3 0 5 20.8 100 28.9 8.8 0 10 44.6 100 88.1 42.8 11.2 15 87.9 100 92 93.9 70 20 100 100 100 95 100 -
TABLE 54 Time pH Change No Ions 0 0 1 0 3 0 5 0 7 7.7 9 17.7 11 100 - The results set forth in Tables 55 and 56 below provide the normalized mean dissolution profiles for a formulation prepared in accordance with Example 59 using the pH change method (0.1M) at 15 and 30 dpm, respectively.
TABLE 55 Normalized mean values using pH change method (0.1 M) at 15 dpm Normalized Normalized Normalized Mean SD % CV 0.0 0.0 #DIV/0! 0.0 0.0 #DIV/0! 0.0 0.0 #DIV/0! 0.0 0.0 #DIV/0! 11.2 0.7 6.0 70.0 36.7 52.4 100.0 0.0 0.0 -
TABLE 56 Normalized mean values using pH change method (0.1 M) at 30 dpm Normalized Normalized Normalized Mean SD % CV 0.0 0.0 #DIV/0! 0.0 0.0 #DIV/0! 0.0 0.0 #DIV/0! 16.7 40.8 244.9 100.0 0.0 0.0 100.0 0.0 0.0 100.0 0.0 0.0 - In Example 62, a 2 mg prednisolone core composition was prepared similarly to Example 24, increasing the amount of prednisolone in the core and decreasing the amount of Prosolve SMCC 90, and having the following formulation listed in Table 57:
TABLE 57 Component Percent amt. (mg) 1. Prednisolone 4 2.0 2. Prosolv SMCC ™ 50 40 20.0 3. Prosolv SMCC ™ 90 45.75 22.875 4. Explotab ® 6 3.0 5. Sodium carboxymethylcellulose 2 1.0 6. Talc 2 1.0 7. Pruv 0.25 0.125 Total 100 50 Core size and shape 3/16 Round SC - In Examples 63-68, the core formulation of Example 62 was coated with coatings prepared in accordance with Examples 11, 12, 13, 14, 15, and 16. The formulations of Examples are listed in Table 58 below:
TABLE 58 Ex. 63 Ex. 64 Ex. 65 Ex. 66 Ex. 67 Ex. 68 amt. amt. amt. amt. amt. amt. Component (mg) (mg) (mg) (mg) (mg) (mg) 1. Core 50 50 50 50 50 50 (Ex. 62) 2. Ex. 11 250 — — — — — coating 3. Ex. 12 — 250 — — — — coating 4. Ex. 13 — — 250 — — — coating 5. Ex. 14 — — — 250 — — coating 6. Ex. 15 — — — — 250 — coating 7. Ex. 16 — — — — — 250 coating Total tablet 300 300 300 300 300 300 weight - Dissolution testing was done on each formulation using USP apparatus 3 with 250 ml of media and 15 dpm. Two dissolution methods using different media (1) DI water and (2) pH change were performed. Table 59 provides the DI water dissolution results, and Table 60 provides the pH change (0.1M) dissolution results.
TABLE 59 Time Ex. 63 Ex. 64 Ex. 65 Ex. 66 Ex. 67 Ex. 68 (hr.) (30% Gums) (22.5% Gums) (20% Gums) (15% Gums) (10% Gums) (7.5% Gums) 0 0 0 0 0 0 0 1 0 1.2 0 0 0 0 1.5 0 33.4 0 30.8 64.5 98.8 2 30.1 98.6 64.5 99.3 98.4 100 2.5 98.7 100 99.5 100 100 100 3 100 100 100 100 100 100 3.5 100 100 100 100 100 100 -
TABLE 60 Time Ex. 63 Ex. 64 Ex. 65 Ex. 66 Ex. 67 Ex. 68 (hr.) (30% Gums) (22.5% Gums) (20% Gums) (15% Gums) (10% Gums) (7.5% Gums) 0 0 0 0 0 0 0 1 0 0.8 0 0 0 3.2 3 5.8 1 0 0.8 0.7 98.9 5 6.7 3.4 3.3 5.6 91.3 100 8 10.8 19.4 11 96.8 100 100 11 15.7 37.6 69.5 100 100 100 14 67.8 93 100 100 100 100 - Analysis of the data allows for the approximation of the lag time based a linear fit of the data obtained. The data demonstrates the lag time can be varied from 0 to 8 hours depending on gum level in the formulation. Table 61 is a summary of the Example and gum ratio (%) used and the approximated lag time before release.
TABLE 61 Approximated Lag Time Before Release Example Number Gum Ratio (%) Lag Time (Hours) 68 7.5 0.7 67 10.0 1.7 66 15.0 3.6 65 20.0 5.0 64 22.5 7.4 63 30.0 9.2 - In Example 69, other formulations were prepared and tested using USP apparatus Type, 3, with 250 ml of the dissolution media and dips per minute as indicated in the Table 62.
- The particular dissolution media are defined as follows:
DI water: USP purified water; pH change NI (“no ion”) pH change method as described in Example 61, or pH change: without the use of ions to change adjust the pH; pH change (0.1 M): pH change method as described in Example 61 with the use of salts to give an ionic strength of 0.1 molar; pH 7.5: dissolution media having a pH of 7.5; pH 7.5 (0.1 M): dissolution media having a pH of 7.5 and ionic strength of 0.1 M; SGI: simulated gastric fluid; Peanut oil pH 7.5: peanut oil with a pH of 7.5; - Other dissolution media indicated would be readily understood by those skilled in the art (e.g, pH 1.5:dissolution media having a pH of 1.5, etc.) in view of the above.
- With respect to certain excipients indicated in the comments section, these excipients have been added to the compression coating prior to coating the cores.
TABLE 62 





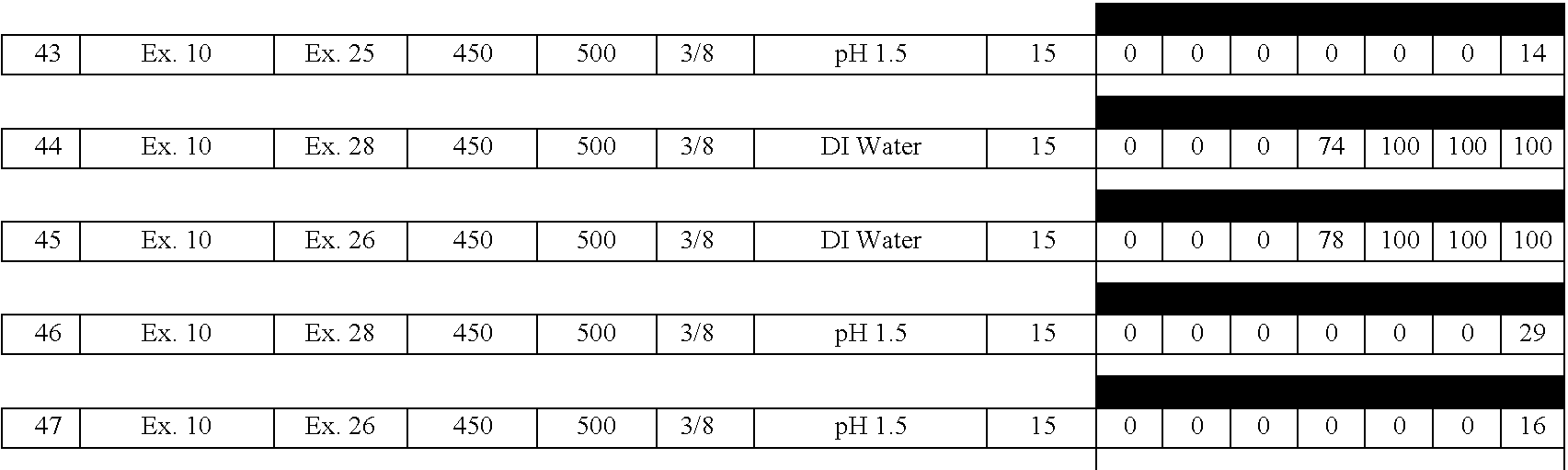
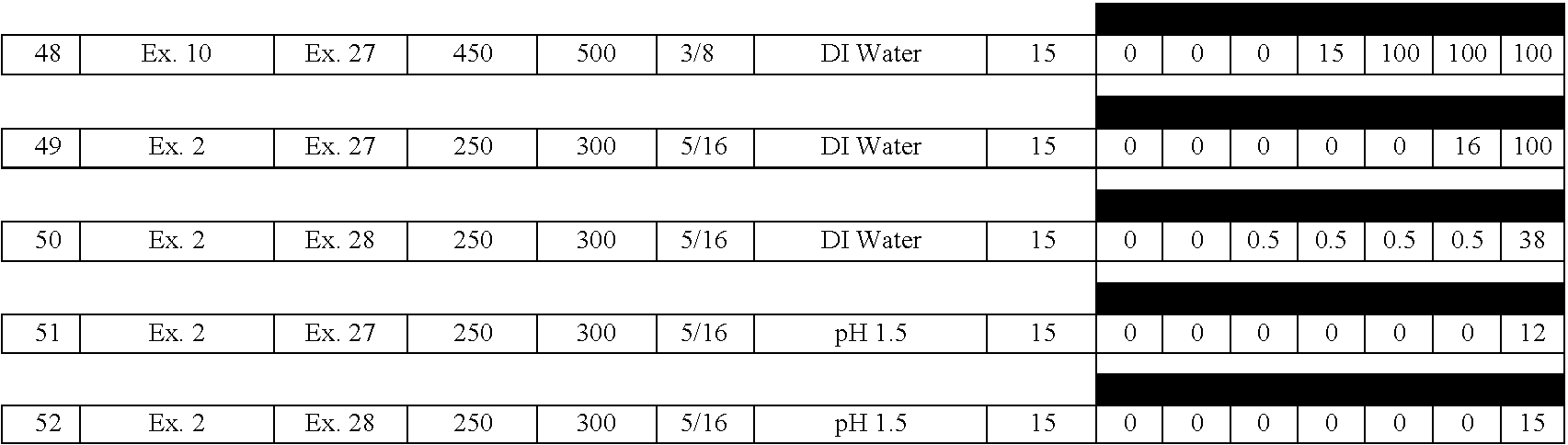





















- The examples provided above are not meant to be exclusive. Many other variations of the present invention would be obvious to those skilled in the art, and are contemplated to be within the scope of the appended claims.
Claims (53)
1. A delayed release oral solid dosage form comprising:
a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto said core, said delayed release material comprising one or more natural or synthetic gums, said compression coating delaying the release of said drug from said dosage form until after a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
2. The delayed release oral solid dosage form of claim 1 , wherein said glucocorticosteroid drug is selected from the group consisting of prednisolone, prednisone, cortisone, hydrocortisone, methylprednisolone, betametasone, dexamethasone, triamcinolone, pharmaceutically acceptable salts thereof, and mixtures thereof.
3. The delayed release oral solid dosage form of claim 1 , wherein said glucocorticosteroid drug is prednisolone.
4. The delayed release oral solid dosage form of claim 1 , wherein said one or more natural or synthetic gums are agglomerated with a saccharide material prior to being compression coated onto said core.
5. The delayed release oral solid dosage form of claim 1 , which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to an aqueous solution.
6. The delayed release oral solid dosage form of claim 1 , wherein said gums comprise a mixture of xanthan gum and locust bean gum.
7. The delayed release oral solid dosage form of claim 1 , wherein said delayed release material further comprises an ionizable gel strength enhancing agent selected from the group consisting of calcium sulfate, sodium chloride, potassium sulfate, sodium carbonate, lithium chloride, tripotassium phosphate, sodium borate, potassium bromide, potassium fluoride, sodium bicarbonate, calcium chloride, magnesium chloride, sodium citrate, sodium acetate, calcium lactate, magnesium sulfate, sodium fluoride, and mixtures thereof.
8. The delayed release oral solid dosage form of claim 7 , wherein said ionizable gel strength enhancing agent is calcium sulfate.
9. The delayed release oral solid dosage form of claim 1 , wherein said delayed release material further comprises a surfactant selected from the group consisting of anionic surfactants, cationic surfactants, amphoteric (amphipathic/amphophilic) surfactants, and non-ionic surfactants.
10. The delayed release oral solid dosage form of claim 1 , wherein said delayed release material further comprises a hydrophobic material.
11. The delayed release oral solid dosage form of claim 10 , wherein said hydrophobic material is selected from the group consisting of an alkylcellulose, a copolymer of acrylic and methacrylic acid esters, waxes, shellac, zein, hydrogenated vegetable oil, and mixtures thereof, in an amount effective to slow the hydration of said gelling agent when exposed to an environmental fluid.
12. The delayed release oral solid dosage form of claim 11 , wherein said hydrophobic material is ethylcellulose.
13. The delayed release oral solid dosage form of claim 4 , wherein said saccharide is selected from the group consisting of sucrose, dextrose, lactose, fructose, mannitol, and mixtures thereof.
14. The delayed release oral solid dosage form of claim 1 , wherein said core further comprises from about 5 to about 20 percent disintegrant, by weight.
15. The delayed release oral solid dosage form of claim 14 , wherein said disintegrant is selected from the group consisting of starch, veegum, crospovidone, cellulose, kaolin, microcrystalline cellulose, crosslinked polyvinyl pyrrolidone, and mixtures thereof.
16. The delayed release oral solid dosage form of claim 14 , wherein said disintegrant is selected from the group consisting of croscarmellose sodium, crospovidone, crosslinked carboxy methyl cellulose, sodium starch glycolate, and mixtures thereof.
17. The delayed release oral solid dosage form of claim 1 , wherein said inner core further comprises an inert diluent selected from the group consisting of sucrose, dextrose, lactose, microcrystalline cellulose, fructose, xylitol, sorbitol, mannitol, starches, mixtures thereof.
18. The delayed release oral solid dosage form of claim 1 , wherein said inner core is an immediate release core.
19. The delayed release oral solid dosage form of claim 1 , wherein said inner core is further comprises a sustained release carrier.
20. A delayed release oral solid dosage form comprising:
a core comprising a therapeutically effective amount of a glucocorticosteroid drug, and an agglomerated delayed release material compression coated onto said core, said agglomerated delayed release material comprising a gum selected from the group consisting of a homopolysaccharide, a heteropolysaccharide, and a mixture of a homopolysaccharide and a heteropolysaccharide, together with a pharmaceutically acceptable excipient, said compression coating delaying the release of said drug from said dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution.
21. The delayed release oral solid dosage form of claim 1 , wherein said heteropolysaccharide gum is in an amount of from about 20 to about 80 percent of the delayed release coating and said homopolysaccharide gum is in an amount of from about 80 to about 20 percent of the delayed release coating.
22. The delayed release oral solid dosage form of claim 21 , wherein said heteropolysaccharide gum is xanthan gum and said homopolysaccharide gum is locust bean gum.
23. The delayed release oral solid dosage form of claim 20 , wherein said glucocorticosteroid is prednisolone.
24. The delayed release oral solid dosage form of claim 20 , wherein said core further comprises an effective amount of disintegrant.
25. A delayed release oral solid dosage form comprising:
a core comprising a therapeutically effective amount of a glucocorticosteroid drug and an effective amount of a disintegrant, and an agglomerated delayed release material compression coated onto said core, said agglomerated delayed release material consisting essentially of one or more natural or synthetic pharmaceutically acceptable gums and one or more optional pharmaceutically acceptable excipients, said compression coating delaying the release of said drug from said dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution.
26. The delayed release oral solid dosage form of claim 26 , wherein said core further comprises from about 5 to about 20 percent disintegrant, by weight.
27. The delayed release oral solid dosage form of claim 27 , wherein said disintegrant is selected from the group consisting of starch, veegum, crospovidone, cellulose, kaolin, microcrystalline cellulose, crosslinked polyvinyl pyrrolidone, and mixtures thereof.
28. The delayed release oral solid dosage form of claim 27 , wherein said disintegrant is selected from the group consisting of croscarmellose sodium, crospovidone, crosslinked carboxy methyl cellulose, sodium starch glycolate, and mixtures thereof.
29. The delayed release oral solid dosage form of claim 25 which delays the release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to an aqueous solution.
30. The delayed release oral solid dosage form of claim 25 , wherein said gums comprise a mixture of xanthan gum and locust bean gum.
31. A delayed release oral solid dosage form comprising:
a core comprising a therapeutically effective amount of a glucocorticosteroid drug and a disintegrant, and a delayed release material compression coated onto said core, said delayed release material comprising one or more natural or synthetic gums, said compression coating delaying the release of said drug from said dosage form for a predetermined period of time after exposure of the dosage form to an aqueous solution, said disintegrant being included in said core in an amount effective to cause the release of at least about 50 percent of said drug into said aqueous solution within one hour after said predetermined period of time.
32. The delayed release oral solid dosage form of claim 31 , wherein said disintegrant comprises from about 5 to about 20 percent of said core, by weight.
33. The delayed release oral solid dosage form of claim 27 , wherein said disintegrant comprises from about about 0.1 to about 5 percent of said oral solid dosage form, by weight.
34. The delayed release oral solid dosage form of claim 32 , wherein said disintegrant is a superdisintegrant.
35. The delayed release oral solid dosage form of claim 34 , wherein said superdisintegrant is selected from the group consisting of croscarmellose sodium, crospovidone, crosslinked carboxy methyl cellulose, sodium starch glycolate, and mixtures thereof.
36. The delayed release oral solid dosage form of claim 34 , wherein said gums comprise a mixture of xanthan gum and locust bean gum.
37. The delayed release oral solid dosage form of claim 36 , wherein said xanthan gum and said locust bean gum are agglomerated with a saccharide material prior to compression coating onto said core.
38. A delayed release oral solid tablet, comprising:
a tablet core comprising a therapeutically effective amount of a glucocorticosteroid drug, and a delayed release material compression coated onto said core, said delayed release material comprising one or more natural or synthetic gums, said gums comprising from about 6.5 percent to about 83 percent of the tablet by weight, said compression coating delaying the release of said drug from said dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
39. The delayed release oral solid tablet of claim 38 , wherein said tablet core further comprises from about 5 to about 20% superdisintegrant.
40. A chronotherapeutic, delayed release oral solid dosage form comprising a core comprising from about 0.01 mg to about 40 mg glucocorticosteroid, and a delayed release material compression coated onto said core, said delayed release material comprising one or more natural or synthetic gums, said compression coating comprising from about 75 to about 94 percent by weight of the oral solid dosage form, and the ratio of the core to gum in said compression coating being from about 1:0.37 to about 1:5, by weight, said compression coating delaying the release of said glucocorticosteroid from said dosage form for a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
41. A method of preparing a chronotherapeutic oral solid dosage form of a glueocorticosteroid, comprising:
preparing a core comprising a therapeutically effective amount of a glucocorticosteroid and from about 5 to about 20% disintegrant, by weight of the core,
preparing a granulate of a delayed release material comprising one or more natural or synthetic gums,
compression coating said granulate onto said core, said compression coating delaying the release of said glucocorticosteroid from said dosage form until after a period of time from about 2 to about 18 hours after exposure of the dosage form to an aqueous solution.
42. The method of claim 41 , further comprising preparing said granulate of delayed release material by wet granulating one or more natural or synthetic gums together with at least one pharmaceutically acceptable excipient, and drying the resultant granulate to obtain agglomerated particles of said delayed release material.
43. The method of claim 42 , further comprising granulating said glucocorticosteroid, said disintegrant, and a pharmaceutically acceptable inert diluent prior to said compression coating step.
44. The method of claim 43 , wherein said disintegrant is a superdisintegrant incorporated into said core in an amount said disintegrant being included in said core in an amount effective to cause the release of at least about 50 percent of said glucocorticosteroid into said aqueous solution within one hour upon completion of the time period for said delayed release.
45. A method of treating early morning inflammation or arthritis comprising: orally administering to a human at bedtime an oral solid dosage comprising a core comprising a therapeutically effective amount of a glucocorticosteroid; and a delayed release compression coating on said core comprising one or more natural or synthetic pharmaceutically acceptable gums; said compression coating delaying the release of said glucocortosteroid from said dosage form for a time period until at least about 4 hours after oral administration.
46. The method of claim 46 , wherein after oral administration of the dosage form, the glucocorticosteroid is not released from the dosage form for about 4 to about 12 hours.
47. The method of claim 46 , wherein after oral administration of the dosage form, the glucocorticosteroid is released over a time period of at least about 4 hours after the period of delay is completed.
48. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 1 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
49. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 20 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
50. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 25 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
51. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 31 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
52. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 38 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
53. A method of chronotherapeutically treating arthritis, comprising orally administering the oral solid dosage form of claim 40 to a human or animal at bedtime, which delays release of said glucocorticosteroid until at least about 4 hours after exposure of the dosage form to gastrointestinal fluid.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/099,462 US20030190360A1 (en) | 2001-03-13 | 2002-03-13 | Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US27538201P | 2001-03-13 | 2001-03-13 | |
| US10/099,462 US20030190360A1 (en) | 2001-03-13 | 2002-03-13 | Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20030190360A1 true US20030190360A1 (en) | 2003-10-09 |
Family
ID=23052055
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/099,462 Abandoned US20030190360A1 (en) | 2001-03-13 | 2002-03-13 | Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment |
| US10/099,461 Abandoned US20030082230A1 (en) | 2001-03-13 | 2002-03-13 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
| US11/001,675 Expired - Fee Related US7887841B2 (en) | 2001-03-13 | 2004-12-01 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
| US12/985,137 Abandoned US20110217336A1 (en) | 2001-03-13 | 2011-01-05 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/099,461 Abandoned US20030082230A1 (en) | 2001-03-13 | 2002-03-13 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
| US11/001,675 Expired - Fee Related US7887841B2 (en) | 2001-03-13 | 2004-12-01 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
| US12/985,137 Abandoned US20110217336A1 (en) | 2001-03-13 | 2011-01-05 | Chronotherapeutic dosage forms and methods of treatment using chronotherapy |
Country Status (15)
| Country | Link |
|---|---|
| US (4) | US20030190360A1 (en) |
| EP (2) | EP1368005B9 (en) |
| JP (3) | JP5005157B2 (en) |
| KR (1) | KR20040047747A (en) |
| CN (1) | CN1499960A (en) |
| AP (1) | AP1748A (en) |
| AU (2) | AU2002244295B2 (en) |
| CA (2) | CA2440641A1 (en) |
| EA (1) | EA008224B1 (en) |
| ES (1) | ES2436523T3 (en) |
| HU (1) | HUP0501071A3 (en) |
| IL (3) | IL157633A0 (en) |
| MX (2) | MXPA03008293A (en) |
| NZ (1) | NZ527832A (en) |
| WO (2) | WO2002072033A2 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050048114A1 (en) * | 2003-08-29 | 2005-03-03 | Burnside Beth A. | Antibiotic product, use and formulation thereof |
| US20050112201A1 (en) * | 2003-09-19 | 2005-05-26 | Penwest Pharmaceuticals Co. | Delayed release dosage forms |
| US20060003001A1 (en) * | 2004-02-11 | 2006-01-05 | John Devane | Chronotherapeutic compositions and methods of their use |
| US20060024368A1 (en) * | 2004-07-30 | 2006-02-02 | Reza Fassihi | Compressed composite delivery system for release-rate modulation of bioactives |
| US20060147511A1 (en) * | 2002-11-24 | 2006-07-06 | Steffen Panzner | Liposomal glucocorticoids |
| US8062672B2 (en) | 2003-08-12 | 2011-11-22 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US20120020951A1 (en) * | 2010-07-20 | 2012-01-26 | Harold Michael Shepard | Adverse side-effects associated with administration of anti-hyaluronan agents and methods for ameliorating or preventing the side-effects |
| US8299052B2 (en) | 2006-05-05 | 2012-10-30 | Shionogi Inc. | Pharmaceutical compositions and methods for improved bacterial eradication |
| US8303988B2 (en) | 2000-10-13 | 2012-11-06 | Shionogi Inc. | Antifungal once-a-day product, use and formulation thereof |
| US8313776B2 (en) | 2003-07-21 | 2012-11-20 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8313775B2 (en) | 2003-07-21 | 2012-11-20 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| RU2471479C1 (en) * | 2012-02-13 | 2013-01-10 | Общество с ограниченной ответственностью "Агентство редких лекарств и технологий" | Tableted pharmaceutical composition containing dexamethasone as active ingredient for oral administration, and method for preparing it |
| US8357394B2 (en) | 2005-12-08 | 2013-01-22 | Shionogi Inc. | Compositions and methods for improved efficacy of penicillin-type antibiotics |
| US8425936B2 (en) | 2003-07-21 | 2013-04-23 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8460710B2 (en) | 2003-09-15 | 2013-06-11 | Shionogi, Inc. | Antibiotic product, use and formulation thereof |
| US8715727B2 (en) | 2004-07-02 | 2014-05-06 | Shionogi Inc. | Tablet for pulsed delivery |
| US8758820B2 (en) | 2003-08-11 | 2014-06-24 | Shionogi Inc. | Robust pellet |
| US8778924B2 (en) | 2006-12-04 | 2014-07-15 | Shionogi Inc. | Modified release amoxicillin products |
| US8889187B2 (en) | 2000-02-24 | 2014-11-18 | Shionogi Inc. | Once a day amoxicillin product comprising immediate and delayed release dosage forms |
Families Citing this family (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030190360A1 (en) * | 2001-03-13 | 2003-10-09 | Baichwal Anand R. | Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment |
| US20030068375A1 (en) | 2001-08-06 | 2003-04-10 | Curtis Wright | Pharmaceutical formulation containing gelling agent |
| GB0128674D0 (en) * | 2001-11-30 | 2002-01-23 | Boots Co Plc | Treatment of sleep disorders and the like |
| CN1777412A (en) * | 2003-04-24 | 2006-05-24 | 佳高泰克有限公司 | Delayed release tablet with defined core geometry |
| ES2335008T3 (en) * | 2003-04-24 | 2010-03-18 | Jagotec Ag | COMPRESSED WITH COLORED NUCLEUS. |
| US20050118267A1 (en) * | 2003-09-19 | 2005-06-02 | Penwest Pharmaceuticals Co. | Chronotherapeutic dosage forms |
| US20050238731A1 (en) * | 2003-12-29 | 2005-10-27 | Stephen Holt | Composition and method for treating the effects of diseases and maladies of the upper digestive tract |
| US20050202079A1 (en) * | 2004-03-15 | 2005-09-15 | Mylan Pharmaceuticals Inc. | Novel orally administrable formulation of nitrofurantoin and a method for preparing said formulation |
| CA2563609C (en) | 2004-04-22 | 2012-11-06 | Duocort Ab | Pharmaceutical compositions for acute glucocorticoid therapy |
| SE0401031D0 (en) * | 2004-04-22 | 2004-04-22 | Duocort Ab | A new glucocorticoid replacement therapy |
| ES2653568T3 (en) | 2004-06-12 | 2018-02-07 | Collegium Pharmaceutical, Inc. | Drug formulations for abuse prevention |
| US20060002999A1 (en) * | 2004-06-17 | 2006-01-05 | Forest Laboratories, Inc. | Immediate release formulations of 1-aminocyclohexane compounds, memantine and neramexane |
| AU2011232809B2 (en) * | 2004-09-10 | 2014-03-20 | Jagotec Ag | Tablets with site and time-controlled gastrointestinal release of active ingredient |
| KR101363563B1 (en) * | 2004-09-10 | 2014-02-14 | 자고텍 아게 | Tablets with site and time-controlled gastrointestinal release of active ingredient |
| DE102004043863A1 (en) * | 2004-09-10 | 2006-03-16 | Nitec Pharma Ag | Tablets with local and time-controlled drug release in the gastrointestinal tract |
| GB0423964D0 (en) * | 2004-10-28 | 2004-12-01 | Jagotec Ag | Dosage form |
| US20090215852A1 (en) * | 2005-03-21 | 2009-08-27 | Chroma Group, Inc. | Compositions and methods for ameliorating cachexia |
| US20080033027A1 (en) * | 2005-03-21 | 2008-02-07 | Vicus Therapeutics Spe 1, Llc | Drug combination pharmaceutical compositions and methods for using them |
| US20070036859A1 (en) * | 2005-08-11 | 2007-02-15 | Perry Ronald L | Sustained release antihistamine and decongestant composition |
| TWI274889B (en) * | 2005-10-06 | 2007-03-01 | Elan Microelectronics Corp | Resistive touch screen measurement system |
| CA2630704C (en) * | 2005-12-20 | 2014-08-19 | Lek Pharmaceuticals D.D. | Pharmaceutical composition |
| SG174026A1 (en) | 2006-08-03 | 2011-09-29 | Nitec Pharma Ag | Delayed-release glucocorticoid treatment of rheumatoid disease |
| US20080081067A1 (en) * | 2006-10-03 | 2008-04-03 | Gupta Manishkumar | Sustained release pharmaceutical compositions of venlafaxine and process for preparation thereof |
| US20080152709A1 (en) * | 2006-12-22 | 2008-06-26 | Drugtech Corporation | Clonidine composition and method of use |
| US8969514B2 (en) | 2007-06-04 | 2015-03-03 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| CA2688161C (en) | 2007-06-04 | 2020-10-20 | Kunwar Shailubhai | Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| US8299122B2 (en) * | 2008-04-14 | 2012-10-30 | Skinvisible Pharmaceuticals, Inc. | Method for stabilizing retinoic acid, retinoic acid containing composition, and method of using a retinoic acid containing composition |
| ES2522968T3 (en) | 2008-06-04 | 2014-11-19 | Synergy Pharmaceuticals Inc. | Guanylate cyclase agonists useful for the treatment of gastrointestinal disorders, inflammation, cancer and other disorders |
| ES2624828T3 (en) | 2008-07-16 | 2017-07-17 | Synergy Pharmaceuticals Inc. | Guanylate cyclase agonists useful for the treatment of gastrointestinal disorders, inflammation, cancer and others |
| EP2374460A4 (en) * | 2008-12-12 | 2013-08-21 | Rhein Siegfried Sa De Cv | Pulsed-release sildenafil composition and method for preparing said composition |
| AU2010211985A1 (en) * | 2009-01-22 | 2011-08-11 | Abbott Healthcare Private Limited | Chronotherapeutic pharmaceutical composition |
| CA2749646A1 (en) | 2009-01-26 | 2010-07-29 | Nitec Pharma Ag | Delayed-release glucocorticoid treatment of asthma |
| CN102883601A (en) | 2009-12-02 | 2013-01-16 | 阿达玛斯医药公司 | Amantadine compositions and methods of use |
| US10668060B2 (en) | 2009-12-10 | 2020-06-02 | Collegium Pharmaceutical, Inc. | Tamper-resistant pharmaceutical compositions of opioids and other drugs |
| RU2574018C2 (en) * | 2010-03-31 | 2016-01-27 | Мотида Фармасьютикал Ко., Лтд. | Easily dosed solid medicinal preparation |
| US9907767B2 (en) | 2010-08-03 | 2018-03-06 | Velicept Therapeutics, Inc. | Pharmaceutical compositions and the treatment of overactive bladder |
| US9616097B2 (en) | 2010-09-15 | 2017-04-11 | Synergy Pharmaceuticals, Inc. | Formulations of guanylate cyclase C agonists and methods of use |
| US8623409B1 (en) | 2010-10-20 | 2014-01-07 | Tris Pharma Inc. | Clonidine formulation |
| CN103370058A (en) | 2010-12-22 | 2013-10-23 | 普渡制药公司 | Encased tamper resistant controlled release dosage forms |
| CN107412173A (en) | 2010-12-23 | 2017-12-01 | 普渡制药公司 | Anti-distort solid oral dosage form |
| JP5976657B2 (en) | 2011-09-30 | 2016-08-24 | 持田製薬株式会社 | Easy-to-use solid preparation |
| GB201202433D0 (en) * | 2012-02-13 | 2012-03-28 | Diurnal Ltd | Controlled drug release |
| US9693962B2 (en) * | 2012-06-05 | 2017-07-04 | Takeda Pharmaceutical Limited | Dry-coated tablet |
| NZ747657A (en) | 2012-11-02 | 2019-12-20 | Murray And Poole Entpr Ltd | Treatment or prevention of cardiovascular events via the administration of a colchicine derivative |
| MX2015010041A (en) | 2013-02-05 | 2015-10-30 | Purdue Pharma Lp | Tamper resistant pharmaceutical formulations. |
| WO2014131024A2 (en) | 2013-02-25 | 2014-08-28 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| CA2902665C (en) * | 2013-02-28 | 2021-03-02 | Lupin Limited | Pharmaceutical compositions of donepezil having specific in vitro dissolution profile or pharmacokinetics parameters |
| EP2961393B1 (en) * | 2013-02-28 | 2020-10-28 | Lupin Limited | Pharmaceutical compositions of donepezil having specific in vitro dissolution profile or pharmacokinetics parameters |
| EP2970384A1 (en) | 2013-03-15 | 2016-01-20 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| US10751287B2 (en) | 2013-03-15 | 2020-08-25 | Purdue Pharma L.P. | Tamper resistant pharmaceutical formulations |
| US9486494B2 (en) | 2013-03-15 | 2016-11-08 | Synergy Pharmaceuticals, Inc. | Compositions useful for the treatment of gastrointestinal disorders |
| MY198452A (en) | 2013-04-16 | 2023-08-29 | Murray And Poole Enterprises Ltd | Sustained-release formulations of colchicine and methods of using same |
| US10154971B2 (en) | 2013-06-17 | 2018-12-18 | Adamas Pharma, Llc | Methods of administering amantadine |
| EP3041463A1 (en) | 2013-09-02 | 2016-07-13 | Sun Pharmaceutical Industries Ltd | Pulsatile-release dosage form |
| EP3054969B1 (en) | 2013-10-10 | 2021-03-10 | Bausch Health Ireland Limited | Agonists of guanylate cyclase useful for the treatment of opioid induced dysfunctions |
| WO2016073179A2 (en) * | 2014-10-23 | 2016-05-12 | The Trustees Of The University Of Pennsylvania | Novel chronotherapy based on circadian rhythms |
| KR20170086659A (en) * | 2014-12-03 | 2017-07-26 | 벨리셉트 테라퓨틱스, 인크. | Compositions and methods of using modified release solabegron for lower urinary tract symptoms |
| FI3365321T3 (en) | 2015-10-23 | 2024-01-02 | B3Ar Therapeutics Inc | Solabegron zwitterion and uses thereof |
| CN108884130B (en) | 2016-01-11 | 2022-09-13 | 博士医疗爱尔兰有限公司 | Formulations and methods for treating ulcerative colitis |
| WO2017210700A1 (en) * | 2016-06-03 | 2017-12-07 | Velicept Therapeutics, Inc. | Compositions and methods of using modified release solabegron for lower urinary tract symptoms |
| US9737530B1 (en) | 2016-06-23 | 2017-08-22 | Collegium Pharmaceutical, Inc. | Process of making stable abuse-deterrent oral formulations |
| CN207648424U (en) | 2017-07-17 | 2018-07-24 | 手持产品公司 | Equipment holding mechanism |
| AU2018320946A1 (en) | 2017-08-24 | 2020-04-16 | Adamas Pharma, Llc | Amantadine compositions, preparations thereof, and methods of use |
| JP7467884B2 (en) * | 2019-10-29 | 2024-04-16 | オムロンヘルスケア株式会社 | Sphygmomanometer, method of operating the same, and program |
| US11918689B1 (en) | 2020-07-28 | 2024-03-05 | Tris Pharma Inc | Liquid clonidine extended release composition |
| CN114588124B (en) * | 2020-12-07 | 2023-10-20 | 江苏恒瑞医药股份有限公司 | Delayed release pharmaceutical composition |
| CA3201720A1 (en) | 2021-01-15 | 2022-07-21 | Galventa Ag | Pulsatile release caffeine formulation |
| CN114712319B (en) * | 2022-03-25 | 2024-01-09 | 北京诺康达医药科技股份有限公司 | Felodipine and propranolol hydrochloride compound preparation and preparation method thereof |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4853249A (en) * | 1985-11-15 | 1989-08-01 | Taisho Pharmaceutical Co., Ltd. | Method of preparing sustained-release pharmaceutical/preparation |
| US4933186A (en) * | 1987-08-11 | 1990-06-12 | Bayer Aktiengesellschaft | Dihydropyridine depot formulation |
| US5077051A (en) * | 1990-04-10 | 1991-12-31 | Warner-Lambert Company | Sustained release of active agents from bioadhesive microcapsules |
| US5302400A (en) * | 1992-06-22 | 1994-04-12 | Digestive Care Inc. | Preparation of gastric acid-resistant microspheres containing digestive enzymes and buffered-bile acids |
| US5316772A (en) * | 1990-12-19 | 1994-05-31 | Solvay & Cie, S.A. (Societe Anonyme) | Bilayered oral pharmaceutical composition with pH dependent release |
| US5378474A (en) * | 1989-01-06 | 1995-01-03 | F. H. Faulding & Co. Limited | Sustained release pharmaceutical composition |
| US5395626A (en) * | 1994-03-23 | 1995-03-07 | Ortho Pharmaceutical Corporation | Multilayered controlled release pharmaceutical dosage form |
| US5468746A (en) * | 1988-07-29 | 1995-11-21 | Zambon Group S.P.A. | Compounds active on the cardiovascular system |
| US5482718A (en) * | 1994-03-23 | 1996-01-09 | Hoffmann-La Roche Inc. | Colon-targeted delivery system |
| US5713852A (en) * | 1995-06-07 | 1998-02-03 | Alza Corporation | Oral dosage and method for treating painful conditions of the oral cavity |
| US5811388A (en) * | 1995-06-07 | 1998-09-22 | Cibus Pharmaceutical, Inc. | Delivery of drugs to the lower GI tract |
| US5858412A (en) * | 1995-01-09 | 1999-01-12 | Edward Mendell Co., Inc. | Sustained release formulations utilizing pharmaceutical excipient having improved compressibility with modified microcrystalline |
| US6024982A (en) * | 1993-11-23 | 2000-02-15 | Euro-Celtique, S.A. | Immediate release tablet cores of insoluble drugs having sustained-release coating |
| US6190692B1 (en) * | 1997-01-29 | 2001-02-20 | Cesare Busetti | Time-specific controlled release capsule formulations and method of preparing same |
| US6500459B1 (en) * | 1999-07-21 | 2002-12-31 | Harinderpal Chhabra | Controlled onset and sustained release dosage forms and the preparation thereof |
Family Cites Families (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3131123A (en) | 1959-03-13 | 1964-04-28 | Lab Francais De Therapeutique | Enteric tablets and manufacture thereof |
| US4248858A (en) | 1979-08-09 | 1981-02-03 | American Home Products Corporation | Sustained release pharmaceutical compositions |
| FR2471186A1 (en) | 1979-12-10 | 1981-06-19 | Roussel Uclaf | NEW COLIC DELITESCENCE TABLETS AND THEIR PREPARATION PROCESS |
| US4851231A (en) | 1982-12-13 | 1989-07-25 | Alza Corporation | System for delivering drug in selected environment of use |
| US4894240A (en) | 1983-12-22 | 1990-01-16 | Elan Corporation Plc | Controlled absorption diltiazem formulation for once-daily administration |
| SE457505B (en) | 1984-01-10 | 1989-01-09 | Lejus Medical Ab | LAMINATED ORAL PHARMACEUTICAL COMPOSITION AND PROCEDURES FOR ITS PREPARATION |
| DK62184D0 (en) | 1984-02-10 | 1984-02-10 | Benzon As Alfred | DIFFUSION COATED POLYDEPOT PREPARATION |
| GB8518301D0 (en) | 1985-07-19 | 1985-08-29 | Fujisawa Pharmaceutical Co | Hydrodynamically explosive systems |
| IL77186A0 (en) | 1985-11-29 | 1986-04-29 | Touitou Elka | Pharmaceutical insulin composition |
| IT1188212B (en) | 1985-12-20 | 1988-01-07 | Paolo Colombo | SYSTEM FOR THE RELEASE SPEED OF ACTIVE SUBSTANCES |
| JPS62195323A (en) | 1986-02-24 | 1987-08-28 | Eisai Co Ltd | Gastric resident particle |
| US4904476A (en) | 1986-03-04 | 1990-02-27 | American Home Products Corporation | Formulations providing three distinct releases |
| IT1204294B (en) | 1986-03-11 | 1989-03-01 | Gentili Ist Spa | METHOD OF MANUFACTURE OF GRANULARS SUITABLE FOR THE PRODUCTION OF COATED TABLETS, FOR ORAL USE, WITH CONTROLLED RELEASE |
| US5238686A (en) | 1986-03-27 | 1993-08-24 | Kinaform Technology, Inc. | Sustained-release pharmaceutical preparation |
| GB2189698A (en) | 1986-04-30 | 1987-11-04 | Haessle Ab | Coated omeprazole tablets |
| GB2189699A (en) | 1986-04-30 | 1987-11-04 | Haessle Ab | Coated acid-labile medicaments |
| US4842867A (en) | 1986-05-09 | 1989-06-27 | Alza Corporation | Pulsed drug delivery of doxylamine |
| IE58401B1 (en) | 1986-06-20 | 1993-09-08 | Elan Corp Plc | Controlled absorption pharmaceutical composition |
| GB8628359D0 (en) | 1986-11-27 | 1986-12-31 | Zyma Sa | Galenical formulation |
| SE460945B (en) | 1987-01-15 | 1989-12-11 | Lejus Medical Ab | A MULTIPLE-UNIT DOS COMPOSITION OF FUROSEMID |
| GB8702411D0 (en) | 1987-02-03 | 1987-03-11 | Zyma Sa | Swellable pellets |
| GB8717168D0 (en) | 1987-07-21 | 1987-08-26 | Roussel Lab Ltd | Controlled-release device |
| US4891223A (en) | 1987-09-03 | 1990-01-02 | Air Products And Chemicals, Inc. | Controlled release delivery coating formulation for bioactive substances |
| US4994276A (en) | 1988-09-19 | 1991-02-19 | Edward Mendell Co., Inc. | Directly compressible sustained release excipient |
| US4841231A (en) | 1987-10-30 | 1989-06-20 | Unisys Corporation | Test probe accessibility method and tool |
| US4971805A (en) | 1987-12-23 | 1990-11-20 | Teysan Pharmaceuticals Co., Ltd. | Slow-releasing granules and long acting mixed granules comprising the same |
| JPH0768125B2 (en) | 1988-05-18 | 1995-07-26 | エーザイ株式会社 | Oral formulation of acid labile compounds |
| US4857337A (en) | 1988-05-24 | 1989-08-15 | American Home Products Corp. (Del) | Enteric coated aspirin tablets |
| US5047246A (en) * | 1988-09-09 | 1991-09-10 | Bristol-Myers Company | Direct compression cyclophosphamide tablet |
| US5169639A (en) | 1988-09-19 | 1992-12-08 | Edward Mendell Co., Inc. | Controlled release verapamil tablets |
| US5128143A (en) | 1988-09-19 | 1992-07-07 | Edward Mendell Co., Inc. | Sustained release excipient and tablet formulation |
| US5135757A (en) | 1988-09-19 | 1992-08-04 | Edward Mendell Co., Inc. | Compressible sustained release solid dosage forms |
| US5334372A (en) | 1988-10-07 | 1994-08-02 | Kao Corporation | Alcohol-modified silicon ester derivative and cosmetic composition containing same |
| IT1230576B (en) | 1988-10-20 | 1991-10-28 | Angeli Inst Spa | ORAL PHARMACEUTICAL FORMULATIONS WITH SELECTIVE LIBERATION IN THE COLON |
| IT1227899B (en) | 1988-12-23 | 1991-05-14 | Poli Ind Chimica Spa | TOTAL OR PARTIAL COATING OF PHARMACEUTICAL ACTIVE SUBSTANCES AND RELATED COMPOSITIONS |
| US5032406A (en) | 1989-02-21 | 1991-07-16 | Norwich Eaton Pharmaceuticals, Inc. | Dual-action tablet |
| US5007790A (en) | 1989-04-11 | 1991-04-16 | Depomed Systems, Inc. | Sustained-release oral drug dosage form |
| US5133974A (en) | 1989-05-05 | 1992-07-28 | Kv Pharmaceutical Company | Extended release pharmaceutical formulations |
| PH27186A (en) | 1989-09-07 | 1993-04-16 | Ciba Geigy Ag | Double-coated granules of disodium pamidronate |
| GB8926639D0 (en) | 1989-11-24 | 1990-01-17 | Agricultural & Food Res | Delayed release formulations |
| EP0441119A3 (en) | 1990-01-09 | 1992-10-14 | Richard D. Levere | The use of l-arginine in the treatment of hypertension and other vascular disorders |
| US5198227A (en) | 1990-01-22 | 1993-03-30 | Mcneil-Ppc, Inc. | Dual subcoated simulated capsule medicament |
| US5229131A (en) | 1990-02-05 | 1993-07-20 | University Of Michigan | Pulsatile drug delivery system |
| US5158777A (en) | 1990-02-16 | 1992-10-27 | E. R. Squibb & Sons, Inc. | Captopril formulation providing increased duration of activity |
| IT1241417B (en) | 1990-03-06 | 1994-01-14 | Vectorpharma Int | THERAPEUTIC COMPOSITIONS WITH CONTROLLED RELEASE OF DRUGS SUPPORTED ON CROSS-LINKED POLYMERS AND COATED WITH POLYMER FILM, AND THEIR PREPARATION PROCESS |
| US5175003A (en) | 1990-04-06 | 1992-12-29 | Biosytes Usa, Inc. | Dual mechanism controlled release system for drug dosage forms |
| IT1246382B (en) | 1990-04-17 | 1994-11-18 | Eurand Int | METHOD FOR THE TARGETED AND CONTROLLED DELIVERY OF DRUGS IN THE INTESTINE AND PARTICULARLY IN THE COLON |
| IE61651B1 (en) | 1990-07-04 | 1994-11-16 | Zambon Spa | Programmed release oral solid pharmaceutical dosage form |
| JP2773959B2 (en) | 1990-07-10 | 1998-07-09 | 信越化学工業株式会社 | Colon release solid preparation |
| US5202338A (en) | 1990-10-31 | 1993-04-13 | Vilmos Bar | Dihydroquinoline derivatives, pharmaceutical compositions and methods of use of dihydroquinoline derivatives as modulators of the arachidonic acid cascade |
| US5232706A (en) | 1990-12-31 | 1993-08-03 | Esteve Quimica, S.A. | Oral pharmaceutical preparation containing omeprazol |
| US5286497A (en) | 1991-05-20 | 1994-02-15 | Carderm Capital L.P. | Diltiazem formulation |
| US5252338A (en) | 1991-06-27 | 1993-10-12 | Alza Corporation | Therapy delayed |
| US5190765A (en) | 1991-06-27 | 1993-03-02 | Alza Corporation | Therapy delayed |
| IT1251153B (en) * | 1991-08-06 | 1995-05-04 | Vectorpharma Int | SOLID PHARMACEUTICAL COMPOSITIONS FOR ORAL ADMINISTRATION HAVING PROHIBITED GASTRIC RESIDENCE |
| US5234947A (en) | 1991-11-07 | 1993-08-10 | New York University | Potassium channel activating compounds and methods of use thereof |
| IT1260505B (en) | 1992-06-01 | 1996-04-09 | Poli Ind Chimica Spa | ORAL PHARMACEUTICAL SYSTEMS WITH DELAYED DELIVERY FOR THE SPECIFIC RELEASE IN THE COLON |
| US5262172A (en) | 1992-06-19 | 1993-11-16 | Digestive Care Inc. | Compositions of gastric acid-resistant microspheres containing buffered bile acids |
| NL9201195A (en) | 1992-07-03 | 1994-02-01 | Tno | PREPARATION FOR THE REGULATED DELIVERY OF AN ACTIVE SUBSTANCE AND METHOD FOR PREPARING SUCH A PREPARATION. |
| US5472711A (en) | 1992-07-30 | 1995-12-05 | Edward Mendell Co., Inc. | Agglomerated hydrophilic complexes with multi-phasic release characteristics |
| US5260069A (en) | 1992-11-27 | 1993-11-09 | Anda Sr Pharmaceuticals Inc. | Pulsatile particles drug delivery system |
| US5330761A (en) | 1993-01-29 | 1994-07-19 | Edward Mendell Co. Inc. | Bioadhesive tablet for non-systemic use products |
| US5650156A (en) | 1993-02-22 | 1997-07-22 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of nutriceuticals and compositions useful therefor |
| JPH072650A (en) | 1993-06-18 | 1995-01-06 | Tanabe Seiyaku Co Ltd | Release part control type preparation |
| WO1995003052A1 (en) | 1993-07-22 | 1995-02-02 | Warner-Lambert Company | Controlled release tacrine drug delivery systems and methods for preparing same |
| US5455046A (en) * | 1993-09-09 | 1995-10-03 | Edward Mendell Co., Inc. | Sustained release heterodisperse hydrogel systems for insoluble drugs |
| US5662933A (en) * | 1993-09-09 | 1997-09-02 | Edward Mendell Co., Inc. | Controlled release formulation (albuterol) |
| US5773025A (en) | 1993-09-09 | 1998-06-30 | Edward Mendell Co., Inc. | Sustained release heterodisperse hydrogel systems--amorphous drugs |
| US5399358A (en) * | 1993-11-12 | 1995-03-21 | Edward Mendell Co., Inc. | Sustained release formulations for 24 hour release of metroprolol |
| US5407687A (en) | 1994-02-22 | 1995-04-18 | Glaxo Inc. | Ranitidine solid dosage form |
| US5399359A (en) * | 1994-03-04 | 1995-03-21 | Edward Mendell Co., Inc. | Controlled release oxybutynin formulations |
| US5374759A (en) | 1994-03-22 | 1994-12-20 | Siltech Inc. | Silicone esters of hydroxy acid |
| US5399362A (en) | 1994-04-25 | 1995-03-21 | Edward Mendell Co., Inc. | Once-a-day metoprolol oral dosage form |
| US5464633A (en) | 1994-05-24 | 1995-11-07 | Jagotec Ag | Pharmaceutical tablets releasing the active substance after a definite period of time |
| US5958458A (en) * | 1994-06-15 | 1999-09-28 | Dumex-Alpharma A/S | Pharmaceutical multiple unit particulate formulation in the form of coated cores |
| US5536507A (en) | 1994-06-24 | 1996-07-16 | Bristol-Myers Squibb Company | Colonic drug delivery system |
| GB9416600D0 (en) | 1994-08-17 | 1994-10-12 | Smithkline Beecham Plc | Pharmaceutical formulation |
| US6103263A (en) | 1994-11-17 | 2000-08-15 | Andrx Pharmaceuticals, Inc. | Delayed pulse release hydrogel matrix tablet |
| US5585115A (en) | 1995-01-09 | 1996-12-17 | Edward H. Mendell Co., Inc. | Pharmaceutical excipient having improved compressability |
| US5612053A (en) | 1995-04-07 | 1997-03-18 | Edward Mendell Co., Inc. | Controlled release insufflation carrier for medicaments |
| US5837284A (en) | 1995-12-04 | 1998-11-17 | Mehta; Atul M. | Delivery of multiple doses of medications |
| AU2068797A (en) | 1996-01-29 | 1997-08-20 | Edward Mendell Co. Inc. | Sustained release excipient |
| US6156340A (en) | 1996-03-29 | 2000-12-05 | Duquesne University Of The Holy Ghost | Orally administrable time release drug containing products |
| JP3148256B2 (en) | 1996-07-08 | 2001-03-19 | エドワード メンデル カンパニー.,インコーポレーテッド | Sustained release matrix for high dose poorly soluble drugs |
| US5792476A (en) | 1996-12-19 | 1998-08-11 | Abigo Medical Ab | Sustained release glucocorticoid pharmaceutical composition |
| US5788987A (en) | 1997-01-29 | 1998-08-04 | Poli Industria Chimica Spa | Methods for treating early morning pathologies |
| US5922352A (en) | 1997-01-31 | 1999-07-13 | Andrx Pharmaceuticals, Inc. | Once daily calcium channel blocker tablet having a delayed release core |
| DE19709532A1 (en) * | 1997-03-10 | 1998-09-17 | Basf Ag | Use of redispersible polymer powders or polymer granules for coating pharmaceutical or agrochemical dosage forms |
| US6210710B1 (en) * | 1997-04-28 | 2001-04-03 | Hercules Incorporated | Sustained release polymer blend for pharmaceutical applications |
| US5840329A (en) | 1997-05-15 | 1998-11-24 | Bioadvances Llc | Pulsatile drug delivery system |
| US5958873A (en) | 1997-06-09 | 1999-09-28 | University Of Cincinnati | Oral formulation for treatment of bacteria-induced diseases of the colon |
| IN186245B (en) | 1997-09-19 | 2001-07-14 | Ranbaxy Lab Ltd | |
| US6056977A (en) | 1997-10-15 | 2000-05-02 | Edward Mendell Co., Inc. | Once-a-day controlled release sulfonylurea formulation |
| US6372254B1 (en) | 1998-04-02 | 2002-04-16 | Impax Pharmaceuticals Inc. | Press coated, pulsatile drug delivery system suitable for oral administration |
| US20010055613A1 (en) | 1998-10-21 | 2001-12-27 | Beth A. Burnside | Oral pulsed dose drug delivery system |
| DK1126826T6 (en) * | 1998-11-02 | 2019-06-24 | Alkermes Pharma Ireland Ltd | Multiparticulate modified release of methylphenidate |
| EP1064938A1 (en) | 1999-06-28 | 2001-01-03 | Sanofi-Synthelabo | Pharmaceutical dosage forms for controlled release producing at least a timed pulse |
| US20030190360A1 (en) * | 2001-03-13 | 2003-10-09 | Baichwal Anand R. | Chronotherapeutic dosage forms containing glucocorticosteroid and methods of treatment |
-
2002
- 2002-03-13 US US10/099,462 patent/US20030190360A1/en not_active Abandoned
- 2002-03-13 JP JP2002570994A patent/JP5005157B2/en not_active Expired - Fee Related
- 2002-03-13 CA CA002440641A patent/CA2440641A1/en not_active Abandoned
- 2002-03-13 US US10/099,461 patent/US20030082230A1/en not_active Abandoned
- 2002-03-13 WO PCT/US2002/007935 patent/WO2002072033A2/en active Application Filing
- 2002-03-13 EP EP02721430.3A patent/EP1368005B9/en not_active Expired - Lifetime
- 2002-03-13 AP APAP/P/2003/002865A patent/AP1748A/en active
- 2002-03-13 IL IL15763302A patent/IL157633A0/en unknown
- 2002-03-13 AU AU2002244295A patent/AU2002244295B2/en not_active Ceased
- 2002-03-13 IL IL15763402A patent/IL157634A0/en unknown
- 2002-03-13 KR KR10-2003-7011643A patent/KR20040047747A/en not_active Application Discontinuation
- 2002-03-13 HU HU0501071A patent/HUP0501071A3/en unknown
- 2002-03-13 MX MXPA03008293A patent/MXPA03008293A/en active IP Right Grant
- 2002-03-13 CN CNA028072324A patent/CN1499960A/en active Pending
- 2002-03-13 EP EP02709836A patent/EP1368000A4/en not_active Withdrawn
- 2002-03-13 ES ES02721430.3T patent/ES2436523T3/en not_active Expired - Lifetime
- 2002-03-13 NZ NZ527832A patent/NZ527832A/en unknown
- 2002-03-13 EA EA200300999A patent/EA008224B1/en unknown
- 2002-03-13 MX MXPA03008292A patent/MXPA03008292A/en unknown
- 2002-03-13 JP JP2002570993A patent/JP2004529902A/en active Pending
- 2002-03-13 WO PCT/US2002/007936 patent/WO2002072034A2/en not_active Application Discontinuation
- 2002-03-13 AU AU2002252364A patent/AU2002252364B2/en not_active Ceased
- 2002-03-13 CA CA2440588A patent/CA2440588C/en not_active Expired - Fee Related
-
2003
- 2003-08-28 IL IL157633A patent/IL157633A/en not_active IP Right Cessation
-
2004
- 2004-12-01 US US11/001,675 patent/US7887841B2/en not_active Expired - Fee Related
-
2009
- 2009-03-31 JP JP2009086332A patent/JP5232062B2/en not_active Expired - Fee Related
-
2011
- 2011-01-05 US US12/985,137 patent/US20110217336A1/en not_active Abandoned
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4853249A (en) * | 1985-11-15 | 1989-08-01 | Taisho Pharmaceutical Co., Ltd. | Method of preparing sustained-release pharmaceutical/preparation |
| US4933186A (en) * | 1987-08-11 | 1990-06-12 | Bayer Aktiengesellschaft | Dihydropyridine depot formulation |
| US5468746A (en) * | 1988-07-29 | 1995-11-21 | Zambon Group S.P.A. | Compounds active on the cardiovascular system |
| US5378474A (en) * | 1989-01-06 | 1995-01-03 | F. H. Faulding & Co. Limited | Sustained release pharmaceutical composition |
| US5077051A (en) * | 1990-04-10 | 1991-12-31 | Warner-Lambert Company | Sustained release of active agents from bioadhesive microcapsules |
| US5316772A (en) * | 1990-12-19 | 1994-05-31 | Solvay & Cie, S.A. (Societe Anonyme) | Bilayered oral pharmaceutical composition with pH dependent release |
| US5302400A (en) * | 1992-06-22 | 1994-04-12 | Digestive Care Inc. | Preparation of gastric acid-resistant microspheres containing digestive enzymes and buffered-bile acids |
| US6024982A (en) * | 1993-11-23 | 2000-02-15 | Euro-Celtique, S.A. | Immediate release tablet cores of insoluble drugs having sustained-release coating |
| US5395626A (en) * | 1994-03-23 | 1995-03-07 | Ortho Pharmaceutical Corporation | Multilayered controlled release pharmaceutical dosage form |
| US5482718A (en) * | 1994-03-23 | 1996-01-09 | Hoffmann-La Roche Inc. | Colon-targeted delivery system |
| US5858412A (en) * | 1995-01-09 | 1999-01-12 | Edward Mendell Co., Inc. | Sustained release formulations utilizing pharmaceutical excipient having improved compressibility with modified microcrystalline |
| US5713852A (en) * | 1995-06-07 | 1998-02-03 | Alza Corporation | Oral dosage and method for treating painful conditions of the oral cavity |
| US5811388A (en) * | 1995-06-07 | 1998-09-22 | Cibus Pharmaceutical, Inc. | Delivery of drugs to the lower GI tract |
| US6190692B1 (en) * | 1997-01-29 | 2001-02-20 | Cesare Busetti | Time-specific controlled release capsule formulations and method of preparing same |
| US6500459B1 (en) * | 1999-07-21 | 2002-12-31 | Harinderpal Chhabra | Controlled onset and sustained release dosage forms and the preparation thereof |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8889187B2 (en) | 2000-02-24 | 2014-11-18 | Shionogi Inc. | Once a day amoxicillin product comprising immediate and delayed release dosage forms |
| US8303988B2 (en) | 2000-10-13 | 2012-11-06 | Shionogi Inc. | Antifungal once-a-day product, use and formulation thereof |
| US20060147511A1 (en) * | 2002-11-24 | 2006-07-06 | Steffen Panzner | Liposomal glucocorticoids |
| US8425936B2 (en) | 2003-07-21 | 2013-04-23 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8313775B2 (en) | 2003-07-21 | 2012-11-20 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8313776B2 (en) | 2003-07-21 | 2012-11-20 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8758820B2 (en) | 2003-08-11 | 2014-06-24 | Shionogi Inc. | Robust pellet |
| US8062672B2 (en) | 2003-08-12 | 2011-11-22 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US9144548B2 (en) | 2003-08-12 | 2015-09-29 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US20050048114A1 (en) * | 2003-08-29 | 2005-03-03 | Burnside Beth A. | Antibiotic product, use and formulation thereof |
| US8246996B2 (en) | 2003-08-29 | 2012-08-21 | Shionogi Inc. | Antibiotic product, use and formulation thereof |
| US8460710B2 (en) | 2003-09-15 | 2013-06-11 | Shionogi, Inc. | Antibiotic product, use and formulation thereof |
| US8389008B2 (en) | 2003-09-19 | 2013-03-05 | Penwest Pharmaceuticals Co. | Delayed release dosage forms |
| US20050112201A1 (en) * | 2003-09-19 | 2005-05-26 | Penwest Pharmaceuticals Co. | Delayed release dosage forms |
| US20060003001A1 (en) * | 2004-02-11 | 2006-01-05 | John Devane | Chronotherapeutic compositions and methods of their use |
| US8715727B2 (en) | 2004-07-02 | 2014-05-06 | Shionogi Inc. | Tablet for pulsed delivery |
| US20060024368A1 (en) * | 2004-07-30 | 2006-02-02 | Reza Fassihi | Compressed composite delivery system for release-rate modulation of bioactives |
| US8357394B2 (en) | 2005-12-08 | 2013-01-22 | Shionogi Inc. | Compositions and methods for improved efficacy of penicillin-type antibiotics |
| US8299052B2 (en) | 2006-05-05 | 2012-10-30 | Shionogi Inc. | Pharmaceutical compositions and methods for improved bacterial eradication |
| US8778924B2 (en) | 2006-12-04 | 2014-07-15 | Shionogi Inc. | Modified release amoxicillin products |
| US20120020951A1 (en) * | 2010-07-20 | 2012-01-26 | Harold Michael Shepard | Adverse side-effects associated with administration of anti-hyaluronan agents and methods for ameliorating or preventing the side-effects |
| KR101809878B1 (en) * | 2010-07-20 | 2017-12-15 | 할로자임, 아이엔씨 | Treatment of the adverse side-effects associated with administration of an anti-hyaluronan agent |
| US9878046B2 (en) * | 2010-07-20 | 2018-01-30 | Halozyme, Inc. | Adverse side-effects associated with administration of an anti-hyaluronan agent and methods for ameliorating or preventing the side-effects |
| US10265410B2 (en) | 2010-07-20 | 2019-04-23 | Halozyme, Inc. | Adverse side-effects associated with administration of an anti-hyaluronan agent and methods for ameliorating or preventing the side-effects |
| RU2471479C1 (en) * | 2012-02-13 | 2013-01-10 | Общество с ограниченной ответственностью "Агентство редких лекарств и технологий" | Tableted pharmaceutical composition containing dexamethasone as active ingredient for oral administration, and method for preparing it |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2002244295B2 (en) | Chronotherapeutic dosage forms containing glucocorticosteroid | |
| AU2002244295A1 (en) | Chronotherapeutic dosage forms containing glucocorticosteroid | |
| US9278076B2 (en) | Chronotherapeutic dosage forms | |
| AU2002252364A1 (en) | Chronotherapeutic dosage forms | |
| US8679535B2 (en) | Sustained release matrix systems for highly soluble drugs | |
| NZ545921A (en) | Delayed released dosage forms | |
| AU2004273956B2 (en) | Delayed release dosage forms | |
| AU2006233161A1 (en) | Sustained Release Matrix Systems for Highly Soluble Drugs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: PENWEST PHARMACEUTICALS COMPANY, NEW YORK Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:BAICHWAL, ANAND R.;WOODCOCK, PAUL;HIGGINS, RAYMOND;AND OTHERS;REEL/FRAME:013387/0711;SIGNING DATES FROM 20020918 TO 20020930 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |