-
Die
Erfindung betrifft Aryl-cycloalkyl substituierte Alkansäurederivate
sowie deren physiologisch verträgliche
Salze und physiologisch funktionelle Derivate.
-
Es
sind bereits strukturähnliche
Verbindungen zur Behandlung von Hyperlipidämie und Diabetes im Stand der
Technik beschrieben (WO 2000/64876 (HOE 1999/S 004)).
-
Der
Erfindung lag die Aufgabe zugrunde, Verbindungen zur Verfügung zu
stellen, die eine therapeutisch verwertbare Triglycerid-senkende
Wirkung entfalten mit günstiger
Beeinflussung des Lipid-und Kohlenhydratstoffwechsels, besonders
bei den Krankheitsbildern der Dyslipidämien, des Diabetes Typ II und
des metabolischen Syndroms/Syndrom X. Insbesondere bestand die Aufgabe
darin, Verbindungen mit verbesserter Wirkung gegenüber den
Verbindungen aus WO 2000/64876 zur Verfügung zu stellen. Dies soll
insbesonders durch eine Aktivierung des PPARα-Rezeptor erreicht werden.
-
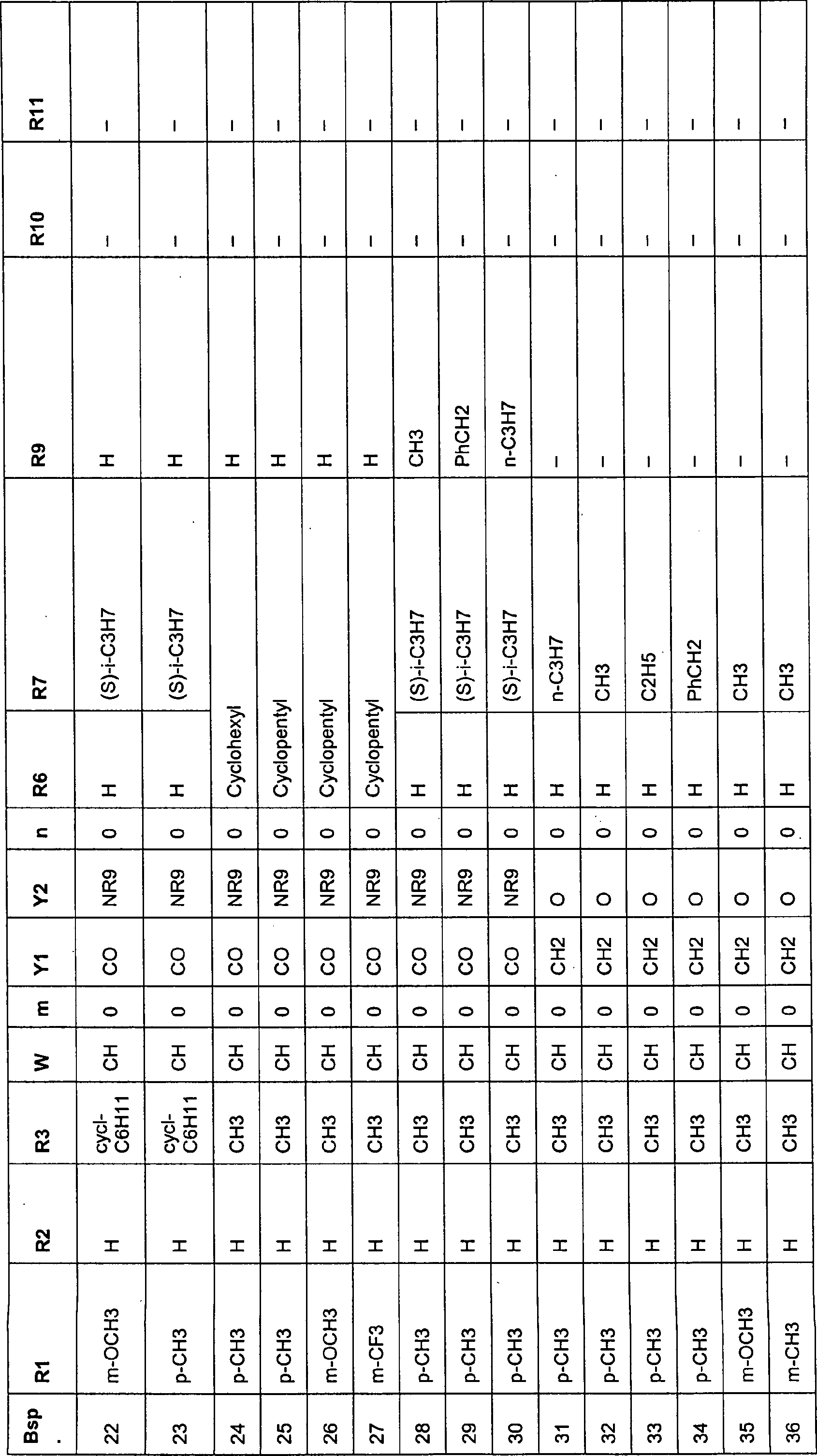
Die
Erfindung betrifft daher Verbindungen der Formel I
worin
bedeuten:
Ring A (C3-C8)-Cycloalkandiyl, (C3-C8)-Cycloalkendiyl,
wobei in den Cycloalkandiyl- oder Cycloalkendiylringen ein oder
mehrere Kohlenstoffatome durch Sauerstoffatome ersetzt sein können;
R1,
R2 unabhängig
voneinander H, F, Br, CF3, OCF3, (C1-C6)-Alkyl, O-(C1-C6)-Alkyl, SCF3,
SF5, OCF2-CHF2, O-Phenyl, OH, NO2,
R3 H, CF3, (C1-C6)-Alkyl,
(C3-C8)-Cycloalkyl, Phenyl;
W CH, falls o = 1;
W O, S,
falls o = 0;
X (C1-C6)-Alkyl, wobei in der Alkandiylgruppe
ein oder mehrere Kohlenstoffatome durch Sauerstoffatome ersetzt
sein können;
m
0 oder 1;
Y1 CO, CH2, Bindung;
Y2 CH2, O, S, SO, SO2,
NR9;
n 0-2;
R4 H, F;
R5 H, F;
R6 H, F, (C1-C6)-Alkyl;
R7
H, F, (C1-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C1-C6)-Alkoxy,
Cyclohexyl, Phenyl, wobei Alkyl und Alkoxy gegebenenfalls substituiert
sein können
durch: Phenyl, (C1-C6)-Alkoxy oder NR10R11 und Phenyl wiederum durch
(C1-C6)-Alkoxy, F oder CF3;
R7 und R9 zusammen Pyrrolidin oder
Piperidin, falls n = 0;
R6 und R7 zusammen (C3-C6)-Cycloalkyl;
Z
O;
R8 H, (C1-C6)-Alkyl;
R9 H, (C1-C6)-Alkyl, (C2-C6)-Alkenyl,
(C2-C6)-Alkinyl, Benzyl, CO-(C1-C6)-Alkyl,
CO-Phenyl, C(O)-O-(C1-C6)-Alkyl, SO2-(C1-C6)-Alkyl, SO2-(C1-C6)-Alkyl-SO2-(C1-C6)-alkyl,
SO2-Phenyl, wobei Phenyl gegebenenfalls substituiert sein kann durch
(C1-C6)-Alkyl, (C1-C6)-Alkoxy,
F, Cl.
R10 (C1-C6)-Alkyl;
R11 (C1-C6)-Alkyl-Phenyl, (C1-C6)-Alkyl;
sowie
deren physiologisch verträgliche
Salze.
-
Bevorzugt
sind Verbindungen der Formel I, in denen
Ring A (C3-C8)-Cycloalkandiyl, (C3-C8)-Cycloalkendiyl, wobei in den Cycloalkandiyl-oder
Cycloalkendiylringen ein Kohlenstoffatom durch Sauerstoffatom ersetzt
sein kann;
X (C1-C6)-Alkyl, wobei in der Alkandiylgruppe das
C1-Kohlenstoffatom durch Sauerstoffatom ersetzt ist.
-
Besonders
bevorzugt sind Verbindungen der Formel I,
worin bedeuten
Ring
A Cyclohexan-1,3-diyl
R1 H, F, Br, CF3, OCF3, (C1-C6)-Alkyl,
O-(C1-C6)-Alkyl;
R2 H;
R3 H, CF3, (C1-C6)-Alkyl, (C3-C8)-Cycloalkyl,
Phenyl;
W CH, falls o = 1;
W O, S, falls o = 0;
X
CH2O
m 0 oder 1;
Y1 CO, CH2, Bindung;
Y2
CH2, O, S, SO, SO2, NR9;
n 0-2;
R4 H;
R5 H;
R6
H, (C1-C6)-Alkyl;
R7 H, (C1-C6)-Alkyl, (C1-C6)-Alkoxy, Cyclohexyl,
Phenyl, wobei Alkyl und Alkoxy gegebenenfalls substituiert sein
können
durch: Phenyl, (C1-C6)-Alkoxy
oder NR10R11 und Phenyl wiederum durch (C1-C6)-Alkyl, Foder CF3;
R7
und R9 zusammen Pyrrolidin, falls n = 0;
R6 und R7 zusammen
(C3-C6)-Cycloalkyl;
Z O;
R8 H, (C1-C6)-Alkyl;
R9
H, (C1-C6)-Alkyl, Benzyl, CO-(C1-C6)-Alkyl, CO-Phenyl, C(O)-O-(C1-C6)-Alkyl, SO2-(C1-C4)-Alkyl, SO2-(C1-C4)-Alkyl-SO2-(C1-C4)-alkyl,
SO2-Tolyl;
R10 (C1-C6)-Alkyl;
R11 (C1-C6)-Alkyl-Phenyl;
sowie
deren physiologisch verträgliche
Salze.
-
Die
Erfindung bezieht sich auf Verbindungen der Formel I, in Form ihrer
Racemate, racemischen Mischungen und reinen Enantiomere sowie auf
ihre Diastereomere und Mischungen davon.
-
Die
Alkylreste in den Substituenten R1, R2, R3, R4, R5, R6, R7, R8,
R9, R10 und R11 können
sowohl geradkettig wie verzweigt sein.
-
Pharmazeutisch
verträgliche
Salze sind aufgrund ihrer höheren
Wasserlöslichkeit
gegenüber
den Ausgangs-bzw. Basisverbindungen besonders geeignet für medizinische
Anwendungen. Diese Salze müssen
ein pharmazeutisch verträgliches
Anion oder Kation aufweisen. Geeignete pharmazeutisch verträgliche Säureadditionssalze
der erfindungsgemäßen Verbindungen
sind Salze anorganischer Säuren,
wie Salzsäure,
Bromwasserstoff-, Phosphor-, Metaphosphor-, Salpeter-und Schwefelsäure sowie
organischer Säuren,
wie z.B. Essigsäure,
Benzolsulfon-, Benzoe-, Zitronen-, Ethansulfon-, Fumar-, Glucon-,
Glykol-, Isethion-, Milch-, Lactobion-, Malein-, Äpfel-, Methansulfon-,
Bernstein-, p-Toluolsulfon-und
Weinsäure.
Geeignete pharmazeutisch verträgliche
basische Salze sind Ammoniumsalze, Alkalimetallsalze (wie Natrium-und
Kaliumsalze) und Erdalkalisalze (wie Magnesium-und Calciumsalze).
-
Salze
mit einem nicht pharmazeutisch verträglichen Anion, wie zum Beispiel
Trifluoracetat, gehören ebenfalls
in den Rahmen der Erfindung als nützliche Zwischenprodukte für die Herstellung
oder Reinigung pharmazeutisch verträglicher Salze und/oder für die Verwendung
in nicht-therapeutischen, zum Beispiel in-vitro-Anwendungen.
-
Der
hier verwendete Begriff "physiologisch
funktionelles Derivat" bezeichnet
jedes physiologisch verträgliche
Derivat einer erfindungsgemäßen Verbindung
der Formel I, z.B. einen Ester, der bei Verabreichung an einen Säuger, wie
z.B. den Menschen, in der Lage ist, (direkt oder indirekt) eine
Verbindung der Formel I oder einen aktiven Metaboliten hiervon zu
bilden.
-
Zu
den physiologisch funktionellen Derivaten zählen auch Prodrugs der erfindungsgemäßen Verbindungen,
wie zum Beispiel in H. Okada et al., Chem. Pharm. Bull. 1994, 42,
57-61 beschrieben. Solche Prodrugs können in vivo zu einer erfindungsgemäßen Verbindung
metabolisiert werden. Diese Prodrugs können selbst wirksam sein oder
nicht.
-
Die
erfindungsgemäßen Verbindungen
können
auch in verschiedenen polymorphen Formen vorliegen, z.B. als amorphe
und kristalline polymorphe Formen. Alle polymorphen Formen der erfindungsgemäßen Verbindungen
gehören
in den Rahmen der Erfindung und sind ein weiterer Aspekt der Erfindung.
-
Nachfolgend
beziehen sich alle Verweise auf "Verbindung(en)
gemäß Formel
I" auf Verbindungen)
der Formel I wie vorstehend beschrieben, sowie ihre Salze, Solvate
und physiologisch funktionellen Derivate wie hierin beschrieben.
-
Die
Menge einer Verbindung gemäß Formel
I, die erforderlich ist, um den gewünschten biologischen Effekt
zu erreichen, ist abhängig
von einer Reihe von Faktoren, z.B. der gewählten spezifischen Verbindung, der
beabsichtigten Verwendung, der Art der Verabreichung und dem klinischen
Zustand des Patienten. Im allgemeinen liegt die Tagesdosis im Bereich
von 0,3 mg bis 100 mg (typischerweise von 3 mg und 50 mg) pro Tag
pro Kilogramm Körpergewicht,
z.B. 3-10 mg/kg/Tag. Eine intravenöse Dosis kann z.B. im Bereich
von 0,3 mg bis 1,0 mg/kg liegen, die geeigneterweise als Infusion
von 10 ng bis 100 ng pro Kilogramm pro Minute verabreicht werden
kann. Geeignete Infusionslösungen
für diese
Zwecke können
z.B. von 0,1 ng bis 10 mg, typischerweise von 1 ng bis 10 mg pro
Milliliter, enthalten. Einzeldosen können z.B. von 1 mg bis 10 g
des Wirkstoffs enthalten. Somit können Ampullen für Injektionen
beispielsweise von 1 mg bis 100 mg, und oral verabreichbare Einzeldosisformulierungen,
wie zum Beispiel Tabletten oder Kapseln, können beispielsweise von 1,0 bis
1000 mg, typischerweise von 10 bis 600 mg enthalten. Zur Therapie
der oben genannten Zustände
können die
Verbindungen gemäß Formel
I selbst als Verbindung verwendet werden, vorzugsweise liegen sie
jedoch mit einem verträglichen
Träger
in Form einer pharmazeutischen Zusammensetzung vor. Der Träger muss
natürlich
verträglich
sein, in dem Sinne, dass er mit den anderen Bestandteilen der Zusammensetzung
kompatibel ist und nicht gesundheitsschädlich für den Patienten ist. Der Träger kann
ein Feststoff oder eine Flüssigkeit oder
beides sein und wird vorzugsweise mit der Verbindung als Einzeldosis
formuliert, beispielsweise als Tablette, die von 0,05% bis 95 Gew.-%
des Wirkstoffs enthalten kann. Weitere pharmazeutisch aktive Substanzen
können
ebenfalls vorhanden sein, einschließlich weiterer Verbindungen
gemäß Formel
I. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen können
nach einer der bekannten pharmazeutischen Methoden hergestellt werden,
die im wesentlichen darin bestehen, dass die Bestandteile mit pharmakologisch verträglichen
Träger-und/oder
Hilfsstoffen gemischt werden.
-
Erfindungsgemäße pharmazeutische
Zusammensetzungen sind solche, die für orale, rektale, topische,
perorale (z.B. sublinguale) und parenterale (z.B. subkutane, intramuskuläre, intradermale
oder intravenöse)
Verabreichung geeignet sind, wenngleich die geeignetste Verabreichungsweise
in jedem Einzelfall von der Art und Schwere des zu behandelnden
Zustandes und von der Art der jeweils verwendeten Verbindung gemäß Formel
I abhängig
ist. Auch dragierte Formulierungen und dragierte Retardformulierungen
gehören
in den Rahmen der Erfindung. Bevorzugt sind säure-und magensaftresistente
Formulierungen. Geeignete magensaftresistente Beschichtungen umfassen
Celluloseacetatphthalat, Polyvinalacetatphthalat, Hydroxypropylmethylcellulosephthalat
und anionische Polymere von Methacrylsäure und Methacrylsäuremethylester.
-
Geeignete
pharmazeutische Verbindungen für
die orale Verabreichung können
in separaten Einheiten vorliegen, wie zum Beispiel Kapseln, Oblatenkapseln,
Lutschtabletten oder Tabletten, die jeweils eine bestimmte Menge
der Verbindung gemäß Formel
I enthalten; als Pulver oder Granulate; als Lösung oder Suspension in einer
wässrigen
oder nicht-wässrigen
Flüssigkeit;
oder als eine Öl-in-Wasser- oder Wasser-in Öl-Emulsion.
Diese Zusammensetzungen können,
wie bereits erwähnt,
nach jeder geeigneten pharmazeutischen Methode zubereitet werden,
die einen Schritt umfasst, bei dem der Wirkstoff und der Träger (der
aus einem oder mehreren zusätzlichen
Bestandteilen bestehen kann) in Kontakt gebracht werden. Im allgemeinen werden
die Zusammensetzungen durch gleichmäßiges und homogenes Vermischen
des Wirkstoffs mit einem flüssigen
und/oder feinverteilten festen Träger hergestellt, wonach das
Produkt, falls erforderlich, geformt wird. So kann beispielsweise
eine Tablette hergestellt werden, indem ein Pulver oder Granulat
der Verbindung verpresst oder geformt wird, gegebenenfalls mit einem
oder mehreren zusätzlichen
Bestandteilen. Gepresste Tabletten können durch tablettieren der
Verbindung in frei fließender
Form, wie beispielsweise einem Pulver oder Granulat, gegebenenfalls
gemischt mit einem Bindemittel, Gleitmittel, inertem Verdünner und/oder
einem (mehreren) oberflächenaktiven/dispergierenden
Mittel in einer geeigneten Maschine hergestellt werden. Geformte
Tabletten können
durch Formen der pulverförmigen,
mit einem inerten flüssigen
Verdünnungsmittel
befeuchteten Verbindung in einer geeigneten Maschine hergestellt
werden.
-
Pharmazeutische
Zusammensetzungen, die für
eine perorale (sublinguale) Verabreichung geeignet sind, umfassen
Lutschtabletten, die eine Verbindung gemäß Formel I mit einem Geschmacksstoff
enthalten, üblicherweise
Saccharose und Gummi arabicum oder Tragant, und Pastillen, die die
Verbindung in einer inerten Basis wie Gelatine und Glycerin oder
Saccharose und Gummi arabicum umfassen.
-
Geeignete
pharmazeutische Zusammensetzungen für die parenterale Verabreichung
umfassen vorzugsweise sterile wässrige
Zubereitungen einer Verbindung gemäß Formel I, die vorzugsweise
isotonisch mit dem Blut des vorgesehenen Empfängers sind. Diese Zubereitungen
werden vorzugsweise intravenös
verabreicht, wenngleich die Verabreichung auch subkutan, intramuskulär oder intradermal
als Injektion erfolgen kann. Diese Zubereitungen können vorzugsweise
hergestellt werden, indem die Verbindung mit Wasser gemischt wird
und die erhaltene Lösung
steril und mit dem Blut isotonisch gemacht wird. Injizierbare erfindungsgemäße Zusammensetzungen
enthalten im allgemeinen von 0,1 bis 5 Gew.-% der aktiven Verbindung.
-
Geeignete
pharmazeutische Zusammensetzungen für die rektale Verabreichung
liegen vorzugsweise als Einzeldosis-Zäpfchen vor. Diese können hergestellt
werden, indem man eine Verbindung gemäß Formel I mit einem oder mehreren
herkömmlichen
festen Trägern,
beispielsweise Kakaobutter, mischt und das entstehende Gemisch in
Form bringt.
-
Geeignete
pharmazeutische Zusammensetzungen für die topische Anwendung auf
der Haut liegen vorzugsweise als Salbe, Creme, Lotion, Paste, Spray,
Aerosol oder Öl
vor. Als Träger
können
Vaseline, Lanolin, Polyethylenglycole, Alkohole und Kombinationen
von zwei oder mehreren dieser Substanzen verwendet werden. Der Wirkstoff
ist im allgemeinen in einer Konzentration von 0,1 bis 15 Gew.-%
der Zusammensetzung vorhanden, beispielsweise von 0,5 bis 2%.
-
Auch
eine transdermale Verabreichung ist möglich. Geeignete pharmazeutische
Zusammensetzungen für
transdermale Anwendungen können
als einzelne Pflaster vorliegen, die für einen langzeitigen engen Kontakt
mit der Epidermis des Patienten geeignet sind. Solche Pflaster enthalten
geeigneterweise den Wirkstoff in einer gegebenenfalls gepufferten
wässrigen
Lösung,
gelöst
und/oder dispergiert in einem Haftmittel oder dispergiert in einem
Polymer. Eine geeignete Wirkstoff-Konzentration beträgt ca. 1% bis 35%, vorzugsweise
ca. 3% bis 15%. Als eine besondere Möglichkeit kann der Wirkstoff,
wie beispielsweise in Pharmaceutical Research, 2(6): 318 (1986)
beschrieben, durch Elektrotransport oder Iontophorese freigesetzt
werden.
-
Die
erfindungsgemäßen Verbindungen
der Formel I können
entsprechend den folgenden Reaktionsschemata erhalten werden:
-
-
Die
Verbindung A-1 wird bei Raumtemperatur in Methanol mit Natriummethanolat
gerührt.
Nach Aufarbeitung wird das Produkt an der Hydroxylgruppe geschützt (SG
= Schutzgruppe), beispielsweise durch Umsetzen mit tert-Butyldiphenylsilylchlorid
und Imidazol in Dimethylformamid bei Raumtemperatur oder mit Methoxymethylchlorid,
Ethyldiisopropylamin in Dichlormethan. Dabei wird die Verbindung
A-2 erhalten.
-
Die
Verbindung A-2 wird in Isopropanol mit Natriumhydroxid 1 Stunde
bei 60°C
gerührt
und aufgearbeitet. Die so erhaltenen Carbonsäure wird in Dimethylformamid
mit dem tert-Butylester einer α-Aminosäure der
allgemeinen Formel A-3, worin R6 und R7 die oben beschriebenen Bedeutungen
haben, Hydroxybenzotriazol, Diisopropylethylamin und O[Cyan(ethoxycarbonyl)methylenamino]-1,1,3,3,-tetramethyluronium tetrafluoroborat
(TOTU) zum Produkt der allgemeinen Formel A-4, worin R9 = H ist,
umgesetzt. In einigen Beispielen wird das Kupplungsprodukt mit Natriumhydrid
und einem Alkyliodid der allgemeinen Formel R9-I, wobei R9 die oben
beschriebene Bedeutung hat – außer R9 =
H –, zur
Verbindung der allgemeinen Formel A-4 umgesetzt.
-
Die
Verbindung A-4 wird nun zur Verbindung A-5 O-entschützt, beispielsweise
mit Tetrabutylammoniumfluorid in Tetrahydrofuran im Falle der tert-Butyldiphenylsilylschutzgruppe
oder mit konzentrierter Salzsäure in
Tetrahydrofuran im Falle der Methoxymethylschutzgruppe.
-
Die
Verbindung der allgemeinen Formel A-5 wird mit Natriumhydrid und
der Verbindung der allgemeinen Formel A-6, worin R1, R2, R3 und
W die oben beschriebenen Bedeutungen haben, in Dimethylformamid umgesetzt.
Das Produkt wird mehrere Stunden in Trifluoressigsäure gerührt, und
anschließend
werden, falls notwendig die Diastereomeren per präparativer
HPLC getrennt. Dabei wird die Verbindung der allgemeinen Formel
A-7 erhalten.
-
Nach
diesem Verfahren können
die Beispiele 1 bis 30 synthetisiert werden.
-
-
Die
Verbindung A-2 (siehe Verfahren A) wird in Diethylether mit Lithiumaluminiumhydrid
zur Verbindung B-1 reduziert. Die Verbindung B-1 wird in einem Zweiphasensystem
aus Toluol und 50%-iger Natriumhydroxidlösung bei 10 °C mit Bromessigsäure-tert-butylester
und Tetrabutylammoniumhydrogensulfat zur Verbindung B-2 umgesetzt.
-
Die
Verbindung B-2 wird in Tetrahydrofuran mit Lithiumdiisopropylamid
und einem Alkyliodid der allgemeinen Formel R6-I, worin R6 die oben
beschriebene Bedeutung hat, umgesetzt. In einigen Beispielen wird die
so erhaltene Verbindung in Tetrahydrofuran mit Lithiumdiisopropylamid
und einem weiteren Alkyliodid der allgemeinen Formel R7-I, worin
R7 die oben beschriebene Bedeutung hat, umgesetzt. Die Schutzgruppe
wird abgespalten, wobei die Verbindung der allgemeinen Formel B-3
erhalten wird.
-
Die
Verbindung B-3 wird in Methyl-tert-butylether oder Dimethylformamid
mit Natriumhydrid und der Verbindung A-6 (siehe Verfahren A), worin
R1, R2, R3 und W die oben beschriebenen Bedeutungen haben, zur Verbindung
B-4 umgesetzt.
-
Das
Produkt B-4 wird mehrere Stunden in Trifluoressigsäure gerührt. Dabei
wird die Verbindung der allgemeinen Formel B-5 erhalten.
-
Nach
diesem Verfahren können
die Beispiele 31 bis 51 synthetisiert werden.
-
-
Die
Verbindung A-2, wobei SG = tert-Butyldimethylsilyl ist, wird mit
Wismuttribromid, Triethylsilan und einer Verbindung der allgemeinen
Formel C1, worin R1, R2, W und R3 die oben beschriebenen Bedeutungen haben,
in Acetonitril bei Raumtemperatur zur Verbindung C-2 umgesetzt.
-
Die
Verbindung C-2 wird mit Lithiumaluminiumhydrid in Diethylether zur
Verbindung C-3 reduziert. Die Verbindung C-3 wird mit Triphenylphosphin
und Iod in Toluol bei Raumtemperatur zur Verbindung C-4 umgesetzt.
-
Die
Verbindung C-4 wird mit der Verbindung der allgemeinen Formel C-5,
wobei R6 und R7 die oben beschriebenen Bedeutungen haben, zur Verbindung
C-6 umgesetzt. Der Ester wird gespalten, indem die Verbindung C-6
mehrere Stunden in einer Mischung aus Methanol und konzentrierter
Kalilauge gerührt
wird. Dabei wird die Verbindung C-7 erhalten.
-
In
einigen Beispielen wird die Verbindung C-7 mit einem Äquivalent
Wasserstoffperoxid in Trifluoressigsäure bei Raumtemperatur zur
Verbindung der allgemeinen Formel C-8, worin R1, R2, R3, R6, W und
R7 die oben beschriebenen Bedeutungen haben, oxidiert.
-
In
einigen Beispielen wird die Verbindung C-7 mit drei Äquivalenten
Wasserstoffperoxid in Trifluoressigsäure bei Raumtemperatur zur
Verbindung der allgemeinen Formel C-9, worin R1, R2, R3, R6, W und
R7 die oben beschriebenen Bedeutungen haben, oxidiert.
-
Nach
diesem Verfahren können
die Beispiele 52 bis 71 synthetisiert werden.
-
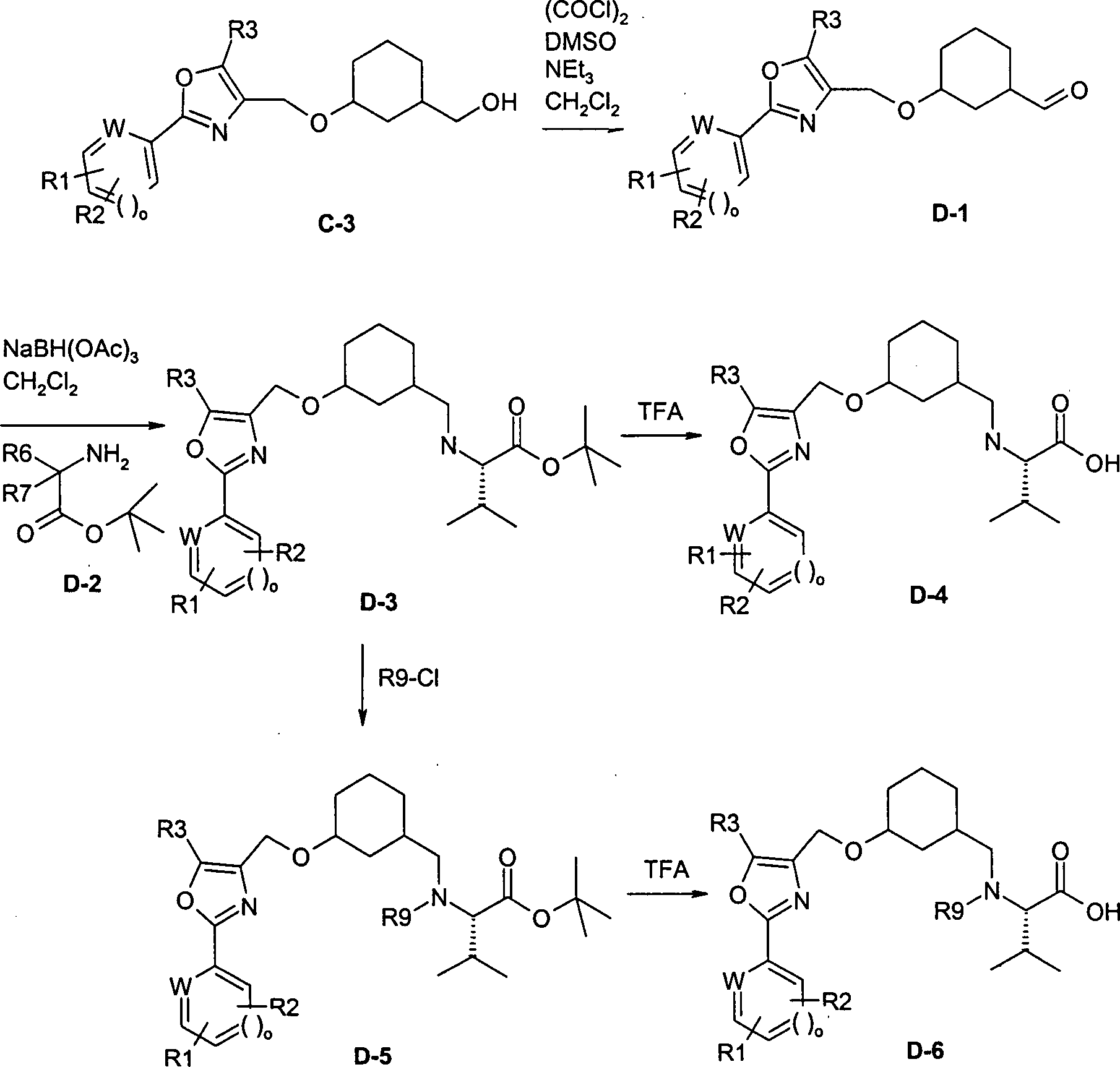
-
Die
Verbindung C-3 (s. Verfahren C) wird mit Oxalylchlorid, Triethylamin
und Dimethylsulfoxid in Dichlormethan bei –78°C zum Aldehyd D-1 oxidiert.
Dieser wird mit Natriumtriacetoxyborhydrid und der Verbindung der
allgemeinen Formel D-2, wobei, R6 und R7 die oben beschriebenen
Bedeutungen haben, zur Verbindung D-3 umgesetzt.
-
Die
Verbindung D-3 wird durch mehrstündiges
Rühren
in Trifluoressigsäure
zur Verbindung D-4 umgesetzt.
-
In
einigen Beispielen wird die Verbindung D-3 mit Acylchloriden, Sulfonylchloriden
oder Chlorameisensäureestern
der allgemeinen Formel R9-Cl, wobei R9 die oben beschriebene Bedeutung
hat, in Dichlormethan in Gegenwart von Pyridin zur Verbindung D-5
umgesetzt. Die Verbindung D-5 wird durch mehrstündiges Rühren in Trifluoressigsäure zur
Verbindung D-6 umgesetzt.
-
Nach
diesem Verfahren können
die Beispiele 72 bis 78 synthetisiert werden.
-
-
Die
Verbindung E1 wird an der Hydroxylgruppe mit einer geeigneten Schutzgruppe
geschützt,
beispielsweise mit der Methoxymethylschutzgruppe. Anschließend wird
die Carboxylgruppe mit Lithiumaluminiumhydrid in Diethylether zur
Verbindung E-2 umgesetzt. Diese wird mit Bromessigsäure-tert-butylester
und Tetrabutylammoniumhydrogensulfat in einem Zweiphasensystem Toluol/50%-ige
Natronlauge zur Verbindung E-3 umgesetzt.
-
Die
Verbindung E-3 wird entschützt
(beispielsweise mit konzentrierter Salzsäure in Tetrahydrofuran im Falle
der Methoxymethylschutzgruppe) und anschließend mit tert-Butyldimethylsilylchlorid
und Imidazol in Dimethylformamid zur Verbindung E-4 umgesetzt.
-
Die
Verbindung E-4 wird mit Lithiumdiisopropylamid in Tetrahydrofuran
bei 0°C
deprotoniert und mit einem Alkyliodid der allgemeinen Formel R6-I,
wobei R6 die oben beschriebene Bedeutung hat, umgesetzt. Anschließend wird
die entstandene Verbindung mit Lithiumdiisopropylamid in Tetrahydrofuran
bei 0°C
deprotoniert und mit einem Alkyliodid der allgemeinen Formel R7-I,
wobei R7 die oben beschriebene Bedeutung hat, zur Verbindung E-5
umgesetzt.
-
Die
Verbindung E-5 wird mit Wismuttribromid, Triethylsilan und der Verbindung
C-1 (siehe Verfahren C)
in Acetonitril bei Raumtemperatur zur Verbindung E-6 umgesetzt.
-
Die
Verbindung E-6 wird durch Rühren
in Trifluoressigsäure
zur Verbindung E-7 umgesetzt.
-
Nach
diesem Verfahren können
die Beispiele 79 und 80 synthetisiert werden.
-
-
Die
Verbindung F-1 wird mit Diisobutylaluminiumhydrid und Isopropanol
in Diethylether zur Verbindung F-2 reduziert. Diese wird mit der
Verbindung der allgemeinen Formel A-6 und Natriumhydrid in Dimethylformamid
zur Verbindung F-3 umgesetzt.
-
Die
Verbindung F-3 wird mit Osmiumtetroxid und Natriumperiodat in Diethylether
zum Aldehyd F-4 umgesetzt. Diese Verbindung wird in einer Horner-Emmons-Wadsworth-Reaktion
mit einem Triethylphosphonoessigsäureester der allgemeinen Formel
F-5, worin R6 die oben beschriebene Bedeutung hat, zur Verbindung F-6
umgesetzt.
-
Die
Verbindung F-6 wird mit Wasserstoff an Palladium/Kohle zur Verbindung
F-7 hydriert, und anschließend
wird der Ester mit Lithiumhydroxid zur Säure F-8 verseift.
-
Nach
diesem Verfahren können
die Beispiele 81 bis 84 synthetisiert werden.
-
-
Die
Verbindung F-2 wird mit tert-Butyldiphenylsilylchlorid und Imidazol
als Base in Dimethylformamid umgesetzt, aufgearbeitet und dann mit
Osmiumtetroxid und Natriumperiodat in Diethylether umgesetzt. Die
so erhaltene Verbindung wird mit Triphenylphosphoranylidenessigsäure-tert-butylester
und nButhyllithium in einer Wittig-Reaktion umgesetzt und anschließend mit
Wasserstoff an Palladium/Kohle zur Verbindung G-1 hydriert.
-
Die
Verbindung G-1 wird mit Lithiumdiisopropylamid in Tetrahydrofuran
bei 0°C
deprotoniert und mit einem Alkyliodid der allgemeinen Formel R6-I,
wobei R6 die oben beschriebene Bedeutung hat, umgesetzt. Anschließend wird
die entstandene Verbindung mit Lithiumdiisopropylamid in Tetrahydrofuran
bei 0°C
deprotoniert und mit einem Alkyliodid der allgemeinen Formel R7-I,
wobei R7 die oben beschriebene Bedeutung hat, zur Verbindung G-2
umgesetzt.
-
Die
Verbindung G-2 wird zur Entschützung
mit Tetrabutylammoniumfluorid in Tetrahydrofuran umgesetzt. Anschließend wird
der so erhaltene Alkohol mit Natriumhydrid und der Verbindung A-6
in Dimethylformamid zur Verbindung G-3 umgesetzt.
-
Der
tert-Butylester wird gespalten, indem die Verbindung G-3 in Trifluoressigsäuremehrere
Stunden gerührt
wird, wobei die Verbindung G-4 erhalten wird.
-
Nach
diesem Verfahren wurden die Beispiele 85 bis 92 synthetisiert.
-
-
Die
Verbindung H-1 wird mit Dibutylzinnoxid in Toluol unter Rückfluss
am Wasserabscheider gekocht. Nach Zugabe von Dimethylformamid, Cäsiumfluorid
und der Verbindung A-6 (siehe Verfahren A) wird die Suspension bei
Raumtemperatur gerührt.
Dabei wird die Verbindung H-2 erhalten. Diese wird mit Chirazym
L-2 in Vinylacetat in das enantiomerenangereicherte Acetat H-3 überführt. Das
Acetat H-3 wird mit Natriumhydroxid in Methanol zum Alkohol H-4
umgesetzt.
-
Die
Verbindung H-4 wird mit Natriumhydrid uns Allylbromid in Dimethylformamid
bei Raumtemperatur zur Verbindung H-5 umgesetzt.
-
Die
Verbindung H-5 wird mit Osmiumtetroxid und Natriumperiodat in Diethylether
zur Verbindung H-6 umgesetzt. Diese wird in einer Horner-Emmons-Wadsworth-Reaktion mit Natriumhydrid
und der Verbindung F-5 zur Verbindung H-7 umgesetzt.
-
Die
Verbindung H-7 wird zur freien Säure
verseift, indem sie mehrere Stunden bei Raumtemperatur mit Natriumhydroxid
in Methanol gerührt
wird. Die dabei erhaltenen Verbindung H-8 wird mit Wasserstoff an Palladium/Kohle
zur Verbindung H-9 hydriert.
-
Nach
diesem Verfahren können
die Beispiele 93 bis 96 synthetisiert werden.
-
-
Die
Verbindung H-5 wird mit Osmiumtetroxid, 1,5-Diazabicyclo[2.2.2]octan
(DABCO) und N-Methylmorpholin-N-oxid zur Verbindung I-1 dihydroxyliert.
Anschließend
wird die primäre
Hydroxylgruppe als tert-Butyldimethylsilylether I-2 geschützt, indem
die Verbindung I-1 mit tert-Butyldimethylsilylchlorid und Imidazol
als Base in Dimethylformamid bei Raumtemperatur gerührt wird.
Anschließend
wird die Verbindung I-2 mit Natriumhydrid und einem Alkyliodid zur
Verbindung I-3, worin R7 die oben beschriebene Bedeutung hat, umgesetzt. Die
Silylschutzgruppe wird mit Tetrabutylammoniumfluorid in Tetrahydrofuran
abgespalten, wobei die Verbindung I-4 erhalten wird.
-
Die
Verbindung I-4 wird mit Dess-Martin-Periodinan (DMP) in Dichlormethan
mehrere Stunden bei Raumtemperatur gerührt, aufgearbeitet und anschließend mit
Natriumchlorit und Wasserstoffperoxid in Acetonitril zur Verbindung
I-5 umgesetzt.
-
Nach
diesem Verfahren können
die Beispiele 97 und 98 synthetisiert werden.
-
-
Die
Verbindung H-2 wird mit Natriumhydrid und 2-Brommethylacrylsäureethylester
in Dimethylformamid bei 0°C
zur Verbindung J-1 umgesetzt.
-
Die
Verbindung J-1 wird nun entweder mit Trimethylsulfoniumiodid und
Natriumhydrid in Dimethylsulfoxid zur Verbindung J-2, oder mit einem
sekundären
Amin NR10R11, worin R10 und R11 die oben beschriebenen Bedeutungen
haben, zur Verbindung J-3 oder mit einem Arylhalogenid und einem
Palladium(0)-Katalysator
in einer Heck-Reaktion zur Verbindung J-4 umgesetzt. Die Verbindung
J-4 wird dann mit Wasserstoff an Palladium auf Kohle zur Verbindung
J-5 hydriert.
-
Die
Verbindungen J-2, J-3 und J-5 werden mit Natriumhydroxid zu Verbindungen
der allgemeinen Formel J-6 umgesetzt, wobei R6 und R7 die oben beschriebenen
Bedeutungen haben.
-
Nach
diesem Verfahren können
die Beispiele 99 bis 103 synthetisiert werden.
-
Verfahren K:
-
Dieses
Verfahren dient zur Synthese des Bausteins A-6, worin R1, R2, W
und R3 die oben genannten Bedeutungen haben.
-
-
Der
Ester K-1, worin R3 die oben genannte Bedeutung hat, wird mit Natriumnitrit
und Salzsäure
zum Oxim K-2 umgesetzt, welches durch Hydrierung mit Wasserstoff
an Palladium/Kohle zum Amin K-3 reduziert wird.
-
Die
Verbindung K-3 wird mit Säurechloriden
der allgemeinen Formel K-4, worin R1, W und R2 die oben genannten
Bedeutungen haben, und Base (beispielsweise Triethylamin) zur Verbindung
K-5 umgesetzt.
-
Die
Verbindung K-5 wird durch Erhitzen in Phosphorylchlorid zur Verbindung
K-6 umgesetzt.
-
Der
Ester K-6 wird mit Lithiumaluminiumhydrid in Diethylether zum Alkohol
K-7 reduziert. Dieser wird mit Iod, Imidazol (ImH) und Triphenylphosphin
in das Iodid A-6 überführt.
-
Alternativ
wird die Verbindung K-7 mit Oxalylchlorid, Dimethylsulfoxid und
Triethylamin in Dichlormethan bei –78°C zum Aldehyd C-1 oxidiert.
-
Verfahren L:
-
Dieses
Verfahren dient zur Synthese des Bausteins A-6, worin R1, R2, W
und R3 die oben genannten Bedeutungen haben.
-
-
Die
Verbindung L-1 wird mit dem Aldehyd L-2, worin R1, R2, W und R3
die oben beschriebenen Bedeutungen haben, in Ethanol mit Chlorwasserstoff
zur Verbindung L-3 umgesetzt.
-
Die
Verbindung L-3 wird in Phosphorylchlorid zum Sieden erhitzt, wobei
die Verbindung L-4 erhalten wird. Diese wird mit Natriumiodid in
Aceton zum Sieden erhitzt. Dabei erhält man die Verbindung A-6.
-
Andere
Verbindungen können
entsprechend den oben genannten Verfahren hergestellt werden.
-
Die
Verbindungen der Formel I zeichnen sich durch günstige Wirkungen auf Stoffwechselstörungen aus.
Sie beeinflussen den Fett-und Zuckerstoffwechsel positiv, sie senken
insbesondere den Triglyceridspiegel und sind zur Prävention
und Behandlung von Typ II Diabetes und Arteriosklerose geeignet.
-
Die
Verbindungen können
allein oder in Kombination mit einer oder mehreren weiteren pharmakologisch
wirksamen Substanzen verabreicht werden, die beispielsweise eine
günstige
Wirkung auf Stoffwechselstörungen
haben und die beispielsweise ausgewählt sind aus Antidiabetika,
Antiadiposita, blutdrucksenkenden Wirkstoffen und Wirkstoffen zur
Behandlung und/oder Prävention
von Komplikationen, die von Diabetes verursacht werden oder mit
Diabetes assoziiert sind.
-
Als
weitere pharmakologisch wirksame Substanzen sind insbesondere geeignet:
Alle
Antidiabetika, die in der Roten Liste 2001, Kapitel 12 genannt sind.
Sie können
mit den erfindungsgemäßen Verbindungen
der Formel I insbesondere zur synergistischen Wirkungsverbesserung
kombiniert werden. Die Verabreichung der Wirkstoffkombination kann
entweder durch getrennte Gabe der Wirkstoffe an den Patienten oder
in Form von Kombinationspräparaten,
worin mehrere Wirkstoffe in einer pharmazeutischen Zubereitung vorliegen,
erfolgen. Die meisten der nachfolgend aufgeführten Wirkstoffe sind in USP
Dictionary of USAN and International Drug Names, US Pharmacopeia,
Rockville 2001, offenbart.
-
Antidiabetika
umfassen Insulin und Insulinderivate, wie z.B. Lantus
® (siehe
www.lantus.com) oder HMR 1964, schnell wirkende Insuline (siehe
US 6,221,633 ), GLP-1-Derivate
wie z.B. diejenigen die in WO 98/08871 von Novo Nordisk A/S offenbart
wurden, sowie oral wirksame hypoglykämische Wirkstoffe.
-
Die
oral wirksamen hypoglykämischen
Wirkstoffe umfassen vorzugsweise Sulphonylfharnstoffe, Biguanidine,
Meglitinide, Oxadiazolidindione, Thiazolidindione, Glukosidase-Inhibitoren,
Glukagon-Antagonisten, GLP-1-Agonisten,
Kaliumkanalöffner,
wie z.B. diejenigen, die in WO 97/26265 und WO 99/03861 von Novo
Nordisk A/S offenbart wurden, Insulin-Sensitizer, Inhibitoren von
Leberenzymen, die an der Stimulation der Glukoneogenese und/oder
Glykogenolyse beteiligt sind, Modulatoren der Glukoseaufnahme, den
Fettstoffwechsel verändernde
Verbindungen wie antihyperlipidämische
Wirkstoffe und antilipidämische
Wirkstoffe, Verbindungen, die die Nahrungsmitteleinnahme verringern,
PPAR-und PXR-Agonisten und Wirkstoffe, die auf den ATP-abhängigen Kaliumkanal
der Betazellen wirken.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem HMGCoA-Reduktase Inhibitor wie Simvastatin, Fluvastatin,
Pravastatin, Lovastatin, Atorvastatin, Cerivastatin, Rosuvastatin
verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Cholesterinresorptionsinhibitor, wie z.B. Ezetimibe, Tiqueside,
Pamaqueside, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem PPAR gamma Agonist, wie z.B. Rosiglitazon, Pioglitazon,
JTT-501, GI 262570, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit PPAR alpha Agonist, wie z.B. GW 9578, GW 7647, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem gemischten PPAR alpha/gamma Agonisten, wie z.B. GW 1536,
AVE 8042, AVE 8134, AVE 0847, oder wie in PCT/US 11833, PCT/US 11490,
DE10142734.4 beschrieben
verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Fibrat, wie z.B. Fenofibrat, Clofibrat, Bezafibrat, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem MTP-Inhibitor, wie z.B. Implitapide, BMS-201038, R-103757, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit Gallensäureresorptionsinhibitor
(siehe z.B.
US 6,245,744 oder
US 6,221,897 ), wie z.B.
HMR 1741, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem CETP-Inhibitor, wie z.B. JTT-705, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem polymeren Gallensäureadsorber,
wie z.B. Cholestyramin, Colesevelam, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem LDL-Rezeptorinducer (siehe
US
6,342,512 ), wie z.B. HMR1171, HMR1586, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem ACAT-Inhibitor, wie z.B. Avasimibe, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Antioxidans, wie z.B. OPC-14117, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Lipoprotein-Lipase Inhibitor, wie z.B. NO-1886, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem ATP-Citrat-Lyase Inhibitor, wie z.B. SB-204990, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Squalen Synthetase Inhibitor, wie z.B. BMS-188494, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Lipoprotein(a) antagonist, wie z.B. CI-1027 oder Nicotinsäure, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit einem Lipase Inhibitor, wie z.B. Orlistat, verabreicht.
-
Bei
einer Ausführungsform
der Erfindung werden die Verbindungen der Formel I in Kombination
mit Insulin verabreicht.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem Sulphonylharnstoff,
wie z.B. Tolbutamid, Glibenclamid, Glipizid oder Glimepirid verabreicht.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem Biguanid, wie
z.B. Metformin, verabreicht.
-
Bei
wieder einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem Meglitinid,
wie z.B. Repaglinid, verabreicht.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem Thiazolidindion,
wie z.B. Troglitazon, Ciglitazon, Pioglitazon, Rosiglitazon oder
den in WO 97/41097 von Dr. Reddy's Research
Foundation offenbarten Verbindungen, insbesondere 5-[[4-[(3,4-Dihydro-3-methyl-4-oxo-2-chinazolinylmethoxy]phenyl]methyl]-2,4-thiazolidindion,
verabreicht.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem α-Glukosidase-Inhibitor,
wie z.B. Miglitol oder Acarbose, verabreicht.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit einem Wirkstoff verabreicht,
der auf den ATP-abhängigen
Kaliumkanal der Betazellen wirkt, wie z.B. Tolbutamid, Glibenclamid, Glipizid,
Glimepirid oder Repaglinid.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit mehr als
einer der vorstehend genannten Verbindungen, z.B. in Kombination
mit einem Sulphonylharnstoff und Metformin, einem Sulphonylharnstoff
und Acarbose, Repaglinid und Metformin, Insulin und einem Sulphonylharnstoff,
Insulin und Metformin, Insulin und Troglitazon, Insulin und Lovastatin,
etc. verabreicht.
-
Bei
einer weiteren Ausführungsform
werden die Verbindungen der Formel I in Kombination mit CARaumtemperatur-Modulatoren
(siehe "Cocaine-amphetamineregulated
transcript influences energy metabolism, anxiety and gastric emptying
in mice" Asakawa,
A, et al., M.:Hormone and Metabolic Research (2001), 33(9), 554-558),
NPY-Antagonisten z.B. Naphthalin-1-sulfonsäure {4-[(4-aminoquinazolin-2-ylamino)-methyl]-cyclohexylmethyl}-amid;
hydrochlorid (CGP 71683A)), MC4-Agonisten (z.B. 1-Amino-1,2,3,4-tetrahydro-naphthalin-2-carbonsäure [2-(3a-benzyl-2-methyl-3-oxo-2,3,3a,4,6,7-hexahydro-pyrazolo[4,3-c]pyridin-5-yl)-1-(4-chloro-phenyl)-2-oxo-ethyl]-amid;
(WO 01/91752)), Orexin-Antagonisten
(z.B. 1-(2-Methyl-benzoxazol-6-yl)-3-[1,5]naphthyridin-4-yl-harnstoff;
hydrochloride (SB-334867-A)), H3-Agonisten (3-Cyclohexyl-1-(4,4-dimethyl-1,4,6,7-tetrahydro-imidazo[4,5-c]pyridin-5-yl)-propan-1-on
Oxalsäuresalz
(WO 00/63208)); TNF-Agonisten, CRF-Antagonisten (z.B. [2-Methyl-9-(2,4,6-trimethylphenyl)-9H-1,3,9-triaza-fluoren-4-yl]-dipropyl-amin
(WO 00/66585)), CRF BP-Antagonisten
(z.B. Urocortin), Urocortin-Agonisten, β3-Agonisten (z.B. 1-(4-Chloro-3-methanesulfonylmethyl-phenyl)-2-[2-(2,3-dimethyl-1H-indol-6-yloxy)-ethylamino]-ethanol;
hydrochloride (WO 01/83451)), MSH (Melanocytstimulierendes Hormon)-Agonisten,
CCK-A Agonisten (z.B. {2-[4-(4-Chloro-2,5-dimethoxy-phenyl)-5-(2-cyclohexyl-ethyl)-thiazol-2-ylcarbamoyl]-5,7-dimethyl-indol-1-yl}-acetic acid
Trifluoressigsäuresalz
(WO 99/15525)); Serotonin-Wiederaufnahme-Inhibitoren
(z.B. Dexfenfluramine), gemischte Sertonin-und noradrenerge Verbindungen
(z.B. WO 00/71549), 5HT-Agonisten z.B. 1-(3-Ethyl benzofuran-7-yl)-piperazin
Oxalsäuresalz
(WO 01/09111), Bombesin-Agonisten, Galanin-Antagonisten, Wachstumshormon
(z.B. humanes Wachstumshormon), Wachstumshormon freisetzende Verbindungen (6-Benzyloxy-1-(2-diisopropylamino-ethylcarbamoyl)-3,4-dihydro-1H-isoquinoline-2-carboxylic
acid tert-butylester (WO 01/85695)), TRH-Agonisten (siehe z.B.
EP 0 462 884 ) entkoppelnde
Protein 2-oder 3-Modulatoren, Leptinagonisten (siehe z.B. Lee, Daniel
W.; Leinung, Matthew C.; Rozhavskaya-Arena, Marina; Grasso, Patricia.
Leptin agonists as a potential approach to the treatment of obesity.
Drugs of the Future (2001), 26(9), 873-881),
-
DA-Agonisten
(Bromocriptin, Doprexin), Lipase/Amylase-Inhibitoren (z.B. WO 00/40569),
PPAR-Modulatoren (z.B. WO 00/78312), RXR-Modulatoren oder TR-β-Agonisten verabreicht.
-
Bei
einer Ausführungsform
der Erfindung ist der weitere Wirkstoff Leptin; siehe z.B. "Perspectives in the
therapeutic use of leptin",
Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert
Opinion on Pharmacotherapy (2001), 2(10), 1615-1622.
-
Bei
einer Ausführungsform
ist der weitere Wirkstoff Dexamphatamin oder Amphetamin.
-
Bei
einer Ausführungsform
ist der weitere Wirkstoff Fenfluramin oder Dexfenfluramin.
-
Bei
noch einer Ausführungsform
ist der weitere Wirkstoff Sibutramin.
-
Bei
einer Ausführungsform
ist der weitere Wirkstoff Orlistat.
-
Bei
einer Ausführungsform
ist der weitere Wirkstoff Mazindol oder Phentermin.
-
Bei
einer Ausführungsform
werden die Verbindungen der Formel I in Kombination mit Ballaststoffen, vorzugsweise
unlöslichen
Ballaststoffen (siehe z.B. Carob/ Caromax® (Zunft
H J; et al., Carob pulp preparation for treatment of hypercholesterolemia,
ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-6.) Caromax ist ein Carob enthaltendes
Produkt der Fa. Nutrinova, Nutrition Specialties &Food Ingredients
GmbH, Industriepark Höchst,
65926 Frankfurt/Main)) verabreicht. Die Kombination mit Caromax® kann
in einer Zubereitung erfolgen, oder durch getrennte Gabe von Verbindungen
der Formel I und Caromax®. Caromax® kann
dabei auch in Form von Lebensmitteln, wie z.B. in Backwaren oder
Müsliriegeln,
verabreicht werden.
-
Es
versteht sich, dass jede geeignete Kombination der erfindungsgemäßen Verbindungen
mit einer oder mehreren der vorstehend genannten Verbindungen und
wahlweise einer oder mehreren weiteren pharmakologisch wirksamen
Substanzen als unter den Schutzbereich der vorliegenden Erfindung
fallend angesehen wird.
-
-
Diese
Erfindung bezieht sich weiterhin auf die Verwendung von Verbindungen
der Formel I und ihren pharmazeutischen Zusammensetzungen als PPAR-Liganden-Rezeptor-Binder.
Die erfindungsgemäßen PPAR-Liganden-Rezeptor-Binder
eignen sich als Agonisten oder Antagonisten des PPAR-Rezeptors.
-
Peroxisom-Proliferator-aktivierte
Rezeptoren (PPAR) können
in die drei Subtypen PPARα,
PPARδ und PPARγ unterteilt
werden. Diese werden von verschiedenen Genen codiert (Motojima,
Cell Structure and Function, 18:267-277, 1993). Darüber hinaus
gibt es zwei Isotope von PPARγ,
PPARγ1 und γ2. Diese beiden Proteine unterscheiden sich
in 30 NH2-terminalen Aminosäuren und
sind das Ergebnis eines alternativen Einsatzes von Promotoren und
einer differenziellen mRNA-Spleißung (Vidal-Puig,
Jiminez, Linan, Lowell, Hamann, Hu, Spiegelman, Flier, Moller, J.
Clin. Invest., 97:2553-2561, 1996).
-
Bei
PPAR-modulierten biologischen Prozessen handelt es sich um solche
Prozesse, die von Rezeptoren oder Kombinationen von Rezeptoren moduliert
werden, die auf die in diesem Patent beschriebenen PPAR-Rezeptor-Liganden
ansprechen. Diese Prozesse umfassen beispielsweise den Plasmalipidtransport und
den Fettsäurekatabolismus,
die Regulierung von Insulinempfindlichkeit und Blutzuckerspiegeln,
die beteiligt sind an Hypoglykämie/Hyperinsulinismus
(die z.B. bedingt sind durch Funktionsstörungen der Pankreas-Betazellen,
insulinsezernierende Tumoren und/oder Autoimmunhypoglykämie infolge
von Autoantikörpern gegen
Insulin, den Insulinrezeptor, oder Autoantikörper, die eine stimulierende
Wirkung auf Pankreas-Betazellen haben), Makrophagen-Differenzierung,
die zur Bildung atherosklerotischer Plaques, zu entzündlichen
Reaktionen, Karzinogenese, Hyperplasie oder Adipozyten-Differenzierung
führt.
-
Adipositas
ist eine übermäßige Ansammlung
von Fettgewebe. Jüngste
Arbeiten auf diesem Gebiet haben aufgezeigt, dass PPARγ eine zentrale
Rolle bei der Genexpression und Differenzierung von Adipozyten spielt. Übermäßiges Fettgewebe
ist assoziiert mit der Entwicklung schwerer Erkrankungen wie beipielsweise nicht-insulinpflichtiger
Diabetes mellitus (NIDDM), Hypertonie, Erkrankungen der Koronararterien,
Hyperlipidämie,
Adipositas und bestimmte maligne Krankheitsbilder. Die Adipozyten
können
sich durch die Bildung von Tumornekrosefaktor α (TNFα) und anderen Molekülen auch
auf die Glukosehomeostase auswirken.
-
Nicht-insulinpflichtiger
Diabetes mellitus (NIDDM) oder Typ-II-Diabetes ist die häufigere
Form von Diabetes. An dieser Form der Krankheit leiden etwa 90-95%
der Hyperglykämie-Patienten.
Bei NIDDM liegen anscheinend eine Reduzierung der Masse der Pankreas-Betazellen,
mehrere verschiedene Störungen
der Insulinsekretion oder eine reduzierte Insulinempfindlichkeit
des Gewebes vor. Die Symptome dieser Form von Diabetes umfassen
Müdigkeit,
häufiges
Wasserlassen, Durst, verschwommenes Sehen, häufige Infektionen und langsames
Heilen von Wunden, diabetische Nervenschädigungen und Nierenerkrankungen.
-
Resistenz
gegen die metabolischen Wirkungen von Insulin ist eines der Hauptmerkmale
von nicht-insulinpflichtigem Diabetes (NIDDM). Insulinresistenz
ist gekennzeichnet durch eine beeinträchtigte Aufnahme und Umsetzung
von Glukose in insulinempfindlichen Zielorganen wie beispielsweise
Adipozyten und Skelettmuskeln, sowie durch eine beeinträchtigte
Hemmung der hepatischen Glukoneogenese. Der funktionelle Insulinmangel
und die fehlende Unterdrückung
der hepatischen Glukoneogenese durch Insulin führt zu Hyperglykämie im nüchternen
Zustand. Die Pankreas-Betazellen kompensieren die Insulinresistenz,
indem sie verstärkt
Insulin sezernieren. Doch die Betazellen können diese hohe Insulinbildung
nicht aufrechterhalten, so dass die Glukose-induzierte Insulinsekretion
zurückgeht
und es zu einer Verschlechterung der Glukosehomeostase und schließlich zur
Entwicklung eines manifesten Diabetes kommt.
-
Hyperinsulinämie steht
ebenfalls in Zusammenhang mit Insulinresistenz, Hypertriglyceridämie und
erhöhten
Plasmakonzentrationen von Lipoproteinen niedriger Dichte. Der Zusammenhang
von Insulinresistenz und Hyperinsulinämie mit diesen Stoffwechselstörungen wurde „Syndrom
X" genannt und wird
stark mit einem erhöten
Risiko von Hypertonie und Erkrankungen der Koronararterien assoziiert.
-
Metformin
ist dem Fachmann zur Behandlung von Diabetes beim Menschen bekannt
(US-Patent Nr. 3,174,901). Metformin bewirkt primär eine reduzierte
Glukosebildung in der Leber. Troglitazon® wirkt
bekanntlich primär
auf die Verbesserung der Fähigkeit
der Skelettmuskeln, auf Insulin zu reagieren und Glukose aufzunehmen.
Es ist bekannt, dass eine Kombinationstherapie von Metformin und
Troglitazon zur Behandlung von Störungen eingesetzt werden kann,
die mit Diabetes einhergehen (DDT 3:79-88, 1998).
-
Es
wurde beobachtet, dass PPARγ-Aktivatoren,
insbesondere Troglitazon®, bei Liposarkomen (Fett-Tumoren)
Krebsgewebe in normale Zellen umwandeln (PNAS 96:3951-3956, 1999).
Ferner wurde vermutet, dass PPARγ-Aktivatoren
zur Behandlung von Brust-und Darmkrebs nützlich sein könnten (PNAS 95:8806-8811, 1998, Nature
Medicine 4:1046-1052, 1998).
-
Darüber hinaus
wurden PPARγ-Aktivatoren
wie beispielsweise Troglitazon® auch zur Behandlung des polyzystischen
Ovarialsyndroms (PCO) eingesetzt. Dieses bei Frauen auftretende
Syndrom ist durch chronische Anovulation und Hyperandrogenismus
gekennzeichnet. Bei Frauen mit diesem Syndrom liegen häufig auch
Insulinresistenz und ein erhöhtes
Risiko der Entwicklung von nichtinsulinpflichtigem Diabetes mellitus
vor (Dunaif, Scott, Finegood, Quintana, Whitcomb, J. Clin. Endocrinol.
Metab., 81:3299, 1996).
-
Ferner
wurde kürzlich
entdeckt, dass PPARγ-Aktivatoren
die Bildung von Progesteron steigern und die Steroidgenese in Granulosa-Zellkulturen
hemmen und sich daher zur Behandlung des Klimakteriums eignen können (US-Patent
Nr. 5,814,647 Urban et al., 29. September 1998; B. Lorke et al.,
Journal of Endocrinology, 159, 429-39, 1998). Klimakterium ist definiert
als das Syndrom der endokrinen, somatischen und psychologischen
Veränderungen,
die zum Ende der fortpflanzungsfähigen
Phase von Frauen auftreten.
-
Peroxisome
sind Zellorganellen, die an der Kontrolle von Redox-Potenzial und
oxidativem Stress von Zellen beteiligt sind, indem sie eine Vielzahl
von Substraten wie beispielsweise Wasserstoffperoxid metabolisieren.
Es gibt eine Reihe von Störungen,
die mit oxidativem Stress assoziiert sind. So gehen beispielsweise entzündliche
Reaktionen auf Gewebeverletzungen, die Pathogenese von Emphysemen,
Ischämie-assoziierte Organschädigungen
(Schock), Doxorubicininduzierte Herzschädigungen, Arzneimittel-induzierte
Hepatotoxizität,
Atherosklerose und durch Hyperoxie bedingte Lungenschädigungen
jeweils mit der Bildung reaktiver Sauerstoff-Spezies und einer Veränderung
der Reduktionsfähigkeit
der Zelle einher. Daher wird erwogen, dass PPARα-Aktivatoren unter anderem das Redox-Potenzial
und den oxidativen Stress in Zellen regulieren und zur Behandlung
dieser Störungen
nützlich
sein könnten
(Poynter et al., J. Biol. Chem. 273, 32833-41, 1998).
-
Es
wurde ebenfalls entdeckt, dass PPARα-Agonisten die NFKB-mediierte
Transkription hemmen und dadurch verschiedene Entzündungsreaktionen
modulieren, wie etwa die Enzympfade der induzierbaren Stickoxid-Synthase
(NOS) und Cyclooxygenase-2 (COX-2) (Pineda-Torra, I. et al., 1999,
Curr. Opinion in Lipidology, 10, 151-9) und daher für therapeutische
Eingriffe bei einer großen
Vielfalt von Entzündungskrankheiten
und anderen pathologischen Zuständen
eingesetzt werden können
(Colville-Nash et al., Journal of Immunology, 161, 978-84, 1998; Staels
et al, Nature, 393, 790-3, 1998).
-
Peroxisom-Proliferatoren
aktivieren PPAR, die wiederum als Transkriptionsfaktoren wirken
und Differenzierung, Zellwachstum und Proliferation von Peroxisomen
verursachen. Es wird auch vermutet, dass PPAR-Aktivatoren eine Rolle
bei Hyperplasie und Carcinogenese spielen und die enzymatischen
Fähigkeiten von
Tierzellen wie beispielsweise Nagerzellen verändern, doch diese PPAR-Aktivatoren
scheinen nur minimale negative Auswirkungen auf menschliche Zellen
zu haben (Green, Biochem. Pharm. 43(3):393, 1992). Die Aktivierung
von PPAR führt
zu einem raschen Anstieg von Gammaglutamyltranspeptidase und -katalase.
-
PPARα wird durch
eine Reihe von Fettsäuren
mittlerer Länge
und langkettigen Fettsäuren
aktiviert und ist an der Stimulierung der β-Oxidation von Fettsäuren in
Geweben wie Leber, Herz, Skelettmuskel und braunes Fettgewebe beteiligt
(Issemann und Green, ibid.; Beck et al., Proc. R. Soc. Lond. 247:83-87,
1992; Gottlicher et al., Proc. Natl. Acad. Sci. USA 89:4653-4657,
1992).
-
Pharmakologische
PPARα-Aktivatoren
wie beispielsweise Fenofibrat, Clofibrat, Genfibrozil und Bezafibrat
sind ebenfalls an der erheblichen Reduzierung von Plasmatriglyceriden
sowie einer mäßigen Reduzierung
von LDL-Cholesterin beteiligt, und sie werden insbesondere zur Behandlung
von Hypertriglyceridämie, Hyperlipidämie und
Adipositas eingesetzt. PPARα ist
bekanntlich auch an entzündlichen
Störungen
beteiligt (Schoonjans, K., Current Opinion in Lipidology, 8, 159-66,
1997).
-
Der
menschliche nukleäre
Rezeptor PPARα wurde
aus einer cDNA-Bibliothek menschlicher Osteosarkomzellen kloniert
und wird bei A. Schmidt et al., Molecular Endocrinology, 6:1634-1641
(1992) vollständig
beschrieben. Der Inhalt dieser Ausführungen wird durch Bezugnahme
in diese Patentschrift aufgenommen. Es sei darauf hingewiesen, dass
PPARδ in
der Literatur auch als PPARβ und
als NUC1 bezeichnet wird, wobei sich jeder dieser Namen auf denselben
Rezeptor bezieht. So wird der Rezeptor beispielsweise bei A. Schmidt et
al., Molecular Endocrinology, 6:1634-1641, 1992 als NUC1 bezeichnet.
PPARα wird
sowohl in embryonalen als auch in adulten Geweben festgestellt.
Es wurde berichtet, dass dieser Rezeptor an der Regulierung der Expression
einiger fettspezifischer Gene beteiligt ist und eine Rolle im Prozess
der Adipogenese spielt (Amri, E. et al., J. Biol. Chem. 270, 2367-71,
1995).
-
Man
weiß,
dass atherosklerotische Erkrankungen durch eine Reihe von Faktoren
verursacht werden wie beispielsweise Hypertonie, Diabetes, geringe
Spiegel von Lipoproteinen hoher Dichte (HDL) und hohe Spiegel von
Lipoproteinen niedriger Dichte (LDL). Zusätzlich zur Reduzierung der
Risiken durch Effekte auf die Konzentration der Plasmalipide und
andere Risikofaktoren haben PPARα-Agonisten direkte
atheroprotektive Wirkungen (Frick, M.H. et al., 1997, Circulation
96:2137-2143, de Faire et al., 1997, Cardiovasc. Drugs Ther. 11
Suppl. 1:257-63).
-
Kürzlich wurde
festgestellt, dass PPARδ-Agonisten
nützlich
sind, um HDL-Spiegel zu erhöhen
und sich daher zur Behandlung atherosklerotischer Erkrankungen eignen
(Leibowitz et al., WO/9728149). Atherosklerotische Erkrankungen
umfassen Gefäßkrankheiten,
koronare Herzkrankheit, zerebrovaskuläre Erkrankungen und Erkrankungen
der peripheren Gefäße. Koronare
Herzkrankheit umfasst Tod durch koronare Herzkrankheit, Myokardinfarkt
und koronare Revaskularisierung. Zerebrovaskuläre Erkrankungen umfassen ischämische oder
hämorrhagische
Infarkte und transiente ischämische
Anfälle.
-
PPARγ-Subtypen
sind an der Aktivierung der Adipozyten-Differenzierung beteiligt
und spielen keine Rolle bei der Stimulierung der Peroxisomproliferation
in der Leber. Die Aktivierung von PPARγ ist an der Adipozyten-Differenzierung
durch die Aktivierung der Adipozyten-spezifischen Genexpression
beteiligt (Lehmann, Moore, Smith-Oliver, Wilkison, Willson, Kliewer,
J. Biol. Chem., 270:12953-12956, 1995). Die DNA-Sequenzen der PPARγ-Subtypen
sind bei Elbrecht et al., BBRC 224; 431-437 (1996) beschrieben.
Obwohl Peroxisom-Proliferatoren einschließlich Fibraten und Fettsäuren die
transkriptorische Aktivität
von PPARs aktivieren, wurden nur Prostaglandin J2-Derivate
wie der Arachidonsäure-Metabolit
15-Deoxy-Delta12, 14-Prostaglandin J2 (15d-PGJ2) als natürliche Liganden identifiziert,
die spezifisch für
den PPARγ-Subtyp
sind, der auch an Thiazolidindione bindet. Dieses Prostaglandin
aktiviert die PPARγ-abhängige Adipogenese,
aktiviert PPARα aber
nur in hohen Konzentrationen (Formann, Tontonoz, Chen, Brun, Spiegelman,
Evans, Cell, 83:803-812, 1995; Kliewer, Lenhard, Wilson, Patel,
Morris, Lehmann, Cell, 83:813-819, 1995). Dies ist ein weiterer
Hinweis darauf, dass die Subtypen der PPAR-Familie sich in ihrer
pharmakologischen Reaktion auf Liganden unterscheiden.
-
Daraus
ergibt sich, dass Verbindungen, die PPARα oder sowohl PPARα als auch
PPARγ aktivieren, wirkungsvolle
hypotriglyceridämische
Arzneimittel sein müssten,
die zur Behandlung von mit Atherosklerose assoziierter Dislipidämie, nicht-insulinpflichtigem
Diabetes mellitus, Syndrom X (Staels, B. et al., Curr. Pharm. Des.,
3 (1), 1-4 (1997)) und familiärer
kombinierter Hyperlipidämie
(FCH) eingesetzt werden können.
Syndrom X ist das Syndrom, das durch ein erstes insulinresistentes
Stadium charakterisiert ist, das Hyperinsulinämie, Dyslipidämie und
eine beeinträchtigte
Glukosetoleranz bewirkt und zu nicht-insulinpflichtigem Diabetes
mellitus (Typ II-Diabetes) progredieren kann, der durch Hyperglykämie gekennzeichnet
ist. FCH ist durch Hypercholesterinämie und Hypertriglyceridämie bei
demselben Patienten und in derselben Familie gekennzeichnet.
-
Die
vorliegende Erfindung betrifft Verbindungen der Formel I, die sich
zur Modulierung von PPAR-Rezeptoren eignen, sowie eine Reihe anderer
damit verbundener pharmazeutischer Anwendungen.
-
Die
Verbindungen der Formel I eignen sich insbesonders zur Behandlung
von Dyslipidämie,
Insulinresistenz, Typ I und Typ II Diabetes, Störungen der Glucose-Toleranz, Syndrom
X, Obesitas, Essstörungen, Thrombosen,
Entzündungen,
Cardiomyopathie sowie zum Beta-Zellen Schutz und Fettsäure-Oxidationsschutz
(siehe z.B. Jean-Charles Fruchart, Bart Staels and Patrick Duriez:
PPARS, Metabolic Disease and Atherrosclerosis, Pharmacological Research,
Vol. 44, No. 5, 2001; Sander Kersten, Beatrice Desvergne & Walter Wahli:
Roles of PPARs in health and disease, NATURE, VOL 405,25 MAY 2000;
Ines Pineda Torra, Giulia Chinetti, Caroline Duval, Jean-Charles
Fruchart and Bart Staels: Peroxisome proliferator-activated receptors: from
transcriptional control to clinical practice, Curr Opin Lipidol
12: 2001, 245-254).
-
Die
Wirksamkeit der Verbindungen wurde wie folgt getestet:
Für die Analyse
der Wirkstärke
von Substanzen, die an humanes PPARalpha binden und es in agonistischer Weise
aktivieren, wird eine stabil transflzierte HEK-Zellinie (HEK= human embryo kidney)
benutzt, die hier als „PPARalpha-Reporterzellinie" bezeichnet wird.
-
Die
Aktivität
von PPARalpha-Agonisten wird in einem 3-Tagestest bestimmt, der
nachfolgend beschrieben ist:
Die PPARalpha-Reporterzellinie
wird bis zu einer 80 %igen Konfluenz in DMEM-Medium (# 41965-039, Life Technologies)
kultiviert, das mit folgenden Zusätzen versehen ist: 10% cs-FKS
(fötales
Kälberserum, #SH-30068.03,
Hyclone), Antibiotika (0,5 mg/ml Zeozin [#R250-01, Invitrogen],
0,5 mg/ml G418 [#10131-019, Life
Technologies], 1% Penicillin-Streptomycin-Lösung [#15140-031, Life Technologies])
und 2 mM L-Glutamin (#25030-032, Life Technologies). Die Kultivierung
erfolgt in Standard-Zellkulturflaschen (# 33111, Becton Dickinson)
in einem Zellkulturbrutschrank bei 37°C und 5% CO2.
Die zu 80% konfluenten Zellen werden einmal mit 30 ml PBS gewaschen
(#14190-094, Life Technologies), mit 2 ml Trypsinlösung (#25300-054,
Life Technologies) für
2 min bei 37°C
behandelt, in 5 ml des oben beschriebenen Mediums aufgenommen und
in einem Zellzählgerät gezählt. Nach
der Verdünnung
auf 500.000 Zellen/ml werden jeweils 100.000 Zellen pro Loch einer
96 Loch-Mikrotiterplatte mit klarem Plastikboden (#3610, Corning
Costar) ausgesät.
Die Platten werden für 24
h in einem Zellkulturbrutschrank bei 37°C und 5% CO2 inkubiert.
-
Zu
testende PPARalpha-Agonisten werden in einer Konzentration von 10
mM in DMSO gelöst.
Diese Stocklösung
wird in phenolrot-freiem DMEM Medium (#21063-029, Life Technologies)
verdünnt,
das mit 5% of cs-FKS (#SH-30068.03, Hyclone), 2 mM L-Glutamin (#25030-032,
Life Technologies) und den bereits unter dem Punkt „Aussaat
der Zellen" beschriebenen
Antibiotika (Zeozin, G418, Penicillin und Streptomycin) versetzt
war.
-
Üblicherweise
werden Testsubstanzen in 11 verschiedenen Konzentrationen getestet
(10 μM;
3.3 μM; 1 μM; 0.33 μM; 0,1 μM; 0,033 μM; 0,01 μM; 0,0033 μM; 0,001 μM; 0,00033 μM; und 0,0001 μM). Potentere Verbindungen
werden in Konzentrationsbereichen von 1 μM bis 10 pM bzw. 100 nM bis
1 pM geprüft.
Das Medium der an Tag 1 ausgesäten
PPARalpha-Reporterzellinie wird vollständig aus jedem Loch abgesaugt
und die in Medium verdünnten
Testsubstanzen sofort zu den Zellen zugegeben. Die Verdünnung und
Zugabe der Substanzen kann mit einem Roboter erfolgen (Beckman Biomek
2000). Das Endvolumen der in Medium verdünnten Testsubstanzen beträgt 100 μl pro Loch
einer 96 Lochplatte. Die DMSO-Konzentration in dem Assay ist immer
unter 0.1 v/v, um zelltoxische Effekte des Lösungsmittels zu vermeiden.
-
Jede
Platte wird mit einem Standard PPARalpha-Agonisten belegt, der ebenfalls
in 11 verschiedenen Konzentrationen verdünnt wird, um die Funktionsfähigkeit
des Assays in jeder Einzelplatte nachzuweisen. Die Testplatten werden
für 24
h in einem Brutschrank bei 37°C
und 5% CO2 inkubiert.
-
Die
mit den Testsubstanzen behandelten PPARalpha-Reporterzellen werden
aus dem Brutschrank entnommen und für 1 h bei –20°C eingefroren, um die Zelllyse
zu verbessern. Nach dem Auftauen der Platten, das über mindestens
30 min. bei Raumtemperatur erfolgt, werden 50 μl Puffer 1 (Luc-Screen kit #LS1000,
PE Biosystems Tropix) zu jedem Loch zupipettiert und die Platten
im Anschluß daran
in ein Lumineszenzmeßgerät mit Pipettiereinheit
(Luminoscan Ascent, LabSystems) überführt. Die
Luziferasereaktion wird in dem Meßgerät durch Zupipettieren von je
50 μl Puffer
2 (Luc-Screen kit #LS1000, PE Biosystems Tropix) zu jedem Loch der 96
Lochplatte gestartet. Die Zugabe des Puffers in jedes einzelne Loch
erfolgt in definierten und gleichen Zeitintervallen nach den Angaben
des Geräteherstellers
(LabSystems). Alle Proben werden exakt 16 min. nach Zugabe von Puffer
2 gemessen. Die Meßzeit
beträgt
10 sec. pro Probe.
-
Die
Rohdaten des Lumineszenzmeßgerätes werden
in ein Microsoft Excel-File transferiert. Dosis-Wirkungskurven,
sowie EC50-Werte werden mit dem Programm
XL.Fit nach Vorgabe des Herstellers (IDBS) berechnet.
-
Die
Ergebnisse für
die Aktivität
der erfindungsgemäßen Verbindungen
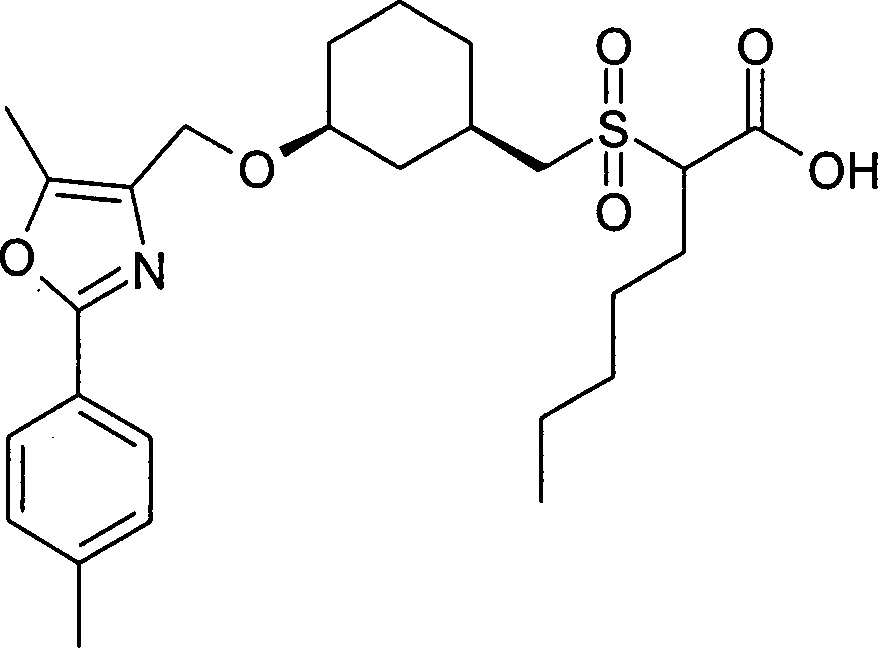
der Formel I sind in der folgenden Tabelle I angegeben:
-
-
Aus
der Tabelle I ist ersichtlich, dass die erfindungsgemäßen Verbindungen
der Formel I den PPARα-Rezeptor
aktivieren und damit analog zu klinisch verwendeten Fibraten im
Organismus eine Triglyceridsenkung bewirken (siehe z.B. J.-Ch. Fruchard
et al.,: PPARS, Metabolic Disease and Atherosclerosis, Pharmacological
Research, Vol. 44, No. 5, 2001; S. Kersten et al.: Roles of PPARs
in health and disease, NATURE, VOL 405,25 MAY 2000;I. Pineda et
al.: Peroxisome proliferator-activated receptors: from transcriptional
control to clinical practice, Curr Opin Lipidol 12: 2001, 245-254).
-
-
-
-
-
-
-
-
-
Bausteinsynthese
nach Verfahren K
-
2-Hydroxyimino-4-methyl-3-oxo-pentansäureethylester
-
42.4
g 4-Methyl-3-oxo-pentansäureethylester
werden in 100 ml Eisessig gelöst
und bei 5°C
mit 21 g Natriumnitrit, gelöst
in 100 ml Wasser, versetzt. Man lässt innerhalb einer Stunde
auf Raumtemperatur erwärmen,
fügt sodann
100 ml Wasser hinzu und rührt
eine weiter Stunde bei Raumtemperatur nach. Man extrahiert dreimal
mit je 150 ml Methyl-tert-butylether, die vereinigten organischen
Phasen werden mit 200 ml Wasser versetzt und durch Zugabe von festem
NaHCO3 neutralisiert. Die organische Phase wird abgetrennt, mit
gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
46 g 2-Hydroxyimino-4-methyl-3-oxo-pentansäure-ethylester
als Öl.
C8H13NO4 (187.20), MS(ESI) = 188 (M + H+).
-
2-Amino-4-methyl-3-oxo-pentansäureethylesterhydrochlorid
-
In
200 ml Ethanol werden 10 g HCl eingeleitet. 46 g 2-Hydroxyimino-4-methyl-3-oxo-pentansäureethylester
werden darin gelöst
und mit 5 g Pd(10% auf Kohle) versetzt und 8 Stunden unter einer
Wasserstoffatmosphäre
(5 bar) gerührt.
Das Reaktionsgemisch wird über
Celite filtriert und das Lösungsmittel
im Vakuum entfernt. Man erhält
45 g 2-Amino-4-methyl-3-oxo-pentansäureethylester hydrochlorid
als weißen
Feststoff. C8H15NO3*HCl (209.5), MS(ESI) = 188 (M + H+).
-
4-Methyl-2-(4-methyl-benzoylamino)-3-oxo-pentansäureethylester
-
10
g 2-Amino-4-methyl-3-oxo-pentansäureethylesterhydrochlorid
und 7.4 g 4-Methyl-benzoylchlorid werden
in 250 ml Dichlormethan gelöst
und bei 0°C
langsam und tropfenweise mit 13.3 ml Triethylamin versetzt. Man
rührt eine
Stunde bei Raumtemperatur nach, dann wird mit Wasser gewaschen,
die organische Phase abgetrennt, über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Man erhält
13 g 4-Methyl-2-(4-methylbenzoylamino)-3-oxo-pentansäureethylester
als Öl.
C16H21NO4 (291.35), MS(ESI) = 292 (M + H+).
-
5-Isopropyl-2-p-tolyl-oxazol-4-carbonsäureethylester
-
13
g 4-Methyl-2-(4-methyl-benzoylamino)-3-oxo-pentansäureethylester
werden in 80 ml Phosphoroxychlorid 2 h unter Rückfluss zum Sieden erhitzt.
Das Phosphoroxychlorid wird im Vakuum entfernt und der resultierende
Rückstand
in 200 ml Dichlormethan gelöst,
dreimal mit gesättigter
NaHCO3-Lösung
gewaschen, über
MgSO4 getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
11 g 5-Isopropyl-2-p-tolyl-oxazol-4-carbonsäureethylester als bräunlichen
Feststoff. C16H19NO3 (273.33), MS(ESI) = 292 (M + H+),
Rf(n-Heptan : Ethylacetat) = 2 : 1) = 0.43.
-
(5-Isopropyl-2-p-tolyl-oxazol-4-yl)-methanol
-
11
g 5-Isopropyl-2-p-tolyl-oxazol-4-carbonsäureethylester werden in 100
ml Tetrahydrofuran gelöst und
bei 0°C
mit 40 ml einer 1 molaren Lösung
von Lithiumaluminiumhydrid in Tetrahydrofuran versetzt. Nach 30
min wird das Reaktionsgemisch mit 50 ml 1N HCl versetzt und fünfmal mit
Ethylacetat extrahiert. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 6
: 1 ⇒ 1
: 1 gereinigt. Man erhält
4.3 g (5-Isopropyl-2-p-tolyl-oxazol-4-yl)-methanol als hellgelben
Feststoff. C14H17NO2 (231.30), MS(ESI) = 232 (M + H+),
Rf(n-Heptan : Ethylacetat) = 1 : 1) = 0.17.
-
4-Iodmethyl-5-isopropyl-2-p-tolyloxazol
-
500
mg (5-Isopropyl-2-p-tolyl-oxazol-4-yl)-methanol werden zusammen
mit 690 mg Triphenylphosphin und 600 mg Imidazol in 20 ml Toluol
gelöst.
Man gibt 715 mg Iod hinzu und rührt
1 Stunde bei Raumtemperatur nach. Dann wird 10 ml gesättigte Natriumcarbonat-Lösung und
500 mg Iod nachgegeben. Nach 10 Minuten wird die organische Phase
abgetrennt und zweimal mit gesättigter
Na2S2O3-Lösung
gewaschen, über
MgSO4 getrocknet und anschließend
die lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 10
: 1 gereinigt. Man erhält
400 mg 4-Iodmethyl-5-isopropyl-2-p-tolyl-oxazol als
weißen
Feststoff. C14H161NO (341.19), MS(ESI): 342 (M + H+),
Rf(n-Heptan : Ethylacetat = 1 : 1) = 0.75.
-
Analog
zur Bausteinsynthese nach Verfahren K wurde aus 2-Amino-4-methyl-3-oxo-pentansäureethylester
hydrochlorid und 3-Methoxy-benzoylchlorid 4-Iodmethyl-2-(3-methoxy-phenyl)-5-isopropyl-oxazol
erhalten.
C14H16INO2 (357.19), MS(ESI):
358 (M + H
+), Rf(n-Heptan : Ethylacetat
= 1 : 1) = 0.60.
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-isopropyl-2-p-tolyl-oxazol
wurde aus 4,4,4-Trifluoro-3-oxo-buttersäureethylester und 3-Methoxybenzoylchlorid
4-Iodmethyl-2-(3-methoxy-phenyl)-5-trifluormethyl-oxazol erhalten.
C12H9F3INO2 (383.11), MS(ESI):
384 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-isopropyl-2-p-tolyl-oxazol
wurde aus 4,4,4-Trifluoro-3-oxo-buttersäureethylester und 3-Trifluormethylbenzoylchlorid
4-Iodmethyl-2-(3-trifluormethyl -phenyl)-5-trifluormethyl-oxazol
erhalten.
C12H6F6INO (421.08), MS(ESI):
422 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-isopropyl-2-p-tolyl-oxazol
wurde aus 4,4,4-Trifluoro-3-oxo-buttersäureethylester und 4-Methyl-benzoylchlorid
4-Iodmethyl-5-trifluormethyl-2-p-tolyl-oxazol
erhalten.
C12H9F3INO (367.11), MS(ESI):
368 (M + H
+).
-
Bausteinsynthese
nach Verfahren L:
-
4-Methyl-5-phenyl-2-p-tolyl-oxazol
3-oxid
-
12.5
g 1-Phenyl-1,2-propandion-2-oxim und 10 ml p-Toluolaldehyd werden
in 50 ml Eisessig gegeben und 30 Minuten unter Eiskühlung HCl
Gas durchgeleitet. Durch Zugabe von Methyl-tert-butylether wird
das Produkt als Hydrochlorid ausgefällt, abgesaugt und der Niederschlag
mit Methyl-tert-butylether gewaschen. Man suspendiert den Niederschlag
in Wasser und stellt mit Ammoniak einen basischen pH-Wert ein. Es
wird dreimal mit je 200 ml Dichlormethan extrahiert, die vereinigten
organischen Phasen werden über
MgSO4 getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
6.4 g 4-Methyl-5-phenyl-2-p-tolyl-oxazol
3-oxid als weißen
Feststoff. C17H15NO2 (265.31), MS(ESI) = 266 (M + H+).
-
4-Chlormethyl-5-phenyl-2-p-tolyl-oxazol
-
6.4
g 4-Methyl-5-phenyl-2-p-tolyl-oxazol 3-oxid werden in 50 ml Chloroform
gelöst,
mit 2.4 ml Phosphoroxychlorid versetzt und 30 Minuten unter Rückfluss
zum Sieden erhitzt. Das Reaktionsgemisch wird auf 0°C abgekühlt, mit
Ammoniak ein schwach alkalischer pH-Wert eingestellt und dreimal
mit je 100 ml Ethylacetat extrahiert. Die vereinigten organischen
Phasen werden mit Wasser gewaschen, über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Man erhält
5.4 g 4-Chlormethyl-5-phenyl-2-p-tolyl-oxazol als gelben Feststoff.
C17H14CINO (283.76), MS(ESI) = 284 (M + H+),
Rf(n-Heptan : Ethylacetat) = 7 : 1) = 0.41.
-
4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol
-
1.8
g 4-Chlormethyl-5-phenyl-2-p-tolyl-oxazol werden zusammen mit 3
g Natriumiodid in 150 ml Aceton 2 Stunden unter Rückfluss
zum Sieden erhitzt. Nach dem Abkühlen
des Reaktionsgemischs wird 300 ml Methyl-tert-butylether zugefügt, das
Gemisch dreimal mit gesättigter
Na2S2O3-Lösung
gewaschen, über MgSO4
getrocknet und anschließend
die Lösungsmittel
im Vakuum entfernt. Man erhält
2.7 g 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol als hellgelben Feststoff.
C17H14INO (375.21), MS(ESI): 376 (M + H+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus 1-Phenyl-1,2-propandion-2-oxim und m-Anisaldehyd 4-Iodmethyl-2-(3-methoxyphenyl)-5-phenyl-oxazol
erhalten.
C17H14INO2 (391.21), MS(ESI):
392 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus 1-Ethyl-1,2-propandion-2-oxim und m-Anisaldehyd 4-Iodmethyl-5-ethyl-2-(3-methoxy-phenyl)-oxazol
erhalten.
C13H14INO2 (343.17), MS(ESI):
344 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus 1-Ethyl-1,2-propandion-2-oxim und p-Toluolaldehyd 4-Iodmethyl-5-ethyl-2-p-tolyloxazol
erhalten.
C13H14INO (327.17), MS(ESI):
328 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus 1-Cyclohexyl-1,2-propandion-2-oxim und m-Anisaldehyd 4-Iodmethyl-5-cyclohexyl-2-(3-methoxy-phenyl)-oxazol
erhalten.
C17H20INO2 (397.26), MS(ESI):
398 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus 1-Cyclohexyl-1,2-propandion-2-oxim und p-Toluolaldehyd 4-Iodmethyl-5-cyclohexyl-2-p-tolyl-oxazol
erhalten.
C17H20INO (381.26), MS(ESI):
382 (M + H+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und p-Toluolaldehyd 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
erhalten.
C12H12INO (313.14), MS(ESI):
314 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und m-Anisaldehyd 4-Iodmethyl-2-(3-methoxy-phenyl)-5-methyl-oxazol erhalten.
C12H12INO2 (329.14), MS(ESI):
330 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 3-Bromo-benzaldehyd 2-(3-Bromo-phenyl)-4-iodmethyl-5-methyl-oxazol erhalten.
C11H9BrINO (377.01/379.01),
MS(ESI): 378/380 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 3-Trifluormethylbenzaldehyd 4-Iodmethyl-5-methyl-2-(3-trifluoromethyl-phenyl)-oxazol
erhalten.
C12H9F3INO (367.11), MS(ESI):
368 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 4-Fluorbenzaldehyd 2-(4-Fluoro-phenyl)-4-iodmethyl-5-methyl-oxazol erhalten.
C11H9FINO (317.10), MS(ESI):
318 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 4-Methoxybenzaldehyd 4-Iodmethyl-2-(4-methoxy-phenyl)-5-methyl-oxazol erhalten.
C12H12INO2 (329.14), MS(ESI):
330 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 3-Trifluormethylbenzaldehyd 4-Iodmethyl-5-methyl-2-(3-trifluoromethyl-phenyl)-oxazol
erhalten.
C12H9F3INO (367.11), MS(ESI):
368 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 4-Trifluormethylbenzaldehyd 4-Iodmethyl-5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol
erhalten.
C12H9F3INO (367.11), MS(ESI):
368 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und m-Toluolaldehyd 4-Iodmethyl-5-methyl-2-m-tolyl-oxazol
erhalten.
C12H12INO (313.14), MS(ESI):
314 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 3-Trifluormethoxybenzaldehyd 4-Iodmethyl-5-methyl-2-(3-trifluoromethoxy-phenyl)-oxazol
erhalten.
C12H9F3INO2 (383.11), MS(ESI):
384 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 5-Methylfuran-2-carbaldehyd 4-Iodmethyl-5-methyl-2-(5-methyl-furan-2-yl)-oxazol
erhalten.
C10H10INO2 (303.11), MS(ESI):
304 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und Thiophen-2-carbaldehyd 4-Iodmethyl-5-methyl-2-thiophen-2-yl-oxazol erhalten.
C9H8INOS (305.14), MS(ESI):
306 (M + H
+).
-
Analog
zur Bausteinsynthese von 4-Iodmethyl-5-phenyl-2-p-tolyl-oxazol wurde
aus Diacetylmonoxim und 4-Isopropylbenzaldehyd 4-Iodmethyl-2-(4-isopropyl-phenyl)-5-methyl-oxazol erhalten.
C14H16INO (341.19), MS(ESI):
342 (M + H
+).
-
Beispiel
1 (S)-3-Methyl-2-{[(1R,3S)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure
-
(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäuremethylester
-
22
g 6-Oxa-bicyclo[3.2.1]octan-7-one werden in 200 ml Methanol gelöst und mit
10%iger Natriummethanolatlösung
versetzt bis ein pH von 10 erreicht ist. Man rührt 30 Minuten bei Raumtemperatur
nach, dann wird durch Zugabe von verdünnter Essigsäure neutralisiert
und das Gemisch im Vakuum eingeengt. Der Rückstand wird in Ethylacetat
gelöst, über MgSO4
getrocknet und anschließend
im Vakuum eingeengt. Man erhält 21
g des Methylesters als farbloses Öl. Dieser wird in 200 ml Dimethylformamid
gelöst
und mit 43 g tert-Butyldiphenylsilylchlorid, 13 g Imidazol und 1
g Dimethylaminopyridin versetzt und 12 h bei Raumtemperatur gerührt. Das
Lösungsmittel
wird im Vakuum entfernt, der Rückstand
in Methy-tert.-butylether aufgenommen und mit Wasser gewaschen.
Die organische Phase wird über
MgSO4 getrocknet und anschließend
das Lösungsmittel
entfernt. Man erhält
56.8 g (cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäuremethylester als gelbes Öl. C24H32O3Si
(396.61), MS(ESI): 397 (M + H+).
-
2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methyl-buttersäure-tert-butylester
-
36.8
g (cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure werden
in 150 ml i-Propanol gelöst
und mit 8 g NaOH, gelöst
in 50 ml Wasser, versetzt. Es wird 1 Stunde auf 60°C erwärmt. Das
Reaktionsgemisch wird abgekühlt
und durch Zugabe von 2N HCl neutralisiert. Das Reaktionsgemisch
wird im Vakuum eingeengt und dreimal mit je 200 ml Etyhlacetat extrahiert.
Die vereinigten organischen Phasen werden über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Man erhält
34 g der freien Säure als
farbloses Öl
(Rf(Ethylacetat) = 0.63). Diese wird in 250 ml Dimethylformamid
gelöst
und mit 18,6 g L-Valin-tert.-butylesterhydrochlorid versetzt. Bei
0°C werden
29,1 g O-[Cyan(ethoxycarbonyl)methylenamino]-1,1,3,3,-tetramethyluroniumtetrafluoroborat
zugegeben. Nach 10 Minuten wird das Eisbad entfernt und 23.9 g Hydroxybenzotriazol
und 61,8 ml Hünigsbase
zugegeben. Man rührt
2 Stunden bei Raumtemperatur nach. Das Lösungsmittel wird im Vakuum
entfernt und der resultierende Rückstand
in Ethylacetat gelöst
und dreimal mit gesättigter NaHCO3-Lösung gewaschen.
Die organische Phase wird über
MgSO4 getrocknet und anschließend
das Lösungsmittel
entfernt. Der Rückstand
wird an Kieselgel mit dem Eluens n-Heptan : Ethylacetat = 2 : 1
gereinigt. Man erhält
43,0 g 2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methylbuttersäure-tert-butylester
als gelbes Öl.
C32H47NO4Si (537,82), MS(ESI): 538.
-
2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methylbuttersäure-tert-butylester
-
43,0
g 2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methyl-buttersäure-tert-butylester
werden in 80 ml Tetrahydrofuran gelöst und mit 80 ml einer 1M Lösung von
Tetrabutylammoniumfluorid in Tetrahydrofuran, versetzt. Man rührt 3 h
bei 60°C
nach, dann wird im Vakuum eingeengt und der Rückstand an Kieselgel mit dem
Eluens n-Heptan : Ethylacetat = 5 : 1 ⇒ 1 : 1 gereinigt. Man erhält 18 g
eines weißen
Feststoffs. Da dieser noch leicht verunreinigt ist werden 8 g nochmals
einer Kieselgelreinigung unterzogen. Man erhält 6,8 g 2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
als weißen
Feststoff. C16H29NO4 (299,41), MS(ESI): 300 (M + H+).
-
2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester kann
durch chirale HPLC getrennt werden. Man erhält 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester (Rt =
4,9 min) und 2-[((1S,3R)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
(Rt = 5,7 min) als farblose Lyophilisate. (Chiralpak AD/34 250 × 4,6; Eluens
n-Heptan : Ethanol : Methanol = 20 : 1 : 1 + 0,1% Trifluoressigsäure).
-
(S)-3-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure
-
4,0
g 2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methylbuttersäure-tert-butylester
und 6,3 g 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol werden in 50 ml
Dimethylformamid gelöst
und portionsweise mit 800 mg Natriumhydrid (60%ig in Paraffinöl) versetzt.
Man rührt
1 Stunde bei Raumtemperatur nach, danach wird das Reaktionsgemisch
durch Zugabe von 200 ml Methyl-tert.-butylether verdünnt und
dreimal mit Wasser gewaschen. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Der resultierende Rückstand wird in 40 ml Dichlormethan
gelöst
und mit 20 ml Trifluoressigsäure
versetzt. Man rührt
5 Stunden bei Raumtemperatur nach. Anschließend werden 100 ml Toluol zugefügt und die
Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Eluens n-Heptan: Ethylacetat = 2 : 1 ⇒ Ethylacetat
gereinigt. Man erhält
1.5 g eines braunen Feststoffes. Dieser wird mittels RP-HPLC weiter
gereinigt. Man erhält
1.0 g des Diastereomerengemisches (S)-3-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure als
farblosen amorphen Feststoff. C24H32N2O5 (428.53), MS(ESI): 429
(M + H+).
-
Das
Diastereomerengemisch (S)-3-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure kann
durch chirale HPLC getrennt werden. Man erhält (S)-3-Methyl-2-{[(1R,3S)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure (Rt
= 6.7 min) und (S)-3-Methyl-2-{[(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure (Rt
= 5,5 min) als farblose Lyophilisate. Chiralcel AD/H259 × 4,6; Eluens
Acetonitril : Ethanol : Methanol = 45 : 4 : 1 + 0,1% Trifluoressigsäure).
-
Beispiel 2
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und (L)-Leucin-tert.-butylesterhydrochlorid
das Diastereomerengemisch (S)-4-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-pentansäure erhalten.
C25H34N2O5 (442.56), MS(ESI):
443 (M + H
+).
-
Beispiel 3
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und (L)-Alanin-terf.-butylesterhydrochlorid
das Diastereomerengemisch (S)-2-{[(1R,3S)/(1S,3)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexane-carbonyl]-amino}-propionsäure erhalten.
C22H28N2O5 (400.48), MS(ESI):
401 (M + H
+).
-
Beispiel 4
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und (L)-Phenylalaninlanin-tert.-butylesterhydrochlorid
das Diastereomerengemisch (S)-2-{[(1R,3S)/(1S,3)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-3-phenyl-propionsäure erhalten.
C28H32N2O5 (476.58), MS(ESI):
477 (M + H
+).
-
Beispiel 5
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und 2-Amino-2-methyl-propionsäure-tert.-butylesterhydrochlorid das
Racemat 2-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-propionsäure erhalten.
C23H30N2O5 (414.51), MS(ESI):
415 (M + H
+).
-
Beispiel 6
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und (D)-Valin-tert.- butylesterhydrochlorid
das Diastereomerengemisch (R)-3-Methyl-2-{[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure erhalten.
C24H32N2O5 (428.53), MS(ESI):
429 (M + H
+).
-
Beispiel 7
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-methoxy-phenyl)-5-methyl-oxazol
und (L)-Prolin-tert.-butylesterhydrochlorid das Diastereomerengemisch
(S)-1-{(1R,3S)/(1S,3R)-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-pyrrolidine-2-carbonsäure erhalten.
C24H30N2O6 (442.52), MS(ESI):
443 (M + H
+).
-
Beisp iel 8
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
und (L)-Prolin-tert.-butylesterhydrochlorid
das Diastereomerengemisch (S)-1-[(1R,3S)/(1S,3R)-3-(5- Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-pyrrolidine-2-carbonsäure erhalten.
C24H30N2O5 (426.52), MS(ESI):
427 (M + H
+).
MS(ESI): 443 (M + H
+).
-
Beispiel 9
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-methoxy-phenyl)-5-methyl-oxazol
und (L)-Valin-tert.-butylesterhydrochlorid das Diastereomerengemisch
(S)-2-({(1R,3S)/(1S,3R)-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C24H32N2O6 (444.53), MS(ESI):
445 (M + H
+).
-
Beispiel 10
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-bromo-phenyl)-5-methyl-oxazol
und (L)- Valin-tert.-butylesterhydrochlorid
das Diastereomerengemisch (S)-2-({(1R,3S)/(1S,3R)-3-[2-(3-Bromo-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-butyrisäure erhalten.
C23H29BrN2O5 (493.40), MS(ESI):
493 (M + H
+).
-
Beispiel 11
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-trifluoromethyl-phenyl)-5-methyl-oxazol
und (L)-Valin-tert.-butylesterhydrochlorid das Diastereomerengemisch (S)-3-Methyl-2-({(1R,3S)/(1S,3R)-3-[5-methyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-buttersäure erhalten.
C24H29F3N2O5 (482.50), MS(ESI):
483 (M + H
+).
-
Beispiel 12
-
Analog
zu Beispiel 1 wurde aus aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(4-methyl-phenyl)-5-isopropyl-oxazol (S)-2-{[(1R,3S)-3-(5-Isopropyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-3-methyl-buttersäure erhalten.
C26H36N2O5 8456.59), MS(ESI):
457 (M + H
+).
-
Beispiel 13
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(3-methoxy-phenyl)-5-isopropyl-oxazol
(S)-2-({(1R,3S)-3-[5-Isopropyl-2-(3-methoxy-phenyl)-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C26H36N2O6 (472.59), MS(ESI):
473 (M + H
+).
-
Beispiel 14
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-methoxy-phenyl)-5-isopropyl-oxazol
und (L)-Valin-tert.-butylesterhydrochlorid das Diastereomerengemisch
(S)-2-({(1R,3S)/(1S,3R)-3-[5-Isopropyl-2-(3-methoxy-phenyl)-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C26H36N2O6 (472.59), MS(ESI):
473 (M + H
+).
-
Beispiel 15
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(3-methoxy-phenyl)-5-trifluormethyl-oxazol (S)-2-({(1R,3S)-3-[2-(3-Methoxy-phenyl)-5-trifluoromethyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C24H29F3N2O6 (498.50), MS(ESI):
499 (M + H
+).
-
Beispiel 16
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(3-trifluormethyl-phenyl)-5-trifluormethyl-oxazol (S)-3-Methyl-2-({(1R,3S)-3-[5-trifluoromethyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-buttersäure erhalten.
C24H26F6N2O5 (536.48), MS(ESI):
537 (M + H
+).
-
Beispiel 17
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(4-methyl-phenyl)-5-trifluormethyl-oxazol
(S)-3-Methyl-2-{[(1R,3S)-3-(2-p-tolyl-5-trifluoromethyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure erhalten.
C24H29F3N2O5 (482.50), MS(ESI):
483 (M + H
+).
-
Beispiel 18
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(4-methyl-phenyl)-5-phenyl-oxazol
(S)-3-Methyl-2-{[(1R,3S)-3-(5-phenyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure erhalten.
C29H34N2O5 (490.60), MS(ESI):
491 (M + H
+).
-
Beispiel 19
-
Analog
zu Beispiel 1 wurde aus cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 4-Iodmethyl-2-(3-methoxy-phenyl)-5-phenyl-oxazol
und (L)-Valin-tert.-butylesterhydrochlorid das Diastereomerengemisch
(S)-2-({cis-3-[2-(3-Methoxy-phenyl)-5-phenyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C29H34N2O6 (506.60), MS(ESI):
507 (M + H
+).
-
Beispiel 20
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(3-methoxy-phenyl)-5-ethyl-oxazol
(S)-2-({(1R,3S)-3-[2-(3-Methoxy-phenyl)-5-ethyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C25H34N2O6 (458.56), MS(ESI):
459 (M + H
+).
-
Beispiel 21
-
Analog
zu Beispiel 1 wurde aus 2-(((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
4a und 4-Iodmethyl-2-(4-methyl-phenyl)-5-ethyl-oxazol
(S)-3-Methyl-2-{[(1R,3S)-3-(5-ethyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure erhalten.
C25H34N2O5 (442.56), MS(ESI):
443 (M + H
+).
-
Beispiel 22
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(3-methoxy-phenyl)-5-cyclohexyl-oxazol (S)-2-({(1R,3S)-3-[2-(3-Methoxy-phenyl)-5-cyclohexyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-3-methyl-buttersäure erhalten.
C29H40N2O6 (512.65), MS(ESI):
513 (M + H
+).
-
Beispiel 23
-
Analog
zu Beispiel 1 wurde aus 2-[((1R,3S)-3-Hydroxy-cyclohexancarbonyl)-amino]-(3S)-methyl-buttersäure-tert-butylester
und 4-Iodmethyl-2-(4-methyl-phenyl)-5-cyclohexyl-oxazol
(S)-3-Methyl-2-{[(1R,3S)-3-(5-cyclohexyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure erhalten.
C29H40N2O5 (496.65), MS(ESI):
497 (M + H
+).
-
Beispiel 24
-
-
Analog
zu Beispiel 1 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 1-Amino-cyclohexancarbonsäuremethylesterhydrochlorid
und 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol das Racemat 1-{[(1R,3S)/(1S,3R)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-cyclohexancarbonsäuremethylester
erhalten. C27H36N2O5(468,60), ), MS(ESI): 469.
-
1-{[(1R,3S)/(1S,3R)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-cyclohexane-carbonsäure
-
500
mg des Racemats 1-{[cis-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-cyclohexancarbonsäuremethylester
werden in 10 ml tert.-Butanol gelöst und mit 1 ml 10 N KOH versetzt. Es
wird 1 Tag unter Rückfluss
zum Sieden erhitzt. Nach Neutralisation mit 2N HCl wird die organische
Phase abgetrennt und die wässrige
Phase dreimal mit je 20 ml Ethylacetat extrahiert. Die vereinigten
organischen Phasen werden über
Magnesiumsulfat getrocknet und anschließend die Lösungsmittel im Vakuum entfernt. Der
resultierende Rückstand
wird durch RP-HPLC gereinigt. Man erhält 170 mg 1-{[(1R,3S)/(1S,3R)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-cyclohexane-carbonsäure als
farbloses Lyophilisat. C26H34N2O5 (454.47), MS(ESI): 455(M + H+).
-
Beispiel 25
-
Analog
zu Beispiel 24 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 1-Amino-cyclopentancarbonsäuremethylesterhydrochlorid
und 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol
das Racemat 1-{[(1R,3S)/(1S,3R)-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-cyclopentancarbonsäure erhalten.
C25H32N2O5 (440.54), MS(ESI):
441(M + H
+).
-
Beispiel 26
-
Analog
zu Beispiel 24 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 1-Amino-cyclopentancarbonsäuremethylesterhydrochlorid
und 4-Iodmethyl-2-(3-methoxy-phenyl)-5-methyl-oxazol
das Racemat 1-({(1R,3S)/(1S,3R)-3-(2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-cyclopentancarbonsäure erhalten.
C25H32N2O6 (456.54), MS(ESI):
457(M + H
+).
-
Beispiel 27
-
Analog
zu Beispiel 24 wurde aus (1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonsäure, 1-Amino-cyclopentancarbonsäuremethylesterhydrochlorid
und 4-Iodmethyl-2-(3-trifluormethyl-phenyl)-5-methyl-oxazol
das Racemat 1-({(1R,3S)/(1S,3R)-3-[5-Methyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexancarbonyl}-amino)-cyclopentancarbonsäure erhalten.
C25H29F3N2O5 (494.52), MS(ESI):
495(M + H
+).
-
-
2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-methyl-amino}-3-methyl-buttersäure-tert-butylester
-
220
mg Natriumhydrid (60%ig in Paraffinöl) werden in 20 ml Dimethylformamid
suspendiert. Zu dieser Suspension werden 2 g des Diastereomerengemisches
2-{[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methyl-buttersäure-tert-butylester
gelöst
in 10 ml Dimethylformamid, zugegeben. Man rührt 15 Minuten bei Raumtemperatur
nach, dann wird 0,5 ml Methyliodid zugetropft. Nach 2 Stunden Rühren bei
Raumtemperatur wird das Reaktionssgemisch mit 200 ml Ethylacetat
verdünnt
und dreimal mit Wasser und gesättigter
NaCl-Lösung
gewaschen. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
2,3 g 2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-methyl-amino}-3-methyl-buttersäure-tert-butylester
als gelbes Öl. C33H49NO4Si
(551,85), MS(ESI): 552(M + H+).
-
2-[(cis-3-Hydroxy-cyclohexancarbonyl)-methyl-amino]-3-methyl-buttersäure-tert-butyl ester
-
2,3
g des Diastereomerengemisches 2-{[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-methyl-amino}-3-methyl-buttersäure-tert-butylester
werden in 10 ml Tetrahydrofuran gelöst und mit 6 ml einer 1M Lösung von
Tetrabutylammoniumfluorid in Tetrahydrofuran, versetzt. Man rührt 2 Stunden
bei 60°C nach,
dann wird im Vakuum eingeengt und der Rückstand an Kieselgel mit dem
Eluens n-Heptan : Ethylacetat = 2 : 1 ⇒ Ethylacetat gereinigt. Man
erhält
970 mg 2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-methyl-amino]-3-methyl-buttersäure-tert-butylester
als gelbes Öl.
C17H31NO4 (313,44), MS(ESI): 314(M + H+), Rf(n-Heptan
: Ethylacetat = 1 : 1) = 0,18.
-
3-Methyl-2-{methyl-[(1R,3S)/(1S,3R)-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure
-
150
mg Natriumhydrid (60%ig in Paraffinöl) werden in 5 ml Dimethylformamid
suspendiert. Zu dieser Suspension werden 970 mg des Diastereomerengemisches
2-[(cis-3-Hydroxy-cyclohexancarbonyl)-methyl-amino]-3-methyl-buttersäure-tert-butylester, gelöst in 10
ml Dimethylformamid, zugegeben. Man rührt 15 Minuten bei Raumtemperatur
nach, dann werden 1.5 g 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol, gelöst in 10
ml Dimethylformamid, zugetropft. Nach 2 Stunden Rühren bei
Raumtemperatur wird das Reaktionsgemisch mit 200 ml Methyl-tert.-butyl-ether
verdünnt
und dreimal mit Wasser und gesättigter
NaCl-Lösung
gewaschen. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der resultierende Rückstand wird in 10 ml Dichlormethan
gelöst
und mit 5 ml Trifluoressigsäure versetzt.
Man rührt
2 Stunden bei Raumtemperatur nach. Dann wird 50 ml Toluol zugegeben
und die Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird mittels RP-HPLC gereinigt. Nach Gefriertrocknung erhält man 105
mg 3-Methyl-2-{methyl-[cis-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-buttersäure als
weißes
Lyophilisat. C25H34N2O5 (442,56), MS(ESI): 443(M + H+).
-
Beispiel 29
-
Analog
zu Beispiel 28 wurde aus dem Diastereomerengemisch 2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methyl-buttersäure-tert-butylester,
Benzylbromid und 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol das Diastereomerengemisch (S)-2-{Benzyl-[(1R,3S)/(1S,3R)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-amino}-3-methyl-buttersäure erhalten.
C31H38N2O5 (518.66), MS(ESI):
519(M + H
+).
-
Beispiel 30
-
Analog
zu Beispiel 28 wurde aus dem Diastereomerengemisch 2-{[(1R,3S)/(1S,3R)-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexancarbonyl]-amino}-(3S)-methyl-buttersäure-tert-butylester
und Allylbromid das Diastereomerengemisch 2-[Allyl-((1R,3S)/(1S,3R)-3-hydroxy-cyclohexancarbonyl)-amino]-3-methyl-buttersäure-tert-butyl ester
erhalten.
C19H33NO4 (339,48), MS(ESI):
340(M + H
+).
-
2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-propyl-amino]-3-methyl-buttersäure-tert-butyl
ester
-
1
g des Diastereomerengemisches 2-[Allyl-((1R,3S)/(1S,3R)-3-hydroxy-cyclohexancarbonyl)-amino]-3-methyl-buttersäure-tert-butylester
werden in 30 ml Methanol gelöst
und mit 100 mg Palladium (10% auf Kohle) versetzt. Es wird 3 h unter
einer Wasserstoffatmosphäre
bei 5 bar gerührt.
Anschließend
wird über
Celite filtriert und das Lösungsmittel
im Vakuum entfernt. Man erhält
1 g 2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-propyl-amino]-3-methyl-buttersäure-tert-butylester
als farbloses Öl.
C19H35NO4 (341,50), MS(ESI): 342(M + H+).
-
Analog
zu Beispiel 28 wurde aus dem Diastereomerengemisch 2-[((1R,3S)/(1S,3R)-3-Hydroxy-cyclohexancarbonyl)-propyl-amino]-3-methyl-buttersäure-tert-butylester und 4-Iodmethyl-5-methyl-2-p-tolyl-oxazol das
Diastereomerengemisch (S)-3-Methyl-2-{[(1R,3S)/(1R,3S)-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl]-propyl-amino}-buttersäure erhalten.
C27H38N2O5 (470.61), MS(ESI):
342(M + H
+).
-
Beispiel
31: 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure:
-
3-(Methoxymethoxy)-cyclohexancarbonsäuremethylester:
-
15
g 6-Oxa-bicyclo[3.2.1]octan-7-on werden in 150 ml Methanol gelöst, mit
13 g Natriummethanolat versetzt und 2 h bei Raumtemperatur gerührt. Dann
werden 13,7 ml Eisessig zugegeben, und das Lösungsmittel wird im Vakuum
weitgehend abdestilliert. Der Rückstand
wird in Wasser aufgenommen und dreimal mit je 100 ml Ethylacetat
extrahiert. Die organischen Phasen werden über MgSO4 getrocknet und anschließend im
Vakuum eingeengt. Man erhält
18,8 g des Methylesters als farbloses Öl. Dieser wird in 150 ml Dichlormethan
gelöst
und mit 19,2 g Methoxymethylchlorid und 23,2 g Diisopropylethylamin
versetzt und 15 h bei Raumtemperatur gerührt. Die Lösung wird mit 250 ml gesättigter
NH4Cl-Lösung
und 200 ml Wasser vesetzt, die organische Phase wird abgetrennt.
Die wäßrige Phase
wird mit Dichlormethan extrahiert, die vereinigten organischen Phasen
werden über
Magnesiumsulfat getrocknet und eingeengt. Man erhält 22,2
g 3-(Methoxymethoxy)-cyclohexancarbonsäuremethylester
als gelbes Öl.
C10H18O4 (202), MS(ESI): 203 (MH+).
-
(3-Methoxymethoxycyclohexyl)-methanol:
-
9,0
g 3-Methoxymethoxy-cyclohexancarbonsäuremethylester werden in 280
ml Diethylether gelöst und
mit 2,2 g LiAlH4 versetzt und bei Raumtemperatur gerührt. Nach
4 h werden bei 0°C
10 ml Ethylacetat und anschließend
15 ml 10N NaOH zugetropft. Die Suspension wird 1 h gerührt und
mit MgSO4 versetzt, über
Celite filtriert, und das Filtrat eingeengt, wobei 7,0 g (3-Methoxymethoxy-cyclohexyl)-methanol als farbloses Öl erhalten
werden. C9H18O3 (174), MS(ESI): 175 (MH+).
-
(3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester:
-
1,0
g (3-Methoxymethoxy-cyclohexyl)-methanol und 3,3 g Bromessigsäure-tert-butylester werden
in 30 ml Toluol gelöst
und mit 0,50 g Tetrabutylammoniumhydrogensulfat versetzt. Die Suspension
wird auf 10°C gekühlt. 10
ml 50% NaOH werden zur Suspension gegeben. Die Mischung wird auf
Raumtemperatur kommen lassen und nach 3 h wird die wäßrige Phase
abgetrennt und mit Methyl-tert-butylether extrahiert. Die vereinigten
organischen Phasen werden über
MgSO4 getrocknet und eingeengt. Nach Flash-Säulenchromatographie an
Kieselgel (Heptan/Ethylacetat 10/1 → 2/1) werden 1,10 g (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester
als farbloses Öl
erhalten. C15H28O5 (288), LCMS(ESI): 306 (M+ +
H2O).
-
2-(3-Hydroxycyclohexylmethoxy)-pentansäure-tert-butylester:
-
200
mg (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester werden
in 5 ml abs. Tetrahydrofuran gelöst
und auf –78°C (Trockeneis/Aceton-Bad) gekühlt. Anschließend werden
0,7 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C gerührt, dann
auf 0°C
erwärmt
(Eisbad) und 20 min bei dieser Temperatur gerührt. Dann werden 600 mg Propyliodid
in 2 ml Tetrahydrofuran zugegeben, und die Lösung wird weitere 2,5 h bei
0°C gerührt. 15
ml gesättigte Ammoniumchloridlösung werden
zugesetzt und die Phasen getrennt. Die wäßrige Phase wird mit Methyl-tert-butylether
extrahiert. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und eingeengt (Ausbeute: 240 mg Rohprodukt). Der Rückstand
wird in 2 ml Tetrahydrofuran aufgenommen, mit 0,5 ml konz. HCl versetzt
und 18 h bei Raumtemperatur gerührt.
Die Mischung wird mit Wasser und Methyl-tert-butylether verdünnt, die
Phasen werden getrennt, und die wäßrige Phase wird mit Methyl-tert-butylether
extrahiert. Die vereinigten organischen Phasen werden mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt. Dabei werden 130 mg 2-(3-Hydroxycyclohexylmethoxy)-pentansäure-tert-butylester
als gelbes Öl
erhalten. NMR(CDCl3) Diastereomerengemisch: 3.55-3.67 (m, 2H), 3.41-3.48
(m, 1H), 3.07-3.18
(m, 1H), 1.91-2.13 (m, 2H), 1.11-1.82 (m, 14H), 1.48 (s, 9H).
-
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure-tert-butylester:
-
130
mg 2-(3-Hydroxycyclohexylmethoxy)-pentansäure-tert-butylester werden
in 3 ml Dimethylformamid gelöst
und mit 20 mg NaH (95%) versetzt. Nach 60 min Rühren werden bei 0°C 350 mg
5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid in 1 ml Dimethylformamid
zugegeben. Die Mischung wird bei Raumtemperatur 2 h gerührt. Anschließend werden
10 ml Methyl-tert-butylether, 5 ml Wasser und 10 ml gesättigte NaCl-Lösung zugegeben.
Die Phasen werden getrennt, die wäßrige Phase wird einmal mit
Methyl-tert-butylether extrahiert, die organsichen Phasen werden über MgSO4
getrocknet und eingeengt. Der Rückstand
wird an Kieselgel chromatographiert (Heptan/Ethylacetat 99/1 → 10/1).
20 mg des rohen 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure-tert-butylesters werden
als gelbes Öl
erhalten. C28H41NO5 (471), LCMS (ESI): 472 (MH+).
-
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure:
-
20
mg 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure-tert-butylesters werden
in 1 ml Trifluoressigsäure über Nacht
gerührt.
Die Lösung
wird vollständig
eingeengt und per HPLC gereinigt, wobei 15 mg 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-pentansäure 7 erhalten
werden. C24H33NO5 (415), MS(ES+) 416 (MH+).
-
Beispiel 32
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Methyliodid
und 5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure. C22H29NO5
(387), LCMS(ES+) 388 (MH+).
-
-
Beispiel 33
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Ethyliodid
und 5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-buttersäure. C23H31NO5
(401), LCMS(ES+) 402 (MH+).
-
-
Beispiel 34
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Benzylbromid
und 5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-3-phenylpropionäure. C28H33NO5
(463), LCMS(ES+) 464 (MH+).
-
-
Beispiel 35
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Methyliodid
und 5-Methyl-2-(3-methoxyphenyl)-oxazol-4-ylmethyliodid 2-[3-(5-Methyl-2-(3-methoxyphenyl)-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure. C22H29NO6
(403), LCMS(ES+) 404(MH+).
-
-
Beispiel 36
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Methyliodid
und 5-Methyl-2-m-tolyl-oxazol-4-ylmethyliodid
2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure. C23H31NO5
(401), LCMS(ES+) 402 (MH+).
-
-
Beispiel 37
-
Analog
zu Beispiel 31 erhält
man aus (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester, Methyliodid
und 5-Methyl-2-(3-trifluormethylphenyl)-oxazol-4-ylmethyliodid 2-[3-(5-Methyl-2-(3-trifluormethylphenyl)-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure. C22H26F3NO5
(441), LCMS(ES+) 442(MH
+).
-
Beispiel
38 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure:
-
2-(3-Hydroxycyclohexylmethoxy)-2-methylpropionsäure-tert-butylester:
-
300
mg (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester werden
in 5 ml abs. Tetrahydrofuran gelöst
und auf –78°C (Trockeneis/Aceton-Bad)
gekühlt.
Anschließend
werden 1,5 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C 90 min
gerührt,
dann auf 0°C
erwärmt
(Eisbad) und mit 1,41 g Methyliodid in 1,5 ml Tetrahydrofuran zugegeben, und
die Lösung
wird 1 h bei 0°C
gerührt.
1 ml HCl (konz.) wird zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt (Ausbeute: 320 mg Rohprodukt). Das
Rohprodukt wird in 5 ml abs. Tetrahydrofuran gelöst und auf –78°C (Trockeneis/Aceton-Bad) gekühlt. Anschließend werden
1,5 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C 90 min
gerührt, dann
auf 0°C
erwärmt
(Eisbad) und mit 1,41 g Methyliodid in 1,5 ml Tetrahydrofuran zugegeben,
und die Lösung
wird 1 h bei 0°C
gerührt.
1 ml HCl (konz.) wird zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt (Ausbeute: 350 mg Rohprodukt). Der
Rückstand
wird in 1 ml Tetrahydrofuran aufgenommen, mit 1 ml konz. HCl versetzt
und 3 d bei Raumtemperatur gerührt.
Die Mischung wird mit Wasser und Ethylacetat verdünnt, die
Phasen werden getrennt, und die wäßrige Phase wird mit Ethylacetat
extrahiert. Die vereinigten organischen Phasen werden mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt. Der Rückstand wird an Kieselgel (Heptan/Ethylacetat
2/1) chromatographiert, wobei 200 mg 2-(3-Hydroxycyclohexylmethoxy)-2-methylpropionsäure-tert-butylester
als gelbes Öl
erhalten werden. C15H28O4 (272,20), MS(ESI): 273,4 (MH+).
-
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylester:
-
200
mg 2-(3-Hydroxycyclohexylmethoxy)-2-methylpropionsäure-tert-butylester
werden in 5 ml Dimethylformamid gelöst und mit 20 mg NaH (95%)
versetzt. Nach 60 min Rühren
bei Raumtemperatur werden bei 0°C
460 mg 5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid
in 1,5 ml Dimethylformamid zugegeben. Die Mischung wird bei Raumtemperatur
2 h gerührt.
Anschließend
werden 10 ml Methyl-tert-butylether
und 10 ml gesättigte NH4Cl-Lösung zugegeben.
Die Phasen werden getrennt, die wäßrige Phase wird einmal mit
Methyl-tert-butylether extrahiert, die organsichen Phasen werden über MgSO4
getrocknet und eingeengt. Der Rückstand
wird an Kieselgel chromatographiert (Heptan/Ethylacetat 5/1 → 1/1). 200
mg des rohen 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylesters
werden als gelbes Öl
erhalten. C27H39NO5 (457), LCMS(ESI): 458 (MH+).
-
Verbesserte
Synthese von 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylester:
-
50
mg 2-(3-Hydroxycyclohexylmethoxy)-2-methylpropionsäure-tert-butylester
werden in 0,5 ml Dimethylformamid gelöst und mit 22 mg NaH (60%)
versetzt. Nach 30 min Rühren
werden bei Raumtemperatur 112 mg 5-Methyl-2-p-tolyl-oxazol-4-ylmethyliodid
zugegeben. Die Mischung wird 10 min ins Ultraschallbad gegeben und
anschließend
3 h bei Raumtemperatur gerührt.
Anschließend
werden 10 ml Methyl-tert-butylether und 10 ml Wasser zugegeben.
Die Phasen werden getrennt, die wäßrige Phase wird einmal mit
Methyl-tert-butylether extrahiert, die organsichen Phasen werden über MgSO4
getrocknet und eingeengt. Der Rückstand
wird an Kieselgel chromatographiert (Heptan/Ethylacetat 5/1 → 1/1 ).
60 mg des rohen 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylesters
werden als gelbes Öl
erhalten. C27H39NO5 (457), LCMS(ESI): 458 (MH+).
-
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure:
-
200
mg 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylesters
werden in 2 ml Trifluoressigsäure
1 h gerührt.
Die Lösung
wird vollständig
eingeengt und flash-chromatographisch gereinigt (Heptan/Ethylacetat
5/1), wobei 66 mg 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-2-methylpropionsäure erhalten
werden. C23H31NO5 (401,51), MS(ES+) 402,29 (MH
+). Beispiel
39: 1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure:
-
2-Allyl-2-(3-methoxymethoxycyclohexylmethoxy)-pent-4-ensäure-tert-butylester:
-
200
mg (3-Methoxymethoxycyclohexylmethoxy)-essigsäure-tert-butylester werden
in 6 ml abs. Tetrahydrofuran gelöst
und auf –78°C (Trockeneis/Aceton-Bad)
gekühlt.
Anschließend
werden 1,05 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C, dann bei
0°C jeweils
20 min gerührt
und bei 0°C
mit 0,85 g Allylbromid in 1,5 ml Tetrahydrofuran versetzt. Die Lösung wird
30 min bei 0°C
gerührt.
1 ml gesättigte
NH4Cl-Lösung
und 5 ml Ethylacetat werden zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt. Der Rückstand wird an Kieselgel chromatographiert (Heptan/Ethylacetat
10/1), wobei 160 mg monoallyliertes Produkt erhalten werden. Dieses
wird in 6 ml abs. Tetrahydrofuran gelöst und auf –78°C (Trockeneis/Aceton-Bad) gekühlt. Anschließend werden
1,05 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78 °C, dann bei
0°C jeweils
20 min gerührt
und bei 0°C
mit 0,85 g Allylbromid in 1,5 ml Tetrahydrofuranversetzt. Die Lösung wird
2 h bei 0°C
gerührt.
1 ml gesättigte
NH4Cl-Lösung
und 5 ml Ethylacetat werden zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt. Der Rückstand wird an Kieselgel chromatographiert (Heptan/Ethylacetat
5/1), wobei 140 mg 2-Allyl-2-(3-methoxymethoxycyclohexylmethoxy)-pent-4-ensäure-tert-butylester als gelbes Öl erhalten
werden. C21H36O5 (368,52), MS(ESI): 296,25 (MH+ – C4H9O).
-
(3-Hydroxycyclohexylmethoxy)-cyclopentancarbonsäure-tert-butylester:
-
140
mg 2-Allyl-2-(3-methoxymethoxycyclohexylmethoxy)-pent-4-ensäure-tert-butylester werden
in 5 ml Dichlormethan gelöst,
unter einer Ar-Atmosphäre
mit 10 mg Grubbs-Katalysator (Cl2(Cy3P)2Ru=CHPh) versetzt
und bei 40°C
48 h gerührt.
10 ml Heptan/Ethylacetat (3/1) werden zugegeben und die Lösung wird über Kieselgel
filtriert. 100 mg 1-(3-Methoxymethoxycyclohexylmethoxy)-cyclopent-3-encarbonsäure-tert-butylester
werden als braunes Öl
erhalten. Dieses wird in 2 ml McOH gelöst, entgast und mit Ar gesättigt. Dann
werden 30 mg Pd/C (10%) zugegeben und erneut entgast. Die Lösung wird
mit Wasserstoff gesättigt
und bei Raumtemperatur über
Nacht gerührt.
Verdünnen
mit 20 ml Ethylacetat und Filtration über Celite liefern 100 mg rohen
1-(3-Methoxymethoxycyclohexylmethoxy)-cyclopentancarbonsäure-tert-butylester.
Dieser wird in 2 ml Tetrahydrofuran aufgenommen, mit 0,5 ml HCl
(konz.) versetzt und über
Nacht bei Raumtemperatur gerührt. Die
Lösung
wird mit gesättigter
NaHCO3-Lösung
neutralisiert und dreimal mit Ethylacetat extrahiert. Die organischen
Phasen werden über
MgSO4 getrocknet und eingeengt. Chromatographie des Rückstandes
an Kieselgel (Heptan/Ethylacetat 10/1 → 1/1) liefert 57 mg 1-(3-Hydroxycyclohexylmethoxy)-cyclopentancarbonsäure-tert-butylesfer als gelbes Öl. Dieses
wird roh für
die nächste
Stufe eingesetzt.
-
1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure-tert-butylester:
-
57
mg 1-(3-Hydroxycyclohexylmethoxy)-cyclopentancarbonsäure-tert-butylester
werden in 3 ml Dimethylformamid gelöst und mit 10 mg NaH versetzt.
Die Suspension wird 30 min bei Raumtemperatur gerührt, dann
auf 0°C
gekühlt
und tropfenweise mit 150 mg Methyl-2-p-tolyl-oxazol-4-ylmethyliodid
in 1 ml Dimethylformamid versetzt. Die Suspension wird 2 h bei Raumtemperatur
gerührt
und mit Methyl-tert-butylether und gesättigter NaCl-Lösung verdünnt. Die
wäßrige Phase
wird abgetrennt und mit Methyl-tert-butylether extrahiert. Die vereinigten
organischen Phasen werden mit gesättigter NaCl-Lösung gewaschen, über MgSO4
getrocknet und eingeengt. Chromatographie des Rückstandes an Kieselgel (Heptan/Ethylacetat
99/ → 10/1)
liefert 20 mg eines Produktgemisches, das den gewünschten
1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure-tert-butylester
It. LCMS enthält.
Dieses Gemisch wird ohne weitere Aufreinigung in der nächsten Stufe
eingesetzt. C29H41NO5 (483,65), LCMS (ESI): 484,2 (MH+).
-
1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure:
-
20
mg verunreinigter 1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure-tert-butylester
wird in 1 ml Trifluoressigsäure
bei Raumtemperatur über
Nacht gerührt.
Die Lösung
wird vollständig
eingeengt und der Rückstand
an Kieselgel chromatographiert (Heptan/Ethylacetat 10/1 → 1/1 → Methyl-tert-butylether),
wobei 7,5 mg 1-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-cyclopentancarbonsäure erhalten
werden. C25H33NO5 (427,55); LCMS 428,2 (MH
+). Beispiel
40:
-
3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-essigsäure-tert-butylester
-
25
g [3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-methanol werden
zusammen mit 40 g Bromessigsäure-tert-butylester
und 6,9 g Tetrabutylammoniumhydrogensulfat in 300 ml Toluol gelöst und anschließend bei 0°C 200 ml
NaOH (50%ig) zugetropft. Bei 0°C
wird 1 h gerührt
und dann auf Raumtemperatur erwärmt. Das
Lösungsmittel
wird entfernt und mit 3 × 100
ml Methyl-tert-butylether extrahiert. Nach der dritten Extraktion wird
die wässrige
Phase angesäuert
und nochmals mit 200 ml Methyl-tert-butylether extrahiert. Die vereinigten organischen
Phasen werden mit gesättigter
Natriumclorid -Lösung
ausgeschüttelt, über Magnesiumsulfat
getrocknet und das Lösungsmittel
im Vakuum entfernt. Man erhält
27,8 g des 3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-essigsäure-tert-butylesters
als gelbes Öl.
C29Ha42O4Si (482.74), MS(ESI): 483 (M + H+)
-
2-[3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-2-methyl-propionsäure- tert-butylester
-
20,0
g des 3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-essigsäure-tert-butylesters werden
in einem ausgeheizten 1 L-Dreihals-Kolben vorgelegt, in 200 ml trockenem
Tetrahydrofuran gelöst,
auf –78°C gekühlt und
83 ml Lithiumdiisopropylamid (2N in Tetrahydrofuran) langsam zugetropft,
so dass die Innentemperatur nicht über –65°C ansteigt. Anschließend wird
auf 0°C
erwärmt
und 1 h gerührt,
wobei sich die Lösung gelb
färbt.
Nach erneutem Abkühlen
auf –70°C werden
35,27 g Methyliodid zugetropft und 3 h bei 0°C gerührt. Reaktionskontrolle (DC
und LCMS) ergibt die Bildung eines neuen Produktes (Monomethyl Verbindung).
-
Das
Reaktionsgemisch wird mit 200 ml gesättigter Ammoniumchlorid-Lösung versetzt
und mit Wasser/Methyl-tert-butylether extrahiert. Man erhält das Rohprodukt
als dunkelrotes öl,
welches ohne Aufreinigung in der gleichen Reaktionssequenz zur geminalen
Dimethylverbindung umgesetzt wird. Das Rohprodukt wird an Kieselgel
(Heptan/Ethylacetat 50 : 1 → 10
: 1) gereinigt. Man erhält
16 g des 2-[3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-2-methyl-propionsäure-tert-butylesters
als hellgelbes Öl.
C31Ha6O4Si
(510.80), MS(ESI): 511 (M + H+)
-
2-(3-Hydroxy-cyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester
-
16
g des 2-[3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexylmethoxy]-2-methyl-propionsäure-tert-butylesters
werden in 100 ml Acetonitril gelöst
und mit 62 ml Tetrabutylammoniumfluorid (1N Lösung in Tetrahydrofuran) versetzt.
Nach 2 h Rühren
bei 60°C
ist die Reaktion beendet und es wird im Vakuum eingeengt. Der Rückstand
wird aus Wasser/Ethylacetat extrahiert. Die vereinigten org. Phasen
werden mit gesättigter
Natriumchlorid-Lösung
ausgeschüttelt, über Magnesiumsulfat
getrocknet und das Lösungsmittel
im Vakuum entfernt. Das Rohprodukt wird an Kieselgel (Heptan/Ethylacetat
15 : 1 → 1
: 1) gereinigt. Man erhält
16 g des Produktes 2-(3-Hydroxy-cyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester
als farbloses Öl.
C15H28O4 (272.39), MS(ESI):
273 (M + H+)
-
2-{3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure-tert-butylester
-
0,05
g des Alkohols 2-(3-Hydroxy-cyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester wird in Methyl-tert-butylether
gelöst
und mit 15 mg Natriumhydrid versetzt. Nach 15 Minuten Rühren bei
Raumtemperatur werden 0.12 g 2-(4-Fluoro-phenyl)-4-iodmethyl-5-methyl-oxazol
zugegeben und 12 h bei Raumtemperatur gerührt. Nach Zugabe von 2 ml 1N
HCl wird mit Ethylacetat (2 × 5
ml) extrahiert, das Lösungsmittel
im Vakuum entfernt und das Rohprodukt anschließend über HPLC gereinigt. Man erhält 0.08
g der Verbindung 2-{3-[2-(4-Fluuoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure-tert-butylester
als farbloses Öl
C26H36FNO5 (461.58), MS(ESI): 462 (M + H+)
-
2-{3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure
-
0,07
g des 2-{3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure-tert-butylesters
wird in 1 ml Dichlormethan gelöst,
mit 1 ml Trifluoressigsäure
versetzt und bei Raumtemperatur gerührt. Reaktionskontrolle (LCMS)
ergibt nach 30 Minuten einen vollständigen Umsatz. Das Reaktionsgemisch
wird mit Wasser/Dichlormethan ausgeschüttelt und nach Entfernen des
Lösungsmittels
im Vakuum mittels präparativer
HPLC gereinigt. Man erhält
0,06 g der Carbonsäure
2-{3-[2-(4-Fluorophenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure als
farbloses Öl.
C22H28FNO5 (405.47), MS(ESI): 406 (M + H+)
-
Beispiel 41:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethyliodid 2-{(1S,3R)-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure.
C
23H
31NO
6 (417,50) MS(ESI):
418 (M + H
+)
-
Beispiel 42:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 2-(3-Trifluormethyl-phenyl)-5-methyl-oxazol-4-ylmethyliodid 2-{(1S,3R)-3-[2-(3-Trifluormethyl-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure.
C
22H
28F
3NO
5 (455,47)
MS(ESI): 456 (M + H
+)
-
Beispiel 43:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 5-methyl-2-(5-methyl-furan-2-yl)-oxazol-4-ylmethyliodid 2-Methyl-2-{(1R,3S)-3-[5-methyl-2-(5-methyl-furan-2-yl)-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-propionsäure.
C
21H
29NO
6 (391,46) MS(ESI):
392 (M + H
+)
-
Beispiel 44:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 2-(3,4-Dimethoxy-phenyl)-5-methyl-oxazol-4-ylmeth yliodid 2-{(1R,3S)-3-[2-(3,4-Dimethoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure.
C
24H
33NO
7 (447,53) MS(ESI):
448 (M + H
+)
-
Beispiel 45:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 5-Phenyl-2-p-tolyl-oxazol-4-ylmethyliodid
2-Methyl-2-[(1R,3S)-3-(5-phenyl-2-p-tolyl-oxazol-4-ymethoxy)-cyclohexylmethoxy]-propionsäure
C
28H
33NO
5 (463,57) MS(ESI):
464 (M + H
+)
-
Beispiel 46:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 5-Methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-ylmethyliodid 2-Methyl-2-{(1R,3S)-3-[5-methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-propionsäure
C
22H
28F
3NO
5 (455,47)
MS(ESI): 456 (M + H
+)
-
Beispiel 47:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 2-(4-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethyliodid 2-{(1R,3S)-3-[2-(4-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexylmethoxy}-2-methyl-propionsäure
C
23H
33NO
6 (417,50) MS(ESI):
418 (M + H
+)
-
Beispiel 48:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 5-Methyl-2-thiophen-2-yl-oxazol-4-ylmethyliodid
2-Methyl-2-[(1R,3S)-3-(5-methyl-2-thiophen-2-yl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure
C
22H
27NO
5S (393,50) MS(ESI):
394 (M + H
+)
-
Beispiel 49:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und -methyl-2-(3-trifluoromethoxy-phenyl)-oxazol-4-ylmethyliodid 2-Methyl-2-[(1R,3S)-3-[5-methyl-2-(3-trifluoromethoxy-phenyl)-oxazol-4-ylmethoxy]-cyclohexylmethoxy]-propionsäure
C
23H
28F
3NO
6 (471,47)
MS(ESI): 472 (M + H
+)
-
Beispiel 50:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methyl-propionsäure-tert-butylester und 5-methyl-2-(4-Isopropyl-phenyl)-oxazol-4-ylmethyliodid 2-Methyl-2-[(1R,3S)-3-(5-methyl-2-(4-Isopropyl-phenyl)-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure.
C
25H
35NO
5 (429,56) MS(ESI):
430 (M + H
+)
-
Beispiel 51:
-
Analog
zu Beispiel 40 erhält
man aus 2-(3-Hydroxycyclohexylmethoxy)-2-methylpropionsäure-tert-butylester
und 5-methyl-2-m-toluyl-oxazol-4-ylmethyliodid 2-Methyl-2-[(1R,3S)-3-(5-methyl-2-m-toluyl-oxazol-4-ylmethoxy)-cyclohexylmethoxy]-propionsäure.
C
23H
31NO
5 (401,50) MS(ESI):
402 (M + H
+) Beispiel
52: 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäure:
-
5-Methyl-2-p-tolyl-oxazol-4-carboxaldehyd:
-
9,3
g LiAIH4 werden in einem trockenen Vierhalskolben mit Rührmotor,
Innenthermometer, Tropftrichter mit Druckausgleich und Rückflusskühler mit
Argonzuleitung (Absaugstück
mit Hahn) mit 600 ml Diethylether überschichtet. Die Suspension
wird auf 0°C
gekühlt.
30 g 5-Methyl-2-p-tolyl-oxazol-4-carbonsäureethylester
werden in 100 ml Diethylether gelöst und der Suspension tropfenweise
zugegeben. Nach einstündigem Rühren bei
Raumtemperatur ist die Reaktion beendet (DC(Heptan/Ethylacetat 1/1
): Rf,Ed ukt = 0.66,
Rf,Prod ukt = 0.18).
80 g MgSO4, 300 ml Methyl-tert-butylether
und 30 ml Ethylacetat werden nacheinander zugegeben, und die Suspension
wird bei Raumtemperatur gerührt.
Dann wird die Mischung auf 0°C
gekühlt,
tropfenweise mit 90 ml 10N KOH versetzt und weitere 60 min gerührt. Die
Feststoffe werden abfiltriert, der Rückstand dreimal mit Ethylacetat
gewaschen, und das Filtrat wird eingeengt, wobei 24 g 5-Methyl-2-p-tolyl-oxazol-4-methanol als
gelber Feststoff erhalten werden. C12H13NO2 (203,24), LCMS(ESI):
204.1 (MH+).
-
Zu
einer Lösung
von 12 ml Oxalylchlorid in 150 ml Dichlormethan werden bei –78 °C 22.2 ml
DMSO in 30 ml Dichlormethan so zugetropft, dass die Temperatur nicht über –70°C steigt.
Anschließend
wird die Lösung
30 min bei dieser Temperatur gerührt.
Dann werden 24 g 5-Methyl-2-p-tolyl-oxazol-4-methanol in 120 ml Dichlormethan/Chloroform
(2/1) zugetropft, wobei die Temperatur nicht über –70 °C steigt. Die Lösung wird
30 min bei dieser Temperatur gerührt.
Anschließend
werden 80 ml NEt3 so zugetropft, dass die
Temperatur nicht über –70°C steigt.
Nach beendeter Zugabe wird das Kühlbad
entfernt und die Lösung
unter Rühren
auf 0°C gebracht.
Bei dieser Temperatur werden 100 ml Wasser zugegeben, und die Mischung
wird heftig bei Raumtemperatur gerührt. Die wäßrige Phase wird abgetrennt
und mit Chloroform extrahiert. Die vereinigten organischen Phasen
werden mit gesättigter
NH4Cl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt, wobei 23,8
g 5-Methyl-2-p-tolyl-oxazol-4-carbaldehyd als gelber Feststoff erhalten
werden. C12H11NO2 (201,23), LCMS(ESI): 202,1 (MH+).
-
[3-(tert-Butyldimethylsilanyloxy)-cyclohexyl]-carbonsäuremethylester:
-
47
g 6-Oxa-bicyclo[3.2.1]octan-7-on werden in 500 ml McOH gelöst und mit
40,5 g NaOMe versetzt. Nach 2,5-stündigem Rühren bei Raumtemperatur werden
135 ml Essigsäure
zugegeben und das Methanol weitgehend abdestilliert. Der Rückstand
wird mit Ethylacetat/Wasser aufgenommen, und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert, die vereinigten organischen Phasen
werden über MgSO4
getrocknet und eingeengt, wobei der Methylester quantitativ als
Rückstand
erhalten wird.
-
10,7
g des Rückstandes
werden in 100 ml Dimethylformamid gelöst und mit 11,2 g tert-Butyldimethylsilylchlorid
versetzt. Bei 0°C
werden 11,5 g Imidazol zugegeben und die Lösung wird bei Raumtemperatur über Nacht
gerührt.
200 ml gesättigte
NaCl-Lösung
werden zugesetzt und die Lösung
wird dreimal mit Methyl-tert-butylether
extrahiert. Die vereinigten organischen Phasen werden mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt. 16,4 g [3-(tert-Butyldimethylsilanyloxy)-cyclohexyl]-carbonsäuremethylester
werden als farbloses Öl
erhalten. C14H28O3Si (272,46), MS(ESI): 273,13 (MH+).
-
3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexancarbonsäuremethylester:
-
1,35
g [3-(tert-Butyldimethylsilanyloxy)-cyclohexyl]-essigsäuremethylester
werden bei Raumtemperatur zu einer Mischung von 4,0 ml HSiEt3 und
1,50 g BiBr3 in 20 ml Acetonitril getropft. Anschließend werden 1,51
g 5-Methyl-2-p-tolyl-oxazol-4-carbaldehyd
in 5 ml Acetonitril zugegeben und die Mischung 4 h bei Raumtemperatur
gerührt.
Die Suspension wird filtriert und eingeengt. Der Rückstand
wird an Kieselgel chromatographiert (Dichlormethan/Methanol 100/1),
wobei 1,20 g 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexancarbonsäuremethylester
als hellgelbes Öl
erhalten werden. C20H25NO4 (343,43), LCMS(ESI): 344,1 (MH+).
-
3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-methanol:
-
1,70
g 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexancarbonsäuremethylester in 5 ml Tetrahydrofuranwerden
bei 0°C
zu einer Suspension von 380 mg LiAIH4 in 50 ml Diethylether getropft
und 2 h bei Raumtemperatur gerührt.
3 g MgSO4, 30 ml Methyl-tert-butylether
und 3 ml Ethylacetat werden nacheinander zugegeben und die Suspension
bei Raumtemperatur gerührt.
Dann wird die Mischung auf 0°C
gekühlt,
tropfenweise mit 1 ml 10N KOH versetzt und weitere 60 min gerührt. Die
Feststoffe werden abfiltriert, der Rückstand dreimal mit Ethylacetat
gewaschen, und das Filtrat wird eingeengt, wobei 1,55 g 3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-methanol als gelbes Öl erhalten
werden. C19H25NO3 (315,42), MS(EI): 315.4 (M+).
-
4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol:
-
Zu
1,55 g 3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-methanol
in 20 ml Toluol werden 1,56 g PPh3, 0,87 g Imidazol und 1,64 g Iod
gegeben und die Mischung 2 h bei Raumtemperatur gerührt. Dann werden
10 ml Dichlormethan zugesetzt und weitere 60 min gerührt. Die
Lösung
wird mit 50 ml Wasser und 50 ml Methyl-tert-butylether verdünnt, die
Phasen werden getrennt, die organische Phase wird über MgSO4
getrocknet und eingeengt. Filtration des Rückstandes über Kieselgel mir Dichlormethan
liefert 1,12 g 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol
als gelben Feststoff. C19H24INO2 (425,31); LCMS (ESI): 426.0 (MH+).
-
3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäureethylester:
-
68
mg Mercaptoessigsäureethylester
in 1,5 ml Dimethylformamid werden mit 50 mg KOtBu
versetzt und 1 h bei Raumtemperatur gerührt. Anschließend werden
120 mg 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol
zugegeben und die Lösung
wird bei Raumtemperatur gerührt.
Nach 1 h werden 20 ml Methyl-tert-butylether,
15 ml gesättigte
NaCl-Lösung
und 15 ml Wasser zugegeben und- die
Phasen getrennt. Die wäßrige Phase
wird mit Methyl-tert-butylether extrahiert, die vereinigten organischen
Phasen werden mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt, wobei 117 mg 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäureethylester
erhalten werden. C23H31NO4S (417,57); LCMS (ESI): 418,1 (MH+).
-
3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäure:
-
117
mg 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäureethylester
in 3 ml Methanol gelöst
werden mit 1 ml 2N KOH versetzt und bei Raumtemperatur über Nacht
gerührt.
Dann werden 2 ml 2N HCl, 10 ml gesättigte NaCl-Lösung, 5
ml Wasser und 20 ml Dichlormethan zugesetzt und die Phasen getrennt.
Die organische Phase wird über
MgSO4 getrocknet und eingeengt, wobei 100 mg 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäure erhalten
werden. C21 H27NO4S (389,52); LCMS(ESI): 390,1 (MH+).
-
Beispiel 53:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercaptopropionsäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-propionsäure als
Diastereomerengemisch. C22H29NO4S (403,54), LCMS(ESI): 404,1 (MH+).
-
-
Beispiel 54:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercaptobuttersäuremethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-buttersäure als
Diastereomerengemisch. C23H31NO4S (417,57), LCMS(ESI): 418,1 (MH+).
-
-
Beispiel 55:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercaptoheptansäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-heptansäure als
Diastereomerengemisch. C26H37NO4S (459,65), MS(ESI): 460,41 (MH+).
-
-
Beispiel 56:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercapto-3-methylbuttersäuresäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-3-methylbuttersäure als
Diastereomerengemisch. C24H33NO4S (431,60), LCMS(ESI): 432.2 (MH+).
-
-
Beispiel 57:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercapto-2-methylpropionsäuresäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-2-methylpropionsäure. C23H31NO4S
(417,57), LCMS(ESI): 418,1 (MH+).
-
-
Beispiel 58:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercapto-2-phenylessigsäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-2-phenylessigsäure als
Diastereomerengemisch. C27H31NO4S (465,62), MS(ESI): 466,39 (MH+).
-
-
Beispiel 59:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercapto-2-cyclohexylessigsäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-2-cyclohexylessigsäure als
Diastereomerengemisch. C27H37NO4S (471,66), LCMS(ESI): 472,2 (MH+).
-
-
Beispiel 60:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 2-Mercaptovaleriansäureethylester
2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-valeriansäure als
Diastereomerengemisch. C24H33NO4S (431,60), MS(ESI): 432,39 (MH+).
-
-
Beispiel 61:
-
Analog
zu Beispiel 52 erhält
man aus 4-(3-Iodmethylcyclohexyloxymethyl)-5-methyl-2-p-tolyl-oxazol und 1-Mercaptocyclobutancarbonsäureethylester
1-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-cyclobutancarbonsäure. C24H31NO4S
(429,58), MS(ESI): 430,35 (MH
+).
-
Bausteinsynthese
der 2-Mercaptobuttersäuremethylester 2-Mercaptobuttersäuremethylester
-
Zu
1,81 g 2-Brombuttersäuremethylester
in 5 ml Dimethylformamid werden 1,43 g KSAc gegeben und die Mischung
12 h bei Raumtemperatur gerührt.
Dann werden 25 ml Methyl-tert-butylether, 10 ml Wasser und 15 ml
gesättigte
NaCl-Lösung
zugesetzt und die Phasen getrennt. Die wäßrige Phase wird mit Methyl-tert-butylether extrahiert,
die vereinigten organischen Phasen werden mit gesättigter
NaCl-Lösung
gewaschen, über MgSO4
getrocknet und eingeengt, wobei 2-Acetylsulfanylbuttersäuremethylester
als gelbes Öl
erhalten werden. Dieses wird in 10 ml Methanol aufgenommen und mit
11 ml einer 1M NaSMe Lösung
in Methanol versetzt und über
Nacht bei Raumtemperatur gerührt.
Das Lösungsmittel
wird im Vakuum vollständig
abdestilliert, der Rückstand
mit 15 ml Methyl-tert-butylether und 20 ml Wasser aufgenommen, die
Phasen werden getrennt, die organische Phase mit gesättigter
NaCl-Lösung
gewaschen und über
MgSO4 getrocknet. Nach Einengen der Lösung im Vakuum werden 1,30
g 2-Mercaptobuttersäuremethylester
als gelbes Öl
erhalten.
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Bromheptansäureethylester
2-Mercaptoheptansäureethylester.
-
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Brom-3-methylbuttersäuresäureethylester
2-Mercapto-3-methylbuttersäuresäureethylester.
-
-
Analog
zur Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Brom-2-methylpropionsäuresäureethylester
2-Mercapto-2-methylpropionsäuresäureethylester.
-
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Brom-2-phenylessigsäureethylester
2-Mercapto-2-phenylessigsäureethylester.
-
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Brom-2-cyclohexylessigsäuremethylester
2-Mercapto-2-cyclohexylessigsäuremethylester.
-
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
2-Bromvaleriansäureethylester
2-Mercaptovaleriansäureethylester.
-
-
Analog
zu Bausteinsynthese der 2-Mercaptobuttersäuremethylester erhält man aus
1-Bromcyclobutancarbonsäureethylester
1-Mercaptocyclobutancarbonsäureethylester.
-
-
3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfinyl]-essigsäure:
65
mg 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäure werden
in 1,5 ml Trifluoressigsäure
gelöst,
bei 0°C
mit 6,3 μl
35% H2O2 versetzt und über
Nacht bei Raumtemperatur gerührt.
Gesättigte
NH4Cl-Lösung und
Methyl-tert-butylether werden zugegeben, die Phasen getrennt, die
wäßrige Phase mit
Methyl-tert-butylether extrahiert, die vereinigten organischen Phasen
mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt. Der Rückstand wird per HPLC gereinigt,
wobei 6,6 mg eines farblosen Feststoffs erhalten wurden. C21H27NO5S
(405,52), LCMS(ESI): 406,1 (MH+).
-
Beispiel 63:
-
Analog
zu Beispiel 62 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-heptansäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfinyl]-heptansäure als Diastereomerengemisch.
C26H37NO5S (475,65), MS(ESI): 476,18 (MH+).
-
-
Beispiel 64:
-
Analog
zu Beispiel 62 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-2-methylpropionsäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfinyl]-2-methylpropionsäure. C23H31NO5S
(433,57), LCMS(ESI): 434,1 (MH+).
-
-
Beispiel 65:
-
Analog
zu Beispiel 62 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-3-methylbuttersäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfinyl]-3-methylbuttersäure als
Diastereomerengemisch. C24H33NO5S (447,60), MS(ESI): 448,43 (MH+).
-
-
Beispiel 66:
-
Analog
zu Beispiel 62 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-valeriansäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfinyl]-valeriansäure als
Diastereomerengemisch. C24H33NO5S (447,60), MS(ESI): 448,14 (MH
+).
-
Beispiel
67: 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfonyl]-essigsäure:
-
65
mg 3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-essigsäure werden
in 1,5 ml Trifluoressigsäure
gelöst,
bei 0°C
mit 21,5 μl
35% H2O2 versetzt und über
Nacht bei Raumtemperatur gerührt.
Gesättigte
NH4Cl-Lösung und
Methyl-tert-butylether werden zugegeben, die Phasen getrennt, die
wäßrige Phase
mit Methyl-tert-butylether extrahiert, die vereinigten organischen
Phasen mit gesättigter
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt. Der Rückstand wird per HPLC gereinigt,
wobei 6,6 mg eines farblosen Feststoffs erhalten wurden. C21H27NO6S
(421,52), LCMS(ESI): 422,1 (MH+).
-
Beispiel 68:
-
Analog
zu Beispiel 67 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-heptansäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfonyl]-heptansäure als Diastereomerengemisch.
C26H37NO6S (491,65), MS(ESI): 492,42 (MH+).
-
-
Beispiel 69:
-
Analog
zu Beispiel 67 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-2-methylbuttersäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfonyl]-2-methylbuttersäure. C23H31NO6S
(449,57), LCMS(ESI): 450,1 (MH+).
-
-
Beispiel 70:
-
Analog
zu Beispiel 67 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-3-methylbuttersäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfonyl]-3-methylbuttersäure als
Diastereomerengemisch. C24H33NO6S (463,60), LCMS(ESI): 464,1 (MH+).
-
-
Beispiel 71:
-
Analog
zu Beispiel 67 erhält
man aus 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfanyl]-valeriansäure 2-[3-(5-Methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethylsulfonyl]-valeriansäure als
Diastereomerengemisch. C24H33NO6S (463,60), MS(ESI): 464,14 (MH
+).
-
Beispiel
72: (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure
-
3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbaldehyd
-
Zu
0,48 ml Oxalylchlorid in 15 ml Dichlormethan werden bei –78°C 089 ml
DMSO in 1 ml Dichlormethan so zugetropft, dass die Temperatur –70°C nicht übersteigt.
Nach beendeter Zugabe wird die Lösung
30 min bei dieser Temperatur gerührt.
Dann werden 1,5 g 3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-methanol
in 2 ml Dichlormethan so zugetropft, dass die Temperatur unter –78°C bleibt.
Die Lösung
wird 30 min bei dieser Temperatur gerührt. Dann werden 3,2 ml NEt3
zugetropft, das Kühlbad
wird entfernt und die Lösung
auf 0°C
erwärmt.
Bei dieser Temperatur werden 10 ml Wasser zugegeben, und die Mischung
wird heftig bei Raumtemperatur gerührt. Die wäßrige Phase wird abgetrennt
und mit Dichlormethan extrahiert. Die vereinigten organischen Phasen
werden mit gesättigter
NH4Cl-Lösung
gewaschen, über
MgSO4 getrocknet und eingeengt, wobei 1,50
g 3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbaldehyd
erhalten werden. C19H23NO3 (313,40); LCMS (ESI): 314,1 (MH+).
-
(S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
-
511
mg 3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexancarbaldehyd,
0,9 ml HOAc und 310 mg (S)-Valin-tert-butylester werden in 5 ml
abs. Dichlormethan gelöst.
Dann werden 500 mg Molsieb 4 Å zugegeben
und die Suspension wird auf 0°C
gekühlt.
414 mg Natriumtriacetoxyborhydrid werden portionsweise zugesetzt.
Dies Suspension wird 2 h bei 0°C
gerührt,
dann mit 3 ml gesättigter
NH4Cl-Lösung
versetzt und weitere 10 min gerührt.
Jeweils 10 ml Wasser und Dichlormethan werden zugegeben, die Phasen
werden getrennt, die wäßrige Phase
wird mit Dichlormethan extrahiert, und die vereinigten organischen
Phasen werden über
MgSO4 getrocknet und eingeengt, wobei 760 mg (S)-3-Methyl-2-{(3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
erhalten werden. C28H42N2O4 (470,66); MS (ESI): 471.50 (MH+).
-
(S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure
-
40
mg (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
werden in 1 ml Ameisensäure
gelöst
und mit 0,5 ml Trifluoressigsäure
versetzt. Die Lösung
wird 18 bei Raumtemperatur gerührt,
dann vollständig
eingeengt. Der Rückstand
wird per HPLC gereinigt, wobei 28,2 mg (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure Trifluoressigsäuresalz
als farbloser Feststoff erhalten werden. C24H34N2O2.C2HF3O2 (414,55); MS(ES-):
413,28 (M
+ – H). Beispiel
73: (S)-2-{Acetyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure
-
(S)-2-{Acetyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure
-
40
mg (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
in 0,5 ml Dichlormethan werden mit 12 μl Acetylchlorid und 22 μl Pyridin
versetzt und 18 h bei Raumtemperatur gerührt. Dann wird die Lösung mit
Wasser und Dichlormethan verdünnt,
die wäßrige Phase
wird abgetrennt und mit Dichlormethan extrahiert, die vereinigten
Phasen werden über
MgSO4 getrocknet und eingeengt. Der Rückstand wird in 0,5 ml Trifluoressigsäure aufgenommen
und bei Raumtemperatur über
Nacht stehen gelassen. Das Lösungsmittel
wird vollständig
abdestilliert, der Rückstand
per HPLC gereinigt, wobei 17 mg (S)-2-{Acetyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure erhalten
werden. C26H36N2O5 (456,59); LCMS (ESI):457,36 (MH+).
-
Beispiel 74:
-
Analog
zu Beispiel 73 erhält
man aus (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
und Benzoylchlorid (S)-2-{Benzoyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure. C31H38N2O5
(518,29); LCMS (ESI): 519,54 (MH+).
-
-
Beispiel 75:
-
Analog
zu Beispiel 73 erhält
man aus (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
und Methylsulfonylchlorid (S)-2-{Methylsulfonyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure. C25H36N2O6S
(492,23); LCMS (ESI): 493,26 (MH+).
-
-
Beispiel 76:
-
Analog
zu Beispiel 73 erhält
man aus (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
und Methylsulfonylchlorid in Triethylamin (S)-2-{Methylsulfonylmethylsulfonyl-[3-(5- methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure. C26H38N2O8S2
(570,21); LCMS (ES-): 569,23 (M+ – H).
-
-
Beispiel 77:
-
Analog
zu Beispiel 73 erhält
man aus (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
und p-Toluolsulfonylchlorid
(S)-2-{p-Toluolsulfonyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure. C31H40N2O6S
(568,26); LCMS (ESI): 569,35 (MH+).
-
-
Beispiel 78:
-
Analog
zu Beispiel 73 erhält
man aus (S)-3-Methyl-2-{[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-buttersäure-tert-butylester
und Chlorameisensäuremethylester
(S)-2-{Methoxycarbonyl-[3-(5-methyl-2-p-tolyloxazol-4-ylmethoxy)-cyclohexylmethyl]-amino}-3-methylbuttersäure. C26H36N2O6 (472,26),
LCMS (ESI): 473,37 (MH
+).
-
Beispiel
79: 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure:
-
(3-Methoxymethoxymethylcyclohexylmethyl)-methanol:
-
1,70g
g 3-Hydroxymethylcyclohexancarbonsäuremethylester werden in 20
ml Dichlormethan gelöst und
mit 1,60 g Methoxymethylchlorid und 2,60 g Diisopropylethylamin
versetzt und 15 h bei Raumtemperatur gerührt. Die Lösung wird mit 50 ml gesättigter
NH4Cl-Lösung
und 50 ml Wasser vesetzt, die organische Phase wird abgetrennt.
Die wäßrige Phase
wird mit Dichlormethan extrahiert, die vereinigten organischen Phasen werden über Magnesiumsulfat
getrocknet und eingeengt. Man erhält 2,0 g 3-Methoxymethoxymethylcyclohexancarbonsäuremethylester
als gelbes Öl.
Dieses wird in 50 ml Diethylether gelöst und mit 350 mg LiAIH4 versetzt
und bei Raumtemperatur gerührt.
Nach 2 h werden bei 0°C
5 ml Ethylacetat, 40 ml Methyl-tert-butylether und
3 g MgSO4 zugegeben. Anschließend
werden 15 ml 10N KOH zugetropft. Die Suspension wird 3 h gerührt, über Celite
filtriert, und das Filtrat eingeengt, wobei 1,65 g (3-Methoxymethoxymethylcyclohexyl)-methanol
als farbloses Öl
erhalten werden. C10H20O3 (188,27), MS(ESI): 189,2 (MH+).
-
[3-(Methoxymethoxymethyl)-cyclohexylmethoxy]-essigsäure-tert-butylester:
-
1,65
g (3-Methoxymethoxymethylcyclohexyl)-methanol und 5,1 g Bromessigsäure-tert-butylester werden
in 20 ml Toluol gelöst
und mit 1,50 g Tetrabutylammoniumhydrogensulfat versetzt. Die Suspension
wird auf 10°C
gekühlt.
20 ml 50% NaOH werden zur Suspension gegeben. Die Mischung wird
6 h bei 10°C
gerührt, dann
wird die wäßrige Phase
abgetrennt und mit Methyl-tert-butylether
extrahiert. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und eingeengt. Nach Flash-Säulenchromatographie an Kieselgel (Heptan/Ethylacetat
10/1 → 2/1)
werden 2,23 g [3-(Methoxymethoxymethyl)-cyclohexylmethoxy]-essigsäure-tert-butylester
als farbloses Öl
erhalten. C16H30O5 (302,41), MS(ESI): 320,30 (M + NH4 +).
-
[3-(tert-Butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-essigsäure-tert-butylester:
-
1,9
g [3-(Methoxymethoxymethyl)-cyclohexylmethoxy]-essigsäure-tert-butylester
werden in 10 ml Tetrahydrofuran gelöst, mit 5 ml konz. HCl versetzt
und 2 h bei Raumtemperatur gerührt.
Anschließend
werden 10 ml ges. NaCl-Lösung,
10 ml Wasser und 30 ml Methyl-tert-butylether zugegeben, die Phasen
getrennt, und die wäßrige Phase
wird mit Methyl-tert-butylether extrahiert. Die vereinigten organischen
Phasen werden über MgSO4
getrocknet und eingeengt. Nach Flash-Chromatographie an Kieselgel (Heptan/Ethylacetat
3/1) werden 600 mg (3-Hydroxymethylcyclohexylmethoxy)-essigsäure-tert-butylester
als farbloses Öl
erhalten (DC(Heptan/Ethylacetat 2/1 ): Rf,E dukt = 0.68, Rf,Prod ukt = 0.18). 260 mg davon werden in 5 ml
Dimethylformamid gelöst
und mit 170 mg tert-Butyldimethylsilylchlorid
versetzt. Dann wird die Lösung
auf 0 °C
gekühlt,
und 160 mg Imidazol werden zugegeben. Die Lösung wird bei Raumtemperatur
15 h gerührt,
dann mit 20 ml ges. NaCl-Lösung,
10 ml Wasser und 30 ml Methyl-tert-butylether versetzt. Die Phasen werden
getrennt, die organische Phase wird mit ges. NaCl-Lösung gewaschen, über MgSO4
getrocknet und eingeengt. Man erhält 350 mg [3-(tert-Butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-essigsäure- tert-butylester als
farbloses Öl. C20H40O4Si
(372,36); LCMS (ESI): 390,3 (M + NH4 +).
-
2-[3-(tert-Butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylester:
-
250
mg [3-(tert-butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-essigsäure-tert-butylester werden in
10 ml abs. Tetrahydrofuran gelöst
und auf –78°C (Trockeneis/Aceton-Bad)
gekühlt.
Anschließend
werden 1,70 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C 20 min
gerührt,
dann auf 0°C
erwärmt
(Eisbad) und mit 950 mg Methyliodid versetzt. Die Lösung wird
1 h bei 0°C
gerührt.
1 ml ges. NH4Cl-Lösung
und 10 ml Wasser werden zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt. Das Rohprodukt wird in 10 ml abs.
Tetrahydrofuran gelöst
und auf –78°C (Trockeneis/Aceton-Bad)
gekühlt:
Anschließend
werden 1,70 ml 2M Lithiumdiisopropylamid-Lösung in Tetrahydrofuran/Hexanfraktion
zugetropft. Die Lösung
wird zunächst
bei –78°C 20 min
gerührt,
dann auf 0°C erwärmt (Eisbad)
und mit 950 mg Methyliodid versetzt. Die Lösung wird 1 h bei 0°C gerührt. 1 ml
ges. NH4Cl-Lösung
und 10 ml Wasser werden zugesetzt und die Phasen werden getrennt.
Die wäßrige Phase
wird mit Ethylacetat extrahiert. Die vereinigten organischen Phasen
werden über
MgSO4 getrocknet und eingeengt. 220 mg 2-[3-(tert-butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylester werden
als hellgelbes Öl
erhalten. DC(Heptan/Ethylacetat 4/1 ): Rf,Edukt =
0,66, Rf,Produkt = 0,80.
-
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure:
-
50
mg 2-[3-(tert-butyldimethylsilanyloxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure-tert-butylester
werden zu einer Mischung von 20 mg BiBr3 und 30 mg HSiEt3 in 0,5 ml Acetonitril gegeben. 38 mg 5-Methyl-2-p-tolyl-oxazol-4-carbaldehyd in 0,2
ml Acetonitril werden zugetropft, und die Mischung wird über Nacht bei
Raumtemperatur gerührt.
Der entstandene schwarze Feststoff wird abfiltriert, das Filtrat
eingeengt und mit 1 ml Trifluoressigsäure aufgenommen. Die Lösung wird über Nacht
bei Raumtemperatur gerührt.
Das Lösungsmittel
wird im Vakuum abdestilliert und der Rückstand per HPLC gereinigt.
3,4 mg 2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxymethyl)-cyclohexylmethoxy]-2-methylpropionsäure werden
als farbloses Öl
erhalten. C24H33NO5 (415,24); MS (ES-): 414,25 (M – H+).
-
Beispiel 80:
-
Analog
zu Beispiel 79 erhält
man aus [3-(Methoxymethoxymethyl)-cyclohexylmethoxy)-essigsäure-tert-butylester,
Methyliodid, Ethyliodid und 5-Methyl-2-p-tolyl-oxazol-4-carbaldehyd
2-[3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxymethyl)-cyclohexylmethoxy]-2-methylbuttersäure. C25H35NO5
(429,25); MS (ES-): 428,22 (M – H
+).
Beispiel
81 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäure
-
-
87
ml einer 1 molaren Lösung
von Lithiumdiisobutylaluminiumhydrid in n-Hexan werden in 100 ml
Diethylether gelöst
und bei 0°C
mit 7 ml Isopropanol versetzt. Nach beendeter Gasentwicklung werden
12.4 g 3-Allylcylohexanon, gelöst
in 50 ml Diethylether, zugegeben. Man rührt 48 Stunden bei Raumtemperatur
nach. Das Reaktionsgemisch wird durch Zugabe 1M HCl abgelöscht, die
wäßrige Phase
mit Natriumchlorid gesättigt und
fünfmal
mit je 200 ml Ethylacetat extrahiert. Die vereinigten organischen
Phasen werden mit 2N NaOH gewaschen, über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Der Rückstand wird
an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 15 : 1 ⇒ 5 : 1
gereinigt. Man erhält
6.8 g cis-3-Allyl-cyclohexanol als Öl. C9H16O (140.23), MS(ESI):
141 (M + H+), Rf(n-Heptan
: Ethylacetat = 2 : 1) = 0.22.
-
4-(cis-3-Allyl-cyclohexyloxymethyl)-2-(3-methoxy-phenyl)-5-methyl-oxazol
-
1.8
g cis-3-Allyl-cyclohexanol werden in 20 ml Dimethylformamid gelöst und mit
770 mg Natriumhydrid (60%ige Suspension in Paraffinöl) versetzt.
Nach 30 Minuten werden 4 g 4-Iodmethyl-5-methyl-2-(3-methoxy-phenyl)--oxazol
gelöst
in 20 ml Dimethylformamid zugetropft. Man rührt 1 Stunde bei Raumtemperatur nach.
Dann wird 200 ml Methy-tert-buthylether zum Reaktionsgemisch gegeben
und dreimal mit Wasser gewaschen. Die organische Phase wird über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 10
: 1 gereinigt. Man erhält
750 mg 4-(cis-3-Allyl-cyclohexyloxymethyl)-2-(3-methoxy-phenyl)-5-methyl-oxazol
als Öl.
C21H27NO3 (341.45), MS(ESI): 342 (M + H+),
Rf(n-Heptan
: Ethylacetat = 2 : 1) = 0.26.
-
{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde
-
750
mg 4-(cis-3-Allyl-cyclohexyloxymethyl)-2-(3-methoxy-phenyl)-5-methyl-oxazol
werden in 20 ml Diethylether gelöst
und mit 1.4 g Natriumperiodat, gelöst in 20 ml Wasser versetzt.
Man gibt bei 0°C
1 ml einer Osmiumtetroxid-Lösung (2.5
Gewichts% in tert-Butanol) hinzu und rührt kräftig bei Raumtemperatur nach. Nach
8 Stunden wird 100 ml Methyl-tert-butyl-ether zugegeben und mit
einer gesättigten
Natriumthiosulfat-Lösung
gewaschen. Die organische Phase wird über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Man erhält
740 mg {cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde
als gelbbraunes Öl.
C20H25NO4 (343.43), MS(ESI): 344 (M + H+),
Rf(n-Heptan : Ethylacetat = 2 : 1) = 0.10.
-
4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester
-
280
mg {cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde werden in
10 ml Dichlormethan gelöst
und mit 370 mg (Triphenylphosphoranylidene)-essigsäureethylester
versetzt. Man rührt
3 Stunden bei Raumtemperatur nach. Das Gemisch wird mit gesättigter
Natriumchlorid-Lösung gewaschen, über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand wird
an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 5 : 1 gereinigt.
Man erhält
190 mg 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester
als Öl. C24H31NO5
(413.52), MS(ESI): 414 (M + H+), Rf(n-Heptan : Ethylacetat = 2 : 1) = 0.30.
-
4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäureethylester
-
190
mg 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester
werden in 25 ml Methanol gelöst
und mit 20 mg Pd (10% auf Aktivkohle) versetzt. Es wird unter einer
Wasserstoffatmosphäre
7 Stunden bei Raumtemperatur gerührt.
Der Katalysator wird über
Celite abfiltriert und das Filtrat im Vakuum eingeengt. Man erhält 110 mg
4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäureethylester
als Öl.
C24H33NO5 (415.53), MS(ESI): 416 (M + H+).
-
4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäure
-
110
mg 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäureethylester
werden in 5 ml einer Mischung aus Tetrahydrofuran und Wasser im
Verhältnis
2 : 1 gelöst
und mit 20 mg Lithiumhydroxid versetzt. Man rührt 12 Stunden bei Raumtemperatur
nach. Durch Zugabe von 1N HCl wird das Gemisch angesäuert und
mit Ethylacetat extrahiert. Die organische Phase wird über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird durch RP-HPLC gereinigt. Nach Gefriertrocknung erhält man 24
mg 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäure als
Lyophilisat. C22H29NO5 (387.48), MS(ESI): 388 (M + H+).
-
Beispiel 82
-
Analog
zu Beispiel 81 wurde aus {cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde
und 2-(Triphenylphosphoranylidene)-propionsäureethylester 4-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-2-methyl-buttersäure erhalten.
C23H31NO5 (401.51), MS(ESI):
402 (M + H
+).
-
Beispiel 83
-
2-Ethyl-4-{cis-3-[2-(3-methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäure
-
2-Ethyl-4-{cis-3-[2-(3-methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester
-
0.4
ml Triethylphosphonobutyrat werden in 5 ml Tetrahydrofuran gelöst und bei –20°C mit 0.5
ml einer 2.5 M n-Butyllithium-lösung
in n-Hexan versetzt. Man rührt
1 Stunde bei –20°C nach, dann
werden 386 mg {cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde,
gelöst
in 4 ml Tetrahydrofuran, zugegeben. Nach 30 Minuten wird das Reaktionsgemisch
langsam auf Raumtemperatur erwärmt,
es werden 0.5 ml Wasser zugegeben, der Rückstand mit Ethylacetat verdünnt, über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
750 mg 2-Ethyl-4-{cis-3-[2-(3-methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester als Öl. C26H35NO5
(441.57), MS(ESI): 442 (M + H+).
-
Analog
zu Beispiel 81 wurde aus 2-Ethyl-4-{cis-3-[2-(3-methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-but-2-ensäure-ethylester
2-Ethyl-4-{cis-3-[2-(3-methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-buttersäure erhalten.
C24H33NO5 (415.53), MS(ESI):
416 (M + H
+).
-
Beispiel 84
-
Analog
zu Beispiel 83 wurde aus {cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-acetaldehyde
und Triethylphosphonopentanoat 2-(2-{cis-3-[2-(3-Methoxy-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-ethyl)-pentansäure erhalten.
C25H35NO5 (429.56), MS(ESI):
430 (M + H
+).
-
Beispiel
85 2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure
-
(cis-3-Allyl-cyclohexyloxy)-tert-butyl-diphenyl-silane
-
6.8
g cis-3-Allyl-cyclohexanol werden mit 15 ml ter-Butyldiphenylsilylchlorid,
5g Imidazol und 200 mg Dimethylaminopyridin in 100 ml Dimethylformamid
gelöst
und 12 h bei Raumtemperatur gerührt.
Es werden 400 ml Methyltert-buthylether zum Reaktionsgemisch gegeben
und dreimal mit Wasser gewaschen. Die organische Phase wird über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
20.5 g (cis-3-Allyl-cyclohexyloxy)-tert-butyl-diphenyl-silane als Öl. C25H34Osi
(378.64), MS(ESI): 379 (M + H+), Rf(n-Heptan
: Ethylacetat = 2 : 1) = 0.93.
-
[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-acetaldehyde
-
5.5
g (cis-3-Allyl-cyclohexyloxy)-tert-butyl-diphenyl-silane werden
in 100 ml Diethylether gelöst
und mit 9.4 g Natriumperiodat, gelöst in 100 ml Wasser, versetzt.
Man gibt bei 0°C
15 ml einer Osmiumtetroxid-Lösung (2.5
Gewichts% in tert-Butanol) hinzu und rührt kräftig bei Raumtemperatur nach.
Nach 5 Stunden werden weiter 5g Natriumperiodat zugegeben und nochmals
3 Stunden bei Raumtemperatur gerührt.
Danach wird das Reaktionsgemisch durch Zugabe von 300 ml Methyl-tert-buthylether
verdünnt
und mit gesättigter
Natriumthiosulfat-Lösung gewaschen.
Die organische Phase wird über
MgSO4 getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Man erhält
6 g [cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-acetaldehyde
als gelbbraunes Öl. C24H32O2Si
(380.61), MS(ESI): 381 (M + H+), Rf(n-Heptan : Ethylacetat = 5 : 1) = 0.44.
-
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-but-2-ensäure-tert-butylester
-
3.4
g [cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-acetaldehyde
werden in 100 ml Dichlormethan gelöst und mit 5g (Triphenylphosphoranylidene)-essigsäuretertbutylester
versetzt. Es wird 1 Stunde unter Rückfluss zum Sieden erhitzt.
Das Gemisch wird mit gesättigter
Natriumchlorid-Lösung
gewaschen, über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 20
: 1 gereinigt. Man erhält
2.4 g 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-but-2-ensäure-tert-butylester
als Öl.
C30H42O3Si (478.75), MS(ESI): 479 (M + H+), Rf(n-Heptan : Ethylacetat = 5 : 1) = 0.56.
-
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-butansäure-tert-butylester
-
2.4
g 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-but-2-ensäure-tert-butylester werden
in 35 ml Methanol gelöst
und mit 200 mg Pd (10% auf Aktivkohle) versetzt. Es wird unter einer
Wasserstoffatmosphäre
7 Stunden bei Raumtemperatur gerührt.
Der Katalysator wird über
Celite abfiltriert und das Filtrat im Vakuum eingeengt. Man erhält 2.3 g
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-butansäure-tert-butylester als Öl. C30H44O3Si
(480.75), MS(ESI): 481 (M + H+).
-
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2,2-dimethyl-buttersäure-tert-butylester
-
2
g 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-butansäure-tert-butylester
werden in 20 ml Tetrahydrofuran gelöst und bei –78°C mit 3.1 ml einer 2M Lösung von
Lithiumdiisopropylamid in Tetrahydrofuran versetzt. Man rührt 2 Stunden
bei –78°C nach, dann
wird das Reaktionsgemisch auf –30°C erwärmt und
mit 1.6 ml Methyliodid versetzt. Innerhalb von 12 Stunden lässt man
auf Raumtemperatur erwärmen.
Danach wird das Reaktionsgemisch durch Zugabe von 150 ml Methyltert-buthylether
verdünnt
und mit gesättigter
NaCl-Lösung
gewaschen. Die organische Phase wird über MgSO4 getrocknet und anschließend das
Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 10
: 1 gereinigt. Man erhält
2.1 g des Monomethylierten Produktes. Dieses Produkt wird in 20
ml Tetrahydrofuran gelöst
und bei –78°C mit 6 ml
einer 2M Lösung
von Lithiumdiisopropylamid in Tetrahydrofuran versetzt. Man rührt 2 Stunden
bei –78°C nach, dann
wird das Reaktionsgemisch auf 0°C
erwärmt
und nach 10 Minuten bei 0°C
mit 2.5 ml Methyliodid versetzt. Innerhalb von 12 Stunden lässt man
auf Raumtemperatur erwärmen.
Danach wird das Reaktionsgemisch durch Zugabe von 150 ml Methyl-tert-buthylether
verdünnt
und mit gesättigter
NaCl-Lösung gewaschen.
Die organische Phase wird über
MgSO4 getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 10
: 1 gereinigt. Man erhält
1.8 g 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2,2-dimethyl-buttersäure-tert-butylester als Öl. C32H48O3Si
(508.82), Rf(n-Heptan : Ethylacetat = 5
: 1) = 0.49.
-
4-(cis-3-Hydroxy-cyclohexyl)-2,2-dimethyl-buttersäure-tert-butylester
-
2
g 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2,2-dimethyl-buttersäure-tert-butylester werden
in 10 ml Tetrahydrofuran gelöst
und mit 8 ml einer 1 M Lösung
von Tetrabutylammoniumfluorid in Tetrahydrofuran versetzt. Man rührt bei
2 Stunden bei 60°C
nach. Das Reaktionsgemisch wird im Vakuum eingeengt und an Kieselgel
mit dem Laufmittel n-Heptan : Ethylacetat = 20 : 1 ⇒ 1 : 1
gereinigt. Man erhält
730 mg 4-(cis-3-Hydroxy-cyclohexyl)-2,2-dimethyl-buttersäure-tert-butylester
als Öl.
C16H30O3 (270.42), Rf(n-Heptan : Ethylacetat
= 5 : 1) = 0.22.
-
2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure-tert-butylester
-
365
mg 4-(cis-3-Hydroxy-cyclohexyl)-2,2-dimethyl-buttersäure-tert-butylester
werden zusammen mit 850 mg 4-Iodmethyl-5-methyl-2-(4 methyl-phenyl)-oxazol
in 5 ml Dimethylformamid gelöst
und mit 110 mg Natriumhydrid (60%ig in Paraffin) versetzt. Nach
1 Stunde Rühren
bei Raumtemperatur wird 100 ml Methyl-tert-butylether zugegeben und das Reaktionsgemisch
dreimal mit Wasser gewaschen. Die organische Phase wird über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand wird
mittels RP-HPLC gereinigt. Man erhält 330 mg 2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure-tert-butylester
als weißen
Feststoff. C28H41NO4 (455.64), MS(ESI): 456 (M + H+).
-
2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure
-
300
mg 2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure-tert-butylester
werden in 20 ml Dichlormethan gelöst und mit 10 ml Trifluoressigsäure versetzt.
Man rührt
1 Stunde bei Raumtemperatur nach. Es werden 200 ml Toluol zugegeben
und dann die Lösungsmittel
im Vakuum eingeengt. Der Rückstand
wird mittels RP-HPLC gereinigt. Man erhält 180 mg 2,2-Dimethyl-4-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure als Öl. C24H33NO4
(399.53), MS(ESI): 400 (M + H+).
-
Beispiel 86
-
Analog
zu Beispiel 85 wurden aus 4-(cis-3-Hydroxy-cyclohexyl)-2,2-dimethyl-buttersäure-tert-butylester
und 4-Iodmethyl-5-methyl-2-(3-trifluormethyl-phenyl)-oxazol 2,2-Dimethyl-4-{3-[5-methyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexyl}-butyric
acid erhalten.
C24H30F3NO4 (453.50), MS(ESI):
454 (M + H
+).
-
Beispiel 87
-
3-Methyl-2-{2-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-butyric acid
-
2-(Diethoxy-phosphoryl)-3-methyl-buttersäure-tert-butylester
-
5.5
ml tert-Butyl-diethylphosphonoacetat werden in 20 ml Dimethylformamid
gelöst
und bei 0°C
portionsweise mit 820 mg Natriumhydrid (60%ig in Paraffinöl) versetzt.
Die Suspension wird 15 Minuten bei 0°C gerührt und dann mit 2.4 ml Isopropyliodid
versetzt. Es wird 12 Stunden bei Raumtemperatur gerührt. Dann werden
250 ml Ethylacetat zugegeben und das Reaktionsgemisch dreimal mit
je 150 ml Wasser gewaschen. Die organische Phase wird über MgSO4
getrocknet und im Vakuum eingeengt. Der Rückstand wird an Kieselgel mit
dem Laufmittel n-Heptan
: Ethylacetat = 5 : 1 gereinigt. Man erhält 4.2 g 2-(Diethoxy-phosphoryl)-3-methyl-buttersäure-tert-butylester
als Öl.
C13H27O5P (294.33), MS(ESI): 239 (M-C4H8 +
H+), Rf(n-Heptan
: Ethylacetat = 1 : 1) = 0.34.
-
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-butyr-2-ensäure-tert-butylester
-
770
mg 2-(Diethoxy-phosphoryl)-3-methyl-buttersäure-tert-butylester werden
in 10 ml Tetrahydrofuran gelöst
und bei –20°C mit 0.73
ml einer 2.7 M Lösung
von n-Butyllithium
in n-Hexan versetzt. Nach 1 Stunde Rühren bei –20°C werden 500 mg [cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-acetaldehyde,
gelöst
in 5 ml Tetrahydrofuran, zugetropft. Das Reaktionsgemisch wird langsam
auf Raumtemperatur erwärmt.
Dann wird 20 ml Wasser zugegeben und dreimal mit je 50 ml Ethylacetat
extrahiert. Die vereinigten organischen Phasen werden über MgSO4
getrocknet und anschließend
das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 30
: 1 gereinigt. Man erhält
340 mg 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-butyr-2-ensäure-tert-butylester
als Öl.
C33H48O3Si (520.83), Rf(n-Heptan : Ethylacetat
= 5 : 1) = 0.70.
-
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-buttersäure
-
1.5
g 4-[cis-3-(tert-Butyl-diphenyl-sifanyloxy)-cyclohexyl]-2-isopropyl-butyr-2-ensäure-tert-butylester werden
in 30 ml Ethylacetat gelöst
und mit 200 mg Perlman's
Catalyst versetzt. Es wird 5 Stunden unter einer Wasserstoffatmosphäre (5 bar)
gerührt.
Der Katalysator wird über
Celite abfiltriert und das Filtrat im Vakuum eingeengt. Der Rückstand
wird in 15 ml Tetrahydrofuran gelöst und mit 3 ml einer 1M Lösung von
Tetrabutylammoniumfluorid in Tetrahydrofuran versetzt. Es wird 2
Stunden bei 60°C
gerührt.
Das Reaktionsgemisch wird im Vakuum eingeengt und an Kieselgel mit
dem Laufmittel n-Heptan : Ethylacetat = 40 : 1 ⇒ 10 : 1 gereinigt. Man erhält 400 mg
4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-buttersäure als Öl. C17H32O3
(284.44), MS(ESI): 211 (M-C4H9O–),
Rf(n-Heptan : Ethylacetat = 10 : 1) = 0.15.
-
Analog
zu Beispiel 90 wurde aus 4-Iodmethyl-5-methyl-2-(4 methyl-phenyl)-oxazol
und 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-buttersäure 3-Methyl-2-{2-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-butyric acid erhalten.
C25H35NO4 (413.56), MS(ESI):
414 (M + H
+).
-
Beispiel 88
-
Analog
zu Beispiel 87 wurde aus 4-Iodmethyl-5-methyl-2-(3-methoxy-phenyl)-oxazol und 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-2-isopropyl-buttersäure 2-(2-{cis-3-[2-(3-Methoxy-phenyl)5-methyl-oxazol-4-ylmethoxy]-cyclohexyl}-ethyl)-3-methyl-buttersäure erhalten.
C25H35NO5 (429.56), MS(ESI):
430 (M + H
+).
-
Beispiel 89
-
2-Benzyl-4-[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure
-
Analog
zu Beispiel 90 wurde aus tert-Butyl-diethylphosphonoacetat und Benzylbromid
2-(Diethoxy-phosphoryl)-3-phenyl-propionsäure-tert-butylester erhalten.
C17H27O5P (342.38), R
f(n-Heptan : Ethylacetat = 1 : 1) = 0.53.
-
Analog
zu Beispiel 88 wurde aus 2-(Diethoxy-phosphoryl)-3-phenyl-propionsäure-tert-butylester, [cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-acetaldehyde
und 4-Iodmethyl-5-methyl-2-(4 methyl-phenyl)-oxazol 2-Benzyl-4-[3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-buttersäure erhalten.
C29H35NO4 (461.61), MS(ESI):
462 (M + H
+).
-
Beispiel 90
-
4-Methyl-2-{2-[cis.3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-pentansäure
-
Analog
zu Beispiel 88 wurde aus 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-butansäure-tert-butylester,
3-Bromo-2-methyl-propene und 4-Iodmethyl-5-methyl-2-(4
methyl-phenyl)-oxazol 4-Methyl-2-{2-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-pent-4-ensäure-tert-butylester
erhalten.
C30H43NO4 (481.68), MS(ESI):
482 (M + H
+).
-
4-Methyl-2-{2-[cis.3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-pentansäure
-
500
mg 4-Methyl-2-{2-[cis-3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]- ethyl}-pent-4-ensäure-tert-butylester
werden in 20 ml Ethylacetat gelöst
und mit 50 mg Palladium (10% auf Aktivkohle) versetzt. Es wird 5
Stunden unter einer Wasserstoffatmosphäre (5 bar) gerührt. Der
Katalysator wird über
Celite abfiltriert und das Filtrat im Vakuum eingeengt. Der Rückstand
wird in 20 ml Dichlormethan gelöst
und mit 10 ml Trifluoressigsäure
versetzt. Man rührt
1 Stunde bei Raumtemperatur nach. Es werden 100 ml Toluol zugegeben
und dann die Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird mittels RP-HPLC gereinigt. Man erhält 100 mg 4-Methyl-2-{2-[cis.3-(5-methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-pentansäure als Öl. C26H37NO4
(427.59), MS(ESI): 428 (M + H+).
-
Beispiel 91
-
2-(2-{cis-3-[5-Methyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexyl}-ethyl)-2-propyl-pentansäure
-
Analog
zu Beispiel 88 wurde aus 4-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-butansäure-tert-butylester
und Allylbromid 2-Allyl-2-{2-[3-(tert-butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-pent-4-ensäure-tert-butylester
erhalten.
C36H52O3Si (560.90), R
f(n-Heptan : Ethylacetat = 20 : 1) = 0.60.
-
Analog
zu Beispiel 90 und Beispiel X wurde aus 2-Allyl-2-{2-[3-(tert-butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-pent-4-ensäure-tert-butylester
und 4-Iodmethyl-5-methyl-2-(3-trifluormethyl-phenyl)-oxazol 2-(2-{cis-3-[5-Methyl-2-(3-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-cyclohexyl}-ethyl)-2-propyl-pentansäure erhalten.
C28H38F3NO4 (509.61), MS(ESI):
510 (M + H
+).
-
Beispiel 92
-
1-{2-[cis-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-cyclopentancarbonsäure
-
1-{2-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-cyclopent-3-enecarbonsäure-tert-butylester
-
2
g 2-Allyl-2-{2-[3-(tert-butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-pent-4-ensäure-tert-butylester werden
in 100 ml Dichlormethan gelöst.
Man leitet 5 Minuten Argon durch die Lösung hindurch. Dann wird 100 mg
Grubbs Katalysator hinzugesetzt. Es wird 2 Stunden bei 40°C gerührt. Danach
wiord das Lösungsmittel
im Vakuum entfernt und der Rückstand
an Kieselgel mit dem Laufmittel n-Heptan : Ethylacetat = 40 : 1
gereinigt. Man erhält
1.4 g 1-{2-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-cyclopent-3-enecarbonsäure-tert-butylester als Öl. C34H48O3Si
(532.85), Rf(n-Heptan : Ethylacetat = 20
: 1) = 0.56.
-
Analog
zu Beispiel 88 wurde aus 1-{2-[cis-3-(tert-Butyl-diphenyl-silanyloxy)-cyclohexyl]-ethyl}-cyclopent-3-enecarbonsäure-tert-butylester
und 4-Iodmethyl-5-methyl-2-(4-methyl-phenyl)-oxazol 1-{2-[cis-3-(5-Methyl-2-p-tolyl-oxazol-4-ylmethoxy)-cyclohexyl]-ethyl}-cyclopentancarbonsäure erhalten.
C26H35NO4 (425.57), MS(ESI):
426 (M + H
+). Beispiel
93 2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-butansäure
-
rac-3-(cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
-
21.7
g 1,3-Cyclohexandiol werden mit 30.3 g Dibutylzinnoxid in 450 ml
Toluol gelöst
und unter Rückfluss
am Wasserabscheider zum Sieden erhitzt. Das Reaktionsvolumen wird
während
der Reaktionsdauer auf die Hälfte
reduziert. Nach 3 Stunden wird das Reaktionsgemisch auf Raumtemperatur
gekühlt
und mit 300 ml Dimethylformamid, 29 g 2-(4-Fluoro-phenyl)-4-iodmethyl-5-methyl-oxazol
und 23.5 g Cäsiumfluorid
versetzt. Man rührt
18 Stunden bei Raumtemperatur nach. Das Reaktionsgemisch wird durch
Zugabe von Ethylacetat verdünnt
und mit gesättigter
Natriumchlorid -Lösung
gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet,
das Lösungsmittel
im Vakuum entfernt und der Rückstand
durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat =
10 : 1 → 1
: 4) gereinigt. Man erhält
58 g rac-3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexanol als gelblichen
Feststoff, der aus n-Heptan/Ethylacetat umkristallisiert wird. C17H20FNO3
(305.35), MS (ESI): 306 (M + H+).
-
3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexanol
-
25
g rac-3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexanol
werden in 320 ml Vinylacetat gelöst
und mit 1,3 g Chirazyme L-2 Lyo (Boehringer Mannheim) versetzt.
Nach ca. dreistündigem
Rühren bei
Raumtemperatur (LC-MS Kontrolle auf 40-45% Umsatz) wird das Enzym
abfiltriert, mit Ethylacetat nachgewaschen und das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat
= 3 : 1) gereinigt. Man erhält
8 g des Acetats als farbloses Öl.
C19H22FNO4 (347.39), MS (ESI): 348 (M + H+).
Man nimmt das Acetat in 170 ml Methanol auf und rührt nach
Zugabe von 27 ml 2N NaOH für
eine Stunde bei Raumtemperatur. Der größte Teil des Lösungsmittels
wird im Vakuum entfernt. Nach Zugabe von je 150 ml Wasser und Ethylacetat,
wird die organische Phase mit Natriumchlorid -Lösung gewaschen. Die organische
Phase wird über
Magnesiumsulfat getrocknet, das Lösungsmittel im Vakuum entfernt.
Man erhält
6,7 g 3-((1R,3S)-cis-3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexanol als
gelblichen Feststoff. C17H20FNO3 (305.35), MS (ESI): 306 (M + H+).
-
4-(3-Allyloxy-cyclohexyloxymethyl)-2-(4-fluoro-phenyl)-5-methyl-oxazol
-
2
g des 3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexanols
werden in 15 ml Dimethylformamid gelöst und mit 0.3 g Natriumhydrid
versetzt. Nach 30 Minuten werden 2.4 g Allylbromid zugetropft. Man
rührt 5
Stunden bei Raumtemperatur nach. Dann wird 15 ml 1N HCl zum Reaktionsgemisch
gegeben und dreimal mit 15 ml Ethylacetat gewaschen. Die organische
Phase wird über
Magnesiumsulfat getrocknet und anschließend das Lösungsmittel im Vakuum entfernt.
Der Rückstand
wird mittels RP-HPLC gereinigt. Man erhält 2.4 g 4-(3-Allyloxy-cyclohexyloxymethyl)-2-(4-fluoro-phenyl)-5-methyl-oxazol
als gelbliches Öl. C20H24FNO3
(345,42) MS(ESI): 346 (M + H+)
-
[3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl]-acetaldehyd
-
2.0
g 4-(3-Allyloxy-cyclohexyloxymethyl)-2-(4-fluoro-phenyl)-5-methyl-oxazol
werden in 50 ml Diethylether gelöst
und mit 3.8 g Natriumperiodat, gelöst in 50 ml Wasser versetzt.
Man gibt bei 0°C
1 ml einer Osmiumtetroxid-Lösung
(2.5 Gewichts% in tert-Butanol) hinzu und rührt kräftig bei Raumtemperatur nach.
Nach 8 h wird 100 ml Methyl-tert-butylether zugegeben und mit einer
gesättigten
Natriumthiosulfat-Lösung
gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet
und das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird an Kieselgel (n-Heptan : Ethylacetat = 1 : 1 → 1 : 5)
gereinigt. Man erhält
1.4 g des [3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyl]-acetaldehyds als
gelbbraunes Öl. C20H25NO4
(343.42), MS(ESI): 344 (M + H+), Rf(n-Heptan : Ethylacetat = 1 : 1) = 0.25.
-
2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäureethylester
-
0,58
g des 2-(Diethoxy-phosphoryl)-butansäureethylesters werden in Tetrahydrofuran
(20 ml) gelöst und
bei 0°C
mit 0,06 g Natriumhydrid versetzt. Die Suspension wird 30 min. bei
0°C und
30 min bei Raumtemperatur gerührt
und anschließend
auf –70°C gekühlt. Nach
Zugabe von 0,4 g 2-((1R,3S)-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-acetaldehyd
(gelöst
in 5 ml Tetrahydrofuran) wird 60 min bei –70°C und anschließend 12
h bei Raumtemperatur gerührt.
Es wird mit 10 ml Wasserversetzt, mit Ethylacetat (3 × 10 ml) extrahiert
und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung (10
ml) gewaschen. Das Lösungsmittel
wird im Vakuum entfernt und der Rückstand durch HPLC gereinigt.
Man erhält
0,32 g des 2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäureethylesters
C25H32FNO5 (445,54) MS(ESI): 446 (M + H+)
-
2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäure
-
0,5
g des 2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäureethylesters
werden in 5 ml Methanol gelöst
und mit 2.5 ml 1N Natronlauge versetzt. Nach 12 h Rühren bei
Raumtemperatur wird mit 3 ml 1N Salzsäure angesäuert und der entstandene Niederschlag
in Ethylacetat aufgenommen. Das Lösungsmittel wird im Vakuum
entfernt und der Rückstand
der Esterverseifung 2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäure als 0,45
g weißer
Feststoff erhalten. C23H28FNO5 (417,48) MS(ESI): 418 (M + H+)
-
2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-butansäure
-
0,3
g der 2-Ethyl-4-{(1R,3S)-3-[2-(4-fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-but-2-ensäure wird
in einem Lösungsmittelgemisch
aus 2 ml Ethylacetat und 1 ml Methanol gelöst und mit 0,05 g Palladium
(10% auf Kohle) versetzt. Anschließend wird 3 h bei 1 bar Wasserstoffdruck
hydriert.
-
Nach
Filtration vom Palladium wird das Lösungsmittelgemisch im Vakuum
entfernt und der Rückstand aus
Acetonitril umkristallisiert. Man erhält 0,25 g der 2-Ethyl-4-{(1R,3S)-3-[2-(4-Fuoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-butansäure als
weißen
Feststoff. C23H30FNO5 (419.49), MS(ESI): 420 (M + H+)
-
Beispiel 94:
-
Analog
zu Beispiel 93 erhält
man aus 2-((1R,3S)-[3-(2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-acetaldehyd
und 2-(Diethoxy-phosphoryl)-pentansäureethylester
2-Propyl-4-[3-(2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-butansäure.
C
24H
32FNO
5 (433,52) MS(ESI):
434 (M + H
+)
-
Beispiel 95:
-
Analog
zu Beispiel 93 erhält
man aus 2-((1R,3S)-[3-(2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-acetaldehyd
und 2-(Diethoxyphosphoryl)-essigsäureethylester
4-{3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}-butansäure
C
21H
26FNO
5 (391,44) MS(ESI):
392 (M + H
+)
-
Beispiel 96:
-
Analog
zu Beispiel 93 erhält
man aus 2-((1R,3S)-[3-(2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-acetaldehyd
und 2-(Diethoxyphosphoryl)-propionsäureethylester
4-{3-[2-(4-Fluoro-phenyl)-5-methyl-oxazol-4-ylmethoxy]-cyclohexyloxy}=2-methyl-butansäure
C22H28FNO5 (405,47) MS(ESI):
406 (M + H
+).
-
Beispiel
97: rac-3-(cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
-
21.7
g 1,3-Cyclohexandiol werden mit 30.3 g Dibutylzinnoxid in 450 ml
Toluol gelöst
und unter Rückfluss
am Wasserabscheider zum Sieden erhitzt. Das Reaktionsvolumen wird
während
der Reaktionsdauer auf die Hälfte
reduziert. Nach 3 Std. wird das Reaktionsgemisch auf Raumtemp. gekühlt und
mit 300 ml Dimethylformamid, 29 g 4-iodmethyl-5-methyl-2-m-tolyl-oxazol
1 und 23.5 g Cäsiumfluorid
versetzt. Man rührt
18 Std. bei Raumtemp. nach. Das Reaktionsgemisch wird durch Zugabe
von Ethylacetat verdünnt
und mit gesättigter NaCl-Lösung gewaschen.
Die organische Phase wird über
Magnesiumsulfat getrocknet, das Lösungsmittel im Vakuum entfernt
und der Rückstand
durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat =
10 : 1 → 1
: 4) gereinigt. Man erhält
58 g rac-3-(cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)- cyclohexanol als
gelblichen Feststoff, der aus n-Heptan/Ethylacetat umkristallisiert
wird. C18H23NO3 (301.39), MS (ESI): 302 (M + H+).
-
3-((1R,3S)-cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
4
-
25
g rac-3-(cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
werden in 320 ml Vinylacetat gelöst
und mit 1,3 g Chirazyme L-2 Lyo (Boehringer Mannheim) versetzt.
Nach ca dreistündigem
Rühren
bei Raumtemp. (LC-MS Kontrolle auf 40–45% Umsatz) wird das Enzym
abfiltriert, mit Ethylacetat nachgewaschen und das Lösungsmittel
im Vakuum entfernt. Der Rückstand
wird durch Flash-Chromatographie
an Kieselgel (n-Heptan/Ethylacetat = 3 : 1) gereinigt. Man erhält 8 g des
Acetats 3 als farbloses Öl.
C20H25NO4 (343.43), MS (ESI): 344 (M + H+),
Man nimmt das Acetat in 170 ml Methanol auf und rührt nach
Zugabe von 27 ml 2N NaOH für
eine Stunde bei Raumtemp. Der größte Teil
des Lösungsmittels
wird im Vakuum entfernt. Nach Zugabe von je 150 ml Wasser und Ethylacetat,
wird die org. Phase mit NaCl-Lösung
gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet,
das Lösungsmittel
im Vakuum entfernt. Man erhält
6,7 g 3-((1R,3S)-cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
als gelblichen Feststoff. C18H23NO3 (301.39), MS (ESI): 302 (M +
H+).
-
4-((1R,3S)-3-Allyloxy-cyclohexyloxymethyl)-5-methyl-2-m-tolyl-oxazol
5
-
Zu
einer Lösung
von 2.2 g 3-((1R,3S)-cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol in 30 ml
Dimethylformamid werden bei Raumtemp. 470 mg 60-proz. Natriumhydrid-Suspension
gegeben und 20 min bei Raumtemperatur gerührt. Anschließend werden
1,36 ml Allylbromid addiert man rührt bei 40°C bis zum vollständigen Umsatz,
eventuell werden weiteres Natriumhydrid und Allybromid zugegeben.
Bei vollständigen Umsatz
(LC-MS Kontrolle) werden 100 ml Ethylacetat und 150 ml ges NaCl-Lösung zugegeben.
Die organische Phase wird über
Magnesiumsulfat getrocknet, die Lösungsmittel im Vakuum entfernt
und der Rückstand durch
Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat = 3 : 1)
gereinigt. Man erhält
2.3 g 4-((1R,3S)-3-Allyloxy-cyclohexyloxymethyl)-5-methyl-2-m-tolyl-oxazol
5 als farbloses Öl.
C21H27NO3 (341.45), MS (ESI): 342 (M + H+).
-
2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propane-1,2-diol
-
Zu
2,8 g 4-((1R,3S)-3-Allyloxy-cyclohexyloxymethyl)-5-methyl-2-m-tolyl-oxazol
in 9 ml Aceton/Wasser 10 : 1 gab man bei 0°C 225 mg DABCO, 1,4 g wasserfreies
N-Methylmorpholin-N-oxid
und 350 μl
Osmiumtetroxid 2,5% in tert Butanol. Nach 24 h Rühren bei Raumtemp. gab man
2,4 g Natriummetabisulfit zu und verdünnte nach 10 min mit 25 ml
CH2Cl2. Man filtrierte und entfernte das Lösungsmittel im Vakuum. Der
Rückstand
wird durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat = 1 : 3) gereinigt.
Man erhält
2.5 g 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-1,2-diol
als farbloses Öl. C21H29NO5 (375.47),
MS (ESI): 376 (M + H+).
-
2S-(1S',3R')-1-(tert-Butyl-dimethyl-silanyloxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-2-ol
-
Zu
2,5 g 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propane-1,2-diol
in 30 ml Dichlormethan gibt man bei 0°C 500 mg Imidazol, 1,02 g tert.Butyldimethylsilylchlorid
und 50 mg Tetrabutylammoniumiodid. Man läßt über 18 h auf Raumtemp. kommen
und gießt
die Mischung auf Eis. Man extrahiert mit Dichlormethan, trocknet über Natriumsulfat,
filtriert und entfernt das Lösungsmittel
im Vakuum. Der Rückstand
wird durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat
= 2 : 1 → 1
: 2) gereinigt. Man erhält
2S-(1S',3R')-1-(tert-Butyl-dimethyl-silanyloxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-2-ol
als farbloses Öl.
C27H43NO5Si (489.73), MS (ESI): 490 (M + H+).
-
2S'-(1S,3R)4-{3-[3-(tert-Butyl-dimethyl-silanyloxy)-2-prop-2-ynyloxy-propoxy]-cyclohexyloxymethyl}-5-methyl-2-m-tolyl-oxazol
-
Zu
245 mg 2S-(1S',3R')-1-(tert-Butyl-dimethyl-silanyloxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-2-ol
in 3 ml Dimethylformamid gibt man 25 mg 60-proz. Natriumhydrid-Suspension und
rührt 20
min bei Raumtemperatur. Anschließend werden 200 mg Propargylbromid
addiert und man rührt bei
Raumtemp bis zum vollständigen
Umsatz. Man nimmt in ges NaCl-Lösung/Ethylacetat
auf, trocknet die organische Phase über Natriumsulfat, filtriert
und entfernt das Lösungsmittel
im Vakuum. Man erhält 2S'-(1S,3R)4-{3-[3-(tert-Butyl-dimethyl-silanyloxy)-2-prop-2-ynyloxy-propoxy]-cyclohexyloxymethyl}-5-methyl-2-m-tolyl-oxazol
als farbloses Öl.
C30H45NO5Si (527.78), MS(ESI): 528 (M + H+).
-
(1R',3S')-2R-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-prop-2-ynyloxy-propan-1-ol
-
Zu
200 mg 2S'-(1S,3R)-{3-[3-(tert-Butyl-dimethyl-silanyloxy)-2-prop-2-ynyloxy-propoxy]-cyclohexyloxymethyl}-5-methyl-2-m-tolyl-oxazol
in 2 ml Tetrahydrofuran gibt man 2 ml Tetrabutylammoniumfluorid
1M in Tetrahydrofuran und rührt
2 h bei Raumtemp. Man nimmt in ges NaCl-Lösung/Ethylacetat auf und trocknet
die organische Phase über
Natriumsulfat, filtriert, entfernt das Lösungsmittel im Vakuum. Man
erhält (1R',3S')-2R-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)- cyclohexyloxy]-2-prop-2-ynyloxy-propan-1-ol
als farbloses Öl.
C24H31NO5 (413.53), MS (ESI): 414 (M + H+).
-
2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-propoxy-propan-1-ol
-
Zum
Rohprodukt (1R',3S')-2R-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-prop-2-ynyloxy-propan-1-ol
gelöst
in 20 ml Methanol gibt man 50 mg Pd/C 10% und rührt unter 1 bar Wasserstoff
für 3 h.
Man filtriert vom Katalysator und entfernt das Lösungsmittel im Vakuum. Man
erhält 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-propoxy-propan-1-ol als farbloses Öl. Der Rückstand
wird durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat
= 1 : 1) gereinigt. C24H35NO5 (417.55), MS (ESI): 418 (M + H+).
-
2R-(1R',3S')-2-(2-propoxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure
-
90
mg 2R-(1R',3S')-2-Propoxy-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-1-ol
werden in 1,5 ml Dichlormethan mit 180 mg Dess- Martin-Periodinan versetzt und 2 h bei
Raumtemperatur gerührt.
Anschließend
werden 41 mg Na2S2O3 in 3 ml 5% NaHCO3-Lösung zugesetzt
und 10 min bei Raumtemperatur gerührt. Die organische Phase wird
abgetrennt, über
MgSO4 getrocknet und eingeengt. Der Rückstand
wird in 2 ml Acetonitril aufgenommen und mit 1,5 ml 0,65 M NaH2PO4-Lösung und
48 μl 35% H2O2-Lösung versetzt.
Bei 0°C
werden 30 mg NaClO2 in 2 ml Wasser über 1 h
zugetropft. Die Mischung wird 3 h bei dieser Temperatur gerührt. Dann
werden Na2SO3-Lösung, 10%
HCl und 10 ml CH2Cl2 zugegeben,
die Phasen getrennt, die organische Phase über MgSO4 getrocknet
und eingeengt. Reinigung des Rückstandes per
HPLC liefert 1,2 mg 2R-(1R',3S')-2-(2-methylpropoxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure. C24H33NO6
(431,53); LCMS (ESI): 432,2 (MH+).
-
Beispiel
98 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-(4-trifluoromethyl-benzyloxy)-propansäure
-
Aus
2S-(1R',3S')-1-(tert-Butyl-dimethyl-silanyloxy)-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propan-2-ol
und 4-Trifluoromethylbenzylbromid erhält man analog zu Beispiel 97 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-(4-trifluoromethyl-benzyloxy)-propan-1-ol
mit dem Molekulargewicht 533.59 (C29H34F3NO5),
MS (ESI): 534 (M + H+).
-
Analog
zu Beispiel 97 erhält
man aus 2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-(4-trifluoromethyl-benzyloxy)-propan-1-ol
2R-(1R',3S')-3-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-2-(4-trifluoromethyl-benzyloxy)-propionsäure. C29H32F3NO6
(547,58), MS (ES+): 548,40 (MH+).
-
-
Beispiel
99 s((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
-
Zu
einer Lösung
von 754 mg 3-((1R,3S)-cis-5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexanol
in 10 ml Dimethylformamid/5 ml Tetrahydrofuran werden bei Raumtemp.
200 mg 60-proz. Natriumhydrid-Suspension gegeben und 20 min bei
Raumtemperatur gerührt.
Anschließend
werden bei 0°C
1 g 2-Bromomethyl-acrylsäureethylester
addiert und rührt
2 h bei dieser Temperatur. Es werden 100 ml Ethylacetat und 150 ml
ges. NaCl-Lösung
zugegeben. Die organische Phase wird über Natriumsulfat getrocknet,
das Lösungsmittel im
Vakuum entfernt und der Rückstand
durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat =
2 : 1) gereinigt. Man erhält
1,18 g ((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
als farbloses Öl.
C24H31NO5 (413.52), MS (ESI): 414 (M + H+).
-
((1R,3S)-1-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-cyclopropancarbonsäureethylester
-
Zu
einer Suspension von 55 mg Trimethylsulfoniumiodid in 2 ml DMSO
werden bei Raumtemp. 12 mg 60-proz. Natriumhydrid-Suspension gegeben
und 20 min bei Raumtemperatur gerührt. Anschließend werden bei
10°C 100
mg ((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
gelöst
in 2 ml DMSO addiert und 90 min bei Raumtemp. gerührt. Man
gießt
auf Eiswasser und extrahiert mit Methyl-tert.-butylether. Die organische
Phase wird über
Natriumsulfat getrocknet, das Lösungsmittel im
Vakuum entfernt und der Rückstand
durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat =
2 : 1) gereinigt. Man erhält
((1R,3S)-1-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy-methyl]-cyclopropancarbonsäureethylester
als farbloses Öl.
C25H33NO5 (427.55), MS (ESI): 428 (M + H+).
-
((1R,3S)-1-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-cyclopropancarbonsäure
-
56
mg ((1R,3S)-1-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-cyclopropancarbonsäureethylester
werden in 3 ml Methanol gelöst
und mit 0,5 ml 5 N NaOH versetzt und 18 h bei Raumtemp. gerührt. Man
entfernt das Lösungsmittel
im Vakuum, säuert
mit Trifluoressigsäure
an und reinigt den Rückstand
durch RP-HPLC. Man erhält
((1R,3S)-1-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyl-oxymethyl]-cyclopropancarbonsäure als
farbloses Öl.
C23H29NO5 (399.49), MS (ESI): 400 (M + H+).
-
Beispiel
100 2RS-((1R',3S')-2-[(Methyl-phenethyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäureethylester
-
50
mg ((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
werden in 5 ml Ethanol gelöst,
mit 95 mg N-Methylhomobenzylamin versetzt und 18 h bei Raumtemp. gerührt. Man
entfernt das Lösungsmittel
im Vakuum und reinigt den Rückstand
durch Flash-Chromatographie an
Kieselgel (n-Heptan/Ethylacetat = 1 : 1 + 3% NEt3)
gereinigt. Man erhält 2RS-((1R',3S')-2-[(Methyl-phenethyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-yl-methoxy)-cyclohexyloxy]-propionsäureethylester
als farbloses Öl.
C33H44N2O5 (548.73), MS (ESI): 549 (M + H+).
-
((1R',3S')-2RS-[(Methyl-phenethyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure
-
65 mg 2RS-((1R',3S')-2-[(Methyl-phenethyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-yl-methoxy)-cyclohexyloxy]-propionsäureethylester
werden in 3 ml Tetrahydrofuran/Methanol 3 : 1 gelöst und mit
0,6 ml 1 N LiOH versetzt und 6 h bei Raumtemp gerührt. Man
entfernt das Lösungsmittel
im Vakuum säuert
mit Trifluoressigsäure an
und reinigt den Rückstand
durch RP-HPLC. Man erhält
2RS-((1R',3S')-[(Methyl-phenethyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyl-oxy]-propionsäure als
farbloses Öl.
C31H40N2O5 (520.67), MS (ESI): 521 (M + H+).
-
Beispiel
101 (1R',3S')-2RS-[(Benzyl-methyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure
-
Aus
((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
und N-Methylbenzylamin erhält
man analog zu Beispiel 100 (1R',3S')-2RS-[(Benzyl-methyl-amino)-methyl]-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure mit
dem Molekulargewicht 506.65 (C30H38N2O5),
MS(ESI): 507.20 (M + H+).
-
Beispiel
102 ((1R',3S')-2RS-Methoxymethyl-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure
-
56
mg ((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
werden in 3 ml Methanol gelöst
und mit 49 mg Natriumcyanid sowie 0,25 ml 2N NaOH versetzt und 18
h bei Raumtemp. gerührt.
Man entfernt das Lösungsmittel
im Vakuum, säuert
mit Trifluoressigsäure
an und reinigt den Rückstand
durch RP-HPLC. Man erhält
((1R',3S')-2RS-Methoxymethyl-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure als
farbloses Öl.
C23H31NO6 (417.51), MS (ESI): 418.15 (M + H+).
-
Beispiel
103 Z-((1R',3S')-3-(4-Fluoro-3-methyl-phenyl)-2-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
-
220
mg Tetrabutylammoniumchlorid und 332 mg Kaliumcarbonat werden in
4 ml Dimethylformamid suspendiert und 20 min innig gerührt. Man
gibt 400 mg des ((1R,3S)-2-[3-(5-Methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]- acrylsäureethylester,
25 mg Triphenylphosphan und 212 mg 4-Fluor-3methyliodbenzol zu, entgast, belüftet mit
Argon und addiert 10 mg Palladiumacetat und 0,2 mol Wasser. Man
erhitzt 4 h auf 60°C.
Nach dem Erkalten werden 20 ml Ethylacetat und 50 ml ges. NaCl-Lösung zugegeben.
Die organische Phase wird über
Natriumsulfat getrocknet, das Lösungsmittel
im Vakuum entfernt und der Rückstand
durch Flash-Chromatographie an Kieselgel (n-Heptan/Ethylacetat =
4 : 1) gereinigt. Man erhält Z-((1R',3S')-3-(4-Fluoro-3-methyl-phenyl)-2-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy-methyl]-acrylsäureethylester
als farbloses Öl.
C31H38FNO5 (523.65), MS (ESI): 524 (M + H+).
-
2RS-(4-Fluoro-3-methyl-benzyl)-(1R',3S')-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäureethylester
-
80 mg Z-((1R',3S')-3-(4-Fluoro-3-methyl-phenyl)-2-(3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxymethyl]-acrylsäureethylester
werden in 15 ml Ethylacetat gelöst
und nach Zugabe von 30 mg Pd/C 10% unter 1 bar H2 24
h gerührt.
Man filtriert vom Katalysator und evaporiert das Lösungsmittel.
Man erhält
2RS-(4-Fluoro-3-methyl-benzyl)-(1R',3S')-3-[3-(5-methyl-2-m-tolyl-oxazol-4- ylmethoxy)-cyclohexyl-oxy]-propionsäureethylester
als farbloses Öl.
C31H38FNO5 (521.63), MS (ESI): 522 (M + H+).
-
(2RS)-(4-Fluoro-3-methyl-benzyl)-(1R',3S')-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure
-
70
mg 2RS-(4-Fluoro-3-methyl-benzyl)-(1R',3S')-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäureethylester
werden in 3 ml Tetrahydrofuran/Methanol 3 : 1 gelöst und mit
0,1 ml 1 N LiOH versetzt und 18 h bei Raumtemp gerührt. Man
entfernt das Lösungsmittel
im Vakuum säuert
mit Trifluoressigsäure
an und reinigt den Rückstand
durch RP-HPLC. Man erhält
2RS-(4-Fluoro-3-methyl-benzyl)-(1R',3S')-3-[3-(5-methyl-2-m-tolyl-oxazol-4-ylmethoxy)-cyclohexyloxy]-propionsäure als
farbloses Öl. C29H34FNO5 (495.60),
MS (ESI): 496.20 (M + H+).