-
Die vorliegende Erfindung bezieht
sich auf ein neues Ethylencopolymeres und ein Verfahren zu seiner Herstellung.
Mehr speziell betrifft die vorliegende Erfindung ein neues Ethylencopolymeres,
umfassend ein Copolymeres von Ethylen mit wenigstens einem Comonomeren,
ausgewählt
aus der Gruppe, umfassend Verbindungen, wiedergegeben durch die
Formel H2C = CHR, worin R eine C1-C20 lineare, verzweigte
oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe darstellt, und einem C4-C20 linearen, verzweigten
oder cyclischen Dien, und das eine spezifische Dichte besitzt und
nicht nur eine charakteristische spezifische Molekulargewichtsverteilung
besitzt, sondern ebenfalls eine spezifische charakteristische Comonomerengehaltverteilung.
-
Die vorliegende Erfindung bezieht
sich ebenfalls auf Mischungszusammensetzungen, umfassend dieses
neue Ethylencopolymere und:
- a) ein zweites
Ethylencopolymeres der vorliegenden Erfindung mit unterschiedlichem
Molekulargewicht oder unterschiedlicher Dichte; oder
- b) ein homogenes Ethyleninterpolymeres mit schmaler Zusammensetzungsverteilung;
- c) ein heterogenes Ethyleninterpolymeres mit breiter Zusammensetzungsverteilung
oder
- d) ein Homopolymeres (hergestellt mit einer anderen Katalysatorkomponente
als diejenige, welche zur Herstellung des Ethylencopolymeren der
vorliegenden Erfindung verwendet wurde); oder
- e) eine Kombination von beliebigen zwei oder mehr von a), b),
c) oder d).
-
Die vorliegende Erfindung betrifft
ebenfalls ein neues Verfahren zur Herstellung der Mischungszusammensetzungen,
welche das Ethylencopolymere der vorliegenden Erfindung umfassen.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat große
Vorteile, welche durch konventionelle Ethylencopolymere nicht geliefert
wurden, d. h. es enthält
keine Verunreinigungen wie ein Wachs, ein Gel. Zusätzlich haben
sowohl das Ethylencopolymere als auch die Mischungszusammensetzungen
hiervon gemäß der vorliegenden
Erfindung ebenfalls ausgezeichnete Eigenschaften, wie hohe Schlagfestigkeit
und ausgezeichnete Rißfestigkeit
gegenüber
Umgebungsspannungen, so daß sie
vorteilhafterweise zur Herstellung von Laminatfolien, blasgeformten
Gegenständen,
Rohren, Überzugsmaterialien
für elektrische Übertragungskabel verwendet
werden können.
-
Ethylencopolymere werden weit verbreitet
in verschiedenen Anwendungsfeldern eingesetzt, beispielsweise bei
der Herstellung von Folien/Filmen, blasgeformten Produkten, Rohren
und Beschichtungsmaterialien für
elektrische Übertragungskabel.
Im Hinblick auf irgendwelche dieser Anwendungen ist es erforderlich,
daß ein
Ethylencopolymeres nicht nur wenig Verunreinigungen, wie Wachs,
Gele, enthält,
sondern ebenfalls ausgezeichnete Eigenschaften, wie hohe Schlagfestigkeit
und hohe Rißfestigkeit
gegenüber
Umgebungsspannungen (im folgenden häufig als "ESCR-Eigenschaften" bezeichnet), zeigt. Jedoch haben Versuche
zum Variieren der Molekülstruktur
eines Polymeren zur Herbeiführung
einer Verbesserung einer solchen Eigenschaft oftmals einen Verlust
der Leistungsfähigkeit
in einer anderen ergeben. Beispielsweise sollten Polymere, die hohe
Steifigkeit und Hitzebeständigkeit
zeigen, hohe Kristallinität
und niedrigen Comonomerengehalt haben, jedoch kann dies einen Verlust
von Zähigkeit,
ESCR, geringe optische Eigenschaften und schlechte Hitzesiegelfähigkeit
bewirken. In gleicher Weise ist es für verbesserte Polymerverarbeitbarkeit
(niedriger Stromverbrauch bei der Extrusion und geringer Rückdruck
und kein Schmelzbruch) erwünscht,
Polymere mit einem niedrigen Molekulargewicht und einer breiten
Molekulargewichtsverteilung mit signifikanten Werten von Langkettenverzweigung
zu verwenden. Jedoch bewirkt breite Molekulargewichtsverteilung,
insbesondere bei niedrigem Polymermolekulargewicht, oftmals den
Aufbau von Wachs an der Düse,
die Entwicklung von Rauch in dem Extruder und Probleme beim Geschmack
und Geruch bei den resultierenden hergestellten Gegenständen.
-
Es ist bekannt, daß die Verbesserung
der Schlagzähigkeit
und der Rißfestigkeit
bei Umgebungsspannung eines Ethylencopolymeren durch Herabsetzung
des Comonomerengehaltes der Fraktion mit niedrigem Molekulargewicht
des Ethylencopolymeren bis auf einen möglichst geringen Wert, während der
Comonomerengehalt der Fraktion mit hohem Molekulargewicht des Ethylencopolymeren
bis zu einem möglichst
hohen Wert erhöht
wird, erreicht werden kann. Ebenfalls wurde gezeigt (beispielsweise
durch Zhou et al, Polymer, Vol. 24, S. 2520 (1993)), daß Eigenschaften
großer
Dehnung wie Zähigkeit,
Reißfestigkeit,
Schlagfestigkeit und ESCR ebenfalls durch die Anwesenheit von "Bindungsmolekülen" in dem Harz verbessert
werden können. Moleküle mit hohem
Molekulargewicht mit dem höchsten
Comonomerengehalt (d. h. dem höchsten
Grad von Kurzkettenverzweigung) sind für die Bildung der meisten der
Bindungsmoleküle
bei Kristallisation verantwortlich.
-
Daher wäre es für ein Copolymeres hoch erwünscht, eine
spezifische charakteristische Verteilung des Comonomerengehaltes
zu haben, bei welchem gemäß einem
Aspekt je niedriger das Molekulargewicht einer Copolymerfraktion
in einer Molekulargewichtsverteilung dieses Copolymeren ist, umso
niedriger der Comonomerengehalt der Copolymerfraktion ist, und,
bei dem anderen Aspekt, je höher
das Molekulargewicht einer Fraktion dieses Copolymeren ist, umso
höher der
Comonomerengehalt der Copolymerfraktion ist.
-
Jedoch ist es in Ethylencopolymeren,
welche unter Verwendung eines konventionellen Ziegler-Natta-Katalysators
(siehe z. B. EP-A-0 117 492; WO-A-94/17112) hergestellt werden,
wahrscheinlich, daß um
so niedriger das Molekulargewicht einer Copolymerfraktion ist, um
so höher
ihr Comonomerengehalt ist. Daher haben solche konventionellen Ethylencopolymere
eine Verteilung des Comonomerengehaltes, welche vollständig im
Gegensatz zu der oben genannten erwünschten Verteilung des Comonomerengehaltes
ist. Daher haben solche konventionellen Ethylencopolymere den Nachteil
hinsichtlich der gewünschten
Eigenschaften, wie verbesserter Schlagfestigkeit und ESCR.
-
Versuche zur Maximierung der Zähigkeit,
des Moduls, der Schlagfestigkeit und der ESCR von Ethylencopolymeren
ergab die Herstellung und Verwendung von Mischungszusammensetzungen,
die aus zwei oder mehr Ethylencopolymerkomponenten von unterschiedlichen
Molekülstrukturen
hergestellt wurden. Zusätzlich
zu getrenntem Mischen von ausgewählten
einzelnen Polymerkomponenten nach ihrer Herstellung und Isolierung
(sogenanntes "off-line"-Mischen), können solche
Zusammensetzungen auch nach einem Verfahren hergestellt werden,
bei welchem eine Copolymerisation von Ethylen mit einem Comonomeren
mittels einer Vielstufenpolymerisation unter Anwendung einer Vielzahl
von unterschiedlichen Polymerisationsreaktoren, die zur Lieferung
von unterschiedlichen Copolymerisationsbedingungen in der Lage sind,
durchgeführt
wird. Dies ermöglicht
sogenannte Herstellung "im
Reaktor" oder "im Verfahren" von Ethylencopolymeren,
umfassend eine Mischung einer Copolymerkomponente mit niedrigem
Molekulargewicht, die einen niedrigen Comonomerengehalt besitzt,
und einer Copolymerkomponente mit hohem Molekulargewicht, die einen
hohen Comonomerengehalt besitzt.
-
Solche Mischungszusammensetzungen,
welche lediglich Produkte mit Ziegler-Katalysatoren enthalten, sind
in einer Anzahl von Patenten beschrieben. Beispielsweise lehrt Nelson (
US 3 280 220 , Phillips Petroleum),
daß eine
Mischung eines Ethylenhomopolymeren von niedrigem Molekulargewicht
(gebildet in einem Lösungsverfahren)
und ein Ethylen/Buten-1-copolymeres von hohem Molekulargewicht (gebildet
in einem teilchenbildenden Verfahren) höhere ESCR liefert, und daß sie vorteilhafter
für Behälter und
Rohre als andere solche Mischungen ist.
-
Hoblitt et al. (
US 3 660 530 , The Dow Chemical Company)
lehrt ein Verfahren, bei welchem ein Teil eines nach einer ersten
Reaktionsstufe erzeugten Homopolymeren 1-Buten ausgesetzt wird.
Der noch aktive Katalysator erzeugt dann ein Blockcopolymeres von
Polyethylen und polymerisiertem Buten-1. Beide Komponenten werden
dann zusammengemischt. Die resultierende Mischung hat verbesserte
ESCR-Eigenschaften.
-
Fukushima et al. (
US 4 438 238 ) beschreibt Mischungen,
bestehend aus Komponenten mit Dichten zwischen 0,910 und 0,49 g/cm
3 und breiten Molekulargewichtsverteilungen,
wobei die Polymere im wesentlichen keine Langkettenverzweigungen
besitzen: Es wurde gefunden, daß diese
Mischungen Verarbeitbarkeit vergleichbar zu derjenigen von Hochdruckpolyethylen
besitzen.
-
Bailey et al. (
US 4 547 551 ) lehrt, daß Ethylenpolymermischungen
mit einem Ethylenpolymerem mit hohem Molekulargewicht, bevorzugt
ein Ethylen/α-Olefincopolymeres,
und einem Ethylenpolymerem mit niedrigem Molekulargewicht, bevorzugt
einem Ethylenhomopolymerem, die beide bevorzugt eine schmale Molekulargewichtsverteilung
und geringe Gehalte an Langkettenverzweigung haben, ausgezeichnete
Film-/Folieneigenschaften und ein besseres Gleichgewicht von Steifigkeit
und Schlagfestigkeit und ESCR besitzen, als dies für Polyethylen
von vergleichbarer Dichte und vergleichbarem Fließen erwartet
würde.
-
Morimoto et al. (
US 5 189 106 und
5 260 384 ) beschreiben Mischungen,
bestehend aus einem Copolymeren mit hohem Molekulargewicht in Kombination
mit einem Hompolymeren mit niedrigem Molekulargewicht, die gute
Verarbeitbarkeit und ausge zeichnete mechanische Eigenschaften bei
niedrigen Temperaturen haben.
-
Boehm et al. (Advanced Materials
4 (1992) Nr. 3, 5.237) beschreiben das Kaskadenpolymerisationsverfahren,
bei welchem das Comonomere in der Fraktion mit hohem Molekulargewicht
des Polymeren eingeführt
wird, was eine größere Menge
von Comonomerem ergibt, das bei derselben Gesamtdichte vorhanden ist.
Dies ergibt wiederum eine Polymerzusammensetzung mit verbesserter
Steifigkeit-Lebensdauer (Versagungszeit), verglichen mit konventionellen
unimodalen Copolymeren. Mehrere Patente wurden ebenfalls ausgegeben,
welche lehrten, daß das
Verfahren solche Materialien in solchen Kaskadenverfahren liefert,
einschließlich
der
EP 0 022 376 (Morita
et al.).
-
Die nicht geprüften japanischen Patentanmeldungen,
offengelegten Spezifikation Nr. 61-221245 und 61-57638 beschreiben
Versuche zur Erhöhung
des Comonomerengehaltes von Copolymerfraktionen mit hohem Molekulargewicht
mittels eines Verfahrens, bei welchem ein Polymeres mit niedrigem
Molekulargewicht, das einen niedrigen Comonomerengehalt besitzt,
und ein Polymeres mit hohem Molekulargewicht, das einen hohen Comonomerengehalt
besitzt, getrennt hergestellt und mittels eines Kneters gemischt
werden, oder ein Verfahren, bei welchem eine Copolymerisation von
Ethylen mit einem Comonomeren durch Vielstufenpolymerisation durchgeführt wird,
wodurch ein Ethylencopolymeres erzeugt wird, das eine Mischung einer
Polymerkomponente mit niedrigem Molekulargewicht, die einen niedrigen
Comonomerengehalt hat, und eine Polymerkomponente mit hohem Molekulargewicht,
die einen hohen Comonomerengehalt hat, erzeugt wird.
-
Schließlich beschreiben Sakurai et
al. (
US 4 230 831 ),
daß es
günstig
ist, Polyethylen mit niedriger Dichte mit verschiedenen Mischungszusammensetzungen
zu vermischen, um das Quellen des Polymeren an der Düse oder
die Schmelzspannung zu verbessern.
-
Bei Einzelkomponenten-Ethylencopolymeren,
hergestellt unter Verwendung eines Ziegler-Katalysators, wird eine
gewisse Verbesserung hinsichtlich der Schlagzähigkeit und den ESCR-Eigenschaften
erreicht. Solche Ethylencopolymere zeigen jedoch von sich aus nicht
nur eine breite Molekulargewichtsverteilung, sondern auch einen
breiten Schwanz an der Seite sowohl bei niedrigem als auch bei hohem
Molekulargewicht der Molekulargewichtsverteilung. Die Anwesenheit
von Material von niedrigem Molekulargewicht kann nachteiligerweise
zur Wachsbildung führen.
Andererseits kann das Material mit hohem Molekulargewicht in nachteiliger Weise
zur Gelbildung führen.
-
Zusätzlich können Mischungszusammensetzungen,
welche eine Mischung von solchen mittels eines Ziegler-Katalysators
erzeugten Ethylencopolymeren sind, Komponentencopolymere umfassen,
welche vollständig
verschieden voneinander hinsichtlich der Eigenschaften sind, d.
h. eine Polymerkomponente mit niedrigem Molekulargewicht, die einen
niedrigen Comonomerengehalt hat, und eine Polymerkomponente mit
hohem Molekulargewicht, die einen hohen Comonomerengehalt hat. Dies
kann dazu führen,
daß die
Komponentenpolymere Phasentrennung erfahren, so daß die Dispersion
der Komponentenpolymere ungleichförmig wird, und daher wird nicht
nur das Ethylencopolymere ungleichmäßig hinsichtlich der Eigenschaften,
sondern es tritt ebenfalls Gelbildung auf.
-
Als eine Alternative zur Verwendung
von Ziegler-Natta-Katalysatoren
wurde in neuerer Zeit die Verwendung von Metallocenkatalysatoren
vorgeschlagen (
DE 3 127 133.2 )
und kommerziell angewandt. Wie beispielsweise in Worldwide Metallocene
Conference (Metcon) '93,
26–28.
Mai, Houston Texas, S. 171–172
und S. 235–244
(1993) und den Proceedings of 5
th International
Business Forum on Specialty Polyolefins '95, 20–22. September, Houston Texas,
S. 341–352
(1995) beschrieben ist, hat ein Ethylencopolymeres, das unter Verwendung
eines solchen Metallocenkatalysators erzeugt wurde, Eigenschaften
derart, daß sowohl
eine Fraktion mit niedrigem Molekulargewicht als auch eine Fraktion
mit hohem Molekulargewicht annähernd
denselben Comonomerengehalt haben, und daß die Verteilung des Comonomerengehaltes
beinahe gleichförmig quer über die
Molekulargewichtsverteilung des Copolymeren ist. Dies bedeutet,
ein Ethylencopolymeres, erzeugt unter Verwendung eines Metallocenkatalysators,
hat eine gleichförmigere
Verteilung des Comonomerengehaltes als bei einem Ethylencopolymeren,
das unter Verwendung eines Ziegler-Natta-Katalysators hergestellt
wurde. Andererseits sind jedoch Ethylencopolymere, erzeugt unter
Verwendung eines Metallocenkatalysators, immer noch nicht zufriedenstellend
hinsichtlich den gewünschten
Verbesserungen der Schlagfestigkeits- und ESCR-Eigenschaften von
Produkten aus solchen Copolymeren.
-
Wiederum, wie im Fall mit Ziegler-Katalysatorprodukten,
schlossen Versuche zur Verbesserung wie der ESCR und der Schlagfestigkeit
von Produkten von Metallocenkatalysatoren ihren Einbau in Mischungszusammensetzungen
ein. Eine Anzahl von Techniken wurden zur Herstellung solcher Mischungen
vorgeschlagen, einschließlich
einem Verfahren, bei welchem zwei oder mehr unterschiedliche Ethylencopolymere
mit unterschiedlichen Comonomerengehalten getrennt hergestellt und
gemischt wurden, und zwar mittels eines Kneters oder eines Verfahrens,
bei welchem ein Ethylencopolymeres, das aus einer Mischung von zwei
oder mehr unterschiedlichen Ethylencopolymerkomponenten mit unterschiedlichen
Comonomerengehalten besteht, mittels Vielstufenpolymerisation hergestellt
wird (siehe beispielsweise
EP
0 447 035 ). Weiterhin wurde ebenfalls vorgeschlagen, ein
Verfahren anzuwenden, bei welchem eine Mischung von zwei oder mehr
unterschiedlichen Typen von Metallocenkatalysatoren zur Herstellung
eines Ethylencopolymeren eingesetzt wird, das aus einer Mischung
von zwei oder mehr unterschiedlichen Ethylencopolymerkomponenten
mit unterschiedlichen Comonomerengehalten besteht (siehe beispielsweise
US-Patente Nr. 4
937 299 und 4 530 914).
-
Jedoch hat ein Ethylencopolymeres,
das unter Verwendung eines Metallocenkatalysators hergestellt wurde,
typischerweise eine sehr schmale Molekulargewichtsverteilung (Mw/Mn) von annähernd 2. Wenn daher zwei unterschiedliche
Typen von Copolymeren, nämlich
ein Copolymeres mit niedrigem Molekulargewicht und ein Copolymeres
mit hohem Molekulargewicht, die extrem unterschiedlich im Molekulargewicht
voneinander sind, unter Verwendung von unterschiedlichen Metallocenkatalysatoren
hergestellt werden, sind die jeweiligen Mengen von Copolymerketten,
welche gemeinsame Molekulargewichte besitzen, sehr gering in den
zwei verschiedenen Copolymeren, so daß die Verträglichkeit zwischen diesen zwei
unterschiedlichen Copolymeren sehr schlecht ist.
-
Um das oben genannte Problem zu lösen, wurde
ein Verfahren vorgeschlagen, bei welchem ein unter Verwendung eines
Metallocenkatalysators hergestelltes Ethylencopolymeres mit einem
unter Verwendung eines Ziegler-Natta-Katalysators hergestellten
Ethylencopolymeren gemischt wird, beispielsweise in den
EP 0 439 964 und
EP 0 435 514 . Weiterhin
wurde ein Verfahren beschrieben, bei welchem ein unter Verwendung eines
Metallocenkatalysators hergestelltes Ethylencopolymeres mit einem
unter Anwendung eines Hochdruckpolymerisationsverfahrens hergestellten
Ethylencopolymeren gemischt wird, beispielsweise in den offengelegten
nicht geprüften
japanischen Patentanmeldungen Nr. 6-207059 und 6-329848.
-
Jedoch bleibt eine Forderung zur
Herstellung eines Ethylencopolymeren, welches nicht nur wenige Verunreinigungen
wie Wachs, Gele enthält,
sondern das ebenfalls ausgezeichnete Eigenschaften, einschließlich hoher
Schlagfestigkeit und ausgezeichnetem ESCR, besitzt. Ebenfalls bleibt
das Erfordernis zur Entwicklung eines Ethylencopolymeren, welches
keine Verunreinigungen wie ein Wachs, ein Gel enthält, während gleichzeitig
die oben genannte gewünschte
Comonomerengehaltverteilung gezeigt wird, nämlich gemäß einem Aspekt, daß um so
niedrigere Molekulargewicht einer Copolymerfraktion, um so niedriger
der Comonomerengehalt der Copolymerfraktion ist, und gemäß dem anderen
Aspekt, je höher
das Molekulargewicht einer Copolymerfraktion ist, um so höher der
Comonomerengehalt der Copolymerfraktion ist.
-
Ebenfalls bleibt das Erfordernis
zur Herstellung von Mischungszusammensetzungen, welche diese Ethylencopolymere
enthalten, welche ebenfalls ausgezeichnete Eigenschaften, wie hohe
Schlagfestigkeit und ausgezeichnete ESCR-Eigenschaften besitzen.
Schließlich
bleibt auch noch ein Erfordernis zur Herstellung von Mischungszusammensetzungen
mit guter Verträglichkeit
zwischen den zwei Komponenten, welche verbesserte Gleichförmigkeit
und Ausgeglichenheit der Eigenschaften aufweisen, während sie
einen niedrigen Gehalt an Wachs und eine niedrige Tendenz zur Gelbildung
aufweisen.
-
Überraschenderweise
wurde gefunden, daß ein
solches gewünschtes
Ethylencopolymeres und eine solche gewünschte Mischungszusammensetzung
unter Verwendung der Polymere und der Anwendung der spezifischen
Polymerisations- und Mischverfahren der vorliegenden Erfindung hergestellt
werden können.
-
Daher ist eine Aufgabe der vorliegenden
Erfindung die Bereitstellung eines neuen Ethylencopolymeren, welches
nicht nur praktisch keine Verunreinigungen wie ein Wachs, ein Gel
enthält,
sondern das ebenfalls eine Comonomerengehaltverteilung zeigt, in
welcher je niedriger das Molekulargewicht einer Fraktion dieses Copolymeren
ist, um so niedriger der Comonomerengehalt ist, und je höher das
Molekulargewicht einer Fraktion dieses Copolymeren ist, um so höher der
Comonomerengehalt ist.
-
Ebenfalls ist es eine Aufgabe der
vorliegenden Erfindung, neue Mischungszusammensetzungen bereitzustellen,
welche dieses Ethylencopolymere umfassen, die ein ausgezeichnetes
Gleichgewicht und eine ausgezeichnete Gleichförmigkeit von Eigenschaften
wie hoher Schlagfestigkeit und ausgezeichnetem ESCR zeigen, während sie
auch einen niedrigen Wachsgehalt und eine reduzierte Tendenz zur
Gelbildung besitzen.
-
Die zuvor genannten und andere Aufgaben,
Merkmale und Vorteile der vorliegenden Erfindung ergeben sich für den Fachmann
auf dem Gebiet aus der folgenden detaillierten Beschreibung und
den anliegenden Ansprüchen.
-
Mehr im einzelnen bezieht sich die
vorliegende Erfindung auf ein Verfahren zur Herstellung eines Ethylencopolymeren,
umfassend ein Copolymeres von Ethylen mit wenigstens einem Comonomeren,
ausgewählt aus
der Gruppe, die besteht aus einer durch die Formel H2C
= CHR wiedergegebenen Verbindung, worin R eine C1-C20 lineare, verzweigte oder cyclische Alkylgruppe
oder eine C6-C20-Arylgruppe
ist, und einem C4-C20 linearen,
verzweigten oder cyclischen Dien, worin dieses Ethylencopolymere
die folgenden Eigenschaften (1) bis (4) hat:
- (1)
eine Dichte d (g/cm3) von 0,870 bis 0,980;
- (2) ein Mw/Mn von
2,5 bis 10, worin Mw und Mn jeweils
ein Gewichtsdurchschnittsmolekulargewicht und ein Zahlendurchschnittsmolekulargewicht,
beide gemessen durch Gelpermeationschromatographie (GPC) sind;
- (3) falls, in Kreuzfraktionschromatographie (CFC) dieses Ethylencopolymeren,
mit Bezug auf Extraktion bei einer willkürlichen Temperatur T(°C), die in
dem Bereich von zwischen einer ersten Temperatur, bei der eine maximale
Menge der Extraktion gezeigt wird, und einer zweiten Temperatur,
welche die niedrigere Temperatur von entweder der Temperatur von
10°C höher als
diese erste Temperatur oder 96°C
ist, fällt,
die Beziehung zwischen dieser willkürlichen Temperatur T(°C) und einem
Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
einer Copolymerfraktion, extrahiert bei dieser willkürlichen
Temperatur T(°C),
wobei bei diesem Punkt im Molekulargewicht dieses Molekulargewichtsverteilungsprofils
der Copolymerfraktion einen Peak mit einer maximalen Intensität zeigt,
durch die Methode der kleinsten Quadrate behandelt wird, um eine
annähernd
gerade Linie innerhalb des Bereiches zwischen dieser ersten Temperatur
und dieser zweiten Temperatur zu erhalten, falls dort die Copoly merfraktion
ist, deren Menge weniger als 1 Gew.-% der Gesamtmenge, ausschließend Spülen, von
Copolymerfraktionen ist, die bei Temperaturen in dem Gesamtbereich
von Extraktionstemperaturen in CFC extrahiert wurden, die Copolymerfraktion aus
der Berechnung für
die annähernd
gerade Linie ausgeschlossen werden kann, die annähernd gerade Linie einen Gradienten
innerhalb des durch die Formel (2) definierten Bereiches hat –1 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2)≤ –0,005 (I)worin:
T1 und T2 zwei verschiedene
willkürliche
Extraktionstemperaturen T(°C)
innerhalb des Bereiches von zwischen dieser ersten Temperatur und
dieser zweiten Temperatur sind, und Mp(T1)
und Mp(T2) jeweils T1 und T2 entsprechende Molekulargewichte auf dieser
annähernd
geraden Linie sind; und
- (4) die Messung dieses Ethylencopolymeren mittels CFC derartige
Eigenschaften zeigt, daß die
Summe der jeweiligen Mengen von Copolymerfraktionen, die bei Temperaturen
extrahiert wurden, welche wenigstens 10°C niedriger als die erste Temperatur,
wie oben definiert sind, 8 Gew.-% oder weniger, bezogen auf die
Gesamtmenge, ausschließlich
Spülen,
von Copolymerfraktionen, die bei Temperaturen in dem Gesamtbereich
von Extraktionstemperaturen in CFC extrahiert wurden, ist, wobei
das Verfahren umfaßt:
das Copolymerisieren dieses Ethylens mit diesem Comonomeren in der
Anwesenheit eines festen Katalysatorsystems, das umfaßt: einen
Träger,
eine Übergangsmetallverbindung
und einen zur Umwandlung der Übergangsmetallverbindung
in einen katalytisch aktiven Übergangsmetallkomplex
fähigen
Aktivator; und bei welchem:
- (i) die Polymerisationsreaktion mittels Aufschlämmungspolymerisation
durchgeführt
wird, so daß das
Produktpolymere nicht geschmolzen wird, sondern den festen Zustand
beibehält
während
der Reaktion;
- (ii) die aktive Spezies des Katalysators fest mit einem Träger verbunden
ist, derart, daß sie
nicht von dem Träger
freigesetzt wird und nicht aus dem herzustellenden Polymeren entweicht;
- (iii) das Übergangsmetall
dieser Übergangsmetallverbindung
aus den Gruppen 3 bis 5 des Periodensystems ausgewählt wird.
-
Gemäß einer zweiten Ausführungsform
bezieht sich die Erfindung auf das nach dem Verfahren der Erfindung,
wie in dem vorhergehenden Absatz beschrieben, erhältliche
Ethylencopolymere, worin dieses Ethylencopolymere die folgenden
Eigenschaften (1) bis (4) hat:
- (1) eine Dichte
d (g/cm3) von 0,870 bis 0,980;
- (2) ein Mw/Mn von
2,5 bis 10, worin Mw und Mn jeweils
ein Gewichtsdurchschnittsmolekulargewicht und ein Zahlendurchschnittsmolekulargewicht,
beide gemessen durch Gelpermeationschromatographie (GPC) sind;
- (3) falls, in Kreuzfraktionschromatographie (CFC) dieses Ethylencopolymeren,
mit Bezug auf Extraktion bei einer willkürlichen Temperatur T(°C), die in
dem Bereich von zwischen einer ersten Temperatur, bei der eine maximale
Menge der Extraktion gezeigt wird, und einer zweiten Temperatur,
welche die niedrigere Temperatur von entweder der Temperatur von
10°C höher als
diese erste Temperatur oder 96°C
ist, fällt,
die Beziehung zwischen dieser willkürlichen Temperatur T(°C) und einem
Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
einer Copolymerfraktion, extrahiert bei dieser willkürlichen
Temperatur T(°C),
wobei bei diesem Punkt im Molekulargewicht dieses Molekulargewichtsverteilungsprofils
der Copolymerfraktion einen Peak mit einer maximalen Intensität zeigt,
durch die Methode der kleinsten Quadrate behandelt wird, um eine
annähernd
gerade Linie innerhalb des Bereiches zwischen dieser ersten Temperatur
und dieser zweiten Temperatur zu erhalten, falls dort die Copolymerfraktion
ist, deren Menge weniger als 1 Gew.-% der Gesamt menge, ausschließend Spülen, von
Copolymerfraktionen ist, die bei Temperaturen in dem Gesamtbereich
von Extraktionstemperaturen in CFC extrahiert wurden, die Copolymerfraktion aus
der Berechnung für
die annähernd
gerade Linie ausgeschlossen werden kann, die annähernd gerade Linie einen Gradienten
innerhalb des durch die Formel (2) definierten Bereiches hat: –1 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2)≤ –0,005 (I)worin:
T1 und T2 zwei verschiedene
willkürliche
Extraktionstemperaturen T(°C)
innerhalb des Bereiches von zwischen dieser ersten Temperatur und
dieser zweiten Temperatur sind, und Mp(T1)
und Mp(T2) jeweils T1 und T2 entsprechende Molekulargewichte auf dieser
annähernd
geraden Linie sind; und
- (4) die Messung dieses Ethylencopolymeren mittels CFC derartige
Eigenschaften zeigt, daß die
Summe der jeweiligen Mengen von Copolymerfraktionen, die bei Temperaturen
extrahiert wurden, welche wenigstens 10°C niedriger als die erste Temperatur,
wie oben definiert sind, 8 Gew.-% oder weniger, bezogen auf die
Gesamtmenge, ausschließlich
Spülen,
von Copolymerfraktionen, die bei Temperaturen in dem Gesamtbereich
von Extraktionstemperatur in CFC extrahiert wurden, ist.
-
In einer dritten Ausführungsform
bezieht sich die Erfindung auf eine Polymermischungszusammensetzung,
umfassend:
- (I) von 1 bis 99 Gew.-%, bezogen
auf das kombinierte Gewicht von Komponenten (I) und (II), des Ethylencopolymeren
entsprechend einem der Ansprüche
10 bis 16; und
- (II) wobei der Rest ist entweder
- (1) ein Ethylenhomopolymeres, das mit einer anderen Katalysatorkomponente,
als derjenigen, die in dem Verfahren der Ansprüche 1–9 verwendet wurde, hergestellt
worden ist; oder
- (2) ein Ethylen/α-Olefincopolymeres,
das von demjenigen von Komponente (I) verschieden ist; oder
- (3) ein Ethylen/α-Olefininterpolymeres,
das einen Zusammensetzungs-Verzweigungsindex von größer als 50
hat, das von demjenigen von Komponente (I) verschieden ist; oder
- (4) ein Ethylen/α-Olefininterpolymeres,
das unter Verwendung von Ziegler-Katalysatoren oder Verwendung von
auf Chrom basierenden Siliziumdioxid-Trägerkatalysatorsystemen hergestellt
worden ist, das von demjenigen von Komponente (I) verschieden ist;
oder
- (5) ein Ethylen/α-Olefincopolymeres
(I), das ein verschiedenes I2, oder eine
verschiedene Dichte oder ein verschiedenes Mw oder
ein Mw/Mn, das von
demjenigen von Komponente (I) verschieden ist, hat; oder
- (6) eine Kombination von beliebigen zwei oder mehr von (II)(2),
(II)(3) oder (II)(4) oder (II)(5).
-
Gemäß einer noch anderen Ausführungsform
bezieht sich die vorliegende Erfindung auf ein Verfahren zur Bildung
einer Polymermischungszusammensetzung, wobei das Verfahren die Stufen
umfaßt
von:
- (A) Herstellen eines Ethylen/α-Olefincopolymeren
(I) nach dem Ethylencopolymerherstellungsverfahren der Erfindung,
- (B) Inkontaktbringen unter Polymerisationsbedingungen einer
Einspeisströmung,
umfassend Ethylen, wahlweise wenigstens ein α-Olefincomonomeres und einen
Ethylenpolymerisationskatalysator, zur Bildung von
- (II) einem Ethylenhomopolymeren oder einem Ethylen/α-Olefincopolymeren,
das mit einer Katalysatorkomponente hergestellt worden ist, die
von dem bei dem Ethylencopolymerherstellungsverfahren der Erfindung verwendeten
verschieden ist; und
- (C) Kombinieren des Ethylen/α-Olefincopolymeren
(2) mit dem Ethylenhomopolymeren oder Ethylen/α-Olefininterpolymeren (II) zur
Bildung (III) der Polymermischungszusammensetzung, wie auch die
nach diesem Verfahren hergestellte Polymermischungszusammensetzung.
-
Zusätzlich bezieht sich die vorliegende
Erfindung auf Sinterpulver, hergestellt aus dem Ethylencopolymeren
der Erfindung. Weiterhin bezieht sich die Erfindung auf einen hergestellten
Gegenstand, hergestellt aus der Polymermischungszusammensetzung
der Erfindung, bevorzugt in Form eines Films/einer Folie, Fasern
oder Platten, oder als das Ergebnis eines Thermoform-, Blasform-,
Spritzgieß-
und Rotationsgießverfahrens.
Bei einer noch anderen bevorzugten Ausführungsform umfaßt dieser
hergestellte Gegenstand der Erfindung Röhren, Schläuche, Kabel- oder Drahtummantelungen,
Rohrbeschichtungen, Geomembrane, thermoverformte Gegenstände, stapelbare
Kunststoffpalleten, blasgeformte Flaschen oder Behälter oder
Umgebungs-Teichauskleidungen.
-
Alle Bezugnahmen hier auf Elemente
oder Metalle, welche zu einer bestimmten Gruppe gehören, beziehen
sich auf das Periodensystem der Elemente, veröffentlicht und mit Copyright
von CRC Press, Inc., 1989. Ebenfalls soll eine beliebige Referenz
auf die Gruppe oder die Gruppen der Gruppe oder den Gruppen entsprechen,
wie sie in diesem Periodensystem der Elemente unter Verwendung des
IUPAC-Systems zur Numerierung von Gruppen verwendet werden.
-
Irgendwelche numerischen Werte, welche
hier angegeben sind, schließen
alle werte von dem niedrigeren Wert bis zu dem oberen Wert in Teilschritten
von einer Einheit ein, vorausgesetzt, daß eine Trennung von wenigstens
2 Einheiten zwischen irgendeinem niedrigeren Wert und irgendeinem
höheren
Wert gegeben ist. Beispielhaft gilt, daß, wenn angegeben ist, daß die Menge
einer Komponente oder eines Wertes einer Verfahrensvariablen, wie
beispielsweise Temperatur, Druck, Zeit, beispielsweise von 1 bis
90, bevorzugt von 20 bis 80, mehr bevorzugt von 30 bis 70 beträgt, dies
bedeuten soll, daß Werte
wie 15 bis 85, 22 bis 68, 43 bis 51, 30 bis 32, etc. aus drücklich in
dieser Beschreibung aufgeführt
sind. Für
werte unterhalb von 1 wird angenommen, daß eine Einheit 0,0001, 0,001,
0,01 oder 0,1, je nach Eignung, ist. Dies sind lediglich Beispiele
davon, was spezifisch beabsichtigt ist, und alle möglichen
Kombinationen von numerischen Werten zwischen dem aufgeführten untersten
Wert und dem aufgeführten
höchsten
Wert sind als ausdrücklich
in dieser Anmeldung in einer ähnlichen
Weise angegeben anzusehen.
-
Der Ausdruck "Hydrocarbyl", wie er hier verwendet wird, bedeutet
beliebige aliphatische, cycloaliphatische, aromatische, aryl-substituierte
aliphatische, aryl-substituierte cycloaliphatische, aliphatisch-substituierte
aromatische oder aliphatisch-substituierte cycloaliphatische Gruppen
oder eine beliebige Kombination hiervon.
-
Der Ausdruck "Hydrocarbyloxy" bedeutet eine Hydrocarbylgruppe, welche
eine Sauerstoffbindung zwischen ihr und dem Kohlenstoffatom, an
welche sie gebunden ist, aufweist.
-
Der Ausdruck "Silyl" bedeutet eine Gruppe mit einer Siliziumbindung
zwischen ihr und dem Kohlenstoffatom, an welche sie gebunden ist.
-
Der Ausdruck "Germyl" bedeutet eine Gruppe, welche eine Germaniumbindung
zwischen ihr und dem Kohlenstoffatom, an welches sie gebunden ist,
hat.
-
Der Ausdruck "substituiertes Cyclopentadienyl" soll ringsubstituierte
oder polynukleare Derivate der Cyclopentadienyleinheit einschließen, in
welchen der Substituent Hydrocarbyl, Hydrocarbyloxy, Hydrocarbylamino,
Cyano, Halogen, Silyl, Germyl, Siloxy oder Mischungen hiervon ist,
oder worin zwei solche Substituenten eine Hydrocarbylengruppe sind,
wobei der Substituent (oder zwei Substituenten zusammen) bis zu
30 Nicht-Wasserstoffatome haben. Spezifische Beispiele von substituierten
Cyclopentadienylen schließen
ein: Indenyl-, Tetrahydroindenyl-, Fluorenyl- und Octahydrofluorenylgruppen.
-
Der Ausdruck "Bronstedsäurekation" bedeutet ein Kation, welches als ein
Protonendonor wirkt.
-
Der Ausdruck "Interpolymeres" wird hier dazu verwendet, um ein Polymeres
zu bezeichnen, in welchem wenigstens zwei unterschiedliche Monomere
zur Herstellung des Interpolymeren polymerisiert sind. Dies schließt Copolymere,
Terpolymere, etc. ein.
-
Die Dichte der Polymerzusammensetzungen
zur Verwendung in der vorliegenden Erfindung wurde entsprechend
ASTM D-792 gemessen.
-
Das Molekulargewicht der Polymerzusammensetzungen
zur Verwendung in der vorliegenden Erfindung ist geeigneterweise
bestimmt unter Anwendung einer Schmelzindexmessung gemäß ASTM D-1238,
Bedingung 190°C/2,16
kg (früher
bekannt als "Bedingung
(E)" und ebenfalls
bekannt als I2) angegeben, wie auch bei
Bedingungen von 190°C
(5 kg, 10 kg und 21,6 kg, bekannt als I5,
I10 bzw. I21. Der
Schmelzindex ist umgekehrt proportional zum Molekulargewicht des
Polymeren. Je höher
daher das Molekulargewicht ist, um so niedriger ist der Schmelzindex,
obwohl die Beziehung nicht linear ist. Schmelzfließverhältnisse
wurden aus einem beliebigen Paar dieser Werte genommen.
-
Andere brauchbare Bestimmungen von
physikalischer Eigenschaft, durchgeführt an den hier beschriebenen
neuen Polymerzusammensetzungen, schließen das Schmelzfließverhältnis (MFR)
ein, gemessen durch Bestimmung von "I10" (gemäß ASTM D-1238,
Bedingung 190°C/10
kg (früher
bekannt als "Bedingung (N)") und Dividieren
des erhaltenen I10 durch den Wert I2. Das Verhältnis dieser zwei Schmelzindexwerte
ist das Schmelzfließverhältnis und
wird als I10/I2 bezeichnet.
Andere gemessene Schmelzfließverhältnisse
schließen
I21,6/I5 und I21,6/I2 ein.
-
Das Molekulargewicht (Mw)
und die Verteilung (Mw/Mn)
der Polymere der vorliegenden Erfindung wurden durch Gelpermeationschromatographie
(GPC) auf einer chromatographischen Hochtemperatureinheit Waters
150C, ausgerüstet
mit Kolonnen von gemischter Porosität und Betrieb bei einer Systemtemperatur
von 140°C
bestimmt. Das Lösungsmittel
war 1,2,4-Trichlorbenzol, aus welchem 0,3 Gew.-%ige Lösungen der
Proben für
die Injektion hergestellt wurden. Die Strömungsgeschwindigkeit betrug
1,0 Milliliter/Minute und die Injektionsgröße war 100 Mikroliter.
-
Die Molekulargewichtsbestimmung wurde
unter Verwendung von Polystyrolstandards mit schmaler Molekulargewichtsverteilung
(von Polymer Laboratories) in Verbindung mit ihren Elutionsvolumina
abgeleitet. Die Äquivalent-Polyethylenmolekulargewichte
wurden unter Verwendung von geeigneten Mark-Houwink-Koeffizienten für Polyethylen
und Polystyrol bestimmt (wie von Williams und Ward in Journal of
Polymer Science, Polymer Letters, Vol. 6 (621) 1968 beschrieben)
zur Ableitung der folgenden Gleichung bestimmt: MPolyethylen = a*(MPolystyrol)b.
-
In dieser Gleichung sind a = 0,4316
und b = 1,0. Das Gewichtsdurchschnittsmolekulargewicht, Mw, und das Zahlendurchschnittsmolekulargewicht,
Mn, wurden in der üblichen Weise entsprechend
der folgenden Gleichung berechnet: Mj =
(Σwi(Mi
j))j worin wi die
Gewichtsfraktion der Moleküle
mit dem Molekulargewicht Mi, die aus der
GPC-Kolonne in Fraktion i eluiert, ist, und j = 1 bei Berechnung
von Mw und j = –1, wenn Mn berechnet
wird.
-
Die Zugeigenschaften der verformten
Materialien wurden entsprechend ASTM D 638-76 gemessen. Die Zugfestigkeit,
die Dehnung, die Zähigkeit
und der 2% Sekantenmodul der Folien wurde entsprechend ASTM D-882
gemessen; das PPT-Reißen
wurde entsprechend ASTM D-2582 gemessen.
-
Der Elastizitätsmodul der Materialien wurde
entsprechend ISO 527 gemessen.
-
Die Viskositätszahl der Materialien in Decalin
wurde entsprechend ISO 1191 gemessen.
-
Die Trübung wurde an einer 0,5 mm
dicken preßverformten
Probe entsprechend ASTM D 1003 gemessen.
-
Die Doppel-V-Kerbschlagfestigkeit
der Materialien wurde entsprechend DIN 53753 (Pendel 1J) gemessen.
-
Die Schlagzähigkeitseigenschaften wurden
entsprechend JIS-K7111 ermittelt.
-
Die kritische Dehnungsenergiefreisetzungsrate
Gc wurde nach der Charpy-Weise entsprechend
der Arbeitsweise gemessen, die von E. Plati und J. G. Williams in
Polymer Engineering and Science, Juni, 1975, Volume 15, No. 6, 5.470
bis 477, beschrieben ist. Für
jede Temperatur wurden wenigstens 6 Proben eingesetzt. Die Probenabmessungen
waren 125 mm × 10
mm × 10
mm. Die Stangen wurden aus dicken preßverformten Platten herausgearbeitet.
Die zum Formen dieser Platten angewandte Arbeitsweise war eine Modifizierung
der Arbeitsweise, die in "A
compression molding technique for thick sheets of thermoplastics" von M. J. Cawood
und G. A. H. Smith in Polymer Testing 1 (1980), 3–7, angegeben
ist.
-
So wurden Polymergranulen und -pulver
in einer 10 mm dicken Form, seitlich isoliert unter Verwendung von
TeflonTM, preßgeformt. Sie wurden bis auf
160°C erhitzt
und bei 6,7 MPa für
3 Minuten gehalten, gefolgt von 3 Zyklen von 1 Minute unter Spannung
Setzen und Freigabe. Überschüssiges Material
wurde entfernt. Das Material wurde dann auf 180°C erhitzt und für etwa 5
Minuten bei 6,7 MPa gehalten, dies wurde ebenfalls unter Spannung
gesetzt und freigegeben für
jeweils 3 Zyklen von 1 Minute. Schließlich wurde die Schmelze unter
einem Druck von 1,7 MPa verfestigt und über Nacht durch Abschalten
des Erhitzens langsam abgekühlt.
-
Der Biege-ESCR-Test wurde in einer
10 Gew.-%igen Lösung
eines Tensids entsprechend JIS-K6760 durchgeführt. Die Testtemperatur war
auf 50°C
oder 80°C.
-
Der Pennsylvania-Kerbtest ist ein
Test mit langsam wachsendem Riß,
durchgeführt
unter Befolgung der Arbeitsweise, die von X. Lu und N. Brown, Polymer
Testing 11 (1992), S.309319, beschrieben ist. Der Test wird bei
2,4 MPa und 80°C
durchgeführt.
Die Probenabmessungen sind 50 mm × 25 mm × 10 mm, und sie werden aus
derselben Platte wie die Gc-Stangen herausgearbeitet.
-
Die Viskositäten wurden an einem mechanischen
Spektrometer Rheometrics bei 190°C
in der Oszillationsweise gemessen.
-
Der Comonomerengehalt wurde unter
Anwendung von Infrarotspektroskopie auf einem Spektrophotometer
Beckman IR2450 gemessen.
-
Das Eigen-Reißen wurde an der preßgeformten
Platte unter Anwendung der Reißmethode
Elmendorf (Typ B), wie in ASTM D-1922
beschrieben, gemessen.
-
Das Ansteigen der Dehnungsverhärtung wird
durch Preßformen
einer Platte aus dem zu testenden Polymeren gemessen. Typischerweise
wird die Platte bei etwa 177°C
für 4 Minuten
unter fast keinem Druck geformt und dann für 3 Minuten unter einem Druck
von etwa 14,66 kg/cm2 (200 psi) gepreßt. Die
Platte wird dann mit etwa 8°C/Minute
abkühlen
gelassen, während
sie immer noch unter 14,66 kg/cm2 (200 psi)
Druck ist. Die geformte Platte hatte eine Dicke von etwa 0,0127
cm (0,005 Zoll). Die Platte wird dann zu einem Teststück mit Hundeknochenform
unter Anwendung eines gesteuerten Stahlwerkzeuges geschnitten. Das
Teststück
hat eine Breite von 0,80 cm (0,315 Zoll) und eine Länge von
2,70 cm (1,063 Zoll). Der Beginn des gekrümmten Abschnittes der Hundeknochenform
beginnt bei 0,80 cm (0,315 Zoll) von jedem Ende der Probe und krümmt sich
sanft (d. h. verjüngt
sich) auf eine Breite von 0,23 cm (0,09 Zoll). Die Kurve endet an
einem Punkt 0,30 cm (0,118 Zoll) von dem Beginn der Krümmung, so
daß der
innere Abschnitt des Hundeknochen-Teststückes eine Breite von 0,23 cm
(0,09 Zoll) und eine Länge
von 0,50 cm (0,197 Zoll) hat.
-
Die Zugeigenschaften der Testprobe
wurden auf einem Instron Zugtester bei einer Kreuzkopfgeschwindigkeit
von 2,54 cm/Minute (1 Zoll/Minute) getestet. Der Anstieg der Dehnungsverhärtung wird
aus der resultierenden Zugkurve durch Ziehen einer Linie parallel
zu dem Dehnungsverhärtungsbereich
der resultierenden Spannungs-/Dehnungskurve berechnet. Der Dehnungsverhärtungsbereich
erfolgt, nachdem die Probe ihre Anfangsbelastung (d. h. Spannung), üblicherweise
mit wenig oder keiner Dehnung während
der Anfangsbelastung) gezogen wurde, und nachdem die Probe eine
Stufe mit schwachem Zug (üblicherweise
mit geringem oder keinem Anstieg der Belastung, jedoch mit zunehmender
Dehnung (d. h. Verformung)) durchlaufen hat. Bei dem Dehnungsverhärtungsbereich
nehmen sowohl die Belastung als auch die Dehnung der Probe fortschreitend
zu. Die Belastungszunahmen in dem Dehnungsverhärtungsbereich bei sehr viel
niedrigerer Geschwindigkeit als während des anfänglichen
Belastungsbereiches und die Dehnung nehmen ebenfalls zu, wiederum
mit einer Geschwindigkeit geringer als sie in dem Ziehbereich gefunden
wurde. Die Neigung der parallelen Linie in dem Dehnungsverhärtungsbereich
wird dann bestimmt.
-
Die Neigung des Dehnungsverhärtungskoeffizienten
(SHC) wird dann nach der folgenden Gleichung berechnet: SHC = (Neigung der Dehnungsverhärtung)* (I2)0,25 worin
I2 = Schmelzindex in Gramm/10 Minuten ist.
-
Bei einer Ausführungsform der vorliegenden
Erfindung wird ein Ethylencopolymeres bereitgestellt, welches ein
Copolymeres von Ethylen mit wenigstens einem Comonomeren umfaßt, ausgewählt aus
der Gruppe, die aus einer Verbindung, wiedergegeben durch die Formel
H2C = CHR, worin R eine C1-C20 lineare, verzweigte oder cyclische Alkylgruppe
oder eine C6-C20 Arylgruppe
ist, und einem C4-C20 linearen,
verzweigten oder cyclischen Dien besteht, das nach einem Verfahren
der Copolymerisation dieses Ethylens mit diesem Comonomeren mittels
Aufschlämmungspolymerisation
in Anwesenheit eines festen Katalysatorsystems hergestellt wurde,
wobei dieses einen Träger,
eine Übergangsmetallverbindung
und einen Aktivator, der zur Umwandlung der Übergangsmetallverbindung in
einen katalytisch aktiven Übergangsmetallkomplex
fähig ist,
umfaßt,
und wobei dieses Ethylencopolymere die folgenden Eigenschaften (1)
bis (5) hat:
- (1) eine Dichte d (g/cm3) von 0,870 bis 0,980;
- (2) ein Mw/Mn von
2,5 bis 10, worin Mw und Mn jeweils
ein Gewichtsdurchschnittsmolekulargewicht und ein Zahlendurch schnittsmolekulargewicht
sind, beide gemessen mittels Gelpermeationschromatographie (GPC),
wobei zusätzlich
Mw/Mn die folgenden Ungleichungen erfüllt: 1,25
log Mw – 2,5 ≤ Mw/Mn ≤ 3.5 logMw – 11,0;
- (3) wenn bei der Kreuzfraktionierungs-Chromatographie (CFC)
des Ethylencopolymeren im Hinblick auf die Extraktion bei einer
willkürlichen
Temperatur T(°C),
welche in den Bereich zwischen einer ersten Temperatur, bei welcher
eine Maximalmenge von Extraktion gezeigt wird, und einer zweiten
Temperatur, welche 10°C
höher als
die erste Temperatur ist, fällt,
die Beziehung zwischen der willkürlichen
Temperatur T(°C) und
einem Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
einer bei der willkürlichen
Temperatur T(°C)
extrahierten Copolymerfraktion an diesem Punkt im Molekulargewicht
des Molekulargewichtsverteilungsprofils der Copolymerfraktion einen
Peak mit einer maximalen Intensität zeigt, nach der Methode der
geringsten Quadrate behandelt wird, um eine annähernd gerade Linie zu erhalten,
die annähernd
gerade Linie einen Gradienten innerhalb des Bereiches hat, der durch
die Formel (I) definiert wird. –1 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2) ≤ –0,005 (I)worin:
T1 und T2 zwei unterschiedliche
willkürliche
Extraktionstemperaturen T(°C)
innerhalb des Bereiches zwischen der ersten Temperatur und der zweiten
Temperatur sind, und Mp(T1) und Mp(T2) jeweils Molekulargewichte sind, welche
T1 und T2 auf der
annähernd
geraden Linie entsprechen; und
- (4) die Messung des Ethylencopolymeren mittels CFC solche Eigenschaften
zeigt, daß die,
Summe der jeweiligen Mengen von Copolymerfraktionen, welche bei
Temperaturen extrahiert wurden, die wenigstens 10°C niedriger
als die erste Temperatur wie definiert sind, oberhalb 8 Gew.-% oder
weniger ist, bezogen auf die Gesamtmenge von, unter Ausschluß von Spülfraktionen,
Copolymerfraktionen, die bei Temperaturen in dem Gesamtbereich von
Extraktionstemperaturen bei der CFC extrahiert wurden;
- (5) innerhalb eines Molekulargewichtsbereiches des Ethylencopolymeren,
welcher durch die Formel (II) definiet wird: log(Mt) – log(Mc) ≤ 0,5 (II)worin:
Mt
ein Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
ist, bei welchem das Profil einen Peak mit einer maximalen Intensität zeigt,
und
Mc ein willkürlicher
Punkt im Molekulargewicht auf dem Molekulargewichtsverteilungsprofil
ist; und
das Molekulargewichtsverteilungsprofil zusammen mit
einem Comonomerengehalt-Verteilungsprofil erhalten wird, indem das
Ethylencopolymere der Gelpermeationschromatographie/Fouriertransformations-Infrarotspektroskopie
(GPC/FT-IR) unterzogen wird, worin
eine annähernd gerade Linie, erhalten
aus dem Comonomerengehalt-Verteilungsprofil nach der Methode der
kleinsten Quadrate, einen Gradienten innerhalb des Bereiches hat,
der durch die Formel (III) definiert ist: 0,0005 ≤ C(Mc1) – C(Mc2)}/(logMc1 – logMc2) ≤ 0,05 (III)worin:
Mc1 und Mc2 zwei unterschiedliche
willkürliche
Punkte (Mc) im Molekulargewicht sind, welche die Formel (II) erfüllen, und
C(Mc1) und C(Mc2) jeweils
Comonomerengehalte sind, welche Mc1 und
Mc2 auf der annähernd geraden Linie entsprechen.
-
Das Ethylencopolymere der vorliegenden
Erfindung, wie oben definiert, ist ein neues Ethylencopolymeres,
das Vorteile dadurch besitzt, daß es nicht nur im wesentlichen
keine Verunreinigungen wie ein Wachs, ein Gel, enthält, sondern
auch ausgezeichnete Eigenschaften wie hohe Schlagfestigkeit und
ausgezeichnete Rißfestigkeit
gegenüber
Umgebungsbeanspruchungen aufweist.
-
Bei einer anderen Ausführungsform
der Erfindung wird ein Verfahren zur Herstellung des Ethylencopolymeren
bereitgestellt, umfassend ein Copolymeres von Ethylen mit wenigstens
einem Comonomeren, ausgewählt
aus der Gruppe, bestehend aus einer Verbindung, wiedergegeben durch
die Formel H2C = CHR, worin R eine C1-C20 lineare, verzweigte
oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe ist, und einem C4-C20 linearen, verzweigten oder cyclischen
Dien, wobei dieses Verfahren die Copolymerisation dieses Ethylens
mit diesem Comonomeren durch Aufschlämmungspolymerisation in Anwesenheit
eines festen Katalysatorsystems, umfassend: einen Träger, eine Übergangsmetallverbindung
und einen zur Umwandlung der Übergangsmetallverbindung
in einen katalytisch aktiven Übergangsmetallkomplex
fähigen
Aktivator, wobei dieses feste Katalysatorsystem umfaßt:
- 1) eine Trägerkatalysatorkomponente,
umfassend (a) ein Trägermaterial,
eine Organometallverbindung, worin das Metall aus den Gruppen 2–13 des
Periodensystems der Elemente, Germanium, Zinn und Blei ausgewählt ist,
und (b) eine Aktivatorverbindung, umfassend (b-1) ein Kation, welches
zur Reaktion mit einer Übergangsmetallverbindung
zur Bildung eines katalytisch aktiven Übergangsmetallkomplexes fähig ist, und
(b-2) ein verträgliches
Anion mit bis zu 100 Nicht-Wasserstoffatomen, und enthaltend wenigstens
einen Substituenten, der eine aktive Wasserstoffeinheit umfaßt; und
- 2) eine Übergangsmetallverbindung.
-
Bei einer noch anderen Ausführungsform
der vorliegenden Erfindung wird ein Verfahren zur Herstellung des
oben definierten Ethylencopolymeren bereitgestellt, bei welchem
die Übergangsmetallverbindung
wenigstens eine cyclische oder nicht-cyclische, π-gebundene anionische Ligandengruppe
enthält.
-
Das Ethylencopolymere der vorliegenden
Erfindung ist ein Copolymeres von Ethylen mit wenigstens einem Comonomeren,
ausgewählt
aus der Gruppe, die aus einer Verbindung besteht, wiedergegeben
durch die Formel H2C = CHR, worin R eine
C1-C20 lineare,
verzweigte oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe
ist, und einem C4-C20 linearen,
verzweigten oder cyclischen Dien.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat eine Dichte d (g/cm3) von
0,870 bis 0,980. Ethylencopolymere mit einer Dichte d von niedriger
als 0,870 g/cm3 können nicht sehr gut mittels
Aufschlämmungspolymerisation
hergestellt werden. Wenn andererseits ein Ethylencopolymeres eine
Dichte d (g/cm3) von höher als 0,980 hat, ist der
Comonomerengehalt eines solchen Copolymeren zu niedrig, so daß es wahrscheinlich
ist, daß das
Copolymere praktisch dieselben Eigenschaften wie diejenigen eines
Ethylenhomopolymeren besitzt, jedoch nicht die verschiedenen ausgezeichneten
Eigenschaften aufweist, welche für
ein Copolymeres mit einer Dichte d (g/cm3)
innerhalb des oben definierten Bereiches charakteristisch sind.
In der vorliegenden Erfindung wird es bevorzugt, daß das Ethylencopolymere
eine Dichte d (g/cm3) von 0,87 bis 0,980, mehr
bevorzugt von 0,890 bis 0,965 und am meisten bevorzugt von 0,915
bis 0,955 hat.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat ein Mw/Mn von 2,5 bis 10,
worin Mw und Mn jeweils das Gewichts durchschnittsmolekulargewicht
und das Zahlendurchschnittsmolekulargewicht, beide gemessen mittels
Gelpermeationschromatographie (GPC), sind. Das Verhältnis Mw/Mn wird als ein Kriterium für die Molekulargewichtsverteilung
benutzt. Wenn ein Ethylencopolymeres ein Mw/Mn
von weniger als 2,5 in der vorliegenden Erfindung besitzt, ist die
Molekulargewichtsverteilung des Copolymeren zu schmal, so daß es schwierig
für das
Ethylencopolymere wird, die spezifische Comonomerengehaltverteilung
aufzuweisen, welche charakteristisch in der vorliegenden Erfindung
definiert ist. Wenn andererseits ein Ethylencopolymeres ein Mw/Mn von größer als 10 hat, ist es wahrscheinlich,
daß die
Schlagzähigkeit
des Copolymeren unvorteilhaft niedrig wird. weiter wird es bei der
vorliegenden Erfindung bevorzugt, daß die Ethylencopolymere ein
Mw/Mn von 2,8 bis 8, mehr bevorzugt von
3 bis 7 haben.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat einen Schmelzindex, (I2),
von 0,0001 bis 10000, bevorzugt von 0,001 bis 5000, mehr bevorzugt
von 0,01 bis 3000 g/10 min.
-
Das Verhältnis I21
,6/I2 des Ethylencopolymeren
der vorliegenden Erfindung beträgt
von 15 bis 65, bevorzugt von 18 bis 55, mehr bevorzugt von 20 bis
50, oder ein Verhältnis
I10/I2 von 5 bis
30, bevorzugt von 5 bis 28, mehr bevorzugt von 5,5 bis 25.
-
Bezüglich des Ethylencopolymeren
der vorliegenden Erfindung gilt, daß, wenn in der Kreuzfraktionierungschromatographie
(CFC) des Ethylencopolymeren der vorliegenden Erfindung mit Bezug
auf die Extraktion bei einer willkürlichen Temperatur T(°C), die in
dem Bereich zwischen einer ersten Temperatur, bei welcher eine Maximalmenge
der Extraktion gezeigt wird, und einer zweiten Temperatur, welche
niedriger als entweder die Temperatur von 10°C höher als diese erste Temperatur
oder 96°C
ist, fällt,
die Beziehung zwischen der willkürlichen
Temperatur T(°C)
und einem Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
einer Copolymerfraktion, die bei der willkürlichen Temperatur T(°C), an welchem
Punkt im Molekulargewicht des Molekulargewichtsverteilungsprofils
der Copolymerfraktion fällt,
einen Peak mit einer maximalen Intensität zeigt, diese mittels der
Methode der kleinsten Quadrate zum Erhalt einer annähernd geraden
Linie innerhalb des Bereiches zwischen dieser ersten Temperatur
und dieser zweiten Temperatur behandelt wird, falls dort die Copolymerfraktion,
deren Menge geringer als 1 Gew.-% der Gesamtmenge, ausschließlich Spülungen, der
bei Temperaturen in dem Gesamtbereich der Extraktionstemperaturen
im CFC beträgt,
die Copolymerfraktion aus der Berechnung der annähernd geraden Linie ausgeschlossen
werden kann. Die annähernd
gerade Linie hat einen Gradienten innerhalb des Bereiches, der durch
die Formel (I) definiert wird: –1 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2) ≤ –0,005 (I)worin:
T1 und T2 zwei unterschiedliche
willkürliche
Extraktionstemperaturen T(°C)
innerhalb des Bereiches zwischen der ersten Temperatur und der zweiten
Temperatur sind, und
Mp(T1) und Mp(T2) jeweils Molekulargewichte sind, welche
T1 und T2 auf dieser
annähernd
geraden Linie entsprechen.
-
In der oben angegebenen Formel (I)
zeigt der Ausdruck {logMp(T1) – logMp(T2)}/(T1 – T2) einen Gradienten der oben genannten annähernd geraden
Linie an.
-
In der vorliegenden Erfindung wird
die Kreuzfraktionschromatographie (CFC) unter Verwendung des CFC
T-150A (hergestellt und vertrieben von Mitsubishi Kagaku Corp.,
Japan) durchgeführt.
Die Messung durch CFC wird wie folgt durchgeführt: 20 mg einer Probe werden
in 20 ml Dichlorbenzol, das eine Temperatur von 140°C hat, aufgelöst, um dadurch
eine Lösung
der Probe zu erhalten. Dann werden 5 ml der erhaltenen Lösung zu
einer TREF-Kolonne (TREF = Elutionsfraktionierung mit ansteigender
Temperatur), die mit Glasperlen gefüllt ist, zugegeben, und die
Lösung
wird auf 0°C
mit einer Geschwindigkeit von 1°C/min
abkühlen
gelassen. Anschließend
wird die Lösung
erhitzt, so daß die
Temperatur der Lösung
mit einer Geschwindigkeit von 1°C/min
erhitzt wird, wodurch Copolymerfraktionen extrahiert werden. Dann
werden die extrudierten Copolymerfraktionen Gelpermeationschromatographie
(GPC) unter Verwendung einer GPC-Kolonne Shodex AD806MS (hergestellt
und vertrieben von Showa Denko K. K., Japan) unterworfen, gefolgt
von Fouriertransformations-Infrarotspektroskopie (FT-IR) unter Verwendung
eines Nicolet Manga-IR-Spektrometers 550 (hergestellt und vertrieben
von Nicolet Co., Ltd., U.S.A.).
-
Hinsichtlich weiterer Einzelheiten
der Methode zur Durchführung
der CFC kann auf den Katalog Bezug genommen werden, der dem oben
genannten Gerät
CFC T-150A beigefügt
ist.
-
Hinsichtlich konventioneller Ethylencopolymere,
hergestellt unter Verwendung eines konventionellen Ziegler-Katalysators,
beträgt
der Gradient {logMp(T1) – logMp(T2)}/(T1 – T2) im allgemeinen 0 oder hat einen positiven
Wert. Hinsichtlich konventioneller Ethylencopolymere, hergestellt
unter Verwendung konventioneller Metallocenkatalysatoren, die neuerlich
in praktische Anwendung genommen wurden, beträgt der Gradient {logMp(T1) – logMp(T2)}/(T1 – T2) fast 0.
-
Wenn, wie oben angegeben, in der
vorliegenden Erfindung bei der Kreuzfraktionierungschromatographie
(CFC) das Ethylencopolymere der vorliegenden Erfindung im Hinblick
auf die Extraktion bei einer willkürlichen Temperatur T(°C), die in
dem Bereich zwischen einer ersten Temperatur, bei welcher eine Maximalmenge
von Extraktion gezeigt wird, und einer zweiten Temperatur, welche
die niedrigere Temperatur von entweder der Temperatur von 10°C höher als
diese erste Temperatur oder 96°C
ist, fällt,
die Beziehung zwischen der willkürlichen
Temperatur T(°C)
und einem Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil
einer Copolymerfraktion, die bei der willkürlichen Temperatur T(°C) extrahiert wurde,
bei welchem Punkt im Molekulargewicht des Molekulargewichtsverteilungsprofils
die Copolymerfraktion einen Peak mit einer maximalen Intensität zeigt,
nach der Methode der kleinsten Quadrate zum Erhalt einer annähernd geraden
Linie behandelt wird, die annähernd
gerade Linie einen Gradienten [d. h. {logMp(T1) – logMp(T2)}/(T1 – T2)], der einen negativen Wert besitzt, hat.
Dies bedeutet, daß die
bei einer niedrigeren Temperatur extrahierte Copolymerfraktion,
d. h. eine Fraktion von Copolymerem niedriger Dichte, das einen
hohen Comonomerengehalt hat, ein höheres Molekulargewicht hat
als diejenige der Fraktion des Copolymeren, welches bei einer hohen
Temperatur extrahiert wurde, d. h. eine Fraktion eines Copolymeren
hoher Dichte mit einem niedrigen Comonomerengehalt.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat einen Gradienten [{logMp(T1) – logMp
(T2)}/(T1 – T2)], der beträchtlich größer hinsichtlich des negativen
Wertes (innerhalb des Bereiches von –,005 bis –1) ist. Dies zeigt deutlich,
daß in
dem Ethylencopolymeren der vorliegenden Erfindung eine Copolymerfraktion,
welche einen hohen Comonomerengehalt besitzt, ein hohes Molekulargewicht
hat, im Gegensatz zu konventionellen Ethylencopolymeren, in denen
eine Copolymerfraktion, die einen hohen Comonomerengehalt besitzt, typischerweise
ein niedriges Molekulargewicht hat.
-
Weiterhin hat das Ethylencopolymere
der vorliegenden Erfindung einen Gradienten [{logMp(T1) – logMp(T2)}/(T1 – T2)] mit negativem Wert innerhalb eines Bereiches
von –0,005
bis –1.
Dies zeigt, daß die
Copolymere, welche Copolymerfraktionen von breit variierenden Comonomerengehalten
und breit variierenden Molekulargewichten haben, innerhalb des oben
genannten Bereiches des Gradienten erhalten werden können, wobei
die Copolymerfraktionen in breitem Maße von einer Copolymerfraktion
mit niedrigem Molekulargewicht, das einen niedrigen Comonmerengehalt
besitzt, d. h. einer Copolymerfraktion mit hoher Dichte mit einem
niedrigen Molekulargewicht, bis zu einer Copolymerfraktion mit hohem
Molekulargewicht, die einen hohen Comonomerengehalt besitzt, d.
h. einer Copolymerfraktion mit niedriger Dichte mit einem hohen
Molekulargewicht, variiert. Die Copolymere der vorliegenden Erfindung,
welche unterschiedliche Comonomerengehalte besitzen, zeigen ausgezeichnete
Mischbarkeit miteinander oder mit einem anderen. Daher können in
der vorliegenden Erfindung die Copolymere mit unterschiedlichen
Comonomerengehalten gemischt werden, so daß ein Copolymeres erhalten
wird, das die gewünschten
Eigenschaften besitzt, und zwar ohne Auftreten von Gelbildung.
-
Wenn der Gradient [{logMp(T1) – logMp(T2)}/(T1 – T2)] jedoch zu klein wird, wird es schwierig,
ein maßgeschneidertes
Copolymeres mit einer gewünschten
Struktur und gewünschten
Eigenschaften zu erhalten. Daher muß der Gradient in der vorliegenden
erfindung –1
oder mehr sein. Weiterhin ist in der vorliegenden Erfindung der
Gradient bevorzugt innerhalb des Bereiches, der durch die Formel
definiert wird: –0,5 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2) ≤ –0,007;bevorzugt–0,1 ≤ {logMp(T1} – logMp(T2)}/(T1 – T2) ≤ –0,01;mehr
bevorzugt, –0,08 ≤ {logMp(T1) – logMp(T2)}/(T1 – T2) ≤ –0,02;worin
T1, T2, Mp(T1) und Mp(T2) wie
für Formel
(I) definiert sind.
-
Wenn das Ethylencopolymere der vorliegenden
Erfindung mittels CFC gemessen wird, zeigt das Ethylencopolymere
solche Merkmale, daß die
Summe von jeweiligen Mengen von Copolymerfraktionen, die bei Temperaturen
extrahiert wurden, welche wenigstens 10°C niedriger als die oben definierte
erste Temperatur ist, 8 Gew.-% oder weniger, bezogen auf die Gesamt menge,
ausschließlich
Spülen,
von Copolymerfraktionen beträgt,
die bei Temperaturen in dem Gesamtbereich von Extraktionstemperatur
bei der CFC extrahiert wurden. In der vorliegenden Erfindung kann
die oben genannte Summe der jeweiligen Mengen von Copolymerfraktionen
aus einer Integralkurve erhalten werden, welche die Mengen von extrahierter
Copolymerfraktion, relativ zu den Extraktionstemperaturen, zeigt.
-
Wenn dagegen konventionelle Ethylencopolymere,
hergestellt unter Verwendung eines Ziegler-Natta-Katalysators, mittels
CFC gemessen werden, zeigen die Ethylencopolymere solche Merkmale,
daß eine
relativ große
Menge von Copolymerfraktionen bei Temperaturen extrahiert wird,
welche wenigstens 10°C
niedriger als die wie oben definierte erste Temperatur sind, wie
durch die Vergleichsbeispiele 4 bis 6 gezeigt wird. Dies zeigt an,
daß solche
Ethylencopolymere eine breite Verteilung der Zusammensetzung haben
und wachsartige Komponenten mit niedrigem Molekulargewicht oder
Copolymerfraktionen mit extrem niedriger Dichte enthalten.
-
Konventionellerweise wurde angenommen,
daß die
unter Verwendung von Metallocenkatalysatoren hergestellten Ethylencopolymere,
welche in neuerer Zeit praktisch angewandt wurden, eine schmale
Verteilung im Comonomerengehalt haben. Wenn jedoch einige dieser
Ethylencopolymere der CFC-Messung unterzogen werden, wird eine beträchtlich
große
Menge von Copolymerfraktionen innerhalb eines breiten Bereiches von
Temperaturen, welche wenigstens 10°C niedriger als die oben definierte
erste Temperatur sind, extrahiert.
-
Im Fall von Ethylencopolymerem der
vorliegenden Erfindung ist die Menge solcher Copolymerfraktionen,
welche bei Temperaturen, die wenigstens 10°C niedriger als die oben definierte
erste Temperatur sind, extrem gering. Spezifisch gilt, wenn das
Ethylencopolymere der vorliegenden Erfindung mittels CFC gemessen
wird, das Ethylencopolymere solche Merkmale zeigt, daß die Summe
von jeweiligen Mengen von Copolymerfraktionen, die bei Temperaturen,
welche wenigstens 10°C niedriger
als die oben definierte erste Temperatur sind, extrahiert wurden,
8 Gew.-% oder weniger, bevorzugt 5 Gew.-% oder weniger, mehr bevorzugt
3,5 Gew.-% oder weniger, bezogen auf die Gesamtmenge von Copolymerfraktionen,
die bei Temperaturen in dem Gesamtbereich der Extraktionstemperaturen
bei CFC extrahiert wurden, ist, wobei jedoch die Spülung ausgeschlossen
wird.
-
Als Folge eines solch extrem kleinen
Gehaltes der Copolymerfraktionen, welche bei Temperaturen extrahiert
wurden, die wenigstens 10°C
niedriger als die oben definierte erste Temperatur sind, hat das
Ethylencopolymere der vorliegenden Erfindung ausgezeichnete Eigenschaften,
beispielsweise das Fehlen von negativen Effekten, welche durch die
Anwesenheit von wachsartigen Komponenten und Copolymerfraktionen
mit niedriger Dichte hervorgerufen werden. Weiterhin ist es gemäß der vorliegenden
Erfindung möglich,
Copolymere mit einer sehr nierigen Dichte und einem sehr niedrigen
Molekulargewicht herzustellen. Solche Copolymere können vorteilhafterweise
gemischt werden, um eine breite Vielzahl von Mischungen bereitzustellen,
wovon jede zwei oder mehr unterschiedliche Copolymerkomponenten
mit unterschiedlichen Comonomerengehalten umfaßt. Daher wird es möglich, verschiedene
Mischungen, welche die gewünschten
Eigenschaften besitzen, durch die Verwendung der oben genannten
Copolymere, welche eine sehr niedrige Dichte und ein sehr niedriges
Molekulargewicht besitzen, maßzuschneidern.
Dies ist unter kommerziellem Gesichtspunkt sehr vorteilhaft.
-
In der vorliegenden Erfindung wird
innerhalb eines Bereiches im Molekulargewicht des Ethylencopolymeren,
welcher durch die Formel (II) definiert wird: log(Mt) – log(Mc) ≤ 0,5 (II)worin:
Mt
ein Punkt im Molekulargewicht auf einem Molekulargewichtsverteilungsprofil,
an welchem das Profil einen Peak mit einer maximalen Intensität zeigt,
und
Mc ein willkürlicher
Punkt im Molekulargewicht auf dem Molekulargewichtsverteilungsprofil
ist,
wobei das Molekulargewichtsverteilungsprofil zusammen
mit einem Comonomerengehalt-Verteilungsprofil dadurch erhalten wird,
daß das
Ethylencopolymere der Gelpermeationschromatographie/Fouriertransformations-Infrarotspektroskopie
(GPC/FT-IR) unterworfen wurde,
eine annähernd gerade Linie erhalten,
die aus dem Comonomerengehalt-Verteilungsprofil nach der Methode der
kleinsten Quadrate erhalten wurde, die einen Gradienten innerhalb
eines Bereiches besitzt, der durch die Formel (III) definiert wird: 0,0005 ≤ {C(Mc1} – C{Me2)}/(logMc1 – logMc2) ≤ 0,05 (III)worin:
Mc1 und Mc2 zwei unterschiedliche
willkürliche
Punkte (Mc) im Molekulargewicht sind, welche die Formel (II) erfüllen, und
C(Mc1) und C(Mc2) jeweils
Comonomerengehalte sind, welche Mc1 und
Mc2 auf der annähernd geraden Linie entsprechen.
-
Wie oben erwähnt, können das Molekulargewichtsverteilungsprofil
und das Comonomerengehaltverteilungsprofil dadurch erhalten werden,
daß das
Ethylencopolymere der Gelpermeationschromatographie/Fouriertransformations-Infrarotspektroskopie
(GPC/FT-IR) unterzogen wird. In der vorliegenden Erfindung wird
die Messung mittels GPC unter Verwendung des Gerätes 150C ALC/GPC (hergestellt
und vertrieben von Waters Assoc. Co. U.S.A.) durchgeführt, in
welchem drei Kolonnen [eine Shodex AT-807S (hergestellt und vertrieben
von Sho- wa Denko K. K., Japan) und zwei TSK-Gel GMH-H6 (hergestellt und
vertrieben von Tosoh Corp., Japan)], welche in Reihe geschaltet
sind, verwendet werden, und die Messung mittels FT-IR durchgeführt wird,
indem 20 bis 30 mg einer Probe in 15 ml Trichlorbenzol mit einer
Temperatur von 140°C aufgelöst und 500
bis 1000 μl
der resultierenden Lösung
einem FT-IR-Gerät
(PERKIN-ELMER 1760X, hergestellt und vertrieben von Perkin Elmer
Cetus, Co., Ltd., U.S.A.) zugeführt
werden.
-
In der vorliegenden Erfindung ist
der Comonomerengehalt als ein Wert definiert, der durch Dividieren der
Anzahl von Comonomereneinheiten relativ zu 1000 Methyleneinheiten,
die in dem Copolymeren enthalten sind, mal 1000 erhalten wurde.
Wenn beispielsweise 5 Comonomereneinheiten relativ auf 1000 Methyleneinheiten
enthalten sind, beträgt
der Comonomerengehalt 0,005. Der Wert des Comonomerengehaltes kann
aus dem Verhältnis
der Intensität
einer Absorptionsgröße, welche
den Comonomereneinheiten zuzuordnen ist, zu der Intensität eines
Absorptionswertes, der den Methyleneinheiten zuzuordnen ist, erhalten
werden, wobei dieses Verhältnis
durch FT-IR erhalten werden kann. Wenn beispielsweise ein lineares α-Olefin als
ein Comonomeres verwendet wird, wird das Verhältnis der Intensität des Absorptionswertes
bei 2960 cm–1,
der den Methylgruppen zuzuordnen ist, zu der Intensität des Absorptionswertes
bei 2925 cm–1,
der der Methylengruppen zuzuordnen ist, mittels FT-IR erhalten.
Aus dem erhaltenen Verhältnis
kann der Comonomerengehalt erhalten werden.
-
Im allgemeinen wird das oben genannte
Comonomerengehaltverteilungsprofil als eine Linie angegeben, welche
Punkte enthält,
die Comonomerengehalte anzeigen. Zur Verbesserung der Genauigkeit
des Profils ist es erwünscht,
eine große
Anzahl von Punkten zu erhalten, welche die Comonomerengehalte durch
wiederholte Durchführung
der Messung des Comonomerengehaltes unter Verwendung derselben Probe
unter denselben Bedingungen anzeigen. In der vorliegenden Erfindung
wird innerhalb des oben definierten Bereiches im Molekulargewicht
des Ethylencopolymeren eine annähernd
gerade Linie aus den erhaltenen Punkten des Comonomerengehaltverteilungsprofils
nach der Methode der kleinsten Quadrate erhalten.
-
In der vorliegenden Erfindung wird
der Gradient der annähernd
geraden Linie, erhalten aus dem Comonomerengehaltverteilungsprofil
durch die folgende Formel definiert: {C(Mc1) – C(Mc1)}/(logMc1 – logMc2)worin:
Mc1 und
Mc2 zwei unterschiedliche willkürliche Punkte
(Mc) im Molekulargewicht sind, welche die Formel (II) erfüllen, und
C(Mc1) und C(Mc2) jeweils
Comonomerengehalte sind, welche Mc1 und
Mc2 auf der annähernd geraden Linie entsprechen.
-
Das Comonomerengehaltverteilungsprofil
zeigt die Comonomerengehalte von Copolymerfraktionen von verschiedenen
Molekulargewichten an, und der Gradient der annähernd geraden Linie, erhalten
aus dem Profil mittels der Methode der kleinsten Quadrate, zeigt
die Veränderung
im Comonomerengehalt relativ zu der Veränderung im Molekulargewicht
der Copolymerfraktion an.
-
Im Hinblick auf die Ethylencopolymere,
welche unter Verwendung eines konventionellen Ziegler-Katalysators
hergestellt wurden, hat der oben genannte Gradient der annähernd geraden
Linie einen negativen Wert. Dies zeigt an, daß solche konventionellen Ethylencopolymere
eine solche Comonomerengehaltverteilung haben, daß, je höher das
Molekulargewicht einer Copolymerfraktion ist, je niedriger der Comonomerengehalt
der Copolymerfraktion ist.
-
Selbst im Fall von Ethylencopolymeren,
hergestellt unter Verwendung konventioneller Metallocenkatalysatoren,
welche in neuerer Zeit in praktische Anwendung genommen wurden,
ist der oben genannte Gradient der annähernd geraden Linie, erhalten
aus dem Comonomerengehaltverteilungsprofil nach der Methode der
kleinsten Quadrate, fast 0. Selbst wenn die Fehler in der Messung
in Betracht gezogen werden, ist der Gradient kleiner als 0,0001.
-
Andererseits hat das Ethylencopolymere
der vorliegenden Erfindung den oben genannten Gradienten [{C(Mc1) – C(Mc2)}/logMc1 – logMc2)] von 0,0005 oder mehr innerhalb des oben
definierten Bereiches im Molekulargewicht des Ethylencopolymeren.
-
Dies zeigt deutlich, daß das Ethylencopolymere
der vorliegenden Erfindung eine spezifische Comonomerengehaltverteilung
derart hat bei einer Ausführungsform,
je niedriger das Molekulargewicht einer Copolymerfraktion ist, um
so niedriger der Comonomerengehalt der Copolymerfraktion ist, und
bei der anderen Ausführungsform,
je höher
das Molekulargewicht einer Copolymerfraktion ist, um so höher der
Comonomerengehalt der Copolymerfraktion ist. Als Folge einer solchen
spezifischen Comonomerengehaltverteilung zeigt das Ethylencopolymere
der vorliegenden Erfindung verschiedene ausgezeichnete Eigenschaften,
wie hohe Schlagfestigkeit und ausgezeichnete ESCR-Eigenschaften, verglichen
mit konventionellen Ethylencopolymeren.
-
In der vorliegenden Erfindung ist
es bevorzugt, daß innerhalb
des oben definierten Bereiches im Molekulargewicht des Ethylencopolymeren
der oben genannte Gradient innerhalb des Bereiches ist, der durch die
Formel (IV) definiert ist: 0,001 ≤ {C(Me1) – C(Me2)}/(logMc1 – logMc2) ≤ 0,02 (IV)worin Mc1, Mc2, C(Mc1) und C(Mc2) wie
für Formel
(III) definiert sind.
-
In der vorliegenden Erfindung wird
ein Verfahren zum Erhalt eines neuen Ethylencopolymeren bereitgestellt.
-
Spezifisch umfaßt das Verfahren das Copolymerisieren
von Ethylen mit wenigstens einem Comonomeren, ausgewählt aus
der Gruppe, die besteht aus einer Verbindung, wiedergegeben durch
die Formel H2C = CHR, worin R eine C1-C20 lineare, verzweigte
oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe ist, und einem C4-C20 linearen, verzweigten oder cyclischen
Dien mittels Aufschlämmungspolymerisation
in Anwesenheit eines festen Katalysatorsystems, welches umfaßt:
einen
Träger,
eine Übergangsmetallverbindung
und einen Aktivator, der zur Umwandlung der Übergangsmetallverbindung in
einen katalytisch aktiven Übergangsmetallkomplex
fähig ist.
-
Die Gründe zum Erhalt der unerwarteten
und überraschenden
Copolymereigenschaften mit Hilfe des Verfahrens der vorliegenden
Erfindung werden als wie folgt angenommen:
-
Wie bereits oben angegeben, hat das
Ethylencopolymere der vorliegenden Erfindung eine spezifische Comonomerengehaltverteilung
derart, daß gemäß einer
Ausführungsform,
je niedriger das Molekulargewicht einer Copolymerfraktion ist, um
so niedriger der Comonomerengehalt der Copolymerfraktion ist, und
gemäß der anderen
Ausführungsform,
je höher
das Molekulargewicht einer Copolymerfraktion ist, um so höher der Comonomerengehalt
der Copolymerfraktion ist.
-
Zusätzlich müssen zur Herstellung des Ethylencopolymeren
der vorliegenden Erfindung die folgenden Anforderungen erfüllt werden:
- (i) das hergestellte Polymere muß in dem
Reaktionsgemisch nicht geschmolzen werden, sondern verbleibt im
festen Zustand;
- (ii) die Polymerisationsgeschwindigkeit bei der aktiven Spezies
des Katalysators ist zufriedenstellend hoch; und
- (iii) die aktive Spezies des Katalysators ist fest mit einem
Träger
verbunden, so daß die
aktive Spezies des Katalysators von dem Träger nicht freigesetzt wird
und nicht aus dem herzustellenden Polymeren entweicht.
-
Je größer die Teilchengröße des herzustellenden
Polymeren ist, wird weiterhin es um so leichter, eine Comonomerengehaltverteilung
zu erreichen, die für
das Ethylencopolymere der vorliegenden Erfindung charakteristisch
ist, nämlich
eine spezifische Comonomerengehaltverteilung derart, daß gemäß einer
Ausführungsform,
je niedriger das Molekulargewicht einer Copolymerfraktion ist, um
so niedriger der Comonomerengehalt der Copolymerfraktion ist, und
gemäß der anderen
Ausführungsform,
je höher
das Molekulargewicht einer Copolymerfraktion ist, um so höher der
Comonomerengehalt der Copolymerfraktion ist.
-
Bei dem Verfahren der vorliegenden
Erfindung zur Herstellung eines Ethylencopolymeren werden die oben
genannten Anforderungen erfüllt,
so daß die
Polymerisationsreaktion fortschreiten kann, wie dies unten erläutert wird.
-
Zuerst, bei dem Verfahren der vorliegenden
Erfindung wird die Polymerisationsreaktion mittels Aufschlämmungspolymerisation
durchgeführt,
so daß das
erzeugte Polymere nicht geschmolzen wird, sondern den festen Zustand
während
der Reaktion beibehält.
Daher ist die Anforderung (i) erfüllt.
-
Zweitens, die bevorzugten Katalysatorsysteme,
welche in der vorliegenden Erfindung eingesetzt werden, enthalten
eine Übergangsmetallverbindung,
d. h. eine Verbindung eines Übergangsmetalls
einer Gruppe, ausgewählt
aus den Gruppen 3 bis 5 des Periodensystems, worin die Verbindung
wenigstens einen, bevorzugt nur einen cyclischen π-gebundenen
anionischen Liganden enthält.
Solche bevorzugten Übergangsmetallverbindungen,
welche nur einen cyclischen π-gebundenen
anionischen Liganden aufweisen, haben einen großen Raum rings um das Übergangsmetall,
verglichen mit dem Raum rings um das Übergangsmetall eines Metallocenkatalysators,
der zwei oder mehr cyclische oder nicht-cyclische, π-gebundene
anionische Liganden enthält.
Daher ist im Hinblick auf eine Übergangsmetallver bindung,
welche nur einen cyclischen oder nicht-cyclischen π-gebundenen anionischen
Liganden aufweist, der Zugang eines raumfüllenden Comonomeren zu dem Übergangsmetall
nicht gehemmt, so daß die
Reaktion glatt voranschreiten kann. Zusätzlich enthält das bevorzugte Katalysatorsystem,
welches bei dem Verfahren der vorliegenden Erfindung eingesetzt
werden soll, eine feste Komponente, welche dazu dient, eine hohe
Polymerisationsgeschwindigkeit zu erreichen. Daher ist bei dem Katalysatorsystem,
welches in der vorliegenden Erfindung eingesetzt werden soll, die
Polymerisationsgeschwindigkeit an den aktiven Spezies des Katalysators
zufriedenstellend hoch. Daher erfüllt das Verfahren der vorliegenden
Erfindung die oben genannte Anforderung (ii).
-
Drittens, in den bevorzugten Katalysatorsystemen,
welche in der vorliegenden Erfindung eingesetzt werden sollen, ist
die aktive Spezies des Katalysators mit einem Träger fest verbunden, so daß die aktive
Spezies des Katalysators nicht von dem Träger freigesetzt wird und nicht
aus dem herzustellenden Polymeren entweicht.
-
Spezifisch gesagt, bei einer bevorzugten
Trägerkatalysatorkomponente,
welche bei dem verfahren der vorliegenden Erfindung eingesetzt werden
soll, kann die aktive Wasserstoffeinheit der Aktivatorverbindung
an die Hydroxylgruppen des Trägermaterials über eine
Organometallverbindung gebunden sein. Dies bedeutet, daß die Aktivatorverbindung
fest an dem Trägermaterial
gebunden und hierauf getragen ist. Bei einer weiteren bevorzugten
Trägerkatalysatorkomponente,
welche in der vorlegenden Erfindung einzusetzen ist, wird Alumoxan
an dem Trägermaterial
durch Erhitzen und/oder Waschbehandlung fixiert, so daß das Alumoxan
im wesentlichen unter harten Bedingungen nicht extrahierbar ist
(Toluol bei 90°C).
Daher ist die obige Anforderung (iii) erfüllt.
-
Das Ethylencopolymere der vorliegenden
Erfindung kann vorteilhafterweise nach dem Verfahren der vorliegenden
Erfindung unter Verwendung des oben beschriebenen Katalysatorsy stems
hergestellt werden, und das katalytische System ist besonders effektiv,
wenn ein Katalysator eine relativ große Teilchengröße hat, und
wenn die Masse des Comonomeren relativ groß ist.
-
Wie oben beschrieben, wird die Herstellung
des Ethylencopolymeren der vorliegenden Erfindung nur möglich, wenn
alle der oben genannten Anforderungen gleichzeitig erfüllt sind.
Die vorliegenden Erfinder haben zum ersten mal unerwarteterweise
die oben genannten Anforderungen für die Herstellung des ausgezeichneten
Ethylencopolymeren der vorliegenden Erfindung gefunden.
-
Im folgenden wird das Verfahren zur
Herstellung des Ethylencopolymeren der vorliegenden Erfindung mehr
im einzelnen erklärt.
-
Das Ethylencopolymere der vorliegenden
Erfindung wird vorteilhafterweise durch Copolymerisieren von Ethylen
mit einem Comonomeren unter Verwendung eines spezifischen festen
Katalysators hergestellt.
-
Geeignete Trägermaterialien zur Verwendung
in der vorliegenden Erfindung schließen poröse harzartige Materialien,
beispielsweise Polyolefine wie Polyethylene und Polypropylene oder
Copolymere von Styrol-Divinylbenzol, sowie feste anorganische Oxide
einschließlich
Oxiden der Metalle der Gruppe 2, 3, 4, 13 oder 14, wie Siliziumdioxid,
Aluminiumoxid, Magnesiumoxid, Titanoxid, Thoriumoxid wie auch Mischoxide
von Siliziumdioxid, ein. Geeignete Mischoxide von Siliziumdioxid
schließen
diejenigen von Siliziumdioxid und einem oder mehreren Oxiden von
Metallen der Gruppe 2 oder 13 ein, wie Siliziumdioxid-Magnesiumoxid-
oder Siliziumdioxid-Aluminiumoxidmischoxide ein. Siliziumdioxid,
Aluminiumoxid und Mischoxide von Siliziumdioxid und einem oder mehreren
Oxiden von Metallen der Gruppe 2 oder 13 sind bevorzugte Trägermaterialien.
Bevorzugte Beispiele solcher Mischoxide sind die Siliziumdioxid-Aluminiumoxidverbindungen.
Das am meisten bevorzugte Trägermaterial
ist Siliziumdioxid. Die Gestalt der Silizium dioxidteilchen ist nicht
kritisch und das Siliziumdioxid kann in Form von Granulen, Kugeln,
in agglomerierter Form, als durch Zersetzung hergestelltes Produkt
oder in anderer Form vorliegen. Geeignete Siliziumdioxide schließen solche
ein, welche von Grace Davison (Division von W. R. Grace & Co.) unter den
Bezeichnungen SD 3216.30, SP-9-10046, Davison SyloidTM 245,
Davison 948 und Davison 952, von Degussa AG unter der Bezeichnung
AerosilTM 812 und von Crossfield unter der
Bezeichnung ES 70X erhältlich
sind.
-
Geeignete Trägermaterialien für die vorliegende
Erfindung haben bevorzugt eine Oberfläche, bestimmt durch Stickstoffporositätsmessung
unter Anwendung der B.E.T.-Methode von 10 bis 1000 m2/g
und bevorzugt von 100 bis 600 m2/g. Das
Porenvolumen des Trägers,
bestimmt durch Stickstoffadsorption, ist typischerweise bis zu 5
cm3/g, vorteilhafterweise zwischen 0,1 und
3 cm3/g, bevorzugt von 0,2 bis 2 cm3/g. Die durchschnittliche Teilchengröße ist nicht
kritisch, jedoch beträgt
sie typischerweise von 0,5 bis 500 μm, bevorzugt von 1 bis 200 μm, mehr bevorzugt
bis 100 μm.
-
Das Trägermaterial kann einer Hitzebehandlung
und/oder einer chemischen Behandlung zum Reduzieren des Wassergehaltes
oder des Hydroxylgehaltes des Trägermaterials
unterzogen werden. Sowohl dehydratisierte Trägermaterialien als auch kleine
Mengen von Wasser enthaltenden Trägermaterialien können verwendet
werden. Typische thermische Vorbehandlungen werden bei einer Temperatur
von 30°C
bis 1000°C
für eine
Dauer von 10 Minuten bis 50 Stunden in einer Inertatmosphäre oder
unter vermindertem Druck durchgeführt. Typische Trägermaterialien
haben einen Oberflächenhydroxylgehalt
von 0,1 Mikromol, bevorzugt von 5 Mikromol, mehr bevorzugt von 0,05
mMol bis nicht mehr als 10 mMol und bevorzugt nicht mehr als 5 mMol Hydroxylgruppen
pro g des festen Trägers,
mehr bevorzugt von 0,5 bis 2 mMol pro Gramm. Der Hydroxylgehalt kann
nach bekannten Arbeitsweisen wie mittels Infrarotspektroskopie und Titrationstechniken
unter Verwendung eines Metallalkyls oder Metallhydroxids bestimmt
werden, beispielsweise durch Zugabe eines Überschusses von Dialkylmagnesium
zu einer Aufschlämmung
des festen Trägers
und Bestimmung der Menge von in der Lösung zurückbleibendem Dialkylmagnesium
nach bekannten Arbeitsweisen. Diese letztgenannte Methode basiert
auf der Reaktion von S-OH + MgR2 → S-OMgR
+ RH, worin
S der feste Träger
ist.
-
Als eine alternative Technik zum
Messen der Menge von Hydroxylgruppen auf der Oberfläche des
anorganischen Feststoffes kann eine Methode, welche die folgenden
Arbeitsschritte umfaßt,
angewandt werden. Ergänzend
angegeben, der anorganische Feststoff wird in einer Stickstoffgasströmung bei
250°C für 10 Stunden
getrocknet, und dann wird das Gewicht des getrockneten anorganischen
Feststoffes gemessen und als Anfangsgewicht, wiedergegeben durch "W1" (Einheit: g) benutzt.
Danach wird der getrocknete anorganische Feststoff auf 1000°C erhitzt,
und dann auf Zimmertemperatur abkühlen gelassen. Das Gewicht
des abgekühlten
anorganischen Feststoffes wird gemessen, und der Unterschied zwischen
dem Anfangsgewicht (W1) und dem Gewicht des abgekühlten anorganischen
Feststoffes wird bestimmt und als ein Gewichtsverlust, wiedergegeben
durch "ΔW" (Einheit: g), gesetzt.
Die Menge der Hydroxylgruppen wurde nach der folgenden Formel berechnet: Hydroxylgruppenmenge = (1000 × ΔW/18,02)/W1
mMol/g (V)
-
Es wird bevorzugt, daß der anorganische
Feststoff, welcher Hydroxylgruppen auf seiner Oberfläche trägt und der
bei dem Verfahren der vorliegenden Erfindung eingesetzt werden soll,
Wasser wie Kristallwasser oder adsorbiertes Wasser nicht enthält.
-
Irgendwelches in dem anorganischen
Feststoff enthaltenes Wasser kann hieraus durch Erhitzen in einer
Stickstoffatmosphäre
oder unter vermindertem Druck auf 250°C oder darüber für 1 Stunde oder mehr entfernt
werden.
-
Geeignete Übergangsmetallverbindungen
zur Verwendung in der vorliegenden Erfindung sind solche, welche
durch eine Aktivatorverbindung (b) umgewandelt werden können, um
hierdurch einen katalytisch aktiven Übergangsmetallkomplex zu bilden.
Die Übergangsmetallverbindungen
können
Derivate von einem beliebigen Übergangsmetall
sein, einschließlich
Lanthaniden, bevorzugt von den Gruppen 3, 4, 5 und 6, mehr bevorzugt
der Gruppe 3 oder 4 der Übergangsmetalle
oder der Lanthaniden, wobei die Übergangsmetalle
im formalen Oxidationszustand +2, +3 oder +4 vorliegen. Die Übergangsmetalle
enthalten bevorzugt wenigstens eine π-gebundene, anionische Ligandengruppe,
wobei dies eine cyclische oder nicht-cyclische, delokalisierte, π-gebundene
anionische Ligandengruppe sein kann. Beispiele solcher π-gebundenen,
anionischen Ligandengruppen sind konjugierte oder nicht-konjugierte,
cyclische oder nichtcyclische Dienylgruppen, Allylgruppen, Arylgruppen,
wie auch substituierte Derivate solcher Gruppen.
-
Der Ausdruck "Derivat", wenn er zur Beschreibung der oben
substituierten, delokalisierten, π-gebundenen
Gruppen verwendet wird, bedeutet, daß jedes Atom in der delokalisierten, π-gebundenen
Gruppe unabhängig
mit einem Rest substituiert sein kann, der aus der Gruppe ausgewählt ist,
die aus Halogen-, Hydrocarbyl-, Halogenhydrocarbyl- und hydrocarbylsubstituierten
Metalloidresten besteht, worin das Metalloid aus Gruppe 14 des Periodensystems
der Elemente ausgewählt
ist. Eingeschlossen innerhalb des Ausdrucks "Hydrocarbyl" sind C1-20 geradkettige,
verzweigtkettige und cyclische Alkylreste, C6-20 aromatische
Reste, C7-20 alkyl-substituierte aromatische
Reste und C7-20 aryl-substituierte Alkylreste.
Zusätzlich
können
zwei oder mehr solcher Reste zusammen ein kondensiertes Ringsystem
oder ein hydriertes kondensiertes Ringsystem bilden. Geeignete hydrocarbyl-substituierte
Organo-Metalloidreste schließen
mono-, di- und tri-substituierete Organo-Metalloidreste von Elementen
der Gruppe 14 ein, worin jede dieser Hydrocarbylgruppen von 1 bis
20 Kohlenstoffatome enthält.
Insbesondere schließen
geeignete hydrocarbyl-substituierte Organo-Metalloidreste Trimethylsilyl,
Triethylsilyl, Ethyldimethylsilyl, Methyldiethylsilyl, Triphenylgermyl,
Trimethylgermyl ein.
-
Bevorzugte anionische, delokalisierte, π-gebundene
Gruppen schließen
Cyclopentadienyl- und substituierte Cyclopentadienylgruppen ein.
Besonders bevorzugt sind Cyclopentadienyl, Indenyl, Fluorenyl, Tetrahydroindenyl,
Tetrahydrofluorenyl und Octahydrofluorenyl. Andere Beispiele von
bevorzugten anionischen Ligandengruppen sind Pentadienyl-, Cyclohexadienyl-,
Dihydroanthracenyl-, Hexahydroanthracenyl- und Decahydroanthracenylgruppen
sowie methyl-substituierte Derivate hiervon.
-
Geeignete Übergangsmetallverbindungen
(c) können
ein Cyclopentadienyl- oder substituiertes Cyclopentadienylderivat
von einem beliebigen Übergangsmetall,
einschließlich
Lanthaniden, jedoch bevorzugt von Übergangsmetallen der Gruppen
3, 4 oder der Lanthanidengruppe sein. Geeignete Übergangsmetallverbindungen
zur Verwendung in der vorliegenden Erfindung sind die verbrückten oder
nicht-verbrückten
Mono-, Bis- und Tricyclopentadienyl- oder substituierten Cyclopentadienylübergangsmetallverbindungen.
-
Geeignete nicht-verbrückte Monocyclopentadienyl-
oder Mono(substituierte-cyclopentadienyl)-übergangsmetallderivate werden
durch die folgende Formel (VI) wiedergegeben: CpMXn (VI)worin Cp
ein Cyclopentadienyl oder ein Derivat hiervon ist, M ein Übergangsmetall
der Gruppe 3, 4 oder 5, das einen forma len Oxidationszustand von
+2, +3 oder +4 hat, ist, X unabhängig
bei jedem Vorkommen eine anionische Ligandengruppe darstellt (verschieden
von einer cyclischen, aromatischen, π-gebundenen, anionischen Ligandengruppe),
ausgewählt
aus der Gruppe von Hydrocarbyl-, Hydrocarbylen-, (einschließlich Hydrocarbadienyl-),
Hydrocarbyloxy-, Hydrid-, Halogen-, Silyl-, Germyl-, Amid- und Siloxyresten,
die bis zu 50 Nicht-Wasserstoffatome haben, und n eine Zahl gleich
1 weniger als der formale Oxidationszustand von M ist, 1,2 oder
3, bevorzugt 3. Bevorzugt ist wenigstens einer der Reste X ein Hydrocarbylrest
mit 1 bis 20 Kohlenstoffatomen, ein substituierter Hydrocarbylrest
mit 1 bis 20 Kohlenstoffatomen, worin eines oder mehrere der Wasserstoffatome
durch ein Halogenatom ersetzt ist/sind, oder ein Organo-Metalloidrest,
der ein Element der Gruppe 14 umfaßt, worin jeder der Hydrocarbylsubstituenten,
die in dem Organoanteil dieses Organo-Metalloids enthalten sind,
unabhängig
von 1 bis 20 Kohlenstoffatome enthält.
-
Geeignete verbrückte Monocyclopentadienyl-
oder Mono-(substituierte-cyclopentadienyl)-übergangsmetallverbindungen
schließen
die sogenannten Komplexe mit gespannter Geometrie ein. Beispiele
solcher Komplexe und Verfahren für
ihre Herstellung sind in der US-Anmeldung Serial No. 545 403, eingereicht
am 3. Juli 1990 (entsprechend der EP-A-416 815), in der US-Anmeldung
Serial No. 241 523, eingereicht am 12. Mai 1994 (entsprechend der
WO-95/00526) wie auch in den US-Patenten 5 055 438, 5 057 475, 5
096 867, 5 064 802, 5 132 380 und 5 374 696 beschrieben, welche
hier durch Bezugnahme eingeschlossen sind.
-
Mehr bevorzugt entsprechen bevorzugte
verbrückte
Monocyclopentadienyl- oder Mono(substituierte-cyclopentadienyl)-übergangsmetallverbindungen
der folgenden Formel (VII):
worin:
M ein Metall
von Gruppen 3–5,
insbesondere ein Gruppe 4 Metall, besonders Titan ist;
Cp*
eine substituierte Cyclopentadienylgruppe, gebunden an Z' und in einer η
5 Bindungsweise an M ist, oder eine solche
Gruppe weiter mit von einem bis vier Substituenten substituiert
ist, ausgewählt
aus der Gruppe, die besteht aus Hydrocarbyl, Silyl, Germyl, Halogen,
Hydrocarbyloxy, Amin und Mischungen hiervon, wobei dieser Substituent
bis zu 20 Nicht-Wasserstoffatome
hat, oder wahlweise zwei solcher weiteren Substituenten zusammen
bewirken, daß Cp*
eine kondensierte Ringstruktur hat;
Z' eine zweiwertige Einheit, verschieden
von einem cyclischen oder nicht-cyclischen π-gebundenen Liganden, ist, wobei
dieses Z' Bor oder
ein Glied von Gruppe 14 des Periodensystems der Elemente und wahlweise
Stickstoff, Phosphor, Schwefel oder Sauerstoff umfaßt, wobei
diese Einheit bis zu 20 Nicht-Wasserstoffatome
hat; und wahlweise Cp* und Z' zusammen
ein kondensiertes Ringsystem bilden;
X unabhängig von
jedem Vorkommen eine anionische Ligandengruppe (verschieden von
einer cyclischen, aromatischen π-gebundenen
anionischen Ligandengruppe) ist, ausgewählt aus der Gruppe von Hydrocarbyl-,
Hydrocarbylen-, (einschließlich
Hydrocarbadienyl-), Hydrocarbyloxy-, Hydrid-, Halogen-, Silyl-,
Germyl-, Amid- und Siloxyresten mit- bis zu 50 Nicht-Wasserstoffatomen
ist, bevorzugt ist X aus der Gruppe von einem Hydridrest, Hydrocarbylrest,
substituierten Hydrocarbylrest oder Organo-Metalloidrest ausgewählt; und
n
= 1 oder 2 ist, abhängig
von der Wertigkeit von M.
-
In Übereinstimmung mit der vorangegangenen
Erläuterung
ist M bevorzugt ein Gruppe 4 Metall, insbesondere Titan; n ist 1
oder 2 und X ist eine einwertige Ligandengruppe mit bis zu 30 Nicht-Wasserstoffatomen,
mehr bevorzugt C1-20 Hydrocarbyl.
-
Wenn n = 1 ist und das Gruppe 3–5 Metall
(bevorzugt das Gruppe 4 Metall) im formalen Oxidationszustand +3
vorliegt, ist X bevorzugt ein stabilisierender Ligand.
-
Unter dem Ausdruck "stabilisierender
Ligand" ist zu verstehen,
daß die
Ligandengruppe den Metallkomplex stabilisiert, entweder durch:
- 1) eine Stickstoff-, Phosphor-, Sauerstoff-
oder Schwefelchelatbindung, oder
- 2) eine η3-Bindung mit einer resonanten, delokalisierten, π-Elektronenstruktur.
-
Beispiele von stabilisierenden Liganden
von Gruppe 1) schließen
ein: Silyl-, Hydrocarbyl-, Amido- oder Phosphidoliganden, substituiert
mit einem oder mehreren aliphatischen oder aromatischen Ether-,
Thioether-, Amin- oder Phosphinfunktionellen Gruppen, insbesondere
solchen Amin- oder Phosphingruppen, welche tertiär-substituiert sind, wobei
dieser stabilisierende Ligand von 3 bis 30 Nicht-Wasserstoffatome
hat. Am meisten bevorzugt sind stabilisierende Liganden der Gruppe
1) 2-Dialkylaminobenzyl- oder 2-(Dialkylaminomethyl)-phenylgruppen, welche
von 1 bis 4 Kohlenstoffatome in den Alkylgruppen enthalten.
-
Beispiele von stabilisierenden Liganden
von Gruppe 2) schließen
ein: C3-10 Hydrocarbylgruppen, enthaltend
ethylenartige Unsättigung,
wie Allyl-, 1-Methylallyl-, 2-Methylallyl-, 1,1-Dimethylallyl- oder
1,2,3-Trimethylallylgruppen.
-
Noch mehr bevorzugt entsprechen solche
Metallkoordinationskomplexe der folgenden Formel (VIII):
worin R' bei jedem Vorkommen unabhängig aus
der Gruppe ausgewählt
ist, die aus Wasserstoff, Hydrocarbyl, Silyl, Germyl, Cyano, Halogen
und Kombinationen hiervon mit bis zu 20 Nicht-Wasserstoffatomen besteht, oder zwei
Gruppen R' zusammen
ein zweiwertiges Derivat hiervon bilden:
X dieselbe Bedeutung
wie für
Formel (VI) definiert hat;
Y eine zweiwertige anionische Ligandengruppe
ist, welche Stickstoff, Phosphor, Sauerstoff oder Schwefel umfaßt und bis
zu 20 Nicht-Wasserstoffatome hat, wobei dieses Y an Z und M über diesen
Stickstoff, Phosphor, Sauerstoff oder Schwefel gebunden ist, und
wahlweise Y und Z zusammen ein kondensiertes Ringsystem bilden;
M
ein Gruppe 4 Metall ist, insbesondere Titan;
Z = SiR*
2, Cr*
2, SiR*
2SiR*
2, CR*
2CR*
2, CR*=CR*, CR*
2SiR*
2, GeR*
2, BR* oder BR*
2 ist,
worin:
R* bei jedem Vorkommen unabhängig aus der Gruppe ausgewählt ist,
die besteht aus Wasserstoff-, Hydrocarbyl-, Silyl-, halogenierten
Alkyl-, halogenierten Arylgruppen mit bis zu 20 Nicht-Wasserstoffatomen
und Mischungen hiervon, oder zwei oder mehr Gruppen R* von Z oder
eine Gruppe R* von Z zusammen mit Y ein kondensiertes Ringsystem
bilden; und
n = 1 oder 2 ist.
-
Mehr bevorzugt ist Y = -O-, -S-,
-NR*-, -PR*-. Am meisten bevorzugt ist Y eine Stickstoff oder Phosphor enthaltende
Gruppe, entsprechend der Formel -N(R')- oder -P(R')-, wor in R' wie zuvor beschrieben ist, d. h. eine
Amido- oder Phosphidogruppe.
-
Am meisten bevorzugte Metallkoordinationskomplexe
entsprechen der folgenden Formel (IX):
worin:
M = Titan ist;
R' bei jedem Vorkommen
unabhängig
aus der Gruppe ausgewählt
ist, die aus Wasserstoff, Silyl, Hydrocarbyl und Kombinationen hiervon
mit bis zu 10 Kohlenstoff- oder Siliziumatomen besteht, oder zwei
Gruppen R' der substituierten
Cyclopentadienyleinheit miteinander verbunden sind;
E = Silizium
oder Kohlenstoff ist;
X unabhängig bei jedem Vorkommen Hydrid,
Alkyl, Aryl mit bis zu 10 Kohlenstoffatomen ist;
m = 1 oder
2 ist; und
n = 1 oder 2 ist.
-
Beispiele der oben genannten am meisten
bevorzugten Metallkoordinationsverbindungen schließen Verbindungen
ein, worin R' an
der Amidogruppe Methyl, Ethyl, Propyl, Butyl, Pentyl, Hexyl (einschließlich Isomeren),
Norbornyl, Benzyl, Phenyl und Cyclododecyl ist; (ER'2)m =
Dimethylsilan oder 1,2-Ethylen ist; R' auf der cyclischen, π-gebundenen
Gruppe unabhängig
bei jedem Vorkommen Wasserstoff, Methyl, Ethyl, Propyl, Butyl, Pentyl,
Hexyl, Norbornyl, Benzyl und Phenyl ist, oder zwei Gruppen R' miteinander unter
Bildung einer In denyl-, Tetrahydroindenyl-, Fluorenyl- oder Octyhydrofluorenyleinheit
verbunden sind; und X = Methyl, Ethyl, Propyl, Butyl, Pentyl, Hexyl,
Norbornyl, Benzyl und Phenyl ist.
-
Übergangsmetallverbindungen,
in denen das Übergangsmetall
im formalen Oxidationszustand +2 vorliegt, schließen solche
Komplexe ein, die eine und nur eine cyclische, delokalisierte, anionische, π-gebundene Gruppe
enthalten, wobei diese Komplexe der folgenden Formel (X) entsprechen:
worin:
M = Titan oder
Zirkonium im formalen Oxidationszustand +2 ist;
L eine Gruppe
ist, die ein cyclisches, delokalisiertes, anionisches, π-System enthält, durch
welches die Gruppe an M gebunden ist, und wobei diese Gruppe ebenfalls
an Z gebunden ist;
Z ist eine Einheit, gebunden an M über eine σ-Bindung,
umfassend Bor, oder ein Glied von Gruppe 14 des Periodensystems
der Elemente, und ebenfalls umfassend Stickstoff, Phosphor, Schwefel
oder Sauerstoff, wobei diese Einheit bis zu 60 Nicht-Wasserstoffatome
hat; und
X* ein neutrales, konjugiertes oder nicht-konjugiertes
Dien ist, wahlweise substituiert mit einer oder mehreren Hydrocarbylgruppen,
wobei dieses X bis zu 40 Kohlenstoffatome hat und einen π-Komplex
mit M bildet.
-
Bevorzugte Übergangsmetallverbindungen
der Formel (X) schließen
solche ein, worin Z, M und X* wie zuvor definiert sind, und L eine
Gruppe C5H4 ist,
gebunden an Z und gebunden in einer η5-Bindungsweise
an M, oder eine solche η5-gebundene Gruppe ist, die mit von 1 bis
4 Substituenten substituiert ist, unabhängig ausgewählt aus Hydrocarbyl, Silyl,
Germyl, Halogen, Cyano und Kombinationen hiervon, wobei dieser Substituent
bis zu 20 Nicht-Wasserstoffatome hat, und wahlweise zwei solcher
Substituenten (ausgenommen Cyano oder Halogen) zusammen eine kondensierte
Ringstruktur bilden.
-
Mehr bevorzugte Übergangsmetallverbindungen
+2 gemäß der vorliegenden
Erfindung entsprechen der folgenden Formel (XI):
worin:
R' bei jedem Vorkommen
unabhängig
ausgewählt
ist aus Wasserstoff, Hydrocarbyl, Silyl, Germyl, Halogen, Cyano
und Kombinationen hiervon, wobei dieses R' bis zu 20 Nicht-Wasserstoffatome hat,
und wahlweise zwei Gruppen R' (wenn
R' nicht Wasserstoff,
Halogen oder Cyano ist) zusammen ein zweiwertiges Derivat hiervon bilden,
das an benachbarte Stellungen des Cyclopentadienylrings zur Bildung
einer kondensierten Ringstruktur gebunden ist;
X* eine neutrale η
4-gebundene Diengruppe mit bis zu 30 Nicht-Wasserstoffatomen
ist, welche einen π-Komplex
mit M bildet;
Y = -O-, -S-, -NR*-, -PR*- ist;
M = Titan
oder Zirkonium im formalen Oxidationszustand +2 ist;
Z* = SiR*
2, CR*
2, SiR*
2SiR*
2, CR*
2CR*
2, CR*=CR*, CR*
2SiR*
2 oder GeR*
2 ist, worin:
X* bei jedem Vorkommen
unabhängig
Wasserstoff oder ein Glied, ausgewählt aus Hydrocarbyl, Silyl,
halogeniertem Alkyl, halogeniertem Aryl und Kombinationen hiervon
ist, wobei dieses R* bis zu 20 Nicht-Wasserstoffatome hat, und wahlweise
zwei Gruppen R* von Z* (wenn R* nicht Wasserstoff ist) oder eine
Gruppe R* von Z* und eine Gruppe R* von Y ein Ringsystem bilden.
-
Bevorzugt ist R' unabhängig bei jedem Vorkommen Wasserstoff,
Hydrocarbyl, Silyl, Halogen und Kombinationen hiervon, wobei R' bis zu 10 Nicht-Wasserstoffatome
hat oder zwei Gruppen R' (wenn
R' nicht Wasserstoff
oder Halogen ist), bilden zusammen ein zweiwertiges Derivat hiervon;
am meisten bevorzugt ist R' Wasserstoff,
Methyl, Ethyl, Propyl, Butyl, Pentyl, Hexyl (einschließlich aller
Isomere, falls möglich),
Cyclopentyl, Cyclohexyl, Norbornyl, Benzyl oder Phenyl, oder zwei
Gruppen R' (mit
Ausnahme von Wasserstoff) sind miteinander verbunden, wobei die
gesamte Gruppe C5R'4 hierdurch
beispielsweise eine Indenyl-, Tetrahydroindenyl-, Fluorenyl-, Tetrahydroflurenyl-
oder Octahydrofluorenylgruppe ist.
-
Weiterhin ist wenigstens einer von
R' oder R* eine
elektronenabgebende Einheit. Unter dem Ausdruck "elektronenabgebend" ist zu verstehen, daß die Einheit
stärker
elektronenabgebend als Wasserstoff ist. So ist Y bevorzugt eine
Stickstoff oder Phosphor enthaltende Gruppe, entsprechend der Formel
-N(R'')- oder -P(R'')-, worin R'' =
C1-10-Hydrocarbyl ist.
-
Beispiele von geeigneten Gruppen
X* schließen
ein: s-trans-η4-1,4-Diphenyl-1,3-butadien; s-trans-η4-3-Methyl-1,3-pentadien; s-trans-η4-1,4-Dibenzyl-1,3-butadien;
s-trans-η4-2,4-Hexadien; s-trans-η4-1,3-Pentadien; s-trans-η4-1,4-Ditolyl-1,3-butadien;
s-trans-η4-1,4-Bis(trimethylsilyl)-1,3-butadien; s-cis-η4-1,4-Diphenyl-1,3-butadien; s-cis-η4-3- Methyl-1,3-pentadien;
s-cis-η4-1,4-Dibenzyl-1,3-butadien; s-cis-η4-2,4-Hexadien; s-cis-η4-1,3-pentadien;
s-cis-η4-1,4-Ditolyl-1,3-butadien
und s-cis-η4-1,4-Bis(trimethylsilyl)-1,3-butadien, wobei diese s-cis-Diengruppe
einen π-Komplex,
wie hier definiert, mit dem Metall bildet.
-
Am stärksten bevorzugte Übergangsmetal
1 + 2-verbindungen sind Amidosilan- oder Amidoalkandiylverbindungen
der Formel (XI), worin:
-Z*-Y- = (ER'''2)m-N(R'') – ist, und
R' bei jedem Vorkommen
unabhängig
ausgewählt
ist aus Wasserstoff, Silyl, Hydrocarbyl und Kombinationen hiervon,
worin R' bis zu
10 Kohlenstoff- oder
Siliziumatome hat, oder solcher Gruppen R' auf
der substituierten Cyclopentadienylgruppe
(wenn R' nicht Wasserstoff
ist) zusammen ein zweiwertiges Derivat hiervon bilden, das an benachbarte
Stellungen des Cyclopentadienylringes gebunden ist;
R'' = C1-10 Hydrocarbyl
ist;
R''' unabhängig bei jedem Vorkommen Wasserstoff
oder C1-10 Hydrocarbyl ist;
E unabhängig bei
jedem Vorkommen Silizium oder Kohlenstoff ist; und
m = 1 oder
2 ist.
-
Beispiele der Metallkomplexe gemäß der vorliegenden
Erfindung schließen
Verbindungen ein, worin R'' = Methyl, Ethyl,
Propyl, Butyl, Pentyl, Hexyl (einschließlich aller Isomeren der zuvor
genannten, falls zutreffend), Cyclododecyl, Norbornyl, Benzyl oder
Phenyl ist; (ER'''2)m =
Dimethylsilan oder Ethandiyl ist, und die cyclische, delokalisierte, π-gebundene
Gruppe Cyclopentadienyl, Tetramethylcyclopentadienyl, Indenyl, Tetrahydroindenyl,
Fluorenyl, Tetrahydrofluorenyl oder Octahydrofluorenyl ist.
-
Geeignete Bis(cyclopentadienyl)-derivate
von Übergangsmetallen
schließen
solche von Titan-, Zirkonium- und Hafnium verbindungen ein, und sie
können
durch die folgenden allgemeinen Formeln (XII) bis (XV) wiedergegeben
werden: (A-Cp)MX1X2 (XII)
(A-Cp)MX'1X'2 (XIII)
(A-Cp)ML (XIV)
(Cp*)(CpR)MX1 (XV)worin:
M ein Metall der Gruppe 4, nämlich
Titan (Ti), Zirkonium (Zr) und Hafnium (Hf) ist; (A-Cp) entweder (Cp)(Cp*)
oder Cp-A'-CP* ist
und Cp und Cp* gleiche oder verschiedene Cyclopentadienylreste sind,
wie auch substituierte Derivate von Cyclopentadienylresten, und
A' eine kovalente
verbrückende
Gruppe ist, die ein Element der Gruppe 14 enthält; L ein Olefin-, Diolefin-
oder Arinligand ist; wenigstens einer von X1 und
X2 ein Hydridrest, Hydrocarbylrest, substituierter
Hydrocarbylrest oder Organo-Metalloidrest ist, wobei der andere
von X1 und X2 ein
Hydridrest, Hydrocarbylrest, substituierter Hydrocarbylrest, Organo-Metalloidrest
oder ein Hydrocarbyloxyrest ist; bevorzugt ist einer oder beide
von X1 und X2 ein
Hydrocarbylrest mit 1 bis 20 Kohlenstoffatomen, ein substituierter
Hydrocarbylrest mit 1 bis 20 Kohlenstoffatomen, worin eines oder
mehrere der Wasserstoffatome durch ein Halogenatom ersetzt ist,
ein Organo-Metalloidrest, umfassend ein Element der Gruppe 14, worin
jeder der Hydrocarbylsubstituenten, der in dem Organoabschnitt dieses
Organo-Metalloids vorliegt, unabhängig von 1 bis 20 Kohlenstoffatome
enthält;
X'1 und
X'2 miteinander
verbunden und an das Metallatom unter Bildung eines Metallzyklus
gebunden sind, worin das Metall, X'1 und X'2 einen
hydrocarbocyclischen Ring bilden, der von 3 bis 20 Kohlenstoffatome
enthält;
und R ein Substituent, bevorzugt ein Hydrocarbylsubstituent, mit
1 bis 20 Kohlenstoffatomen auf einem der Cyclopentadienylreste ist,
der ebenfalls an das Metallatom gebunden ist.
-
Wenn nicht beide X1 und
X2 ein Hydridrest, Hydrocarbylrest, substituierter
Hydrocarbylrest oder Organo-Metalloidrest sind, kann einer von diesen
ein Hydrocarbyloxyrest mit 1 bis 20 Kohlenstoffatomen sein. Geeignete
Beispiele von Hydrocarbyloxyresten schließen ein: Alkyloxy-, Aryloxy-,
Aralkyloxy- und Alkaryloxyreste mit 1 bis 20 Kohlenstoffatomen,
mehr bevorzugt Alkylreste mit 1 bis 6 Kohlenstoffatomen und Aryl-,
Aralkyl- und Alkarylreste mit 6 bis 10 Kohlenstoffatomen, noch mehr
bevorzugt Isopropyloxy, n-Butyloxy oder t-Butyloxy.
-
Beispiele von solchen Bis(cyclopentadienyl)-derivaten
von übergangsmetallen
und Verfahren zu ihrer Herstellung sind im US-Patent 5 384 299 (entsprechend
der EP-A-277 004) und der US-Anmeldung Serial No. 459 921, eingereicht
am 2. Januar 1990 (entsprechend der WO-91/09882) angegeben, auf
welche hiermit Bezug genommen wird.
-
Geeignete Tri-cyclopentadienyl- oder
substituierte Cyclopentadienylübergangsmetallverbindungen schließen solche
ein, die eine verbrückende
Gruppe enthalten, welche zwei Cyclopentadienylgruppen miteinander
verbindet, und solche ohne diese verbrückenden Gruppen.
-
Geeignete nicht-verbrückte Tri-cyclopentadienylübergangsmetallderivate
werden durch die folgende Formel (XVI) wiedergegeben: Cp3MXn'' (XVI)worin
Cp, M und X wie für
die Formel (VI) definiert sind, und
n'' 3
weniger als der formale Oxidationszustand von M ist und = 0 oder
1, bevorzugt = 1 ist. Bevorzugte Ligandengruppen X sind Hydrocarbyl,
Hydrocarbyloxy, Hydrid, Halogen, Silyl, Germyl, Amid und Siloxy.
-
Gemäß einer bevorzugten Ausführungsform
umfaßt
der feste (oder getragene) Katalysator:
eine Trägerkatalysatorkomponente,
umfassend (a) ein Trägermaterial
und eine Organometallverbindung, worin das Metall aus den Gruppen
2–13 des
Periodensystems der Elemente, Germanium, Zinn und Blei ausgewählt ist,
und (b) eine Aktivatorverbindung, umfassend (b-1) ein Kation, das
zur Reaktion mit einer Übergangsmetallverbindung
zur Bildung eines katalytisch aktiven Übergangsmetallkomplexes fähig ist,
und (b-2) ein verträgliches
Anion, das bis zu 100 Nicht-Wasserstoffatome hat und wenigstens
einen Substituenten, der eine aktive Wasserstoffeinheit umfaßt, enthält; und
eine Übergangsmetallverbindung.
-
Das Trägermaterial wird typischerweise
mit der Organometallverbindung behandelt. Geeignete Organometallverbindungen
sind solche, welche umfassen: Metalle von Gruppen 2–13, Germanium,
Zinn und Blei sowie wenigstens zwei Substituenten, ausgewählt aus
Hydrid, Hydrocarbylresten, Trihydrocarbylsilylresten und Trihydrocarbylgermylresten.
Zusätzliche
Substituenten umfassen bevorzugt einen oder mehrere Substituenten,
ausgewählt
aus Hydrid, Hydrocarbylresten, trihydrocarbyl-substituierten Silylresten,
trihydrocarbyl-substituierten Germylresten und hydrocarbyl-, trihydrocarbylsilyl-
oder trihydrocarbylgermyl-substituierten Metalloidresten.
-
Die Angabe "Metalloid", wie hier verwendet, schließt Nicht-Metalle
wie Bor, Phosphor ein, welche Halbmetalleigenschaften zeigen.
-
Beispiele solcher Organometallverbindungen
schließen
ein: Organomagnesium-, Organozink-, Organobor-, Organoaluminium-,
Organogermanium-, Organozinn- und Organobleiverbindungen und Mischungen
hiervon. Weitere geeignete Organometallverbindungen sind Alumoxane.
Bevorzugte Beispiele sind Alumoxane und Verbindungen, welche durch
die folgenden Formeln wiedergegeben werden: MgR1
2, ZnR1
2, BR1
xR2
y, AlR1
xR2
y, worin R1 unabhängig
bei jedem Vorkommen ist: Hydrid, ein Hydrocarbylrest, ein Trihydrocarbylsilylrest,
ein Trihydrocarbylgermylrest oder ein trihydrocarbyl-, trihydrocarbylsilyl-
oder trihydrocarbylgermyl-substituierter Metalloidrest; R2 unabhängig
dasselbe wie R1 ist; x = 2 oder 3 ist; y
= 0 oder 1 ist, und die Summe von x und y = 3 ist; sowie Mischungen
hiervon. Beispiele von geeigneten Hydrocarbyleinheiten sind solche
mit 1 bis 20 Kohlenstoffatomen im Hydrocarbylabschnitt hiervon,
wie Alkyl, Aryl, Alkaryl oder Aralkyl. Bevorzugte Reste schließen Methyl,
Ethyl, n- oder i-Propyl, n-, s- oder t-Butyl, Phenyl und Benzyl
ein. Bevorzugt wird die Aluminiumkomponente aus der Gruppe ausgewählt, die
aus Alumoxan und Aluminiumverbindungen der Formel AlR1
x besteht, worin R1 bei
jedem Vorkommen unabhängig
Hydrid oder ein Hydrocarbylrest mit 1 bis 20 Kohlenstoffatomen ist,
und x = 3 ist. Geeignete Trihydrocarbylaluminiumverbindungen sind
Trialkyl- oder Triarylaluminiumverbindungen, worin jede Alkyl- oder
Arylgruppe von 1 bis 10 Kohlenstoffatome hat, oder Mischungen hiervon,
und bevorzugt Trialkylaluminiumverbindungen wie Trimethyl-, Triethyl-,
Triisobutylaluminium.
-
Alumoxane (auch bezeichnet als Aluminoxane)
sind oligomere oder polymere Aluminiumoxyverbindungen, enthaltend
Ketten von alternierenden Aluminium- und Sauerstoffatomen, wobei
das Aluminium einen Substituenten, bevorzugt eine Alkylgruppe, trägt. Es wird
angenommen, daß die
Struktur von Alumoxan durch die folgende allgemeine Formel (-Al(R)-O)m für
ein cyclisches Alumoxan und R2Al-O(-Al(R)-O)m-AlR2 für eine lineare
Verbindung wiedergegeben wird, worin R unabhängig bei jedem Vorkommen ein
C1-C10-Hydrocarbyl, bevorzugt
Alkyl, oder Halogenid ist, und m eine ganze Zahl im Bereich von
1 bis 50, bevorzugt wenigstens 4, ist. Alumoxane sind typischerweise
die Reaktionsprodukte von Wasser und einem Aluminiumalkyl, welches
zusätzlich
zu einer Alkylgruppe Halogenid- oder Alkoxidgruppen enthalten kann.
Die Umsetzung von mehreren unterschiedlichen Aluminiumalkylverbindungen,
wie beispielsweise Trimethylaluminium und Triisobutylaluminium,
mit Wasser ergibt sogenannte modifizierte oder gemischte Alumoxane.
Bevorzugte Alumoxane sind Methylalumoxan und mit kleineren Mengen
von anderen niederen Alkylgruppen, wie Isobutyl, modifiziertes Methylalumoxan.
Alumoxane enthalten im allgemeinen kleinere bis wesentliche Mengen
von Ausgangs-Aluminiumalkylverbindung.
-
Die Art und Weise, in welcher das
Alumoxan hergestellt wird, ist nicht kritisch. Wenn es durch Reaktion zwischen
Wasser und Aluminiumalkyl hergestellt wird, kann das Wasser mit
dem Aluminiumalkyl in verschiedenen Formen, wie als Flüssigkeit,
als Dampf oder als Feststoff, kombiniert werden, beispielsweise
in Form von Kristallisationswasser. Besondere Arbeitsweisen für die Herstellung
von Verbindungen von Alumoxantyp durch Inkontaktbringen einer Aluminiumalkylverbindung
mit einem Kristallisationswasser enthaltenden anorganischen Salz
sind im US-Patent 4 542 199 angegeben. Bei einer besonders bevorzugten
Ausführungsform wird
eine Aluminiumalkylverbindung mit einer regenerierbaren wasserhaltigen
Substanz wie hydratisiertem Aluminiumoxid, Siliziumdioxid oder einer
anderen Substanz in Kontakt gebracht. Dies ist in der europäischen Patentanmeldung
Nr. 338 044 beschrieben.
-
Der Trägerkatalysator gemäß dieser
Ausführungsform
umfaßt
im allgemeinen ein Trägermaterial, kombiniert
oder behandelt mit der Organometallverbindung und enthaltend wenigstens
0,1 Mikromol Organometallverbindung pro g Trägermaterial, typischerweise
wenigstens 5 Mikromol pro g Trägermaterial,
vorteilhafterweise wenigstens 0,5 Gew.-% des Metalls, bevorzugt
Aluminium, ausgedrückt
in Gramm von Metallatomen pro g von Trägermaterial. Bevorzugt beträgt die Metallmenge
wenigstens 2 Gew.-% und im allgemeinen nicht mehr als 40 Gew.-%
und mehr bevorzugt nicht mehr als 30 Gew.-%. Zu hohe Mengen von
Metall machen den Trägerkatalysator
kostspielig. Bei zu niedrigen Mengen fällt die Effizienz des Katalysators
unterhalb annehmbarer Werte.
-
Die Trägerkatalysatoren enthalten
bevorzugt ein behandeltes Trägermaterial
(a), umfassend ein Trägermaterial
und ein Alumoxan, worin nicht mehr als 10% Aluminium in dem behandelten
Trägermaterial
bei einer Extraktion von 1 Stunde mit Toluol von 90°C unter Anwendung
von 10 ml Toluol pro Gramm von vorbehandeltem Trägermaterial extrahierbar sind.
Mehr bevorzugt sind nicht mehr als 9% des in der Trägerkata torkomponente
vorhandenen Aluminiums extrahierbar, und am meisten bevorzugt nicht
mehr als 8%. Dies ist besonders vorteilhaft, wenn der Trägerkatalysator
bei einem Polymerisationsverfahren eingesetzt wird, bei welchem
ein Verdünnungsmittel
oder Lösungsmittel
verwendet wird, das nicht-fixiertes Alumoxan von dem Trägermaterial
extrahieren könnte.
Es wurde gefunden, daß bei
Mengen von Extrahierbarem unterhalb der oben angegebenen Werte die
Menge von Alumoxan in das Polymerisationslösungsmittel, falls dieses verwendet wird,
diffundieren kann, so niedrig ist, daß keine nennenswerte Menge
von Polymerem in dem Verdünnungsmittel
gebildet wird, verglichen mit dem auf dem Trägermaterial gebildeten Polymeren.
Falls zu viel Polymeres in dem Verdünnungsmittel gebildet wird,
nimmt die Polymerdichte auf unterhalb annehmbare Werte ab, und es können Probleme
der Reaktorverstopfung auftreten.
-
Der Toluolextraktionstest wird wie
folgt durchgeführt:
Etwa 1 g Trägerkatalysatorkomponente
oder getragener Katalysator mit einem bekannten Aluminiumgehalt
wird zu 10 ml Toluol zugesetzt, und das Gemisch wird dann auf 90°C unter einer
inerten Atmosphäre
erhitzt. Die Suspension wird bei dieser Temperatur für 1 Stunde
gut gerührt.
Dann wird die Suspension unter Anwendung von vermindertem Druck
zur Unterstützung der
Filtrationsstufe filtriert. Die Feststoffe werden zweimal mit etwa
3 bis 5 ml Toluol von 90°C
pro Gramm von Feststoffen gewaschen. Die Feststoffe werden dann
bei 120°C
für 1 Stunde
getrocknet, und anschließend
wird der Aluminiumgehalt der Feststoffe gemessen. Der Unterschied
zwischen dem anfänglichen
Aluminiumgehalt und dem Aluminiumgehalt nach der Extraktion, dividiert
durch den anfänglichen
Aluminiumgehalt und multipliziert mit 100 ergibt die Menge von extrahierbarem
Aluminium.
-
Der Aluminiumgehalt kann durch Aufschlämmen von
etwa 0,5 g Trägerkatalysatorkomponente
oder Trägerkatalysator
in 10 ml Hexan bestimmt werden. Die Aufschlämmung wird mit 10 bis 15 ml
6 N Schwefelsäure
behandelt, gefolgt von Zugabe eines be kannten Überschusses von EDTA. Die Überschußmenge von EDTA
wird dann mit Zinkchlorid zurücktitriert.
-
Ohne an irgendeine Theorie gebunden
zu sein, wird angenommen, daß die
Aktivatorverbindung gemäß dieser
Ausführungsform
mit der Organometallverbindung über
den aktiven Wasserstoff enthaltenden Substituenten reagiert. Es
wird angenommen, daß eine
Gruppe R1 der Organometallverbindung mit
der aktiven Wasserstoffeinheit der Aktivatorverbindung kombiniert,
um eine neutrale organische Verbindung, beispielsweise ein Alkan,
oder Wasserstoffgas freizusetzen, wodurch das Metallatom mit dem
Rest der Aktivatorverbindung chemisch gekuppelt wird. Es wird daher
angenommen, daß der
Aktivator an das Trägermaterial chemisch
gebunden wird, sobald das Trägermaterial
mit der Organometallverbindung oder dem Addukt von Organometallverbindung
und Aktivatorverbindung behandelt wurde. Bei Zugabe der Übergangsmetallverbindung
wird ein Trägerkatalysator
mit verbesserten Eigenschaften gebildet.
-
Die in der vorliegenden Erfindung
brauchbare Aktivatorverbindung enthält ein verträgliches
Anion mit bis zu 100 und bevorzugt mit bis zu 50 Nicht-Wasserstoffatomen
und hat wenigstens einen Substituenten, der eine aktive Wasserstoffeinheit
umfaßt.
Bevorzugte Substituenten, welche eine aktive Wasserstoffeinheit
umfassen, entsprechen der Formel (XVII): Gq(T-H)r (XVII)worin
G ein polyvalenter Kohlenwasserstoffrest ist, T = 0, 2, NR oder
PR, worin R ein Hydrocarbylrest, ein Trihydrocarbylsilylrest, ein
Trihydrocarbylgermylrest oder Wasserstoff ist, q = 0 oder 1 und
bevorzugt 1 ist, und r eine ganze Zahl von 1 bis 3, bevorzugt 1
ist. Der polyvalente Kohlenwasserstoffrest G hat r + 1 Wertigkeiten, eine
Wertigkeit ist mit einem Metall oder Metalloid der Gruppen 5–15 des
Periodensystems der Elemente in dem verträglichen Anion verbunden, die
andere Wertigkeit oder die anderen Wertigkeiten von G sind an r
Gruppen T-H gebunden. Bevorzugte Beispiele von G schließen zweiwertige
Kohlenwasserstoffreste wie Alkylen-, Arylen-, Aralkylen oder Alkarylenreste
mit 1 bis 20 Kohlenstoffatomen, mehr bevorzugt 2 bis 12 Kohlenstoffatomen,
ein. Geeignete Beispiele von G schließen Phenylen, Biphenylen, Naphthylen,
Methylen, Ethylen, 1,3-Propylen, 1,4-Butylen, Phenylmethylen (-C6H4-CH2-)
ein. Der polyvalente Hydrocarbylanteil G kann weiter mit Resten,
welche die Kupplungsfunktion der aktiven Wasserstoffeinheit nicht
stören,
substituiert sein. Bevorzugte Beispiele von solchen nicht-störenden Substituenten
sind Alkyl-, Aryl-, alkyl- oder aryl-substituierte Silyl- und Germylreste
und Fluorsubstituenten.
-
Die Gruppe T-H in der zuvor genannten
Formel kann daher eine Gruppe -OH, -SH, -NRH oder -PRH sein, worin
R bevorzugt ein C1-18, bevorzugt ein C1-10 Hydrocarbylrest oder Wasserstoff ist
und H = Wasserstoff ist. Bevorzugte Gruppen R sind Alkyle, Cycloalkyle,
Aryle, Arylalkyle oder Alkylaryle von 1 bis 18 Kohlenstoffatomen,
mehr bevorzugt solche von 1 bis 12 Kohlenstoffatomen. Die Gruppen
-OH, -SH, -NRH oder -PRH können
Teil einer größeren Funktionalität sein,
wie beispielsweise C(O)-OH, C(S)-SH, C(O)-NRH und C(O)-PRH. Am meisten
bevorzugt ist die Gruppe T-H eine Hydroxygruppe, -OH, oder eine
Aminogruppe, -NRH.
-
Sehr bevorzugte Substituenten Gq(T-H)r, die eine
aktive Wasserstoffeinheit umfassen, schließen hydroxy- und aminosubstituierte
Aryl-, Aralkyl-, Alkaryl- oder Alkylgruppen ein, und am meisten
bevorzugt sind die Hydroxyphenylgruppen, insbesondere die 3- und
4-Hydroxyphenylgruppen, Hydroxytolylgruppen, Hydroxybenzylgruppen
(Hydroxymethylphenyl), Hydroxybiphenylgruppen, Hydroxynaphthylgruppen,
Hydroxycyclohexylgruppen, Hydroxymethylgruppen und Hydroxypropylgruppen,
sowie die entsprechenden amino-substituierten Gruppen, insbesondere
solche, die mit -NRH substituiert sind, worin R ein Alkyl- oder Arylrest mit
1 bis 10 Kohlenstoffatomen, beispielsweise Methyl, Ethyl, Propyl,
i-Propyl, n-, i- oder t-Butyl, Pentyl, Hexyl, Heptyl, Octyl, Nonyl
und Decyl, Phenyl, Benzyl, Tolyl, Xylyl, Naphthyl und Biphenyl ist.
-
Das verträgliche Anion, welches den Substituenten
enthält,
der eine aktive Wasserstoffeinheit aufweist, kann weiterhin ein
Einzelelement der Gruppe 5–15
oder eine Vielzahl von Elementen der Gruppe 5–15 umfassen, bevorzugt ist
es jedoch ein Einzelkoordinationskomplex, der einen ladungstragenden
Metall- oder Metalloidkern umfaßt,
wobei dieses Anion raumfüllend
ist. Ein verträgliches
Anion bezieht sich spezifisch auf ein Anion, welches bei Funktionieren
als ein die Ladung ausgleichendes Anion in dem Katalysatorsystem
dieser Erfindung, einen anionischen Substituenten oder ein Fragment
hiervon zu dem Übergangsmetallkation nicht
unter Bildung einer neutralen Übergangsmetallverbindung
und eines neutralen Metallnebenproduktes überträgt. "Verträgliche Anionen" sind Anionen, welche
nicht bis zur Neutralität
abgebaut werden, wenn der anfänglich
gebildete Komplex sich zersetzt, und welche die gewünschten
nachfolgenden Polymerisationen nicht stören.
-
Bevorzugte Anionen sind solche, welche
einen Einzelkoordinationskomplex enthalten, der einen ladungstragenden
Metall- oder Metalloidkern, der eine aktive Wasserstoffeinheit enthaltenden
Substituenten trägt,
wobei dieses Anion relativ groß (raumfüllend),
zur Stabilisierung der aktiven Katalysatorspezies (des Übergangsmetallkations),
das gebildet wird, wenn die Aktivatorverbindung und die Übergangsmetallverbindung
kombiniert werden, ist, und wobei dieses Anion ausreichend labil
ist, um durch olefinische, diolefinische und acetylenartig ungesättigte Verbindungen
oder andere neutrale Lewisbasen, wie Ether, Nitrile, verdrängt zu werden.
Geeignete Metalle für
die Anionen von Aktivatorverbindungen schließen ein, sind jedoch nicht
beschränkt
auf Aluminium, Gold, Platin. Geeignete Metalloide schließen ein,
sind jedoch nicht beschränkt
auf Bor, Phosphor, Silizium. Aktivatorverbindungen, welche Anionen
enthalten, die einen Koordinationskomplex umfassen, der ein einzelnes
Boratom und einen eine aktive Wasserstoffeinheit umfassenden Substituenten enthalten,
sind bevorzugt.
-
Bevorzugt können verträgliche Anionen, welche einen
eine aktive Wasserstoffeinheit umfassenden Substituenten enthalten,
durch die folgende allgemeine Formel (XVIII) wiedergegeben werden: [M'm+Qn(Gq(T-H)r)z]d– (XVIII)worin:
M' ein Metall oder
Metalloid, ausgewählt
aus den Gruppen 5– 15
des Periodensystems der Elemente ist;
Q unabhängig bei
jedem Vorkommen aus der Gruppe ausgewählt ist, die aus Hydrid-, Dihydrocarbylamido-, bevorzugt
Dialkylamido-, Halogenid-, Hydrocarbyloxid-, bevorzugt Alkoxid-
und Aryloxid-, Hydrocarbyl- und substituierten Hydrocarbylresten,
einschließlich
halogen-substituierten Hydrocarbylresten und hydrocarbyl- und halogenhydrocarbyl-substituierten
Organo-Metalloidresten
besteht, wobei der Hydrocarbylantei 1 bis 20 Kohlenstoffatome hat,
mit der Maßgabe,
daß bei
nicht mehr als einem Vorkommen Q = Halogenid ist;
G ein polyvalenter
Kohlenwasserstoffrest, der r + 1 Wertigkeiten besitzt und bevorzugt
zweiwertig ist, gebunden an M' und
T ist;
T = O, S, NR oder PR ist, worin R ein Kohlenwasserstoffrest,
ein Trihydrocarbylsilylrest, ein Trihydrocarbylgermylrest oder Wasserstoff
ist;
m = eine ganze Zahl von 1 bis 7, bevorzugt 3 ist;
n
= eine ganze Zahl von 0 bis 7, bevorzugt 3 ist;
q = eine Ganze
Zahl von 0 oder 1, bevorzugt 1 ist;
r = eine ganze Zahl von
1 bis 3, bevorzugt 1 ist;
z = eine ganze Zahl von 1 bis 8,
bevorzugt 1 ist;
d = eine ganze Zahl von 1 bis 7, bevorzugt
1 ist; und n + 2 – m
= d.
-
Bevorzugte Bor enthaltende Anionen,
welche besonders brauchbar in der vorliegenden Erfindung sind, können durch
die folgende allgemeine Formel (XIX) wiedergegeben werden: [BQ4-z'(Gq(T-H)r)z']d– (XIX)worin:
B
= Bor in einem Wertigkeitszustand von 3 ist;
z' eine ganze Zahl
von 1–4,
bevorzugt 1 ist;
d = 1 ist; und
Q, G, T, H, q und r wie
für Formel
(XVIII) definiert sind. Bevorzugt sind: z' = 1, q = 1 und r = 1.
-
Erläuternde, jedoch nicht einschränkende Beispiele
von Anionen von Aktivatorverbindungen, welche in der vorliegenden
Erfindung eingesetzt werden, sind Bor enthaltende Anionen wie: Triphenyl(hydroxyphenyl)borat,
Diphenyl-di(hydroxyphenyl)borat, Triphenyl(2,4-dihydroxyphenyl)borat,
Tri(p-tolyl)-(hydroxyphenyl)borat,
Tris(pentafluorphenyl)(hydroxyphenyl)-borat, Tris(2,4-dimethylphenyl)(hydroxyphenyl)borat, Tris-(3,5-dimethylphenyl)(hydroxyphenyl)borat,
Tris(3,5-di-tri-fluormethylphenyl)(hydroxyphenyl)borat, Tris(pentafluorphenyl)(2-hydroxyethyl)borat,
Tris(pentafluorphenyl)(4-hydroxybutyl)borat, Tris(pentafluorphenyl)(4-hydroxycyclohexyl)-borat, Tris(pentafluorphenyl)(4-(4'-hydroxyphenyl)phenyl)-borat, Tris(pentafluorphenyl)(6-hydroxy-2-naphthyl)borat.
Ein hoch bevorzugter Aktivatorkomplex ist Tris(pentafluorphenyl)-(4-hydroxyphenyl)borat.
Andere bevorzugte Anionen von Aktivatorverbindungen sind die oben
genannten Borate, in denen die Hydroxyfunktionalität durch
eine Amino-NHR-Funktionalität,
worin R bevorzugt Methyl, Ethyl oder t-Butyl ist, ersetzt ist.
-
Der kationische Anteil (b-1) der
Aktivatorverbindung, welche in Verbindung mit dem verträglichen
Anion (b-2) verwendet werden soll, kann ein beliebiges Kation sein,
das zur Reaktion mit der Übergangsmetallverbindung
unter Bildung ei nes katalytisch aktiven Übergangsmetallkomplexes, insbesondere
eines kationisches Übergangsmetallkomplexes,
in der Lage ist. Die Kationen (b-1) und die Anionen (b-2) werden
in solchen Verhältnissen
eingesetzt, daß eine
neutrale Aktivatorverbindung erhalten wird. Bevorzugt wird das Kation
aus der Gruppe ausgewählt,
die aus Bronstedsäurekationen,
Carboniumkationen, Silyliumkationen und kationischen oxidierenden
Mitteln besteht.
-
Bronstedsäurekationen können durch
die folgende allgemeine Formel wiedergegeben werden: (L-H)+, worin:
L
eine neutrale Lewisbase, bevorzugt eine Stickstoff, Phosphor oder
Schwefel enthaltende Lewisbase ist, und (L-H)+ eine
Bronstedsäure
ist. Es wird angenommen, daß die
Bronstedsäurekationen
mit der Übergangsmetallverbindung
durch Übertragung
eines Protons auf dieses Kation reagieren, wobei dieses Proton mit
einem der Liganden auf der Übergangsmetallverbindung
unter Freisetzung einer neutralen Verbindung kombiniert.
-
Erläuternde, jedoch nicht beschränkende Beispiele
von Bronstedsäurekationen
von Aktivatorverbindungen, welche in der vorliegenden Erfindung
verwendet werden, sind trialkylsubstituierte Ammoniumkationen wie
Triethylammonium, Tripropylammonium, Tri(n-butyl)ammonium, Trimethylammonium,
Tributylammonium und Tri(n-octyl)ammonium. Ebenfalls geeignet sind
N,N-Dialkylaniliniumkationen wie N,N-Dimethylanilinium, N,N-Diethylanilinium,
N,N-2,4,6-Pentamethylanilinium, N,N-Dimethylbenzylammonium; Dialkylammoniumkationen
wie Di-(i-propyl)ammonium, Dicyclohexylammonium sowie Triarylphosphoniumkationen
wie Triphenylphosphonium, Tri(methylphenyl)phosphonium, Tri(dimethylphenyl)phosphonium,
Dimethylsulfonium, Diethylsulfonium und Diphenylsulfonium.
-
Ein zweiter Typ von geeignetem Kation
entspricht der Formel:
©+
worin ©+ ein stabiles Carbonium- oder Silyliumion
ist, das bis zu 30 Nicht-Wasserstoffatome enthält, wobei das Kation zur Reaktion
mit einem Substituenten von der Übergangsmetallverbindung
und zur Umwandlung hiervon in einen katalytisch aktiven Übergangsmetallkomplex,
insbesondere einen kationischen Übergangsmetallkomplex,
fähig ist.
Geeignete Beispiele von Kationen schließen Tropyllium, Triphenylmethylium,
Benzol(diazonium) ein. Silyliumsalze sind zuvor allgemein in J.
Chem. Soc. Chem. Comm., 1993, 383–384, wie auch in Lambert,
J. B. et al., Organometallics, 1994, 13, 2430–2443, beschrieben. Bevorzugte
Silyliumkationen sind Triethylsilylium und Trimethylsilylium und
ether-substituierte Addukte hiervon.
-
Ein anderer geeigneter Typ von Kationen
umfaßt
ein kationisches Oxidationsmittel, wiedergegeben durch die Formel: Oxe+
worin Oxe+ ein kationisches Oxidationsmittel mit
einer Ladung e+ ist und e eine ganze Zahl von 1 bis 3 ist.
-
Beispiele von kationischen Oxidationsmitteln
schließen
ein: Ferrocenium, hydrocarbyl-substituiertes Ferrocenium, Ag+ und Pb2+.
-
Die Menge von Aktivatorverbindung
in der Trägerkatalysatorkomponente
und dem Trägerkatalysator ist
nicht kritisch, jedoch reicht sie typischerweise von 0,1, bevorzugt
von 1 bis 2000 Mikromol Aktivatorverbindung pro Gramm von behandeltem
Trägermaterial.
Bevorzugt enthält
der Trägerkatalysator
oder die Komponente von 10 bis 1000 Mikromol Aktivatorverbindung
pro Gramm von behandeltem Trägermaterial.
-
Im allgemeinen beträgt das Verhältnis von
Mol Aktivatorverbindung (b) zu Grammatomen von Übergangsmetall in Verbindung
(c) in dem Trägerkatalysator
von 0,05 : 1 bis 100 : 1, bevorzugt von 0,5 : 1 bis 20 : 1 und am
meisten bevorzugt von 1 : 1 bis 5 : 1 Mol Aktivatorverbindung pro
Grammatom von Übergangsmetall in
der Übergangsmetallverbindung.
Bei zu niedrigen Verhältnissen
ist der Trägerkatalysator
nicht sehr aktiv, während
bei zu hohen Verhältnissen
der Katalysator weniger wirtschaftlich als Folge der relativ hohen
Kosten, die mit der Verwendung von großen Mengen von Aktivatorverbindung
verbunden sind, wird.
-
Der Trägerkatalysator gemäß dieser
Ausführungsform
kann durch Kombinieren des Trägermaterials mit
der Organometallverbindung und der Aktivatorverbindung hergestellt
werden. Die Reihenfolge der Zugabe ist nicht kritisch. Die Organometallverbindung
kann entweder zuerst mit dem Trägermaterial
oder mit der Aktivatorverbindung kombiniert werden, und anschließend kann
die Aktivatorverbindung oder das Trägermaterial zugegeben werden.
Eine bevorzugte Ausführungsform
umfaßt
die Behandlung des Trägermaterials
zuerst mit der Organometallverbindung durch Kombinieren der Organometallverbindung
in einem geeigneten Lösungsmittel,
wie einem Kohlenwasserstofflösungsmittel,
mit dem Trägermaterial.
Die Temperatur, der Druck und die Kontaktzeit für diese Behandlung sind nicht
kritisch, jedoch variieren sie im allgemeinen von –20°C bis 150°C, von unteratmosphärischem
Druck bis 10,50 kg/cm2 (10 bar), mehr bevorzugt
bei atmosphärischem
Druck, für 5
Minuten bis 48 Stunden. Üblicherweise
wird die Aufschlämmung
gerührt.
Nach dieser Behandlung werden die Feststoffe typischerweise von
dem Lösungsmittel
abgetrennt. Irgendein Überschuß von Organometallverbindung
könnte
dann nach auf dem Fachgebiet bekannten Arbeitsweisen entfernt werden.
Dieses Verfahren ist besonders zum Erhalt von Trägermaterial mit relativ niedrigen
Metallbeladungen geeignet.
-
Gemäß einer bevorzugten Ausführungsform
wird das Trägermaterial
zuerst einer thermischen Behandlung bei 100°C bis 1000°C, bevorzugt bei 200°C bis 850°C, unterzogen.
Typischerweise wird diese Behandlung für 10 Minuten bis 72 Stunden,
bevorzugt von 0,5 Stunden bis 24 Stunden, durchgeführt. Dann wird das
thermisch behandelte Trägermaterial
mit der Organometallverbindung, bevorzugt AlR'3, worin R' die zuvor definierte
Bedeutung hat, in einem geeigneten Verdünnungsmittel oder Lösungsmittel,
bevorzugt in einem Mittel, in welchem die Organometallverbindung
löslich
ist, kombiniert. Typische Lösungsmittel
sind Kohlenwassertofflösungsmittel
mit 5 bis 12 Kohlenstoffatomen, bevorzugt aromatische Lösungsmittel
wie Toluol und Xylole oder aliphatische Lösungsmittel mit 6 bis 10 Kohlenstoffatomen
wie Hexan, Heptan, Octan, Nonan, Decan sowie Isomere hiervon, cycloaliphatische
Lösungsmittel
mit 6 bis 12 Kohlenstoffatomen wie Cyclohexan oder Mischungen von
beliebigen hiervon.
-
Das Trägermaterial wird mit der Organometallverbindung
bei einer Temperatur von –20°C bis 150°C, bevorzugt
bei 20°C
bis 100°C,
kombiniert. Die Kontaktzeit ist nicht kritisch und kann von 5 Minuten
bis 72 Stunden variieren, und bevorzugt beträgt sie von 0,5 Stunden bis
36 Stunden. Bevorzugt wird Rühren
angewandt. Das so behandelte Trägermaterial
wird dann bevorzugt mit der Aktivatorverbindung in Kontakt gebracht.
-
Eine alternative Behandlung des Trägermaterials,
welche zum Erhalt von an das Trägermaterial
gebundenen Alumoxanbeladungen geeignet ist, beinhaltet eine oder
beide der folgenden Stufen A und B:
- A. Erhitzen
eines Alumoxan enthaltenden Trägermaterials
unter einer Inertatmosphäre
für eine
ausreichende Zeit und bei einer ausreichenden Temperatur zum Fixieren
des Alumoxans an das Trägermaterial;
- B. Unterziehen des Alumoxan enthaltenden Trägermaterials einer oder mehreren
Waschstufen zur Entfernung von nicht an dem Trägermaterial fixiertem Alumoxan;
wobei
die Bedingungen in der Erhitzungsstufe A und der Waschstufe B so
ausgewählt
sind, daß ein
behandeltes Trägermaterial
gebildet wird, in welchem nicht mehr als 10% des in dem behandelten
Trägermaterial
enthaltenden Aluminiums bei einer Extraktion von 1 Stunde mit Toluol
von 90°C
unter Ver wendung von 10 ml Toluol pro Gramm von Trägerkatalysatorkomponente
extrahierbar ist. Hohe Mengen von an das Trägermaterial gebundenen Alumoxan
werden unter Anwendung erster Erhitzungsstufe A, wahlweise gefolgt
von Waschstufe B, erhalten.
-
Bei diesem Verfahren kann das mit
Alumoxan behandelte Trägermaterial
dadurch erhalten werden, daß in
einem Verdünnungsmittel
ein Alumoxan mit einem Trägermaterial
kombiniert wird, welches von null bis nicht mehr als 20 Gew.-% Wasser,
bevorzugt von null bis nicht mehr als 6 Gew.-% Wasser, bezogen auf
das Gesamtgewicht von Trägermaterial
und Wasser, enthält.
Das Alumoxan wird wünschenswerterweise
in einer aufgelösten
Form eingesetzt.
-
Alternativ kann das mit Alumoxan
behandelte Trägermaterial
dadurch erhalten werden, daß in
einem Verdünnungsmittel
ein Trägermaterial,
welches von 0,5 bis 50 Gew.-% Wasser, bevorzugt von 1 bis 20 Gew.-% Wasser,
bezogen auf das Gesamtgewicht von Trägermaterial und Wasser enthält, mit
einer Verbindung der Formel R''n*AlX''3-n*, worin
R'' unabhängig bei
jedem Vorkommen ein Hydrocarbylrest ist, X'' Halogen
oder Hydrocarbyloxy ist und n* eine ganze Zahl von 1 bis 3 ist,
kombiniert wird. Bevorzugt ist n* = 3. R'' ist
unabhängig bei
jedem Vorkommen bevorzugt ein Alkylrest, vorteilhaft ein Rest mit
einem Gehalt von 1 bis 12 Kohlenstoffatomen. Bevorzugte Alkylreste
sind Methyl, Ethyl, Propyl, Isopropyl, n-Butyl, Isobutyl, tert-Butyl,
Pentyl, Isopentyl, Hexyl, Isohexyl, Heptyl, Octyl und Cyclohexyl.
Hoch bevorzugte Verbindungen der Formel R''n*AlX''3-n* sind
Trimethylaluminium, Triethylaluminium und Tri-isobutylaluminium.
Wenn das Alumoxan in situ durch Umsetzen der Verbindung der Formel
R''n*AlX''3-n* mit Wasser
hergestellt wird, beträgt
das Molverhältnis
von R''n*AlX''3-n* zu Wasser
typischerweise 10 : 1 bis 1 : 1, bevorzugt von 5 : 1 bis 1 : 1.
-
Das Trägermaterial wird zu dem Alumoxan
oder der Verbindung der Formel R''n*AlX''3-n*, bevorzugt aufgelöst in einem
Lösungsmittel,
am meisten bevorzugt einem Kohlenwasserstofflösungsmittel, zugesetzt, oder
die Lösung
von Alumoxan oder die Verbindung der Formel R''n*AlX''3-n*
wird zu dem Trägermaterial
zugesetzt. Das Trägermaterial
kann als solches in trockener Form oder aufgeschlämmt in einem
Kohlenwasserstoffverdünnungsmittel
verwendet werden. Sowohl aliphatische als auch aromatische Kohlenwasserstoffe
können
verwendet werden. Geeignete aliphatische Kohlenwasserstoffe schließen beispielsweise
Pentan, Isopentan, Hexan, Heptan, Octan, Isooctan, Nonan, Isononan,
Decan, Cyclohexan, Methylcyclohexan und Kombinationen von zwei oder
mehr solcher Verdünnungsmittel
ein. Geeignete Beispiele von aromatischen Verdünnungsmitteln sind Benzol,
Toluol, Xylol und andere alkyl- oder halogensubstituierte aromatische
Verbindungen. Am meisten bevorzugt ist das Verdünnungsmittel ein aromatischer
Kohlenwasserstoff, insbesondere Toluol. Geeignete Konzentrationen
von festem Träger
in dem Kohlenwasserstoffmedium reichen von 0,1 bis 15, bevorzugt
von 0,5 bis 10, mehr bevorzugt von 1 bis 7 Gew.-%. Die Kontaktzeit
und die Temperatur sind nicht kritisch. Bevorzugt beträgt die Temperatur
von 0°C
bis 60°C,
mehr bevorzugt von 10°C
bis 40°C.
Die Kontaktzeit beträgt
von 15 Minuten bis 40 Stunden, bevorzugt von 1 bis 20 Stunden.
-
Bevor das mit Alumoxan behandelte
Trägermaterial
der Erhitzungsstufe oder der Waschstufe unterzogen wird, wird das
Verdünnungsmittel
oder Lösungsmittel
bevorzugt zum Erhalt eines freifließenden Pulvers entfernt. Dies
wird typischerweise durch Anwenden einer Arbeitsweise durchgeführt, welche
lediglich die Flüssigkeit
entfernt und die Aluminiumverbindungen auf dem Feststoff zurückläßt, beispielsweise
durch Anwendung von Wärme,
reduziertem Druck, Verdampfen oder einer Kombination hiervon. Gewünschtenfalls
kann die Entfernung von Verdünnungsmittel
zusammen mit der Erhitzungsstufe kombiniert werden, obwohl Sorge
getragen werden sollte, daß das
Verdünnungsmittel
allmählich
entfernt wird.
-
Die Erhitzungsstufe und/oder die
Waschstufe werden in einer solchen Weise durchgeführt, daß ein großer Anteil
(mehr als etwa 90 Gew.-%) des Alumoxans, welches auf dem Trägermaterial
verbleibt, fixiert ist. Bevorzugt wird eine Erhitzungsstufe angewandt,
mehr bevorzugt wird eine Erhitzungsstufe, gefolgt von einer Waschstufe
angewandt. Bei Verwendung in der bevorzugten Kombination wirken
beide Stufen zusammen, so daß in
der Erhitzungsstufe das Alumoxan auf dem Trägermaterial fixiert wird, während in
der Waschstufe das Alumoxan, welches nicht fixiert ist, in einem
wesentlichen Ausmaß entfernt
wird. Die obere Temperatur für
die Hitzebehandlung liegt bevorzugt unterhalb der Temperatur, bei
welcher das Trägermaterial
zu agglomerieren beginnt und Klumpen bildet, welche schwierig wieder
dispergiert werden können,
und unterhalb der Alumoxan-Zersetzungstemperatur. Wenn die Übergangsmetallverbindung
c) vor der Hitzebehandlung zugesetzt wird, sollte die Erhitzungstemperatur
unterhalb der Zersetzungstemperatur der Übergangsmetallverbindung sein.
Bevorzugt wird die Erhitzungsbehandlung bei einer Temperatur von
90°C bis
250°C für eine Zeitspanne von
15 Minuten bis 24 Stunden durchgeführt. Mehr bevorzugt wird die
Erhitzungsbehandlung bei einer Temperatur von 160°C bis 200°C für eine Zeitspanne
von 30 Minuten bis 4 Stunden durchgeführt. Gute Ergebnisse wurden
während
Erhitzen für
8 Stunden auf 100°C
wie auch während
Erhitzen für
2 Stunden auf 175°C
erhalten. Aufgrund von Vorversuchen ist ein Fachmann auf dem Gebiet
in der Lage, die Bedingungen der Hitzebehandlung, welche das gewünschte Ergebnis
liefern, zu definieren. Ebenfalls ist darauf hinzuweisen, daß, je länger die
Erhitzungsbehandlung durchgeführt
wird, um so höher
die Menge von an dem Trägermaterial
fixierten Alumoxan sein wird. Die Hitzebehandlung wird bei reduziertem
Druck oder unter einer Inertatmosphäre, wie Stickstoffgas, oder
unter beidem, jedoch bevorzugt bei reduziertem Druck durchgeführt. In Abhängigkeit von
den Bedingungen der Erhitzungsstufe kann das Alumoxan an dem Trägermaterial
in einem so hohen Grad fixiert werden, daß eine Waschstufe ausgelassen
werden kann.
-
In der Waschstufe ist die Anzahl
der Waschvorgänge
und das Lösungsmittel
derart, daß ausreichende Mengen
von nichtfixiertem Alumoxan entfernt werden. Die Waschbedingungen
sollten derart sein, daß nicht-fixiertes
Alumoxan in dem Waschlösungsmittel
löslich
ist. Das Alumoxan enthaltende Trägermaterial,
das bevorzugt bereits einer Hitzebehandlung unterworfen wurde, wird
bevorzugt einer bis fünf
Waschstufen unter Anwendung eines aromatischen Kohlenwasserstofflösungsmittels
bei einer Temperatur von 0°C
bis 110°C
unterworfen. Mehr bevorzugt beträgt
die Temperatur von 20°C
bis 100°C.
Bevorzugte Beispiele von aromatischen Lösungsmitteln schließen Toluol,
Benzol und Xylole ein. Mehr bevorzugt ist das aromatische Kohlenwasserstofflösungsmittel
Toluol. Am Ende der Waschbehandlung wird das Lösungsmittel mittels einer Arbeitsweise entfernt,
welche ebenfalls das in dem Lösungsmittel
aufgelöste
Alumoxan entfernt, beispielsweise durch Filtration oder Dekantieren.
Bevorzugt wird das Waschlösungsmittel
unter Lieferung eines freifließenden
Pulvers entfernt.
-
Das mir Organometallverbindung behandelte
Trägermaterial
wird dann typischerweise in einem geeigneten Verdünnungsmittel
erneut aufgeschlämmt
und mit der Aktivatorverbindung kombiniert. Die Aktivatorverbindung
wird bevorzugt in einem Verdünnungsmittel
angewandt. Geeignete Verdünnungsmittel
schließen
Kohlenwasserstoffverdünnungsmittel
und Verdünnungsmittel
von halogeniertem Kohlenwasserstoff ein. Ein beliebiger Typ von
Lösungsmittel
oder Verdünnungsmittel,
welcher nicht mit den Katalysatorkomponenten in einer solchen Weise
unter negativer Beeinträchtigung
der katalytischen Eigenschaften reagiert, kann verwendet werden.
Bevorzugte Verdünnungsmittel
sind aromatische Kohlenwasserstoffe wie Toluol, Benzol und Xylole, sowie
aliphatische Kohlenwasserstoffe wie Hexan, Heptan und Cyclohexan.
Bevorzugte halogenierte Kohlenwasserstof fe schließen Methylenchlorid
und Tetrachlorkohlenstoff ein. Die Temperatur ist nicht kritisch,
variiert jedoch im allgemeinen zwischen –20°C und der Zersetzungstemperatur
des Aktivators. Typische Kontaktzeiten variieren von wenigen Minuten
bis zu mehreren Tagen. Rühren
des Reaktionsgemisches ist bevorzugt. Vorteilhafterweise ist die
Aktivatorverbindung aufgelöst,
wobei Wärme
zur Unterstützung
des Auflösens,
falls gewünscht,
angewandt wird. Es kann wünschenswert
sein, das Inkontaktbringen zwischen dem mit Organometall behandelten
Trägermaterial
und der Aktivatorverbindung bei erhöhten Temperaturen durchzuführen. Bevorzugt
betragen solche erhöhten
Temperaturen von 45°C
bis 120°C.
-
Statt zuerst das Trägermaterial
mit der Organometallverbindung, bevorzugt der Aluminiumkomponente,
zu behandeln und anschließend
die Aktivatorverbindung zuzusetzen, kann die Organometallverbindung, bevorzugt
die Aluminiumkomponente, und die Aktivatorverbindung in einem geeigneten
Verdünnungsmittel kombiniert
werden, bevor das Reaktionsgemisch zu dem oder mit dem Trägermaterial
zugesetzt oder kombiniert wird.
-
Ohne an irgendeine Theorie gebunden
zu sein, wird angenommen, daß eine
Organogruppe der Organometallverbindung mit der aktiven Wasserstoffeinheit,
die in dem Aktivatoranion (b-2)
enthalten ist, unter Bildung eines Reaktions- oder Kontaktproduktes
(im folgenden bezeichnet als "Addukt") reagiert. Wenn
beispielsweise die Organometallverbindung Trialkylaluminium, AlR3, ist, und die aktiven Wasserstoff enthaltende
Einheit durch G-OH wiedergegeben wird, wird angenommen, daß das Reaktionsprodukt
G-O-AlR2 umfaßt, wobei weiterhin ein Alkan
als Nebenprodukt RH gebildet wird. Es wird angenommen, daß dieses
Addukt G-O-AlR2, wenn es mit dem Hydroxylgruppen
enthaltenden Trägermaterial,
Si-OH im Fall eines Siliziumdioxidträgermaterials, kombiniert wird,
Si-O-Al(R)-O-G bildet, zusammen mit Alkan RH als Nebenprodukt. Es
wurde gefunden, daß dieses
Verfahren zur Herstellung der Trägerkatalysatorkomponente
sehr glatt abläuft
und Katalysatoren und Katalysa torvorläufer oder Komponenten mit den
gewünschten
Eigenschaften liefert. Typische Verhältnisse, welche bei dieser
Reaktion angewendet werden sollen, sind von 1 : 1 bis 20 : 1 Mol
Organometallverbindung zu Moläquivalenten
von aktiven Wasserstoffeinheiten, die in dem Aktivatoranion (b-2)
enthalten sind.
-
Die Menge von Addukt, gebildet durch
Kombinieren der Organometallverbindung mit der Aktivatorverbindung,
welche mit dem Trägermaterial
kombiniert werden soll, ist nicht kritisch. Bevorzugt ist die Menge
nicht höher
als diejenige, welche an dem Trägermaterial
fixiert werden kann. Typischerweise wird dies durch die Menge von
Hydroxylgruppen des Trägermaterials
bestimmt. Die Menge von anzuwendendem Addukt beträgt bevorzugt
nicht mehr als die Äquivalentmenge
solcher Hydroxylgruppen. Weniger als die Äquivalentmenge wird bevorzugt
angewandt, mehr bevorzugt beträgt
das Verhältnis
zwischen Mol von Addukt zu Mol von oberflächenaktiven Gruppen wie Hydroxylgruppen
zwischen 0,01 und 1, noch mehr bevorzugt zwischen 0,02 und 0,8.
Vor der Zugabe der Übergangsmetallverbindung
wird es bevorzugt, insbesondere wenn weniger als eine Äquivalentmenge
von Addukt, bezogen auf die oberflächenaktiven Gruppen, zugesetzt
wird, eine Zusatzmenge von Organometallverbindung zu dem Reaktionsprodukt
von Trägermaterial
und dem Addukt zuzusetzen, um irgendwelche verbleibenden oberflächenaktiven
Gruppen zu entfernen, welche sonst mit dem Übergangsmetall reagieren könnten und
auf diese Weise höhere
Mengen hiervon zum Erzielen von gleicher katalytischer Aktivität erfordern
würden.
Vor der Kombination hiervon mit der Übergangsmetallverbindung kann
die Trägerkatalysatorkomponente
gewünschtenfalls
gewaschen werden, um irgendeinen Überschuß von Addukt oder Organometallverbindung
zu entfernen.
-
Die Trägerkatalysatorkomponente, welche
das Trägermaterial,
die Organometallverbindung und den Aktivator umfaßt, kann
isoliert werden, um ein freifließendes Pulver zu erhal ten,
indem das Flüssigkeitsmedium
unter Anwendung bevorzugt von Filtrations- oder Verdampfungstechniken
entfernt wird.
-
Obwohl die Übergangsmetallverbindung mit
der Aktivatorverbindung oder das Addukt der Organometallverbindung
und der Aktivatorverbindung vor dem Kombinieren der Aktivatorverbindung
oder ihres Adduktes mit dem Trägermaterial
kombiniert werden kann, ergibt dies reduzierte Katalysatorwirksamkeit.
Bevorzugt wird das Übergangsmetall
zuerst mit dem Trägermaterial,
das mit der Organometallkomponente behandelt wurde, und vor Zugabe
der Aktivatorverbindung kombiniert, oder das Übergangsmetall wird zugesetzt,
nachdem das behandelte Trägermaterial
und der Aktivator kombiniert worden sind, oder nachdem das Aktivatoraddukt
und das Trägermaterial
kombiniert wurden. Mehr bevorzugt wird die Übergangsmetallverbindung (c)
zu dem Reaktionsprodukt des Trägermaterials,
das mit der Organometallverbindung und der Aktivatorverbindung behandelt
wurde, oder nachdem das Aktivatoraddukt und das Trägermaterial
kombiniert worden sind, zugesetzt.
-
Die Übergangsmetallverbindung wird
bevorzugt in einem geeigneten Lösungsmittel
aufgelöst
eingesetzt, wie in einem Kohlenwasserstofflösungsmittel, vorteilhafterweise
einem C5-10 aliphatischen oder cycloaliphatischen
Kohlenwasserstoff oder einem C6-10 aromatischen
Kohlenwasserstoff. Die Kontakttemperatur ist nicht kritisch, vorausgesetzt
sie liegt unterhalb der Zersetzungstemperatur des Übergangsmetalls
und des Aktivators. Gute Ergebnisse werden in einem Temperaturbereich
von 0°C
bis 100°C
erhalten. Alle Stufen in dem vorliegenden Verfahren sollten bei
Abwesenheit von Sauerstoff und Feuchtigkeit durchgeführt werden.
-
Beim Kombinieren der Übergangsmetallverbindung
mit der Trägerkatalysatorkomponente
ist die überstehende
Flüssigkeit
typischerweise farblos, was anzeigt, daß die Übergangsmetallverbindung, deren
Lösung typischerweise
gefärbt
ist, im wesentlichen zusammen mit dem festen Trägerkatalysator verbleibt.
-
Gemäß einer alternativen bevorzugten
Ausführungsform
umfaßt
der feste Katalysator (oder getragene Katalysator):
eine Trägerkatalysatorkomponente,
umfassend ein Trägermaterial
und ein Alumoxan, worin nicht mehr als 10% des in der Trägerkatalysatorkomponente
vorliegenden Aluminiums bei einer Extraktion von 1 Stunde mit Toluol
von 90°C
unter Verwendung von 10 ml Toluol pro Gramm von Trägerkatalysatorkomponente
extrahierbar ist;
und eine Übergangsmetallverbindung.
-
Dieser feste Katalysator gemäß dieser
Ausführungsform
kann bei Abwesenheit der Aktivatorverbindung (b) verwendet werden,
umfassend (b-1) ein Kation, das zur Reaktion mit einer Übergangsmetallverbindung
unter Bildung eines katalytisch aktiven Übergangsmetallkomplexes fähig ist,
und (b-2) ein verträgliches Anion,
das bis zu 100 Nicht-Wasserstoffatome hat und wenigstens einen eine
aktive Wasserstoffeinheit umfassenden Substituenten enthält.
-
Gemäß dieser alternativen Ausführungsform
beträgt
das Molverhältnis
von Aluminiumatomen (aus der Alumoxankomponente) zu Übergangsmetallatomen
in dem Trägerkatalysator
im allgemeinen von 1 bis 5000, bevorzugt von 25 bis 1000 und am
meisten bevorzugt von 50 bis 500.
-
Die Menge von Übergangsmetallverbindung in
dem Trägerkatalysator
der vorliegenden Erfindung ist nicht kritisch, jedoch reicht sie
bevorzugt von 0,1 bis 1000 Mikromol Übergangsmetallverbindung pro
Gramm von Trägermaterial.
Bevorzugt enthält
der Trägerkatalysator
von 1 bis 250 Mikromol Übergangsmetallverbindung
pro Gramm von Trägermaterial.
-
Der Trägerkatalysator gemäß dieser
Ausführungsform
ist durch Erhitzen und/oder Waschen eines Alumoxan enthaltenden
Trägermaterials
unter einer inerten Atmosphäre
für eine
ausreichende Zeitspanne und bei einer ausreichenden Temperatur zum
Fixieren von Alumoxan an dem Trägermaterial,
wie oben angegeben, erhältlich.
-
Es kann vorteilhaft sein, bei dem
vorliegenden Verfahren den festen Katalysator in Verbindung mit Fängern von
Verunreinigungen anzuwenden, welche dazu dienen, den festen Katalysator
vor Katalysatorgift wie Wasser, Sauerstoff und polaren Verbindungen
zu schützen.
Bevorzugte Verbindungen für
diesen Zweck schließen
eine Organoaluminiumverbindung ein, welche durch die folgende Formel
wiedergegeben wird:
RnAlX3-n
worin
R eine C
1-C
20-Hydrocarbylgruppe
ist, X ein Halogenatom oder eine C
1-C
20-Hydrocarbyloxygruppe ist und n eine positive
ganze Zahl, ausgewählt
von 1 bis 3, ist, oder eine Organoaluminiumoxyverbindung, wiedergegeben
durch die folgende Formel:
worin R eine C
1-C
20-Hydrocarbylgruppe ist und n eine positive
ganze Zahl, ausgewählt
aus 5 bis 50, ist.
-
Durch die Behandlung mit der Organoaluminiumverbindung
oder der Organoaluminiumoxyverbindung kann die Beständigkeit
des festen Katalysatorsystems gegenüber Verunreinigungen wie Wasser,
Sauerstoff, welche in dem festen Katalysatorsystem vorliegen, verbessert
werden, und das feste Katalysatorsystem kann für eine verlängerte Zeitdauer aufbewahrt
werden.
-
Bei der oben genannten Behandlung
wird die Organoaluminiumverbindung oder die Organoaluminiumoxyverbindung
bevorzugt in einer Menge von 0,1 bis 100 Mol, ausgedrückt als
Aluminium, mehr bevorzugt in einer Menge von 1 bis 30 Mol pro Mol Übergangsmetallverbindung,
welche in dem festen Katalysatorsystem vorhanden ist, angewandt.
Es ist darauf hinzuweisen, daß die
Organoaluminiumoxyverbindung bevorzugt nicht in einer Menge verwendet
werden sollte, welche die Desorption der Übergangsmetallverbindung von
dem festen Katalysator bewirken könnte.
-
Das bei dem Verfahren der vorliegenden
Erfindung einzusetzende feste Katalysatorsystem kann in Form einer
Aufschlämmung
hiervon in einem inerten Kohlenwasserstofflösungsmittel oder getrocknet
und aufbewahrt in seiner festen Form gelagert werden.
-
Wenn eine Copolymerisationsreaktion
zur Herstellung des Ethylencopolymeren der vorliegenden Erfindung
unter Verwendung des festen Katalysatorsystems durchgeführt wird,
ist es wesentlich, daß die
Copolymerissationsreaktion unter solchen Bedingungen durchgeführt wird,
daß die
Reaktionsgeschwindigkeit durch unabhängige Diffusion von jedem der
Polymerisationsteilnehmer (wie Wasserstoff, Ethylen und wenigstens
einem Comonomeren) in das zu bildende Ethylencopolymere beschränkt wird.
Für diesen
Zweck muß die
Copolymerisationsreaktion unter solchen Bedingungen durchgeführt werden,
daß das
um das feste Katalysatorsystem gebildete Ethylencopolymere nicht
geschmolzen oder in dem Reaktionssystem aufgelöst wird.
-
Zur Realisierung der oben genannten
Bedingungen der Polymerisationsreaktion wird die Copolymerisationsreaktion
mittels Aufschlämmungspolymerisation
durchgeführt.
-
Bei Durchführung von Aufschlämmungspolymerisation
unter den oben genannten Reaktionsbedingungen wird, so lange die
Reaktionsbedingungen geeignet gesteuert werden, das rings um das
feste Katalysatorsystem zu bildende Ethylencopolymere nicht geschmolzen
oder während
der Polymerisationsreaktion aufgelöst, sondern verbleibt in pulverförmiger Form
(wobei diese pulverförmige
Form durch Verwendung des oben genannten spezifischen Katalysatorsystems
erreicht wird) während
der Reaktion, so daß eine
der oben genannten Erfordernisse zur Ermöglichung des Fortschreitens
der Polymerisationsreaktion in einer durch Diffusion beschränkten Geschwindigkeit
befriedigt werden kann, so daß das
erzeugte Copolymere nicht in dem Reaktionsgemisch geschmolzen wird,
sondern im festen Zustand verbleibt.
-
Wenn eine Copolymerisationsreaktion
durch Aufschlämmungspolymerisation
durchgeführt
wird, beträgt
ein Polymerisationsdruck im allgemeinen von 1,03 bis 102,9 kg/cm2 (1 bis 100 atm), bevorzugt von 3,09 bis
308,8 kg/cm2 (3 bis 30 atm) und eine Polymerisationstemperatur
beträgt
im allgemeinen von 20 bis 115°C, bevorzugt
von 50 bis 105°C.
Jedoch ist die obere Grenze der Polymerisationstemperatur eine Temperatur,
welche die höchste
unter den Temperaturen ist, bei welchen das erzeugte Ethylencopolymere
im wesentlichen in einem pulverförmigen
Zustand verbleiben kann. Eine solche höchste Temperatur variiert mit
der Dichte des erzeugten Ethylencopolymeren und dem Typ des verwendeten
Lösungsmittels.
-
Als ein für die Aufschlämmungspolymerisation
zu verwendendes Lösungsmittel
können
in geeigneter Weise inerte Lösungsmittel,
welche oben in Verbindung mit der Herstellung des festen Katalysatorsystems
erwähnt
werden, verwendet werden. Insbesondere sind Isobutan, Isopentan,
Heptan, Hexan und Octan bevorzugt.
-
Wie oben angegeben, ist es bei der
vorliegenden Erfindung wichtig, daß das erzeugte Ethylencopolymere
in einem pulverförmigen
Zustand während
der Polymerisationsreaktion verbleibt. Daher ist der obere Grenzwert
der Polymerisationstemperatur extrem wichtig.
-
Bei dem Verfahren der vorliegenden
Erfindung wird Ethylen, wie oben erwähnt, mit wenigstens einem Comonomeren
copolymerisiert. Typische Reaktoren für die Polymerisation können Aufschlämmungs-Schleifenreaktoren
oder Autoklaven einschließen.
-
Wie oben angegeben, wird wenigstens
ein bei dem Verfahren der vorliegenden Erfindung zu verwendendes
Comonomeres aus der Gruppe ausgewählt, die eine durch die Formel
H2C = CHR wiedergegebene Verbindung umfaßt, worin
R eine C1-C20 lineare,
verzweigte oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe und ein C4-C20 lineares, verzweigtes oder cyclisches
Dien ist. Illustrative Beispiele der durch die For mel H2C
= CHR wiedergegebenen Verbindungen schließen ein: Propylen, 1-Buten,
1-Penten, 1-Hexen, 4-Methyl-1-penten, 1-Octen, 1-Decen, 1-Dodecen, 1-Tetradecen,
1-Hexadecen, 1-Octadecen,
1-Eicosen, Vinylcyclohexen und Styrol. Illustrative Beispiele von
C4-C20 linearen,
verzweigten und cyclischen Dienen schließen ein: 1,3-Butadien, 1,4-Pentadien,
1,5-Hexadien, 1,4-Hexadien und Cyclohexadien. Von diesen sind Propylen, 1-Buten,
1-Penten, 1-Hexen, 4-Methyl-1-penten, 1-Octen, 1-Decen, 1-Dodecen,
1-Tetradecen, 1-Hexadecen, 1-Octadecen und 1-Eicosen besonders bevorzugt.
-
Bei der Herstellung des Ethylencopolymeren
kann das Molekulargewicht des erzeugten Ethylencopolymeren durch
Veränderung
des Gehaltes von Wasserstoff in dem Reaktionssystem oder durch Veränderung der
Polymerisationstemperatur gesteuert werden, wie in der
DE 3127133.2 beschrieben.
-
In der vorliegenden Erfindung kann
das feste Katalysatorsystem zusätzlich
zu den oben genannten Komponenten verschiedene Zusatzstoffe enthalten,
welche für
die Ethylencopolymerisation bekannterweise nützlich sind.
-
Die vorliegende Erfindung bezieht
sich ebenfalls auf Mischungszusammensetzungen, umfassend dieses
neue Ethylencopolymere und:
- a) ein zweites
Ethylencopolymeres der vorliegenden Erfindung von unterschiedlichem
Molekulargewicht oder unterschiedlicher Dichte; oder
- b) ein homogenes Ethylen/α-Olefininterpolymeres
mit schmaler Zusammensetzungsverteilung;
- c) ein heterogenes Ethylen/α-Olefininterpolymeres
mit breiter Verteilung; oder
- d) ein Homopolymeres (hergestellt mittels einer Katalysatorkomponente,
die von derjenigen zur Herstellung des Ethylencopolymeren der vorliegenden
Erfindung verwendeten verschieden ist); oder
- e) eine Kombination von beliebigen von zwei oder mehr von a),
b), c) oder d).
-
Bei Verwendung als erste Komponente
(Komponente I) in allen zuvor genannten Mischungszusammensetzungen
der vorliegenden Erfindung mit einer zweiten Komponente (Komponente
II) hat das Ethylencopolymere die folgenden Eigenschaften:
-
Die Menge des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten ersten Ethylencopolymeren
beträgt
von 1 bis 99, bevorzugt von 10 bis 90, mehr bevorzugt von 25 bis
75 und am meisten bevorzugt von 35 bis 65%, in Gewicht bezogen auf
die kombinierten Gewichte von Komponenten I und II.
-
Die Dichte des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten ersten Ethylencopolymeren
beträgt
im allgemeinen von allgemein von 0,870 bis 0,980, bevorzugt von
0,890 bis 0,965, mehr bevorzugt von 0,915 bis 0,955 g/cm3.
-
Der Schmelzindex (I2)
für das
in die Mischungszusammensetzung der vorliegenden Erfindung eingebaute
erste Ethylencopolymere beträgt
im allgemeinen von 0,0001 bis 10000, bevorzugt von 0,001 bis 5000, mehr
bevorzugt von 0,01 bis 3000 g/10 min.
-
Das Verhältnis I21,6/I2 des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten ersten Ethylencopolymeren beträgt von 15
bis 65, bevorzugt von 18 bis 55, mehr bevorzugt von 20 bis 50, oder das
Verhältnis
I10/I2 beträgt von 5
bis 30, bevorzugt von 5 bis 28, mehr bevorzugt von 5,5 bis 25.
-
Das Verhältnis Mw/Mn des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten ersten Ethylencopolymeren beträgt von 2,5
bis 10, bevorzugt von 2,8 bis 8 und mehr bevorzugt von 3 bis 7.
-
Mischungszusammensetzungen, welche
das Ethylencopolymere mit einem zweiten Ethylencopolymeren der vorliegenden
Erfindung von unterschiedlichem Molekulargewicht oder unterschied licher
Dichte umfassen, sind eine andere Ausführungsform der vorliegenden
Erfindung. So lange das Verfahren der vorliegenden Erfindung im
wesentlichen auf die Herstellung eines Ethylencopolymeren angewandt
wird, kann ein beliebiges Verfahren, bei welchem ein oder mehrere
unterschiedliche Ethylencopolymere dieser Erfindung mit jeweils
ausgezeichneten ESCR-Eigenschaften und unterschiedlichen Comonomerengehalten
getrennt unter Anwendung dieses Verfahrens hergestellt und mittels
eines Kneters (im folgenden häufig
bezeichnet als "Mischungs- und Knetverfahren") gemischt werden,
und ein Verfahren, bei welchem ein Ethylencopolymeres, das eine
Mischung von zwei oder mehr unterschiedlichen Ethylencopolymerkomponenten
mit unterschiedlichen Comonomerengehalten mittels einer Mehrstufenpolymerisation
oder unter Verwendung einer Vielzahl von unterschiedlichen Typen
von Katalysatoren, welche in der vorliegenden Erfindung einzusetzen
sind, hergestellt wird, in sehr vorteilhafter Weise angewandt werden.
Weiterhin kann ein Ethylencopolymeres, bestehend aus einer Mischung
von zwei oder mehr unterschiedlichen Ethylencopolymerkomponenten
mit unterschiedlichen Comonomerengehalten, unter Anwendung einer
Vielzahl von unterschiedlichen Typen von Katalysatoren, welche in
der vorliegenden Erfindung einzusetzen sind, hergestellt werden,
wobei nicht nur die Schlagfestigkeit und die ESCR-Eigenschaften
weiter verbessert werden können,
sondern ebenfalls ein merklich verbessertes Gleichgewicht von verschiedenen
Eigenschaften, wie Schlagfestigkeit, Steifigkeit, Schmelzfließeigenschaften, erreicht
werden kann.
-
Die Menge des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten zweiten Ethylencopolymeren
beträgt
von 1 bis 99, bevorzugt von 10 bis 90, mehr bevorzugt von 25 bis
75 und am meisten bevorzugt von 35 bis 65%, in Gewicht bezogen auf
die kombinierten Gewichte der Komponenten I und II.
-
Die Dichte des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten zweiten Ethylencopolymeren beträgt im allgemeinen
von allgemein von 0,915 bis 0,985, bevorzugt von 0,935 bis 0,983, mehr
bevorzugt von 0,955 bis 0,980 und am meisten bevorzugt von 0,960
bis 0,978 g/cm3.
-
Der Schmelzindex (I2)
für das
in die Mischungszusammensetzung der vorliegenden Erfindung eingebaute
zweite Ethylencopolymere beträgt
im allgemeinen von 0,0001 bis 10000, bevorzugt von 0,001 bis 5000, mehr
bevorzugt von 0,01 bis 3000, am meisten bevorzugt von 10 bis 1000
g/10 min.
-
Das Verhältnis I10/I2 des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten zweiten Ethylencopolymeren beträgt von 5
bis 30, bevorzugt von 5,3 bis 28, mehr bevorzugt ist es von 5,5
bis 25, oder das Verhältnis
I21,6/I2 des in
die Mischungszusammensetzung der vorliegenden Erfindung eingebauten zweiten
Ethylencopolymeren beträgt
von 15 bis 55, bevorzugt von 18 bis 55, mehr bevorzugt ist es von
20 bis 50, am meisten bevorzugt von 22 bis 35.
-
Das Verhältnis Mw/Mn des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten zweiten Ethylencopolymeren beträgt von 2,5
bis 10, bevorzugt von 2,8 bis 8 und am meisten bevorzugt von 3 bis
7.
-
Mischungszusammensetzungen, welche
das Ethylencopolymere mit homogenen Interpolymeren mit schmaler
Zusammensetzung, am meisten bevorzugt die im wesentlichen linearen
Ethylen/α-Olefininterpolymere,
umfassen, sind eine andere Ausführungsform
der vorliegenden Erfindung. Die homogenen Interpolymerkomponenten
der Mischungszusammensetzungen werden hier so definiert, wie sie
in der USP 3 645 992 (Elston) definiert sind, wobei auf diese Druckschrift
hiermit Bezug genommen wird. Daher sind homogene Interpolymere solche,
in denen das Comonomere statistisch innerhalb eines vorgegebenen
Interpolymermoleküls
verteilt ist, und worin im wesentlichen alle der Interpolymermoleküle dasselbe
Verhältnis
Ethylen/Comonomeres innerhalb dieses Interpolymeren haben. Solche
Interpolymere sind von den typischen gemäß Ziegler katalysierten Interpolymeren
verschieden, wobei dies bekanntermaßen heterogene In terpolymere
sind, und solche sind, in denen die Interpolymermoleküle nicht
dasselbe Verhältnis
Ethylen/Comonomeres haben. Die homogenen Polymere sind ebenfalls
von LDPE, das mittels freiradikalisch katalysierter Hochdruck-Ethylenpolymerisation
hergestellt wurde, verschieden, wobei dies hoch verzweigtes Polyethylen
ergibt, das, wie dem Fachmann auf dem Gebiet bekannt, zahlreiche
Langkettenverzweigungen aufweist.
-
Der Ausdruck "schmale Zusammensetzungsverteilung", wie er hier verwendet
wird, beschreibt die Comonomerenverteilung für homogene Interpolymere und
bedeutet, daß die
homogenen Interpolymere nur einen einzelnen Schmelzpeak, gemessen
durch Differentialabtastkalorimetrie (DSC) und im wesentlichen das
Fehlen einer meßbaren "linearen" Polymerfraktion
haben.
-
Die homogenen Interpolymere mit schmaler
Zusammensetzungsverteilung können
ebenfalls durch ihr SCBDI (Kurzkettenverzweigungs-Verteilungsindex)
oder CDBI (Zusammensetzungsverteilungs-Verzweigungsindex) charakterisiert
werden, wobei dies als der Gewichtsprozentsatz der Polymermoleküle definiert
ist, die einen Comonomerengehalt innerhalb 50% des mittleren gesamtmolaren
Comonomerengehaltes haben. Der CDBI eines Polymeren ist leicht aus
den Werten berechenbar, die aus auf dem Fachgebiet bekannten Techniken
erhalten wurden, wie beispielsweise der Elutionsfraktionierung bei
ansteigender Temperatur (abgekürzt
hier als "TREF"), wie beispielsweise
beschrieben in Wild et al., Journal of Polymer Science, Poly. Phys. Ed.,
Vol. 20, 5.441 (1982), im US-Patent 4 798 081 (Hazlitt et al.),
oder wie dies in der USP 5 008 204 (Stehling) beschrieben ist, wobei
die Angaben hiervon unter Bezugnahme aufgenommen sind. Die Technik
zur Berechnung von CDBI ist in der USP 5 322 728 (Davey et al.)
und der USP 5 246 783 (Spenadel et al.) oder im US-Patent 5 089
321 (Chum et al.) beschrieben, wobei sämtliche Angaben hiervon unter
Bezugnahme aufgenommen werden. Der SCBDI oder der CDBI für Ethylen/α-Olefininterpolymere
mit schmaler Zusammensetzung, die in der vorliegenden Erfindung
verwendet werden, beträgt im
allgemeinen größer als
50%, insbesondere größer als
70%, am meisten bevorzugt größer als
90%.
-
Den Mischungskomponenten in Form
von homogenen Interpolymeren mit schmaler Zusammensetzungsverteilung
dieser Erfindung fehlt im wesentlichen eine meßbare Fraktion mit "hoher Dichte" (oder Homopolymeres),
gemessen mittels der TREF-Technik. Die homogenen Interpolymere und
Polymere haben einen Verzweigungsgrad von weniger als oder gleich
2 Methylgruppen/ 1000 Kohlenstoffe in 15 Gew.-% oder weniger, bevorzugt
weniger als 10 Gew.-% und insbesondere weniger als 5 Gew.-%.
-
Bevorzugte Komponenten der Mischungen
der vorliegenden Erfindung sind die im wesentlichen linearen Ethylen/α-Olefininterpolymere.
Die im wesentlichen linearen Ethylen/α-Olefininterpolymere sind hierin
definiert wie in den US-Patenten Nr. 5 272 236 und 5 278 272 von
Lai et al. (
US 5 272 236 und
5 278 272 ), deren hierin
enthaltene Lehren in ihrer Gesamtheit unter Bezugnahme aufgenommen
sind.
-
Die im wesentlichen linearen Ethylen/α-Olefininterpolymere
sind ebenfalls homogene Interpolymere, da das Comonomere statistisch
innerhalb eines vorgegebenen Interpolymermoleküls verteilt ist und im wesentlichen
alle Interpolymermoleküle
dasselbe Verhältnis
Ethylen/Comonomeres innerhalb dieses Interpolymeren haben.
-
Jedoch bedeutet der Ausdruck "im wesentlichen lineares" Ethylen/α-Olefininterpolymeres,
daß das Polymere
ebenfalls Langkettenverzweigung enthält. Als Ergebnis der gespannten
Geometrie des Katalysators können
im wesentlichen lineare Ethyleninterpolymere Langkettenverzweigung
enthalten, welche, wie in LDPE, in starkem Maße die Verarbeitbarkeit (gemessen
durch den Verarbeitungsindex (PI) oder dem Beginn von Schmelzbruch
oder die Scherungsverdünnungsfähigkeit)
im Vergleich zu anderen Polymeren mit etwa demselben I2 und
Mw/Mn verbessern
können.
Langkettenverzweigung ist hier als eine Kettenlänge von wenigstens einem Kohlenstoffatom
mehr als zwei Kohlenstoffatome weniger als die Gesamtanzahl von
Kohlenstoffen in dem Comonomeren definiert, beispielsweise ist die
Langkettenverzweigung eines im wesentlichen linearen Ethyleninterpolymeren
aus Ethylen/Octen wenigstens sieben (7) Kohlenstoffatome in der
Länge (d.
h. 8 Kohlenstoffatome weniger 2 ergibt 6 Kohlenstoffatome plus eins
ergibt sieben Kohlenstoffatome als Länge der Langkettenverzweigung).
Die Langkettenverzweigung kann so lang wie etwa dieselbe Länge wie
die Länge des
Polymerrückgrates
sein. Langkettenverzweigung wird unter Anwendung von 13C
kernmagnetischer Resonanz(NMR)-Spektroskopie
bestimmt, und sie wird unter Anwendung der Methode von Randall (Rev.
Macromol. Chem. Phys., C29 (2&3),
S. 285–297)
quantifiziert, wobei die Angaben hiervon unter Bezugnahme aufgenommen
sind. Langkettenverzweigung ist selbstverständlich von Kurzkettenverzweigungen,
welche lediglich von dem Einbau des Comonomeren resultieren, zu
unterschieden, beispielsweise die Kurzkettenverzweigung eines im
wesentlichen linearen Polymeren von Ethylen/Octen beträgt sechs
Kohlenstoffatome in der Länge,
während
die Langkettenverzweigung für
dasselbe Polymere wenigstens sieben Kohlenstoffatome in der Länge ist.
-
Mehr spezifisch ist das Polymerrückgrat eines
im wesentlichen linearen Ethylen/α-Olefininterpolymeren
mit 0,01 Langkettenverzweigungen/1000 Kohlenstoffatome bis 3 Langkettenverzweigungen/1000
Kohlenstoffatome, mehr bevorzugt mit von 0,01 Langkettenverzweigungen/1000
Kohlenstoffatome bis 1 Langkettenverzweigungen/1000 Kohlenstoffatome
und speziell von 0,05 Langkettenverzweigungen/1000 Kohlenstoffatome
bis 1 Langkettenverzweigungen/1000 Kohlenstoffatome substituiert.
-
Die in dieser Erfindung nützlichen,
im wesentlichen linearen Ethylen/α-Olefininterpolymere
haben ausgezeichnete Verarbeitbarkeit, obwohl sie relativ schmale
Molekulargewichtsverteilungen aufweisen. Die im wesentlichen linearen Ethylen/α-Olefininterpolymere
haben eine Molekulargewichtsverteilung, Mw/Mn, definiert durch die Gleichung: Mw/Mn ≤ (I10/I2) – 4,63.
-
Noch mehr überraschend kann das Schmelzfließverhältnis (I10/I2) der im wesentlichen
linearen Ethylen/α-Olefininterpolymere
im wesentlichen unabhängig
von dem Polydispersitätsindex
(d. h. der Molekulargewichtsverteilung (Mw/Mn)) variiert werden. Dies steht im Gegensatz
zu konventionellen heterogen verzweigten linearen Polyethylenharzen,
welche solche rheologischen Eigenschaften besitzen, daß bei Anstieg
des Polydispersitätsindex
der Wert I10/I2 ebenfalls
ansteigt.
-
Für
die in den Zusammensetzungen der Erfindung verwendeten im wesentlichen
linearen Ethylen/α-Olefinpolymere
zeigt das Verhältnis
I10/I2 das Ausmaß der Langkettenverzweigung,
d. h. je höher
das Verhältnis
I10/I2 ist, um so
mehr Langkettenverzweigung liegt in dem Polymeren vor.
-
Der "rheologische Verarbeitungsindex" (PI) ist die scheinbare
Viskosität
(in kPoise) eines Polymeren, gemessen mittels eines Gasextrusionsrheometers
(GER). Das Gasextrusionsrheometer ist von M. Shida, R. N. Shroff
und L. V. Cancio in Polymer Engineering Science, Vol. 17, Nr. 11,
5.770 (1977) und in "Rheometers for
Molten Plastics" von
John Dealy, veröffentlicht
durch Van Nostrand Reinhold Co. (1982) auf Seite 97–99 beschrieben,
wobei beide Veröffentlichungen
in ihrer Gesamtheit hier unter Bezugnahme aufgenommen sind. Alle
GER-Experimente
werden bei einer Temperatur von 190°C, einem Stickstoffdruck zwischen
385 bis 36,7 kg/cm2 (5250 bis 500 psig) Überdruck
unter Verwendung einer Düse
von 0,075 cm (0,0296 Zoll) Durchmesser mit L/D = 20 : 1 mit einem
Eintrittswinkel von 180° durchgeführt. Für die hier
beschriebenen im wesentlichen linearen Ethylen/α-Olefinpolymere ist der PI die
scheinbare Viskosität
in Pa·sec
eines Materials, gemessen mittels GER bei einer scheinbaren Scherspannung
von 21,5 N/cm2 (2,15 × 106 dyn/cm2). Die hier beschriebenen im wesent lichen
linearen Ethylen/α-Olefininterpolymere
haben bevorzugt ein PI im Bereich von 10 Pa·sec (0,01 kPoise) bis 50.000
Pa·sec
(50 kPoise), bevorzugt von 15.000 Pa·sec (15 kPoise) oder weniger.
Die hier beschriebenen im wesentlichen linearen Ethylen/α-Olefinpolymere
haben einen PI weniger als oder gleich 70% des PI eines vergleichbaren
linearen Ethylen/α-Olefinpolymeren,
welches keine Langkettenverzweigung enthält, jedoch etwa denselben I2 und Mw/Mn hat.
-
Eine Auftragung der scheinbaren Scherspannung
gegen scheinbare Schergeschwindigkeit wird zur Identifizierung der
Schmelzbruchphänomene
benutzt. Entsprechend Ramamurthy in Journal of Rheology, 30(2),
337–357,
1986, kann oberhalb einer kritischen Fließgeschwindigkeit die beobachteten
Unregelmäßigkeiten
des Extrudates breit in zwei Haupttypen klassifiziert werden: Oberflächenschmelzbruch
und grober Schmelzbruch.
-
Oberflächenschmelzbruch tritt unter
anscheinend gleichmäßigen Fließbedingungen
und -bereichen im Detail von dem Verlust von Spiegelglanz bis zu
den schwereren Formen von "Haifischhaut" auf. In der vorliegenden
Beschreibung ist der Beginn von Oberflächenschmelzbruch (OSMF) als
der Beginn des Verlustes des Glanzes des Extrudates, bei welchem
die Oberflächenrauhigkeit
des Extrudates nur bei einer Vergrößerung von 40X festgestellt
werden kann, charakterisiert. Die kritische Scherrate beim Beginn
von Oberflächenschmelzbruch
für die
im wesentlichen linearen Ethylen/α-Olefininterpolymere
ist wenigstens 50% größer als die
kritische Scherrate beim Beginn von Oberflächenschmelzbruch eines linearen
Ethylen/α-Olefinpolymeren, welches
keine Langkettenverzweigung enthält,
jedoch etwa dieselben I2 und Mw/Mn besitzt, worin "etwa dieselben", wie hier verwendet, bedeutet, daß jeder
Wert innerhalb von 10% des Vergleichswertes des linearen Vergleichs-Ethylenpolymeren
liegt.
-
Grober Schmelzbruch tritt bei nicht
gleichförmigen
Fließbedingungen
auf und reicht im Detail von regelmäßigen (alternierende rauhe
und glatte, spiralförmige,
etc.) bis zu statistischen Verziehungen. Für die kommerzielle Annehmbarkeit
(beispielsweise bei Produkten aus geblasenen Folien) sollten Oberflächendefekte
minimal oder sogar fehlend sein. Die kritische Scherrate beim Beginn
von Oberflächenschmelzbruch (OSMF)
und bei Beginn von grobem Schmelzbruch (OGMF) wird hier basierend
auf den Veränderungen
der Oberflächenrauhigkeit
und der Konfigurationen der mittels GER extrudierten Extrudate benutzt.
-
Die homogene Interpolymerkomponente
der Mischung ist bevorzugt ein Interpolymeres von Ethylen mit wenigstens
einem Comonomeren, ausgewählt
aus der Gruppe, welche Verbindungen umfaßt, die durch die Formel H2C = CHR wiedergegeben werden, worin R eine
C1-C18 lineare,
verzweigte oder cyclische Alkylgruppe oder eine C6-C20 Arylgruppe ist, und einem C4-C20 linearen, verzweigten oder cyclischen
Dien. Illustrative Beispiele der durch die Formel H2C
= CHR wiedergegebenen Verbindungen schließen ein: Propylen, 1-Buten,
1-Penten, 1-Hexen, 4-Methyl-1-penten,
1-Octen, 1-Decen, 1-Dodecen, 1-Tetradecen, 1-Hexadecen, 1-Octadecen,
1-Eicosen, Vinylcyclohexen und Styrol.Illustrative Beispiele von
C4-C20 linearen,
verzweigten und cyclischen Dienen schließen ein: 1,3-Butadien, 1,4-Pentadien,
1,5-Hexadien, 1,4-Hexadien und Cyclohexadien. von diesen sind Propylen,
1-Buten, 1-Penten, 1-Hexen, 4-Methyl-1-penten, 1-Octen, 1-Decen, 1-Dodecen,
1-Tetradecen, 1-Hexadecen, 1-Octadecen und 1-Eicosen besonders bevorzugt.
-
Die Menge des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten homogenen Ethylen/α-Olefininterpolymere
mit schmaler Zusammensetzungsverteilung beträgt von 1 bis 99, bevorzugt von
10 bis 90, mehr bevorzugt von 25 bis 75 und am meisten bevorzugt
von 35 bis 65%, in Gewicht basierend auf den kombinierten Gewichten
der Komponenten I und II.
-
Die Dichte des in die Mischungszusammensetzung
der vorliegenden Erfindung eingebauten homogenen Ethylen/α-Olefininterpolymeren
mit schmaler Zusammensetzungsverteilung beträgt im allgemeinen von allgemein
von 0,915 bis 0,985, bevorzugt von 0,935 bis 0,983, mehr bevorzugt
von 0,955 bis 0,980 g/cm3.
-
Der Schmelzindex (I2)
des in die Mischungszusammensetzung der vorliegenden Erfindung eingebauten
homogenen Ethylen/α-Olefininterpolymeren
mit schmaler Zusammensetzungsverteilung beträgt im allgemeinen von 0,0001
bis 10000, bevorzugt von 0,001 bis 5000, mehr bevorzugt von 0,01
bis 3000 g/10 min.
-
Das Verhältnis I10/I2 des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten homogenen Ethylen/α-Olefininterpolymeren
mit schmaler Zusammensetzungsverteilung beträgt von 5 bis 25, bevorzugt
von 5,3 bis 25, mehr bevorzugt ist es von 5,5 bis 20, oder das Verhältnis I21,6/I2 des in die
Mischungszusammensetzung der vorliegenden Erfindung eingebauten
homogenen Ethylen/α-Olefininterpolymeren
mit schmaler Zusammensetzungsverteilung beträgt von 10 bis 50, bevorzugt
von 12 bis 45, mehr bevorzugt von 15 bis 40.
-
Das Verhältnis Mw/Mn des in die Mischungszusammensetzung der
vorliegenden Erfindung eingebauten homogenen Ethylen/α-Olefininterpolymeren
mit schmaler Zusammensetzungsverteilung (einschließlich des
im wesentlichen linearen Ethylen/α-Olefininterpolymeren)
beträgt
weniger als 3.
-
Die homogene Ethylen/α-Olefininterpolymerkomponente
mit schmaler Zusammensetzungsverteilung kann unter Anwendung der
zuvor beschriebenen Übergangsmetallkatalysatoren
hergestellt werden. Die Herstellung der homogenen, im wesentlichen
linearen Ethylen/α-Olefinpolymere
mit schmaler Zusammensetzungsverteilung erfordert die Verwendung
der zuvor beschriebenen Übergangsmetallverbindungen
und von Einzelplatzkatalysatoren mit gespannter Geometrie. Die aktivierenden
Cokatalysatoren und die Aktivierungstechniken wurden zuvor mit Bezug
auf verschiedene Metallkomplexe in den folgenden Druckschriften
gelehrt: Europäisches
Patent EP-A-277 003, US-A-5 153 157, US-A-5 064 802, Europäische Patente EP-A-468 651 und
EP-A-520 732 (äquivalent
zu U.S. Serial No. 07/876 268, eingereicht am 1. Mai 1992) und US-A-5
350 723, deren Lehren hiermit unter Bezugnahme aufgenommen werden.
-
Geeignete aktivierende Cokatalysatoren,
welche in Kombination mit der Einzelplatzkatalysatorkomponente brauchbar
sind, sind solche Verbindungen, die zur Abgabe eines Substituenten
X hieraus unter Bildung eines inerten, nichtstörenden Gegenions fähig sind,
oder welche ein Zwitterionderivat der Katalysatorkomponente bilden.
Geeignete aktivierende Cokatalysatoren zur Verwendung hierfür schließen ein:
perfluorierte Tri(aryl)borverbindungen und am meisten bevorzugt
Tris(pentafluorphenyl)boran, nicht-polymere, verträgliche, nicht-koordinierende
ionenbildende Verbindungen (einschließlich der Verwendung solcher
Verbindungen unter oxidierenden Bedingungen), insbesondere die Verwendung
von Ammonium-, Phosphonium-, Oxonium-, Carbonium-, Silylium- oder
Sulfoniumsalzen von verträglichen,
nicht-koordinierenden Anionen und Ferroceniumsalze von verträglichen,
nicht-koordinierenden Anionen. Geeignete Aktivierungstechniken schließen die
Anwendung von Massenelektrolyse ein. Eine Kombination der zuvor
genannten aktivierenden Cokatalysatoren und Aktivierungstechniken
kann ebenfalls angewandt werden.
-
Mehr speziell umfassen geeignete
ionenbildende Verbindungen, welche als Cokatalysatoren nützlich sind,
ein Kation, das eine Bronstedsäure
ist, die zur Abgabe eines Protons fähig ist, und ein verträgliches, nicht-koordinierendes
Anion, A". Wie hier
verwendet, bedeutet der Ausdruck "nicht-koordinierend" ein Anion oder eine Substanz, welche
entweder nicht zu dem das Metall der Gruppe 4 enthaltenden Vorläuferkomplex und
des hieraus abstammenden katalytischen Derivates koordi niert, oder
welcher nur schwach koordiniert ist, so daß solche Komplexe hierdurch
ausreichend labil bleiben, um durch eine neutrale Lewisbase verschoben zu
werden. "Verträgliche Anionen" sind Anionen, welche
nicht zur Neutralität
abgebaut werden, wenn der anfänglich
gebildete Komplex sich zersetzt, und welche bei der gewünschten
nachfolgenden Polymerisation oder anderen Anwendungen des Komplexes
nicht stören.
-
Bevorzugte Anionen sind solche, welche
einen einzelnen Koordinationskomplex enthalten, umfassend einen
ladungstragenden Metall- oder Metalloidkern, dessen Anion zum Ausgleich
der Ladung der aktiven Katalysatorspezies (dem Metallkation) fähig ist,
welches gebildet werden kann, wenn die zwei Komponenten zusammengegeben
werden. Weiterhin sollte dieses Anion ausreichend labil sein, um
durch olefinische, diolefinische und acetylenartig ungesättigte Verbindungen
oder andere neutrale Lewisbasen wie Ether oder Nitrile verdrängt zu werden.
Geeignete Metalle schließen
ein, sind jedoch nicht beschränkt
auf: Aluminium, Gold und Platin. Geeignete Metalloide schließen ein,
sind jedoch nicht beschränkt
auf: Bor, Phosphor und Silizium. Verbindungen, welche Anionen enthalten,
die ein einzelnes Metall- oder Metalloidatom enthaltende Koordinationskomplexe
umfassen, sind selbstverständlich
wohlbekannt, und zahlreiche dieser Verbindungen, insbesondere solche
Verbindungen, welche ein einzelnes Boratom in dem Anionenanteil
enthalten, sind kommerziell erhältlich.
-
Bevorzugt können solche Cokatalysatoren
durch die folgende allgemeine Formel wiedergegeben werden: (L*-H)+
d(A)d–
worin:
L*
eine neutrale Lewisbase ist;
(L*-H)+ eine
Bronstedsäure
ist;
Ad– ein
nicht-koordinierendes, verträgliches
Anion ist, das eine Ladung von d– hat, und
d eine ganze
Zahl von 1 bis 3 ist.
-
Mehr bevorzugt entspricht Ad– der
Formel : [M'Q4]–, worin:
M' = Bor oder Aluminium
im formalen Oxidationszustand 3 ist; und
Q unabhängig von
jedem Vorkommen ausgewählt
ist aus: Hydrid-, Dialkylamido-, Halogenid-, Hydrocarbyl-, Hydrocarbyloxid-,
halogensubstituierten Hydrocarbyl-, halogensubstituierten Hydrocarbyloxy-
und halogensubstituierten Silylhydrocarbylresten (einschließlich perhalogenierten
Hydrocarbyl-, perhalogenierten Hydrocarbyloxy- und perhalogenierten
Silylhydrocarbylresten), wobei Q bis zu 20 Kohlenstoffatome hat,
mit der Maßgabe,
daß in
nicht mehr als bei einem Vorkommen Q = Halogenid ist. Beispiele
von geeigneten Hydrocarbyloxidgruppen Q sind im US-Patent 5 296
433 angegeben, dessen Lehren hier unter Bezugnahme aufgenommen sind.
-
Bei einem mehr bevorzugten Beispiel
ist d = 1, d. h. das Gegenion hat eine negative Einzelladung und ist
A–.
Bor umfassende aktivierende Cokatalysatoren, welche bei der Herstellung
der Katalysatoren dieser Erfindung besonders nützlich sind, können durch
die folgende allgemeine Formel wiedergegeben werden: (L*-H)+(BQ4)–, worin:
L*
wie zuvor definiert ist;
B = Bor in einem formalen Oxidationszustand
von 3 ist; und
Q eine Hydrocarbyl-, Hydrocarbyloxy-, fluorierte
Hydrocarbyl-, fluorierte Hydrocarbyloxy- oder fluorierte Silylhydrocarbylgruppe
mit bis zu 20 Nicht-Wasserstoffatomen ist, mit der Maßgabe, daß bei nicht
mehr als einem Fall Q = Hydrocarbyl ist.
-
Am meisten bevorzugt ist Q bei jedem
Vorkommen eine fluorierte Arylgruppe, insbesondere eine Pentafluorphenylgruppe.
-
Erläuternde, jedoch nicht beschränkende Beispiele
von Borverbindungen, welche als ein aktivierender Cokatalysator für die vorliegende
Erfindung verwendet werden können,
sind tri-substituierte Ammoniumsalze wie:
Trimethylammoniumtetrakis(pentafluorphenyl)borat,
Triethylammoniumtetrakis(pentafluorphenyl)borat,
Tripropylammoniumtetrakis(pentafluorphenyl)borat,
Tri(n-butyl)ammoniumtetrakis(pentafluorphenyl)borat,
Tri(sec-butyl)ammoniumtetrakis(pentafluorphenyl)borat,
N,N-Dimethyl-N-dodecylammoniumtetrakis(pentafluorphenyl)-borat,
N,N-Dimethyl-N-octadecylammoniumtetrakis(pentafluorphenyl)-borat,
N-Methyl-N,N-didodecylammoniumtetrakis(pentafluorphenyl)-borat,
N-Methyl-N,N-dioctadecylammoniumtetrakis(pentafluorphenyl)-borat,
N,N-Dimethylaniliniumtetrakis(pentafluorphenyl)borat,
N,N-Dimethylanilinium-n-butyltris(pentafluorphenyl)borat,
N,N-Dimethylaniliniumbenzyltris(pentafluorphenyl)borat,
N,N-Dimethylaniliniumtetrakis(-(t-butyldimethylsilyl)-2,3,5,6-tetrafluorphenyl)borat,
N,N-Dimethylaniliniumtetrakis(4-(triisopropylsilyl)-2,3,5,6-tetrafluorphenyl)borat,
N,N-Dimethylaniliniumpentafluorphenoxytris(pentafluorphenyl)-borat,
N,N-Diethylaniliniumtetrakis(pentafluorphenyl)borat,
N,N-Dimethyl-2,4,6-trimethylaniliniumtetrakis(pentafluorphenyl)borat,
Trimethylammoniumtetrakis(2,3,4,6-tetrafluorphenyl)borat,
Triethylammoniumtetrakis(2,3,4,6-tetrafluorphenyl)borat,
Tripropylammoniumtetrakis(2,3,4,6-tetrafluorphenyl)borat,
Tri(n-butyl)ammoniumtetrakis(2,3,4,6-tetrafluorphenyl)borat,
Dimethyl(t-butyl)ammoniumtetrakis(2,3,4,6-tetrafluorphenyl)-borat,
N,N-Dimethylaniliniumtetrakis(2,3,4,6-tetrafluorphenyl)borat,
N,N-Diethylaniliniumtetrakis(2,3,4,6-tetrafluorphenyl)borat
und
N,N-Dimethyl-2,4,6-trimethylaniliniumtetrakis(2,3,4,6-tetrafluorphenyl)borat;
disubstituierte
Ammoniumsalze wie:
Di-(i-propyl)ammoniumtetrakis(pentafluorphenyl)borat
und
Dicyclohexylammoniumtetrakis(pentafluorphenyl)borat;
trisubstituierte
Phosphoniumsalze wie:
Triphenylphosphoniumtetrakis(pentafluorphenyl)borat,
Tri-(o-tolyl)phosphoniumtetrakis(pentafluorphenyl)borat
und
Tri(2,6-dimethylphenyl)phosphoniumtetrakis(pentafluorphenyl)-borat;
disubstituierte
Oxoniumsalze wie:
Diphenyloxoniumtetrakis(pentafluorphenyl)borat,
Di(o-tolyl)oxoniumtetrakis(pentafluorphenyl)borat
und
Di(2,6-dimethylphenyl)oxoniumtetrakis(pentafluorphenyl)borat;
disubstituierte
Sulfoniumsalze wie:
Diphenylsulfoniumtetrakis(pentafluorphenyl)borat,
Di(o-tolyl)sulfoniumtetrakis(pentafluorphenyl)borat
und
Bis(2,6-dimethylphenyl)sulfoniumtetrakis(pentafluorphenyl)-borat.
-
Bevorzugte Kationen (L*-H)– sind
N,N-Dimethylanilinium, Tributylammonium, N-Methyl-N,N-didodecylammonium,
N-Methyl-N,N-dioctadecylammonium
und Mischungen hiervon.
-
Ein anderes zur Bildung des aktivierenden
Cokatalysators geeignetes Ion umfaßt ein Salz eines kationischen
Oxidationsmittels und ein nicht-koordinierendes, verträgliches
Anion, wiedergegeben durch die Formel: (Oxg+
d( Ad–)e, worin
Oxe–,
Ad– und
d wie zuvor definiert sind.
-
Beispiele von kationischen Oxidationsmitteln
schließen
ein: Ferrocenium, hydrocarbyl-substituiertes Ferrocenium, Ag+ oder Pb+2. Bevorzugte
Ausführungsformen
von Ad– sind
solche Anionen, welche zuvor mit Bezug auf die Bronstedsäure enthal tenden
aktivierenden Cokatalysatoren definiert wurden, insbesondere Tetrakis(pentafluorphenyl)borat.
-
Ein anderer geeigneter ionenbildender,
aktivierender Cokatalysator umfaßt eine Verbindung, welche ein
Salz eines Carbeniumions und eines nicht-koordinierenden, verträglichen
Anions ist, wiedergegeben durch die Formel: ©+A–
worin:
©+ und A– wie zuvor definiert
sind.
-
Ein bevorzugtes Carbeniumion ist
das Tritylkation, d. h. Triphenylmethylium.
-
Ein weiterer geeigneter ionenbildender
aktivierender Cokatalysator umfaßt eine Verbindung, welche ein
Salz eines Silyliumions und eines nicht-koordinierenden, verträglichen
Anions ist, das durch die Formel wiedergegeben wird: R*3Si+A–
worin:
R*
= C1-10-Hydrocarbyl ist und A– wie
zuvor definiert ist.
-
Bevorzugte aktivierende Cokatalysatoren
in Form eines Silyliumsalzes sind Trimethylsilyliumtetrakispentafluorphenylborat,
Triethylsilyliumtetrakispentafluorphenylborat und ether-substituierte
Addukte hiervon. Die Verwendung der oben genannten Silyliumsalze
als aktivierende Cokatalysatoren für Katalysatoren zur Additionspolymerisation
sind in der USSN 08/304 314, eingereicht am 12. September 1994,
beansprucht.
-
Bestimmte Komplexe von Alkoholen,
Mercaptanen, Silanolen und Oximen mit Tris(pentafluorphenyl)boran
sind ebenfalls effektive Katalysatoraktivatoren, und sie können für die vorliegende
Erfindung verwendet werden. Solche Cokatalysatoren sind in der USP
5 296 433 beschrieben, und die Lehren hiervon sind hierin unter
Bezugnahme aufgenommen.
-
Die am meisten bevorzugten aktivierenden
Cokatalysatoren sind Trispentafluorphenylboran und N,N-Dioctadecyl-N-methylammoniumtetrakispentafluorphenylborat.
Die letztgenannte Ver bindung ist die Hauptkomponente einer Mischung
von Boratsalzen, abstammend von Bis(hydrierten-talg)methylammoniumverbindungen,
deren Mischungen als aktivierender Cokatalysator hier verwendet
werden können.
-
Das bevorzugt verwendete Molverhältnis von
Metallkomplex: aktivierender Cokatalysator reicht von 1 : 10 bis
2 : 1, mehr bevorzugt von 1 : 5 bis 1,5 : 1, am meisten bevorzugt
von 1 : 5 bis 1 : 1.
-
Andere Aktivatoren schließen die
zuvor genannten Aluminoxane ein. Bevorzugte Aluminoxane schließen Methylaluminoxan,
Propylaluminoxan, Isobutylaluminoxan, Kombinationen hiervon ein.
Sogenanntes modifiziertes Methylaluminoxan (MMAO) ist ebenfalls
zur Verwendung als Cokatalysator geeignet. Eine Arbeitsweise zur
Herstellung eines solchen modifizierten Alumoxans ist im US-Patent
Nr. 4 960 878 (Crapo et al.) angegeben, die Angaben hiervon sind
hier unter Bezugnahme aufgenommen. Aluminoxane können ebenfalls hergestellt
werden, wie in den US-Patenten Nr. 4 544 762 (Kaminsky et al.),
5 015 749 (Schmidt et al.), 5 041 583 (Sangokoya), 5 041 584 (Crapo
et al.) und 5 041 585 (Deavenport et al.) angegeben, wobei die Angaben hiervon
sämtlich
hier durch Bezugnahme aufgenommen sind. Wenn Aluminoxane als der
aktivierende Cokatalysator verwendet werden, reicht das Molverhältnis von Übergangsmetallkomplex
: Aluminium bevorzugt von 1 : 2000 bis 2 : 1, mehr bevorzugt von
1 : 1000 bis 1,5 : 1, am meisten bevorzugt von 1 : 500 bis 1 : 1.
-
Im allgemeinen kann die Polymerisation
bei Bedingungen durchgeführt
werden, die aus dem Stand der Technik für Polymerisationsreaktionen
vom Ziegler-Natta- oder Kaminsky-Sinn-Typ bekannt sind, d. h. Temperaturen
von 0–250°C, bevorzugt
30 bis 200°C,
und Drücke
von atmosphärischem
Druck bis 30.000 Atmosphären
oder höher.
Polymerisationen in Suspension, in Lösung, als Aufschlämmung, in
der Gasphase, im festen Zustand als Pulver oder unter anderen Verfahrensbedingungen
können
gewünschtenfalls
angewandt werden. Eine feste Komponente (anders als diejenige, welche
zur Herstellung des Ethylenhomopolymeren der vorliegenden Erfindung
verwendeten Katalysators angewandt wurde), kann verwendet werden,
insbesondere Siliziumdioxid, Aluminiumoxid oder ein Polymeres (insbesondere
Poly(tetrafluorethylen) oder ein Polyolefin), und wird wünschenswerterweise
verwendet, wenn die Katalysatoren in einem Gasphasen-Polymerisationsverfahren
eingesetzt werden. Der Träger
wird bevorzugt in einer Menge verwendet, um ein Gewichtsverhältnis von
Katalysator (basierend auf Metall): Träger von 1 : 100.000 bis 1 :
10, mehr bevorzugt von 1 : 50.000 bis 1 : 20 und am meisten bevorzugt
von 1 : 10.000 bis 1 : 30 zu erhalten.
-
Bei den meisten Polymerisationsreaktionen
beträgt
das angewandte Molverhältnis
von Katalysator: polymerisierbaren Verbindungen von 10–12 :
1 bis 10–1 :
1, mehr bevorzugt von 10–9 : 1 bis 10–5 :
1.
-
Geeignete Lösungsmittel für die Polymerisation
sind inerte Flüssigkeiten.
Beispiele schließen
ein: gerade oder verzweigtkettige Kohlenwasserstoffe wie Isobutan,
Butan, Pentan, Hexan, Heptan, Octan und Mischungen hiervon; cyclische
und alicyclische Kohlenwasserstoffe wie Cyclohexan, Cycloheptan,
Methylcyclohexan, Methylcycloheptan und Mischungen hiervon; perfluorierte
Kohlenwasserstoffe wie perfluorierte C4-10-Alkane; und aromatische
und alkyl-substituierte aromatische Verbindungen wie Benzol, Toluol,
Xylol, Ethylbenzol. Geeignete Lösungsmittel
schließen
ebenfalls flüssige
Olefine ein, welche als Monomere oder Comonomere wirken können, einschließlich Ethylen,
Propylen, Butadien, Cyclopenten, 1-Hexen, 1-Hexan, 4-Vinylcyclohexen, Vinylcyclohexan,
3-Methyl-1-penten,
4-Methyl-1-penten, 1,4-Hexadien, 1-octen, 1-Decen, Styrol, Divinylbenzol,
Allylbenzol, Vinyltoluol (einschließlich aller Isomeren alleine
oder in Mischung). Mischungen der zuvor genannten Lösungsmittel
sind ebenfalls geeignet.
-
Mischungszusammensetzungen, welche
das Ethylencopolymere mit den heterogenen Ethyleninterpolymeren
mit breiter Zusam mensetzungsverteilung umfassen, sind eine andere
Ausführungsform
der vorliegenden Erfindung. In der Definition der heterogenen Interpolymere,
wie sie hier verwendet werden, sind solche eingeschlossen, welche
unter Verwendung von Ziegler-Katalysatoren
hergestellt wurden, und ebenfalls solche, welche mittels der auf
Chrom basierenden auf Siliziumdioxid getragenen Systeme, die üblicherweise
als Katalysatoren vom Phillips-Typ bekannt, sind hergestellt wurden.
-
Der Ausdruck heterogen beschreibt
Interpolymere, in denen die Interpolymermoleküle nicht das gleiche Verhältnis Ethylen/Comonomeres
haben. Der hier verwendete Ausdruck "breite Zusammensetzungsverteilung" beschreibt die Comonomerenverteilung
für heterogene
Interpolymere und bedeutet, daß die
heterogenen Interpolymere eine "lineare" Fraktion haben,
und daß die
heterogenen Interpolymere bei der DSC Mehrfachschmelzpeaks haben
(d. h. wenigstens zwei unterscheidbare Schmelzpeaks zeigen). Die
heterogenen Interpolymere und Polymere haben einen Verzweigungsgrad
von weniger als oder gleich 2 Methyle/1000 Kohlenstoffe in 10% (in
Gewicht) oder mehr, bevorzugt mehr als 15% (in Gewicht) und insbesondere
mehr als 20% (in Gewicht). Die heterogenen Interpolymere haben ebenfalls
einen Verzweigungsgrad, der gleich oder größer als 25 Methyle/1000 Kohlenstoffe
ist in 25% oder weniger (in Gewicht), bevorzugt weniger als 15%
(in Gewicht) und insbesondere weniger als 10% (in Gewicht).
-
Die heterogene Interpolymerkomponente
der Mischung kann ebenfalls ein Ethylenhomopolymeres oder ein Interpolymeres
von Ethylen mit wenigstens einem Comonomeren sein, welches aus der
Gruppe ausgewählt
ist, die aus einer Verbindung besteht, wiedergegeben durch die Formel
H2C = CHR, worin R eine C1-C18 lineare, verzweigte oder cyclische Alkylgruppe
oder eine C6-C20 Arylgruppe
ist, und einem C4-C20 linearen,
verzweigten oder cyclischen Dien. Illustrative Beispiele der durch
die Formel H2C = CHR wiedergegebenen Verbindungen
schließen
Propylen, 1-Buten, 1-Penten, 1-Hexen, 4-Methyl-1-penten, 1- Octen, 1-Decen, 1-Dodecen,
1-Tetradecen, 1-Hexadecen, 1-Octadecen,
1-Eicosen, Vinylcyclohexen und Styrol ein. Illustrative Beispiele
von C4-C20 linearen,
verzweigten und cyclischen Dienen schließen 1,3-Butadien, 1,4-Pentadien, 1,5-Hexadien, 1,4-Hexadien
und Cyclohexadien ein. Von diesen sind Propylen, 1-Buten, 1-Penten,
1-Hexen, 4-Methyl-1-penten, 1-Octen,
1-Decen, 1-Dodecen, 1-Tetradecen, 1-Hexadecen, 1-Octadecen und 1-Eicosen besonders bevorzugt.
Heterogene Interpolymere von Ethylen und 1-Buten, 1-Penten, 1-Hexen
und 1-Octen sind
am meisten bevorzugt.
-
Die Menge des in die gemischte Zusammensetzung
der vorliegenden Erfindung eingebauten heterogenen Ethylen/α-Olefininterpolymeren
mit breiter Zusammensetzungsverteilung beträgt von 1 bis 99, bevorzugt
von 10 bis 90, mehr bevorzugt von 25 bis 75 und am meisten bevorzugt
von 35 bis 65 Gew.-%, bezogen auf die kombinierten Gewichte von
Komponenten I und II.
-
Die Dichte des in die gemischte Zusammensetzung
der vorliegenden Erfindung eingebauten heterogenen Ethylen/α-Olefininterpolymeren
mit breiter Zusammensetzungsverteilung beträgt im allgemeinen von allgemein
0,915 bis 0,985, bevorzugt von 0,935 bis 0,983, mehr bevorzugt von
0,955 bis 0,980 g/cm3.
-
Der Schmelzindex (I2)
für das
in die gemischte Zusammensetzung der vorliegenden Erfindung eingebaute
heterogene Ethylen/α-Olefininterpolymere
mit breiter Zusammensetzungsverteilung ist allgemein von 0,0001
bis 10000, bevorzugt von 0,001 bis 5000, mehr bevorzugt von 0,011
bis 3000 g/10 min.
-
Das Verhältnis I10/I2 des heterogenen Ethylen/α-Olefininterpolymeren
oder Homopolymeren mit breiter Zusammensetzungsverteilung, hergestellt
durch denselben Katalysator und unter denselben Verfahrensbedingungen
wie dieses in die gemischte Zusammensetzung der vorliegenden Erfindung
eingebaute Interpolymere beträgt
von 5 bis 40, bevorzugt von 5,3 bis 35, mehr bevorzugt von 5,5 bis
30, oder das Verhältnis
I21,6/I2 des in
die gemischte Zusammensetzung der vorliegenden Erfindung eingebaute
Ethylen/α-Interpolymere
mit breiter Zusammensetzungsverteilung beträgt von 15 bis 80, bevorzugt
von 20 bis 70, mehr bevorzugt von 25 bis 60.
-
Das Verhältnis Mw/Mn des in die gemischte Zusammensetzung der
vorliegenden Erfindung eingebauten heterogenen Ethylen/α-Olefininterpolymeren
mit breiter Zusammensetzungsverteilung beträgt im allgemeinen von 3 bis
12, bevorzugt von 3,5 bis 10, mehr bevorzugt von 4 bis 9.
-
Zur Herstellung der heterogenen Komponente
der Polymermischung können
Ziegler-Natta-Katalysatoren verwendet werden. Bevorzugte Ziegler-Natta-Katalysatoren
schließen
Katalysatoren auf Magnesiumalkoxidbasis ein, wie sie in der USP
4 526 943, USP 4 426 316, 4 661 465, USP 4 783 512 und USP 4 544
647 angegeben sind, wobei die Angaben von jeder hiervon durch Bezugnahme
aufgenommen sind. Solche Katalysatoren sind besonders nützlich,
falls die heterogene Polymerkomponente unter Aufschlämmungsverfahrensbedingungen
hergestellt werden soll.
-
Zusätzliche Beispiele von Katalysatoren
vom Ziegler-Typ, welche für
die Herstellung der heterogenen Polymermischungskomponente nützlich sind,
die unter hohen Polymerisationstemperaturen des Lösungsverfahrens
hergestellt werden sollen, schließen Katalysatoren ein, die
von Organomagnesiumverbindungen, Alkylhalogeniden oder Aluminiumhalogeniden
oder Chlorwasserstoff sowie einer Übergangsmetallverbindung abstammen.
Beispiele solcher Katalysatoren sind in den US-Patenten Nr. 4 314 912 (Lowery, Jr.
et al.), 4 547 475 (Glass et al.) und 4 612 300 (Coleman, III) beschrieben,
wobei die Angaben hiervon durch Bezugnahme aufgenommen sind.
-
Besonders geeignete Organomagnesiumverbindungen
schließen
beispielsweise kohlenwasserstofflösliches Dihydrocarbylmagnesium,
wie die Magnesiumdialkyle und die Magnesiumdiaryle, ein. Beispiele
von geeigneten Magnesiumdialkylen schließen insbesondere n-Butyl-sec-butylmagnesium,
Diisopropylmagnesium, Di-n-hexylmagnesium, Isopropyl-n-butylmagnesium, Ethyl-n-hexylmagnesium,
Ethyl-n-butylmagnesium, Di-n-octylmagnesium und andere, worin der
Alkylrest von 1 bis 20 Kohlenstoffatome hat, ein. Beispiele von
geeigneten Magnesiumdiarylen schließen Diphenylmagnesium, Dibenzylmagnesium
und Ditolylmagnesium ein. Geeignete Organomagnesiumverbindungen
schließen
Alkyl- und Arylmagnesiumalkoxide und -aryloxide und Aryl- und Alkylmagnesiumhalogenide
ein, wobei die halogenfreien Organomagnesiumverbindungen stärker erwünscht sind.
-
Zu den Halogenidquellen, welche hier
verwendet werden können,
gehören
die aktiven nicht-metallischen Halogenide, metallischen Halogenide
und Chlorwasserstoff.
-
Geeignete nicht-metallische Halogenide
werden durch die Formel R'X
wiedergegeben, worin R' Wasserstoff
oder ein aktiver einwertiger organischer Rest ist und X ein Halogen
ist. Besonders geeignete nicht-metallische Halogenide schließen beispielsweise
Halogenwasserstoff und aktive organische Halogenide wie t-Alkylhalogenide,
Allylhalogenide, Benzylhalogenide und andere aktive Hydrocarbylhalogenide,
worin das Hydrocarbyl wie zuvor definiert ist, ein. Unter einem
aktiven organischen Halogenid ist ein Hydrocarbylhalogenid zu verstehen,
welches ein labiles Halogen wenigstens als aktiv enthält, d. h.
das leicht an eine andere Verbindung verloren gehen kann, wie das
Halogen von sec-Butylchlorid, bevorzugt ebenso aktiv wie t-Butylchlorid.
Zusätzlich
zu den organischen Monohalogeniden ist zu verstehen, daß organische
Dihalogenide, Trihalogenide und andere Polyhalogenide, welche wie
hier zuvor definiert aktiv sind, ebenfalls geeigneterweise eingesetzt
werden können.
Beispiele von bevorzugten nichtmetallischen Halogeniden schließen Chlorwasserstoff,
Bromwasserstoff, t-Butylchlorid, t-Amylbromid, Allylchlorid, Benzylchlorid,
Crotylchlorid, Methylvinylcarbinylchlorid, α-Phenylethylbromid, Diphenylmethylchlorid
ein. Am meisten bevorzugt sind Chlorwasserstoff, t-Butylchlorid, Allylchlorid
und Benzylchlorid.
-
Geeignete metallische Halogenide,
welche hier verwendet werden können,
schließen
solche ein, welche durch die Formel wiedergegeben werden: MRy-aXa, worin:
M
ein Metall der Gruppen IIB, IIIA oder IVA des Periodensystems der
Elemente nach Mendeleev ist,
R ein einwertiger organischer
Rest ist,
x ein Halogen ist,
Y einen Wert hat, der der
Wertigkeit von M entspricht, und
a einen Wert von 1 bis y hat.
-
Bevorzugte metallische Halogenide
sind Aluminiumhalogenide der Formel AlR3-aXa, worin:
jedes
R unabhängig
Hydrocarbyl, wie zuvor definiert ist, wie Alkyl,
X ein Halogen
ist, und
a eine Zahl von 1 bis 3 ist.
-
Am meisten bevorzugt sind Alkylaluminiumhalogenide
wie Ethylaluminiumsesquichlorid, Diethylaluminiumchlorid, Ethylaluminiumdichlorid
und Diethylaluminiumbromid, wobei Ethylaluminiumdichlorid besonders bevorzugt
ist. Alternativ kann ein Metallhalogenid wie Aluminiumtrichlorid
oder eine Kombination von Aluminiumtrichlorid mit einem Alkylaluminiumhalogenid
oder eine Trialkylaluminiumverbindung geeigneterweise verwendet
werden.
-
Es ist dies so zu verstehen, daß die organischen
Einheiten des zuvor genannten Organomagnesiums, beispielsweise R'', und die organischen Einheiten der
Halogenidquelle, beispielsweise R und R', geeigneterweise ein beliebiger anderer
organischer Rest sind, vorausgesetzt, daß sie keine funktionelle Gruppen
enthalten, welche konventionelle Ziegler-Katalysatoren vergiften.
-
Das Magnesiumhalogenid kann aus der
Organomagnesiumverbindung und der Halogenidquelle vorgebildet werden,
oder es kann in situ gebildet werden, wobei in diesem Fall der Katalysator
bevorzugt durch Mischen in einem geeigneten Lösungsmittel oder Reaktionsmedium
von (1) der Organomagnesiumkomponente und (2) der Halogenidquelle,
gefolgt von den anderen Katalysatorkomponenten hergestellt wird.
-
Beliebige der konventionellen Ziegler-Natta-Übergangsmetallverbindungen
können
nützlicherweise als
die Übergangsmetallkomponente
bei der Herstellung der Trägerkatalysatorkomponente
verwendet werden. Typischerweise ist die Übergangsmetallkomponente eine
Verbindung eines Metalls der Gruppe IVB, VB oder VIB. Die Übergangsmetallkomponente
wird im allgemeinen durch die Formeln wiedergegeben:
TrX'4-q(OR1)q, TrX'4-qR2
q, VOX'3 und VO(OR1)3.
Tr ist
ein Metall der Gruppe IVB, VB oder VIB, bevorzugt ein Metall der
Gruppe IVB oder VB, bevorzugt Titan, Vanadium oder Zirkonium,
q
ist 0 oder eine Zahl gleich bis oder weniger als 4,
X' ist ein Halogen,
und
R1 ist eine Alkylgruppe, Arylgruppe
oder Cycloalkylgruppe mit von 1 bis 20 Kohlenstoffatomen, und
R2 ist eine Alkylgruppe, Arylgruppe, Aralkylgruppe,
substituierte Aralkyle. Das Aryl, die Aralkyle und substituierten
Aralkyle enthalten 1 bis 20 Kohlenstoffatome, bevorzugt 1 bis 10
Kohlenstoffatome. Wenn die Übergangsmetallverbindung
eine Hydrocarbylgruppe, R2, enthält, die
eine Alkyl-, Cycloalkyl-, Aryl- oder Aralkylgruppe ist, enthält die Hydrocarbylgruppe
bevorzugt kein H-Atom in der beta-Stellung zu der Bindung Metall-Kohlenstoff.
Erläuternde,
jedoch nichtbeschränkende
Beispiele von Aralkylgruppen sind Methyl, Neopentyl, 2,2-Dimethylbutyl,
2,2-Dimethylhexyl; Arylgruppen wie Benzyl; Cycloalkylgruppen wie
1-Norbornyl. Mischun gen dieser Übergangsmetallverbindungen
können
gewünschtenfalls
verwendet werden.
-
Illustrative Beispiele der Übergangsmetallverbindungen
schließen
ein: TiCl4, TiBr4,
Ti(OC2H5)3Cl, Ti(OC2H5)Cl3, Ti(OC4H9)3Cl,
Ti(OC3H7)2Cl2, Ti(OC6H13)2Cl2, Ti(OC8H17)2Br2 und
Ti(OC12H25)Cl3, Ti(O-i-C3H7)4 und Ti(O-n-C4H9)4
-
Illustrative Beispiele von Vanadiumverbindungen
schließen
ein : VCl4, VOCl3,
VO(OC2H5)3 und VO(OC4H9)3.
-
Illustrative Beispiele von Zirkoniumverbindungen
schließen
ein: ZrCl4, ZrCl3(OC2H5), ZrCl2(OC2H5)2, ZrCl(OC2H5)3, Zr(OC2H5)4,
ZrCl3(OC4H9), ZrCl2(OC4H9)2 und
ZrCl(OC4H9)3.
-
Wie oben angegeben, können geeigneterweise
Mischungen der Übergangsmetallverbindungen
verwendet werden, wobei keine Einschränkungen durch die Anzahl der Übergangsmetallverbindungen,
welche mit dem Träger
in Kontakt gebracht werden können,
gegeben sind. Beliebige Halogenid- und Alkoxidübergangsmetallverbindungen
oder Mischungen hiervon können
vorteilhafterweise verwendet werden. Die zuvor aufgeführten Übergangsmetallverbindungen
sind besonders bevorzugt, wobei Vanadiumtetrachlorid, Vanadiumoxychlorid,
Titantetraisopropoxid, Titantetrabutoxid und Titantetrachlorid am
meisten bevorzugt sind.
-
Geeignete Ziegler-Katalysatormaterialien
können
ebenfalls von inerten Oxidträgern
und Übergangsmetallverbindungen
abstammen. Beispiele solcher Zusammensetzungen, welche für die Verwendung
bei dem Lösungspolymerisationsverfahren
geeignet sind, sind im US-Patent Nr. 5 420 090 (Spencer, et al.)
beschrieben, wobei die Lehren hiervon hier durch Bezugnahme aufgenommen
sind.
-
Die Menge des mit einem Katalysator
hergestellten Homopolymeren, der verschieden von dem ist, der zur
Herstellung des in die gemischte Zusammensetzung der vorliegenden
Erfindung eingebauten Ethylencopolymeren der vorliegenden Erfindung
verwendet wurde, beträgt
von 1 bis 99, bevorzugt von 10 bis 90, mehr bevorzugt von 25 bis
75 und am meisten bevorzugt von 35 bis 65 Gew.-%, bezogen auf die
kombinierten Gewichte von Komponenten I und II.
-
Der Schmelzindex (I2)
für das
Homopolymere, das mit einem Katalysator und nach einem Verfahren hergestellt
wurde, die von demjenigen verschieden sind, die zur Herstellung
des in die gemischte Zusammensetzung der vorliegenden Erfindung
eingebauten Ethylencopolymeren der vorliegenden Erfindung verwendet wurde,
beträgt
im allgemeinen von 0,0001 bis 10000, bevorzugt von 0,001 bis 5000,
mehr bevorzugt von 0,01 bis 3000 g/10 min.
-
Das Verhältnis I21,6/I2 des Homopolymeren, der mit einem Katalysator
und nach einem Verfahren hergestellt wurde, die von demjenigen verschieden
sind, die zur Herstellung des in die gemischte Zusammensetzung der
vorliegenden Erfindung eingebauten Ethylencopolymeren der vorliegenden
Erfindung verwendet wurde, beträgt
von 15 bis 80, bevorzugt von 18 bis 70, mehr bevorzugt von 20 bis
60, oder das I10/I2 beträgt von 5
bis 40, bevorzugt von 5,3 bis 35, mehr bevorzugt von 5,5 bis 30.
-
Das Verhältnis Mw/Mn des Homopolymeren, das mit einem Katalysator
und einem Verfahren hergestellt wurde, die von demjenigen verschieden
sind, welche zur Herstellung des in die gemischte Zusammensetzung
der vorliegenden Erfindung eingebauten Ethylencopolymeren der vorliegenden
Erfindung verwendet wurde, beträgt
im allgemeinen von 2,5 bis 12, bevorzugt von 2,8 bis 10, mehr bevorzugt
von 3 bis 9.
-
Falls Mischungen des Ethylencopolymeren
der vorliegenden Erfindung, wie hier beschrieben, mit weiteren Ethyleninterpolymeren
erforderlich sind, kann jede Komponente getrennt in unterschiedlichen
Reaktoren hergestellt und anschließend miteinander gemischt werden.
-
Die Mischungszusammensetzungen können ebenfalls
mittels eines kontinuierlichen gesteuerten Polymerisationsverfahrens
(im Gegensatz zu einem ansatzweisen oder halb-ansatzweisen Arbeiten)
unter Verwendung wenigstens eines Reaktors hergestellt werden. Bevorzugt
werden jedoch das Ethylencopolymere der vorliegenden Erfindung und
die zusätzlichen
Ethyleninterpolymerkomponenten der Mischungszusammensetzungen in
einer Mehrfachreaktoranordnung hergestellt, die entweder parallel
oder in Reihe betrieben wird, wie sie im USP 3 914 342 (Mitchell)
und der WO 94/00500 beschrieben sind, wobei die Lehren hiervon hierin
durch Bezugnahme aufgenommen sind. Beispielsweise können wenigstens
zwei Reaktoren, die in Reihe betrieben werden, d. h. einer nach
dem anderen, verwendet werden. Alternativ können die Reaktoren parallel
betrieben werden, d. h. unter Durchführung der Polymerisationsstufen
A und B in getrennten Reaktoren und anschließendes Kombinieren der Schmelzströmungen zum
Erhalt einer Verbundproduktmischung. Bei der Mehrfachreaktoranordnung
erzeugt wenigstens einer der Reaktoren das Ethylencopolymere der
vorliegenden Erfindung unter Verwendung des hier beschriebenen Träger-Metallocenkatalysators
unter Aufschlämmungsverfahrensbedingungen,
und wenigstens einer der Reaktoren erzeugt die zusätzlichen
Komponenten der Mischung unter Anwendung der erforderlichen Einzel-
oder Mehrfachkatalysatoren bei Polymerisationstemperaturen, -drücken und
-einspeisungskonzentrationen, die zur Erzeugung des Polymeren mit
den gewünschten
Eigenschaften erforderlich sind.
-
So wird bei einer Ausführungsform
das Ethylencopolymere der vorliegenden Erfindung unter Verwendung
des hier beschriebenen Träger-Metallocenkatalysators
unter Aufschlämmungsverfahrensbedingungen
in einem ersten Reaktor in Stufe A hergestellt, und der Inhalt des
ersten Reaktors wird zu einem zweiten Reaktor geführt, in
welchem die Einspeisungskonzentrationen und die Temperatur so eingestellt
werden, um unter Aufschlämmungsverfahrensbedingungen
in Stufe B ein zweites Ethylencopolymeres der vorliegenden Erfindung,
das ein unterschiedliches Molekulargewicht oder eine unterschiedliche
Dichte hat, zu bilden.
-
Bei einer weiteren Ausführungsform
wird das Ethylencopolymere der vorliegenden Erfindung unter Verwendung
des hier beschriebenen Träger-Metallocenkatalysators
unter Aufschlämmungsverfahrensbedingungen
in einem ersten Reaktor in Stufe A hergestellt, und der Inhalt des
ersten Reaktors wird zu einem zweiten Reaktor geführt, in
welchem die Einspeisungskonzentrationen und die Temperatur eingeregelt
werden, und einer oder mehrere der hier beschriebenen Ziegler-Katalysatoren
zugesetzt werden, um in Stufe B unter Aufschlämmungsverfahrensbedingungen
die heterogene Ethyleninterpolymer- oder Homopolymerkomponente der
Polymermischung mit den gewünschten
Eigenschaften zu bilden.
-
Bei einer weiteren Ausführungsform
wird das Ethylencopolymere der vorliegenden Erfindung unter Verwendung
des hier beschriebenen Träger-Metallocenkatalysators
unter Aufschlämmungsverfahrensbedingungen
in einem ersten Reaktor in Stufe A hergestellt, und der Inhalt des
ersten Reaktors tritt in einen zweiten Reaktor ein, in welchem die
Einspeisungskonzentrationen und Temperaturen eingeregelt werden,
und einer der hier beschriebenen Metallocenkatalysatoren zur Bildung
der homogenen Ethyleninterpolymer- oder Homopolymerkomponente der
Polymermischung mit den gewünschten
Eigenschaften in Stufe B unter Lösungsverfahrensbedingungen
zugesetzt wird.
-
Bei einer weiteren Ausführungsform
wird das Ethylencopolymere der vorliegenden Erfindung unter Verwendung
des hier beschriebenen Träger-Metallocenkatalysators
unter Aufschlämmungsverfahrensbedingungen
in einem ersten Reaktor in Stufe A hergestellt, und der Inhalt des
ersten Reaktors wird zu einem zweiten Reaktor überführt, in welchem die Temperatur
und die Einspeisungskonzentrationen eingestellt sind, und worin
ein oder mehrere der hier beschriebenen Ziegler-Katalysatoren zugesetzt
werden, um in Stufe B unter Lösungsverfahrensbedingungen
die heterogene Ethyleninterpolymer- oder Homopolymerkomponente der
Polymermischung mit den gewünschten
Eigenschaften zu bilden.
-
Zusätze wie Antioxidantien (beispielsweise
gehinderte Phenolverbindungen (beispielsweise IrganoxTM 1010),
Phosphite (beispielsweise IrgafosTM 168)),
Verzahnungszusätze
(beispielsweise PIB), Antiblockzusätze, Pigmente, Füllstoffe
können
ebenfalls in die Formulierungen in dem Ausmaß eingegeben werden, daß sie die
durch die Anmelder aufgefundenen verbesserten Formulierungseigenschaften
nicht beeinträchtigen.
Sowohl IrganoxTM als auch IrgafosTM werden hergestellt und sind Marken von
Ciba Geigy Corporation. IrgafosTM 168 ist
ein Phosphitstabilisator, und IrganoxTM 1010
ist ein Stabilisator in Form eines gehinderten Polyphenols (beispielsweise
Tetrakis[methylen-3(3,5-di-t-butyl-4-hydroxyphenylpropionat)]-methan.
-
Die Dichte der endgültigen Mischungszusammensetzungen
der vorliegenden Erfindung beträgt
im allgemeinen von 0,870 bis 0,980, bevorzugt von 0,915 bis 0,975,
mehr bevorzugt von 0,935 bis 0,970 und am meisten bevorzugt von
0,945 bis 0,968 g/cm3.
-
Der Schmelzindex, I2,
der fertigen Mischungszusammensetzungen der vorliegenden Erfindung
beträgt im
allgemeinen von 0,0001 bis 10000, bevorzugt von 0,001 bis 5000 und
am meisten bevorzugt von 0,01 bis 3000 g/10 min.
-
Das Verhältnis I10/I2 der fertigen Mischungszusammensetzungen
der vorliegenden Erfindung beträgt von
5 bis 100, bevorzugt von 5 bis 90, mehr bevorzugt von 5 bis 80,
oder das Verhältnis
I21,6/I2 der fertigen
Mischungszusammensetzungen der vorliegenden Erfindung beträgt von 20
bis 200, bevorzugt von 30 bis 180, mehr bevorzugt von 40 bis 150
und am meisten bevorzugt von 50 bis 130.
-
Das Verhältnis Mw/Mn der fertigen Mischungszusammensetzungen
der vorliegenden Erfindung beträgt von
2,5 bis 50, bevorzugt von 3 bis 45 und mehr bevorzugt von 5 bis
40.
-
Das Ethylencopolymere der vorliegenden
Erfindung hat ein spezifisches Verteilungsmerkmal des Comonomerengehaltes,
bei welchem bei einer Ausführungsform
je niedriger das Molekular gewicht einer Copolymerfraktion in einer
Molekulargewichtsverteilung eines Ethylencopolymeren ist, um so
niedriger der Comonomerengehalt der Copolymerfraktion ist, und,
bei der anderen Ausführungsform,
je höher
das Molekulargewicht einer Copolymerfraktion ist, um so höher der
Comonomerengehalt der Copolymerfraktion ist. Als Folge dieses Verteilungsmerkmals
des Comonomerengehaltes hat das Ethylencopolymere der vorliegenden
Erfindung ausgezeichnete Eigenschaften, wie hohe Schlagfestigkeit
und ausgezeichnete Rißfestigkeit
gegenüber
Umgebungsspannungen (ESCR). Weiterhin zeigt das Ethylencopolymere
der vorliegenden Erfindung keine breite Schwanzbildung sowohl auf
der Seite des niedrigen Molekulargewichtes als auch auf der Seite
des hohen Molekulargewichtes, so daß das Ethylencopolymere im
wesentlichen keine Verunreinigungen wie ein Wachs, ein Gel enthält. Sofern
das Verfahren der vorliegenden Erfindung im wesentlichen auf die
Herstellung eines Ethylencopolymeren angewandt wird, kann weiter
ein beliebiges Verfahren, bei welchem zwei oder mehr unterschiedliche
Ethylencopolymere mit unterschiedlichen Comonomerengehalten getrennt
erzeugt und mittels eines Kneters gemischt werden, und ein Verfahren,
bei welchem ein Ethylencopolymeres, das eine Mischung von zwei oder
mehr unterschiedlichen Ethylencopolymerkomponenten mit unterschiedlichen
Comonomerengehalten mittels einer Vielstufenpolymerisation oder
unter Verwendung einer Vielzahl von unterschiedlichen Typen von
in der vorliegenden Erfindung zu verwendenden Katalysatoren erzeugt
wird, sehr vorteilhaft bei einer bevorzugten Ausführungsform
des Verfahrens der vorliegenden Erfindung angewandt werden. Die
Mischung der nach dem oben genannten bevorzugten Verfahren der vorliegenden
Erfindung hergestellten Ethylencopolymere kann eine Verteilung des
Comonomerengehaltes aufweisen, in welcher der Comonomerengehalt
kontinuierlich in Übereinstimmung
mit einer Zunahme des Molekulargewichtes des Copolymeren variiert, im
Gegensatz zu demjenigen des Gemisches von konventionellen Ethylencopolymeren.
Daher können
gemäß dem oben genannten
Aufschlämmungsverfahren
der vorliegenden Erfindung ausgezeichnete Eigenschaften erhalten
werden hinsichtlich der Verteilung des Comonomerengehaltes des Gemisches
der Ethylencopolymere, welche durch den Stand der Technik nicht
erreicht werden konnten.
-
Als Folge der oben genannten ausgezeichneten
Eigenschaften und Merkmale können
das Ethylencopolymere und die Mischungszusammensetzungen der vorliegenden
Erfindung vorteilhafterweise für
die Herstellung von geblasenen Folien, gegossenen Folien bzw. Filmen,
Laminatfolien, blasverformten Gegenständen, spritzgegossenen Gegenständen, Rohren/Schläuchen, Beschichtungsmaterialien
für elektrische Übertragungskabel
verwendet werden.
-
BEISPIELE
-
Die vorliegende Erfindung wird im
folgenden mehr im einzelnen unter Bezugnahme auf die folgenden Beispiele
und Vergleichsbeispiele erläutert,
welche jedoch nicht als den Umfang der vorliegenden Erfindung einschränkend ausgelegt
werden sollen.
-
Beispiel 1
-
20 g Siliziumdioxid SP-9-10046 (hergestellt
und vertrieben von Grace GmbH, Deutschland), welche bei 250°C für 2 Stunden
unter Vakuum behandelt worden waren, wurden in 250 ml Toluol aufgeschlämmt. Die resultierende
Aufschlämmung
wurde zu einer Lösung
von 20 ml (0,11 Mol) Triethylaluminium in 100 ml Toluol zugegeben.
Das resultierende Gemisch wurde für 2 Stunden gerührt, filtriert,
mit zwei Portionen von 100 ml frischem Toluol gewaschen und unter
Vakuum getrocknet. Die resultierenden 22 g des getrockneten Gemisches
wurden in 300 ml Toluol aufgeschlämmt und auf 70°C erhitzt,
um eine Aufschlämmung
hierdurch zu erhalten. Zu dieser Aufschlämmung wurde eine Lösung von
1,25 g (1,77 mMol) Triethylammonium-tris(pentafluorphenyl)(4-hydroxyphenyl)borat
in 200 ml Toluol zugesetzt, welche auf 70°C erhitzt worden war, und bei
70°C für 30 Minuten
gehalten. Bei der Zugabe wurde das Erhitzen unterbrochen, und das
resultierende Gemisch wurde für
3 Stunden gerührt.
Danach wurde eine Teilmenge von 12,3 ml einer dunkelviolett gefärbten 0,0714 M
Lösung
von Titan(N-1,1-dimethylethyl)dimethyl[1-(1,2,3,4,5-eta)-2,3,4,5-tetramethyl-2,4-cyclopentadien-1-yl]silanaminato[(2-)N]-η4-1,3-pentadien) in ISOPARTM E
(hergestellt und vertrieben von Exxon Chemical Co., USA) zu dem
Gemisch zugegeben, und das resultierende Gemisch wurde für 2 Stunden
gerührt,
wodurch ein grün
gefärbtes festes
Katalysatorsystem erhalten wurde.
-
Isopentan, Ethylen, 1-Buten, Wasserstoff
und das feste Katalysatorsystem wurden kontinuierlich in einen kontinuierlichen
gerührten,
mit Mantel versehenen 10 Liter Tankreaktor eingespeist. Die Strömungsgeschwindigkeiten
von Isopentan, Ethylen, 1-Buten und Wasserstoff waren jeweils 2500
g/h, 700 g/h, 20 g/h und 0,3 Liter/h. Das gebildete Aufschlämmungsprodukt
wurde kontinuierlich aus dem Reaktor abgezogen. Der Gesamtdruck
des Reaktors war 15,4 kg/cm2 (15 atm) und
die Innentemperatur des Reaktors wurde auf 70°C gehalten. Die abgezogene Aufschlämmung wurde
zu einem Abdampftank eingespeist, um das Verdünnungsmittel zu entfernen,
und das trockene freifließende
Ethylencopolymerpulver wurde erhalten.
-
Das so erhaltene Ethylencopolymere
hatte die folgenden Eigenschaften: eine Dichte gemäß ASTM D-792
von 0,929 g/cm3; einen Schmelzindex von
0,50 g/10 Minuten, gemessen gemäß ASTM D-1238
bei 190°C
unter einer Belastung von 2,16 kg; ein Mw von 152.000 und ein Mw/Mn von 4,5, beide
gemessen mittels GPC; ein Mt (ein Punkt im Molekulargewicht auf
einem Molekulargewichtsverteilungsprofil, gemessen mittels GPC,
bei welchem das Profil einen Peak mit einer maximalen Intensität zeigte)
von 69.000; eine annähernd gerade
Linie, erhalten aus dem Comonomerengehalt-Verteilungsprofil) hatte
einen Gradienten von 0,0013 innerhalb des Bereiches von 22.000 bis
220.000 in Werten eines Molekulargewichtes Mc, welches die Formel erfüllt log(69.000) – log(Mc) ≤ 0,5;eine
Temperatur (bei welcher eine Maximalmenge von Extraktion gezeigt
wurde) von 86°C,
gemessen mittels CFC; in der CFC wurde eine annähernd gerade Linie mit einem
Gradienten von –0,053
erhalten aus der Beziehung zwischen einer willkürlichen Temperatur, die innerhalb
des Bereiches von zwischen 86°C
und 96°C fällt, und
einem Punkt auf einem Molekulargewichts-Verteilungsprofil einer
Copolymerfraktion, die bei der willkürlichen Temperatur extrahiert
wurde, wobei dieser Punkt eine Spitze mit einer maximalen Intensität hatte; und
die Gesamtmenge von Copolymerfraktionen, die bei Temperaturen von
76°C oder
weniger bei der CFC extrahiert wurden, betrug 3,1 Gew.-%.
-
Beispiele 2 und 3
-
25 g Siliziumdioxid SP-9-10046 (hergestellt
und vertrieben von Grace GmbH, Deutschland) mit einem Wassergehalt
von 3,5 Gew.-% wurden zu 508 g 10%iger Methylalumoxanlösung in
Toluol (hergestellt und vertrieben von Witco GmbH, Deutschland)
unter kontinuierlichem Rühren
zugegeben. Das Gemisch wurde für weitere
2 Stunden gerührt,
und dann wurde das Lösungsmittel
unter vermindertem Druck bei 20°C
zum Erhalt eines freifließenden
Pulvers entfernt. Das erhaltene freifließende Pulver wurde dann auf
175°C für 2 Stunden unter
Vakuum erhitzt. Das resultierende Pulver wurde in 700 ml Toluol
erneut aufgeschlämmt,
und das Gemisch wurde mit 2 Portionen von frischem Toluol bei 100°C gewaschen.
Der Träger
wurde dann unter Vakuum bei 120°C
für 1 Stunde
getrocknet. 63,9 g Träger
mit einem Aluminiumgehalt von 26,4 Gew.-% wurden erhalten.
-
Zu 60 Gramm des Trägers wurde
eine Teilmenge von 33,6 ml einer dunkelvioletten 0,0714 M Lösung von
Titan (N-1,1-dimethylethyl)dimethyl[1-(1,2,3,4,5-eta)-2,3,4,5-tetramethyl-2,4-cyclopentadien-1-yl]silanaminato[(2-)N]-(eta4-1,3-penta dien) in ISOPARTM E
(hergestellt und vertrieben von EXXON Chemical Co., USA) zugesetzt, und
das Gemisch wurde für
mehrere Stunden zum Erhalt eines grün gefärbten Trägerkatalysators gerührt.
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in Beispiel 1 wurden mit der Ausnahme wiederholt, daß die Strömungsgeschwindigkeiten
von Isopentan, Ethylen, 1-Buten und Wasserstoff wie in Tabelle 1
angegeben verändert
wurden. Die Ergebnisse der Reaktionen sind in Tabelle 1 gezeigt.
-
Beispiele 4 bis 6
-
Im wesentlichen dieselben Arbeitsweisen
wie in den Beispielen 2 und 3 wurden zur Herstellung des Trägerkatalysators
mit der Ausnahme angewandt, daß ein
unterschiedliches mit MAO behandeltes Siliziumdioxid verwendet wurde,
worin das MAO ebenfalls auf dem Siliziumdioxid immobilisiert war,
wobei jedoch eine unterschiedliche Methode angewandt wurde.
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in Beispiel 1 wurden mit der Ausnahme angewandt, daß die Polymerisationstemperaturen
und die Strömungsgeschwindigkeiten
von Isopentan, 1-Buten und Wasserstoff wie in Tabelle 1 angegeben
verändert
wurden. Der Gradient und die Werte für CFC 1 und CFC 2 für die Beispiele
4–6 sind
extrapolierte Werte, basierend auf der Dichte aus den Ergebnissen
der Beispiele 1–3.
-
Die Ergebnissen der Reaktionen sind
in Tabelle 1 gezeigt.
-
Beispiel 7
-
6,2 g (8,8 mMol) von Triethylammonium-tris(pentafluorphenyl)(4-hydroxyphenyl)borat
wurden in 4 Liter Toluol, welches auf 90°C erhitzt und hierauf gehalten
worden war, für
30 Minuten erhitzt. Zu diesem Lösungsmittel
wurden eine Teilmenge von 40 ml einer 1 M Lösung von Trihexylaluminium
in Toluol zugegeben. Das resultierende Gemisch wurde für 1 Minute
bei 90°C
gerührt.
In einem zweiten Behälter
wurden 100 g Silizi umdioxid P-10 (hergestellt und vertrieben von
Fuji silysia, Japan), welche bei 500°C für 3 Stunden in strömendem Stickstoff
behandelt worden waren, in 1,7 Liter Toluol aufgeschlämmt. Diese
Siliziumdioxidaufschlämmung
wurde auf 90°C
erhitzt. Zu dieser Siliziumdioxidaufschlämmung wurde dieses Gemisch
von Triethylammonium-tris(pentafluorphenyl)(4-hydroxyphenyl)borat und Trieexylaluminium,
welches sich auf 90°C
befand, zugegeben, und die resultierende Aufschlämmung wurde für 3 Stunden
bei 90°C
gerührt.
Eine Teilmenge von 206 ml einer 1 M Lösung von Trihexylaluminium
in Toluol wurde zugegeben. Das resultierende Gemisch in etwa 5,9
Liter Toluol wurde bei 90°C
für 1 Stunde
gerührt.
Dann wurde die überstehende
Flüssigkeit
des resultierenden Gemisches mittels Dekantiermethode unter Verwendung
von 90°C
Toluol zur Entfernung von überschüssigem Trihexylaluminium
entfernt. Das Dekantieren wurde 5 mal wiederholt. Danach wurde eine
Teilmenge von 20 ml einer dunkelviolett gefärbten 0,218 M Lösung von Titan-(N-1,1-dimethylethyl)dimethyl[1-(1,2,3,4,5-eta)-2,3,4,5-tetramethyl-2,4-cyclopentadien-1-yl]silanaminato[(2-)N]-(η4-1,3-pentadien)
in ISOPARTM E (hergestellt und vertrieben
von Exxon Chemical Co., USA) zu dem Gemisch zugesetzt, und das resultierende
Gemisch wurde für
3 Stunden gerührt,
um hierdurch ein grün
gefärbtes
festes Katalysatorsystem zu erhalten.
-
Hexan, Ethylen, 1-Buten, Wasserstoff
und das feste Katalysatorsystem wurden kontinuierlich in einen kontinuierlich
gerührten
Tankreaktor eingespeist. Die Strömungsgeschwindigkeiten
von Hexan, Ethylen und Wasserstoff waren jeweils 46,2 kg/h, 0,15
kg/h, 0,15 kg/h. Die Strömungsgeschwindigkeiten
von 1-Buten waren 0,11 kg/h (Beispiel 7). Das gebildete Aufschlämmungsprodukt
wurde kontinuierlich aus dem Reaktor abgezogen. Der Gesamtdruck
des Reaktors war 10 atm und die Innentemperatur des Reaktors wurde
auf 80°C gehalten.
Die abgezogene Aufschlämmung
wurde zu einem Verdampfungstank geführt, um das Verdünnungsmittel
zu entfernen, und das trockene, freifließende Ethylencopolymerpulver
wurde erhalten. Die Eigenschaften der so erhaltenen Ethylencopolymere
sind in Tabelle 2 gezeigt.
-
Beispiel 8
-
200 g von Siliziumdioxid P-10 (hergestellt
und vertrieben von Fuji silysia, Japan), welche bei 500°C für 3 Stunden
in strömendem
Stickstoff behandelt worden waren, wurden in 5 Liter Hexan aufgeschlämmt. Zu
der resultierenden Aufschlämmung
wurde eine Teilmenge von 400 ml einer 1 M Lösung von Triethylaluminium
in Hexan zugegeben. Das resultierende Gemisch wurde für 0,5 Stunden
bei Zimmertemperatur gerührt.
Zu dieser Aufschlämmung
wurde eine Lösung
von 20,1 g (17,6 mMol) von Bis(hydriertem-talgalkyl)methylammonium-tris(pentafluorphenyl)(4-hydroxyphenyl)borat
in 296 ml Toluol zugegeben. Das resultierende Gemisch wurde für 0,5 Stunden
bei Zimmertemperatur gerührt.
Danach wurde eine Teilmenge von 60 ml einer dunkelviolett gefärbten 0,218
M Lösung
von Titan(N-1,1-dimethylethyl)dimethyl[1-(1,2,3,4,5-eta)-2,3,4,5-tetramethyl-2,4-cyclopentadien-1-yl]silanaminato[(2-)N]-(η4-1,3-pentadien) in ISOPARTM E
(hergestellt und vertrieben von Exxon Chemical Co., USA) zu dem Gemisch
zugegeben, und das resultierende Gemisch wurde für 3 Stunden bei Zimmertemperatur
gerührt,
um auf diese Weise ein grün
gefärbtes
festes Katalysatorsystem zu erhalten.
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in Beispiel 7 wurden mit der Ausnahme wiederholt, daß die Strömung von
1-Buten wie in Tabelle 1 angegeben verändert wurde.
-
Die Ergebnisse der Reaktionen sind
in Tabelle 2 gezeigt.
-
Vergleichsbeispiel 1
-
Ein 1000 ml Kolben wurde mit 508
g einer 10 eigen Lösung
von Methylaluminoxan in Toluol (hergestellt und vertrieben von Witco
GmbH, Deutschland) beschickt, und dann wurden 25 g Siliziumdioxid
SD 3216.30 silica (hergestellt und vertrieben von Grace GmbH, Deutschland)
mit einem Wassergehalt von etwa 3,5 Gew.-% zu dem Kolben unter kontinuierlichem
Rühren
zugegeben. Das resultierende Gemisch wurde für weitere 2 Stunden gerührt, und
dann wurde das Lösungsmittel
unter vermindertem Druck bei 20°C
entfernt, um hierdurch ein freifließendes Pulver zu erhalten.
Das erhaltene Pulver wurde dann auf 175°C für 2 Stunden unter Vakuum erhitzt.
Das Pulver wurde dann in 700 ml Toluol erneut aufgeschlämmt. Das
resultierende Gemisch wurde für
1 Stunde erhitzt und unter Rückfluß gekocht.
Das Gemisch wurde filtriert, mit 2 Portionen von frischem Toluol
bei 100°C
gewaschen und unter Vakuum bei 120°C für 1 Stunde getrocknet. Als
Ergebnis wurden 63,9 g des getrockneten Gemisches mit einem Aluminiumgehalt
von 23,8 Gew.-% erhalten.
-
Es wurde ein festes Katalysatorsystem
durch Aufschlämmung
von 0,5 g des oben erhaltenen getrockneten Gemisches in 20 ml ISOPARTM E (hergestellt und vertrieben von Exxon
Chemical Co., USA) hergestellt, und nach Rühren für wenige Minuten zum Dispergieren
des getrockneten Gemisches wurde eine Teilmenge von 0,142 ml einer
dunkelorange-braun gefärbten
Lösung
von [(tert-Butylamido)(dimethyl)(tetramethyl-η5-cyclopentadienyl)silandimethyltitan
zugegeben. Das resultierende Gemisch wurde für wenige Minuten gerührt, um auf
diese Weise ein gelb-orange gefärbtes
festes Katalysatorsystem zu erhalten.
-
Ein gerührter 3 Liter Reaktor wurde
mit 1.191 ml ISOPARTM E (hergestellt und
vertrieben von Exxon Chemical Co., USA), 309 ml 1-Octen und 0,3
Liter Wasserstoff beschickt. Die Reaktorinhalte wurden auf 80°C erhitzt,
und Ethylen wurde zu dem Reaktor in einer ausreichenden Menge zugegeben,
um den Gesamtdruck des Reaktors auf etwa 31,9 kg/cm2 (31
atm) zu bringen. Das feste Katalysatorsystem, welches 1,5 μMol Titan enthielt,
wurde zu dem Reaktor zugegeben, um die Polymerisationsreaktion zu
starten. Ethylen wurde zu dem Reaktor konti nuierlich je nach Bedarf
angeliefert. Nach 21 Minuten wurde die Ethylenleitung blockiert,
und die Reaktorinhalte wurden in einen Probenbehälter abgelassen. Das resultierende
Copolymere wurde über
Nacht getrocknet. Als Ergebnis wurden 41 g eines Ethylencopolymeren
erhalten.
-
Das so erhaltene Ethylencopolymere
hatte die folgenden Eigenschaften: eine Dichte von 0,883 g/cm3; ein MFR von 0,35 g/10 Minuten, gemessen
bei 190°C
unter einer Belastung von 2,16 kg; ein Mw von 130.000 und ein Mw/Mn von 3,5, beide
gemessen mittels GPC; ein Mt (ein Punkt im Molekulargewicht auf
einem Molekulargewichtsverteilungsprofil, gemessen mittels GPC,
bei welchem das Profil einen Peak mit maximaler Intensität zeigte)
von 96.000; eine annähernd
gerade Linie, erhalten aus dem Comonomerengehalt-Verteilungsprofil)
hatte einen Gradienten von 0,0030 innerhalb des Bereiches von 30.000
bis 304.000 in Werten eines Molekulargewichtes Mc, welches die Formel
log (96.000) – log(Mc) ≤ 0,5 erfüllte; eine
Temperatur (bei welcher eine Maximalmenge der Extraktion gezeigt
wurde) von 37°C,
gemessen mittels CFC; bei der CFC wurde eine annähernd gerade Linie mit einem
Gradienten von –0,010
aus der Beziehung zwischen einer willkürlichen Temperatur, die in
den Bereich zwischen 37°C
und 47°C
fällt,
und einem Punkt auf einem Molekulargewichts-Verteilungsprofil einer
Copolymerfraktion, die bei der willkürlichen Temperatur extrahiert
wurde, wobei dieser Punkt einen Peak mit einer maximalen Intensität hatte,
erhalten, und die Gesamtmenge von Copolymerfraktionen, extrahiert
bei Temperaturen von 27°C
oder weniger bei der CFC, war 18,5 Gew.-%.
-
Vergleichsbeispiel 2
-
Ein 200 ml Glaskolben, der vollständig mit
Stickstoffgas gespült
worden war, wurde mit 4,0 g Siliziumdioxid (hergestellt und vertrieben
von Fuji Silysia Chemical Ltd., Japan) und 40 ml Toluol beschickt.
Das resultierende Gemisch wurde zum Erhalt einer Suspension gerührt. Die
erhaltene Suspension wurde auf –10°C abgekühlt. Zu
der abgekühlten
Suspension wurden tropfenweise 30 ml einer Lösung von Methylaluminoxan (hergestellt
und vertrieben von Albemarle Corporation, USA) in Toluol (Al-Konzentration:
1 Mol/Liter) während
1 Stunde in einer Stickstoffatmosphäre unter Halten der Temperatur
der Suspension auf –10°C zugegeben.
Das resultierende Gemisch wurde auf 0°C für 1 Stunde und dann bei Zimmertemperatur
für 1 Stunde
in der Stickstoffatmosphäre
gehalten. Danach wurde die Temperatur des Gemisches weiter auf 110°C erhöht, so daß das Gemisch
für 3 Stunden
in der Stickstoffatmosphäre
unter Rückfluß gehalten
wurde. Während
einer Reihe der oben genannten Arbeitsvorgänge wurde Bildung von Methangas
aus dem Gemisch beobachtet. Dann wurde das Gemisch auf 20°C abgekühlt, so
daß eine
Suspension von Siliziumdioxid, welche Methylaluminoxan hierauf trug,
erhalten wurde.
-
Ein 1,6 Liter Autoklav aus rostfreiem
Stahl, der vollständig
mit Stickstoffgas gespült
worden war, wurde mit 0,8 Liter Hexan beschickt, und dann wurden
0,2 mMol Triisobutylaluminium zu dem Hexan in dem Autoklaven zugegeben.
Zu der resultierenden Mischung wurde die oben erhaltene Suspension
von Siliziumdioxid in einer Menge von 0,3 mMol, in Werten des Aluminiums
des auf dem Siliziumdioxid getragenen Methylaluminoxans, zugegeben.
Ethylen wurde zu dem Autoklaven in einer ausreichenden Menge zugeführt, um
den Gesamtdruck des Autoklaven auf 7 kg/cm2-G
zu bringen. Die Innentemperatur des Autoklaven wurde auf 65°C eingestellt.
-
Eine Lösung von Bis(n-butylcyclopentadienyl)zirkoniumdichlorid
(welches als ein Metallocen bekannt ist) in Toluol wurde zu dem
Autoklaven in einer Menge von 1,0 μMol, angegeben als Zirkonium,
zugesetzt, und die Innentemperatur des Autoklaven wurde auf 70°C erhöht, um auf
diese Weise die Polymerisationsreaktion des Ethylens zu starten.
Das Ethylen wurde zu dem Autoklaven je nach Bedarf kontinuierlich
zugeführt.
-
Unter Halten des Gesamtdruckes des
Autoklaven auf 7 kg/cm2-G und Halten der
Innentemperatur des Autoklaven auf 70°C wurde die Polymerisationsreaktion
durchgeführt,
so daß der
Gesamtverbrauch des Ethylens 1,5 kg/cm2-G
wurde.
-
Nach Abschluß der Polymerisationsreaktion
wurde der Inhalt im Autoklaven in einen Behälter aus rostfreiem Stahl,
welcher Methanol enthielt, abgelassen. Das resultierende Gemisch
wurde filtriert, um hierdurch ein Polymeres zu erhalten. Das erhaltene
Polymere wurde bei 50°C über Nacht
getrocknet. Als Ergebnis wurde ein Ethylencopolymeres erhalten.
-
Der Autoklav wurde geöffnet, und
die Innenseite hiervon wurde untersucht. Es wurde kein an der Innenwand
des Autoklaven anhaftendes Polymeres beobachtet.
-
Das so erhaltene Ethylencopolymere
hatte die folgenden Eigenschaften: eine Dichte von 0,926 g/cm3; ein MFR von 2,1 g/10 Minuten, gemessen
bei 190°C
unter einer Belastung von 2,16 kg; ein Mw von 88.000 und ein Mw/Mn von 2,6, beide
gemessen mittels GPC; ein Mt (ein Punkt im Molekulargewicht auf
einem Molekulargewichts-Verteilungsprofil, gemessen mittels GPC,
bei welchem das Profil einen Peak mit einer maximalen Intensität zeigte)
von 63.000; eine annähernd
gerade Linie, erhalten aus dem Comonomeren-Verteilungsprofil) hatte
einen Gradienten von –0,00005
innerhalb des Bereiches von 20.000 bis 199.000 in Werten eines Molekulargewichtes
Mc, welches die Formel log(63.000) – log(Mc) ≤ 0,5 erfüllt; eine Temperatur (bei welcher eine
maximale Menge der Extraktion gezeigt wurde) von 85°C, gemessen
mittels CFC; bei der CFC wurde eine annähernd gerade Linie mit einem
Gradienten von –0,006
aus der Beziehung zwischen einer willkürlichen Temperatur, die innerhalb
des Bereiches von zwischen 85°C
und 95°C
fällt,
und einem Punkt auf einem Molekulargewichts-Verteilungsprofil einer
bei der willkürlichen
Temperatur extrahierten Copolymerfraktion, wobei dieser Punkt einen
Peak mit einer maximalen Intensität hatte, erhalten; und die
Gesamtmenge von bei Temperaturen von 75°C oder weniger extrahierten
Copolymerfraktionen bei der CFC war 10,8 Gew.-%.
-
Vergleichsbeispiel 3
-
Ein gerührter 3 Liter Reaktor wurde
mit 1.388 ml ISOPARTM E (hergestellt und
vertrieben von Exxon Chemical Co., USA), 128 ml 1-Octen und 0,3
Liter Wasserstoff beschickt. Der Reaktorinhalt wurde auf 130°C erhitzt,
und Ethylen wurde zu dem Reaktor in einer ausreichenden Menge zugegeben,
um den Gesamtdruck des Reaktors auf etwa 31,9 kg/cm2 (31
atm) zu bringen.
-
Ein festes Katalysatorsystem, enthaltend
0,625 μMol
Titan, das im wesentlichen in derselben Weise wie in Beispiel 1
hergestellt worden war, wurde zu dem Reaktor zugegeben, um hierdurch
eine Polymerisationsreaktion zu starten. Ethylen wurde zu dem Reaktor
kontinuierlich je nach Bedarf zugeführt. Nach 10 Minuten wurde
die Ethylenleitung abgeschaltet, und der Reaktorinhalt wurde in
einen Probenbehälter
abgelassen. Das resultierende Polymere wurde über Nacht getrocknet, um auf
diese Weise 30 g eines Ethylencopolymeren zu erhalten.
-
Das so erhaltene Ethylencopolymere
hatte die folgenden Eigenschaften: eine Dichte von 0,912 g/cm3, ein MFR von 4,5 g/10 Minuten, gemessen
bei 190°C
unter einer Belastung von 2,16 kg; ein Mw von 130.000 und ein Mw/Mn von 2,5, beide
gemessen mittels GPC; ein Mt (ein Punkt im Molekulargewicht auf
einem Molekulargewichts-Verteilungsprofil, gemessen mittels GPC,
bei welchem das Profil einen Peak mit einer maximalen Intensität zeigte)
von 50.000; eine annähernd
gerade Linie, erhalten aus dem Comonomerengehalt-Verteilungsprofil)
hatte einen Gradienten von 0,00003 innerhalb des Bereiches von 16.000
bis 158.000 in Werten eines Molekulargewichtes Mc, welches die Formel
log(50.000) – log(mc) ≤ 0,5 erfüllt; eine
Temperatur (bei welcher eine maximale Menge von Extraktion gezeigt
wurde) von 74°C,
gemessen mittels CFC; bei der CFC wurde eine annähernd gerade Linie mit einem
Gradienten von –0,033
aus der Beziehung zwischen einer willkürlichen Temperatur, die innerhalb
des Bereiches von zwischen 74°C
und 84°C fällt, und
einem Punkt auf einem Molekulargewichts-Verteilungsprofil einer
bei der willkürlichen
Temperatur extrahierten Copolymerfraktion, wobei dieser Punkt einen
Peak mit einer maximalen Intensität hat, erhalten; und die Gesamtmenge
der Copolymerfraktionen, extrahiert bei Temperaturen von 64°C oder weniger
bei der CFC, war 13,4 Gew.-%.
-
Vergleichsbeispiel 4
-
Ein kommerziell erhältliches
Ethylencopolymeres, EXACTTM 3029 (hergestellt
und vertrieben von Exxon Chemical Co., USA), wurde analysiert. Die
Ergebnisse der Analyse sind in Tabelle 2 gezeigt.
-
Vergleichsbeispiel 5
-
Ein kommerziell erhältliches
Ethylencopolymeres, SP 2040 (hergestellt und vertrieben vom Mitsui
Petrochemical Industries, Ltd., Japan), wurde analysiert. Die Ergebnisse
der Analyse sind in Tabelle 2 gezeigt.
-
Vergleichsbeispiel 6
-
Ein kommerziell erhältliches
Ethylencopolymeres, PL 1880 (hergestellt und vertrieben von The
Dow Chemical Co., USA), wurde analysiert. Die Ergebnisse der Analyse
sind in Tabelle 2 gezeigt.
-
In Tabelle 2 sind die Eigenschaften
des Ethylencopolymeren der vorliegenden Erfindung, das in Beispiel
1 erhalten wurde, zusammen mit denjenigen der Ethylencopolymere,
welche einzeln in den Vergleichsbeispielen 1 bis 3 erhalten wurden
und diejenigen der kommerziell erhältlichen Ethylencopolymere
in den Vergleichsbeispielen 4 bis 6 angegeben. Es ist offensichtlich,
daß beliebige
der Ethylencopolymere in den Vergleichsbeispielen 1 bis 6 nicht
die Eigenschaften besitzen, welche mit denjenigen der Ethylencopolymere
der vorliegenden Erfindung übereinstimmen.
-
Vergleichsbeispiel 7
-
Ein auf Magnesiumchlorid getragener
Ziegler-Natta-Katalysator
mit etwa 2 Gew.-% Ti auf der Oberfläche des Trägers wurde für die Polymerisation
verwendet.
-
Hexan, Ethylen, 1-Buten, Wasserstoff
und das feste Katalysatorsystem wurden kontinuierlich in einen kontinuierlich
gerührten
Tankreaktor, im wesentlichen wie in Beispiel 1 beschrieben, eingespeist,
um das Copolymerpulver zu erzeugen, dessen Eigenschaften in Tabelle
2 gezeigt sind.
-
Beispiele 9–12
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in Beispiel 1 wurden mit der Ausnahme angewandt, daß der zur
Herstellung der Komponenten 1 und 2 verwendete Übergangsmetallkomplex [(tert-Butylamido)(dimethyl)(tetramethyl-η5-cyclopentadienyl)silandimethyltitan
war, und daß die
Strömungsgeschwindigkeiten
von Isopentan, 1-Buten und Wasserstoff verändert wurden, um die Polymereigenschaften
für die
in Tabelle 3 aufgeführten
Komponenten I und II zu ergeben. Die Mischungen wurden in einem
Mischer Winkworth 2Z-blade hergestellt. Dieser Mischer ist mit zwei
Mischblättern,
die bei unterschiedlichen Upm laufen, ausgerüstet: die vordere Schnecke
rotiert mit 52 Upm, die rückwärtige Schnecke
mit 30 Upm. Die Arbeitskapazität ist
1,2 Liter.
-
Die Pulver wurden zuerst mit 2000
ppm Irganox® B225,
erhältlich
von Ciba Geigy, trockengemischt. Beladungen von 350 g des Gemisches
mit der gewünschten
Zusammensetzung wurden dann eingefüllt und für 10 Minuten bei 190°C gemischt.
Nach dem Mischen wurde das Polymere entfernt und in einer Mahleinrichtung
Heinrich Dreher S20 gemahlen. Das gemahlene Polymere war dann für die Preßverformung
fertig. Die Ergebnisse des Tests der verschiedenen Mischungszusammensetzungen
sind in Tabelle 4 gezeigt.
-
Beispiele 13–16
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in den Beispielen 7 und 8 wurden wiederholt, um die Ethylencopolymere,
welche als Komponente II eingesetzt wurden, mit der Ausnahme herzustellen,
daß die
Strömungsgeschwindigkeit
von Hexan, Ethylen, 1-Buten, Wasserstoff zusammen mit der Polymerisationstemperatur
eingeregelt wurden. Die in den Beispielen 7 und 8 verwendeten Ethylencopolymere wurden
als Komponente I verwendet. Die Polymereigenschaften für Komponente
I und Komponente II und die Mischungsformulierung sind in Tabelle
5 zusammengefaßt.
Die Mischungen wurden unter Verwendung des Doppelschneckenextruders
(PCM-45, hergestellt von IKEGAI, Co., Ltd., Japan) hergestellt.
Die Schnecke rotierte mit 100–200
Upm. Die Schneckenzylinder-Temperatur betrug 220°C.
-
Die Pulver wurden zuerst mit 2000
ppm Irganox® 1076,
erhältlich
von Ciba Geigy, 600 ppm Calciumstearat und 1000 ppm P-EPQ®,
erhältlich
von Sando, trockengemischt. Die Ergebnisse des Tests der verschiedenen
Mischungszusammensetzungen sind in Tabelle 6 gezeigt.
-
Vergleichsbeispiele 8–10
-
Im wesentlichen dieselben Polymerisationsarbeitsweisen
wie in Vergleichsbeispiel 7 wurden angewandt, um die Ethylenhomopolymere,
die für
Komponente II verwendet wurden, herzustellen. Ebenfalls wurde das
in Vergleichsbeispiel 7 erzeugte Ethylencopolymere als Komponente
2 verwendet. Die Polymereigenschaften für Komponente I und Komponente
II und die Mischungsformulierung sind in Tabelle 5 zusammengefaßt. Die
Mischungen wurden unter Anwendung derselben Arbeitsweise wie in
den Beispielen 13–16
hergestellt. Die Ergebnisse des Tests der verschiedenen Mischungszusammensetzungen
sind in Tabelle 6 gezeigt.
-
Die Beispiele 9–16 zeigen das ausgezeichnete
Gleichgewicht der Eigenschaften wie der Schlagfestigkeit bei niedriger
Temperatur (gemessen mittels Gc bei 0 und –20°C und mittels
Charpy-Schlagfestigkeit –20°C), die Verarbeitbarkeit
(gemessen durch V bei 100 l/s und durch I21,6)
und ESCR (gemessen durch PENT und Biegen-ESCR-TEST) für die verschiedenen
Mischungszusammensetzungen, und daß ein solches Gleichgewicht
am besten dadurch erreicht wird, daß das Comonomere bevorzugt
in der Komponente mit hohem Molekulargewicht vorliegt. Weiterhin
wird deutlich, daß die
Beispiele 13–16 überlegenere
Eigenschaften zeigen, verglichen mit denjenigen der Vergleichsbeispiele
8–10.
-
-
-
-
-
-
-












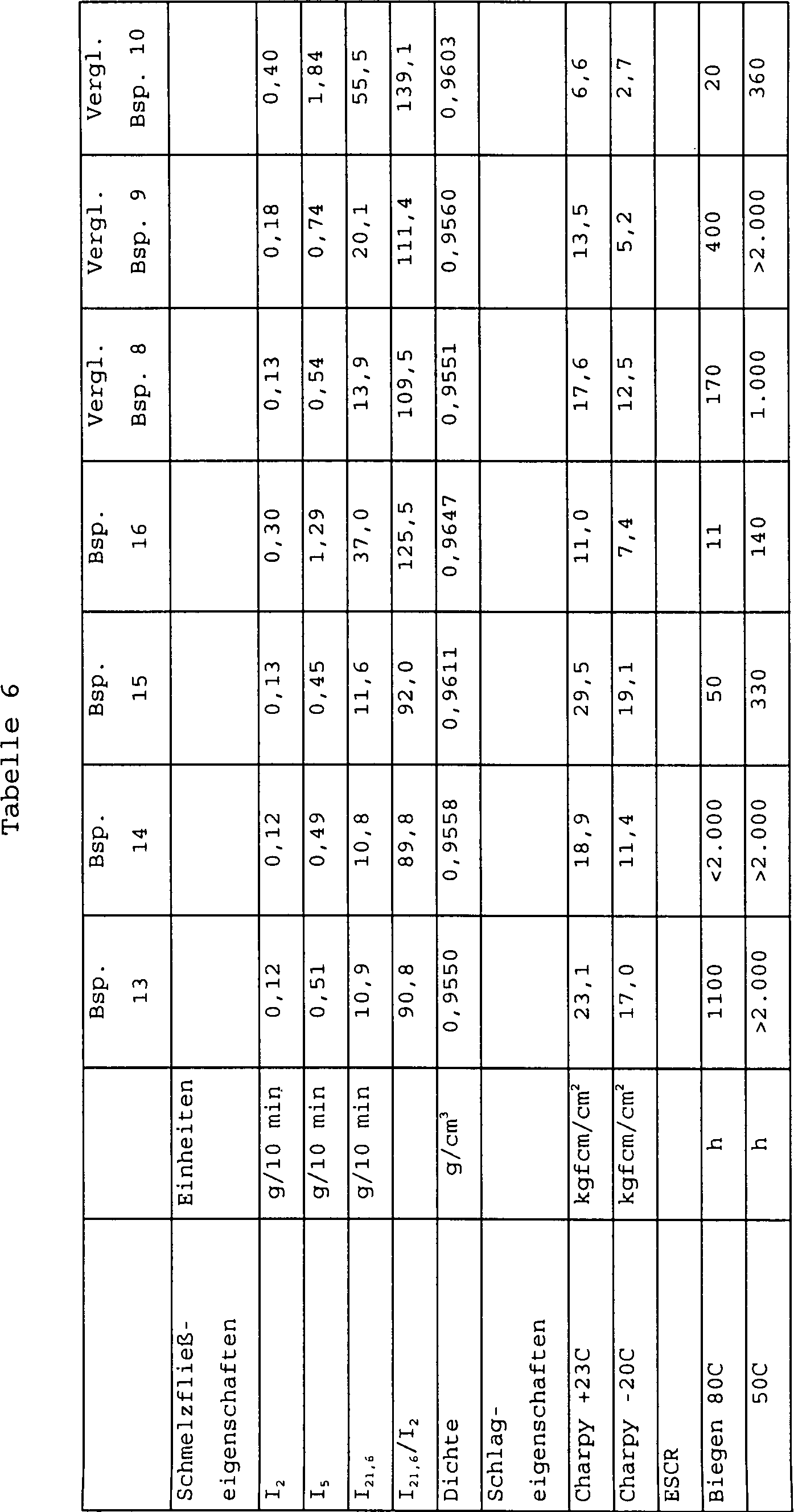
 worin: M Titan oder Zirkonium im formalen Oxidationszustand +2 ist; L eine Gruppe ist, die ein cyclisches, delokalisiertes, anionisches π-System enthält, durch welches die Gruppe an M gebunden ist, und diese Gruppe ebenfalls an Z gebunden ist; Z eine Einheit ist, gebunden an M über eine σ-Bindung, umfassend Bor oder ein Glied von Gruppe 14 des Periodensystems der Elemente, und ebenfalls umfassend Stickstoff, Phosphor, Schwefel oder Sauerstoff, wobei diese Einheit bis zu 60 Nicht-Wasserstoffatome hat; und X* ein neutrales, konjugiertes oder nicht-konjugiertes Dien ist, wahlweise substituiert mit einer oder mehreren Hydrocarbylgruppen, wobei dieses X bis zu 40 Kohlenstoffatome hat und einen π-Komplex mit M bildet.
worin: M Titan oder Zirkonium im formalen Oxidationszustand +2 ist; L eine Gruppe ist, die ein cyclisches, delokalisiertes, anionisches π-System enthält, durch welches die Gruppe an M gebunden ist, und diese Gruppe ebenfalls an Z gebunden ist; Z eine Einheit ist, gebunden an M über eine σ-Bindung, umfassend Bor oder ein Glied von Gruppe 14 des Periodensystems der Elemente, und ebenfalls umfassend Stickstoff, Phosphor, Schwefel oder Sauerstoff, wobei diese Einheit bis zu 60 Nicht-Wasserstoffatome hat; und X* ein neutrales, konjugiertes oder nicht-konjugiertes Dien ist, wahlweise substituiert mit einer oder mehreren Hydrocarbylgruppen, wobei dieses X bis zu 40 Kohlenstoffatome hat und einen π-Komplex mit M bildet.