CN100515492C - 包含聚亚烷基二醇的降钙素药物-低聚物缀合物的混合物、其应用和制备方法 - Google Patents
包含聚亚烷基二醇的降钙素药物-低聚物缀合物的混合物、其应用和制备方法 Download PDFInfo
- Publication number
- CN100515492C CN100515492C CNB028153022A CN02815302A CN100515492C CN 100515492 C CN100515492 C CN 100515492C CN B028153022 A CNB028153022 A CN B028153022A CN 02815302 A CN02815302 A CN 02815302A CN 100515492 C CN100515492 C CN 100515492C
- Authority
- CN
- China
- Prior art keywords
- mixture
- calcitonin
- oligomer
- conjugate
- polyalkylene glycol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 715
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 title claims abstract description 700
- 102000055006 Calcitonin Human genes 0.000 title claims abstract description 566
- 108060001064 Calcitonin Proteins 0.000 title claims abstract description 566
- 229960004015 calcitonin Drugs 0.000 title claims abstract description 564
- 229920001515 polyalkylene glycol Polymers 0.000 title claims abstract description 101
- 238000004519 manufacturing process Methods 0.000 title 1
- 239000003814 drug Substances 0.000 claims abstract description 232
- 125000003827 glycol group Chemical group 0.000 claims abstract description 104
- 229940079593 drug Drugs 0.000 claims abstract description 67
- 210000002966 serum Anatomy 0.000 claims abstract description 33
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims abstract description 30
- 239000011575 calcium Substances 0.000 claims abstract description 30
- 229910052791 calcium Inorganic materials 0.000 claims abstract description 30
- 230000008878 coupling Effects 0.000 claims description 157
- 238000010168 coupling process Methods 0.000 claims description 157
- 238000005859 coupling reaction Methods 0.000 claims description 157
- 150000001875 compounds Chemical class 0.000 claims description 156
- 229920001223 polyethylene glycol Polymers 0.000 claims description 129
- 229960003773 calcitonin (salmon synthetic) Drugs 0.000 claims description 111
- 108010068072 salmon calcitonin Proteins 0.000 claims description 111
- 239000002202 Polyethylene glycol Substances 0.000 claims description 99
- 238000006243 chemical reaction Methods 0.000 claims description 88
- 230000015556 catabolic process Effects 0.000 claims description 86
- 238000006731 degradation reaction Methods 0.000 claims description 86
- 108090000631 Trypsin Proteins 0.000 claims description 55
- 102000004142 Trypsin Human genes 0.000 claims description 55
- 239000012588 trypsin Substances 0.000 claims description 55
- 239000006185 dispersion Substances 0.000 claims description 50
- 238000000034 method Methods 0.000 claims description 50
- 229920000642 polymer Polymers 0.000 claims description 47
- 230000004071 biological effect Effects 0.000 claims description 45
- 238000009826 distribution Methods 0.000 claims description 42
- 108090000227 Chymases Proteins 0.000 claims description 30
- 102000003858 Chymases Human genes 0.000 claims description 30
- 230000000269 nucleophilic effect Effects 0.000 claims description 24
- 230000004913 activation Effects 0.000 claims description 20
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 238000002360 preparation method Methods 0.000 claims description 16
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 12
- HOGDNTQCSIKEEV-UHFFFAOYSA-N n'-hydroxybutanediamide Chemical compound NC(=O)CCC(=O)NO HOGDNTQCSIKEEV-UHFFFAOYSA-N 0.000 claims description 12
- 230000000903 blocking effect Effects 0.000 claims description 7
- 208000020084 Bone disease Diseases 0.000 claims description 6
- 208000010392 Bone Fractures Diseases 0.000 claims description 5
- 208000001132 Osteoporosis Diseases 0.000 claims description 4
- 150000001768 cations Chemical class 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 208000006386 Bone Resorption Diseases 0.000 claims description 3
- 208000037147 Hypercalcaemia Diseases 0.000 claims description 3
- 230000024279 bone resorption Effects 0.000 claims description 3
- 230000000148 hypercalcaemia Effects 0.000 claims description 3
- 208000030915 hypercalcemia disease Diseases 0.000 claims description 3
- 125000005647 linker group Chemical group 0.000 claims description 3
- 230000004044 response Effects 0.000 claims description 3
- 125000003277 amino group Chemical group 0.000 claims 1
- 230000029087 digestion Effects 0.000 abstract description 7
- 230000000968 intestinal effect Effects 0.000 abstract description 4
- 238000010874 in vitro model Methods 0.000 abstract 1
- 230000000694 effects Effects 0.000 description 98
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 62
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 60
- 239000000243 solution Substances 0.000 description 59
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 57
- 108090000765 processed proteins & peptides Proteins 0.000 description 55
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 53
- 150000004665 fatty acids Chemical class 0.000 description 52
- -1 Polyethylene Polymers 0.000 description 50
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 50
- 239000000178 monomer Substances 0.000 description 49
- 239000000376 reactant Substances 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- 230000015572 biosynthetic process Effects 0.000 description 43
- 238000003786 synthesis reaction Methods 0.000 description 43
- 238000001542 size-exclusion chromatography Methods 0.000 description 42
- 239000003153 chemical reaction reagent Substances 0.000 description 37
- 239000003921 oil Substances 0.000 description 37
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 36
- 238000005406 washing Methods 0.000 description 36
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 35
- 125000000217 alkyl group Chemical group 0.000 description 33
- 235000001014 amino acid Nutrition 0.000 description 33
- 229940024606 amino acid Drugs 0.000 description 33
- 150000003333 secondary alcohols Chemical group 0.000 description 33
- 150000001413 amino acids Chemical class 0.000 description 32
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 32
- 150000002148 esters Chemical class 0.000 description 31
- 108090000317 Chymotrypsin Proteins 0.000 description 30
- 238000001035 drying Methods 0.000 description 28
- 230000008569 process Effects 0.000 description 27
- 229920006395 saturated elastomer Polymers 0.000 description 27
- 229920001184 polypeptide Polymers 0.000 description 25
- 102000004196 processed proteins & peptides Human genes 0.000 description 25
- 238000003756 stirring Methods 0.000 description 25
- 239000002253 acid Substances 0.000 description 24
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol group Chemical group [C@@H]1(CC[C@H]2[C@@H]3CC=C4C[C@@H](O)CC[C@]4(C)[C@H]3CC[C@]12C)[C@H](C)CCCC(C)C HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 24
- 230000021615 conjugation Effects 0.000 description 24
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 22
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 21
- 235000014113 dietary fatty acids Nutrition 0.000 description 21
- 235000019439 ethyl acetate Nutrition 0.000 description 21
- 229930195729 fatty acid Natural products 0.000 description 21
- 239000000194 fatty acid Substances 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 20
- 125000004432 carbon atom Chemical group C* 0.000 description 20
- 239000000460 chlorine Substances 0.000 description 20
- 238000001727 in vivo Methods 0.000 description 20
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 20
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 18
- 230000002194 synthesizing effect Effects 0.000 description 18
- 239000012141 concentrate Substances 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- 238000000746 purification Methods 0.000 description 17
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 16
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 16
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 125000003545 alkoxy group Chemical group 0.000 description 15
- 210000000988 bone and bone Anatomy 0.000 description 15
- 229910052799 carbon Inorganic materials 0.000 description 15
- 231100000673 dose–response relationship Toxicity 0.000 description 15
- 239000012043 crude product Substances 0.000 description 14
- UWHCKJMYHZGTIT-UHFFFAOYSA-N tetraethylene glycol Chemical compound OCCOCCOCCOCCO UWHCKJMYHZGTIT-UHFFFAOYSA-N 0.000 description 14
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 13
- 239000004472 Lysine Substances 0.000 description 13
- 150000001721 carbon Chemical group 0.000 description 13
- 239000000741 silica gel Substances 0.000 description 13
- 229910002027 silica gel Inorganic materials 0.000 description 13
- 102000017631 Calcitonin-like Human genes 0.000 description 12
- 108050005865 Calcitonin-like Proteins 0.000 description 12
- 229920001451 polypropylene glycol Polymers 0.000 description 12
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 11
- 239000000539 dimer Substances 0.000 description 11
- 238000005516 engineering process Methods 0.000 description 11
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 239000011734 sodium Substances 0.000 description 11
- 229910000104 sodium hydride Inorganic materials 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 10
- 150000001408 amides Chemical class 0.000 description 10
- 235000012000 cholesterol Nutrition 0.000 description 10
- 238000005336 cracking Methods 0.000 description 10
- 229910052739 hydrogen Inorganic materials 0.000 description 10
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- 150000003335 secondary amines Chemical class 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 239000001257 hydrogen Substances 0.000 description 9
- 239000000825 pharmaceutical preparation Substances 0.000 description 9
- 150000003839 salts Chemical class 0.000 description 9
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 8
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 8
- 238000001212 derivatisation Methods 0.000 description 8
- 229940126534 drug product Drugs 0.000 description 8
- 229940088598 enzyme Drugs 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 239000003446 ligand Substances 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 239000000546 pharmaceutical excipient Substances 0.000 description 8
- 229920000233 poly(alkylene oxides) Polymers 0.000 description 8
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 8
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 7
- 208000035126 Facies Diseases 0.000 description 7
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 238000004364 calculation method Methods 0.000 description 7
- 239000004519 grease Substances 0.000 description 7
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 7
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 7
- 239000000377 silicon dioxide Substances 0.000 description 7
- 235000012239 silicon dioxide Nutrition 0.000 description 7
- 235000011121 sodium hydroxide Nutrition 0.000 description 7
- 239000013638 trimer Substances 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 6
- 125000003275 alpha amino acid group Chemical group 0.000 description 6
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 238000005227 gel permeation chromatography Methods 0.000 description 6
- 238000002169 hydrotherapy Methods 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- YJCJVMMDTBEITC-UHFFFAOYSA-N omega-Oxy-caprinsaeure Natural products OCCCCCCCCCC(O)=O YJCJVMMDTBEITC-UHFFFAOYSA-N 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 239000012312 sodium hydride Substances 0.000 description 6
- 125000006850 spacer group Chemical group 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- 241000972773 Aulopiformes Species 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 5
- 102000005593 Endopeptidases Human genes 0.000 description 5
- 108010059378 Endopeptidases Proteins 0.000 description 5
- 101800003632 Katacalcin Proteins 0.000 description 5
- 102400000112 Katacalcin Human genes 0.000 description 5
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 5
- 238000012300 Sequence Analysis Methods 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 230000017531 blood circulation Effects 0.000 description 5
- 125000005587 carbonate group Chemical group 0.000 description 5
- 230000004087 circulation Effects 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 230000009089 cytolysis Effects 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 230000002255 enzymatic effect Effects 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- BOARIOLZPFSAQJ-NQSKQZERSA-N katacalcin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](OC)C(=O)N[C@@H](CCSC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O)C(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)C(CC=1NC=NC=1)NC(=O)C(CC(O)=O)NC(=O)C(CCCNC(N)=N)NC(=O)C(CCC(O)=O)NC(=O)C(CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)C(CO)NC(=O)C(CCSC)NC(=O)[C@@H](N)CC(O)=O)C1=CN=CN1 BOARIOLZPFSAQJ-NQSKQZERSA-N 0.000 description 5
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 5
- 230000010534 mechanism of action Effects 0.000 description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 235000019515 salmon Nutrition 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000011877 solvent mixture Substances 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 238000013268 sustained release Methods 0.000 description 5
- 239000012730 sustained-release form Substances 0.000 description 5
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 5
- 238000001291 vacuum drying Methods 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 241001597008 Nomeidae Species 0.000 description 4
- 108010038807 Oligopeptides Proteins 0.000 description 4
- 102000015636 Oligopeptides Human genes 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- CGIHPACLZJDCBQ-UHFFFAOYSA-N acibenzolar Chemical compound SC(=O)C1=CC=CC2=C1SN=N2 CGIHPACLZJDCBQ-UHFFFAOYSA-N 0.000 description 4
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 4
- 150000003973 alkyl amines Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 4
- 229940073608 benzyl chloride Drugs 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 239000003613 bile acid Substances 0.000 description 4
- SHZIWNPUGXLXDT-UHFFFAOYSA-N caproic acid ethyl ester Natural products CCCCCC(=O)OCC SHZIWNPUGXLXDT-UHFFFAOYSA-N 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 230000002508 compound effect Effects 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 239000000693 micelle Substances 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 4
- 229920000768 polyamine Polymers 0.000 description 4
- 230000002035 prolonged effect Effects 0.000 description 4
- 229960004889 salicylic acid Drugs 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- CUZKCNWZBXLAJX-UHFFFAOYSA-N 2-phenylmethoxyethanol Chemical compound OCCOCC1=CC=CC=C1 CUZKCNWZBXLAJX-UHFFFAOYSA-N 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- NKTKZONTXDRJQI-UHFFFAOYSA-N CS(O)(=O)=O.C(OCc1ccccc1)c1ccccc1 Chemical class CS(O)(=O)=O.C(OCc1ccccc1)c1ccccc1 NKTKZONTXDRJQI-UHFFFAOYSA-N 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- KJYFPAAKGJJPJH-UHFFFAOYSA-N O(CCCCCCCCCCCCCCCCCCCCCCCCCCC)O Chemical class O(CCCCCCCCCCCCCCCCCCCCCCCCCCC)O KJYFPAAKGJJPJH-UHFFFAOYSA-N 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- 239000004473 Threonine Substances 0.000 description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 235000003704 aspartic acid Nutrition 0.000 description 3
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 3
- 230000000975 bioactive effect Effects 0.000 description 3
- 230000008827 biological function Effects 0.000 description 3
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 3
- 229960000310 isoleucine Drugs 0.000 description 3
- 229920001427 mPEG Polymers 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 229920002959 polymer blend Polymers 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 159000000000 sodium salts Chemical class 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 239000004474 valine Substances 0.000 description 3
- 150000004818 1,2-dichlorobenzenes Chemical class 0.000 description 2
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 2
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 2
- BMVXCPBXGZKUPN-UHFFFAOYSA-N 1-hexanamine Chemical compound CCCCCCN BMVXCPBXGZKUPN-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- VVBQKDDPSXBMMZ-UHFFFAOYSA-N 2-[2-[2-[2-[2-(2-phenylmethoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound OCCOCCOCCOCCOCCOCCOCC1=CC=CC=C1 VVBQKDDPSXBMMZ-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- 101000741445 Homo sapiens Calcitonin Proteins 0.000 description 2
- 101000999322 Homo sapiens Putative insulin-like growth factor 2 antisense gene protein Proteins 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 208000029725 Metabolic bone disease Diseases 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 102100036485 Putative insulin-like growth factor 2 antisense gene protein Human genes 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000001785 acacia senegal l. willd gum Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 150000005829 chemical entities Chemical class 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 229940125900 compound 59 Drugs 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- JQVDAXLFBXTEQA-UHFFFAOYSA-N dibutylamine Chemical compound CCCCNCCCC JQVDAXLFBXTEQA-UHFFFAOYSA-N 0.000 description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 238000003810 ethyl acetate extraction Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- IIRDTKBZINWQAW-UHFFFAOYSA-N hexaethylene glycol Chemical compound OCCOCCOCCOCCOCCOCCO IIRDTKBZINWQAW-UHFFFAOYSA-N 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 229940045644 human calcitonin Drugs 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 230000001788 irregular Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- XGFDHKJUZCCPKQ-UHFFFAOYSA-N nonadecan-1-ol Chemical class CCCCCCCCCCCCCCCCCCCO XGFDHKJUZCCPKQ-UHFFFAOYSA-N 0.000 description 2
- GLZWNFNQMJAZGY-UHFFFAOYSA-N octaethylene glycol Chemical compound OCCOCCOCCOCCOCCOCCOCCOCCO GLZWNFNQMJAZGY-UHFFFAOYSA-N 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 2
- 239000004533 oil dispersion Substances 0.000 description 2
- 230000001009 osteoporotic effect Effects 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 229920001281 polyalkylene Polymers 0.000 description 2
- 229920001083 polybutene Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- NGEZPLCPKXKLQQ-VOTSOKGWSA-N (e)-4-(3-methoxyphenyl)but-3-en-2-one Chemical compound COC1=CC=CC(\C=C\C(C)=O)=C1 NGEZPLCPKXKLQQ-VOTSOKGWSA-N 0.000 description 1
- HCUOEKSZWPGJIM-YBRHCDHNSA-N (e,2e)-2-hydroxyimino-6-methoxy-4-methyl-5-nitrohex-3-enamide Chemical compound COCC([N+]([O-])=O)\C(C)=C\C(=N/O)\C(N)=O HCUOEKSZWPGJIM-YBRHCDHNSA-N 0.000 description 1
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical class CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 1
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 1
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical class O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 description 1
- HCXJFMDOHDNDCC-UHFFFAOYSA-N 5-$l^{1}-oxidanyl-3,4-dihydropyrrol-2-one Chemical group O=C1CCC(=O)[N]1 HCXJFMDOHDNDCC-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- UHBXGHADNVDXMM-UHFFFAOYSA-N C(C)(C)N(C(C)C)CC[Li] Chemical compound C(C)(C)N(C(C)C)CC[Li] UHBXGHADNVDXMM-UHFFFAOYSA-N 0.000 description 1
- OREVSZRHNJJVEX-UHFFFAOYSA-N C1(=CC=CC=C1)OC=O.[Cl] Chemical compound C1(=CC=CC=C1)OC=O.[Cl] OREVSZRHNJJVEX-UHFFFAOYSA-N 0.000 description 1
- KSIYPKPZIBBUFR-LJNLPFSOSA-N CSCC[C@H](NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1c[nH]c2ccccc12)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1)C(C)C)C(=O)NCC(=O)N[C@@H](Cc1ccccc1)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(N)=O Chemical compound CSCC[C@H](NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1c[nH]c2ccccc12)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1)C(C)C)C(=O)NCC(=O)N[C@@H](Cc1ccccc1)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(N)=O KSIYPKPZIBBUFR-LJNLPFSOSA-N 0.000 description 1
- 108010001789 Calcitonin Receptors Proteins 0.000 description 1
- 102100038520 Calcitonin receptor Human genes 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 1
- 108010069941 DNA receptor Proteins 0.000 description 1
- 206010011878 Deafness Diseases 0.000 description 1
- 206010011968 Decreased immune responsiveness Diseases 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 101000741447 Gallus gallus Calcitonin Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 208000013038 Hypocalcemia Diseases 0.000 description 1
- 208000029663 Hypophosphatemia Diseases 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- 208000032912 Local swelling Diseases 0.000 description 1
- 241000222065 Lycoperdon Species 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 241000768494 Polymorphum Species 0.000 description 1
- NPYPAHLBTDXSSS-UHFFFAOYSA-N Potassium ion Chemical compound [K+] NPYPAHLBTDXSSS-UHFFFAOYSA-N 0.000 description 1
- 244000184734 Pyrus japonica Species 0.000 description 1
- 101000910301 Rattus norvegicus Calcitonin Proteins 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 101000910302 Sus scrofa Calcitonin Proteins 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 208000009205 Tinnitus Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- 206010047513 Vision blurred Diseases 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 238000005815 base catalysis Methods 0.000 description 1
- 125000001743 benzylic group Chemical group 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 230000037182 bone density Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- NCMHKCKGHRPLCM-UHFFFAOYSA-N caesium(1+) Chemical compound [Cs+] NCMHKCKGHRPLCM-UHFFFAOYSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- SKCNIGRBPJIUBQ-UHFFFAOYSA-N chloroform;ethyl acetate Chemical compound ClC(Cl)Cl.CCOC(C)=O SKCNIGRBPJIUBQ-UHFFFAOYSA-N 0.000 description 1
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 231100000895 deafness Toxicity 0.000 description 1
- 238000006264 debenzylation reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 231100000223 dermal penetration Toxicity 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000016354 hearing loss disease Diseases 0.000 description 1
- FHHGCKHKTAJLOM-UHFFFAOYSA-N hexaethylene glycol monomethyl ether Chemical compound COCCOCCOCCOCCOCCOCCO FHHGCKHKTAJLOM-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- 210000001981 hip bone Anatomy 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000000705 hypocalcaemia Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000003446 memory effect Effects 0.000 description 1
- KNWQLFOXPQZGPX-UHFFFAOYSA-N methanesulfonyl fluoride Chemical compound CS(F)(=O)=O KNWQLFOXPQZGPX-UHFFFAOYSA-N 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000011164 ossification Effects 0.000 description 1
- 210000002997 osteoclast Anatomy 0.000 description 1
- 210000004409 osteocyte Anatomy 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 210000004197 pelvis Anatomy 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 210000004224 pleura Anatomy 0.000 description 1
- 229920001197 polyacetylene Polymers 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000007151 ring opening polymerisation reaction Methods 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- TYWMIZZBOVGFOV-UHFFFAOYSA-N tetracosan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCO TYWMIZZBOVGFOV-UHFFFAOYSA-N 0.000 description 1
- 229910052716 thallium Inorganic materials 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 231100000886 tinnitus Toxicity 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 229940099259 vaseline Drugs 0.000 description 1
- 239000007762 w/o emulsion Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Images














Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/23—Calcitonins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/585—Calcitonins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
Abstract
公开了共轭物的混合物,其中混合物中每一共轭物包含与包括聚亚烷基二醇部分的寡聚体偶联的降钙素药物。混合物可以降低个体血清钙水平达10、15或20%或以上。而且,混合物可以比非共轭的降钙素更加有效地在体外肠消化模型中存活。此外,混合物可以比非共轭的降钙素表现更高的生物利用度。
Description
发明领域
本发明涉及药物-低聚物缀合物,更确切地涉及降钙素药物-低聚物缀合物。
发明背景
降钙素是一种天然存在的激素,半衰期短,据信它直接作用于破骨细胞(经由细胞表面上的降钙素受体)。这种作用可以直接抑制破骨性骨吸收,这可以引起血钙过少和/或血磷酸盐过少性血清效应。降钙素可以用于治疗各种骨障碍,包括但不限于骨质疏松和佩吉特氏病。
骨质疏松是这样一种骨疾病,其中骨组织被正常地矿质化,但是骨量减少,小梁状骨的结构完整性被削弱了。密质骨变得更加多孔和更薄。这使骨更脆弱,更容易骨折。在美国,约21%的绝经后妇女患有骨质疏松(低骨密度),约16%已经发生过骨折。在80岁以上的妇女中,约40%已经经历过髋骨、椎骨、手臂或骨盆的骨折。老年男性与妇女的人口一直在增加,因此患有骨质疏松的人数也在增加。
作为皮下注射给药的降钙素已经显示可显著提高骨密度;不过,已有报道说副作用的发生率高,包括注射部位的疼痛、面红和恶心,这可能限制药物的应用。
骨的佩吉特氏病是一种未知来源的代谢性骨障碍,它通常影响老年人。该疾病导致骨生成增加和不规则,因为负责溶解机体旧骨并用新骨代替的骨细胞变得失去控制。经过一段时间,变形了的新骨变得更大、更脆弱,具有比正常骨更多的血管。与正常的骨不同,其结构是不规则的,所以也是更脆弱的,使它具有即使微小损伤也会骨折的倾向。
在其最轻微的形式中,该疾病没有症状。在更严重的情况下,疼痛可能会很强烈。疾病的无情进展可以导致骨弯成弓形,头颅的大小增加,和脊柱弯曲。随着骨增大,它们可以压迫附近的神经,这可能导致肌肉萎弱。在严重头颅增大的情况下,这种压迫可能导致耳聋、视力模糊、头晕和耳鸣。
降钙素可以有效治疗骨骼变形增加的障碍,例如佩吉特氏病。在治疗佩吉特氏病时,降钙素的长期应用可以引起症状的长期减少;不过,降钙素给药的副作用可以包括恶心、手胀、荨麻疹和肠痉挛。
多份参考文献已经提出了共轭多肽,例如降钙素-聚乙二醇或含聚乙二醇聚合物的多分散混合物。例如,Ekwuribe的美国专利No.5,359,030提出了共轭多肽,例如降钙素-聚乙二醇改性糖脂聚合物的多分散混合物和聚乙二醇改性脂肪酸聚合物的多分散混合物。由每种组合所生成的聚合物的数均分子量优选地在约500至约10,000道尔顿范围内。
Ekwuribe所述聚合物混合物和缀合物的多分散性同样是在聚合物合成中使用多分散聚乙二醇的结果。PEG通常是由碱催化的环氧乙烷的开环聚合所生成的。该反应是通过向乙二醇加入环氧乙烷而引发的,以氢氧化钾作为催化剂。该过程生成聚乙二醇聚合物的多分散聚合物,数均分子量在给定的分子量范围内。例如,由Sigma-Aldrich ofMilwaukee,Wisconsin供应的PEG产品是以多分散混合物的形式提供的,例如PEG 400(Mn 380-420);PEG 1,000(Mn 950-1,050);PEG1,500(Mn 1,400-1,600);和PEG 2,000(Mn 1,900-2,200)。
需要提供降钙素药物-低聚物缀合物的非多分散混合物,其中该低聚物包含聚乙二醇。
发明概述
已经意外地发现,根据本发明实施方式的包含聚乙二醇的降钙素-低聚物缀合物混合物可以降低血清钙水平达10、15或者甚至20%或以上。而且,根据本发明实施方式的包含聚乙二醇的降钙素-低聚物缀合物混合物可以比非共轭的降钙素更加有效地在体外肠消化模型中存活。此外,根据本发明实施方式的包含聚乙二醇的降钙素-低聚物缀合物混合物可以比非共轭的降钙素表现更高的生物利用度。
按照本发明的实施方式,提供了缀合物的基本单分散混合物,各自包含与包含聚乙二醇部分的低聚物偶联的降钙素药物。聚乙二醇部分优选地具有至少2、3或4个聚乙二醇亚单位,最优选地具有至少7个聚乙二醇亚单位。低聚物优选地进一步包含亲脂性部分。降钙素药物优选地是鲑鱼降钙素。低聚物优选地偶联在鲑鱼降钙素的Lys11和Lys18上。缀合物优选地是两亲性平衡的,以便缀合物是水溶性的,并且能够穿透生物膜。
按照本发明的其他实施方式,提供了缀合物的基本单分散混合物,其中每一缀合物包括鲑鱼降钙素,在鲑鱼降钙素的Lys11上共价偶联第一低聚物的羧酸部分,该第一低聚物包含在羧酸部分远端与以甲基封端的聚乙二醇部分共价偶联的辛酸,该聚乙二醇部分具有至少7个聚乙二醇亚单位,并且在鲑鱼降钙素的Lys18上共价偶联第二低聚物的羧酸部分,该第二低聚物包含在羧酸部分远端与以甲基封端的聚乙二醇部分共价偶联的辛酸,该聚乙二醇部分具有至少7个聚乙二醇亚单位。
按照本发明的其他实施方式,提供了缀合物的基本单分散混合物,其中每一缀合物包含与包含聚乙二醇部分的低聚物偶联的降钙素药物,该混合物能够降低个体血清钙水平达至少5%。
按照本发明的其他实施方式,提供了缀合物的基本单分散混合物,其中每一缀合物包含与包含聚乙二醇部分的低聚物偶联的降钙素药物,与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解的抗性相比,该混合物增加了对胰凝乳蛋白酶和/或胰蛋白酶降解的抗性。
按照本发明的其他实施方式,提供了缀合物的基本单分散混合物,其中每一缀合物包含与包含聚乙二醇部分的低聚物偶联的降钙素药物,该混合物具有比没有与低聚物偶联的降钙素药物更高的生物功效。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物包括与包含聚乙二醇部分的低聚物偶联的降钙素药物,该混合物分子量分布的标准偏差小于22道尔顿。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物包括与包含聚乙二醇部分的低聚物偶联的降钙素药物,该混合物具有大于10,000的分散系数(DC),其中
其中:
n是样本中不同分子的数量;
Ni是样本中第i个分子的数量;
Mi是样本中第i个分子的质量。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物包括与低聚物偶联的降钙素药物,并且具有相同的聚乙二醇亚单位数。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物具有相同的分子量,并且具有下式:
降钙素药物-[-B-Lj-Gk-R-G′m-R′-G″n-T]p (A)
其中:
B是键合部分;
L是连接基团;
G、G′和G″是各自选择的间隔基团;
R是亲脂性基团,且R′是聚亚烷基二醇基团,或者R′是亲脂性基团,且R是聚亚烷基氧基团;
T是封端基团;
j、k、m和n各自是0或1;
p是1至降钙素药物上亲核残基数的整数。
还提供了包含本发明缀合物混合物的药物组合物,以及治疗需要这类治疗的个体骨质疏松的方法,该方法给以有效量的这类药物组合物。另外,提供了合成这类缀合物混合物的方法。
根据本发明实施方式的降钙素-低聚物缀合物混合物可以降低血清钙水平达20%或以上。而且,与非共轭的降钙素相比,这类缀合物可以降低肠酶的降解作用和/或增加生物利用度。
附图的简要说明
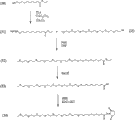
图1阐述合成根据本发明实施方式的包含聚乙二醇部分和脂肪酸部分的活化聚合物混合物的一般流程;
图2阐述合成根据本发明实施方式的mPEG混合物的流程;
图3阐述合成根据本发明实施方式的活化mPEG7-己基低聚物混合物的流程;
图4阐述合成根据本发明实施方式的活化mPEG7-辛基低聚物混合物的流程;
图5阐述合成根据本发明实施方式的活化mPEG7-癸基低聚物混合物的流程;
图6阐述合成根据本发明实施方式的活化硬脂酸酯-PEG6低聚物混合物的流程;
图7阐述合成根据本发明实施方式的活化硬脂酸酯-PEG8低聚物混合物的流程;
图8阐述合成根据本发明实施方式的活化PEG3低聚物混合物的流程;
图9阐述合成根据本发明实施方式的活化棕榈酸酯-PEG3低聚物混合物的流程;
图10阐述合成根据本发明实施方式的活化PEG6低聚物混合物的流程;
图11阐述合成根据本发明实施方式的各种丙二醇单体的流程;
图12阐述合成根据本发明实施方式的各种丙二醇聚合物的流程;
图13阐述合成根据本发明实施方式的各种丙二醇聚合物的流程;
图14阐述根据本发明实施方式的各种降钙素-低聚物缀合物混合物与非共轭降钙素的平均AUC对比,这仅供对比,不构成本发明的一部分;
图15阐述根据本发明实施方式的mPEG7-辛基-降钙素二缀合物混合物的剂量-响应曲线和降钙素的剂量-响应曲线,这仅供对比,不是本发明的一部分;
图16阐述根据本发明实施方式的mPEG7-辛基-降钙素二缀合物混合物口服给药后的剂量-响应曲线;
图17阐述根据本发明实施方式的mPEG7-辛基-降钙素二缀合物混合物皮下给药后的剂量-响应曲线;
图18阐述鲑鱼降钙素皮下给药后的剂量-响应曲线,这仅供对比,不是本发明的一部分。
优选实施方式的详细说明
现在将参照本文所述优选实施方式描述本发明。不过应当意识到的是这些实施方式仅供阐述发明,不被解释为限制由权利要求所限定的发明范围。
本文所用的术语“非多分散的”用于描述具有与Ekwuribe的美国专利No.5,359,030所述多分散混合物相反的分散性的化合物混合物。
本文所用的术语“基本单分散的”用于描述这样的化合物混合物,其中在混合物中至少约95%的化合物具有相同的分子量。
本文所用的术语“单分散的”用于描述这样的化合物混合物,其中在混合物中约100%的化合物具有相同的分子量。
本文所用的术语“基本纯粹单分散的”用于描述这样的化合物混合物,其中在混合物中至少约95%的化合物具有相同的分子量,并且具有相同的分子结构。因而,基本纯粹单分散混合物是基本单分散混合物,但是基本单分散混合物不一定是基本纯粹单分散混合物。
本文所用的术语“纯粹单分散的”用于描述这样的化合物混合物,其中在混合物中约100%的化合物具有相同的分子量,并且具有相同的分子结构。因而,纯粹单分散混合物是单分散混合物,但是单分散混合物不一定是纯粹单分散混合物。
本文所用的术语“重均分子量”被定义为
本文所用的术语“重均分子量”被定义为在混合物中给定分子的重量份数乘以混合物中每一分子的分子质量的结果之和。“重均分子量”用符号Mw代表。
本文所用的术语“数均分子量”被定义为混合物的总重量除以混合物中的分子数,用符号Mn代表。
本文所用的术语“分散系数”(DC)是由下式定义的:
其中:
n是样本中不同分子的数量;
Ni是样本中第i个分子的数量;
Mi是样本中第i个分子的质量。
本文所用的术语“个体内差异”表示在不同时间向该个体给以相同剂量药物或药物组合物时在同一个体内存在的的活性差异。
本文所用的术语“个体间差异”表示向每一个体给以相同剂量的给定药物或药物制剂时在两个或多个个体之间的活性差异。
本文所用的术语“生物功效”表示药物或药物缀合物与体内一种或多种所需受体发生相互作用的能力。
本文所用的术语“降钙素药物”表示具有降钙素的全部或一些生物活性的药物。
本文所用的术语“降钙素”表示鸡降钙素、鳗鱼降钙素、人降钙素、猪降钙素、大鼠降钙素或鲑鱼降钙素,由天然、合成或遗传工程来源所提供。
本文所用的术语“降钙素类似物”表示这样的降钙素,其中一个或多个氨基酸已被置换,而保留了降钙素的一些或全部活性。在描述类似物时,用置换位置上标注明置换氨基酸,继之以降钙素的说明。例如,“Pro2降钙素,人”意味着通常见于人降钙素2位的甘氨酸已被脯氨酸置换。
降钙素类似物可以借助各种手段获得,这将为本领域技术人员所理解。例如,降钙素结构中的某些氨基酸可以被取代为其他氨基酸,而与诸如抗体的抗原结合区或底物分子上的结合位点等结构的相互结合能力不会有可察觉的降低。由于降钙素相互作用的能力和性质决定了其生物学功能活性,可以在氨基酸序列中进行某些氨基酸序列的取代并仍然保留具有相似性质的多肽。
在进行这类取代时,可以考虑氨基酸的水疗指数。水疗氨基酸指数在赋予多肽以相互作用性生物功能中的重要性是本领域公知的。公认的是氨基酸的相对水疗性有助于所得多肽的二级结构,这继而决定了多肽与其它分子的相互作用,例如酶、底物、受体、DNA、抗体、抗原等等。已经根据其疏水性和电荷特性而指定了每种氨基酸的水疗指数如下:异亮氨酸(+4.5);缬氨酸(+4.2);亮氨酸(+3.8);苯丙氨酸(+2.8);半胱氨酸/胱氨酸(+2.5);甲硫氨酸(+1.9);丙氨酸(+1.8);甘氨酸(-0.4);苏氨酸(-0.7);丝氨酸(-0.8);色氨酸(-0.9);酪氨酸(-1.3);脯氨酸(-1.6);组氨酸(-3.2);谷氨酸(-3.5);谷氨酰胺(-3.5);天冬氨酸(-3.5);天冬酰胺(-3.5);赖氨酸(-3.9);和精氨酸(-4.5)。正如将为本领域技术人员所理解的,某些氨基酸可以被具有相似水疗指数或得分的其他氨基酸取代,而仍然生成具有相似生物活性的多肽,也就是说仍然得到生物功能上等价的多肽。在进行这类变化时,优选其水疗指数在彼此±2内的氨基酸的取代,特别优选在彼此±1内的那些,进而更特别优选在彼此±0.5内的那些。
本领域还已知可以根据亲水性有效进行相似氨基酸的取代。美国专利4,554,101提出,受相邻氨基酸的亲水性支配,蛋白质的最大局部平均亲水性与蛋白质的生物学性质有相互关系。正如美国专利4,554,101所详细描述的,已经对氨基酸残基指定了下列亲水性值:精氨酸(+3.0);赖氨酸(±3.0);天冬氨酸(+3.0±1);谷氨酸(+3.0±1);丝氨酸(+0.3);天冬酰胺(+0.2);谷氨酰胺(+0.2);甘氨酸(0);苏氨酸(-0.4);脯氨酸(-0.5±1);丙氨酸(-0.5);组氨酸(-0.5);半胱氨酸(-1.0);甲硫氨酸(-1.3);缬氨酸(-1.5);亮氨酸(-1.8);异亮氨酸(-1.8);酪氨酸(-2.3);苯丙氨酸(-2.5);色氨酸(-3.4)。正如本领域技术人员所理解的,氨基酸可以被取代为具有相似亲水性值的另一氨基酸,并仍然得到生物学等效、特别是免疫学等效的多肽。在这类变化中,优选其亲水性值在彼此±2内的氨基酸的取代,特别优选在彼此±1内的那些,进而更特别优选在彼此±0.5内的那些。
如上所述,氨基酸取代一般因此是基于氨基酸侧链取代基的相对相似性的,例如它们的疏水性、亲水性、电荷、大小等。考虑各种上述特征的示范性取代(也就是可以互换而不显著改变多肽生物活性的氨基酸)是本领域技术人员熟知的,例如包括:精氨酸与赖氨酸;谷氨酸与天冬氨酸;丝氨酸与苏氨酸;谷氨酰胺与天冬酰胺;和缬氨酸、亮氨酸与异亮氨酸。
本文所用的术语“降钙素片段”表示见于降钙素中的氨基酸序列的节段,它保留降钙素的一些或全部活性。
本文所用的术语“降钙素片段类似物”表示见于降钙素分子中的氨基酸序列的节段,其中该节段中的一个或多个氨基酸已被置换,同时保留降钙素的一些或全部活性。
本文所用的术语“PEG”指的是直链或支链聚乙二醇聚合物,包括聚乙二醇的一甲醚(mPEG)。术语“PEG亚单位”和聚乙二醇亚单位指的是单一的聚乙二醇单位,即-(CH2CH2O)-。
本文所用的术语“亲脂性”表示溶解在脂质中的能力和/或穿透生物膜、与生物膜相互作用和/或跨越生物膜的能力,术语“亲脂性部分”或“亲脂物”表示这样一种部分,它是亲脂性的,和/或在与另一化学实体连接时,它增加这类化学实体的亲脂性。亲脂性部分的实例但不限于烷基、脂肪酸、脂肪酸酯、胆固醇基、金刚烷基等。
本文所用的术语“低级烷基”指的是具有1至5个碳原子的取代或未取代的烷基部分。
本文所用的术语“高级烷基”指的是具有6个或以上碳原子的取代或未取代的烷基部分。
在本发明的实施方式中,提供了降钙素药物-低聚物缀合物的基本单分散混合物。单分散混合物中每一降钙素药物-低聚物缀合物包括与包含聚乙二醇部分的低聚物偶联的降钙素药物。优选地,混合物中至少约96、97、98或99%的缀合物具有相同的分子量。更优选地,混合物是单分散混合物。进而更优选地,混合物是基本纯粹单分散混合物。进而更优选地,混合物中至少约96、97、98或99%的缀合物具有相同的分子量,并且具有相同的分子结构。最优选地,混合物是纯粹单分散混合物。
降钙素药物优选地是降钙素。更优选地,降钙素药物是鲑鱼降钙素。不过,可以理解的是降钙素药物可以选自本领域已知的各种降钙素药物,例如包括降钙素前体肽、降钙素、降钙素类似物、降钙素片段和降钙素片段类似物。降钙素前体肽包括但不限于katacalcin(PDN-21)(C--前降钙素)和N-proCT(氨基末端前降钙素裂解肽),人。降钙素类似物可以如上所述借助降钙素中一个或多个氨基酸的取代而得。降钙素片段包括但不限于降钙素1-7,人;和降钙素8-32,鲑鱼。降钙素片段类似物可以如上所述借助降钙素片段中一个或多个氨基酸的取代而得。
低聚物可以是包含聚乙二醇部分的各种低聚物,这将为本领域技术人员所理解。优选地,低聚物的聚乙二醇部分具有至少2、3或4个聚乙二醇亚单位。更优选地,聚乙二醇部分具有至少5或6个聚乙二醇亚单位,最优选地,聚乙二醇部分具有至少7个聚乙二醇亚单位。
低聚物可以包含一个或多个其他部分,这将为本领域技术人员所理解,包括但不限于另外的亲水性部分、亲脂性部分、间隔部分、连接部分和终端部分。低聚物中的各种部分是通过可水解的或不可水解的键彼此共价偶联的。
低聚物可以进一步包含一个或多个另外的亲水性部分(也就是除了聚乙二醇部分以外的部分),包括但不限于糖、聚亚烷基氧和聚胺/PEG共聚物。由于聚乙二醇是一种聚亚烷基氧,另外的亲水性部分可以是聚乙二醇部分。
如果通过醚键偶联,相邻的聚乙二醇部分将被视为相同的部分。例如,-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-是具有六个聚乙二醇亚单位的单一聚乙二醇部分。如果该部分是低聚物中唯一的亲水性部分,那么该低聚物将不含有另外的亲水性部分。如果通过除醚键以外的键偶联,相邻的聚乙二醇部分将被视为不同的部分。例如,
是具有四个聚乙二醇亚单位的聚乙二醇部分和具有两个聚乙二醇亚单位的另外亲水性部分。优选地,根据本发明实施方式的低聚物包含聚乙二醇部分,没有另外的亲水性部分。
低聚物可以进一步包含一个或多个亲脂性部分,这将为本领域技术人员所理解。亲脂性部分优选地是饱和或不饱和的、直链或支链烷基部分或者饱和或不饱和的、直链或支链脂肪酸部分。当亲脂性部分是烷基部分时,它优选地是直链的、饱和或不饱和的烷基部分,具有1至28个碳原子。更优选地,烷基部分具有2至12个碳原子。当亲脂性部分是脂肪酸部分时,它优选地是天然脂肪酸部分,它是直链的、饱和或不饱和的,具有2至18个碳原子。更优选地,脂肪酸部分具有3至14个碳原子。最优选地,脂肪酸部分具有至少4、5或6个碳原子。
低聚物可以进一步包含一个或多个间隔部分,这将为本领域技术人员所理解。间隔基团例如可以用于分隔亲水性部分与亲脂性部分,分隔亲脂性部分或亲水性部分与降钙素药物,分隔第一亲水性或亲脂性部分与第二亲水性或亲脂性部分,或者分隔亲水性部分或亲脂性部分与连接部分。间隔部分优选地选自由糖、胆固醇和甘油部分组成的组。
低聚物可以进一步包含一个或多个连接部分,用于使低聚物与降钙素药物偶联,这将为本领域技术人员所理解。连接部分优选地选自由烷基和脂肪酸部分组成的组。
低聚物可以进一步在低聚物不与降钙素药物偶联的一个或多个末端包含一个或多个终端部分。终端部分优选地是烷基或烷氧基部分,更优选地是低级烷基或低级烷氧基部分。最优选地,终端部分是甲基或甲氧基。尽管终端部分优选地是烷基或烷氧基部分,不过可以理解的是终端部分可以是将为本领域技术人员所理解的各种部分,包括但不限于糖、胆固醇、醇和脂肪酸。
低聚物优选地是与降钙素药物共价偶联的。在一些实施方式中,利用可水解的键(例如酯或碳酸酯键)使降钙素药物与低聚物偶联。可水解的偶联可以提供充当前体药物的降钙素药物-低聚物缀合物。在某些情形中,例如其中降钙素药物-低聚物缀合物是无活性的(也就是说,缀合物缺乏通过降钙素药物的主要作用机理来影响机体的能力),可水解的偶联可以提供定时释放或控制释放的效果,随着一个或多个低聚物从它们各自的降钙素药物-低聚物缀合物上裂解下来以提供活性药物,在给定时间阶段内给以降钙素药物。在其他实施方式中,利用不可水解的键(例如氨基甲酸酯、酰胺或醚键)使降钙素药物与低聚物偶联。当需要使降钙素药物-低聚物缀合物在血流中长时间循环时,优选至少2小时,不可水解的键的使用可能是优选的。当低聚物与降钙素药物共价偶联时,低聚物进一步包含一个或多个键合部分,用于使低聚物与降钙素药物共价偶联,这将为本领域技术人员所理解。键合部分优选地选自由共价键、酯部分、碳酸酯部分、氨基甲酸酯部分、酰胺部分和仲胺部分组成的组。低聚物上一种以上的部分可以与降钙素药物共价偶联。
尽管低聚物优选地是与降钙素药物共价偶联的,不过可以理解的是,低聚物可以与降钙素药物非共价偶联,生成非共价共轭的降钙素药物-低聚物配合物。正如将为本领域技术人员所理解的,非共价偶联包括但不限于氢键合、离子键合、范德华键合和胶束或脂质体包封。按照本发明的实施方式,低聚物可以被适当地改造、改性和/或适当地官能化,以按所选择的方式赋予非共价共轭的能力(例如赋予氢键合能力),这将为本领域技术人员所理解。按照本发明的其他实施方式,低聚物可以用各种化合物衍生化,包括但不限于氨基酸、寡肽、肽、胆汁酸、胆汁酸衍生物、脂肪酸、脂肪酸衍生物、水杨酸、水杨酸衍生物、氨基水杨酸、和氨基水杨酸衍生物。所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价偶联(配位化合)。所得配合物优选地具有平衡的亲脂性和亲水性。按照本发明的其他实施方式,低聚物可以用胺和/或烷基胺衍生化。在适合的酸性条件下,所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价生成共轭配合物。从这类配位化合作用所得产物优选地具有平衡的亲脂性和亲水性。
一种以上的低聚物(即大量低聚物)可以与降钙素药物偶联。这些低聚物优选地是相同的。不过可以理解的是,这些低聚物可以是彼此不同,或者这些低聚物中的一些可以是相同的,另一些可以是不同的。当大量低聚物与降钙素药物偶联时,可以优选的是利用可水解的键使一个或多个低聚物与降钙素药物偶联,利用不可水解的键使一个或多个低聚物与降钙素药物偶联。或者,所有使大量低聚物与降钙素药物偶联的键都是可水解的,但是具有不同程度的可水解性,以便例如一个或多个低聚物在体内被水解作用迅速从降钙素药物上除去,一个或多个低聚物在体内被水解作用缓慢从降钙素药物上除去。
低聚物可以与降钙素药物偶联在该降钙素药物的各种亲核残基上,包括但不限于亲核性羟基官能和/或氨基官能。当降钙素药物是多肽时,亲核性羟基官能例如可以见于丝氨酸和/或酪氨酸残基,亲核性氨基官能例如可以见于组氨酸和/或赖氨酸残基、和/或多肽的一个或多个N-末端。当低聚物与降钙素多肽的一个或多个N-末端偶联时,偶联作用优选地生成仲胺。当降钙素药物是鲑鱼降钙素时,例如,低聚物可以与鲑鱼降钙素的氨基官能度偶联,包括Lys11、Lys18和/或N-末端的氨基官能度。尽管一个或多个低聚物可以与鲑鱼降钙素偶联,不过当低聚物与Lys11和Lys18的氨基官能度偶联时,在二共轭的鲑鱼降钙素中观察到更高的生物功效,例如提高了的血清钙降低能力。
本发明的降钙素药物-低聚物缀合物的基本单分散混合物可以借助各种方法合成。例如,由羧酸和聚乙二醇组成的低聚物的基本单分散混合物是这样合成的,使羧酸的基本单分散混合物与聚乙二醇的基本单分散混合物在足以提供低聚物的基本单分散混合物的条件下接触。然后活化低聚物的基本单分散混合物,以便它们能够与降钙素药物反应,得到降钙素药物-低聚物缀合物。用于提供低聚物的基本单分散混合物的一种合成途径实施方式阐述在下列图3和实施例11-18中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图4和实施例19-24中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图5和实施例25-29中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图6和实施例30-31中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图7和实施例32-37中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图8和实施例38中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图9和实施例39中。用于提供低聚物的基本单分散混合物的另一种合成途径实施方式阐述在下列图10和实施例40中。
可以使活化低聚物的基本单分散混合物与降钙素药物的基本单分散混合物在足以提供降钙素药物-低聚物缀合物混合物的条件下反应。优选的合成描述在下列实施例41中。正如将为本领域技术人员所理解的,可以控制反应条件(例如选择摩尔比、溶剂混合物和/或pH),以便从活化低聚物的基本单分散混合物与降钙素药物的基本单分散混合物的反应所得降钙素药物-低聚物缀合物混合物是一种基本单分散混合物。例如,通过维持反应溶液的pH低于赖氨酸pKa,可以抑制赖氨酸氨基官能度上的共轭作用。或者,例如利用HPLC可以分离和离析降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物、例如单-、二-或三-缀合物的基本单分散混合物。所离析的特定缀合物的共轭程度(例如所离析的分子是单-、二-还是三-缀合物)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于质谱法。特定缀合物结构(例如低聚物位于鲑鱼降钙素单缀合物的Lys11、Lys18还是N-末端上)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于序列分析、肽绘图、选择性酶裂解和/或内肽酶裂解。
正如将为本领域技术人员所理解的,降钙素药物上一个或多个反应位点例如可以这样保护,使降钙素药物与适合的保护试剂反应,例如N-叔丁氧羰基(t-BOC)或N-(9-芴基甲氧羰基)(N-FMOC)。例如当降钙素药物是一种多肽,并且需要生成在该多肽N-末端上具有低聚物的不饱和缀合物(也就是其中不是所有的亲核残基都被共轭的缀合物)时,这种方法可能是优选的。在这类保护之后,可以使被保护的降钙素药物的基本单分散混合物与活化低聚物的基本单分散混合物反应,得到降钙素药物-低聚物缀合物混合物,具有与一个或多个亲核残基偶联的低聚物,并且具有与其他亲核残基偶联的保护部分。在共轭反应后,可以使降钙素药物-低聚物缀合物去保护,这将为本领域技术人员所理解。如果必要的话,然后可以如上所述分离降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物的基本单分散混合物。或者,可以在去保护之前分离降钙素药物-低聚物缀合物混合物。
与常规混合物相比,根据本发明实施方式的降钙素药物-低聚物缀合物的基本单分散混合物优选地具有提高了的性质。例如,降钙素药物-低聚物缀合物的基本单分散混合物优选地能够降低血清钙水平达至少5%。优选地,缀合物混合物能够降低血清钙水平达至少10、11、12、13或14%。更优选地,缀合物混合物能够降低血清钙水平达至少15、16、17、18或19%,最优选地,缀合物混合物能够降低血清钙水平达至少20%。
作为另一个实例,分别与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性。对胰凝乳蛋白酶或胰蛋白酶的抗性相当于利用下列实施例51所述操作,当供试分子在可用的酶中被消化时所剩余的百分比。优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约15%。最优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约20%。优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约20%。最优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约30%。
作为另一个实例,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有比没有与低聚物偶联的降钙素药物的生物功效更高的生物功效。特定化合物的生物功效相当于它的曲线下面积(AUC)值。优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约5%。更优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约10%。
作为另一个实例,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体内活性更大的体内活性,该多分散混合物具有与基本单分散混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMERCHEMISTRY 394-402(2d.ed.,1991)所述。
作为另一个实例,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体外活性更大的体外活性,该多分散混合物具有与基本单分散混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,与多分散降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性,该多分散混合物具有与基本单分散混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,降钙素药物-低聚物缀合物的基本单分散混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的个体间差异更小的个体间差异,该多分散混合物具有与基本单分散混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。个体间差异可以借助各种方法测量,这将为本领域技术人员所理解。个体间差异优选地是如下计算的。测定每一个体的剂量响应曲线下面积(AUC)(也就是剂量响应曲线与基线值之间的面积)。将每一个体的AUC求和,再除以个体数,测定所有个体的平均AUC。然后测定每一个体的个别AUC与平均AUC之间差异的绝对值。然后将所得差异的绝对值求和,得到代表个体间差异的数值。数值越低,代表个体间差异越低,数值越高,代表个体间差异越高。
根据本发明实施方式的降钙素药物-低聚物缀合物的基本单分散混合物优选地具有两个或多个上述提高了的性质。更优选地,根据本发明实施方式的降钙素药物-低聚物缀合物的基本单分散混合物具有三个或多个上述提高了的性质。最优选地,根据本发明实施方式的降钙素药物-低聚物缀合物的基本单分散混合物具有四个或多个上述提高了的性质。
在根据本发明的实施方式中,提供了分子量分布的标准偏差小于约22道尔顿的缀合物混合物。混合物中每一缀合物包括与包含聚乙二醇部分的低聚物偶联的降钙素药物。标准偏差优选地小于约14道尔顿,更优选地小于约11道尔顿。分子量分布可以借助本领域技术人员已知的方法加以测定,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMERCHEMISTRY 394-402(2d.ed.,1991)所述。分子量分布的标准偏差然后可以借助将为本领域技术人员所理解的统计学方法加以测定。
降钙素药物优选地是降钙素。更优选地,降钙素药物是鲑鱼降钙素。不过,可以理解的是降钙素药物可以选自本领域已知的各种降钙素药物,例如包括降钙素前体肽、降钙素、降钙素类似物、降钙素片段和降钙素片段类似物。降钙素前体肽包括但不限于katacalcin(PDN-21)(C-前降钙素)和N-proCT(氨基末端前降钙素裂解肽),人。降钙素类似物可以如上所述借助降钙素中一个或多个氨基酸的取代而得。降钙素片段包括但不限于降钙素1-7,人;和降钙素8-32,鲑鱼。降钙素片段类似物可以如上所述借助降钙素片段中一个或多个氨基酸的取代而得。
低聚物可以是包含聚乙二醇部分的各种低聚物,这将为本领域技术人员所理解。优选地,低聚物的聚乙二醇部分具有至少2、3或4个聚乙二醇亚单位。更优选地,聚乙二醇部分具有至少5或6个聚乙二醇亚单位,最优选地,聚乙二醇部分具有至少7个聚乙二醇亚单位。
低聚物可以包含一个或多个其他部分,这将为本领域技术人员所理解,包括但不限于另外的亲水性部分、亲脂性部分、间隔部分、连接部分和终端部分。低聚物中的各种部分是通过可水解的或不可水解的键彼此共价偶联的。
低聚物可以进一步包含一个或多个另外的亲水性部分(也就是除了聚乙二醇部分以外的部分),包括但不限于糖、聚亚烷基氧和聚胺/PEG共聚物。由于聚乙二醇是一种聚亚烷基氧,另外的亲水性部分可以是聚乙二醇部分。
如果通过醚键偶联,相邻的聚乙二醇部分将被视为相同的部分。例如,-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-是具有六个聚乙二醇亚单位的单一聚乙二醇部分。如果该部分是低聚物中唯一的亲水性部分,那么该低聚物将不含有另外的亲水性部分。如果通过除醚键以外的键偶联,相邻的聚乙二醇部分将被视为不同的部分。例如,

是具有四个聚乙二醇亚单位的聚乙二醇部分和具有两个聚乙二醇亚单位的另外亲水性部分。优选地,根据本发明实施方式的低聚物包含聚乙二醇部分,没有另外的亲水性部分。
低聚物可以进一步包含一个或多个亲脂性部分,这将为本领域技术人员所理解。亲脂性部分优选地是饱和或不饱和的、直链或支链烷基部分或者饱和或不饱和的、直链或支链脂肪酸部分。当亲脂性部分是烷基部分时,它优选地是直链的、饱和或不饱和的烷基部分,具有1至28个碳原子。更优选地,烷基部分具有2至12个碳原子。当亲脂性部分是脂肪酸部分时,它优选地是天然脂肪酸部分,它是直链的、饱和或不饱和的,具有2至18个碳原子。更优选地,脂肪酸部分具有3至14个碳原子。最优选地,脂肪酸部分具有至少4、5或6个碳原子。
低聚物可以进一步包含一个或多个间隔部分,这将为本领域技术人员所理解。间隔基团例如可以用于分隔亲水性部分与亲脂性部分,分隔亲脂性部分或亲水性部分与降钙素药物,分隔第一亲水性或亲脂性部分与第二亲水性或亲脂性部分,或者分隔亲水性部分或亲脂性部分与连接部分。间隔部分优选地选自由糖、胆固醇和甘油部分组成的组。
低聚物可以进一步包含一个或多个连接部分,用于使低聚物与降钙素药物偶联,这将为本领域技术人员所理解。连接部分优选地选自由烷基和脂肪酸部分组成的组。
低聚物可以进一步在低聚物不与降钙素药物偶联的一个或多个末端包含一个或多个终端部分。终端部分优选地是烷基或烷氧基部分,更优选地是低级烷基或低级烷氧基部分。最优选地,终端部分是甲基或甲氧基。尽管终端部分优选地是烷基或烷氧基部分,不过可以理解的是终端部分可以是将为本领域技术人员所理解的各种部分,包括但不限于糖、胆固醇、醇和脂肪酸。
低聚物优选地是与降钙素药物共价偶联的。在一些实施方式中,利用可水解的键(例如酯或碳酸酯键)使降钙素药物与低聚物偶联。可水解的偶联可以提供充当前体药物的降钙素药物-低聚物缀合物。在某些情形中,例如其中降钙素药物-低聚物缀合物是无活性的(也就是说,缀合物缺乏通过降钙素药物的主要作用机理来影响机体的能力),可水解的偶联可以提供定时释放或控制释放的效果,随着一个或多个低聚物从它们各自的降钙素药物-低聚物缀合物上裂解下来以提供活性药物,在给定时间阶段内给以降钙素药物。在其他实施方式中,利用不可水解的键(例如氨基甲酸酯、酰胺或醚键)使降钙素药物与低聚物偶联。当需要使降钙素药物-低聚物缀合物在血流中长时间循环时,优选至少2小时,不可水解的键的使用可能是优选的。当低聚物与降钙素药物共价偶联时,低聚物进一步包含一个或多个键合部分,用于使低聚物与降钙素药物共价偶联,这将为本领域技术人员所理解。键合部分优选地选自由共价键、酯部分、碳酸酯部分、氨基甲酸酯部分、酰胺部分和仲胺部分组成的组。低聚物上一种以上的部分可以与降钙素药物共价偶联。
尽管低聚物优选地是与降钙素药物共价偶联的,不过可以理解的是,低聚物可以与降钙素药物非共价偶联,生成非共价共轭的降钙素药物-低聚物配合物。正如将为本领域技术人员所理解的,非共价偶联包括但不限于氢键合、离子键合、范德华键合和胶束或脂质体包封。按照本发明的实施方式,低聚物可以被适当地改造、改性和/或适当地官能化,以按所选择的方式赋予非共价共轭的能力(例如赋予氢键合能力),这将为本领域技术人员所理解。按照本发明的其他实施方式,低聚物可以用各种化合物衍生化,包括但不限于氨基酸、寡肽、肽、胆汁酸、胆汁酸衍生物、脂肪酸、脂肪酸衍生物、水杨酸、水杨酸衍生物、氨基水杨酸、和氨基水杨酸衍生物。所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价偶联(配位化合)。所得配合物优选地具有平衡的亲脂性和亲水性。按照本发明的其他实施方式,低聚物可以用胺和/或烷基胺衍生化。在适合的酸性条件下,所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价生成共轭配合物。从这类配位化合作用所得产物优选地具有平衡的亲脂性和亲水性。
一种以上的低聚物(即大量低聚物)可以与降钙素药物偶联。这些低聚物优选地是相同的。不过可以理解的是,这些低聚物可以是彼此不同,或者这些低聚物中的一些可以是相同的,另一些可以是不同的。当大量低聚物与降钙素药物偶联时,可以优选地利用可水解的键使一个或多个低聚物与降钙素药物偶联,利用不可水解的键使一个或多个低聚物与降钙素药物偶联。或者,所有使大量低聚物与降钙素药物偶联的键都是可水解的,但是具有不同程度的可水解性,以便例如一个或多个低聚物在体内被水解作用迅速从降钙素药物上除去,一个或多个低聚物在体内被水解作用缓慢从降钙素药物上除去。
低聚物可以与降钙素药物偶联在该降钙素药物的各种亲核残基上,包括但不限于亲核性羟基官能和/或氨基官能。当降钙素药物是多肽时,亲核性羟基官能例如可以见于丝氨酸和/或酪氨酸残基,亲核性氨基官能例如可以见于组氨酸和/或赖氨酸残基、和/或多肽的一个或多个N-末端。当低聚物与降钙素多肽的一个或多个N-末端偶联时,偶联作用优选地生成仲胺。当降钙素药物是鲑鱼降钙素时,例如,低聚物可以与鲑鱼降钙素的氨基官能度偶联,包括Lys11、Lys18和/或N-末端的氨基官能度。尽管一个或多个低聚物可以与鲑鱼降钙素偶联,不过当低聚物与Lys11和Lys18的氨基官能度偶联时,在二共轭的鲑鱼降钙素中观察到更高的生物功效,例如提高了的血清钙降低能力。
分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物可以借助各种方法合成。例如,由羧酸和聚乙二醇组成的分子量分布的标准偏差小于约22道尔顿的低聚物混合物是这样合成的,使分子量分布的标准偏差小于约22道尔顿的羧酸混合物与分子量分布的标准偏差小于约22道尔顿的聚乙二醇混合物在足以提供分子量分布的标准偏差小于约22道尔顿的低聚物混合物的条件下接触。然后活化分子量分布的标准偏差小于约22道尔顿的低聚物混合物,以便它们能够与降钙素药物反应,得到降钙素药物-低聚物缀合物。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的一种合成途径实施方式阐述在下列图3和实施例11-18中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图4和实施例19-24中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图5和实施例25-29中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图6和实施例30-31中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图7和实施例32-37中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图8和实施例38中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图9和实施例39中。用于提供分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物的另一种合成途径实施方式阐述在下列图10和实施例40中。
使分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物与分子量分布的标准偏差小于约22道尔顿的降钙素药物混合物在足以提供降钙素药物-低聚物缀合物混合物的条件下反应。优选的合成描述在下列实施例41中。正如将为本领域技术人员所理解的,可以控制反应条件(例如选择摩尔比、溶剂混合物和/或pH),以便从分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物与分子量分布的标准偏差小于约22道尔顿的降钙素药物混合物的反应所得降钙素药物-低聚物缀合物混合物是一种分子量分布的标准偏差小于约22道尔顿的混合物。例如,通过维持反应溶液的pH低于赖氨酸pKa,可以抑制赖氨酸氨基官能度上的共轭作用。或者,例如利用HPLC可以分离和离析降钙素药物-低聚物缀合物混合物,得到分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物、例如单-、二-或三-缀合物的混合物。所离析的特定缀合物的共轭程度(例如所离析的分子是单-、二-还是三-缀合物)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于质谱法。特定缀合物结构(例如低聚物位于鲑鱼降钙素单缀合物的Lys11、Lys18还是N-末端上)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于序列分析、肽绘图、选择性酶裂解和/或内肽酶裂解。
正如将为本领域技术人员所理解的,降钙素药物上一个或多个反应位点例如可以这样保护,使降钙素药物与适合的保护试剂反应,例如N-叔丁氧羰基(t-BOC)或N-(9-芴基甲氧羰基)(N-FMOC)。例如当降钙素药物是一种多肽,并且需要生成在该多肽N-末端上具有低聚物的不饱和缀合物(也就是其中不是所有的亲核残基都被共轭的缀合物)时,这种方法可能是优选的。在这类保护之后,可以使被保护的分子量分布的标准偏差小于约22道尔顿的降钙素药物混合物与分子量分布的标准偏差小于约22道尔顿的活化低聚物混合物反应,得到降钙素药物-低聚物缀合物混合物,具有与一个或多个亲核残基偶联的低聚物,并且具有与其他亲核残基偶联的保护部分。在共轭反应后,可以使降钙素药物-低聚物缀合物去保护,这将为本领域技术人员所理解。如果必要的话,然后可以如上所述分离降钙素药物-低聚物缀合物混合物,得到分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物。或者,可以在去保护之前分离降钙素药物-低聚物缀合物混合物。
与常规混合物相比,根据本发明实施方式的分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有提高了的性质。例如,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地能够降低血清钙水平达至少5%。优选地,缀合物混合物能够降低血清钙水平达至少10、11、12、13或14%。更优选地,缀合物混合物能够降低血清钙水平达至少15、16、17、18或19%,最优选地,缀合物混合物能够降低血清钙水平达至少20%。
作为另一个实例,分别与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性。对胰凝乳蛋白酶或胰蛋白酶的抗性相当于利用下列实施例51所述操作,当供试分子在可用的酶中被消化时所剩余的百分比。优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约15%。最优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约20%。优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约20%。最优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约30%。
作为另一个实例,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有比没有与低聚物偶联的降钙素药物的生物功效更高的生物功效。特定化合物的生物功效相当于它的曲线下面积(AUC)值。优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约5%。更优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约10%。
作为另一个实例,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体内活性更大的体内活性,该多分散混合物具有与分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMER CHEMISTRY 394-402(2d.ed.,1991)所述。
作为另一个实例,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体外活性更大的体外活性,该多分散混合物具有与分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,与多分散降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性,该多分散混合物具有与分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的个体间差异更小的个体间差异,该多分散混合物具有与分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。个体间差异可以借助各种方法测量,这将为本领域技术人员所理解。个体间差异优选地是如下计算的。测定每一个体的剂量响应曲线下面积(AUC)(也就是剂量响应曲线与基线值之间的面积)。将每一个体的AUC求和,再除以个体数,测定所有个体的平均AUC。然后测定每一个体的个别AUC与平均AUC之间差异的绝对值。然后将所得差异的绝对值求和,得到代表个体间差异的数值。数值越低,代表个体间差异越低,数值越高,代表个体间差异越高。
根据本发明实施方式的分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物优选地具有两个或多个上述提高了的性质。更优选地,根据本发明实施方式的分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物具有三个或多个上述提高了的性质。最优选地,根据本发明实施方式的分子量分布的标准偏差小于约22道尔顿的降钙素药物-低聚物缀合物混合物具有四个或多个上述提高了的性质。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物包括与包含聚乙二醇部分的低聚物偶联的降钙素药物,该混合物具有大于10,000的分散系数(DC),其中
其中:
n是样本中不同分子的数量;
Ni是样本中第i个分子的数量;
Mi是样本中第i个分子的质量。
缀合物混合物优选地具有大于100,000的分散系数。更优选地,缀合物混合物的分散系数大于500,000,最优选地,分散系数大于10,000,000。变量n、Ni和Mi可以借助各种将为本领域技术人员所理解的方法加以测定,包括但不限于如下实施例49所述方法。
降钙素药物优选地是降钙素。更优选地,降钙素药物是鲑鱼降钙素。不过,可以理解的是降钙素药物可以选自本领域已知的各种降钙素药物,例如包括降钙素前体肽、降钙素、降钙素类似物、降钙素片段和降钙素片段类似物。降钙素前体肽包括但不限于katacalcin(PDN-21)(C-前降钙素)和N-proCT(氨基末端前降钙素裂解肽),人。降钙素类似物可以如上所述借助降钙素中一个或多个氨基酸的取代而得。降钙素片段包括但不限于降钙素1-7,人;和降钙素8-32,鲑鱼。降钙素片段类似物可以如上所述借助降钙素片段中一个或多个氨基酸的取代而得。
低聚物可以是包含聚乙二醇部分的各种低聚物,这将为本领域技术人员所理解。优选地,低聚物的聚乙二醇部分具有至少2、3或4个聚乙二醇亚单位。更优选地,聚乙二醇部分具有至少5或6个聚乙二醇亚单位,最优选地,聚乙二醇部分具有至少7个聚乙二醇亚单位。
低聚物可以包含一个或多个其他部分,这将为本领域技术人员所理解,包括但不限于另外的亲水性部分、亲脂性部分、间隔部分、连接部分和终端部分。低聚物中的各种部分是通过可水解的或不可水解的键彼此共价偶联的。
低聚物可以进一步包含一个或多个另外的亲水性部分(也就是除了聚乙二醇部分以外的部分),包括但不限于糖、聚亚烷基氧和聚胺/PEG共聚物。由于聚乙二醇是一种聚亚烷基氧,另外的亲水性部分可以是聚乙二醇部分。
如果通过醚键偶联,相邻的聚乙二醇部分将被视为相同的部分。例如,-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-是具有六个聚乙二醇亚单位的单一聚乙二醇部分。如果该部分是低聚物中唯一的亲水性部分,那么该低聚物将不含有另外的亲水性部分。如果通过除醚键以外的键偶联,相邻的聚乙二醇部分将被视为不同的部分。例如,
是具有四个聚乙二醇亚单位的聚乙二醇部分和具有两个聚乙二醇亚单位的另外亲水性部分。优选地,根据本发明实施方式的低聚物包含聚乙二醇部分,没有另外的亲水性部分。
低聚物可以进一步包含一个或多个亲脂性部分,这将为本领域技术人员所理解。亲脂性部分优选地是饱和或不饱和的、直链或支链烷基部分或者饱和或不饱和的、直链或支链脂肪酸部分。当亲脂性部分是烷基部分时,它优选地是直链的、饱和或不饱和的烷基部分,具有1至28个碳原子。更优选地,烷基部分具有2至12个碳原子。当亲脂性部分是脂肪酸部分时,它优选地是天然脂肪酸部分,它是直链的、饱和或不饱和的,具有2至18个碳原子。更优选地,脂肪酸部分具有3至14个碳原子。最优选地,脂肪酸部分具有至少4、5或6个碳原子。
低聚物可以进一步包含一个或多个间隔部分,这将为本领域技术人员所理解。间隔基团例如可以用于分隔亲水性部分与亲脂性部分,分隔亲脂性部分或亲水性部分与降钙素药物,分隔第一亲水性或亲脂性部分与第二亲水性或亲脂性部分,或者分隔亲水性部分或亲脂性部分与连接部分。间隔部分优选地选自由糖、胆固醇和甘油部分组成的组。
低聚物可以进一步包含一个或多个连接部分,用于使低聚物与降钙素药物偶联,这将为本领域技术人员所理解。连接部分优选地选自由烷基和脂肪酸部分组成的组。
低聚物可以进一步在低聚物不与降钙素药物偶联的一个或多个末端包含一个或多个终端部分。终端部分优选地是烷基或烷氧基部分,更优选地是低级烷基或低级烷氧基部分。最优选地,终端部分是甲基或甲氧基。尽管终端部分优选地是烷基或烷氧基部分,不过可以理解的是终端部分可以是将为本领域技术人员所理解的各种部分,包括但不限于糖、胆固醇、醇和脂肪酸。
低聚物优选地是与降钙素药物共价偶联的。在一些实施方式中,利用可水解的键(例如酯或碳酸酯键)使降钙素药物与低聚物偶联。可水解的偶联可以提供充当前体药物的降钙素药物-低聚物缀合物。在某些情形中,例如其中降钙素药物-低聚物缀合物是无活性的(也就是说,缀合物缺乏通过降钙素药物的主要作用机理来影响机体的能力),可水解的偶联可以提供定时释放或控制释放的效果,随着一个或多个低聚物从它们各自的降钙素药物-低聚物缀合物上裂解下来以提供活性药物,在给定时间阶段内给以降钙素药物。在其他实施方式中,利用不可水解的键(例如氨基甲酸酯、酰胺或醚键)使降钙素药物与低聚物偶联。当需要使降钙素药物-低聚物缀合物在血流中长时间循环时,优选至少2小时,不可水解的键的使用可能是优选的。当低聚物与降钙素药物共价偶联时,低聚物进一步包含一个或多个键合部分,用于使低聚物与降钙素药物共价偶联,这将为本领域技术人员所理解。键合部分优选地选自由共价键、酯部分、碳酸酯部分、氨基甲酸酯部分、酰胺部分和仲胺部分组成的组。低聚物上一种以上的部分可以与降钙素药物共价偶联。
尽管低聚物优选地是与降钙素药物共价偶联的,不过可以理解的是,低聚物可以与降钙素药物非共价偶联,生成非共价共轭的降钙素药物-低聚物配合物。正如将为本领域技术人员所理解的,非共价偶联包括但不限于氢键合、离子键合、范德华键合和胶束或脂质体包封。按照本发明的实施方式,低聚物可以被适当地改造、改性和/或适当地官能化,以按所选择的方式赋予非共价共轭的能力(例如赋予氢键合能力),这将为本领域技术人员所理解。按照本发明的其他实施方式,低聚物可以用各种化合物衍生化,包括但不限于氨基酸、寡肽、肽、胆汁酸、胆汁酸衍生物、脂肪酸、脂肪酸衍生物、水杨酸、水杨酸衍生物、氨基水杨酸、和氨基水杨酸衍生物。所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价偶联(配位化合)。所得配合物优选地具有平衡的亲脂性和亲水性。按照本发明的其他实施方式,低聚物可以用胺和/或烷基胺衍生化。在适合的酸性条件下,所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价生成共轭配合物。从这类配位化合作用所得产物优选地具有平衡的亲脂性和亲水性。
一种以上的低聚物(即大量低聚物)可以与降钙素药物偶联。这些低聚物优选地是相同的。不过可以理解的是,这些低聚物可以是彼此不同,或者这些低聚物中的一些可以是相同的,另一些可以是不同的。当大量低聚物与降钙素药物偶联时,可以优选地利用可水解的键使一个或多个低聚物与降钙素药物偶联,利用不可水解的键使一个或多个低聚物与降钙素药物偶联。或者,所有使大量低聚物与降钙素药物偶联的键都是可水解的,但是具有不同程度的可水解性,以便例如一个或多个低聚物在体内被水解作用迅速从降钙素药物上除去,一个或多个低聚物在体内被水解作用缓慢从降钙素药物上除去。
低聚物可以与降钙素药物偶联在该降钙素药物的各种亲核残基上,包括但不限于亲核性羟基官能和/或氨基官能。当降钙素药物是多肽时,亲核性羟基官能例如可以见于丝氨酸和/或酪氨酸残基,亲核性氨基官能例如可以见于组氨酸和/或赖氨酸残基、和/或多肽的一个或多个N-末端。当低聚物与降钙素多肽的一个或多个N-末端偶联时,偶联作用优选地生成仲胺。当降钙素药物是鲑鱼降钙素时,例如,低聚物可以与鲑鱼降钙素的氨基官能度偶联,包括Lys11、Lys18和/或N-末端的氨基官能度。尽管一个或多个低聚物可以与鲑鱼降钙素偶联,不过当低聚物与Lys11和Lys18的氨基官能度偶联时,在二共轭的鲑鱼降钙素中观察到更高的生物功效,例如提高了的血清钙降低能力。
分散系数大于10,000的降钙素药物-低聚物缀合物混合物可以借助各种方法合成。例如,由羧酸和聚乙二醇组成的分散系数大于10,000的低聚物混合物是这样合成的,使分散系数大于10,000的羧酸混合物与分散系数大于10,000的聚乙二醇混合物在足以提供分散系数大于10,000的低聚物混合物的条件下接触。然后活化分散系数大于10,000的低聚物混合物,以便它们能够与降钙素药物反应,得到降钙素药物-低聚物缀合物。用于提供分散系数大于10,000的活化低聚物混合物的一种合成途径实施方式阐述在下列图3和实施例11-18中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图4和实施例19-24中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图5和实施例25-29中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图6和实施例30-31中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图7和实施例32-37中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图8和实施例38中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图9和实施例39中。用于提供分散系数大于10,000的活化低聚物混合物的另一种合成途径实施方式阐述在下列图10和实施例40中。
使分散系数大于10,000的活化低聚物混合物与分散系数大于10,000的降钙素药物混合物在足以提供降钙素药物-低聚物缀合物混合物的条件下反应。优选的合成描述在下列实施例41中。正如将为本领域技术人员所理解的,可以控制反应条件(例如选择摩尔比、溶剂混合物和/或pH),以便从分散系数大于10,000的活化低聚物混合物与分散系数大于10,000的降钙素药物混合物的反应所得降钙素药物-低聚物缀合物混合物是一种分散系数大于10,000的混合物。例如,通过维持反应溶液的pH低于赖氨酸pKa,可以抑制赖氨酸氨基官能度上的共轭作用。或者,例如利用HPLC可以分离和离析降钙素药物-低聚物缀合物混合物,得到分散系数大于10,000的降钙素药物-低聚物缀合物、例如单-、二-或三-缀合物的混合物。所离析的特定缀合物的共轭程度(例如所离析的分子是单-、二-还是三-缀合物)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于质谱法。特定缀合物结构(例如低聚物位于鲑鱼降钙素单缀合物的Lys11、Lys18还是N-末端上)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于序列分析、肽绘图、选择性酶裂解和/或内肽酶裂解。
正如将为本领域技术人员所理解的,降钙素药物上一个或多个反应位点例如可以这样保护,使降钙素药物与适合的保护试剂反应,例如N-叔丁氧羰基(t-BOC)或N-(9-芴基甲氧羰基)(N-FMOC)。例如当降钙素药物是一种多肽,并且需要生成在该多肽N-末端上具有低聚物的不饱和缀合物(也就是其中不是所有的亲核残基都被共轭的缀合物)时,这种方法可能是优选的。在这类保护之后,可以使被保护的分散系数大于10,000的降钙素药物混合物与分散系数大于10,000的活化低聚物混合物反应,得到降钙素药物-低聚物缀合物混合物,具有与一个或多个亲核残基偶联的低聚物,并且具有与其他亲核残基偶联的保护部分。在共轭反应后,可以使降钙素药物-低聚物缀合物去保护,这将为本领域技术人员所理解。如果必要的话,然后可以如上所述分离降钙素药物-低聚物缀合物混合物,得到分散系数大于10,000的降钙素药物-低聚物缀合物混合物。或者,可以在去保护之前分离降钙素药物-低聚物缀合物混合物。
与常规混合物相比,根据本发明实施方式的分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有提高了的性质。例如,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地能够降低血清钙水平达至少5%。优选地,缀合物混合物能够降低血清钙水平达至少10、11、12、13或14%。更优选地,缀合物混合物能够降低血清钙水平达至少15、16、17、18或19%,最优选地,缀合物混合物能够降低血清钙水平达至少20%。
作为另一个实例,分别与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性。对胰凝乳蛋白酶或胰蛋白酶的抗性相当于利用下列实施例51所述操作,当供试分子在可用的酶中被消化时所剩余的百分比。优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约15%。最优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约20%。优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约20%。最优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约30%。
作为另一个实例,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有比没有与低聚物偶联的降钙素药物的生物功效更高的生物功效。特定化合物的生物功效相当于它的曲线下面积(AUC)值。优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约5%。更优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约10%。
作为另一个实例,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体内活性更大的体内活性,该多分散混合物具有与分散系数大于10,000的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMER CHEMISTRY394-402(2d.ed.,1991)所述。
作为另一个实例,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体外活性更大的体外活性,该多分散混合物具有与分散系数大于10,000的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,与多分散降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性,该多分散混合物具有与分散系数大于10,000的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的个体间差异更小的个体间差异,该多分散混合物具有与分散系数大于10,000的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。个体间差异可以借助各种方法测量,这将为本领域技术人员所理解。个体间差异优选地是如下计算的。测定每一个体的剂量响应曲线下面积(AUC)(也就是剂量响应曲线与基线值之间的面积)。将每一个体的AUC求和,再除以个体数,测定所有个体的平均AUC。然后测定每一个体的个别AUC与平均AUC之间差异的绝对值。然后将所得差异的绝对值求和,得到代表个体间差异的数值。数值越低,代表个体间差异越低,数值越高,代表个体间差异越高。
根据本发明实施方式的分散系数大于10,000的降钙素药物-低聚物缀合物混合物优选地具有两个或多个上述提高了的性质。更优选地,根据本发明实施方式的分散系数大于10,000的降钙素药物-低聚物缀合物混合物具有三个或多个上述提高了的性质。最优选地,根据本发明实施方式的分散系数大于10,000的降钙素药物-低聚物缀合物混合物具有四个或多个上述提高了的性质。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物包括与低聚物偶联的降钙素药物,并且具有相同的聚乙二醇亚单位数。
降钙素药物优选地是降钙素。更优选地,降钙素药物是鲑鱼降钙素。不过,可以理解的是降钙素药物可以选自本领域已知的各种降钙素药物,例如包括降钙素前体肽、降钙素、降钙素类似物、降钙素片段和降钙素片段类似物。降钙素前体肽包括但不限于katacalcin(PDN-21)(C-前降钙素)和N-proCT(氨基末端前降钙素裂解肽),人。降钙素类似物可以如上所述借助降钙素中一个或多个氨基酸的取代而得。降钙素片段包括但不限于降钙素1-7,人;和降钙素8-32,鲑鱼。降钙素片段类似物可以如上所述借助降钙素片段中一个或多个氨基酸的取代而得。
低聚物可以是包含聚乙二醇部分的各种低聚物,这将为本领域技术人员所理解。优选地,低聚物的聚乙二醇部分具有至少2、3或4个聚乙二醇亚单位。更优选地,聚乙二醇部分具有至少5或6个聚乙二醇亚单位,最优选地,聚乙二醇部分具有至少7个聚乙二醇亚单位。
低聚物可以包含一个或多个其他部分,这将为本领域技术人员所理解,包括但不限于另外的亲水性部分、亲脂性部分、间隔部分、连接部分和终端部分。低聚物中的各种部分是通过可水解的或不可水解的键彼此共价偶联的。
低聚物可以进一步包含一个或多个另外的亲水性部分(也就是除了聚乙二醇部分以外的部分),包括但不限于糖、聚亚烷基氧和聚胺/PEG共聚物。由于聚乙二醇是一种聚亚烷基氧,另外的亲水性部分可以是聚乙二醇部分。
如果通过醚键偶联,相邻的聚乙二醇部分将被视为相同的部分。例如,-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-O-C2H4-是具有六个聚乙二醇亚单位的单一聚乙二醇部分。如果该部分是低聚物中唯一的亲水性部分,那么该低聚物将不含有另外的亲水性部分。如果通过除醚键以外的键偶联,相邻的聚乙二醇部分将被视为不同的部分。例如,

是具有四个聚乙二醇亚单位的聚乙二醇部分和具有两个聚乙二醇亚单位的另外亲水性部分。优选地,根据本发明实施方式的低聚物包含聚乙二醇部分,没有另外的亲水性部分。
低聚物可以进一步包含一个或多个亲脂性部分,这将为本领域技术人员所理解。亲脂性部分优选地是饱和或不饱和的、直链或支链烷基部分或者饱和或不饱和的、直链或支链脂肪酸部分。当亲脂性部分是烷基部分时,它优选地是直链的、饱和或不饱和的烷基部分,具有1至28个碳原子。更优选地,烷基部分具有2至12个碳原子。当亲脂性部分是脂肪酸部分时,它优选地是天然脂肪酸部分,它是直链的、饱和或不饱和的,具有2至18个碳原子。更优选地,脂肪酸部分具有3至14个碳原子。最优选地,脂肪酸部分具有至少4、5或6个碳原子。
低聚物可以进一步包含一个或多个间隔部分,这将为本领域技术人员所理解。间隔基团例如可以用于分隔亲水性部分与亲脂性部分,分隔亲脂性部分或亲水性部分与降钙素药物,分隔第一亲水性或亲脂性部分与第二亲水性或亲脂性部分,或者分隔亲水性部分或亲脂性部分与连接部分。间隔部分优选地选自由糖、胆固醇和甘油部分组成的组。
低聚物可以进一步包含一个或多个连接部分,用于使低聚物与降钙素药物偶联,这将为本领域技术人员所理解。连接部分优选地选自由烷基和脂肪酸部分组成的组。
低聚物可以进一步在低聚物不与降钙素药物偶联的一个或多个末端包含一个或多个终端部分。终端部分优选地是烷基或烷氧基部分,更优选地是低级烷基或低级烷氧基部分。最优选地,终端部分是甲基或甲氧基。尽管终端部分优选地是烷基或烷氧基部分,不过可以理解的是终端部分可以是将为本领域技术人员所理解的各种部分,包括但不限于糖、胆固醇、醇和脂肪酸。
低聚物优选地是与降钙素药物共价偶联的。在一些实施方式中,利用可水解的键(例如酯或碳酸酯键)使降钙素药物与低聚物偶联。可水解的偶联可以提供充当前体药物的降钙素药物-低聚物缀合物。在某些情形中,例如其中降钙素药物-低聚物缀合物是无活性的(也就是说,缀合物缺乏通过降钙素药物的主要作用机理来影响机体的能力),可水解的偶联可以提供定时释放或控制释放的效果,随着一个或多个低聚物从它们各自的降钙素药物-低聚物缀合物上裂解下来以提供活性药物,在给定时间阶段内给以降钙素药物。在其他实施方式中,利用不可水解的键(例如氨基甲酸酯、酰胺或醚键)使降钙素药物与低聚物偶联。当需要使降钙素药物-低聚物缀合物在血流中长时间循环时,优选至少2小时,不可水解的键的使用可能是优选的。当低聚物与降钙素药物共价偶联时,低聚物进一步包含一个或多个键合部分,用于使低聚物与降钙素药物共价偶联,这将为本领域技术人员所理解。键合部分优选地选自由共价键、酯部分、碳酸酯部分、氨基甲酸酯部分、酰胺部分和仲胺部分组成的组。低聚物上一种以上的部分可以与降钙素药物共价偶联。
尽管低聚物优选地是与降钙素药物共价偶联的,不过可以理解的是,低聚物可以与降钙素药物非共价偶联,生成非共价共轭的降钙素药物-低聚物配合物。正如将为本领域技术人员所理解的,非共价偶联包括但不限于氢键合、离子键合、范德华键合和胶束或脂质体包封。按照本发明的实施方式,低聚物可以被适当地改造、改性和/或适当地官能化,以按所选择的方式赋予非共价共轭的能力(例如赋予氢键合能力),这将为本领域技术人员所理解。按照本发明的其他实施方式,低聚物可以用各种化合物衍生化,包括但不限于氨基酸、寡肽、肽、胆汁酸、胆汁酸衍生物、脂肪酸、脂肪酸衍生物、水杨酸、水杨酸衍生物、氨基水杨酸、和氨基水杨酸衍生物。所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价偶联(配位化合)。所得配合物优选地具有平衡的亲脂性和亲水性。按照本发明的其他实施方式,低聚物可以用胺和/或烷基胺衍生化。在适合的酸性条件下,所得低聚物可以与药物分子、药物产品和/或药物赋形剂非共价生成共轭配合物。从这类配位化合作用所得产物优选地具有平衡的亲脂性和亲水性。
一种以上的低聚物(即大量低聚物)可以与降钙素药物偶联。这些低聚物优选地是相同的。不过可以理解的是,这些低聚物可以是彼此不同,或者这些低聚物中的一些可以是相同的,另一些可以是不同的。当大量低聚物与降钙素药物偶联时,可以优选地利用可水解的键使一个或多个低聚物与降钙素药物偶联,利用不可水解的键使一个或多个低聚物与降钙素药物偶联。或者,所有使大量低聚物与降钙素药物偶联的键都是可水解的,但是具有不同程度的可水解性,以便例如一个或多个低聚物在体内被水解作用迅速从降钙素药物上除去,一个或多个低聚物在体内被水解作用缓慢从降钙素药物上除去。
低聚物可以与降钙素药物偶联在该降钙素药物的各种亲核残基上,包括但不限于亲核性羟基官能和/或氨基官能。当降钙素药物是多肽时,亲核性羟基官能例如可以见于丝氨酸和/或酪氨酸残基,亲核性氨基官能例如可以见于组氨酸和/或赖氨酸残基、和/或多肽的一个或多个N-末端。当低聚物与降钙素多肽的一个或多个N-末端偶联时,偶联作用优选地生成仲胺。当降钙素药物是鲑鱼降钙素时,例如,低聚物可以与鲑鱼降钙素的氨基官能度偶联,包括Lys11、Lys18和/或N-末端的氨基官能度。尽管一个或多个低聚物可以与鲑鱼降钙素偶联,不过当低聚物与Lys11和Lys18的氨基官能度偶联时,在二共轭的鲑鱼降钙素中观察到更高的生物功效,例如提高了的血清钙降低能力。
其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物可以借助各种方法合成。例如,由羧酸和聚乙二醇组成的其中混合物中每一缀合物具有相同聚乙二醇亚单位数的低聚物混合物是这样合成的,使其中混合物中每一缀合物具有相同聚乙二醇亚单位数的羧酸混合物与其中混合物中每一缀合物具有相同聚乙二醇亚单位数的聚乙二醇混合物在足以提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的低聚物混合物的条件下接触。然后活化其中混合物中每一缀合物具有相同聚乙二醇亚单位数的低聚物混合物,以便它们能够与降钙素药物反应,得到降钙素药物-低聚物缀合物。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的一种合成途径实施方式阐述在下列图3和实施例11-18中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图4和实施例19-24中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图5和实施例25-29中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图6和实施例30-31中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图7和实施例32-37中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图8和实施例38中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图9和实施例39中。用于提供其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物的另一种合成途径实施方式阐述在下列图10和实施例40中。
使其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物与降钙素药物混合物在足以提供降钙素药物-低聚物缀合物混合物的条件下反应。优选的合成描述在下列实施例41中。正如将为本领域技术人员所理解的,可以控制反应条件(例如选择摩尔比、溶剂混合物和/或pH),以便从其中混合物中每一缀合物具有相同聚乙二醇亚单位数的活化低聚物混合物与降钙素药物混合物的反应所得降钙素药物-低聚物缀合物混合物是一种其中混合物中每一缀合物具有相同聚乙二醇亚单位数的混合物。例如,通过维持反应溶液的pH低于赖氨酸pKa,可以抑制赖氨酸氨基官能度上的共轭作用。或者,例如利用HPLC可以分离和离析降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物、例如单-、二-或三-缀合物的混合物,其中混合物中每一缀合物具有相同的聚乙二醇亚单位数。所离析的特定缀合物的共轭程度(例如所离析的分子是单-、二-还是三-缀合物)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于质谱法。特定缀合物结构(例如低聚物位于鲑鱼降钙素单缀合物的Lys11、Lys18还是N-末端上)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于序列分析、肽绘图、选择性酶裂解和/或内肽酶裂解。
正如将为本领域技术人员所理解的,降钙素药物上一个或多个反应位点例如可以这样保护,使降钙素药物与适合的保护试剂反应,例如N-叔丁氧羰基(t-BOC)或N-(9-芴基甲氧羰基)(N-FMOC)。例如当降钙素药物是一种多肽,并且需要生成在该多肽N-末端上具有低聚物的不饱和缀合物(也就是其中不是所有的亲核残基都被共轭的缀合物)时,这种方法可能是优选的。在这类保护之后,可以使被保护的降钙素药物混合物与活化低聚物混合物反应,其中混合物中每一缀合物具有相同的聚乙二醇亚单位数,得到降钙素药物-低聚物缀合物混合物,具有与一个或多个亲核残基偶联的低聚物,并且具有与其他亲核残基偶联的保护部分。在共轭反应后,可以使降钙素药物-低聚物缀合物去保护,这将为本领域技术人员所理解。如果必要的话,然后可以如上所述分离降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物混合物,其中混合物中每一缀合物具有相同的聚乙二醇亚单位数。或者,可以在去保护之前分离降钙素药物-低聚物缀合物混合物。
与常规混合物相比,根据本发明实施方式的其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有提高了的性质。例如,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地能够降低血清钙水平达至少5%。优选地,缀合物混合物能够降低血清钙水平达至少10、11、12、13或14%。更优选地,缀合物混合物能够降低血清钙水平达至少15、16、17、18或19%,最优选地,缀合物混合物能够降低血清钙水平达至少20%。
作为另一个实例,分别与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性。对胰凝乳蛋白酶或胰蛋白酶的抗性相当于利用下列实施例51所述操作,当供试分子在可用的酶中被消化时所剩余的百分比。优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约15%。最优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约20%。优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约20%。最优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约30%。
作为另一个实例,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有比没有与低聚物偶联的降钙素药物的生物功效更高的生物功效。特定化合物的生物功效相当于它的曲线下面积(AUC)值。优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约5%。更优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约10%。
作为另一个实例,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体内活性更大的体内活性,该多分散混合物具有与其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMER CHEMISTRY 394-402(2d.ed.,1991)所述。
作为另一个实例,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体外活性更大的体外活性,该多分散混合物具有与其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,与多分散降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性,该多分散混合物具有与其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的个体间差异更小的个体间差异,该多分散混合物具有与其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。个体间差异可以借助各种方法测量,这将为本领域技术人员所理解。个体间差异优选地是如下计算的。测定每一个体的剂量响应曲线下面积(AUC)(也就是剂量响应曲线与基线值之间的面积)。将每一个体的AUC求和,再除以个体数,测定所有个体的平均AUC。然后测定每一个体的个别AUC与平均AUC之间差异的绝对值。然后将所得差异的绝对值求和,得到代表个体间差异的数值。数值越低,代表个体间差异越低,数值越高,代表个体间差异越高。
根据本发明实施方式的其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物优选地具有两个或多个上述提高了的性质。更优选地,根据本发明实施方式的其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物具有三个或多个上述提高了的性质。最优选地,根据本发明实施方式的其中混合物中每一缀合物具有相同聚乙二醇亚单位数的降钙素药物-低聚物缀合物混合物具有四个或多个上述提高了的性质。
按照本发明的其他实施方式,提供了缀合物的混合物,其中每一缀合物具有相同的分子量,并且具有式A结构:
降钙素药物-[-B-Lj-Gk-R-G′m-R′-G″n-T]p (A)
其中:
B是键合部分;
L是连接基团;
G、G′和G″是各自选择的间隔基团;
R是亲脂性基团,且R′是聚亚烷基二醇基团,或者R′是亲脂性基团,且R是聚亚烷基氧基团;
T是封端基团;
j、k、m和n各自是0或1;
p是1至降钙素药物上亲核残基数的整数。
降钙素药物优选地是降钙素。更优选地,降钙素药物是鲑鱼降钙素。不过,可以理解的是降钙素药物可以选自本领域已知的各种降钙素药物,例如包括降钙素前体肽、降钙素、降钙素类似物、降钙素片段和降钙素片段类似物。降钙素前体肽包括但不限于katacalcin(PDN-21)(C-前降钙素)和N-proCT(氨基末端前降钙素裂解肽),人。降钙素类似物可以如上所述借助降钙素中一个或多个氨基酸的取代而得。降钙素片段包括但不限于降钙素1-7,人;和降钙素8-32,鲑鱼。降钙素片段类似物可以如上所述借助降钙素片段中一个或多个氨基酸的取代而得。
按照本发明的这些实施方式,低聚物的聚亚烷基二醇部分优选地具有至少2、3或4个聚亚烷基二醇亚单位。更优选地,聚亚烷基二醇具有至少5或6个聚亚烷基二醇亚单位,最优选地,聚亚烷基二醇具有至少7个聚亚烷基二醇亚单位。聚亚烷基二醇部分优选地是低级聚亚烷基二醇部分,例如聚乙二醇部分、聚丙二醇部分或聚丁二醇部分。更优选地,聚亚烷基二醇部分是聚乙二醇部分或聚丙二醇部分。最优选地,聚亚烷基二醇部分是聚乙二醇部分。当聚亚烷基二醇部分是聚丙二醇部分时,该部分优选地具有均匀的(也就是非随机的)结构。具有均匀结构的示范性聚丙二醇部分如下:

这种均匀的聚丙二醇结构可以被描述为在聚丙二醇链中仅有一个甲基取代的碳原子与每一氧原子相邻。这类均匀的聚丙二醇部分可以表现亲脂性和亲水性特征,因而可用于提供两亲性降钙素药物-低聚物缀合物,无需使用亲脂性聚合物部分。此外,使聚丙二醇部分的仲醇部分与降钙素药物偶联,可以为降钙素药物(例如鲑鱼降钙素)提供提高了的对酶降解作用的抗性,例如见于肠中的胰蛋白酶和胰凝乳蛋白酶。
根据本发明实施方式的均匀聚丙二醇优选地是如图11至13所述合成的,现在将描述之。正如图11所述,使1,2-丙二醇53与伯醇保护试剂反应,得到仲醇延长单体54。伯醇保护试剂可以是将为本领域技术人员所理解的各种伯醇保护试剂,包括但不限于甲硅烷基氯化合物,例如叔丁基二苯基甲硅烷基氯和叔丁基二甲基甲硅烷基氯,和酯化试剂,例如Ac2O。优选地,伯醇保护试剂是与仲醇基本无反应性的伯醇保护试剂,例如叔丁基二苯基甲硅烷基氯或叔丁基二甲基甲硅烷基氯。可以使仲醇延长单体(54)与甲磺酰氯(MeSO2Cl)反应,得到伯醇延长单体甲磺酸酯55。
或者,可以使仲醇延长单体54与仲醇保护试剂反应,得到化合物56。仲醇保护试剂可以是将为本领域技术人员所理解的各种仲醇保护试剂,包括但不限于苄基氯。可以使化合物56与B1去保护试剂反应,以除去保护部分B1,得到伯醇延长单体57。B1去保护试剂可以选自将为本领域技术人员所理解的各种去保护试剂。当通过成酯来保护伯醇时,B1去保护试剂是一种去酯化试剂,例如一种碱(例如碳酸钾)。当用一种甲硅烷基氯保护伯醇时,B1去保护试剂优选地是氟化四丁铵(TBAF)。可以使伯醇延长单体57与甲烷磺酰氯反应,得到仲醇延长单体甲磺酸酯58。
伯醇延长单体54和仲醇延长单体57可以如下被封端。可以使仲醇延长单体54与封端试剂反应,得到化合物59。封端试剂可以是将为本领域技术人员所理解的各种封端试剂,包括但不限于烷基卤,例如甲基氯。可以使化合物59与B1去保护试剂反应,得到伯醇封端单体60。可以使伯醇封端单体60与甲磺酰氯反应,得到仲醇封端单体甲磺酸酯61。可以使伯醇延长单体57与封端试剂反应,得到化合物62。封端试剂可以是如上所述的各种封端试剂。可以使化合物62与B2去保护试剂反应,以除去保护部分B2,得到仲醇封端单体63。B2去保护试剂可以是将为本领域技术人员所理解的各种去保护试剂,包括但不限于在钯/活性碳催化剂的存在下的H2。可以使仲醇封端单体与甲磺酰氯反应,得到伯醇封端单体甲磺酸酯64。尽管图11所述实施方式显示了封端单体的合成,不过可以理解的是可以进行相似的反应得到封端聚合物。
一般而言,链延长可以这样进行,使伯醇延长单体或聚合物、例如伯醇延长单体57与伯醇延长单体或聚合物甲磺酸酯、例如伯醇延长单体甲磺酸酯55反应,得到各种均匀的聚丙烯链,或者使仲醇延长单体或聚合物、例如仲醇延长单体54与仲醇延长单体或聚合物甲磺酸酯、例如仲醇延长单体甲磺酸酯58反应。
例如,在图13中,使伯醇延长单体甲磺酸酯55与伯醇延长单体57反应,得到二聚化合物65。或者,可以使仲醇延长单体甲磺酸酯58与仲醇延长单体54反应,得到二聚化合物65。二聚化合物65上的B1保护部分可以用如上所述的B1去保护试剂除去,得到伯醇延长二聚物66。可以使伯醇延长二聚物66与甲磺酰氯反应,得到仲醇延长二聚物甲磺酸酯67。或者,二聚化合物65上的B2保护部分可以用如上所述的B2去保护试剂除去,得到仲醇延长二聚物69。可以使仲醇延长二聚物69与甲磺酰氯反应,得到伯醇延长二聚物甲磺酸酯70。
正如将为本领域技术人员所理解的,可以重复链延长过程,以达到各种其他链长。例如,如图13所述,可以使伯醇延长二聚物66与伯醇延长二聚物甲磺酸酯70反应,得到四聚化合物72。进一步如图13所述,一般的链延长反应流程牵涉使伯醇延长单体或聚合物73与伯醇延长单体或聚合物甲磺酸酯74反应,得到均匀的聚丙烯聚合物75。m和n的值可以各自为0至1000或以上。优选地,m和n各自为0至50。尽管图13所述实施方式显示了伯醇延长单体和/或聚合物是与伯醇延长单体和/或聚合物甲磺酸酯反应的,不过可以理解的是可以使用仲醇延长单体和/或聚合物和仲醇延长单体和/或聚合物甲磺酸酯来进行相似的反应。
可以使伯醇延长单体或聚合物的末端或伯醇延长单体或聚合物甲磺酸酯的末端分别与伯醇封端单体或聚合物甲磺酸酯或伯醇封端单体或聚合物反应,得到被封端的均匀的聚丙烯链。例如,如图12所述,使伯醇延长二聚物甲磺酸酯70与伯醇封端单体60反应,得到被封端/被保护的伯醇延长三聚物71。正如将为本领域技术人员所理解的,可以除去B1保护部分,可以使所得封端伯醇延长三聚物与伯醇延长单体或聚合物甲磺酸酯反应,以延长被封端的三聚物71的链。
可以使仲醇延长单体或聚合物的末端或仲醇延长单体或聚合物甲磺酸酯的末端分别与仲醇封端单体或聚合物甲磺酸酯或仲醇封端单体或聚合物反应,得到被封端的均匀的聚丙烯链。例如,如图12所述,使仲醇延长二聚物甲磺酸酯67与仲醇封端单体63反应,得到被封端/被保护的伯醇延长三聚物68。可以如上所述除去B2保护部分,可以使所得被封端的仲醇延长三聚物与仲醇延长链节甲磺酸酯反应,以延长被封端的三聚物68的链。尽管图12所述实施方式显示了二聚物与封端单体反应得到三聚物,不过可以理解的是可以在均匀聚丙二醇部分合成中的任何点上进行封端过程,或者可以提供未被封端的均匀聚丙二醇部分。尽管图12所述实施方式显示了聚丁烯低聚物借助封端单体合成而封端,不过可以理解的是可以如上图11所述使用封端试剂将本发明的聚丁烯低聚物直接封端(也就是无需加入封端单体)。
可以使根据本发明实施方式的聚丙二醇部分与降钙素药物、亲脂性部分(例如羧酸)和/或各种其他部分偶联,借助将为本领域技术人员所理解的各种方法,包括但不限于本文关于聚乙二醇部分所述那些。
按照本发明的这些实施方式,亲脂性部分是将为本领域技术人员所理解的亲脂性部分。亲脂性部分优选地是饱和或不饱和的直链或支链烷基部分或者饱和或不饱和的直链或支链脂肪酸部分。当亲脂性部分是烷基部分时,它优选地是直链的、饱和或不饱和烷基部分,具有1至28个碳原子。更优选地,烷基部分具有2至12个碳原子。当亲脂性部分是脂肪酸部分时,它优选地是天然脂肪酸部分,它是直链的、饱和或不饱和的,具有2至18个碳原子。更优选地,脂肪酸部分具有3至14个碳原子。最优选地,脂肪酸部分具有至少4、5或6个碳原子。
按照本发明的这些实施方式,间隔部分G、G′和G″是将为本领域技术人员所理解的间隔部分。间隔部分优选地选自由糖、胆固醇和甘油部分组成的组。优选地,这些实施方式的低聚物不包括间隔部分(也就是说,k、m和n优选地是0)。
按照本发明的这些实施方式,连接部分L可以用于使低聚物与药物偶联,这将为本领域技术人员所理解。连接部分优选地选自由烷基和脂肪酸部分组成的组。
按照本发明的这些实施方式,终端部分优选地是烷基或烷氧基部分,更优选地是低级烷基或低级烷氧基部分。最优选地,终端部分是甲基或甲氧基。尽管终端部分优选地是烷基或烷氧基部分,不过终端部分可以是将为本领域技术人员所理解的各种部分,包括但不限于糖、胆固醇、醇和脂肪酸。
按照本发明的这些实施方式,使由式A结构的括号部分代表的低聚物与降钙素药物共价偶联。在一些实施方式中,利用可水解的键(例如酯或碳酸酯键)使降钙素药物与低聚物偶联。可水解的偶联可以提供充当前体药物的降钙素药物-低聚物缀合物。在某些情形中,例如其中降钙素药物-低聚物缀合物是无活性的(也就是说,缀合物缺乏通过降钙素药物的主要作用机理来影响机体的能力),可水解的偶联可以提供定时释放或控制释放的效果,随着一个或多个低聚物从它们各自的降钙素药物-低聚物缀合物上裂解下来以提供活性药物,在给定时间阶段内给以降钙素药物。在其他实施方式中,利用不可水解的键(例如氨基甲酸酯、酰胺或醚键)使降钙素药物与低聚物偶联。当需要使降钙素药物-低聚物缀合物在血流中长时间循环时,优选至少2小时,不可水解的键的使用可能是优选的。当低聚物与降钙素药物共价偶联时,低聚物进一步包含一个或多个键合部分,用于使低聚物与降钙素药物共价偶联,这将为本领域技术人员所理解。键合部分优选地选自由共价键、酯部分、碳酸酯部分、氨基甲酸酯部分、酰胺部分和仲胺部分组成的组。低聚物上一种以上的部分可以与降钙素药物共价偶联。
变量p是1至降钙素药物上亲核残基数的整数。当p大于1时,一个以上低聚物(即大量低聚物)与药物偶联。按照本发明的这些实施方式,这些低聚物都是相同的。当大量低聚物与降钙素药物偶联时,可以优选的是利用可水解的键使一个或多个低聚物与降钙素药物偶联,利用不可水解的键使一个或多个低聚物与降钙素药物偶联。或者,所有使大量低聚物与降钙素药物偶联的键都是可水解的,但是具有不同程度的可水解性,以便例如一个或多个低聚物在体内被水解作用迅速从降钙素药物上除去,一个或多个低聚物在体内被水解作用缓慢从降钙素药物上除去。
低聚物可以与降钙素药物偶联在该降钙素药物的各种亲核残基上,包括但不限于亲核性羟基官能和/或氨基官能。当降钙素药物是多肽时,亲核性羟基官能例如可以见于丝氨酸和/或酪氨酸残基,亲核性氨基官能例如可以见于组氨酸和/或赖氨酸残基、和/或多肽的一个或多个N-末端。当低聚物与降钙素多肽的一个或多个N-末端偶联时,偶联作用优选地生成仲胺。当降钙素药物是鲑鱼降钙素时,例如,低聚物可以与鲑鱼降钙素的氨基官能度偶联,包括Lys11、Lys18和/或N-末端的氨基官能度。尽管一个或多个低聚物可以与鲑鱼降钙素偶联,不过当低聚物与Lys11和Lys18的氨基官能度偶联时,在二共轭的鲑鱼降钙素中观察到更高的生物功效,例如提高了的血清钙降低能力。
其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物可以借助各种方法合成。例如,由羧酸和聚乙二醇组成的低聚物混合物是这样合成的,使羧酸混合物与聚乙二醇混合物在足以提供低聚物混合物的条件下接触。然后活化低聚物混合物,以便它们能够与降钙素药物反应,得到降钙素药物-低聚物缀合物。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的一种合成途径实施方式阐述在下列图3和实施例11-18中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图4和实施例19-24中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图5和实施例25-29中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图6和实施例30-31中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图7和实施例32-37中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图8和实施例38中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图9和实施例39中。用于提供其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物的另一种合成途径实施方式阐述在下列图10和实施例40中。
可以使其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物与其中混合物中每一药物具有相同分子量的降钙素药物混合物在足以提供降钙素药物-低聚物缀合物混合物的条件下反应。优选的合成描述在下列实施例41中。正如将为本领域技术人员所理解的,可以控制反应条件(例如选择摩尔比、溶剂混合物和/或pH),以便从其中每一低聚物具有相同分子量且具有式A低聚物结构的活化低聚物混合物与降钙素药物混合物的反应所得降钙素药物-低聚物缀合物混合物是缀合物的混合物,其中每一缀合物具有相同分子量且具有式A结构。例如,通过维持反应溶液的pH低于赖氨酸pKa,可以抑制赖氨酸氨基官能度上的共轭作用。或者,例如利用HPLC可以分离和离析降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物、例如单-、二-或三-缀合物的混合物,其中混合物中每一缀合物具有相同分子量且具有式A结构。所离析的特定缀合物的共轭程度(例如所离析的分子是单-、二-还是三-缀合物)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于质谱法。特定缀合物结构(例如低聚物位于鲑鱼降钙素单缀合物的Lys11、Lys18还是N-末端上)可以利用各种将为本领域技术人员所理解的技术加以测定和/或验证,包括但不限于序列分析、肽绘图、选择性酶裂解和/或内肽酶裂解。
正如将为本领域技术人员所理解的,降钙素药物上一个或多个反应位点例如可以这样保护,使降钙素药物与适合的保护试剂反应,例如N-叔丁氧羰基(t-BOC)或N-(9-芴基甲氧羰基)(N-FMOC)。例如当降钙素药物是一种多肽,并且需要生成在该多肽N-末端上具有低聚物的不饱和缀合物(也就是其中不是所有的亲核残基都被共轭的缀合物)时,这种方法可能是优选的。在这类保护之后,可以使被保护的降钙素药物混合物与活化低聚物混合物反应,其中混合物中每一低聚物具有相同分子量且具有式A低聚物结构,得到降钙素药物-低聚物缀合物混合物,具有与一个或多个亲核残基偶联的低聚物,并且具有与其他亲核残基偶联的保护部分。在共轭反应后,可以使降钙素药物-低聚物缀合物去保护,这将为本领域技术人员所理解。如果必要的话,然后可以如上所述分离降钙素药物-低聚物缀合物混合物,得到降钙素药物-低聚物缀合物混合物,其中混合物中每一缀合物具有相同分子量且具有式A结构。或者,可以在去保护之前分离降钙素药物-低聚物缀合物混合物。
与常规混合物相比,根据本发明实施方式的其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有提高了的性质。例如,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地能够降低血清钙水平达至少5%。优选地,缀合物混合物能够降低血清钙水平达至少10、11、12、13或14%。更优选地,缀合物混合物能够降低血清钙水平达至少15、16、17、18或19%,最优选地,缀合物混合物能够降低血清钙水平达至少20%。
作为另一个实例,分别与没有与低聚物偶联的降钙素药物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性。对胰凝乳蛋白酶或胰蛋白酶的抗性相当于利用下列实施例51所述操作,当供试分子在可用的酶中被消化时所剩余的百分比。优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约15%。最优选地,降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰凝乳蛋白酶降解作用的抗性约20%。优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约10%。更优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约20%。最优选地,降钙素药物-低聚物缀合物混合物对胰蛋白酶降解作用的抗性大于没有与低聚物共轭的降钙素药物混合物对胰蛋白酶降解作用的抗性约30%。
作为另一个实例,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有比没有与低聚物偶联的降钙素药物的生物功效更高的生物功效。特定化合物的生物功效相当于它的曲线下面积(AUC)值。优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约5%。更优选地,混合物的生物功效大于没有与低聚物偶联的降钙素药物的生物功效约10%。
作为另一个实例,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体内活性更大的体内活性,该多分散混合物具有与其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法,例如凝胶渗透色谱法,例如H.R.Allcock&F.W.Lampe,CONTEMPORARY POLYMER CHEMISTRY 394-402(2d.ed.,1991)所述。
作为另一个实例,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的体外活性更大的体外活性,该多分散混合物具有与其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,与多分散降钙素药物-低聚物缀合物混合物对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性相比,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有增加了的对胰凝乳蛋白酶和/或胰蛋白酶降解作用的抗性,该多分散混合物具有与其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。
作为另一个实例,其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有比多分散降钙素药物-低聚物缀合物混合物的个体间差异更小的个体间差异,该多分散混合物具有与其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物相同的数均分子量。正如将为本领域技术人员所理解的,混合物的数均分子量可以借助各种方法测量,包括但不限于尺寸排阻色谱法。个体间差异可以借助各种方法测量,这将为本领域技术人员所理解。个体间差异优选地是如下计算的。测定每一个体的剂量响应曲线下面积(AUC)(也就是剂量响应曲线与基线值之间的面积)。将每一个体的AUC求和,再除以个体数,测定所有个体的平均AUC。然后测定每一个体的个别AUC与平均AUC之间差异的绝对值。然后将所得差异的绝对值求和,得到代表个体间差异的数值。数值越低,代表个体间差异越低,数值越高,代表个体间差异越高。
根据本发明实施方式的其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物优选地具有两个或多个上述提高了的性质。更优选地,根据本发明实施方式的其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物具有三个或多个上述提高了的性质。最优选地,根据本发明实施方式的其中混合物中每一缀合物具有相同分子量且具有式A结构的降钙素药物-低聚物缀合物混合物具有四个或多个上述提高了的性质。
还提供了根据本发明实施方式的包含缀合物混合物的药物组合物。可以按照已知技术将上述降钙素药物-低聚物缀合物混合物配制成在药物载体中给药的形式。例如参见Remington,The Science AndPractice of Pharmacy(9th ed.1995)。在根据本发明实施方式的药物组合物的制备中,一般将降钙素药物-低聚物缀合物混合物尤其与药学上可接受的载体混合。当然,载体在与药物组合物中任何其他成分可相容的意义上必须是可接受的,并且不应有害于患者。载体可以是固体或液体,或者二者皆是,并且优选地与降钙素药物-低聚物缀合物混合物配制成单位剂量制剂,例如片剂,可以含有约0.01或0.5%至约95%或99%重量的降钙素药物-低聚物缀合物混合物。药物组合物可以借助任何熟知的药学技术加以制备,包括但不限于混合各组分,可选地包括一种或多种辅助成分。
根据本发明实施方式的药物组合物包括适合于口服、直肠、局部、吸入(例如经由气雾剂)、颊(例如舌下)、阴道、肠胃外(例如皮下、肌内、真皮内、关节内、胸膜内、腹膜内、脑内、动脉内或静脉内)、局部(也就是皮肤和粘膜表面,包括气道表面)和透皮给药的那些,不过最适合的途径在任何给定情况下将取决于所治疗病症的性质与严重性和所用特定降钙素药物-低聚物缀合物混合物的性质。
适合于口服给药的药物组合物可以以离散单元的形式存在,例如胶囊剂、扁囊剂、锭剂或片剂,各自含有预定量的降钙素药物-低聚物缀合物混合物;可以为粉剂或颗粒剂;可以为在水性或非水性液体中的溶液剂或混悬剂;或者可以为水包油或油包水乳剂。这类制剂可以借助任何适合的药学方法制备,包括将降钙素药物-低聚物缀合物混合物与适合载体(可以含有一种或多种辅助成分,如上所述)缔合的步骤。一般而言,根据本发明实施方式的药物组合物是这样制备的,将降钙素药物-低聚物缀合物混合物与液体或微细粉碎的固体载体或二者皆是均匀紧密地混合,然后如果必要的话,使所得混合物成型。例如,片剂可以这样制备,压制或模制含有降钙素药物-低聚物缀合物混合物和可选的一种或多种辅助成分的粉末或颗粒。压制片可以这样制备,在适合的机器中压制自由流动形式的混合物,例如粉末或颗粒,可选地混有粘合剂、润滑剂、惰性稀释剂和/或表面活性剂/分散剂。模制片可以这样制备,在适合的机器中模制用惰性液体粘合剂润湿的粉状化合物。
适于颊(舌下)给药的药物组合物包括锭剂,在矫味的基质中包含降钙素药物-低聚物缀合物混合物,基质通常为蔗糖和阿拉伯胶或黄蓍胶;和软锭剂,在惰性基质中包含降钙素药物-低聚物缀合物混合物,基质例如明胶和甘油或蔗糖和阿拉伯胶。
适合于肠胃外给药的根据本发明实施方式的药物组合物包含降钙素药物-低聚物缀合物混合物的无菌水性与非水性注射溶液,该制剂优选地与打算接受者的血液是等渗的。这些制剂可以含有抗氧化剂、缓冲剂、制菌剂和赋予组合物与打算接受者的血液以等渗性的溶质。水性与非水性的无菌混悬液可以包括悬浮剂和增稠剂。组合物可以存在于单位剂量或多剂量容器中,例如密封的安瓿和小瓶,并且可以贮存在冷冻-干燥(冻干)条件下,仅需在使用之前立即加入无菌的液体载体,例如盐水或注射用水。临时的注射溶液和混悬液可以从前述类型的无菌粉剂、颗粒剂和片剂制备。例如,可以提供位于密封容器中的可注射、稳定无菌组合物的单位剂量形式,其中包含降钙素药物-低聚物缀合物混合物。可以提供降钙素药物-低聚物缀合物混合物的冻干产物形式,它能够与适合的药学上可接受的载体再生为适合向个体注射的液体组合物。单位剂量形式一般包含约10mg至约10克降钙素药物-低聚物缀合物混合物。当降钙素药物-低聚物缀合物混合物基本上是水不溶性的时,可以采用生理学上可接受的足量乳化剂,使降钙素药物-低聚物缀合物混合物在水性载体中乳化。一种这类有用的乳化剂是磷脂酰胆碱。
适合于直肠给药的药物组合物优选地是以单位剂量栓剂的形式存在的。它们可以这样制备,混合降钙素药物-低聚物缀合物混合物与一种或多种常规固体载体,例如椰子油,然后使所得混合物成型。
适合于皮肤局部用药的药物组合物优选地采取软膏剂、霜剂、洗剂、糊剂、凝胶剂、喷雾剂、气雾剂或油剂的形式。可以使用的载体包括凡士林、羊毛脂、聚乙二醇、醇、透皮增强剂和两种或多种的组合。
适合于透皮给药的药物组合物可以以离散贴剂的形式存在,适于与接受者的表皮保持长时间的紧密接触。适合于透皮给药的组合物还可以借助离子电渗疗法来释放(例如参见Pharmaceutical Research3(6):318(1986)),一般采取降钙素药物-低聚物缀合物混合物的可选被缓冲的水溶液的形式。适合的制剂包含枸橼酸盐或bis\tris缓冲剂(pH 6)或乙醇/水,含有0.1至0.2M活性成分。
还提供了治疗需要这类治疗的个体骨障碍的方法,该方法给以有效量的这类药物组合物。骨障碍优选地是以过量破骨性骨吸收和/或血钙过多性血清效应为特征的。
其应用属于本发明范围的任何降钙素药物-低聚物缀合物混合物的有效量将因混合物和患者而异,将取决于一些因素,例如患者的年龄与病症和释放的途径。这类剂量可以按照本领域技术人员已知的常规药理学操作加以确定。作为一般性建议,约0.1至约50mg/kg的剂量将具有治疗功效,所有重量都是基于降钙素药物-低聚物缀合物混合物的重量来计算的。在较高剂量下的毒性考虑可能限制静脉内剂量至较低的水平,例如最高约10mg/kg,所有重量都是基于降钙素药物-低聚物缀合物混合物的重量来计算的。约10mg/kg至约50mg/kg的剂量可以用于口服给药。一般而言,约0.5mg/kg至5mg/kg的剂量可以用于肌内注射。给药的频率通常为每天一、二或三次,或者可以根据控制病症的需要。或者,药物-低聚物缀合物可以通过连续输注给药。治疗的持续时间取决于所治疗的骨障碍类型,可以长达患者的一生。
还提供了合成根据本发明实施方式的缀合物混合物的方法。尽管下列合成途径实施方式涉及单分散混合物的合成,不过可以利用相似的合成途径来合成根据本发明实施方式的其他降钙素药物-低聚物缀合物混合物。
如反应1所述得到包含聚乙二醇部分的基本单分散聚合物混合物:
R1(OC2H4)nO-X++R2(OC2H4)mOMs→R2(OC2H4)m+nOR1 1
(I) (II) (III)
R1是H或亲脂性部分。R1优选地是H、烷基、芳基烷基、芳族部分、脂肪酸部分、脂肪酸部分的酯、胆固醇基或金刚烷基。R1更优选地是H、低级烷基或芳族部分。R1最优选地是H、甲基或苄基。
式I中,n是1至25。n优选地是1至6。
X+是阳离子。X+优选地是能够使PEG上羟基部分电离的化合物、例如强碱中的任何阳离子。阳离子的实例包括但不限于钠离子、钾离子、锂离子、铯离子和铊离子。
R2是H或亲脂性部分。R2优选地是直链或支链烷基、芳基烷基、芳族部分、脂肪酸部分或脂肪酸部分的酯。R2更优选地是低级烷基、苄基、具有1至24个碳原子的脂肪酸部分或具有1至24个碳原子的脂肪酸部分的酯。R2最优选地是甲基、具有1至18个碳原子的脂肪酸部分或具有1至18个碳原子的脂肪酸部分的酯。
式II中,m是1至25。m优选地是1至6。
Ms是甲磺酸酯部分(即CH3S(O2)-)。
如反应1所述,使具有式I结构的化合物的混合物与具有式II结构的化合物的混合物反应,得到包含聚乙二醇部分并具有式III结构的聚合物的混合物。具有式I结构的化合物的混合物是基本单分散混合物。优选地,式I化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量,更优选地,式I化合物混合物是单分散混合物。式II化合物的混合物是基本单分散混合物。优选地,式II化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量,更优选地,式II化合物混合物是单分散混合物。式III化合物的混合物是基本单分散混合物。优选地,式III化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量。更优选地,式III化合的混合物是单分散混合物。
反应1优选地是在约0℃与约40℃之间进行的,更优选地是在约15℃与约35℃之间进行的,最优选地是在室温(大约25℃)下进行的。
反应1可以进行将为本领域技术人员所理解的各种时间阶段。反应1优选地进行约0.25、0.5或0.75小时至约2、4或8小时。
反应1优选地是在质子惰性溶剂中进行的,例如但不限于N,N-二甲基乙酰胺(DMA)、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、六甲基磷酸三酰胺、四氢呋喃(THF)、二噁烷、二乙醚、甲基叔丁醚(MTBE)、甲苯、苯、己烷、戊烷、N-甲基吡咯烷酮、四氢萘、十氢萘、1,2-二氯苯、1,3-二甲基-2-咪唑烷酮或它们的混合物。更优选地,溶剂是DMF、DMA或甲苯。
式I化合物与式II化合物的摩尔比优选地大于约1∶1。摩尔比更优选为至少约2∶1。通过提供过量的式I化合物,可以确保基本上所有的式II化合物都进行了反应,这有助于式III化合物的回收,正如下文所讨论的。
式I化合物优选地是如反应2所述制备的:
能够使式IV
R1(OC2H4)nOH+PEG部分上羟基部分→R1(OC2H4)nO-X+ 2
电离的化合物
(IV) (I)
R1和X+是如上所述的,式IV化合物混合物是基本单分散的;优选地,式IV化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量;更优选地,式IV化合物混合物是单分散混合物。
各种能够使式IV化合物PEG部分上羟基部分电离的化合物将为本领域技术人员所理解。能够使羟基部分电离的化合物优选地是强碱。更优选地,能够使羟基部分电离的化合物选自由氢化钠、氢化钾、叔丁醇钠、叔丁醇钾、丁基锂(BuLi)和二异丙氨基化锂组成的组。能够使羟基部分电离的化合物更优选地是氢化钠。
能够使式IV化合物PEG部分上羟基电离的化合物与式IV化合物的摩尔比优选为至少约1∶1,更优选为至少约2∶1。通过提供过量的能够使羟基部分电离的化合物,确保了基本上所有的式IV化合物都进行了反应,得到式I化合物。因此,可以避免了分离困难,这出现于式IV化合物和式I化合物都存在于反应产物混合物中。
反应2优选地是在约0℃与约40℃之间进行的,更优选地是在约0℃与约35℃之间进行的,最优选地是在约0℃与室温(大约25℃)之间进行的。
反应2可以进行将为本领域技术人员所理解的各种时间阶段。反应1优选地进行约0.25、0.5或0.75小时至约2、4或8小时。
反应2优选地是在质子惰性溶剂中进行的,例如但不限于N,N-二甲基乙酰胺(DMA)、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、六甲基磷酸三酰胺、四氢呋喃(THF)、二噁烷、二乙醚、甲基叔丁醚(MTBE)、甲苯、苯、己烷、戊烷、N-甲基吡咯烷酮、二氯甲烷、氯仿、四氢萘、十氢萘、1,2-二氯苯、1,3-二甲基-2-咪唑烷酮或它们的混合物。更优选地,溶剂是DMF、二氯甲烷或甲苯。
式II化合物优选地是如反应3所述制备的:

R2和Ms是如上所述的,式V化合物是以式V化合物的基本单分散混合物的形式存在的;式V化合物混合物中优选至少约96、97、98或99%的化合物具有相同的分子量;更优选地,式V化合物混合物是单分散混合物。
Q是卤化物,优选氯化物或氟化物。
CH3S(O2)Q是甲磺酰卤。甲磺酰卤优选地是甲磺酰氯或甲磺酰氟。更优选地,甲磺酰卤是甲磺酰氯。
甲磺酰卤与式V化合物的摩尔比优选地大于约1∶1,更优选为至少约2∶1。通过提供过量的甲磺酰卤,确保了基本上所有的式V化合物都进行了反应,得到式II化合物。因此,可以避免了分离困难,这出现于式V化合物和式II化合物都存在于反应产物混合物中。
反应3优选地是在约-10℃与约40℃之间进行的,更优选地是在约0℃与约35℃之间进行的,最优选地是在约0℃与室温(大约25℃)之间进行的。
反应3可以进行将为本领域技术人员所理解的各种时间阶段。反应1优选地进行约0.25、0.5或0.75小时至约2、4或8小时。
反应3优选地是在脂族胺的存在下进行的,包括但不限于一甲胺、二甲胺、三甲胺、一乙胺、二乙胺、三乙胺、单异丙胺、二异丙胺、单正丁胺、二正丁胺、三正丁胺、单环己胺、二环己胺或它们的混合物。更优选地,脂族胺是叔胺,例如三乙胺。
正如将为本领域技术人员所理解的,各种式V化合物的基本单分散混合物是商业上可得到的。例如,当R2是H或甲基时,式V化合物分别是PEG或mPEG化合物,在商业上可从Aldrich of Milwaukee,Wisconsin;Fluka of Switzerland和/或TC1 America of Portland,Oregon获得。
当R2是亲脂性部分时,例如高级烷基、脂肪酸、脂肪酸的酯、胆固醇基或金刚烷基,式V化合物可以借助将为本领域技术人员所理解的各种方法制得。式V化合物优选地是如下制得的:
R2-OMs+R3(OC2H4)m-O-X2 +→R3(OC2H4)m-OR2 4
(VI) (VII) (VIII)
R3(OC2H4)m-OR2→H(OC2H4)m-OR2 5
(VIII) (V)
R2是亲脂性部分,优选高级烷基、脂肪酸酯、胆固醇基或金刚烷基,更优选脂肪酸的低级烷基酯,最优选具有1至18个碳原子的脂肪酸乙基酯。
R3是H、苄基、三苯甲基、四氢吡喃或将为本领域技术人员所理解的其他醇保护基团。
X2 +是如上关于X+所述的阳离子。
m的值是如上所述的。
关于反应4,使式VI化合物混合物与式VII化合物混合物在与上面关于反应1所述相似的条件下反应。式VI化合物混合物是基本单分散混合物。优选地,式VI化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量。更优选地,式VI化合物混合物是单分散混合物。式VII化合物混合物是基本单分散混合物。优选地,式VII化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量。更优选地,式VII化合物混合物是单分散混合物。
关于反应5,VIII化合物可以被将为本领域技术人员所理解的各种方法水解,转化R3部分为醇。当R3是苄基或三苯甲基时,水解作用优选地是这样进行的,利用H2,在钯-木炭催化剂的存在下,这将为本领域技术人员所理解。当然,当R3是H时,反应5是不必要的。
式VI化合物可以是商业上可得到的或者如上关于反应3所述制得。式VII化合物可以如上关于反应2所述制得。
可以使包含PEG部分且具有上式III结构的基本单分散聚合物混合物进一步与其他包含PEG部分的基本单分散聚合物反应,目的是延长PEG链。例如,可以采用下列流程:

R2(OC2H4)m+n-OMs+R4(OC2H4)p-O-X2 +→R2(OC2H4)m+n+p-OR4
(IX) (X) (XI)
Ms、m和n是如上反应1所述的;p与n和m相似,X2 +与如上关于反应1所述X+相似。Q是如上反应3所述的。R2是如上反应1所述的,优选低级烷基。R1是H。反应6优选地是按照如上关于反应3所述相似的方式进行的。反应7优选地是按照如上关于反应1所述相似的方式进行的。优选地,式III化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量,更优选地,式III化合物混合物是单分散混合物。式X化合物混合物是基本单分散混合物。优选地,式X化合物混合物中至少约96、97、98或99%的化合物具有相同的分子量,更优选地,式X化合物混合物是单分散混合物。
图1所示流程阐述了根据本发明实施方式的方法,现将描述之。基本单分散的含聚乙二醇低聚物的合成开始于基本单分散聚乙二醇一苄醚(1)的制备。使过量的商业上可得到的基本单分散聚乙二醇与苄基氯在含水氢氧化钠的存在下反应,如Coudert et al(SyntheticCommunications,16(1):19-26(1986))所述。然后加入NaH制备1的钠盐,使这种钠盐与从羟基链烷酸(2)的酯合成的甲磺酸酯反应。经由催化氢化作用使甲磺酸酯置换产物(3)脱苄基化,得到醇(4)。可以通过加入甲磺酰氯制备这种醇的甲磺酸酯(5),用作亲电试剂与基本单分散聚乙二醇衍生物的一甲醚的钠盐反应,从而延长低聚物的聚乙二醇部分至所需长度,得到延长了的酯(6)。酯可以在水性基质中水解为酸(7),借助与碳二亚胺和N-羟基琥珀酰亚胺的反应转化为活化酯(8)。尽管图1所述低聚物是用N-羟基琥珀酰亚胺活化的,不过可以理解的是各种其他试剂也可以用于活化本发明的低聚物,包括但不限于活性氯甲酸苯基酯,例如氯甲酸对硝基苯基酯、氯甲酸苯基酯、3,4-苯基二氯甲酸酯和3,4-苯基二氯甲酸酯;tresylation;和缩醛生成。
参照图1,q是1至24。优选地,q是1至18,q更优选地是4至16。R4是能够经历水解作用得到羧酸的部分。R4优选地是低级烷基,更优选乙基。变量n和m是如上关于反应1所述的。
用在本文所述操作中的所有原料都是商业上可得到的,或者可以使用商业上可得到的原料借助本领域已知的方法加以制备。
现将参照下列实施例描述本发明。应当意识到的是这些实施例是用来阐述本发明各方面的,并不限制由权利要求所限定的发明范围。
实施例
实施例1至10
实施例1至10中的反应是在氮下、用磁搅拌进行的,另有指定除外。“逐步处理(Work-up)”表示用有机溶剂萃取、有机相用饱和NaCl溶液洗涤、干燥(MgSO4)、蒸发(旋转蒸发器)。薄层色谱法是利用预先涂有硅胶60°F-254的Merck玻璃板进行的,斑点是用碘蒸气显色的。所有质谱法都是用Macromolecular Resources Colorado StateUNiversity,CO测定的,是按m/z、(相对强度)的顺序报告的。元素分析和熔点测定是用Galbraith Laboratories,Inc.,Knoxville,TN进行的。实施例1-10涉及图2所述流程。
实施例1
8-甲氧基-1-(甲磺酰基)氧基-3,6-二氧杂辛烷(9)
将非多分散三甘醇一甲醚分子(4.00ml,4.19g,25.5mmol)与三乙胺(4.26ml,3.09g,30.6mmol)的无水二氯甲烷(50ml)溶液在冰浴中冷却,置于氮气氛下。从加液漏斗滴加甲磺酰氯(2.37ml,3.51g,30.6mmol)的无水二氯甲烷(20ml)溶液。氯化物加入完成10分钟后,除去反应混合物的冰浴,使其恢复至室温。将混合物搅拌另外1小时,此时TLC(以含15%MeOH的CHCl3作为洗脱剂)显示没有剩余的三甘醇一甲醚。将反应混合物用另外75ml二氯甲烷稀释,连续用饱和NaHCO3、水和盐水洗涤。将有机相经Na2SO4干燥,过滤,在真空中浓缩,得到化合物9的非多分散混合物,为澄清的油(5.31g,86%)。
实施例2
乙二醇一甲醚(10)(m=4,5,6)
在N2下,向搅拌着的非多分散化合物11(35.7mmol)的无水DMF(25.7ml)溶液分批加入60%NaH的矿物油分散体,将混合物在室温下搅拌1小时。向该盐12一次性加入非多分散甲磺酸酯9(23.36mmol)的无水DMF(4ml)溶液,将混合物在室温下搅拌3.5小时。用TLC(12%CH3OH-CHCl3)监测反应进程。将反应混合物用等量1N HCl稀释,用乙酸乙酯萃取(2x20ml),弃去。萃取水溶液并逐步处理,得到非多分散聚合物10(收率82-84%)。
实施例3
3,6,9,12,15,18,21-七氧杂二十二烷醇(10)(m=4)
油状物;Rf 0.46(甲醇∶氯仿=3∶22);MS m/z C15H32O8的计算值为340.21(M++1),实测值为341.2。
实施例4
3,6,9,12,15,18,21,24-八氧杂二十五烷醇(10)(m=5)
油状物;Rf 0.43(甲醇∶氯仿=6∶10);MS m/z C17H36O9的计算值为384.24(M++1),实测值为385.3。
实施例5
3,6,9,12,15,18,21,24,27-九氧杂二十八醇(10)(m=6)
油状物;Rf 0.42(甲醇∶氯仿=6∶10);MS m/z C19H40O10的计算值为428.26(M++1),实测值为429.3。
实施例6
20-甲氧基-1-(甲磺酰基)氧基-3,6,9,12,15,18-六氧杂二十烷(14)
如9所述从醇13(m=13)和甲磺酰氯以定量收率得到非多分散化合物14,为油状物;Rf 0.4(乙酸乙酯∶乙腈=1∶5);MS m/z C17H37O10的计算值为433.21(M++1),实测值为433.469。
实施例7
乙二醇一甲醚(15)(m=3,4,5)
利用上述化合物10操作从二醇制备非多分散化合物15。
实施例8
3,6,9,12,15,18,21,24,27,30-十氧杂二十一醇(15)(m=3)
油状物;Rf 0.41(甲醇∶氯仿=6∶10);MS m/z C21H44O11的计算值为472.29(M++1),实测值为472.29。
实施例9
3,6,9,12,15,18,21,24,27,30,33-十一氧杂三十四醇(15)(m=4)
油状物;Rf 0.41(甲醇∶氯仿=6∶10);MS m/z C23H48O12的计算值为516.31(M++1),实测值为516.31。
实施例10
3,6,9,12,15,18,21,24,27,30,33,36-十二氧杂三十七醇(15)(m=5)
油状物;Rf 0.41(甲醇∶氯仿=6∶10);MS m/z C25H52O13的计算值为560.67(M++1),实测值为560.67。
实施例11至18涉及图3所述流程。
实施例11
六甘醇一苄醚(16)
将3.99g(100mmol)NaOH溶于4ml水,向非多分散六甘醇(28.175g,25ml,100mmol)加入该氢氧化钠水溶液。加入苄基氯(3.9g,30.8mmol,3.54ml),将反应混合物加热至100℃,搅拌18小时。然后将反应混合物冷却,用盐水(250ml)稀释,用二氯甲烷萃取(200mlx2)。合并有机层,用盐水洗涤一次,经Na2SO4干燥,过滤,在真空中浓缩,得到暗褐色油。粗产物混合物经由快速色谱纯化(硅胶,梯度洗脱:乙酸乙酯至9/1乙酸乙酯/甲醇),得到8.099g(70%)非多分散的16,为黄色的油。
实施例12
乙基6-甲磺酰氧基己酸酯(17)
将非多分散乙基6-羟基己酸酯(50.76ml,50.41g,227mmol)的无水二氯甲烷(75ml)溶液在冰浴中冷却,置于氮气氛下。加入三乙胺(34.43ml,24.99g,247mmol)。从加液漏斗滴加甲磺酰氯(19.15ml,28.3g,247mmol)的无水二氯甲烷(75ml)溶液。将混合物搅拌三个半小时,随着冰浴融化,缓慢恢复至室温。通过硅胶过滤混合物,滤液连续用水、饱和NaHCO3、水和盐水洗涤。有机相经Na2SO4干燥,过滤,在真空中浓缩,得到淡黄色油。最后,粗产物经过快速色谱纯化(硅胶,1/1己烷/乙酸乙酯),得到非多分散产物(46.13g,85%),为澄清无色的油。FAB MS:m/e 239(M+H),193(M-C2H5O).
实施例13
6-{2-[2-(2-{2-[2-(2-苄氧基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸乙酯(18)
将氢化钠(3.225g或60%油分散体,80.6mmol)悬浮在80ml无水甲苯中,置于氮气氛下,在冰浴中冷却。向NaH悬液加入非多分散醇16(27.3g,73.3mmol)的80ml无水甲苯溶液。将混合物在0℃下搅拌30分钟,使其恢复至室温,搅拌另外5小时,在此期间混合物变为澄清褐色溶液。向NaH/醇混合物加入非多分散甲磺酸酯17(19.21g,80.6mmol)的80ml无水甲苯溶液,将合并了的溶液在室温下搅拌3天。将反应混合物用50ml甲醇猝灭,通过碱性氧化铝过滤。在真空中浓缩滤液,经过快速色谱纯化(硅胶,梯度洗脱:3/1乙酸乙酯/己烷至乙酸乙酯),得到非多分散产物,为淡黄色油(16.52g,44%)。FAB MS:m/e 515(M+H).
实施例14
6-{2-[2-(2-{2-[2-(2-羟基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸乙酯(19)
将非多分散苄基醚18(1.03g,2.0mmol)溶于25ml乙醇。向该溶液加入270mg 10%Pd/C,将混合物置于氢气氛下,搅拌4小时,此时TLC显示原料完全消失。通过C盐545过滤反应混合物以除去催化剂,在真空中浓缩滤液,得到非多分散标题化合物,为澄清的油(0.67g,79%)。FAB MS:m/e 425(M+H),447(M+Na).
实施例15
6-{2-[2-(2-{2-[2-(2-甲磺酰基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸乙酯(20)
将非多分散醇19(0.835g,1.97mmol)溶于3.5ml无水二氯甲烷,置于氮气氛下。加入三乙胺(0.301ml,0.219g,2.16mmol),将混合物在冰浴中冷却。2分钟后,加入甲磺酰氯(0.16ml,0.248g,2.16mmol)。将混合物在0℃下搅拌15分钟,然后在室温下搅拌2小时。通过硅胶过滤反应混合物以除去氯化三乙铵,滤液连续用水、饱和NaHCO3、水和盐水洗涤。有机相经Na2SO4干燥,过滤,在真空中浓缩。残余物经过柱色谱纯化(硅胶,9/1乙酸乙酯/甲醇),得到非多分散化合物20,为澄清的油(0.819g,83%)。FAB MS:m/e 503(M+H).
实施例16
6-{2-[2-(2-{2-[2-(2-甲氧基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸乙酯(21)
在N2下,将NaH(88mg 60%油分散体,2.2mmol)悬浮在无水甲苯(3ml)中,冷却至0℃。加入非多分散二甘醇一甲醚(0.26ml,0.26g,2.2mmol),后者已经经由甲苯共沸蒸馏干燥。使反应混合物恢复至室温,搅拌4小时,在此期间浑浊的灰色悬液变为澄清的黄色,再变为褐色。加入甲磺酸酯20(0.50g,1.0mmol)的2.5ml无水甲苯溶液。在室温下搅拌过夜后,加入2ml甲醇猝灭反应,通过硅胶过滤所得溶液。在真空中浓缩滤液,FAB MS:m/e 499(M+H),521(M+Na)。另外经过制备型色谱纯化(硅胶,19/3氯仿/甲醇),得到非多分散产物,为澄清黄色的油(0.302g,57%)。FAB MS:m/e 527(M+H),549(M+Na).
实施例17
6-{2-[2-(2-{2-[2-(2-甲氧基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸(22)
将非多分散酯21(0.25g,0.46mmol)在0.71ml 1N NaOH中搅拌18小时。18小时后,在真空中浓缩混合物以除去醇,将残余物溶于另外10ml水。将水溶液用2N HCl酸化至pH 2,产物用二氯甲烷萃取(30mlx2)。合并有机相,用盐水洗涤(25mlx2),经Na2SO4干燥,过滤,在真空中浓缩,得到非多分散标题化合物,为黄色的油(0.147g,62%)。FAB MS:m/e 499(M+H),521(M+Na).
实施例18
6-{2-[2-(2-{2-[2-(2-甲氧基乙氧基)乙氧基]乙氧基}乙氧基)乙氧基]乙氧基}己酸2,5-二氧代吡咯烷-1-基酯(23)
将非多分散酸22(0.209g,0.42mmol)溶于4ml无水二氯甲烷,在N2气氛下加入到已经含有NHS(N-羟基琥珀酰亚胺)(57.8mg,0.502mmol)和EDC(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐)(98.0mg,0.502mmol)的干燥烧瓶内。将溶液在室温下搅拌过夜,通过硅胶过滤以除去过量试剂和从EDC生成的脲。在真空中浓缩滤液,得到非多分散产物,为暗黄色油(0.235g,94%)。FAB MS:m/e 596(M+H),618(M+Na).
实施例19至24涉及图4所述流程。
实施例19
三甘醇一甲醚的甲磺酸酯(24)
向在冰浴中冷却至0℃的CH2Cl2溶液(100ml)加入非多分散三甘醇一甲醚(25g,0.15mol)。然后加入三乙胺(29.5ml,0.22mol),将溶液在0℃下搅拌15分钟,然后滴加甲磺酰氯(13.8ml,0.18mol,溶于20ml CH2Cl2)。将反应混合物在0℃下搅拌30分钟,使其温热至室温,然后搅拌2小时。将粗反应混合物通过C盐过滤(用CH2Cl2~200ml洗涤),然后用H2O(300ml)、5% NaHCO3(300ml)、H2O(300ml)、饱和NaCl(300ml)洗涤,经MgSO4干燥,蒸发至干。然后将油置于真空管道上~2小时以确保干燥,得到非多分散标题化合物,为黄色的油(29.15g,收率80%)。
实施例20
七甘醇一甲醚(25)
向非多分散四甘醇(51.5g,0.27mol)的THF(1L)溶液加入叔丁醇钾(14.8g,0.13mol,历经~30分钟分小批量加入)。然后将反应混合物搅拌1小时,然后滴加24(29.15g,0.12mol)的THF(90ml)溶液,将反应混合物搅拌过夜。将粗反应混合物通过C盐过滤(用CH2Cl2~200ml洗涤),蒸发至干。然后将油溶于HCl(250ml,1N),用乙酸乙酯(250ml)洗涤以除去过量的24。为了除去残留的24可能需要另外的乙酸乙酯(125ml)洗涤。将含水相用CH2Cl2(125ml)反复洗涤,直至大多数25已经从含水相中除去。第一萃取液将含有24、25和双偶联的副产物,应当用HCl(125ml,1N)反萃取。合并有机层,蒸发至干。然后将所得油溶于CH2Cl2(100ml),用H2O(50ml)反复洗涤,直至除去25。合并含水部分,总体积为500ml,加入NaCl,直至溶液变为浑浊,然后用CH2Cl2洗涤(2x500ml)。合并有机层,经MgSO4干燥,蒸发至干,得到非多分散标题化合物,为一种油(16.9g,收率41%)。为了确保高纯度,可能需要重复一步或多步纯化。
实施例21
8-溴辛酸酯(26)
向8-溴辛酸(5.0g,22mmol)的乙醇(100ml)溶液加入H2SO4(0.36ml,7.5mmol),将反应物加热至回流,搅拌3小时。将粗反应混合物冷却至室温,用H2O(100ml)、饱和NaHCO3(2x100ml)、H2O(100ml)洗涤,经MgSO4干燥,蒸发至干,得到澄清的油(5.5g,收率98%)。
实施例22
MPEG7-C8酯(27)的合成
向非多分散化合物25(3.0g,8.8mmol)的乙醚(90ml)溶液加入叔丁醇钾(1.2g,9.6mmol),将反应混合物搅拌1小时。然后滴加非多分散化合物26(2.4g,9.6mmol)的乙醚(10ml)溶液,将反应混合物搅拌过夜。将粗反应混合物通过C盐过滤(用CH2Cl2~200ml洗涤),蒸发至干。将所得油溶于乙酸乙酯,用H2O洗涤(2x200ml),经MgSO4干燥,蒸发至干。进行柱色谱(二氧化硅,乙酸乙酯至乙酸乙酯/甲醇10∶1),得到非多分散标题化合物,为澄清的油(0.843g,收率19%)。
实施例23
MPEG7-C8酸(28)
向非多分散化合物27(0.70g,1.4mmol)的油加入1N NaOH(2.0ml),将反应混合物搅拌4小时。将粗反应混合物浓缩,酸化(pH~2),用NaCl饱和,用CH2Cl2洗涤(2x50ml)。合并有机层,用饱和NaCl洗涤,经MgSO4干燥,蒸发至干,得到非多分散标题化合物,为澄清的油(0.35g,收率53%)。
实施例24
MPEG7-C8酸的活化(29)
将非多分散mPEG7-C8酸28(0.3g,0.64mmol)溶于3ml无水二氯甲烷,然后加入N-羟基琥珀酰亚胺(0.079g,0.69mmol)与EDCI.HCl(135.6mg,0.71mmol)的无水二氯甲烷溶液。将反应物搅拌若干小时,然后用1N HCl、水洗涤,经MgSO4干燥,过滤,浓缩。粗产物经过柱色谱纯化,浓缩,得到非多分散标题化合物,为澄清的油,经由真空干燥。
实施例25至29涉及图5所述流程。
实施例25
10-羟基癸酸酯(30)
向非多分散10-羟基癸酸(5.0g,26.5mmol)的乙醇(100ml)溶液加入H2SO4(0.43ml,8.8mmol),将反应物加热至回流,搅拌3小时。将粗反应混合物冷却至室温,用H2O(100ml)、饱和NaHCO3(2x100ml)、H2O(100ml)洗涤,经MgSO4干燥,蒸发至干,得到非多分散标题化合物,为澄清的油(6.9g,收率98%)。
实施例26
10-羟基癸酸酯甲磺酸酯(31)
向CH2Cl2溶液(27ml)加入非多分散10-羟基癸酸酯30(5.6g,26mmol),在冰浴中冷却至0℃。然后加入三乙胺(5ml,37mmol),将反应混合物在0℃下搅拌15分钟。然后加入甲磺酰氯(2.7ml,24mmol)的CH2Cl2(3ml)溶液,将反应混合物在0℃下搅拌30分钟,除去冰浴,将反应物在室温下搅拌另外2小时。将粗反应混合物通过C盐过滤(用CH2Cl2 80ml洗涤),将滤液用H2O(100ml)、5%NaHCO3(2x100ml)、H2O(100ml)、饱和NaCl(100ml)洗涤,经MgSO4干燥,蒸发至干,得到非多分散标题化合物,为浅黄色油(7.42g,收率97%)。
实施例27
MPEG7-C10酯(32)
向非多分散七甘醇一甲醚25(2.5g,7.3mmol)的四氢呋喃(100ml)溶液加入氢化钠(0.194g,8.1mmol),将反应混合物搅拌1小时。然后滴加非多分散10-羟基癸酸酯甲磺酸酯31(2.4g,8.1mmol)的四氢呋喃(10ml)溶液,将反应混合物搅拌过夜。将粗反应混合物通过C盐过滤(用CH2Cl2~200ml洗涤),蒸发至干。将所得油溶于乙酸乙酯,用H2O洗涤(2x200ml),经MgSO4干燥,蒸发至干,经过色谱纯化(二氧化硅,乙酸乙酯/甲醇10∶1)和色谱纯化(二氧化硅,乙酸乙酯),得到非多分散标题化合物,为澄清的油(0.570g,收率15%)。
实施例28
MPEG7-C10酸(33)
向非多分散mPEG7-C10酯32的油(0.570g,1.1mmol)加入1N NaOH(1.6ml),将反应混合物搅拌过夜。将粗反应混合物浓缩,酸化(pH~2),用NaCl饱和,用CH2Cl2洗涤(2x50ml)。合并有机层,用饱和NaCl洗涤(2x50ml),经MgSO4干燥,蒸发至干,得到非多分散标题化合物,为澄清的油(0.340g,收率62%)。
实施例29
MPEG7-C10酸(34)的活化
利用与上面实施例24所述相似的操作活化非多分散酸33。
实施例30和31涉及图6所述流程。
实施例30
C18(PEG6)低聚物(36)的合成
向PEG6(5g,17.7mmol)与吡啶(0.97g,12.4mmol)在苯中的混合物缓慢加入非多分散硬脂酰氯35(0.7g,2.31mmol)。将反应混合物搅拌若干小时(~5)。然后进行TLC,用乙酸乙酯/甲醇作为展开溶剂。然后将反应混合物用水洗涤,经MgSO4干燥,浓缩,经由真空干燥。用FABMS分析经过纯化的非多分散化合物36:m/e 549 M+H。
实施例31
C18(PEG6)低聚物的活化
非多分散C18(PEG6)低聚物的活化是分两步完成的:
1)将非多分散硬脂酰-PEG6 36(0.8g,1.46mmol)溶于甲苯,加入到用冰浴冷却的光气溶液中(10ml,20%甲苯溶液)。将反应混合物在0℃下搅拌1小时,然后在室温下搅拌3小时。然后蒸馏除去光气和甲苯,剩余的非多分散硬脂酰PEG6氯甲酸酯37经P2O5干燥过夜。
2)向非多分散硬脂酰PEG6氯甲酸酯36(0.78g,1.27mmol)与TEA(128mg,1.27mmol)的无水二氯甲烷溶液加入N-羟基琥珀酰亚胺(NHS)的二氯甲烷溶液。将反应混合物搅拌16小时,然后用水洗涤,经MgSO4干燥,过滤,浓缩,经由真空干燥,得到非多分散活化C18(PEG6)低聚物38。
实施例32至37涉及图7所述流程。
实施例32
四甘醇一苄醚(39)
向非多分散四甘醇的油(19.4g,0.10mol)加入NaOH溶液(4.0g,4.0ml),将反应物搅拌15分钟。然后加入苄基氯(3.54ml,30.8mmol),将反应混合物加热至100℃,搅拌过夜。将反应混合物冷却至室温,用饱和NaCl(250ml)稀释,用CH2Cl2洗涤(2x200ml)。合并有机层,用饱和NaCl洗涤,经MgSO4干燥,经过色谱纯化(二氧化硅,乙酸乙酯),得到非多分散标题化合物,为黄色的油(6.21g,收率71%)。
实施例33
四甘醇一苄醚甲磺酸酯(40)
向CH2Cl2溶液(20ml)加入非多分散四甘醇一苄醚39(6.21g,22mmol),在冰浴中冷却至0℃。然后加入三乙胺(3.2ml,24mmol),将反应混合物在0℃搅拌15分钟。然后加入甲磺酰氯(1.7ml,24mmol)的CH2Cl2(2ml)溶液,将反应混合物在0℃下搅拌30分钟,除去冰浴,将反应物在室温下搅拌另外2小时。将粗反应混合物通过C盐过滤(用CH2Cl2 80ml洗涤),将滤液用H2O(100ml)、5%NaHCO3(2x100ml)、H2O(100ml)、饱和NaCl(100ml)洗涤,经MgSO4干燥。所得黄色的油在含有活性碳(10g)的二氧化硅垫上经过色谱纯化,得到非多分散标题化合物,为澄清的油(7.10g,收率89%)。
实施例34
八甘醇一苄醚(41)
向含有氢化钠(0.43g,18mmol)的四氢呋喃(140ml)溶液滴加非多分散四甘醇(3.5g,18mmol)的四氢呋喃(10ml)溶液,将反应混合物搅拌1小时。然后滴加非多分散四甘醇一苄醚甲磺酸酯40(6.0g,16.5mmol)的四氢呋喃(10ml)溶液,将反应混合物搅拌过夜。将粗反应混合物通过C盐过滤(用CH2Cl2 250ml洗涤),将滤液用H2O洗涤,经MgSO4干燥,蒸发至干。所得油经过色谱纯化(二氧化硅,乙酸乙酯/甲醇10∶1)和色谱纯化(二氧化硅,氯仿/甲醇25∶1),得到非多分散标题化合物,为澄清的油(2.62g,收率34%)。
实施例35
硬脂酸酯PEG8-苄基(43)的合成
向搅拌着的冷却的非多分散八甘醇一苄醚41(0.998g,2.07mmol)与吡啶(163.9mg,2.07mmol)溶液加入非多分散硬脂酰氯42(627.7mg,2.07mmol)的苯溶液。将反应混合物搅拌过夜(18小时)。第二天,将反应混合物用水洗涤,经MgSO4干燥,浓缩,经由真空干燥。粗产物然后经过快速硅胶柱色谱纯化,10%甲醇/90%氯仿洗脱。合并含有产物的部分,浓缩,经由真空干燥,得到非多分散标题化合物。
实施例36
硬脂酸酯-PEG8-苄基的氢解
向非多分散硬脂酸酯-PEG8-Bzl 43(0.854g,1.138mmol)的甲醇溶液加入Pd/C(10%)(10%wt披钯活性碳)。将反应混合物在氢下搅拌过夜(18小时)。然后将溶液过滤,浓缩,经过快速柱色谱纯化,用10%甲醇/90%氯仿洗脱,收集Rt=0.6的部分,浓缩,干燥,得到非多分散酸44。
实施例37
C18(PEG8)低聚物的活化
如上实施例31硬脂酸酯-PEG6所述进行非多分散硬脂酸酯-PEG8低聚物的两步活化,得到非多分散活化C18(PEG8)低聚物45。
实施例38
活化三甘醇一甲基低聚物的合成
下列说明涉及图8所述流程。将含有20%光气的甲苯溶液(100ml,约18.7g,189mmol光气)在N2气氛下冷却至0℃。将非多分散mTEG(三甘醇,一甲醚,7.8g,47.5mmol)溶于25ml无水乙酸乙酯,加入到冷却的光气溶液中。将混合物在0℃下搅拌1小时,然后使其温热至室温,搅拌另外两个半小时。经由真空蒸馏除去残留的光气、乙酸乙酯和甲苯,得到非多分散mTEG氯甲酸酯46,为澄清的油状残余物。
将非多分散残余物46溶于50ml无水二氯甲烷,向其中加入TEA(三乙胺,6.62ml,47.5mmol)和NHS(N-羟基琥珀酰亚胺,5.8g,50.4mmol)。将混合物在室温干燥气氛下搅拌20小时,在此期间有大量白色沉淀出现。过滤混合物以除去该沉淀,在真空中浓缩。将所得油47溶于二氯甲烷,用冷去离子水洗涤两次,用1N HCl洗涤两次,用盐水洗涤一次。有机相经MgSO4干燥,过滤,浓缩,得到非多分散标题化合物,为澄清的浅黄色油。如果必要的话,NHS酯可以进一步经过硅胶快速色谱纯化,使用EtOAc作为洗脱剂。
实施例39
活化棕榈酸酯-TEG低聚物的合成
下列说明涉及图9所述流程。将非多分散棕榈酸酐(5g;10mmol)溶于无水THF(20ml),在室温下搅拌。向搅拌中的溶液加入3mol过量吡啶,然后加入非多分散三甘醇(1.4ml)。将反应混合物搅拌1小时(用TLC监测反应进程;乙酸乙酯-氯仿;3∶7)。反应结束时,除去THF,将产物与10% H2SO4混合,用乙酸乙酯萃取(3x30ml)。合并萃取液,依次用水、盐水洗涤,经MgSO4干燥,蒸发,得到非多分散产物48。向非多分散产物48(1mmol)的10ml无水DMF溶液加入N,N′-二琥珀酰亚氨基碳酸酯(3mmol)的DMF(~10ml)溶液,同时搅拌。向反应混合物缓慢加入氢化钠(3mmol)。将反应混合物搅拌若干小时(例如5小时)。加入二乙醚,使活化低聚物沉淀。该过程重复3次,最后将产物干燥。
实施例40
活化六甘醇一甲基低聚物的合成
下列说明涉及图10所述流程。类似于上述实施例39非多分散三甘醇的方法制备非多分散活化六甘醇一甲醚。将20%光气的甲苯溶液(35ml,6.66g,67.4mmol光气)在N2气氛下、在冰/盐水浴中冷却。将非多分散六甘醇50(1.85ml,2.0g,6.74mmol)溶于5ml无水EtOAc,经由注射器加入到光气溶液中。将反应混合物在冰浴中持续搅拌1小时,除去冰浴,在室温下搅拌另外2.5小时。通过真空蒸馏除去光气、EtOAc和甲苯,得到非多分散化合物51,为澄清的油状残余物。
将非多分散残余物51溶于20ml无水二氯甲烷,置于干燥的惰性气氛下。加入三乙胺(0.94ml,0.68g,6.7mmol),然后加入NHS(N-羟基琥珀酰亚胺,0.82g,7.1mmol),将反应混合物在室温下搅拌18小时。将混合物通过硅胶过滤以除去白色沉淀,在真空中浓缩。将残余物溶于二氯甲烷,用冷水洗涤两次,用1N HCl洗涤一次,用盐水洗涤一次。有机相经Na2SO4干燥,过滤,浓缩。经由快速色谱(硅胶,EtOAc)进行最后的纯化,得到UV活性非多分散NHS酯52。
实施例41
将150mg鲑鱼降钙素(MW 3432,0.043mmol)溶于30ml无水DMF。然后加入TEA(35μl)与实施例24活化低聚物(42mg,0.067mmol)的无水THF(2ml)溶液。将反应物搅拌1小时,然后用2ml 0.1%TFA水溶液猝灭。用HPLC监测反应。然后将反应混合物浓缩,经过制备型HPLC纯化(RC Vydac C18蛋白质和肽,1x25柱,含0.1%TFA的水/乙腈,检测波长280nm)。解析到两个峰,相当于单-与二-缀合物。用MALDI-MS分析样本。关于PEG7-辛基-sCT单缀合物的MS:3897。关于PEG7-辛基-sCT二缀合物的MS:4361。
实施例42
利用实施例41的操作使鲑鱼降钙素与实施例29活化低聚物共轭。关于PEG7-癸基-sCT单缀合物的MS:3926。关于PEG7-癸基-sCT二缀合物的MS:4420。
实施例43
利用实施例41的操作使鲑鱼降钙素与实施例31活化低聚物共轭。关于硬脂酸酯-PEG6-sCT单缀合物的MS:4006。关于硬脂酸酯-PEG6-sCT二缀合物的MS:4582。
实施例44
利用实施例41的操作使鲑鱼降钙素与实施例37活化低聚物共轭。关于硬脂酸酯-PEG8-sCT单缀合物的MS:4095。
实施例45
利用实施例41的操作使鲑鱼降钙素与实施例18活化低聚物共轭。
实施例46
利用实施例41的操作使鲑鱼降钙素与实施例38活化低聚物共轭。
实施例47
利用实施例41的操作使鲑鱼降钙素与实施例39活化低聚物共轭。
实施例48
利用实施例41的操作使鲑鱼降钙素与实施例40活化低聚物共轭。
实施例49
鲑鱼降钙素-低聚物缀合物混合物的分散系数测定
鲑鱼降钙素-低聚物缀合物混合物的分散系数是如下测定的。制得鲑鱼降钙素-低聚物缀合物混合物,例如上面实施例41所述。经由HPLC纯化第一份混合物样本,以分离和离析样本中的各种鲑鱼降钙素-低聚物缀合物。假定每一所离析的部分含有缀合物的纯粹单分散混合物,“n”等于所收集的部分数。混合物可以包括一种或多种下列缀合物,通过规定共轭位置继之以共轭程度加以描述:Lys11单缀合物;Lys18单缀合物;N-末端单缀合物;Lys11,18二缀合物;Lys11,N-末端二缀合物;Lys18,N-末端二缀合物;和/或Lys11,18,N-末端三缀合物。经由质谱法分析混合物每一所离析的部分,以测定该部分的质量,这可以将每一所离析的部分分为单-、二-或三-缀合物,并且为样本中每一缀合物的变量“Mi”赋值。
经由HPLC分析第二份混合物样本,以提供HPLC痕量。假定摩尔吸收性不因共轭作用而改变,混合物中特定缀合物的重量百分比是由对应于特定缀合物的HPLC痕量的峰下面积占所有HPLC痕量峰下总面积的百分比所提供的。收集样本,冷冻干燥,测定样本的无水重量克数。样本的重量克数乘以样本中每一组分的重量百分比,以测定样本中每一缀合物的重量克数。特定缀合物(第i个缀合物)的变量“Ni”是这样测定的,样本中特定缀合物的重量克数除以特定缀合物的质量,商再乘以阿弗伽德罗常数(6.02205x1023mol-1)、如上测定的Mi,得到样本中特定缀合物的分子数Ni。然后利用n、根据每一缀合物所测定的Mi和根据每一缀合物所测定的Ni计算分散系数。
实施例50
细胞传感器 研究
研究
将T-47D细胞(输乳管癌细胞系,得自美国典型菌种保藏中心)按1x107细胞/ml的密度悬浮在工作缓冲液中(低缓冲的、无血清的、无碳酸氢盐的RPMI 1640培养基,来自Molecular Devices ofSunnyvale,California)。然后将大约100,000个细胞固定在琼脂糖细胞包埋培养基中,呈10μl小滴,在细胞传感器套杯中夹在两个3μm聚碳酸酯膜之间。细胞传感器套杯置于细胞传感器 微量生理仪上的传感腔内,再固定在非常靠近pH敏感性检测器处。然后泵入工作缓冲液穿过细胞,速率100μl/mi n,不过有30秒间隔,此时停止流动,测量传感腔内工作缓冲液的酸化作用。每2分钟测定酸化率。传感腔的温度为37℃。在实验开始之间使细胞在传感腔内平衡2-3小时,在此期间监测基础酸化率。然后使细胞暴露于按不同nM浓度稀释在工作缓冲液中的供试化合物(鲑鱼降钙素或辛基-二降钙素)。细胞暴露于供试化合物发生在每次2分钟泵入周期的前40秒,该周期重复进行总计20分钟。这使细胞充分暴露于供试化合物,引发细胞代谢中的受体介导反应,然后流过不含化合物的工作缓冲液达约50秒。该操作在测量酸化率之前清洗除去传感腔内的供试溶液(它已具有略低于单独工作缓冲液的pH)。因而,酸化率仅仅是细胞活性的量度。利用相似的操作得到关于PEG7-辛基-sCT,单缀合物(辛基-Mono);PEG7-癸基-sCT,单缀合物(癸基-Mono);PEG7-癸基-sCT,二缀合物(癸基-Di);硬脂酸酯-PEG6sCT,单缀合物(PEG6 St.Mono);和硬脂酸酯-PEGS-sCT,单缀合物(PEG8 St.Mono)的数据。就这些数据分析化合物的相对活性,计算每一细胞传感器腔酸化率图的曲线下面积(AUC),作成图14所述条形图,显示在相同实验条件下进行多次实验,由此测量平均AUC。
微量生理仪上的传感腔内,再固定在非常靠近pH敏感性检测器处。然后泵入工作缓冲液穿过细胞,速率100μl/mi n,不过有30秒间隔,此时停止流动,测量传感腔内工作缓冲液的酸化作用。每2分钟测定酸化率。传感腔的温度为37℃。在实验开始之间使细胞在传感腔内平衡2-3小时,在此期间监测基础酸化率。然后使细胞暴露于按不同nM浓度稀释在工作缓冲液中的供试化合物(鲑鱼降钙素或辛基-二降钙素)。细胞暴露于供试化合物发生在每次2分钟泵入周期的前40秒,该周期重复进行总计20分钟。这使细胞充分暴露于供试化合物,引发细胞代谢中的受体介导反应,然后流过不含化合物的工作缓冲液达约50秒。该操作在测量酸化率之前清洗除去传感腔内的供试溶液(它已具有略低于单独工作缓冲液的pH)。因而,酸化率仅仅是细胞活性的量度。利用相似的操作得到关于PEG7-辛基-sCT,单缀合物(辛基-Mono);PEG7-癸基-sCT,单缀合物(癸基-Mono);PEG7-癸基-sCT,二缀合物(癸基-Di);硬脂酸酯-PEG6sCT,单缀合物(PEG6 St.Mono);和硬脂酸酯-PEGS-sCT,单缀合物(PEG8 St.Mono)的数据。就这些数据分析化合物的相对活性,计算每一细胞传感器腔酸化率图的曲线下面积(AUC),作成图14所述条形图,显示在相同实验条件下进行多次实验,由此测量平均AUC。
实施例51
酶稳定性
将化合物的冻干粉末重新悬浮在10mM磷酸盐缓冲液pH 7.4中,然后借助HPLC进行浓度测定。磷酸盐缓冲液用于生成这样一种溶液,其pH对每一特定肠酶的活性而言是最佳的。将如此制备的化合物试样转移至1.7ml微量离心管内,在37℃水浴中摇动15分钟,使化合物与温度平衡。15分钟后,向每支试管加入2μl适当浓缩的肠酶,以达到所需的最终浓度。将胰凝乳蛋白酶和胰蛋白酶重新悬浮在1mM HCl中。而且,作为对照,将化合物用2μl 1mM HCl处理。加入后立即从对照管中取出100μl样本,用25μl胰凝乳蛋白酶/胰蛋白酶猝灭溶液(1∶11% TFA∶异丙醇)猝灭。该样本将充当T=0分钟。在不同的时间间隔重复取样操作,这取决于所用的肠酶。胰凝乳蛋白酶具有15、30和60分钟的样本。胰蛋白酶具有30、60、120和180分钟的样本。一旦已经获得所有时间点的样本,从对照管中取出最后一份样本,以确定所观察的降解作用与温度或缓冲液无关。胰凝乳蛋白酶和胰蛋白酶样本可以被直接收集到HPLC小瓶内。利用RP-HPLC(乙腈梯度)测定每份样本的AUC,基于T=0分钟对照计算%降解作用。下表1至4提供了结果。
表1
0.5U/ml胰凝乳蛋白酶消化后,PEG7-辛基-鲑鱼降钙素二缀合物的残留%

表2
0.5U/ml胰凝乳蛋白酶消化后,鲑鱼降钙素的残留%(仅供对比,不是本发明的一部分)

表3
1U/ml胰蛋白酶消化后,PEG7-辛基-鲑鱼降钙素二缀合物的残留%

表4
1U/ml胰蛋白酶消化后,鲑鱼降钙素的残留%(仅供对比,不是本发明的一部分)

实施例52
活性和个体间差异
将体重20-25g的雄性CF-1小鼠(Charles River,Raleigh,NC)笼养在Nobex动物园中,控制房间的光照(L∶D周期为12∶12,早6时开始光照)、温度(21-23℃)和湿度(40-60%相对湿度)。允许动物自由获取实验室饲料(PMI Nutrition)和自来水。在实验当天之前,使小鼠适应笼养条件达48-72小时。
给药之前,使小鼠禁食过夜,可随意饮水。将小鼠随机分组,每一时间点五只,给以单一口服剂量的根据本发明的PEG7-辛基-sCT二缀合物(辛基-Di)或供对比的鲑鱼降钙素(sCT或降钙素)。口服剂量是利用管饲针头(Popper#18,从插孔至斜面5cm)给药的,剂量为10ml/kg的下列0.2μg/ml磷酸盐缓冲的PEG7-辛基-sCT二缀合物制剂:
| 成分 | 量 |
| PEG7-辛基-sCT二缀合物 | 20μg |
| 胆酸钠 | 2.5g |
| 脱氧胆酸钠 | 2.5g |
| 磷酸钠缓冲液100mM,pH 7.4 | 适量至100g |
缓冲制剂是这样制备的,在清洁的涂以焦油的玻璃烧杯内加入80ml磷酸盐缓冲液。向磷酸盐缓冲液缓慢加入胆酸钠,同时搅拌,直至溶解。然后加入脱氧胆酸盐,继续搅拌,直至溶解。加入相当于20μg的PEG7-辛基-sCT二缀合物溶液。最后,加入其余磷酸盐缓冲液,达到100g的最终重量。在全部实验中使用载体对照小鼠。利用药物给药后的单一时间点60分钟构造剂量-响应曲线。这些曲线如图15-18所述。
在适当的时间点,将小鼠用乙醚麻醉,使腔静脉外置,经由装有25度量针头的注射器获得血样。使血样在22℃下凝固1小时,除去血清,吸移至清洁的容器内。利用经过校准的Vitros DT60II分析仪测定每只动物的总血清钙。
将血清钙数据作图,利用SigmaPlot软件(4.1版)经由曲线适配技术测定药动学参数。计算平均和标准偏差(或标准误差),作图,以测定给药组之间的效应差异。下表5提供了各种缀合物的平均血清钙数据。
表5
| 缀合物 | 分散性 | 在2.0μg/kg剂量下的基线钙下降% |
| PEG7-辛基-sCT二缀合物 | 单分散混合物 | 21.0 |
| 硬脂酸酯-PEG6-sCT二缀合物 | 单分散混合物 | 16.0 |
| PEG7-癸基-sCT单缀合物 | 单分散混合物 | 11.5 |
| 硬脂酸酯-PEG8-sCT二缀合物 | 单分散混合物 | 11.0 |
| PEG7-癸基-sCT二缀合物 | 单分散混合物 | 8.3 |
尽管如上实施例50所测定的体外活性可能不与PEG7-辛基-sCT与PEG7-癸基-sCT单-与二-缀合物、硬脂酸酯-PEG6-sCT二缀合物和硬脂酸酯-PEG8-sCT二缀合物相当,不过体内活性似乎与PEG7-辛基-sCT与PEG7-癸基-sCT单-与二-缀合物的体内活性相当(得到上表5中基线钙下降%的证明)。尽管不希望局限于特定的理论,不过含有硬脂酸酯的缀合物提高了的体内活性可能表明这些缀合物经历体内水解作用,得到活性鲑鱼降钙素或活性鲑鱼降钙素-PEG缀合物。
本说明书已经公开了本发明的典型优选实施方式,尽管采用了专用术语,不过它们仅仅是在一般性的和说明性的意义上使用的,而不是限制性的,发明的范围是由下列权利要求所阐明的。
Claims (21)
1.缀合物混合物,每一个缀合物包含通过降钙素的胺官能团与至少一个低聚物偶联的降钙素,其中该混合物是基本单分散混合物,该低聚物包含聚乙二醇部分。
2.根据权利要求1的混合物,其中该混合物是基本纯粹单分散混合物。
3.根据权利要求1的混合物,其中该混合物是纯粹单分散混合物。
4.根据权利要求1-3任一项的混合物,其中该聚乙二醇部分具有至少2个聚乙二醇亚单位。
5.根据权利要求1-3任一项的混合物,其中该混合物具有选自如下的特征:(a)降低血清钙水平至少5%的能力,(b)与没有与低聚物偶联的降钙素对胰凝乳蛋白酶或胰蛋白酶降解的抗性相比,增加了对胰凝乳蛋白酶或胰蛋白酶降解的抗性,(c)生物功效大于没有与低聚物偶联的降钙素的生物功效,和这些特征的组合。
6.根据权利要求1-3任一项的混合物,其中该降钙素是通过可水解的键、不可水解的键或二者而与该低聚物以共价方式偶联的。
7.根据权利要求1-3任一项的混合物,其中该降钙素是与该低聚物的聚乙二醇部分以共价方式偶联的。
8.根据权利要求1-3任一项的混合物,其中该低聚物进一步包含亲脂性部分,和/或每一低聚物具有相同的分子结构。
9.根据权利要求8的混合物,其中该降钙素和/或聚乙二醇部分是与该亲脂性部分以共价方式偶联的。
10.药物组合物,其包含根据权利要求1-3任一项的混合物;和药学上可接受的载体。
11、缀合物的基本单分散混合物,各自包含在鲑鱼降钙素的Lys11与羧酸的羧酸部分以共价方式偶联的鲑鱼降钙素,该羧酸在该羧酸部分的远端与以甲基封端的聚乙二醇部分以共价方式偶联,该聚乙二醇部分具有至少7个聚乙二醇亚单位,并且在该鲑鱼降钙素的Lys18与羧酸的羧酸部分以共价方式偶联,该羧酸在该羧酸部分的远端与以甲基封端的聚乙二醇部分以共价方式偶联,该聚乙二醇部分具有至少7个聚乙二醇亚单位。
12、根据权利要求11的混合物,其中该缀合物各自由在鲑鱼降钙素的Lys11与辛酸的羧酸部分以共价方式偶联的鲑鱼降钙素组成,该辛酸在其羧酸部分的远端与以甲基封端的聚乙二醇部分以共价方式偶联,该聚乙二醇部分具有7个聚乙二醇亚单位,并且在该鲑鱼降钙素的Lys18与辛酸的羧酸部分以共价方式偶联,该辛酸在其羧酸部分的远端与以甲基封端的聚乙二醇部分以共价方式偶联,该聚乙二醇部分具有7个聚乙二醇亚单位。
13.权利要求1、2、3或9的缀合物混合物在药物制备中的用途,该药物用于治疗以过度破骨性骨吸收或血钙过多性血清效应为特征的骨障碍。
14.根据权利要求13的用途,其中该骨障碍是骨质疏松、佩吉特氏病或高钙血症。
15.缀合物的基本单分散混合物,其中每一个缀合物包含与低聚物偶联的降钙素,该低聚物包含具有至少4个聚乙二醇亚单位的聚乙二醇部分,所述混合物的分子量分布的标准偏差小于22道尔顿。
16.缀合物的基本单分散混合物,各自包含与包含聚乙二醇部分的低聚物偶联的降钙素,其中该混合物的分散系数大于10,000,其中
其中:
n是样本中不同分子的数量;
Ni是样本中第i个分子的数量;和
Mi是样本中第i个分子的质量。
17.根据权利要求16的缀合物混合物,其中该分散系数大于100,000。
18、缀合物的基本单分散混合物,其中每一缀合物具有相同的分子量,并且具有下式结构:
降钙素-[-B-Lj-Gk-R-G′m-R′-G″n-T]p (A)
其中:
B是键合部分;
L是连接基团;
G、G′和G″是各自选择的间隔部分;
R是亲脂性部分,且R′是聚亚烷基二醇部分,或者R′是亲脂性部分,且R是聚亚烷基二醇部分;
T是封端部分;
j、k、m和n各自是0或1;和
p是1至降钙素上亲核残基数的整数。
19、合成缀合物的基本单分散混合物的方法,每一缀合物包含与包含聚乙二醇部分的低聚物偶联的降钙素,所述方法包含:
使包含具有式I结构的化合物的基本单分散混合物
R1(OC2H4)m-O-X+ (I)
其中R1是H或亲脂性部分;m是1至25;X+是阳离子,
与包含具有式II结构的化合物的基本单分散混合物
R2(OC2H4)n-OMs (II)
其中R2是H或亲脂性部分;n是1至25,
在足以提供包含具有式III结构的聚合物的基本单分散混合物的条件下反应,
R2(OC2H4)m+n-OR1 (III);
活化包含式III聚合物的基本单分散混合物,得到能够与降钙素反应的活化聚合物的基本单分散混合物;
使该活化聚合物的基本单分散混合物与降钙素在足以提供各自包含与包含具有m+n个亚单位的聚乙二醇部分的低聚物偶联的降钙素的缀合物的基本单分散混合物的条件下反应。
20、根据权利要求19的方法,其中该基本单分散混合物的活化包含使式III聚合物的基本单分散混合物与N-羟基琥珀酰亚胺反应,得到能够与降钙素反应的活化聚合物。
21、根据权利要求19的方法,其中该降钙素是鲑鱼降钙素,其中该活化聚合物的基本单分散混合物与鲑鱼降钙素的反应包含:
使该活化聚合物的基本单分散混合物与该鲑鱼降钙素的Lys11和Lys18反应,得到二缀合物的基本单分散混合物,该二缀合物各自包含与两个各自包含具有m+n个亚单位的聚乙二醇部分的低聚物偶联的鲑鱼降钙素。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/873,777 | 2001-06-04 | ||
| US09/873,777 US6713452B2 (en) | 2001-06-04 | 2001-06-04 | Mixtures of calcitonin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1538851A CN1538851A (zh) | 2004-10-20 |
| CN100515492C true CN100515492C (zh) | 2009-07-22 |
Family
ID=25362290
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028153022A Expired - Fee Related CN100515492C (zh) | 2001-06-04 | 2002-06-04 | 包含聚亚烷基二醇的降钙素药物-低聚物缀合物的混合物、其应用和制备方法 |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US6713452B2 (zh) |
| EP (1) | EP1404360B1 (zh) |
| JP (1) | JP4272510B2 (zh) |
| KR (1) | KR100930606B1 (zh) |
| CN (1) | CN100515492C (zh) |
| AT (1) | ATE454160T1 (zh) |
| CA (1) | CA2449686A1 (zh) |
| CY (1) | CY1109932T1 (zh) |
| DE (1) | DE60235010D1 (zh) |
| DK (1) | DK1404360T3 (zh) |
| ES (1) | ES2339224T3 (zh) |
| MX (1) | MXPA03011283A (zh) |
| WO (1) | WO2002098451A1 (zh) |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6191105B1 (en) * | 1993-05-10 | 2001-02-20 | Protein Delivery, Inc. | Hydrophilic and lipophilic balanced microemulsion formulations of free-form and/or conjugation-stabilized therapeutic agents such as insulin |
| US6703381B1 (en) * | 1998-08-14 | 2004-03-09 | Nobex Corporation | Methods for delivery therapeutic compounds across the blood-brain barrier |
| US6638906B1 (en) * | 1999-12-13 | 2003-10-28 | Nobex Corporation | Amphiphilic polymers and polypeptide conjugates comprising same |
| US8394813B2 (en) | 2000-11-14 | 2013-03-12 | Shire Llc | Active agent delivery systems and methods for protecting and administering active agents |
| US6828305B2 (en) | 2001-06-04 | 2004-12-07 | Nobex Corporation | Mixtures of growth hormone drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US7713932B2 (en) * | 2001-06-04 | 2010-05-11 | Biocon Limited | Calcitonin drug-oligomer conjugates, and uses thereof |
| WO2005004792A2 (en) * | 2003-06-24 | 2005-01-20 | Nobex Corporation | Mixtures of calcitonin drug-oligomer conjugates and methods of use in pain treatment |
| US20090281023A9 (en) * | 2001-06-04 | 2009-11-12 | Nobex Corporation | Mixtures Of Calcitonin Drug-Oligomer Conjugates And Methods Of Use In Pain Treatment |
| US6858580B2 (en) * | 2001-06-04 | 2005-02-22 | Nobex Corporation | Mixtures of drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6713452B2 (en) * | 2001-06-04 | 2004-03-30 | Nobex Corporation | Mixtures of calcitonin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6828297B2 (en) * | 2001-06-04 | 2004-12-07 | Nobex Corporation | Mixtures of insulin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6835802B2 (en) | 2001-06-04 | 2004-12-28 | Nobex Corporation | Methods of synthesizing substantially monodispersed mixtures of polymers having polyethylene glycol moieties |
| US7169752B2 (en) * | 2003-09-30 | 2007-01-30 | New River Pharmaceuticals Inc. | Compounds and compositions for prevention of overdose of oxycodone |
| US20060014697A1 (en) * | 2001-08-22 | 2006-01-19 | Travis Mickle | Pharmaceutical compositions for prevention of overdose or abuse |
| US7030082B2 (en) * | 2001-09-07 | 2006-04-18 | Nobex Corporation | Pharmaceutical compositions of drug-oligomer conjugates and methods of treating disease therewith |
| US7196059B2 (en) * | 2001-09-07 | 2007-03-27 | Biocon Limited | Pharmaceutical compositions of insulin drug-oligomer conjugates and methods of treating diseases therewith |
| US6770625B2 (en) * | 2001-09-07 | 2004-08-03 | Nobex Corporation | Pharmaceutical compositions of calcitonin drug-oligomer conjugates and methods of treating diseases therewith |
| WO2004043396A2 (en) * | 2002-11-09 | 2004-05-27 | Nobex Corporation | Modified carbamate-containing prodrugs and methods of synthesizing same |
| EP1569683B1 (en) * | 2002-11-26 | 2010-03-03 | Biocon Limited | Modified natriuretic compounds, conjugates, and uses thereof |
| US7648962B2 (en) * | 2002-11-26 | 2010-01-19 | Biocon Limited | Natriuretic compounds, conjugates, and uses thereof |
| US8133881B2 (en) | 2003-01-13 | 2012-03-13 | Shire Llc | Carbohydrate conjugates to prevent abuse of controlled substances |
| EP1594513B1 (en) * | 2003-01-13 | 2011-08-10 | Shire LLC | Carbohydrate conjugates to prevent abuse of controlled substances |
| WO2004073620A2 (en) * | 2003-02-14 | 2004-09-02 | Quanta Biodesign, Ltd | The selective and specific preparation of discrete peg compounds |
| GB0305977D0 (en) * | 2003-03-15 | 2003-04-23 | Koninkl Philips Electronics Nv | Control of a conditional access mechanism |
| US20060182692A1 (en) | 2003-12-16 | 2006-08-17 | Fishburn C S | Chemically modified small molecules |
| EP1694363B1 (en) * | 2003-12-16 | 2014-01-22 | Nektar Therapeutics | Monodisperse PEGylated naloxol compositions |
| US8329958B2 (en) | 2004-07-02 | 2012-12-11 | Biocon Limited | Combinatorial synthesis of PEG oligomer libraries |
| RU2393168C2 (ru) * | 2004-07-19 | 2010-06-27 | Биокон Лимитед | Инсулин-олигомерные конъюгаты, их препараты и применения |
| US20080207505A1 (en) * | 2005-01-12 | 2008-08-28 | James Kenneth D | Bna Conjugates and Methods of Use |
| KR101234540B1 (ko) * | 2007-10-16 | 2013-02-19 | 바이오콘 리미티드 | 경구 투여가능한 고형 약제학적 조성물 및 그 제조방법 |
| BR112012012945A2 (pt) | 2009-11-25 | 2020-12-29 | Arisgen Sa | Composição de liberação mucosal, seu método de produção, complexo de peptídeo pré-formado, kit e uso de um agente ativo de peptídeo |
| MY173890A (en) | 2010-09-30 | 2020-02-26 | Astrazeneca Ab | Crystalline naloxol-peg conjugate |
| JP6329486B2 (ja) | 2011-10-21 | 2018-05-23 | シーチェイド ファーマシューティカルズ,インコーポレーテッド | 医薬組成物及びそれらの使用 |
| CN102875427B (zh) * | 2012-09-18 | 2014-02-19 | 安徽世华化工有限公司 | 一种2-甲氧基甲磺酸甲酯的合成方法 |
| CN113087623A (zh) * | 2021-04-13 | 2021-07-09 | 苏州昊帆生物股份有限公司 | 一种8-溴辛酸乙酯的合成方法 |
Family Cites Families (163)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3256153A (en) | 1963-02-08 | 1966-06-14 | Smith Kline French Lab | Method of stabilizing wax-fat coating materials and product thereof |
| US4003792A (en) | 1967-07-01 | 1977-01-18 | Miles Laboratories, Inc. | Conjugates of acid polysaccharides and complex organic substances |
| US3950517A (en) | 1970-05-08 | 1976-04-13 | National Research Development Corporation | Insulin derivatives |
| GB1381274A (en) | 1971-01-28 | 1975-01-22 | Nat Res Dev | Insulin derivatives |
| US3919411A (en) | 1972-01-31 | 1975-11-11 | Bayvet Corp | Injectable adjuvant and compositions including such adjuvant |
| US4044196A (en) | 1972-03-30 | 1977-08-23 | Bayer Aktiengesellschaft | Crosslinked copolymers of α,β-olefinically unsaturated dicarboxylic anhydrides |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| FR2408387A2 (fr) | 1975-06-30 | 1979-06-08 | Oreal | Compositions a base de dispersions aqueuses de spherules lipidiques |
| US4093574A (en) | 1977-02-02 | 1978-06-06 | Eli Lilly And Company | Somatostatin analogs and intermediates thereto |
| US4087390A (en) | 1977-02-02 | 1978-05-02 | Eli Lilly And Company | Somatostatin analogs and intermediates thereto |
| GB1492997A (en) | 1976-07-21 | 1977-11-23 | Nat Res Dev | Insulin derivatives |
| US4223163A (en) | 1976-12-10 | 1980-09-16 | The Procter & Gamble Company | Process for making ethoxylated fatty alcohols with narrow polyethoxy chain distribution |
| JPS53116315A (en) | 1977-03-17 | 1978-10-11 | Ueno Seiyaku Oyo Kenkyujo Kk | Powder or granular containing improved sorbinic acid |
| US4100117A (en) | 1977-04-21 | 1978-07-11 | Eli Lilly And Company | Somatostatin analogs and intermediates thereto |
| US4253998A (en) | 1979-03-09 | 1981-03-03 | American Home Products Corporation | Peptides related to somatostatin |
| JPS54148722A (en) | 1978-05-12 | 1979-11-21 | Takeda Chem Ind Ltd | Nonapeptide and its preparation |
| US4277394A (en) | 1979-04-23 | 1981-07-07 | Takeda Chemical Industries, Ltd | Tetrapeptidehydrazide derivatives |
| GB2051574B (en) | 1979-05-10 | 1984-01-18 | Kyoto Pharma Ind | Adjuvant for promoting absorption of pharmacologically active substances through the rectum |
| US4348387A (en) | 1979-07-31 | 1982-09-07 | The Rockefeller University | Method and system for the controlled release of biologically active substances to a body fluid |
| US4469681A (en) | 1979-07-31 | 1984-09-04 | The Rockefeller University | Method and system for the controlled release of biologically active substances to a body fluid |
| FR2465486A1 (fr) | 1979-09-21 | 1981-03-27 | Roussel Uclaf | Nouvelle application utilisant la lh-rh ou des agonistes |
| JPS5692846A (en) | 1979-12-27 | 1981-07-27 | Takeda Chem Ind Ltd | Tetrapeptide derivative and its preparation |
| US4554101A (en) | 1981-01-09 | 1985-11-19 | New York Blood Center, Inc. | Identification and preparation of epitopes on antigens and allergens on the basis of hydrophilicity |
| US4698264A (en) | 1982-08-02 | 1987-10-06 | Durkee Industrial Foods, Corp. | Particulate composition and process for making same |
| IL68769A (en) | 1983-05-23 | 1986-02-28 | Hadassah Med Org | Pharmaceutical compositions containing insulin for oral administration |
| US4662392A (en) | 1983-07-29 | 1987-05-05 | Intevep, S.A. | Check valve |
| US4585754A (en) | 1984-01-09 | 1986-04-29 | Valcor Scientific, Ltd. | Stabilization of proteins and peptides by chemical binding with chondroitin |
| US4684524A (en) | 1984-03-19 | 1987-08-04 | Alza Corporation | Rate controlled dispenser for administering beneficial agent |
| US4717566A (en) | 1984-03-19 | 1988-01-05 | Alza Corporation | Dosage system and method of using same |
| US4849405A (en) | 1984-05-09 | 1989-07-18 | Synthetic Blood Corporation | Oral insulin and a method of making the same |
| US4963367A (en) | 1984-04-27 | 1990-10-16 | Medaphore, Inc. | Drug delivery compositions and methods |
| US4963526A (en) | 1984-05-09 | 1990-10-16 | Synthetic Blood Corporation | Oral insulin and a method of making the same |
| US4839341A (en) | 1984-05-29 | 1989-06-13 | Eli Lilly And Company | Stabilized insulin formulations |
| US4797288A (en) | 1984-10-05 | 1989-01-10 | Warner-Lambert Company | Novel drug delivery system |
| US4946828A (en) | 1985-03-12 | 1990-08-07 | Novo Nordisk A/S | Novel insulin peptides |
| US5157021A (en) | 1985-03-15 | 1992-10-20 | Novo Nordisk A/S | Insulin derivatives and pharmaceutical preparations containing these derivatives |
| US4917888A (en) | 1985-06-26 | 1990-04-17 | Cetus Corporation | Solubilization of immunotoxins for pharmaceutical compositions using polymer conjugation |
| SE457326B (sv) | 1986-02-14 | 1988-12-19 | Lejus Medical Ab | Foerfarande foer framstaellning av en snabbt soenderfallande kaerna innehaallande bl a mikrokristallin cellulosa |
| US4801575A (en) | 1986-07-30 | 1989-01-31 | The Regents Of The University Of California | Chimeric peptides for neuropeptide delivery through the blood-brain barrier |
| ZA877505B (en) | 1986-10-14 | 1989-05-30 | Lilly Co Eli | Process for transforming a human insulin precursor to human insulin |
| GB8706313D0 (en) | 1987-03-17 | 1987-04-23 | Health Lab Service Board | Treatment & prevention of viral infections |
| US5093198A (en) | 1987-06-19 | 1992-03-03 | Temple University | Adjuvant-enhanced sustained release composition and method for making |
| DE3721721C1 (de) | 1987-07-01 | 1988-06-09 | Hoechst Ag | Verfahren zur Umhuellung von Granulaten |
| US5080891A (en) | 1987-08-03 | 1992-01-14 | Ddi Pharmaceuticals, Inc. | Conjugates of superoxide dismutase coupled to high molecular weight polyalkylene glycols |
| US4774976A (en) | 1987-09-23 | 1988-10-04 | Applied Power Inc. | Modulating hydraulic pressure control valve and assembly method therefor |
| JPH01308231A (ja) | 1988-06-03 | 1989-12-12 | Takeda Chem Ind Ltd | 安定化された医薬組成物および製造法 |
| US5055300A (en) | 1988-06-17 | 1991-10-08 | Basic Bio Systems, Inc. | Time release protein |
| DK336188D0 (da) | 1988-06-20 | 1988-06-20 | Nordisk Gentofte | Propeptider |
| US5349052A (en) | 1988-10-20 | 1994-09-20 | Royal Free Hospital School Of Medicine | Process for fractionating polyethylene glycol (PEG)-protein adducts and an adduct for PEG and granulocyte-macrophage colony stimulating factor |
| US5306500A (en) | 1988-11-21 | 1994-04-26 | Collagen Corporation | Method of augmenting tissue with collagen-polymer conjugates |
| US5162430A (en) | 1988-11-21 | 1992-11-10 | Collagen Corporation | Collagen-polymer conjugates |
| AU641631B2 (en) | 1988-12-23 | 1993-09-30 | Novo Nordisk A/S | Human insulin analogues |
| US4994439A (en) | 1989-01-19 | 1991-02-19 | California Biotechnology Inc. | Transmembrane formulations for drug administration |
| US5089261A (en) | 1989-01-23 | 1992-02-18 | Cetus Corporation | Preparation of a polymer/interleukin-2 conjugate |
| US5182258A (en) | 1989-03-20 | 1993-01-26 | Orbon Corporation | Systemic delivery of polypeptides through the eye |
| US5122614A (en) | 1989-04-19 | 1992-06-16 | Enzon, Inc. | Active carbonates of polyalkylene oxides for modification of polypeptides |
| US5324844A (en) | 1989-04-19 | 1994-06-28 | Enzon, Inc. | Active carbonates of polyalkylene oxides for modification of polypeptides |
| US5286637A (en) | 1989-08-07 | 1994-02-15 | Debiopharm, S.A. | Biologically active drug polymer derivatives and method for preparing same |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| DE3937797A1 (de) | 1989-11-14 | 1991-05-16 | Basf Ag | Verfahren zur herstellung von polyetherglykolen |
| US5650388A (en) | 1989-11-22 | 1997-07-22 | Enzon, Inc. | Fractionated polyalkylene oxide-conjugated hemoglobin solutions |
| US5312808A (en) | 1989-11-22 | 1994-05-17 | Enzon, Inc. | Fractionation of polyalkylene oxide-conjugated hemoglobin solutions |
| CA2030174C (en) | 1990-01-10 | 1996-12-24 | Anthony H. Cincotta | Process for the long term reduction of body fat stores, insulin resistance, hyperinsulinemia and hypoglycemia in vertebrates |
| US5545618A (en) | 1990-01-24 | 1996-08-13 | Buckley; Douglas I. | GLP-1 analogs useful for diabetes treatment |
| US5126324A (en) | 1990-06-07 | 1992-06-30 | Genentech, Inc. | Method of enhancing growth in patients using combination therapy |
| IE912365A1 (en) | 1990-07-23 | 1992-01-29 | Zeneca Ltd | Continuous release pharmaceutical compositions |
| DD297249A5 (de) | 1990-08-07 | 1992-01-02 | Veb Mineralwollewerk Flechtingen Bereich F/E Mineralwolle,De | Verfahren zur automatischen ueberwachung des aushaertegrades an materialbahnen |
| IL99699A (en) | 1990-10-10 | 2002-04-21 | Autoimmune Inc | Drug with the option of oral, intra-intestinal, or inhaled dosing for suppression of autoimmune response associated with type I diabetes |
| US5595732A (en) | 1991-03-25 | 1997-01-21 | Hoffmann-La Roche Inc. | Polyethylene-protein conjugates |
| WO1992018147A1 (en) | 1991-04-19 | 1992-10-29 | Affinity Biotech, Inc. | Convertible microemulsion formulations |
| FR2675807B1 (fr) | 1991-04-23 | 1994-07-01 | Medgenix Group Sa | Conjugue de calcitonine et de polyethylene glycol. |
| US5304473A (en) | 1991-06-11 | 1994-04-19 | Eli Lilly And Company | A-C-B proinsulin, method of manufacturing and using same, and intermediates in insulin production |
| ZA925581B (en) | 1991-07-26 | 1993-05-14 | Smithkline Beecham Corp | Pharmaceutical microemulsions. |
| US5206219A (en) | 1991-11-25 | 1993-04-27 | Applied Analytical Industries, Inc. | Oral compositions of proteinaceous medicaments |
| US5693769A (en) | 1991-12-13 | 1997-12-02 | Transcell Technologies, Inc. | Glycosylated steroid derivatives for transport across biological membranes and process for making and using same |
| DE59309678D1 (de) | 1992-01-17 | 1999-08-05 | Alfatec Pharma Gmbh | Verfahren zur Herstellung von Wirkstoff enthaltenden Pulvern, Granulaten oder Pellets mit einem Gerüst aus hydrophilen Makromolekülen und ihre Verwendung |
| GB9212511D0 (en) | 1992-06-12 | 1992-07-22 | Cortecs Ltd | Pharmaceutical compositions |
| US5262172A (en) | 1992-06-19 | 1993-11-16 | Digestive Care Inc. | Compositions of gastric acid-resistant microspheres containing buffered bile acids |
| US5415872A (en) | 1992-06-22 | 1995-05-16 | Digestive Care Inc. | Compositions of gastric acid-resistant microspheres containing salts of bile acids |
| US6093391A (en) | 1992-10-08 | 2000-07-25 | Supratek Pharma, Inc. | Peptide copolymer compositions |
| GB9316895D0 (en) | 1993-08-13 | 1993-09-29 | Guy S And St Thomas Hospitals | Hepatoselective insulin analogues |
| US5298643A (en) | 1992-12-22 | 1994-03-29 | Enzon, Inc. | Aryl imidate activated polyalkylene oxides |
| US5349001A (en) | 1993-01-19 | 1994-09-20 | Enzon, Inc. | Cyclic imide thione activated polyalkylene oxides |
| US5321095A (en) | 1993-02-02 | 1994-06-14 | Enzon, Inc. | Azlactone activated polyalkylene oxides |
| US5298410A (en) | 1993-02-25 | 1994-03-29 | Sterling Winthrop Inc. | Lyophilized formulation of polyethylene oxide modified proteins with increased shelf-life |
| US5359030A (en) | 1993-05-10 | 1994-10-25 | Protein Delivery, Inc. | Conjugation-stabilized polypeptide compositions, therapeutic delivery and diagnostic formulations comprising same, and method of making and using the same |
| US6191105B1 (en) | 1993-05-10 | 2001-02-20 | Protein Delivery, Inc. | Hydrophilic and lipophilic balanced microemulsion formulations of free-form and/or conjugation-stabilized therapeutic agents such as insulin |
| US5681811A (en) | 1993-05-10 | 1997-10-28 | Protein Delivery, Inc. | Conjugation-stabilized therapeutic agent compositions, delivery and diagnostic formulations comprising same, and method of making and using the same |
| US5621039A (en) | 1993-06-08 | 1997-04-15 | Hallahan; Terrence W. | Factor IX- polymeric conjugates |
| WO1995000162A1 (en) | 1993-06-21 | 1995-01-05 | Enzon, Inc. | Site specific synthesis of conjugated peptides |
| US5747445A (en) | 1993-06-24 | 1998-05-05 | Astra Aktiebolag | Therapeutic preparation for inhalation |
| TW402506B (en) | 1993-06-24 | 2000-08-21 | Astra Ab | Therapeutic preparation for inhalation |
| US5830853A (en) | 1994-06-23 | 1998-11-03 | Astra Aktiebolag | Systemic administration of a therapeutic preparation |
| IS1796B (is) | 1993-06-24 | 2001-12-31 | Ab Astra | Fjölpeptíð lyfjablanda til innöndunar sem einnig inniheldur eykjaefnasamband |
| US5506203C1 (en) | 1993-06-24 | 2001-02-06 | Astra Ab | Systemic administration of a therapeutic preparation |
| US6342225B1 (en) | 1993-08-13 | 2002-01-29 | Deutshces Wollforschungsinstitut | Pharmaceutical active conjugates |
| CA2171424C (en) | 1993-09-17 | 2002-06-04 | Svend Havelund | Acylated insulin |
| US5919455A (en) | 1993-10-27 | 1999-07-06 | Enzon, Inc. | Non-antigenic branched polymer conjugates |
| US5605976A (en) | 1995-05-15 | 1997-02-25 | Enzon, Inc. | Method of preparing polyalkylene oxide carboxylic acids |
| US5643575A (en) | 1993-10-27 | 1997-07-01 | Enzon, Inc. | Non-antigenic branched polymer conjugates |
| US5951974A (en) | 1993-11-10 | 1999-09-14 | Enzon, Inc. | Interferon polymer conjugates |
| EP0736041B1 (en) | 1993-11-17 | 2006-02-08 | Athena Neurosciences, Inc. | Transparent liquid for encapsulated drug delivery |
| GB9406094D0 (en) | 1994-03-28 | 1994-05-18 | Univ Nottingham And University | Polymer microspheres and a method of production thereof |
| US5504188A (en) | 1994-06-16 | 1996-04-02 | Eli Lilly And Company | Preparation of stable zinc insulin analog crystals |
| US5461031A (en) | 1994-06-16 | 1995-10-24 | Eli Lilly And Company | Monomeric insulin analog formulations |
| US6165976A (en) | 1994-06-23 | 2000-12-26 | Astra Aktiebolag | Therapeutic preparation for inhalation |
| US5730990A (en) | 1994-06-24 | 1998-03-24 | Enzon, Inc. | Non-antigenic amine derived polymers and polymer conjugates |
| GB9417524D0 (en) | 1994-08-31 | 1994-10-19 | Cortecs Ltd | Pharmaceutical compositions |
| US5738846A (en) | 1994-11-10 | 1998-04-14 | Enzon, Inc. | Interferon polymer conjugates and process for preparing the same |
| US5646242A (en) | 1994-11-17 | 1997-07-08 | Eli Lilly And Company | Selective acylation of epsilon-amino groups |
| US5693609A (en) | 1994-11-17 | 1997-12-02 | Eli Lilly And Company | Acylated insulin analogs |
| CA2206852A1 (en) | 1994-12-07 | 1996-06-13 | Novo Nordisk A/S | Polypeptide with reduced allergenicity |
| GB9424902D0 (en) | 1994-12-09 | 1995-02-08 | Cortecs Ltd | Solubilisation Aids |
| SE9404468D0 (sv) | 1994-12-22 | 1994-12-22 | Astra Ab | Powder formulations |
| US5843866A (en) | 1994-12-30 | 1998-12-01 | Hampshire Chemical Corp. | Pesticidal compositions comprising solutions of polyurea and/or polyurethane |
| US5932462A (en) | 1995-01-10 | 1999-08-03 | Shearwater Polymers, Inc. | Multiarmed, monofunctional, polymer for coupling to molecules and surfaces |
| US5907030A (en) | 1995-01-25 | 1999-05-25 | University Of Southern California | Method and compositions for lipidization of hydrophilic molecules |
| KR0150565B1 (ko) | 1995-02-15 | 1998-08-17 | 김정재 | 유전자 조환에 의한 사람 인슐린 전구체의 제조 및 이를 이용한 인슐린의 제조방법 |
| US6251856B1 (en) | 1995-03-17 | 2001-06-26 | Novo Nordisk A/S | Insulin derivatives |
| YU18596A (sh) | 1995-03-31 | 1998-07-10 | Eli Lilly And Company | Analogne formulacije monomernog insulina |
| US5606038A (en) | 1995-04-10 | 1997-02-25 | Competitive Technologies, Inc. | Amphiphilic polyene macrolide antibiotic compounds |
| DE741187T1 (de) | 1995-05-05 | 1997-04-30 | Hoffmann La Roche | Rekombinante Obesitäts-(OB)-Proteine |
| US5824638A (en) | 1995-05-22 | 1998-10-20 | Shire Laboratories, Inc. | Oral insulin delivery |
| US5700904A (en) | 1995-06-07 | 1997-12-23 | Eli Lilly And Company | Preparation of an acylated protein powder |
| US5631347A (en) | 1995-06-07 | 1997-05-20 | Eli Lilly And Company | Reducing gelation of a fatty acid-acylated protein |
| GB9516268D0 (en) | 1995-08-08 | 1995-10-11 | Danbiosyst Uk | Compositiion for enhanced uptake of polar drugs from the colon |
| DE122006000003I2 (de) | 1995-09-21 | 2011-01-13 | Genentech Inc | Varianten des menschlichen wachstumshormons |
| WO1997014740A1 (en) * | 1995-10-19 | 1997-04-24 | Receptagen Corporation | Discrete-length polyethylene glycols |
| US5766620A (en) | 1995-10-23 | 1998-06-16 | Theratech, Inc. | Buccal delivery of glucagon-like insulinotropic peptides |
| US5639705A (en) | 1996-01-19 | 1997-06-17 | Arco Chemical Technology, L.P. | Double metal cyanide catalysts and methods for making them |
| US5948751A (en) | 1996-06-20 | 1999-09-07 | Novo Nordisk A/S | X14-mannitol |
| US5866538A (en) | 1996-06-20 | 1999-02-02 | Novo Nordisk A/S | Insulin preparations containing NaCl |
| GB9613858D0 (en) | 1996-07-02 | 1996-09-04 | Cortecs Ltd | Hydrophobic preparations |
| US5905140A (en) | 1996-07-11 | 1999-05-18 | Novo Nordisk A/S, Novo Alle | Selective acylation method |
| US5856369A (en) | 1996-07-30 | 1999-01-05 | Osi Specialties, Inc. | Polyethers and polysiloxane copolymers manufactured with double metal cyanide catalysts |
| DE19632440A1 (de) | 1996-08-12 | 1998-02-19 | Basf Ag | Verfahren zur Herstellung von aus Polykationen aufgebauten, geformten Mischhydroxiden |
| US5874111A (en) | 1997-01-07 | 1999-02-23 | Maitra; Amarnath | Process for the preparation of highly monodispersed polymeric hydrophilic nanoparticles |
| US6011008A (en) | 1997-01-08 | 2000-01-04 | Yissum Research Developement Company Of The Hebrew University Of Jerusalem | Conjugates of biologically active substances |
| US5830918A (en) | 1997-01-15 | 1998-11-03 | Terrapin Technologies, Inc. | Nonpeptide insulin receptor agonists |
| US6310038B1 (en) | 1997-03-20 | 2001-10-30 | Novo Nordisk A/S | Pulmonary insulin crystals |
| US5898028A (en) | 1997-03-20 | 1999-04-27 | Novo Nordisk A/S | Method for producing powder formulation comprising an insulin |
| US6043214A (en) | 1997-03-20 | 2000-03-28 | Novo Nordisk A/S | Method for producing powder formulation comprising an insulin |
| CO4750643A1 (es) | 1997-06-13 | 1999-03-31 | Lilly Co Eli | Formulacion estable de la insulina que contiene l-arginina y protamina |
| IL134901A0 (en) | 1997-10-24 | 2001-05-20 | Lilly Co Eli | Insoluble insulin compositions |
| ZA989744B (en) | 1997-10-31 | 2000-04-26 | Lilly Co Eli | Method for administering acylated insulin. |
| US5985263A (en) | 1997-12-19 | 1999-11-16 | Enzon, Inc. | Substantially pure histidine-linked protein polymer conjugates |
| US5981709A (en) | 1997-12-19 | 1999-11-09 | Enzon, Inc. | α-interferon-polymer-conjugates having enhanced biological activity and methods of preparing the same |
| US6703381B1 (en) | 1998-08-14 | 2004-03-09 | Nobex Corporation | Methods for delivery therapeutic compounds across the blood-brain barrier |
| US6211144B1 (en) | 1998-10-16 | 2001-04-03 | Novo Nordisk A/S | Stable concentrated insulin preparations for pulmonary delivery |
| DE19908041A1 (de) | 1999-02-24 | 2000-08-31 | Hoecker Hartwig | Kovalent verbrückte Insulindimere |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6309633B1 (en) * | 1999-06-19 | 2001-10-30 | Nobex Corporation | Amphiphilic drug-oligomer conjugates with hydroyzable lipophile components and methods for making and using the same |
| KR100345214B1 (ko) * | 1999-08-17 | 2002-07-25 | 이강춘 | 생체적합성 고분자가 수식된 펩타이드의 비점막 전달 |
| US6323311B1 (en) | 1999-09-22 | 2001-11-27 | University Of Utah Research Foundation | Synthesis of insulin derivatives |
| US6867183B2 (en) | 2001-02-15 | 2005-03-15 | Nobex Corporation | Pharmaceutical compositions of insulin drug-oligomer conjugates and methods of treating diseases therewith |
| US7060675B2 (en) | 2001-02-15 | 2006-06-13 | Nobex Corporation | Methods of treating diabetes mellitus |
| US6835802B2 (en) | 2001-06-04 | 2004-12-28 | Nobex Corporation | Methods of synthesizing substantially monodispersed mixtures of polymers having polyethylene glycol moieties |
| US6858580B2 (en) * | 2001-06-04 | 2005-02-22 | Nobex Corporation | Mixtures of drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6828297B2 (en) | 2001-06-04 | 2004-12-07 | Nobex Corporation | Mixtures of insulin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6828305B2 (en) | 2001-06-04 | 2004-12-07 | Nobex Corporation | Mixtures of growth hormone drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6713452B2 (en) * | 2001-06-04 | 2004-03-30 | Nobex Corporation | Mixtures of calcitonin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same |
| US6770625B2 (en) | 2001-09-07 | 2004-08-03 | Nobex Corporation | Pharmaceutical compositions of calcitonin drug-oligomer conjugates and methods of treating diseases therewith |
| US6913903B2 (en) | 2001-09-07 | 2005-07-05 | Nobex Corporation | Methods of synthesizing insulin polypeptide-oligomer conjugates, and proinsulin polypeptide-oligomer conjugates and methods of synthesizing same |
-
2001
- 2001-06-04 US US09/873,777 patent/US6713452B2/en not_active Expired - Fee Related
-
2002
- 2002-06-04 CA CA002449686A patent/CA2449686A1/en not_active Abandoned
- 2002-06-04 CN CNB028153022A patent/CN100515492C/zh not_active Expired - Fee Related
- 2002-06-04 EP EP02732030A patent/EP1404360B1/en not_active Expired - Lifetime
- 2002-06-04 WO PCT/US2002/017575 patent/WO2002098451A1/en active Application Filing
- 2002-06-04 DE DE60235010T patent/DE60235010D1/de not_active Expired - Lifetime
- 2002-06-04 MX MXPA03011283A patent/MXPA03011283A/es active IP Right Grant
- 2002-06-04 KR KR1020037015912A patent/KR100930606B1/ko not_active IP Right Cessation
- 2002-06-04 DK DK02732030.8T patent/DK1404360T3/da active
- 2002-06-04 JP JP2003501489A patent/JP4272510B2/ja not_active Expired - Fee Related
- 2002-06-04 AT AT02732030T patent/ATE454160T1/de active
- 2002-06-04 ES ES02732030T patent/ES2339224T3/es not_active Expired - Lifetime
-
2004
- 2004-03-23 US US10/806,523 patent/US7084121B2/en not_active Expired - Fee Related
-
2010
- 2010-03-31 CY CY20101100300T patent/CY1109932T1/el unknown
Non-Patent Citations (2)
| Title |
|---|
| oral delivery of calcitionin by conjugation with amphiphilicoligomers. radha krishnana B. 等.proceedings of the controllled release society,No.26. 1999 |
| oral delivery of calcitionin by conjugation with amphiphilicoligomers. radha krishnana B. 等.proceedings of the controllled release society,No.26. 1999 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1538851A (zh) | 2004-10-20 |
| EP1404360A4 (en) | 2006-04-12 |
| CY1109932T1 (el) | 2014-09-10 |
| DK1404360T3 (da) | 2010-05-03 |
| CA2449686A1 (en) | 2002-12-12 |
| ATE454160T1 (de) | 2010-01-15 |
| DE60235010D1 (de) | 2010-02-25 |
| US7084121B2 (en) | 2006-08-01 |
| WO2002098451A1 (en) | 2002-12-12 |
| JP2004534782A (ja) | 2004-11-18 |
| KR20040004694A (ko) | 2004-01-13 |
| ES2339224T3 (es) | 2010-05-18 |
| EP1404360B1 (en) | 2010-01-06 |
| US20040180831A1 (en) | 2004-09-16 |
| US20030060606A1 (en) | 2003-03-27 |
| KR100930606B1 (ko) | 2009-12-09 |
| MXPA03011283A (es) | 2004-03-26 |
| JP4272510B2 (ja) | 2009-06-03 |
| EP1404360A1 (en) | 2004-04-07 |
| US6713452B2 (en) | 2004-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100515492C (zh) | 包含聚亚烷基二醇的降钙素药物-低聚物缀合物的混合物、其应用和制备方法 | |
| US7268210B2 (en) | PEG-LHRH analog conjugates | |
| US6828305B2 (en) | Mixtures of growth hormone drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same | |
| CA2449426C (en) | Mixtures of insulin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same | |
| DK2279007T3 (en) | Pegylated recombinant relations of human growth hormone | |
| JP2003104913A (ja) | ポリアルキレングリコールを含む薬物−オリゴマー結合体の混合物、その使用及びその製造方法 | |
| US8030269B2 (en) | Calcitonin drug-oligomer conjugates, and uses thereof | |
| US6914121B2 (en) | PEG-LHRH analog conjugates | |
| JP4829783B2 (ja) | カルシトニン薬−オリゴマーコンジュゲートの混合物および疼痛治療における使用方法 | |
| EP1588717B1 (en) | PEG-LHRH analog conjugates | |
| AU2002303961A1 (en) | Mixtures of calcitonin drug-oligomer conjugates comprising polyalkylene glycol, uses thereof, and methods of making same | |
| AU2002310278A1 (en) | Mixtures of growth hormone drug-oligomer conjugates compromising polyalkylene glycol, uses thereof, and methods of making same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: BIOCON OY Free format text: FORMER OWNER: NOBEX CORP. Effective date: 20060908 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20060908 Address after: bangalore Applicant after: Biocon Ltd. Address before: North Carolina Applicant before: Nobecos Inc. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20090722 Termination date: 20150604 |
|
| EXPY | Termination of patent right or utility model |