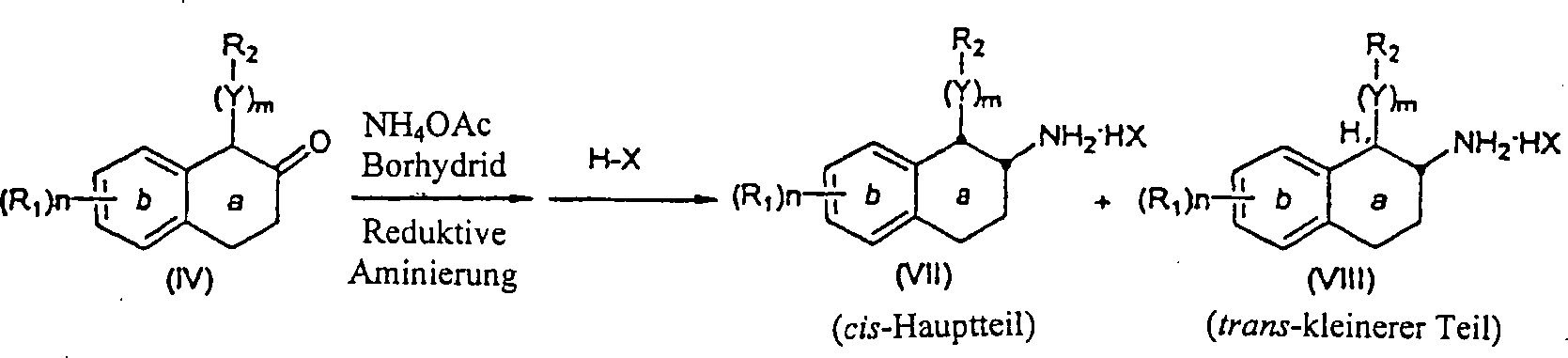-
BEREICH DER
ERFINDUNG
-
Diese
Erfindung betrifft eine Reihe von p-Aminotetralinderivaten, pharmazeutische
Zusammensetzungen, die diese enthalten und bei deren Herstellung
verwendete Intermediate. Die Verbindungen der Erfindung sind Liganden
für den
Neuropeptid Y Y5 (NPY5)-Rezeptor, einem Rezeptor, der mit einer
Zahl von Erkrankungen des zentralen Nervensystems und affektiven
Zuständen
assoziiert ist. Zusätzlich
verringern viele der Verbindungen der Erfindung die Nahrungsaufnahme
in einem Fütterungs-Nagermodell.
-
HINTERGRUND
DER ERFINDUNG
-
Die
Regulation und Funktion des Säuger-zentralen
Nervensystems wird durch eine Reihe von zusammenhängenden
Rezeptoren, Neuronen, Neurotransmittern und Proteinen bestimmt.
Die Neuronen spielen eine vitale Rolle in diesem System, dadurch
daß, wenn
sie extern oder intern stimuliert werden, sie durch die Freisetzung
von Neurotransmittern reagieren, die an spezifische Proteine binden.
Herkömmliche
Beispiel von endogenen Neurotransmittern niedrigen Molekulargewichts,
wie zum Beispiel Acetylcholin, Adrenalin, Norepinephrin, Dopamin,
Serotonin, Glutamat und gamma-Aminobuttersäure sind gut bekannt, genau
wie die spezifischen Rezeptoren, die diese Verbindungen als Liganden
erkennen ("The Biochemical
Basis of Neuropharmacology",
Sixth Edition, Cooper, J. R.; Bloom, F. E.; Roth, R. H. Eds., Oxford
University Press, New York, NY 1991).
-
Zusätzlich zu
den endogenen Neurotransmittern niedrigen Molekulargewichts gibt
es zunehmende Hinweise, daß Neuropeptide
eine integrale Rolle in den neuronalen Funktionen spielen. Von Neuropeptiden wird
nun geglaubt, daß sie
mit vielleicht mehr als einer Hälfte
der 100 Milliarden Neuronen des menschlichen zentralen Nervensystems
co-lokalisiert sind. Zusätzlich
zu Menschen wurden Neuropeptide in einer Zahl von Tierarten entdeckt.
In einigen Fällen
ist die Zusammensetzung dieser Peptide zwischen den Arten bemerkenswert
homogen. Dieses Ergebnis läßt vermuten,
daß die
Funktion von Neuropeptiden vital ist und gegenüber evolutionären Veränderungen
stabil war. Weiterhin werden Neuropeptide, anders als Neurotransmitter
niedrigen Molekulargewichts, typischerweise durch die neuronalen
Ribosomen synthetisiert. In einigen Fällen werden die aktiven Neuropeptide
als Teil eines größeren Proteins
produziert, das enzymatisch prozessiert wird, um die aktive Substanz
zu ergeben. Basierend auf diesen Unterschieden, verglichen mit Neurotransmittern niedrigen
Molekulargewichts, können
Neuropeptid-basierte Strategien neue Therapien für CNS Erkrankungen und Abweichungen
bieten. Genauer sind Mittel, die die Bindung von Neuropeptiden an
ihre jeweiligen Rezeptoren beeinflussen oder Antworten abschwächen, die
durch Neuropeptide vermittelt werden, potentielle Therapien für Erkrankungen,
die mit Neuropeptiden assoziiert sind.
-
Es
gibt eine Zahl von Beeinträchtigungen,
die mit dem komplexen zusammenhängenden
System von Rezeptoren und Liganden innerhalb des zentralen Nervensystems
assoziiert sind; diese schließen
neurodegenerative Erkrankungen, affektive Abweichungen, wie zum
Beispiel Angstzustände,
Depressionen, Schmerzen und Schizophrenie ein, und affektive Zustände die
eine Stoffwechselkomponente einschließen, nämlich Fettsucht. Solche Zustände, Abweichungen
und Erkrankungen wurden mit Substanzen niedrigen Molekulargewichts
und Peptiden behandelt, die die neuronalen Antworten auf endogene
Neurotransmitter modulieren.
-
Ein
Beispiel für
die Klasse der Neuropeptide ist Neuropeptid Y (NPY). NPY wurde zuerst
aus dem Schweinehirn isoliert (Tatemoto, K. et al. Nature 1982,
296,659) und es wurde von ihm gezeigt, daß es zu anderen Mitgliedern
der pankreatischen Polypeptid-(PP)-familie strukturell ähnlich ist,
wie zum Beispiel Peptid YY, das hauptsächlich durch endokrine Zellen
im Darm synthetisiert wird und pankreatisches Polypeptid, das durch
den Pankreas synthetisiert wird. Neuropeptid Y ist ein Einzelpeptid-Protein,
das aus sechsunddreißig Aminosäuren besteht,
wobei es einen amidierten C-Terminus aufweist. Wie andere Mitglieder
der pankreatischen Polypeptidfamilie weist NPY eine distinkte Konformation
auf, die aus einer N-terminalen Polyprolin-helikalen Region und
einer amphiphilischen α-Helix
besteht, die durch a charakteristische PP-Faltung zusammengefügt sind
(Vladimir, S. et. al. Biochemistry 1990, 20, 4509). Weiterhin wurden
NPY Sequenzen aus einer Zahl Tierarten aufgeklärt, und alle zeigen ein hohes
Ausmaß an
Aminosäurehomologie
zu dem menschlichen Protein (> 94%
in Ratte, Hund, Kaninchen, Schwein, Rind, Schaf) (siehe Larhammar,
D. in "The Biology
of Neuropeptid Y und Related Peptides", Colmers, W. F. und Wahlestedt, C.
Eds., Humana Press, Totowa, NJ 1993).
-
Endogene
Rezeptorproteine, die NPY und verwandte Peptide als Liganden binden
wurden identifiziert und unterschieden, und mehrere solcher Proteins
wurden kloniert und exprimiert. Sechs verschiedene Rezeptorsubtypen
[Y1, Y2, Y3, Y4 (PP), Y5, Y6 (früher
als Y5 Rezeptor bezeichnet)] werden heutzutage anhand ihres Bindungsprofils,
Pharmakologie und/oder Zusammensetzung unterschieden, falls die
Identität
bekannt ist (Wahlestedt, C. et. al. Ann. NYAcad. Sci. Larhammar,
D. et. al. J. Biol. Chem. 1992,267,10935; Wahlestedt, C. et. al.
Regul. Pept. Fuhlendorff, J. U. et. al. Proc. Natl. Acad. Sci. USA
1990,87, 182; Grundemar, L. et. al. J. Pharmacol. Exp. Ther. Laburthe,
M. et. al. Endocrinology 1986, 118, 1910; Castan, I. et. al. Endocrinology 1992,
131,1970; Gerald, C. et. al. Nature 1996,382,168; Weinberg, D. H.
et. al. Journal of Biological Chemistry Gehlert, D. et. al. Current
Pharmaceutical Design Lundberg, J. M. et. al. Trends in Pharmaceutical
Sciences). Die meisten und möglicherweise
alle NPY Rezeptorproteine gehören
zur Familie der so genannten G-Protein gekoppelten Rezeptoren (GPCR).
Der Neuropeptid-Y5-Rezeptor, ein putativer GPCR, ist über die
Wirkung der Adenylatcyclase negativ an die zellulären cyclisches
Adenosinmonophosphat-(cAMP)-spiegel gekoppelt (Gerald, C. et. al.
Nature 1996,382,168; Gerald, C. et. al. PCT WO 96/16542). Zum Beispiel
inhibiert NPY die Forskolin-stimulierte cAMP Produktion/Spiegel
in einer Neuroblastom-Zellinie. Ein Y5 Ligand, der NPY auf diese
Weise nachahmt ist ein Agonist, wohingegen einer, der kompetetiv
die NPY Inhibierung der Forskolin-stimulierten cAMP Produktion revertiert,
ein Antagonist ist.
-
Neuropeptid
Y selber ist das archetypische Substrat für die NPY Rezeptoren, und seine
Bindung kann eine Vielzahl von pharmakologischen und biologischen
Effekten in vitro und in vivo hervorrufen. Wenn in das Hirn von
lebenden Tieren (intracerebroventriculär (icv) oder in die Amygdalae)
verabreicht, produziert NPY angstlösende Effekte in etablierten
Tiermodellen von Angst, wie zum Beispiel dem erhöhten Plus-Irrgarten, Vogel-bestraftes
Trinken und Geller-Seifter's
Hebeldruck Konfliktparadigmen (Heilig, M. et. al. Psychopharmacology
1989, 98, 524; Heilig, M. et. al. Reg. Peptides Heilig, M. et. al.
Neuropsycho-pharmacology 1993, 8, 357). Daher werden Verbindungen,
die NPY nachahmen, als brauchbar für die Behandlung von angstlösenden Abweichungen
postuliert.
-
Die
Immunreaktivität
von Neuropeptid Y ist in der Cerebrospinalflüssigkeit von Patienten mit
schwerer Depression und derjenigen von Selbstmordopfern deutlich
verringert (Widdowson, P. S. et. al. Journal of Neurochemistry 1992,
59, 73), und mit tricyclischen Antidepressiva behandelte Ratten
zeigen signifikante Anstiege von NPY, relativ zu einer Kontrollgruppe (Heilig,
M. et. al. European Journal of Pharmacology 1988, 147, 465). Diese
Ergebnisse lassen vermuten, daß eine
ungenügende
NPY Antwort eine Rolle in einigen depressiven Erkrankungen spielen
kann, und daß Verbindungen
die das NPY-erge System regulieren, für die Behandlung von Depression
brauchbar sein können.
-
Neuropeptid
Y verbessert die Erinnerungs- und Performance-Werte in Tiermodellen
der Lernens (Flood, J. F. et. al. Brain Research 1987, 421, 280)
und könne
daher als ein Wahrnehmungs-Verbesserer für die Behandlung von neurodegenerative
Erkrankungen wie zum Beispiel Alzheimer Erkrankung (AD) sowie mit AIDS
zusammenhängender
und seniler Demenz dienen.
-
Erhöhte Plasmaspiegel
von NPY sind in Tieren und Menschen vorhanden, die Episoden von
hoher sympathischer Nervenaktivität erfahren, wie zum Beispiel
Chirurgie, Geburt und Hämorrhagie
(Morris, M. J. et. al. Journal of Autonomic Nervous System 1986,
17, 143). Daher können
chemische Substanzen die das NPY-erge System verändern, für die Linderung des Zustands
von Streß brauchbar
sein.
-
Neuropeptid
Y vermittelt auch endokrine Funktionen, wie zu Beispiel die Freisetzung
von luteinisierendem Hormon (LH) in Nagern (Kalra, S. P. et. al.
Frontiers in Neuroendrocrinology 1992, 13, 1). Da LH vital für die Säugerovulation
ist, könnte
eine Verbindung, die die Wirkung von NPY nachahmt für der Behandlung
von Unfruchtbarkeit brauchbar sein, besonders bei Frauen mit sogenannten
lutealer Phase Defekten.
-
Neuropeptid
Y ist ein starkes Stimulans der Nahrungsaufnahme; so wenig wie ein
Billionstel eines Gramms, wenn direkt in das ZNS injiziert bewirkt,
daß satte
Ratten sich überfressen
(Clark, J. T. et. al. Endocrinology Levine, A. S. et. al. Peptides
1984,5,1025; Stanley, B. G. et. al. Life Sci. 1984, 35, 2635; Stanley,
B. G. et. al. Proc. Nat. Acad. Sci. USA 1985, 82, 3940). Daher ist
NPY in Nagern orexigen, jedoch nicht angstlösend, wenn intracerebroventrikulär gegeben,
und so können
Antagonisten von Neuropeptid Rezeptoren für die Behandlung von Eßstörungen,
wie zum Beispiel Fettsucht, Anorexia nervosa und Bulimia nervosa
brauchbar sein.
-
Seit
einigen Jahren werden eine Vielzahl von potenten, strukturell distinkten
Y1 Antagonisten niedrigen Molekulargewichts entdeckt und entwickelt
(Hipskind, P. A. et. al. Annu. Rep. Med. Chem. 1996, 31, 1–10; Rudolf,
K. et. al. Eur. J. Pharmacol. 1994, 271, R11; Serradeil-Le Gal, C. et. al.
FEBS Lett. 1995, 362, 192; Wright, J. et. al. Bioorg. Med. Chem.
Lett. 1996,6,1809; Poindexter, G. S. et. al. United States Patent 5,668,151;
Peterson, J. M. et. al. WO 96/14307 (1996)). Trotz jedoch einer
behaupteten Aktivität
in Nagermodellen der Fütterung
ist es unklar, ob die Inhibierung einer Fütterungsantwort einem Antagonismus
des Y1 Rezeptors zugeordnet werden kann.
-
Mehrere
richtungsweisende Untersuchungen lassen stark vermuten, daß ein "atypischer Y1" Rezeptor und/oder
der Y5 Rezeptor, anders als der klassische Y1 Rezeptor, für das Hervorrufen
NPY-stimulierter Nahrungsaufnahme in Tieren verantwortlich ist.
Es wurde gezeigt, daß das
NPY Fragment NPY2–36 ein potenter Induzierer
der Fütterung
trotz schlechter Bindung an den klassischer Y1 Rezeptor ist (Stanley,
B. G. et. al. Peptides 1992, 13,5 81). Umgekehrt wurde ein potenter
und selektiver Y1 Agonist als inaktiv bei der Stimulierung der Fütterung
in Tieren berichtet (Kirby, D. A. et. al. J. Med. Chem. 1995, 38,
4579). Relevanter für
die hier beschriebene Erfindung wurde [D-Trp32]
NPY, ein selektiver Y5 Rezeptor Aktivator, als die Nahrungsaufnahme stimulierend
beschrieben, wenn in den Hypothalamus von Ratten injiziert (Gerald,
C. et. al. Nature 1996, 382, 168). Da [D-Trp32]
NPY ein voller Agonist des Y5 Rezeptors mit keiner wesentlichen
Y1 Aktivität
zu sein scheint, wird der Y5 Rezeptor hypothetisch als verantwortlich
für die
Nahrungs-Antwort angenommen. Demzufolge sollten Verbindungen, die
Antagonisten des Y5 Rezeptors sind, effektiv bei der Inhibierung
der Nahrungsaufnahme, insbesondere der durch NPY stimulierten, sein.
-
Auch
für die
hier beschriebene Erfindung bedeutend sind Beschreibungen von Arylsulfonamiden,
die als Y5 Antagonisten wirken. In der PCT WO 97/19682 sind Arylsulfonamide
und Sulfamide abgeleitet von Arylalkylaminen als Y5 Antagonisten
beschrieben und als den Nahrungsverbrauch in Tieren verringernd
berichtet. In PCT WO 97/20820, PCT WO 97/20822 und PCT WO 97/20823
werden Sulfonamide enthaltende heterocyclische Systeme, wie zum
Beispiel Chinazolin-2,4-diazirin, ähnlich als Y5 Antagonist beansprucht
und als die Fütterung
verringernd berichtet. Es gibt keine Beschreibung eines α-substituierten β-Aminotetralins
in irgendeiner dieser Publikationen. Die in dieser Anmeldung beschriebenen
N-substituierten Aminotetraline sind neue molekulare Gruppen, die
Bindungsmotive aufweisen können,
die anders als diese sind, und andere Y5 Liganden, die in Patentanmeldungen
oder Publikationen beschrieben wurden, und dennoch an eine ähnlichen
Region des Y5 Rezeptors binden.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft Verbindungen der Formel 1
worin
R
1 unabhängig voneinander
ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff; Hydroxy; Halo; C
1-8Alkyl; substituiertes C
1-8Alkyl,
wobei der Substituent ausgewählt
ist aus Halo, wie zum Beispiel Chlor, Brom und Fluor; C
1-8Alkoxy;
substituiertes C
1- 8Alkoxy,
wobei der Substituent ausgewählt
ist aus Halo, wie zum Beispiel Chlor, Brom, Fluor und Jod; Trifluoralkyl;
C
1-8Alkylthio und substituiertes C
1-8Alkylthio, wobei der Substituent ausgewählt ist
aus Halo, wie zum Beispiel Chlor, Brom, Fluor und Jod, Trifluoralkyl
und C
1-8Alkoxy; C
3-6Cycloalkyl;
C
3-8Cycloalkoxy; Nitro; Amino; C
1-6Alkylamino; C
1-6Dialkylamino;
C
4-6Cycloalkylamino; Cyano; Carboxy; C
1- 5Alkoxycarbonyl; C
1-5Alkylcarbonyloxy;
Formyl; Carbamoyl; Phenyl; substituiertes Phenyl, wobei der Substituent
ausgewählt
aus Halo, Hydroxyl, Nitro, Amino und Cyano;
n 0–2 ist
B
2 ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff; C
1-5Alkyl;
substituiertes C
1-
5Alkyl, wobei
der Substituent Halogen ist;
B
2 kann
entweder eine cis- oder trans-stereochemische Orientierung im Hinblick
auf B
1 aufweisen; beide Enantiomere jedes
diastereomerischen Sets sind Bestandteile dieser Erfindung.
Y
Methylen ist
m 0–3
R
2 ausgewählt
ist aus der Gruppe bestehend aus Hydroxy; C
1-6Alkyl;
C
1-6Alkenyl; Halo, wie zum Beispiel Fluor und
Chlor; C
3-7Cycloalkyl; Phenyl; substituiertes
Phenyl, wobei der Substituent ausgewählt ist aus Halo, C
1-6Alkyl, C
1-6Alkoxy,
TrifluorC
1-6alkyl; Cyano, Nitro, Amino,
C
1-6Alkylamino und C
1-6Dialkylamino;
Naphthyl; Phenoxy; substituiertes Phenoxy, wobei der Substituent
ausgewählt
ist aus Halo, C
1-6Alkyl, C
1- 6Alkoxy, TrifluorC
1-6Alkyl, Cyano und Nitro; Phenylthio und
substituiertes Phenylthio, wobei der Substituent ausgewählt ist
aus Halo, C
1-6Alkyl, Nitro und Amino; eine
Heteroarylgruppe, wie zum Beispiel Pyridyl, Pyrimidyl, Furyl, Thienyl
und Imidazolyl; substituiertes Heteroaryl, wobei der Substituent
ausgewählt
ist aus C
1-6Alkyl und Halo; und Heterocycloalkyl,
wie zum Beispiel Pyrrolidino oder Piperidino;
B
1 ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff; C
1-5Alkyl;
substituiertes C
1-
5Alkyl, wobei
der Substituent Halo ist;
B
1 kann entweder
eine cis- oder trans- stereochemische Orientierung im Hinblick auf
B
2 aufweisen; beide Enantiomere jedes diastereomerischen
Sets sind Bestandteile dieser Erfindung.
L ist ausgewählt aus
der Gruppe bestehend aus C
1-8Alkylen; C
2-10Alkenylen; C
2- 10Alkinylen; C
1-4Alkylen
3-7cycloalkylen;
C
1-4Alkylen
3-7cycloalkyl
1-4alkylen; C
2- 4Alkenylen
3-7cycloalkyl
2-4alkenylen; C
2-4Alkinylen
3-7cycloalkyl
2-4alkinylen; C
2- 4Alkylenaryl
1-4Alkylen; und C
2-4Alkenylenaryl
2-4Alkenylen;
R
3 ist
ausgewählt
aus C
1-8Alkyl; substituiertem C
1-8Alkyl,
wobei der Substituent ausgewählt
ist aus Alkoxy und Halo; Cycloalkyl; substituiertes Cycloalkyl,
wobei der Substituent ausgewählt
ist aus Alkoxy und Halo; Phenyl, substituiertes Phenyl, wobei der
Substituent ausgewählt
ist aus C
1-8Alkyl, Halo, Nitro, Amino, Alkylamino,
Alkylsulfonyl, Alkoxy und Cyano; Naphthyl; substituiertes Naphthyl,
wobei der Substituent ausgewählt
ist aus Halo, Amino und Cyano; Heteroaryl, wobei die Heteroaryl-Gruppe
ausgewählt
ist aus Pyridyl, Pyrimidyl, Furyl, Thienyl und Imidazolyl; und substituiertes
Heteroaryl, wobei der Substituent ausgewählt ist aus Halo, Nitro, Amino
und Cyano;
und Enantiomere, Diastereomere und pharmazeutisch
akzeptable Salze davon.
-
Wie
hier verwendet, wenn nicht anders angegeben, schließen die
Ausdrücke "Alkyl" und "Alkoxy", ob allein oder
als Teil einer Substituentengruppe verwendet, gerade und verzweigte
Ketten ein, die 1–3
Kohlenstoffatome aufweisen. Zum Beispiel schließen Alkylreste Methyl, Ethyl,
Propyl, Isopropyl, Butyl, Isobutyl, sec-Butyl, t-Butyl, Pentyl,
2-Methyl-3-butyl, 1-Methylbutyl,
2-Methylbutyl, Neopentyl, Hexyl, 1-Methylpentyl und 3-Methylpentyl
ein. Alkoxyreste sind Oxyether, gebildet aus den vorhergehend beschriebenen
geraden oder verzweigten Alkylkettengruppen. Der Ausdruck "Aryl" ist als Phenyl und
Naphthyl einschließend
gedacht. Der Ausdruck "Halo", wenn nicht anders
angegeben, schließt
Brom, Chlor, Fluor und Jod ein. Der Ausdruck "Cycloalkyl" ist als Cycloalkylgruppen einschließend gedacht,
die 3–7
Kohlenstoffatome aufweisen. Mit Bezug auf Substituenten bedeutet
der Ausdruck "unabhängig", daß, wenn
mehr als einer eines solchen Substituenten möglich ist, können diese
Substituenten dieselben oder verschieden voneinander sein.
-
Diejenigen
Verbindungen der vorliegenden Erfindung, die eine basische Gruppe
enthalten, können durch
dem Fachmann im Stand der Technik bekannte Techniken in die entsprechenden
Säureadditionsalze überführt werden.
Geeignete Säuren,
die für
diesen Zweck verwendet werden können,
schließen
Hydrochlor-, Hydrobrom-, Hydrojod-, Perchlor-, Schwefel-, Salpeter-,
Phosphor-, Essig-, Propion-, Glycol-, Milch-, Brennztrauben-, Oxal-,
Malon-, Succinin-, Malein-, Fumar-, Äpfel-, Tartar-, Zitronen-,
Benzoe-, Zimt-, Mandel-, Methansulfon, p-Toluolsulfon, Cyclohexansulfam-, Salicyl-,
2-Phenoxybenzoe-, 2-Acetoxybenzoe- oder Saccharinsäure ein.
Im allgemeinen kann das Säureadditionsalz
durch reagieren der freien Base von Verbindungen der Formel 1 mit
der Säure
und isolieren des Salzes hergestellt werden.
-
Pharmazeutische
Zusammensetzungen, die eine oder mehrere der Verbindungen der hier
beschriebenen Erfindung als den aktiven Inhaltsstoff enthalten,
können
durch enges Vermischen der Verbindung oder Verbindungen mit einem
pharmazeutischen Träger
gemäß herkömmlicher
pharmazeutischer Verbindungstechniken hergestellt werden. Der Träger kann
in Abhängigkeit
der gewünschten
Route der Verabreichung (e. g., oral, parenteral) eine große Vielzahl
von Formen annehmen. Daher schließen für flüssige orale Präparationen
wie zum Beispiel Suspensionen, Elixiere und Lösungen, geeignete Träger und
Additive Wasser, Glycole, Öle,
Alkohole, Aromastoffe, Konservierungsmittel, Stabilisatoren und
Farbstoffe ein; für
feste orale Präparationen,
wie zum Beispiel Pulver, Kapseln und Tabletten, schließen geeignete
Träger
und Additive Stärken,
Zucker, Verdünnungsmittel,
granulierende Mittel, Gleitmittel, Bindemittel und Sprengmittel
ein. Feste orale Präparationen
kann auch mit Substanzen wie zum Beispiel Zucker oder enterisch
beschichtet werden, um so den Hauptort der Absorption zu modulieren.
Für die
parenterale Verabreichung wird der Träger gewöhnlich aus sterilem Wasser
bestehen, und andere Inhaltsstoffe können hinzugefügt werden,
um die Löslichkeit
oder Konservierung zu erhöhen.
Injizierbare Suspensionen oder Lösungen
können
auch unter der Verwendung von wäßrigen Trägern, zusammen
mit geeigneten Additiven, hergestellt werden.
-
Für die Behandlung
von Abweichungen des zentralen Nervensystems wird die hier beschriebene pharmazeutische
Zusammensetzungen typischerweise von 1 bis 1000 mg des aktiven Inhaltsstoffs
pro Dosis enthalten; eine oder mehr Dosen pro Tag können verabreicht
werden. Die Bestimmung der optimalen Dosis und der Frequenz der
Dosierung für
einen bestimmten Erkrankungszustand oder Erkrankung liegt innerhalb der
experimentellen Fähigkeiten
des Fachmanns in der Behandlung von Abweichungen des zentralen Nervensystems.
Der bevorzugte Dosierungsbereich ist 1–100 mg/kg.
-
Als
Modulatoren des NPY5 Rezeptors sind die Verbindungen der Formel
1 für die
Behandlung von Eßstörungen,
wie zum Beispiel Fettsucht, Anorexia nervosa und Bulimia nervosa
und abnormalen Zuständen,
wie zum Beispiel Epilepsie, Depression, Angstzuständen und
sexuellen/reproduktiven Abweichungen, bei denen eine Modulation
des NPY5 Rezeptors sinnvoll sein kann, brauchbar. Die Verbindungen
sind mit den endogenen Liganden NPY und PYY und möglicherweise
nicht-endogenen Liganden kompetetiv, und binden an den NPY5 Rezeptor.
Zusätzlich
zeigen die Verbindungen Antagonistenaktivität durch Antagonisieren der
Wirkung von NPY nach Bindung an den Y5 Rezeptor.
-
Die
hier beschriebenen Verbindungen sind Liganden des NPY5 Rezeptor,
sind jedoch nicht nötiger weise
aufgrund lediglich der Bindung an diesen oder andere Neuropeptide,
Neurotransmitter oder G-Protein gekoppelte Rezeptoren in ihren pharmakologischen
oder biologi- schen Wirkungen beschränkt. Zum Beispiel können die
beschriebenen Verbindungen auch Bindung an Dopamin- oder Serotoninrezeptoren
unterzogen werden. Die beschriebenen Verbindungen sind potentiell
bei der Regulation von metabolischen und endokrinen Funktionen brauchbar,
insbesondere denjenigen, die mit der Nahrungsaufnahme assoziiert
sind, und als solche können
sie für
die Behandlung der Fettsucht brauchbar sein. Zusätzlich sind die hier beschriebenen Verbindungen
potentiell für
die Modulation anderer endokriner Funktionen brauchbar, insbesondere
denjenigen, die durch die pituitären
und Hypothalamusdrüsen
kontrolliert werden und können
daher für
die Behandlung der Inovulation/Unfruchtbarkeit aufgrund von unzureichender
Freisetzung von luteinisierendem Hormon (LH) brauchbar sein.
-
Die
vorliegende Erfindung umfaßt
pharmazeutische Zusammensetzungen, die eine oder mehrere der Verbindungen
der Formel 1 enthalten, Amidvorläufer
der Verbindungen der Formel 1 sind ebenfalls neu und werden als
Teil der Erfindung angesehen.
-
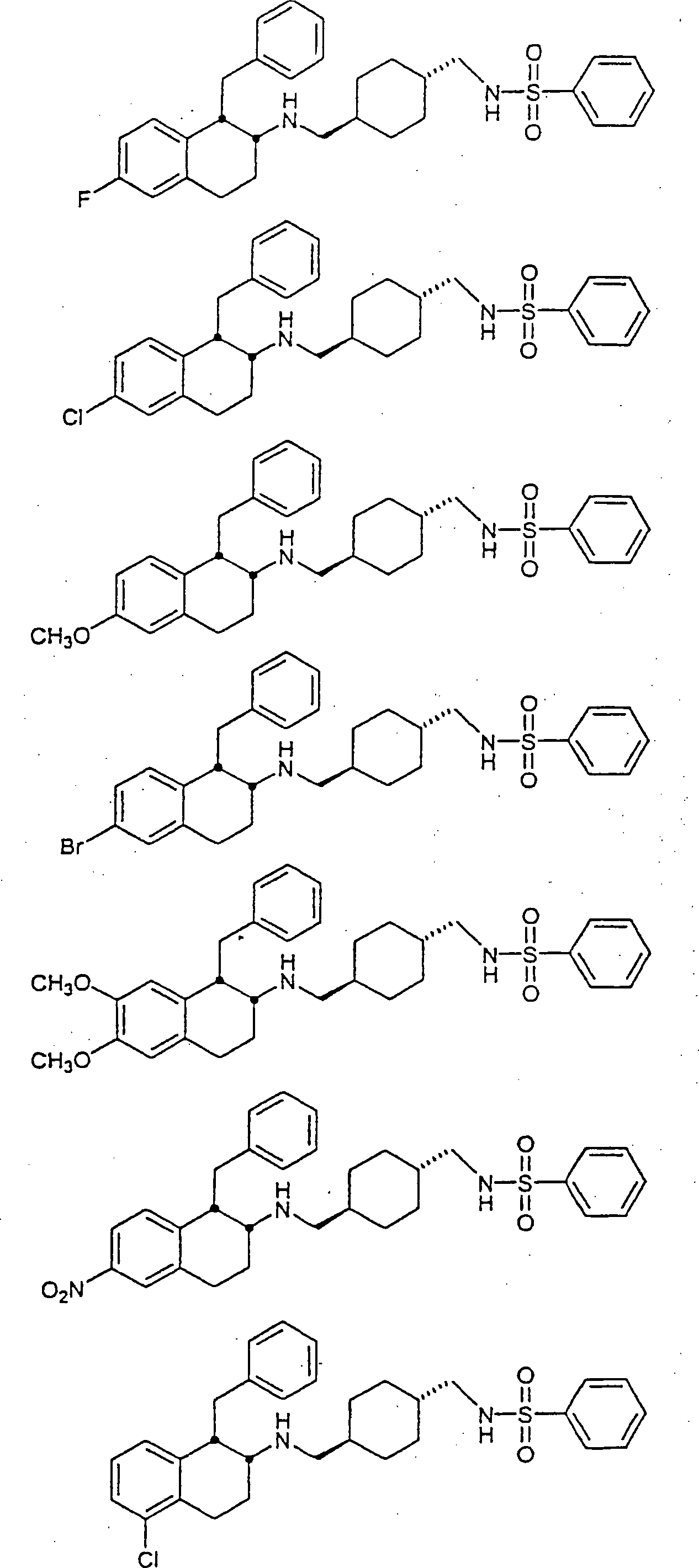
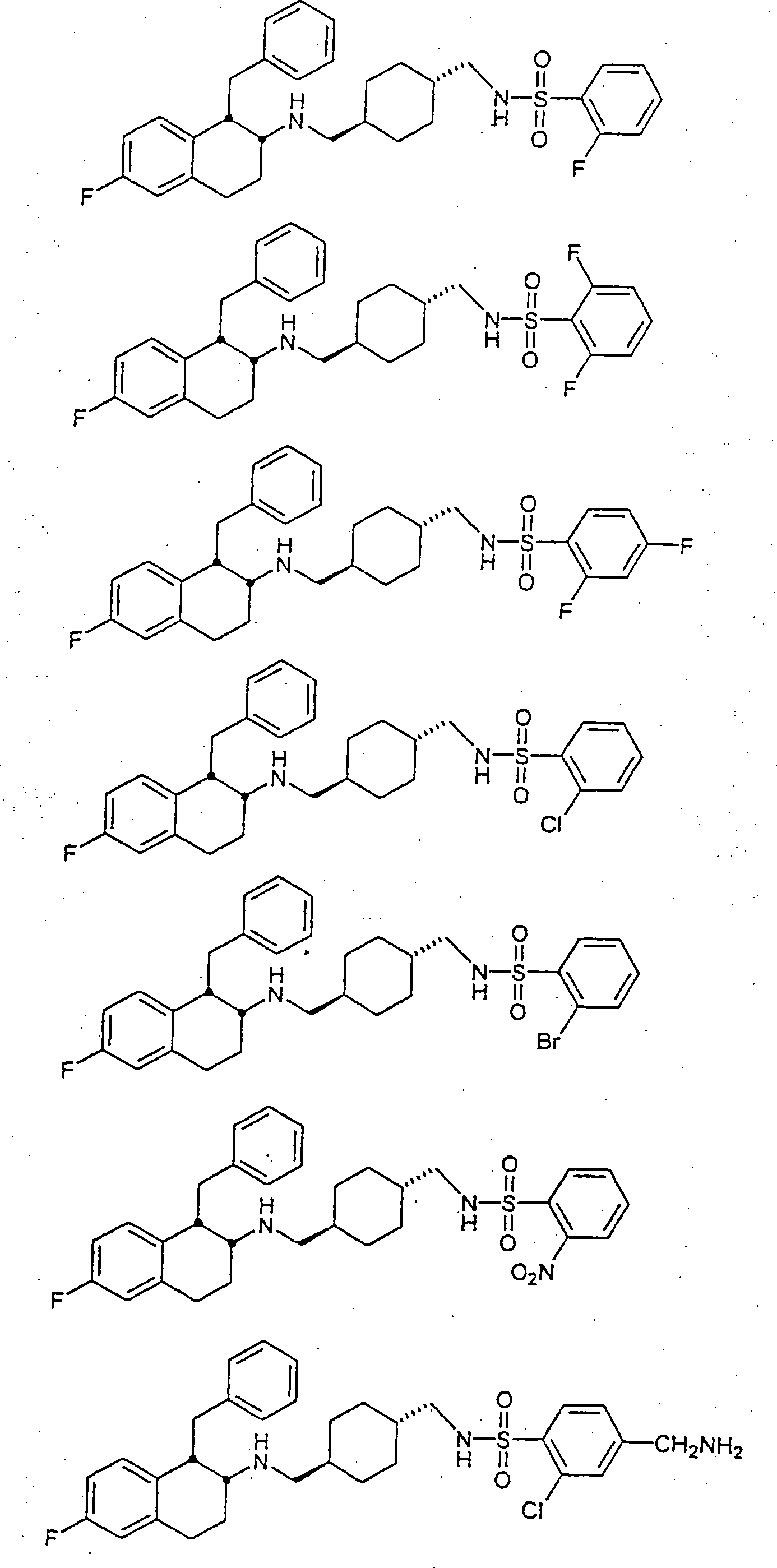
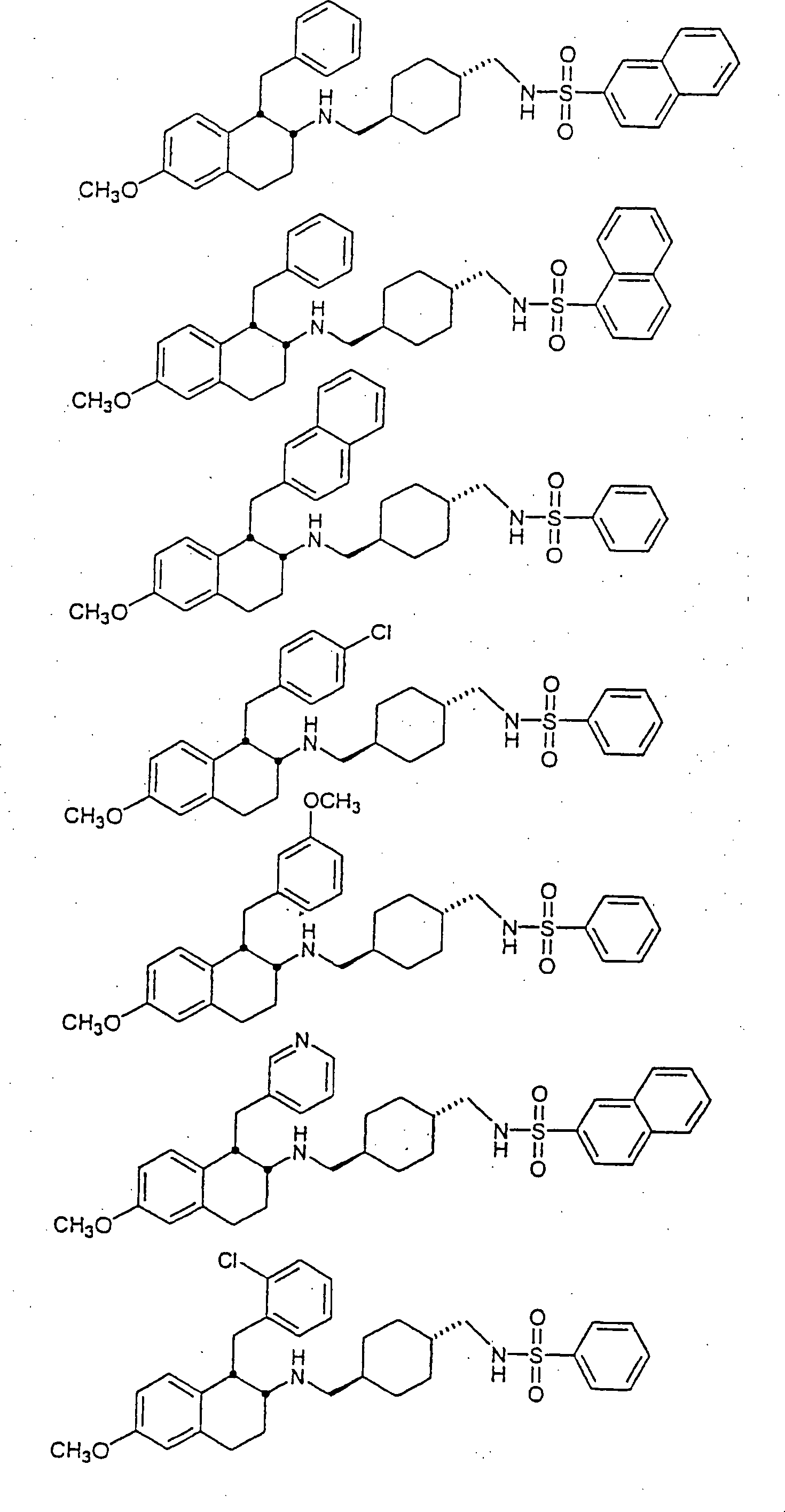
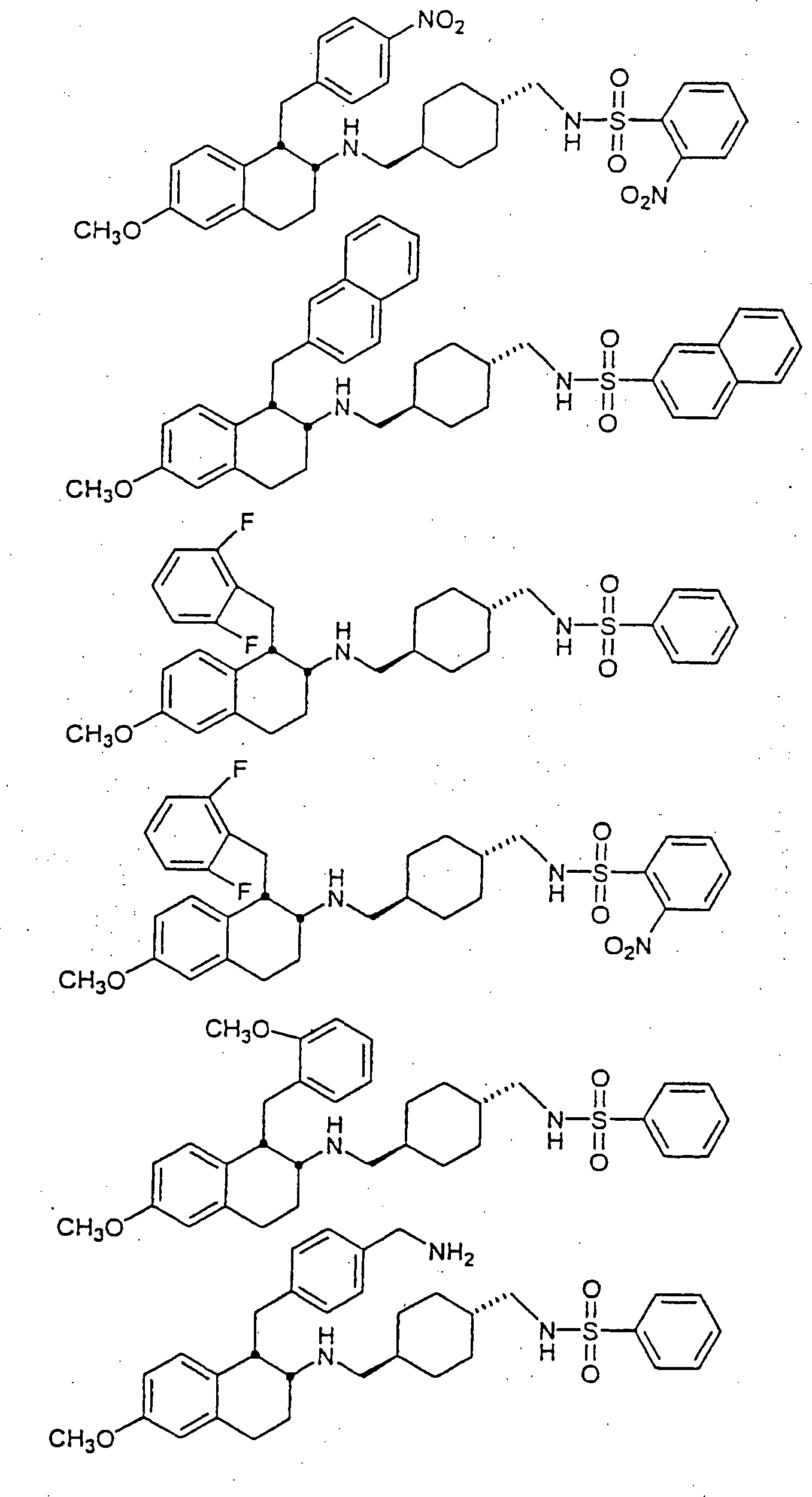
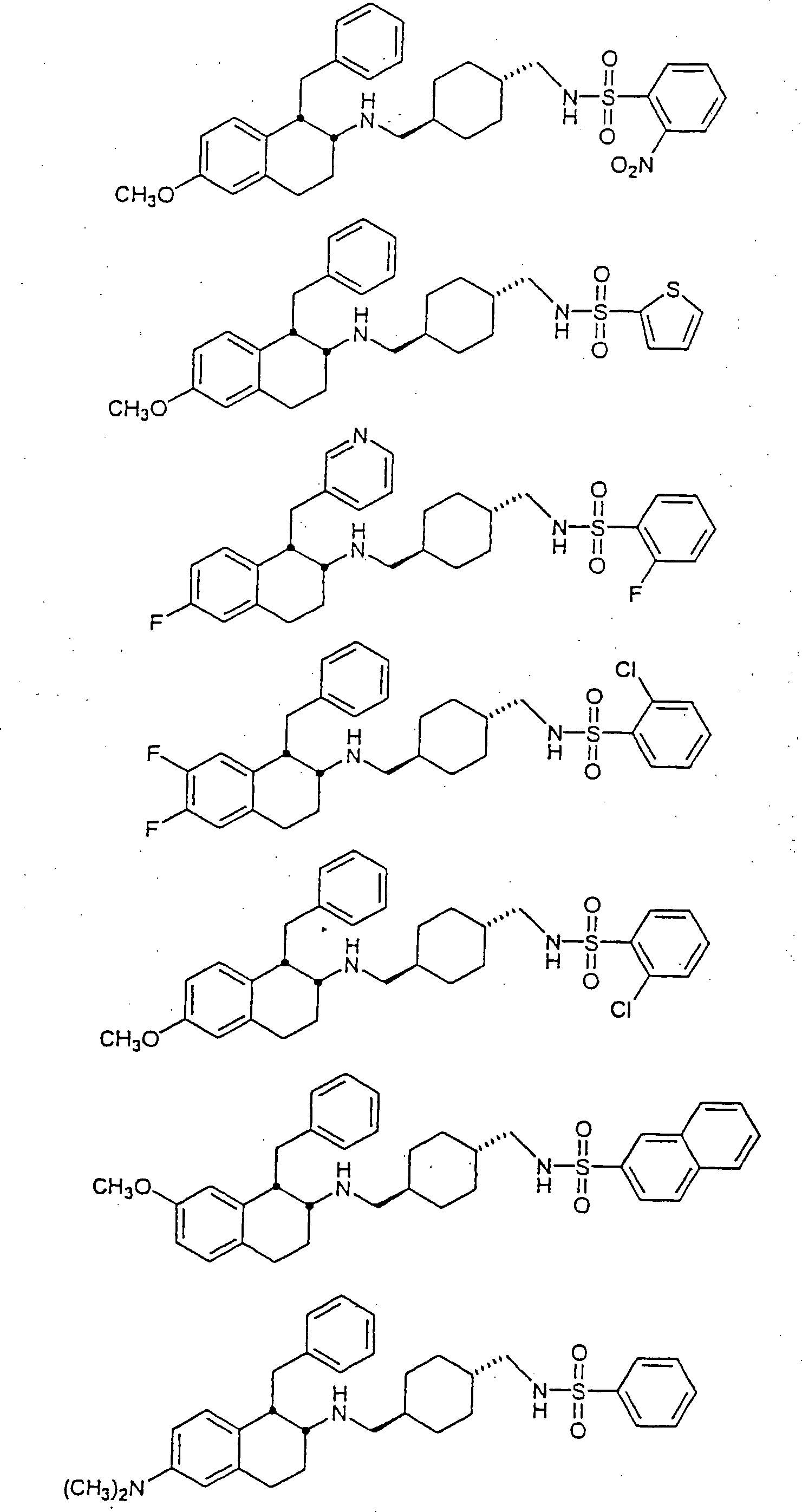
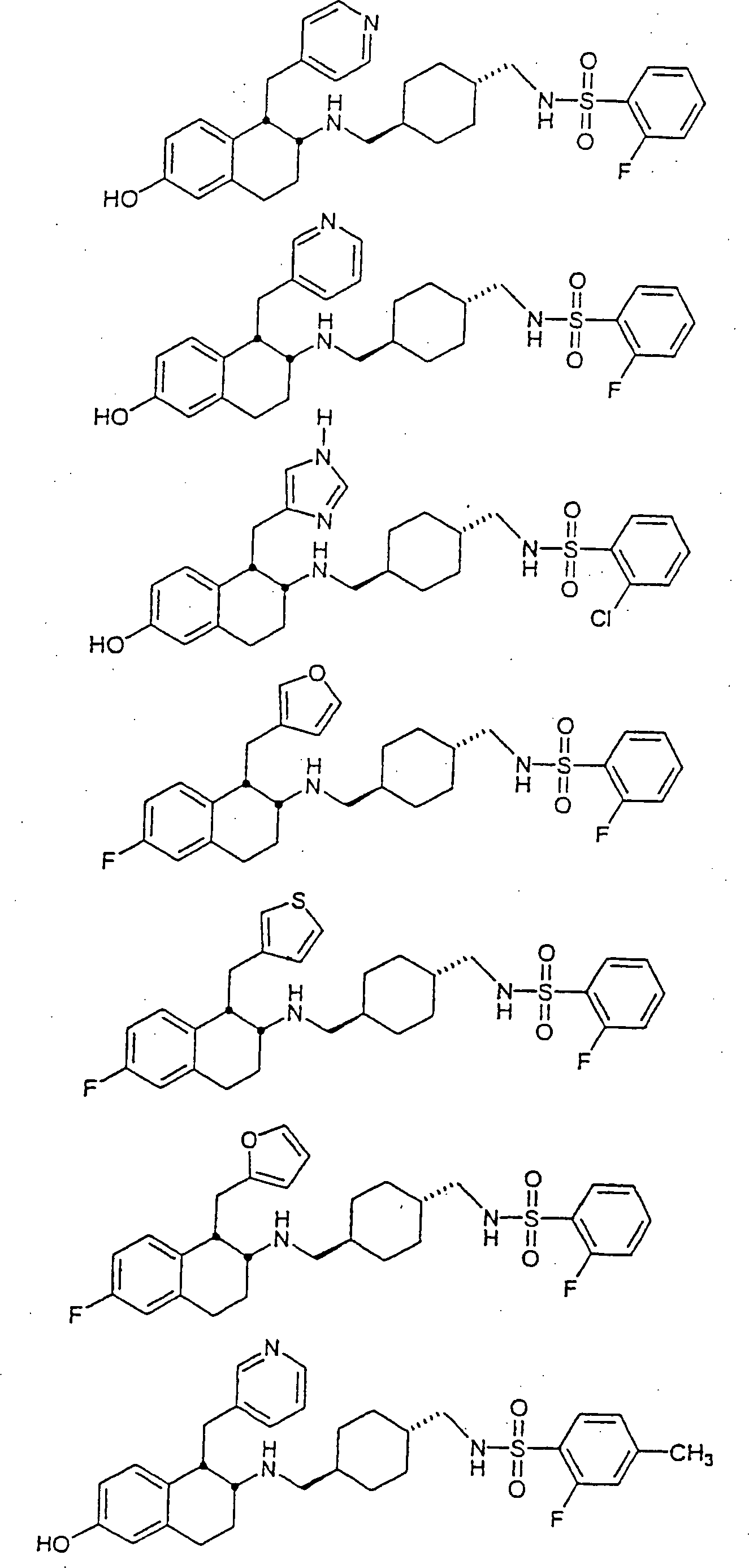
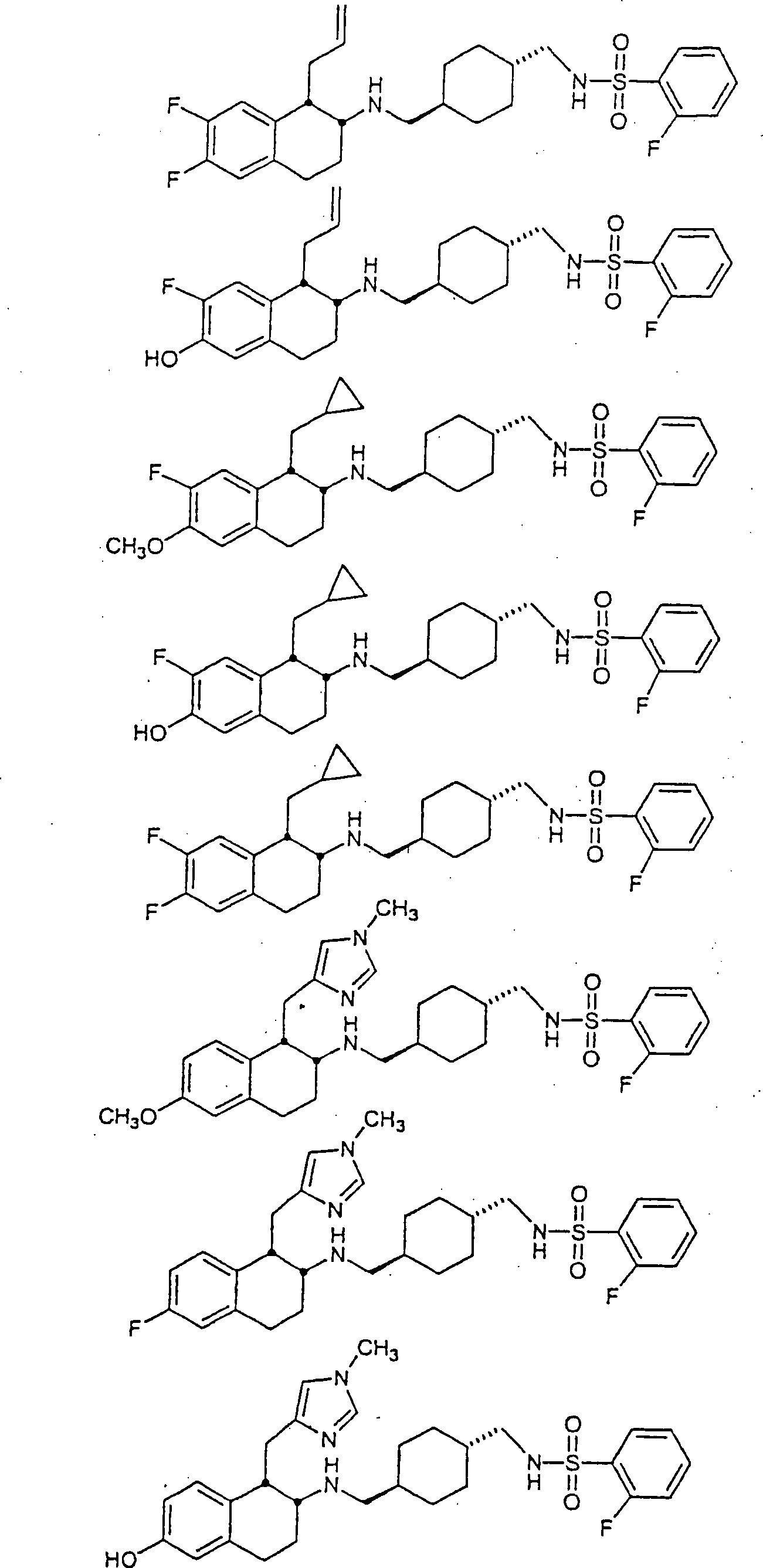
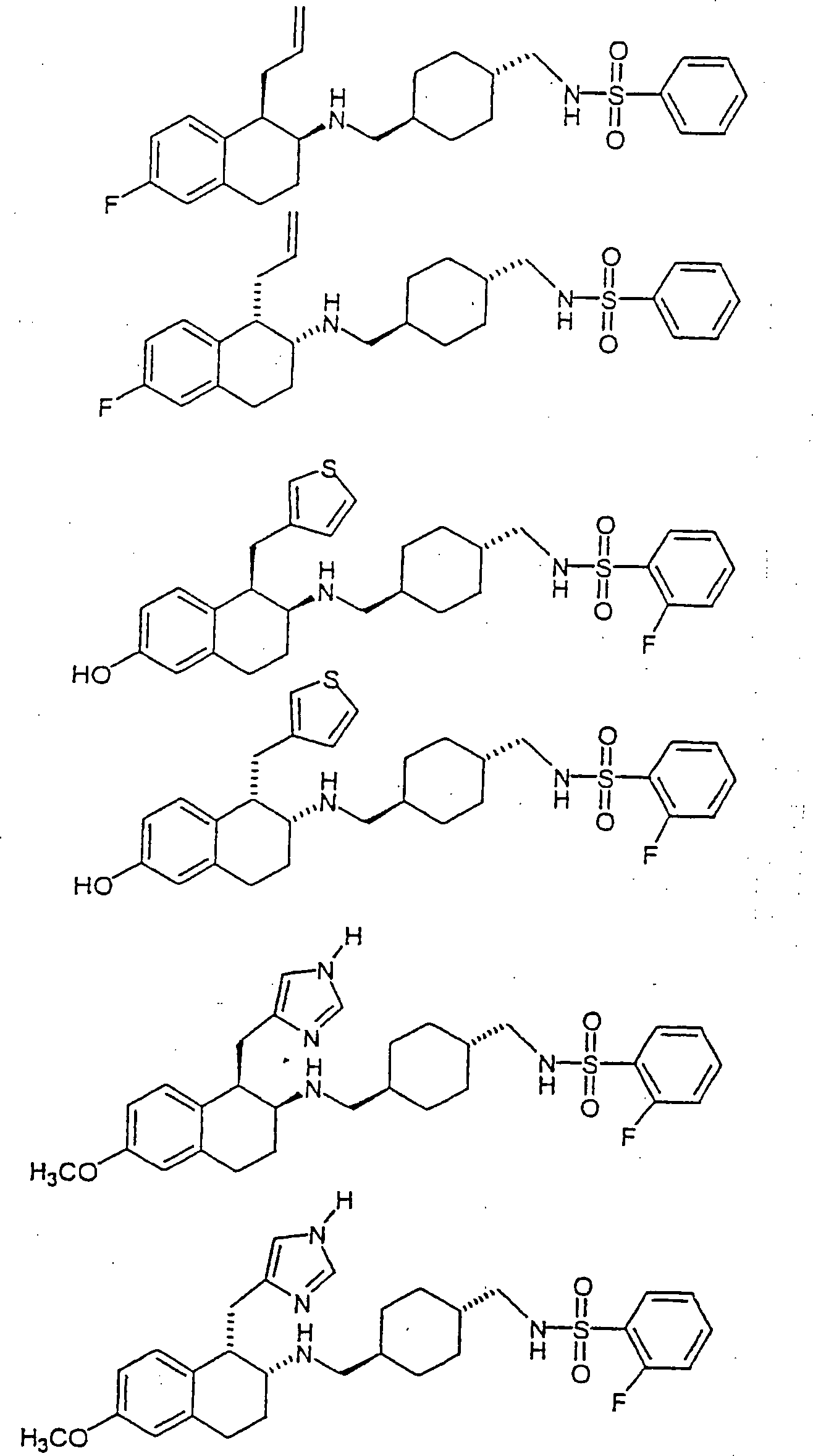
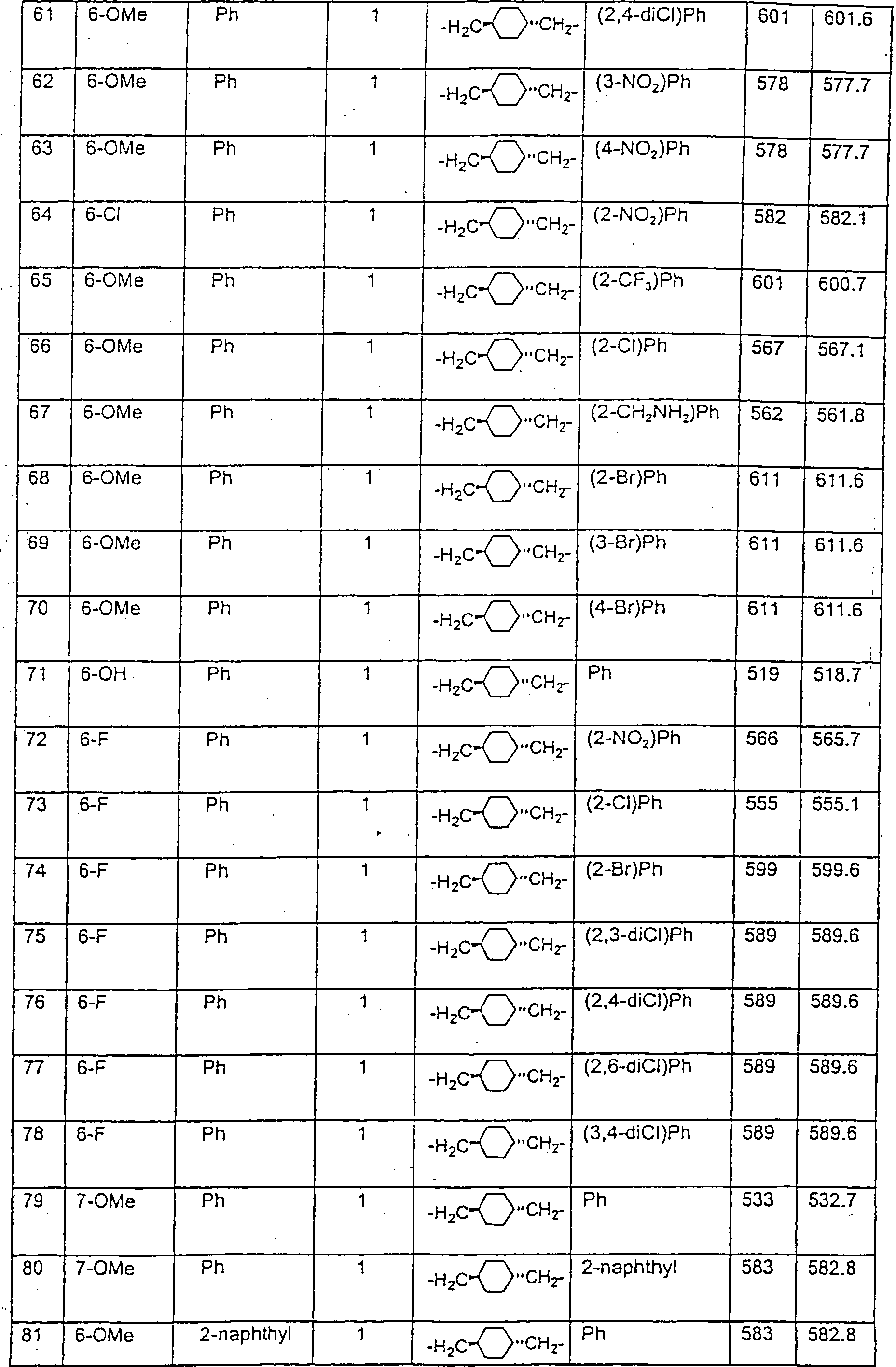
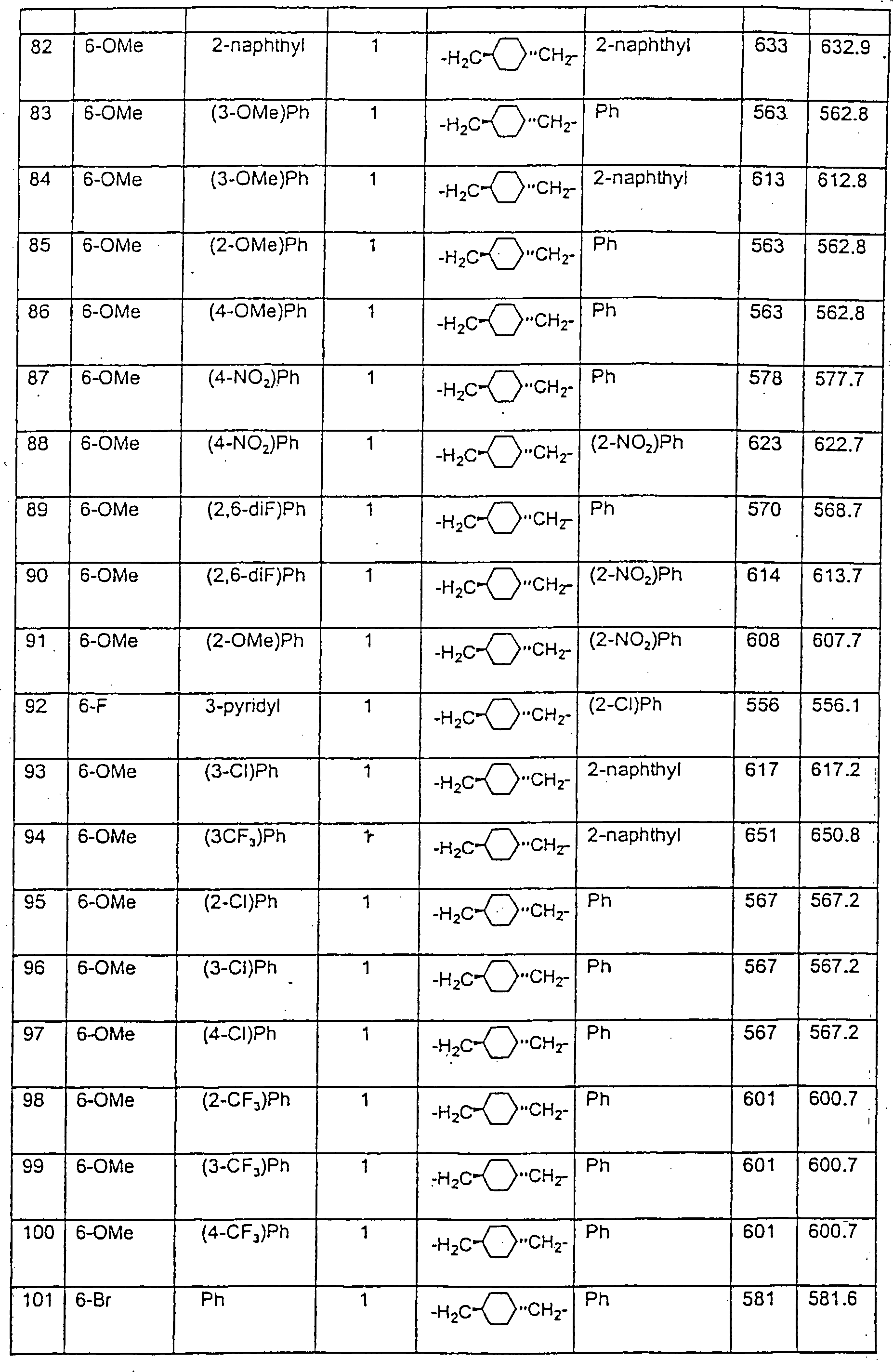
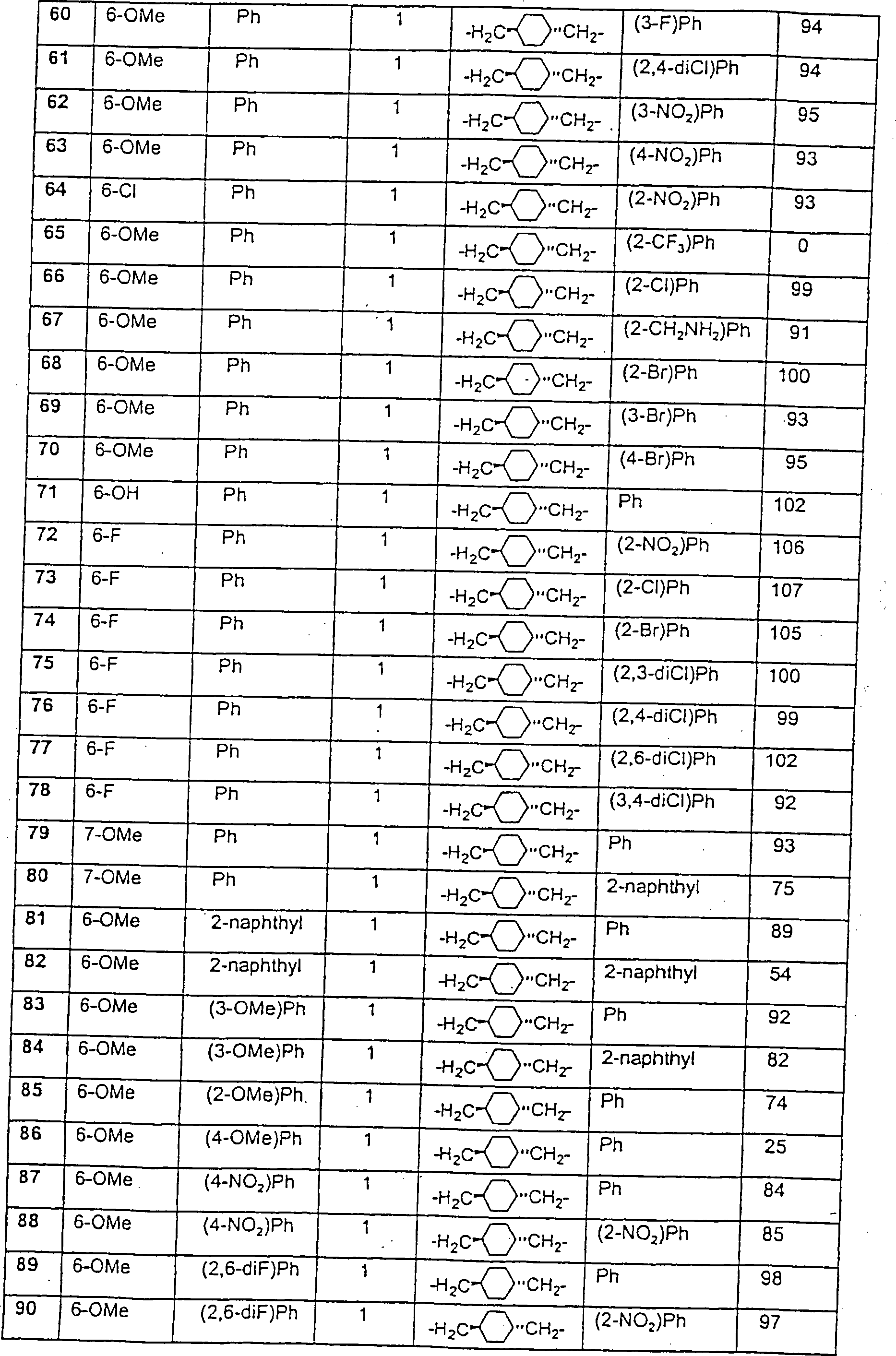
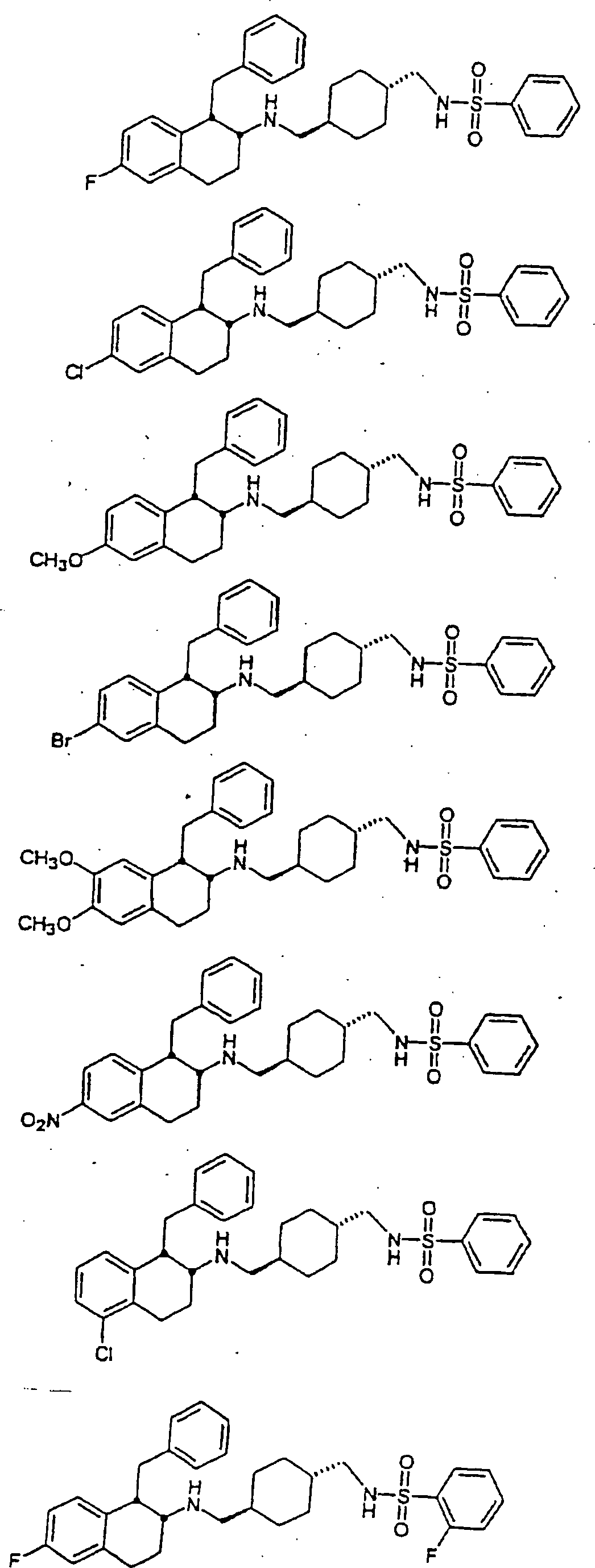
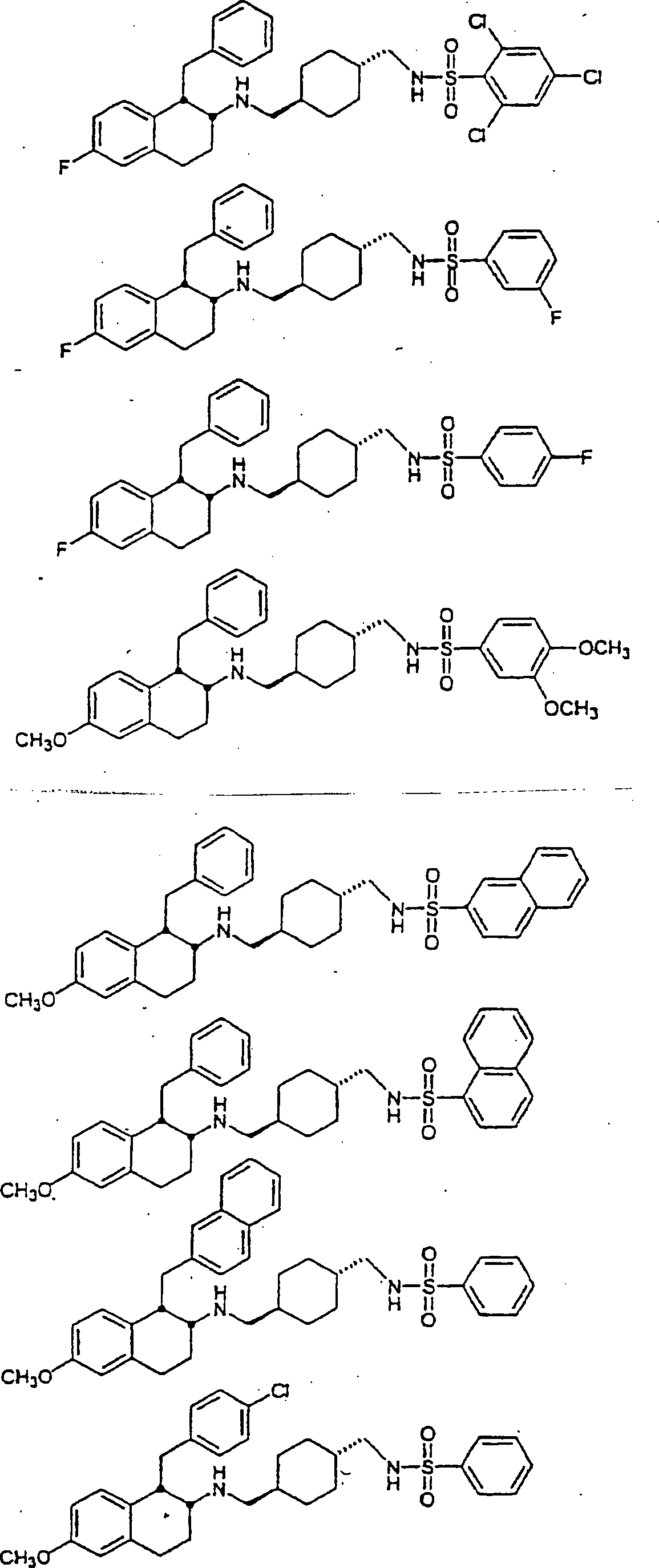
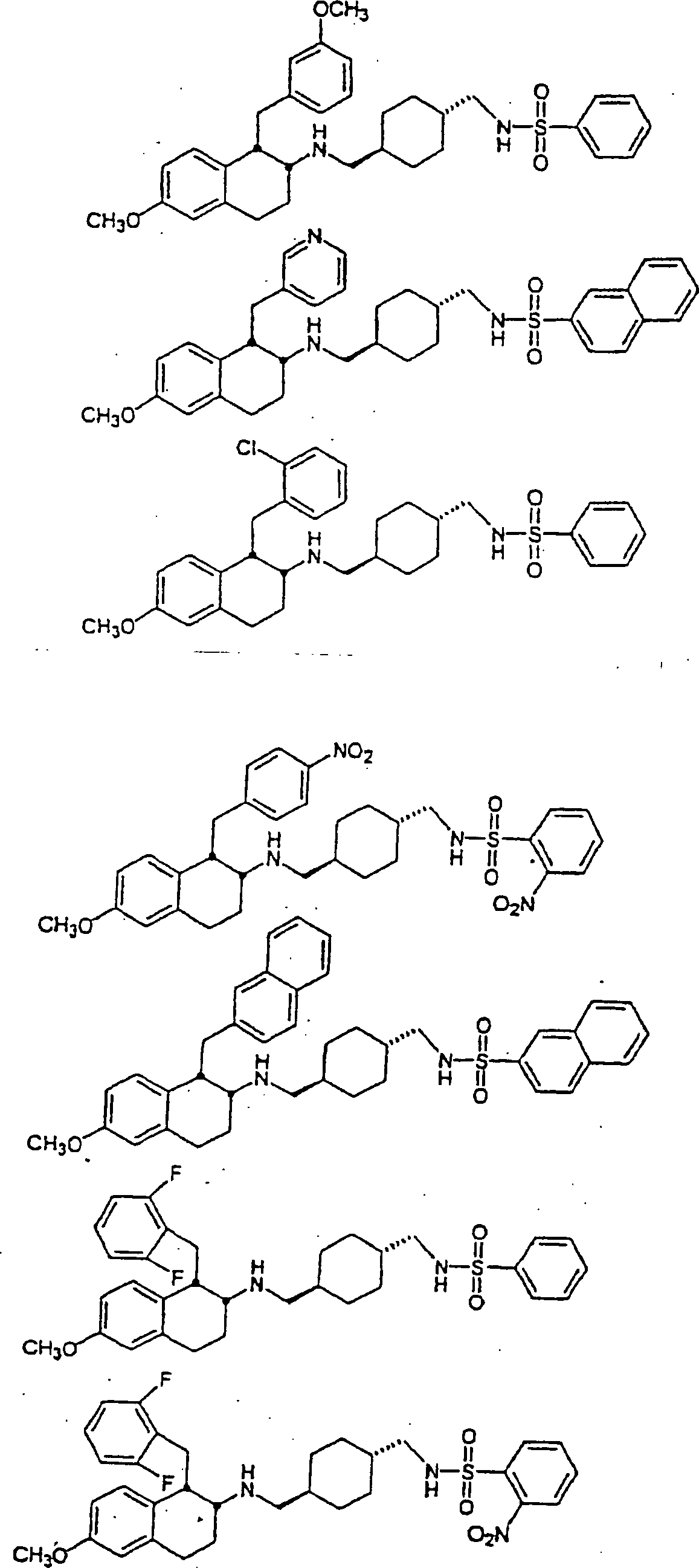
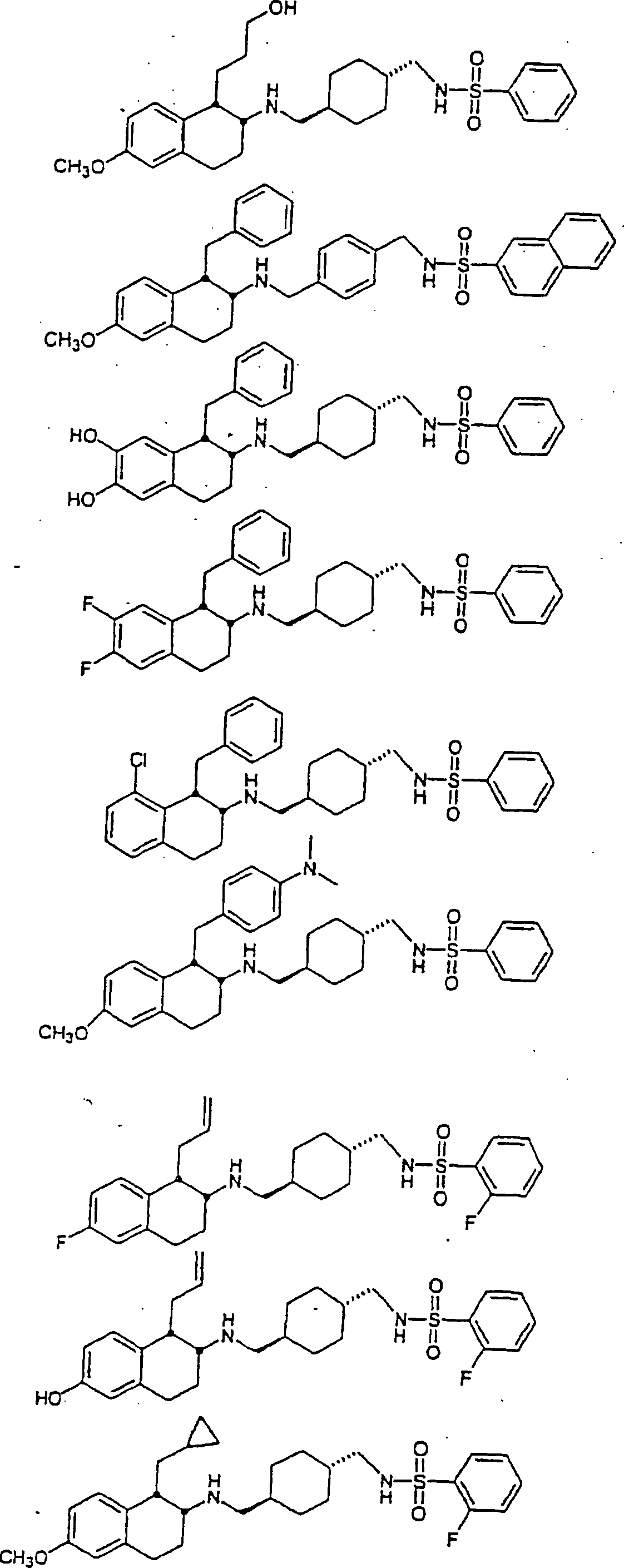
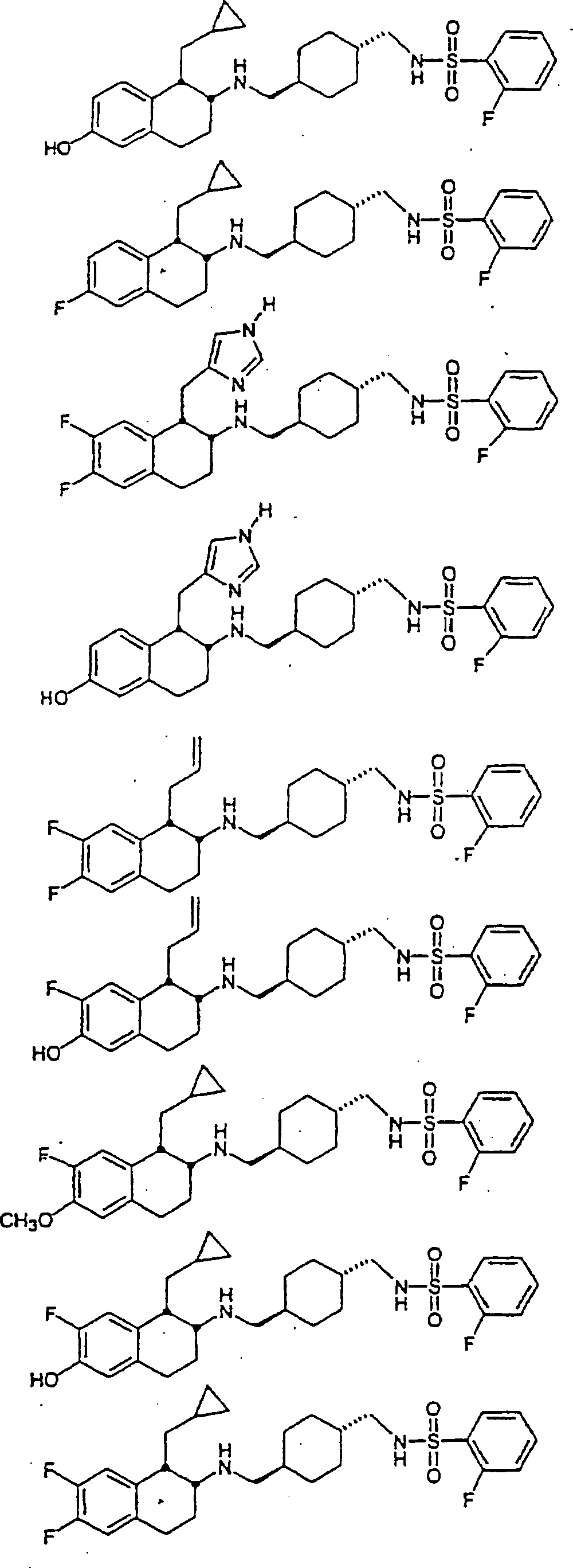
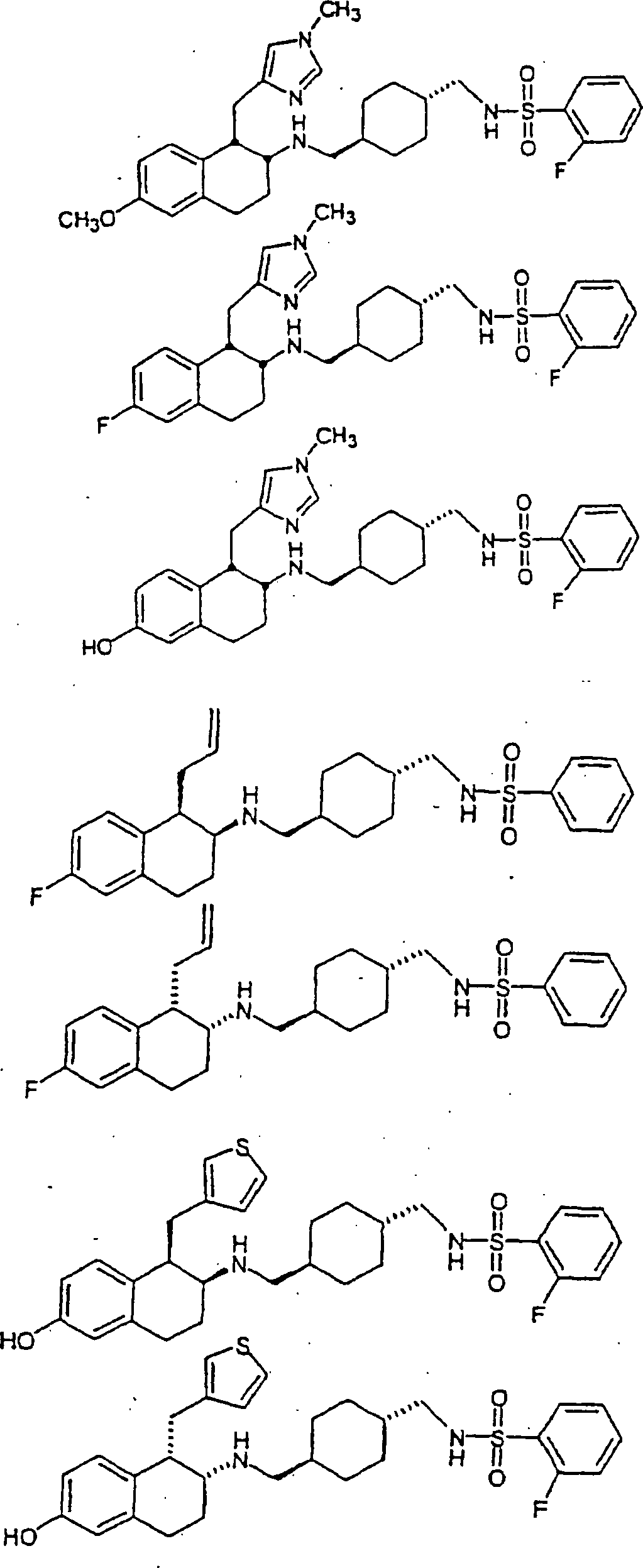
Beispiele
von besonders bevorzugten Verbindungen der Formel 1 schließen ein:
-
-
-
-
-
-
-
-
-
-
-
-
-
-
GENAUE BESCHREIBUNG
DER ERFINDUNG
-
Die
N-substituierten Aminotetraline der Formel 1, die diese Erfindung
umfaßt,
werden über
mehrere distinkte chemische Synthesen synthetisiert, wie in Schemata
1–5 dargestellt;
wobei jede synthetische Route aus mehreren aufeinander folgenden
chemischen Handlungen besteht, die wie unten beschrieben verallgemeinert
werden können:
- – Einführung des α-Substituenten
auf den Tetralonnukleus;
- – Umwandlung
in das entsprechende α-substituierte-β-Aminotetralins;
- – Acylierung
des Aminotetralins oder reduktive Aminierung des α-substituierten-D-Tetralons;
- – Reduktion,
um das Aminotetralin-System zu regenerieren (falls erforderlich)
und/oder Sulfonylierung (falls erforderlich)
(Schutzgruppen-Manipulationen
können
zu verschiedenen Phasen erforderlich sein)
-
Es
ist allgemein bevorzugt, daß das
jeweilige Produkt jedes Verfahrensschritts von anderen Komponenten
des Reaktionsgemischs abgetrennt wird und einer Reinigung unterzogen
wird, bevor es als ein Ausgangsmaterial in einem anschließenden Schritt
verwendet wird. Abtrenntechniken schließen typischerweise Evaporation,
Extraktion, Ausfällen
und Filtration ein. Reinigungstechniken schließen typischerweise Säulenchromatographie
(Still, W. C. et. al., J. Org. Chem. 1978, 43, 2921), Dünnschichtchromatographie,
Kristallisierung und Destillation ein. Die Strukturen der finalen
Produkten, Zwischenprodukte und Ausgangsmaterialien werden durch
spektroskopische, spektrometrische und analytische Verfahren einschließlich Kernmagnetresonanz
(NMR), Massenspektrometrie (MS) und Flüssigkeitschromatographie (HPLC)
bestätigt.
In den Beschreibungen für
die Präparation
der Verbindungen dieser Erfindung sind Ethylether, Tetrahydrofuran
und Dioxan herkömmliche
Beispiele eines etheralen Lösungsmittels;
Benzol, Toluol, Hexan und Cyclohexan sind typische Hydrocarbon-Lösungsmittel und Dichlormethan
und Dichlorethan sind repräsentative
Halohydrocarbon-Lösungsmittel.
In den Fällen,
wobei das Produkt als das Säureadditionssalz
isoliert wird, wird die freie Base durch Techniken erhalten, die
dem Fachmann bekannt sind.
-
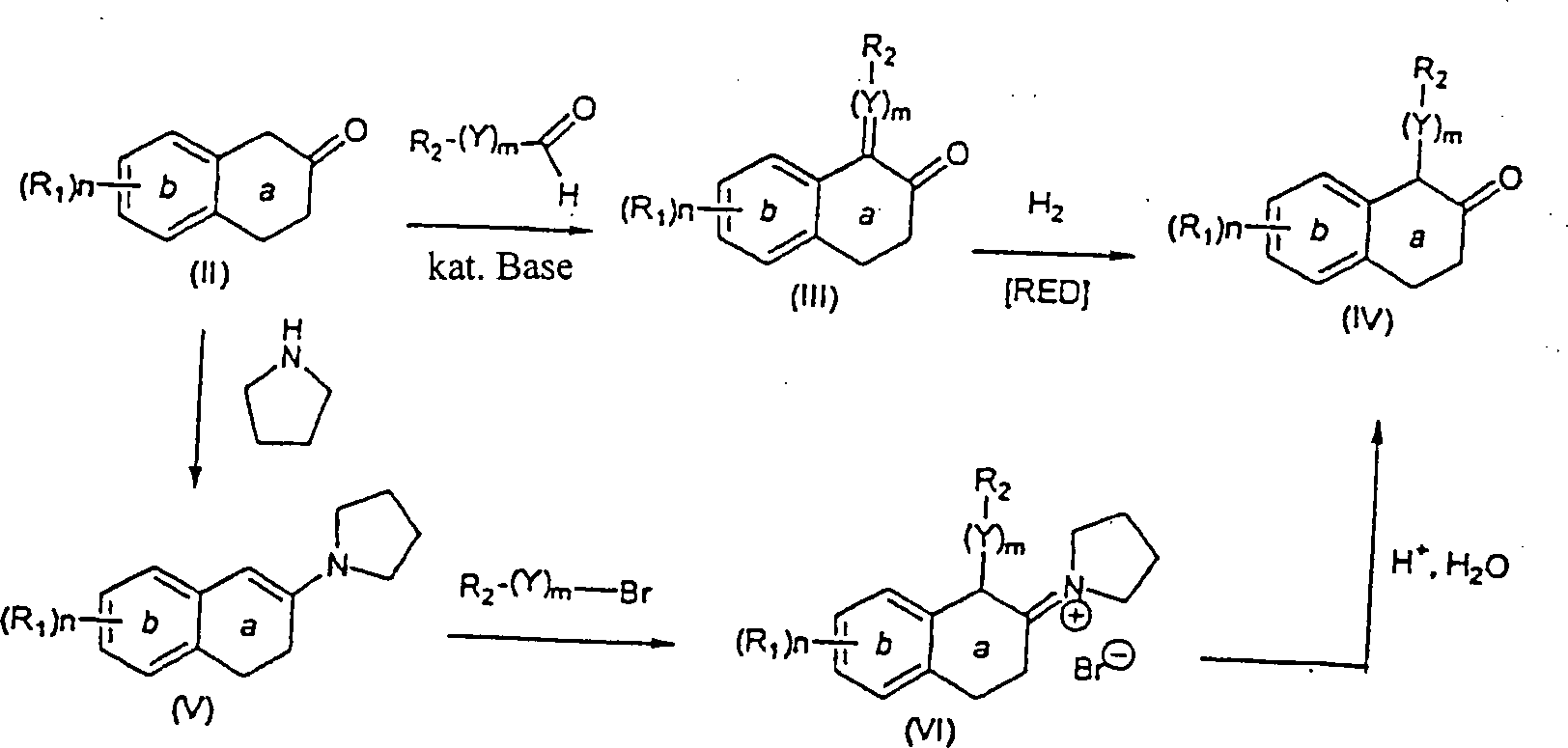
Spezifisch,
wird ein geeignet substituiertes β-Tetralon
(II) mit einem Aryl- oder Heteroarylaldehyd in der Anwesenheit einer
Base, wie zum Beispiel Piperidin, in einem inerten Halohydrocarbon-,
etheralen oder Hydrocarbon-Lösungsmittel,
wie zum Beispiel Benzol, von Umgebungstemperatur bis Reflux reagiert,
um das entsprechende α-Benzylidenyl-β-tetralon
oder α-Heteroarylmethylidenyl-β-tetralon
(III) zu erhalten. Das β-Tetralon
(III) wird in einem inerten Hydrocarbon, etheralem, Ester- oder
Alkohol-Lösungsmittel,
wie zum Beispiel Methanol, gelöst
und mit Wasserstoffgas von Umgebungsdruck bis zu ungefähr 100 psi
in der Anwesenheit eines geeigneten Katalysators, wie zum Beispiel
Palladium auf Kohle, reagiert. Die Reaktion wird bei einer Temperatur
von Umgebungstemperatur bis Reflux durchgeführt, um das gewünschte α-substituierte-β-Tetralonprodukt
(IV) zu ergeben (Schema 1).
-
Ein
alternatives Verfahren zur Herstellung der α-substituierten-β-Tetralone
(IV) schließt
die Reaktion eines geeignet substituierten β-Tetralons (II) mit einer Base,
wie zum Beispiel Pyrrolidin in einem inerten Halohydrocarbon-Lösungsmittel,
wie zum Beispiel Dichlormethan oder Hydrocarbon-Lösungsmittel,
wie zum Beispiel Benzol, unter Dean-Stark Bedingungen (Entfernung
von Wasser) oder in einem alkoholischen Lösungsmittel, wie zum Beispiel
Methanol, bei einer Temperatur von Umgebungstemperatur bis Reflux
ein, um Enamin (V) zu erhalten. Die Alkylierung des Enamins (V)
wird durch Reaktion mit einem benzylischen, heterocyclischen Alkyl-
oder einem allylischen Halid in einem inerten Lösungsmittel, wie zum Beispiel
Acetonitril, bei einer Temperatur von Umgebungstemperatur bis Reflux
erreicht, um das α-substituierte-β-Iminiumsalz
(VI) zu erhalten. Hydrolyse des Salzes (VI), um das gewünschte α-substituierte-β-Tetralonprodukt
(IV) herzustellen, wird durch Reaktion von (VI) mit Wasser und einer
anorganischen oder organischen Säure,
wie zum Beispiel Hydrochlor- oder
Eisessigsäure
in einem inerten Hydrocarbon-, etheralem, alkoholischem oder Halohydrocarbon-Lösungsmittel,
oder einem Gemisch davon, wie zum Beispiel Methanol und Dichlormethan
erreicht (Schema 1).
-
-
Die α-substituierten-β-Tetralone
(IV) werden in die entsprechenden Aminotetraline über Reaktion
mit einem Ammoniumsalz, wie zum Beispiel Ammoniumacetat in der Anwesenheit
eines Reduktionsmittels, wie zum Beispiel Natriumcyanoborhydrid,
zum Beispiel in einem inerten Halohydrocarbon-, Hydrocarbon-, etheralem
oder alkoholischem Lösungsmittel,
wie zum Beispiel Methanol, überführt, um
das cis-Aminotetralin herzustellen. In einigen Fällen wird das trans-Aminotetralin
(VIII) auch als ein Nebenprodukt gebildet. Die cis-Aminotetraline können auch
als Säureadditionssalze
durch Behandlung mit einer organischen oder anorganischen Säure isoliert
werden, wie zum Beispiel Trifluoressigsäure oder Hydrochlorsäure (Schema
2).
-
-
Ein
alternatives Verfahren zur Herstellung der α-substituierten-β-Aminotetraline
(VII) besteht aus der Reaktion eines geeignet α-substituierten-β-Tetralons
mit Dibenzylamin in einem inerten Halohydrocarbon- etheralem, alkoholischem
oder Hydrocarbon-Lösungsmittel,
wie zum Beispiel Benzol, unter Dean-Stark Bedingungen (Entfernung
von Wasser) ein, um Enamin (IX) zu erhalten. Die Alkylierung des
Enamins (IX) wird durch Reaktion mit einem benzylischen oder heterocyclischen
Halid in einem inerten Lösungsmittel,
wie zum Beispiel Acetonitril, bei einer Temperatur von Umgebungstemperatur
bis Reflux bewirkt, um das α-substituierte-β-Iminiumsalz
(X) zu erhalten. Das Iminiumsalz (X) wird in einem inerten Hydrocarbon-,
etheralem oder Ester-Lösungsmittel,
wie zum Beispiel Ethylacetat oder alkoholischen Lösungsmittel,
wie zum Beispiel Methanol, gelöst und
mit Wasserstoffgas bei einem Druck von Umgebungsdruck bis 6,89 bar
(100 psi) in der Anwesenheit eines geeigneten Katalysators, wie
zum Beispiel Palladium auf Kohle, bei einer Temperatur von Umgebungstemperatur
bis Reflux reagiert, um das gewünschte β-Aminotetralin
(VII) zu ergeben (Schema 3).
-
-
Die
oben beschriebenen β-Aminotetraline
werden über
geeignete Amidierungsverfahren (siehe Gross und Meienhofer, Eds., "The Peptides", Vols. 1–3, Academic
Press, New York, NY, 1979–1981)
acyliert. Eine Carbonsäure
wird über
Peptid-Kopplungsverfahren, die dem Fachmann bekannt sind, in einen
aktivierten Ester überführt, und
anschließend
mit einem Aminotetralin (VII) reagiert, um das entsprechende Amidprodukt
zu erhalten. Zum Beispiel wird eine Carbonsäure, wie zum Beispiel trans-4-(2-Naphthylsulfonamido)methylcyclohexancarbonsäure oder
4-(tert-Butoxycarbonyl)aminomethylcyclohexancarbonsäure mit
HBTU (2-(1H-Benzotrazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorphosphat
und einem β-Aminotetralin
(VII) in der Anwesenheit einer Base, wie zum Beispiel Diisopropylethylamin,
in einem inerten Lösungsmittel,
wie zum Beispiel N,N-Dimethylformamid, bei einer Temperatur von
Umgebungstemperatur bis Reflux reagiert, um jeweils Amid (XI) oder
(XII) zu erhalten. Abspaltung der BOC (Butoxycarbonyl) Schutzgruppe
mit Trifluoressigsäure
produziert das freie Amin, das sulfonyliert wird, um Amid (XI) zu
ergeben.
-
Alternativ
wird die Sulfonamidocarbonsäure
mit einer Aminbase, wie zum Beispiel Triethylamin, in einem inerten
Hydrocarbon-, etheralem oder Halohydrocarbon-Lösungsmittel, wie zum Beispiel
Dichlorethan behandelt, und anschließend mit Isobutylchlorformat
bei einer Temperatur von ungefähr –20°C bis 80°C reagiert. Dieses
Gemisch wird dann mit β-Aminotetralin (VII)
in einem geeigneten inerten Lösungsmittel,
wie zum Beispiel Dichlormethan, bei einer Temperatur von ungefähr –20°C bis Reflux
reagiert, um das Tetralinamid (XI) zu erhalten.
-
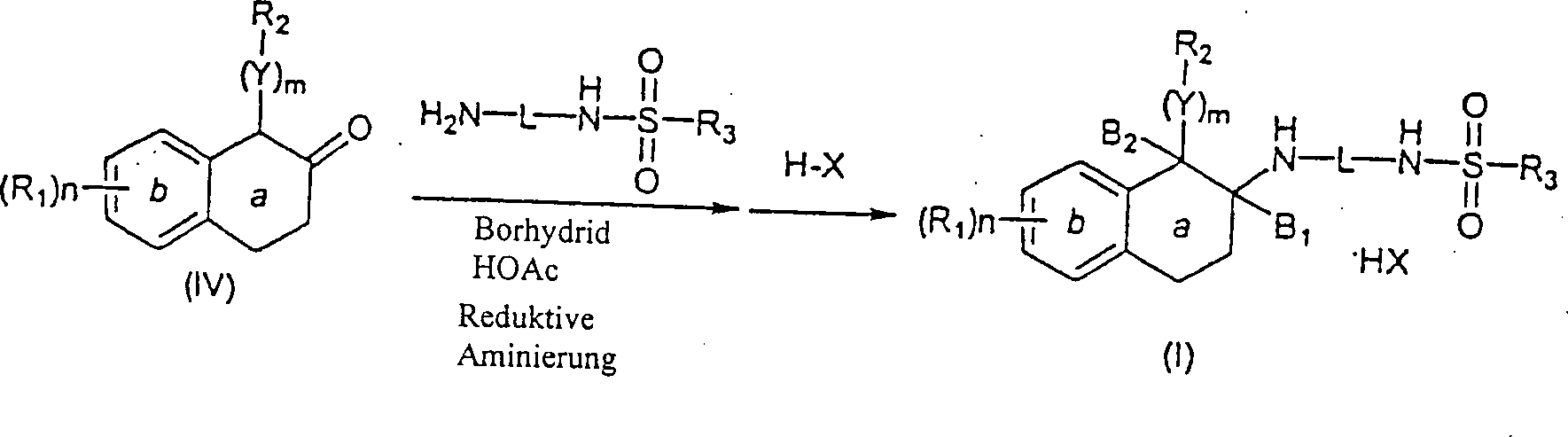
Die
N-substituierten Aminotetralin-Verbindungen (I) der Erfindung werden über Reduktion
von Tetralinamid (XI) durch Reaktion mit einem geeigneten Reduktionsmittel,
wie zum Beispiel Bor-Tetrahydrofurankomplex oder Lithiumaluminiumhydrid
in einem inerten Hydrocarbon-Lösungsmittel,
wie zum Beispiel Toluol oder etheralem Lösungsmittel, wie zum Beispiel
Tetrahydrofuran, bei einer Temperatur von Umgebungstemperatur bis
Reflux hergestellt. Das finale Produkt kann als ein Säureadditionssalz
nach Behandlung mit einer geeigneten organischen Säure, wie
zum Beispiel Trifluoressigsäure
oder anorganischen Säure,
wie zum Beispiel Hydrochlorsäure,
isoliert werden (Schema 4).
-
-
Ein
alternatives Verfahren zur Synthese von N-substituierten Aminotetralinen
(I) umfaßt
die Reaktion eines geeignet α-substituierten β-Tetralons
(IV) mit einem Amin (H2N-L-NHSO2-R3)
in der Anwesenheit eines Reduktionsmittels, wie zum Beispiel Natriumborhydrid,
oder zum Beispiel Natriumtriacetoxyborhydrid, in einem inerten etheralen,
Halohydrocarbon- oder alkoholischen Lösungsmittel, wie zum Beispiel
jeweils Dichlormethan oder Methanol, bei einer Temperatur von Umgebungstemperatur
bis Reflux, um das gewünschte
N-substituierte Aminotetralinprodukt (I) zu ergeben (Schema 5).
-
-
In
den obigen Reaktionsschemata ist X Halo, wie zum Beispiel Chlor,
Brom und Jod und Ph ist Phenyl.
-
BEISPIELE
-
Die
folgenden Beispiele beschreiben die Erfindung genauer. Alle Verbindungen
wurden durch eine Vielzahl von Verfahren, einschließlich Kernmagnetresonanz-Spektroskopie,
Massenspektrometrie und, in einigen Fällen, Infrarotspektroskopie
und Elementaranalyse identifiziert. Kernmagnetresonanz (300 MHz NMR)-Daten
sind in Teilen pro Million downfield von Tetramethylsilan angegeben.
Die Massenspektrum-Daten sind in Masse/Ladungs-(m/z) Einheiten angegeben.
Wenn nicht anders angegeben, wurden die in den Beispielen verwendeten
Materialien von leicht erhältlichen
kommerziellen Quellen erhalten oder durch Standardverfahren synthetisiert,
die dem Fachmann bekannt sind.
-
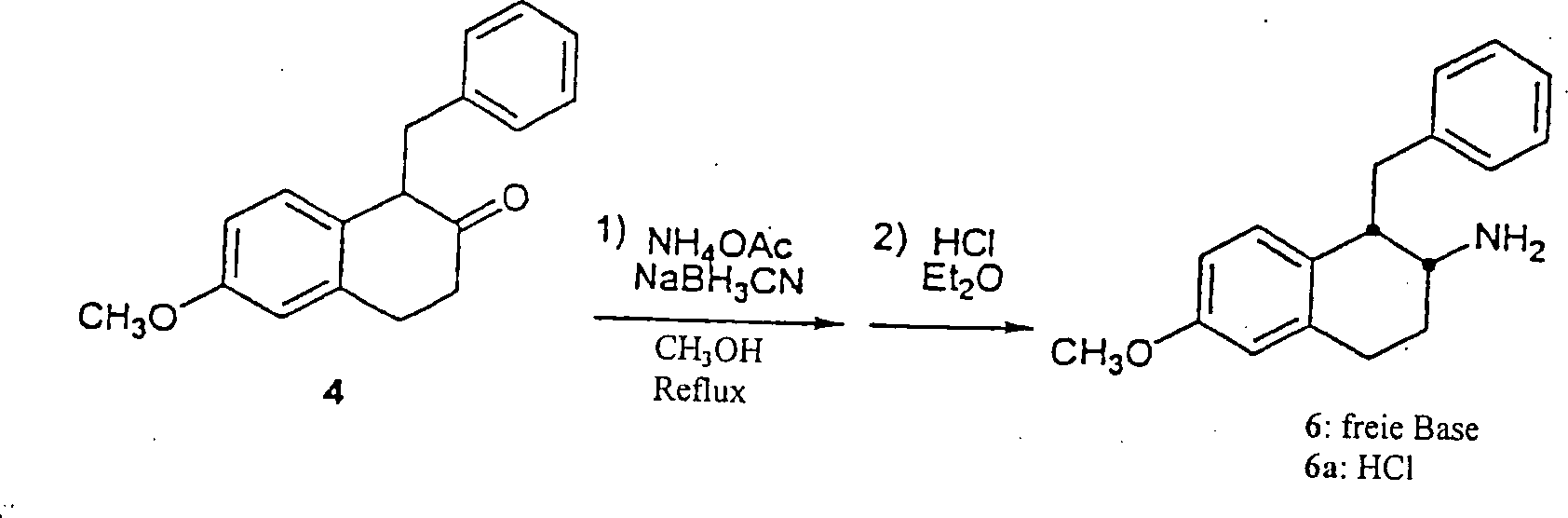
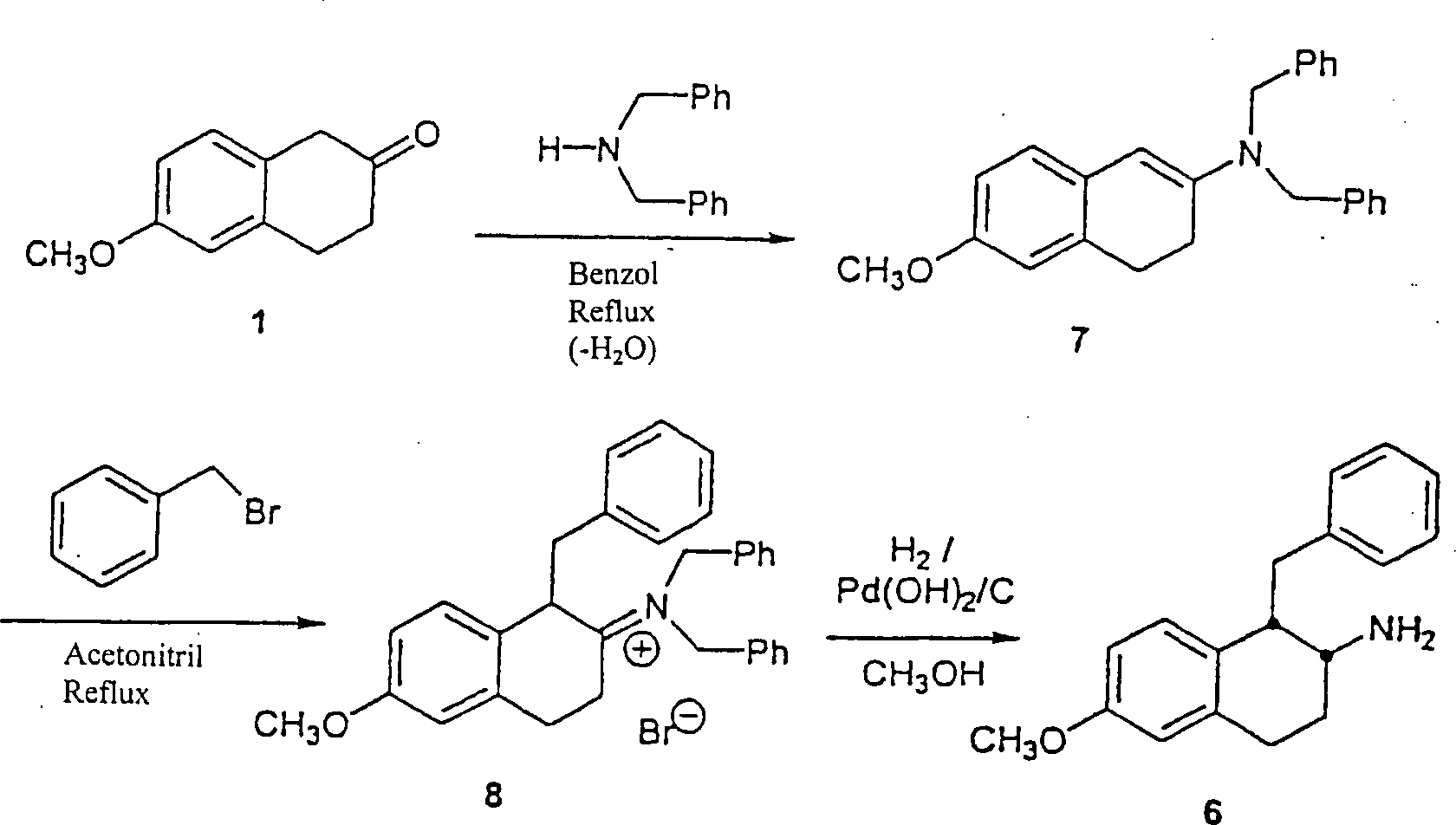
BEISPIEL 1
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]2-naphthalinsulfonamid
(10)
-
A.
6-Methoxy-β-tetralon
1 (3,0 g, 17,0 mmol) wurde in eine 250 ml Rundboden Flasche plaziert
und in Benzol (90 ml) gelöst.
Pyrrolidin (2,4 ml, 28,8 mmol) wurde unter Rühren hinzugefügt und die
Flasche wurde mit Argon gespült.
Eine Dean-Stark Falle und ein Refluxkondensator wurden angebracht,
und die Lösung
wurde für
67 Stunden auf Reflux erhitzt. Nach Abkühlen wurde das Lösungsmittel
in vacuo entfernt, um Enamin 2 als einen orangen glasar tigen Feststoff
zu ergeben, der in anschließenden
Reaktionen ohne weitere Reinigung verwendet wurde. MS (MH+) 230; 1H NMR (CDCl3) δ 1,92
(m, 4H), 2,45 (t, 2H), 2,84 (t, 2H), 3,26 (m, 4H), 3,79 (s, 3H),
5,11 (s, 1H), 6,65 (m, 2H), 6,81 (m, 1H).
-
B.
Enamin 2 wurde in Acetonitril (90 ml) in einer 250 ml Rundboden
Flasche gelöst
und Benzylbromid (3,4 ml, 29 mmol) wurde zu dieser Lösung unter
Rühren
hinzugefügt.
Die Flasche wurde mit Argon gespült
und ein Refluxkondensator wurde angebracht. Die Lösung wurde
bei Reflux für
19 Stunden erhitzt. Nach Abkühlen wurde
das Lösungsmittel
in vacuo entfernt und der sich ergebende orange glasartige Feststoff
mit Ethylether tituriert und wiederholt filtriert bis alle Spuren
des Benzylbromids entfernt waren. Das sich ergebende Iminiumsalz
3 wurde im nächsten
Schritt ohne weitere Reinigung verwendet. MS (MH–)
320.
-
C.
Das Iminiumsalz 3 aus der vorherigen Reaktion wurde in eine 500
ml Erlenmeyer Flasche transferiert, und Methanol (100 ml), Dichlormethan
(50 ml), Wasser (50 ml), und Eisessig (3 ml) wurden hinzugefügt. Das
sich ergebende Gemisch wurde mit Stickstoff gespült, zugedeckelt und für 14 Stunden
gerührt.
Die Lösungsmittel
wurden in vacuo entfernt. Das sich ergebende Öl wurde in Ethylacetat (250
ml) gelöst
und mit Wasser (4 × 100
ml) gewaschen. Der organische Extrakt wurde über Magnesiumsulfat getrocknet,
filtriert, und die Lösungsmittel
in vacuo entfernt, um ein öliges
Rohprodukt zu ergeben. Dieses Material wurde über Chromatographie gereinigt
(Silikagelsäule
(Abmessungen 2,5 × 27
cm); 25% Ethylacetat: 75% Hexane (v/v) als dem Eluenten). Nach Evaporation
der geeigneten Fraktionen wurde 3,4-Dihydro-6-methoxy-1-(phenylmethyl)-2(1H)-naphthalinon
4 als ein dickes gelbes Öl
(2,13 g, 8,0 mmol). erhalten. MS (MH+) 267; 1H NMR (CDCl3) δ 2,43–2,60 (m,
3H), 2,75 2,81 (m, 1H), 3,18 (dd, 1H), 3,68 (dd, 2H), 3,79 (s, 3H),
6,58–6,91
(m, 5H), 7,15 (m, 3H). (Figur 1).
-
-
Alternativ
wird 3,4-Dihydro-6-methoxy-1-(phenylmethyl)-2(1H)-naphthalinon 4
wie folgt präpariert:
-
6-Methoxy-β-tetralon
1 (1,0 g, 5,7 mmol) wurde in Benzol (25 ml) unter Rühren in
einer 50 ml Rundboden-Flasche gelöst. Zu dieser Lösung wurde
Benzaldehyd (0,60 ml, 5,9 mmol) hinzugefügt, gefolgt durch katalytisches
Piperidin (0,014 ml, 0,14 mmol). Die Flasche wurde mit Argon gespült und ein
Refluxkondensator, ausgerüstet
mit einer Dean-Stark Falle wurde angebracht. Die Lösung wurde
auf Reflux für
28 Stunden erhitzt und dann auf Raumtemperatur abgekühlt. Das
Lösungsmittel
wurde in vacuo entfernt, um ein dunkeloranges Öl zu ergeben. Dieses Rohprodukt
wurde in Ethylether (100 ml) gelöst
und dann mit 3 N HCl (2 × 50
ml), Wasser (1 × 50
ml), und zuletzt mit gesättigter
Lauge (1 × 50
ml) gewaschen. Der organische Extrakt wurde über Magnesiumsulfat getrocknet,
filtriert, und die Lösungsmittel
in vacuo entfernt. Das sich ergebende Öl wurde über Säulenchromatographie (Silikagelsäule (Abmessungen
5 × 25
cm); 25% Ethylacetat: 75% Hexane (v/v) als dem Eluenten) gereinigt.
Nach Evaporation der geeigneten Fraktionen wurde 3,4-Dihydro-6-methoxyl-(phenylmethylidenyl)-2-naphthalinon
5 als ein blaßgelbes Öl (0,70
g, 2,6 mmol) erhalten, das sich bei Lagerung in einem Kühlschrank
verfestigte. MS (MH+) 265; 1H
NMR (CDCl3) δ 2,54 (t, 2H), 2,98 (t, 2H),
3,79 (s, 3H), 6,63 (dd, 1H), 6,96 (d, 1H), 7,12 (d, 1H), 7,29 (m,
3H), 7,40–7,48
(m, 3H).
-
Verbindung
5 (0,464 g, 1,8 mmol) wurde in einer 250 ml Parr Schüttelflasche
plaziert und in Ethylacetat (25 ml) gelöst. Getrennt wurde 10% Palladium
auf Kohlenstoff (0,029 g) in einem Gefäß plaziert und dazu wurde Methanol
(25 ml) hinzugefügt,
um eine Schlämme
herzustellen. Diese Material wurde dann sorgfältig zu dem Parr Gefäß hinzugefügt und das
Gemisch wurde unter einem Druck von ungefähr 50 psi für 19 Stunden hydrogeniert.
Die Reaktionslösung
wurde über
ein Kissen Celite filtriert. Die Lösungsmittel wurden in vacuo entfernt
und das sich ergebende Öl
wurde durch Säulenchromatographie
(Silikagelsäule
(Abmessungen 2,5 × 26
cm); 25% Ethylacetat: 75% Hexane (v/v) als dem Eluenten) gereinigt.
Nach Evaporation der geeigneten Fraktionen wurde 3,4-Dihydro-6-methoxy-1-(phenylmethyl)-2(1H)-naphthalinon 4 als
ein cremefarbenes Öl (0,40
g, 1,50 mmol) (Figur 2) erhalten.
-
-
D.
Ammoniumacetat (10,7 g, 138 mmol) wurde zu einer Lösung von
3,4-Dihydro-6-methoxy-1-(phenylmethyl)-2-(1H)-naphthalinon
4 (3,64 g, 13,6 mmol) in Methanol (530 ml) in einer 1 1 Rundboden-Flasche unter
starkem Rühren
hinzugefügt.
Natriumcyanborhydrid (4,29 g, 68,3 mmol) wurde hinzugefügt und die
Flasche wurde mit Argon gespült.
Ein Kondensator wurde angebracht, und die Lösung wurde bei Reflux für 21 Stunden
erhitzt. Die Lösung
wurde auf Raumtemperatur abgekühlt,
und das Lösungsmittel
wurde in vacuo entfernt. Der cremefarbene Feststoff wurde in einem
Gemisch von Ethylether (600 ml) und 0,1 M Natriumhydroxid Lösung (225
ml) gelöst.
Die wäßrige Phase
wurde entfernt und die organischen Reste wurden durch Zugabe von
0,1 M Natriumhydroxid Lösung
(1 × 225
ml) und dann mit Wasser (1 × 200
ml) gewaschen. Die vereinigten wäßrigen Extrakte
wurden mit Ethylether (3 × 100
ml) rückextrahiert.
Die vereinigten organischen Extrakte wurden über Magnesiumsulfat getrocknet,
filtriert, und das Lösungsmittel
wurden in vacuo entfernt, um cis-1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinamin
6 zu ergeben. Das Rohprodukt wurde in Ethylether (75 ml) gelöst, und
ein Überschuß von 1
M Hydrogenchlorid in Ethylether wurde hinzugefügt. Dieses führte zu
einem Ausfallen des Produkts als ein HCl-Salz. Der Ethylether wurde
in vacuo entfernt und jegliche großen Stücke wurden mit einem Spatel
zerstoßen.
Ethylacetat (25 ml) wurde hinzugefügt, die sich ergebende Schlämme wurde
auf Reflux erhitzt und dann auf Raumtemperatur abgekühlt. Die
Feststoffe wurden abfiltriert und mit ei nem kleinen Teil von Ethylacetat
und dann mit Ethylether gespült
und über
Ausblasen getrocknet, um cis-1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinaminhydrochlorid
6a als ein cremefarbenes Pulver (2,13 g, 7,0 mmol) zu ergeben. MS
(MH+) 268; 1H NMR
(CDCl3) δ 2,05–2,30 (m,
2H), 2,50–2,60
(m, 1H), 2,83–3,03
(m, 3H), 3,30–3,40
(m, 2H), 3,71 (s, 3H), 6,00 (d, 1H), 6,35 (dd, 1H), 6,60 (d, 1H),
7,02–7,16
(m, 5H), 8,53 (bs, 1H), 8,96 (bs, 2H) (Figur 3).
-
-
Alternativ
wird cis-1,2,3,4-tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinamin
6 wie folgt hergestellt:
-
6-Methoxy-2-tetralon
1 (2,0 g, 11,3 mmol) wurde in Benzol (60 ml) in einer 100 ml Rundboden-Flasche unter
Rühren
gelöst.
N,N-Dibenzylamin (2,4 ml, 12,5 mmol) wurde hinzugefügt, und
die Flasche wurde mit Argon gespült.
Eine Dean Stark Falle und ein Kondensator wurden angebracht und
die Lösung
wurde für
19 Stunden auf Reflux erhitzt. Nach Abkühlen wurde das Lösungsmittel
in vacuo entfernt, um Enamin 7 zu ergeben, das ohne weitere Reinigung
verwendet wurde. MS (MH+) 356.
-
Enamin
7 wurde in Acetonitril (60 ml) in einer 100 ml Rundboden-Flasche
gelöst
und Benzylbromid (1,5 ml, 12,6 mmol) wurde hinzugefügt. Die
Flasche wurde mit Argon gespült
und ein Refluxkondensator wurde angebracht. Die Lösung wurde
für 14
Stunden auf Reflux erhitzt. Nach Abkühlen wurden die Lösungsmittel
in vacuo entfernt, um das Iminiumsalz 8 als eine durchscheinenden
orangen Feststoff zu ergeben, der ohne weitere Reinigung verwendet
wurde. MS (MH–)
446.
-
Ungefähr die Hälfte des
Iminiumsalzes aus der vorherigen Reaktion wurde in eine 250 ml Parr
Schüttelflasche
zusammen mit Methanol (50 ml) transferiert. Getrennt wurde 10% Palladiumhydroxid
auf Kohle (0,30 g) in einem Gefäß plaziert
und Methanol (50 ml) wurde sorgfältig
hinzugefügt,
um eine Schlämme
zu bilden. Dieses Material wurde zu der Iminiumsalz-Lösung hinzugefügt und das
Gemisch wurde unter einem Druck von ungefähr 3,45 bar (50 psi) für 17 Stunden
hydrogeniert. Die Reaktionslösung
wurde über
ein Kissen von Celite filtriert, um den Katalysator zu entfernen.
Das Lösungsmittel
wurde in vacuo entfernt. Das sich ergebende Öl wurde in Ethylacetat (300
ml) gelöst
und diese Lösung
wurde mit 0,2 M Natriumhydroxid Lösung (2 × 125 ml) und dann mit Wasser
(1 × 100
ml) gewaschen. Die wäßrigen Phasen
wurden mit Ethylacetat (1 × 50
ml) rückextrahiert.
Die vereinigten organischen Extrakte wurden über Magnesiumsulfat getrocknet,
filtriert, und das Lösungsmittel
wurde in vacuo entfernt. Das sich ergebende Öl wurde über Chromatographie gereinigt
(Silikagelsäule
(Abmessungen 5 × 28
cm) wobei zuerst mit Dichlormethan (400 ml) und dann mit Dichlormethan/Aceton/Methanol
(50 : 50 : 5) (v/v) eluiert wurde. Nach Evaporation der geeigneten
Fraktionen, wurde cis-1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinamin
6 als ein braunes Öl
(0,37 g, 1,4 mmol) erhalten. MS (MH+) 268; 1H NMR (CDCl3) δ 1,45 (bs,
2H), 1,86 (m, 2H), 2,80–3,07
(m, 5H), 3,20 (m, 1H), 3,75 (s, 3H), 6,52–6,67 (m, 3H), 7,10–7,30 (m,
5H). (Figur 4).
-
-
E.
trans-4-(2-Naphthylsulfonamido)methylcyclohexancarbonsäure (0,394
g, 1,13 mmol) wurde in einer 50 ml Rundboden-Flasche plaziert und
in Dichlormethan (10 ml) suspendiert. Triethylamin (0,32 ml, 2,3
mmol) wurde hinzugefügt,
was zu einer Lösung
führte.
Isobutylchlorformat (0,29 ml, 2,3 mmol) wurde langsam hinzugefügt und das
Gemisch wurde für
1 Stunde gerührt,
wobei vermutlich die Anhydridspezies gebildet wurde. cis-1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinamin
6 (0,364 g, 1,36 mmol) wurde in Dichlormethan (10 ml) gelöst, und
diese Lösung
wurde zu der oben präparierten
Lösung
hinzugefügt.
Das Reaktionsgemisch wurde für
3 Stunden bei Umgebungstemperatur gerührt und zu dieser Zeit wurde
zusätzliches
Dichlormethan (50 ml) hinzugefügt.
Dieses Gemisch wurde mit 0,25 M Natriumhydroxid Lösung (35
ml) gewaschen. Die organische Lage wurde abgetrennt und die wäßrige Lage
wurde mit zusätzlichem
Dichlormethan (2 × 25
ml) extrahiert. Die organischen Extrakte wurden vereinigt und mit
Lauge (1 × 25
ml) gewaschen. Die organischen Stoffe wurden über Magnesiumsulfat getrocknet,
filtriert, und das Lösungsmittel
wurden in vacuo entfernt. Der erhaltene Rest wurde über Chromatographie
gereinigt (Silikagelsäule
(Abmessungen 2,5 × 26 cm)
wobei mit einem Gradienten von: 100% Dichlormethan (100 ml), 98
: 2 Dichlormethan/Aceton (100 ml), 96 : 4 Dichlormethan/Aceton (100
ml), 94 : 6 Dichlormethan/Aceton (100 ml), 92 : 8 Dichlormethan/Aceton
(100 ml) und dann der Rest mit 90 : 10 Dichlormethan/Aceton (100
ml) eluiert wurde. Nach Evaporation der geeigneten Fraktionen wurde [1α,2α(trans)]-4-[[(2-Naphthalinylsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinyl]-cyclohexancarboxamid
9 (0,314 g, 0,526 mmol) als ein cremefarbenes Pulver erhalten. MS
(MH+) 597; 1H NMR
(DMSOd6) δ 0,71–0,85 (m,
2H), 1,25–1,38
(m, 3H), 1,69 (m, 5H), 1,90 (m, 1H), 2,10 (m, 1H), 2,43–2,63 (m,
3H), 2,79–2,96
(m, 3H), 3,11 (s, 1H), 3,65 (s, 3H), 3,82–3,92 (m, 1H), 6,31 (d, 1H),
6,45 (dd, 1H), 6,63 (d, 1H), 6,97 (app d, 2H), 7,13–7,26 (m,
3H), 7,68 (m, 3H), 7,85 (app d, 2H), 8,05 (app d, 1H), 8,15 (m, 2H),
8,44 (s, 1H).
-
F.
Das Amid 9 von der vorhergehenden Reaktion (0,282 g, 0,473 mmol)
wurde in Tetrahydrofuran (20 ml) in einer 50 ml Rundboden-Flasche
suspendiert und Lithiumaluminumhydrid Lösung (1,4 ml einer 1 M Lösung in
THF) wurde hinzugefügt.
Die Flasche wurde mit Argon gespült
und ein Kondensator wurde angebracht. Das Reaktionsgemisch wurde
auf Reflux erhitzt und während
des Verlaufs der Reaktion wurde mehr LAH Lösung hinzugefügt (2,5
ml) und mehr THF wurde hinzugefügt
(20 ml). Nach einer Refluxperiode von 50 Stunden wurde die Reaktion
auf Raumtemperatur abgekühlt
und Überschuß Ethylacetat
wurde hinzugefügt,
um das verbleibende LAH abzulöschen.
Die Lösung
wurde über
Celite filtriert, um die anorganischen Salze zu entfernen. Die Lösungsmittel
wurden in vacuo entfernt. Das Rohprodukt wurde in Ethylacetat (150
ml) gelöst
und mit 1 M Hydrochlorsäure
(2 × 50
ml) gewaschen. Der organische Extrakt wurde über Magnesiumsulfat getrocknet, filtriert,
und das Lösungsmittel
wurde in vacuo entfernt. Überschuß etherales
Hydrogenchlorid (ca. 15 ml einer 1 M Lösung) wurde hinzugefügt und die
Lösungsmittel
und Überschuß HCl wurden
in vacuo entfernt. Das Produkt wurde aus Ethylacetat (15 ml)/Aceton
(19 ml) rekristallisiert, um [1α,2α (trans)]-N-[[[[(1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-naphthalinsulfonamidhydrochlorid
10a (0,082 g, 0,132 mmol) als ein weißes Pulver zu ergeben. MS (MH+) 583; 1H NMR (DMSO-d6) δ 0,82–1,07 (m,
4H), 1,39 (m, 1H), 1,64–1,96
(m, 5H), 2,17 (m, 2H), 2,45 (m, 1H), 2,65 (m, 2H), 2,83–3,12 (m,
6H), 3,47 (m, 1H), 3,64 (s, 3H), 5,84 (d, 1H), 6,31 (d, 1H), 6,68
(app s, 1H), 7,06 (m, 2H), 7,27 (m, 4H), 7,70 (m, 2H), 7,83 (app
d, 1H), 8,05 (app d, 1H), 8,16 (m, 2H), 8,43 (s, 1H), 8,95 (bs,
2H). (Figur 5).
-
-
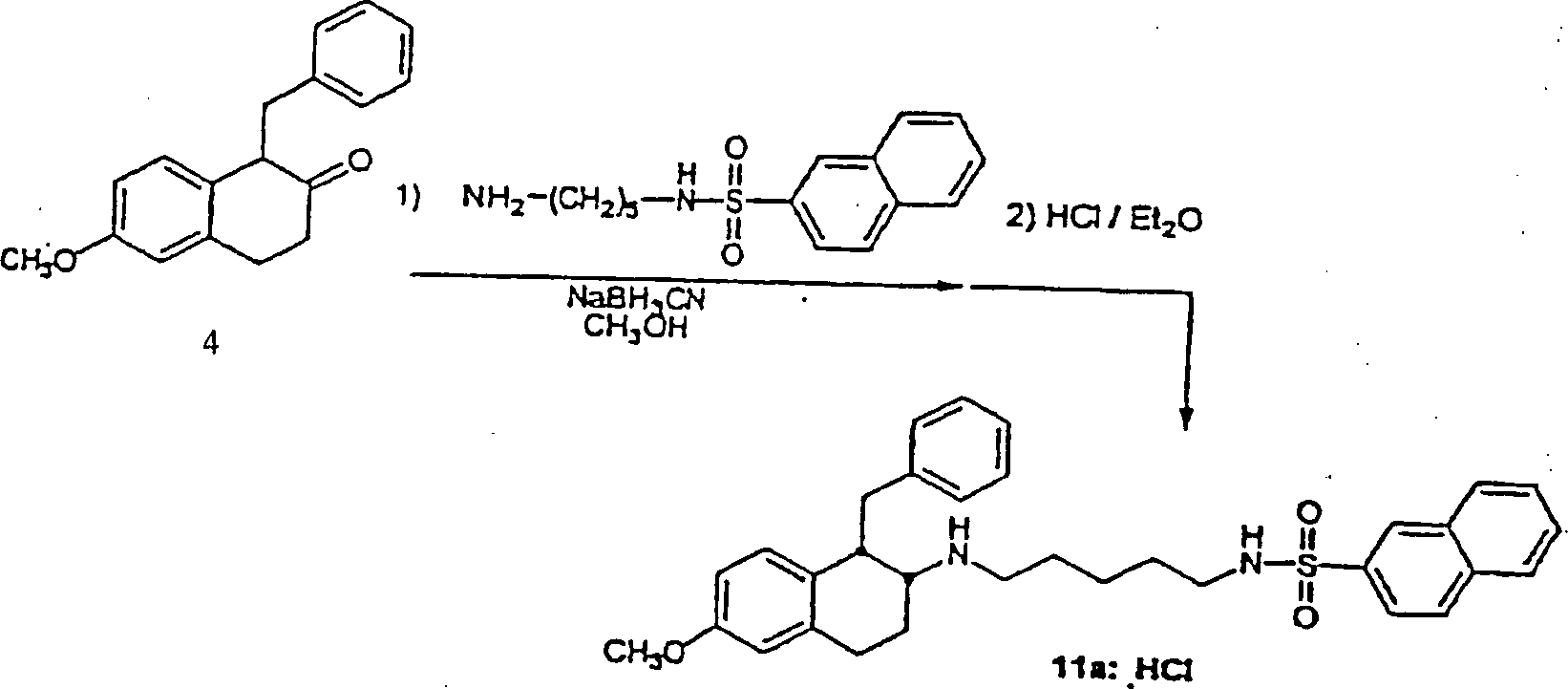
BEISPIEL 2
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinyl]amino]-5-pentyl]-2-naphthalinsulfonamid
(11)
-
3,4-Dihydro-6-methoxy-1-(phenylmethyl)-2(1H)-naphthalinon
4 (0,136 g, 0,511 mmol) wurde in Methanol (5 ml) in einem 20 ml
Schraubdeckelgefäß, ausgerüstet mit
einem Rührstab,
gelöst.
Nach Lösung
wurde 1-Amino-5-(2-naphthalinylsulfonamido)pentanhydrochloridsalz
(0,170 g, 0,517 mmol) hinzugefügt,
gefolgt von Natriumcyanoborhydrid (0,098 g, 1,60 mmol). Das Gefäß wurde
mit Stickstoff gespült
und zugedeckelt. Das Rühren
wurde für
17 Stunden fortgeführt,
wonach Dichlormethan (25 ml) und gesättigtes Natriumbicarbonat (25
ml) hinzugefügt
wurde. Die organischen Stoffe wurden entfernt und die wäßrige Lage
wurde mit Dichlormethan (2 × 25
ml) extrahiert. Die organischen Extrakte wurden vereinigt und mit
Lauge (1 × 25
ml) gewaschen, über
Magnesiumsulfat getrocknet, filtriert, und das Lösungsmittel wurde in vacuo
entfernt. Das Rohprodukt wurde über
Chromatographie (Silikagelsäule
(Abmessungen 2,5 × 17
cm); 25% Dichlormethan: 75% Aceton (v/v) als dem Eluenten) gereinigt.
Nach Evaporation der geeigneten Fraktionen, wurde das Produkt in
Ethylether gelöst
und 1 M Hydrogenchlorid in Ethylether wurde hinzugefügt, um [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(phenylmethyl)-2-naphthalinyl]amino]-5-pentyl]-2-naphthalinsulfonamidhydrochlorid
11a (0,036 g, 0,062 mmol) als ein cremefarbenes Pulver auszufällen. MS
(MH+) 543, (Figur 7)
-
-
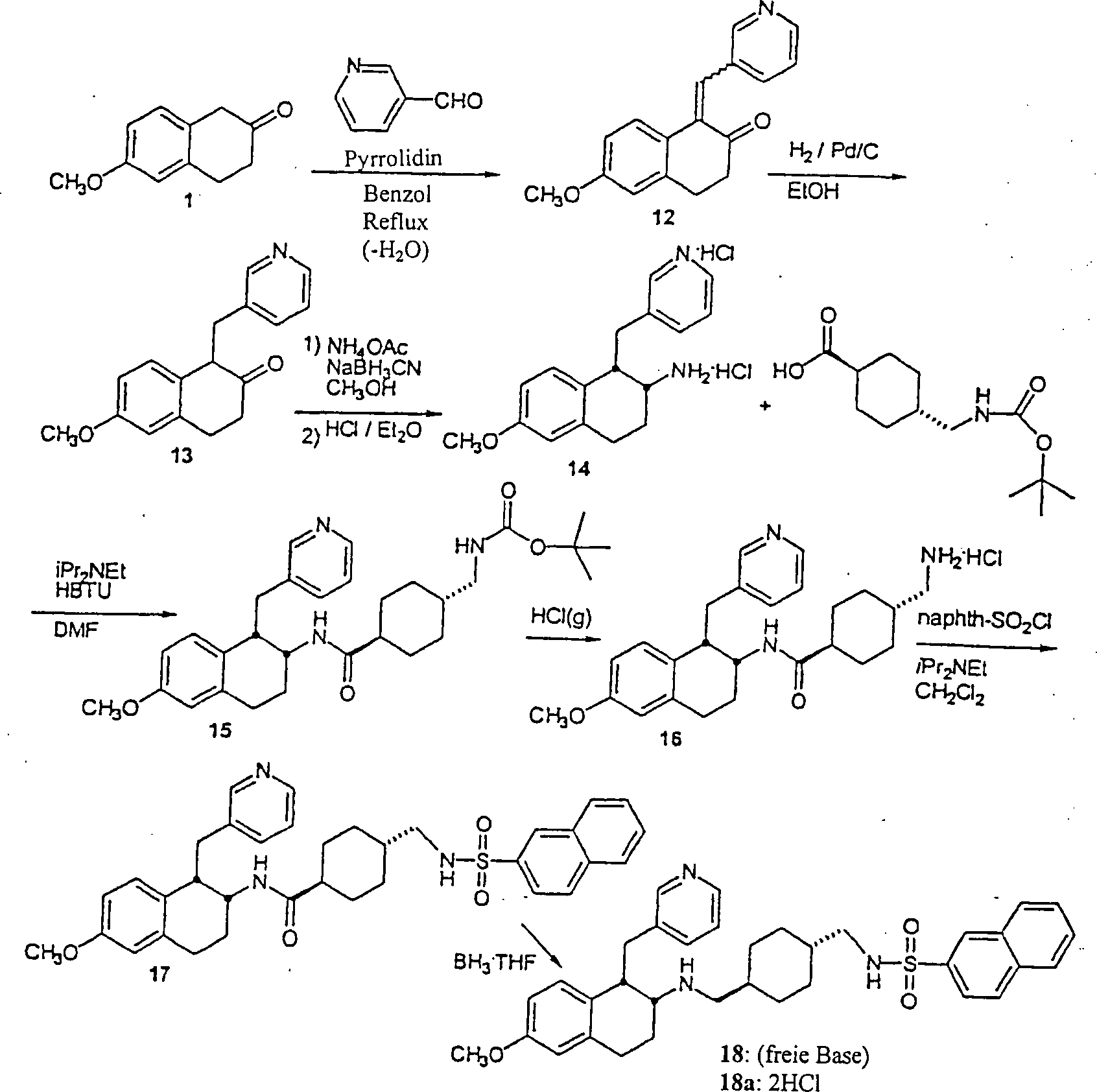
BEISPIEL 3
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-naphthalinsulfonamid
(18)
-
- A. 6-Methoxy-β-tetralon 1 (2,0 g, 11,3 mmol)
und Diisopropylethylamin (0,20 ml, 1,1 mmol) wurden in Benzol (60
ml) unter Rühren
in einer 100 ml Rundboden-Flasche gelöst. 3-Pyridylcarboxaldehyd (1,1 ml, 11,7 mmol)
wurde hinzugefügt,
und das Reaktionsgefäß wurde
mit Argon gespült
und eine Dean-Stark Falle mit Reflux Kondensator wurde angebracht.
Das Gemisch wurde für
19 Stunden auf Reflux erhitzt. Nach Abkühlen zeigte die HPLC Analyse,
daß sich
keine Produkte gebildet hatten. Piperidin (0,094 ml, 1,1 mmol) wurde zu
dieser Zeit hinzugefügt
und Erhitzen bei Reflux wurde für
23 Stunden fortgeführt.
Das Lösungsmittel
wurde in vacuo entfernt, um einen glasartigen orangefarbenen Feststoff
zu ergeben. Eine chromatographische Reinigung (Silikagelsäule (Abmessungen
5 × 29
cm), wobei mit einem Gradienten eluiert wurde von: 100% Hexan (400
ml), 75%/25% Hexan/Ethylacetat (v/v) (400 ml), 50%/50% Hexan/Ethylacetat
(v/v) (400 ml), 25%/75% Hexan/Ethylacetat (v/v) (400 ml), und zuletzt
mit 100% Ethylacetat) wurde durchgeführt. Nach Evaporation der geeigneten
Fraktionen wurde 3,4-Dihydro-6-methoxy-1-((3-pyridinyl)methylidenyl)-2-naphthalinon
12 (1,484 g, 5,59 mmol) als ein orangefarbenes Öl erhalten, das sich nach Stehen
im Kühlschrank
verfestigte. MS (MH+) 266. 1H
NMR (CDCl3) δ 2,67 (t, 2H), 3,02 (t, 2H),
3,83 (s, 3H), 6,60 (dd, 1H), 6,82 (d, 1H), 7,19 (m, 2H), 7,51 (s,
1H). 7,71 (d, 1H), 8,49 (dd, 1H), 8,65 (d, 1H).
- B. Das Naphthalin-2-on 12 (1,442 g, 5,44 mmol), wie oben erhalten,
wurde in absolutem Ethanol (50 ml) gelöst und in eine 250 ml Parr
Hydrogenierungsflasche transferiert. Getrennt wurde sorgfältig Ethanol
zu 10% Palladium auf Kohle (0,020 g) hinzugefügt und diese Schlämme wurde
zu der Parr Flasche hinzugefügt.
Das Gemisch wurde unter einem Druck von 3,45 bar (50 psi) für 16 Stunden
hydrogeniert. Der Katalysator wurde durch Filtration über Celite
entfernt. Spektroskopische Untersuchung zeigte die Anwesenheit von
einigem Ausgangsmaterial und so wurde mehr Palladiumkatalysator
zu der Ethanollösung
hinzugefügt und
die Hydrogenierung wurde für
20 Stunden wiederholt. Der Katalysator wurde durch Filtration über Celite
entfernt. Entfernung der Lösungsmittel
in vacuo ergab 3,4-Dihydro-6-methoxy-1-(3-pyridinykmethyl)-2(1H)-naphthalinon
13 als ein orangefarbenes Öl,
das in dem nächsten
Schritt ohne weitere Reinigung verwendet wurde. MS (MH+)
268.
- C. Das Naphthalin-2-on 13, wie oben erhalten, wurde in Methanol
(275 ml) in einer 11 Rundboden-Flasche gelöst. Ammoniumacetat (4,27 g;
55,4 mmol) wurde zu der gerührten
Methanollösung
hinzugegeben und es wurde dieser ermöglicht, sich vor dem Fortschreiten
vollständig
zu lösen.
Natriumcyanborhydrid (1,703 g, 27,5 mmol) wurde dann zu der Methanollösung hinzugegeben.
Das Reaktionsgefäß wurde
mit Stickstoff gespült
und die Lösung
für 18
Stunden Reflux-behandelt. Die Lösungsmittel
wurden dann in vacuo entfernt um einen gelben Feststoff zu ergeben,
der in Ethylether (500 ml) und 0,1 M Natriumhydroxidlösung (275 ml)
gelöst
wurde. Die organische Phase wurde entfernt und mit einer weiteren
0,1 M Natriumhy droxidlösung (275
ml) und mit Wasser (250 ml) gewaschen. Die vereinigten Waschungen
wurden mit Ethylether (3 × 100 ml)
rückextrahiert.
Die organischen Extrakte wurden vereinigt und über Natriumsulfat getrocknet.
Die Lösungsmittel
wurden in vacuo entfernt und der Rest wurde in Ethylether und eine
minimale Menge an Dichlormethan aufgenommen. Ein Überschuß von 1
M Hydrogenchlorid in Ethylether wurde hinzugefügt und ein dunkelbraunes Präzipitat
bildete sich. Das Lösungsmittel
wurde in vacuo entfernt und der sich ergebende Feststoff wurde mit
Ether tituriert und in einem Vakuumofen getrocknet, um 1,2,3,4-Tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinaminbishydrochlorid
14 als einen braun-orangen Feststoff (1,208 g, 3,54 mmol) zu ergeben.
MS (MH+) 269; 1H
NMR (DMSO-d6) δ 1,95–2,20 (m,
2H), 2,68–3,29
(m, 4H), 3,30–3,48
(m, 2H), 3,69 (s, 3H), 5,98 (d, 1H), 6,41 (dd, 1H), 6,75 (d, 1H),
7,98 (dd, 1H), 8,36 (d, 1H), 8,68–8,89 (m, 5H).
- D. Das 2-Naphthalinamin 14 (1,193 g, 3,50 mmol) wurde in N,N-Dimethylformamid
(30 ml) in einer 100 ml Rundboden-Flasche gelöst und Diisopropylethylamin
(2,0 ml, 11,5 mmol) wurde zu der Lösung hinzugefügt. N-(tert-Butoxycarbonyl)aminomethylcyclohexancarbonsäure (0,912
g, 3,54 mmol) wurde hinzugefügt,
gefolgt von HBTU (1,336 g, 3,52 mmol). Das Reaktionsgemisch wurde
für 2 Stunden
gerührt
und dann in Wasser (400 ml) gegossen. Ein feines Präzipitat
bildete sich, das durch Zentrifugation, gefolgt von Dekantieren, Hinzufügen von
frischem Wasser und Re-Zentrifugation, gefolgt von einem finalen
Dekantieren, abgetrennt wurde. Das verbleibende Material wurde in
einem Vakuumofen getrocknet und dann über Chromatographie gereinigt
(Silikagelsäule
(5 × 17
cm) wobei mit einem Gradienten von: 75% Hexan/Ethylacetat (v/v)
(300 ml), 50% Hexan/Ethylacetat (300 ml), 25% Hexan/Ethylacetat
(300 ml), und zuletzt mit 100% Ethylacetat eluiert wurde. Nach Evaporation
der geeigneten Fraktionen wurde der sich ergebende gelbe Feststoff
mit Ethylether tituriert und dann in einem Vakuumofen getrocknet,
um [1α,2α(trans)]-4-[[(tert-Butoxycarbonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinyl]-cyclohexancarboxamid
15 (0,629 g, 1,24 mmol) zu ergeben. MS (MH+)
508; 1H NMR (DMSOd6) δ 0,81–1,04 (m,
2H), 1,31–1,54
(m, 13H), 1,70–2,02
(m, 7H), 2,80–3,04
(m, 6H), 3,35 (m, 1H), 3,79 (s, 3H), 4,27 (m, 1H), 4,59 (m, 1H),
5,42 (d, 1H), 6,58–6,77
(m, 3H), 7,47 (d, 1H), 8,34 (s, 1H), 8,48 (d, 1H).
- E. Das oben erhaltene Carboxamid 15 (0,603 g, 1,19 mmol) wurde
in 100 ml Dioxan in einer 250 ml Rundboden-Flasche suspendiert.
Während
Abkühlung
mit einem Eisbad wurde bis zur Sättigung
Hydrogenchloridgas in die Lösung
geblasen. Das Lösungsmittel
wurde in vacuo entfernt und das sich ergebende Material wurde in
Methanol gelöst,
und ein Überschuß von etheralem
Hydrogenchlorid wurde hinzugefügt.
Das Lösungsmittel
wurde in vacuo entfernt und das sich ergebende Produkt wurde mit
Ethylether tituriert und filtriert. Der sich ergebende hygroskopische
cremefarbene Feststoff wurde bei 40°C in einem Vakuumofen getrocknet,
um [1α,2α(trans)]-4-(Aminomethyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinyl]-cyclohexancarboxamid-bis-hydrochlorid
16 (0,502 g, 1,04 mmol) zu erhalten. MS (MH+)
408; 1H NMR (DMSO-d6) δ 0,80–1,03 (m,
2H), 1,19–1,42
(m, 2H), 1,44–1,89
(m, 6H), 1,93 (m, 1H), 2,10 (m, 1H), 2,56–2,70 (m, 2H), 2,71–3,01 (m,
3H), 3,09 (m, 1H), 3,34 (m, 1H), 3,70 (s, 3H), 3,91 (m, 1H), 6,58–6,63 (m,
2H), 6,71 (s, 1H), 7,87–8,11
(m, 5H), 8,22 (d, 1H), 8,59 (s, 1H), 8,75 (d, 1H).
- F. Das oben erhaltene Aminhydrochlorid 16 (0,102 g, 0,212 mmol)
wurde mit Dichlormethan (13 ml) und Diisopropylethylamin (0,125
ml, 0,718 mmol) gemischt. 2-Napthylsulfonylchlorid
(0,048 g, 0,212 mmol), gelöst
in Dichlormethan (12 ml), wurde zu dem Gemisch hinzugefügt. Die
sich ergebende Lösung
wurde für 1
Stunde gerührt,
wonach das Lösungsmittel
in vacuo entfernt wurde. Der Rest wurde in Dichlormethan (75 ml)
aufgenommen und dieses Gemisch wurde mit 0,1 M Natriumhydroxid Lösung (2 × 55 ml)
und Wasser (1 × 50
ml) gewaschen. Die organischen Stoffe wurden über Magnesiumsulfat getrocknet
und das Lösungsmittel
wurde in vacuo entfernt, um [1α,2α(trans)]-4-[[(2-Naphthalinylsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinyl]-cyclohexancarboxamid
17 (0,126 g, 0,211 mmol) zu ergeben. MS (MH+)
598.
- G. Das oben erhaltene Carboxamid 17 (0,119 g, 0,199 mmol) wurde
in Tetrahydrofuran (15 ml) in einer 100 ml Rundboden-Flasche gelöst. Bortetrahydrofuran
(2,00 ml einer 1 M Lösung,
2,00 mmol) wurde hinzugefügt.
Das sich ergebende Gemisch wurde für 3 Stunden bei Raumtemperatur
gerührt,
wobei zu dieser Zeit gefunden wurde, daß die Reaktion sehr langsam
fortschritt (HPLC). Ein Reflux-Kondensator wurde angebracht und
die, Lösung
wurde bei Reflux für
1 Stunde erhitzt. Nachdem die Lösung
abgekühlt
war, wurde Wasser (2 ml) hinzugefügt, um den Überschuß Bor abzulöschen. Die Lösungsmittel
wurden dann in vacuo entfernt. Hydrochlorsäure (15 ml einer 6 M Lösung) wurde
zu dem Rest hinzugefügt
und dieses Gemisch wurde bei Reflux für 30 Minuten erhitzt. Die Lösung wurde
abgekühlt
und Dichlormethan (100 ml) und 1 M Natriumhydroxid Lösung (100
ml) wurden hinzugefügt.
Der organische Extrakt wurde entfernt und die wäßrige Lage wurde mit Dichlormethan
(2 × 100 ml)
gewaschen. Die organischen Extrakte wurden vereinigt und über Magnesiumsulfat
getrocknet, und das Lösungsmittel
wurde in vacuo entfernt. Ethylether (100 ml) wurde zusammen mit
genug Methanol hinzugefügt,
um die freie Base zu lösen.
Ein Überschuß von etheralem Hydrogenchlorid
wurde hinzugefügt
und das Lösungsmittel
wurde in vacuo entfernt. Das Produkt wurde mit Ethylether tituriert
und in einem Vakuumofen getrocknet, um [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(3-pyridinylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-naphthalinsulfonamid-bis-hydrochlorid
18a (0,110 g, 0,167 mol) zu ergeben. MS (MH+) 584. 1H NMR (DMSOd6) δ 0,70–1,03 (m,
4H), 1,19–1,44
(m, 2H), 1,65–1,87
(m, 3H), 1,88–2,02
(m, 2H), 2,07–2,30
(m, 2H), 2,64 (dd, 2H), 2,69–3,19
(m, 4H), 3,33–3,62
(m, 3H), 3,65 (s, 3H), 5,82 (d, 1H), 6,35 (dd, 1H), 6,72 (dd, 1H),
7,63–7,88
(m, 4H), 7,93 (dd, 1H), 8,05 (d, 1H), 8,16 (m, 2H), 8,30 (d, 1H),
8,42 (s, 1H), 8,71 (s, 1H), 8,75 (d, 1H), 9.08 (br, 1H), 9.53 (br,
1H) (Figur 8).
-
-
BEISPIEL 4
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-fluor-1-(3-phenylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-fluorbenzolsulfonamid
(26)
-
- A. 3,4-Dihydro-6-fluor-2-(1H)-naphthalinon
wurde unter der Verwendung eines modifizierten Verfahrens von Stjernlof,
P.; et. al. (J. Med. Chem. 1995, 38, 2202) präpariert. Eine Lösung von
4-Fluorphenylessigsäure
(10,0 g, 64,9 mmol) und Thionylchlorid (11,8 ml, 0,162 mol) in 1,2-Dichlorethan
(150 ml) wurde bei Reflux für
4 h in einer 500 ml Rundboden-Flasche
erhitzt. Das Lösungsmittel
wurde in vacuo evaporiert. Der Rest wurde in 1,2-Dichlorethan gelöst und das Lösungsmittel
in vacuo evaporiert (um den Überschuß Thionylchlorid
zu entfernen). Der Rest wurde in Dichlormethan (50 ml) gelöst, und
die Lösung
wurde tropfenweise über
20 min zu einer abgekühlten
Suspension von Aluminiumchlorid (21,6 g, 162 mmol) in Dichlormethan (250
ml) bei –10
bis –5°C hinzugefügt. Die
Suspension wurde für
10 min bei –10°C gerührt. Ethylen
wurde für
20 min bei –10
bis 5°C
schnell durch die Suspension geblasen. Das Durchblasen wurde bei
einer sehr langsamen Rate für
die nächsten
2 h fortgeführt,
während
eine Temperatur von –5°C beibehalten
wurde. Das Reaktionsgemisch wurde mit Eis (100 g) gelöscht, und
die organische Lage wurde abgetrennt und zweimal mit Wasser und
einmal mit einer gesättigten
wäßrigen Natriumbicarbonat-Lösung gewaschen.
Die organische Lösung
wurde über
Magnesiumsulfat getrocknet und das Lösungsmittel wurde in vacuo
evaporiert, um das ungereinigte Tetralon (13,2 g), als einen gelben
Feststoff zu ergeben. Das Tetralon wurde ohne Reinigung in der anschließenden Reaktion
verwendet, obwohl ein Teil des Rohprodukts aus Hexan rekristallisiert
wurde, um gereinigtes 3,4-Dihydro-6-fluor-2-(1H)-naphthalinon als
einen farblosen Feststoff (~50% Ausbeute) zu ergeben. 1H
NMR (CDCl3) δ 2,55 (t, 2H), 3,05 (t, 2H),
3,54 (s, 2H), 6,85–6,97
(m, 2H) und 7,05–7,12
(m, 1H).
- B. Pyrrolidin (1,78 ml, 21,4 mmol) wurde zu einer Lösung von
3,4-Dihydro-6-fluor-2-(1H)-naphthalinon
(3,2 g, 19,5 mmol) in Benzol (40 ml) in einer 100 ml Rundboden-Flasche
hinzugefügt,
und die sich ergebende Lösung
wurde bei Raumtemperatur für
1 h gerührt.
Das Lösungsmittel
wurde in vacuo evaporiert. Der Rest wurde in 1,2-Dichlorethan gelöst und das
Lösungsmittel
wurde in vacuo evaporiert (um Überschuß Pyrrolidin
zu entfernen). Das Roh produkt, 6-Fluor-2-(pyrrolidin-1-yl)-3,4-dihydronaphthalin
19 wurde ohne Reinigung in dem anschließenden Schritt verwendet.
- C. Benzylbromid (2,8 ml, 23,4 mmol) wurde zu einer Lösung des
Roh-Enamins 19 (19,5 mmol) in Acetonitril (60 ml) in einer 100 ml
Rundboden-Flasche hinzugefügt,
und die sich ergebende Lösung
wurde bei Raumtemperatur für
1,5 h gerührt.
Das Lösungsmittel
wurde in vacuo evaporiert und der Rest wurde aus heißem Tetrahydrofuran
kristallisiert. Die Suspension wurde abgekühlt und das Iminiumsalz 20
wurde durch Filtration gesammelt, um einen weißen Feststoff, 4,4 g (58%),
zu ergeben. MS m/e (M+) 308. 1H
NMR (DMSO-d6): δ 1,70–2,03 (m, 4H), 2,91–3,13 (m,
3H), 3,17–3,29
(m, 2H), 3,38–3,61
(m, 2H), 3,81–3,93
(m, 1H), 3,96–4,07
(m, 1H), 4,13–4,27
(m, 1H), 4,52 (t, 1H), 6,87–7,02
(m, 2H), 7,09–7,17
(m, 2H) und 7,20–7,32
(m, 2H).
- D. Das Iminiumsalz 20 (4,4 g, 11,33 mmol) wurde mit Essigsäure (5 ml,
87,3 mmol), Dichlormethan (50 ml), Wasser (50 ml) und Methanol (100
ml) in einer 500 ml Rundboden-Flasche gemischt und bei Raumtemperatur
für 16
h gerührt.
Eine organische Lage bildete sich und wurde abgetrennt, und die
wäßrige Lage
wurde mit Dichlormethan extrahiert. Die organischen Extrakte wurden
vereinigt, zweimal mit Wasser und einmal mit einer gesättigten
Lösung
von wäßrigem Natriumbicarbonat
gewaschen, und dann über
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde in vacuo
evaporiert, um 3,4-Dihydro-6-fluor-1-(phenylmethyl)-2-(1H)-naphthalinon
21 als ein braunes Öl,
3,0 g (100%) zu ergeben. Dieses Material wurde ohne weitere Reinigung
in dem anschließenden
Schritt verwendet.
- E. Eine Lösung
von 3,4-Dihydro-6-fluor-1-(phenylmethyl)-2-(1H)-naphthalinon 21
von oben (2,9 g, 11,4 mmol) wurde in Methanol (50 ml) in einer 250
ml Rundboden-Flasche gelöst.
Ammoniumacetat (13,2 g, 0,171 mol) wurde hinzugefügt, und
das Gemisch wurde bei Raumtemperatur für 10 min gerührt. Natriumcyanborhydrid
(3,58 g, 57 mmol) wurde hinzugefügt
und die sich ergebende Lösung
wurde auf Reflux für
1 h erhitzt. Das Lösungsmittel
wurde in vacuo evaporiert, und der Rest wurde mit wäßrigem Natriumhydroxid (50
ml einer 1 N Lösung)
behandelt. Das Produkt wurde in Dichlormethan (2 × 50 ml)
extrahiert und zweimal mit Wasser gewaschen und über Natriumsulfat getrocknet.
Das Lösungsmittel
wurde in vacuo evaporiert, und der Rest wurde in Diethylether (50
ml) gelöst
und mit etheraler Hydrochlorsäure
(15 ml einer 1 N Lösung)
behandelt, was zu der Präzipitation
eines Feststoffs führte.
Dieses Material wurde durch Filtration gesammelt, mit Diethylether
gewaschen und in vacuo getrocknet, um cis-1,2,3,4-Tetrahydro-6-fluor-1-(phenylmethyl)-2-naphthalinaminhydrochlorid
22 als einem blaßrosa
Feststoff (1,6 g, 48%) zu ergeben. MS m/e (MH+)
256, 1H NMR (DMSO-d6): δ 1,96–2,13 (m,
2H), 2,40 (t, 1H), 2,82–3,12
(m, 2H), 3,17 (dd, 1H), 3,28–3,37
(m; 1H), 3,47–3,60
(br m, 1H), 5,98 (m, 1H), 6,62 (m, 1H), 6,98 (m, 1H), 7,08 (d, 2H),
7,18–7,30 (m,
3H), 8,64 (br s, 3H).
- F. cis-1,2,3,4-Tetrahydro-6-fluor-1-(phenylmethyl)-2-naphthalinaminhydrochlorid
22 (0,96 g, 3,29 mmol) wurde in N,N-Dimethylformamid (50 ml) in
einer 250 ml Rundboden-Flasche unter Rühren gelöst. Diisopropylethylamin (1,30
ml, 7,46 mmol) wurde hinzugefügt,
gefolgt von 4-(tert-Butoxycarbonyl)aminomethylcyclohexancarbonsäure (0,85
g, 3,31 mmol). Zu dieser gerührten
Lösung
wurde langsam HBTU (1,25 g, 3,29 mmol) hinzugefügt. Die Flasche wurde mit Argon
gespült,
zugedeckelt, und es ihr erlaubt, für 3 Stunden zu rühren. Zu
dieser Zeit wurde die Reaktionslösung
in 500 ml Wasser gegossen. Ein Präzipitat bildete sich sofort,
und diese Schlämme
wurde über
Nacht gerührt.
Der Feststoff wurde dann filtriert und mit zusätzlichen Teilen an Wasser gespült. Luft
wurde über
den Feststoff fast bis zur Trocknung gezogen. Dieser Feststoff wurde
zu Methanol (15 ml) hinzugefügt,
und das Feststoff-Flüssigkeits-Gemisch
wurde für
mehrere Minuten auf Reflux erhitzt. Nach Abkühlen der Lösung auf Raumtemperatur wurde
der weiße
Feststoff von der orange-braunen Flüssigkeit abfiltriert. Das Filtrat
wurde leicht evaporiert, um zweite Charge an weißem Feststoff zu erhalten,
die wie oben filtriert wurde und mit der ersten Charge vereinigt
wurde. Dieser weiße Feststoff
wurde in vacuo getrocknet, um [1α,2α(trans)]-4-[[(tert-Butoxycarbonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-fluor-1-(3-phenylmethyl)-2-naphthalinyl]-cyclohexancarboxamid
23 (1,28 g, 2,59 mmol) zu erhalten. MS m/e (MH+)
256, 1H NMR (CDCl3): δ 0,79–1,00 (m,
2H), 1,23–1,53
(m, 12H), 1,70–2,08
(m, 7H), 2,75–3,03
(m, 6H), 3,37 (m, 1H), 4,29 (m, 1H), 4,55 (m, 1H), 5,33 (d, 1H),
6,67–6,87
(m, 3H), 7,12 (d, 2H), 7,37–7,18
(m, 3H).
- G. Das oben erhaltene Carboxamid 23 (1,28 g, 2,58 mmol) wurde
in Dioxan (150 ml) in einer 250 ml Rundboden-Flasche gelöst und in
einem Eisbad abgekühlt. Überschuß an gasförmigem Hydrogenchlorid
wurde zu dem sich ergebenden Fest-Flüssig-Gemisch bis zur Sättigung
hinzugefügt.
Die klare Lösung
wurde dann auf Raumtemperatur erwärmt und gerührt, bis das Ausgangsmaterial
vollständig
verbraucht wurde (HPLC). Das Lösungsmittel
wurde in vacuo entfernt und der sich ergebende Feststoff mit Diethylether
tituriert, um einen weißen
Feststoff zu ergeben, der nach Filtration und Trocknen in vacuo
[1α,2α(trans)]-4-(Aminomethyl)-N- [1,2,3,4-tetrahydro-6-fluor-1-(phenylmethyl)-2-naphthalinyl]-cyclohexancarboxamidhydrochlorid
24 ergab. MS m/e (MH+) 395; 1H
NMR (DMSO-d6): δ 0,84–1,05 (m, 2H), 1,28–1,49 (m,
2H), 1,50–1,62
(m, 1H), 1,65–2,04
(m, 6H), 2,09–2,27
(m, 1H), 2,51–2,59
(m, 1H), 2,60–2,73
(m, 2H), 2,77–3,04
(m, 3H), 3,12–3,26 (m,
1H), 3,92 (m, 1H), 6,41 (dd, 1H), 6,73 (dt, 1H), (m, 3H), 7,13–7,32 (m,
3H), 7,88 (br, 3H), 7,97 (d, 1Η).
- H. Das Naphthalinylcarboxamid 24 (0,087 g, 0,20 mmol) wurde
in einer Dichlormethan Lösung
(15 ml) von Diisopropylethylamin (0,080 ml, 0,46 mmol) unter Rühren in
einer 100 ml Rundboden-Flasche gelöst. Eine Lösung von 2-Fluorbenzolsulfonylchlorid
(0,045 g, 0,23 mmol) in Dichlormethan (15 ml) wurde hinzugefügt. Dem
Reaktionsgemisch wurde es erlaubt, über Nacht bei Raumtemperatur
zu rühren.
Das Lösungsmittel wurde
in vacuo entfernt, um ein glasartiges farbloses Material zu ergeben.
Dieses Material wurde in Dichlormethan (100 ml) gelöst und die
Lösung
wurde mit 0,1 M Natriumhydroxid Lösung (2 × 55 ml) und dann mit Wasser
(1 × 50
ml) gewaschen. Die organischen Stoffe wurden über Magnesiumsulfat getrocknet,
filtriert, und das Lösungsmittel
in vacuo entfernt, um [1α,2α(trans)]-4-[[(2-Fluorbenzolsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-fluor-1-(phenylmethyl)-2-naphthalinyl]-cyclohexancarboxamid
25 (0,110 g, 0,199 mmol) als ein braunes Pulver zu ergeben. MS m/e (MH+) 553; 1H NMR (DMSO-d6): δ 0,71–0,91 (m,
2H), 1,18–1,43
(m, 3H), 1,61–1,81
(m, 5H), 1,85–2,00
(m, 1H), 2,03–2,19
(m, 1H), 2,51 (m, 1H, verdeckt durch DMSO), 2,71 (t, 2H), 2,79–3,03 (m,
3H), 3,083,24 (m, 1H), 3,91 (m, 1H), 6,42 (dd, 1H), 6,72 (dt, 1H),
6,86–7,02
(m, 3H), 7,08–7,29
(m, 3H), 7,33–7,52
(m, 2H), 7,65–7,77
(m, 1H), 7,79 (dt, 1H), 7,84–7,99
(m, 2H).
- I. Das oben erhaltene Carboxamid 25 (0,110 g, 0,199 mmol) wurde
in THF (15 ml) gelöst
und unter Rühren wurde
eine Lösung
an Bor-Tetrahydrofurankomplex-Lösung
(1 M in THF, 2,0 ml, 2,0 mmol) hinzugefügt. Die Lösung wurde mit Stickstoff gespült und dann
für ungefähr 1 Stunde
auf Reflux erhitzt. Nach Abkühlen
auf Raumtemperatur wurde zu der Lösung unter Rühren tropfenweise
Wasser (2 ml) hinzugefügt,
und das Lösungsmittel
wurde in vacuo entfernt, um einen weißen Film zu ergeben. Hydrochlorsäure (15
ml einer 6 M Lösung)
wurde zu diesem Material hinzugefügt, und das Gemisch wurde für ungefähr 30 Minuten
auf Reflux erhitzt. Nach Abkühlen
auf Raumtemperatur wurde Natriumhydroxid (100 ml einer 1 N Lösung) hinzugefügt. Dieses
wäßrige Gemisch
wurde mit Dichlormethan (3 × 100
ml) extrahiert. Die organischen Extrakte wurden vereinigt und über Magnesiumsulfat
getrocknet, filtriert, und das Lösungsmittel
wurde in vacuo entfernt. Der Rest wurde in THF (4 ml) gelöst und etherales
Hydrogenchlorid (2 ml einer 1 M Lösung) wurde hinzugefügt. Die
Lösungsmittel
wurden in vacuo entfernt, um einen weißen gelatinösen Feststoff zu ergeben. Methanol
und Dichlormethan wurden hinzugefügt, um den Feststoff aufzubrechen
und dann in vacuo entfernt, um ein weißes Pulver zu ergeben. Isopropanol
(4 ml) wurde hinzugefügt
und die Schlämme
wurde kurz auf Reflux erhitzt und dann abgekühlt. Das Lösungsmittel wurde entfernt
und das feuchte Produkt wurde unter Vakuum getrocknet, um [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-fluor-1-(3-phenylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-fluorbenzolsulfonamid
26 (0,087 g, 0,151 mmol) als ein weißes Pulver zu erhalten. MS
m/e (MH+) 539; 1H
NMR (DMSO-d6): δ 0,71–1,03 (m, 4H), 1,24–1,43 (m,
1H), 1,61–1,97
(m, 5H), 2,03–2,25
(m, 2H), 2,44 (m, 1H), 2,73 (t, 2H), 2,83–3,18 (m, 5H), 3,40–3,59 (m,
2H), 5,96 (dd, 1H), 6,59 (dt, 1H), 6,98 (dd, 1H), 7,07 (d, 2H),
7,17–7,32
(m, 3H), 7,36–7,53
(m, 2H), 7,73 (q, 1H), 7,81 (dt, 1H), 7,98 (t, 1H), 8,85 (br, 2H)
(Figur 9).
-
-
BEISPIEL 5
-
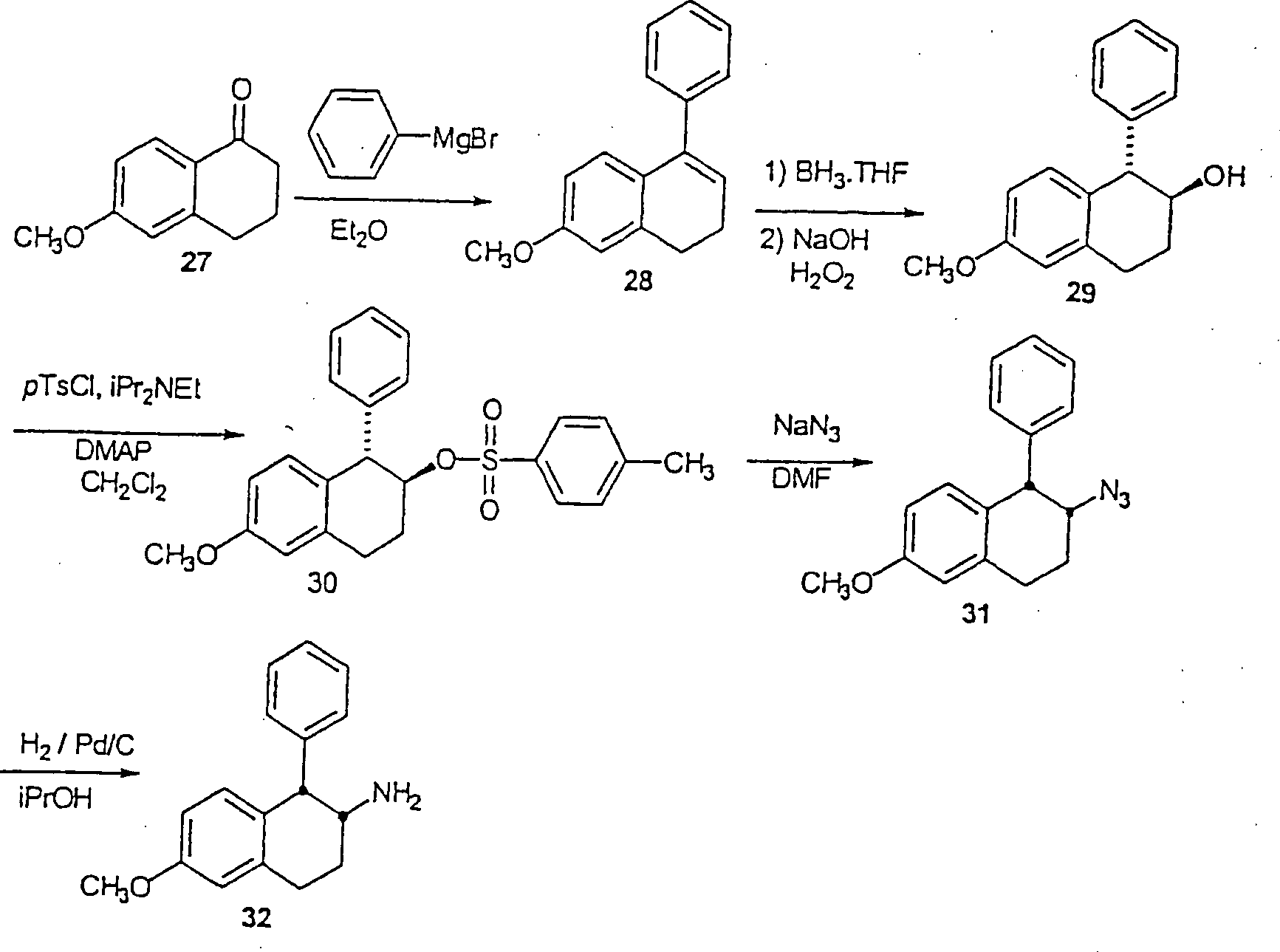
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-fluor-1-phenyl-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-naphthalinsulfonamid
(34)
-
- A. Eine Lösung
von Phenylmagnesiumbromid in Diethylether (3,0 M, 23 ml, 69 mmol)
wurde zu einer Lösung
von 6-Methoxy-1,2,3,4-tetrahydronaphthalin-1-on 27 (10,0 g, 56,7
mmol) in Diethylether (100 ml) bei Raumtemperatur tropfenweise hinzugefügt. Das
Reaktionsgemisch wurde für
1,5 h auf Reflux erhitzt. Ein zusätzlicher Teil von Phenylmagnesiumbromid-Lösung (10 ml, 60 mmol) wurde
zu dem abgekühlten
Reaktionsgemisch hinzugefügt,
und das sich ergebende Gemisch wurde für weitere 2,5 h auf Reflux
erhitzt. Das abgekühlte
Gemisch wurde in eine gesättigte
Lösung
von Ammoniumchlorid (200 ml) gegossen und für 15 min gerührt. Die
organische Lage wurde abgetrennt, mit einer gesättigten Lösung an Natriumchlorid gewaschen
und über
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde in vacuo
evaporiert, und das sich ergebende Öl wurde mit einer Lösung von
Schwefelsäure
(8 ml) in Essigsäure
(30 ml) bei Raumtemperatur für
1,5 h behandelt. Eiswasser (300 ml) wurde zu der Lösung hinzugefügt, und
das Produkt wurde in Dichlormethan (200 ml) extrahiert, mit Wasser
und einer gesättigten
wäßrigen Lösung von
Natriumbicarbonat gewaschen und über
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde in vacuo
evaporiert und der Rest wurde durch Chromatographie mittleren Drucks
unter der Verwendung von Ethylacetat 0 bis 3% in Hexanen als dem
Eluenten gereinigt, um das 6-Methoxy-1-phenyl-3,4-dihydronaphthalon
28 in zwei Anteilen 4,0 g (29%) und einer unreinen Fraktion 5,02
g (37%) als ein Öl
zu ergeben.
- B. Bor in Tetrahydrofuran (34 ml einer 1 M Lösung, 34 mmol) wurde zu Tetrahydrofuran
(50 ml) hinzugefügt und
die sich ergebende Lösung
wurde auf 0°C
abgekühlt.
Eine Lösung
von 6-Methoxy-1-phenyl-3,4-dihydronaphthalin 28 (5,0 g, 21,2 mmol)
in Tetrahydrofuran (10 ml) wurde hinzugefügt. Das sich ergebende Gemisch
wurde bei Umgebungstemperatur für
18 h gerührt.
Ein Lösung
von Wasser (5 ml) in Tetrahydrofuran (20 ml) wurde langsam zu der
abgekühlten
Lösung
hinzugefügt,
was zu beträchtlichem
Schäumen
führte. Weiteres
Wasser (10 ml) wurde hinzugefügt
gefolgt von 10% wäßrigem Natriumhydroxid
(15 ml) und 30% Wasserstoffperoxid (30 ml). Das sich ergebende Gemisch
wurde bei Raumtemperatur für
6 h gerührt.
Die organische Lage wurde abgetrennt und die wäßrige Lage wurde mit Diethylether
(2 × 50
ml) extrahiert. Die vereinigten organische Lösungen wurden mit gesättigtem
wäßri gem Natriumchlorid
gewaschen und über Magnesiumsulfat
getrocknet. Das Lösungsmittel
wurde in vacuo evaporiert, und der Rest wurde durch Flashchromatographie
unter der Verwendung von 30 bis 40% Ethylacetat in Hexan als dem
Eluenten gereinigt, um trans-6-Methoxy-1-phenyl-1,2,3,4-tetrahydronaphthalin-2-ol
29 als ein Öl
(2,0 g, 37%) zu erhalten. 1H NMR (CDCl3) δ 1,77
(d, 1H), 1,83–1,97
(m, 1H), 2,13–2,22
(m, 1H), 2,94–3,05
(m, 2H), 3,78 (s, 3H), 3,90 (d, 1H), 3,98–4,07 (m, 1H), 6,59–6,70 und
7,16–7,39
(m, 8H).
- C. Eine Lösung
von para-Toluolsulfonylchlorid (1,8 g, 9,43 mmol) in Dichlormethan
(10 ml) wurde zu einer Lösung
von trans-6-Methoxy-1-phenyl-1,2,3,4-tetrahydronaphthalin-2-ol 29
(2,0 g, 7,86 mmol), N,N-Diisopropylethylamin (4,8 ml, 27,5 mmol)
und 4-Dimethylaminopyridin
(1,15 g, 9,43 mmol) in Dichlormethan (40 ml) bei 0°C hinzugefügt. Die
sich ergebende Lösung
wurde bei Umgebungstemperatur für
16 h gerührt.
Die Lösung
wurde nacheinander mit 1 N wäßrigem Natriumhydroxid
(× 2)
und einer gesättigten
wäßrigen Lösung an
Natriumchlorid gewaschen, dann über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde in vacuo evaporiert, um rohes trans-6-Methoxy-1-phenyl-2-(4-methylbenzolsulfonyl)oxy-1,2,3,4-tetrahydronaphthalin
30 (3,47 g) als einen blaßgelben
Feststoff zu ergeben, der ohne weitere Reinigung in dem anschließenden Schritt
verwendet wurde. 1H NMR (CDCl3) δ 1,90–2,04 (m,
1H), 2,14–2,24
(m, 1H), 2,42 (s, 3H), 2,83–3,07
(m, 2H), 3,77 (s, 3H), 4,16 (d, 1H), 4,80–4,87 (m, 1H), 6,60–6,67 (m,
3H), 6,82–6,90
(m, 2H), 7,13–7,23
(m, 5H), 7,58 (d, 2H).
- D. Eine N,N-Dimethylformamid-Lösung (50 ml) von ungereinigtem
trans-6-Methoxy-1-phenyl-2-(4-methylbenzolsulfonyl)oxy-1,2,3,4-tetrahydronaphthalin
30 (3,4 g), Natriumazid (3,78 g, 58,3 mmol) und 15-Kronen-5 (6,61
ml, 33,2 mmol) wurde auf 75°C
für 7 h
erhitzt. Das Reaktionsgemisch wurde in Eiswasser (200 ml) gegossen
und das Produkt wurde in Diethylether extrahiert (3 × 50 ml).
Die organischen Extrakte wurden kombiniert und nacheinander mit
Wasser (4 × 100
ml) und einer gesättigten
wäßrigen Lösung von
Natriumchlorid gewaschen und über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde in vacuo evaporiert, und der verbleibende Rest wurde durch
Chromatographie mittleren Drucks unter der Verwendung von 3% Ethylacetat
in Hexanen als dem Eluenten gereinigt, um ungereinigtes cis-2-Azido-6-methoxy-1-phenyl-1,2,3,4-tetrahydronaphthalin
31 (1,35 g, ca. 62%) als ein Öl
zu ergeben, das in dem anschließenden Schritt
ohne Reinigung verwendet wurde.
- E. Das oben erhaltene Azido-tetrahydronaphthalin 31 (1,3 g)
wurde in Isopropanol (50 ml) gelöst
und diese Lösung
wurde bei 50 psi über
10% Palladium auf Kohle (0,2 g) bei Raumtemperatur für 18 h hydrogeniert. Der
Katalysator wurde durch Filtration entfernt und das Lösungsmittel
wurde in vacuo evaporiert, um ungereinigtes cis-1,2,3,4-Tetrahydro-6-methoxy-1-phenyl-2-naphthalinamin
32 als ein Öl
zu erhalten, das in dem anschließenden Schritt ohne Reinigung
verwendet wurde (Figur 10). MS m/e (MH+)
254.
- F. Eine Lösung
von Isobutylchlorformat (0,88 ml, 6,76 mmol) in Dichlormethan (5
ml) wurde tropfenweise zu einer Lösung von trans-4-(2-Napthylsulfonamido)methylcyclohexancarbonsäure (1,12
g, 3,22 mmol) und Triethylamin (1,35 ml, 9,66 mmol) in Dichlormethan
(30 ml) bei 0°C
hinzugefügt.
Die sich ergebende Lösung
wurde bei Raumtemperatur für
1 h gerührt.
Eine Lösung
von Naphthalinamin 32 (4,65 mmol) in Dichlormethan wurde tropfenweise
zu dem Reaktionsgemisch bei 0°C
hinzugefügt.
Die Reaktion wurde bei Umgebungstemperatur für 16 h gerührt, wonach zu dieser Zeit
das Lösungsmittel
und andere flüchtige
Materialien in vacuo evaporiert wurden. Der sich ergebende Rest
wurde mit einer Lösung
von 1 N wäßrigem Natriumhydroxid
(20 ml) und Tetrahydrofuran (20 ml) für 30 min behandelt. Die Lösung wurde
in vacuo konzentriert und mit 1 N wäßriger Hydrochlorsäure (30
ml) angesäuert.
Das Produkt wurde in 10% Isopropanol in Dichlormethan (2 × 50 ml)
extrahiert. Das Lösungsmittel
wurde in vacuo evaporiert und der Rest wurde durch Chromatographie
mittleren Drucks unter der Verwendung von 2% Methanol in Dichlormethan
als dem Eluenten gereinigt, um [1α,2α(trans)]-4-[[(2-Naphthalinylsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-phenyl-2-naphthalinyl]-cyclohexancarboxamid
33 (0,95 g, 52%) als ein Glas zu ergeben, das aus Dithylether kristallisiert wurde,
um einen farblosen Feststoff (0,38 g, 20%) zu ergeben. MS m/e (MH+) 583; 1H NMR (DMSO-d6) δ 0,68–0,83 (m,
2H), 1,20–1,37
(m, 3H), 1,50–1,77
(m, 6H), 1,87–1,99
(m, 1H), 2,60 (t, 2H), 2,90–3,02
(m, 2H), 3,73 (s, 3H), 3,94–4,07
(m, 1H), 4,40 (d, 1H), 6,61–6,81
(m, 5H), 7,14–7,32
(m, 4H), 7,63–7,75
(m, 3H), 7,81 (d, 1H), 8,04 (d, 1H), 8,14 (t, 2H) und 8,43 (s, 1H).
- G. Eine 1 M Lösung
von Bor in Tetrahydrofuran (5 ml, 5 mmol) wurde tropfenweise zu
einer Lösung
von Carboxamid 33 (0,25 g, 0,43 mmol) in Tetrahydrofuran (15 ml)
hinzugefügt
und bei Umgebungstemperatur für
5 h gerührt.
Wasser (5 ml) in Tetrahydrofuran (15 ml) wurde tropfenweise zu der
Lösung
bei 0°C über 10 min
hinzugefügt.
Hydrochlorsäure
(5 ml einer 4 N Lösung)
wurde zu der Lösung
hinzugefügt,
und das sich ergebende Gemisch wurde bei 0°C für 16 h gerührt. Das Reaktionsgemisch wurde
in vacuo konzentriert, und mit wäßrigem Natriumbicarbonat
neutralisiert. Das Produkt wurde in Dichlormethan (2 × 50 ml) extrahiert.
Die organischen Extrakte wurden vereinigt und das Lösungsmittel
wurde in vacuo evaporiert. Der sich ergebende Rest wurde durch präparative
reverse-Phase HPLC unter der Verwendung von 0,1% Trifluoressigsäure in Acetonitril
und Wasser als dem Eluenten gereinigt. Das Produkt eluierte bei
55% Acetonitril, um das Trifluoressigsäuresalz von [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-phenyl-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-naphthalinsulfonamid
34 als einen farblosen Feststoff (0,125 g, 43%) zu erhalten. MS
(MH+) 569; 1H NMR
(DMSO-d6) δ 0,73–0,90 (m, 4H), 1,23–1,36 (m,
1H), 1,40–1,57
(m, 1H), 1,60–1,75
(m, 4H), 1,83–2,09 (m,
2H), 2,60 (t, 2H, fällt
zusammen mit D2O zu d), 2,64–2,77 (m,
1H), 2,87–3,16
(m, 3H), 3,62–3,76
(m, 1H), 3,73 (s, 3H), 4,58 (d, 1H), 6,68 (dd, 1H), 6,76–6,80 (m,
2H), 7,13 (d, 2H), 7,24–7,37
(m, 3H), 7,67–7,77 (m,
3H, fällt
zusammen 2H mit D2O), 7,83 (d, 1H), 7,87–8,00 (br
s, 1H, ausgetauscht mit D2O), 8,06 (d, 1H), 8,10–8,26 (m,
3H) und 8,43 (s, 1H). (Figur 11).
-
-
-
BEISPIEL 6
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(1-propen-3-yl)-2-naphthalinyl]
amino]methyl]-4-cyclohexyl]methyl]benzolsulfonamid (39)
-
- A. 3,4-Dihydro-6-methoxy-2-(pyrrolidin-1-yl)naphthalin
2 wurde durch Reagieren einer Lösung
von 6-Methoxy-β-tetralon
(4,73 g, 26,8 mmol) in Methanol (50 ml) mit Pyrrolidin (2,35 ml,
28,18 mmol) in einer 100 ml Rundboden-Flasche bei Raumtemperatur
für 30
min präpariert.
Das Lösungsmittel
wurde in vacuo evaporiert, um das gewünschte Enamin 2 als einen gelben
Feststoff (eine einzelne Komponente durch reverse Phase HPLC) zu
erhalten, die ohne Reinigung in dem anschließenden Schritt verwendet wurde.
- B. Enamin 2 (26,8 mmol) wurde in Acetonitril (50 ml) in einer
100 ml Rundboden-Flasche gelöst
und Allylbromid (2,55 ml, 29,5 mmol) wurde hinzugefügt. Nach
Rühren
bei Raumtemperatur für
18 h wurde das Lösungsmittel
in vacuo evaporiert und der sich ergebende Rest wurde mit Tetrahydrofuran
tituriert. Das entsprechende Iminiumsalz 35 wurde durch Filtration
als ein gummiartiger Feststoff gesammelt und ohne weitere Reinigung
in dem anschließenden
Schritt verwendet. Das Produkt ist eine einzelne Komponente durch reverse
Phase HPLC. MS (M+) 270, 1H
NMR (CDCl3): δ 2,00–2,15 (m, 4H), 2,33–2,47 (m,
1H), 2,60–2,70 (m,
1H), 2,90–3,34
(m, 2H), 2,90–3,34
(m, 4H), 3,76 (m, 3H), 3,84–4,16
(m, 3H), 4,23–4,39
(m, 2H), 5,04 (d, 1H), 5,07 (s, 1H), 5,70–5,83 (m, 1H), 6,84 (d, 1H),
6,92 (s, 1H) und 7,14 (d, 1H).
- C. Das Iminiumsalz 35 von oben (26,8 mmol) wurde mit Essigsäure (4 ml),
Dichlormethan (40 ml), Methanol (80 ml) und Wasser (40 ml) in einer
250 ml Rundboden-Flasche gemischt und bei Raumtemperatur für 18 h gerührt. Das
Gemisch trennte sich in zwei Phases und die organische Phase wurde
abgetrennt. Die wäßrige Phase
wurde mit Dichlormethan extrahiert. Die organischen Extrakte wurden
vereinigt, zweimal mit Wasser und einmal mit gesättigtem wäßrigem Natriumbicarbonat gewaschen
und über
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde in vacuo
evaporiert, um 3,4-Dihydro-6-methoxy-1-(1-propen-3-yl)-2-(1H)naphthalinon
36 als ein Öl,
eine einzelne Komponente durch HPLC (> 95%), 2,5 g (46% von 2), zu ergeben. 1H NMR (CDCl3): δ 2,52–2,70 (m,
4H), 2,92–3,15
(m, 2H), 3,45 (t, 1H), 3,81 (s, 3H), 4,97 (s, 1H), 5,03 (d, 1H),
5,65–5,81
(m, 1H), 6,74–6,82
(m, 2H) und 7,08 (d, 1 H).
- D. Eine Lösung
von ungereinigtem Naphthalinon 36 (2,5 g, 11,6 mmol) in Methanol
(50 ml) wurde mit Ammoniumacetat (13,4 g, 0,173 mol) in einer 100
ml Rundboden-Flasche behandelt und bei Raumtemperatur für 10 min
gerührt.
Natriumcyanborhydrid (3,58 g, 57 mmol) wurde hinzugefügt und die
sich ergebende Lösung
wurde auf Reflux für
3 h erhitzt. Das Lösungsmittel
wurde in vacuo evaporiert, und der Rest wurde mit wäßrigem Natriumhydroxid
(50 ml einer 1 N Lösung)
behandelt. Das Produkt wurde in Dichlormethan (2 × 50 ml)
extrahiert. Die organischen Extrakte wurden vereinigt, mit Wasser
gewaschen und über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde in vacuo evaporiert, und der Rest wurde in Diethylether (50
ml) und Methanol (1–2
ml) gelöst.
Die sich ergebende Lösung
wurde mit etheraler Hydrochlorsäure
(14 ml einer 1 N Lösung)
behandelt, um einen gummiartigen Feststoff zu produzieren, der die
Seiten der Flasche beschichtete. Das Lösungsmittel wurde abgegossen
und zusätzliche
50 ml an Ether wurden hinzugefügt,
wobei sich der Rest zu diesem Punkt verfestigte. Das Produkt wurde
durch Filtration gesammelt, mit Diethylether gewaschen und in vacuo
konzentriert, um cis-1,2,3,4-Tetrahydro-6-methoxy-1-(1-propen-3-yl)-2-naphthalinaminhydrochlorid
37a als einen violetten Feststoff zu ergeben (1,75 g, ein Gemisch von
2 Komponenten 3 : 1 durch HPLC). MS m/e (MH+)
218.
- E. Eine Lösung
von trans-4-[(Benzolsulfonamido)methyl]cyclohexancarbonsäure (0,924
g, 3,31 mmol), 2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorphosphat
(HBTU), (1,26 g, 3,31 mmol) und N,N-Diisopropylethylamin (1,7 ml,
9,77 mmol) in N,N-Dimethylformamid
(10 ml) wurde bei Raumtemperatur in einer 50 ml Rundboden-Flasche
für 15
min gerührt.
Naphthalinaminhydrochlorid 37a (0,80 g, 3,15 mmol) wurde zu der
Lösung
hinzugefügt.
Das Rühren
wurde für
eine weitere Stunde fortgeführt,
und die sich ergebende Lösung
wurde in Wasser (100 ml) gegossen. Ein gummiartiger Feststoff bildete
sich auf den Seiten der Flasche. Ethanol wurde hinzugefügt und das
Produkt kristallisierte bei Erhitzen. Das Gemisch wurde auf Raumtemperatur
abgekühlt
und das Produkt wurde durch Filtration gesammelt und in vacuo getrocknet, um [1α,2α(trans)]-4[[(Benzulsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(1-propen-3-yl)-2-naphthalinyl]-cyclohexancarboxamid
38, einen hellgrauen Feststoff, 0,67 g (43%) zu ergeben, eine einzelne Komponente
durch HPLC. MS (MH+) 497; 1H
NMR (CDCl3): δ 0,71–0,97 (m, 2H), 1,33–1,50 (m,
3H), 1,74–1,98
(m, 7H), 2,24–2,53
(m, 2H), 2,76–2,87
(m, 4H), 3,00–3,09
(m, 1H), 3,78 (s, 3H), 4,34–4,43
(m, 1H), 4,62 (t, H), 5,02 (s, 1H), 5,07 (d, 1H), 5,48 (d, 1H),
5,81–5,96
(m, H), 6,63 (s, 1H), 6,72 (d, 1H), 7,02 (d, 1H), 7,47–7,62 (m,
3H) und 7,84 (d, 2H).
- F. Eine Lösung
von Lithiumaluminumhydrid in Tetrahydrofuran (4 ml einer 1,0 M Lösung, 4
mmol) wurde sorgfältig
zu einer Lösung
von Carboxamid 38 (0,21 g, 0,422 mmol) in Tetrahydrofuran (10 ml)
in einer 50 ml Rundboden-Flasche hinzugefügt. Die sich ergebende Lösung wurde
für 24
h auf Reflux erhitzt. Die Lösung
wurde in einem Wasserbad abgekühlt,
und der Überschuß Hydrid
wurde durch sorgfältige
Zugabe von Wasser (0,16 ml) in Tetrahydrofuran (5 ml), gefolgt von
15% wäßrigem Natriumhydroxid
(0,16 ml) in Tetrahydrofuran (5 ml) und zuletzt Wasser (0,5 ml)
gelöscht.
Die anorganischen Feststoffe wurden durch Filtration entfernt und
großzügig mit
Tetrahydrofuran gewaschen. Das Filtrat wurde in vacuo evaporiert
und der sich ergebende Rest wurde in Ethanol gelöst und mit einer gesättigten
Lösung
von Hydrochlorsäure
in Ethanol (2 ml) behandelt. Evaporation und Tituration mit Diethylether
ergab [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(1-propen-3-yl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-benzolsulfonamidhydrochlorid
39a als einen farblosen Feststoff, 0,118 g (54%), eine einzelne Komponente
durch HPLC (> 95%).
MS (MH+) 483; 1H
NMR (DMSO-d6) δ 0,75–1,04 (m, 4H), 1,23–1,40 (m, 1H),
1,57–2,16
(m, 7H), 2,43–2,62
(m, 2H), 2,79–3,00
(m, 4H), 3,133,23 (m, 1H), 3,35–3,44
(m, 2H), 3,73 (s, 3H), 4,91 (d, 1H), 5,03 (d, 1H), 5,735,88 (m,
1H), 6,65–6,72
(m, 2H), 6,93 (d, 1H), 7,57–7,70
(m, 4H), 7,84 (d, 2H), 8,70 (br s, 1H, wechselwirkt mit D2O), und 9,07 (br s, 1H, wechselwirkt mit
D2O). (Figur 12)
-
-
BEISPIEL 7
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(3-hydroxypropyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]benzolsulfonamid
(40)
-
Eine
Lösung
von Bor (3,5 ml einer 1,0 M Lösung,
3,5 mmol) in Tetrahydrofuran wurde zu einer Lösung von Carboxamid 38 (0,25
g, 0,503 mmol) in Tetrahydrofuran (10 ml) in einer 100 ml Rundboden-Flasche
hinzugefügt.
Das sich ergebende Gemisch wurde auf Reflux für 1 h erhitzt. Wasser (1,5
ml) wurde sorgfältig
hinzugefügt,
und das Gemisch wurde auf Reflux für 1 h erhitzt. Wäßriges Natriumhydroxid
(50%, 0,5 ml) wurde hinzugefügt,
gefolgt von Wasserstoffperoxid (30%, 1,0 ml). Das Zweiphasensystem
wurde stark für
2 h gerührt. Die organische
Lage wurde abgetrennt, und die wäßrige Lage
wurde mit Dichlormethan extrahiert. Die organischen Extrakte wurden
vereinigt, über
Natriumsulfat getrocknet und das Lösungsmittel wurde in vacuo
evaporiert. Der Rest wurde in Ethanol gelöst und mit einer gesättigten
Lösung
von Hydrogenchlorid in Ethanol (2 ml) behandelt. Das Lösungsmittel
wurde evaporiert und der Rest wurde mit Diethylether tituriert,
um [1α,2α(trans)-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(3-hydroxypropyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]benzolsulfonamidhydrochlorid
40 als einen weißen
Feststoff 0,242 g (90%) zu ergeben. Die Reinheit durch HPLC ist
80–90%.
MS (MH+) 501; 1H
NMR (DMSO-d6) δ 0,75–0,97 (m,
4H), 1,02–1,13
(m, 1H), 1,16–1,51
(m, 5H), 1,58–2,18
(m, 8H), 2,54–2,63
(m, 2H), 2,73–3,12
(m, 4H), 3,28–3,46
(m, 3H), 3,72 (s, 3H), 6,64–6,73
(m, 2H), 6,97 (d, 1H), 7,54–7,69
(m, 4H), 7,80 (d, 2H), 8,57 (br s, 1H, wechselwirkt mit D2O), und 8,93 (br s, 1H, wechselwirkt mit
D2O)
(Figur 13)
-
BEISPIEL 8
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(n-propyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-benzolsulfonamid
(42)
-
- A. Das Carboxamid 38 (0,4 g, 0,805 mmol) wurde
in Methanol/Dioxan (20 ml/20 ml) gelöst und über 10% Palladium auf Kohle
(katalytisch) für
18 h hydrogeniert (55 psi). Der Katalysator wurde durch Filtration
entfernt, und das Lösungsmittel
wurde in vacuo evaporiert, um [1α,2α(trans)]-4[[(Benzolsulfonyl)amino]methyl]-N-[1,2,3,4-tetrahydro-6-methoxy-1-(n-propyl)2-naphthalinyl]-cyclohexancarboxamid
41 als einen cremefarbenen Feststoff (0,5 g, eine Komponente durch
HPLC) zu ergeben. MS (MH+) 499. 1H (DMSO-d6) δ 0,75–0,92 (m,
7H), 1,22–2,17
(m, 11H), 2,13 (m, 1H), 2,57 (t, 2H), 2,67–2,88 (m, 3H), 3,70 (s, 3H),
3,90–4,03
(m, 1H), 4,16 (d, 1H), 6,62–6,73
(m, 2H), 6,97 (d, 1H), 7,56–7,74 (m,
4H) und 7,80 (d, 2H); NMR zeigt auch eine nicht identifizierte Verunreinigung.
Dieses Material wurde ohne Reinigung in dem anschließenden Schritt
verwendet.
- B. Eine Lösung
von Bor in Tetrahydrofuran (4,0 ml einer 1,0 M Lösung, 4,0 mmol) wurde zu einer
Lösung des
ungereinigten Carboxamids 41 (0,43 g, 0,86 mmol) in Tetrahydrofuran
(10 ml) hinzugefügt.
Das sich ergebende Gemisch wurde auf Reflux für 1 h erhitzt. Wasser (1,5
ml) wurde zu der abgekühlten
Lösung langsam
hinzugefügt,
was zu beträchtlichem
Schäumen führte. Konzentriertes
wäßriges Hydrogenchlorid (0,75
ml) wurde hinzugefügt
und die Lösung
wurde auf Reflux für
1 h erhitzt. Die Lösung
wurde konzentriert und der pH auf 7–8 mit wäßrigem Natriumhydroxid (1 N)
eingestellt. Der sich ergebende Feststoff wurde durch Filtration
gesammelt und mit Wasser gewaschen. Dieses Material wurde in Ethanol
gelöst
und mit einer gesättigter
Lösung
von Hydrogenchlorid in Ethanol behandelt. Das Hydrogenchloridsalz
des Produkts kristallisierte aus der Lösung und wurde durch Filtration
gesammelt, mit Diethylether gewaschen und in vacuo getrocknet, um [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-methoxy-1-(n-propyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]benzolsulfonamid
42 als einen farblosen Feststoff (0,147 g, eine einzelne Komponente
durch HPLC) zu ergeben. Die Mutterflüssigkeiten wurden evaporiert,
und der sich ergebende Rest mit Diethylether tituriert, um zusätzliche
0,120 g an Produkt zu ergeben. MS (MH+)
485; 1H NMR (DMSO-d6) δ 0,75–0,96 (m, 7H),
1,12–1,37
(m, 4H), 1,56–2,16
(m, 7H), 2,58 (t, 2H), 2,54–2,63
(m, 2H), 2,72–3,09
(m, 5H), 3,24–3,36 (m,
1H), 3,71 (s, 3H), 6,65–6,74
(m, 2H), 6,96 (d, 1H), 7,56–7,68
(m, 4H), 7,80 (d, 2H), 8,56 (br s, 1H, wechselwirkt mit D2O), und 8,95 (br s, 1H, wechselwirkt mit
D2O) (Figur 13).
-
-
BEISPIEL 9
-
rac-[1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-hydroxy-1-(3-pyridinylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexylmethyl]-2-fluorbenzolsulfonamid
(44)
-
Das
bis-Aminsalz des Ausgangsmaterials 43 (0,109 g, 0,174 mmol) wurde
zusammen mit 30 ml Dichlormethan in eine 100 ml Rundboden-Flasche
gefüllt.
Zu dieser gerührten
Lösung
wurde Diisopropylethylamin (0,067 ml, 0,385 mmol) hinzugefügt, was
zu der Lösung
des Ausgangsmaterials führte.
Diese gerührte
Lösung
wurde auf einem Eisbad abgekühlt.
Bortribromid in Dichlormethan (1,74 ml einer 1 M Lösung, 1,74
mmol) wurde zu der Aminlösung
hinzugefügt
und ein Präzipitat
bildete sich. Diese Lösung
wurde für
ungefähr
2 Stunden gerührt,
während
sie auf dem Eisbad belassen wurde, wobei zu dieser Zeit 4 ml Methanol
hinzugefügt
wurden, um den Überschuß Bortribromid
abzulöschen.
Die Lösungsmittel
wurden dann in vacuo entfernt und der Rest in 100 ml Dichlormethan
gelöst.
Der organische Extrakt wurde zweimal mit 100 ml 0,02 M Natriumhydroxid
gewaschen. Eine Emulsion bildete sich, die durch die Zugabe von
festem Natriumchlorid aufgebrochen wurde. Die organischen Extrakte
wurden einmal mit 100 ml Lauge gewaschen und dann über Magnesiumsulfat getrocknet,
gefolgt durch das Entfernen der Lösungsmittel in vacuo. Der Rest
wurde in Methanol gelöst
und ethanolisches Hydrogenchlorid wurde hinzugefügt. Die Lösungsmittel wurden in vacuo
entfernt, um das ungereinigte Produkt als einen Feststoffilm zu
ergeben. Dieses Material wurde weiter durch kurzes Erhitzen in Isopropanol
gereinigt, wobei des dem Feststoff ermöglicht wurde, sich abzutrennen,
gefolgt von Filtration, und dann Trocknen unter Vakuum um [1α,2α(trans)]-N-[[[[[1,2,3,4-Tetrahydro-6-hydroxy-1-(3-pyridinylmethyl)-2-naphthalinyl]amino]methyl]-4-cyclohexyl]methyl]-2-fluorbenzolsulfonamid-bis-hydrochlorid
44 als ein braunes Pulver (0,054 g, 0,088 mmol) zu ergeben. MS (MH+) 538; 1H NMR (DMSO-d6) δ 0,69–1,12 (m,
4H), 1,22–1,46
(m, 1H), 1,61–2,32
(m, 7H), 2,60–3,12
(m, 7H), 3,293,61 (m, 3H), 5,67 (d, 1H), 6,18 (dd, 1H), 6,52 (s,
1H), 7,30–7,51
(m, 2H), 7,637,84 (m, 2H), 7,85–8,02
(m, 2H), 8,27 (d, 1H), 8,62–8,84
(m, 2H), 9,03 (br, 1H), 9,45 (br, 1H) (Figur 14).
-
-
-
Andere
Verbindungen dieser Erfindung, die die Formel 1 aufweisen, können unter
der Verwendung der hier beschriebenen Verfahren präpariert
werden. Es gibt über
eintausend Verbindungen, die eine Phenylessigsäuregruppe enthalten, die käuflich erhältlich sind,
und viele mehr, die bekannt sind, und diese Verbindungen können in
die entsprechenden β-Tetralone
unter der Verwendung der in BEISPIEL 4 beschriebenen Chemie überführt werden.
Diese Zwischenprodukte können
in Produkte der Formel 1, die eine große Vielzahl von (R1)n Gruppen enthalten, unter der Verwendung
der in BEISPIEL 4 beschriebenen Chemie überführt werden. In einigen Fällen kann
die Verwendung von Schutzgruppen erforderlich sein, und diese Manipulationen
sind dem Fachmann bekannt. Zum Beispiel kann Aminophenylessigsäure in das
entsprechende Phthalimid nach Reaktion mit phthalischem Anhydrid
oder N-Carbethoxyphthalimid überführt werden.
Unter der Verwendung der in BEISPIEL 4A beschriebenen Chemie können Phthalimido-β-tetralone
durch Substitution von (Phthalimido)phenylessigsäuren für 4-Fluoressigsäure hergestellt
werden, und diese Materialien können
anschließend in
Produkte der Formel 1 überführt werden
wobei, nach Phthalimid-Spaltung, (R1)n Amino (NH2) ist.
Alkylamino-(-NHR) und Dialkylamino-(-NR'R'')-Analoga können auch
aus dem Phthalimido-β-tetralon
hergestellt werden.
-
Die
Verwendung von alpha-substituierten Phenylessigsäure-Ausgangsmaterialien um
Verbindungen der Formel 1 zu erhalten, worin B2 Alkyl
oder substituiertes Alkyl und nicht Wasserstoff ist.
-
Verbindungen
dieser Erfindung der Formel 1 die einen Pyrimidyl-, Imidazolyl-,
Thienyl- oder Furyl-Substituenten als R2 aufweisen,
können
unter der Verwendung von der in BEISPIEL 3 beschriebenen Chemie
hergestellt werden, wobei ein β-Tetralon
mit einem Heteroarylaldehyd reagiert wird. Zum Beispiel kann für 3-Pyridylcarboxaldehyd
in BEISPIEL 3A Furan- und Thienylcarboxaldehyd substituiert werden
und mit β-Tetralonen reagiert
werden, und diese Zwischenprodukte können anschließend in
Produkte der Formel 1 überführt werden,
worin R2 2-Furyl oder 3-Furyl oder 2-Thienyl
oder 3-Thienyl ist und Y Methylen ist und m = 1, Ähnlich kann N-Tritylimidazolcarboxaldehyd
verwendet werden, um Verbindungen der Formel 1 herzustellen, in
denen R2 2-Imidazolyl oder 4(5)-Imidazolyl
ist und Y Methylen ist und m = 1, Verbindungen der Formel 1, in
denen der R2-Substituent Cyclopropyl ist
und Y = Methylen und m = 1, unter der Verwendung von der in BEISPIEL
1 beschriebenen Chemie, wobei Cyclopropylmethylbromid für Benzylbromid
substituiert wird. Verbindungen der Formel 1, in denen der R2-Substituent Phenoxy oder Thiophenyl ist,
können
durch substituieren von Chlormethylphenylether oder einem Chlormethylphenylsulfid
für Benzylbromid
in BEISPIEL 1 hergestellt werden.
-
Verbindungen
der Formel 1, worin der R2-Substituent Piperidin
ist, können
durch reduzieren des entsprechenden Pyridylanalogons, wie zum Beispiel
dem in BEISPIEL 3 Beschriebenen, unter der Verwendung von katalytischen
Hydrogenierungsbedingungen (d. h., Platinoxid auf Kohle), hergestellt
werden.
-
Verbindungen
der Formel 1, worin der R3-Substituent Heteroaryl
ist, können
durch substituieren eines Pyridinyl-, Thienyl- oder Furylsulfonylchlorids
für 2-Naphthylsulfonamid
in BEISPIEL 3F hergestellt werden. N-Alkylimidazolylsulfonylchloride
können
verwendet werden, um Verbindungen der Formel 1 herzustellen, worin
der R3-Substituent Imidazolyl ist.
-
Weitere
Verbindungen dieser Erfindung, die unter der Verwendung von den
oben beschriebenen experimentellen Protokollen hergestellt wurden,
schließen
ein:
-
Massenspektrumdaten
der Verbindungen (1)
-
-
-
-
IN VITRO TESTS
-
NPY5 HTS Zentrifugationstest
-
Die
in dieser Erfindung beschriebenen Verbindungen wurden auf die Bindung
an den menschlichen Neuropeptid Y5 Rezeptor untersucht.
-
Stabile Transfection
-
Die
menschliche NPY5 Rezeptor cDNA (Genbank Zugangsnummer U66275) wurde
in dem Vektor pClneo (Invitrogen) inseriert und in menschliche embryonale
Nierenzellen (HEK-293) über
das Calciumphosphatverfahren (Cullen 1987) transfiziert. Stabil
transfizierte Zellen wurden mit G-418 (600 μg/ml) selektiert. Die stabil
transfizierten Zellen dienten als die Quelle für die Membranen für den NPY5
Rezeptorbindungstest.
-
Membranpräparation
-
NPY5-transfizierte
HEK293 Zellen wurden bis zur Konfluenz in 150 cm2 Kulturschalen
angezogen. Die Zellen wurden einmal mit Phosphat-gepufferter Kochsalzlösung (Gibco
Katalog-#; 14040-133) gewaschen. Die Zellen wurden dann in Phosphat-gepufferter
Kochsalzlösung
ohne Calcium und ohne Magnesium, ergänzt mit 2 mM EDTA, inkubiert.
Die Zellen wurden für
10 Minuten bei Raumtemperatur inkubiert und die Zellen wurden durch
wiederholtes Pipettieren gesammelt. Die Zellen wurden in Pellets
geformt und dann bei –80°C bis Bedarf eingefroren.
Die gefrorenen Pellets wurden mit einem Poltron bei voller Geschwindigkeit
für 12
Sekunden in einem Homogenisierungspuffer (20 mM Tris HCl, 5 mM EDTA,
pH 7,4) homogenisiert. Die Homogenate wurden für 5 Minuten bei 4°C bei 200
g zentrifugiert. Die Überstände wurden
in Corexröhrchen
transferiert und für 25
Minuten bei 28,000 g zentrifugiert. Die Pellets wurden in Bindungspuffer
(20 mM HEPES, 10 mM NaCl, 0,22 mM KH2PO4, 1,3 mM CaCl2,
0,8 mM MgSO4, pH 7,4) resuspendiert. Die
Membranen wurden bis zur Verwendung auf Eis gehalten.
-
Ein
kompetetiver Bindungstest, der dem Fachmann bekannt ist, wurde verwendet,
in denen Aminotetraline (I) mit 125J-PYY
um die Bindung an Zellmembranen kompetetiv sind. Einfach gesagt,
impliziert weniger an die Membranen gebundenes 125J-PYY,
daß eine
Verbindung ein guter Inhibitor (Kompetitor) ist. Das gebundene 125J-PYY wird durch die Zentrifugation von
Membranen, Ausblasen des Überstands,
Abwaschen von restlichem 125J-PYY und anschließend Zählen der
gebundenen Probe in einem gamma-Zähler bestimmt.
-
Verfahren für Radioliganden-Bindungstest
-
Die
zu testenden Verbindungen wurden als 10× Vorratslösungen in Bindungspuffer hergestellt
und zuerst zu Teströhrchen
(RIA Gefäße, Sarstedt)
hinzugefügt.
Zwanzig (20) μl
jeder 10× Verbindungs-Vorratslösung wird
in Gefäße pipettiert
und 80 μl
von 125J-PYY (NEN Katalog Nummer NEX240),
das auf eine Konzentration von 200 pM in 0,25% BSA in Bindungspuffer
verdünnt
wurde, wird zu dem Verbindungsröhrchen
(finale Konzentration von 125J-PYY ist 80
pM) hinzugefügt.
Zu jedem Röhrchen
werden 100 μl
von Membranen hinzugefügt
und das Gemisch wird durch zweimaliges Pipettieren gemischt. Die
Proben werden für
1 h bei Raumtemperatur inkubiert. Gegossene Aluminiumplatten (Sarstedt),
die die Gefäße enthalten,
werden dann für
10 Minuten bei 3200 U/min in einer Sorvall RT6000 zentrifugiert.
Der Überstand
wird dann ausgeblasen. Zu jedem Gefäß werden 400 μl PBS hinzugefügt und dieses
wird dann nochmals ausgeblasen. Die Gefäße werden dann in Träger-Polypropylen
12 × 75-Röhrchen hineingestellt
und in einem gamma-Zähler
(Packard) gezählt.
Die nichtspezifische Bindung wird in Anwesenheit von 300 nM NPY
bestimmt. Die Prozent Inhibierung der 125J-PYY Bindung
wird durch subtrahieren der nicht-spezifischen Bindung von den Testproben
(Verbindung (I)), hernehmen dieser Zählungen und dividieren durch
die Gesamt- bindung und multiplizieren mit 100 berechnet. Die inhibitorischen
Konzentrationswerte (IC50) von Verbindungen,
die eine annehmbare Inhibierung der 125J-PYY Bindung
zeigen, werden durch erhalten der Prozent Inhibierung von 125J-PYY Bindungswerten bei verschiedenen
Konzentrationen der Testverbindung berechnet, und unter der Verwendung
eines graphischen Programms, wie zum Beispiel GraphPad Prism (San
Diego, CA), um die Konzentration an Testverbindung, die fünfzig Prozent
der 125J-PYY Bindung inhibiert (Tabelle
4), zu berechnen. Diese Schritte sind dem Fachmann bekannt.
-
Bindungsaffinitäten von
Verbindungen ein (1) für
den menschlichen NPY Y5 Rezeptor
(ausgedrückt als % Inhibierung der
125J-PYY Bindung)
-
-
-
IN VIVO TESTS
-
Nagermodell der Fütterung
-
Messung der Futteraufnahme
in Futter-Entzug Ratten
-
Männliche
Long-Evans Ratten (180–200
Gramm) werden einzeln gehalten und werden auf einem einmal täglich Fütterungsplan
(d. h. 10 Uhr bis 16 Uhr) für
fünf Tage
gehalten, gefolgt von Quarantäne,
um es den Tieren zu ermöglichen,
sich an die Fütterung
mit Trockenfutter (Kat#; 5002 PMI Certified Rodent Meal) während der
zugeordneten Zeit zu gewöhnen.
Das Futter wird in einem offenen Behälter erhältlich gemacht der in dem Käfig mit
einem Draht verankert ist, mit einem Metallverfolger, der das Futter
bedeckt, um ein Verstreuen zu minimieren. Wasser ist erhältlich ad
libitum.
-
Die
Tiere werden für
18 Stunden vor dem Testen fasten gelassen. Am Ende der Fastenperiode,
werden den Tieren entweder Verbindungen der Erfindung oder Vehikel
verabreicht. Vehikel und Testverbindungen werden entweder oral (5
ml/kg) 60 Minuten vor dem Experiment verabreicht oder 30 Minuten
vorher, wenn subkutan (1 ml/kg) oder intraperitoneal (1 ml/kg) verabreicht.
Verbindungen der Erfindung werden oral als eine Suspension in wäßrigem 0,5%
Methylcellulose-0,4% Tween 80, oder intraperitoneal als eine Lösung oder
Suspension in PEG 200 verabreicht; die Verbindungskonzentrationen
reichen typischerweise von 1 mg/kg bis 100 mg/kg, bevorzugt von
10–30
mg/kg. Die Futteraufnahme wird bei 2, 4 und 6 Stunden nach Verabreichung durch
Wiegen des Spezialbehälters,
der das Futter enthält,
vor dem Experiment und zu den angegebenen Zeiten gemessen. Nach
Vervollständigung
des Experiments wird allen Tieren eine einwöchige Ausscheidungszeit vor
dem nochmaligen Testen gegeben.
-
Die
Prozent Verringerung der Futteraufnahme wird durch Subtrahieren
der Gramm an Futter, das durch die behandelte Gruppe verbraucht
wurde von den Gramm an Futter, das durch die Kontrollgruppe verbraucht
wurde, geteilt durch die Gramm an Futter das durch die Kontrollgruppe
verbraucht wurde, multipliziert mit 100, berechnet
-
-
Ein
negativer Wert zeigt eine Verringerung der Futteraufnahme und ein
positiver Wert zeigt eine Erhöhung
der Futteraufnahme an.
-